Note: Descriptions are shown in the official language in which they were submitted.
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
METHODS FOR THE PREVENTION AND TREATMENT OF INFECTIONS
USING ANTI-C3b(i) ANTIBODIES
1. INTRODUCTION
The present invention relates to methods of treating and preventing viral
infection,
microbial infection, or septic shock in an animal comprising administrating to
said animal
antibodies specific for C3b(i). The present invention also relates to methods
of treating and
preventing viral infection, microbial infection, or septic shoclc in an animal
comprising
administering to said animal IgG antibodies, IgM antibodies and/or complement
components in combination with antibodies immunospecific for C3b(i). The
present
invention also relates methods of treating and preventing viral infection or
microbial
infection in an animal comprising administrating to said animal antibodies
that
irnmunospecifically bind to one or more viral antigens or microbial antigens,
respectively,
in combination with antibodies immunospecific for C3b(i). The present
invention also
relates methods of treating and preventing septic shock in an animal
comprising
administrating said anmal antibodies that immunospecif cally bind to
lipopolysaccharide,
an endotoxin or a constituent of the outer wall of a gram negative bacteria in
combination
with antibodies immunospecific for C3b(i). The present invention further
relates to
pharmaceutical compositions for the treatment and prevention of viral
infection, microbial
infection, and septic shock comprising antibodies immunospecific for C3b(i).
2. BACKGROUND OF THE INVENTION
The complement system which is composed of some 21 plasma proteins plays an
important role in the human immune system, both in the resistance to
infections and in the
pathogenesis of tissue injury. The activated products of the complement system
attract
phagocytic cells and greatly facilitate the uptake and destruction of foreign
particles by
opsouzation. There are two distinct pathways for activating complement, the
classical
pathway and the alternate pathway, that result in conversion of C3 to C3b and
subsequent
responses (e.g., the formation of the membrane attack complex ("MAC")).
Activation of
the classical pathway is initiated by antigen-antibody complexes or by
antibody bound to
cellular or particulate antigens. The alternate pathway is activated
independent of antibody
by complex polysaccharides in pathogens such as bacterial wall constituents,
bacterial
lipopolysaccharides (LPS), and cell wall constituents of yeast (zymosan).
The classic complement pathway is initiated by the binding of C1 to immune
complexes containing IgG or IgM antibodies. Activated CI cleaves C2 and C4
into active
components, C2a and C4b. The C4b2a complex is an active protease called C3
convertase,
-1-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
and acts to cleave C3 into C3a and C3b. C3b forms a complex with C4b2a to
produce
C4b2a3b, which cleaves CS into CSa and CSb. CSb combines with C6, and the CSb6
complex combines with C7 to form the ternary complex CSb67. The CSb67 complex
binds
C8 to form the CSb678 complex which in turn binds C9 and results in the
generation of the
$ CS-C9 MAC. The insertion of the MAC into the cell membrane results the
formation of a
transmembrane channel that causes cell lysis.
In the alternative pathway, conversion of C3 to C3b (or C3i) produces a
product that
can combine with factor B, giving C3bB (or C3iB). These complexes are acted
upon by
factor D to generate C3bBb, which is a C3 convertase capable of cleaving more
C3 to C3b,
leading to more C3bBb and even more C3 conversion. Under certain circumstances
the
C3bBb complex is stabilized by association with the positive regulator
properdin (P) by
association of C3b and Bb. The C3 convertases can associate with an additional
C3b
subunit to form the CS convertase, C3bBbC3b, which is active in the production
of the
CS-C9 MAC.
In both the classical and alternative pathways, the critical step in the
activation of
complement is the proteolytic conversion of C3 to the fragments C3b and C3a.
C3a is an
anaphylatoxin that attracts mast cells to the site of challenge, resulting in
local release of
histamine, vasodilation and other inflammatory effects. The nascent C3b has an
ability to
bind to surfaces around its site of generation and functions as a ligand for
C3 receptors
mediating, for example, phagocytosis.
Endogenous cell surfaces normally exposed to complement are protected by
membrane-bound regulators such as decay accelerating factor ("DAF"), C59
("protectin"),
MCP, and the soluble C1 inhibitor or C1NH. DAF and MCP are responsible for
limiting
production of C3b and insure the generation of inactive forms of C3b, C3bi and
C3dg from
C3b. CD59 prevents attack of the MAC, which would otherwise destroy the cancer
cell.
C1 inhibitor binds to the active subcomponents of C1, Clr and Cls, and
inhibits their
activity.
2.1. CANCER TREATMENT
Despite advances in prevention and early detection, refinements in surgical
30 technique, and improvements in adjuvant radiotherapy and chemotherapy, the
ability to cure
many patients of cancer remains elusive. This is especially pertinent to
prostate cancer,
which remains the most prevalent visceral tumor in American men, with
approximately
180,000 new cases and nearly 40,000 deaths expected in 1999 (Landis et al.,
1999, Cancer J
Clin 49: 8-31). The continuing challenge of prostate cancer treatment is the
successful
35 management and eradication of recurrent, metastatic, and hormone-refractory
disease,
-2-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
which accounts for the vast majority of prostate cancer-specific morbidity and
mortality
(Small, 1998, Drugs and Aging13:71-81).
Many treatment modalities currently under investigation for prostate and other
cancers depend upon tissue-specific delivery of anti-neoplastic agents. One
S immunotherapeutic approach involves conjugating cytotoxic agents to
monoclonal
antibodies (mAbs) specific for a particular cancer cell epitope. In this
manner, the
therapeutic agents can be delivered at a high therapeutic dose directly, and
selectively, to
the tumor site, thereby minimizing injury to healthy tissue (Bach et al.,
1993, Tm_m__unol
Today 14:421-S; Reithmuller et al., 1993, Cur. Op. Immunol 5:732-9; and Gruber
et
a1.,1996, Spring Sem Immunopath 18:243-S1). This method first requires the
identification
of specific epitopes for each cancer type. Such candidate epitopes must be
expressed at
high levels on the cancer cells compared to normal tissue. Second, this method
requires the
development of high affinity mAbs specific for these epitopes and these mAbs
must show
minimal cross-reactivity with self tissue. The biological mechanism of killing
with mAbs
1 S will be variable, depending upon the epitopes identified on the cancer
cells, and the effector
functions of the specific mAb isotype. However, due to antigenic modulation
and/or
mutation, the cancer cells may reduce the available levels of the target
epitope per cell, or
eliminate it from their surface altogether. Thus, the use of mAbs in cancer
diagnosis and
treatment remains problematic.
A more widely applicable approach to treatment of cancer with mAbs would be to
identify a ubiquitous antigenic site, present on virtually all cancer cells,
and then to develop
a panel of mAbs specific for this antigen. A voluminous literature reveals
that cancer cells
share certain common characteristics. Many types of human cancer cells are
characterized
by substantial abnormalities in the glycosylation patterns of their cell-
surface proteins and
2S lipids (Halcomori et. al., 1996, Canc Res. S6:S309-18; Castronovo et
a1.,1989, J Nat Canc
Inst 81:212-6; Springer et a1.,1984, Science 224:1198-206; and Springer et
al., 1997, J Mol
Med 75:594-602). These differences have led to the identification of antigenic
determinants
on cancer cells which are expressed at far lower levels on normal cells.
Natural IgM
antibodies to these epitopes are present in the circulation, and the
interaction of such IgM
antibodies with these cancer cell surface antigens leads to activation of
complement and
covalent coupling of complement activation products (C3b and its fragments,
collectively
referred to as C3b(i)) to the tumor cells (Okada et al., 1974, Nature 248:521-
2S; Irie et. al.,
1974, Science 186:454-456; Desai et al., 1995, J Immunol Methods 188:175-8S;
Vetvicka
et al., 1996, J Clin Invest 98:50-61; Vetvicka et al., 1997, J Iminunol
1S9:S99-605; and
3S Vetvicka et al., 1999, Clin Exp Immunol 115:229-3S). Although relatively
large amounts
of C3b(i) can be deposited on cancer cells, the concomitant expression of high
levels of
-3-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
membrane-associated complement control proteins (e.g., decay accelerating
factor ("DAF"),
membrane cofactor protein ("MCP"), and, in particular, "protectin" i.e., CD59)
usually
prevents complement-mediated lysis (Cheung et al., 1988, J Clin Invest 81:1122-
8; Gorter
et al., 1996, Lab Invest 74:1039-49; Maenpaa et al., 1996, Am J Path 148:1139-
52; and Li
et a1.,1997, Int J Canc 71:1049-55). Further, several investigators have
established that in
most cases, cancer patients have substantially lowered levels of the
potentially protective
IgM antibodies. Thus, in many cases the cancer cells cannot easily be killed
by
complement activation because of the reduced levels of protective IgM antibody
and the
increased expression of human complement control proteins on their surface.
2.2. ROLE OF COMPLEMENT IN INFECTIONS
Innate immunity allows humans and most animals to respond to foreign organisms
even before the initiation of the immune response. One of the most important
aspects of
innate immunity involves the complement system (Cooper, N. R. Complement and
viruses.
Volanalcis, J. B. and Frank, M. M. The Human Complement System in Health and
Disease.
18, 393-408. (1998) New York, Marcel Dekker, Inc.; and Petry, F. and Loos, M.
Bacteria
and complement. Volanakis, I. E. and Frank, M. M. The Human Complement System
in
Health and Disease. 171, 375-392. (1998) New York, Marcel Dekker, Inc.). It is
now well-
documented that a wide variety of bacteria, viruses and other microorganisms
activate
complement in the immunologically naive individual (Peterson et al., 1978,
Infect. Immun.
19:943; Verbrugh et al., 1982, J. Tt'nmunol. 129:1681; Newman et al., 1985, J.
Exp. Med.
161:1414; Seelen et al., 1995, Immunol. 84:653; Wagner et al., 1998, J.
Immunol.
160:1936; Bakker et al.,. 1992, AIDS 6:35; Hiavacek et al., 1999, Proc. Nat!.
Acad. Sci.
96:14681; Schmitz et al., 1994, J. Immunol. 153:1352; Tacnet-Delorme et al.,
1999, J.
I~unol. 162:4088; Joling et al.,. 1993, J. Immunol. 150:1065; Dominguez et
al., 1999, J.
Exp. Med. 189:25; and Washbum et a1.,1991, Molec. Immunol. 28:465). These
include, but
are not limited to, both gram positive and gram negative bacteria, and the
virus which
causes AIDS, HIV. As a consequence of complement activation, these organisms
are
covalently labeled with complement activation fragments, and in particular
with C3b(i).
Moreover, in individuals with previous exposure to these pathogens (either
through
immunization or previous infection), specific antibodies will bind to the
pathogens and also
activate complement and promote C3b(i) deposition. Although in many instances
this
complement activation ultimately leads to the clearance and destruction of the
microorganism, in some cases the C3b(i)-labeled invader remains in the
circulation and can
still infect susceptible tissues and organs. In fact, there are many examples
of
microorganisms which actually use these covalently attached C3b(i) molecules
to gain entry
-4-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
into cells (which have receptors for C3bCi)) and establish productive
infections (Bakker et
al., 1992, AIDS 6:35; Tacnet-Delorme et al., 1999, J. Immunol. 162:4088;
Cooper, N. R.
Complement and viruses. Volanakis, J. B. and Frank, M. M. The Human Complement
System in Health and Disease. 18, 393-408. 1998. New York, Marcel Dekker,
Inc.; and
Petry, F. and Loos, M. Bacteria and complement. Volanakis, I. E. and Frank, M.
M. The
Human Complement System in Health and Disease. 171, 375-392. (1998) New York,
Marcel Dekker, Inc.). Several lines of evidence indicate that C3b(i)-opsonized
HIV can
persist in the body bound to the surface of dendritic cells fox very long
periods of time, and
this cell-bound HIV may represent one of the main obstacles to the permanent
elimination
of HIV from the body (Hiavacek et al., 1999, Proc. Nat!. Acad. Sci. 96:14681;
and Schmitz,
et al.,. 1994,. J. Immunol. 153:1352).
Citation of a reference in this section or any section of this application
shall not be
construed as an admission that such reference is prior art to the present
invention.
3. SUMMARY OF THE INVENTION
The present invention encompasses compositions comprising antibodies
immunospecific for C3b(i) and methods for the treatment and prevention of
viral infection,
microbial infection and septic shock in an animal comprising administering
such
compositions to said animal. In particular, the present invention provides
methods for
treating or preventing viral infection, microbial infection, and septic shock
in animal
comprising administering to said animal a therapeutically or prophylactically
effective
amount of an antibody immunospecific for C3b(i), an antibody immunospecific
for C3b(i)
linked (e.g., covalently linked) to a second molecule (e.g., an IgM antibody,
an IgG
antibody, a glycoprotein or a glycolipid), a nucleic acid sequence encoding an
antibody
immlospecific for C3b(i), or a nucleic acid sequence encoding an antibody
immunospecific for C3b(i) linked (e.g., covalently linked) to a second
molecule. The
present invention also provides methods for the treatment or prevention of
viral infection,
microbial infection or septic shock in an animal comprising administering to
said animal
IgG antibodies, IgM antibodies and/or complement components in combination
with
anti-C3b(i) antibodies. The present invention also provides methods for the
treatment or
prevention of viral infection or microbial infection in an animal comprising
administering to
said animal antibodies immunospecific for one or more viral antigens or
microbial antigens,
respectively, in combination with anti-C3b(i) antibodies. The present
invention also
provides methods for the treatment or prevention of septic shock in an animal
comprising
administering to said animal antibodies immunospecific for lipopolysaccharide,
an
-5-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
endotoxin, or a constituent of the outer wall of a gram negative bacteria
(e.g., a constituent
of the outer wall of SlZigella or E. coli) in combination with anti-C3b(i)
antibodies.
The present invention also provides compositions comprising one or more
antibodies immunospecific for C3b(i) or C3b(i) linked to a second molecule, in
an amount
effective for the treatment or prevention of a viral infection, a microbial
infection, or septic
shock in an animal. The present invention also provides compositions
comprising one or
more nucleic acid molecules encoding one or more antibodies immunospecific for
C3b(i) or
C3b(i) linked to a second molecule, in an amount effective for the treatment
or prevention
of a viral infection, a microbial infection, or septic shock in an animal. The
present
invention further provides a composition comprising one or more bispecific
antibodies
which are immunospecific for C3b(i) or C3b(i) linked to a second molecule and
an effector
cell receptor or antigen, in an amount effective for the treatment or
prevention of a viral
infection, a microbial infection or septic shock in an animal.
3.1. DEFINITIONS
The term "C3b(i)" as used herein refers to C3b and its fragments, including,
but not
limited to, C3b(i), C3b, and C3d.
As used herein, the terms "antibody immunospecific for C3b(i)", "antibodies
immunospecific for C3b(i)", "C3b(i) specific antibodies", "anti-C3b(i)
antibodies" and the
like refer one or more antibodies that specifically bind to C3b(i), a fragment
of C3b(i),
C3b(i) linked (e.g., covalently linked) to a second molecule, or a fragment of
C3b(i) linked
(e.g., covalently linked) to a second molecule and do not non-specifically
bind to
polypeptides unrelated to C3b(i). Anti-C3b(i) antibodies may cross-react with
other, non-
C3b(i) antigens. Preferably, anti-C3b(i) antibodies do not cross-react with
other antigens.
Anti-C3b(i) antibodies can be identified, for example, by immunoassays or
other techniques
known to those of skill in the art. In a preferred embodiment, anti-C3b(i)
antibodies
immunospecifically bind to C3b(i) covalently linked to a second molecule. In
another
preferred embodiment, anti-C3b(i) antibodies preferentially bind to C3b(i)-
opsonized
cancer cells, and not to C3b(i) in the milieu. In another preferred
embodiment, anti-C3b(i)
30 antibodies preferentially bind to C3b(i) deposited on viruses or microbes,
and not C3b(i) in
the milieu.
In certain embodiments, the "second molecule" that C3b(i) is linked to is IgG
or
IgM. In certain other embodiments, the "second molecule" that C3b(i) is linked
to is a
cancer cell antigen such as, e.g., a protein, glycoprotein, peptide,
polypeptide, or glycolipid
35 differentially or preferentially expressed by cancer cells. In certain
other embodiments, the
-6-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
"second molecule" that C3b(i) is linked to is a microbial or viral antigen.
Preferably, C3b(i)
is covalently linked to a second molecule.
The term "antibodies immunospecific for one or more cancer antigens, viral
antigens, or microbial antigens" and the like as used herein refer to
antibodies thereof that
immunospecifically bind to cancer antigens, viral antigens, or microbial
antigens and do not
non-specifically bind to other polypeptides. Antibodies that
immunospecifically bind to
cancer antigens, viral antigens, or microbial antigens may have cross-
reactivity with other
antigens. Preferably, antibodies that immunospecifically bind to cancer
antigens, viral
antigens, or microbial antigens do not cross-react with other antigens.
Antibodies that
i~~ospecifically bind to cancer antigens, viral antigens, or microbial
antigens can be
identified, for example, by immunoassays or other techniques lcnown to those
of skill in the
art.
The term "fragment" as used herein refers to a peptide or polypeptide
comprising an
amino acid sequence of at least 5 contiguous amino acid residues, at least 10
contiguous
amino acid residues, at least 15 contiguous amino acid residues, at least 20
contiguous
amino acid residues, at least 25 contiguous amino acid residues, at least 40
contiguous
amino acid residues, at least 50 contiguous amino acid residues, at least 60
contiguous
amino residues, at least 70 contiguous amino acid residues, at least
contiguous 80 amino
acid residues, at least contiguous 90 amino acid residues, at least contiguous
100 amino acid
residues, at least contiguous 125 amino acid residues, at least 150 contiguous
amino acid
residues, at least contiguous 175 amino acid residues, at least contiguous 200
amino acid
residues, or at least contiguous 250 amino acid residues of the amino acid
sequence of a
cancer cell peptide or polypeptide (e.g., a peptide or polypeptide
preferentially or
differentially expressed on a cancer cell), a viral peptide or polypeptide, a
microbial peptide
or polypeptide or an antibody that immunospecifically binds to C3b(i).
The term "fusion protein" as used herein refers to a polypeptide that
comprises an
amino acid sequence of an antibody or fragment thereof and an amino acid
sequence of a
heterologous polypeptide (i. e., an unrelated polypeptide).
The term "host cell" as used herein refers to the particular subject cell
transfected
with a nucleic acid molecule and the progeny or potential progeny of such a
cell. Progeny
of such a cell may not be identical to the paxent cell transfected with the
nucleic acid
molecule due to mutations or environmental influences that may occur in
succeeding
generations or integration of the nucleic acid molecule into the host cell
genome.
An "isolated" or "purified" antibody or polypeptide is substantially free of
cellular
material or other contaminating proteins from the cell or tissue source from
which the
protein is derived, or substantially free of chemical precursors or other
chemicals when
_7_
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
chemically synthesized. The language "substantially free of cellular material"
includes
preparations of an antibody or a polypeptide in which the antibody or
polypeptide is
separated from cellular components of the cells from which it is isolated or
recombinantly
produced. Thus, an antibody or a polypeptide that is substantially free of
cellular material
includes preparations of antibody or polypeptide having less than about 30%,
20%, 10%, or
5% (by dry weight) of heterologous protein (also referred to herein as a
"contaminating
protein"). When the antibody or polypeptide is recombinantly produced, it is
also
preferably substantially free of culture medium, i.e., culture medium
represents less than
about 20%, I O%, or 5% of the volume of the protein preparation. When the
antibody or
polypeptide is produced by chemical synthesis, it is preferably substantially
free of
chemical precursors or other chemicals, i. e., it is separated from chemical
precursors or
other chemicals which are involved in the synthesis of the protein.
Accordingly such
preparations of the antibody or polypeptide have less than about 30%, 20%,
10%, 5% (by
dry weight) of chemical precursors or compounds other than the antibody or
polypeptide
fragment of interest. In a preferred embodiment, antibodies of the invention
are isolated or
purified.
An "isolated" nucleic acid molecule is one which is separated from other
nucleic
acid molecules which are present in the natural source of the nucleic acid
molecule.
Moreover, an "isolated" nucleic acid molecule, such as a cDNA molecule, can be
substantially free of other cellular material, or culture medium when produced
by
recombinant techniques, or substantially free of chemical precursors or other
chemicals
when chemically synthesized. In a preferred embodiment, nucleic acid molecules
encoding
antibodies of the invention are isolated or purified.
In certain embodiments of the invention, a "prophylactically effective amount"
is the
amount of a composition of the invention that reduces the incidence of cancer,
viral
infection, microbial infection, or septic shock in an animal. Preferably, the
incidence of
cancer, viral infection, microbial infection or septic shock in an animal is
reduced by at least
2.5 %, at least 5 %, at least 10 %, at least 15%, at least 25 %, at least 35
%, at least 45%, at
least 50 %, at least 75%, at Ieast 85 %, by at least 90 %, at least 95 %, or
at least 99 % in an
a~mal administered a composition of the invention relative to an animal or
group of
animals (e.g., two, three, five, ten or more animals) not administered a
composition of the
invention.
In certain embodiments of the invention, a "therapeutically effective amount"
is the
amount of a composition of the invention that reduces the severity, the
duration and/or the
symptoms associated with cancer, viral infection, microbial infection, or
septic shock in an
animal. In certain other embodiments of the invention, a "therapeutically
effective amount"
_g_
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
is the amount of a composition of the invention that results in a reduction in
viral titer or
microbial titer by at least 2.5 %, at least 5 %, at least 10 %, at least 15%,
at least 25 %, at
least 35 %, at least 45%, at least 50 %, at least 75%, at least 85 %, by at
least 90 %, at least
95 %, or at least 99 % in an animal administered a composition of the
invention relative to
the viral titer or microbial titer in an animal or group of animals (e.g.,
two, three, five, ten or
more animals) not administered a composition of the invention. In certain
other
embodiments, a "therapeutically effective amount" is the amount of a
composition of the
invention that results in a reduction of the growth or spread of cancer by at
least 2.5 %, at
least 5 %, at least 10 %, at least 15%, at least 25 %, at least 35 %, at least
45%, at least 50
%, at least 75%, at least 85 %, by at least 90 %, at least 95 %, or at least
99 % in an animal
administered a composition of the invention relative to the growth or spread
of cancer in an
animal or group of animals (e.g., two, three, five, ten or more animals) not
administered a
composition of the invention.
4. BRIEF DESCRIPTION OF THE FIGURES
FIG. 1 (A-D). Representative flow cytometry data from a study with serum from
a
normal donor (A, B) and a cancer patient (C, D). Measurement of C3b(i) (A, C)
and IgM
(B, D) deposition on C4-2 human prostate cancer cells is shown. Abundant
C3b(i) is
deposited on C4-2 cancer cells in response to the addition of normal human
senun; this
opsonization appears to be facilitated by both the classical and alternative
complement
pathways. After opsonization with serum from a prostate cancer patient,
significantly less
C3b(i) and IgM are deposited on the tumor cells (C, D). C3b(i) deposition via
the
alternative pathway (serum with Mg-EGTA), however, is comparable for both the
normal
and cancer patient serum, suggesting that the alternative pathway of the
complement system
remains intact in prostate cancer patient serum.
FIG. 2 (A-B). Flow cytometry (A) and radioimmunoassay (B) data demonstrating
that removal of IgM from AB-positive serum results in a large reduction in the
amount of
C3b(i) that is deposited on LNCaP (A) or C4-2 (B) cells. C3b(i) deposition can
be restored
with either whole normal human plasma (A, B) (e.g., plasma / IgM-depleted
serum), which
provides a source of human IgM, or with purified IgM/IgM-depleted serum (B).
FIG. 3 (A-B). Radioimmunoassay data demonstrating that complement activation
generates between 50,000 and 500,000 C3b(i) epitopes/opsonized cancer cell
(net binding,
background subtracted), as defined by binding of both'ZSI-labeled mAb 7C12 and
3E7. In
panel A, an AB-positive serum was used for opsonization and, after three
washes, cells were
probed with differing amounts of the two mAb. Alternatively, mAb 3E7 was added
to the
cells just before the serum, and was present during opsonization. In panel B,
AB-positive
-9-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
serum was used in conjunction with the 1-~,g/ml and 10-~,g/ml probes, and an
AB-positive
citrated plasma (different donor from that in A) was used for the 3-~.g/ml
probes
FIG. 4. Flow cytometry results from surveys of sera from normal donors and
patients with prostate cancer. Binding of human immunoglobulin to LNCaP and C4-
2
prostate cancer cells was measured. Significant differences were determined by
t-tests.
FIG. 5 (A-B). Immunohistochemical staining of normal (A) and neoplastic (B)
human prostate tissue after incubation with anti-C3b(i) mAb.
FIG. 6. Rosetting experiment using erythrocytes and opsonized LNCaP prostate
cancer cells incubated in plasma in the presence of an anti-CRl X anti-C3b(i)
bispecific
monoclonal antibody complex (7G9 X 3E7).
FIG. 7 (A-B). In vitro killing of LNCaP (A) and C4-2 (B) prostate cancer cells
using'3'I_labeled mAbs. Dashed line (----) delineates normal serum opsonized
cells treated
with'31I-labeled irrelevant mAbs; dotted line (....) delineates non-opsonized
cells treated
with'3'I-anti-Cb3(i) mAbs; solid line (-) delineates normal serum opsonized
cells treated
1 S with'3'I-labeled anti-C3b(i) mAbs. Measured as cell proliferation relative
to non-treated
cells.
FIG. 8. The schematic illustrates the steps of the invention, all of which
occur on
the cell surface of tumor cells within the body of the cancer patient. In the
first step, human
IgM (either endogenous, or infused into the patient) binds to specific sites
on the cancer
cell. In the second step, complement (either endogenous, or infused into the
patient as fresh
plasma) is activated, and the resulting proteolytic fragment C3b(i) is
deposited on the
surface of the cancer cell. In the third step, a mAb specific for the C3b(i)
epitope is
administered. The mAb can be associated with a toxic, enzymatic, genetic,
differentiating,
and/or imaging agent (therefore it is an "effector mAb"), which results in the
destruction or
imaging of the cancer cell.
FIG. 9. Red cell binding experiment using erythrocytes and opsonized Rajii
cells.
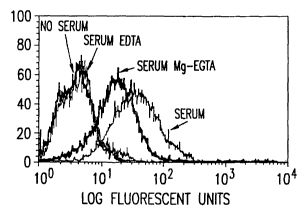
Rajii cells treated with: serum alone; EDTA treated serum alone; serum and
Rituximab;
EDTA treated serum and Rituximab; serum and anti-C3b(i) X anti-CRl bispecific
monoclonal antibody complexes; EDTA treated serum and anti-C3b(i) X anti-CRl
bispecific monoclonal antibody complexes; serum, Rituximab, and anti-C3b(i) X
anti-CR1
bispecific monoclonal antibody complexes; and EDTA treated serum Rituximab,
and anti-
C3b(i) X anti-CRl bispecific monoclonal antibody complexes.
FIG. 10. Red cell binding experiment using erythrocytes and opsonized Rajii
cells.
Rajii cells treated with: washed whole blood reconstituted serum alone; washed
whole
blood reconstituted serum and Rituximab; washed whole blood reconstituted
serum and
anti-C3b(i) X anti-CRl bispecific monoclonal antibody complexes; and washed
whole
-10-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
blood reconstituted serum, Rituximab, and anti-C3b(i) X anti-CRl bispecific
monoclonal
antibody complexes; washed whole blood reconstituted serum, Rituximab (15'),
and anti-
C3b(i) X anti-CRl bispecific monoclonal antibody complexes; and washed whole
blood
reconstituted serum, anti-C3b(i) X anti-CRl bispecific monoclonal antibody
(15'), and
Rituximab.
5. DETAILED DESCRIPTION OF THE INVENTION
The present invention encompasses compositions comprising one or more
antibodies
immunospecific for C3b(i) and the use of such compositions in the treatment
and prevention
of cancer, viral infection, microbial infection, and septic shock. The present
invention
provides methods for treating or preventing cancer, viral infection, microbial
infection, or
septic shock in an animal, said methods comprising administering to said
animal a
therapeutically or prophylactically effective amount of one or more antibodies
immunospecific for C3b(i) or C3b(i) linked to a second molecule. The present
invention
provides methods for treating or preventing cancer, viral infection, microbial
infection, or
septic shock in an animal, said methods comprising administering to said
animal IgG
antibodies, IgM antibodies, and/or one or more complement components in
combination
with a therapeutically or prophylactically effective amount of one or more
antibodies
immunospecific for C3b(i) or C3b(i) linked to a second molecule. The present
invention
also provides methods for treating or preventing cancer, viral infection, or
microbial
infection in an animal, said methods comprising administering to said animal
one or more
antibodies immunospecific for one or more cancer antigens, viral antigens, or
microbial
antigens, respectively, in combination with one or more antibodies
immunospecific for
C3b(i) or C3b(i) linked to a second molecule. The present invention further
provides
methods for treating or preventing septic shock in an animal, said methods
comprising
administering to said animal one or more antibodies immunospecific for
lipopolysaccharide
(LPS), an endotoxin, or a constituent of the outer wall of a gram negative
bacteria (e.g., the
outer wall of E. coli) in combination with one or more antibodies
irrununospecific for
C3b(i) or C3b(i) lii~lced to a second molecule.
In accordance with the present invention, antibodies immunospecific for C3b(i)
are
administered to an animal, preferably a mammal and most preferably a human, to
treat or
prevent cancer, viral infection, microbial infection, or septic shock. The
antibodies of the
present invention comprise monoclonal antibodies, polyclonal antibodies,
bispecific
antibodies, humanized antibodies, human antibodies, chimeric antibodies,
single chain
antibodies, sFvs, idiotypic antibodies, Fab fragments, and F(ab') fragments,
fragments
produced by a Fab expression library, and epitope-binding fragments. In a
specific
embodiment, monoclonal antibodies immunospecific for C3b(i) are administered
to an
-11-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
animal, preferably a mammal and most preferably a human, to treat or prevent
cancer, viral
infection, microbial infection, or septic shock. In a preferred embodiment,
monoclonal
antibodies immunospecific for C3b(i) covalently linked to a second molecule
(e.g., an IgM
antibody, an IgG antibody, a glycoprotein or glycolipid) are administered to
an animal,
preferably a mammal and most preferably a human, to treat or prevent cancer,
viral
infection, microbial infection, or septic shock. In another embodiment,
monoclonal
antibodies immunospecific for C3b(i) are conjugated to a therapeutic moiety
such as an
antibiotic, a chemotherapeutic cytotoxin, e.g., a cytostatic or cytocidal
agent (e.g.,
paclitaxol, cytochalasin B or diphtheria toxin), a thrombotic or anti-
angiogenic agent or a
radioactive label. In another embodiment, monoclonal antibodies immunospecific
for
C3b(i) are conjugated to a detectable substrate such as, e.g., an enzyme,
fluorescent marker,
luminescent material, bioluminescent material, or radioactive material. In yet
another
embodiment, the valency of monoclonal antibodies immunospecific for C3b(i) are
increased
to that, for example, of a dimer or an IgM-like pentamer.
In a preferred embodiment, bispecific antibodies which are immunospecific for
C3b(i) and an effector cell receptor or antigen are administered to an animal,
preferably a
mammal and most preferably a human, to treat, or prevent cancer, viral
infection, microbial
infection, or septic shock. The term "effector cell" as used herein refers to
a cell which is
involved in a cell-mediated immune response. Examples of effector cells
include, but are
not limited to, monocytes, macrophages, dendritic cells, neutrophils, natural
killer cells,
lymphocytes and erythrocytes. In one embodiment, anti-C3b(i) heteropolymer
constructs
(bispecific monoclonal complexes) bound ex vivo to an effector cell via a cell
surface
receptor are administered to an animal, preferably a mammal and most
preferably a human,
to treat or prevent cancer, viral infection or microbial infection. Cell
surface receptors
include, but are not limited to, CRl, CR2, CR3, CR4, human Fc~y receptors
CDlb, CD32
and CD64, and the Fc receptor for IgA, CD89. In a preferred embodiment, anti-
C3b(i)
heteropolymer constructs bound ex vivo to erythrocytes via CRl are
administered to an
animal, preferably a mammal and most preferably a human, to treat, inhibit or
prevent
cancer, viral infection, microbial infection, or septic shock.
In a preferred embodiment, bispecific diabodies which are antibody fragments
immunospecific for C3b(i) and a complement component are administered to an
animal,
preferably a mammal and most preferably a human, to treat or prevent cancer,
viral
infection, microbial infection, or septic shock. In accordance with this
embodiment, the
diabodies are capable of recruiting complement components. In another
embodiment,
bispecific diabodies which are ixnmunospecific for C3b(i) and Clq are
administered to an
animal, preferably a mammal and most preferably a human, to treat or prevent
cancer, viral
-12-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
infection, microbial infection, or septic shock. Methods of preparing
diabodies are taught in
U.S. Patent No. 5,37,242, which is incorporated herein in its entirety.
The present invention encompasses the administration of IgG antibodies and/or
IgM
antibodies in combination with one or more anti-C3b(i) antibodies to animals
to facilitate
the opsonization of cancer cells, viruses, and microbes. In one embodiment,
IgG antibodies
and/or IgM antibodies are administered to an animal prior to, subsequent to,
or
concomitantly with the administration of one or more antibodies immunospecific
for C3b(i)
to treat or prevent cancer, viral infection, microbial infection, or septic
shock. In a preferred
embodiment, IgG antibodies and/or IgM antibodies are administered to an animal
prior to
the administration of one or more antibodies immunospecific for C3b(i) to
treat or prevent
cancer, viral infection, microbial infection or septic shock. In a specific
embodiment, one
or more antibodies immunospecific for one or more cancer cell antigens are
administered to
an animal prior to, subsequent to, or concomitantly with the administration of
one or more
antibodies immunospecific for C3b(i), in an amount effective for the treatment
or
prevention cancer. In a preferred embodiment, one or more antibodies
immunospecific for
one or more cancer cell antigens are administered to an animal prior to the
administration of
one or more antibodies immunospecific for C3b(i), in an amount effective for
the treatment
or prevention cancer. Examples of cancer cell antigens include, but are not
limited to,
improperly glycosylated cell-surface proteins and lipids.
In a specific embodiment, one or more antibodies used in cancer immunotherapy
are
administered to an animal prior to, subsequent to, or concomitantly with the
administration
of one or more anti-C3b(i) antibodies, in an amount effective for the
treatment or prevention
cancer. In a preferred embodiment, one or more antibodies used in cancer
immunotherapy
are administered to an animal prior to the administration of one or more anti-
C3b(i)
antibodies, in an amount effective for the treatment or prevention cancer.
Examples of
antibodies used in cancer immunotherapy include, but are not limited to:
Herceptin~
(Trastuzumab; Genetech, CA) which is a humanized anti-HER2 monoclonal antibody
for
the treatment of patients with metastatic breast cancer; Retuxan~ (rituximab;
Genentech)
which is a chimeric anti-CD20 monoclonal antibody for the treatment of
patients with non-
Hodgkin's lymphoma; OvaRex (AltaRex Corporation, MA) which is a marine
antibody for
the treatment of ovarian cancer; Panorex (Glaxo Wellcome, NC) which is a
marine IgGZa
antibody for the treatment of colorectal cancer; BEC2 (ImClone Systems Inc.,
NY) which is
marine IgG antibody for the treatment of lung cancer; IMC-C225 (Imclone
Systems Inc.,
N~ which is a chimeric IgG antibody for the treatment of head and neck cancer;
Vitaxin
(MedImmune, Inc., MD) which is a humanized antibody for the treatment of
sarcoma;
Campath I/H (Leukosite, MA) which is a humanized IgGI antibody for the
treatment of
chronic lymphocytic leukemia (CLL); Smart MI95 (Protein Design Labs, Inc., CA)
which is
-13-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
a humanized IgG antibody for the treatment of acute myeloid leukemia (AML);
LyrnphoCide (Immunomedics, Inc., NJ) which is a humanized IgG antibody for the
treatment of non-Hodgkin's lymphoma; Smart I D 10 (Protein Design Labs, Inc.,
CA) which
is a humanized antibody for the treatment of non-Hodgkin's lymphoma; Oncolym
(Techniclone, Inc., CA) which is a marine antibody for the treatment of non-
Hodgkin's
lymphoma; the monoclonal antibody 7E11 which immunospecif cally binds to
prostate-
specific membrane antigen (PSMA; Lin et al., 1997, Cancer Res. 57:3629); and
the anti-
CD20 monoclonal antibody sold by Beckman Coulter, Inc., CA.
In a specific embodiment, one or more antibodies immunospecific for one or
more
viral or microbial antigens are administered to an animal prior to, subsequent
to, or
concomitantly with the administration of one or more antibodies immunospecific
for
C3b(i), in an amount effective for the treatment or prevention of viral or
microbial infection.
Examples of antibodies used for the treatment or prevention of viral or
microbial infection
include, but are not limited to: Synagis~ (Medlmmune, Inc., MD) which is a
humanized
anti-respiratory syncytial virus (RSV) monoclonal antibody fox the treatment
of patients
with RSV infection; PR0542 (Progenics) which is a CD4 fusion antibody for the
treatment
of HIV infection; Ostavir (Protein Design Labs, Inc., CA) which is a human
antibody for
the treatment of hepatitis B virus; Protovir (Protein Design Labs, Inc., CA)
wluch is a
humanized IgGI antibody for the treatment of cytomegalovirus (CMV); WIN1 which
i~~ospecifically binds to LPS (Novardis) and mouse monoclonal anti-Legionella
Pneumonia LPS (Research Diagnostics, Inc. NJ).
The present invention encompasses the administration of plasma as a source of
IgG
and/or IgM antibodies and one or more anti-C3b(i) antibodies for the treatment
or
prevention of cancer, viral infection, and microbial infection. In a specific
embodiment,
plasma or selectively enriched IgG and/or IgM antibodies is administered to an
animal,
preferably a mammal and most preferably a human, prior to, subsequent to, or
concomitantly with the administration of one or more anti-C3b(i) antibodies in
an amount
effective for the treatment or prevention of cancer, viral infection or
microbial infection.
The plasma or selectively enriched IgG and/or IgM antibodies may be obtained
from
normal animals or from animals with a particular type of cancer, a particular
type viral
infection, or a particular type of microbial infection.
Preferably, the plasma or selectively enriched IgG and/or IgM is obtained from
an
animal of the same species which receives the administration. The plasma may
or may not
be treated with EDTA, citrate or heparin to block the complement pathways. In
a specific
embodiment, plasma or selectively enriched IgG and/or IgM antibodies from an
animal,
which contains antibodies immunospecific for cancer cell antigens (e.g.,
improperly
glycosylated proteins or lipids) are administered to an animal prior to the
administration of
- 14-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
anti-C3b(i) antibodies. In another specific embodiment, plasma or selectively
enriched IgG
and/or IgM antibodies from an animal, which contains antibodies immunospecific
for a
viral antigen or a microbial antigen (e.g., anti-HIV gp 120 antibodies or anti-
LPS antibodies)
are administered to an animal prior to the administration of anti-C3b(i)
antibodies.
The present invention encompasses the administration of recombinant complement
components or plasma as a source of complement components and one or more anti-
C3b(i)
antibodies for the treatment or prevention of cancer, viral infection,
microbial infection, and
septic shock In a specific embodiment, one or more complement components or
recombinant complement components are administered to an animal, preferably a
mammal
~d most preferably a human, prior to, subsequent to, or concomitantly with the
administration of antibodies immunospecific for C3b(i) in an amount effective
for the
treatment or prevention of cancer, viral infection, microbial infection, or
septic shock. In
another embodiment, nonnal plasma as a source of complement components is
administered to an animal prior to, subsequent to, or concomitantly with the
administration
of antibodies immunospecific for C3b(i) in an amount effective for the
treatment or
prevention of cancer, viral infection, microbial infection, or septic shock.
In a specific embodiment, a source of IgG and/or IgM antibodies and complement
components (e.g., normal plasma) is administered to an animal to insure
efficient
opsonization prior to the administration of antibodies specific for C3b(i). In
accordance
with the invention, the administration of C3b(i) immunospecific antibodies in
combination
with IgG antibodies, IgM antibodies and/or complement components will initiate
a chain
reaction which results in increased complement activity a~ld ultimately the
killing of
cancerous cells.
In a preferred embodiment, the endogenous levels of IgG antibodies, IgM
antibodies
and/or complement components are analyzed to determine whether an animal,
preferably a
mammal and most preferably a human, requires the administration of IgG
antibodies, IgM
antibodies and/or complement components. Standard techniques known to those of
skill in
the art can be utilized to measure the endogenous levels of IgG antibodies,
IgM antibodies
and complement components in an animal's sera. For example, the level of IgM
antibodies
or IgG antibodies in sera can be determined by titration of the sera against
cell lines such as
comparable cancer cell lines. Further, the level of complement components and
complement activity can be determined by, for example, i~ vitro tests for the
ability to
interact with complement proteins and the ability to lyse target cells
opsonized with specific
antibodies (Complement: A Practical Approach, Dodds and Sim, Oxford University
Press
1997; Makrides et al., 1992, J. Biol. Chem. 264:24754-2476; and Weisman, H.
F., et al.,
1990, Science, 244:146-151). The present invention also provides compositions
comprising
-15-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
one or more anti-C3b(i) antibodies for use in the treatment, prevention,
detection or
diagnosis of cancer, viruses, and microbes.
The present invention provides methods and kits for depleting cancerous cells
ih
vitro utilizing C3b(i) specific antibodies. The invention also provides
methods and kits for
the detection, imaging, and diagnosis of cancer utilizing antibodies specific
for C3b(i).
Further, the invention provides pharmaceutical compositions comprising
antibodies specific
for C3b(i).
5.1. I~G AND IgM ENRICHMENT
I O In accordance with certain embodiments of the present invention, the
levels of IgG
antibodies, IgM antibodies, and complement components in the sera or plasma of
an animal
are measured prior to the administration of anti-C3b(i) antibodies. In one
embodiment,
animals determined to have low levels of IgG antibodies and/or IgM antibodies
are
administered normal plasma containing IgG antibodies and/or IgM antibodies
(preferably,
15 IgG antibodies or IgM antibodies that immunospecifically bind to one or
more cancer cell
epitopes (e.g., improperly glycosylated proteins or lipids expressed by cancer
cells), viral
epitopes, or microbial epitopes) prior to (e.g., 1 minute, 1 S minutes, 30
minutes, 45 minutes,
1 hour, 2 hours, 4 hours, 6 hours, 8 hours, 12 hours, 24 hours, 2 days, or 1
week before),
subsequent to (e.g., 1 minute, 15 minutes, 30 minutes, 45 minutes, 1 hour, 2
hours, 4 hours,
20 6 hours, 8 hours, 12 hours, 24 hours, 2 days, or 1 week after), or
concomitantly with the
administration of anti-C3b(i) antibodies for the treatment or prevention of
cancer, viral
infection, or microbial infection. In accordance with this embodiment, the
plasma is
obtained from an animal of the same species that receives the plasma. In
another
embodiment, animals determined to have low levels of IgM antibodies are
administered
25 plasma enriched for IgM antibodies prior to (e.g., 1 minute, 15 minutes, 30
minutes, 45
minutes, I hour, 2 hours, 4 hours, 6 hours, 8 hours, 12 hours, 24 hours, 2
days, or 1 week
before), subsequent to (e.g., 1 minute, 15 minutes, 30 minutes, 45 minutes, 1
hour, 2 hours,
4 hours, 6 hours, 8 hours, 12 hours, 24 hours, 2 days, or 1 week after), or
concomitantly
with the administration of anti-C3b(i) antibodies for the treatment or
prevention of cancer,
30 viral infection, or microbial infection. In another embodiment, animals
determined to have
low levels of IgG antibodies are administered plasma enriched for IgG
antibodies prior to
(e.g., 1 minute, 15 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours,
6 hours, 8
hours, 12 hours, 24 hours, 2 days, or 1 week before), subsequent to (e.g., 1
minute, 15
minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 8 hours,
12 hours, 24
35 hours, 2 days, or 1 week after), or concomitantly with the administration
of anti-C3b(i)
antibodies for the treatment or prevention of cancer, viral infection, or
microbial infection.
In accordance with these embodiments, IgM antibodies or IgG antibodies are
selectively
-16-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
enriched utilizing standard techniques known to those of skill in the art.
Such techniques
include, but are not limited to, chromatography, centrifugation, and
differential solubility.
In a particular embodiment of the invention, native or recombinant IgG
antibodies or
IgM antibodies known to immunospecifically bind to cancer cells, viruses or
microbes are
administered to an animal prior to (e.g., 1 minute, 15 minutes, 30 minutes, 45
minutes, I
hour, 2 hours, 4 hours, 6 hours, 8 hours, 12 hours, 24 hours, 2 days, or 1
week before),
subsequent to (e.g., 1 minute, 15 minutes, 30 minutes, 45 minutes, 1 hour, 2
hours, 4 hours,
6 hours, 8 hours, 12 hours, 24 hours, 2 days, or 1 week after), or
concomitantly with the
administration of anti-C3b(i) antibodies for the treatment or prevention of
cancer. IgG
antibodies or IgM antibodies that immunospecifically bind to one or more
cancer cell
antigens (e.g., improperly glycosylated proteins or lipids expressed by cancer
cells), viral
antigens (e.g., glycoprotein F of RSV and gp120 of HIV), or microbial antigens
(e.g., LPS)
can be purchased from a company or purified utilizing standard protein
purification
techniques known to those of skill in the art. Examples of standard protein
purification
techniques include, but are not limited to, gel purification, chromatography
(e.g., ion
exchange, affinity, particularly by affinity for the specific antigen after
Protein A, and sizing
column chromatography), centrifugation, and differential solubility.
Recombinant IgG
antibodies and IgM antibodies can be produced utilizing standard teclnuques
known to
those of skill in the art.
In one embodiment, plasma as a source of IgG and IgM antibodies is
administered
to an animal the same day as the animal is administered antibodies
immunospecific for
C3b(i) or C3b(i) covalently linked to a second molecule for the treatment or
prevention of
cancer, viral infections or microbial infections. In another embodiment,
plasma as a source
of IgG and IgM antibodies is adminstered to an animal a few minutes or hours
(e.g., 15
minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 8 hours,
12 hours)
before the antibodies immunospecific for C3b(i) or C3b(i) covalently linked to
a second
molecule are administered to the animal for the treatment or prevention of
cancer, viral
infections or microbial infections.
In a preferred embodiment, IgG antibodies and/or IgM antibodies are
administered
to an animal the same day as the animal is administered antibodies
immunospecific for
C3b(i) or C3b(i) covalently linlced to a second molecule for the treatment or
prevention of
cancer, viral infections or microbial infections. In another preferred
embodiment, IgG
antibodies and/or IgM antibodies are administered to an animal a few minutes
or hours
(e.g., 5 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 8
hours or 12
hours) before administering antibodies immunospecific for C3b(i) or C3b(i)
covalently
linked to a second molecule for the treatment or prevention of cancer, viral
infections or
microbial infections.
-17-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
5.2. COMPLEMENT COMPONENTS
In a preferred embodiment, animals determined to have low levels of
complement,
particularly C3, are infused with normal plasma prior to (e.g., 1 minute, 15
minutes, 30
minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 8 hours, 12 hours, 24
hours, 2 days,
or I week before), subsequent to (e.g., 1 minute, 15 minutes, 30 minutes, 45
minutes, I
hour, 2 hours, 4 hours, 6 hours, 8 hours, 12 hours, 24 hours, 2 days, or 1
week after), or
concomitantly with the administration of anti-C3b(i) antibodies for the
treatment or
prevention of cancer, viral infection, or microbial infection. In accordance
with this
embodiment, the plasma is obtained from an animal of the same species that
receives the
plasma. In another preferred embodiment, animals determined to have low levels
of
complement are administered native or recombinant complement proteins (e.g.,
C3) prior to
(e.g., 1 minute, 15 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours,
6 hours, 8
hours, 12 hours, 24 hours, 2 days, or 1 week before), subsequent to (e.g., 1
minute, 15
minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 8 hours,
12 hours, 24
hours, 2 days, or 1 week after), or concomitantly with the administration of
anti-C3b(i)
antibodies for the treatment or prevention of cancer, viral infection, or
microbial infection.
In a specific embodiment, one or more complement components are administered
to an
aiumal the same day as the animal is administered anti-C3b(i) antibodies for
the treatment
or prevention of cancer, viral infection, or microbial infection. In another
embodiment, one
or more complement components are administered to an animal a few hours (e.g.,
1 hour, 2
hours, 4 hours, 6 hours, 8 hours or 12 hours) before administering antibodies
anti-C3b(i)
antibodies for the treatment or prevention of cancer, viral infection, or
microbial infection.
Complement components, in particular complement component C3, can be
purchased from a company or purified utilizing standaxd protein purification
techniques
down to those of skill in the art. Examples of such purification techniques
include, but are
not limited to, gel purification, chromatography (e.g., ion exchange,
affinity, particularly by
affinity for the specific antigen after Protein A, and sizing column
chromatography),
centrifugation, and differential solubility. Recombinant complement components
(e.g., C3)
can be produced utilizing standard techniques known to those of shill in the
art. In
accordance with the invention, the nucleic acid sequences encoding complement
components can be obtained from available sequence databases, e.g., GenBank.
Both
cDNA and genomic sequences can be cloned and expressed. Further, in accordance
with
the invention, the native or recombinant complement components administered to
an animal
for the treatment or prevention of cancer, viral infection, or microbial
infection retain the
ability to function in the classical and/or alternative complement pathways.
The nucleotide sequence encoding complement components or a functionally
active
analogs or other derivatives thereof (e.g., C3) can be inserted into an
appropriate expression
-18-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
vector, i.e., a vector which contains the necessary elements for the
transcription and
translation of the inserted protein-coding sequence. For example, the
nucleotide sequence
encoding human C3 as disclosed in Genbank Accession Numbers NM 000064 and
K02765 can be inserted into an appropriate expression vector. In another
example, the
nucleotide sequence encoding human C1 subcomponents, human C2 or human C2 as
disclosed in Genbank Accession Numbers NM 000063, NM OOI734, J04080, and
AF019413, respectively, can be inserted into an appropriate expression vector.
The
necessary transcriptional and translational signals can be supplied by the
native complement
component genes or its flanking regions. A variety of host-vector systems may
be utilized
I O to express the protein-coding sequence. These include, but are not limited
to, mammalian
cell systems infected with virus (e.g., vaccinia virus, adenovirus, etc.);
insect cell systems
infected with virus (e.g., baculovirus); and microorganisms such as yeast
containing yeast
vectors, or bacteria transformed with bacteriophage, DNA, plasmid DNA, or
cosmid DNA.
The expression elements of vectors vary in their strengths and specificities.
Depending on
15 the host-vector system utilized, any one of a number of suitable
transcription and translation
elements may be used. In specific embodiments, the human complement component
genes
or sequences encoding functionally active portions of the human complement
components
are expressed.
Any of the methods known to one of skill in the art for the insertion of DNA
2p fragments into a vector may be used to construct expression vectors
containing a chimeric
gene consisting of appropriate transcriptional and translational control
signals and the
protein coding sequences. These methods may include i» vitz~o recombinant DNA
and
synthetic techniques and irz vivo recombinants (genetic recombination). The
expression of a
nucleic acid sequence encoding a complement component or fragments thereof may
be
25 regulated by a second nucleic acid sequence so that the complement
component or
fragments thereof are expressed in a host transformed with the recombinant DNA
molecule.
The expression of complement components (e.g., C3) may be controlled by any
promoter or
enhancer element known to one of skill in the art. In particular, the
expression of a
complement component may regulated by any constitutive promoter, inducible
promoter,
30 tissue specific promoter, or native complement component promoter known to
one of skill
in the art.
Promoters which may be used to regulate the expression of a complement
component (e.g., C3) include, but are not limited to, viral promoters such as
the SV40 early
promoter region (Bernoist and Chambon, 1981, Nature 290:304-310), the promoter
35 contained in the 3' long terminal repeat of Rous sarcoma virus (Yamamoto,
et al., 1980, Cell
22:787-797), the herpes thymidine kinase promoter (Wagner et al., 1981, Proc.
Natl. Acad.
Sci. USA 78:1441-1445), the regulatory sequences of the metallothionein gene
(Brinster et
-19-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
al., 1982, Nature 296:39-42); prokaryotic promoters such as the (3-lactamase
promoter
(Villa-Kamaroff et al., 1978, Proc. Natl. Aced. Sci. USA 75:3727-3731), or the
tac
promoter (DeBoer et al., 1983, Proc. Natl. Aced. Sci. USA 80:21-25); see also
"Useful
proteins from recombinant bacteria" in Scientific American, 1980, 242:74-94;
plant
promoters such as the nopaline synthetase promoter region (Henrera-Estrella et
al., Nature
303:209-213) or the cauliflower mosaic virus 35S RNA promoter (Gardner et al.,
1981,
Nucl. Acids Res. 9:2871), and the promoter of the photosynthetic enzyme
ribulose
biphosphate carboxylase (Herrera-Estrella et al., 1984, Nature 310:115-120);
promoter
elements from yeast or other fungi such as the Gal 4 promoter, the ADC
(alcohol
dehydrogenase) promoter, PGK (phosphoglycerol kinase) promoter, alkaline
phosphatase
promoter; and the following animal transcriptional control regions, which
exhibit tissue
specificity and have been utilized in transgenic animals: elastase I gene
control region
which is active in pancreatic acinar cells (Swift et al., 1984, Cell 38:639-
646; Ornitz et al.,
1986, Cold Spring Harbor Symp. Quart. Biol. 50:399-409; MacDonald, 1987,
Hepatology
7:425-515), insulin gene control region wluch is active in pancreatic beta
cells (Hanahan,
1985, Nature 315:1 I S-I22), immunoglobulin gene control region which is
active in
lymphoid cells (Grosschedl et al., 1984, Cell 38:647-658; Adames et al., 1985,
Nature
318:533-538; Alexander et al., 1987, Mol. Cell. Biol. 7:1436-1444), mouse
mammary
tumor virus control region which is active in testicular, breast, lymphoid and
mast cells
(Leder et al., 1986, Cell 45:485-495), albumin gene control region which is
active in liver
(Pinkert et al., 1987, Genes and Devel. 1:268-276), alpha-fetoprotein gene
control region
which is active in liver (Krumlauf et aL, I985, Mol. Cell. Biol. 5:1639-1648;
Hammer et al.,
1987, Science 235:53-58), alpha 1-antitrypsin gene control region which is
active in the
liver (Kelsey et al., 1987, Genes and Devel. 1:161-171), beta-globin gene
control region
which is active in myeloid cells (Mogram et al., 1985, Nature 3I5 :338-340;
Kollias et al.,
1986, Cell 46:89-94), myelin basic protein gene control region which is active
in
oligodendrocyte cells in the brain (Readhead et al., 1987, Cell 48:703-712),
myosin light
chain-2 gene control region wluch is active in skeletal muscle (Sari, 1985,
Nature
314:283-286), and gonadotropic releasing hormone gene control region which is
active in
the hypothalamus (Mason et al., 1986, Science 234:1372-1378). In certain
embodiments,
the promoter regulating the expression of the complement component is a
constitutive
promoter. In certain other embodiments, the promoter regulating the expression
of the
complement component is an inducible or tissue-specific promoter.
In a specific embodiment, the vector used to express a complement component
comprises a promoter operably linked to a complement component (e.g., C3)-
encoding
nucleic acid, one or more origins of replication, and, optionally, one or more
selectable
markers (e.g., an antibiotic resistance gene).
-20-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
Expression vectors containing nucleotide sequences encoding one or more
complement components can be identified by three general approaches: (a)
nucleic acid
hybridization; (b) presence or absence of "marker" gene functions; and (c)
expression of
inserted sequences. In the first approach, the presence of a nucleotide
sequence encoding a
complement component (e.g., C3) in an expression vectors) can be detected by
nucleic acid
hybridization using probes comprising sequences that are homologous to the
inserted
nucleotide sequence. In the second approach, the recombinant vector/host
system can be
identified and selected based upon the presence or absence of certain "marker"
gene
functions (e.g., thymidine kinase activity, resistance to antibiotics,
transformation
phenotype, occlusion body formation in baculovirus, etc.) caused by the
insertion of the
nucleotide sequence encoding the complement component in the vector(s). For
example, if
a nucleotide sequence encoding C3 is inserted witlun the marker gene sequence
of the
vector, recombinant vectors containing the nucleotide sequence can be
identified by the
absence of the marker gene function. In the third approach, recombinant
expression vectors
can be identified by assaying for RNA or polypeptides encoded by the
nucleotide sequence
for the complement component. Such assays can be based, for example, on the
physical or
functional properties of the complement component in ih vitro assay systems,
e.g., binding
of C3 with anti-C3 antibody.
A host cell can be transfected with a nucleotide sequence encoding a
complement
component or a vector comprising a nucleotide sequence encoding a complement
component using techniques known to those of skill in the art such as, for
example,
microinjection, electroporation, lipofection, and calcium phosphate
precipitation. Host cells
may be stably or transiently transfected with a nucleotide sequence encoding a
complement
component or a vector comprising a nucleotide sequence encoding a complement
component.
For long-term, high-yield production of one or more recombinant complement
components, stable expression is preferred. For example, cell lutes which
stably express
one or more complement components may be engineered. Rather than using
expression
vectors which contain viral origins of replication, host cells can be
transformed with DNA
controlled by appropriate expression control elements (e.g., promoter,
enhancer, sequences,
transcription terminators, polyadenylation sites, etc.), and a selectable
marlcer. Following
the introduction of the foreign DNA, engineered cells may be allowed to grow
for 1-2 days
in an enriched media, and then are switched to a selective media. The
selectable marker in
the recombinant plasmid confers resistance to the selection and allows cells
to stably
integrate the plasmid into their chromosomes and grow to form foci which in
turn can be
cloned and expanded into cell lines.
-21-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
A number of selection systems may be used, including but not limited to the
herpes
simplex virus thymidine kinase (Wigler, et al., 1977, Cell 11:223),
hypoxanthine-guanine
phosphoribosyltransferase (Szybalska & Szybalski, 1962, Proc. Natl. Acad. Sci.
USA
48:2026), and adenine phosphoribosyltransferase (Lowy et al., 1980, Cell
22:817) genes can
be employed in tk-, hgprt- or aprt- cells, respectively. Also, antimetabolite
resistance can be
used as the basis of selection for dhfr, which confers resistance to
methotrexate (Wigler et
al., 1980, Natl. Acad. Sci. USA 77:3567; O'Hare et al., 1981, Proc. Natl.
Acad. Sci. USA
78:1527); gpt, which confers resistance to mycophenolic acid (Mulligan & Berg,
1981,
Proc. Natl. Acad. Sci. USA 78:2072); neo, which confers resistance to the
aminoglycoside
G-418 (Colberre-Garapin et al., 1981, J. Mol. Biol. 150:1); and hygro, which
confers
resistance to hygromycin (Santerre et al., 1984, Gene 30:147) genes.
Once a suitable host system and growth conditions are established, recombinant
expression vectors can be propagated and prepared in quantity. A host cell
strain can be
chosen which modulates the expression of the inserted sequences in the
specific fashion
desired. Appropriate cell lines or host systems can be chosen to ensure the
desired
modification and processing of the complement protein expressed. Different
host cells have
characteristic and specific mechanisms for the translational and post-
translational
processing and modification (e.g., glycosylation and phosphorylation) of
proteins. For
example, expression in a bacterial system can be used to produce an
unglycosylated core
protein product. Expression in yeast will produce a glycosylated product.
Expression in
mammalian cells can be used to ensure "native" glycosylation of a heterologous
protein.
Such mammalian host cells include, but are not limited to, 293T, CHO, VERY,
BHK, Hela,
COS, MDCK, 293, 3T3, MT2, U937, WI38, BT483, Hs578T, HTB2, BT20, T47D,
CRL7030, Hs578Bst, lymphocytes and fibroblasts. Furthermore, different
vector/host
expression systems may effect processing reactions to different extents.
5.3. ANTIBODIES
Antibodies of the invention include, but are not limited to, polyclonal,
monoclonal,
bispecific, human, humanized or chimeric antibodies, single chain antibodies,
sFvs, Fab
fragments, F(ab') fragments, fragments produced by a Fab expression library,
anti-
idiotypic (anti-Id) antibodies, and epitope-binding fragments of any of the
above which
immunospecifically bind to cancer cell antigens, viral antigens, microbial
antigens, C3b(i)
or fragments thereof, or C3b(i) linked to a second molecule. The term
"antibody" as used
herein refers to immunoglobulin molecules and immunologically active portions
of
i~~oglobulin molecules, i. e., molecules that contain an antigen binding site
which
irnmunospecifically binds a cancer cell antigen, a viral antigen, a microbial
antigen, C3b(i),
or C3b(i) linked (e.g., covalently linked) to a second molecule. The
immunoglobulin
-22-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
molecules of the invention can be of any type (e.g., IgG, IgE, IgM, IgD and
IgA ), class, or
subclass of immunoglobulin molecule.
Polyclonal antibodies which may be used in the methods of the invention are
heterogeneous populations of antibody molecules derived from the sera of
immunized
animals. Various procedures well known in the art may be used for the
production of
polyclonal antibodies to an antigen-of interest. For example, for the
production of
polyclonal antibodies, various host animals can be immunized by injection with
an antigen
of interest or derivative thereof, including but not limited to rabbits, mice,
rats, etc. Various
adjuvants may be used to increase the immunological response, depending on the
host
species, and including but not limited to Freund's (complete and incomplete),
mineral gels
such as aluminum hydroxide, surface active substances such as lysolecithin,
pluronic
polyols, polyanions, peptides, oil emulsions, keyhole limpet hemocyanins,
dinitrophenol,
and potentially useful htunan adjuvants such as BCG (bacille Calinette-Guerin)
and
corynebacterium parvum. Such adjuvants are also well lcnown in the art.
Monoclonal antibodies which may be used in the methods of the invention are
homogeneous populations of antibodies to a particular antigen (e.g., a cancer
cell antigen, a
viral antigen, a microbial antigen, C3b(i) or C3b(i) covalently linked to a
second molecule).
A monoclonal antibody (mAb) to an antigen-of interest can be prepared by using
any
technique known in the art which provides for the production of antibody
molecules by
continuous cell lines in culture. These include, but are not limited to, the
hybridoma
technique originally described by Kohler and Milstein (1975, Nature 256, 495-
497), the
more recent human B cell hybridoma technique (Kozbor et al., 1983, Immunology
Today 4:
72), and the EBV-hybridoma technique (Cole et al., 1985, Monoclonal Antibodies
and
Cancer Therapy, Alan R. Liss, Inc., pp. 77-96). Such antibodies may be of any
i~n~oglobulin class including IgG, IgM, IgE, IgA, and IgD and any subclass
thereof.
The hybridoma producing the mAbs of use in this invention may be cultivated ih
vitro or iya
vavo.
The monoclonal antibodies which may be used in the methods of the invention
include, but are not limited to, human monoclonal antibodies or chimeric human-
mouse (or
other species) monoclonal antibodies. Human monoclonal antibodies may be made
by any
of numerous techniques known in the art (e.g., Teng et al., 1983, Proc. Natl.
Aced. Sci.
U.S.A. 80, 7308-7312; Kozbor et al., 1983, Immunology Today 4, 72-79; and
Olsson et al.,
1982, Meth. Enzymol. 92, 3-16).
The invention further provides for the use of bispecific antibodies. Methods
for
making bispecific antibodies are known in the art. Traditional production of
full length
bispecific antibodies is based on the coexpression of two immunoglobulin heavy
chain-light
chain pairs, where the two chains have different specificities (Milstein et
al., 1983, Nature
-23-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
305:537-539). Because of the random assortment of immunoglobulin heavy and
light
chains, these hybridomas (quadromas) produce a potential mixture of 10
different antibody
molecules, of which only one has the correct bispecific structure.
Purification of the correct
molecule, which is usually done by affinity chromatography steps, is rather
cumbersome,
and the product yields are Iow.
Similar procedures are disclosed in PCT Publication No. WO 93/08829, published
13 May 1993, and in Traunecker et al., 1991, EMBO J. 10:3655-3659 .
According to a different and more preferred approach, antibody variable
domains
with the desired binding specificities (antibody-antigen combining sites) are
fused to
i~~oglobulin constant domain sequences. The fusion preferably is with an
immunoglobulin heavy chain constant domain, comprising at least part of the
hinge, CH2,
and CH3 regions. It is preferred to have the first heavy-chain constant region
(CH1)
containing the site necessary for light chain binding, present in at least one
of the fusions.
DNAs encoding the immunoglobulin heavy chain fusions and, if desired, the
i~unoglobulin light chain, are inserted into separate expression vectors, and
are co-
transfected into a suitable host organism. This provides for great flexibility
in adjusting the
mutual proportions of the three polypeptide fragments in embodiments when
unequal ratios
of the three polypeptide chains used in the construction provide the optimum
yields. It is,
however, possible to insert the coding sequences for two or all three
polypeptide chains in
2p one expression vector when the expression of at least two polypeptide
chains in equal ratios
results in high yields or when the ratios are of no particular significance.
In a preferred embodiment of this approach, the bispecific antibodies are
composed
of a hybrid immunoglobulin heavy chain with a first binding specificity in one
arm, and a
hybrid immunoglobulin heavy chain-light chain pair (providing a second binding
specificity) in the other arm. It was found that this asymmetric structure
facilitates the
separation of the desired bispecific compound from unwanted immunoglobulin
chain
combinations, as the presence of an immunoglobulin light chain in only one
half of the
bispecific molecule provides for a facile way of separation. This approach is
disclosed in
PCT Publication No. WO 94/04690 published March 3,1994, which is incorporated
herein
by reference in its entirety.
For further details of generating bispecific antibodies see, for example,
Suresh et al.,
Methods in Enzymology,1986, 121:210. Using such techniques, a bispecific
molecule
which combines anti-C3b(i) antibody and an antibody specific for an effector
cell receptor
or antigen can be prepared for use in the treatment or inhibition of disease
as defined herein.
The invention provides for the use of functionally active fragments,
derivatives or
analogs of antibodies which immunospecifically bind to cancer cell antigens,
viral antigens,
and microbial antigens. Functionally active means that the fragment,
derivative or analog is
-24-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
able to elicit anti-anti-idiotype antibodies that recognize the same antigen
that the antibody
from which the fragment, derivative or analog is derived recognized.
Specifically, in a
preferred embodiment the antigenicity of the idiotype of the immunoglobulin
molecule may
be enhanced by deletion of framework and CDR sequences that are C-terminal to
the CDR
sequence that specifically recognizes the antigen. To determine which CDR
sequences bind
the antigen, synthetic peptides containing the CDR sequences can be used in
binding assays
with the antigen by any binding assay method known in the art (e.g., the BIA
core assay)
Other embodiments of the invention include fragments of the antibodies of the
invention such as, but not limited to, F(ab')2 fragments, which contain the
variable region,
l0 the light chain constant region and the CH1 domain of the heavy chain can
be produced by
pepsin digestion of the antibody molecule, and Fab fragments, which can be
generated by
reducing the disulfide bridges of the F(ab')2 fragments. The invention also
provides heavy
chain and light chain dimers of the antibodies of the invention, or any
minimal fragment
thereof such as Fvs or single chain antibodies (SCAB) (e.g., as described in
U.S. Patent
15 4,946,778; Bird, 1988, Science 242:423-42; Huston et al., 1988, Proc. Natl.
Aced. Sci. USA
85:5879-5883; and Ward et al., 1989, Nature 334:544-54), or any other molecule
with the
same specificity as the antibody of the invention.
Additionally, recombinant antibodies, such as chimeric and humanized
monoclonal
antibodies, comprising both human and non-human portions, which can be made
using
20 standard recombinant DNA techniques, are within the scope of the invention.
A chimeric
antibody is a molecule in which different portions are derived from different
animal species,
such as those having a variable region derived from a marine monoclonal and a
human
immunoglobulin constant region. (See, e.g., Cabilly et al., U.S. Patent No.
4,816,567; and
Boss et al., U.S. Patent No. 4,816397, which are incorporated herein by
reference in their
25 entirety.) Humanized antibodies are antibody molecules from non-human
species having
one or more complementarily determining regions (CDRs) from the non-human
species
and a framework region from a human immunoglobulin molecule. (See, e.g.,
Queen, U.S.
Patent No. 5,585,089, which is incorporated herein by reference in its
entirety.) Such
chimeric and humanized monoclonal antibodies caaz be produced by recombinant
DNA
30 techniques known in the art, for example using methods described in PCT
Publication No.
WO 87/02671; European Patent Application 184,187; European Patent Application
171,496; European Patent Application 173,494; PCT Publication No. WO 86/01533;
U.S.
Patent No. 4,816,567; European Patent Application 125,023; Better et al.,
1988, Science
240:1041-1043; Liu et al., 1987, Proc. Natl. Aced. Sci. USA 84:3439-3443; Liu
et al.,
35 1987, J. Tm_m__unol. 139:3521-3526; Sun et al., 1987, Proc. Natl. Aced.
Sci. USA 84:214-
218; Nishimura et al., 1987, Canc. Res. 47:999-1005; Wood et al., 1985, Nature
314:446-
449; and Shaw et al., 1988, J. Natl. Cancer Inst. 80:1553-1559; Morrison,
1985, Science
-25-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
229:1202-1207; Oi et al., 1986, Bio/Techniques 4:214; U.S. Patent 5,225,539;
Jones et al.,
1986, Nature 321:552-525; Verhoeyan et al. (1988) Science 239:1534; and
Beidler et al.,
1988, J. Tmmunol. 14I :4053-4060; each of which is incorporated herein by
reference n its
entirety.
Completely human antibodies are particularly desirable for therapeutic
treatment of
human patients. Such antibodies can be produced using transgenic mice which
are
incapable of expressing endogenous immunoglobulin heavy and light chains
genes, but
which can express human heavy and light chain genes. The transgenic mice are
immunized
in the normal fashion with a selected antigen, e.g., all or a portion of a
polypeptide of the
invention. Monoclonal antibodies directed against the antigen can be obtained
using
conventional hybridoma technology. The human immunoglobulin transgenes
harbored by
the transgenic mice rearrange during B cell differentiation, and subsequently
undergo class
switching and somatic mutation. Thus, using such a technique, it is possible
to produce
therapeutically useful IgG, IgA, IgM and IgE antibodies. For an overview of
this
technology for producing human antibodies, see Lonberg and Huszar (1995, Tnt.
Rev.
Immunol. 13:65-93). For a detailed discussion of this technology for producing
human
antibodies and human monoclonal antibodies and protocols for producing such
antibodies,
see, e.g., U.S. Patent 5,625,126; U.S. Patent 5,633,425; U.S. Patent
5,569,825; U.S. Patent
5,661,016; and U.S. Patent 5,545,806; each of which is incorporated herein by
reference in
its entirety. In addition, companies such as Abgenix, Inc. (Freemont, CA) and
Genpharm
(San Jose, CA) can be engaged to provide human antibodies directed against a
selected
antigen using technology similar to that described above.
Completely human antibodies which recognize a selected epitope can be
generated
using a technique referred to as "guided selection." In this approach a
selected non-human
monoclonal antibody, e.g., a mouse antibody, is used to guide the selection of
a completely
human antibody recognizing the same epitope. (Jespers et al. (1994)
Biotechnology
12:899-903).
In other embodiments, the invention provides fusion proteins of the antibodies
of the
invention (or functionally active fragments thereof), for example in which the
antibody is
fused via a covalent bond (e.g., a peptide bond), at either the N-terminus or
the C-terminus
to an amino acid sequence of another protein (or portion thereof, preferably
at least 10, 20
or 50 amino acid portion of the protein) that is not the antibody. Preferably,
the antibody or
fragment thereof is covalently linked to the other protein at the N-terminus
of the constant
domain.
The antibodies of the invention include analogs and derivatives that are
either
modified, i.e, by the covalent attachment of any type of molecule as long as
such covalent
attachment does not prevent the antibody from generating an anti-idiotypic
response. For
-26-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
example, but not by way of limitation, the derivatives and analogs of the
antibodies include
those that have been further modified, e.g., by glycosylation, acetylation,
pegylation,
phosphylation, amidation, derivatization by known protecting/blocking groups,
proteolytic
cleavage, linkage to a cellular ligand or other protein, etc. Any of numerous
chemical
modifications may be carned out by known techniques, including, but not
limited to
specific chemical cleavage, acetylation, formylation, metabolic synthesis of
tunicamycin,
etc. Additionally, the analog or derivative may contain one or more non-
classical amino
acids.
The antibodies of the invention include antibodies with modifications (e.g.,
substitutions, deletions or additions) in amino acid residues that interact
with Fc receptors.
In particular, the antibodies of the invention include antibodies with
modifications in amino
acid residues identified as involved in the interaction between the Fc domain
and the FcRn
receptor (see, e.g., PCT Publication No. WO 97/34631, which is incorporated
herein by
reference in its entirety).
5.3.1. ANTI-C3b(i) ANTIBODIES
In a specific embodiment, anti-C3b(i) antibodies are immunospecific for C3b(i)
or a
fragment thereof (e.g., at least 5, at least 10, at least 15, at least 25, or
more contiguous
amino acid residues of C3b(i)). In another embodiment, anti-C3b(i) antibodies
are
inununospecific for C3b(i) covalently linked to a second molecule. In a
preferred
embodiment, anti-C3b(i) antibodies are monoclonal antibodies or Fab fragments.
In
another preferred embodiment, anti-C3b(i) antibodies are immunospecific for
C3b(i) bound
to IgG or IgM antibodies and said anti-C3b(i) antibodies have minimal cross-
reactivity with
C3b(i) in the media or milieu. In another preferred embodiment, anti-C3b(i)
antibodies are
immunospecific for C3b(i) bound to a cancer cell or C3b(i) bound to a virus or
microbe; and
said anti-C3b(i) antibodies have minimal cross-reactivity with C3b(i) in the
media or
milieu. In yet another preferred embodiment, anti-C3b(i) antibodies
administered to
humans are humanized or human monoclonal antibodies.
Anti-C3b(i) antibodies caal be obtained from any organization (e.g., a
university
30 scientist or a company such as Research Diagnostics Inc. in New Jersey) or
produced by
any method known to one of skill in the art. For example, anti-C3b(i)
antibodies can be
produced by chemical synthesis or recombinant expression techniques. The
nucleotide
sequence encoding an anti-C3b(i) antibodies can be obtained, e.g., from the
GenBank
database or a database like it, the literature publications, or by routine
cloning and
35 sequencing.
In one embodiment, an anti-C3b(i) antibody is monoclonal antibody 7C12, 2H11,
8E11 or 3E7 (see, e.g., Ferguson et al., 1995, Arthritis Rheum. 38:190; Taylor
et al., 1989,
-27-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
J. Immunology 143:3626; Tosic et al., 1989, J. Ixnmunol. Methods 120:241; and
Sokoloff et
al., 2000, Cancer Tmmunol. Tm_m__unother. 49:551-562, each of which is
incorporated herein
by reference in their entirety). In another embodiment, an anti-C3b(i)
antibody is an
antibody that competes with 7C12, 2H11, 8E11 or 3E7 for binding to C3b(i) or a
fragment
thereof, as determined by an immunoassay such as, e.g., an ELISA. In a
preferred
embodiment, an anti-C3b(i) antibody is monoclonal antibody 3E7. In another
preferred
embodiment, an anti-C3b(i) antibody is an antibody that competes with
monoclonal 3E7 for
binding to C3b(i) or a fragment thereof, as determined by an immunoassay such
as, e.g., an
ELISA. In yet another preferred embodiment, an anti-C3b(i) antibody is a
bispecific
monoclonal antibody comprising monoclonal antibody 3E7 and monoclonal antibody
7G9
(see, e.g., Sokoloff et al., 2000, Cancer hnmunol. Irnmunother. 49:551-562).
5.3.2. ANTIBODIES IMMUNOSPECIFIC
FOR CANCER CELLS
~ a specific embodiment, antibodies immunospecific for a cancer cell antigen
for
use in accordance with methods of the invention are monoclonal antibodies or
Fab
fragments. Preferably, antibodies immunospecific for a cancer cell antigen
which are
administered to humans are humanized or human monoclonal antibodies. As used
herein,
the term "cancer cell antigen" refers to an antigen (e.g., a protein,
glycoprotein, polypeptide,
peptide, or glycolipid) that is preferentially or differentially expressed on
cancer cells
relative to non-cancerous cells, preferably normal cells. Examples of cancer
cell antigens
include, but are not limited, to improperly glycosylated proteins and lipids,
CD20, Her-2,
and PSMA.
Antibodies immunospecific fox a cancer cell antigen can be obtained from any
°rg~ization (e.g., a university scientist or a company such as
Genentech) or produced by
any method known to one of skill in the art such as, e.g., chemical synthesis
or recombinant
expression techniques. The nucleotide sequence encoding antibodies
immunospecific for a
cancer cell antigen can be obtained, e.g., from the GenBank database or a
database like it,
the literature publications, or by routine cloning and sequencing.
~ a specific embodiment, known antibodies for the treatment or prevention
cancer
are used in accordance with the methods of the invention. Examples of
antibodies available
for the treatment of cancer include, but are not limited to, Herceptin~
(Trastuzumab;
Genetech, CA) which is a humanized anti-HER2 monoclonal antibody for the
treatment of
patients with metastatic breast cancer; Retuxan~ (rituximab; Genentech) which
is a
chimeric anti-CD20 monoclonal antibody for the treatment of patients with non-
Hodgkin's
lymphoma; OvaRex (AltaRex Corporation, MA) which is a marine antibody for the
treatment of ovarian cancer; Panorex (Glaxo Wellcome, NC) which is a marine
IgG2a
-28-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
antibody for the treatment of colorectal cancer; BEC2 (ImClone Systems Inc.,
NY) which is
marine IgG antibody for the treatment of lung cancer; IMC-C225 (Imclone
Systems Inc.,
N~ which is a chimeric IgG antibody for the treatment of head and neck cancer;
Vitaxin
(MedTmmune, Inc., MD) which is a humanized antibody for the treatment of
sarcoma;
Campath I/H (Leukosite, MA) which is a humanized IgGI antibody fox the
treatment of
chronic lymphocytic leukemia (CLL); Smart MI95 (Protein Design Labs, Inc., CA)
which is
a humanized IgG antibody for the treatment of acute myeloid leukemia (AML);
LymphoCide (Immunomedics, Inc., NJ) which is a humanized IgG antibody for the
treatment of non-Hodgkin's lymphoma; Smart I D10 (Protein Design Labs, Inc.,
CA) which
is a humanized antibody for the treatment of non-Hodgkin's lymphoma; and
Oncolym
(Techniclone, Inc., CA) which is a marine antibody for the treatment of non-
Hodgkin's
lymphoma.
5.3.3. ANTIBODIES TO VIRAL AND
MICROBIAL ANTIGENS
~ a specific embodiment, antibodies immunospecific for a viral or microbial
antigen for use in accordance with methods of the invention are monoclonal
antibodies.
Preferably, antibodies immunospecific for a viral antigen or microbial antigen
which are
administered to humans are humanized or human monoclonal antibodies. As used
herein,
the term "viral antigen" includes, but is not limited to, any viral peptide,
polypeptide protein
(e.g., HIV gp120, HIV nef, RSV F glycoprotein, influenza virus neuraminidase,
influenza
virus hemagglutinin, HTLV tax, herpes simplex virus glycoprotein (e.g., gB,
gC, gD, and
gE) and hepatitis B surface antigen) which is capable of eliciting an immune
response. As
used herein, the term "microbial antigen" includes, but is not limited to, any
microbial
peptide, polypeptide, protein, saccharide, polysaccharide, or lipid molecule
(e.g., a bacterial,
~gl' pathogenic protozoa, or yeast polypeptide including, e.g., LPS and
capsular
polysaccharide 5/8) which is capable of eliciting an immune response.
Antibodies immunospecific for a viral or microbial antigen can be obtained
from
any organization (e.g., a university scientist or a company such as Genentech)
or produced
by any method known to one of skill in the art such as, e.g., chemical
synthesis or
recombinant expression techniques. The nucleotide sequence encoding antibodies
immunospecific for a viral or microbial antigen can be obtained, e.g., from
the GenBank
database or a database like it, the literature publications, or by routine
cloning and
sequencing.
In a specific embodiment, known antibodies for the treatment or prevention
viral or
microbial infection are used in accordance with the methods of the invention.
Examples of
antibodies available for the treatment of viral infection or microbial
infection include, but
are not limited to, Synagis~ (Medlmmune, Inc., MD) which is a humanized anti-
respiratory
-29-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
syncytial virus (RSV) monoclonal antibody for the treatment of patients with
RSV
infection; PR0542 (Progenics) which is a CD4 fusion antibody for the treatment
of HIV
infection; Ostavir (Protein Design Labs, Inc., CA) which is a human antibody
for the
treatment of hepatitis B virus; Protovir (Protein Design Labs, Inc., CA) which
is a
humanized IgGI antibody for the treatment of cytomegalovirus (CMV); and anti-
LPS
antibodies.
5.4. PRODUCTION OF RECOMBINANT ANTIBODIES
The antibodies of the invention can be produced by any method known in the art
for
the synthesis of antibodies, in particular, by chemical synthesis or by
recombinant
expression, and are preferably produced by recombinant expression techniques.
Recombinant expression of the antibodies of the invention, or fragment,
derivative
or analog thereof, requires construction of a nucleic acid that encodes the
antibody. If the
nucleotide sequence of the antibody is known, a nucleic acid encoding the
antibody may be
assembled from chemically synthesized oligonucleotides (e.g., as described in
Kutmeier et
al., 1994, BioTechniques 17:242), which, briefly, involves the synthesis of
overlapping
oligonucleotides containing portions of the sequence encoding the antibody,
annealing and
ligation of those oligonucleotides, and then amplification of the ligated
oligonucleotides by
PCR.
Alternatively, a nucleic acid molecule encoding an antibody may be generated
from
a suitable source. If a clone containing the nucleic acid encoding the
particular antibody is
not available, but the sequence of the antibody is known, a nucleic acid
encoding the
antibody may be obtained from a suitable source (e.g., an antibody cDNA
library, or cDNA
library generated from any tissue or cells expressing the immunoglobulin) by
PCR
amplification using synthetic primers hybridizable to the 3' and 5' ends of
the sequence or
by cloning using an oligonucleotide probe specific for the particular gene
sequence.
If an antibody that specifically recognizes a particular antigen is not
available (or a
source for a cDNA library for cloning a nucleic acid encoding such an
irnmunoglobulin),
antibodies specific for a particular antigen may be generated by any method
known in the
~, for example, by immunizing an animal, such as a rabbit, to generate
polyclonal
antibodies or, more preferably, by generating monoclonal antibodies, e.g., as
described by
Kohler and Milstein (1975, Nature 256:495-497) or, as described by Kozbor et
al. (1983,
Immunology Today 4:72) or Cole et al. (1985 in Monoclonal Antibodies and
Cancer
Therapy, Alan R. Liss, Inc., pp. 77-96). Alternatively, a clone encoding at
least the Fab
potion of the antibody may be obtained by screening Fab expression libraries
(e.g., as
described in Huse et al., 1989, Science 246:1275-1281) for clones of Fab
fragments that
-30-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
bind the specific antigen or by screening antibody libraries (See, e.g.,
Clackson et al., 1991,
Nature 352:624; Hane et al., 1997 Proc. Natl. Acad. Sci. USA 94:4937).
Once a nucleic acid sequence encoding at least the variable domain of the
antibody
is obtained, it may be introduced into a vector containing the nucleotide
sequence encoding
the constant regions of the antibody (see, e.g., PCT Publication No.WO
86/05807; PCT
Publication No. WO 89/01036; and U.S. Patent No. 5,122,464). Vectors
containing the
complete light or heavy chain that allow for the expression of a complete
antibody molecule
are available. Then, the nucleic acid encoding the antibody can be used to
introduce the
nucleotide substitutions or deletion necessary to substitute (or delete) the
one or more
viable region cysteine residues participating in an intrachain disulfide bond
with an amino
acid residue that does not contain a sulfhydyl group. Such modifications can
be carried out
by any method known in the art for the introduction of specific mutations or
deletions in a
nucleotide sequence, for example, but not limited to, chemical mutagenesis and
in vitro site
directed mutagenesis (Hutchinson et al., 1978, J. Biol. Chem. 253:6551).
In addition, techniques developed for the production of "chimeric antibodies"
(Morrison et al., 1984, Proc. Natl. Acad. Sci. 81:851-855; Neuberger et al.,
1984, Nature
312:604-608; Takeda et al., 1985, Nature 314:452-454) by splicing genes from a
mouse
antibody molecule of appropriate antigen specificity together with genes from
a human
antibody molecule of appropriate biological activity can be used. As described
supra, a
c~meric antibody is a molecule in which different portions are derived from
different
animal species, such as those having a variable region derived from a marine
monoclonal
antibody and a human immunoglobulin constant region, e.g., humanized
antibodies.
Alternatively, techniques described for the production of single chain
antibodies
(IJ.S. Patent 4,694,778; Bird, 1988, Science 242:423-42; Huston et al., 1988,
Proc. Natl.
Acad. Sci. USA 85:5879-5883; and Ward et al., 1989, Nature 334:544-54) can be
adapted
to produce single chain antibodies. Single chain antibodies are formed by
linking the heavy
and light chain fragments of the Fv region via an amino acid bridge, resulting
in a single
chain polypeptide. Techniques for the assembly of functional Fv fragments in
E. coli may
also be used (Skerra et al., 1988, Science 242:1038-1041).
Antibody fragments which recognize specific epitopes may be generated by known
techniques. For example, such fragments include but are not limited to: the
F(ab')2
fragments which can be produced by pepsin digestion of the antibody molecule
and the Fab
fragments which can be generated by reducing the disulfide bridges of the
F(ab')2
fragments.
Once a nucleic acid sequence encoding an antibody of the invention has been
obtained, the vector for the production of the antibody may be produced by
recombinant
DNA technology using techniques well known in the art. Methods which are well
known to
-31-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
those skilled in the art can be used to construct expression vectors
containing the antibody
coding sequences and appropriate transcriptional and translational control
signals. These
methods include, for example, in vitYO recombinant DNA techniques, synthetic
techniques,
and ih vivo genetic recombination. See, fox example, the techniques described
in Sambrook
et al. (1990, Molecular Cloning, A Laboratory Manual, 2d Ed., Cold Spring
Harbor
Laboratory, Cold Spring Harbor, NYC and Ausubel et al. (eds., 1998, Current
Protocols in
Molecular Biology, John Wiley & Sons, N~.
An expression vector comprising the nucleotide sequence of an antibody or the
nucleotide sequence of an antibody can be transferred to a host cell by
conventional
techniques (e.g., electroporation, liposomal transfection, and calcium
phosphate
precipitation) and the transfected cells are then cultured by conventional
techniques to
produce the antibody of the invention. In specific embodiments, the expression
of the
antibody is regulated by a constitutive, an inducible or a tissue, specific
promoter.
The host cells used to express the recombinant antibody of the invention may
be
either bacterial cells such as Esclaerichia coli, or, preferably, eukaryotic
cells, especially for
the expression of whole recombinant immunoglobulin molecule. In particular,
mammalian
cells such as Chinese hamster ovary cells (CHO), in conjunction with a vector
such as the
major intermediate early gene promoter element from human cytomegalovirus is
an
effective expression system for immunoglobulins (Foecking et al., 198, Gene
45:101;
Cockett et al., 1990, Bio/Technology 8:2).
A variety of host-expression vector systems may be utilized to express the
immunoglobulin molecules of the invention. Such host-expression systems
represent
vehicles by which the coding sequences of the antibody may be produced and
subsequently
purified, but also represent cells which may, when transformed or transfected
with the
appropriate nucleotide coding sequences, express the immunoglobulin molecule
of the
invention in situ. These include, but are not limited to, microorganisms such
as bacteria
(e.g., E. coli and B. subtilis) transformed with recombinant bacteriophage
DNA, plasmid
DNA or cosmid DNA expression vectors containing immunoglobulin coding
sequences;
yeast (e.g., Saccharomyces Pichia) transformed with recombinant yeast
expression vectors
containing immunoglobulin coding sequences; insect cell systems infected with
recombinant virus expression vectors (e.g., baculovirus) containing the
immunoglobulin
coding sequences; plant cell systems infected with recombinant virus
expression vectors
(e.g., cauliflower mosaic virus (CaMV) and tobacco mosaic virus (TMV)) or
transformed
with recombinant plasmid expres-sion vectors (e.g., Ti plasmid) containing
i~~oglobulin coding sequences; or mammalian cell systems (e.g., COS, CHO, BHK,
293, 293T, 3T3 cells) harboring recombinant expression constructs containing
promoters
-32-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
derived from the genome of mammalian cells (e.g., metallothionein promoter) or
from
mammalian viruses (e.g., the adenovirus late promoter; the vaccinia virus 7.5K
promoter).
In bacterial systems, a number of expression vectors may be advantageously
selected depending upon the use intended for the antibody being expressed. For
example,
when a large quantity of such a protein is to be produced, for the generation
of
pharmaceutical compositions of an antibody, vectors which direct the
expression of high
levels of fusion protein products that are readily purified may be desirable.
Such vectors
include, but are not limited, to the E. coli expression vector pUR278 (Ruther
et al., 1983,
EMBO J. 2:1791), in which the antibody coding sequence may be ligated
individually into
the vector in frame with the lac Z coding region so that a fusion protein is
produced; pIN
vectors (Inouye & Inouye, 1985, Nucleic Acids Res. 13:3101-3109; Van Heeke &
Schuster,
1989, J. Biol. Chem. 24:5503-5509); and the like. pGEX vectors may also be
used to
express foreign polypeptides as fusion proteins with glutathione S-transferase
(GST). In
general, such fusion proteins are soluble and can easily be purified from
lysed cells by
adsorption and binding to a matrix glutathione-agarose beads followed by
elution in the
presence of free gluta-thione. The pGEX vectors are designed to include
thrombin or factor
Xa protease cleavage sites so that the cloned target gene product can be
released from the
GST moiety.
In an insect system, Autog~apha califo~nica nuclear polyhedrosis virus (AcNPV)
is
used as a vector to express foreign genes. The virus grows in Spodoptera
frugiperda cells.
The antibody coding sequence may be cloned individually into non-essential
regions (for
example the polyhedrin gene) of the virus and placed under control of an AcNPV
promoter
(for example the polyhedrin promoter).
In mammalian host cells, a number of viral-based expression systems may be
utilized. In cases where an adenovirus is used as an expression vector, the
antibody coding
sequence of interest may be ligated to an adenovirus transcription/translation
control
complex, e.g., the late promoter and tripartite leader sequence. This chimeric
gene may
then be inserted in the adenovirus genome by in vitro or in vivo
recombination. Insertion in
a non-essential region of the viral genome (e.g., region El or E3) will result
in a
recombinant virus that is viable and capable of expressing the immunoglobulin
molecule in
infected hosts. (e.g., see Logan & Shenk, 1984, Proc. Natl. Acad. Sci. USA
81:355-359).
Specific initiation signals may also be required for efficient translation of
inserted antibody
coding sequences. These signals include the ATG initiation colon and adjacent
sequences.
Furthermore, the initiation colon must be in phase with the reading frame of
the desired
coding sequence to ensure translation of the entire insert. These exogenous
translational
control signals and initiation colons can be of a variety of origins, both
natural and
synthetic. The efficiency of expression may be enhanced by the inclusion of
appropriate
-33-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
transcription enhancer elements, transcription terminators, etc. (see Bittner
et al., 1987,
Methods ih Enzymol. 153:51-544).
In addition, a host cell strain may be chosen which modulates the expression
of the
inserted sequences, or modifies and processes the gene product in the specific
fashion
desired. Such modifications (e.g., glycosylation) and processing (e.g.,
cleavage) of protein
products may be important for the function of the protein. Different host
cells have
characteristic and specific mechanisms for the post-translational processing
and
modification of proteins and gene products. Appropriate cell lines or host
systems can be
chosen to ensure the correct modification and processing of the foreign
protein expressed.
To this end, eukaryotic host cells which possess the cellular machinery for
proper
processing of the primary transcript, glycosylation, and phosphorylation of
the gene product
may be used. Such mammalian host cells include but are not limited to CHO,
VERY,
BHK, Hela, COS, MDCK, 293, 293T, 3T3, WI38, BT483, Hs578T, HTB2, BT20 and
T47D, CRL7030 and Hs578Bst.
For long-term, high-yield production of recombinant proteins, stable
expression is
preferred. For example, cell lines which stably express an antibody may be
engineered.
Rather than using expression vectors which contain viral origins of
replication, host cells
can be transformed with DNA controlled by appropriate expression control
elements (e.g.,
promoter, enhancer, sequences, transcription terminators, polyadenylation
sites, etc.), and a
selectable marker. Following the introduction of the foreign DNA, engineered
cells may be
allowed to grow fox 1-2 days in an enriched media, and then are switched to a
selective
media. The selectable marker in the recombinant plasmid confers resistance to
the selection
and allows cells to stably integrate the plasmid into their chromosomes and
grow to form
foci which in turn can be cloned and expanded into cell lines. This method may
advantageously be used to engineer cell lines which express the antibody Such
engineered
cell lines may be particularly useful in screening and evaluation of compounds
that interact
directly or indirectly with the antibody.
A number of selection systems may be used, including but not limited to the
herpes
simplex virus thymidine kinase (Wigler et al., 1977, Cell 11:223),
hypoxanthine-guanine
phosphoribosyltransferase (Szybalska & Szybalski, 192, Proc. Natl. Acad. Sci.
USA
48:202), and adenine phosphoribosyltransferase (Lowy et al., 1980, Cell
22:817) genes can
be employed in tk-, hgprt- or aprt- cells, respectively. Also, antimetabolite
resistance can be
used as the basis of selection for the following genes: dhfr, which confers
resistance to
methotrexate (Wigler et al., 1980, Natl. Acad. Sci. USA 77:357; O'Hare et al.,
1981, Proc.
Natl. Acad. Sci. USA 78:1527); gpt, which confers resistance to mycophenolic
acid
(Mulligan & Berg, 1981, Proc. Natl. Acad. Sci. USA 78:2072); neo, which
confers
resistance to the aminoglycoside G-418Clinical Pharmacy 12:488-505; Wu and Wu,
1991,
-34-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
Biotherapy 3:87-95; Tolstoshev, 1993, Ann. Rev. Pharmacol. Toxicol. 32:573-
596;
Mulligan, 1993, Science 260:926-932; and Morgan and Anderson, 1993, Ann. Rev.
Biochem. 62:191-217; May, 1993, TIB TECH 11(5):155-215). Methods commonly
known
in the art of recombinant DNA technology which can be used are described in
Ausubel et al.
(eds.), 1993, Current Protocols in Molecular Biology, John Wiley & Sons, NY;
Kriegler,
1990, Gene Transfer and Expression, A Laboratory Manual, Stockton Press, NY;
and in
Chapters 12 and 13, Dracopoli et al. (eds), 1994, Current Protocols in Human
Genetics,
John Wiley & Sons, NY.; Colberre-Garapin et al., 1981, J. Mol. Biol. 150:1;
and hygro,
which confers resistance to hygromycin (Santerre et al., 1984, Gene 30:147).
The expression levels of an antibody can be increased by vector amplification
(for a
review, see Bebbington and Hentschel, The use of vectors based on gene
amplification fog
the expression of cloned genes in mammalian cells in DNA cloning, Vol.3.
(Academic
Press, New York, 1987)). When a marker in the vector system expressing an
antibody is
amplifiable, increase in the level of inhibitor present in culture of host
cell will increase the
number of copies of the marker gene. Since the amplified region is associated
with the
nucleotide sequence of the antibody, production of the antibody will also
increase (Crouse
et al., 1983, Mol. Cell. Biol. 3:257).
The host cell may be co-transfected with two expression vectors of the
invention, the
first vector encoding a heavy chain derived polypeptide and the second vector
encoding a
light chain derived polypeptide. The two vectors may contain identical
selectable markers
which enable equal expression of heavy and light chain polypeptides.
Alternatively, a
single vector may be used which encodes both heavy and light chain
polypeptides. In such
situations, the light chain should be placed before the heavy chain to avoid
an excess of
toxic free heavy chain (Proudfoot, 1986, Nature 322:52; Kohler, 1980, Proc.
Natl. Acad.
Sci. USA 77:2197). The coding sequences for the heavy and light chains may
comprise
cDNA or genomic DNA.
Once the antibody has been recombinantly expressed, it may be purified by any
method known in the art for purification of an antibody, for example, by
chromatography
(e.g., ion exchange, affinity, particularly by aff nity for the specif c
antigen after Protein A,
~d sizing column chromatography), centrifugation, differential solubility, or
by any other
standard technique for the purification of proteins.
-35-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
5.5. ANTIBODY CONJUGATES
In certain embodiments, one or more antibodies immunospecific for one or more
cancer cell antigens, viral antigens or microbial antigens are conjugated to a
therapeutic
agent. In a preferred embodiment, anti-C3b(i) antibodies are conjugated to a
diagnostic or
therapeutic agent. The antibodies can be used diagnostically to, for example,
monitor the
development or progression of a tumor as part of a clinical testing procedure
to, e.g.,
determine the efficacy of a given treahnent regimen. Detection can be
facilitated by
coupling the antibody to a detectable substance. Examples of detectable
substances include
various enzymes, prosthetic groups, fluorescent materials, luminescent
materials,
bioluminescent materials, radioactive materials, positron emitting metals
using various
positron emission tomographies, and nonradioactive paramagnetic metal ions.
See
generally U.S. Patent No. 4,741,900 for metal ions which can be conjugated to
antibodies
for use as diagnostics according to the present invention. Examples of
suitable enzymes
include horseradish peroxidase, alkaline phosphatase, beta-galactosidase, or
acetylcholinesterase; examples of suitable prosthetic group complexes include
streptavidin/biotin and avidinlbiotin; examples of suitable fluorescent
materials include
umbelliferone, fluorescein, fluorescein isothiocyanate, rhodamine,
dichlorotriazinylamine
fluorescein, dansyl chloride or phycoerythrin; an example of a luminescent
material
includes Iuminol; examples of bioluminescent materials include luciferase,
luciferin, and
aequorin, and examples of suitable radioactive material include'zSI,131I, "'In
or 99Tc.
Further, an antibody may be conjugated to a therapeutic moiety such as a
cytotoxin,
e.g., a cytostatic or cytocidal agent, a therapeutic agent or a radioactive
metal ion. A
cytotoxin or cytotoxic agent includes any agent that is detrimental to cells.
Examples
include paclitaxol, cytochalasin B, gramicidin D, ethidium bromide, emetine,
mitomycin,
2S etoposide, tenoposide, vincristine, vinblastine, colchicin, doxorubicin,
daunorubicin,
dihydroxy anthracin dione, mitoxantrone, mithramycin, actinomycin D,
1-dehydrotestosterone, glucocorticoids, procaine, tetracaine, lidocaine,
propranolol, and
puromycin and analogs or homologs thereof. Therapeutic agents include, but are
not
limited to, antimetabolites (e.g., methotrexate, 6-mercaptopurine, 6-
thioguanine,
cytarabine, 5-fluorouracil decarbazine), alkylating agents (e.g.,
mechlorethamine, thioepa
chlorambucil, melphalan, carmustine (BSNU) and lomustine (CCNU),
cyclothosphamide,
busulfan, dibromomannitol, streptozotocin, mitomycin C, and cis-
dichlorodiamine platinum
(II) (DDP) cisplatin), anthracyclines (e.g., daunorubicin (formerly
daunomycin) and
doxorubicin), antibiotics (e.g., dactinomycin (formerly actinomycin),
bleomycin,
mithramycin, anthramycin (AMC)), and anti-mitotic agents (e.g., vincristine
and
vinblastine).
-36-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
In one embodiment, anti-C3b(i) antibodies are conjugated to cobra venom
factor. In
accordance with the invention, C3b(i) specific antibodies conjugated to cobra
venom factor
are utilized in vitro to deplete cancerous cells from, e.g., bone marrow
obtained from an
animal, preferably a mammal and most preferably a human, with cancer. Methods
of
S conjugating antibodies to cobra venom factor are taught in U.S. Patent No.
5,773,243,
which is incorporated herein by reference in its entirety.
The conjugates of the invention can be used for modifying a given biological
response; thus, the therapeutic agent or drug moiety is not to be construed as
limited to
classical chemical therapeutic agents. For example, the drug moiety may be a
protein or
polypeptide possessing a desired biological activity. Such proteins may
include, for
example, a toxin such as abrin, ricin A, pseudomonas exotoxin, or diphtheria
toxin; a
protein such as tumor necrosis factor, ec-interferon, (3-interferon,~y-
interferon, nerve growth
factor, platelet derived growth factor, tissue plasminogen activator, a
thrombotic agent or an
anti-angiogenic agent, e.g., angiostatin or endostatin; or, biological
response modifiers such
1S as, for example, lymphol~ines, IL-1, IL-2, IL-4, IL-S ,IL-6, IL-7, Il-8, Il-
9, Il-10, IL-12, IL
1S, granulocyte macrophase colony stimulating factor ("GM-CSF"), granulocyte
colony
stimulating factor ("G-CSF"), or other growth factors.
Techniques for conjugating therapeutic moieties to antibodies are well known,
see,
e.g., Arnon et al., "Monoclonal Antibodies For Immunotargeting Of Drugs In
Cancer
Therapy", in Monoclonal Antibodies And Cancer Therapy, Reisfeld et al. (eds.),
pp. 243-S6
(Alan R. Liss, Inc. 1985); Hellstrom et al., "Antibodies For Drug Delivery",
in Controlled
Drug Delivery (2nd Ed.), Robinson et al. (eds.), pp. 623-S3 (Marcel Dekker,
Inc. 1987);
Thorpe, "Antibody Carriers Of Cytotoxic Agents In Cancer Therapy: A Review",
in
Monoclonal Antibodies '84: Biological And Clinical Applications, Pinchera et
al. (eds.), pp.
47S-S06 (1985); "Analysis, Results, And Future Prospective Of The Therapeutic
Use Of
Radiolabeled Antibody In Cancer Therapy", in Monoclonal Antibodies For Cancer
Detection And Therapy, Baldwin et al. (eds.), pp. 303-16 (Academic Press
1985), and
Thorpe et al., "The Preparation And Cytotoxic Properties Of Antibody-Toxin
Conjugates",
Immunol. Rev., 62:119-S8 (1982); each of which is incorporated herein by
reference.
Alternatively, an antibody can be conjugated to a second antibody to form an
antibody heteroconjugate as described by Segal 11 U.S. Patent No. 4,676,980,
which is
incorporated herein by reference.
An antibody with or without a therapeutic moiety conjugated to it can be used
as a
therapeutic and administered alone or in combination with cytotoxic factors)
andlor
3S cytokine(s).
-37-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
5.6. DEPLETION OF CANCEROUS CELLS IN VITRO
The invention provides for methods of depleting cancerous cells from non-
cancerous
tissues and/or cells ih vitro (or ex vivo). W particular, the invention
provides for methods of
depleting cancerous cells by killing them or by separating them from non-
cancerous cells.
In one embodiment, anti-C3b(i) antibodies alone or in combination with plasma,
are
combined ih vitro with tissues and/or cells obtained from an animal,
preferably a mammal
and most preferably a human. In another embodiment, anti-C3b(i) antibodies
alone or in
combination with IgG antibodies, IgM antibodies and/or one or more complement
components, are combined in vitro with tissues and/or cells obtained from an
animal,
preferably a mammal and most preferably a human. In another embodiment, anti-
C3b(i)
antibodies alone or in combination with antibodies inununospecific for cancer
cell antigens
are combined in vitro with tissues and/or cells obtained from an animal,
preferably a
mammal and most preferably a human. Preferably, the tissue obtained from an
animal for
the in vitro depletion of cancerous cells from non-cancerous cells is bone
marrow and /or
peripheral blood.
In a specific embodiment, monoclonal antibodies inununospecific for C3b(i) are
incubated in vitro with tissues and/or cells obtained from an animal,
preferably a mammal
and most preferably a human. In a preferred embodiment, the monoclonal
antibodies are
immunospecific for C3b(i) covalently linked to IgM or IgG antibody which is
bound to the
cancer cells. In another preferred embodiment, the monoclonal antibodies are
immunospecific for C3b(i) covalently linked to a glycoprotein or or glycolipid
on the cancer
cells, CD20, Her2 or PSMA.
In a preferred embodiment, bispecific antibodies which are immunospecific for
C3b(i) and an effector cell receptor or antigen axe incubated ira vitro with
tissues and/or cells
obtained from an animal, preferably a mammal and most preferably a human. W
another
preferred embodiment, bispecific antibodies which are immunospecific for
C3b(i) and a
complement component (e.g., Clc~ are incubated in vitro with tissues and/or
cells obtained
from an animal, preferably a mammal and most preferably a human. In a
particular
embodiment, bispecific diabodies which are antibodies fragments immunospecific
for
30 C3b(i) and a complement component (e.g., C1~ are incubated iya vitro with
tissues and/or
cells obtained from an animal, preferably a mammal and most preferably a
human. In
accordance with this embodiment, the bispecific diabodies facilitate
complement mediated
lysis of the cancer cells.
In accordance with the invention, the anti-C3b(i) antibodies used in the in
vitro
35 depletion of cancer cells from tissues can be conjugated to detectable
substances (e.g.,
various enzymes, fluorescent materials, luminescent materials, bioluminescent
materials,
and radioactive materials) or therapeutic agents (e.g., cytostatic and
cytocidal agents), which
-38-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
are disclosed in section 5.5. For example, anti-C3b(i) antibodies may be
conjugated to
cobra venom factor in order to use enhanced complement activation to lyse the
cancer cells.
In a specific embodiment, tissues and/or cells thus depleted of cancerous
cells are
administered to an animal, preferably a mammal and most preferably a human. In
a
preferred embodiment, the tissues and/or cells are obtained from an animal
with cancer
prior to treatment for cancer, and tissues and/or cells depleted of cancerous
cells are
administered to the animal after the treatment. In yet another preferred
embodiment, the in
vitro depletion of cancerous cells using anti-C3b(i) antibodies is done prior
to
administration of autologous bone marrow peripheral blood, or peripheral blood
stem cells.
Anti-C3b(i) antibodies conjugated to detectable substances can be utilized to
sort
cancerous cells from non-cancerous cells by methods known to those of skill in
the art. In
one embodiment, cancerous cells are sorted using a fluorescence activated cell
sorter
(FAGS). Fluorescence activated cell sorting (FACS) is a well-knoum method for
separating particles, including cells, based on the fluorescent properties of
the particles
(Kamarch, 1987, Methods Enzymol, 151:150-165). Laser excitation of fluorescent
moieties
in the individual particles results in a small electrical charge allowing
electromagnetic
separation of positive and negative particles from a mixture.
In one embodiment, cells, particularly bone marrow cells or peripheral blood
stem
cells, obtained from an animal, preferably a mammal and most preferably a
human, are
incubated with fluorescently labeled C3b(i) specific antibodies for a time
sufficient to allow
the labeled antibodies to bind to the cells (e.g., 10 minutes, 20 minutes, 30
minutes, 45
minutes, 60 minutes, 90 minutes, 120 minutes or more), preferably between 10
to 60
minutes. In an alternative embodiment, cells, particularly bone marrow cells
or peripheral
blood stem cells, obtained from an animal preferably a mammal and most
preferably a
h~n~, are incubated with C3b(i) specific antibodies, the cells are washed, and
the cells are
incubated with a second labeled antibody that recognizes the C3b(i) specific
antibodies. hl
accordance with these embodiments, the cells are washed and processed through
the cell
sorter, allowing separation of cells that bind both antibodies to be separated
from hybrid
cells that do not bind both antibodies. FACS sorted particles may be directly
deposited into
individual wells of 96-well or 384-well plates to facilitate separation.
In another embodiment, magnetic beads can be used to separate cancerous cells
from
non-cancerous cells. Cancerous cells may be sorted using a magnetic activated
cell sorting
(MACS) technique, a method for separating particles based on their ability to
bind magnetic
beads (0.5-100 nm diameter) (Dynal, 1995). A variety of useful modifications
can be
performed on the magnetic microspheres, including covalent addition of
antibody which
immunospecifically recognizes C3b(i). A magnetic field is then applied, to
physically
-39-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
manipulate the selected beads. The beads are then mixed with the cells to
allow binding.
Cells are then passed through a magnetic field to separate out cancerous
cells.
5.7. THERAPEUTIC AND PROPHYLACTIC
USE OF ANTI-C3b~) ANTIBODIES
The invention provides for treatment or prevention of cancer, including, but
not
limited to, neoplasms, tumors, metastases, or any disease or disorder
characterized by
uncontrolled cell growth, by the administration of therapeutically or
prophylactically
effective amounts of anti-C3b(i) antibodies or nucleic acid molecules encoding
said
~tibodies. Examples of types of cancer and proliferative disorders include,
but are not
limited to, leukemia (e.g., myeloblastic, promyelocytic, myelomonocytic,
monocytic,
erythroleukemia, chronic myelocytic (granulocytic) leukemia, and chronic
lymphocytic
leukemia), lymphoma (e.g., Hodgkin's disease and non-Hodgkin's disease),
fibrosarcoma,
myxosarcoma, liposarcoma, chondrosarcoma, osteogenic sarcoma, angiosarcoma,
endotheliosaxcoma, Ewing's tumor, colon carcinoma, pancreatic cancer, breast
cancer,
ovarian cancer, prostate cancer, squamous cell carcinoma, basal cell
carcinoma,
adenocarcinoma, renal cell carcinoma, hepatoma, Wilms' tumor, cervical cancer,
uterine
cancer, testicular tumor, lung carcinoma, small cell lung carcinoma, bladder
carcinoma,
epithelial carcinoma, glioma, astrocytoma, oligodendroglioma, melanoma,
neuroblastoma,
retinoblastoma, dysplasia and hyperplasia. In a particular embodiment,
therapeutic
compounds of the invention are administered to men with prostate cancer (e.g.,
prostatitis,
benign prostatic hypertrophy, benign prostatic hyperplasia (BPH), prostatic
paraganglioma,
prostate adenocarcinoma, prostatic intraepithelial neoplasia, prostato-rectal
fistulas, and
atypical prostatic stromal lesions). The treatment and/or prevention of cancer
includes, but
is not limited to, alleviating one or more symptoms associated with cancer,
the inhibition or
reduction of the progression of cancer, the promotion of the regression of
cancer, and/or the
promotion of the immune response. In one embodiment, commercially available or
naturally occurring anti-C3b(i) antibodies, functionally active fragments or
derivatives
thereof are used in the present invention.
30 ~ certain embodiments, a composition of the invention is administered to an
animal
to ameliorate one or more symptoms associated with cancer. In certain other
embodiments,
a composition of the invention is administered to an animal to inhibit or
reduce the
progression of cancer. In certain other embodiments, a composition of the
invention is
administered to an animal to promote the regression of cancer.
35 ~ a specific embodiment, a composition comprising a therapeutically
effective
amount of one or more anti-C3b(i) antibodies is administered to an animal in
order to
ameliorate one or more symptoms associated with cancer. In another embodiment,
a
-40-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
composition comprising a therapeutically effective amount of one or more anti-
C3b(i)
antibodies is administered to an animal in order to promote of the regression
of cancer
and/or the inhibit or reduce the progression of cancer in an aiumal. In
another embodiment,
a composition comprising a therapeutically effective amount of one or more
anti-C3b(i)
antibodies and one or more antibodies immunospecific for one or more cancer
cell antigens
is administered to an animal in order to ameliorate one or more symptoms
associated with
cancer. In yet another embodiment, a composition comprising a therapeutically
effective
amount of one or more anti-C3b(i) antibodies and one or more antibodies
immunospecific
for one or more cancer cell antigens is administered to an animal in order to
promote of the
regression of cancer andlor the inhibit or reduce the progression of cancer in
an animal.
Anti-C3b(i) antibodies may be administered alone or in combination with other
types of cancer treatments (e.g., radiation therapy, chemotherapy, hormonal
therapy,
immunotherapy and anti-tumor agents). Examples of anti-tumor agents include,
but axe not
limited to, cisplatin, ifosfamide, paclitaxel, taxanes, topoisomerase I
inhibitors (e.g.,
CPT-11, topotecan, 9-AC, and GG-211), gemcitabW e, vinorelbine, oxaliplatin, 5-
fluorouracil (5-FLT, leucovorin, vinorelbine, temodal, and taxol. In one
embodiment, one
or more anti-C3b(i) antibodies are administered to an animal, preferably a
mammal and
most preferably a human, after surgical resection of cancer. In another
embodiment, one or
more anti-C3b(i) antibodies are administered to an animal, preferably a mammal
and most
preferably a human, in conjugation with chemotherapy or radiotherapy. In
another
embodiment, one or more anti-C3b(i) antibodies are administered to an animal,
preferably a
mammal and most preferably a human, for the prevention or treatment of cancer
prior to
(e.g., 1 minute, 15 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours,
6 hours, 8
hours, 12 hours, 24 hours, 2 days, or 1 week before), subsequent to (e.g., 1
minute, 15
minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 8 hours,
12 hours, 24
hours, 2 days, or 1 week after), or concomitantly with the administration of
plasma to the
animal.
hi a preferred embodiment, one or more anti-C3b(i) antibodies are administered
to
an animal, preferably a mammal and most preferably a human, for the prevention
or
~'eatrnent of cancer prior to (e.g., 1 minute, 15 minutes, 30 minutes, 45
minutes, 1 hour, 2
hours, 4 hours, 6 hours, 8 hours, 12 hours, 24 hours, 2 days, or 1 week
before), subsequent
to (e.g., 1 minute, 15 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4
hours, 6 hours, 8
hours, 12 hours, 24 hours, 2 days, or 1 week after), or concomitantly with the
administration
of IgG antibodies, IgM antibodies and/or one or more complement components to
the
a~mal. In another preferred embodiment, one or more anti-C3b(i) antibodies are
administered to an animal, preferably a mammal and most preferably a human,
prior to
(e.g., 1 minute, 15 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours,
6 hours, 8
-41-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
hours, 12 hours, 24 hours, 2 days, or 1 week before), subsequent to (e.g., 1
minute, 15
minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 8 hours,
12 hours, 24
hours, 2 days, or 1 week after), or concomitantly with the administration of
antibodies
immunospecific for one or more cancer cell antigens. In yet another preferred
embodiment,
one or more anti-C3b(i) antibodies are administered to an animal, preferably a
mammal and
most preferably a human, for the prevention or treatment of cancer prior to
(e.g., 1 minute,
15 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 8
hours, 12 hours, 24
hours, 2 days, or 1 week before), subsequent to (e.g., 1 minute, 15 minutes,
30 minutes, 45
minutes, 1 hour, 2 hours, 4 hours, 6 hours, 8 hours, 12 hours, 24 hours, 2
days, or 1 week
after), or concomitantly with the administration of antibodies currently used
for the
treatment of cancer. Examples of such antibodies include, but are not limited
to,
Herceptin~ , Retuxan~, OvaRex, Panorex, BEC2, IMC-C225, Vitaxin, Campath I/H,
Smart MI95, LymphoCide, Smart I D10, and Oncolym.
In a specific embodiment, men with prostate cancer are administered anti-
C3b(i)
antibodies in conjugation with androgen ablation therapy. In another specific
embodiment,
non-Hodgkin's lymphoma patients are treated with Retuxan~ prior to the
administration of
anti-C3b(i) antibodies. In certain embodiments, animals with increased rislc
of cancer are
administered a composition of the invention. Examples of such animals include,
but are not
limited to, humans prone to breast cancer.
The invention provides methods for the treatment or prevention of viral
infections
in an animal, preferably a mammal and most preferably a human, said methods
comprising
the administration of a therapeutically or prophylactically effective amount
of anti-C3b(i)
antibodies or nucleic acid molecules encoding said antibodies. Examples of
viral infections
which can be treated or prevented in accordance with this invention include,
but are limited
to, viral infections caused by retroviruses (e.g., human T-cell lymphotrophic
virus (HTLV)
types I and II and human immunodeficiency virus (HIV)), herpes viruses (e.g.,
herpes
simplex virus (HSV) types I and II, Epstein-Barr virus and cytomegalovirus),
arenaviruses
(e.g., lassa fever virus), paramyxoviruses (e.g., morbillivirus virus, human
respiratory
syncytial virus, and pneumovirus), adenoviruses, bunyaviruses (e.g.,
hantavirus),
cornaviruses, filoviruses (e.g., Ebola virus), flaviviruses ( e.g., hepatitis
C virus (HCV),
yellow fever virus, and Japanese encephalitis virus), hepadnaviruses (e.g.,
hepatitis B
viruses (HBV)), orthomyoviruses (e.g., Sendai virus and influenza viruses A, B
and C),
papovaviruses (e.g., papillomavirues), picornaviruses (e.g., rhinoviruses,
enteroviruses and
hepatitis A viruses), poxviruses, reoviruses (e.g., rotavirues), togaviruses
(e.g., rubella
virus), and rhabdoviruses (e.g., rabies virus). The treatment and/or
prevention of a viral
infection includes, but is not limited to, alleviating one or more symptoms
associated with
-42-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
said infection, the inhibition, reduction or suppression of viral replication,
and/or the
enhancement of the immune response.
In certain embodiments, a composition of the invention is administered to an
animal
to ameliorate one or more symptoms associated with a viral infection or a
disease or
disorder resulting, directly or indirectly, from a viral infection. In a
specific embodiment, a
composition of the invention is administered to a human to ameliorate one or
more
symptoms associated with AIDS. In certain other embodiments, a composition of
the
invention is administered to reduce the titer of a virus in an aiumal. In
certain other
embodiments, a composition of the invention is administered to an animal to
enhance or
promote the immune response.
In a specific embodiment, a composition comprising a therapeutically effective
amount of one or more anti-C3b(i) antibodies is administered to an animal in
order to
ameliorate one or more synptoms associated with a viral infection. In another
embodiment,
a composition comprising a therapeutically effective amount of one or more
anti-C3b(i)
antibodies is administered to an animal in order to reduce the titer of a
virus in an animal.
In another embodiment, a composition comprising a therapeutically effective
amount of one
or more anti-C3b(i) antibodies and one or more antibodies immunospecific for
one or more
viral antigens is administered to an animal in order to ameliorate one or more
symptoms
associated with a viral infection. In yet another embodiment, a composition
comprising a
therapeutically effective amount of one or more anti-C3b(i) antibodies and one
or more
antibodies immunospecific for one or more viral antigens is administered to an
animal in
order to reduce the titer of a virus in an animal.
Anti-C3b(i) antibodies may be administered alone or in combination with other
types of anti-viral agents. Examples of anti-viral agents include, but are not
limited to:
cytolcines (e.g., IFN-a, IFN-(3, and IFN-'y); inhibitors of reverse
transcriptase (e.g., AZT,
3TC, D4T, ddC, ddI, d4T, 3TC, adefovir, efavirenz, delavirdine, nevirapine,
abacavir, and
other dideoxynucleosides or dideoxyfluoronucleosides); inhibitors of viral
mRNA capping,
such as ribavirin; inhibitors of proteases such HIV protease inhibitors (e.g.,
amprenavir,
indinavir, nelfinavir, ritonavir, and saquinavir,); amphotericin B;
castanospermine as an
inhibitor of glycoprotein processing; inhibitors of neuraminidase such as
influenza virus
neuraminidase inhibitors (e.g., zanamivir and oseltamivir); topoisomerase I
inhibitors (e.g.,
camptothecins and analogs thereof); amantadine; and rimantadine. Such anti-
viral agents
may be administered to an animal, preferably a mammal and most preferably a
human, for
the prevention or treatment of a viral infection prior to (e.g., 1 minute, 15
minutes, 30
minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, ~ hours, 12 hours, 24
hours, 2 days,
or 1 week before), subsequent to (e.g., 1 minute, 15 minutes, 30 minutes, 45
minutes, 1
-43-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
hour, 2 hours, 4 hours, 6 hours, 8 hours, 12 hours, 24 hours, 2 days, or 1
week after), or
concomitantly with the administration of anti-C3b(i) antibodies to the animal.
In a specific embodiment, one or more anti-C3b(i) antibodies are adminstered
to an
animal, preferably a mammal and most preferably a human, for the prevention or
treatment
of a viral infection prior to (e.g., 1 minute, 15 minutes, 30 minutes, 45
minutes, 1 hour, 2
hours, 4 hours, 6 hours, 8 hours, 12 hours, 24 hours, 2 days, or 1 week
before), subsequent
to (e.g., 1 minute, 15 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4
hours, 6 hours, 8
hours, 12 hours, 24 hours, 2 days, or 1 week after), or concomitantly with the
administration
of plasma to the animal.
In a preferred embodiment, one or more anti-C3b(i) antibodies are administered
to
an animal, preferably a mammal and most preferably a human, for the prevention
or
treatment of a viral infection prior to (e.g., 1 minute, 15 minutes, 30
minutes, 45 minutes, 1
hour, 2 hours, 4 hours, 6 hours, 8 hours, 12 hours, 24 hours, 2 days, or 1
week before),
subsequent to (e.g., 1 minute, 15 minutes, 30 minutes, 45 minutes, 1 hour, 2
hours, 4 hours,
6 hours, 8 hours, 12 hours, 24 hours, 2 days, or 1 week after), or
concomitantly with the
administration of IgG antibodies, IgM antibodies and/or one or more complement
components to the anmal. In another preferred embodiment, anti-C3b(i)
antibodies are
administered to an animal, preferably a mannnal and most preferably a human,
for the
prevention or treatment of a viral infection prior to (e.g., 1 minute, 15
minutes, 30 minutes,
45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 8 hours, 12 hours, 24 hours, 2
days, or 1 week
before), subsequent to (e.g., 1 minute, 15 minutes, 30 minutes, 45 minutes, 1
hour, 2 hours,
4 hours, 6 hours, 8 hours, 12 hours, 24 hours, 2 days, or 1 week after), or
concomitantly
with the administration of antibodies immunospecific for one or more viral
antigens.
Example of antibodies immunospecific for viral antigens include, but are not
limited to,
Synagis~, PR0542, Ostavir, and Protovir.
The invention provides methods for the treatment or prevention of microbial
infections in an animal, preferably a marmnal and most preferably a human,
said methods
comprising the administration of a therapeutically or prophylactically
effective amount of
anti-C3b(i) antibodies or nucleic acid molecules encoding said antibodies.
Examples of
microbial infections which can be treated or prevented in accordance with this
invention
include, but are not limited to, yeast infections, fungal infections,
protozoan infections and
bacterial infections. Bacteria which cause microbial infections include, but
are not limited
to, Streptococcus pyogenes, Streptococcus pneumoniae, Neisseria gonorrhoea,
Neisse~ia
meningitidis, Corynebacteniuna diplatheriae , ClostYidium botulinum,
Clostridium
peyfy'ingens, Clostridium tetani, Haemophilus iy~uenzae, Klebsiella
pneumoniae, Klebsiella
ozaenae, Klebsiella rhinoscleromotis, Staphylococcus au~eus, Tlib~io
c7ZOlenae, EschericlZia
coli, Pseudomonas aeruginosa, Campylobacte~ (Vibrio) fetus, Campylobacter
jejuni,
-44-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
Ae~omonas layd~oplaila, Bacillus ce~eus, Edwa~dsiella tarda, Ye~sinia
ente~ocolitica,
Ye~sinia pestis, Yet~sinia pseudotuberculosis, Shigella dysente~iae, Shigella
flexneYi,
Shigella sonnei, Salmonella typhimurium, Treponema palliduna, Treponema
peYtenue,
Ti~epoyaema carateneum, Bof°Yelia vincentii, Bor~elia burgdo~feri,
Leptospif~a
icte~ohemo~rhagiae, Mycobacterium tuberculosis, Toxoplastna gondii,
Pneumocystis
carinii, F~aracisella tula~eszsis, B~ucella abo~tus, B~ucella suis, B~ucella
melitensis,
Mycoplasma spp., Rickettsia prowazeki, Rickettsia tsutsugunauslZi, Chlatnydia
spp., and
Helicobacter pyloni. The treatment and/or prevention of a microbial infection
includes, but
is not limited to, alleviating one or more symptoms associated with said
infection, the
inhibition, reduction or suppression of microbial replication, and/or the
enhancement of the
immune response.
In certain embodiments, a composition of the invention is administered to an
animal
to ameliorate one or more symptoms associated with a microbial infection or a
disease or
disorder resulting, directly or indirectly, from a microbial infection. In
certain other
embodiments, a composition of the invention is administered to reduce the
number or
microbes in an animal.
In a specific embodiment, a composition comprising a therapeutically effective
amount of one or more anti-C3b(i) antibodies is adminstered to an animal in
order to
ameliorate one or more symptoms associated with a microbial infection. In
another
embodiment, a composition comprising a therapeutically effective amount of one
or more
anti-C3b(i) antibodies is administered to an animal in order to reduce the
number of
microbes in an animal. In another embodiment, a composition comprising a
therapeutically
effective amount of one or more anti-C3b(i) antibodies and one or more
antibodies
immunospecific for one or more microbial antigens is administered to an animal
in order to
ameliorate one or more symptoms associated with a microbial infection. In yet
another
embodiment, a composition comprising a therapeutically effective amount of one
or more
anti-C3b(i) antibodies and one or more antibodies immunospecific for one or
more
microbial antigens is administered to an animal in order to reduce the number
of microbes
in an animal.
Anti-C3b(i) antibodies may be administered alone or in combination with other
types of anti-microbial agents. Examples of anti-microbial agents include, but
are not
limited to: antibiotics such as penicillin, amoxicillin, ampicillin,
carbenicillin, ticarcillin,
piperacillin, cepalospolin, vancomycin, tetracycline, erythromycin,
amphotericin B,
nystatin, metronidazole, ketoconazole, and pentamidine. Such anti-microbial
agents may be
administered to an animal, preferably a mammal and most preferably a human,
for the
prevention or treatment of a microbial infection prior to (e.g., 1 minute, 15
minutes, 30
minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 8 hours, 12 hours, 24
hours, 2 days,
- 45 -
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
or 1 week before), subsequent to (e.g., 1 minute, 15 minutes, 30 minutes, 45
minutes, 1
hour, 2 hours, 4 hours, 6 hours, 8 hours, 12 hours, 24 hours, 2 days, or 1
week after), or
concomitantly with the administration of anti-C3b(i) antibodies to the animal.
In a specific embodiment, anti-C3b(i) antibodies are administered to an
animal,
preferably a mammal and most preferably a human, for the prevention or
treatment of a
microbial infection prior to (e.g., 1 minute, 15 minutes, 30 minutes, 45
minutes, 1 hour, 2
hours, 4 hours, 6 hours, 8 hours, 12 hours, 24 hours, 2 days, or 1 week
before), subsequent
to (e.g., 1 minute, 15 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4
hours, 6 hours, 8
hours, 12 hours, 24 hours, 2 days, or 1 week after) or concomitantly with the
administration
of plasma to the animal.
In a preferred embodiment, one or more anti-C3b(i) antibodies are administered
to
an animal, preferably a mammal and most preferably a htunan, for the
prevention or
treatment of a microbial infection prior to (e.g., 1 minute, 15 minutes, 30
minutes, 45
minutes, 1 hour, 2 hours, 4 hours, 6 hours, 8 hours, 12 hours, 24 hours, 2
days, or 1 week
before), subsequent to (e.g., 1 minute, 15 minutes, 30 minutes, 45 minutes, 1
hour, 2 hours,
4 hours, 6 hours, 8 hours, 12 hours, 24 hours, 2 days, or 1 week after), or
concomitantly
with the administration of IgG antibodies, IgM antibodies and/or one or more
complement
components to the animal. In another preferred embodiment, one or more anti-
C3b(i)
antibodies are administered to an animal, preferably a mammal and most
preferably a
h~nan, for the prevention or treatment of a microbial infection prior to
(e.g., 1 minute, 15
minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 8 hours,
12 hours, 24
hours, 2 days, or 1 week before), subsequent to (e.g., 1 minute, 15 minutes,
30 minutes, 45
minutes, 1 hour, 2 hours, 4 hours, 6 hours, 8 hours, 12 hours, 24 hours, 2
days, or 1 week
after), or concomitantly with the administration of antibodies immunospecific
for one or
more microbial antigens. Example of antibodies immunospecific for microbial
antigens
include, but are not limited to, antibodies immunospecific for LPS and
capsular
polysaccharide 5/8. W certain embodiments, animals with increased risk of a
viral or
bacterial infection are administered a composition of the invention. Examples
of such
animals include, but are not limited to human burn patients, infants,
irninunocompromised
or immunodeficient humans, and the elderly.
The present invention provides methods for the treatment or prevention of
septic
shock caused by lipopolysaccharide (LPS), an endotoxin, or a constituent of
the outer cell
wall of a gram negative bacteria, which can be released into the circulation.
In a specific
embodiment, anti-C3b(i) antibodies are administered to an animal, preferably a
mammal
and most preferably a human, to treat or prevent septic shock. In certain
embodiments,
animals with increased risk of septic shock are administered a composition of
the invention.
-46-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
Examples of such animals include, but are not limited to human burn patients,
infants,
immunocompromised or immunodeficient humans, and the elderly.
In a specific embodiment, one or more anti-C3(i) antibodies are administered
to an
animal, preferably a mammal and most preferably in human, prior to (e.g., 15
minuted, 30
minutes, 45 minutes, 1 hour, 2 hours, 6 hours, 12 hours, or 24 hours before),
subsequent to
(e.g., 15 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 6 hours, 12 hours,
or 24 hours
after) or concomitantly with any known technique for the treatment or
prevention of septic
shock in said animal. Examples of known techtuques for the treatment or
prevention of
septic shoclc include, but are not limited to, antithrombin, intravenous
immunoglobulins,
cytokine antagonists (e.g., anti-tumor necrosis factor (TNF) antibodies,
soluble TNF
receptor, anti-interleukin-1 (IL-1) antibodies, and soluble IL-1 receptor),
antibiotics, and
anti-inflammatory agents. The treatment and/or prevention of septic shock
includes, but is
not limited to, alleviating one or more symptoms associated with one or more
symptoms
with septic shook and the enhancement of the immune response.
In a specific embodiment, a composition comprising a therapeutically effective
amount of one or more anti-C3b(i) antibodies is administered to an animal in
order to
ameliorate one or more symptoms associated with septic shock. In another
embodiment, a
composition comprising a therapeutically effective amount of one or more anti-
C3b(i)
antibodies and IgG and/or IgM enriched plasma is administered to an animal in
order to
ameliorate one or more symptoms associated with septic shock. In another
embodiment, a
composition comprising a therapeutically effective amount of one or more anti-
C3b(i)
antibodies and one or more complement components is administered to an animal
in order
to ameliorate one or more symptoms associated with septic shock. In yet
another
embodiment, a composition comprising a therapeutically effective amount of one
or more
anti-C3b(i) antibodies and one or more antibodies immunospecific for
lipopolysaccharide is
administered to an animal in order to ameliorate one or more symptoms
associated with
septic shock.
Generally, administration of products of a species origin or species
reactivity (in the
case of antibodies) that is the same species as that of the patient is
preferred. Thus, in a
preferred embodiment, human anti-C3b(i) antibodies, derivatives, analogs, or
nucleic acids,
are administered to a human patient for therapy or prophylaxis.
-47-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
5.7.1. GENE THERAPY
The present invention provides methods for the treatment or prevention of
cancer,
viral infection, and microbial infection comprising administering nucleic acid
molecules
encoding one or more anti-C3b(i) antibodies. In a specific embodiment, nucleic
acids
comprising sequences encoding one or more anti-C3b(i) antibodies are
administered to treat
or prevent cancer, viral infection or microbial infection, by way of gene
therapy. In another
embodiment, nucleic acids comprising sequences encoding one or more anti-
C3b(i)
antibodies and one or more antibodies immunospecific for one or more cancer
antigens are
administered to treat or prevent cancer, by way of gene therapy. In another
embodiment,
nucleic acids comprising sequences encoding one or more anti-C3b(i) antibodies
and one or
more antibodies immunospecific for one or more viral or microbial antigens are
administered to treat or prevent viral or microbial infection, by way of gene
therapy. In yet
another embodiment, nucleic acids comprising sequences encoding one or more
anti-C3b(i)
antibodies and one or more complement components are administered to treat or
prevent
cancer, viral infection, or microbial infection, by way of gene therapy. Gene
therapy refers
to therapy performed by the administration to a subject of an expressed or
expressible
nucleic acid. In this embodiment of the invention, the nucleic acids produce
their encoded
protein that mediates a prophylactic or therapeutic effect.
Any of the methods for gene therapy available in the art can be used according
to the
present invention. Exemplary methods are described below.
For general reviews of the methods of gene therapy, see Goldspiel et al.,
1993,
Cliucal Pharmacy 12:488-505; Wu and Wu, 1991, Biotherapy 3:87-95; Tolstoshev,
1993,
Ann. Rev. Pharmacol. Toxicol. 32:573-596; Mulligan, 1993, Science 260:926-932;
and
Morgan and Anderson, 1993, Ann. Rev. Biochem. 62:191-217; May, 1993, TIBTECH
11 (5):155-215). Methods commonly known in the art of recombinant DNA
technology
which can be used are described in Ausubel et al. (eds.), 1993, Current
Protocols in
Molecular Biology, John Wiley & Sons, NY; and I~riegler, 1990, Gene Transfer
and
Expression, A Laboratory Manual, Stockton Press, NY.
In one aspect, a composition comprising nucleic acid sequences encoding one or
more anti-C3b(i) antibodies, one or more antibodies imrnunospecific for cancer
antigens,
viral antigens or microbial antigens, and/or one or more component components
in an
expression vector are admiiustered to suitable hosts. The expression of
nucleic acid
sequences encoding anti-C3b(i) antibodies, antibodies immunospecific for
cancer antigens,
viral antigens or microbial antigens, and/or component components may be
regulated by
any inducible, constitutive, or tissue-specific promoter known to those of
skill in the art. In
a specific embodiment, the nucleic acid to be introduced for purposes of gene
therapy
comprises an inducible promoter operably linked to the coding region, such
that expression
- 48 -
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
of the nucleic acid is controllable by controlling the presence or absence of
the appropriate
inducer of transcription.
In a particular embodiment, nucleic acid molecules encoding the coding regions
of
anti-C3b(i) antibodies, antibodies immunospecific for cancer antigens, viral
antigens or
microbial antigens, and/or component components and any other desired
sequences are
flanked by regions that promote homologous recombination at a desired site in
the genome,
thus providing for intrachromosomal expression of said coding regions (Koller
and
Smithies, 1989, Proc. Natl. Acad. Sci. USA 86:8932-8935; Zijlstra et al.,
1989, Nature
342:435-438).
Delivery of the nucleic acids into a patient may be either direct, in which
case the
patient is directly exposed to the nucleic acid or nucleic acid-carrying
vectors, or indirect, in
which case, cells are first transformed with the nucleic acids ih vitro, then
transplanted into
the patient. These two approaches are known, respectively, as in vivo or ex
vivo gene
therapy.
In a specific embodiment, the nucleic acid sequences are directly administered
ih
vivo, where it is expressed to produce the encoded product. This can be
accomplished by
any of numerous methods known in the art, e.g., by constructing them as part
of an
appropriate nucleic acid expression vector and administering it so that they
become
intracellular, e.g., by infection using defective or attenuated retrovirals or
other viral vectors
(see U.S. Patent No. 4,980,286), or by direct injection of naked DNA, or by
use of
microparticle bombardment (e.g., a gene gun; Biolistic, Dupont), or coating
with lipids or
cell-surface receptors or transfecting agents, encapsulation in liposomes,
microparticles, or
microcapsules, or by administering them in linkage to a peptide which is known
to enter the
nucleus, by administering it in linkage to a ligand subject to receptor-
mediated endocytosis
(see, e.g., Wu and Wu, 1987, J. Biol. Chem. 262:4429-4432) (which can be used
to target
cell types specifically expressing the receptors), etc. In another embodiment,
nucleic
acid-ligand complexes can be formed in which the ligand comprises a fusogenic
viral
peptide to disrupt endosomes, allowing the nucleic acid to avoid lysosomal
degradation. In
yet another embodiment, the nucleic acid can be targeted ih vivo for cell
specific uptake and
expression, by targeting a specific receptor (see, e.g., PCT Publications WO
92/06180 dated
April 16, 1992 (Wu et al.); WO 92/22635 dated December 23, 1992 (Wilson et
al.);
W092/20316 dated November 26, 1992 (Findeis et al.); W093/14188 dated July 22,
1993
(Clarke et al.), WO 93/20221 dated October 14, 1993 (Young)). Alternatively,
the nucleic
acid can be introduced intracellularly and incorporated within host cell DNA
for expression,
by homologous recombination (Koller and Smitlues, 1989, Proc. Natl. Acad. Sci.
USA
86:8932-8935; Zijlstra et al., 1989, Nature 342:435-438).
-49-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
In a specific embodiment, viral vectors are used to express nucleic acid
sequences.
For example, a retroviral vector can be used (see Miller et al., 1993, Meth.
Enzymol.
217:581-599). These retroviral vectors have been to delete retroviral
sequences that are not
necessary for packaging of the viral genome and integration into host cell
DNA. The
nucleic acid sequences encoding the antibodies and/or complement components to
be used
in gene therapy are cloned into one or more vectors, which facilitates
delivery of the gene
into a patient. More detail about retroviral vectors can be found in Boesen et
al., 1994,
Biotherapy 6:291-302, which describes the use of a retroviral vector to
deliver the mdrl
gene to hematopoietic stem cells in order to make the stem cells more
resistant to
chemotherapy. Other references illustrating the use of retroviral vectors in
gene therapy
are: Clowes et al., 1994, J. Clin. Invest. 93:644-651; Kiem et al., 1994,
Blood
83:1467-1473; Salmons and Gunzberg, 1993, Human Gene Therapy 4:129-141; and
Grossman and Wilson, 1993, Curr. Opin. in Genetics and Devel. 3:110-114.
Adenoviruses are other viral vectors that can be used in gene therapy.
Adenoviruses
are especially attractive vehicles for delivering genes to respiratory
epithelia. Adenoviruses
naturally infect respiratory epithelia where they cause a mild disease. Other
targets for
adenovirus-based delivery systems are liver, the central nervous system,
endothelial cells,
and muscle. Adenoviruses have the advantage of being capable of infecting non-
dividing
cells. Kozarsky and Wilson, 1993, Current Opinion in Genetics and Development
3:499-503 present a review of adenovirus-based gene therapy. Bout et al.,
1994, Human
Gene Therapy 5:3-10 demonstrated the use of adenovirus vectors to transfer
genes to the
respiratory epithelia of rhesus monkeys. Other instances of the use of
adenoviruses in gene
therapy can be found in Rosenfeld et al., 1991, Science 252:431-434; Rosenfeld
et al., 1992,
Cell 68:143-155; Mastrangeli et al., 1993, J. Clin. Invest. 91:225-234; PCT
Publication
W094/12649; and Wang, et al., 1995, Gene Therapy 2:775-783. In a preferred
embodiment, adenovirus vectors are used to deliver antibodies and/or
complement
components to an animal in accordance with methods of the invention.
Adeno-associated virus (AAV) has also been proposed for use in gene therapy
(Walsh et al., 1993, Proc. Soc. Exp. Biol. Med. 204:289-300; U.S. Patent No.
5,436,146).
In one embodiment, AAV vectors are used to delivery antibodies and/or
complement
components to an animal in accordance with methods of the invention.
Another approach to gene therapy involves transferring a gene to cells in
tissue
culture by such methods as electroporation, lipofection, calcium phosphate
mediated
transfection, or viral infection. Usually, the method of transfer includes the
transfer of a
selectable marker to the cells. The cells are then placed under selection to
isolate those cells
that have taken up and are expressing the transferred gene. Those cells are
then delivered to
a patient.
-50-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
In this embodiment, the nucleic acid is introduced into a cell prior to
administration
in vivo of the resulting recombinant cell. Such introduction can be carried
out by any
method known in the art, including but not limited to transfection,
electroporation,
microinjection, infection with a viral or bacteriophage vector containing the
nucleic acid
sequences, cell fusion, chromosome-mediated gene transfer, microcell-mediated
gene
transfer, spheroplast fusion, etc. Numerous techniques are known in the art
for the
introduction of foreign genes into cells (see, e.g., Loeffler and Behr, 1993,
Meth. Enzymol.
217:599-618; Cohen et al., 1993, Meth. Enzymol. 217:618-644; Cline, 1985,
Pharmac.
Ther. 29:69-92) and may be used in accordance with the present invention,
provided that the
necessary developmental and physiological functions of the recipient cells are
not disrupted.
The technique should provide for the stable transfer of the nucleic acid to
the cell, so that
the nucleic acid is expressible by the cell and preferably heritable and
expressible by its cell
progeny.
The resulting recombinant cells can be delivered to a patient by various
methods
known in the art. Recombinant blood cells (e.g., hematopoietic stem or
progenitor cells) are
preferably administered intravenously. The amount of cells envisioned for use
depends on
the desired effect, patient state, etc., and can be determined by one skilled
in the art.
Cells into which a nucleic acid can be introduced for purposes of gene therapy
encompass any desired, available cell type, and include, but are not limited
to, epithelial
cells, endothelial cells, keratinocytes, fibroblasts, muscle cells,
hepatocytes, blood cells
(e.g., T lymphocytes, B lymphocytes, monocytes, macrophages, neutrophils,
eosinophils,
megakaryocytes, and granulocytes) and various stem or progenitor cells, in
particular
hematopoietic stem or progenitor cells, e.g., as obtained from bone marrow,
umbilical cord
blood, peripheral blood, fetal liver, etc.
In a preferred embodiment, the cell used for gene therapy is autologous to the
patient.
In an embodiment in which recombinant cells are used in gene therapy, nucleic
acid
sequences encoding anti-C3b(i) antibodies, antibodies immunospecific for
cancer antigens,
viral antigens or microbial antigens, and/or component components are
introduced into the
cells such that they are expressible by the cells or their progeny, and the
recombinant cells
are then administered in vivo for therapeutic effect. In a specific
embodiment, stem or
progenitor cells are used. Any stem and/or progenitor cells which can be
isolated and
maintained ifz vitro can potentially be used in accordance with this
embodiment of the
present invention (see e.g. PCT Publication WO 94/08598, dated April 28, 1994;
Stemple
and Anderson, 1992, Cell 71:973-985; Rheinwald, 1980, Meth. Cell Bio. 21A:229;
and
Pittelkow and Scott, 1986, Mayo Clinic Proc. 61:771).
-51-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
5.8. DEMONSTRATION OF THERAPEUTIC OR
PROPHYLACTIC UTILITY
The compositions of the invention are preferably tested ih vitro, and then ih
vivo for
the desired therapeutic or prophylactic activity, prior to use in humans. For
example, ih
vitro assays to demonstrate the therapeutic or prophylactic utility of a
composition include,
the effect of a composition on a cell line, particularly one characteristic of
a specific type of
cancer, or a patient tissue sample. The effect of the composition on the cell
line and/or
tissue sample can be determined utilizing techniques known to those of skill
in the art
including, but not limited to, rosette formation assays and cell lysis assays.
Test
compositions can be tested for their ability to augment activated immune cells
by contacting
activated immune cells with a test composition or a control composition and
determining
the ability of the test composition to modulate the biological activity of the
activated
immune cells. The ability of a test composition to modulate the biological
activity of
activated immune cells can be assessed by detecting the expression of
cytokines or antigens,
detecting the proliferation of immune cells, detecting the activation of
signaling molecules,
detecting the effector function of immune cells, or detecting the
differentiation of immune
cells. Techniques known to those of skill in the art can be used for measuring
these
activities. For example, cellular proliferation can be assayed by 3H-thymidine
incorporation
assays and trypan blue cell counts. Cytokine and antigen expression can be
assayed, for
example, by immunoassays including, but are not limited to, competitive and
non-competitive assay systems using techniques such as western blots,
immunohistochemistry radioimmunoassays, ELISA (enzyme linked immunosorbent
assay),
"sandwich" immunoassays, immunoprecipitation assays, precipitin reactions, gel
diffusion
precipitin reactions, immunodiffusion assays, agglutination assays, complement-
fixation
assays, immunoradiometric assays, fluorescent immunoassays, protein A
immunoassays
and FAGS analysis. The activation of signaling molecules can be assayed, for
example, by
lcinase assays and electromobility shift assays (EMSAs). The effector function
of T-cells
can be measured, for example, by a SlCr-release assay (see, e.g., Palladino et
al., 1987,
Cancer Res. 47:5074--5079 and Blachere et al., 1993, J. Immunotherapy 14:352-
356).
Test composition can be tested for their ability to reduce tumor formation in
patients
(i.e., animals) suffering from cancer. Test compositions can also be tested
for their ability
to reduce viral load or bacterial numbers ifa vitro and ira vivo (e.g., in
patients suffering from
an infectious disease) utilizing techniques known to one of skill in the art.
Test
compositions can also be tested for their ability to alleviate of one or more
symptoms
associated with cancer or an infectious disease (e.g., a viral or microbial
infection). Test
compositions can also be tested for their ability to decrease the time course
of the infectious
disease. Further, test compositions can be tested for their ability to
increase the survival
-52-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
period of patients suffering from cancer or an infectious disease (e.g., a
viral or microbial
infection). Techniques known to those of skill in the art can be used to
analyze or test the to
function of the test compositions in patients.
In various embodiments, with the invention, in vitro assays which can be
used to determine whether administration of a specific composition is
indicated, include iyz
vitro cell culture assays in which a patient tissue sample is grown in
culture, and exposed to
or otherwise administered a composition, and the effect of such composition
upon the tissue
sample is observed.
Compositions for use in therapy can be tested in suitable animal model
systems prior to testing in humans, including but not limited to rats, mice,
chicken, cows,
monkeys, rabbits, etc. For ih vivo testing, prior to administration to humans,
any animal
model system known in the art may be used.
5.9. THERAPEUTIC/PROPHYLACTIC
MINISTRATION AND COMPOSITION
The invention provides methods of preventing and treating cancer, viral
infection,
microbial infection, and septic shock by administrating to an animal (e.g.,
cows, pigs,
horses, chickens, cats, dogs, humans, etc.) an effective amount of a
composition of the
invention. In certain embodiments, compositions of the invention are
administered to
human burn patients, infants, immunocompromised or immunodeficient humans, or
the
elderly. Compositions of the invention include any one of the following or a
combination
of any of the following: one or more anti-C3b(i) antibodies, one or more
antibodies
imrnunospecific for one or more cancer antigens, one or more antibodies
immunospecific
for one or more viral antigens, one or more antibodies irmnunospecific for one
or more
microbial antigens, one or more complement components, nucleic acid sequences
encoding
one or more anti-C3b(i) antibodies, nucleic acids encoding one or more
antibodies
immunospecific for one or more cancer cell antigens; nucleic acids encoding
one or more
antibodies immunospecific for one or more viral antigens; nucleic acids
encoding one or
more complement components, nucleic acids encoding one or more microbial
antigens, one
or more antibodies immunospecific for lipopolysaccharide, an endotoxin, or a
constituent of
the outer wall of a gram negative bacteria, and/or nucleic acids encoding one
or more
antibodies immunospecific for lipopolysaccharide, an endotoxin, or a
constituent of the
outer wall of a gram negative bacteria. In a preferred aspect, a composition
of the invention
is substantially purified (e.g., substantially free from substances that limit
its effect or
produce undesired side-effects).
Various delivery systems are known and can be used to administer a composition
of
the invention, e.g., encapsulation in liposomes, microparticles,
microcapsules, recombinant
-53-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
cells capable of expressing the compound, receptor-mediated endocytosis (see,
e.g., Wu and
Wu, 1987, J. Biol. Chem. 262:4429-4432), construction of a nucleic acid as
part of a
retroviral or other vector, etc. Methods of introduction include, but are not
limited to,
intradermal, intramuscular, intraperitoneal, intravenous, subcutaneous,
intranasal, epidural,
and oral routes. The compositions may be administered by any convenient route,
for
example by infusion or bolus inj ection, by absorption through epithelial or
mucocutaneous
linings (e.g., oral mucosa, rectal and intestinal mucosa, etc.) and may be
administered
together with other biologically active agents. Achninistration can be
systemic or local. In
addition, it may be desirable to introduce the compositions of the invention
into the central
nervous system by any suitable route, including intraventricular and
intrathecal inj ection;
intraventricular injection may be facilitated by an intraventricular catheter,
for example,
attached to a reservoir, such as an Ommaya reservoir. Pulmonary administration
can also
be employed, e.g., by use of an inhaler or nebulizer, and formulation with an
aerosolizing
agent.
In a specific embodiment, it may be desirable to administer the compositions
of the
invention locally to the area in need of treatment; this may be achieved by,
for example, and
not by way of limitation, local infusion during surgery, topical application,
e.g., in
conjunction with a wound dressing after surgery, by injection, by means of a
catheter, by
means of a suppository, or by means of an implant, said implant being of a
porous,
non-porous, or gelatinous material, including membranes, such as sialastic
membranes, or
fibers.
In another embodiment, the composition can be delivered in a vesicle, in
particular a
liposome (see Langer, 1990, Science 249:1527-1533; Treat et al., in Liposomes
in the
Therapy of Infectious Disease and Cancer, Lopez-Berestein and Fidler (eds.),
Liss, New
York, pp. 353-365 (1989); Lopez-Berestein, ibid., pp. 317-327; see generally
ibid.)
In yet another embodiment, the composition can be delivered in a controlled
release
or sustained release system. In one embodiment, a pump may be used (see
Langer, supra;
Sefton, 1987, CRC Crit. Ref. Biomed. Eng. 14:201; Buchwald et al., 1980,
Surgery 88:507;
Saudek et al., 1989, N. Engl. J. Med. 321:574). In another embodiment,
polymeric
materials can be used in a controlled release system (see Medical Applications
of
Controlled Release, Langer and Wise (eds.), CRC Pres., Boca Raton, Florida
(1974);
Controlled Drug Bioavailability, Drug Product Design and Performance, Smolen
and Ball
(eds.), Wiley, New York (1984); Ranger and Peppas, J., 1983, Macromol. Sci.
Rev.
Macromol. Chem. 23:61; see also Levy et al., 1985, Science 228:190 ; During et
al., 1989,
Ate. Neurol. 25:351; Howard et al., 1989, J. Neurosurg. 71:105). In yet
another
embodiment, a controlled release system can be placed in proximity of the
therapeutic target
(e.g., the brain, kidney, stomach, pancreas, and lung), thus requiring only a
fraction of the
-54-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
systemic dose (see, e.g., Goodson, in Medical Applications of Controlled
Release, supra,
vol. 2, pp. 115-138 (1984)).
Other controlled release systems are discussed in the review by Langer (1990,
Science 249:1527-1533).
In a specific embodiment where the composition of the invention is a nucleic
acid
encoding a protein, the nucleic acid can be administered ih vivo to promote
expression of its
encoded protein, by constructing it as part of an appropriate nucleic acid
expression vector
and administering it so that it becomes intracellular, e.g., by use of a
retroviral vector (see
U.S. Patent No. 4,980,286), or by direct injection, or by use of microparticle
bombardment
(e.g., a gene gun; Biolistic, Dupont), or coating with lipids or cell-surface
receptors or
transfecting agents, or by administering it in linkage to a homeobox-like
peptide which is
known to enter the nucleus (see e.g., Joliot et al., 1991, Proc. Natl. Acad.
Sci. USA
88:1864-1868), etc. Alternatively, a nucleic acid can be introduced
intracellularly and
incorporated within host cell DNA for expression, by homologous recombination.
The present invention also provides pharmaceutical compositions. Such
compositions comprise a prophylactically or therapeutically effective amount
of one or
more anti-C3b(i) antibodies, one or more antibodies immunospecific for one or
more cancer
antigens, one or more antibodies immunospecific for one or more viral
antigens, one or
more antibodies immunospecific for one or more microbial antigens, one or more
complement components, or a combination thereof, and a pharmaceutically
acceptable
carrier. In a specific embodiment, the term "pharmaceutically acceptable"
means approved
by a regulatory agency of the Federal or a state government or listed in the
U.S.
Pharmacopeia or other generally recognized pharmacopeia for use in animals,
and more
particularly in humans. The term "carrier" refers to a diluent, adjuvant,
excipient, or vehicle
with which the therapeutic is administered. Such pharmaceutical earners can be
sterile
liquids, such as water and oils, including those of petroleum, animal,
vegetable or synthetic
origin, such as peanut oil, soybean oil, mineral oil, sesame oil and the like.
Water is a
preferred carrier when the pharmaceutical composition is administered
intravenously.
Saline solutions and aqueous dextrose and glycerol solutions can also be
employed as liquid
carriers, particularly for injectable solutions. Suitable pharmaceutical
excipients include
starch, glucose, lactose, sucrose, gelatin, malt, rice, flour, chalk, silica
gel, sodium stearate,
glycerol monostearate, talc, sodium chloride, dried skim milk, glycerol,
propylene, glycol,
water, ethanol and the like. The composition, if desired, can also contain
minor amounts of
wetting or emulsifying agents, or pH buffering agents. These compositions can
take the
form of solutions, suspensions, emulsion, tablets, pills, capsules, powders,
sustained-release
formulations and the like. The composition can be formulated as a suppository,
with
traditional binders and carriers such as triglycerides. Oral formulation can
include standard
-55-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
carriers such as pharmaceutical grades of mannitol, lactose, starch, magnesium
stearate,
sodium saccharine, cellulose, magnesium carbonate, etc. Examples of suitable
pharmaceutical Garners are described in "Remington's Pharmaceutical Sciences"
by E.W.
Martin. Such compositions will contain a therapeutically effective amount of
the
$ compound, preferably in purified form, together with a suitable amount of
carrier so as to
provide the form for proper administration to the patient. The formulation
should suit the
mode of administration.
In a preferred embodiment, the composition is formulated in accordance with
routine procedures as a pharmaceutical composition adapted for intravenous
administration
to human beings. Typically, pharmaceutical compositions for intravenous
administration
are solutions in sterile isotonic aqueous buffer. Where necessary, the
pharmaceutical
composition may also include a solubilizing agent and a local anesthetic such
as lignocaine
to ease pain at the site of the injection. Generally, the ingredients are
supplied either
separately or mixed together in unit dosage form, for example, as a dry
lyophilized powder
or water free concentrate in a hermetically sealed container such as an
ampoule or sachette
indicating the quantity of active agent. Where the composition is to be
administered by
infusion, it can be dispensed with an infusion bottle containing sterile
pharmaceutical grade
water or saline. Where the composition is administered by injection, an
ampoule of sterile
water for injection or saline can be provided so that the ingredients may be
mixed prior to
administration.
The pharmaceutical compositions of the invention can be formulated as neutral
or
salt forms. Pharmaceutically acceptable salts include those formed with anions
such as
those derived from hydrochloric, phosphoric, acetic, oxalic, tartaric acids,
etc., and those
formed with rations such as those derived from sodium, potassium, ammonium,
calcium,
ferric hydroxides, isopropylamine, triethylamine, 2-ethylamino ethanol,
histidine, procaine,
etc.
The amount of a composition of the invention which will be effective in the
treatment or prevention of cancer, viral infection, or microbial infection can
be determined
by standard clinical techniques. In addition, in vitro assays may optionally
be employed to
help identify optimal dosage ranges. The precise dose to be employed in the
formulation
will also depend on the route of administration, and the seriousness of the
disease or
disorder, and should be decided according to the judgment of the practitioner
and each
patient's circumstances. However, suitable dosage ranges for intravenous
administration are
generally about 20-500 micrograms of active compound per kilogram body weight.
Suitable dosage ranges for intranasal administration axe generally about 0.01
pg/kg body
weight to 1 mg/kg body weight. Effective doses may be extrapolated from dose-
response
curves derived from in vitro or animal model test systems.
-56-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
Suppositories generally contain active ingredient in the range of 0.5% to 10%
by
weight; oral formulations preferably contain 10% to 95% active ingredient.
For antibodies, the preferred dosage is 0.1 mg/kg to 100 mg/kg of body weight
(generally 10 mg/kg to 20 mg/kg). If the antibody is to act in the brain, a
dosage of 50
mg/kg to 100 mg/kg is usually appropriate. Generally, partially human
antibodies and fully
human antibodies have a longer half life within the human body than other
antibodies.
Accordingly, lower dosages and less frequent administration is often possible.
Modifications such as lipidation can be used to stabilize antibodies and to
enhance uptalce
and tissue penetration (e.g., into the brain). A method for lipidation of
antibodies is
described by Cruikshank et al., 1997, J. Acquired Immune Deficiency Syndromes
and
Human Retrovirology 14:193).
The invention also provides a pharmaceutical pack or kit comprising one or
more
containers filled with one or more of the ingredients of the pharmaceutical
compositions of
the invention. Optionally associated with such containers) can be a notice in
the form
prescribed by a governmental agency regulating the manufacture, use or sale of
pharmaceuticals or biological products, which notice reflects approval by the
agency of
manufacture, use or sale for human administration.
5.10. DIAGNOSIS AND IMAGING OF CANCER
Labeled antibodies, derivatives and analogs thereof, which specifically bind
to
C3b(i) can be used for diagnostic purposes to detect, diagnose, or monitor
cancer. In a
preferred embodiment, anti-C3b(i) antibodies which immunospecifically bind to
C3b(i)
covalently bound to IgG or IgM antibodies, or C3b(i) covalently bound to a
cancer cell are
used for diagnostic purposes to detect, diagnosis, or monitor cancer. In a
preferred
embodiment, cancer is detected in the patient. The patient is an animal,
preferably a
mammal and most preferably a human.
In one embodiment, diagnosis is carried out by: a) administering to an animal
an
effective amount of a labeled molecule which immunospecifically binds to
C3b(i) or C3b(i)
covalently linked to a second molecule; b) waiting for a time interval
following the
administering for permitting the labeled molecule to preferentially
concentrate at any
cancerous site in the animal (and for unbound labeled molecule to be cleared
to background
level); c) determining background level; and d) detecting the labeled molecule
in the
subject, such that detection of labeled molecule above the background level
indicates the
presence of cancer. In one embodiment, diagnosis is carried out by: a)
administering to an
animal an effective amount of a labeled molecule which immunospecifically
binds to C3b(i)
or C3b(i) covalently linked to a second molecule prior to, subsequent to, or
concomitantly
with the administration of IgG antibody, IgM antibody, plasma, one or more
complement
-57-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
components, and/or one or more antibodies which immunospecifically bind to a
cancer cell
antigen; b) waiting for a time interval following the administering for
permitting the labeled
molecule to preferentially concentrate at any cancerous site in the animal
(and for unbound
labeled molecule to be cleared to background level); c) determining background
level; and
d) detecting the labeled molecule in the subject, such that detection of
labeled molecule
above the background level indicates the presence of cancer. Background level
can be
determined by various methods including, comparing the amount of labeled
molecule
detected to a standard value previously determined for a particular system.
The invention provides methods for the diagnosis or detection of cancer in an
animal, said methods comprising imaging said animal at a time interval after
administering
to said animal an effective amount of a labeled antibody which
immunospecifically binds to
C3b(i) or C3b(i) covalently linked to a second molecule, said time interval
being sufficient
to permit the labeled antibody to preferentially concentrate at any cancerous
site in said
animal, wherein detection of the labeled antibody localized at the site in the
subject
indicates the presence of cancer. The invention also provides methods for the
diagnosis or
detection of cancer in an animal, said methods comprising imaging said animal
at a time
interval after administering to said animal an effective amount of a labeled
antibody which
immunospecifically binds to C3b(i) or C3b(i) covalently linked to a second
molecule prior
to, subsequent to, or concomitantly with the administration of IgG antibody,
IgM antibody,
plasma, one or more complement components, and/or one or more antibodies which
immunospecifically bind to a cancer cell antigen, said time interval being
sufficient to
permit the labeled antibody to preferentially concentrate at any cancerous
site in said
animal, wherein detection of the labeled antibody localized at the site in the
subj ect
indicates the presence of cancer.
Depending on several variables, including the type of label used and the mode
of
achninistration, the time interval following the administering for permitting
the labeled
molecule to preferentially concentrate at any cancerous site in the subject
and for unbound
labeled molecule to be cleared to background level is 6 to 48 hours or 6 to 24
hours or 6 to
12 hours. In another embodiment the time interval following administration is
S to 20 days
or 5 to 10 days.
In a specific embodiment, monitoring of the cancer is carried out by repeating
the
method for diagnosing the cancer, for example, one month after initial
diagnosis, six
months after initial diagnosis, one year after initial diagnosis, etc.
In another specific embodiment of the invention, the density of a tumor
facilitates
the detection of said tumor using anti-C3b(i) antibodies in accordance with
the methods of
the invention.
-58-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
U.S. patent application Serial No. 60/181,143, filed February 8, 2000, in
particular
page 2 of said application, is incorporated herein by reference in its
entirety.
5.10.1. METHODS OF DETECTION AND IMAGING
Presence of the labeled molecule can be detected in the patient using methods
known in the art for ih vivo scanning. These methods depend upon the type of
label used.
Skilled artisans will be able to determine the appropriate method for
detecting a particular
label. Methods and devices that may be used in the diagnostic methods of the
invention
include but are not limited to: computed tomography (CT), whole body scan such
as
position emission tomography (PET), magnetic resonance imaging (MRI), and
sonography.
In a specific embodiment, the molecule is labeled with a radioisotope and is
detected
in the patient using a radiation responsive surgical instrument (Thurston et
al., U.S. Patent
5,441,050). In another embodiment, the molecule is labeled with a fluorescent
compound
and is detected in the patient using a fluorescence responsive scanning
instrument. In
another embodiment, the molecule is labeled with a positron emitting metal and
is detected
in the patent using positron emission-tomography. In yet another embodiment,
the
molecule is labeled with a paramagnetic label and is detected in a patient
using magnetic
resonance imaging (MRI).
5.11. HITS
The present invention also provides kits that can be used in the above
methods. In
one embodiment, a kit comprises one or more antibodies immunospecific for
C3b(i) or
C3b(i) linked (e.g., covalently linked) to a second molecule, in one or more
containers. In
another embodiment, a kit comprises one or more antibodies irmnunospecific for
C3b(i) or
C3b(i) linked (e.g., covalently linlced) to a second molecule and IgM
antibody, in one or
more containers. In another embodiment, a kit comprises one or more antibodies
irmnunospecific for C3b(i) or C3b(i) linked (e.g., covalently linked) to a
second molecule
and IgG antibody, in one or more containers. In another embodiment, a kit
comprises one
or more antibodies immunospecific for C3b(i) or C3b(i)linked (e.g., covalently
linked) to a
second molecule and one or more complement components, in one or more
containers. In
another embodiment, a kit comprises one or more antibodies irnrnunospecific
for C3b(i) or
C3b(i) linked (e.g., covalently linlced) to a second molecule and one or more
antibodies
immunospecific for one or more cancer cell antigens, viral antigens, or
microbial antigens,
in one or more containers. In another embodiment, a kit comprises one or more
antibodies
~ospecific for C3b(i) or C3b(i) linked (e.g., covalently linked) to a second
molecule,
IgM or IgG antibody, and one or more complement components in one or more
containers.
In yet another embodiment, a kit comprises one or more antibodies
immunospecific for
-59-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
C3b(i) or C3b(i) linked (e.g., covalently linked) to a second molecule, one or
more
antibodies immunospecific for one or more cancer cell antigens, viral antigens
or microbial
antigens, and one or more complement components, in one or more containers.
Preferably, the kits of the present invention further comprise a control
antibody
which is not immunospecific for C3b(i) or C3b(i) linked (e.g., covalently
linked) to a
second molecule. In a specific embodiment, the kits of the present invention
contain a
labeled C3b(i) immunospecific antibody. In a preferred embodiment, the kits of
the
invention contain a C3b(i) immunospecific antibody conjugated to a therapeutic
agent. In
another preferred embodiment, the kits of the present invention contain a
C3b(i) specific
antibody conjugated to a diagnostic agent. In yet another preferred
embodiment, the kits of
the present invention contain a purified C3b(i) immunospecific antibody. In
yet another
embodiment, the kits of the present invention contain anti-C3b(i) antibodies
which
immunospecifically bind to C3b(i) covalently bound to IgG or IgM antibody, or
C3b(i)
covalently bound to a cancer cell, virus, or microbe.
In certain embodiments, the lcits of the invention contain instructions for
the use of
the antibodies for the treatment, prevention or diagnosis of cancer, viral
infections, or
microbial infections.
6. EXAMPLE: C3b(i) AS A TUMOR-SPECIFIC ANTIGEN
The following example demonstrates that after opsonization of prostate tumor
cells,
C3b(i) can function as a tumor-specific antigen. Antibodies specific for
C3b(i) can be
utilized to target tumor cells for the delivery of therapeutic or diagnostic
agents, including
cytotoxic, chemotherapeutic, immune-enhancing drugs, radioactive compounds,
genetic
material and immune effector cells.
LNCaP and lineage-derived C4-2 human prostate cancer cell lines were utilized
in
this example to demonstrate the use of C3b(i) as a target for immunotherapy.
The
LNCaP/C4-2 progression model recapitulates progression of human neoplastic
prostate
disease from an androgen-responsive and minimally metastatic (LNCaP cells) to
an
androgen-refractory (defined as being able to proliferate in castrate hosts)
and highly
aggressive phenotype (C4-2 subline) (Thalmann et al., 1994, Caxlc. Res.
54:2577-81;
Chung et al., 1996, Urol. Oncol. 2:99-128; Hyytinen et al., 1997, Br. J. Cane.
75:190-5). It
shares remarkable similarities with clinical human prostate cancer both in its
genotypic and
phenotypic changes. Furthermore, the LNCaP/C4-2 progression model has been
shown to
be a powerful tool for evaluating anti-prostate cancer therapeutic approaches
both ih vitYo
and in vivo (Chung et al., 1997, Acta Urol. Jap. 43:815-20), especially with
regard to
hormone-refractory disease, for which few effective or durable treatment
options currently
exist (Scher et al., 1994, Sem Oncol 21:630-56).
-60-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
6.1. MATERIALS AND METHODS
Cell lines and Serum Specimens
LNCaP (American Type Culture Collection, Rockville, MD) and C4-2 (Urocor,
Oklahoma City, OK) human prostate cancer cell lines were maintained in T-media
with 5%
heat inactivated fetal bovine serum (FBS; Gibco, Grand Island, NY'. Cultures
were
maintained at 37°C in humidified 5% COz, split and harvested at 80 to
90% confluence, and
treated, if applicable, at 25% confluence. Cells were collected using either
phosphate
buffered saline (PBS) with 2.5 mM ethylenediitrilotetraacetic acid (EDTA;
Sigma, St.
Louis, MO) or trypsin (Gibco, Grand Island, NY) diluted 1:10 in phosphate
buffered saline
(PBS). Samples were then washed twice in PBS by centrifugation at 450 g for 2
minutes
and resuspended at a final concentration of 0.2-1 X 10' cellshnl in PBS with
1% bovine
serum albumin (BSA-PBS).
Serum samples were obtained with written informed consent from normal male and
female volunteers (University of Virgiua, Charlottesville, VA) and from men
being
followed for prostate disease (University of Virginia and Eastern Virginia
Medical School,
Norfolk, VA). Prostate disease patients had pathologic documentation of either
benign or
neoplastic prostate disease. Blood was drawn into SST gel and clot activator
Vacutainer
tubes (Becton Dickinson, Franklin Lakes, NJ), held at room temperature for 30
min, and
then centrifuged for 20 min at 700 X g to obtain serum which was stored at -80
°C.
Serum Opsonization of Tumor Cells
Harvested LNCaP and C4-2 tumor cells (1 X 10' cells/ml in BSA-PBS) were mixed
with an equal volume of freshly thawed serum and gently shalcen for 20 mm at
37°C. The
opsonized cells were washed twice and brought to a final concentration of 1 X
10' cells/ml
in BSA-PBS. Alternative opsonization procedures included addition of 10 mM
EDTA to
sera to block all complement activation (or use of EDTA-containing plasma),
addition of 10
mM ethylene glycol tetraacetic acid (EGTA) and 5 mM Mg (Mg-EGTA) to allow only
alternative pathway activation, use of purified IgM (1 mg/ml, Sigma, St.
Louis, MO), or use
of IgM-depleted serum. In this case, IgM was removed from normal human sera
(NHS) by
incubating 2.5 ml of serum with 1.65 ml (settled volume) anti-human IgM
agarose (Sigma,
St. Louis, MO) on ice for 1 hr with gentle shaking. The depleted serum was
separated from
the agarose by centrifugation at 1600 X g and then stored at -80 ° C.
ELISA determinations
(not shown) demonstrated that >90% of the human IgM was specifically removed
from the
serum by this procedure, but the level of human IgG was reduced by less than
10%.
-61-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
Monoclonal Antibodies
IgGl mAb 7C12, 2H11, and 8E11, specific for C3b(i); IgGl mAb HB57, specific
for human lgM; and IgG2a mAb 7G9, specific for human complement receptor 1
(CRl),
have been previously reported (Ferguson et al., 1995, Arthritis Rheum 38:190;
Taylor et al.,
1989, J. Immunol. 143:3626; and Tosic et al., 1989, J. Tmmunol. Methods
120:241) and
were used in parallel with isotype-matched controls. Anti-C3b(i) mAb 3E7
(IgGl), which
bound to a different epitope and was not blocked by the other anti-C3b(i) mAb,
was
prepared by our previously described methods (Tosic et al., 1989, J. Immunol.
Methods
120:241). The specificity of mAb 3E7 for C3b(i) was confirmed by indirect flow
cytometry
as follows. Both mAb 3E7 and 7C12 bound to serum-opsonized pig erythrocytes,
as
revealed by fluorescein-isothiocyanate(FITC)-labeled anti-mouse IgG. In the
presence of 5
~g/ml purified, heat-treated (56°C) C3 (Quidel, San Diego, CA), binding
of the mAb to
opsouzed pig erythrocytes was reduced by 88% and 62% respectively (results not
shown).
Radiolabeling of mAb with'zsI was performed by the Iodogen procedure (Edberg
et al.,
1988, J. Immunol. 141:4258; and Fraker et al., 1978, Biochem. Biophys. Res.
Commun.
80:849). A bispecific mAb complex (a heteropolymer) was prepared by cross-
linking mAb
3E7 with mAb 7G9 (Segal et al., 1995, Curr Protocol Immunol 2:131; and Taylor
et al.,
1997, J. Immunol. 97:842).
Serum o sonization of tumor cells
Harvested LNCaP and C4-2 tumor cells (approx. 5 x 106 cells/ml in BSA-PBS)
were
mixed with an equal volume of freshly thawed serum and gently shaken for 20-30
mm at
37°C. The opsonized cells were washed twice and brought to a final
concentration of 5 x
106 cells/ ml in BSA/PBS. Alternative opsonization procedures included
addition of 10
mM EDTA to sera or the use of EDTA-containing plasma to block all complement
activation, addition of 10 mM [ethylenebis (oxonitrilo)~-N,N,N ;N'-tetraacetic
acid (EGTA)
and 5 mM Mg+z (MgEGTA) to allow only alternative pathway activation, the use
of purified
IgM (1 mg/ml, Sigma, St. Louis, MO), or the use of IgM-depleted serum. In the
latter case,
IgM was removed from NHS by incubating 2.5 ml serum with 1.65 ml (settled
volume)
anti-(human 1gM) linked to agarose (Sigma, St. Louis, Mo.) on ice for 1 h with
gentle
shaking. The depleted serum was separated from the agarose by centrifugation
at 1,600 g
and then stored at -80°C. Enzyme-linlced immunosorbent assay
determinations (not shown)
demonstrated that more than 90% of the human IgM was specifically removed from
the
serum by this procedure, but the level of human IgG was reduced by less than
10%.
-62-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
Flow c, ometry and radioimmunoassa~
Opsonized cancer cells were probed with FITC-labeled goat anti-(human IgM)
FcS~,
(Pierce, Rockford, Ill.), FITC-labeled goat anti-human IgG Fc (Accurate,
Westbury, N.Y.),
or a cocktail of the anti-C3b(i) mAb 7C12 and 8E11 (typically 200 ng each
mAb/106 cells)
followed by a secondary FITC-labeled goat anti-mouse IgG (Sigma, St. Louis,
MO). All
incubations were at 37°C for 20 min in BSA-PBS. Controls included non-
opsonized cells
and irrelevant isotype-matched mAb. In selected cases, cells were stained with
propidium
iodide (Sigma, St. Louis, MO) used at a final concentration of 2 ~,g/ml in
BSA/PBS for
5 min, in the dark, on ice, to ascertain IgM or C3b-opsonization of the viable
cell
populations only. Viability was usually above 75%. One- or two-color
fluorescence
analysis was performed with CellQuest software on a FACSCalibur flow cytometer
(Becton
Dickinson, San Jose, CA).
Studies of the binding of lasl_labeled anti-C3b(i) and anti-human IgM mAb to
cancer
cells followed previously published procedures (Edberg et al., 1988, J.
Immunol. 141:4258;
~d Taylor et al., 1989, J. hmnunol. 143:3626). Briefly, after opsonization, 5
x 106 cancer
cells were incubated for 20-30 min with'ZSI-labeled mAb 7C12, 8E11, 3E7, or
HB57 (final
concentrations between 0.1 ~,g/ml and 10 ~g/ml) or matched-isotype controls.
As a
separate control for background binding, naive cancer cells were incubated
with the'ZSI-
Iabeled mAb. The level of binding of the mAb to the cancer cells was then
determined by
centrifuging the sample through oil and measuring radioactive counts in the
cell pellets
(Ross et al., 1985, J. Immunol. 135:2005). Alternatively, in order to
determine if the anti-
C3b(i) mAb could bind to cancer cells during complement opsonization, 'ZSI-
labeled mAb
3E7 or 7C12 was added directly to the incubation mixture containing cancer
cells and NHS.
After incubation at 37°C, the cells were spun through oil to measure
mAb binding.
Rosette experiments
Samples of 10 ~,1 50% suspension of human erythrocytes (approximately 5 x 10'
cells) in either BSA/PBS or plasma were incubated with 2 x 104 LNCaP cells
(either non
opsonized, or serum- opsonized as described above) in the presence or absence
of 20 ng
anti-CRl x anti-C3b(i)(7G9 x 3E7) heteropolymer. After 30 min at 37°C,
the cell mixtures
were resuspended in BSA-PBS at a final concentration of 1% erythrocytes. Light
microscopy was used to evaluate the presence and extent of erythrocyte/tumor-
cell
rosetting.
-63-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
Irnmunohistochemistry
Paraffinized tissue sections with defined histopathology were deparaffinized
treated
with 3% hydrogen peroxide, blocked with Super Block (Scytek Laboratories,
Logan, Utah),
and then treated with Avidin/Biotin Block (Vector Laboratories Inc.,
Burlingame, Calif.).
Fixed sections were incubated with 4 ~glmI anti-C3b(i) IgGImAb 7C12 and 8E11
overnight
at 4°C, followed by biotinylated goat anti-mouse IgG and peroxidase-
conjugated
streptavidin (Biogenex Laboratories, San Ramon, CA). Subsequently, 3-amino-9-
ethylcarbazole/HZOZ was used as the peroxidase substrate. Mouse IgGl was used
as a
negative control for staiiung. The presence and extent of immunohistochemical
staining
was evaluated by light microscopy.
Ma etic ur ins of cancer cells: anal s~y radioimmunoassay (RIA)
Anti-C3b(i) mAb 2H1 I (which recognizes the same epitope bound by mAb 7C12
(Tosic et al., 1989, J. Immunol. Methods 120:241) or mAb 3E7 was covalently
coupled to
amine-terminated BioMag particles (Polysciences Inc., Warrington, PA)
following the
manufacturer's instructions. Cancer cells were labeled with SICr (Ferguson et
al., 1995,
Arthritis Rheum 38:190), opsonized with an equal volume of NHS, washed three
times and
then subjected to the magnetic purging protocols (see below). For experiments
with blood
cells in serum, blood was collected into EDTA, the cells were washed three
times and re-
constituted into serum (or into plasmalEDTA as a control), and then 5'Cr-
labeled cancer
cells were added. Alternatively, in certain experiments, SICr-labeled cancer
cells (approx.
50,000 cells) were first mixed with aliquots of whole blood anti-coagulated in
EDTA
(complement activation is prevented) or in citrate (partially permissive for
complement
activation). After opsonization the samples were washed three times. More than
80% of
the 5'Cr label was retained by the cancer cells in all opsonization
procedures.
After the opsonization steps, 25 ~,1 containing 5 x 104 cancer cells
corresponding to
approx. 50,000 cpm were incubated with 100 ~,1 BioMag particles containing
approx. 100
~,g anti-C3b(i) mAb, or a control irrelevant IgG antibody (goat anti-(mouse
IgG) or sheep
anti-FITC). After an incubation of 30 min on ice, the mixtures were placed in
the
Polysciences BioMag 15-ml separation unit, and 7 ml BSA/PBS was added to
increase the
volume of the separated (unbound) supernatant. This separation procedure was
repeated
two more times. Material bound to the magnetic particles was counted for SICr
and, after a
centrifugation step, pelleted material from the unbound supernatant
(representing intact but
unbound cells) was counted for SICr as well, in order to calculate the
percentage of intact
cells bound to the beads. Approximately 90%-95% of the initial input level of
cell-
associated 5lCr was found in either the pelleted supernatants or was directly
bound to the
particles.
-64-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
Effects of pur_~ing on normal cells: analyses by flow c. ometrX
In order to determine the effects of purging on normal cells, the same
procedures,
which included opsonization, washing, and reaction with the BioMag particles,
were
performed on aliquots of whole blood and on blood cells reconstituted in
serum. In these
experiments the aliquots were not mixed with 5'Cr-labeled cancer cells in
order to avoid
possible contamination of the flow cytometer with radioactivity. After the
whole blood
samples had been processed, they were assayed by flow cytometry with a number
of
standard markers (see below). Finally, we obtained blood samples that were
enriched for
CD34-positive cells from a leukophoresis of an oncology patient at the
University of
Virginia who was being treated with granulocyte-colony-stimulating factor in
preparation
for autologous stem cell transplantation. These samples were diluted into
blood-type-
matched whole blood from a normal individual (containing citrate or EDTA), or
were added
to washed blood cells reconstituted with serum, and subjected to the same
opsonization and
purging protocols.
After purging, unbound cellular fractions were reconstituted to the starting
volumes
and examined by flow cytometry following the manufacturers' directions for use
of the
developing reagents. The Becton Dickinson Multitest reagent (CN 340499, CD3
FITC/CD8 PE/CD45 PerCp/CD4 APC) was used to analyze for CD3/CD4-and CD3/CD8-
positive lymphocytes. Light-scattering profiles of CD45-positive cells were
used in the
same experiment to count granulocytes.. In separate experiments we measured
CD34-
positive cells by dual gating on CD45 (PerCp, BD 347474) and CD34 (APC, BD
340441,
isotype control BD 340442). CD14 positive cells were identified by dual gating
on CD45
and CD14 (FITC, Caltag, Burlingame, Calif., MHCDI401, isotype control MG2a01).
In
these measurements FluorCount Fluorospheres (Coulter, Miami, Fla., PN 2547053)
were
used to normalize sample volumes.
Radioimmunothera~~, otoxicity studies
The cytotoxic effects of'3'I-labeled anti-C3b(i) (7C12 and 8E11) mAbs on the
LNCaP and C4-2 prostate cancer cell lines were evaluated as follows. 1 X 106
cells of each
prostate cancer cell line were opsonized with 25% NHS (diluted in BSA-PBS) or
maintained in BSA-PBS at 37°C for 30 min. After washing twice with PBS,
either 2 ug or
200 ng of'31I-labeled 7C12+8E11 or'3'I-labeled irrelevant mAb (diluted in BSA-
PBS) was
added to each set of cells and incubated at room temperature for 30 min. The
cells were
washed twice with PBS, and plated in triplicate in 24-well tissue culture
plates (Fisher
Scientific, Pittsburgh, PA) in T-media + 5% FBS at 3 X104 cells per well. The
plates were
then placed in a humidified environment at 37°C with 5% CO2. A single
media change was
performed on day 3. On 5 (LNCaP) and 6 (C4-2) subsequent days, beginning 24 hr
after
-65-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
mAb treatment, the triplicate wells were harvested to evaluate cell killing by
comparing
differences in 3-(4,5-dimethylthiazol-2-yl)-2,5-Biphenyl-tetrazolium-bromide
(MTT)
(Sigma, St. Louis, MO) assay results.
6.2. RESULTS
C3b(i) and IBM are deposited on prostate cancer cells
Opsonization of C4-2 prostate cancer cells (and LNCaP, see below) with normal
human serum (NHS) results in deposition of substantial amounts of C3b(i) on
the cells. In
the representative flow-cytometry experiment displayed in FIG. 1, the effect
of C3b(i)
opsonization by NHS on C4-2 cells is shown in the top panel (FIG. 1A). Similar
results
were obtained with sera from 8 other normal individuals (data not shown).
C3b(i)
deposition appears to be facilitated by activation of both the classical and
alternative
complement pathways, but considerably less C3b(i) is demonstrable when Mg-
EGTA,
w~ch allows for alternative pathway activation only, is added to the serum.
Moreover,
opsonization with NHS provides a source of IgM specific for the cancer cells
(Fig. 1B).
IgM is more readily revealed on the cancer cells when the experiment is
conducted under
conditions (Mg-EGTA) that block the classical pathway of complement
activation, as C3b
deposition via the classical pathway seems to partially block epitopes on IgM
(see Table 1,
below). The flow cytometry results also demonstrate that, after opsonization
with serum
from a prostate cancer patient, significantly less C3b(i) and IgM are
deposited on the tumor
cells (FIG. 1C, D). It is noteworthy, however, that C3b(i) deposition via the
alternative
pathway (Mg-EGTA-treated serum) is comparable for both the normal and cancer
patient
serum, suggesting that the alternative pathway of complement activation
remains intact in
serum from prostate cancer patients.
IBM binding_promotes robust cancer cell opsonization with C3b(i)
On the basis of classic studies of the mechanisms of antibody-mediated
complement
activation (Borsos et al., 1965, Science 150:505; Circolo et al., 1985, Mol.
Immunol.
22:207; and Schreiber et al., 1972, J. Clin. Invest. 51:583), it was
hypothesized that the
observed complement activation on the cancer cells was predominantly
facilitated by the
binding of serum IgM to these cells. To isolate the effects of IgM, affinity
chromatography
was used to remove IgM from NHS under conditions that preserve the complement
activity
of the serum. Both RIA and flow cytometry demonstrate that, when IgM-depleted
serum is
used to opsonize LNCaP or C4-2 cells, substantially less C3b(i) is deposited
on the cancer
cells (FIG. 2). Normal levels of C3b(i) deposition can be restored, however,
when cancer
cells are first incubated with whole normal human plasma containing EDTA,
which blocks
-66-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
both classical and alternative complement pathways. Thus, the intact plasma
provides a
source of human IgM sufficient to allow for robust deposition of C3b(i) on the
cancer cells
after they have been washed and subsequently reacted with the IgM-depleted
serum, which
serves as a source of complement. RIA analysis further confirms that treatment
of the
cancer cells with purified IgM followed by treatment with IgM-depleted serum
as a
complement source also results in enhanced deposition of C3b(i) on the cancer
cells (FIG.
2B).
Next, the number of available epitopes on serum-opsonized cancer cells that
can be
targeted by anti-C3b(i) mAb was measured. Dose-response studies were performed
under
several conditions to estimate the number of C3b(i) sites that are generated
on the cells after
opsonization with NHS. The results displayed in FIG. 3 indicate that
complement
activation can produce in excess of 100,000 C3b(i) epitopes per cell after
correction for
background. It is well-known that large amounts of C3b and its breakdown
products are
also generated in solution during complement activation by immune complexes
even if they
are on cell surfaces or particulates (Circolo et al., 1984, Mol. Immunol.
21:191; Edberg et
al., 1988, J. Immunol. 141 4258; and Gadd et al., 1981, Biochem J. 195:471),
and it would
be reasonable to anticipate that these fragments could compete for binding to
the anti-C3b(i)
mAb, depending upon the exact specificity of these mAb. In fact, in most
experiments
binding of'zsI_labeled mAb 7C12 was reduced, within experimental error, to
background
levels when it was present during the opsonization step. For example,
radioactivity bound
(used to calculate molecules bound) was 6,700 + 100 cpm for 3 ~g/ml mAb 7C12
added to
the washed cells; the corresponding value for the "during" protocol was 90 +
100 cpm.
Background labeling (no opsonization) was 900 + 100 cpm. However,
'zsI_labelled mAb
3E7 is still able to bind to the cancer cells even when it is present during
the opsonization
step. Moreover, in certain cases, at high input concentrations (FIG. 3B;
10~g/ml), this mAb
appears to enhance the net deposition of C3b fragments on the cancer cells.
Inhibition
studies (see materials and methods) with purified heat-treated C3 confirmed
the specificity
of mAb 3E7 for C3b(i). These findings indicate that mAb 3E7 binds
preferentially to
C3b(i) located on the opsonized cancer cell, and is less susceptible than mAb
7C12 to
inhibition by C3b(i) fragments in the medium.
-67-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
Table 1. C3b(i) deposition on C4-2 cells by sera from two different normal
donors partially
blocks the detection of human IgM by both flow cytometry and radioimmunoassay
Treatment Log mean Number of
fluorescence bound'ZSI-mAb
intensity*
Anti-C3b(i)Anti-(human Anti-C3b(i) Anti-human
IgM) IgM)
mAb 8E11 mAb HB57
(mean SD) (mean SD)
No serum 7.5 4.7 1,200 + 270 850 + 30
Serum 266 14.8 27,700 + 70 3,100 + 60
Serum + EDTA 9.1 36.8 880 + 100 7,900 + 30
Serum + MgEGTA55.6 29.3 12,000 + 50 7,300 + 330
*
Same
data
presented
in
Fig.
1
The data strongly suggests that natural human IgM binds to surface antigens on
cancer cells and facilitates activation of the classical pathway, thus
allowing for deposition
of large amounts of human C3b(i) on the cells. However, following complement
activation
~d C3b(i) deposition, relatively diminished levels of cancer-cell-bound IgM
can be
demonstrated by flow cytometry and RIA (FIG. 1 and Table 1). This is probably
due to the
fact that once C3b(i) becomes covalently linlced to IgM, epitopes on the IgM
molecule are
obstructed by C3b(i), thereby preventing the binding of anti-IgM antibodies
used for flow
cytometry and RIA. Deposition of C3b fragments on human IgM in immune
complexes has
been demonstrated in several reports (Mehta et al., 1986, J. Immunol.
136:1765; Taylor et
al., 1989, J. Immunol. 143:3626; and Thornton et al., 1996, Clin. Exp.
Immunol. 104:531).
Therefore, some C3b(i) is complexed to the IgM on the cancer cell, and it is
likely that
C3b(i) is also covalently attached to glycoproteins and glycolipids on the
cancer cell.
The representative data in FIG. 1 indicates that the serum from a man with
prostate
cancer was less effective than NHS in depositing C3b(i) on cancer cells.
Several studies
have previously demonstrated that the amount of IgM that can bind to cancer
cells is
reduced in the sera of cancer patients (Desai et al., 1995, J. Immunol.
Methods 188:175;
Dillman, R.O., 1994, J. Clin. Oncol. 12:1497; Gross et al., 1988, Eur. J.
Cancer Clin. Oncol.
24:363; Higuchi et al., 1980, J. Clin. Lab. Immunol. 4:141; and Seegal et al.,
1976, Int.
tech. Allergy Appl. Immunol. 52:205). To independently confirm these findings,
sera from
a number of normal individuals and men with prostate cancer were surveyed to
evaluate
differences in the levels of anti-tumor IgM. The experiments were conducted
with sera
containing 0.01 M EDTA to remove the presumed confounding and blocking effect
of
C3b(i) in detecting cancer-cell-bound IgM. The results, displayed in FIG. 4,
indicate that in
two of three experiments the level of IgM bound by cancer cells was
significantly greater in
normal sera than in that from prostate cancer patients. The third experiment
approaches
-68-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
significance and might have reached it if not for the small number of samples
in the control
group. In one of the surveys, we assayed for cancer-cell bound IgG in addition
to IgM. As
shown in FIG. 4, little if any IgG in NHS is bound to the cancer cells.
However, sera from
some of the cancer patients show a notable titer, revealed by the large
standard deviation in
the patients' group. Although the numbers are too small to draw definite
conclusions, these
results do suggest the possibility of an active antitumor immune response in
some of the
cancer patients. Elevated tumor-specific IgG in patients with cancer has been
previously
reported (Vetvicka et al., 1997, J. Immunol. 159:599).
Chi) deposition is tumor cell-specific
To determine the cancer-tissue specificity of the C3b(i) epitope, frozen-
sectioned
prostate tissue specimens with anti-C3b(i) mAb were immunohistochemically
stained. The
surgical specimens from 2 men undergoing transurethral resection for benign
prostatic
hypertrophy were used as a control. Neither control demonstrated
immunohistochemical
evidence of anti-C3b(i) mAb binding (FIG. 5A). Conversely, of the thirty
specimens from
men with prostate cancer, eight (61%) stained positively for anti-C3b(i) mAb
(FIG. 5B).
Furthermore, in these eight specimens, only areas of malignancy were stained;
regions
containing a predominance of beiugn cells remained negative. In only 2
specimens was
staining of extremely high intensity, implying that, although complement is
deposited on
2p prostate cancer cells, inherent host complement deposition by itself
provides suboptimal
opsonization and systemic infusions with IgM (in the form of plasma) from
normal donors
may be of benefit.
Erythrocytes rosette with opsonized tumor cells in the
presence of specific anti-CRl x anti-C3b(i) heteropol~
One current application for mAb in cancer immunotherapy involves the
generation
of bispecific reagents in which a mAb specific for a cancer cell antigen is
cross-linked with
a mAb specific for an effector site (e.g., Fc receptors on
monocytes/macrophages,
granulocytes, or natural filler cells) (Renner et al., 1995, Immunol. Rev.
145:179; and Segal
DM, Bakacs T, Jost CR, Kuruez I, Sconocchia G, Titus JA (1995) T-cell targeted
cytotoxicity. In: Fanger MW (ed) Bispecific antibodies. Landis. Austin, Tex, p
27-42). In
this approach, immunocompetent cells can be delivered directly, and
specifically, to a tumor
via the guidance of the anti tumor mAb. A prototype for this approach was
examined by
testing whether human erythrocytes could bind to C3b(i)-opsonized cancer cells
through
bispecific mAb complexes (heteropolymers) specific for C3b(i) and the primate
erythrocyte
complement receptor (CRl). As demonstrated in FIG. 6, rosettes consisting of
erythrocytes
completely surrounding the opsonized tumor cells are formed in normal human
plasma after
-69-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
addition of the heteropolymer. In contrast, in the absence of anti-C3b(i)-
specific
heteropolymer, the majority of opsonized tumor cells bind no erythrocytes and
a small
percentage bind only two or three erythrocytes, because of a small amount of
CRl-mediated
immune adherence (not shown) (Okada et al., 1974, Nature 248:521). Finally, no
rosettes
were obtained if erythrocytes and heteropolymer were incubated with naive
cancer cells
(not shown).
Complement-opsonized cancer cells can be paged by immunomay etic methods
One of the more recent immunotherapeutic approaches to treatment of many forms
of cancer involves the purging of cancer cells from autologous bone marrow or
peripheral
stem cells before autologous transplantation (Tyer et al., 1996, Clin. Cancer
Res. 2:81).
Although this method is not currently used as a therapy for prostate cancer,
the use of
prostate cancer cells is a reasonable test of the potential feasibility of
using complement and
anti-C3b(i) mAb to remove cancer cells selectively from solution. This
approach is
particularly attractive because opsonized cancer cells can be washed before
addition of
coupled mAb, eliminating solution-phase competition by C3b(i) fragments. As
shown in
Table 2, opsonization of both LNCaP and C4-2 cells with NHS followed by
treatment with
anti-C3b(i) mAb of different specificities bound to BioMag particles leads to
very efficient
binding and removal of the cancer cells. It is noteworthy that mAb 3E7, which
binds to the
2p cancer cells during serum opsonization, did not block purging with mAb 2H11
(Table 2).
Table 2 Removal of C3b(i) opsonized cancer cells by magnetic purging with anti-
C3b(i)
mAb. Results are means + SD
Cell type Cells removed (%) Conditions
Naive Opsonized
LNCaP 13 + 4 99.0 + 0.2 AB+ serum, 2H 11 beads
C4-2 4 + 2 98.0 + 0.3 AB's serum, 2H11 beads
C4-2 15 + 1 98.0 + 0.2 O' serum, 2H11 beads
C4-2 11 + 1 96.0 + 0.1 O- serum, 2H11 beads
mAb 3E7 present
during opsonization
LNCaP 18 + 3 98 + 1 O~ serum, 3E7 beads
LNCaP 21 + 2 99.0 + 0.3 O+ serum, 3E7 beads
2% hematocrit~
LNCaP 20 + 5 96 + 1 O~ serum, 3E7 beads
50% hematocritb
a Whole blood was washed three times, and the cells reconstituted to 2%
hematocrit with 50°/~ serum
-70-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
b Whole blood was washed three times, and the cells reconstituted to 50%
hematocrit with serum
Robust and specific binding is demonstrable when the experiment is conducted
in a
reconstituted system that approximates whole blood and allows for complement
activation
(Table 3). When the whole-blood experiment is conducted in EDTA, which blocks
complement activation, binding of the C4-2 cells to the anti-C3b(i) beads is
reduced to
background levels. Experiments conducted with whole blood under the two
conditions that
allow complement activation (serum, or citrated plasma) demonstrated a high
level of
binding of the C4-2 cells to the mAb 3E7 magnetic beads (Table 3). Similar
experiments
with reconstituted blood samples spiked with CD34-positive cells to facilitate
detection by
flow cytometry revealed that CD34-positive cells are not removed by the anti-
C3b(i)-coated
beads (3E7) after complement activation (Table 3), although there was
apparently some
non-specific binding of the cells to both types of bead for all conditions.
Flow-cytometric
analyses (Table 4) of the effect of anti-C3b magnetic bead purging on normal
cells indicate
no specific removal of CD4+ or CD8~ T lymphocytes, or granulocytes. CD14+
monocytes
treated with serum showed some reduction compared to controls. This reduction
was not
observed in citrated plasma.
Table 3 Effects of purging on cancer and progenitor cells in whole blood. The
results for
targeting of C4-2 cancer cells are based on SICr-labeled cells. The retention
was determined
by flow-cytometric analysis of samples spiked with CD34+ cells. Results are
means + SD.
ConditionsTargeting Retention
of C4-2 of CD34'"
cancer cells
cells: after
removal purging
(%) cells/ul)
3E7 beads IgG beads 3E7 beads IgG beads No beads
2$ 50%sennna 98+1 3+0.3 6.5+1 6.5+2 17+5
50% Serums8 + 1 5 + 0.5
Citrateb 84 + 8 9 + 3 11 + 2 8 + 1 21 + 1
EDTAb 10+5 13+3 8+2 11+3 20+2
a Whole blood was washed three times, and the cells reconstituted to 50~
hematocrit with serum or plasma
b Whole blood anti-coagulated with citrate or EDTA
35
-71 -
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
Table 4 Flow-cytometric analyses of the effects of purging on populations of T
lymphocytes, granulocytes and monocytes. Whole blood anti-coagulated with
citrate or
EDTA was used or washed blood cells were reconstituted to 50% hematocrit in
serum. 3E7
anti-C3b(i)-coated magnetic beads, IgG beads coated with a control irrelevant
antibody.
ConditioCell
population
(cells/~1)
ns
CD3/CD4 CD3/CD8 Granulocytes CD14
3E7 Ig None 3E7 IgG None3E7 Ig None 3E7 IgG None
Bead: G G
50% 730 65 730 370 330 370 2,900 3,3 3,00 280 400 620
serum 750 0 900 380 340 470 3,400 00 0 500 530 540
Citrate,+ 66 800 + ND 560 + 500 3,5 3,50 ND ND ND
60 30
expt.l 600 0 910 370 370 480 2,700 00 0 370 410 580
Citrate,790 N 760 420 ND 490 3,000 ND 2,90 ND ND ND
expt.2 630 D 420 3,500 3,2 0
EDTA, 68 50 3,30
expt.l 0 ND 0
EDTA, N 3,40
expt.2 D 0
By flow-cytometry experiments several other cancer cell lines, including
astrocytoma (LT-87 MG; ATCC), breast cancer (MDA-MB-231;ATCC), colon cancer
(LoVo; ATCC), lung cancer (CCD-l6Lu; ATCC), and osteosarcoma (MG-63; ATCC)
were
efficiently opsonized with C3b(i) by the AB-positive serum used to obtain the
results shown
in FIG. 1A. More than 94% of the cells registered as positive after
opsonization, compared
to a background level (before opsonization) of less than 5% (not shown).
Radiolabeled anti-C3b(i) mAbs can lcill prostate cancer cells ih vitro
Another application of cancer-specific mAbs involves the coupling of
radioactive
agents to the mAbs to allow for the imaging or destruction of tumors (Glennie
MJ, French
~, T~geting drugs, toxins, and radionuclides with bispecific antibodies. In:
Fanger MW,
editor. Bispecific Antibodies. ed. Austin: RG Landis Co.; 1995, p. 107-20).
The potential
of this approach was examined by labeling anti-C3b(i) mAbs with'31I, and then
testing their
effectiveness in killing cancer cells in culture. After serum opsonization and
reaction with
the radiolabeled mAbs in solution phase (see Methods), the cells were plated.
In all cases
the experiments included both serum-opsonized and naive cells, as well as
radiolabeled
isotype-matched irrelevant mAbs. Although the level of cytotoxicity was
modest,
-72-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
progressive killing of serum opsonized LNCaP and C4-2 cells by the'3'I-labeled
anti-C3b(i)
mAbs over a period of 3 to 6 days was demonstrated (FIG. 7). Cell death was
not observed
in control cultures consisting of either nonopsonized tumor cells or'31I-
labeled irrelevant
mAb-treated cells. LNCaP and C4-2 prostate cancer cells opsonized and treated
with mAbs
after being plated in tissue culture wells demonstrated similar patterns of
killing (data not
shown).
6.3. DISCUSSION
It has long been recognized that C3b and its fragments can deposit on the
surface of
cancer cells in patients with tumors (Okada et al., 1974, Nature 248:521-25;
Irie et al.,
1974, Science 186:454-456.;Vetvicka et al., 1996, J. Clin. Invest. 98:50-61;
Vetvicka V et
al., 1997, J. Immunol. 159:599-605; and Vetvicka et al., 1999, Clin. Exp.
Irmnunol.
115:229-35). This reaction is facilitated by natural IgM. Investigations by
Springer and
others suggest that the natural IgM repertoire recognizes cancer cell-
associated carbohydrate
epitopes which are not found on normal tissue (Hakomori et al., 1996, Canc.
Res. 56:5309-
18; Castronovo et al., 1989, J. Nat. Canc. Inst. 81:212-6; Springer et al.,
1984, Science
224:1198-206; Springer et al., 1997, J. Mol. Med. 75;594-602; and Desai et
al., 1995, J.
Immunol. Methods 188:175-85). In fact, several investigators are using
carbohydrate
epitopes as vaccines to induce an active immune response to certain cancers (
Springer,
1984, Science 224:1198-206; Springer, 1997, J. Mol. Med. 75;594-602;
Livingston et al.,
1997, Canc. Immunol. Immunotherapy 43 :324-30; and Zhang et al., 1998, Canc.
Res.
58:2844-9). The findings presented herein demonstrate the utility of deposited
C3b(i) as a
tumor-associated membrane antigen with which to design a general diagnostic
and
therapeutic modality.
2$ Large amounts of C3b(i) have been shown to specifically deposit on cancer
cells
after opsonization with NHS (FIGS. 1, 2 and 3 and Table 1). As indicated in
Figures l and
4, the level of the presumably protective IgM is often reduced in cancer
patients, including
those with breast tumors (Desai et al., 1995, J. Immunol. Methods 188:175-85;
Seegal et al.,
1976, Int. Arch. Allergy App. Immunol. 52:205-11; Higuchi et al., 1980, J. Lab
Chin.
~~ol. 5:407-18; and Gross et al., 1988, Eur. J. Canc. Chin. Oncol. 24:363-7).
Therefore, the inftision of normal human plasma in some cancer patients will
help to restore
or enhance C3b(i) opsonization of tumor sites accessible to the bloodstream.
However,
even if normal human plasma deposits a large quantity of C3b(i) on the cancer
cell surface,
it is unlilcely that this action alone will be sufficient to eradicate a
tumor, since cancer cells
often express high levels of complement control proteins (Goner et al., 1996,
Lab. W vest.
74:1039-49; Maenpaa et al., 1996, Am J Path 148:1139-52; and Li et al.., 1997,
Int. J. Canc.
71:1049-55). For example, the expression of CD59 ("protectin") by cancer cells
blocks the
- 73 -
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
action of the membrane attack complex which might otherwise lyse the cancer
cell. The
results presented herein demonstrate that one approach to treating cancer is
to infuse a
patient with normal human plasma (to supply IgM and, if necessary, complement)
and to
then deliver systemically anti-neoplastic agents to the cancer cells by
conjugating the agents
to anti-C3b(i) mAbs, which would circulate through the body and home to sites
of
opsonized tumor cells.
To ensure that a sufficient quantity of therapeutic agent is delivered in
close
proximity to the tumor cell, mAb-based immunotherapy for cancer requires a
very high
level of selective and high avidity binding of the mAb to the tumor. The
results indicate
that at least 50,000 C3b(i) epitopes are available on opsonized prostate
cancer cells and,
based on the i~c vitro killing studies, this level of cancer-associated
antigen should be
sufficient for specific targeting of the cancer cell, enabling the delivery of
abundant
therapeutic or diagnostic agents.
Tumor tissue-specific delivery of therapeutic agents is crucial to avoid
undesirable
injury to healthy tissue. In the case of C3b(i) as a target, it is important
that complement
activation be limited to tumor cells. Except for a few relatively rare disease
conditions
(Rosse et al., 1995, Blood 86;3277-86; Morgan BP. Complement: clinical aspects
and
relevance to disease. ed. London: Harcourt Brace Jovanovich; 1990), the
complement
system is highly regulated and C3b(i) is not deposited on normal tissue.
Moreover, C3b(i)
deposition has been shown to be confined to areas of malignancy in human
prostate tissue
specimens, and is absent in benign (FIG. 5A) and hyperplastic regions (not
shown). These
data confirm earlier studies on breast cancer, which established a similar
tumor tissue-
specific pattern of opsonization (Vetvicka et al., 1997, J. Immunol. 159:599-
605; Howard et
al., 1979, Cancer 43:2279-87; and Niculescu et al., 1992, Am. J. Path.
140:1039-43).
Due to normal turnover, a small fraction of circulating C3 expresses antigenic
epitopes similar to C3b(i), and this endogenous C3b(i)-like molecule might
block the action
of the anti-C3b(i) mAbs (Mollnes et al., 1987, J. Immunol. Methods 101;201-7;
Petronis et
al., 1998, Clin. Nuc. Med. 23:672-7). Generation of C3b fragments in the
solution phase
during complement activation would also tend to block binding of the mAb to
cancer cells,
~d indeed mAb 7C12 binds to the C3b-opsonized cancer cells only after they are
washed
(not shown). C3b(i) covalently linked directly to IgM or to a carbohydrate on
the cancer
cell should, however, contain unique and specific antigenic determinants
different from
those expressed by C3b fragments in solution, and it should be possible to
generate mAb
specific for such cell-associated molecules. Indeed mAb 3E7, which was able to
bind to the
cancer cells during the opsonization step (Fig. 3), did not block purging with
the 2H11
beads (Table 2). It is likely that other mAb can be developed that will
demonstrate even
greater binding specificity for C3b(i) covalently bound to the cancer cells.
-74-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
The feasibility of an ex vivo application in which anti-C3b(i) mAb can be used
to
target and purge complement-opsonized cancer cells, which are first washed to
remove
potentially competing soluble C3b(i) fragments has been demonstrated (Tables 2
and 3).
The procedure appears to spare normal cells including CD34-positive cells
(Tables 3 and 4),
which are most representative of progenitor cells (Bensinger, W.L, 1998, Bone
Marrow
Transplant 21:113; LaCasse et al., 1999, Blood 94:2901; and Vescio et al.,
1999, Blood
93:1858). The therapeutic efficacy of purging for autologous transplantation
remains
controversial (Bensinger, W.L, 1998, Bone Marrow Transplant 21:113). It is
reasonable to
believe that contaminating cancer cells may indeed promote tumor resurgence
after
autologous transplantation (Schneiderman et al., 1998, Radiat Res 149:147; and
Vescio et
al., 1999, Blood 93:1858) and, as noted by Bensiger (1998, Bone Marrow
Transplant
21:113), carefully controlled trials will be required to determine the
efficacy of purging
protocols. Several other new methods for purging cancer cells from bone marrow
or stem
cell preparations have recently been reported (LaCasse et al., 1999, Blood
94:2901; Lazzaro
et al., 1995, Exp. Hematol. 23:1347; and Schneiderman et al., 1998, Radiat Res
149:147),
and C3b(i) targeting may provide a powerful orthogonal approach. Use of these
procedures
in tandem with C3b(i)-based targeting may allow for highly efficacious purging
of cancer
cells from bone marrow or peripheral blood stem cells before autologous
transplantation.
The potential use of anti-C3b(i) mAb in bispecific mAb complexes bound to
either
e~~'ocytes or immune effector cells (Gast et al., 1997, Cancer Immunol.
Immunother.
45:121; Renner et al., 1995, Immunol. Rev. 145:179; Segal DM, Bakacs T, Jost
CR, Kuruez
I, Sconocchia G, Titus JA (1995) T-cell targeted cytotoxicity. In: Fanger MW
(ed)
Bispecific antibodies. Landis. Austin, Tex, p 27-42; and Taylor et al., 1997,
Cancer
Immunol. Immunother. 45:152) is illustrated by our rosetting data (FIG. 6). In
the presence
of the anti-C3b(i) x anti-CRl heteropolymer, erythrocytes completely encircled
prostate
tumor cells opsonized with human serum. Circulating "micrometastatic" prostate
tumor
cells have been demonstrated in men with prostate cancer of varying grades and
stages
(Sokoloff et al., 1996, Cancer 77:1862). In one possible therapeutic approach,
anti-C3b(i)
heteropolymer constructs could be infused into men with prostate cancer in
conjunction
with, or soon after an infusion of normal serum, in order to target the tumor
cells.
Once the tumor cells are opsonized, anti-C3b(i) mAbs coupled with toxic agents
or
radioisotopes can be administered to individuals. The potential use of this
approach is
illustrated in FIG. 7. When LNCaP and C4-2 cells were treated with'3'I_labeled
specific
anti-C3b(i) mAbs, only those cells that had been opsonized with NHS prior to
treatment
with the 1311-anti-C3b(i) mAbs were killed (FIG. 7). This approach can also be
utilized for
diagnostic imaging purposes, similar to the PROSTASCINTTM scan, when tumor
cell
-75-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
deposits are effectively opsonized and then targeted with anti-C3b(i) mA.b-
conjugated
compounds (Petronis et al., 1998, Clin. Nuc. Med. 23:672-7).
The results herein demonstrate that while opsonization with normal human serum
results in the deposition of large amounts of IgM and C3b(i) on prostate
cancer cells,
opsonization with sera from most men with prostate cancer leads to
substantially
diminished levels of cell-associated IgM and C3b(i). This deficiency can be
restored by the
infusion of normal plasma as a source of human IgM which will ultimately allow
for the
opsonization of cancer cells with C3b(i). These opsonized cells will therefore
present
unique and specific antigenic determinants for targeting by appropriate C3b(i)
mAbs.
7. EXAMPLE: EFFECT OF ANTIGEN SPECIFIC ANTIBODIES
ON THE ABILITY OF ANTI-C3b(i) ANTIBODIES
TO BIND TO RED BLOOD CELLS
The following example demonstrates that the presence of an antibody that
i~unospecifically binds to a cancer antigen increases the ability of opsonized
tumor cells
to bind to erythrocytes in the presence of bispecific monoclonal antibody
(heteropolymer)
for C3b(i) and the complement receptor CRl.
Materials & Methods
In certain experiments, SICr-labeled Raji cells (model for B cell lymphoma)
were
dispersed in either serum or in serum containing 0.01 M EDTA (which blocks
complement
activation). They were incubated for 30 minutes at 37°C in the presence
or absence of 5
~,g/ml Rituximab (Genentech, CA), to allow C3b(i) deposition. The cells were
then washed
three times and redispersed in normal human plasma containing human red cells
at a 50
hematocrit. In two cases the incubation mixture also contained a bispecific
monoclonal
antibody for C3b(i) and the complement component CRl (HP7G9X3E7; "HP") at a
concentration of 3 ~,ghnl. After another 30 minute incubation, 100 ~,1
aliquots of the
samples were layered on 1 ml of ficoll-hypaque and centrifuged at 500 X g for
15 minutes.
The SICr counts associated with the Raji cellls were then measured in the E
pellet, and in the
supernatant, which is the region in which free Raji cells are found.
In other experiments, SICr-labeled Raji cells were added to washed whole blood
which was reconstituted in fresh serum. The samples were incubated for 30
minutes at
37°C in the presence of varying combinations of reagents: 5 ~,g/ml
Rituximab; 3 ~.g/ml HP;
Rituximab plus HP incubated together; Rituximab added 15 minutes before HP;
and HP
added 15 minutes before Rituximab. After another 30 minute incubation,
duplicate 200 ~.l
aliquots of the samples were layered on 1 ml of ficoll-hypaque and centrifuged
at 500 X g
-76-
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
for 15 minutes. The 5'Cr counts associated with the Raji cellls were then
measured in the E
pellet, and in the supernatant, which is the region in which free Raji cells
are found.
Results
As shown in FIGS. 9 and 10, a larger percentage Rajii cells bound to
erythrocytes
when the Rajii cells were opsonized by serum or whole blood reconstituted in
serum and
Rituximab in the presence of the heteropolymer than when the Rajii cells were
opsonized by
serum or whole blood reconstituted in serum and Rituximab in the absence of
the
heteropolymer. Also, a larger percentage Rajii cells bound to erythrocytes
when the Rajii
cells were opsonized by serum or whole blood reconstituted in serum and
Rituximab in the
presence of the heteropolymer than when the Rajii cells were opsonized with
serum alone or
whole blood reconstituted in serum alone in the presence of the heteropolymer
(FIGS. 9 and
10). Further, as shown in FIG. 10, comparable numbers of Rajii cells bound to
erythrocytes
when the Rajii cells were opsonized with whole blood reconstituted in serum
and Rituximab
in presence of the heteropolymer regardless of whether the Rituximab and
heteropolymer
were added to the Rajii cells concomitantly or either of them was added first.
Similar
results were obtained with whole blood that has not been washed. These results
suggest
that the addition of an antibody irninunospecific for a cancer cell antigen
may enhance the
therapeutic effect that anti-C3b(i) antibodies in cancer patients.
8. EXAMPLE: SERUM-MEDIATED BINDING OF
BACTERIA TO ERYTHROCYTES
Two gram negative bacteria, EsclZerichia coli and Pseudornonas aeruginosa,
both of
which can cause sepsis, were used to assess the ability of bacteria in the
presence of serum
to bind to erythrocytes. As shown in Table 2, in the presence of normal serum,
these
bacteria can activate complement, covalently capture C3b, and then bind to
primate
erythrocytes via their complement receptor 1 (CRl) which is immunospecific for
C3b, the
first complement activation product. Binding in sample buffers (1% BSA in
phosphate
buffered saline) or in serum or plasma containing EDTA (to bloclc complement
activation)
averaged <15% for E. coli, and <12% for P. aeYUginosa. It is important to note
that only a
fraction of the bacteria bind to the erythrocytes, and with time binding
actually decreases
(see Table 5). One possible explanation for the decrease is that C3b bound to
the bacteria is
converted to further breakdown products, C3bi and C3dg, and the primate
erythrocyte
complement receptor does not bind these ligands with any measurable affinity.
_77_
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
Table 5. Serum-Mediated Binding of Bacteria to Erythrocytes at 37°C is
Time-Dependent
Percentage
Of Bacteria
Bound to
Erythrocytes
Time E. coli P. ae~uginosa
(mins)
5 50 34
20 75 85
60 48 39
120 22 15
9. EXAMPLE: THE EFFECT OF BISPECIFIC MONOCLONAL
ANTIBODIES ON THE ABILITY OF
P. AERUGlNOSA TO BIND TO ERYTHROCYTES
The following example demonstrates that bispecific antibodies for CRl and
C3b(i)
enhance the ability of Pseudofyaouas ae~ugihosa to bind to erythrocytes in the
presence of
serum.
Green fluorescent (GP) Pseudomonas aeYUginosa (P. ae~uginosa) was incubated
for
varying periods of time at 37°C with 7G9x2H4 (a bispecific monoclonal
antibody for
complement receptor CRl and a Pseudomoyzas aeruginosa antigen), 7G9x7C12 (a
bispecific monoclonal antibody for complement receptor CRl and C3b(i)),
7G9x3E7 (a
bispecific monoclonal antibody for complement receptor CRl and C3b(i)), or no
heteropolymer (HP) and normal human serum containing human erythrocytes at a
30-50
hematocrit. Following an incubation period, aliquots of the samples were
diluted 100 fold
~d the binding of GP P. aerugiraosa to erythrocytes was detected by flow
cytometry. The
percentage of bound erythrocytes was then calculated based upon the number of
bound and
free GP P. ae~ugifaosa detected using flow cytometry. In experiment 1, a
concentration of 8
~g/ml of 7G9x2H4, 7G9x7C12, or 7G9x3E7 was incubated with GP P. aerugihosa and
erythrocytes. In this experiment, erythrocytes outnumbered the GP P.
aerugifZOSa 50:1. In
experiment 2, a concentration of 1.3 ~g/ml of 7G9x2H4 or 7G9x3E7 was incubated
with
GP P. aerugihosa and erythrocytes. In this experiment, erythrocytes
outnumbered the GP
P. aerugifaosa 10:1. In experiment 3, a concentration of 1.3 ~g/ml of 7G9x2H4
or
7G9x3E7 was incubated with GP P. aeYUgifaosa and erythrocytes. In this
experiment,
erythrocytes outnumbered the GP P. aerugiraosa 5,000:1.
As shown in Table 6 if~fra, a larger percentage of GP P. aerugihosa bound to
erythrocytes when incubated together for 15 minutes or more in the presence of
the
_78_
CA 02400488 2002-08-07
WO 01/58483 PCT/USO1/04020
bispecific monoclonal antibodies than when incubated together in the absence
of
heteropolymer. Also, the percentage of GP P. aerugihosa bound to erythrocytes
in the
presence of 7G9x2H4 was comparable to the percentage of GP P. aeYUgifaosa
bound to
erythrocytes in the presence of 7G9x3E7. These results suggest that an
antibody
immunospecific for C3b(i) is capable of binding to microbes such as P.
aeYUginosa in
serum containing complement components as well as or almost as well as an
antibody
immunospecific for a particular microbial antigen. Thus, these results suggest
the
therapeutic usefulness of anti-C3b(i) antibodies in treatment of microbial
infections.
Table 6. Heteropolymers Specific for P. aeu~uginosa or Cb(i)
Enhance Binding to Erythrocytes in the Presence of Serum
Percentage
of P. aerugiraosa
Bound to
Erythrocytes
Time (mins) No HP 7G9x2H4 7G9x7C12 7G9x3E7
* 10 87 83 84 93
*30 81 98 92 97
*90 47 97 82 98
** 15 65 82 ND 82
**90 32 70 ND 81
*** 10 98 98 ND 98
***60 91 93 ND 98
(*) Experiment 1; (**) Experiment 2; (***) Experiment 3; (ND) not determined
The present invention is not to be limited in scope by the exemplified
embodiments,
which are intended as illustrations of single aspects of the invention.
Indeed, various
modifications of the invention in addition to those shown and described herein
will become
apparent to those skilled in the art from the foregoing description and
accompanying
~a~'~'ings. Such modifications are intended to fall within the scope of the
appended claims.
All publications cited herein are incorporated by reference in their entirety.
_79_