Note: Descriptions are shown in the official language in which they were submitted.
CA 02399598 2007-10-09
25771-748
1
Process for preparing aryl-iminomethyl-carbamino acid esters
The invention relates to a process which can be used on an industrial scale
for the
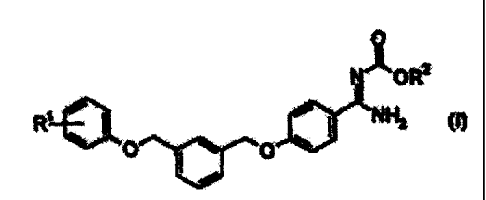
production of compounds of general formula (I)
0
NOR2
R~ N H2
O O
(I)
wherein the groups R' and R2 have the meanings described herein.
Background of the ira-vention
From Intemational Patent Application WO 96/02497, benzamidines and aryl-
iminomethylcarbamino acid esters are known which are highly effective as
pharmaceutical compositions with an LTB4-antagonistic activity. Compounds of
general formula (f) are of particular importance.
The problem of the present invention is to provide a process which can be used
for
industrial-scale synthesis of compounds of general formula(I) in high yields
and with
end products of high purity.
Detailed description of the invention
To solve the problem mentioned above, the invention proposes a process for
preparing compounds of general formula (I)
O
NOR2
I
R1 ao I NH2
O
(I)
wherein
Rl denotes a group selected from among methyl, ethyl, propyl, cyclopentyl,
cyclohexyl, phenyl, benzyl and -C(Me2)phenyl, each of which may be mono-,
di- or trisubstituted by hydroxy;
R2 denotes a group selected from among,methyl, ethyl, propyl and benzyl,
- CA 02399598 2002-07-11
2
characterised in that a compound of general formula (II)
R~, i I O, CN
~ ~ ~
O I ~
~ (II)
wherein
R1 ' denotes a group selected from among methyl, ethyl, propyl, cyclopentyl,
cyclohexyl, phenyl, benzyl and -C(Me2)phenyl, each of which may be mono-,
di- or trisubstituted by a group -0-PG, the group -0-PG denoting a protected
hydroxyl function selected from among methoxymethyloxy, 2-
methoxyethoxymethyloxy, 1-ethoxyethyloxy, 2-tetrahydropyranyloxy, 1-
butoxyethyloxy, tert.-butyloxy, benzyloxy and 4-methoxybenzyloxy,
is first reacted in an ethereal or aromatic solvent with an alkali metal
hexaalkyldisilazane and is then treated with a compound of formula (III)
R2-O-COX' (III)
wherein
R2 is as hereinbefore defined and
X' denotes chlorine, bromine or -O-R2,
after working up using an acid of formula HY a compound of formula (IV)
O
HN~OR2
RI a ~ I NH x HY
o o ~
(IV)
wherein the groups R1 and R2 are as hereinbefore defined and Y denotes any
desired acid group,
is isolated and from this the compound of formula (I) is liberated.
The compounds of formulae (I) and (IV) also include the corresponding
tautomers of
formulae (I-T) and (IV-T):
CA 02399598 2002-07-11
3
0
HNIfl, ORZ
R~ NH
o ~
0
~
(I-T)
O
N'k OR2
I
R1 ao 0~ I NH2 x HY
~
(IV-T)
The term "alkali metal hexaalkyldisilazane" as used above and hereinafter to
denote
the reagent which is reacted with the compound of formula (II) generally
designates
a compound of formula (VIII)
3 3
RSf\R3
Met+ N_ R
3 (VIII)
R3~Si-R
R3
wherein
Met denotes an alkali metal, preferably lithium, sodium or potassium,
particularly
lithium, and
R3 independently in each case denotes a C,-4alkyl group, preferably methyl or
ethyl,
especially methyl.
Most particularly preferred are lithium hexamethyldisilazane, sodium
hexamethyidisilazane and potassium hexamethyldisilazane, particularly lithium
hexamethyldisilazane.
The term "subsequent reaction" with a compound of formula (III) covers both
procedures in which the product of the reaction of the compound of formula
(II) with
the alkali metal hexamethyidisilazane is reacted with the compound of formula
(III)
directly, without any further intermediate reaction, and also procedures in
which the
free amidine base is liberated in the mean time from the product formed.
Preferably,
CA 02399598 2002-07-11
4
the product of the reaction of the compound of formula (II) with the alkali
metal
hexamethyldisilazane is reacted with the compound of formula (111) directly,
especially in a "one-pot synthesis".
A preferred process for preparing compounds of general formula (I) is one
wherein
R1 denotes a group selected from among phenyl, benzyl and -C(Me2)phenyl,
each of which may be mono- or disubstituted, preferably monosubstituted by
hydroxy;
R2 denotes a group selected from among ethyl, propyl and benzyl,
io characterised in that a compound of general formula (II)
wherein
R1 ' denotes a group selected from among phenyl, benzyl and -C(Me2)phenyl,
each of which may be mono- or disubstituted, preferably monosubstituted by a
group -0-PG, the group -0-PG denoting a protected hydroxyl function
selected from among methoxymethyloxy, 2-methoxyethoxymethyloxy, 1-
ethoxyethyloxy, 2-tetrahydropyranyloxy, 1-butoxyethyloxy, tert.-butyloxy,
benzyloxy and 4-methoxybenzyloxy, preferably 2-tetrahydropyranyloxy,
is first reacted in an ethereal or aromatic solvent with an alkali metal
hexaalkyldisilazane and then treated with a compound of formula (III)
R2-0-COX' (111)
wherein
R2 is as hereinbefore defined and
X' denotes chlorine, bromine or -O-R2,
after working up with aqueous hydrochloric acid a compound of formula (IVA)
O
HN'k OR2
R~ ~ ~ J(NH x HCI
~ O
(IVA)
wherein the groups R1 and R2 are as hereinbefore defined is isolated and from
this
the compound of formula (I) is liberated.
Particularly preferred is a process for preparing compounds of general formula
(I)
wherein
CA 02399598 2002-07-11
R1 denotes -C(Me2)phenyl which may optionally be monosubstituted by hydroxy
and
R2 denotes ethyl,
characterised in that a compound of general formula (II)
5 wherein
R1 denotes -C(Me2)phenyl, which may optionally be monosubstituted by a group
-0-PG, the group -0-PG denoting a protected hydroxyl function selected from
among methoxymethyloxy, 2-tetrahydropyranyloxy , 1 -butoxyethyloxy,
tert.-butyloxy, benzyloxy and 4-methoxybenzyloxy, preferably
2-tetrahydropyranyloxy,
is first reacted in an ethereal or aromatic solvent with an alkali metal
hexaalkyldisilazane and then treated with a compound of formula (III)
R2-0-COX' (III)
wherein
R2 is as hereinbefore defined and
X' denotes chlorine, bromine or -0-R2, preferably chlorine,
after working up with aqueous hydrochloric acid a compound of formula (IVA)
wherein the groups R1 and R2 are as hereinbefore defined is isolated and from
this
the compound of formula (I) is liberated.
In a particularly preferred embodiment of the process according to the
invention, the
compound of formula (II) is prepared in a process comprising the following
steps:
(a) reacting C,.a-a{kyf 3-halomethylbenzoates with 4-hydroxybenzonitrile in
the
manner of a Wilkinson ether synthesis;
(b) reductively converting the resulting alkyl 3-(4-cyano-phenoxy)benzoates of
formula (VII)
, I CN
0
R'O '~ O ~
i (VII)
wherein R' denotes C,,-alkyl, into a compound of formula (V)
~ X O
(V)
CA 02399598 2002-07-11
6
wherein X denotes hydroxy;
(c) optionally treating the compound of formula (V) wherein X denotes hydroxy
with a halogenating reagent or a sulphonic acid chloride;
(d) reacting the compound of formula (V) wherein X denotes hydroxy,
chlorine, bromine, mesylate, triflate or tosylate, with a phenol derivative of
formula (VI)
R1. ~ I
~ OH
(VI)
wherein R1 ' has the meanings given in claims 1 to 4; optionally in the form
of the
corresponding sodium or potassium phenoxides, under basic reaction conditions,
preferably in a polar organic solvent.
The hydrochlorides of formula (IVA) are of central importance in the process
according to the invention for preparing the compounds of general formula (I).
They
are obtained directly in high yields as readily crystallising salts, from
which by-
products and/or impurities can easily be removed by crystallisation.
Accordingly, one
aspect of the present invention relates to intermediate products of general
formula
(IVA)
O
HN'J~ OR2
R1~ I NH x HCI
~ o
(IVA)
wherein the groups R1 and R2 may be as hereinbefore defined.
Of the compounds of general formula (IVA) the compound ethyl {[4-(3-{4-[1-(4-
hydroxy-phenyl)-1-methyl-ethyl]-phenoxymethyl}-benzyloxy)-phenyl]-imino-
methyl}-
carbaminate hydrochloride is particularly preferred.
The compounds of general formula (II)
R~, i I O, CN
~ ~ ~
O ( ~
~ (II)
wherein R1 ~ may be as hereinbefore defined are obtained according to the
invention
by reacting a compound of formula (V)
CA 02399598 2002-07-11
7
X p, CN
~ I
1105:~ (V)
wherein X denotes hydroxy, chlorine, bromine, mesylate, triflate,
benzenesulphonate
or tosylate, with a compound of formula (VI)
R1, i I
OH
(VI)
wherein Rl' may be as hereinbefore defined and wherein the compound of formula
(VI) may optionally also be used in the form of the sodium and potassium
phenoxides thereof, under basic reaction conditions in a polar organic
solvent.
It is preferred to prepare compounds of general formula (II) wherein Rl' may
be as
io hereinbefore defined by reacting a compound of formula (V) wherein X
denotes
hydroxy, chlorine or mesylate, more preferably hydroxy or chlorine, most
preferably
chlorine, with a compound of formula (VI) wherein Rl'may be as hereinbefore
defined and wherein the compound of formula (VI) is used in the form of the
alkali
metal phenoxides thereof, preferably in the form of the sodium phenoxides
thereof.
When the compounds of general formula (I) are synthesised according to the
invention, the intermediate products of general formula (V) are of central
importance.
In another aspect, therefore, the present invention relates to the compounds
of
general formula (V)
X O, CN
~ I
I ~
(V)
as such, wherein X may be as hereinbefore defined and most preferably may
denote
hydroxy or chlorine.
The compound of formula (VII), one of the starting compounds, is also of
particular
importance in the synthesis according to the invention of the compounds of
general
formula (I). Therefore, in another aspect, the present invention relates to
the
compounds of formula (VII)
p , I CN
R'O I O ~
(VII)
CA 02399598 2002-07-11
8
as such, wherein R' denotes C1-4-a1kyl, preferably methyl or ethyl, most
preferably
methyl.
In order to perform the process for preparing the compounds of general formula
(I)
according to the invention starting from the nitriles of general formula (II)
the
following method is used:
A compound of general formula (II) is slowly metered into a solution of the
alkali
metal hexaalkyldisilazane, preferably lithium bis(trimethylsilyl)-amide,
sodium
1o bis(trimethylsilyl)-amide, most preferably lithium bis(trimethylsilyl)-
amide, in an
ethereal or aromatic organic solvent, preferably in a solvent selected from
among
tetrahydrofuran, toluene, dioxane, more preferably in tetrahydrofuran or
dioxane,
most preferably in tetrahydrofuran, preferably with cooling, especially at a
temperature between -50 C and 30 C, more especially at -20 C to 10 C, most
especially at about 0 C. The quantity of the alkali metal hexaalkyldisilazane
used is
determined by the amount of nitrile of formula (II) used. At least 1 mol,
preferably
1.01 to 1.15 mol of alkali metal hexaalkyldisilazane is used per mol of
nitrile of
formula (II). The quantity of ethereal solvent used is between 0.7 and 1.5,
preferably
0.9 to 1.3 kg per mol of compound of formula (li) used.
After all the compound of formula (II) has been added the resulting suspension
is
stirred at constant temperature, optionally at a temperature of up to 40 C,
preferably
at about 20-25 C over a period of 6 to 24 hours, preferably 8 to 18 hours. The
stirring is preferably continued for 10 to 12 hours. During this time the
solid which
was initially in suspension may go into solution.
The mixture can then optionally be diluted either with additional ethereal
solvent or
with a nonpolar organic solvent, preferably with an aromatic organic solvent.
A
solvent selected from among toluene, benzene, cyclohexane, methylcyclohexane
or
xylene is preferably used, of which toluene and xylene are particularly
preferred,
toluene being most preferred. If the mixture is diluted, it is sufficient to
add up to 0.5
L, preferably up to 0.3 L of solvent per mol of compound of formula (II) used.
The reaction mixture is heated to a reaction temperature of between -50 C and
20 C, more especially -20 C to 10 C, most preferably -10 to 0 C before the
addition
of the compound of general formula (III). Then the compound of formula (III)
is
added, in an amount of at least 1 mol, preferably 1.05 to 1.3 mol, more
particularly
1.1 to 1.2 mol, per mol of compound (II) used.
CA 02399598 2002-07-11
9
After the reaction is complete, the product is hydrolysed by the addition of
an acid of
formula HX, preferably an inorganic or organic acid such as hydrochloric acid,
sulphuric acid, phosphoric acid, acetic acid, trifluoroacetic acid, oxalic
acid and
fumaric acid, particularty aqueous hydrochloric acid. About 1 mol of acid,
particulariy hydrochloric acid, is used per mol of the compound of formula
(II)
originally used. According to the invention, it is preferable to add dilute
hydrochloric
acid (preferably 8-15%, more especially 10-12% strength).
After a period of about 10 minutes to 1 hour the aqueous lower phase is
separated
off and an organic solvent selected from among acetone, methylisobutylketone,
lo methylethylketone, optionally a mixture of two of the above solvents, most
preferably
a mixture of acetone and methylisobutylketone in a ratio of 3-1 : 1, most
preferably in
a ratio of 2.5-1.5 : 1, is added to the organic phase. Crystallisation of the
compounds
of formula (IVA) is initiated by the addition of aqueous hydrochloric acid.
About 1 to
1.2 mol of hydrochloric acid is used per mole of the compound of formula (11)
originally used. It is preferred according to the invention to add preferably
32-37%,
most preferably 37% hydrochloric acid. The compounds of formula (IV) are
separated from the reaction mixture by conventional methods, e.g. by
centrifugation,
washed with an organic solvent selected from among acetone,
methylisobutylketone,
methylethylketone or a carboxylic acid ester, preferably acetone, and dried.
The release of the compounds of formula (I) from the acid addition salts of
formula
(IV), particularly from the hydrochlorides of formula (IVA), is generally
carried out
with basic reactants under reaction conditions which are as neutral as
possible,
preferably in the presence of buffer systems, according to the method
described
hereinafter:
An organic solvent selected from among acetone, methylisobutylketone,
methylethylketone, tetrahydrofuran or a carboxylic acid ester, preferably
acetone,
and then a compound of formula (IV) are added to a solution of trisodium
citrate
dihydrate, sodium hydroxide, potassium hydroxide, alkali or alkaline earth
metal salts
of organic or inorganic weak acids, preferably trisodium citrate dihydrate,
trisodium
citrate or sodium hydroxide, most preferably trisodium citrate dihydrate in
water at 0-
C, preferably at 20-25 C, particularly at about 20 C. About 1-2 mol,
preferably
about 1.5 mol of the sodium or potassium citrate used and about 1 to 3 L of
the
35 abovementioned organic solvent, preferably about 2 L, are used per mol of
compound of formula (IV) put in. The mixture is stirred at constant
temperature over
a period of 20 minutes to 2 hours, preferably 1-1.2 hours.
CA 02399598 2002-07-11
When strong bases such as sodium hydroxide are used, the method of addition
may
be reversed if desired. The crystalline product is separated off by
filtration, for
example, washed with water to remove any salt, and with the abovementioned
organic solvent and finally dried.
5
The compounds of general fomnula (II) may be obtained by reacting a compound
of
formula (V) with a compound of formula (VI), as already mentioned. According
to the
invention, the following procedure may be used.
The compound of formula (V) wherein X denotes hydroxy is dissolved in an
organic
io solvent, if possible an aprotic-polar organic solvent, preferably N,N-
dimethylacetamide, acetone, methylethylketone, methylisobutylketone, N-
methyl pyrrol i done, N,N-dimethylformamide, tetraalkylurea, most preferably
in N,N-
dimethylacetamide. 0.5 to 1.0, preferably about 0.7 L of solvent are used at
this
point per mol of starting compound according to the invention. Then the
solution thus
obtained is cooled to a temperature of <10 C, preferably to a temperature
between
+5 C and -20 C, most preferably to about -10 C to 0 C. Then a suitably
substituted
sulphonic acid chloride, optionally the abovementioned organic solvent, an
organic
base, optionally the abovementioned organic solvent and the aqueous solution
of an
inorganic base are added one after the other. A suitably substituted sulphonic
acid
chloride according to the invention might be methanesulphonic acid chloride,
para-
toluenesulphonic acid chloride, benzenesulphonic acid chloride or
trifluoromethanesulphonic acid chloride. Preferably, methanesulphonic acid
chloride
is used. The organic bases may be, for example, dimethylaminopyridine,
pyridine,
methylpyridine, tert. amines, e.g. trimethylamine, triethylamine,
diisopropylethylamine, or cyclic amines such as N-methylpyrrolidine or DBU
(diazabicycloundecene). Preferred organic amines are N-methylpyrrolidine,
trimethylamine, triethylamine or diisopropylethylamine, most preferably
triethylamine.
The organic base is used in at least a stoichiometric amount, based on the
starting
compound of formula (V). Preferably, the organic base is used in an excess of
10-50
mol%, most preferably in about a 30% excess in relation to the compound of
formula
(V) used. The aqueous solution of an organic base used will usually be an
alkali
metal or alkaline earth metal hydroxide solution, the alkali metal hydroxide
solutions
being preferred. The aqueous solutions of potassium hydroxide and sodium
hydroxide are particularly important according to the invention. Usually, 20-
50%
solutions of the abovementioned inorganic hydroxides are used. More
concentrated
solutions such as, for example, 45% solutions are preferred according to the
invention. Based on the compound of formula (V) used, the inorganic base is
used in
at least a stoichiometric amount, preferably in an excess of 50-100 mol%. The
CA 02399598 2002-07-11
11
inorganic base is particularly preferably used in an excess of about 75 mol% ,
based
on the compound of formula (V) added.
If desired, the reaction mixture may be diluted after the addition of the
suitably
substituted sulphonic acid chloride or the organic base by the addition of the
abovementioned organic solvent. In this case, 2-10%, preferably about 5% of
the
quantity of solvent put in initially is added.
In any case, after all the aqueous solution of the inorganic base has been
added, the
reaction mixture is diluted with the organic solvent mentioned above. About
0.5 to
io 1.0 L, preferably between 0.7 and 0.8 L of the solvent used are added, per
mol of
starting compound of formula (V) used. Then alkoxides or metal salts of
formula (VI)
are added. The sodium and potassium phenoxides which may be derived from the
compounds of formula (VI) are preferably used. According to the invention,
stoichiometric quantities, optionally substoichiometric quantities or an
excess of the
compound (VI) may be added, based on the educt of formula (V). After all the
compound (VI) has been added, the reaction is continued for a period of about
1-3
hours, preferably about 1.5 to 2 hours at a temperature of 5-35 C, preferably
at
about 25 C, and finally stirred for a period of about 1-3 hours, preferably
about 1.5-2
hours at a temperature of 50-100 C, preferably at about 70-90 C. After the
reaction
2o has ended the product of formula (II) is crystallised by the addition of a
suitable polar
solvent selected from among the lower alcohols and water.
In order to obtain high yields of particularly pure products, it has proved
preferable
according to the invention to add, for the crystallisation, a solvent mixture
consisting
of a nonpolar organic solvent, preferably xylene or toluene, most preferably
toluene,
a polar organic solvent, preferably a lower alcohol such as methanol, ethanol
,
butanol or isopropanol, especially isopropanol and water. The ratio by volume
of
nonpolar to polar organic solvent to water can vary within the range from 1: 7
- 10 :
5 - 8, preferably 1: 8 - 9 : 6 - 7. By cooling to below 50 C, preferably to
about 30 C,
the crystallisation of the product of formula (II) is completed. After
isolation, the
crystallised product is optionally washed with the abovementioned lower
alcohol and
with water.
If the compounds of formula (II) are to be obtained from the compounds of
formula
(V) wherein X has a meaning other than hydroxy, the following procedure may be
used according to the invention.
The sodium or potassium phenoxide derived from the compounds of formula (VI)
is
taken up in water together with a compound of formula (V), mixed with a
nonpolar
organic solvent and optionally reacted under phase transfer conditions. The
phase
CA 02399598 2002-07-11
12
transfer catalysts which may be used according to the invention include the
quaternary ammonium salts, preferably the halides, sulphates or hydroxides of
tetradecyltrimethylammonium, hexadecyltrimethylammonium, tetrabutyiammoniurn,
tributylmethylammonium or triethylbenzylammonium . The nonpolar organic
solvent
may be a chlorinated hydrocarbon such as methylene chloride or preferably,
according to the invention, an aromatic hydrocarbon such as benzene, toluene,
xylene, preferably toluene. The compounds of formula (V) and (VI) are used in
a
virtually stoichiometric ratio, and if desired one of the two reactants may
also be
used in a slight excess (e.g. 15%). The amount of solvent to be used depends
on the
io quantity of educt put in. Between 1 and 2 L of water and between 0.3 and
1.0 L of
the organic solvent, preferably between 1.5 and 1.8 L of water and 0.5 to 0.7
L of the
organic solvent are used per mol of compound of formula (VI) put in.
The reaction is carried out with intensive stirring over a period of 3 to 9,
preferably 5
to 7 hours at a temperature of 50 to100 C, preferably at 70 to 80 C. Then, to
crystallise the product, a polar organic solvent, preferably a lower alcohol,
most
preferably isopropanol is added to the separated organic phase. By cooling to
below
50 C, preferably to about 30 C, the crystallisation of the product of formula
(li) is
completed. After being isolated, the crystallised product of formula (II) is
optionally
washed with the abovementioned lower alcohol and with water.
The starting compounds of formula (V)
X O ~ CN
~ (
(V)
which, as already mentioned, relate to one aspect of the present invention,
may be
prepared analogously to methods of synthesis known per se. The compound of
formula (V) wherein X denotes hydroxy may be prepared by reacting methyl 3-
halomethyl-benzoates with 4-hydroxybenzonitrile, for example, in a Wilkinson
ether
synthesis. The methyl 3-(4-cyano-phenoxy)-benzoate (VII) thus obtained can be
reductively converted into the compound of general formula (V) wherein X =
hydroxy
(= 4-(3-hydroxymethyl-benzyloxy)-benzonitrile) by the analogous use of current
standard procedures.
The compounds of formula (V) wherein X denotes chlorine or bromine may be
prepared analogously to methods of synthesis known per se from the compound of
formula (V) wherein X denotes hydroxy, using common halogenation reagents such
as, for example, thionylchloride, phosphorus oxychloride or phosphorus
CA 02399598 2002-07-11
13
pentachloride, methanesulphonic acid chloride, benzenesulphochloride,
preferably
thionylchloride or methanesulphonic acid chloride.
The compounds of formula (V) wherein X denotes mesylate, triflate or tosylate
may
be prepared analogously to methods of synthesis known per se from the compound
of formula (V) wherein X denotes hydroxy, by reacting with the appropriate
sulphonic
acid chlorides in aprotic, preferably polar organic solvents, preferably
selected from
among dichloromethane, N,N-dimethylacetamide, dimethylformamide, acetonitrile,
N-methylpyrrolidone, tetraalkylurea in the presence of organic bases,
preferably
selected from among dimethylaminopyridine, pyridine, methylpyridine, N-
lo methylpyrrolidine, trimethylamine, triethylamine, diisopropylethylamine and
DBU
(diazabicycloundecene).
The following Examples serve to illustrate methods of synthesis according to
the
invention, carried out by way of example, for preparing the compound of
formula (I).
They must be considered as possible methods given by way of example, without
restricting the invention to their content.
Example 1: Methyl 3-(4-cyano-phenoxymethyl)-benzoate:
10.00 kg (43.6mol) of methyl 3-(bromomethyl)benzoate and 5.21 kg (43.74mo1) of
4-
2o hydroxybenzonitriie are dissolved in 100 litres of acetone and stirred with
8.4 kg
(60.7mol) of potassium carbonate in the presence of 0.1 kg of sodium iodide
for
about 4 h under reflux conditions. Then 35 litres of acetone are distilled off
and 100
litres of water are added at reflux conditions. The reaction mixture is cooled
to 20 C
and the crystallisation is completed by the addition of another 30 litres of
water. The
crystals formed are separated off, washed with 50 litres of water and dried in
vacuo.
Yield: 11.1 kg (95%) of methyl 3-(4-cyano-phenoxymethyl)-benzoate;
Melting point 109...112 C, white solid,
TLC (silica gel 60 F254 -ready-made plate (Merck): Rf=0.5 (toluene :
acetone=9:1)
3o Example 2: 4-(3-hydroxymethyl-benzyloxy)-benzonitrile:
20.05 kg (26.7 mol) of methyl 3-(4-cyano-phenoxymethyl)-benzoate are dissolved
in
100 litres of THF and 40 litres of methanol. At 40 to 45 C, 8.51 kg of sodium
boranate are added in batches. The reaction is completed by stirring the
reaction
mixture at 61 to 63 C for about 5 hours. The reaction mixture is then cooled
to 25 C
and 90 litres of a 15% sodium hydroxide solution are added. After stirring,
the
aqueous supernatant is separated off and mixed with 30 litres of a 22.5%
sodium
hydroxide solution. After stirring, the aqueous supernatant is separated off
and from
this about 100 litres of solvent are distilled off at a sump temperature of 63
to 75 C.
CA 02399598 2002-07-11
14
The distillation residue is crystallised by the addition of 20 litres of
isopropanol at 50
to 60 C and 150 litres of water at 40 to 50 C. After the suspension has been
cooled
to 20 to 30 C the crystals are separated off, washed with 60 to 100 litres of
water
and batchwise with 25 litres of cold isopropanol and dried in vacuo.
Yield: 15.8 kg (88%) of 4-(3-hydroxymethyl-benzyloxy)-benzonitrile;
Melting point (DSC): 110-115 C, white solid
IR: 3444 /cm (OH band); 2229 /cm (CN band)
Example 3: 4-(3-Chloromethyl-benzyloxy)-benzonitrile:
Variant A:
7.18 g (30 mmol) of 4-(3-hydroxymethyl-benzyloxy)-benzonitrile are dissolved
in 80
ml of dichloromethane, mixed with 4.13g (35 mmol) of thionylchloride and 0.1 g
of
DMF and stirred while heating to 40 C until the development of gas has ceased.
After cooling, the organic reaction mixture is washed successively with water
and
dilute sodium hydroxide solution and crystallised by evaporation.
Yield: 6.8g (88%) of 4-(3-chloromethyl-benzyloxy)-benzonitrile;
TLC (silica gel 60 F254 -ready-made plate (Merck): Rf = 0.9 (toluene-acetone =
9:1),
Rf = 0.44 (toluene)
Variant B:
7.18 g (30 mmol) of 4-(3-hydroxymethyl-benzyloxy)-benzonitrile are dissolved
in 22
ml of N,N-dimethylacetamide, mixed with 4.47g (39 mmol) of methanesulphonic
acid
chloride and 3.95g (39mmol) of triethylamine and stirred for 10 hours at 20-30
C.
Then the triethylammonium chloride precipitated is filtered off, the filtrate
is mixed
with 30m1 of isopropanol and the desired 4-(3-chloromethyl-benzyloxy)-
benzonitrile is
crystallised by the metered addition of 30 ml of water. The suspension is
stirred for
15 min at 10 C and filtered. The crystals are washed with a mixture of 5 ml of
isopropanol and 20 ml water and dried at 20 C in vacuo.
Yield: 6.8g (88%) of 4-(3-chloromethyl-benzyloxy)-benzonitrile
Melting point: 65-68 C
Example 4: Sodium-4-{1-methyl-1-(4-tetrahydro-pyran-2-yloxy)-pheny1-ethyl}-
phenoxide
121.8 kg of bisphenol A are suspended in 480 1 of toluene and 461 of THF.
After the
addition of the catalyst (1.3 kg of 37% hydrochloric acid) , 44.9 kg of 3,4-
dihydro-
2H-pyran are metered in so as not to exceed a temperature of 40 C. The solid
then
goes into solution. Then the reaction mixture is mixed with 26. 4 kg of 45%
sodium
I~ I
CA 02399598 2002-07-11
hydroxide solution and 260 I of water. The organic upper phase is separated
off and
about 50 I of solvent are eliminated by distillation. At 30 to 40 C the
organic phase is
washed several times with dilute sodium hydroxide solution so that sufficient
purity
can be achieved (monitored by TLC). If the aqueous lower phase is in the pH
range
5 11.8 to 12.2, excess bisphenol A can easily be separated off.
The toluene phase purified by extraction is mixed with 11 litres of
isopropanol and 80
litres of water and heated to 50 to 55 C. By the addition of 47.4 kg of 45%
sodium
hydroxide solution and cooling the reaction mixture to 20 to 25 C, a crystal
suspension is obtained. The crystals are separated by filtration, washed with
about
io 160 I of toluene and then dried in vacuo.
Yield: 96.5 kg (54%) (as the tetra-hydrate)
Example 5: Synthesis of 4-f3-(4-{1-methyl-1-[4-(tetrahydro-pyran-2-yloxy)-
phenLril-
ethyl}-phenoxymethyl)-benzyloxyl-benzonitrile:
Variant A: (starting from Example 2)
28 kg (244 moi) of methanesulphonic acid chloride, 6 litres of N,N-
dimethylacetamide, 24.7 kg (244 mol) of triethylamine, 6 litres of N,N-
dimethylacetamide, 29.4 kg of 45% sodium hydroxide solution, 143 litres of N,N-
dimethylacetamide, 59.7 kg (178.5 mol) of Example 4(- sodium-4-{1-methyl-1-[4-
tetrahydro-pyran-2-yloxy)-phenyl]-ethyl}-phenoxide, as the tetrahydrate) are
metered
successively into a solution of 45 kg (188mo1) of 4-(3-hydroxymethyl-
benzyloxy)-
benzonitrile (Example 2) in 133 litres of N,N-dimethylacetamide at about -10
to 0 C.
Then the reaction mixture is stirred for 2 h at 25 C and for another 1.5 h at
75 to
80 C. After the addition of 32 litres of toluene, 255 litres of isopropanol
and 200
litres of water crystallisation begins which is completed by cooling to 30 C.
The
crystalline product is separated off by filtration, washed with isopropanol
and water
and then dried in vacuo. Yield: 85 kg (90%) of 4-[3-(4-{1-methyl-1-[4-
(tetrahydro-
pyran-2-yloxy)-phenyl]-ethyl}-phenoxymethyl)-benzyloxy]-benzonitrile;
Variant B: (starting from Example 3)
19.4 kg (50 moi) of Example 4(- sodium-4-{1-methyl-1-[4-tetrahydro-pyran-2-
yloxy)-
phenyl]-ethyl}-phenoxide, as the tetrahydrate) and 12.2 kg (47.5 mol) of
Example 3
(- 4-(3-chloromethyl-benzyloxy)-benzonitrile) are mixed with 85 litres of
water, a
phase transfer catalyst (e.g.: 2.1 kg (2.5 mol) of a 40% aqueous solution of
tetradecyltrimethylammonium bromide and 32 litres of toluene and intensively
stirred
for 6 h at about 80 C. Then 44 litres of isopropanol are metered into the
separated
organic upper phase, at 50 to 70 C, the crystal suspension obtained is cooled
to
CA 02399598 2002-07-11
16
about 25 C and filtered. The crystals separated off are washed twice with 25
litres of
cold isopropanol and dried in vacuo.
Yield: 22.8 kg (90%) of 4-[3-(4-{1-methyl-1-[4-(tetrahydro-pyran-2-yloxy)-
phenyl]-
ethyl}-phenoxymethyl )-benzyloxy]-benzonitrile;
Example 6: Ethyl {f4-(3-{4-f 1-(4-hydroxy-phenvl)-1-methyl-ethyll
phenoxymethyl}-
benzyloxy)-phenyll-imino-methyl}-carbaminate hydrochloride
132 kg (247 mol) of 4-(3-{4-[1-(4-tetrahydropyranyl-phenyl)-1-methyl-ethyl]-
phenoxymethyl}-benzyloxy)-benzonitrile (Example 5)are metered into a solution
of
io 45.5 kg (272mo1) of lithium-bis(trimethylsilyl-)amide in 266 kg of THF at
about 0 C.
The resulting suspension is stirred for about 10 h at about 25 C. The solid
then goes
into solution. After the addition of 68 litres of toluene the reaction mixture
is cooled
to -10 to 0 C and at this temperature 30.8 kg (284mo1) of ethyl chloroformate
are
added to the reaction vessel. Once the reaction has finished completely 24.3
kg of
37% hydrochloric acid (diluted with 50 litres of water) are metered in and
about 20
min later the aqueous lower phase is separated off. Crystallisation of the
intended
product is initiated by the subsequent addition of 106 litres of acetone, 48
litres of
methylisobutylketone and 24.3 kg of 37% hydrochloric acid.
123 kg (87%) of ethyl {[4-(3-{4-[1-(4-hydroxy-phenyl)-1-methyl-ethyl]-
phenoxymethyl}-benzyloxy)-phenyl]-imino-methyl}-carbaminate hydrochloride are
obtained after centrifuging, washing with acetone and drying in vacuo.
Melting point: 170-175 C.
Example 7: Ethyl {f4-(3-(4-f1-(4-hydroxy-pheny)-1-methyl-ethyl]-phenoxymethyl}-
benzyloxy)-phenyl]-imino-methyl}-carbaminate:
466 litres of acetone and 142 kg of ethyl {[4-(3-{4-[1-(4-hydroxy-phenyl)-1-
methyl-
ethyl]-phenoxymethyl}-benzyloxy)-phenyl]-imino-methyl}-carbaminate
hydrochloride
(Example 6) are added to a solution of 109 kg of trisodium citrate dihydrate
at 20 C.
After one hour's stirring the crystalline product is separated off by
filtration, washed
with water to remove any salts, washed again with about 100 litres of acetone
and
finally dried in vacuo.
116 kg (90%) of ethyl {[4-(3-{4-[1-(4-hydroxy-phenyl)-1-methyl-ethyl]-
phenoxymethyl}-benzyloxy)-phenyl]-imino-methyl}-carbaminate are obtained.