Note: Descriptions are shown in the official language in which they were submitted.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-1-
METHOD FOR PREPARING ALPHA-SULFONYL
HYDROXAMIC ACID DERIVATIVES
The invention relates to a novel method of producing alpha-sulfonyl
hydroxamic acid derivatives that can be important as matrix metalloproteinase
(MMP) and TNF-alpha converting enzyme (TACE) inhibitors, phosphodiesterase
inhibitors, renin inhibitors, antithrombotics, and 5-lipoxygenase inhibitors.
This
invention also relates to new alpha-sulfonyl hydroxamic acid derivatives and
their
preparation by the new process and pharmaceutical compositions containing them
BACKGROUND OF THE INVENTION
Matrix metalloproteinases are a family of structurally related zinc-containing
enzymes that mediate the breakdown of the extracellular matrix proteins.
Members
of this family, which include collagenases, stromelysins, and gelatinases are
involved
in the normal tissue remodeling process such as wound-healing, angiogenesis,
and
pregnancy. In these pathological processes, the MMP activity is tightly
regulated by
the endogenous tissue inhibitors of matrix metalloproteinases (TIMPS). In
pathological conditions, this fine balance between MMP-TIMP can be disrupted
leading to several disease states including rheumatoid and osteoarthritis,
atherosclerosis, tumor growth, metastasis, and fibrosis. Therapeutic
inhibition of
MMPs is a promising approach for treatment of these diseases and therefore the
MMPs are attractive targets for rational drug design.
TALE is also a new member of metalloproteinase family, which catalyses the
formation of tumor necrosis factor-alpha precursor protein. TNF-alpha was
selected
as one of the early targets leading to the succesful cloning and sequencing of
human
TNF-alpha in 1984 by Goeddel and collegues. TNF-alpha is a very powerful
proinflammatory mediator produced by activated macrophages, blood monocytes,
and mast cells. In addition to its anti-tumor properties, TNF-alpha is a
proinflammatory cytokine that has a central role in rheumatoid arthritis, and
Crohn's
disease. Animal models and association studies in humans have indicated a
potential
role for TNF in insulin resistance, multiple sclerosis, organ failure,
pulmonary
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-2-
fibrosis, and HIV infection. Therefore, the inhibition of TNF-alpha has been
the
focus of drug discovery.
The lipoxygenases are a family of enzymes, which c atalyze the oxygenation
of arachidonic acid leading to the production of leukotrienes. Leukotrienes
have been
implicated as important mediators in asthma, rheumatoid arthritis, gout,
psoriasis,
allergic rhinitis, adult respiratory distress syndrome, Crohn's disease,
endotoxin
shock, and inflammatory bowel disease. It is believed that inhibition of these
enzymes will provide effective systematic treatment of these diseases. Renin
inhibitors can be used to control or prevent high blood pressure and cardiac
insufficiency.
Alpha-sulfonyl hydroxamic acids of the general formula I have been disclosed
as potent MMP and TALE inhibitors (Venkatesan, A.M.; Grosu, G.T.; Davis, J.M.;
Baker, J.L.; Levin, J.I. PCT Int. Appl. WO 9942436; Barta, T.E.; Becker, D.P.;
Boehm, T.L.; De Crescenzo, G.A.; Villamil, C.L; McDonald, J.J.; Freskos, J.N.;
Getman, D.P. PCT Int. Appl. 9925687; Almstead, N.G.; Bookland, R.G.; Taiwo,
Y.O.; Bradley, R.S.; Bush, R.D.; De B.; Natchus, M.G.; Pikul, S. PCT Int.
Appl.
9906340; Venkatesan, A.M.; Grosu, G.T.; Davis, J.M.; Baker, J.L.; Hu, B.;
O'Dell,
M.J.; Cole, D.C.; Jacobson, M.P., PCT Int. Appl. WO 9838163; Venkatesan, A.M.;
Grosu, G.T.; Davis, J.M.; Baker, J.L. PCT Int. Appl. WO 9837877; Levin, J.L;
Venkatesan, A.M.; Zask, A.; Sandanayaka, V.P.; PCT Int. Appl. WO 0001864;
Zook,
S.E.; Dagnino, R.; Deason, M.E.; Bender, S.L.; Melnick, M. PCT Int. Appl. WO
9720824), renin inhibitors (Branca, Q.; Heitz, M.P.; Neidhart, W.; Stadler,
H.; Vieira,
E.; Wostl, W. EP 509354), 5-lipoxygenase inhibitors (Brooks, D.W.; Summers,
J.B.;
Rodriques, K.E.; Maki, R.G.; Dellaria, J.F.; Holms, J.H.; Moore, J.L. US
5250565),
and antithrombotics (Nakane, M.; Reid, J. US 4734425).
General preparation of alpha-sulfonylhydroxamates in the above literature
involves first, the alkylation of appropriately substituted mercaptan
derivative with
either substituted or unsubstituted alpha-bromoacetic acid ester to give alpha-
thio
ester followed by oxidation of sulfur to sulfone to provide alpha-sulfonyl
ester. This
alpha-sulfonyl ester is converted to the corresponding hydroxamic acid
derivative via
the carboxylic acid. Alternatively, the enolate of the carbonyl compound is
treated
with the appropriately substituted disulfide to obtain the alpha-thio ester,
which is
then oxidized to the corresponding sulfone. The alpha-sulfonyl ester is
converted to
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-3-
the hydroxamic acid derivative as mentioned above. In either case, the
preparation of
the thiol or the disulfide requires multiple steps that involve sulfonyl
chloride,
protected thiols, or disulfides as intermediates and the oxidation step to
convert
alpha-thio ester to alpha-sulfonyl ester.
It is the object of this invention to provide a novel method for preparing
alpha-sulfonyl hydroxamic acid derivatives, which provides the target
molecules in a
highly convergent and efficient manner.
SUMMARY OF THE INVENTION
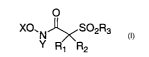
In accordance with the present invention is provided a method of preparing
alpha-sulfonyl hydroxamic acid derivatives of the formula I:
O
X0. N ~S02R3
Y R1 R2
I
wherein
X is hydrogen, alkyl of 1-6 carbon atoms, benzyl, hydroxyethyl, t-
butyldimethylsilyl,
trimethylsilyl or tetrahydropyranyl;
Y is hydrogen, alkyl of 1-6 carbon atoms, aryl of 6 to 10 carbon atoms, 5-10
membered heteroaryl having 1-3 heteroatoms selected from N, NR4, O and S,
cycloalkyl of 3-6 carbon atoms, 5-10 membered cycloheteroalkyl; wherein said
alkyl,
aryl, heteroaryl, cycloalkyl and cycloheteroalkyl group of Y is optionally
substituted
on any atom capable of substitution, with 1 to 3 substituents selected from
the group
consisting of halogen, alkyl of 1-6 carbon atoms; alkenyl of 2-6 carbon atoms
having
from 1 to 3 double bonds; alkynyl of 2-6 carbon atoms having from 1 to 3
triple
bonds, cycloalkyl of 3-6 carbon atoms, -ORS, =O, -CN, -CORS perfluoroalkyl of
1-4
carbon atoms, -O-perfluoroalkyl of 1-4 carbon atoms, -CONRSR6,-S(O)nRs,
-OPO(ORS)OR6, -PO(ORS)R6, -OC(O)ORS, -ORSNRSR6, -OC(O)NRSR6, -C(O)NRSOR6,
-COORS, -S03H, -NRSR6, -N[(CHz)~]~NRS, -NR5COR6, -NRSCOOR6, SO~NRSR6, -NOz,
-N(RS)SOZR6, -NRSCONRSR6, -NRSC(=NR6)NRSR6, -NRSC(=NR6)N(SO,RS)R6.
-NRSC(=NR6)N(C=ORS)R6, -tetrazol-5-yl, -SOZNHCN, -S02NHCONRSR6, phenyl,
heteroaryl and 5-10 membered cycloheteroalkyl;
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-4-
R, and R2 are each, independently, hydrogen; aryl of 6 to 10 carbon atoms; 5-
10
membered heteroaryl having 1-3 heteroatoms selected from N, NR~, O and S;
cycloalkyl of 3-6 carbon atoms; 5-10 membered cycloheteroalkyl; alkyl of 1-18
carbon atoms; alkenyl of 2-18 carbon atoms having 1 to 3 double bonds; alkynyl
of 2-
18 carbon atoms having from 1 to 3 triple bonds; or R, and R, taken together
with the
carbon atom to which they are attached form a cycloalkyl ring of 3-8 carbon
atoms or
a 5-10 membered cycloheteroalkyl ring; and wherein the aryl, heteroaryl,
cycloalkyl,
cycloheteroalkyl, alkyl, alkenyl, and alkynyl, may be optionally substituted
on any
atom capable of substitution with from 1 to 3 substituents selected from
halogen,
alkyl of 1-6 carbon atoms; alkenyl of 2-6 carbon atoms having from 1 to 3
double
bonds; alkynyl of 2-6 carbon atoms having from 1 to 3 triple bonds, cycloalkyl
of 3-6
carbon atoms, -ORS, =O, -CN, -CORS perfluoroalkyl of 1-4 carbon atoms, -O-
perfluoroalkyl of 1-4 carbon atoms, -CONRSR6, -S(O)nRS, -OPO(ORS)OR6,
-PO(ORS)R6, -OC(O)ORS, -ORSNRSR6, -OC(O)NRSR6, -C(O)NRSOR6, -COORS,
-S03H, -NRSR6, -N[(CHZ)2]zNRs, -NRSCOR6, -NRSCOOR6, SOzNR5R6, -NO2,
-N(RS)SOzRb, -NRSCONRSR6, -NRSC(=NR6)NRSR6, -NRSC(=NR6)N(SOZRS)R6,
-NRSC(=NR6)N(C=ORS)R6, -tetrazol-5-yl, -SOZNHCN, -S02NHCONRSR6, phenyl,
heteroaryl and 5-10 membered cycloheteroalkyl;
R3 is alkyl of 1-18 carbon atoms, alkenyl of 2-18 carbon atoms having 1 to 3
double
bonds, alkynyl of 2-18 carbon atoms having from 1 to3 triple bonds, cycloalkyl
of 3-6
carbon atoms, 5-10 membered cycloheteroalkyl, aryl of 6 to 10 carbon atoms, 5-
6
membered heteroaryl having 1-3 heteroatoms selected from N, NR4, O, and S;
wherein said alkyl, alkenyl, alkynyl, cycloalkyl, cycloheteroalkyl, aryl and
heteroaryl
of R3 may optionally be substituted on any atom capable of substitution with
from 1
to 3 substituents selected from halogen, alkyl of 1-6 carbon atoms; alkenyl of
2-6
carbon atoms having from 1 to 3 double bonds; alkynyl of 2-6 carbon atoms
having
from 1 to 3 triple bonds, cycloalkyl of 3-6 carbon atoms, -ORS, =O, -CN, -CORS
perfluoroalkyl of 1-4 carbon atoms, -O-perfluoroalkyl of 1-4 carbon atoms,
-CONRSR6,-S(O)nRS, -OPO(ORS)OR6, -PO(ORS)R6, -OC(O)ORS, -ORSNRSR6,
-OC(O)NRSR6, -C(O)NRSOR6, -COORS, -S03H, -NRSR6, -N[(CHZ)z]ZNRS, -NRSCOR6,
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-5-
-NRSCOOR6, SOZNRSR6, -NOz, -N(RS)SOZR6, -NRSCONRSR6, -NRSC(=NR6)NRSR6,
-NRSC(=NR6)N(SOZRS)R6,-NRSC(=NR6)N(C=ORS)R6, -tetrazol-5-yl, -SO=NHCN,
-S02NHCONRSR6, phenyl, heteroaryl and 5-10 membered cycloheteroalkyl;
R4 is hydrogen; aryl; aralkyl, heteroaryl; heteroaralkyl, alkyl of 1-6 carbon
atoms;
cycloalkyl of 3-6 carbon atoms; -C(O)~RS, -CONRSR6 or SO2R5;
RS and R6 are each independently hydrogen, optionally substituted aryl; 4-8
membered heteroaryl having 1-3 heteroatoms selected from N, NR4, O and S;
cycloalkyl of 3-6 carbon atoms; 5-10 membered cycloheteroalkyl; alkyl of 1-18
carbon atoms; alkenyl of 2-18 carbon atoms or alkynyl of 2-18 carbon atoms; or
R5
and R6 taken together with the nitrogen atom to which they are attached may
form a
5-10 membered cycloheteroalkyl ring; and
n is 1 or 2; or pharmaceutical salts thereof,
comprising the steps of reacting a sulfonyl fluoride of the formula III
R3'S02F
III
wherein R3' is as hereinabove defined for R3 with the proviso that R3' does
not contain
a group that can form an anion under basic conditions;
with a carbonyl compound of the formula IV:
O
Z~H
R1 R2
IV
wherein Z is H, OH, YNOX, or ORS, and X, Y, R,, Rz, R, and R6 are as
hereinabove
defined; in the presence of a metal hydride or amide base in an ether organic
solvent
at temperatures from about -78°C to about room temperature (eg up to
about 15°C to
about 30°C, preferably up to about 20-25°C) to produce an alpha-
sulfonyl carbonyl
compound of formula V:
0
Z~S02R3'
R~ R2
V
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-6-
wherein Z, R,, R=and R3' are as hereinabove defined; and converting compound
of
formula V into a hydroxamic acid derivative.
Compounds of Formula I may also be prepared by reacting a sulfonyl fluoride of
formula III:
R3'S02F
III
wherein R3' is as hereinabove defined for R3 with the proviso that R3' does
not contain
a group that can form an anion under basic conditions; with an enol ether of
formula
VIII:
ORS
Z
R1 R2
VIII
wherein Z is H, OH, YNOX, or ORS, and R, and Rz, are as hereinabove defined;
R, is cycloalkyl of 3-6 carbon atoms; 5-10 membered cycloheteroalkyl; alkyl
of 1-18 carbon atoms; alkenyl of 2-18 carbon atoms having from 1 to 3 double
bonds;
alkynyl of 2-18 carbon atoms having from 1 to 3 triple bonds; or -SiR8R9R,o;
and
Rg, R9, and R,o are each, independently, aryl; 4-8 membered heteroaryl having
1-3 heteroatoms selected from N, NR4, O and S; cycloalkyl of 3-6 carbon atoms;
5-10
membered cycloheteroalkyl; alkyl of 1-18 carbon atoms; alkenyl of 2-18 carbon
atoms having from 1 to 3 double bonds; alkynyl of 2-18 carbon atoms having
from 1
to 3 triple bonds; or two of R8, R9, and R,o taken together with the silicon
atom to
which they are attached form a heterocyclic ring of 5 or 6 members;
in the presence of a Lewis acid or fluoride reagent in an ether organic
solvent
at temperatures ranging from about -78°C to about room temperature ( eg
up to about
15-30°C to produce an alpha-sulfonyl carbonyl compound of formula V:
O
Z ~S02R3~
Ri R2
V
wherein Z, R,, R., and R3' are as hereinabove defined, and converting the
compound of
Formula V into a hydroxamic acid derivative.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
In other embodiments of the present invention are provided methods of
preparing
alpha-sulfonyl hydroxamic acid derivatives of the general formula I:
XO O
~N S02R3
Y
N
i
Ra
Ia
wherein
X is hydrogen, or alkyl of 1-6 carbon atoms;
Y is hydrogen, alkyl of 1-6 carbon atoms, aryl of 6 to 10 carbon atoms, 5-10
membered heteroaryl having 1-3 heteroatoms selected from N, NR4, O and S,
cycloalkyl of 3-6 carbon atoms, 5-10 membered cycloheteroalkyl; wherein said
alkyl,
aryl, heteroaryl, cycloalkyl and cycloheteroalkyl group of Y is optionally
substituted
on any atom capable of substitution, with 1 to 3 substituents selected from
the group
consisting of halogen, alkyl of 1-6 carbon atoms; alkenyl of 2-6 carbon atoms
having
from 1 to 3 double bonds; alkynyl of 2-6 carbon atoms having from 1 to 3
triple
bonds, cycloalkyl of 3-6 carbon atoms, -ORS, =O, -CN, -CORS perfluoroalkyl of
1-4
carbon atoms, -O-perfluoroalkyl of 1-4 carbon atoms, -CONRSR6,-S(O)nRS,
-OPO(ORS)OR6, -PO(ORS)R6, -OC(O)ORS, -ORSNRSR6, -OC(O)NRSR6, -C(O)NRSOR6,
-COORS, -S03H, -NRSR6, -N[(CHz)2],NRS, -NR5COR6, -NRSCOOR~, SO,NRSR6, -NO~,
-N(R5)SOZRb, -NRSCONRSR6, -NRSC(=NR6)NRSR6, -NRSC(=NR6)N(SOzRS)Rb.
-NRSC(=NR6)N(C=ORS)R6, -tetrazol-5-yl, -SOZNHCN, -S02NHCONRSR6, phenyl,
heteroaryl and 5-10 membered cycloheteroalkyl;
R3 is alkyl of 1-18 carbon atoms, alkenyl of 2-18 carbon atoms having 1 to 3
double
bonds, alkynyl of 2-18 carbon atoms having from 1 to3 triple bonds, cycloalkyl
of 3-6
carbon atoms, 5-10 membered cycloheteroalkyl, aryl of 6 to 10 carbon atoms, 5-
6
membered heteroaryl having 1-3 heteroatoms selected from N, NR4, O, and S;
wherein said alkyl, alkenyl, alkynyl, cycloalkyl, cycloheteroalkyl, aryl and
heteroaryl
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-g-
of R3 may optionally be substituted on any atom capable of substitution with
from 1
to 3 substituents selected from halogen, alkyl of 1-6 carbon atoms; alkenyl of
2-6
carbon atoms having from 1 to 3 double bonds; alkynyl of 2-6 carbon atoms
having
from 1 to 3 triple bonds, cycloalkyl of 3-6 carbon atoms, -ORS, =O, -CN, -CORS
perfluoroalkyl of 1-4 carbon atoms, -O-perfluoroalkyl of 1-4 carbon atoms,
-CONRSR6,-S(O)nRs, -OPO(ORS)OR6, -PO(ORS)R6, -OC(O)ORS, -ORSNRSR6,
-OC(O)NRSR6, -C(O)NRSOR6, -COORS, -S03H, -NRSR6, -N[(CHZ)z]zNRS, -NR5COR6,
-NRSCOOR6, SO,NRSR6, -NO2, -N(RS)SOzRb, -NRSCONRSR6, -NRSC(=NR6)NRSR6,
-NRSC(=NR6)N(SOZRS)R6,-NRSC(=NR6)N(C=ORS)R6, -tetrazol-5-yl, -SOZNHCN,
-S02NHCONRSR6, phenyl, heteroaryl and 5-10 membered cycloheteroalkyl;
R4 is hydrogen; aryl; aralkyl, heteroaryl; heteroaralkyl, alkyl of 1-6 carbon
atoms;
cycloalkyl of 3-6 carbon atoms; -C(O)"R5, -CONR5R6 or SOZRS;
RS and R6 are each independently hydrogen, optionally substituted aryl; 4-8
membered heteroaryl having 1-3 heteroatoms selected from N, NR4, O and S;
cycloalkyl of 3-6 carbon atoms; 5-10 membered cycloheteroalkyl; alkyl of 1-18
carbon atoms; alkenyl of 2-18 carbon atoms or alkynyl of 2-18 carbon atoms; or
RS
and R6 taken together with the nitrogen atom to which they are attached may
form a
5-10 membered cycloheteroalkyl ring; and
n is 1 or 2; or pharmaceutical salts thereof, comprising the steps of
a) treating a compound of formula
~O O
Ra
with diisopropylamide or lithium hexamethyldisilazide to form an enolate;
b) reacting the enolate with a sulfonyl fluoride of formula III:
R3S02F
III
CA 02398561 2002-07-24
WO 01/55112 PCT/USOl/02669
-9-
to form a compound
c) hydrolyzing the compound of step b) to produce
O
HO S02Rs
NJ
R4 , and
d) reacting compound of step c) with hydroxylamine or hydroxylamine derivative
of
the formula VII:
XONHY
VII
in the presence of coupling reagent and polar organic solvent at temperatures
rangW g
from 0°C to about room temperature, eg up to about 15-30°C.
In other aspects of the invention are provided methods of preparing compounds
of
Formula 8
° °,.°
HOHN \S \ ~ OR~2
NJ
i
R4 8
wherein R4 is hydrogen; aryl; aralkyl, heteroaryl; heteroaralkyl, alkyl of 1-6
carbon
atoms; cycloalkyl of 3-6 carbon atoms; -C(O)~RS, -CONRSR6 or S02R5;
RS and R6 are each independently hydrogen, optionally substituted aryl; 4-8
membered heteroaryl having 1-3 heteroatoms selected from N, NR4, O and S;
cycloalkyl of 3-6 carbon atoms; 5-10 membered cycloheteroalkyl; alkyl of 1-18
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
- 10-
carbon atoms; alkenyl of 2-18 carbon atoms or alkynyl of 2-18 carbon atoms: or
RS
and R~ taken together with the nitrogen atom to which they are attached may
form a
5-10 membered cycloheteroalkyl ring; and
R,= is methyl, n-butyl, 2-butynyl, or p-chlorophenyl;
and n is 1 or 2; or pharmaceutical salts thereof,
comprising the steps of
a) treating a compound of formula
~O O
NJ
Ra
with diisopropylamide or lithium hexamethyldisilazide to form an enolate;
b) reacting the enolate with a sulfonyl fluoride of Formula 2:
R o ~ \ ~s
~2 ~ F
to form a compound of Formula 13
O O\ O _
Et0 S \ ~ ORi2
NJ
R4 13
c) hydrolyzing compound of Formula 13 with lithium hydroxide to produce
compound of Formula 14
o O
O~ "
HO S \ ~ OR12
NJ
R4 14 ; and
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-11-
d) treating the compound of Formula 14 with oxalyl chloride, triethylamine,
and
hydroxylamine hydrochloride at temperatures ranging from 0°C to about
roo
temperature, eg up to about 15-30°C.
In some aspects of the present invention compounds of formula V are prepared
O
Z~S02R3~
Ri R2
V
wherein
R,, RZ and R3' are as previously defined and Z is H, OH, YNOX, ORS or NRSR~,
comprising reacting a sulfonyl fluoride of the formula III
R3'S02F
III
wherein R3' is as hereinabove defined;
with a carbonyl compound of the formula IV:
O
Z~H
R~ R2
IV
wherein Z, R, and RZ are as previously defined, in the presence of a metal
hydride or
amide base in an ether organic solvent at temperatures from about -78°C
to about
room temperature (eg up to from about 15°C to about 30°C).
Alternatively, compounds of Formula V are prepared
O
Z ~S02R3~
Ri R2
V
wherein
R,, RZ and R3' are as previously defined and Z is H, OH, YNOX, ORS or NRSR6,
comprising reacting a sulfonyl fluoride of the formula III
R3'S02F
III
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-12-
wherein R3' is as hereinabove defined with an enol ether of Formula VIII
ORS
Z
R1 R2
VIII
wherein Z is H, OH, YNOX, OR 5 or NR5R6, and R, and R" are as hereinabove
defined;
R, is cycloalkyl of 3-6 carbon atoms; 5-10 membered cycloheteroalkyl; alkyl
of 1-18 carbon atoms; alkenyl of 2-18 carbon atoms having from 1 to 3 double
bonds;
alkynyl of 2-18 carbon atoms having from 1 to 3 triple bonds; or -SiRgRgR,o;
and
R8, Rg, and R,o are each, independently, aryl; 4-8 membered heteroaryl having
1-3 heteroatoms selected from N, NR4, O and S; cycloalkyl of 3-6 carbon atoms;
5-10
membered cycloheteroalkyl; alkyl of 1-18 carbon atoms; alkenyl of 2-18 carbon
atoms having from 1 to 3 double bonds; alkynyl of 2-18 carbon atoms having
from 1
to 3 triple bonds; or two of R8, R9, and R,o taken together with the silicon
atom to
which they are attached form a heterocyclic ring of 5 or 6 members;
in the presence of a Lewis acid or fluoride reagent in an ether organic
solvent at
temperatures ranging from about -78 °C to about room temperature (eg up
to from
about 15°C to about 30°C) to produce an alpha-sulfonyl carbonyl
compound of
formula V.
When Z is ORS, compounds of Formula V may be converted to a hydroxamic
acid derivative of Formula I in accordance with the steps of
reacting the alpha-sulfonyl carbonyl compound of the formula V with an
alkali metal hydroxide in the presence of water, and/or ether organic solvent
or
alcohol at temperatures ranging from about 0 °C to about 100°C
to produce a
carboxylic acid of the formula VI:
O
SO2R3
HO~
R1 R2
VI
wherein, R,, RZ, and R3 are as hereinabove defined; and
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-13-
reacting a carboxylic acid of formula VI with a hydroxylamine or
hydroxylamine derivative of the formula VII:
XONHY
VII
wherein X and Y are as hereinabove defined;
in the presence of suitable coupling reagent and polar organic solvent to
produce a
hydroxamic acid derivative of the formula I:
O
XO.N~S02R3
Y R~ R2
I
wherein X, Y, R,, R2, and R3 are as hereinabove defined.
In other embodiments of the present invention, when Z is OH, compounds of
Formula V may be converted to a hydroxamic acid derivative by reacting the
alpha-
sulfonyl carbonyl compound of formula V:
with a hydroxylamine or hydroxylamine derivative of the formula VII:
XONHY
VII
wherein X and Y are as hereinabove defined; in the presence of a coupling
reagent
and polar organic solvent at temperatures ranging from about 0°C to
about room
temperature (eg up to from about 15°C to about 30°C).
Further, in accordance with the present invention sulfonyl fluoride compounds
of Formula III can be prepared by reacting a sulfonyl chloride of formula II
R3'S02C1
II
wherein R3' is as hereinabove defined for R 3 the proviso that R 3' does not
contain a
group that can form an anion under basic conditions, with a fluorinating agent
in the
presence of a polar organic solvent at room temperature (eg at about 15
°C to about
30°C) to produce a sulfonyl fluoride of formula III.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-14-
Further chemical transformations can be carned out before or after each step
for compounds of the formula I, V, and VI in cases where R ,, R, or R3 of the
product
differs from R,, R, or R3' of the starting compound.
Groups which may form an anion under basic conditions of the invention and
thus are excluded from the definition of R3', include, but are not limited to -
OH, -NH,
-SH, -COCH, -SO,CH, -CHNO~, CHCN. Accordingly, during the sulfonylation of
the carbonyl compound, such substituents at R3 should be avoided or protected
and
released later by deprotection, as designated by R3'.
Sulfonyl chloride compounds of the present invention are commercially
available or can be prepared by those skilled in the art in accordance with
procedures
described in the literature such as Kende, A.S.; Medoza, J.S., J. Org. Chern.,
1990,
55, 1125-1126.
Appropriate fluorinating agents are taught, for example by Clark, J.H.; Hyde,
A.J.; Smith, D.K. J.Chem.Soc.Chem.Comm. 1986, 791-792; Ichihara, J.; Matsuo,
T.;
Hanafusa, T.; Ando, T. J. Chem.Soc. Chem. Comm. 1986, 793-794 and include but
are
not limited to potassium fluoride, potassium fluoride-calcium fluoride
mixture, or
cesium fluoride.
Preferred ether organic solvents of the present invention are those known to
those skilled in the art including, but not limited to tetrahydrofuran,
diethylether or
dioxane.
Bases used in methods of the present invention are those known to those
skilled in the art, preferably metal hydride or amide bases, such as, but not
limited to
of lithiumdiisopropylamide, lithiumhexamethyldisilazide, and sodium hydride.
Lewis acids and fluoride reagents used in methods of the present invention are
known to those skilled in the art and include, but are not limited to
borontribromide,
tetrabutylammonium and sodium hydride.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-15-
Polar organic solvents useful in methods of the present invention are known to
those skilled in the art and include, but are not limited to acetonitrile,
tetrahydrofuran
and dimethylformamide.
Alkali metal hydroxides used in preferred methods of the present invention
are known to those skilled in the art and include, but are not limited to
lithium
hydroxide and sodium hydroxide.
Alcohols used in some methods of the present invention are known to those
skilled in the art and include, but are not limited to methanol and ethanol.
Coupling reagents of the present invention are those known to those skilled in
the art including, but not limited one or more of 1-(3-dimethylaminopropyl)-3-
ethylcarbodimide hydrochloride, N-hydroxybenzotriazole, N-methylmorpholine and
oxalylchloride and triethylamine.
The present invention further relates to low molecular weight, non-peptide
inhbitors of matrix metalloproteinases (MMPs) and TNF-alpha converting enzyme
(TACE) for the treatment of rheumatoid arthritis, tumor metastasis, tissue
ulceration,
abnormal wound healing, periodontal disease, bone disease, diabetes and HIV
infection.
Thus, in accordance with the present invention is provided compounds of
Formula IX
XO,N 0283
Y
Ra
IX
wherein:
X is hydrogen and alkyl of 1-6 carbon atoms; and
O
S
1
N
Y, R3 and R; are as previously defined, and pharmaceutical salts thereof.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
- 16-
Particularly preferred is 1-Benzyl-3-(4-methoxybenzenesulfonyl)piperidine-
3-carboxylic acid hydroxamide, or pharmaceutical salts thereof.
Certain compounds prepared by the novel method of the present invention
contain one or more asymmetric carbon atoms, giving rise to enantiomeric and
diastereomeric forms of the compounds. In addition, certain compounds of this
invention contain a carbon-carbon double bond, giving rise to cis- and trans-
geometric isomers. It is to be understood that the invention encompasses the
enantiomers, diastereomers, and geometrical isomers as well as mixtures
thereof
including racemic mixtures.
Alkyl, as used herein, refers to branched and straight chain alkyl groups,
preferably having from 1 to 18 carbon atoms, and more preferably from 1 to 6
carbon
atoms. Exemplary alkyl groups include methyl, ethyl, propyl, i-propyl, butyl,
t-butyl,
pentyl, hexyl, n-heptyl, octyl and the like.
Alkenyl, as used herein, refers to alkenyl groups, preferably having from 2-18
carbon atoms and more preferably from 2 to 6 carbon atoms, and having from 1
to 3
sites of alkenyl unsaturation (double bond).
Alkynyl, as used herein, refers to alkynyl groups, preferably having from 2-18
carbon atoms and more preferably from 2 to 6 carbon atoms, and having from 1
to 3
sites of alkynyl unsaturation (triple bond).
Cycloalkyl refers to cyclic alkyl groups of from 3 to 8 carbon atoms, and
more preferably from 3-6 carbon atoms, including, by way of example,
cyclopropyl,
cyclobutyl, cyclopentyl, cyclohexyl, cyclooctyl and the like.
Heteroaryl, as used throughout, is a 5-10 membered mono- or bicyclic
aromatic carbocyclic ring having from 1-3 heteroatoms selected from N, NR4, S
and
O within the ring. Such heteroaryl groups can have a single ring (e.g. pyridyl
or
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-17-
furyl) or multiple condensed rings (e.g. benzothienyl), which condensed ring
may or
may not contain a heteroatom. Heteroaryl is preferably
~K ~ ' ~K ~N
K K N
N
NN
N / / N~ , N
N R . R4
4
~: N> ~: ,N cN:
~K
N , , , N
K N ~ ~ N ~ N~
/ ~ , ~ ~~
N
C~~ , , N
N
wherein K is defined as O, S or -NR4, and R4 is as hereinabove defined. More
preferred heteroaryl rings include pyrrole, furan, thiophene, pyridine,
pyrimidine,
pyridazine, pyrazine, triazole, pyrazole, imidazole, isothiazole, thiazole,
isoxazole,
oxazole, indole, isoindole, benzofuran, benzothiophene, quinoline,
isoquinoline,
quinoxaline, quinazoline, benzotriazole, indazole, benzimidazole,
benzothiazole,
benzisoxazole, and benzoxazole. Heteroaryl groups of the present invention may
have from 1 to 3 substituents, and more preferably may have one or two
substituents.
5-10 Membered cycloheteroalkyl is a saturated or unsaturated group having a
single
ring or multiple condensed rings, from 2 to 10 carbon atoms and from 1 to 3
heteroatoms selected from S, N, O, or NR, within the ring, wherein, in fused
ring
systems, one or more of the rings can be aryl or heteroaryl.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-18-
Preferred cycloheteroalkyl are
K N > >
R K
4
K
Ra ~K~ ,
K _~K~
~N
R ~ K ~ N ~ Ra
4
wherein K is O, N, S or NR4; and R~ is as hereinabove defined. The rings above
shown as mono-radicals may also be illustrated as di-radicals eg when R, and
R:
together form a cycloheteroalkyl ring.
Preferred cycloheteroalkyl rings include piperidine, piperazine, morpholine,
tetrahydropyran, tetrahydrofuran or pyrrolidine. Cycloheteroalkyl groups of
the
present invention may optionally be mono-, di- or tri substituted.
Aryl, as used herein refers to an unsaturated, aromatic carbocyclic group of
from 6 to 10 carbon atoms having a single ring (phenyl) or multiple condensed
rings
(naphthyl), which condensed rings may or may not be aromatic. Preferred aryls
include phenyl and naphthyl. Aryl groups may optionally be mono-, di- or tri-
substituted.
Alkyl, alkenyl, alkynyl, and perfluoroalkyl include both straight chain as
well
as branched moieties. Alkyl, alkenyl, alkynyl, and cycloalkyl groups may be
unsubstituted (carbons bonded to hydrogen or other carbons in the chain or
ring) or
may be mono- or poly-substituted.
Halogen means bromine, chlorine, fluorine, and iodine.
Suitable substituents of aryl, aralkyl, heteroaryl, heteroaralkyl, alkyl,
alkenyl,
alkynyl and cycloalkyl include, but are not limited to halogen, alkyl of 1-6
carbon
atoms; alkenyl of 2-6 carbon atoms; alkynyl of 2-6 carbon atoms, cycloalkyl of
3-6
carbon atoms, -ORS, -CN, -CORS perfluoroalkyl of 1-4 carbon atoms, -O-
perfluoroalkyl
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-19-
of 1-4 carbon atoms, -CONRSR6,-S(O)nRs, -OPO(ORS)OR6, -PO(ORS)R6, -OC(O)ORS,
-OC(O)NRSR~, -C(O)NRSOR6, -COORS, -SO,H, -NRSR6, -N[(CHz)z],NRS, -NRSCOR6,
-NRSCOOR~, SO~NR5R6, -NO~, -N(RS)SO,R~, -NRSCONRSR~, -NRSC(=NR6)NRSR~,
-NRSC(=NR6)N(SO,RS)R6, -NRSC(=NR6)N(C=ORS)R6, -tetrazol-5-yl, -SO ,NHCN,
-S02NHCONR5R6, phenyl, heteroaryl or 5-10 membered cycloheteroalkyl; and RS
and
R6 are as hereinabove defined; -NR SR6 may form a cycloheteroalkyl ring such
as
pyrrolidine, piperidine, morpholine, thiomorpholine, oxazolidine,
thiazolidine,
pyrazolidine, piperazine or azetidine ring.
Pharmaceutically acceptable salts are those derived from pharmaceutically
acceptable organic and inorganic acids such as lactic, citric, acetic,
tartaric, succinic,
malefic, malonic, hydrochloric, hydrobromic, phosphoric, nitric, sulfuric,
methane-
sulfonic, and similarly known acceptable acids.
In the compounds referred to above, examples of Z are preferably OH or ORS
for example where RS is alkyl (preferably 1-6 carbon atoms, e.g. methyl,
ethyl,
propyl, isopropyl, butyl and pentyl).
R3 is preferably an optionally substituted aryl group, e.g. a phenyl group,
most
preferably a 4-substituted phenyl group. The aryl group is preferably
substituted by
one or more -ORS groups, e.g. where RS is is alkyl (preferably 1-6 carbon
atoms, eg
methyl, ethyl, propyl, isopropyl, butyl or pentyl), alkynyl (preferably 2-7
carbon
atoms) or optionally substituted aryl, eg where the substituents are selected
from C ~-
C6-alkyl, C1-C6-alkoxy and halogen, such as chlorine.
R1 and RZ together with the carbon atoms to which they are attached
preferably form a 5-10 membered heteroalkyl ring, eg having 1-3 heteroatoms
selected from N, NR4, O and S, most preferably a ring containing a single NR4
group,
e.g. a six membered piperidine ring. They preferably form a 3,3 di-
substituted, 4,4-
di-substituted, 1,3,3 tri-substituted or 1,4,4-tri-substituted piperidine.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-20-
Examples of R4 are hydrogen, alkyl of 1-6 carbon atoms, -CORS, -COORS,
-SOZRS and optionally substituted benzyl (eg 4-chlorobenzyl, 4-methoxybenzyl
or 4-
(2-piperidin-1-yl-ethoxy)benzyl).
Examples of RS are an optionally substituted alkyl of 1-18 carbon atoms (such
as methyl, trifluoromethyl), an optionally substituted alkenyl of 2-18 carbon
atoms,
an optionally substituted aryl (such as phenyl), an optionally substituted 4-8
membered heteroaryl (such as pyridyl, thienyl) or an optionally substituted 5-
10
membered cycloheteroalkyl (such as pyrrolyl); preferably RS is methyl, ethyl,
n-butyl,
t-butyl, but-2-ynyl, 4-chlorophenyl, 4-methoxyphenyl, 1-pyrrolidinyl, 3,
pyridinyl, 2-
thienyl, 2,2,5-trimethyl-1,3-dioxan-5-yl or 2-hydroxy-1-(hydroxymethyl)-1-
methyl-
ethyl.
Typical optional substituents as used herein include C1-C6alkyl, C1-C6 alkoxy,
C1-C6haloalkyl and halogen.
The present invention accordingly provides a pharmaceutical composition
which comprises a compound of this invention in combination or association
with a
pharmaceutically acceptable Garner. In particular, the present invention
provides a
pharmaceutical composition which comprises an effective amount of compound of
this invention and a pharmaceutically acceptable Garner.
The compositions are preferably adapted for oral administration. However,
they may be adapted for other modes of administration, for example, parenteral
administration for patients.
In order to obtain consistency of administration, it is preferred that a
composition of the invention is in the form of a unit dose. Suitable unit dose
forms
include tablets, capsules, and powders in sachets or vials. Such unit dose
forms may
contain from 0.1 to 100 mg of a compound of the invention. The compounds of
the
present invention can be administered orally at a dose range of about 0.01 to
100 mg
per kg. Such composition may be administered from 1 to 6 times a day, more
usually
from 1 to 4 times a day.
The compositions of the invention may be formulated with conventional
excipients, such as fillers, a disintegrating agent, a binder, a lubricant, a
flavoring
agent, and the like. They are formulated in conventional manner.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-21 -
DETAILED DESCRIPTION OF THE INVENTION
Several synthetic routes can be employed to prepare the compounds of
formula I, using alpha-sulfonylation of the enolisable carbonyl compound as
the key
step in the process. Several preferred routes for the preparation of these
compounds
are described in schemes I-III. Although, each sequence is illustrated with a
compound of formula I, wherein X and Y are hydrogen, R 3 is aryl, and R , and
R
taken together with the carbon atom to which they are attached form a 6-
membered
cycloheteroalkyl ring containing NR4, additional compounds of this invention
can be
prepared in the same manner using the appropriate starting materials and
routes as
would be appreciated by one skilled in the art and illustrated by the specific
examples. The reagents and the solvents for the individual step are given for
illustrative purposes only and may be replaced by other reagents and solvents
known
to those skilled in the art.
Scheme I
KF-CaF2
CI02S \ ~ OR~2 ~ FO2S \ ~ OR~2
CH3CN
1
step 1
O O
C02H Me0 O Me0 O S \ / OR~2
Mel/K2C03
N
acetone N step 2 Boc
Boc Boc
4
3
O O
O\ "
TFA Me0 O O\ O \ ~ OR~2 R4L Me0 S \ ~ OR~2
base
CF3CH20H N
H
step 3 . TFA step 4
5
LiOH/THF/H20/MeOH O
HO \S \ ~ OR~2 HOBT/EDC/NMM HOHN \S \ ~ OR~Z
step 5 N J aq. NH20H N
step 6 Ra
1~ 7
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-22-
In Scheme I, step 1, sulfonyl chloride 1, wherein R,~ is methyl, n-butyl, 2-
butynyl, or p-chlorophenyl, is treated with a potassium fluoride-calcium
fluoride
mixture (either commercially available or prepared according to the procedure
by
Ichihara) in acetonitrile at room temperature to obtain the sulfonyl fluoride
2.
In step 2, the enolate prepared from the ester 3 (prepared by treating
commercially available Boc-isonipecotic acid with methyl iodide/potassium
carbonate ) and lithium diisopropylamide (LDA) (prepared in situ using n-butyl
lithium and diisopropylamine) is treated with compound 2 at -78 °C-
25°C to obtain
the compound 4.
In step 3, the protecting group, t-butoxycarbonyl, is cleaved with
trifluoroacetic acid in trifluoroethanol to obtain the compound 5 as a salt.
In step 4, R~, as hereinabove defined, is introduced by treating compound 5
with R~L, wherein L is a leaving group such as but not limited to halogen, in
the
presence of other reagents such as triethylamine and the solvents known to
those
skilled in the art, to obtain compound 6.
In step 5, the ester 6 is hydrolyzed with lithium hydroxide at 50 °C or
sodium
hydroxide for 15 hours to obtain acid 7.
In step 6, compound 7 is treated with N-hydroxybenzotriazole, 1-(3-dimethyl
aminopropyl)-3-ethylcarbodimide hydrochloride, N-methylmorpholine, and aqueous
hydroxylamine to obtain the desired hydroxamic acid 8.
Scheme II
O O\ O _ O O\ 0
LiOHITHF/H20 HO \S \ ~ OR~2 (COCI)2, DMF HOHN \S \ ~ OR~2
J J
MeOH N Et3N, NH20H.HCI
Boc Boc
step 1 g step 2
O O\ O
4M HCI HOHN \S \ ~ OR~2 R4~
J
dioxane
H.HCI base
step 3 11 step 4
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-23-
The target compounds can also be obtained by changing the order of
transformations carried out for compound 4 as shown in the Scheme II.
In Scheme II, step l, the compound 4 is treated with lithium hydroxide as in
Scheme I, step 5, to obtain the N-protected carboxylic acid 9.
In step 2, the acid 9 is treated with oxalyl chloride, triethylamine, and
hydroxylamine hydrochloride in dimethylformamide to obtain N-protected
hydroxamic acid 10, which is then deprotected with 4M hydrochloric acid in
dioxane
to give the salt 11 in step 3.
In step 4, R4 as hereinabove defined, is introduced selectively using the
conditions in Scheme I, step 4 to obtain the hydroxamic acid 8.
Scheme III
O O KF or CsF
8120 ~ \ 'S~ R~20 S,F
~CI
CH3CN or THF
2
1 step 1
O ~O O O O°
O Et0 S \ ~ OR~2
R4L
N
N~ LDA or LiHMDS
base R R4 13
4
step 2 step 3
12
O O ~\
O~S~ORi2
LiOH HO \~~ (COCI)2, DMF
J
THF/MeOH/H20 N Et3N, NH20H.HC1
R4 14
step 4 step 5
Alternatively, the target compounds can be obtained by following the
synthetic sequence of Scheme III.
In Scheme III, step 1, sulfonyl fluoride 2 is obtained by treating sulfonyl
chloride 1 with either potassium fluoride or cesium fluoride in acetonitrile.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-24-
Alternatively, this reaction is carried out in tetrahydrofuran and the
resulting solution
is used for the next step without isolation of the sulfonyl fluoride.
In step 2, R4 as hereinabove defined, is introduced early in the sequence by
treating starting material such as ethyl isonipecotate R4L, wherein L is a
leaving
group such as but not limited to halogen, in the presence of appropriate
reagents such
as triethylamine with commercially available ethyl isonipecotate.
In step 3, enolate prepared by reacting the compound 12 with lithium
diisopropylamide or lithium hexamethyldisilazide, is treated with the fluoride
2 to
obtain the compound 13.
In step 4 ester 13 is hydrolyzed with lithium hydroxide to Qive acid 14.
Alternatively, step 3 and step 4 are carried out sequentially as a one-pot
process
without isolation of the ester 13.
In step 5, acid 14 is treated with oxalyl chloride, triethylamine, and
hydroxylamine hydrochloride as in the Scheme II, step 2, to obtain the
compound 8.
In the following examples, there are described several preferred embodiments
to illustrate the invention. However, it should be understood that the
invention is not
intended to be limited to the specific embodiments.
General procedure for the preparation of sulfonyl fluorides from sulfonyl
chlorides
Method A To a solution of the sulfonyl chloride (1 equiv) in acetonitrile was
added
potassium fluoride-calcium fluoride mixture (2 equiv with respect to potassium
fluoride) and the resulting mixture was stirred for 4 hours at room
temperature. The
reaction mixture was filtered and the filtrate was concentrated. The crude
product
was dissolved in ethyl acetate and washed with water. The organic layer was
dried
over anhydrous sodium sulfate and the solvent was removed in vacuo to obtain
the
product.
Method B : To a solution of the sulfonyl chloride ( 1 equiv) in acetonitrile
was added
potassium fluoride (2 equiv). The resulting suspension was stirred for 18
hours at
20-25 °C. The suspension was filtered and the solid was washed with
diethylether.
The mother liquor was concentrated in vacuo and resulting oil was seeded to
give the
product as a white crystalline solid.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-25-
Method C: To a solution of the sulfonyl chloride (1 equiv) in acetonitrile was
added
cesium fluoride (2 equiv). The resulting suspension was stirred for 18 hours
at 24-
25°C. The suspension was filtered and the solid washed with
diethylether. The
mother liquor was concentrated in vacuo and resulting oil was seeded to
produce the
product as a white crystalline solid.
Method D : The solution of sulfonyl chloride (1 equiv) in tetrahydrofuran was
mixed with potassium fluoride (2 equiv) and stirred for 30 hours at 20-
°C. The
suspension was filtered and the solid was washed with tetrahydrofuran. This
solution
was used for the next step without isolation.
General procedure for alpha-sulfonylation of the carbonyl compound (step 1)
To a solution of lithium diisopropylamide ( 1 equiv)(either commercially
available or freshly prepared from n-butyllithium and diisopropylamine) in
tertahydrofuran cooled to -78°C, was added a solution of the carbonyl
compound (1
equiv) in tetrahydrofuran and the resulting mixture was stirred for 0.5-1 hour
at that
temperature. A solution of the sulfonyl fluoride ( 1.1 equiv) in
tetrahydrofuran was
then added to the mixture and the resulting mixture was stirred for 4-15 hours
at room
temperature, quenched with saturated aqueous ammonium chloride solution and
extracted with ethyl acetate. The organic layer was washed with brine and
dried over
anhydrous sodium sulfate. The crude product was purified by either
recrystallization
or silica gel chromatography to obtain the desired product.
General procedure for the preparation of the carboxylic acid from the ester
(step 31
A solution of the ester ( 1 equiv) and lithium hydroxide or sodium hydroxide
(1.5-2 equiv) in tetrahydrofuran/methanol/water (3:3:2) mixture was stirred at
room
temperature or heated at 55°C for 15 hours. The mixture was
concentrated, acidified
to pH 3-5 with 1N aqueous hydrochloric acid, and extracted with ethyl acetate.
The
organic layer was washed with brine and dried over anhydrous sodium sulfate.
Removal of the solvent under vacuuo gave the product.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-26-
General procedure for the preparation of hydroxamic acid from the carboxylic
acid
ste 4
Method A;
To a solution of the acid (1 equiv) in dimethylformamide was added
hydroxybenzotriazol (1.2 equiv) followed by 1-(3-dimethylaminopropyl)-3-
ethylcarbodimide hydrochloride (1.4 equiv) and N-methylmorpholine (1.5 equiv).
The resulting mixture was stirred for 1 h at room temperature when 50% aqueous
hydroxylamine solution (5 equiv) was added and the mixture was stirred for 15
h at
that temperature. The solvent was removed in vacuo and ethyl acetate/water was
added to the crude product. The organic layer was separated and washed
successively with 1N aqueous hydrochloric acid, water, saturated aqueous
sodium
bicarbonate, and water. The organic layer was dried over anhydrous sodium
sulfate
and the solvent was removed in vacuo to obtain the product.
Method B;
To a solution of oxalyl chloride in methylene chloride was added dimethyl-
formamide followed by the acid ( 1 equiv) in methylene chloride at 0°C
and the
mixture was stirred for 1 hour at room temperature. This mixture was added to
a
solution containing hydroxylamine hydrochloride ( 10 equiv) and triethyl amine
( 15
equiv) in tetrahydrofuran/water (5:1) that had been stirring for 0.25-1 hour
at 0°C.
The reaction was allowed to warm to room temperature and stirred for 15-24 h
at that
temperature. The reaction mixture was concentrated and the residue was taken
up in
ethyl acetate. The organic layer was washed with saturated aqueous sodium
bicarbonate and water and dried over anhydrous sodium sulfate. The solvent was
removed in vacuo and the crude product was purified by triturating or silica
gel
chromatography to obtain the product.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-27-
Example 1
4-But-2-yn~oxybenzenesulfonyl fluoride
F02S ~ ~ O
The general procedure for the preparation of sulfonyl fluorides was followed
using 4-but-2-ynyloxybenzenesulfonyl chloride (2.0g, 8.18 mmol) in
acetonitrile ( 10
ml) and potassium fluoride-calcium fluoride mixture to obtain 1.5g(80%) of the
product as a solid.
IR: 2925, 2242, 1596, 1579, 1406, 1261, 997 cm-';
'H NMR(300 MHz, CDC13):8 1.87(t, 3H, J= 1.8 Hz), 4.76(q, 2H, J= 1.8 Hz),
7.14(d,
2H, J= 6.6 Hz), 7.95(d, 2H, J= 6.6 Hz);
'3C NMR(75 MHz, CDC13):8 3.6, 56.9, 72.4, 85.4, 115.8, 130.8, 163.3.
Example 2
1-(tert-Butyl) 4-methyl 1.4-piperidinecarboxylate
Me0 O
N
Boc
A mixture of N-t-butoxycarbonyl isonipecotic acid (20g, 0.087 mmol), methyl
iodide (62g, 0.435 mmol), and potassium carbonate (120g, 0.87 mmol) was
stirred for
2 days. The mixture was filtered and the solvent was removed in vacuo. The
crude
product was dissolved in methylene chloride, washed with water and dried over
anhydrous sodium sulfate. Removal of the solvent gave 20g (95%) of the product
as
a white solid.
CA 02398561 2002-07-24
WO 01/55112 PCT/LTSO1/02669
-28-
Example 3
4-(4-But-2-ynyloxybenzenesulfonyl)-piperidine-1,4-dicarboxylic acid tert-but 1
methyl ester
O O
O~ "
Me0 S \ ~ O
N
i
Boc
The general procedure for step 1 was followed using lithium diisopropylamide
(70
mmol), product from Example 2 (15.5g, 64 mmol), and the product from Example 1
(70 mmol) to obtain 24.5g(85%) of the product as a white solid.
IR: 2978, 2242, 1740, 1697, 1594, 1418, 1301, 1002, 908cm-';
'H NMR(300 MHz, CDC13):8 1.44(s, 9H), 1.87(m, 3H), 1.98(m, 2H). 2.32(m, 2H),
2.62(m, 2H), 3.74(s, 3H), 4.17(m, 2H), 4.74(m, 2H), 7.09(d, 2H, J= 7.2 Hz),
7.71(d,
2H, J= 7.2 Hz); '3C NMR(75 MHz, CDCl3):8 4.0, 28.2, 28.7, 53.5, 57.2, 72.9,
73.1,
80.5, 85.4, 115.3, 127.0, 132.6, 154.7, 162.9, 167.8;
HR-MS: Calculated for CzzH29NO,S (M+Na) 474.1557; Found 474.1547.
Example 4
1-(tent-Butoxycarbonyl)-4-( f4-(2-butynyloxy)phen~llsulfonyl ~-4-
piperidinecarboxylic acid
O O\SO
HO
~ O
N \
O O
The general procedure for step 3 was followed using the product from
Example 3 (15g, 33.2 mmol) in water (100 ml), methanol (50 ml),
tetrahydrofuran
(50 ml) and lithium hydroxide hydrate (2.73g, 66.4 mmol) at reflux temperature
for 8
hours to obtain 14.5g (100%) of the acid as a white powder.
'H NMR(300 MHz, DMSO-db):8 1.38 (s, 9H), 1.7 - 1.8 (m, 2H), 1.85 (t, 3H, J =
2.2
Hz), 2.2 - 2.3 (m, 2H), 2.5 - 2.7 (m, 2H), 3.95 - 4.05 (m, 2H), 4.89 (q, 2H, J
= 2.2
Hz), 7.1 - 7.8 (m, 4H); MS -ES: m/z 482 (M-H);
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-29-
Analysis for C~,HZ~NO,S): Calculated: C, 57.65; H, 6.22; N, 3.20;
Found: C, 57.59; H, 6.49; N, 3.20.
Example 5
tert-Butyl4-(f4-(2-but_ynylox )~phenyllsulfonyll-4-f(hydroxyamino)carbonyll-1-
piperidinecarboxalate
O O..SO
HO,N
H
O
N \
O O
The general procedure for step 4 was followed using dimethylformamide
(3.53 ml, 46 mmol), oxalyl chloride (22.9 ml of a 2.0M solution in
dichloromethane),
the product from Example 4 (10g, 22.9 mmol), hydroxylamine hydrochloride (16g,
229 mmol), and triethylamine (48 m, 344 mmol) to obtain the product as a white
powder 6.3g (61 °70).
'H NMR(300 MHz, DMSO-db):8 1.38 (s, 9H), 1.6 - 1.7 (m, 2H), 1.85 (t, 3H, J =
2.2
Hz), 2.2 - 2.3 (m, 2H), 2.5 - 2.7 (m, 2H), 3.9 - 4.0 (m, 2H), 4.87 (q, 2H, J =
2.2 Hz),
7.1 - 7.7 (m, 4H);
MS-ES: m/z 453 (M+H)+.
Example 6
4-; f4-(2-Butynyloxy)nhenyllsulfonyll-N-hydroxy-4-piperidinecarboxamide
hydochloride
HO,
N
H
To a solution of product from Example 5 (6.3g, 13.9 mmol) in methylene
chloride was added 4N hydrochloric acid in dioxane. After 6 hours the reaction
mixture was concentrated in vacuo. Methanol was added and the resulting
mixture
was concentrated in vacuo. Methylene chloride was added and removed in vacuo
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-30-
(2X). Trituration with diethyl ether gave the product as a white powder 5.148.
'H NMR(300 MHz, DMSO-db):8 1.86 (t, 3H, J = 2.2 Hz), 2.0 - 2.7 (m, 8H), 4.89
(q,
2H, J = 2.2 Hz), 7.1 - 7.8 (m, 4H), 8.8 - 11.0 (m, 4H);
MS - ES: m/z 353 (M+H)+.
Example 7
4-(4-Chlorophenox )~phenylsulfonyl fluoride
CI / \ O / \ OS
The general procedure for the preparation of sulfonyl fluorides was followed
using 4-(4-Chlorophenoxy)phenylsulfonyl fluoride (770 mg, 2.54 mmol) and
potassium fluoride-calcium fluoride mixture (1.478. 2 equiv) to obtain 660 mg
(91%)
of the product.
IR: 1599, 1579, 1484, 1395, 1258, 1210, 1183, 768 cm-'; 'H NMR(300 MHz,
CDC13):8 7.03-7.13 (m, 4H), 7.38-7.43 (m, 2H), 7.93-8.00 (m, 2H);
'3C NMR(75 MHz, CDC13):8 117.6, 117.7, 122.0, 129.7, 130.5, 131.1, 152.9,
163.7;
MS - ES: m/z 285.9.
Example 8
1-(tert-Butyl) 4-methyl 4- ( f 4-(4-chlorophenoxy)phenyll sulfonyl ) -1,4-
piperidinedicarboxylate
O\ "
Me0 O O \ / O \ / CI
N
i
Boc
The general procedure for step 1 was followed using lithium diisopropylamide
(2.31 mmol), the product from Example 1 (510 mg, 2.1 mmol), and the product
from
Example 7 (600 mg, 2.2 mmol) to obtain 520 mg (49%) of the product as a solid.
IR: 1727, 1682, 1485, 1427, 1252, 1153 cm';
'H NMR(300 MHz, CDCl3):8 1.44 (s, 9H), 1.97-2.07 (m, ZH), 2.29-2.33 (m, 2H),
2.62 (br s, 2H), 3.76 (s, 3H), 4.08-4.15 (m, 2H), 7.01-7.07 (m, 4H), 7.36-7.42
(m,
2H), 7.68-7.73 (m, 2H);
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-31-
'3C NMR(75 MHz, CDCI~):8 28.3, 53.3, 72.6, 80.2, 117.1, 121.9, 128.5, 130.4,
130.7, 132.6, 153.2, 154.4, 162.8, 167.4;
HR - MS: m/z Calculated for C,QH~8C1NO,S (M+Na) 532.1167; Found 532.1152.
Example 9
1-(tert-Butoxycarbonyl)-4-( [4-(4-chlorophenox~phenyllsulfonyl ~-4-piperidine
carboxxlic acid
O~"
HO O O \ / O \ / CI
N
i
Boc
The general procedure for step 3 was followed using the product from
Example 8 (450 mg, 0.88 mmol) and lithium hydroxide (32 mg, 1.32 mmol) in
tetrahydrofuran (3 ml)/methanol (3 ml)/water (2 ml) at 55°C for 15
hours to obtain
375 mg (86%) of the product.
IR: 3438, 2976, 1693, 1627, 1484, 1248, 1139 cm~';
'H NMR(300 MHz, DMSO-db):8 1.38 (s, 9H), 1.55-1.64 (m, 2H), 2.09 (s, 1H), 2.13
(s, 1H), 2.68 (br s, 2H), 3.39 (br s, 1H), 3.90 (m, 2H), 7.06 (d, 2H, J = 9.0
Hz), 7.16
(d, 2H, J= 12.0 Hz), 7.52 (d, 2H, J = 12.0 Hz), 7.70 (d, 2H, J = 9.0 Hz);
'3C NMR(75 MHz, DMSO-d~):8 116.6, 121.9, 128.8, 130.2, 131.2, 132,7, 153.7,
153.8, 160.7, 165.2;
HR - MS: m/z Calculated for C~3H26C1NO,S(2M + H) 991.2311; Found 991.2273.
Example 10
1-(tert-Butyl)-4-( [4-(4-chlorophenoxy)phenyllsulfonyl l-4-
[(hydro~amino)carbonyll-1-piperidinecarbox
O O\ ~O
HO,N S \ / O \ / CI
N
i
Boc
The general procedure for step 4 was followed using the product from
Example 9 (350 mg, 0.71 mmol) in dimethylformamide (7 ml), hydroxybenzotriazol
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-32-
(114 mg, 0.85 mmol), 1-(3-dimethylaminopropyl)-3-ethylcarbodimide
hydrochloride
(190 mg, 0.99 mmol), N-methylmorpholine (117 uL, 1.06 mmol), and 50°70
aqueous
hydroxylamine (217 uL, 3.55 mmol) to obtain 150 mg (41 %) of the product.
IR: 3739, 3382, 2931, 1664, 1484, 1249, 1150 cm'.
'H NMR(300 MHz, DMSO-db):8 1.38 (s, 9H), 1.68 (m, 2H), 2.14 (m, 2H), 2.51 (m,
2H), 3.95 (m, 2H), 7.14 (d, 2H, J = 9 Hz), 7.20 (d, 2H, J = 9 Hz), 7.55 (d,
2H, J = 9
Hz), 8.01 (d, 2H, J = 9 Hz), 9.20 (s, 1H), 11.02 (s, 1H);
'3C NMR(75 MHz, DMSO-db):8 27.9, 70.0, 79.2, 117.3, 122.2, 128.4, 129.2,
130.4,
132.8, 153.3, 153.7, 160.2, 161.8;
HR - MS:m/z Calculated for C,3Hz,CINzO,S(2M + H) 1021.2527; Fund 1021.2523.
Example 11
4-( f4-(4-Chlorophenoxy)phenyllsulfon~)-N-hydroxy-4-piperidinecarboxamide
HO.N O O~SO ~ / O ~ / CI
H
N
H .HCI
To a solution of product from Example 10 ( 105 mg, 0.21 mmol) in methylene
chloride (20 ml) was added a 4M hydrochloric acid solution (258 ~,L, 1.03
mmol) and
the resulting mixture was stirred for 4 hours at room temperature. The solvent
was
removed and diethyl ether was added. The precipitated solid was filtered and
dried to
obtain 80 mg (85°10) of the product.
IR: 3392, 3214, 2875, 1664, 1484, 1250, 1142, 1087 cm';
'H NMR(300 MHz, DMSO):8 2.13 (m, 2H), 2.46 (m, 2H), 2.59 (m, 2H), 3.33 (m,
2H), 7.19 (m, 4H), 7.52 (d, 2H, J = 9.0 Hz), 7.72 (d, 2H, J = 9.0 Hz), 9.19
(br s, 1H),
9.56 (br s, 1H);
HR-MS: m/z Calculated for C,$H,9C1NZOSS(M + H)) 411.0776; Found 411.0777.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-33-
Example 12
Piperidine-1 3-dicarboxylic acid 1-tert-butyl 3-ethyl ester
0 0
~O N~O
To a stirred solution of ethyl nipecotate (5.1g, 33 mmol) in methylene
chloride (75 ml) and triethylamine (3.7g, 36 mmol) was added in portions di-t-
butyldicarbonate (7.1g, 33 mmol). The reaction mixture was stirred at room
temperature for 18 hours, quenched with ice water and extracted with
chloroform.
The organic layer was dried over sodium sulfate, filtered, concentrated and
chromatographed on a silica-gel column with 20:80 ethyl acetate:hexane.
Piperidine
l,3dicarboxylic acid 1-tert-butyl ester-3-ethyl ester was isolated as a waxy
solid, 6.86
g ( 82%).
'H NMR(300 MHz, CDC13):8 1.26 (t, 3H), 1.46 (s, 9H), 1.63 (m, 2H), 2.03 (m,
1H),
2.41 (m, 1H), 2.76 (m, 2H), 3.89 (m, 1H), 4.14 (m, 2H);
MS - ES:m/z 258.2 (M+H)+;
Analalysis for C,3H23NO4, Calculated: C, 60.68; H, 9.08; N, 5.44
Found: C, 60.60; H, 9.10; N, 5.38.
Example 13
4-Methoxyphenylsulfonyl fluoride
Me0 ~ ~ S02F
The general procedure for the preparation of sulfonyl fluorides was followed
using 4-methoxyphenylsulfonyl chloride (ll.Og, 53 mmol) and potassium fluoride-
calcium fluoride mixture (17.0g) in acetonitrile (100 ml) to obtain
10.0g(100%) of
the product.
MS - ES: m/z 187.0(M-H)-.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-34-
Example 14
1-(tert-Butyl)-3-ethyl-3-f(4-methoxyphen~)sulfon 1y 1-1 3-piperidine
dicarboxylate
~O N~O
02 O
The general procedure for step 1 was followed using lithium diisopropyl-
amide (28 mmol), product from Example 12 (5.3g, 28 mmol), and product from
Example 13 (5.3g, 28 mmol) to obtain 7.2g (60%) of the product.
'H NMR(300 MHz, DMSO-d~):8 1.15 (t, 3H), 1.44 (s, 9H), 1.69 (m, 2H), 2.14 (m,
2H), 3.17 (m, 2H), 3.35 (d, 2H), 3.8 (s, 3H), 4.06 (m, 2H), 7.19 (d, 2H), 7.69
(d, 2H);
MS - ES: m/z 428.5 (M+H)+;
Analysis for CzoHZ9NO,S Calculated: C, 56.19; H, 6.84; N, 3.28
Found: C, 56.84; H, 7.20; N, 3.48.
Example 15
Ethyl 3-f (4-methoxy~henyl)sulfonyll-3-piperidinecarboxylate
~O / NH
02 O
To a stirred solution of product from Example 14 ( 1.728, 4.0 mmol) in
methylene chloride(25 ml) at 0° C was added a saturated solution of
hydrogen
chloride in methylene chloride (25 ml). After 5 hours the solution was
concentrated
to afford 1.238 (84.5%) of the product.
MS - ES: m/z 328.3 (M+H)';
Analalysis for C,SHz,NOSS Calculated: C, 49.51; H, 6.09; N, 3.85
Found: C, 47.91; H, 7.08; N, 4.16;
'H NMR(300 MHz, DMSO-db ):8 1.09 (t, 3H), 2.29 (d, 2H), 2.99 (m, 2H), 3.07 (m,
2H), 3.72 (d, 2H), 3.89 (s, 3H), 4.11 (m, 4H), 7.22 (d, 2H), 7.72 (d, 2H).
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-35-
Example 16
EthXl 1-benzyl-3-f (4-methoxv)sulfon~ll-3-piperidinecarbox~ate
i
,O , N
~ ~ S O~
02 O
A solution of product from Example 15 (1.23g, 3.4 mmol), benzyl bromide
(0.64g, 3.7 mmol) and dry powdered potassium carbonate (3.8g) in dry acetone
(60
ml) was heated at reflux temperature for 18 hours. The mixture was cooled and
the
potassium salts were removed by filtration and the filtrated was concentrated.
The
residue was dissolved in chloroform, washed with water, dried over sodium
sulfate
and concentrated to afford 1.8g (94%) of the product as a yellow oil.
'H NMR(300 MHz, DMSO-db):8 1.04 (t, 3H), 2.71 (m, 2H), 3.39 (m, 3H), 3.54
(m, 2H), 3.38 (m, 4H), 3.92 (s, 3H), 4.02 (m, 4H), 4.54 (s, 2H); 7.13 (d, 2H),
7.21 (d,
2H) , 7.29 (d, 2H), 7.62 (d, 2H);
MS - ES: m/z418.5 (M+H)'.
Example 17
1-Benzyl-3-(4-methoxybenzenesulfonyl)-piperidine-3-carboxylic acid
~O / N
I S OH
~2 0
The general procedure for step 3 was followed using the product from
Example 16 (1.7g, 4.0 mmol), sodium hydroxide (10N, 3m1), methanol (10 ml) and
tetrahydrofuran (10 ml) at 50°C for 2 hours to obtain 1.13 g (67%) of
the product, mp
103°C.
'H NMR(300 MHz, DMSO-db):8 1.04 (t, 3H), 2.71 (m, 2H), 3.36 (m, 6H), 3.55 (m,
2H), 3.85 (3, 3H), 7.12 (d, 2H), 7.27 (d, 2H) , 7.64 (d, 2H), 7.77 (d, 2H);
MS - ES: m/z 344.4 (M-H) -COZ.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-36-
Example 18
1-Benzyl-3-(4-methoxybenzenesulfonyl)piperidine-3-carboxylic acid hydroxamide
I
~O / N
\ I S NHOH
02 O
To a stirred solution of product from Example 17 (1.g, 2.9 mmol) and
dimethylformamide ( 5 ml) in methylene chloride (30 ml) at O°C was
added,
dropwise, oxalyl chloride ( 1.8gm, 14.5 mmol). After the addition, the
reaction
mixture was stirred at room temperature for 1 hour. Simultaneously, in a
separate
flask a mixture of hydroxylamine hydrochloride ( 1.6gm, 23 mmol) and
triethylamine
(3 ml, excess) was stirred in tetrahydrofuran:water (5:1, 30 ml) at O°C
for 1 hour. At
the end of 1 hour, the oxalyl chloride reaction mixture was concentrated and
the pale
yellow residue was dissolved in 10 ml of methylene chloride and added slowly
to the
hydroxylamine solution at O°C. The reaction mixture was stirred at room
temperature for 24 hours and concentrated. The residue obtained was extracted
with
chloroform and washed well with water. The product obtained was purified by
silica
gel column chromatography; eluted with 2°Io methanol:chloroform. The
product was
converted to the hydrochloride salt by dissolving in methanol (10 ml) at
5°C and
adding saturated hydrogen chloride in methanol (5m1). 1-Benzyl-3-(4-methoxy-
benzenesulfonyl)-piperidine-3-carboxylic acid hydroxamide propionamide was
isolated as a white solid, 1.17g,.(91°Io), mp 132.9°C.
'H NMR(300 MHz, DMSO-db):8 1.08 (m, 23H), 2.49 (m, 2H), 3.87(s, 3H), 4.25 (d,
2H),
7.10 (d, 2H), 7.44 (s, 5H), 7.58 (d, 2H), 8.85 (s, 1H), 9.45 (s, 1H);
MS - ES: m/z 405.3(M+H)+.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-37-
Example 19
1-(tert-Butyl) 4-ethyl4-(f4-2-butynyloxy~~henyllsulfonyl)-1,4-piperidine
dicarboxylate
~O N~O
OZ O
The general procedure for the preparation of sulfonyl fluorides was
followed using lithium diisopropylamide (20 mmol), product from Example 1
(4.4g,
19.5 mmol), and product from Example 12 (5.0g, 19.5 mmol) to obtain 10.97g
(76°Io)
of the product, mp 103.4 °C.
'H NMR (300 MHz, DMSO-db):8 1.07 (t, 3H), 1.34 (s, 9H), 3.31 (s, 3H), 3.84 (m,
2H), 4.00 (m, 4H), 4.53 (d, 2H), 4.91 (m, 4H), 7.22 (d, 2H), 7.71 (d, 2H);
MS - ES: m/z 466.4(M+H)+;
Analysis for C23H3,C1NO,S ) Calcuclated: C, 59.34, H, 6.71; N, 3.01
Found: C, 59.49; H, 6.84; N, 3.16.
ExamRle 20
Ethyl 3-( f4-(2-butynyloxy)phenyllsulfonyl l-3-piperidinecarboxylate
~O , NH
02 O
Following the procedure of Example 15, using the product from Example 19
(5.458, 11.7 mmol) in dissolved in methylene chloride, the desired product was
obtained as a white solid 3.47g (74°Io). The solid is very hydroscopic
and is store
under nitrogen.
'H NMR(300 MHz, DMSO-db):8 1.08 (t, 3H), 2.30 (bd, 1H), 2.96 (t, 2H), 3.07 (m,
2H), 3.33 (s, 3H), 3.38 (m, 4H), 4.09 (m, 2H), 4.93 (s, 2H), 7.26 (d, 2H),
7.74 (d,
2H);
MS - ES:m/z 366.2 (M+H)+;
Analysis for C,gH2305S Calculated: C, 53.79; H, 6.02; N, 3.49
Found: C, 52.34; H, 6.17; N, 3.52.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-38-
Example 21
Ethyl 3-( f4-(2-butynyloxy)pheny11su1fonXl )-1-ethyl-3-piperidinecarboxylate
o , NJ
02 0
Following the procedure of Example 16, using the product from Example 20
(2.97g, 8.0 mmol) in dry acetone (50 ml), the desired product was isolated as
an
amber gum, 3.47g (99%).
'H NMR(300 MHz, DMSO-db):8 0.89 (t, 3H), 1.05 (t, 3H), 2.72 (d 2H), 3.28 (m,
2H), 3.31 (s, 3H), 4.01 (m, 4H), 4.91 (m, 2H), 7.19 (d, 2H), 7.70 (d, 2H);
MS - ES: m/z 394.3 (M+H)'.
Example 22
3-(f4-(2-Butyn~oxy)phenyllsulfonyl)-1-ether-3-piperidinecarboxylicacid
NJ
i S off
p
The general procedure for step 3 was followed using the product from
Example 21 (3.2g, 8.0 mmol) in tetrahydrofuran:methanol (15:25 ml), and sodium
hydroxide (15 ml) at 50°C for 2 hours to obtain 2.11g (71%) of the
product as a white
solid: mp 159.2°C.
'H NMR (300 MHz, DMSO-db):8 1.02 (t, 3H), 2.70 (m, 4H), 2.92 (d, 2H), 3.47 (d,
2H), 4.865 (m, 2H), 7.09 (d, 1H), 7.17 (d, 1H) , 7.60 (d, 1H), 7.68 (d, 1H);
MS- ES: m/z 366.3 (M+H)';
Analysis for C,gHz305 Calculated: C, 59.16; H, 6.34; N, 3.83
Found: C, 59.2; H, 6.45; N, 3.67.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-39-
Example 23
3- ( f 4-(2-Butynyloxy)phenyll sulfonyl ~ -1-ethyl-N-h~drox~-3-piperidi
necarboxamide
~O , NJ
NHOH
02 O
Following the procedure of Example 18, using the product from Example 22
(2.0g, 5.5 mmol), 0.193g (10%) of the desired product was isolated as a white
solid,
mp 190°C.
'H NMR(300 MHz, DMSO-db):8 1.18 (m, 3H), 1.97 (m, 2H), 2.55 (m, 2H), 3.21(m,
SH), 3.52 9S, 3H), 3.82 (d, 1H), 4.91 (m, 2H), 7.19 (d, 2H), 7.51 (s, SH),
8.67 (s, 1H),
9.48 (s, 1H); MS - ES: m/z 405.3 (M+H)'.
Analysis for C'8Hz4N205S Calculated: C. 51.86; H, 6.04; N, 6.72
Found: C, 50.03; H, 6.33; N, 6.42.
Example 24
Ethyl 3-( f4-(2-butynyloxy)phenyllsulfon~)-1-(4-chlorobenzyl)-3-
piperidinecarboxylate
ci
I
~O ~ ~N
~ I S Ow/
02 O
Following the procedure of Example 16, using the product from Example 20
(2.97g, 8.0 mmol) in dry acetone (50 ml), 1.66g (99%) of the product was
isolated as
a brown oil.
MS - ES: m/z 491.3 (M+H)'.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-40
Example 25
3-~(f4-(2-Butynyloxy)phenyllsulfonyl)-1-(4-chlorobenz l~)-3-piperidinecarbox
acid
ci
I
=~O ~ ~N
OH
02 O
The general procedure for step 3 was followed using the product from
Example 24 (1.64g, 8.0 mmol) in tetrahydrofuran:methanol (15:50 ml) and sodium
hydroxide (15 ml) at 50°C for 2 hours to obtain 1.1 1g (75%) of the
product as a white
solid, mp 115.2°C.
'H NMR(300 MHz, DMSO-d 6):8 2.33 (d, 2H), 2.7 (d, 2H), 3.29 (s, 32H), 3.33 9m,
2H), 3.52 (q, 2H), 4.47 (s, 2H), 4.81 (s, 2H), 7.16 (d, 2H), 7.27 (d, 2H) ,
7.34 (d, 2H),
7.67 (d, 2H);
MS-ES: m/z 462.1 (M+H)';
Analysis for C23Hz4C1NO5S Calculated: C, 59.16; H, 6.34; N, 3.83
Found: C, 59.64; H, 5.65; N, 2.66.
Example 26
3-( f4-(2-Butyn~oxy)phen~lsulfonyl )-1-(4-chlorobenzyl)-N-hvdrox
piperidinecarboxamide
ci
I
~O ~ ~N
NHOH
~2 0
Following the procedure of Example 18, using the product from Example 25
(2.0g, 5.5 mmol), 0.48g (43%) of the product was isolated as a white solid, mp
124.4°C.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-41 -
'H NMR(300 MHz, DMSO-db):8 2.0 (m, 2H), 339 (m, 5H), 4.27 (d, 2H), 4.89 (m,
2H), 7.14 (d, 42H), 7.15 (m, 45H), 7.61 (d, 2H), 8.95 (s, 1H), 9.46 (s, 1H);
MS - ES: m/z 477.1 (M+H)+;
Analysis for C,BH,,N~OSS Calculated: C, 53.8; H, 5.10; N, 5.46
Found: C, 51.4; H, 5.42; N, 6.32.
Example 27
Sodium-4-butox~benzenesulfonic acid
ors o
0
Na0
To a suspension of sodium-4-hydroxybenzenesulfonic acid (40.0g, 0.172
mol) in 2-propanol (300 ml) was added 1N sodium hydroxide ( 190 ml, 0.189 mol
j.
After 10 minutes n-butylbromide (38.98, 0.28 mol) was added and the hazy
solution
was heated at reflux temperature. The reaction mixture was partially
evaporated,
filtered, washed with diethyl ether, and dried to give 38.6g (88.4°1o)
of the product.
'H NMR(300 MHz, DMSO-db): b 7.5 (d, 2H, J=8.7Hz), 6.83 (d, 2H, J=8.7Hz), 3.96
(t, 2H, J=6.5Hz), 1.68 (m, 2H), 1.42 (m, 2H), 0.92 (t, 3H, J=7.4Hz);
HPLC: 99.94% area; LC-MS: consistent.
Example 28
4-n-Butoxybenzenesulfonyl chloride
oso
0
m
To the product from Example 27 (34.0g, 0.134 mol) was added phosphorous
oxychloride (60 ml, 0.643 mol) and the heterogeneous mixture was heated at
reflux
temperature ( 105 ° C) for 4 hours. After 4 hours the reaction mixture
was cooled to
ambient temperature and ice water (600 ml) was added while stirring. The
mixture
was extracted with diethyl ether. The organic layer was washed with water (200
ml),
saturated sodium bicarbonate solution (200 ml) and water (200 ml). The organic
layer was dried over anhydrous sodium sulfate and the solvent was removed to
give
33.3g (99.3°1o) of the product as a colorless liquid.
CA 02398561 2002-07-24
WO 01/55112 PCT/USOl/02669
-42-
'H NMR(300 MHz, CDCl3):8 7.96 (d, 2H, J=6.0 Hz), 7.02 (d, 2H, J=6.0 Hz), 4.07
(t,
2H, J=4.3 Hz), 1.82 (m, 2H), 1.51 (m, 2H), 0.99 (t, 3H, J=4.9 Hz);
GC-MS: 100% pure.
Example 29
4-n-Butoxybenzenesulfonyl fluoride
o~ ,o
~S ~ ~ o
F
To a solution of product from Example 28 (33.3g, 0.134 mol) in acetonitrile
(200 ml) was added potassium fluoride on calcium fluoride (85.8g, 0.298 mol)
and
the resulting heterogeneous mixture was stirred at ambient temperature for 20
hours.
The reaction mixture was filtered, washed with acetonitrile (20 ml x 2) and
evaporated. The oily residue was dissolved in ethyl acetate (200 ml), washed
with
saturated sodium chloride solution (200 ml), dried over anhydrous sodium
sulfate and
concentrated to give 30.4g, (98%) of the product as a clear colorless liquid.
'H NMR(300 MHz, CDC13):8 7.92 (d, 2H, J=6.0 Hz), 7.04 (d, 2H, J=6.0 Hz), 4.06
(t,
2H, J=4.3 Hz), 1.82 (m, 2H), 1.5 (m, 2H), 0.99 (t, 3H, J=4.9 Hz);
GC-MS: 97.6% pure, 2.4% starting material.
Example 30
1-f4-(2-Piperidin-1-yl-ethox )~benzyllpiperidine ~-carboxylic acid meth liter
'N
H3C02C ~ O~ N
A mixture of methyl isonipecotate (5.0g, 34.9 mmol), 4-piperidine ethoxy
benzyl chloride hydrochloride ( 10.13g, 34.9 mmol) and potassium carbonate (
10.68,
76.6 mmol, -325 mesh) in acetone was heated at reflux temperature for 24
hours.
After cooling to room temperature, the reaction was filtered, washed with
acetone (25
ml x 3), and evaporated to give a light brown oil. The oil was dissolved in
ethyl
acetate ( 100 ml), washed with water ( 100 ml x 2), saturated sodium chloride
solution
(100 ml), dried over sodium sulfate and concentrated to afford 9.0g (72%) of
the
product as a light brown oil.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
- 43 -
'H NMR(300 MHz, CDCl3):8 7.19 (d, 2H, J=5.7 Hz), 6.84 (d, 2H, J=5.7 Hz), 4.09
(t,
2H, J=4.06 Hz), 3.66 (s, 3H), 3.41 (s, 2H), 2.8 (m, 4H), 2.51 (m, 4H), 2.3 (m,
1H),
2.02 (m, 2H), 1.87 (m, 2H), 1.76 (m, 2H), 1.51 (m, 4H), 1.45 (m, 2H);
GC-MS: 94.1 % pure.
Example 31
4-(4-Butoxybenzenesulfon~)-1-f4-(2-piperidin-1-yl-ethox )b~enz-yllpiperidine-4-
carboxylic acid methyl ester
H3COOC o
NJ
N
O'~
To a O°C solution of diisopropylamine (0.67 ml, 4.8 mmol) in
tetrahydrofuran (6m1) at was added n-butyllithium (2.0 ml, 2.5M in hexane).
The
resulting mixture was stirred for 20 minutes, cooled to -78°C and a
solution of
product from Example 30 (1.5g, 4.16 mmol) in tetrahydrofuran (6ml)was added
dropwise. After 1 hour at-78°C, the product from Example 3(1.01g, 4.36
mmol) in
tetrahydrofuran (4m1) was added in one portion and the mixture warmed to
ambient
temperature. After 3 hours, the reaction mixture was quenched with saturated
ammonium chloride solution (8ml)and extracted with ethyl acetate (20 ml x 2).
The
organic layer was washed with water (30 ml) and saturated sodium chloride
solution
(30 ml), dried over sodium sulfate and concentrated to give 2.24g of the crude
product as a brown syrup.
MS - ES: m/z 573 (M+H)'.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-44-
Example 32A
1-Benz~piperidine-4-carboxylic acid ethyl ester
COOEt
NJ
Ph
To a solution of ethyl isonipecotate (72.8g, 0.45 mol) in ethanol (150 ml) was
added benzyl bromide (lOlg, 0.59 mol), dropwise, at 0 to 10 °C,
followed by
triethylamine (68.8g, 0.68 mol). The resulting suspension was warmed to
ambient
temperature and stirred for 6 hours. The reaction mixture was diluted with
water
(200 ml) and extracted with ethyl acetate (3 X 150 ml). The organic layer was
dried
over anhydrous magnesium sulfate, filtered through silica pad, and
concentrated to
afford 98.6g (89 %) of the product as a yellow viscous liquid.
'H-NMR(CDC13):S 7.9-7.0 (m, 5H), 4.4-4.1(q, 2H), 3.5 (s,2H), 2.9-2.8 (m, 4H),
2.6-
2.3 (m, 1H), 2.1-1.6 (m, 4H), 1.3-1.2 (t, 3H);
GC-MS: 99.4 % pure.
Example 32B
1-Benzxlpiperidine-4-carboxylic acid methxl ester
COOMe
NJ
Ph
The above named compound was prepared from methyl isonipecotate in
methanol using the procedure of Example 32A (yield 97 %);
'H-NMR(CDC13):8 7.7-6.9 (m, 5H), 3.7 (s, 3H), 3.5 (s, 2H), 3.0-2.8 (m, 4H),
2.4-2.2
(m, 1H), 2.1-1.6 (m, 4H);
GC - MS: 92 % pure.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
- 45 -
Example 33
4-f4-(4-Chlorophenoxy)benzenesulfonyll-1-benzylpiperidine-4-carboxylic acid
methyl ester
O
CI / / S02 COOMe
NJ
Ph
Prepared with LDA as base:
Freshly distilled diisopropylamine (1.58g, 15.6 mmol) was dissolved in
tetrahydrofuran (18 ml) and cooled to 0°C. A solution of 2.5M n-butyl
lithiumi in
hexane (S ml, 12.5 mmol) was added at a temperature below 5°C and the
resulting
yellow solution was stirred for 0.5 hour while cooling to -20°C. A
solution of product
from Example 32B ( 1.46g, 6.25 mmol) in tetrahydrofuran (5 ml) was added,
dropwise, at -20°C and the resulting mixture was stirred for 2 hours. A
solution of
product from Example 6 in tetrahydrofuran (5 ml) was added at -20 to -
25°C and the
dark yellow reaction mixture was stirred for lhour at -20°C. The
mixture was
quenched with saturated ammonium chloride (20 ml) and extracted with ethyl
acetate
(3 X 15 ml). The organic solution was dried with magnesium sulfate, filtered
through
silica pad and concentrated to a small residual volume. The residue was
triturated
with isopropyl ether (10 ml) to produce 1.73g (69%) of the product as yellow
gummy
crystals.
'H-NMR(CDC13):8 7.8-7.0 (m, 13H), 3.7 (s, 3H), 3.4 (s, 2H), 3.0-1.8 (m, 8H);
HPLC: 87 % pure.
Prepared with LiHMDS as base:
A solution of product from Example 32B (2g, 6 mmol) in tetrahydrofuran (15
ml) was cooled to -20 to -22°C under a nitrogen atmosphere. A solution
of lithium
hexamethyldisilazide (LiHMDS) (1.0M in THF, 7.2 ml, 7.2 mmol) was added,
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
- 46 -
dropwise, maintaining the temperature at -20 to -22°C. After the
addition, the
solution was stirred at -20 to -22°C for 2 hours. A solution of product
from Example
7 (2.26g, 8 mmol) in tetrahydrofuran (10 ml) was added, dropwise, at -20 to -
22°C.
The reaction was stirred for an additional 2.5 hours while maintaining the low
temperature. The mixture was quenched with saturated ammonium chloride ( 15
ml)
and extracted with ethyl acetate (3 X 10 ml). The organic extract was dried
over
anhydrous magnesium sulfate, filtered through silica pad and concentrated to a
small
residual volume. n-Heptane (10 ml) was add and the solution was left overnight
at
room temperature to produce 2.4g (69%) of the product as white crystals.
HPLC: 90 % pure.
Example 34
4-f4-(4-Chlorophenoxy)benzenesulfonyll-1-benzylpiperidine-4-carboxylic acid
CI
N
Ph
To a solution of product from Example 33 (30.7g, HPLC 99.4 area %, 123
mmol) in t-butylmethylether (100 ml) at -25°C was added a 2M lithium
diisopropyl-
amide solution (136 ml, 272 mmol) over a period of 15-20 minutes maintaining
the
temperature between -20 and -25°C. The yellow solution was stirred at
this
temperature for 2 hours. A solution of product from Example 7 (108 ml, 136
mmol)
in tetrahydrofuran was added over a period of 15 minutes at -20°C and
the reaction
was stirred for an additional hour while maintaining the low temperature. The
reaction progress was monitored by thin layer chromatography, showing the
formation of intermediate ester, Example 8. The reaction mixture was quenched
with
water and warmed to 20 - 25°C while stirring for 0.5 hour. The organic
solvent was
removed by distillation (50 mm Hg, 35°C) forming an oily layer on the
bottom of the
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-47-
flask. Lithium hydroxide (15.5g, 370 mmol) and methanol (150 ml) were added
and
the reaction mixture was heated at reflux temperature overnight (70
°C). The reaction
mixture was clarified by filtration through filter paper to remove a small
amount of
gel-like insoluble material. The clarified solution was acidified with acetic
acid (30
ml) at 20-25 °C to pH = 5. Resulting slurry was stirred for 1 hour at
ambient
temperature and filtered. The solid residue was washed with water, slurried
with
ethanol (500 ml) for 0.5 hour, filtered and dried in vacuo at 40°C to
afford 36.48
(61 % HPLC) of the desired product as a yellow solid.
MS - ES: m/z 486 (M+H)'.
Example 35
4-f4-(4-ChlorophenoxX)benzenesulfonyll-1-ben~lpiperidine-4-carboxylic acid
hvdroxamide
HOHNOC ~ S ~ ~ ~ o
N / CI
To a stirred suspension of the product from Example 34 (122.0g, 0.251 mol)
in acetonitrile ( 1.0 L) with a catalytic amount of dimethylformamide ( 1.0
ml) at 0°C
(ice bath) was added oxalyl chloride (55.1g, 0.402 mol) over a period of 30
minutes
(CAUTION: Gas evolution). The cooling bath was removed and the mixture was
stirred at room temperature for 5 hour. (The reaction was monitored for
completion
by adding an aliquot of the reaction mixture to an excess of methanol followed
by
TLC, MS or HPLC). The acid chloride suspension was added, over a 20 minute
period, to a cooled solution of powdered hydroxylamine hydrochloride ( 175.0g,
2.51
mol) and triethylamine (330.9g, 3.27 mol) in acetonitrile (2.5 L), which had
been
stirnng for 3-5 hours at room temperature. The reaction temperature should not
exceed ~8° C. After stirnng at room temperature for 18 hours, the
reaction mixture
was concentrated to afford an off-white residue. To the residue ethyl acetate
(2.0 L)
and water (2.0 L) were added, and the mixture was stirred for 15-20 minutes.
The
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-48-
ethyl acetate layer was separated, filtered through anhydrous sodium
sulfateand
concentrated to give 130.48 (crude yield 103%) as a semisolid product.
'H NMR(DMSO-db, 300MHz):b 10.9 (br s, 1H); 9.1 (br s, 1H); 7.71 (d, 2H, J =
8.8
Hz); 7.52 (d, 2H, J = 8.8 Hz); 7.33-7.19 (m, 7H); 7.14 (d, 2H, J = 8.8 Hz);
3.4 (s, 2H);
2.7 (m, 2H); 2.28 (m, 2H); 1.95-1.8 (m, 4H);
HPLC: 94.06% product, (0.3% carboxylic acid and 2.88% mixed anhydride).
Example 36
4-f4-(4-Chloropheno~)benzenesulfonyll-1-benzX~iperidine-4-carboxylic acid
hydroxamide hydrochloride
HOHNOC o~s ~ ~ ~ o
N ' CI
HC1
The crude product from Example 35 (130.48, 0.260 mol) was dissolved in
ethyl acetate (350 ml) and conncentrated hydrochloric acid (31.3 ml, 0.313
mol) was
added over a 20 minute period. Salts precipitated out of solution and the
mixture was
cooled in an ice bath at 2°C for 30 minutes. The mixture was filtered,
washed with
cold (0°C) ethyl acetate (50 ml x 2), dried in an oven for 18 hours to
give the product
118.68, (85%). This compound was recrystallized as follows:
A 5-L flask fitted with reflux condenser, thermometer/controller, and
mechanical stirrer, was charged with ethanol (2.3 L, 200 proof) and the above
crude
product (118.6 g). The contents of the flask were heated at reflux
temperature, then
water (850 ml) was added over 60 minutes. The solution was clarified by
filtration
and reheated to boiling. The heating mantle was removed and the reaction
mixture
was cooled. Crystallization started at 60°C. The reaction was gradually
cooled in an
ice bath and kept at 2-4°C for 30 minutes. The white crystals were
collected, washed
with cold ethanol (100 ml x 2), dried in vacuo at 60°C with a nitrogen
bleed for
l8hours to give 89.238, (75%) of the desired product as crystals, m.p. 233-
235°C.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-49-
HPLC: 98.5% pure; 'H NMR(DMSO-db, 300 MHz):8 11.2 (s, 1H); 10.9 (br s, 1H);
9.35 (s, 1H); 7.73 (d, 2H, J = 8.8 Hz); 7.52 (m, 4H0; 7.44 (br s, 3H); 7.23
(d, 2H, J =
8.8 Hz); 7.17 (d, 2H, J = 8.8 Hz); 4.26 (s, 2H0; 2.78 (m, 2H); 2.30 (m, 2H);
IR (KBr pellet): 3700-3300, 3156, 2931, 2543, 1677, 1483, 1244, 1144, 1087,
598
cm'.
Example 37
4-(4-but-2-ynyloxy-benzenesulfon~l)-niperidine-4-carboxylic acid methyl ester
O O
O~"
Me0 S \ ~ O
N
H
To a solution of product from Example 3 (500 mg, 1.11 mmol) in methylene
chloride (10 ml) was added 4M HCl (2 ml) and the resulting mixture was stirred
for 2
hours at room temperature. The solid was filtered, washed with ether to obtain
410mg(95%) of the product as a solid.
IR: 3096, 2741, 2242, 1726, 1668, 1590, 1144, 836 cm-';
'H NMR(300 MHz, CDCl3):8 1.86(m, 3H), 2.52(m, 4H), 2.89(m, 2H), 3.52(m, 2H),
3.74(s, 3H), 4.74(m, 2H), 7.10(d, 2H, J= 8.7 Hz), 7.69(d, 2H, J= 8.7 Hz);
'3C NMR(75 MHz, CDCI,):8 3.6, 25.2, 41.2, 53.8, 56.9, 69.7, 72.6, 85.2, 115.4,
125.7, 132.3, 163.1, 166.6;
HR-MS:m/z Calculated for C"H~,NOSS 352.121; Found 352.1207.
Example 38
1-Acetyl-4-(4-but-2-ynyloxybenzenesulfon~piperidine-4-carboxylic acid methyl
ester
O O\ ~O
Me0 S \ ~ O
N
O~CH3
To a solution of product from Example 37 ( 105 mg, 0.23 mmol) in methylene
chloride ( 1 ml) was added triethylamine (93 mg, 0.92 mmol), acetyl chloride(
18 mg,
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-50-
0.23 mmol) followed by a catalytic amount of dimethylaminopyridine. The
resulting
mixture was stirred for 8 hours at room temperature, quenched with water and
extracted with methylene chloride. The organic layer was dried over anhydrous
sodium sulfate and concentrated to give 75 mg (80%) of the product as a solid.
IR: 2928, 2242, 1726, 1636, 1595, 1451, 1302, 1150, 996 cm';
'H NMR(300 MHz, CDC13):8 1.87(t, 3H, J= 2.4 Hz), 1.97-2.13(m, 2H), 2.09(s,
3H),
2.22-2.51(m, 3H), 3.02(m, 1H), 3.76(s, 3H), 3.89(m, 1H), 4.63(m, 1H), 4.74(q,
2H,
J= 2.4 Hz), 7.08(d, 2H, J= 7.5 Hz), 7.14(d, 2H, J= 7.5 Hz);
'3C NMR(75 MHz, CDCI~):8 4.1, 21.7, 28.4, 28.5, 38.9, 43.9, 53.7, 57.2, 72.7,
73.1,
85.5, 115.5, 126.9, 132.6, 163.1, 167.8, 169.2;
MS-ES: m/z 393.9 (M+H)'.
Example 39
1-Acetyl-4-(4-but-2-yn~~benzenesulfon~piperidine-4-carboxylic acid
O O
O~ "
HO S \ ~ O
N
O~CH3
General procedure for step 3 was followed using product from Example 38
(240 mg, 0.61 mmol) in 4 ml of tetrahydrofuran: water (3:1), and lithium
hydroxide
(18 mg, 0.75 mmol) to obtain 200 mg(87%) of the acid.
IR: 2923, 2246, 1713, 1591, 1575, 1494, 1232, 994 cm-'; 'H NMR(300 MHz,
acetone-db):8 1.84(t, 3H, J= 2.8 Hz), 1.90-2.05(m, 2H), 2.06(s, 3H), 2.25-
2.51(m,
3H), 3.06(m, 1H), 4.04(m, 1H), 4.63(m, 1H), 4.86(q, 1H, J= 2.4 Hz), 7.18(d,
2H, J=
8.4 Hz), 7.80(d, 2H, J= 8.4 Hz);
'3C NMR(75 MHz, CDCI,):8 3.3, 21.3, 28.7, 39.0, 44.0, 57.4, 72.8, 74.2, 85.0,
11.8,
128.3, 133.4, 163.5, 168.4, 169.0;
HR - MS: m/z Calculated for C,$HZ,NO6S 380.1162; Found 380.1160.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-51-
Example 40
1-Acetyl-4-(4-but-2-~nyloxvbenzenesulfonyl)piperidine-4-carboxylic acid
hydroxamide
O O\ O
HOHN \S \ ~ O
N
O~Me
The general procedure for step 4 was followed using product from Example
39 (180 mg, 0.48 mmol) in dimethylformamide (4 ml), 1-hydroxybenzotriazole (77
mg, 0.57 mmol), 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride
(127 mg, 0.66 mmol), N-methylmorpholine (0.078 ml, 0.71 mmol), and
hydroxylamine (0.145 ml, 2.37 mmol) to obtain 100 mg (53°70) of the
product as a
solid.
'H NMR(300 MHz, CDC13):8 1.64(m, 1H), 1.85(m, 3H), 1.99(s, 3H), 2.31(m, 4H),
2.83(m, 1H), 3.88(m, 1H), 4.41(m, 1H), 4.88(m, 2H), 7.16(d, 2H, J= 9.0 Hz),
7.66(d,
2H, J= 9.0 Hz), 9.20(m, 1H), 11.00(m, 1H); '3C NMR(75 MHz, CDC13):8 3.5, 21.5,
36.1, 56.8, 70.2, 74.3, 84.7, 115.3, 126.7, 132.6, 162.3, 168.6;
MS-ES: m/z395.2 (M+H)'.
Example 41
1-Benzoyl-4-(4-but-2-ynyloxybenzenesulfon~piperidine-4-carboxylic acid methyl
ester
O O\ ~O
Me0 S \ ~ O
N
o'
To a solution of product from Example 37 (400 mg, 1.03 mmol) m
chloroform ( 10 ml) was added triethylamine (416 mg, 4.12 mmol), benzoyl
chloride(144 p,1, 1.24 mmol) followed by a catalytic amount of dimethylamino-
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-52-
pyridine. The resulting mixture was stirred for 15 hours at room temperature,
quenched with water and extracted with methylene chloride. The organic layer
was
dried over anhydrous sodium sulfate and concentrated to give 375 mg (80%) of
the
product as a solid.
MS-ES: m/z 456.1 (M+H)+.
Example 42
1-Benzoyl-4-(4-but-2- n~yloxXbenzenesulfonyl)piperidine-4-carboxylic acid
O O _
O~"
HO S \ ~ O
N
O
The general procedure for step 3 was followed using product from Example
41 (300 mg, 0.66 mmol) in 4 ml of tetrahydrofuran:water (3:1), and lithium
hydroxide (18 mg, 0.75 mmol) to obtain 250 mg(86°Io) of the acid.
HR - MS: m/z Calculated for C23Hz3NO6S 442.1319; Found 442.1317.
Example 43
1-Benzoyl-4-(4-but-2-ynyloxybenzenesulfon~piperidine-4-carboxylic acid
h~roxamide
O O\ ~O -
HOHN S \ ~ O
N
O
The general procedure for step 4 was followed using product from Example
42 (100 mg, 0.23 mmol) in dimethylformamide (2 ml), 1-hydroxybenzotriazole (36
mg, 0.27 mmol), 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-53-
(62 mg, 0.32 mmol), N-methylmorpholine (0.038 ml, 0.35 mmol) and hydroxylamine
(0.083 ml, 1.15 mmol) to obtain 40 mg (38%) of the product as a solid.
MS-ES: m/z 457.2 (M+H)+.
Example 44
1-(4-Methoxybenzoyl)-4-(4-but-2-~~ybenzenesulfonyl)piperidine-4-carboxylic
acid methyl ester
O O
O~"
Me0 S \ ~ O
N
O'
OMe
To a solution of product from Example 37 (260 mg, 0.77 mmol) in
chloroform (7 ml) was added triethylamine (311 mg, 3.08 mmol), 4-
methoxybenzoyl
chloride (158 mg, 0.92 mmol) followed by a catalytic amount of dimethylamino-
pyridine. The resulting mixture was stirred for 15 hours at room temperature,
quenched with water and extracted with methylene chloride. The organic layer
was
dried over anhydrous sodium sulfate and concentrated to give 280 mg (75%) of
the
product as a solid.
HR - MS: m/z Calculated for Cz5H2,NO,S 486.1581; Found 486.1576.
Example 45
1-(4-Methoxybenzo~l)-4-(4-but-2-ynyloxybenzenesulfonyl)piperidine-4-carboxylic
acid
O O -
O~"
HO S \ ~ O
N
O
OMe
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-54-
The general procedure for step 3 was followed using product from Example
44 (250 mg, 0.52 mmol) in 4 ml of tetrahydrofuran:methanol (1:1) and 1N sodium
hydroxide (1.03 ml, 1.03 mmol) to obtain 150 mg(62%) of the acid.
HR - MS: m/z Calculated for CZ~H~SNO,S 472.1425; Found 472.1426.
Example 46
1-(4-Methoxybenzoyl)-4-(4-but-2-~~oxybenzenesulfonyl)piperidine-4-carboxylic
acid hvdroxamide
O O
O~"
HOHN S \ ~ O
N
O
OMe
The general procedure for step 4 was followed using product from Example
45 (90 mg, 0.19 mmol) in dimethylformamide (2 ml), 1-hydroxybenzotriazole (31
mg, 0.23 mmol), 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride
(51 mg, 0.27 mmol), N-methylmorpholine (0.031 ml, 0.28 mmol) and hydroxylamine
(0.068 ml, 0.95 mmol) to obtain 70 mg (76%) of the product as a solid.
HR - MS: m/z Calculated for Cz4HzbNzO,S 487.1534; Found 487.1531.
Example 47
4-(4-But-2-ynyloxybenzenesulfonyl)-1-(~yrrolidine-1-carbonyl)piperidine-4-
carboxylic acid methyl ester
O O
O~ "
Me0 S \ ~ O
N
O' _ N
To a solution of product from Example 37 (400 mg, 1.03 mmol) in
chloroform (10 ml) was added triethylamine (208 mg, 2.06 mmol), pyrrolidine-
carbonyl chloride (206 mg, 1.54 mmol) followed by a catalytic amount of
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-55-
dimethylaminopyridine. The resulting mixture was stirred for 15 hours at room
temperature, quenched with water and extracted with methylene chloride. The
organic layer was dried over anhydrous sodium sulfate and concentrated to give
400
mg (87°70) of the product as a solid.
MS-ES: m/z 449.3 (M+H)'.
Example 48
4-(4-but-2-yn~ybenzenesulfon l~~yrrolidine-1-carbonyl)-piperidine-4-
carboxylic acid
O O
O~ "
HO S \ ~ O
N
O' _ N
The general procedure for step 3 was followed using product from Example
47 (400 mg, 0.89 mmol) in 4 ml of tetrahydrofuran: methanol; water (1:1:0.5)
and
lithium hydroxide (48 mg, 2.0 mmol) to obtain 300 mg(78°Io) of the
acid.
MS-ES: m/z 435.2 (M+H)'.
Example 49
4-(4-But-2~nyloxybenzenesulfon 1y )-N-hydroxv-1-(pyrrolidine-1-carbon l~)-4-
piperidinecarboxamide
O O\ ~O
HOHN S \ ~ O
N
O' _ N
The general procedure for step 4 was followed using product from Example
48 (255 mg, 0.23 mmol) in dimethylformamide (6 ml), 1-hydroxybenzotriazole (96
mg, 0.71 mmol), 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride
( 157 mg, 0.82 mmol), N-methylmorpholine (0.099 ml, 0.84 mmol) and
CA 02398561 2002-07-24
WO 01/55112 PCT/US01102669
-56-
hydroxylamine (0.181 ml, 2.8 mmol) to obtain 150 mg (60%) of the product as a
solid.
HR - MS: m/z Calculated for CZ,H=,NjO6S 450.1693; Found 450.1692.
Example 50
1-Ethyl 4-methyl 4-(4-but-2-ynyloxybenzenesulfonyl)-1,4-
piperidinedicarboxylate
O O\ O
Me0 S \ ~ O
N
O' 'OEt
To a solution of product from Example 37 (400 mg, 1.03 mmol) in
chloroform (10 ml) was added sodium bicarbonate (865 mg, 10.3 mmol) and
ethylchloroformate (0.147 ml, 1.54 mmol). The resulting mixture was stirred
for 15
hours at room temperature, quenched with water and extracted with methylene
chloride. The organic layer was dried over anhydrous sodium sulfate and
concentrated to give 425 mg (98°Io) of the product as a solid.
MS-ES: m/z 424.4 (M+H)'.
Example 51
1-(Ethylcarbonyl)-4-(4-but-2-ynyloxybenzenesulfonyl)-1-piperidinecarboxylic
acid
O O\ ~O
HO S \ ~ O
N
O~O Et
The general procedure for step 3 was followed using product from Example
50 (400 mg, 0.95 mmol) in 8 ml of tetrahydrofuran: methanol: water (1:1:0.5)
and
lithium hydroxide (50 mg, 2.04 mmol) to obtain 340 mg(88°Io) of the
acid.
HR - MS: m/z Calculated for C,9H,3NO,S 408.1122; Found 408.1126.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-57-
Example 52
Ethyl 4-(4-but-2-~nyloxybenzenesulfonyl)-4-f (hydroxyamino)carbonyll-1-
piperidinecarbox
O O\ ~O
HOHN S \ ~ O
N
O' _OEt
The general procedure for step 4 was followed using product from Example
51 (225 mg, 0.55 mmol) in dimethylformamide (5 ml), 1-hydroxybenzotriazole (89
mg, 0.66 mmol), 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride
( 148 mg, 0.77 mmol), N-methylmorpholine (0.091 ml, 0.86 mmol) and
hydroxylamine (0.168 ml, 2.75 mmol) to obtain 150 mg (64%) of the product as a
solid.
HR - MS: m/z Calculated for C,9Hz4NZO,S 425.1377; Found 425.1375.
Example 53
Methyl 4-(4-but-2-ynyloxybenzenesulfonyl)-1-f (trifluoromethyl)sulfonyll-4-
piperidinecarboxylate
O O\ ~O
Me0 S \ ~ O
N
i
O O .CFs
To a solution of product from Example 37 (350 mg, 0.90 mmol) in
chloroform ( 10 ml) was added triethylamine ( 182 mg, 1.81 mmol) and
trifluoromethanesulfonyl chloride(0.125 ml, 1.17 mmol) followed by a catalytic
amount of dimethylaminopyridine. The resulting mixture was stirred for 15
hours at
room temperature, quenched with water and extracted with methylene chloride.
The
organic layer was dried over anhydrous sodium sulfate and concentrated to give
245
mg (56°Io) of the product as a solid.
HR - MS: m/z Calculated for C,gH2oF3NO,Sz 484.0706; Found 484.0700.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-58-
Example 54
4-(4-But-2-vnyloxybenzenesulfonyl)-1-f (trifluoromethyl)sulfonyll-4-
piperidinecarbox~ic acid
O O\ O
HO \S \ ~ O
N
O O ~CF3
The general procedure for step 3 was followed using product from Example
53 (225 mg, 0.47 mmol) in 5 ml of tetrahydrofuran: methanol; water (1:1:0.5)
and
lithium hydroxide (24 mg, 0.98 mmol) to obtain 175 mg (80°10) of the
acid.
MS-ES: m/z 468.1 (M-H)-.
Example 55
4-(4-But-2-yn.~ybenzenesulfonyl)-N-hydroxy-1-f(trifluoromethvl)sulfonvll-4-
~peridinecarboxamide
O O\ O
HOHN S \ ~ O
N
O O .CF3
The general procedure for step 4 was followed using product from Example
54 (145 mg, 0.31 mmol) in dimethylformamide (3 ml), 1-hydroxybenzotriazole (50
mg, 0.37 mmol), 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride
(83 mg, 0.47 mmol), N-methylmorpholine (0.051 ml, 0.47 mmol) and hydroxylamine
(0.095 ml, 1.55 mmol) to obtain 90 mg(60°Io) of the product as a solid.
HR - MS: m/z Calculated for C"H,9F3N20,Sz 485.0659; Found 485.0666.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-59-
Example 56
Methyl 4-(4-but-2-~~ybenzenesulfon~)-1-(3-p r~ylcarbonyl)- 4-
piperidinecarboxYlate
O O\ ~O
Me0 \S \ ~ O
N
O
~N
To a solution of product from Example 37 (500 mg, 1.29 mmol) in methylene
chloride ( 10 ml) was added triethylamine (443 mg, 4.39 mmol) and nicotinyl
chloride
(276 ml, 1.55 mmol) followed by a catalytic amount of dimethylaminopyridine.
The
resulting mixture was stirred for 15 hours at room temperature, quenched with
water
and extracted with methylene chloride. The organic layer was dried over
anhydrous
sodium sulfate and concentrated to give 460 mg (78°Io) of the product
as a solid.
HR - MS: m/z Calculated for C_3HZQNzO6S 457.1428; Found 457.1428.
Example 57
4-(4-But-2-ynyloxybenzenesulfonyl)-1-(3-pyridin~carbonyl)- 4-
piperidinecarboxylic
acid
O O
O~ i,
HO S \ ~ O
N
O
N
The general procedure for step 3 was followed using product from Example
56 (430 mg, 0.94 mmol) in 8 ml of tetrahydrofuran: methanol (1:l) and 1N
sodium
hydroxide (1.89 ml, 1.89 mmol) to obtain 235 mg(57°l0) of the acid.
HR - MS: m/z Calculated for C~~HZZN206S 443.1271; Found 443.1270.
CA 02398561 2002-07-24
WO 01/55112 PCT/USOi/02669
-60-
Example 58
4-(4-But-2-ynyloxybenzenesulfonyl)-N-hydrox -~pyridinylcarbonyl)- 4-
piperidinecarboxamide
O O
O~ "
HOHN S \ ~ O
N
O ~ ~N
The general procedure for step 4 was fo Mowed using product from Example
57 (195 mg, 0.44 mmol) in dimethylformamide (4 ml), 1-hydroxybenzotriazole (72
mg, 0.53 mmol), 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride
( 119 mg, 0.62 mmol), N-methylmorpholine (0.072 ml, 0.66 mmol) and
hydroxylamine (0.135 ml, 2.2 mmol) to obtain 65 mg (32%) of the product as a
solid.
HR - MS: m/z Calculated for CZZH23N306S 458.1380; Found 458.1373.
Example 59
Methyl 4-(4-but-2~nyloxybenzenesulfonyl)-1-(2-thienylcarbonyl)- 4-
piperidinecarboxylate
O O
O~ "
Me0 S \ ~ O
N
O S
To a solution of product from Example 37 (500 mg, 1.29 mmol) in methylene
chloride (10 ml) was added triethylamine (261 mg, 2.58 mmol) and
thiophenylcarbonyl chloride (227 mg, 1.55 mmol) followed by a catalytic amount
of
dimethylaminopyridine. The resulting mixture was stirred for 15 hours at room
temperature, quenched with water and extracted with methylene chloride. The
organic layer was dried over anhydrous sodium sulfate and concentrated to give
480
mg (81%) of the product as a solid.
HR - MS: m/z Calculated for CZZHz3NO6SZ 462.1040; Found 462.1039.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-61
Example 60
4-(4-But-2-yn~ybenzenesulfonyl)-1-(2-thienylcarbonyl)- 4-piperidinecarboxvlic
acid
O O
O~ "
HO S \ ~ O
N
O S
The general procedure for step 3 was followed using product from Example
59 (435 mg, 0.94 mmol) in 8 ml of tetrahydrofuran: methanol (1:1) and 1N
sodium
hydroxide (1.89 ml, 1.89 mmol) to obtain 360 mg(86%) of the acid.
HR - MS: m/z Calculated for C2,Hz,NO6S2 448.0883; Found 448.0882.
Example 61
4-(4-but-2-ynyloxybenzenesulfonyl)-N-hydroxy-1-(2-thienylcarbonyl)- 4-
piperidinecarboxamide
O O
O~"
HOHN S \ ~ O
N
O S
The general procedure for step 4 was followed using product from Example
60 (335 mg, 0.75 mmol) in dimethylformamide (7 ml), 1-hydroxybenzotriazole
(121
mg, 0.90 mmol), 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride
(201 mg, 1.05 mmol), N-methylmorpholine (0.124 ml, 1.13 mmol) and hydroxyl-
amine (0.229 ml, 3.75 mmol) to obtain 216 mg (62°Io) of the product as
a solid.
HR - MS: m/z Calculated for CZ,HZZN206Sz 463.0992; Found 463.0988.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-62-
Example 62
Methyl4-(4-but-2-ynyloxybenzenesulfonyl)-1-((4-methoxyphenyl)sulfon 1
~peridinecarbox,
O O
O. ~~
Me0 S \ ~ O
N
O;S
O
OMe
To a solution of product from Example 37 (500 mg, 1.29 mmol) in methylene
chloride(10 ml) was added triethylamine (261 mg, 2.58 mmol) and 4-
methoxyphenyl-
sulfonyl chloride(320 mg, 1.55 mmol) followed by a catalytic amount of
dimethyl-
aminopyridine. The resulting mixture was stirred for 15 hours at room
temperature,
quenched with water and extracted with methylene chloride. The organic layer
was
dried over anhydrous sodium sulfate and concentrated to give 590 mg
(88°70) of the
product as a solid.
HR - MS: m/z Calculated for Cz4H,=NO$Sz 522.1251; Found 522.1252.
Example 63
4-(4-but-2-~yloxybenzenesulfonXl)-1-f(4-methoxyphenyl)sulfonyll-4-
piperidinecarboxylic acid
O O\ ~O
HO S \ ~ O
N
O;S
O
OMe
The general procedure for step 3 was followed using product from Example
62 (545 mg, 1.04 mmol) in 8 ml of tetrahydrofuran: methanol (1:1) and 1N
sodium
hydroxide (2.09 ml, 2.09 mmol) to obtain 446 mg (85%) of the acid.
HR - MS: m/z Calculated for Cz3Hz5N08S, 508.1094; Found 508.1073.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-63-
Example 64
4-(4-but-2-~~ybenzenesulfon l~ydroxy-1-f(4-methoxyphenyl>sulfon 1
piperidinecarboxamide
O O _
O~"
HOHN \S \ ~ O
N
O;S
O
OMe
The general procedure for step 4 was followed using product from Example
63 (402 mg, 0.79 mmol) in dimethylformamide (8 ml), 1-hydroxybenzotriazole
(128
mg, 0.95 mmol), 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride
(212 mg, 1.11 mmol), N-methylmorpholine (0.130 ml, 1.19 mmol) and hydroxyl-
amine (0.242 ml, 3.95 mmol) to obtain 396 mg (96°Io) of the product as
a solid.
HR - MS: m/z Calculated for CZjHz6NzO8Sz 523.1203; Found 523.1198.
Example 65
Metl~l 4-(4-but-2-ynyloxybenzenesulfonyl)-1-f (2,2,5-trimethyl-1.3-dioxan-5-
yl)carbonyll-4-piperidinecarboxylate
O O
O~"
Me0 S \ ~ O
N
O~O
The general procedure for step 4 was followed using product from Example
37 (500 mg, 1.29 mmol) in dimethylformamide (10 ml), (2,2,5-trimethyl-1,3-
dioxan-
5-yl)carboxylic acid (224 mg, 1.29 mmol), 1-hydroxybenzotriazole (209 mg, 1.56
mmol), 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (346 mg,
1.81 mmol) and N-methylmorpholine (0.212 ml, 1.94 mmol) to obtain 385 mg (59%)
of the product as a solid.
HR - MS: m/z Calculated for CZSH33N08S 508.2000; Found 508.1998.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-64-
Example 66
4-(4-But-2-~.~ybenzenesulfonyl)-1-f(2 2 5-trimeth~l-1,3-dioxan-5-yl)carbonyll-
4-piperidinecarbox~ic acid
O O
O~ "
HO S \ ~ O
N
O
O
O
The general procedure for step 3 was followed using product from Example
65 (335 mg, 0.66 mmol) in 4 ml of tetrahydrofuran: methanol (1:1) and 1N
sodium
hydroxide (1.3 ml, 1.3 mmol) to obtain 315 mg (97%) of the acid.
HR - MS: m/z Calculated for CZQH3,NO8S 494.1843; Found 494.1835.
Example 67
4-(4-but-2-ynyloxybenzenesulfon l~ydrox~ 1-f(2,2,5-trimethyl-1,3-dioxan-5-
y1 carbonyll-4-piperidinecarboxamide
O O
O~"
HOHN S \ ~ O
N
O~O
O'
The general procedure for step 4 was followed using product from Example
66 (280 mg, 0.57 mmol) in dimethylformamide (6 ml), 1-hydroxybenzotriazole (92
mg, 0.68 mmol), 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride
(153 mg, 0.80 mmol), N-methylmorpholine (0.094 ml, 0.85 mmol) and hydroxyl-
amine (0.174 ml, 2.85 mmol) to obtain 180 mg (62%) of the product as a solid.
HR - MS: m/z Calculated for Cz4H3,N208S 531.1771; Found 531.1768.
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-65-
Example 68
4-(4-but-2-ynyloxybenzenesulfonyl)-N-hydroxy-1-f 3-hydroxy-2-(hydroxymethyl)-2-
meths rod panoyll-4-Riperidinecarboxamide
O O
O\ "
HOHN S \ ~ O
N
O
~OH
OH
To a solution of product from Example 67 ( 150 mg, 0.29 mmol) in
tetrahydrofuran (2 ml) was added 1N aqueous hydrochloric acid (2 ml) and the
resulting mixture was stirred for 4 hours. The organic layer was washed with
sodium
bicarbonate, saturated sodium chloride solution and dried over anhydrous
sodium
sulfate. The organic solvent was concentrated to obtain 40 mg (29°Io)
of the product.
HR - MS: m/z Calculated for Cz,H2gN208S 469.1639; Found 469.1637.
Example 69
Methyl ((4-(f4-(2-butyn~loxy)phenyllsulfonyl~-4-f(hydroxyamino)carbonyll-1-
piperidin l~)met~l)benzoate hydrochloride
To a solution of product from Example 6 (2.5g, 6.43 mmol) and methyl 4-
(bromomethyl)benzoate ( 1.62g, 7.07 mmol) in methanol ( 100 ml) at 50
°C was added
triethylamine (2.25 ml, 16.1 mmol). After 30 minutes additional methanol (50
ml)
was added. The reaction mixture was stirred for 18 hours, concentrated in
vacuo and
1N aqueous hydrochloric acid (10 ml) and water were added. The resulting solid
was
isolated and methanol (20 ml) and 1N hydrochloric acid in diethyl ether (15
ml) were
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-66-
added. Additional diethyl ether was added followed by trituration of the
precipitate to
give the desired product as a white powder (2.4g).
'H NMR (DMSO-db, 300 MHz):8 1.85 (t, 3H, CH3, J = 2.2 Hz), 2.1 - 3.5 (m, 8H),
3.87 (S, 3H), 4.40 (bd s, 2H), 4.89 (q, 2H, J = 2.2 Hz), 7.1 - 8.1 (m, 8H),
9.3 - 11.2
(m, 3H); MS-ES: m/z 501.5 (M+H)'.
The subject compounds of the present invention may be tested for biological
activity
according to the following procedures.
In Vitro Gelatinase Assay
The assay is based on the cleavage of the thiopeptide substrate ((Ac-Pro-Leu-
Gly(2
mercapto-4 methyl-pentanoyl)-Leu-Gly-OEt), Bachem Bioscience) by the enzyme,
gelatinase, releasing the substrate product which reacts colorimetrically with
DTNB
((5,5'-dithio-bis(2-nitro-benzoic acid)). The enzyme activity is measured by
the rate
of the color increase.
The thiopeptide substrate is made up fresh as a 20 mM stock in 100% DMSO and
the
DTNB is dissolved in 100% DMSO as a 100 mM stock and stored in dark at room
temperature. Both the substrate and DTNB are diluted together to 1 mM with
substrate buffer (50 mM HEPES pH 7.5, 5 mM CaCI 2) before use. The stock of
human neutrophil gelatinase B is diluted with assay buffer (50 mM HEPES pH
7.5, 5
mM CaCl2, 0.02% Brij) to a final concentration of 0.15 nM.
The assay buffer, enzyme, DTNB/substrate (500 ~M final concentration) and
vehicle
or inhibitor are added to a 96 well plate (total reaction volume of 2001) and
the
increase in color is monitored spectrophotometrically for 5 minutes at 405 nm
on a
plate reader.
The increase in OD 405 is plotted and the slope of the line is calculated
which
represents the reaction rate.
The linearity of the reaction rate is confirmed (r2 >0.85). The mean (x ~ sem)
of the
control rate is calculated and compared for statistical significance (p <0.0~)
with
drug-treated rates using Dunnett's multiple comparison test. Dose-response
relationships can be generated using multiple doses of drug and ICSO values
with 95%
CI are estimated using linear regression (IPRED, HTB).
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-67-
References: Weingarten, H and Feder, J., Spectrophotometric assay for
vertebrate
collagenase, Anal. Biochem. 147, 437-440 (1985).
In Vitro Colla~enase Assay
The assay is based on the cleavage of a peptide substrate ((Dnp-Pro-Cha-Gly-
Cys(Me)-His-Ala-Lys(NMa)-NH2), Peptide International, Inc.) by collagenase
releasing the fluorescent NMa group which is quantitated on the fluorometer.
Dnp
quenches the NMa fluorescence in the intact substrate. The assay is run in
HCBC
assay buffer (50 mM HEPES, pH 7.0, 5 mM Ca +2, 0.02% Brij, 0.5% Cysteine),
with
human recombinant fibroblast collagenase (truncated, mw=18,828, WAR, Radnor).
Substrate is dissolved in methanol and stored frozen in 1 mM aliquots.
Collagenase
is stored frozen in buffer in 25 ~M aliquots. For the assay, substrate is
dissolved in
HCBC buffer to a final concentration of 10 ~M and collagenase to a final
concentration of 5 nM. Compounds are dissolved in methanol, DMSO, or HCBC.
The methanol and DMSO are diluted in HCBC to < 1.0%. Compounds are added to
the 96 well plate containing enzyme and the reaction is started by the
addition of
substrate.
The reaction is read (excitation 340 nm, emission 444 nm) for 10 min. and the
increase in fluorescence over time is plotted as a linear line. The slope of
the line is
calculated and represents the reaction rate.
The linearity of the reaction rate is confirmed (r2 >0.85). The mean (x ~ sem)
of the
control rate is calculated and compared for statistical significance (p <0.05)
with
drug-treated rates using Dunnett's multiple comparison test. Dose-response
relationships can be generated using multiple doses of drug and ICSO values
with 95%
CI are estimated using linear regression (IPRED, HTB) .
References: Bickett, D. M. et al., A high throughput fluorogenic substrate for
interstitial collagenase (MMP-1) and gelatinise (MMP-9), Anal. Biochem. 212,58-
64
(1993).
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-68-
Procedure for Measuring TACE Inhibition
Using 96-well black microtiter plates, each well receives a solution composed
of
~tL, TACE (Immunex, final concentration lpg/mL), 70NL Tris buffer, pH 7.4
containing 10°Io glycerol (final concentration 10 mM), and 10 pI, of
test compound
5 solution in DMSO (final concentration l~tM, DMSO concentration <1%) and
incubated for 10 minutes at room temperature. The reaction is initiated by
addition of
a fluorescent peptidyl substrate (final concentration 100 E.~M) to each well
and then
shaking on a shaker for 5 sec.
The reaction is read (excitation 340 nm, emission 420 nm) for 10 min. and the
10 increase in fluorescence over time is plotted as a linear line. The slope
of the line is
calculated and represents the reaction rate.
The linearity of the reaction rate is confirmed (r 2 >0.85). The mean (x~sem)
of the
control rate is calculated and compared for statistical significance (p<0.05)
with drug-
treated rates using Dunnett's multiple comparison test. Dose-response
relationships
can be generate using multiple doses of drug and IC 50 values with 95% CI are
estimated using linear regression.
The compound of Example 18 was found to inhibit MMPs and TACE as
follows: TACE inhibition (atlOuM) : 54%;
MMP1 (1C50): l.3uM;
MMP9 (IC50): 0.732uM;
MMP13 (IC50): 0.14uM.
Thus compounds of the present invention are useful inhibitors of MMPs and
TACE.
Pharmaceutical Composition
Compounds of this invention may be administered neat or with a
pharmaceutical carrier to a patient in need thereof. The pharmaceutical earner
may
be solid or liquid.
Applicable solid earners can include one or more substances which may also
act as flavoring agents, lubricants, solubilizers, suspending agents, fillers,
glidants,
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-69-
compression aids, binders or tablet-disintegrating agents or an encapsulating
material.
In powders, the Garner is a finely divided solid which is in admixture with
the finely
divided active ingredient. In tablets, the active ingredient is mixed with a
Garner
having the necessary compression properties in suitable proportions and
compacted
in the shape and size desired. The powders and tablets preferably contain up
to 99%
of the active ingredient. Suitable solid Garners include, for example, calcium
phosphate, magnesium stearate, talc, sugars, lactose, dextrin, starch,
gelatin,
cellulose, methyl cellulose, sodium carboxymethyl cellulose,
polyvinylpyrrolidine,
low melting waxes and ion exchange resins.
Liquid carriers may be used in preparing solutions, suspensions, emulsions,
syrups and elixirs. The active ingredient of this invention can be dissolved
or
suspended in a pharmaceutically acceptable liquid carrier such as water, an
organic
solvent, a mixture of both or pharmaceutically acceptable oils or fat. The
liquid
carrier can contain other suitable pharmaceutical additives such a
solubilizers,
emulsifiers, buffers, preservatives, sweeteners, flavoring agents, suspending
agents,
thickening agents, colors, viscosity regulators, stabilizers or osmo-
regulators.
Suitable examples of liquid Garners for oral and parenteral administration
include
water (particularly containing additives as above, e.g., cellulose
derivatives,
preferable sodium carboxymethyl cellulose solution), alcohols (including
monohydric
alcohols and polyhydric alcohols, e.g., glycols) and their derivatives, and
oils (e.g.,
fractionated coconut oil and arachis oil). For parenteral administration the
carrier can
also be an oily ester such as ethyl oleate and isopropyl myristate. Sterile
liquid
carriers are used in sterile liquid form compositions for parenteral
administration.
Liquid pharmaceutical compositions which are sterile solutions or
suspensions can be utilized by, for example, intramuscular, intraperitoneal or
subcutaneous injection. Sterile solutions can also be administered
intravenously.
Oral administration may be either liquid or solid composition form.
The compounds of this invention may be administered rectally in the form of
a conventional suppository. For administration by intranasal or intrabronchial
inhalation or insufflation, the compounds of this invention may be formulated
into an
aqueous or partially aqueous solution, which can then be utilized in the form
of an
aerosol. The compounds of this invention may also be administered
transdermally
through the use of a transdermal patch containing the active compound and a
Garner
CA 02398561 2002-07-24
WO 01/55112 PCT/USO1/02669
-70-
that is inert to the active compound, is non-toxic to the skin, and allows
delivery of
the agent for systemic absorption into the blood stream via the skin. The
carrier may
take any number of forms such as creams and ointments, pastes, gels, and
occlusive
devices. The creams and ointments may be viscous liquid or semi-solid
emulsions of
either the oil in water or water in oil type. Pastes comprised of absorptive
powders
dispersed in petroleum or hydrophilic petroleum containing the active
ingredient may
also be suitable. A variety of occlusive devices may be used to release the
active
ingredient into the blood stream such as a semipermeable membrane covering a
reservoir containing the active ingredient with or without a carrier, or a
matrix
containing the active ingredient. Other occlusive devices are known in the
literature.
The dosage to be used in the treatment of a specific patient suffering from a
disease or condition in which MMPs and TALE are involved must be subjectively
determined by the attending physician. The variables involved include the
severity of
the dysfunction, and the size, age, and response pattern of the patient.
Treatment will
generally be initiated with small dosages less than the optimum dose of the
compound. Thereafter the dosage is increased until the optimum effect under
the
circumstances is reached. Precise dosages for oral, parenteral, nasal, or
intrabronchial administration will be determined by the administering
physician
based on experience with the individual subject treated and standard medical
principles.
Preferably the pharmaceutical composition is in unit dosage form, e.g., as
tablets or capsules. In such form, the composition is sub-divided in unit dose
containing appropriate quantities of the active ingredient; the unit dosage
form can be
packaged compositions, for example packed powders, vials, ampoules, prefilled
syringes or sachets containing liquids. The unit dosage form can be, for
example, a
capsule or tablet itself, or it can be the appropriate number of any such
compositions
in package form.