Note: Descriptions are shown in the official language in which they were submitted.
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
PEPTIDE EXTENDED GLYCOSYLATED POLYPEPTIDES
FIELD OF THE INVENTION
The present invention relates to novel glycosylated polypeptides as well as
means and methods
for their preparation.
BACKGROUND OF THE INVENTION
to
Polypeptides, including proteins, are used for a wide range of applications,
including industrial
uses and human or veterinary therapy.
One generally recognized drawback associated with polypeptides is that they do
not
have a sufficiently high stability, are immunogenic or allergenic, have a
reduced serum half-
life, are susceptible to clearance, are susceptible to proteolytic
degradation, and the like.
One method for improving properties of polypeptides has been to attach non-
peptide moieties
to the polypeptide to improve properties thereof. For instance, polymer
molecules such as PEG
has been used for reducing immunogenicity and/or increasing serum half-life of
therapeutic
polypeptides and for reducing allergenicity of industrial enzymes.
Glycosylation has been
2o suggested as another convenient route for improving properties of
polypeptides such as
stability, half-life, etc.
Machamer and Rose, J. Biol. Chem., 1988, 263, 5948-5954 and 5955-5960,
disclose
modified glycoprotein G of vesicular stomatitis virus that is glycosylated at
additional N-
glycosylation sites introduced in the polypeptide backbone.
US 5,218,092 discloses physiologically active polypeptides with at least one
new or
additional carbohydrate attached thereto. The additional carbohydrate
molecules) is/are
provided by adding one or more additional N-glycosylation sites to the
polypeptide backbone,
and expressing the polypeptide in a glycosylating host cell.
US 5,041,376 discloses a method of identifying or shielding epitopes of a
transportable
protein, in which method an N-glycosylation site is introduced on the exposed
surface of the
protein baclcbone (using oligonucleotide-directed mutagenesis of the
nucleotide sequence
encoding the protein), the resulting protein is expressed, glycosylated and
assayed for protein
activity and for shielded epitopes.
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
2
WO 00/26354 discloses a method of reducing the allergenicity of proteins by
including
an additional glycosylation site in the protein backbone and glycosylating the
resulting protein
variant.
Guan et al., Cell, 1985, Vol. 42, 489-496 disclose glycosylated fusion protein
variants
comprising a rat growth hormone backbone C-terminally extended with
transmembrane and
cytoplasmic domains of the vesicular stomatitis virus glycoprotein, which
growth hormone
backbone has been modified to incorporate two additional N-glycosylation
sites.
WO 97/04079 discloses lipolytic enzymes modified to by an N- or C-terminal
peptide
extension capable of conferring improved performance, in particular wash
performance to the
enzyme.
Matsuura et al., Nature Biotechnology, 1999, Vol. 17, 58-61 disclose the use
of random
elongation mutagenesis for improving thermostability of a non-glycosylated
microbial catalase.
The random elongation mutagenesis is conducted in the C-terminal end of the
catalase.
US 5,338,835, entitled CTP extended forms of FSH, describe the use of the C-
terminal
portion of the CG beta subunit or a variant thereof for extension of the C-
terminal of CG, FSH
and LH. Said C-terminal portion may comprise O-glycosylation sites. It is
speculated that a
similar approach may be used for other proteins.
US 5,508,261 discloses alpha, beta-heterodimeric polypeptide having binding
affinity
to vertebrate luteinizing hormone (LH) receptors and vertebrate follicle
stimulating hormone
(FSH) receptors comprising a glycoprotein hormone alpha-subunit polypeptide
and a specified
non-naturally occurring beta-subunit polypeptide.
WO 95/05465 discloses EPO analogs which have one or more amino acids extending
from the C-terminal end of EPO, the C-terminal extention having at least one
additional
carbohydrate site. The 28 amino acid C-terminal part of CG (having four O-
glycosylation sites)
is mentioned as an example.
WO 97/30161 discloses hybrid proteins comprising two coexpressed amino acid
sequences forming a dimes, each comprising a) at least one amino acid sequence
selected from
a homomeric receptor, a chain of a heteromeric receptor, a ligand, and
fragments theref; and b)
a subunit of a heterodimeric proteinaceous hormone or fragments thereof; in
which a) and b)
3o are bonded directly or though a peptide linker, and, in each couple, the
two subunits (b) are
different and capable of aggregating to form a dimes complex.
In none of the above reference it has been disclosed or indicated that a
polypeptide of
interest can be modified to include additional glycosylation sites by N-
terminally extending
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
3
said polypeptide with a~ peptide sequence comprising one or more additional
glycosylation
sites. The present invention is based on this finding.
BRIEF DESCRIPTION OF THE INVENTION
Accordingly, in a first aspect the invention relates to a glycosylated
polypeptide comprising the
primary structure,
NH2-X-Pp-COOH
wherein
X is a peptide addition comprising or contributing to a glycosylation site,
and
Pp is a polypeptide of interest.
The introduction of additional glycosylation sites by means of a peptide
addition is an
elegant way of providing additional glycosylation sites in a polypeptide of
interest. More
specifically, the invention has the advantage that polypeptides with altered
glycosylation
pattern are more easily obtained, e.g. the variants can be designed without
detailed knowledge
or use of structural and/or functional properties of the polypeptide. Also,
the utilization of
glycosylation sites introduced by a peptide addition has been found to be
improved relative to
glycosylation sites introduced within a structural part of the polypeptide Pp.
Also other
properties of the peptide extended polypeptide, such as uptake in specific
cells, may be
improved relative to a polypeptide modified with glycosylation sites in a
structural part (and
not being subjected to peptide extension).
In a second aspect the invention relates to a glycosylated polypeptide
comprising the
primary structure NH2-PX X-Py-COOH, wherein
PX is an N-terminal part of a polypeptide Pp of interest,
Py is a C-terminal part of said polypeptide Pp, and
X is a peptide addition comprising or contributing to a glycosylation site.
In other aspects the invention relates to a nucleotide sequence encoding a
polypeptide
of the invention, an expression vector comprising said nucleotide sequence and
methods of
preparing a polypeptide of the invention.
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
4
In a further aspect the invention relates to a method of improving (a)
selected
property/ies of a polypeptide Pp of interest, which method comprises a)
preparing a nucleotide
sequence encoding a polypeptide comprising the primary structure
NHZ-X-Pp-COOH,
wherein
X is a peptide addition comprising or contributing to a glycosylation site,
the peptide addition
being capable of conferring the selected improved property/ies to the
polypeptide Pp,
b) expressing the nucleotide sequence of a) in a suitable host cell under
conditions ensuring
attachment of an oligosaccharide moiety thereto, optionally
c) conjugating the expressed polypeptide of b) to a second non-peptide moiety,
and
d) recovering the polypeptide resulting from step c).
DRAWINGS
Figure 1 is a dosis response curve for uptake of glucocerebrosidase wildtype
and modified
according to the invention into J774E macrophages. The activity is measured by
the GCB
activity assay.
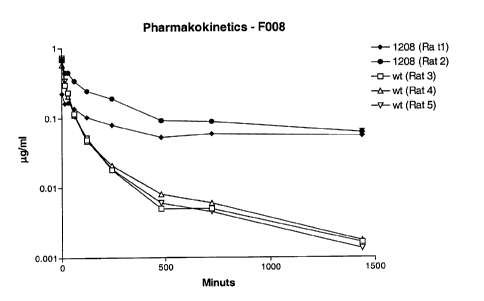
Figure 2 illustrates the pharmakokinetics of a FSH polypeptide produced
according to the
invention.
DETAILED DISCLOSURE OF THE INVENTION
DEFINTTIONS
In the context of the present application and invention the following
definitions apply:
The term "conjugate" is used about the covalent attachment of of one or more
polypeptide(s) to one or more non-peptide moieties. The term covalent
attachment means that
the polypeptide and the non-peptide moiety are either directly covalently
joined to one another,
or else are indirectly covalently joined to one another through an intervening
moiety or
moieties, such as a bridge, spacer, or linkage moiety or moieties.
The term "non-peptide moiety" is intended to indicate a molecule, different
from a
peptide polymer composed of amino acid monomers and linked together by peptide
bonds,
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
S
which molecule is capable of conjugating to an attachment group of the
polypeptide of the
invention. Preferred examples of such molecule include polymers, e.g.
polyalkylene oxide
moieties lipophilic groups, e.g. fatty acids and ceramides. The term "polymer
molecule" is
defined as a molecule formed by covalent linkage of two or more monomers and
may be used
interchangeably with "polymeric group". Except where the number of non-peptide
moieties,
such as polymeric groups, attached to the polypeptide is expressly indicated,
every reference to
"non-peptide moiety " referred to herein is intended as a reference to one or
more non-peptide
moieties attached to the polypeptide.
The term "oligosaccharide moiety" is intended to indicate a carbohydrate-
containing molecule
to comprising one or more monosaccharide residues, capable of being attached
to the polypeptide
(to produce a glycosylated polypeptide) by way of ih vivo or ifZ vitro
glycosylation. Except
where the number of oligosaccharide moieties attached to the polypeptide is
expressly
indicated, every reference to "oligosaccharide moiety" referred to herein is
intended as a
reference to one or more such moieties attached to the polypeptide.
The term "ifz vivo glycosylation" is intended to mean any attachment of an
oligosaccharide moiety occurring ire vivo, i.e. during posttranslational
processing in a
glycosylating cell used for expression of the polypeptide, e.g. by way of N-
linked and O-linlced
glycosylation. Usually, the N-glycosylated oligosaccharide moiety has a common
basic core
structure composed of five monosaccharide residues, namely two N-
acetylglucosamine
residues and three mannose residues. The exact oligosaccharide structure
depends, to a large
extent, on the glycosylating organism in question and on the specific
polypeptide. Depending
on the host cell in question the glycosylation is classified as a high mannose
type, a complex
type or a hybrid type. The term "if2 vitro glycosylation" is intended to refer
to a synthetic
glycosylation performed ifa vitro, normally involving covalently linking an
oligosaccharide
moiety to an attachment group of a polypeptide, optionally using a cross-
linlung agent. In vivo
and ih vitro glycosylation are discussed in detail further below.
An "N-glycosylation site" has the sequence N-X'-S/T/C-X", wherein X' is any
amino
acid residue except proline, X" any amino acid residue that may or may not be
identical to X'
and preferably is different from proline, N asparagine and S/T/C either
serine, threonine or
3o cysteine, preferably serine or threonine, and most preferably threonine.
The oligosaccharide
moiety is attached to the N-residue of such site. An "O-glycosylation site" is
the OH-group of a
serine or threonine residue. An "i~z vitro glycosylation site" is, e.g.,
selected from the group
consisting of the N-terminal amino acid residue of the polypeptide, the C-
terminal residue of
the polypeptide, lysine, cysteine, arginine, glutamine, aspartic acid,
glutamic acid, serine,
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
6
tyrosine, histidine, phenylalanine and tryptophan. Of particular interest is
an in vitro
glycosylation site that is an epsilon-amino group, in particular as part of a
lysine residue.
The term "peptide addition" is intended to indicate one or more consecutive
amino acid
residues that are added to the amino acid sequence of the polypeptide Pp of
interest. Normally,
the peptide addition is linked to the amino acid sequence of the polypeptide
Pp by a peptide
linkage.
The term "attachment group" is intended to indicate a functional group of the
polypeptide, in particular of an amino acid residue thereof or an
oligosaccharide moiety
attached to the polypeptide, capable of attaching a non-peptide moiety of
interest. Useful
to attachment groups and their matching non-peptide moieties are apparent from
the table below.
AttachmentAmino acidExamples of non-Conjugation Reference
group peptide moiety method/Activate
d PEG
-NH2 N-terminal,Polymer, e.g. mPEG-SPA Shearwater
PEG, Inc.
Lys with amide or Tresylated Delgado et
imine al,
group mPEG critical
reviews
in Therapeutic
Drug Carner
Lipophilic Systems
substituent 9(3,4):249-304
(1992)
WO 97/31022
-COOH C-term, Polymer, e.g. mPEG-Hz Shearwater
Asp, PEG, Inc
Glu with ester or
amide
group
-SH Cys Polymer, e.g. PEG- Shearwater
PEG, Inc
with disulfide, vinylsulphoneDelgado et
al,
maleimide or PEG-maleimidecritical
vinyl reviews
sulfone group in Therapeutic,
Drug Carrier
Systems
9(3,4):249-304
(1992)
-OH Ser, Thr, PEG with ester,
OH-, Lys ether, carbamate,
carbonate
-CONH~
Polymer, e.g.
PEG
Aldehyde Oxidized Polymer, e.g. PEG-hydrazideAndresz et
PEG, al.,
Ketone oligosacchari 1978,
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
7
de Makromol.
Chem.
179:301,
WO
92/16555,
WO
00/23114
The term "comprising an attachment group" is intended to mean that the
attachment
group is present on an amino acid residue of the relevant peptide or
polypeptide or on an
oligosaccharide moiety attached to said peptide or polypeptide.
The term "contributing to a glycosylation site" as used in connection with the
peptide
addition X is intended to cover the situation, where a glycosylation site is
formed from more
than one amino acid residue (as is the case with an N-glycosylation site), and
where at least one
such amino acid residue originates from the peptide X and at least one amino
acid residue
to originates from the polypeptide Pp, whereby the glycosylation site can be
considered to bridge
X and Pp (or, where relevant, PX or Py).
The term "non-structural part" as used about a part of the polypeptide Pp of
interest is
intended to indicate a part of either the C- or N-terminal end of the folded
polypeptide (e.g.
protein) that is outside the first structural element, such as an a-helix or a
(3-sheet structure.
The non-structural part can easily be identified in a three-dimensional
structure or model of the
polypeptide. If no structure or model is available, a non-structural part
typically comprises or
consists of the first or last 1-20 (1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13,
14, 15, 16,.17, 18, 19 or
20) amino acid residues, such as 1-10 amino acid residues of the amino acid
sequence
constituting the mature form of the polypeptide of interest.
Amino acid names and atom names (e.g. CA, CB, NZ, N, O, C, etc) are used as
defined
by the Protein DataBank (PDB) (www.pdb.org) which are based on the ILTPAC
nomenclature
(IUPAC Nomenclature and Symbolism for Amino Acids and Peptides (residue names,
atom
names e.t.c.), Eur. J. Bioche»z.,138, 9-37 (1984) together with their
corrections in Eur. J.
Biochezzz., 152, 1 (1985). The term "amino acid residue" is intended to
indicate an amino acid
residue contained in the group consisting of alanine (Ala or A), cysteine (Cys
or C), aspartic
acid (Asp or D), glutamic acid (Glu or E), phenylalanine (Phe or F), glycine
(Gly or G),
histidine (His or H), isoleucine (Ile or I), lysine (Lys or K), leucine (Leu
or L), methionine
(Met or M), asparagine (Asn or N), proline (Pro or P), glutamine (Gln or Q),
arginine (Arg or
R), serine (Ser or S), threonine (Thr or T), valine (Val or V), tryptophan
(Trp or W), and
3o tyrosine (Tyr or Y) residues. The terminology used for identifying amino
acid
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
8
positions/mutations is illustrated as follows: A15 (indicates an alanine
residue in position 15 of
the polypeptide), A15T (indicates replacement of the alanine residue in
position 15 with a
threonine residue), A15[T/S] (indicates replacement of the alanine residue in
position 15 with a
threonine residue or a serine residue). Multiple substitutions are indicated
with a "+", e.g.
A15T+F57S means an amino acid sequence which comprises a substitution of the
alanine
residue in position 15 for a threonine residue and a substitution of the
phenylalanine residue in
position 57 for a serine residue.
The term "nucleotide sequence" is intended to indicate a consecutive stretch
of two or
more nucleotides. The nucleotide sequence can be of genomic, cDNA, RNA,
semisynthetic,
1o synthetic origin, or any combinations thereof.
"Cell", "host cell", "cell line" and "cell culture" are used interchangeably
herein and all
such terms should be understood to include progeny resulting from growth or
culturing of a
cell. "Transformation" and "transfection" are used interchangeably to refer to
the process of
introducing DNA into a cell.
"Operably linked" refers to the covalent joining of two or more nucleotide
sequences in
such a manner that the normal function of the sequences can be performed. For
example, the
nucleotide sequence encoding a presequence or secretory leader is operably
linked to a
nucleotide sequence for a polypeptide if it is expressed as a preprotein that
participates in the
secretion of the polypeptide: a promoter or enhancer is operably linked to a
coding sequence if
2o it affects the transcription of the sequence.
"Introduction" or "removal" of a glycosylation site or an attachment group for
a non-
peptide moiety is normally achieved by introducing or removing an amino acid
residue
comprising or contributing to such site or group to/from the relevant amino
acid sequence,
conveniently by suitable modification of the encoding nucleotide sequence. For
instance, when
an N-glycosylation site is to be introduced/removed this can be done by
introducing/removing
a codon for the amino acid residues) required for a functional N-glycosylation
site. When an
attachment group for a PEG molecule is to be introduced/removed, it will be
understood that
this be done by introducing/removing a codon for an amino acid residue, e.g. a
lysine residue,
comprising such group tolfrom the encoding nucleotide sequence. The term
"introduce" is
3o primarily intended to include substitution of an existing amino acid
residue, but can also mean
insertion of additional amino acid residue. The term "remove" is primarily
intended to include
substitution of the amino acid residue to be removed for another amino acid
residue, but can
also mean deletion (without substitution) of the amino acid residue to be
removed.
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
9
The term "epitope" is used in its conventional meaning to indicate one or more
amino
acid residues) displaying specific 3D and/or charge characteristics at the
surface of the
polypeptide, which is/are capable of giving rise to an immune response in a
mammal and/or
specifically binding to an antibody raised against said epitope or which
is/are capable of giving
rise to an allergic response.
The term "unshielded epitope" is intended to indicate that the epitope is not
shielded
and therefore has the above properties. The term "shielded epitope" is
intended to indicate that
the non-peptide moiety shields, and thus inactivates the epitope, whereby it
is no longer
capable of giving rise to any substantial immune response in a mammal, e.g.
due to
1o inappropriate processing and/or presentation in the antigen presenting
cells, andlor of reacting
with an antibody raised against the unshielded epitope. The shielding should
thus be effective
in both the naive mammal and mammals that already produce antibodies reacting
with the
unshielded epitope.
The degree of shielding of epitopes can be determined as reduced
immunogenicity
and/or reduced antibody reactivity and/or reduced reactivity with monoclonal
antibodies raised
against the epitope(s) in question using methods known in the art. The degree
of shielding of
allergenic epitopes can be determined, e.g., as described in WO 00/26354.
The term "reduced" as used about an immunogenic or allergic response is
intended to
indicate that a given molecule gives rise to a measurably lower immune or
allergic response
2o than a reference molecule, when determined under comparable conditions.
Preferably, the
relevant response is reduced by at least 25%, such as at least 50%, such as
preferably by at
least 75%, such as by at least 90% or even at least 100%.
The term "serum half. life" is used in its normal meaning, i.e. the time in
which half of
the relevant molecules circulate in the plasma or bloodstream prior to being
cleared.
Alternatively used terms include "plasma half-life", "circulating half-life",
"serum clearance",
"plasma clearance" and "clearance half life". The term "functional ih vivo
half-life" is the time
in which 50% of a given function (such as biological activity) of the relevant
molecule is
retained, when tested in vivo (such as the time at which 50% of the biological
activity of the
molecule is still present in the body/target organ, or the time at which the
activity of the
3o polypeptide is 50% of the initial value). The molecule is normally cleared
by the action of one
or more of the reticuloendothelial systems (RES), kidney (e.g. by glomerular
filtration), spleen
or liver, or receptor-mediated elimination, or degraded by specific or
unspecific proteolysis.
Normally, clearance depends on size or hydrodynamic volume (relative to the
cut-off for
glomerular filtration), shape/rigidity, charge, attached carbohydrate chains,
and the presence of
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
cellular receptors for the molecule. The term "increased" as used about serum
half-life or
functional ifi vivo half life is used to indicate that the relevant half life
of the relevant molecule
is statistically significantly increased relative to that of the reference
molecule as determined
under comparable conditions. For instance, the relevant half life is increased
by at least 25%,
5 such as by at least 50%, by at least 100% or by at least 1000%.
The term "function" is intended to indicate one or more specific functions of
the
polypeptide of interest and is to be understood qualitatively (i.e. having a
similar function as
the polypeptide of interest) and not necessarily quantitatively (i.e. the
magnitude of the
function is not necessarily similar). Typically, a given polypeptide has many
different
to functions, examples of which are given further below in the section
entitled "Screening for or
measurement of function". For therapeutically useful polypeptides an important
"function" is
biological activity, e.g. in vitro or in vivo bioactivity. For enzymes, an
important function is
biological activity such as catalytic activity.
The interchangeably used terms "measurable function" and "functional" are
intended to
indicate that the relevant function (preferably reflecting the intended use)
of a polypeptide of
the invention is above detection limit when measured by standard methods known
in the art,
e.g. as an in vitro bioactivity and/or in vivo bioactivity. For instance, if
the polypeptide is a
hormone and the function of interest is the hormone's affinity towards a
specific receptor a
measurable function is defined to be a detectable affinity between the hormone
modified in
2o accordance with the invention and the receptor as determined by the normal
methods used for
measuring such affinity. If the polypeptide is an enzyme and a function of
interest is the
catalytic activity a measurable function is the enzyme's ability to catalyze a
reaction involving
the normal substrates for the enzyme as measured by the normal methods for
determining the
enzyme activity in question. Typically, if not otherwise stated herein, a
measurable function is
at least 2%, such as at least 5% of that of the unmodified polypeptide Pp, as
determined under
comparable conditions, e.g. in the range of 2-1000%, such as 2-500% or 2-100%,
such as 5-
100% of that of the unmodified polypeptide.
The term "functional site" is intended to indicate one ox more amino acid
residues
which is/are essential for or otherwise involved in the function or
performance of the
polypeptide, i.e. the amino acid residues) that mediates) a desired biological
activity of the
polypeptide Pp. Such amino acid residues are "located at" the functional site.
For instance, the
functional site can be a binding site (e.g. a receptor-binding site of a
hormone or growth factor
or a ligand-binding site of a receptor), a catalytic site (e.g. of an enzyme),
an antigen-binding
site (e.g. of an antibody), a regulatory site (e.g. of a polypeptide subject
to regulation), or an
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
11
interaction site (e.g. for a regulatory protein or an inhibitor). The
functional site can be
determined by methods known in the art and is conveniently identified by
analysing a three-
dimensional or model structure of the polypeptide complexed to a relevant
ligand.
The term "polypeptide" is intended to indicate any structural form (e.g. the
primary,
secondary or tertiary form (i.e. protein form)) of an amino acid sequence
comprising more than
5 amino acid residues, which may or may not be post-translationally modified
(e.g. acetylated,
carboxylated, phosphorylated, lipidated, or acylated). The interchangeably
used terms "native"
and "wild-type" are used about a polypeptide which has an amino acid sequence
that is
identical to one found in nature. The native polypeptide is typically isolated
from a naturally
1o occurring source, in particular a mammalian or microbial source, such as a
human source, or is
produced recombinantly by use of a nucleotide sequence encoding the naturally
occurring
amino acid sequence. The term "native" is intended to encompass allelic
variants of the
polypeptide in question. A "variant" is a polypeptide, which has an amino acid
sequence that
differs from that of a native polypeptide in one or more amino acid residues.
The variant is
typically prepared by modification of a nucleotide sequence encoding the
native polypeptide
(e.g. to result in substitution, deletion or truncation of one or more amino
acid residues of the
polypeptide or by introduction (by addition or insertion) of one or more amino
acid xesidues
into the polypeptide) so as to modify the amino acid sequence constituting
said native
polypeptide. A "fragment" is a part of a parent native or variant polypeptide,
typically differing
2o from such parent in one or more removed C-terminal or N-terminal amino acid
residues or
removal of both types of such residues. Normally, the variant or fragment has
retained at least
one of the functions of the corresponding parent polypeptide (e.g. a
biological function such as
enzyme activity or receptor binding capability). Normally, the polypeptide Pp
is a full length
protein or a variant or fragment thereof.
The term "antibody" includes single monoclonal antibodies (including agonist
and
antagonist antibodies) and antibody compositions with polyepitopic specificity
(also termed
polyclonal antibodies).
The term "monoclonal antibody" is used in its conventional meaning to indicate
a
population of substantially homogeneous antibodies. The individual antibodies
comprised in
3o the population have identical binding affinities and vary structurally only
to a limited extent.
Monoclonal antibodies are highly specific, being directed against a single
epitope.
Furthermore, in contrast to conventional (polyclonal) antibody preparations
that typically
include different antibodies directed against different epitopes, each
monoclonal antibody is
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
12
directed against a single epitope on the antigen. The antibody to be modified
is preferably a
human or humanized monoclonal antibody.
"Antibody fragment" is defined as a portion of an intact antibody comprising
the
antigen binding site or the entire or part of the variable region of the
intact antibody, wherein
the portion is free of the constant heavy chain domains (i.e. CH2, CH3, and
CH4, depending on
antibody isotype) of the Fc regions of the intact antibody. Examples of
antibody fragments
include Fab, Fab', Fab'-SH, F(ab')2, and Fv fragments; diabodies; any antibody
fragment that
is a polypeptide having a primary structure consisting of one uninterrupted
sequence of
contiguous amino acid residues (which may also be termed a single chain
antibody fragment or
1o a single chain polypeptide). '
Polypeptide of the invention
In its first aspect the invention relates to a glycosylated polypeptide
comprising the primary
structure,
NH2-X-Pp-COOH,
wherein
X is a peptide addition comprising or contributing to a glycosylation site,
and Pp is a
polypeptide of interest.
In one embodiment the polypeptide consists essentially of or consists of a
polypeptide
with the primary structure NH2-X-Pp-COOH.
The peptide addition according to this aspect is preferably one, which has
less than 90%
identity to a native full length protein. The identity is determined on the
basis of an alignment
of the peptide addition to the entire amino acid sequence of the full length
native protein, the
alignment being made to ensure the highest possible degree of identity between
amino acid
residues. For instance, the program CLUSTALW version 1.74 using default
parameters
(Thompson et al., 1994, CLUSTAL W: improving the sensitivity of progressive
multiple
sequence alignment through sequence weighting, position-specific gap penalties
and weight
matrix choice, Nucleic Acids Research, 22:4673-4680) can be used.
Usually, the peptide addition is fused to the N-terminal end of the
polypeptide Pp as
reflected in the above shown structure so as to provide an N-terminal
elongation of the
polypeptide Pp. However, it is also possible to insert the peptide addition
within the amino acid
sequence of the polypeptide Pp. This is reflected in the polypeptide according
to the second
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
13
aspect of the invention, wherein the polypeptide comprises the primary
structure NHZ-PX X-Py-
COOH, wherein
PX is an N-terminal part of a polypeptide Pp of interest,
Py is a C-terminal part of said polypeptide Pp, and
X is a peptide addition comprising or contributing to a glycosylation site.
In one embodiment the polypeptide consists essentially of or consists of a
polypeptide
with the primary structure NH2-PX X-Py-COOH.
In order to minimize structural changes effected by the insertion of the
peptide addition
within the sequence of the polypeptide Pp, it is desirable that it be inserted
in a non-structural
to part thereof. For instance, PX is a non-structural N-terminal part of a
mature polypeptide Pp,
and Py is a structural C-terminal part of said mature polypeptide, or PX is a
structural N-terminal
part of a mature polypeptide Pp, and Py is a non-structural C-terminal part of
said mature
polypeptide. Preferably, when the glycosylation site to be introduced is an N-
glycosylation site,
Px is a non-structural N-terminal part since, in general, the best N-
glycosylation is obtained in
the N-terminal part of a polypeptide.
When the peptide addition comprises only few amino acid residues, e.g. 1-5
such as 1-3
amino acid residues, and in particular 1 amino acid residue, the peptide
addition can be inserted
into a loop structure of the polypeptide Pp and thereby elongate said loop.
When the peptide
addition is constituted by one amino acid residue it will be understood that
this is selected so as
2o to ensure that a functional glycosylation site is introduced.
Polypeptides of the invention are glycosylated polypeptides. Normally, the
peptide
addition part of the polypeptide of the invention has attached at least one
oligosaccharide
moiety. The polypeptide Pp part of the polypeptide may or may not have
attached at least one
oligosaccharide moiety. Glycosylation can be achieved as described in the
section entitled
"Glycosylation"
Preferably, the polypeptide of the invention has properties such as size,
charge,
molecular weight and/or hydrodynamic volume that are sufficient to reduce or
escape clearance
by any of the clearance mechanisms disclosed herein, in particular renal
clerance. Such
properties are, e.g., determinable by the nature and number of oligosaccharide
and second non-
peptide moieties attached thereto. Tn one embodiment, the polypeptide of the
invention has a
molecular weight of at least 67 kDa, in particular at least 70 lcDa as
measured by SDS-PAGE
according to Laemmli, U.K., Nature Vol 227 (1970), p680-85. This is of
particular relevance
when the polypeptide of interest is a therapeutically useful protein, the
functional in vivo half-
life of which is to be prolonged. A molecular weight of at least 67 kDa is
obtainable by
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
14
introduction of a sufficient number of glycosylation sites to obtain a
glycosylated polypeptide
with such Mw, or by conjugating the glycosylated polypeptide to a sufficient
number and type
of a second non-peptide moiety to obtain such Mw. For instance, for a
glycosylated
polypeptide of interest having a molecular weight of at least 25 kDa linked to
a peptide
addition of 2 kDa, the combined extended polypeptide having at least two PEG-
attachment
groups, conjugation to two or more PEG molecules each having a molecular
weight of 20 kDa
results in a total molecular weight of at least 67 kDa.
Preferably, the polypeptide of the invention has at least one of the following
properties
relative to the polypeptide Pp, the properties being measured under comparable
conditions:
ire vitro bioactivity which is at least 25%, such as at least 30% or at least
45% of that of the
polypeptide Pp as measured under comparable conditions, increased affinity for
a mannose
receptor, a mannose-6-phosphate receptor or other carbohydrate receptors,
increased serum
half life, increased functional in vivo half-life, reduced renal clearance,
reduced
immunogenicity, increased resistance to proteolytic cleavage, improved
targeting to lysosomes,
macrophages and/or other subpopulations of human cells, improved stability in
production,
improved shelf life, improved formulation, e.g. liquid formulation, improved
purification,
improved solubility, and/or improved expression.
Improved properties are determined by conventional methods known in the art
for determining
such properties. The improvement is of a magnitude that is within detection
limits.
Improved affinity for or uptake by the mannose receptor is expected to result
in increased
uptake in phagocytic cells, preferably monocytes, macrophages (e.g. Kupffer
cells,
glia/mikroglia, alveolar phagocytes, reticulum cells, or other peripheral
macrophages) or
macrophage like cells (for instance osteoclasts, dendritic cells, or
astrocytes) in increased
uptake of the polypeptide in phagocytic cells (e.g. macrophages). This is of
particular relevance
when the polypeptide of interest is one for which such uptake is required for
the polypeptide to
exert its biological activity. Such polypeptide is e.g. an antigen intended
for use for vaccine
purposes or a lysosomal enzyme.
Polypeptide of interest
The present invention can be applied broadly. Thus, the polypeptide of
interest can have any
function and be of any origin. Accordingly, the polypeptide can be a protein,
in particular a
mature protein or a precursor form thereof or a functional fragment thereof
that essentially has
retained a biological activity of the mature protein. Furthermore, the
polypeptide can be an
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
I5
oligopeptide that contains in the range of 30 to 4500 amino acids, preferably
in the range of 40
to 3000 amino acids.
The polypeptide can be a native polypeptide or a variant thereof. For
instance, the
polypeptide is a variant that comprises at least one introduced and/or at
least one removed
glycosylation site as compared to the corresponding native polypeptide. The
variant has
retained at least one function of the corresponding native polypeptide, in
particular a biological
activity thereof.
The polypeptide can be a therapeutic polypeptide useful in human or veterinary
therapy, i.e. a polypeptide that is physiologically active when introduced
into the circulatory
1o system of or otherwise administered to a human or an animal; a diagnostic
polypeptide useful
in diagnosis; or an industrial polypeptide useful for industrial purposes,
such as in the
manufacture of goods wherein the polypeptide constitutes a functional
ingredient or wherein
the polypeptide is used for processing or other modification of raw
ingredients during the
manufacturing process.
The polypeptide can be of mammalian origin, e.g. of human, porcine, ovine,
urcine,
murine, rabbit, donkey, or bat origin, of microbial origin, e.g. of fungal,
yeast or bacterial
origin, or can be derived from other sources such as venom, leech, frog or
mosquito origin.
Preferably, the industrial polypeptide of interest is of microbial origin and
the therapeutic
polypeptide of human origin.
2o Specific examples of groups of polypeptides to be modified according to the
invention
include: an antibody or antibody fragment, an irnmunoglobulin or
immunoglobulin fragment, a
plasma protein, an erythrocyte or thrombocyte protein, a cytol~ine, a growth
factor, a
profibrinolytic protein, a binding protein, a protease inhibitor, an antigen,
an enzyme, a ligand,
a receptor, or a hormone. Of particular interest is a polypeptide that
mediates its biological
effect by binding to a cellular receptor, when administered to a patient. The
antibody can be a
polyclonal or monoclonal antibody, and can be of any origin including human,
rabbit and
murine origin. Preferably, the antibody is a human or humanized monoclonal
antibody.
Immunoglobulins of interest include IgG, TgE, IgM, IgA, and IgD and fragments
thereof, e.g.
Fab fragments. Specific antibodies and fragments thereof are those reactive
with any of the
3o proteins mentioned immediately below.
The non-antibody polypeptide of interest can be i) a plasma protein, e.g. a
factor from
the coagulation system, such as Factor VII, Factor VIII, Factor IX, Factor X,
Factor XIII,
thrombin, protein C, antithrombin III or heparin co-factor II, Tissue factor
inhibitor (e.g. 1 or
2), endothelial cell surface protein C receptor, a factor from the
fibrinolytic system such as pro-
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
16
urokinase, urokinase, tissue plasminogen activator, plasminogen activator
inhibitor 1 (PAI-1)
or plasminogen activator inhibitor 2 (PAI-2), the Von Willebrand factor, or an
a-1-proteinase
inhibitor, ii) a erythrocyte or thrombocyte protein, e.g. hemoglobin,
thrombospondin or platelet
factor 4, iii) a cytokine, e.g. an interleukin such as IL-1 (e.g. IL-la or 1L-
1(3), IL-2, IL-4, IL-5,
IL-6, IL-9, IL-10, IL-11, lL-12, TL,-13, IL-15, 1L.-16, IL-17, IL-18,1L-19, IL-
20, IL-21, IL-22,
IL-23, a cytokine-related polypeptide, such as IL-lRa, an interferon such as
interferon-a,
interferon-(3 or interferon-'y, a colony-stimulating factor such as GM-CSF or
G-CSF, stem cell
factor (SCF), a binding protein, a member of the tumor necrosis factor family
(e.g TNF-oc,
lymphotoxin-a, lymphotoxin-Vii, FasL, CD40L, CD30L, CD27L, Ox40L, 4-1BBL,
RANKL,
1o TRAIL, TWEAK, LIGHT, TRANCE, APRIL, THANK or TALL-1), iv) a growth factor,
e.g
platelet-derived growth factor (PDGF), transforming growth factor a (TGF-a),
transforming
growth factor (3 (TGF-(3), epidermal growth factor (EGF), vascular endothelial
growth factor
(VEGF), somatotropin (growth hormone), a somatomedin such as insulin-like
growth factor I
(IGF-I) or insulin-like growth factor II (IGF-II), erythropoietin (EPO),
thrombopoietin (TPO)
or angiopoietin, v) a profibrinolytic protein, e.g. staphylokinase or
streptokinase, vi) a protease
inhibitor, e.g. aprotinin or CI-2A, vii) an enzyme, e.g. superoxide dismutase,
catalase, uricase,
bilirubin oxidase, trypsin, papain, asparaginase, arginase, arginine
deiminase, adenosin
deaminase, ribonuclease, alkaline phosphatase, (3-glucuronidase, purine
nucleoside
phosphorylase or batroxobin, viii) an opioid, e.g. endorphins, enkephalins or
non-natural
opioids, ix) a hormone or neuropeptide, e.g. insulin, calcitonin, glucagons,
adrenocorticotropic
hormone (ACTH), somatostatin, gastrins, cholecystokinins, parathyroid hormone
(PTH),
luteinizing hormone (LH), follicle-stimulating hormone (FSH), gonadotropin-
releasing
hormone, chorionic gonadotropin, corticotropin-releasing factor, vasopressin,
oxytocin,
antidiuretic hormones, thyroid-stimulating hormone, thyrotropin-releasing
hormone, relaxin,
glucagon-like peptide 1 (GLP-1), glucagon-like peptide 2 (GLP-2), prolactin,
neuropeptide Y,
peptide YY, pancreatic polypeptide, leptin, orexin, CART (cocaine and
amphetamine regulated
transcript), a CART-related peptide, melanocortins (melanocyte-stimulating
hormones),
melanin-concentrating hormone, natriuretic peptides, adrenomedullin,
endothelin, exendin,
secretin, amylin (IAPP;islet amyloid polypeptide precursor), vasoactive
intestinal peptide
(VlP), pituitary adenylate cyclase activating polypeptide (PACAP), agouti and
agouti-related
peptides or somatotropin-releasing hormones, or x) another type of protein or
peptide such as
thymosin, bombesin, bombesin-like peptides, heparin-binding protein, soluble
CD4,
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
17
pigmentary hormones, hypothalamic releasing factor, malanotonins,
phospholipase activating
protein, a detoxifying enzyme such as acyloxyacyl hydrolase, or an
antimicrobial peptide.
One group of polypeptides of particular interest in the present invention is
selected
from the group of lysosomal enzymes (as defined in US 5,929,304) such as those
responsible
for or otherwise involved in a lysosomal storage disease, i.e. enzymes that
have a therapeutical
effect on patients with a lysosomal storage disease. Such enzymes, e.g.
include
glucocerebrosidase, a-L-iduronidase, acid oc-glucosidase, a-galactosidase,
acid
sphingomyelinase, galactocerebrosidase, arylsulphatase A, sialidase, and
hexosaminidase.
Also, other proteins involved in lysosomal storage diseases such as Saposin A,
B, C or D
to (Nakano et al., J. Biochem. (Tokyo) 105, 152-154, 1989; Gavrieli-Rorman and
Grabowski,
Genomics 5, 486-492, 1989) can be modified as described herein. Preferably,
these
polypeptides are of human origin.
The present inventors have shown that providing such enzymes with additional N-
linked oligosaccharide moieties considerably improve properties thereof, such
as stability,
targeting, expression, and in vivo activity and targeting. Accordingly, in one
embodiment the
polypeptide of the invention is a glycosylated lysosomal enzyme comprising a
peptide addition
comprising or contributing to a glycosylation site.
The industrial polypeptide is typically an enzyme, in particular a microbial
enzyme, and
can be used in products or in the manufacture of products such as detergents,
household
2o articles, personal care products, agrochemicals, textile, food products, in
particular bakery
products, feed products, or in industrial processes such as hard surface
cleaning. The industrial
polypeptide is normally not intended for internal administration to humans or
animals. Specific
examples include hydrolases, such as proteases, lipases or cutinases,
oxidoreductases, such as
lactase and peroxidase, transferases such as transglutaminases, isomerases,
such as protein
disulphide isomerase and glucose isomerase, cell wall degrading enzymes such
as cellulases,
xylanases, pectinases, mannanases, etc., amylolytic enzymes such as
endoamylases, e.g. alpha-
amylases, or exo-amylases, e.g. beta-amylases or amyloglucosidases, etc.
Further specific
examples are those listed in WO 00/26354, the contents of which are
incorporated herein by
reference. Normally, an enzyme modified according to the present invention has
one or more
3o improved properties selected from the group consisting of increased
stability (in particular
against proteolytic degradation or thermal degradation) leading to, e.g.,
improved shelf life and
improved performance in use; improved production, e.g. in terms of improved
expression (e.g.
as a consequence of improved secretion and/or increased stability of the
expressed enzyme)
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
1$
and improved purification, decreased allergenicity, increased activity in the
relevant industrial
process in which it is used, and improved properties with respect to
immobilization.
When the polypeptide Pp is an industrial enzyme the N-terminal peptide
addition may
comprise or contribute to a glycosylation site. However, it is also within the
scope of the
present invention to provide a polypeptide comprising an industrial enzyme
arid a C-terminal
or N-terminal peptide addition comprising an attachment group for a second non-
peptide
moiety being a polymer, e.g. PEG. The peptide addition may or may not comprise
a
glycosylation site. The peptide addition is preferably as described herein.
For instance, such
attachment group can be provided by a lysine or cysteine residue.
In one embodiment the polypeptide of the invention comprises a personal care
enzyme
(i.e. an enzyme useful for personal care applications), which polypeptide is
incapable of
passing the mucous membrane of a mammal in particular a human exposed to the
polypeptide.
Thereby, allergenicity can be reduced or avoided. Furthermore, stability of
such enzyme can be
increased. The polypeptide according to this embodiment comprises an N-
terminal or C-
terminal peptide addition comprising or contributing to a glycosylation site
and/or an
attachment group for a second non-peptide moeity, e.g. a polymer such as PEG.
In another embodiment the polypeptide comprises a lipase as disclosed in WO
97/04079, in particular a Humicola lanugiuosa lipase, wherein the N- or C--
terminal peptide
addition comprises a glycosylation site and/or at least one attachment group
for a second non-
2o peptide moeity, e.g. a polymer such as PEG. Thereby, the N- or C-terminal
peptide addition is
shielded from degradation and/or increased expression, including secretion, of
the enzyme is
likely to be obtained. In connection with this embodiment the N-terminal
peptide addition can
comprise any of the peptide additions disclosed in WO 97/04079.
In yet another embodiment the polypeptide Pp is an amyloglucosidase and the N-
or C-
terminal peptide addition comprises or contributes to a glycosylation site
and/or an attachment
group for a second non-peptide moeity, e.g. a polymer such as PEG. When the
peptide addition
is N-terminal the modification of such enzyme is contemplated to result in
reduced or no
degradation of the N-terminus of said enzyme (an otherwise well known problem
associated
with the recombinant production of amyloglucosidase). In other words the N-
terminus of the
enzyme is protected by the non-peptide moiety attached to the N-terminal
peptide addition of
the amyloglucosidase.
In yet another embodiment the polypeptide Pp is an antigen, in particular an
antigen
intended for use in eliciting an immune response (for vaccine purposes). It is
contemplated to
be advantageous to add N-terminal glycosylation sites) to antigens in
accordance with the
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
19
invention in that the risk of changing antigenicity is thereby reduced.
Antigens are recognized
by a wide range of target cells, including antigen presenting cells (APC), and
taken up by those
cells for efficient intracellular processing and presentation to other cells
of the immune system,
such as, e.g., T cells, to induce or elicit desired immune responses. Antigens
(and fragments
thereof, e.g., antigen peptides) can be modified by a peptide addition and non-
peptide moieties
according to the invention. Such modifications facilitate and/or optimize
uptake and/or
targeting to processing compartment of the antigen by such target cells. For
example, N-
terminally extended antigen polypeptides of the invention are taken up by the
target cells more
efficiently and/or at an enhanced or improved rate (when the non-peptide
moiety is one
to involved in such uptake). Such efficient, improved, or enhanced uptake of
modified antigens
by the target cells increases the kinetics and potency of the immune response
to the
immunizing antigen. These modifications to antigens also improve the affinity
of the antigens
for particular cellular receptors on target cells, including, e.g., mannose
receptors and other
carbohydrate receptors (in particular when the non-peptide moiety is an
oligosaccharide
moiety).
Antigen polypeptides of the invention include, but are not limited to those,
for which an
improved, enhanced or altered uptake of antigens in the following type of
target cells is
desired: antigen-presenting and antigen-processing cells, such as monocytes, B
cells, antigen-
presenting macrophages, marginal zone macrophages, follicular dendritic cells,
dendritic cells,
2o Langerhans cells, keratinocytes, M-cells (e.g., M-cells of the gut),
myocytes for intramuscular
immunization or epithelial cells for mucosal immunization, Kuppfer cells in
the liver, and the
like. A number of other cells, including capillary endothelium and some
endocrine cells, can
present antigen in some circumstances; the cells develop MHC class II
molecules that confer
antigen-presenting function. Furthermore, MHC class I molecules are expressed
on the surface
of most nucleated cells, including, for example, muscle cells, and therefore
these cells can also
present antigens to CD8+ T cells. Activated T cells, which release IFN-gamma
actively induce
expression of MHC molecules on some tissue cells. Such cells are also of use
with the novel
polypeptides of the invention. Preferably, such cells are of mammalian origin,
in particular
human (for use in immunization of a human) or animal (for veterinary
purposes).
A wide range of antigens can be modified according to the invention. Examples
are as follows:
Caf~cer antigens
Examples of cancer antigens that can be modified according to the invention
include,
but are not limited to: bullous pemphigoid antigen 2, prostate mucin antigen
(PMA) (Beckett
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
and Wright (1995) 1>2t. J. Cancer 62: 703-710), tumor associated Thomsen-
Friedenreich
antigen (Dahlenborg et al. (1997) Iht. J. Caf~cer 70: 63-71), prostate-
specific antigen (PSA)
(Dannull and Belldegrun (1997) Br. J. Urol. 1: 97-103), EpCam/KSA antigen,
luminal
epithelial antigen (LEA.135) of breast carcinoma and bladder transitional cell
carcinoma
5 (TCC) (Jones et al. (1997) Arzticazzcer Res. 17: 685-687), cancer-associated
serum antigen
(CASA) and cancer antigen 125 (CA 125) (Kierkegaard et al. (1995) Gyrzecol.
Oyzcol. 59: 251-
254), the epithelial glycoprotein 40 (EGP40) (Kievit et al. (1997) Iht. J.
Cancer 71: 237-245),
squamous cell carcinoma antigen (SCC) (Lozza et al. (1997) Ayztica>zcer Res.
17: 525-529),
cathepsin E (Mota et al. (1997) Am. J. Pathol. 150: 1223-1229), tyrosinase in
melanoma
10 (Fishman et al. (1997) Cafzcer 79: 1461-1464), cell nuclear antigen (PCNA)
of cerebral
cavernomas (Notelet et al. (1997) Surg. Neurol. 47: 364-370), DF3/MLTC1 breast
cancer
antigen (Apostolopoulos et al. (1996) Immu>zol. Cell. Biol. 74: 457-464;
Pandey et al. (1995)
Cancer Res. 55: 4000-4003), carcinoembryonic antigen (Paone et al. (1996) J.
Cancer Res.
Cli>z. Oucol. 122: 499-503; Schlom et al. (1996) Breast Caucer Res. Treat. 38:
27-39), tumor-
15 associated antigen CA 19-9 (Tolliver and O'Brien (1997) South Med. J. 90:
89-90; Tsuruta et
al. (1997) Urol. hzt. 58: 20-24), human melanoma antigens MART-1/Melan-A27-35
and gp100
(Kawakami and Rosenberg (1997) hzt. Rev. lyzzuzurzol. 14: 173-192; Zajac et
al. (1997) Ifzt. J.
Cancer 71: 491-496), the T and Tn pancarcinoma (CA) glycopeptide epitopes
(Springer (1995)
Crit. Rev. Ozzcog. 6: 57-85), a 35 kD tumor-associated autoantigen in
papillary thyroid
20 carcinoma (Lucas et al. (1996) Antica~zcer Res. 16: 2493-2496), KH-1
adenocarcinoma antigen
(Deshpande and Danishefsky (1997) Nature 387: 164-166), the A60 mycobacterial
antigen
(Maes et al. (1996) J. Cancer Res. Clin. Oyzcol. 122: 296-300), heat shock
proteins (HSPs)
(Blachere and Srivastava (1995) Semiyz. Cayzcer Biol. 6: 349-355), and MAGE,
tyrosinase,
melan-A and gp75 and mutant oncogene products (e.g., p53, ras, and HER-2/neu
(Bueler and
Mulligan (1996) Mol. Med. 2: 545-555; Lewis and Houghton (1995) Semifz. Cancer
Biol. 6:
321-327; Theobald et al. (1995) Proc. Nat'l. Acad. Sci. USA 92: 11993-11997);
TAG-72, a
mucin ag expressed in most human adenocarcinomas (McGuinness et al. (1999) Hum
Gene
Ther 10:165-73.
Bacterial antigeyzs
Bacterial antigens that can be modified according to the invention include,
but are not
limited to, Helicobacter pylori antigens CagA and VacA (Blaser (1996)
Alirzzefzt. Plaamzacol.
Ther. 1: 73-7; Blaser and Crabtree (1996) Am. J. Clin. Pathol. 106: 565-7;
Censini et al. (1996)
Proc. Nat'l. Acad. Sci. USA 93: 14648-14643). Other suitable H. pylori
antigens include, for
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
21
example, four immunoreactive proteins of 45-65 kDa as reported by Chatha et
al. (1997)
Ihdiarz J. Med. Res. 105: 170-175 and the H. pylori GroES homologue (HspA)
(Kansau et al.
(1996) Mol. Microbiol. 22: 1013-1023. Other suitable bacterial antigens
include, but are not
limited to, the 43-kDa and the fimbrilin (41 kDa) proteins of P. girzgivalis
(Boutsl et al. (1996)
Oral Microbiol. Immurzol. 11: 236-241); pneumococcal surface protein A (Briles
et al. (1996)
Arzrz. NYAcad. Sci. 797: 118-126); Chlarrzydia psittaci antigens, 80-90 kDa
protein and 110
kDa protein (Buendia et al. (1997) FEMS Microbiol. Lett. 150: 113-9); the
chlamydial
exoglycolipid antigen (GLXA) (Whittum-Hudson et al. (1996) Nature Med. 2: 1116-
1121);
Chlamydia przeumorziae species-specific antigens in the molecular weight
ranges 92-98, 51-55,
l0 43-46 and 31.5-33 kDa and genus-specific antigens in the ranges 12, 26 and
65-70 kDa (Halme
et al. (1997) Scahd. J. Immurzol. 45: 378-84); Neisseria gorzorrhoeae (GC) or
Escherichia coli
phase-variable opacity (Opa) proteins (Chen and Gotschlich (1996) Proc. Nat'l.
Acad. Sci.
USA 93: 14851-14856), any of the twelve immunodominant proteins of Schistosoma
marzsoni
(ranging in molecular weight from 14 to 208 kDa) as described by Cutts and
Wilson (1997)
Parasitology 114: 245-55; the 17-kDa protein antigen of Brucella abortus (De
Mot et al.
(1996) Curr. Microbiol. 33: 26-30); a gene homolog of the 17-kDa protein
antigen of the
Gram-negative pathogen Brucella abortus identified in the nocardioform
actinomycete
Rhodococcus sp. NI86/21 (De Mot et al. (1996) Curr. Microbiol. 33: 26-30); the
staphylococcal enterotoxins (SEs) (Wood et al. (1997) FEMS Immurzol. Med.
Microbiol. 17: 1-
10), a 42-kDa M. hyopneurzzorziae NrdF ribonucleotide reductase R2 protein or
15-kDa subunit
protein of M. lzyoprzeurnorziae (Fagan et al. (1997) Infect. Immurz. 65: 2502-
2507), the
meningococcal antigen PorA protein (Feavers et al. (1997) Clin. Diagrz. Lab.
Immurzol. 3: 444-
50); pneumococcal surface protein A (PspA) (McDaniel et al. (1997) Gerze Ther.
4: 375-377);
F. tularerzsis outer membrane protein FopA (Fulop et al. (1996) FEMS Immunol.
Med.
Microbiol. 13: 245-247); the major outer membrane protein within strains of
the genus
Actinobacillus (Hartmann et al. (1996) Zerztralbl. Bakteriol. 284: 255-262);
p60 or listeriolysin
(HIy) antigen of Listeria rrzozzocytogerzes (Hess et al. (1996) Proc. Nat'l.
Acad. Sci. USA 93:
1458-1463); flagellar (G) antigens observed on Salrrzorzella erzteritidis and
S. pullorum (Holt
and Chaubal (1997) J. Clin. Microbiol. 35: 1016-1020); Bacillus anthracis
protective antigen
3o (PA) (Ivins et al. (1995) Vaccine 13: 1779-1784); Echirzococcus grarzulosus
antigen 5 (Jones et
al. (1996) Parasitology 113: 213-222); the rol genes of Slzigella dysenteriae
1 and Escherichia
coli K-12 (Klee et al. (1997) J. Bacteriol. 179: 2421-2425); cell surface
proteins Rib and alpha
of group B streptococcus (Larsson et al. (1996) Infect. Imrnurz. 64: 3518-
3523); the 37 kDa
secreted polypeptide encoded on the 70 kb virulence plasmid of pathogenic
Yersinia spp.
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
22
(teary et al. (1995) Contrib. Microbiol. Imrnunol. 13: 216-217 and Roggenkamp
et al. (1997)
Infect. Immure. 65: 446-51); the OspA (outer surface protein A) of the Lyme
disease spirochete
Borrelia burgdorferi (ti et al. (1997) Proc. Nat'l. Acad. Sci. USA 94: 3584-
3589, Padilla et al.
(1996) J. Infect. Dis. 174: 739-746, and Wallich et al. (1996) Infection 24:
396-397); the
Brucella melitensis group 3 antigen gene encoding Omp28 (Lindler et al. (1996)
Infect.
Immun. 64: 2490-2499); the PAc antigen of Streptococcus mutans (Murakami et
al. (1997)
Infect. Immurz: 65: 794-797); pneumolysin, Pneumococcal neuraminidases,
autolysin,
hyaluronidase, and the 37 kDa pneumococcal surface adhesin A (Paton et al.
(1997) Microb.
Drug Resist. 3: 1-10); 29-32, 41-45, 63-71 x 10(3) MW antigens of Salmonella
typhi (Perez et
l0 al. (1996) Immunology 89: 262-267); K-antigen as a marker of Klebsiella
przeumoniae
(Priamukhina and Morozova (1996) Klin. Lab. Diagn. 47-9); nocardial antigens
of molecular
mass approximately 60, 40, 20 and 15-10 kDa (Prokesova et al. (1996) Int. J.
Immunophannacol. 18: 661-668); Staphylococczzs aureus antigen ORF-2 (Rieneck
et al.
(1997) Biochim Biophys Acta 1350: 128-132); GIpQ antigen of Borrelia hermsii
(Schwan et al.
(1996) J. Clin. Microbiol. 34: 2483-2492); cholera protective antigen (CPA)
(Sciortino (1996)
J. Diarrlzoeal Dis. Res. 14: 16-26); a 190-kDa protein antigen of
Streptococcus mutans
(Senpuku et al. (1996) Oral Microbiol. Irnmunol. 11: 121-128); Anthrax toxin
protective
antigen (PA) (Sharma et al. (1996) Protein Expr. Purif. 7: 33-38); Clostridium
perfringerzs
antigens and toxoid (Strom et al. (1995) Br. J. Rheumatol. 34: 1095-1096); the
SEF14 fimbrial
antigen of Salrraonella enteritidis (Thorns et al. (1996) Microb. Pathog. 20:
235-246); the
Yersinia pestis capsular antigen (Fl antigen) (Titball et al. (1997) Infect.
Inzrnun. 65: 1926-
1930); a 35-kilodalton protein of Mycobacterium leprae (Triccas et al. (1996)
Infect. Immun.
64: 5171-5177); the major outer membrane protein, CD, extracted from Moraxella
(Branhamella) catarrhalis (Yang et al. (1997) FEMS Immurzol. Med. Microbiol.
17: 187-199);
pH6 antigen (PsaA protein) of Yersirzia pestis (Zav'yalov et al. (1996) FEMS
Immunol. Med.
Microbiol. 14: 53-57); a major surface glycoprotein, gp63, of Leislamarzia
major (Xu and Liew
(1994) Vaccine 12: 1534-1536; Xu and Liew (1995) Immunology 84: 173-176);
mycobacterial
heat shock protein 65, mycobacterial antigen (Mycobacterium leprae hsp65)
(Lowrie et al.
(1994) Vaccirze 12: 1537-1540; Ragno et al. (1997) Arthritis Rheurn. 40: 277-
283; Silva (1995)
Braz. J. Med. Biol. Res. 28: 843-851); Mycobacterium tz~berculosis antigen 85
(Ag85) (Huygen
et al. (1996) Nat. Med. 2: 893-898); the 45/47 kDa antigen complex (APA) of
Mycobacterium
tuberculosis, M. bovis and BCG (Horn et al. (1996) J. Inzmunol. Methods 197:
151-159); the
mycobacterial antigen, 65-kDa heat shock protein, hsp65 (Tascon et al. (1996)
Nat. Med. 2:
888-892); the mycobacterial antigens MPB64, MPB70, MPB57 and alpha antigen
(Yamada et
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
23
al. (1995) Kekkaku 70: 639-644); the M. tuberculosis 38 kDa protein
(Vordermeier et al.
(1995) Vaccine 13: 1576-1582); the MPT63, MPT64 and MPT-59 antigens from
Mycobacterium tuberculosis (Manca et al. (1997) Infect. Immun. 65: 16-23;
Oettinger et al.
(1997) Scand. J. Immunol. 45: 499-503; Wilcke et al. (1996) Tuber. Lung Dis.
77: 250-256);
the 35-kilodalton protein of Mycobacterium leprae (Triccas et al. (1996)
Infect. Imnaun. 64:
5171-5177); the ESAT-6 antigen of virulent mycobacteria (Brandt et al. (1996)
J. Immunol.
157: 3527-3533; Pollock and Andersen (1997) T. Infect. Dis. 175: 1251-1254);
Mycobacterium
tuberculosis 16-kDa antigen (Hsp16.3) (Chang et al. (1996) .l. Biol. Chem.
271: 7218-7223);
and the 18-kilodalton protein of Mycobacterium leprae (Baumgart et al. (1996)
Infect. ImnZUn.
l0 64: 2274-2281); protective antigen (PA) of B. anthracis; V antigen from
Yersinia pestis, Y.
enterocolitica, and Y. pseudotuberculosis; antigens against bacterium Vibrio
cholerae, cholera
toxin B subunit, and heat-labile enterotoxins (LT) from enterotoxigenic E.
coli strains.
Viral pathogens
Polypeptides or proteins corresponding to or associated with various viral
pathogens,
including, but not limited to, e.g., hanta virus (e.g., hanta virus
glycoproteins), flaviviruses,
such as, e.g., Dengue viruses (e.g., envelope proteins), Japanese, St. Louis
and Murray Valley
encephalitis viruses, tick-borne encephalitis viruses can be modified
according to the invention.
Viral antigens that can be modified according to the invention include, but
are not
2o limited to, influenza A virus N2 neuraminidase (Kilbourne et al. (1995)
Vaccine 13: 1799-
1803); Dengue virus envelope (E) and premembrane (prM) antigens (Feighny et
al. (1994) Am.
J. Trop. Med. Hyg. 50: 322-328; Putnak et al. (1996) Am. J. Trop. Med. Hyg.
55: 504-10); HIV
antigens Gag, Pol, Vif and Nef (Vogt et al. (1995) Vaccine 13: 202-208); HIV
antigens gp120
and gp160 (Achour et al. (1995) Cell. Mol. Biol. 41: 395-400; Hone et al.
(1994) Dev. Biol.
Stand. 82: 159-162); gp41 epitope of human immunodeficiency virus (Eckhart et
al. (1996) J.
Gen. Virol. 77: 2001-2008); rotavirus antigen VP4 (Mattion et al. (1995) J.
Virol. 69: 5132-
5137); the rotavirus protein VP7 or VP7sc (Emslie et al. (1995) J. Virol. 69:
1747-1754; Xu et
al. (1995) J. Gen. Virol. 76: 1971-1980); herpes simplex virus (HSV)
glycoproteins gB, gC,
gD, gE, gG, gH, and gI (Fleck et al. (1994) Med. Microbiol. Imrnunol. (Berl)
183: 87-94
[Mattion, 1995]; Ghiasi et al. (1995) Invest. Ophthalmol. Vis. Sci. 36: 1352-
1360; McLean et
al. (1994) J. Infect. Dis. 170: 1100-1109); immediate-early protein ICP47 of
herpes simplex
virus-type 1 (HSV-1) (Banks et al. (1994) Virology 200: 236-245); immediate-
early (1E)
proteins ICP27, ICPO, and ICP4 of herpes simplex virus (Manickan et al. (1995)
J. Virol. 69:
4711-4716); influenza virus nucleoprotein and hemagglutinin (Deck et al.
(1997) Vaccine 15:
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
24
71-78; Fu et al. (1997) J. Virol. 71: 2715-2721); B19 parvovirus capsid
proteins VPl (Kawase
et al. (1995) Virology 211: 359-366) or VP2 (Brown et al. (1994) Virology 198:
477-488);
Hepatitis B virus core and a antigen and capsid protein (Schodel et al. (1996)
Izztervirology 39:
104-106); hepatitis B surface antigen (Shiau and Murray (1997) J. Med. Virol.
51: 159-166);
hepatitis B surface antigen fused to the core antigen of the virus (Id.);
Hepatitis B virus core-
preS2 particles (Nemeckova et al. (1996) Acta Virol. 40: 273-279); HBV preS2-S
protein
(Kutinova et al. (1996) Vaccine 14: 1045-1052); VZV glycoprotein I (Kutinova
et al. (1996)
Vaccine 14: 1045-1052); rabies virus glycoproteins (Xiang et al. (1994)
Virology 199: 132-
140; Xuan et al. (1995) Virus Res. 36: 151-161) or ribonucleocapsid (Hooper et
al. (1994)
Proc. Nat'l. Acad. Sci. USA 91: 10908-10912); human cytomegalovirus (HCMV)
glycoprotein
B (UL55) (Britt et al. (1995) J. Infect. Dis. 171: 18-25); the hepatitis C
virus (HCV)
nucleocapsid protein in a secreted or a nonsecreted form, or as a fusion
protein with the middle
(pre-S2 and S) or major (S) surface antigens of hepatitis B virus (HBV)
(Inchauspe et al.
(1997) DNA Cell Biol. 16: 185-195; Major et al. (1995) J. Virol. 69: 5798-
5805); the hepatitis
C virus antigens: the core protein (pC); El (pEl) and E2 (pE2) alone or as
fusion proteins
(Saito et al. (1997) Gastroenterology 112: 1321-1330); the gene encoding
respiratory syncytial
virus fusion protein (PFP-2) (Falsey and Walsh (1996) Vaccine 14: 1214-1218;
Piedra et al.
(1996) Pediatr. Infect. Dis. J. 15: 23-31); the VP6 and VP7 genes of
rotaviruses (Choi et al.
(1997) Virology 232: 129-138; Jin et al. (1996) Arch. Virol. 141: 2057-2076);
the E1, E2, E3,
2o E4, E5, E6 and E7 proteins of human papillomavirus (Brown et al. (1994)
Virology 201: 46-
54; Dillner et al. (1995) Cancer Detect. Prev. 19: 381-393; Krul et al. (1996)
Cancer lmuznno1.
Immunother. 43: 44-48; Nakagawa et al. (1997) J. Infect. Dis. 175: 927-931); a
human T-
lymphotropic virus type I gag protein (Porter et al. (1995) J. Med. Virol. 45:
469-474); Epstein-
Barr virus (EBV) gp340 (Mackett et al. (1996) J. Med. Virol. 50: 263-271); the
Epstein-Barr
virus (EBV) latent membrane protein LMP2 (Lee et al. (1996) Eur. J. Iznnzunol.
26: 1875-
1883); Epstein-Barr virus nuclear antigens 1 and 2 (Chen and Cooper (1996) J.
Virol. 70:
4849-4853; Khanna et al. (1995) Virology 214: 633-637); the measles virus
nucleoprotein (N)
(Fooks et al. (1995) Virology 210: 456-465); and cytomegalovirus glycoprotein
gB (Marshall
et al. (1994) J. Med. Virol. 43: 77-83) or glycoprotein gH (Rasmussen et al.
(1994) J. Infect.
3o Dis. 170:673-677).
Parasites
Antigens from parasites can also be modified according to the invention. These
include, but are not limited to, the schistosome gut-associated antigens CAA
(circulating
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
anodic antigen) and CCA (circulating cathodic antigen) in Schistosonza
nzazzsozZi, S.
laaenzatobium or S. japonicuzn (Deelder et al. (1996) Parasitology 112: 21-
35); a multiple
antigen peptide (MAP) composed of two distinct protective antigens derived
from the parasite
Sclaistosoma mansoni (Ferro et al. (1997) Parasite Immunol. 19: 1-11);
Leishmania parasite
5 surface molecules (Lezama-Davila (1997) Arch. Med. Res. 28: 47-53); third-
stage larval (L3)
antigens of L. loa (Akue et al. (1997) J. Infect. Dis. 175: 158-63); the
genes, Tams1-1 and
Tamsl-2, encoding the 30-and 32-kDa major merozoite surface antigens of
Theileria annulata
(Ta) (d'Oliveira et al. (1996) Gene 172: 33-39); Plasmodium falciparum
merozoite surface
antigen 1 or 2 (al-Yaman et al. (1995) Trans. R. Soc. Trop. Med. Hyg. 89: 555-
559; Beck et al.
l0 (1997) J. Infect. Dis. 175: 921-926; Rzepczyk et al. (1997) Infect. Immun.
65: 1098-1100);
circumsporozoite (CS) protein-based B-epitopes from Plasmodium berghei,
(PPPPNPND)2
and Plasmodiuzn yoelii, (QGPGAP)3QG, along with a P. berghei T-helper epitope
KQIRDSITEEWS (Reed et al. (1997) Vaccine 15: 482-488); NYVAC-Pf7 encoded
Plasmodium falciparunz antigens derived from the sporozoite (circumsporozoite
protein and
15 sporozoite surface protein 2), liver (liver stage antigen 1), blood
(merozoite surface protein 1,
serine repeat antigen, and apical membrane antigen 1), and sexual (25-kDa
sexual-stage
antigen) stages of the parasite life cycle were inserted into a single NYVAC
genome to
generate NYVAC-Pf7 (Tine et al. (1996) Infect. Immun. 64: 3833-3844);
Plasmodium
falciparum antigen Pfs230 (Williamson et al. (1996) Mol. Biochenz. Parasitol.
78: 161-169);
20 Plasnzodiuzzz falciparuzzz apical membrane antigen (AMA-1) (Lal et al.
(1996) Infect. Inznzun.
64: 1054-1059); Plasmodium falciparum proteins Pfs28 and Pfs25 (puffy and
Kaslow (1997)
Izzfect. Immun. 65: 1109-1113); Plasnzodiunz falciparum merozoite surface
protein, MSP1 (Hui
et al. (1996) Infect. Izzzmun. 64: 1502-1509); the malaria antigen Pf332
(Ahlborg et al. (1996)
Immunology 88: 630-635); Plasmodium falciparum erythrocyte membrane protein 1
(Baruch et
25 al. (1995) Proc. Nat'l. Acad. Sci. USA 93: 3497-3502; Baruch et al. (1995)
Cell 82: 77-87);
Plasmodium falcaparum merozoite surface antigen, PfMSP-1 (Egan et al. (1996)
J. Infect. Dis.
173: 765-769); Plasmodium falciparum antigens SERA, EBA-175, RAP1 and RAP2
(Riley
(1997) J. Pharnz. Pharnzacol. 49: 21-27); Schistosozna japonicum paramyosin
(Sj97) or
fragments thereof (Yang et al. (1995) Biochem. Biophys. Res. Comnzun. 212:
1029-1039); and
Hsp70 in parasites (Maresca and Kobayashi (1994) Experientia 50: 1067-1074).
Allergen antigens
Allergen antigens that can be modified according to the invention, include,
but are not
limited to those of animals, including the mite (e.g., Dermatophagoides
pterozzyssinus,
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
26
Dennatoplaagoides farinae, Blomia tropicalis), such as the allergens der p1
(Scobie et al.
(1994) Biochem. Soc. Traps. 22: 4485; Yssel et al. (1992) J. Immunol. 148: 738-
745), der p2
(Chua et al. (1996) Clin. Exp. Allergy 26: 829-837), der p3 (Smith and Thomas
(1996) Clin.
Exp. Allergy 26: 571-579), der p5, der p V (Lip et al. (1994) J. Allergy Clin.
Immunol. 94: 989-
996), der p6 (Bennett and Thomas (1996) Clin. Exp. Allergy 26: 1150-1154), der
p 7 (Shen et
al. (1995) Clin. Exp. Allergy 25: 416-422), der f2 (Yuuki et al. (1997) Int.
Arch. Allergy
Immunol. 112: 44-48), der f3 (Nishiyama et al. (1995) FEBS Lett. 377: 62-66),
der f7 (Shen et
al. (1995) Clin. Exp. Allergy 25: 1000-1006); Mag 3 (Fujikawa et al. (1996)
Mol. Immunol. 33:
311-319). Also of interest as antigens are the house dust mite allergens Tyr
p2 (Erilcsson et al.
to (1998) Eur. J. Biochem. 251: 443-447), Lep dl (Schmidt et al. (1995) FEBS
Lett. 370: 11-14),
and glutathione S-transferase (O'Neill et al. (1995) Imrnunol Lett. 48: 103-
107); the 25,589 Da,
219 amino acid polypeptide with homology with glutathione S-transferases
(O'Neill et al.
(1994) Biochifn. BioplZys. Acta. 1219: 521-528); Blo t 5 (Arruda et al. (1995)
Int. Arch. Allergy
Immunol. 107: 456-457); bee venom phospholipase A2 (Carballido et al. (1994)
J. Allergy
Clin. Immunol. 93: 758-767; Jutel et al. (1995) J. Immunol. 154: 4187-4194);
bovine
dermal/dander antigens BDA 11 (Rautiainen et al. (1995) J. Invest. Dennatol.
105: 660-663)
and BDA20 (Mantyjarvi et al. (1996) J. Allergy Clin. Immunol. 97: 1297-1303);
the major
horse allergen Equ c1 (Gregoire et al. (1996) J. Biol. Chem. 271: 32951-
32959); Jumper ant M.
pilosula allergen Myr p I and its homologous allergenic polypeptides Myr p2
(Donovan et al.
(1996) Biochem. Mol. Biol. Int. 39: 877-885); 1-13, 14, 16 kD allergens of the
mite Blornia
tropicalis (Caraballo et al. (1996) J. Allergy Clin. InZmunol. 98: 573-579);
the cockroach
allergens Bla g Bd90K (Helm et al. (1996) J. Allergy Clin. Immunol. 98: 172-
80) and Bla g 2
(Arruda et al. (1995) J. Biol. Claeyn. 270: 19563-19568); the cockroach Cr-PI
allergens (Wu et
al. (1996) J. Biol. Chem. 271: 17937-17943); fire ant venom allergen, Sol i 2
(Schmidt et al.
(1996) J. Allergy Clin. InZmunol. 98: 82-88); the insect Chironomus thummi
major allergen Chi
t 1-9 (Kipp et al. (1996) Int. Arch. Allergy Immunol. 110: 348-353); dog
allergen Can f 1 or cat
allergen Fel d 1 (Ingrain et al. (1995) J. Allergy Clin. Immunol. 96: 449-
456); albumin,
derived, for example, from horse, dog or cat (Goubran Botros et al. (1996)
Immufaology 88:
340-347); deer allergens with the molecular mass of 22 kD, 25 kD or 60 kD
(Spitzauer et al.
(1997) Clin. Exp. Allergy 27: 196-200); and the 20 kd major allergen of cow
(Ylonen et al.
(1994) J. Allergy Clin. Irnmunol. 93: 851-858).
Pollen and grass allergens can also be modified according to the invention.
Such
allergens include, for example, Hor v9 (Astwood and Hill (1996) Gene 182: 53-
62, Lig v1
(Batanero et al. (1996) Clin. Exp. Allergy 26: 1401-1410); Lol p 1 (Muller et
al. (1996) Int.
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
27
Arch. Allergy Immunol. 109: 352-355), Lol p II (Tamborini et al. (1995) Mol.
Immunol. 32:
505-513), Lol pVA, Lol pVB (Ong et al. (1995) Mol. Immunol. 32: 295-302), Lol
p 9 (Blaher
et al. (1996) J: Allergy Clin. Imnzunol. 98: 124-132); Par J I (Costa et al.
(1994) FEBS
Lett. 341: 182-186; Sallusto et al. (1996) J. Allergy Clin. Imnzunol. 97: 627-
637), Par j 2.0101
(Duro et al. (1996) FEBS Lett. 399: 295-298); Bet v1 (Faber et al. (1996) J.
Biol. Chem. 271:
19243-19250), Bet v2 (Rihs et al. (1994) Int. Arclz. Allergy Immunol. 105: 190-
194); Dac g3
(Guerin-Marchand et al. (1996) Mol. Irnmunol. 33: 797-806); Phl p 1 (Petersen
et al. (1995) J.
Allergy Clin. Immunol. 95: 987-994), Phl p 5 (Muller et al. (1996) Int. Arcla.
Allergy Immunol.
109: 352-355), Phl p 6 (Petersen et al. (1995) Int. Arch. Allergy Immunol.
108: 55-59); Cry j I
l0 (Sone et al. (1994) Bioclaem. Bioplzys. Res. Commuf2. 199: 619-625), Cry j
II (Namba et al.
(1994) FEBS Lett. 353: 124-128); Cor a 1 (Schenk et al. (1994) Eur. J.
Biochem. 224: 717-
722); cyn d1 (Smith et al. (1996) J. Allergy Clin. Immunol. 98: 331-343), cyn
d7 (Suphioglu et
al. (1997) FEBS Lett. 402: 167-172); Pha a 1 and isoforms of Pha a 5
(Suphioglu and Singh
(1995) Clirz. Exp. Allergy 25: 853-865); Cha o 1 (Suzuki et al. (1996) Mol.
Immunol. 33: 451-
460); profilin derived, e.g, from timothy grass or birch pollen (Valenta et
al. (1994) Biochem.
Biophys. Res. Commun. 199: 106-118); P0149 (Wu et al. (1996) Plant Mol. Biol.
32: 1037-
1042); Ory s1 (Xu et al. (1995) Gene 164: 255-259); and Amb a V and Amb t 5
(Kim et al.
(1996) Mol. Immunol. 33: 873-880; Zhu et al. (1995) J. Immunol. 155: 5064-
5073).
Food allergens that can be modified according to the invention include, for
example,
profilin (Rihs et al. (1994) Int. Arcla. Allergy Inzmunol. 105: 190-194); rice
allergenic cDNAs
belonging to the alpha-amylase/trypsin inhibitor gene family (Alvarez et al.
(1995) Biochim
Biophys Acta 1251: 201-204); the main olive allergen, Ole a I (Lombardero et
al. (1994) Clirz
Exp Allergy 24: 765-770); Sin a 1, the major allergen from mustard (Gonzalez
De La Pena et
al. (1996) Eur J Biochem. 237: 827-832); parvalbumin, the major allergen of
salmon
(Lindstrom et al. (1996) Scand. J. Immunol. 44: 335-344); apple allergens,
such as the major
allergen Mal d 1 (Vanek-Krebitz et al. (1995) Biochem. Biophys. Res. Commun.
214: 538-
551); and peanut allergens, such as Ara h I (Burks et al. (1995) J. Clin.
lyzvest. 96: 1715-1721).
Fungal allergens that can be modified according to the invention include, but
are not
limited to, the allergen, Cla h III, of Cladosporium herbarum (Zhang et al.
(1995) J. Inzmunol.
154: 710-717); the allergen Psi c 2, a fungal cyclophilin, from the
basidiomycete Psilocybe
cubensis (Homer et al. (1995) Int. Arch. Allergy Immunol. 107: 298-300); hsp
70 cloned from a
cDNA library of Cladosporiunz herbarum (Zhang et al. (1996) Clin Exp Allergy
26: 88-95); the
68 kD allergen of Penicillium notatum (Shen et al. (1995) Clin. Exp. Allergy
26: 350-356);
aldehyde dehydrogenase (ALDH) (Achatz et al. (1995) Mol Immunol. 32: 213-227);
enolase
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
28
(Achatz et al. (1995) Mol. Immunol. 32: 213-227); YCP4 (Id.); acidic ribosomal
protein P2
(Id.).
Other allergens that can be modified include latex allergens, such as a major
allergen
(Hev b 5) from natural rubber latex (Akasawa et al. (1996) J. Biol. Clzem.
271: 25389-25393;
Slater et al. (1996) J. Biol. Chem. 271: 25394-25399).
Antigens associated with autoimmune diseases and iyzflammatory conditions
Autoantigens that can be modified according to the invention include, but are
not
limited to, myelin basic protein (Stinissen et al. (1996) J. Neurosci. Res.
45: 500-511) or a
1o fusion protein of myelin basic protein and proteolipid protein (Elliott et
al. (1996) J. Clin.
hzvest. 98: 1602-1612), proteolipid protein (PLP) (Rosener et al. (1997) J.
Neuroimmunol. 75:
28-34), 2',3'-cyclic nucleotide 3'-phosphodiesterase (CNPase) (Rosener et al.
(1997) J.
Neuroinzmunol. 75: 28-34), the Epstein Barr virus nuclear antigen-1 (EBNA-1)
(Vaughan et al.
(1996) J. Neuroimmurzol. 69: 95-102), HSP70 (Salvetti et al. (1996) J.
Neuroimmunol. 65: 143-
53; Feldmann et al. (1996) Cell 85: 307).
Antigens that can be modified according to the invention and used to treat
scleroderma,
systemic sclerosis, and systemic lupus erythematosus include, for example, (-2-
GPI, 50 kI~a
glycoprotein (Blank et al. (1994) J. Autoimnzun. 7: 441-455), Ku (p70/p80)
autoantigen, or its
80-lcd subunit protein (Hong et al. (1994) Invest. Ophthalmol. Vis. Sci. 35:
4023-4030; Wang et
al. (1994) J. Cell Sci. 107: 3223-3233), the nuclear autoantigens La (SS-B)
and Ro (SS-A)
(Huang et al. (1997) J. Clin. Immunol. 17: 212-219; Igarashi et al. (1995)
Autoimrnunity 22:
33-42; Keech et al. (1996) Clin. Exp. Immuuol. 104: 255-263; Manoussakis et
al. (1995) J.
Autoimmun. 8: 959-969; Topfer et al. (1995) Proc. Nat'l. Acad. Sci. USA 92:
875-879),
proteasome (-type subunit C9 (Feist et al. (1996) J. Exp. Med. 184: 1313-
1318), Scleroderma
antigens Rpp 30, Rpp 38 or Scl-70 (Eder et al. (1997) Proc. Nat'l. Acad. Sci.
USA 94: 1101-
1106; Hietarinta et al. (1994) Br. J. Rheurrzatol. 33: 323-326), the
centrosome autoantigen
PCM-1 (Bao et al. (1995) Autoimmunity 22: 219-228), polymyositis-scleroderma
autoantigen
(PM-Scl) (Kho et al. (1997) J. Biol. Chem. 272: 13426-13431), scleroderma (and
other
systemic autoimmune disease) autoantigen CENP-A (Muro et al. (1996) Clin.
Immunol.
Imnzunopathol. 78: 86-89), U5, a small nuclear ribonucleoprotein (snRNP)
(Okano et al.
(1996) Clin. Immunol. Imyrzunopathol. 81: 41-47), the 100-kd protein of PM-Scl
autoantigen
(Ge et al. (1996) Arthritis Rlzeum. 39: 1588-1595), the nucleolar U3- and Th(7-
2)
ribonucleoproteins (Verheijen et al. (1994) J. Immunol. Methods 169: 173-182),
the ribosomal
protein L7 (Neu et al. (1995) Clin. Exp. Inzmunol. 100: 198-204), hPop1
(Lygerou et al. (1996)
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
29
EMBO J. 15: 5936-5948), and a 36-kd protein from nuclear matrix antigen (Deng
et al. (1996)
Arthritis Rheum. 39: 1300-1307).
Antigens useful in treatment of hepatic autoimmune disorders can also be
modified;
these include the cytochromes P450 and UDP-glucuronosyl-transferases
(Obermayer-Straub
and Manns (1996) Baillieres Clue. Gastroerzterol. 10: 501-532), the
cytochromes P450 2C9 and
P450 IA2 (Bourdi et al. (1996) Chem. Res. Toxicol. 9: 1159-1166; Clemente et
al. (1997) J.
Clin. Eudocrihol. Metab. 82: 1353-1361), LC-1 antigen (Klein et al. (1996) J.
Pediatr.
Gastroehterol. Nutr. 23: 461-465), and a 230-kDa Golgi-associated protein
(Funaki et al.
(1996) Cell Struct. Funct. 21: 63-72).
1o Antigens useful for treatment of autoimmune disorders of the skin that can
be modified
according to the invention include, but are not limited to, the 450 kD human
epidermal
autoantigen (Fujiwara et al. (1996) J. Invest. Dermatol. 106: 1125-1130), the
230 kD and 180
kD bullous pemphigoid antigens (Hashimoto (1995) Keio J. Med. 44: 115-123;
Murakami et
al. (1996) J. Dennatol. Sci. 13: 112-117), pemphigus foliaceus antigen
(desmoglein I),
pemphigus vulgaris antigen (desmoglein 3), BPAg2, BPAgl, and type VII collagen
(Batteux et
al. (1997) J. Clin. Imrnunol. 17: 228-233; Hashimoto et al. (1996) J.
Dermatol. Sci. 12: 10-17),
a 168-kDa mucosal antigen in a subset of patients with cicatricial pemphigoid
(Ghohestani et
al. (1996) J. luvest. Dermatol. I07: 136-139), and a 218-kd nuclear protein
(218-kd Mi-2)
(Seelig et al. (1995) Artlaritis Rheum. 38: 1389-1399).
Antigens for treating insulin dependent diabetes mellitus can also be
modified; these,
include, but are not limited to, insulin, proinsulin, GAD65 and GAD67, heat-
shock protein 65
(hsp65), and islet-cell antigen 69 (ICA69) (French et al. (1997) Diabetes 46:
34-39; Roep
(1996) Diabetes 45: 1147-1156; Schloot et al. (1997) Diabetologia 40: 332-
338), viral proteins
homologous to GAD65 (Jones and Crosby (1996) Diabetologia 39: 1318-1324),
islet cell
antigen-related protein-tyrosine phosphatase (PTP) (Cui et al. (1996) J. Biol.
Chem. 271:
24817-24823), GM2-1 ganglioside (Cavallo et al. (1996) J. Endocrinol. 150: 113-
120; Dotta et
al. (1996) Diabetes 45: 1193-l I96), glutamic acid decarboxylase (GAD) (Nepom
(1995) Curr.
Opih. Immuhol. 7: 825-830; Panina-Bordignon et al. (1995) J. Exp. Med. 181:
1923-1927), an
islet cell antigen (ICA69) (Karges et al. (1997) Biochim. Bioplays. Acta 1360:
97-101; Roep et
al. (I996) Eur. J. Immunol. 26: 1285-1289), Tep69, the single T cell epitope
recognized by T
cells from diabetes patients (Karges et al. (1997) Biochim. Biophys. Acta
1360: 97-101), ICA
512, an autoantigen of type I diabetes (Solimena et al. (1996) EMBO J. 15:
2102-2114), an
islet-cell protein tyrosine phosphatase and the 37-kDa autoantigen derived
from it in type 1
diabetes (including IA-2, IA-2) (La Gasse et al. (1997) Mol. Med. 3: I63-I73),
the 64 kDa
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
protein from In-111 cells or human thyroid follicular cells that is
immunoprecipitated with sera
from patients with islet cell surface antibodies (ICSA) (Igawa et al. (1996)
Endocr. J. 43: 299-
306), phogrin, a homologue of the human transmembrane protein tyrosine
phosphatase, an
autoantigen of type 1 diabetes (Kawasaki et al. (1996) Biochenz. Biophys. Res.
Conzmun. 227:
5 440-447), the 40 kDa and 37 kDa tryptic fragments and their precursors IA-2
and IA-2 in
IDDM (Lampasona et al. (1996) J. Irnmunol. 157: 2707-2711; Notkins et al.
(1996) J.
Autoimmuyz. 9: 677-682), insulin or a cholera toxoid-insulin polypeptide
(Bergerot et al. (1997)
Proc. Nat'). Acad. Sci. USA 94: 4610-4614), carboxypeptidase H, the human
homologue of
gp330, which is a renal epithelial glycoprotein involved in inducing Heymann
nephritis in rats,
10 and the 38-kD islet mitochondria) autoantigen (Arden et al. (1996) J. Clin.
Invest. 97: 551-561.
Useful antigens for rheumatoid arthritis treatment that can be modified
according to the
invention include, but are not limited to, the 45 kDa DEK nuclear antigen, in
particular onset
juvenile rheumatoid arthritis and iridocyclitis (Murray et al. (1997) J.
Rheumatol. 24: 560-
567), human cartilage glycoprotein-39, an autoantigen in rheumatoid arthritis
(Verheijden et al.
15 (1997) Arthritis Rlaeum. 40: 1115-1125), a 68k autoantigen in rheumatoid
arthritis (B)ass et al.
(1997) Ann. Rheum. Dis. 56: 317-322), collagen (Rosloniec et al. (1995) J.
Imnzunol. 155:
4504-4511), collagen type II (Cook et al. (1996) Arthritis Rheum. 39: 1720-
1727; Trentham
(1996) Ann. N. Y. Acad. Sci. 778: 306-314), cartilage link protein
(Guerassimov et al. (1997) J.
Rheumatol. 24: 959-964), ezrin, radixin and moesin, which are auto-immune
antigens in
2o rheumatoid arthritis (Wagatsuma et al. (1996) Mol. Immunol. 33: 1171-1176),
and
mycobacterial heat shock protein 65 (Ragno et al. (1997) Arthritis Rheunz. 40:
277-283).
Antigens useful for treatment are autoimmune thyroid disorders that can be
modified
include, for example, thyroid peroxidase and the thyroid stimulating hormone
receptor (Tandon
and Weetman (1994) J. R. Col). Physicians Lond. 28: 10-18), thyroid peroxidase
from human
25 Graves' thyroid tissue (Gardas et al. (1997) Biochem. Biophys. Res. Commun.
234: 366-370;
Zimmer et al. (1997) Histoclzern. Cell. Biol. 107: 115-120), a 64-kDa antigen
associated with
thyroid-associated ophthalmopathy (Zhang et al. (1996) Clin. Imnzunol.
Immunopathol. 80:
236-244), the human TSH receptor (Nicholson et al. (1996) J. Mol. Endocrinol.
16: 159-170),
and the 64 kDa protein from In-111 cells or human thyroid follicular cells
that is
30 immunoprecipitated with sera from patients with islet cell surface
antibodies (ICSA) (Igawa et
al. (1996) Endocr. J. 43: 299-306).
Other associated antigens that can be modified include, but are not limited
to, Sjogren's
syndrome (-fodrin; Haneji et al. (1997) Science 276: 604-607), myastenia
gravis (the human
M2 acetylcholine receptor or fragments thereof, specifically the second
extracellular loop of
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
31
the human M2 acetylcholine receptor; Fu et al. (1996) Clin. Immuhol.
Immuyaopathol. 78: 203-
207), vitiligo (tyrosinase; Fishman et al. (1997) Cancer 79: 1461-1464), a 450
kD human
epidermal autoantigen recognized by serum from individual with blistering skin
disease, and
ulcerative colitis (chromosomal proteins HMG1 and HMG2; Sobajima et al. (1997)
Clirc. Exp.
Immuyaol. 107: 135-140).
Sperm Antigens
Sperm antigens which can be used in the genetic vaccines include, for example,
lactate
dehydrogenase (LDH-C4), galactosyltransferase (GT), SP-10, rabbit sperm
autoantigen (RSA),
to guinea pig (g)PH-20, cleavage signal protein (CS-1), HSA-63, human (h)PH-
20, and AgX-1
(Zhu and Naz (1994) Arch. Androl. 33: 141-144), the synthetic sperm peptide,
PlOG (O'Rand
et al. (1993) J. Reprod. Immuhol. 25: 89-102), the 135kD, 95kD, 65kD, 47kD,
4lkD and 23kD
proteins of sperm, and the FA-1 antigen (Naz et al. (1995) Arch. Androl. 35:
225-231), and the
35 kD fragment of cytokeratin 1 (Lucas et al. (1996) AyZticancer Res. 16: 2493-
2496).
Also, examples of antigens are set forth in Punnonen et al. (1999) WO
99/41369; Punnonen et
al. (1999) WO 99/41383; Punnonen et al. (1999) WO 99/41368; and Punnonen et
al. (1999)
WO 99/41402), the contents of all of which are incorporated herein by
reference in their
entirety for all purposes. Other useful antigens have been described in the
literature or can be
discovered using genomics approaches.
Peptide addition
In principle the peptide addition X can be any stretch of amino acid residues
ranging from a
single amino acid residue to a large protein, e.g. a mature protein. Usually,
the peptide addition
X comprises 1-500 amino acid residues, such as 2-500, normally 2-50 or 3-50
amino acid
residues, such as 3-20 amino acid residues. The length of the peptide addition
to be used for
modification of a given polypeptide is dependent of or determined on the basis
of a number of
factors including the type of polypeptide of interest and the desired effect
to be achieved by the
modification. Normally, the peptide addition has less than 90% identity to the
amino acid
sequence of a native full length polypeptide, in particular less than 80%
identity, such as less
3o than 70% identity or even lower degree of identity to a full length
protein. In one embodiment
the peptide addition may constitute a part of a full length protein (e.g. 1-50
amino acid residues
thereof.
The peptide addition may be designed by a site-specific or random approach,
e.g as out-
lined in further detail in the Methods section below. This section also
comprises a set of
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
32
guidelines useful for preparing a peptide addition for use in the present
invention are described.
It will be understood that those guidelines are intended for illustration
purposes only and that a
person skilled in the art will be aware of alternative useful routes for
design of peptide
addition. Thus, the method of designing a peptide addition for use herein
should not be
considered limited to that described in the Materials section.
The number of glycosylation sites should be sufficient to provide the desired
effect.
Typically, the peptide addition X comprises 1-20, such as 1-10 glycosylation
sites. For
instance, the peptide addition X comprises 1, 2, 3, 4, 5, 6, 7, 8, 9 or 10
glycosylation sites. It is
well known that one frequently occurring consequence of modifying an amino
acid sequence
l0 of, e.g., a human protein is that new epitopes are created by such
modification. In order to
shield any new epitopes created by the peptide addition, it is desirable that
sufficient
glycosylation sites are present to enable shielding of all epitopes introduced
into the sequence.
This is e.g. achieved when the peptide addition X comprises at least one
glycosylation site
within a stretch of 30 contiguous amino acid residues, such as at least one
glycosylation site
within 20 amino acid residues or at least one glycosylation site within 10
amino acid residues,
in particular 1-3 glycosylation sites within a stretch of 10 contiguous amino
acid residues in the
peptide addition X.
Thus, in one embodiment the peptide addition X comprises at least two
glycosylation
sites, wherein two of said sites are separated by at most 10 amino acid
residues, none of which
2o comprises a glycosylation site. Furthermore, the polypeptide Pp can
comprise at Ieast one
introduced glycosylation site, in particular 1-5 introduced glycosylation
sites. Analogously, the
polypeptide Pp can comprise at least one removed glycosylation site, in
particular 1-5 removed
glycosylation sites.
The glycosylation site of the peptide addition may be an iyz vivo or in vitro
glycosylation site. Prefererably, the glycosylation site is an in vivo
glycosylation site, in
particular an N-glycosylation site since glycosylation of such site is more
easy to control than
to an O-glycosylation site. Accordingly, in a preferred embodiment the peptide
addition X
comprises at least one N-glycosylation site, typically at least two N-
glycosylation sites. For
instance, the peptide addition X has the structure Xl-N-X2-[T/S]/C-Z, wherein
Xi is a peptide
3o comprising at least one amino acid residue or is absent, X2 is any amino
acid residue different
from Pro, and Z is absent or a peptide comprising at least one amino acid
residue. For instance,
Xl is absent, X2 is an amino acid residue selected from the group consisting
of I, A, G, V and S
(all relatively small amino acid residues), and Z comprises at least 1 amino
acid residue.
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
33
For instance, Z can be a peptide comprising 1-50 amino acid residues and,
e.g., 1-10
glycosylation sites.
In another polypeptide of the invention Xl comprises at least one amino acid
residue,
e.g. 1-50 amino acid residues, X2 is an amino acid residue selected from the
group consisting
of I, A, G, V and S, and Z is absent. For instance, Xl comprises 1-10
glycosylation sites.
For instance, the peptide addition for use in the present invention can
comprise a peptide
sequence selected from the group consisting of INA[TlS], GNI[T/S], VNI[T/S],
SNI[T/S],
ASNI[T/S], NI[T/S], SPINA[T/S], ASPINA[TlS], ANI[T/S]ANI[T/S]ANI,
ANI[T/S]GSNI[T/S]GSNI[T/S], FNI[T/S]VNI[T/S]V
to YNI[T/S]VNI[T/S]V, AFNI[T/S]VNI[T/S]V, AYNI[T/S]VNI[T/S]V,
APND[T/S]VNI[T/S]V,
ANI[T/S], ASNS[T/S]NNG[T/S]LNA[T/S], ANH[TlS]NE[T/S]NA[T/S], GSPINA[T/S],
ASPINA[T/S]SPINA[T/S], ANN[T/S]NY[T/S]NW[T/S], ATNI[T/S]LNY[T/S]AN[T/S]T,
AANS[T/S]GNI[T/S]ING[T/S], AVNW[T/S]SND[T/S]SNS[T/S], GNA[T/S],
AVNW[T/S]SND[T/S]SNS[T/S], ANN[T/S]NY[T/S]NS[TlS], ANNTNYTNWT,
ANI[T/S]VNI[TlS]V, ND[T/S]VNF[T/S] and NI[T/S]VNI[T/S]V wherein [T/S] is
either a T
or an S residue, preferably a T residue. Other non-limiting examples include a
peptide addition
comprising the sequence NSTQNATA, which corresponds to positions 231 to 238 of
the
human calcium activated channel 2 precursor (to add two N-glycosylation
sites), or the
sequence ANLTVRNLTRNVTV, which corresponds to positions 538 to 551 of the
human G
2o protein coupled receptor 64 (to add three N-glycosylation sites).
The peptide addition can comprise one or more of these peptide sequences, i.e.
at least
two of said sequences either directly linked together or separated by one or
more amino acid
residues, or can contain two or more copies of any of these peptide sequence.
It will be
understood that the above specific sequences are given for illustrative
purposes and thus do not
constitute an exclusive list of peptide sequences of use in the present
invention.
In a more specific embodiment the peptide addition X is selected from the
group
consisting of lNA[T/S], GNI[T/S], VNI[T/S], SNI[T/S], ASNI[T/S], NI[T/S],
SP1NA[T/S],
ASPINA[T/S], ANI[T/S]ANI[T/S]ANI, and ANI[T/S]GSNI[T/S]GSNI[T/S], wherein
[T/S] is
either a T or an S residue, preferably a T residue.
3o As stated further above the polypeptide Pp can be a native polypeptide that
may or may
not comprise one or more glycosylation sites. In order to further modify the
glycosylation of
the polypeptide Pp of interest (in terms of the number of oligosaccharide
moieties attached to
the polypeptide), the polypeptide Pp can be a variant of a native polypeptide
that differs from
said polypeptide in at least one introduced or at least one removed
glycosylation site.
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
34
For instance, the polypeptide Pp comprises at least one introduced
glycosylation site, in
particular 1-5 introduced glycosylation sites, such as 2-5 introduced
glycosylation sites.
In order to affect the total glycosylation of the polypeptide of interest the
glycosylation
site is introduced so that the N residue of said glycosylation site is exposed
at the surface of the
polypeptide, when folded in its active form. Likewise, a glycosylation site to
be removed is
selected from those having an N residue exposed at the surface of the
polypeptide.
In one embodiment, the peptide addition X has an N residue in position -2 or -
1, and
the polypeptide Pp or Px has a T or an S residue in position +1 or +2,
respectively, the residue
numbering being made relative to the N-terminal amino acid residue of Pp or
PX, whereby an
1o N-glycosylation site is formed.
Glycosylatio~z
The polypeptide of the invention is glycosylated (i.e. comprises an in vivo
attached N- or O-
linked oligosaccharide moiety or if2 vitro attached oligosaccharide moiety)
and furthermore has
an altered glycosylation profile as compared to that of the polypeptide Pp.
For instance, the
altered glycosylation profile is a consequence of an altered, normally
increased, number of
attached oligosaccharide moieties and/or an altered type or distribution of
attached
oligosaccharide moieities.
Furthermore, for polypeptides intended for therapeutic or veterinary uses or
to which a
2o human or animal is otherwise exposed, the type of oligosaccharide moiety to
be attached
should normally be one that does not lead to increased immunogenicity of the
polypeptide as
compared to that of the polypeptide Pp. The coupling of an oligosaccharide
moiety may take
place ifz vivo or iyz vitro. In order to achieve in vivo glycosylation of a a
nucleotide sequence
encoding the polypeptide should be inserted in a glycosylating, eucaryotic
expression host. The
expression host cell may be selected from fungal (filamentous fungal or
yeast), insect,
mammalian cells or transgenic plant cells as disclosed in further detail in
the section entitled
"Methods of preparing a polypeptide of the invention" . Also, the
glycosylation may be
achieved in the human body when using a nucleotide sequence encoding the
polypeptide of the
invention in gene therapy.
In vitro glycosylation can be achieved by attaching chemically synthesized
oligosaccharide structures to the polypeptide using a variety of different
chemistries e.g. the
chemistries employed for attachment of PEG to proteins, wherein the
oligosaccharide is linked
to a functional group, optionally via a short spacer (see the section entitled
Conjugation to a
Non-Oligosaccharide Macromolecular Moiety). The irz vitro glycosylation can be
carried out
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
in a suitable buffer at pH 4-7 in protein concentrations of 0.5-2 mg/ml and a
volume of 0.02-2
ml. The activated mannose compound is present in 2-200 fold molar excess, and
reactions are
incubated at 4-25°C for periods of 0.1-3 hours. h2 vitro glycosylated
GCB polypeptides are
purified by dialysis and standard chromatographic techniques.
Other i~ vitro glycosylation methods are described, for example in WO
87/05330, by
Aplin etl al., CRC Crit Rev. Biochem., pp. 259-306, 1981, by Lundblad and
Noyes, Chemical
Ragents for Protein Modification, CRC Press Inc. Boca Raton, FI, by Yan and
Wold,
Biochemistry, 1984, Jul. 31: 23(I6): 3759-65, and by Doebber et al., J. Biol.
Chem., 257,
pp2193-2199, 1982.
10 Furthermore, in vitro glycosylation to protein- and peptide-bound Gln-
residues can be
carried out by transglutaminases (TGases). Transglutaminases catalyse the
transfer of donor
amine-groups to protein- and peptide-bound Gln-residues in a so-called cross-
linking reaction.
The donor-amine groups can be protein- or peptide-bound e.g. as the E-amino-
group in Lys-
residues or it can be part of a small or large organic molecule. An example of
a small organic
15 molecule functioning as amino-donor in TGase-catalysed cross-linking is
putrescine (1,4-
diaminobutane). An example of a larger organic molecule functioning as amino-
donor in
TGase-catalysed cross-linking is an amine-containing PEG (Sato et al.,
Biochemistry 35, 1996,
13072-13080).
TGases, in general, are highly specific enzymes, and not every Gln-residues
exposed on
2o the surface of a protein is accessible to TGase-catalysed cross-linking to
amino-containing
substances. In order to render a protein susceptible to TGase-catalysed cross-
linking reactions
stretches of amino acid sequence known to function very well as TGase
substrates are inserted
at convenient positions in the amino acid sequence encoding a GCB polypeptide.
Several
amino acid sequences are known to be or to contain excellent natural TGase
substrates e.g.
25 substance P, elafin, fibrinogen, fibronectin, a,~-plasmin inhibitor, a-
caseins, and (3-caseins and
may thus be inserted into and thereby constitute part of the amino acid
sequence of a
polypeptide of the invention.
The nature and number of oligosaccharide moieties of a glycosylated
polypeptide of the
invention may be determined by a number of different methods known in the art
e.g.by lectin
3o binding studies (Reddy et al., 1985, Biochem. Med. 33: 200-210; Cummings,
1994, Meth.
Enzymol. 230: 66-86; Protein Protocols (Walker ed.), 1998, chapter 9); by
reagent array
analysis method (RAAM) sequencing of released oligosaccharides (Edge et aL,
I992, Proc.
Natl. Acad. Sci. USA 89: 6338-6342; Prime et al., 1996, J. Chrom. A 720: 263-
274); by
RAAM sequencing of released oligosaccharides in combination with mass
spectrometry
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
36
(Klausen, et al., 1998, Molecular Biotechnology 9: 195-204); or by combining
proteolytic
degradation, glycopeptide purification by HPLC, exoglycosidase degradations
and mass
spectrometry (Krogh et al, 1997, Eur. J. Biochem. 244: 334-342). Specific
methods for
determining the glycosylation profile is described in the examples section
hereinafter.
Normally, the glycosylated polypeptide of the invention comprises 1-15
oligosaccharide
moieties, such as 1-10 or 1-6 oligosachharide moieties. Usually, at least one
of these is
attached to the peptide addition and further oligosaccharide structures are
attached to the
peptide addition or the polypeptide Pp.
Polypeptide of the if2vention corzjugated to a second non peptide moiety
It can be advantageous that the glycosylated polypeptide of the invention
further comprises at
least one second non-peptide moiety. The term "second non-peptide moeity" is
intended to
indicate a non-peptide moiety different from an oligosaccharide moiety, e.g. a
polymer
molecule, a lipophilic compound and an organic derivatizing agent.
For this purpose the polypeptide must comprise at least one attachment group
for the
second non-peptide moiety. The attachment group can be one present on an amino
acid residue,
e.g., selected from the group consisting of the N-terminal or C-terminal amino
acid residue of
the polypeptide of the invention, lysine, cysteine, arginine, glutamine,
aspartic acid, glutamic
acid, serine, tyrosine, histidine, phenylalanine and tryptophan, or on an
oligosaccharide moiety
attached to the polypeptide. For instance, the attachment group for the non-
peptide moiety is an
epsilon-amino group.
It will be understood that an attachment group for the second non-peptide
moiety may
be provided by the N-terminal peptide addition, within the polypeptide Pp,
andlor as a C-
terminal peptide addition (having similar properties to those described above
for the peptide
addition X). In one embodiment, the peptide addition X comprising or
contributing to an
attachment site further comprises an attachment group for a second non-peptide
moeity. For
instance, the peptide addition may comprise 1-20, such as 1-10 attachment
groups for a second
non-peptide moiety. Such attachment groups may be distributed in a similar
manner as that
described immediately above for glycosylation sites. Also, the peptide
addition X can comprise
at least two attachment groups for the second non-peptide moiety.
Also, the polypeptide Pp can be a variant of a native polypeptide, which as
compared to
said native polypeptide, comprises at least one introduced andlor at least one
removed
attachment group for the second non-peptide moiety. For instance, the
polypeptide Pp
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
37
comprises at least one introduced attachment group, in particular 1-5
introduced attachment
groups, such as 2-5 introduced attachment groups.
The attachment group is preferably located in a position that is exposed at
the surface of
the folded protein and thus accessible for conjugation to the polymer
molecule. For instance,
attachment to one or more polymer molecules increases the molecular weight of
the
polypeptide and can further serve to shield one or more epitopes thereof. The
polymer
molecule may be any of the molecules mentioned in the section entitled
"Conjugation to a
polymer molecule", but is preferably selected from the group consisting of
linear or branched
polyethylene glycol or polyalkylene oxide. Most preferably, the polymer
molecule is mPEG-
lo SPA, mPEG-SCM, mPEG-BTC from Shearwater Polymers, Inc, SC-PEG from Enzon,
Inc.,
tresylated mPEG (US 5,880,255) or oxycarbonyl-oxy-N-dicarboxyimide PEG (US
5,122,614)
(and the relevant attachment group is one present on a lysine or N-terminal
residue).
Alternatively, the polymer molecule is an activated PEG molecule reactive with
a cysteine
residue, e.g. VS-PEG from Shearwater Polymers.
Especially, when the polypeptide Pp is an' industrial enzyme, the second non-
peptide
moiety may be one which is capable of cross-linking and thereby of being
immobilized on a
suitable solid support. Such cross-linking polymers are available from
Shearwater Polymers,
Inc. It will be understood that the peptide addition of the polypeptide
according to this
embodiment comprises an attachment group for the cross-linking polymer in
question. In
2o connection with this embodiment the polypeptide Pp is preferably an
amyloglucosidase, an
alpha-amylase, a glucose isomerase, an amidase, or a lipolytic enzyme.
In the following sections "Conjugation to a lipophilic compound", "Conjugation
to a
polymer molecule", and "Conjugation to an organic derivatizing agent"
conjugation to specific
types of non-peptide moieties is described.
It will be understood that a conjugation step of any method of the invention
only finds
relevance when a non-polypeptide moiety other than an ire vivo attached
oligosaccharide
moiety is to be conjugated to the polypeptide, since in vivo glycosylation
takes place during the
expression step when using an appropriate glycosylating host cell as
expression host.
Accordingly, whenever a conjugation step occurs in the present invention this
is intended to be
3o conjugation to a non-polypeptide moiety other than an oligosaccharide
moiety attached by in
vivo glycosylation during expression in a glycosylating organism. In vitro
glycosylation
methods are described in the section entitled "glycosylation".
Conjugation to a lipophilzc compound
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
38
The polypeptide and the lipophilic compound can be conjugated to each other,
either directly
or by use of a linker. The lipophilic compound can be a natural compound such
as a saturated
or unsaturated fatty acid, a fatty acid diketone, a terpene, a prostaglandin,
a vitamine, a
carotenoide or steroide, or a synthetic compound such as a carbon acid, an
alcohol, an amine
and sulphonic acid with one or more alkyl-, aryl-, alkenyl- or other multiple
unsaturated
compounds. Furthermore, the lipophilic compound may be any of the lipophilic
substituents
disclosed in WO 97/31022, the contents of which are incorporated herein by
reference. The
conjugation between the polypeptide and the lipophilic compound, optionally
through a linker
can be done according to methods known in the art, e.g. as described by
Bodanszky in Peptide
1o Synthesis, John Wiley, New York, 1976 and in WO 96/12505 and further as
described in WO
97!31022.
Cofijugatiora to a polywer molecule
The polymer molecule to be coupled to the polypeptide of the invention can be
any suitable
polymer molecule, such as a natural or synthetic homo-polymer or
heteropolymer, typically
with a molecular weight in the range of 300-100,000 Da, such as 300-20,000 Da,
more
preferably in the range of 500-10,000 Da, even more preferably in the range of
500-5000 Da.
Examples of homo-polymers include a polyol (i.e. poly-OH), a polyamine (i.e.
poly-
NH~) and a polycarboxylic acid (i.e. poly-COOH). A hetero-polymer is a polymer
that
2o comprises different coupling groups, such as a hydroxyl group and an amine
group.
Examples of suitable polymer molecules include polymer molecules selected from
the
group consisting of polyalkylene oxide (PAO), including polyalkylene glycol
(PAG), such as
polyethylene glycol (PEG) and polypropylene glycol (PPG), branched PEGs, poly-
vinyl
alcohol (PVA), poly-carboxylate, poly-(vinylpyrolidone), polyethylene-co-
malefic acid
anhydride, polystyrene-co-malic acid anhydride, dextran, including
carboxymethyl-dextran, or
any other biopolymer suitable for the intended purpose, such as for reducing
immunogenicity
and/or increasing functional ih vivo half-life and/or serum half life, or for
providing
immobilization properties to the polypeptide (as discussed in the section
entitled "Polypeptide
of interest". Another example of a polymer molecule is human albumin or
another abundant
3o plasma protein. Generally, polyalkylene glycol-derived polymers are
biocompatible, non-toxic,
non-antigenic, non-immunogenic, have various water solubility properties, and
are easily
excreted from living organisms.
PEG is the preferred polymer molecule for reducing immunogenicity,
allergenicity
andlor increasing half-life, since it has only few reactive groups capable of
cross-linking
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
39
compared, e.g., to polysaccharides such as dextran, and the like. In
particular, monofunctional
PEG, e.g. methoxypolyethylene glycol (mPEG), is of interest since its coupling
chemistry is
relatively simple (only one reactive group is available for conjugating with
attachment groups
on the polypeptide). Consequently, the risk of cross-linking is eliminated,
the resulting
polypeptide conjugates are more homogeneous and the reaction of the polymer
molecules with
the polypeptide is easier to control.
To effect covalent attachment of the polymer molecules) to the polypeptide,
the
hydroxyl end groups of the polymer molecule must be provided in activated
form, i.e. with
reactive functional groups. Suitable activated polymer molecules are
commercially available,
e.g. from Shearwater Polymers, Inc., Huntsville, AL, USA. Alternatively, the
polymer
molecules can be activated by conventional methods known in the art, e.g. as
disclosed in WO
90/13540. Specific examples of activated linear or branched polymer molecules
for use in the
present invention are described in the Shearwater Polymers, Inc. 1997 and 2000
Catalogs
(Functionalized Biocompatible Polymers for Research and pharmaceuticals,
Polyethylene
Glycol and Derivatives, incorporated herein by reference). Specific examples
of activated PEG
polymers include the following linear PEGs: NHS-PEG (e.g. SPA-PEG, SSPA-PEG,
SBA-
PEG, SS-PEG, SSA-PEG, SC-PEG, SG-PEG, and SCM-PEG), and NOR-PEG), BTC-PEG,
EPOX-PEG, NCO-PEG, NPC-PEG, CDI-PEG, ALD-PEG, TRES-PEG, VS-PEG, IODO-PEG,
and MAL-PEG, and branched PEGs such as PEG2-NHS and those disclosed in US
5,932,462
2o and US 5,643,575, both of which are incorporated herein by reference.
Furthermore, the
following publications, incorporated herein by reference, disclose useful
polymer molecules
and/or PEGylation chemistries: US 5,824,778, US 5,476,653, WO 97/32607, EP
229,108, EP
402,378, US 4,902,502, US 5,281,698, US 5,122,614, US 5,219,564, WO 92/16555,
WO
94/04193, WO 94/14758, WO 94/17039, WO 94/18247, WO 94128024, WO 95/00162, WO
95/11924, W095/13090, WO 95/33490, WO 96/00080, WO 97/18832, WO 98/41562, WO
98/48837, WO 99/32134, WO 99/32139, WO 99/32140, WO 96/40791, WO 98/32466, WO
95/06058, EP 439 508, WO 97/03106, WO 96/21469, WO 95/13312, EP 921 131, US
5,736,625, WO 98/05363, EP 809 996, US 5,629,384, WO 96/41813, WO 96/07670, US
5,473,034, US 5,516,673, EP 605 963, US 5,382,657, EP 510 356, EP 400 472, EP
183 503
3o and EP 154 316.
The conjugation of the polypeptide and the activated polymer molecules is
conducted
by use of any conventional method, e.g. as described in the following
references (which also
describe suitable methods for activation of polymer molecules): R.F. Taylor,
(1991), "Protein
immobilisation. Fundamental and applications", Marcel Dekker, N.Y.; S.S. Wong,
(1992),
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
"Chemistry of Protein Conjugation and Crosslinking", CRC Press, Boca Raton;
G.T.
Hermanson et al., (1993), "Immobilized Affinity Ligand Techniques", Academic
Press, N.Y.).
The skilled person will be aware that the activation method and/or conjugation
chemistry to be
used depends on the attachment groups) of the polypeptide (examples of which
are given
5 further above), as well as the functional groups of the polymer (e.g. being
amine, hydroxyl,
carboxyl, aldehyde, sulfydryl, succinimidyl, maleimide, vinysulfone or
haloacetate). The
PEGylation can be directed towards conjugation to all available attachment
groups on the
polypeptide (i.e. such attachment groups that are exposed at the surface of
the polypeptide) or
can be directed towards one or more specific attachment groups, e.g. the N-
terminal amino
to group (US 5,985,265). Furthermore, the conjugation can be achieved in one
step or in a
stepwise manner (e.g. as described in WO 99/55377).
It will be understood that the PEGylation is designed so as to produce the
optimal
molecule with respect to the number of PEG molecules attached, the size and
form of such
molecules (e.g. whether they are linear or branched), and where in the
polypeptide such
15 molecules are attached. For instance, the molecular weight of the polymer
to be used can be
chosen on the basis of the desired effect to be achieved. For instance, if the
primary purpose of
the conjugation is to achieve a polypeptide having a high molecular weight
(e.g. to reduce renal
clearance) it is usually desirable to conjugate as few high Mw polymer
molecules as possible to
obtain the desired molecular weight. When a high degree of epitope shielding
is desirable this
2o can be obtained by use of a sufficiently high number of low molecular
weight polymer
molecules (e.g. with a molecular weight of about 5,000 Da) to effectively
shield all or most
epitopes of the polypeptide. For instance, 2-8, such as 3-6 such polymers can
be used.
In connection with conjugation to only a single attachment group on the
protein (as
described in US 5,985,265), it can be advantageous that the polymer molecule,
which can be
25 linear or branched, has a high molecular weight, e.g. about 20 kDa.
Normally, the polymer conjugation is performed under conditions aiming at
reacting all
available polymer attachment groups with polymer molecules. Typically, the
molar ratio of
activated polymer molecules to polypeptide is up to about 1000-1, in
particular 200-1,
preferably 100-1, such as 10-1 or 5-1, but also equimolar ratios can be used
in order to obtain
30 optimal reaction.
It is also contemplated according to the invention to couple the polymer
molecules to
the polypeptide through a linker. Suitable linkers are well known to the
skilled person. A
preferred example is cyanuric chloride (Abuchowski et al., (1977), J. Biol.
Chem., 252,
3578-3581; US 4,179,337; Shafer et al., (1986), J. Polym. Sci. Polym. Chem.
Ed., 24, 375-378.
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
41
Subsequent to the conjugation residual activated polymer molecules are blocked
according to methods known in the art, e.g. by addition of primary amine to
the reaction
mixture, and the resulting inactivated polymer molecules are removed by a
suitable method.
In a specific embodiment, the polypeptide of the invention is one that
comprises one or
more PEG molecules attached to the peptide addition, but not to the
polypeptide P. For
instance, the PEG molecule is attached to one or more cysteine residues
present in the peptide
addition X and, if necessary, one or more cysteine residues have been removed
from the
polypeptide P of interest in order to avoid conjugation thereto.
In another specific embodiment, the polypeptide of the invention comprises at
least one
l0 PEG molecule attached to a lysine residue of the peptide addition X, in
particular a linear or
branched PEG molecule with a molecular weight of at least 5kDa.
Methods of preparing a polypeptide of the ifZVeratioyz
The invention further comprises a method of producing the polypeptide of the
invention, which
method comprises culturing a host cell transformed or transfected with a
nucleotide sequence
encoding the polypeptide under conditions permitting the expression of the
polypeptide, and
recovering the polypeptide from the culture.
Apart from recombinant production, polypeptides of the invention may be
produced,
albeit less efficiently, by chemical synthesis or a combination of chemical
synthesis and
recombinant DNA technology.
The nucleotide sequence of the invention encoding a polypeptide of the
invention may
be constructed by isolating or synthesizing a nucleotide sequence encoding the
parent
polypeptide and fusing a nucleotide sequence encoding the relevant peptide
addition in
accordance with established technologies. To the extent amino acid
modifications are to be
made in the parent polypeptide, these are conveniently done by mutagenesis,
e.g. using site-
directed mutagenesis in accordance with well-known methods, e.g. as described
in Nelson and
Long, Analytical Biochemistry 180, 147-151, 1989, random mutagenesis or
shuffling.
The nucleotide sequence may be prepared by chemical synthesis, e.g. by using
an
oligonucleotide synthesizer, wherein oligonucleotides are designed based on
the amino acid
sequence of the desired polypeptide, and preferably selecting those codons
that are favoured in
the host cell in which the recombinant polypeptide will be produced. For
example, several
small oligonucleotides coding for portions of the desired polypeptide may be
synthesized and
assembled by polymerase chain reaction (PCR), ligation or ligation chain
reaction (LCR). The
individual oligonucleotides typically contain 5' or 3' overhangs for
complementary assembly.
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
42
Once assembled (by synthesis, site-directed mutagenesis or another method),
the
nucleotide sequence encoding the polypeptide may be inserted into a
recombinant vector and
operably linked to control sequences necessary for expression of thereof in
the desired
transformed host cell.
It should of course be understood that not all vectors and expression control
sequences
function equally well to express the nucleotide sequence encoding the
polypeptide part of the
invention. Neither will all hosts function equally well with the same
expression system.
However, one of skill in the art may make a selection among these vectors,
expression control
sequences and hosts without undue experimentation. For example, in selecting a
vector, the
l0 host must be considered because the vector must replicate in it or be able
to integrate into the
chromosome. The vector's copy number, the ability to control that copy number,
and the
expression of any other proteins encoded by the vector, such as antibiotic
markers, should also
be considered. In selecting an expression control sequence, a variety of
factors should also be
considered. These include, for example, the relative strength of the sequence,
its controllability,
and its compatibility with the nucleotide sequence encoding the polypeptide,
particularly as
regards potential secondary structures. Hosts should be selected by
consideration of their
compatibility with the chosen vector, the toxicity of the product coded for by
the nucleotide
sequence, their secretion characteristics, their ability to fold the
polypeptide correctly, their
fermentation or culture requirements, and the ease of purification of the
products coded for by
the nucleotide sequence.
The recombinant vector may be an autonomously replicating vector, i.e. a
vector
existing as an extrachromosomal entity, the replication of which is
independent of
chromosomal replication, e.g. a plasmid. Alternatively, the vector is one
which, when
introduced into a host cell, is integrated into the host cell genome and
replicated together with
the chromosomes) into which it has been integrated.
The vector is preferably an expression vector, in which the nucleotide
sequence
encoding the polypeptide of the invention is operably linked to additional
segments required
for transcription of the nucleotide sequence. The vector is typically derived
from plasmid or
viral DNA. A number of suitable expression vectors for expression in the host
cells mentioned
3o herein are commercially available or described in the literature. Useful
expression vectors for
eukaryotic hosts, include, for example, vectors comprising expression control
sequences from
SV40, bovine papilloma virus, adenovirus and cytomegalovirus. Specific vectors
are, e.g.,
pCDNA3.1(+)~Iiyg (Invitrogen, Carlsbad, CA, USA) and pCI-neo (Stratagene, La
Jolla, CA,
USA). Useful expression vectors for yeast cells include the 2~, plasmid and
derivatives thereof,
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
43
the POT1 vector (US 4,931,373), the pJSO37 vector described in (Okkels, Ann.
New York
Acad. Sci. 782, 202-207, 1996) and pPICZ A, B or C (Invitrogen, Carlsbad, CA,
USA). Useful
vectors for insect cells include pVL941, pBG311 (Gate et al., "Isolation of
the Bovine and
Human Genes for Mullerian Inhibiting Substance And Expression of the Human
Gene In
Animal Cells", Cell, 45, pp. 685-98 (1986), pBluebac 4.5 and pMelbac (both
available from
Invitrogen, Carlsbad, CA, USA).
Other vectors for use in this invention include those that allow the
nucleotide sequence
encoding the polypeptide of the invention to be amplified in copy number. Such
amplifiable
vectors are well known in the art. They include, for example, vectors able to
be amplified by
to DHFR amplification (see, e.g., Kaufman, U:S. Pat. No. 4,470,461, Kaufman
and Sharp,
"Construction Of A Modular Dihydrafolate Reductase cDNA Gene: Analysis Of
Signals
Utilized For Efficient Expression", Mol. Cell. Biol., 2, pp. 1304-19 (1982))
and glutamine
synthetase ("GS") amplification (see, e.g., US 5,122,464 and EP 338,841).
The recombinant vector may further comprise a DNA sequence enabling the vector
to
replicate in the host cell in question. An example of such a sequence (when
the host cell is a
mammalian cell) is the SV40 origin of replication. When the host cell is a
yeast cell, suitable
sequences enabling the vector to replicate are the yeast plasmid 2~,
replication genes REP 1-3
and origin of replication.
The vector may also comprise a selectable marker, e.g. a gene the product of
which
2o complements a defect in the host cell, such as the gene coding for
dihydrofolate reductase
(DHFR) or the Schizosaccharomyces pombe TPI gene (described by P.R. Russell,
Gene 40,
1985, pp. 125-130), or one which confers resistance to a drug, e.g.
ampicillin, kanamycin,
tetracyclin, chloramphenicol, neomycin, hygromycin or methotrexate. For
filamentous fungi,
selectable markers include amdS, pyre, arcB, niaD, sC.
The term "control sequences" is defined herein to include all components,
which are
necessary or advantageous for the expression of the polypeptide of the
invention. Each control
sequence may be native or foreign to the nucleic acid sequence encoding the
polypeptide. Such
control sequences include, but are not limited to, a leader, polyadenylation
sequence,
propeptide sequence, promoter, enhancer or upstream activating sequence,
signal peptide
sequence, and transcription terminator. At a minimum, the control sequences
include a
promoter operably linked to the nucleotide sequence encoding the polypeptide.
"Operably linked" refers to the covalent joining of two or more nucleotide
sequences,
by means of enzymatic ligation or otherwise, in a configuration relative to
one another such
that the normal function of the sequences can be performed. For example, the
nucleotide
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
44
sequence encoding a presequence or secretory leader is operably linked to a
nucleotide
sequence for a polypeptide if it is expressed as a preprotein that
participates in the secretion of
the polypeptide: a promoter or enhancer is operably linked to a coding
sequence if it affects the
transcription of the sequence; a ribosome binding site is operably linked to a
coding sequence
if it is positioned so as to facilitate translation. Generally, "operably
linked" means that the
nucleotide sequences being linked are contiguous and, in the case of a
secretory leader,
contiguous and in reading phase. Linking is accomplished by ligation at
convenient restriction
sites. If such sites do not exist, then synthetic oligonucleotide adaptors or
linkers are used, in
conjunction with standard recombinant DNA methods.
1o A wide variety of expression control sequences may be used in the present
invention.
Such useful expression control sequences .include the expression control
sequences associated
with structural genes of the foregoing expression vectors as well as any
sequence known to
control the expression of genes of prokaryotic or eukaryotic cells or their
viruses, and various
combinations thereof.
Examples of suitable control sequences for directing transcription in
mammalian cells
include the early and late promoters of SV40 and adenovirus, e.g. the
adenovirus 2 major late
promoter, the MT-1 (metallothionein gene) promoter, the human cytomegalovirus
immediate-
early gene promoter (CMV), the human elongation factor la (EF-la) promoter,
the
Drosoplaila minimal heat shock protein 70 promoter, the Rous Sarcoma Virus
(RSV) promoter,
2o the human ubiquitin C (UbC) promoter, the human growth hormone terminator,
SV40 or
adenovirus Elb region polyadenylation signals and the Kozak consensus sequence
(Kozak, M.
J Mol Biol 1987 Aug 20;196(4):947-50).
In order to improve expression in mammalian cells a synthetic intron may be
inserted in
the 5' untranslated region of the nucleotide sequence encoding the polypeptide
of the
invention. An example of a synthetic intron is the synthetic intron from the
plasmid pCI-Neo
(available from Promega Corporation, WI, USA).
Examples of suitable control sequences for directing transcription in insect
cells include
the polyhedrin promoter, the P10 promoter, the Autographa califonZica
polyhedrosis virus
basic protein promoter, the, baculovirus immediate early gene 1 promoter and
the baculovirus
39K delayed-early gene promoter, and the SV40 polyadenylation sequence.
Examples of suitable control sequences for use in yeast host cells include the
promoters
of the yeast a-mating system, the yeast triose phosphate isomerase (TP~
promoter, promoters
from yeast glycolytic genes or alcohol dehydogenase genes, the ADH2-4c
promoter and the
inducible GAL promoter.
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
Examples of suitable control sequences for use in filamentous fungal host
cells include
the ADH3 promoter and terminator, a promoter derived from the genes encoding
Aspergillus
oryzae TAKA amylase triose phosphate isomerase or alkaline protease, an A.
niger a-amylase,
A. Niger or A. nidulafZS glucoamylase, A. >zidulans acetamidase, Rhizomucor
nzielzei aspartic
5 proteinase or lipase, the TPI1 terminator and the ADH3 terminator.
The nucleotide sequence of the invention may or may not also include a
nucleotide
sequence that encode a signal peptide. The signal peptide is present when the
polypeptide is to
be secreted from the cells in which it is expressed. Such signal peptide, if
present, should be
one recognized by the cell chosen for expression of the polypeptide. The
signal peptide may be
l0 homologous (e.g. be that normally associated with the parent polypeptide in
question) or
heterologous (i.e. originating from another source than the parent
polypeptide) to the
polypeptide or may be homologous or heterologous to the host cell, i.e. be a
signal peptide
normally expressed from the host cell or one which is not normally expressed
from the host
cell. Accordingly, the signal peptide may be prokaryotic, e.g. derived from a
bacterium, or
15 eukaryotic, e.g. derived from a mammalian, or insect, filamentous fungal or
yeast cell.
The presence or absence of a signal peptide will, e.g., depend on the
expression host
cell used for the production of the polypeptide, the protein to be expressed
(whether it is an
intracellular or extracelluar protein) and whether it is desirable to obtain
secretion. For use in
filamentous fungi, the signal peptide may conveniently be derived from a gene
encoding an
20 Aspergillus sp. amylase or glucoamylase, a gene encoding a Rhizomucor
nziehei lipase or
protease or a Humicola lazzuginosa lipase. The signal peptide is preferably
derived from a gene
encoding A. oryzae TAKA amylase, A. zziger neutral oc-amylase, A. niger acid-
stable amylase,
or A. rziger glucoamylase. For use in insect cells, the signal peptide may
conveniently be
derived from an insect gene (cf. WO 90/05783), such as the lepidopteran
Manduca sexta
25 adipokinetic hormone precursor, (cf. US 5,023,328), the honeybee melittin
(Invitrogen,
Carlsbad, CA, USA), ecdysteroid UDPglucosyltransferase (egt) (Murphy et al.,
Protein
Expression and Purification 4, 349-357 (1993) or human pancreatic lipase (hpl)
(Methods in
Enzymology 284, pp. 262-272, 1997).
Specific examples of signal peptides for use in mammalian cells include that
of human
30 glucocerebrosidase apparent from the examples hereinafter or the murine Ig
kappa light chain
signal peptide (Coloma, M (1992) J. Imrn. Methods 152:89-104). For use in
yeast cells suitable
signal peptides have been found to be the a-factor signal peptide from S.
cereviczae. (cf. US
4,870,008), the signal peptide of mouse salivary amylase (cf. O. Hagenbuchle
et al.,
Nature 289, 1981, pp. 643-646), a modified carboxypeptidase signal peptide
(cf. L.A.
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
46
Valls et al., Cell 48, 1987, pp. 887-897), the yeast BAR1 signal peptide (cf.
WO 87/02670),
and the yeast aspartic protease 3 (YAP3) signal peptide (cf. M. Egel-Mitani et
al., Yeast 6,
1990, pp. 127-137).
Any suitable host may be used to produce the polypeptide of the invention,
including
bacteria, fungi (including yeasts), plant, insect, mammal, or other
appropriate animal cells or
cell lines, as well as transgenic animals or plants. When a non-glycosylating
organism such as
E. coli is used, and the polypeptide is to be a glycosylated polypeptide, the
expression in E. coli
is preferably followed by suitable izz vitro glycosylation.
Examples of bacterial host cells include grampositive bacteria such as strains
of
to Bacillus, e.g. B. brevis or B. subtilis, Pseudomozzas or Streptomyces, or
gramnegative bacteria,
such as strains of E. coli. The introduction of a vector into a bacterial host
cell may, for
instance, be effected by protoplast transformation (see, e.g., Chang and
Cohen, 1979,
Molecular Gefzeral Gezzetics 168: 111-115), using competent cells (see, e.g.,
Young and
Spizizin, 1961, Journal of Bacteriology 81: 823-829, or Dubnau and Davidoff-
Abelson, 1971,
Journal of Molecular Biology 56: 209-221), electroporation (see, e.g.,
Shigekawa and Dower,
1988, Biotechzziques 6: 742-751), or conjugation (see, e.g., Koehler and
Thorne, 1987, Jounzal
of Bacteriology 169: 5771-5278).
Examples of suitable filamentous fungal host cells include strains of
Aspergillus, e.g. A.
oryzae, A. sZiger, or A. z2idulans, Fusarium or Trichodenzza. Fungal cells may
be transformed
by a process involving protoplast formation, transformation of the
protoplasts, and regeneration
of the cell wall in a manner known per se. Suitable procedures for
transformation of
Aspergillus host cells are described in EP 238 023 and US 5,679,543. Suitable
methods for
transforming Fusarium species are described by Malardier et al., 1989, Gene
78: 147-156 and
WO 96/00787. Yeast may be transformed using the procedures described by Becker
and
Guarente, In Abelson, J.N. and Simon, M.L, editors, Guide to Yeast Ge>zetics
and Molecular
Biology, Methods i>z Eszzyuzology, Volume 194, pp 182-187, Academic Press,
Inca, New York;
Ito et al., 1983, Jounzal of Bacteriology 153: 163; and Hinnen et al., 1978,
Proceedi>zgs of the
NatiofZal Academy of Sciences USA 75: 1920.
When the polypeptide of the invention is to be in vivo glycosylated, the host
cell is
3o selected from a group of host cells capable of generating the desired
glycosylation of the
polypeptide. Thus, the host cell may advantageously be selected from a yeast
cell, insect cell,
or mammalian cell.
Examples of suitable yeast host cells include strains of Saccharomyces, e.g.
S. cerevisiae,
Schizosacclzaronzyces, Klyveronzyces, Pichia, such as P. pastoris or P.
methanolica,
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
47
Haf2senula, such as H. polymorpha or yarrowia. Of particular interest are
yeast glycosylation
mutant cells, e.g. derived from S. cereviciae, P. pastoris or Hahsenula spp.
(e.g. the S.
cereviciae glycosylation mutants ochl, ochi mnml or ochl mnml alga described
by Nagasu et
al. Yeast 8, 535-547, 1992 and Nakanisho-Shindo et al. J. Biol. Chem. 268,
26338-26345,
1993). Methods for transforming yeast cells with heterologous DNA and
producing
heterologous polypeptides therefrom are disclosed by Clontech Laboratories,
Inc, Palo Alto,
CA, USA (in the product protocol for the Yeastmaker~ Yeast Tranformation
System Kit), and
by Reeves et al., FEMS Microbiology Letters 99 (1992) 193-198, Manivasakam and
Schiestl,
Nucleic Acids Research, 1993, Vol. 21, No. 18, pp. 4414-4415 and Ganeva et
al., FEMS
to Microbiology Letters 121 (1994) 159-164.
Examples of suitable insect host cells include a Lepidoptora cell line, such
as
Spodoptera frugiperda (Sf9 or Sf21) or Trichoplusia ~i cells (High Five) (US
5,077,214).
Transformation of insect cells and production of heterologous polypeptides
therein may be
performed as described by Invitrogen, Carlsbad, CA, USA.
Examples of suitable mammalian host cells include Chinese hamster ovary (CHO)
cell lines,
(e.g. CHO-K1; ATCC CCL-61), Green Monkey cell lines (COS) (e.g. COS 1 (ATCC
CRL-
1650), COS 7 (ATCC CRL-1651)); mouse cells (e.g. NS/O), Baby Hamster Kidney
(BHK) cell
lines (e.g. ATCC CRL-1632 or ATCC CCL-10), and human cells (e.g. HEK 293 (ATCC
CRL-
1573)), as well as plant cells in tissue culture. Additional suitable cell
lines are known in the art
and available from public depositories such as the American Type Culture
Collection,
Rockville, Maryland. Of interest for the present purpose are a mammalian
glycosylation mutant
cell line, such as CHO-LEC1, CHOL-LEC2 or CHO-LEC18 (CHO-LEC1: Stanley et al.
Proc.
Natl. Acad. USA 72, 3323-3327, 1975 and Grossmann et al., J. Biol. Chem. 270,
29378-29385,
1995, CHO-LEC18: Raju et al. J. Biol. Chem. 270, 30294-30302, 1995).
Methods for introducing exogeneous DNA into mammalian host cells include
calcium
phosphate-mediated transfection, electroporation, DEAF-dextran mediated
transfection,
liposome-mediated transfection, viral vectors and the transfection method
described by Life
Technologies Ltd, Paisley, UK using Lipofectamin 2000. These methods are well
known in the
art and e.g. described by Ausbel et al. (eds.), 1996, Current Protocols in
Molecular Biology,
3o John Wiley & Sons, New York, USA. The cultivation of mammalian cells are
conducted
according to established methods, e.g. as disclosed in (Animal Cell
Biotechnology, Methods
and Protocols, Edited by Nigel Jenkins, 1999, Human Press Inc, Totowa, New
Jersey, USA
and Harrison MA and Rae 1F, General Techniques of Cell Culture, Cambridge
University Press
I997). .
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
48
In the production methods of the present invention, cells are cultivated in a
nutrient
medium suitable for production of the polypeptide using methods known in the
art. For
example, cells are cultivated by shake flask cultivation, small-scale or large-
scale fermentation
(including continuous, batch, fed-batch, or solid state fermentations) in
laboratory or industrial
fermenters performed in a suitable medium and under conditions allowing the
polypeptide to
be expressed and/or isolated. The cultivation takes place in a suitable
nutrient medium
comprising carbon and nitrogen sources and inorganic salts, using procedures
known in the art.
Suitable media are available from commercial suppliers or may be prepared
according to
published compositions (e.g., in catalogues of the American Type Culture
Collection). If the
to polypeptide is secreted into the nutrient medium, the polypeptide can be
recovered directly
from the medium. If the polypeptide is not secreted, it can be recovered from
cell lysates.
The resulting polypeptide may be recovered by methods known in the art. For
example, the polypeptide may be recovered from the nutrient medium by
conventional
procedures including, but not limited to, centrifugation, filtration,
extraction, spray drying,
evaporation, or precipitation.
The polypeptides may be purified by a variety of procedures known in the art
including,
but not limited to, chromatography (e.g., ion exchange, affinity, hydrophobic,
chromatofocusing, and size exclusion), electrophoretic procedures (e.g.,
preparative isoelectric
focusing), differential solubility (e.g., ammonium sulfate precipitation), SDS-
PAGE, or
2o extraction (see, e.g., Proteizz Purification, J-C Janson and Lars Ryden,
editors, VCH
Publishers, New York, 1989).
Other methods of the ifzveyztiofz
In accordance with a specific aspect a nucleotide sequence encoding the
polypeptide of the
invention is prepared by a method comprising
a) subjecting a nucleotide sequence encoding the polypeptide Pp to elongation
mutagenesis,
b) expressing the mutated nucleotide sequence obtained in step a) in a
suitable host cell,
optionally
c) conjugating polypeptides expressed in step b) to a second non-peptide
moiety,
d) selecting polypeptides of step b) or c) which comprises at least one
oligosaccharide moiety
and optionally second non-peptide moiety attached to the peptide addition part
of the
polypeptide, and
e) isolating a nucleotide sequence encoding the polypeptide selected in step
d).
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
49
In the present context the term "elongation mutagenesis" is intended to
indicate any
manner in which the nucleotide sequence encoding the parent polypeptide Pp can
be extended
to further encode the peptide addition. For instance, a nucleotide sequence
encoding a peptide
addition of a suitable length may be synthesized and fused to a nucleotide
sequence encoding
the polypeptide Pp: The resulting fused nucleotide sequence may then be
subjected to further
modification by any suitable method, e.g. one which involves gene shuffling,
other
recombination between nucleotide sequences, random mutagenesis, random
elongation
mutagenesis or any combination of these methods. Such methods are further
described in the
Methods section herein.
l0 The expression and optional conjugation steps are conducted as described in
further
detail elsewhere in the present application, and the selection step d) using
any suitable method
available in the art.
In one embodiment the above method further comprises screening polypeptides
resulting from step b) or c) for at least one improved property, in particular
any of those
improved properties listed herein, prior to the selection step, and wherein
the selection step d)
further comprises selecting polypeptides having such improved property.
Furthermore, in the above method the elongation mutagenesis can be conducted
so as to
enrich for codons encoding a glycosylation site and/or an amino acid residue
comprising an
attachment group for a second non-peptide moiety., in particular an ifz vivo
glycosylation site.
2o Still further, the above method can comprise subjecting the part of the
nucleotide
sequence encoding the polypeptide Pp of interest to mutagenesis to remove
and/or introduce
glycosylation sites) and/or amino acid residues) comprising an attachment
group for the
second non-peptide moiety. The nucleotide sequence may be subjected to any
type of
mutagenesis, e.g. any of those described herein. The mutagenesis of the
nucleotide sequence
encoding the polypeptide Pp of interest can be conducted prior to assembling
the sequence
with that encoding the peptide addition, concomitantly with or after any
mutagenesis of the
peptide addition part of the assembled nucleotide sequence.
In a further aspect, the invention relates to a method of producing a
glycosylated
polypeptide encoded by a nucleotide sequence of the invention prepared by the
above method,
wherein the nucleotide sequence encoding the polypeptide selected in step c)
is expressed in a
glycosylating host cell and the resulting glycosylated expressed polypeptide
is recovered.
In a still further aspect the invention relates to a method of improving one
or more
selected properties of a polypeptide Pp of interest, which method comprises
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
a) preparing a nucleotide sequence encoding a polypeptide comprising or
consisting
essentially of the primary structure
NH2-X-Pp-COOH,
wherein
X is a peptide addition comprising or contributing to a glycosylation site
and/or an attachment
group for a second non-peptide moiety that is capable of conferring the
selected improved
propertylies to the polypeptide Pp,
1o b) expressing the nucleotide sequence of a) in an suitable host cell,
optionally
c) conjugating the expressed polypeptide of b) to a second non-peptide moiety,
and
d) recovering the polypeptide resulting from step b) or c).
For instance, the polypeptide is any of those described herein. For instance
the
nucleotide sequence of step a) is prepared by subjecting a nucleotide sequence
encoding the
15 polypeptide Pp to elongation mutagenesis, e.g. to enrich for codons
encoding an amino acid
residue comprising or contributing to a glycosylation site and/or an
attachment group for a
second non-peptide moiety, in particular an irZ vivo glycosylation site. Also,
in the preparation
of the nucleotide. sequence of a), the part of the nucleotide sequence
encoding the polypeptide
Pp can be subjected to mutagenesis to remove and/or introduce glycosylation
sites) and/or
2o attachment groups) for a second non-peptide moiety.
The method according to this aspect can further comprise a screening step
(after step
c)), wherein the polypeptide resulting from step b) or c) is screened for one
or more improved
properties, in particular any of those improved properties which are described
hereinabove.
Usually, when a polypeptide has been selected in a screening step of a method
of the
25 invention the nucleotide sequence encoding the polypeptide is isolated and
used for expression
of larger amounts of the polypeptide. The amino acid sequence of the resulting
polypeptide is
determined and the polypeptide may be subjected to conjugation in a larger
scale.
Subsequently, the polypeptide is assayed with respect to the property to be
improved.
3o Uses of a poly~eptide of the invention
It will be understood that polypeptides of the invention can be used for a
variety of purposes,
depending on the type and nature of polypeptide. For instance, it is
contemplated that a
polypeptide of the invention prepared from a therapeutic polypeptide is useful
for the same
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
51
therapeutic purposes as the parent polypeptide, i.e. for the treatment of a
particular disease.
Accordingly, the polypeptide of the invention may be formulated into a
pharmaceutical
composition. Also, when the polypeptide of the invention is an i~c vivo
glycosylated
polypeptide which does not comprise any other type of non-peptide moiety, a
nucleotide
sequence encoding the polypeptide can be used in gene therapy in accordance
with established
principles. When the polypeptide Pp is an antigen the polypeptide of the
invention may be
provided in the form of a vaccine.
METHODS
Nucleotide sequef2ee modifieatioyz methods
For example, a peptide addition may be constructed from two or more nucleotide
sequences
encoding a polypeptide of interest with a peptide addition, the sequences
being sufficiently
homologous to allow recombination between the sequences, in particular in the
part thereof
encoding the peptide addition. The combination of nucleotide sequences or
sequence parts is
conveniently conducted by methods known in the art, for instance methods which
involve
homologous cross-over such as disclosed in US 5,093,257, or methods which
involve gene
shuffling, i.e., recombination between two or more homologous nucleotide
sequences resulting
2o in new nucleotide sequences having a number of nucleotide alterations when
compared to the
starting nucleotide sequences. In order for homology based nucleic acid
shuffling to take place
the relevant parts of the nucleotide sequences are preferably at least 50%
identical, such as at
least 60% identical, more preferably at least 70% identical, such as at least
80% identical. The
recombination can be performed in vitro or in vivo. Examples of suitable in
vitro gene
shuffling methods are disclosed by Stemmer et al (1994), Proc. Natl. Acad.
Sci. USA; vol. 91,
pp. 10747-10751; Stemmer (1994), Nature, vol. 370, pp. 389-391; Smith (1994),
Nature vol.
370, pp. 324-325; Zhao et al., Nat. Biotechnol. 1998, Mar; 16(3): 258-61; Zhao
H. and Arnold,
FB, Nucleic Acids Research, 1997, Vol. 25. No. 6 pp. 1307-1308; Shao et al.,
Nucleic Acids
Research 1998, Jan 15; 26(2): pp. 681-83; and WO 95/17413. Example of a
suitable in vivo
shuffling method is disclosed in WO 97/07205.
Furthermore, a peptide addition can be constructed by preparing a randomly
mutagenized library, conveniently prepared by subjecting a nucleotide sequence
encoding the
polypeptide of the invention or the peptide addition to random mutagenesis to
create a large
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
52
number of mutated nucleotide sequences. While the random mutagenesis can be
entirely
random, both with respect to where in the nucleotide sequence the mutagenesis
occurs and with
respect to the nature of mutagenesis, it is preferably conducted so as to
randomly mutate only
the part of the sequence that encode the peptide addition. Also, the random
mutagenesis can be
directed towards introducing certain types of amino acid residues, in
particular amino acid
residues containing an attachment group, at random into the polypeptide
molecule or at random
into peptide addition part thereof. Besides substitutions, random mutagenesis
can also cover
random introduction of insertions or deletions. Preferably, the insertions are
made in reading
frame, e.g., by performing multiple introduction of three nucleotides as
described by Hallet et
to al., Nucleic Acids Res. 1997, 25(9):1866-7 and Sondek and Shrotle, Proc
Natl. Acad. Sci USA
1992, 89(8):3581-5.
The random mutagenesis (either of the whole nucleotide sequence or more
preferably
the part thereof encoding the peptide addition) can be performed by any
suitable method. For
example, the random mutagenesis is performed using a suitable physical or
chemical
mutagenizing agent, a suitable oligonucleotide, PCR generated mutagenesis or
any
combination of these mutagenizing agentsand/or other methods according to
state of the art
technology, e.g. as disclosed in WO 97!07202.
Error prone PCR generated mutagenesis, e.g. as described by J.O. Deshler
(1992),
GATA 9(4): 103-106 and Leung et al., Technique (1989) Vol. 1, No. 1, pp. 11-
15, is
particularly useful for mutagenesis of longer peptide stretches (corresponding
to nucleotide
sequences containing more than 100 bp) or entire genes, and are preferably
performed under
conditions that increase the misincorporation of nucleotides.
Random mutagenesis based on doped or spiked oligonucleotides or by specific
sequence oligonucleotides, is of particular use for mutagenesis of the part of
the nucleotide
sequence encoding the peptide addition.
Random mutagenesis of the part of the nucleotide sequence encoding the peptide
addition can be performed using PCR generated mutagenesis, in which one or
more suitable
oligonucleotide primers flanking the area to be mutagenized are used. In
addition, doping or
spiking with oligonucleotides can be used to introduce mutations so as to
remove or introduce
3o attachment groups for the relevant non-peptide moiety. State of the art
knowledge and
computer programs (e.g. as described by Siderovski DP and Mak TW, Comput.
Biol. Med.
(1993) Vol. 23, No. 6, pp. 463-474 and Jensen et al. Nucleic Acids Research,
1998, Vol. 26,
No. 3) can be used for calculating the most optimal nucleotide mixture for a
given amino acid
preference. The oligonucleotides can be incorporated into the nucleotide
sequence encoding the
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
53
peptide addition by any published technique using e.g. PCR, LCR or any DNA
polymerase or
ligase.
According to a convenient PCR method the nucleotide sequence encoding the
polypeptide of the invention and in particular the peptide addition thereof is
used as a template
and, e.g., doped or specific oligonucleotides are used as primers. In
addition, cloning primers
localized outside the targetted region can be used. The resulting PCR product
can either
directly be cloned into an appropriate expression vector or gel purified and
amplified in a
second PCR reaction using the cloning primers and cloned into an appropriate
expression
vector.
1o In addition to the random mutagenesis methods described herein, it is
occasionally
useful to employ site specific mutagenesis techniques to modify one or more
selected amino
acids in the peptide addition, in particular to optimise the peptide addition
with respect to the
number of attachment groups.
Furthermore, random elongation mutagenesis as described by Matsuura et al, op
cit can
be used to construct a nucleotide sequence encoding a polypeptide having a C-
terminal peptide
addition. Construction of a nucleotide sequence encoding the polypeptide of
the invention
having an N-terminal peptide addition can be constructed in an analogous way.
Also, the methods disclosed in WO 97/04079, the contents of which are
incorporated
herein by reference, can be used for constructing a nucleotide sequence
encoding the
2o polypeptide of the invention.
The nucleotide sequences) or nucleotide sequence regions) to be mutagenized is
typically present on a suitable vector such as a plasmid or a bacteriophage,
which as such is
incubated with or otherwise exposed to the mutagenizing agent. The nucleotide
sequences) to
be mutagenized can also be present in a host cell either by being integrated
into the genome of
said cell or by being present on a vector harboured in the cell.
Alternatively, the nucleotide
sequence to.be mutagenized is in isolated form. The nucleotide sequence is
preferably a DNA
sequence such as a cDNA, genomic DNA or synthetic DNA sequence.
Subsequent to the incubation with or exposure to the mutagenizing agent, the
mutated
nucleotide sequence, normally in amplified form, is expressed by culturing a
suitable host cell
3o carrying the nucleotide sequence under conditions allowing expression to
take place. The host
cell used for this purpose is one, which has been transformed with the mutated
nucleotide
sequence(s), optionally present on a vector, or one which carried the
nucleotide sequence
during the mutagenesis, or any kind of gene library.
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
54
Design of peptide addition
One example of a useful guide for designing an N-terminal peptide addition
containing N-
glycosylation sites is characterized by the following formula:
Xl(NX2[T/S])X3(NX2[T/S])n~.-I'1~
wherein each of Xl, X3 and X4 independently is absent or 1, 2, 3 or 4 amino
acid residues of
any type, X2 a single amino acid residue of any type except for proline, n any
integer between 0
and 6, [T/S] a threonine or serine residue, preferably a threonine residue,
and N and Pp has the
meaning defined elsewhere herein. It has been found that sometimes the nature
of the amino
acid residue occupying position -1 to -4 relative to the N-residue of an N-
glycosylation site
to may be important for the degree to which said N-glycosylation site is used.
Accordingly, Xl,
X3, and X4 may be chosen so as to obtain an increased utilization of the
relevant site (as
determined by a trial and error type of experiment). In a first step about 10
different muteins
are made that has the above formula. For instance, the about 10 muteins are
designed on the
basis that each of Xl, X3 and X4independently is 1 or 2 alanine residues or is
absent, Z any
integer between 0 and 5, [T/S] threonine, and Alanine. Based on, e.g., in
vitro bioactivity and
half-life results obtained with these muteins (or any other relevant
property), optimal
numbers) of amino acids and glycosylation(s) can be determined and new muteins
can be
constructed based on this information. The process is repeated until an
optimal glycosylated
polypeptide is obtained.
2o Alternatively, random mutagenesis may be used for creating N-terminally
extended
polypeptides. For instance, a random mutagenized library is made on the basis
of the above
formula. Doped oligonucleotides are synthesized coding for one amino acid
residue in position
B (the amino acid residue being different from proline), each of Xl, X3, and
X4 independently is
0, 1 or 2 amino acid residues of any type, n is 2 and T is threonine and used
for constructing
the random mutagenized library.
One example of a useful guide for designing an N-terminal peptide addition
containing
a PEGylation attachment group is characterized by the following formula using
a lysine residue
as an example of a PEGylation site. It will be understood that peptide
additions with other
attachment groups can be designed in an analogous way.
Y1(K)Y2(K)nY3-I'P
wherein each of Yl, Y2 and Y3 independently is 0, 1, 2, 3 or 4 amino acid
residues of any type
except lysine, n an integer between 0 and 6, K lysine, and Pp is as defined
elsewhere herein.
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
In a first step about 10 different muteins are made that has the above
formula. For
instance, the about 10 muteins are designed on the basis that each of Yl, YZ
and Y3
independently is 1 or 2 alanine residues or is absent, n any integer between 0
and 5. The
muteins are then PEGylated withl0 kDa PEG (e.g. using mPEG-SPA). Based on,
e.g., izz vitro
5 bioactivity and half-life results obtained with these muteins (or any other
relevant property),
optimal numbers) of amino acids and PEGylation sites can be determined and new
muteins
can be constructed based on this information. The process is repeated until an
optimal
PEGylated polypeptide is obtained.
Alternatively, random mutagenesis may be performed by making a random
l0 mutagenized library based on the above formula. Doped oligonucleotides are
synthesized
coding for one amino acid residue in position Yl, Y2° and/or Y3
independently is 0, 1 or 2
amino acid residues of any type, and n is 2 and used for constructing the
random mutagenized
library.
15 Glucocerebrosidase (GCB) Activity Assay using PNP-glucopyranoside substrate
The enzymatic activity of recombinant GCB is measured using p-nitrophenyl-~3-D-
glucopyranoside (PNP-Glu) as a substrate. Hydrolysis of the PNP-Glu substrate
generates p-
nitrophenyl, which can be quantified by measuring absorption at 405 nm using a
spectrophotometer, as previously described (Friedmann et al., 1999, Blood 93;
2807-2816).
20 The assay is carried out under conditions which partially inhibit non-GCB
glucosidase
activities, such conditions being achieved by using a phosphate/citrate buffer
pH=5.5, 0.25 %
Triton X-100 and 0.25 % taurocholate.
The assay is run in a final volume of 200 ~l, containing GCB Activity Assay
Buffer and
4 mM PNP-Glu. The enzymatic hydrolysis is initiated by adding GCB and the
reaction is
25 allowed to proceed for 1 hour at 37°C before being stopped by adding
50 ~l 1 M NaOH and
measuring absorption at 405 nm. A reference standard curve of p-nitrophenyl,
assayed in
parallel, is used to quantify concentrations of GCB in samples to be tested.
Irt vitro uptake and stability of GCB polypeptide in fzzacrophages
30 The murine monocyte/macrophage cells line, J774E (Mukhopadhyay and Stahl,
Arch
Biochem Biophys 1995 Dec 1;324(1):78-84 and Diment et al., JLeukoc Biol 1987
Nov;42(5):485-90) is used to study the uptake and stability of GCB
polypeptides. Cells are
grown in alpha-MEM (supplemented with 10 % fetal calf serum, 1X Pen/Strep, and
60 ~.M 6-
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
56
thioguanine), seeded (200,000 cells pr. well) in the above-mentioned media
containing 10 p,M
conditol B epoxide, CBE (an irreversible GCB inhibitor) and incubated for 24
hr at 37°C.
Before starting the uptake assay, cells are washed in 0.5 ml HBSS (Hanks
balanced salt
solution). The uptake is done in a 200 ~l volume, containing the appropriate
concentration of
GCB polypeptide (a dosis response curve is made with GCB concentrations in the
range of 25
400 mU/ml). As a control, yeast mannan (final concentration 1.4 mg/ml) is
added to inhibit the
uptake through the macrophage mannose receptor. The cells are incubated for 1
hr at 37°C and
washed three times with 0.5 ml cold HBSS.
To measure the amount of GCB taken up by the J774E cells, cells are lyzed in
200 p1
1o GCB Activity Assay Buffer with 4 mM PMP-Glu and incubated for 1 hr at
37°C. Then, the
hydrolysis is stopped by addition of 50 ~.1 1M NaOH and OD405 is measured. The
data are
analysed by non-linear regression using GraphPad Prizm 2.0 (GraphPad Software,
San Diego,
CA)
To study the stability of GCB polypeptides in J774E cells, CBE treated cells
are
incubated with 400 mU/mI GCB for 1 hr at 37°C. Then, cells are washed 3
times in HBSS to
remove extracellular GCB and incubated in HBSS. A time-course study is done by
lyzing the
cells after 30 min, 1 hr, 2 hr, 3hr, 4 hr, and 5 hr in 200 ~l GCB Activity
Assay Buffer with
4mM PNP-Glu and incubating the samples for 1 hr at 37°C before stopping
the hydrolysis with
50 ~1 1 M NaOH and measuring OD405. The data are analysed by non-linear
regression using
2o GraphPad Prizm 2.0 (GraphPad Software, San Diego, CA).
Site-directed muta~enesis
Constructions of site-directed mutations were performed using PCR with
oligonucleotides
containing the desired amino acid exchanges or additions (e.g. to introduce
glycosylation sites).
The resulting PCR fragment was cloned into the GCB expression vector using
approparite
restriction enzymes and subsequently DNA sequenced in order to confirm that
the construct
contained the desired exchanges.
3o MATERIALS
GCB Activity Assay Buffer:
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
57
120 mM phosphate/citrate buffer, pH=5.5, 1 mM EDTA, pH=8.0, 0.25 % Triton X-
100, 0.25
% taurocholate, 4 mM ~3-mercaptoethanol
pGC-12 vector
pVL1392 (Pharmingen, USA) with GCB wt cDNA sequence (SEQ m NO 2) inserted
between
EcoRV and XbaI.
Table 1
Sequence of primers used for cloning the wt GCB coding region and inserting
signal peptides
into the pGCBmat plasmid as described in Example 1.
5049 (WT-sp-BgllI): 5'-CGCAGATCTGATGGCTGGCAGCCTCACAGGATTGC-3'
5050 (WT-stop-EcoRl): 5'-CCGGAATTCCCATCACTGGCGACGCCACAGGTAGGTG-3'
5051 (WT-mature-Sacl): 5'-ACGCGAGCTCGCCCCTGCATCCCTAAAAGCTTCGG-3'
5052 (SPegt-NheIlSacI-as): 5'-
GCGTTGACGGCAGTCAGAGTTGACAGAAGGGCCAGCCAGCAAAGGATAGTCATG-
3'
S053 (SPegt-NheI/SacI-s): 5'-
CTAGCATGACTATCCTTTGCTGGCTGGCCCTTCTGTCAACTCTGACTGCCGTCAACG
2o CAGCT-3'
5054 (SPegt-Nhei/SacI-as): 5'-
CCTGCTACTGCTCCCAGCAGCAGTGAAAGAGTCCAAAGTGGCAGCATG-3'
5055 (SPegt-NheI/SacI-s): 5'-
CTAGCATGCTGCCACTTTGGACTCTTTCACTGCTGCTGGGAGCAGTAGCAGGAGCT
-3'
Cerezynae was kindly provided by Dr. E. Beutler, Scripps Institute, CA, USA.
J774E was kindly provided by G. Grabowski, Cincinnati, Ohio, US
EXAMPLE 1
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
58
PRODUCTION OF WT GCB
Cloning_and Expression in Insect Cells
A human fibroblast cDNA library was obtained from Clontech (Human fibroblast
skin cDNA
cloned in lambda-gtll, cat# HL1052b). Lambda DNA was prepared from the library
by
standard methods and used as a template in a PCR reaction with either 5049 and
5050 as
primer (amplifies the GCB coding region with the human signal peptide from the
second ATG)
or S050 and 5051 as primer (amplifies the mature part of the GCB coding
region) (see Table
1 in the Materials section).
to The PCR products were reamplified with the same primers and agarose gel
purified.
Subsequently the 5049/50 PCR product was digested with BglII and EcoRI and
cloned into
the pBlueBac 4.5 vector (InVitrogenInvitrogen, Carlsbad, CA, USA, Carlsbad,
CA, USA)
digested with BamHI and EcoRI. Sequencing confirmed that the insert is
identical to the
wtGCB sequence as given in SEQ ID NO 2. The resulting plasmid was used for
infection of
insect cells with the GCB being partly secreted from the cells due to the
human signal sequence
as described in Martin et al., DNA 7, pp. 99-106, 1988. The 5050/51 PCR
product was
digested with SacI and EcoRI and cloned into the pBlueBac 4.5 vector
(InVitrogenInvitrogen,
Carlsbad, CA, USA) digested with the same enzymes resulting in the pGCBmat
plasmid. Two
different signal sequences were inserted upstream of the mature GCB codons in
order to
2o increase the secreted amount of enzyme. The baculovirus ecdysteroid
UDPglucosyltransferase
(egt) signal sequence (Murphy et al., Protein Expression and Purification 4,
349-357, 1993)
was inserted by annealling 5052 and 5053 (Table 1) and the human pancreatic
lipase signal
sequence (Lowe et al., J. Biol. Chem. 264, 20042, 1989) was inserted by
annealling 5054 and
5055 (Table 1) and cloning them into the NheI and SacI digested pGCBmat
plasmid. Infection
of Spodoptera frugiperda (Sf9) cells of the resulting plasmid was done
according to the
protocols from InVitrogenInvitrogen, Carlsbad, CA, USA.
Purificatio>z of GCB polypeptides produced in insect cells
Polypeptides with GCB activity were purified as described in US 5,236,838,
with some
modifications. Cells were removed from the culture medium by centrifugation
(10 min at 4000
rpm in a Sorvall RCSC centrifuge) and the supernatant microfiltrated using a
0.22 ~m filter
prior to purification. DTT was added to 1 mM and the culture supernatant was
ultrafiltrated to
approximately 1/10 of the starting volume using a Vivaflow 200 system
(Vivascience). The
concentrated media was centrifuged to remove possible aggregates before
application on a
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
59
Toyopearl Buty1650C resin (TosoHaas) previously equilibrated in 50 mM sodium
citrate, 20 %
(v/v) ethylene glycol, 1 mM DTT, pH 5Ø This chromatographic step was
performed at room
temperature. The resin was washed with at least 3 column volumes of 50 mM
sodium citrate,
20 % (v/v) ethylene glycol, 1 mM DTT, pH 5.0 (until the absorbance at 280 nm
reaches
baseline level) and GCB was eluted with a linear gradient from 0% to 100% 50
mM sodium
citrate, 80% (v/v) ethylene glycol, 1 mM DTT, pH 5Ø Fractions were collected
and assayed
for GCB activity using the GCB Activity Assay. Usually, wt GCB starts to elute
at approx.
70% (v/v) ethylene glycol. a
The subsequent purification was done by either of the following two methods.
#2
method results in GCB of a higher purity.
Method #1
GCB enriched fractions from the first process step were pooled and diluted
approx. 4 times
with a buffer containing 50 mM sodium citrate, 5 mM DTT, pH 5.0 to reduce the
ethylene
glycol content to 20% (or lower). In the second HIC purification step the
diluted and partially
purified GCB was applied on a Toyopearl phenyl resin (TosoHaas) equilibrated
in 50 mM
sodium citrate, I mM DTT, pH 5.0 (Buffer A) before use. After application, the
resin was
washed with at least 3 column volumes of 50 mM sodium citrate, pH 5 (until the
absorbance at
280 nm reaches baseline level) and GCB was then eluted with a linear ethanol
gradient from
0% to 100% buffer B (50 mM sodium citrate, 50% (v/v) ethanol, 1 mM DTT, pH
5.0). Highly
purified fractions of GCB (wildtype >_ 95% pure), identified using the GCB
Activity Assay,
start to elute at approx. 40% ethanol. The purified GCB bulls product was
dialyzed against 50
mM sodium citrate, 0.2 M mannitol, 0.09% tween80, pH 6.1 to retain the GCB
activity upon
subsequent storage at 4-8°C or at -80°C.
Method #2
GCB enriched fractions eluted from the Toyopearl buty1650C resin were pooled
and applied at
4°C on a SP sepharose resin (Amersham Pharmacia Biotech) previously
equilibrated in 25
mM sodium citrate, 1 mM DTT, 10% ethylene glycol, pH 5Ø After application,
the resin was
3o washed with 25 mM sodium citrate, 1 mM DTT, 10% ethylene glycol, pH 5.0
(until absorption
at 280 nm reached baseline level) and GCB was then eluted with a linear
gradient from 0
to100% 0.25 M sodium citrate, 1 mM DTT, 10% ethylene glycol, pH 5Ø GCB
begins to elute
around 0.15 M sodium citrate. Fractions containing GCB were pooled and applied
at room
temperature onto a Phenyl sepharose High Performance (Pharmacia Biotech)
previously
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
equilibrated in 25 mM sodium citrate 1 mM DTT, pH 5Ø After application, the
resin was
washed with 25 mM sodium citrate 1 mM DTT, pH 5.0 until absorption at 280 nm
reached
baseline level, and GCB was then eluted with a linear ethanol gradient from 0
to100% 25 mM
sodium citrate 1 mM DTT 50 % ethanol pH 5Ø GCB typically elutes around 35 %
ethanol.
5 The purified GCB bulk product was dialyzed against either 50 mM sodium
citrate, 1 mM DTT,
pH 5.0 or 50 mM sodium citrate, 0.2 M mannitol, 1 mM DTT, pH 6.1 to retain the
GCB
activity upon subsequent storage. The purified GCB was concentrated and
sterilfiltrered before
storage at 4 - 8°C or at -80°C. Typically, GCB purified by this
method is >95% pure.
EXAMPLE 2
Preparatiofi of GCB witla N tenninal peptide additions usifZg a site-directed
or randoy2
mutagefaesis approach
Nucleotide sequences encoding the following N-terminal peptide additions were
added to the
nucleotide sequence shown in SEQ ID NO 2 encoding wtGCB: (A-4)+(N-3)+(I-2)+(T-
1)
(representing an extension to the N-terminal of the amino acid sequence shown
in SEQ ID NO
1 with the amino acid residues ANIT), and (A-7)+(S-6)+(P-5)+(I-4)+(N-3)+(A-
2)+(T-1)
(ASPINAT).
A nucleotide sequence encoding the N-terminal peptide addition (A-4)+(N-3)+(I-
2)+(T-1) was prepared by PCR using the following conditions:
PCR 1:
Template: 10 ng pBlueBac5 with wt GCB cDNA sequence
primer 5060: 5'-CAGCTGGCCATGGGTACCCGG-3' and
primer 5085:
5'-TGGGCATCAGGTGCCAACATTACAGCCCGCCCCTGCATCCCTAAAAGC-3'
BIO-X-ACTS DNA polymerase (Bioline, London, U.K.)
lxOptiBuffer~ (Bioline, London, U.K.)
30 cycles of 96°C 30s, 55°C 30s, 72°C 1 min
PCR 2:
Template: 10 ng pBlueBac5 with wt GCB,
Baculo virus forward primer: 5'-TTTACTGTTTTCGTAACAGTTTTG-3' and
PrimerS086:
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
61
5'-GCAGGGGCGGGCTGTAATGTTGGCACCTGATGCCCACGACACTGCCTG-3'
BIO-X-ACTS DNA polymerase (Bioline, London, U.K.)
lxOptiBuffer~ (Bioline, London, U.K.)
30 cycles of 96°C 30s, 55°C 30s, 72°C 1 min
PCR 3:
3 ~1 of agarose gel purified PCR1 and PCR2 products (app. 10 ng)
Baculo virus forward primer: 5'-TTTACTGTTTTCGTAACAGTTTTG-3' and primer 5060.
BIO-X-ACTS DNA polymerase (Bioline, London, U.K.)
lxOptiBuffer~ (Bioline, London, U.K.)
30 cycles of 96°C 30s, 55°C 30s, 72°C 1 min
PCR 3 was agarose gel purified and digested with NheI and NcoI and cloned into
pBluebac4.5+wtGCB digested with NheI and NcoI.
After confirmation of the correct mutations by DNA sequencing the plasmid was
transfected into insect cells using the Bac-N-Blues transfection lcit from
Invitrogen, Carlsbad,
CA, USA. Expression of the muteins was tested by western blotting and by
activity
measurement of the muteins using the GCB Activity Assay.
Enzymatic activity of wtGCB (SEQ ID NO 1) expressed in the expression vector
pVL1392 in insect cells (Sf9) using an analogous method to that described in
Example 1 gave
13 units/L, while the N-terminal peptide addition ASPINAT gave 28.5 units/L.
Construction of libraries of GCB with N-terminal p~tide addition
Using random mutagenesis two different libraries were constructed on the basis
of GCB
polypeptides with an N-terminal extension - library A with an N-terminal
extension encoding
the following amino acid sequence AXNXTXNXTXNXT, and library B with an N-
terminal
extension encoding ANXTNXTNXT.
Primers for library A were designed:
50167: 5'-
GTGTCGTGGGCATCAGGTGCCNN(G/C)AA(C/T)(T/A/G)N(G/C)AC(A/T/C)(T/A/G)N(G/
3o C)AA(C/T)(T/A/G)N(G/C)AC(A/T/C)(T/A/G)N(G/C)AA(C/T)(T/A/G)N(G/C)AC(A/T/C)GC
CCGCCCCTGCATCCCTAAAAGC
SO 168 : 5'-GGCACCTGATGCCCACGACACTGCCTG
Primers for library B were designed using trinucleotides in the random
positions.
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
62
X is a mixture of trinucleotide codons for all natural amino acid residues,
except proline. The
trinucleotide codons used were the same as described by Kayushin et al.,
Nucleic Acids
Research, 24, 3748-3755, 1996.
50165: 5'-
CGTGGGCATCAGGTGCCAAC(X)AC(A/T/C)AA(C/T)(X)AC(A/T/C)AA(C/T)(X)AC(A/T/
C)GCCCGCCCCTGCATCCCTAAAAGC
50166:5'-GTTGGCACCTGATGCCCACGACACTGCCTG
l0 For both libraries:
5060 and pBRlO: 5'- TTT ACT GTT TTC GTA ACA GTT TTG
In all PCR reactions BIO-X-ACTS DNA polymerase (Bioline, London, U.K.) and
1*Optibuffer~ (Bioline, London, U.K.) were used. The PCR conditions were 30
cycles of
94°C 30s, 55°C 1 min, and 72°C 1 min.
Templates and primers used for preparing a nucleotide sequence encoding the N-
terminal
extension by the above PCR were as follows:
PCR 1A:
Template: pGC 12
Primers: 5060 + 50167
PCR 1B:
Template: pGCl2
Primers: S060 + 50165
PCR 2A:
Template: pGCl2
Primers: 50168 + pBRlO
PCR 2B:
Template: pGCl2
Primers: 50166 + pBRlO
PCR 3A:
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
63
Template: 1 ~1 of agarose gel purified PCR 1A and 2A products
Primers: 5060 + pBRlO
PCR 3B:
Template: 1 ~,1 of agarose gel purified PCR 1B and 2B products
Primers: 5060 + pBRlO
PCR 3A and 3B were agarose gel purified and digested with NheI and NcoI and
ligated into
pGC-12 digested with NheI and NcoI. The ligation mixture is transformed into
competent E.
to coli. The diversity of the library was examined by DNA sequencing of
different E. coli clones
and gave rise to the following amino acid sequences:
Library A:
l: AFNXTLNKTWN(F/L)T
2: TMNNTWNWTWNWT
3: -EXT wt
4: ALNSTGNLTVDGT
5: ASNSTFNLTENLT
6: TRNVTINCTUNST
7: -EXT wt
8: ALNWTYNGTKNVT
9: AANWTVNF"TGNFT
10: -EXT wt
11: AXNXTVNSTUNVT
12: ANNFTFNGTLNLT
13: AGNWTANVTVNVT
14: AGNSTSNVTGNWT
15: AVNST1~~VIHAIPP ( 1 deletion - nonsens)
16: AGNGTVNGTINGT
17: AVNSTGNXTGNWT
18: AGNGTLTNGTSNLT
19: -EXT wt
20: AMNSTKNSTLNIT
21: AFNYTSKNST
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
64
22: -EXT wt
23: AVNATMNWTANGT
24: ASNSTNNGTLNAT
25: ARNKTKNFTINLT
26: APNITUNDTVNMT
27: AQNKTFNFTMNCT
28: ALNVTWNCTLNLT
29: ALNTTWTNLT
to Library B:
1: ANTTNFTNET
2: ANWTNRTNCT
3: ANWTNFTNWT
4: PTGLIGTNFT
5: ANWTNKTNFT
6: ANNTNLTNAT
7: ANYTNWTNFT
8: ANTTNQTNDT
9: - EXT wt
10: ANRTNWTNTT
11: PTATNHTNST
12: - EXT wt
13: ANWTNQTNQT
14: ANWTNWTNAT
15: ANFTNKTNMT
16: ANHTNETNAT
17: AN(C/W)TNFTNET
18: ANLDKLHKUH (insertion - nonsens)
19: ANCFTNQTNFT
20: ANWTNWTNEWT
21: ANCTNWTNCT
22: - EXT wt
23: - EXT wt
24: CHPYNWTNWT
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
25: ANETNYTNET
26: ANWTNWT
27: AKPYKSYKFY (insertion - nonsens)
28: ANITNKTNWT
5 29: ANWTNMTNIT
30: ANNTNRTNFT
31: ANWTNWTNWT
32: ANWRTNHTNKT
33: - EXT wt
l0 34: ANQTNITNWT
Library B was transfected into insect cells using the Bac-N-Blues transfection
kit
from Invitrogen, Carlsbad, CA, USA. First, 96 plaques from Library B were
picked and tested
by activity measurement (GCB Activity Assay). Plaques were selected as
follows: 3 with high
15 activity, 3 with medium activity and 3 with low or no activity, and virus
was purified for DNA
sequencing resulting in the following amino acid sequences:
High activity:
1-1: Mixed sequence
1-2: ANFTNVATNQT
20 1-3: (A)(N)TTXLTN(K)T
Medium activity:
2-1: ANKTN(S/C)TNIT
2-2: Mixed sequence
25 2-3: ANWTNCTN(I)T
Low activity:
3-1: ANWTN(F/L)TNWT
3-2: CQLDURSTNET
30 3-3: No sequence
From both libraries 96 plaques were picked and tested by activity measurement
(GCB Activity
Assay). From each library 6 plaques with high activity were selected and virus
were purified
for DNA sequencing. The amino acid sequence encoded by the different clones
were:
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
66
Library A:
l: Mixed sequence
2: Mixed sequence
3: Mixed sequence
4: WT
5: ANNTNYTNWT
6: ANNTNYTNWT
Library B:
1: AANDTUNWTVNCT
2: ATNITLNYTANTT
3: WT
4: AANSTGNITINGT
5: AVNWTSNDTSNST
GCB polypeptides of the invention were tested for various properties,
including GCB activity,
stability in J774E cells and uptake in J774E cells. Unless otherwise stated
the properties were
tested by use of the methods described in the Methods section herein.
2o In the below table the GCB activity of various GCB polypeptides of the
invention is
listed together with the activity of the positives from Library A and B after
plaque purification.
Table 2
Activity
after
# Glycosylation
Plaque
Isolation
Plasmid VectorMutations sires
introduced
(u~L)
pGC-1 PBlueBac4.5 0 6
Wt
pGC-6 pBlueBac4.5 N-termANIT 1 3
~
pGC-12 pVL1392Wt 0 13
pGC-13 pVL1392N-termASPINAT 1 29
pGC-36 pVL1392N-term: ASPINATSPINAT 2 16
pGC-38 pVL1392N-term: ASPINAT,K194N, 3 16
K321N
pGC-40 pVL1392N-term: ASPINAT,T132N, 3 3.5
K293N, V295T
pGC-47 pVL1392N-term: AGNGTVNGTINGT 3 30
pGC-48 pVL1392N-term: ASNSTNNGTLNAT 3 36
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
67
pGC-56 pVL1392N-term: ASPINATSPINAT, K194N,4 24
K321N
pGC-57 pVL1392N-term: ASPINAT, T132N, K194N,4 20
K321N
pGC-58 pVL1392N-term: ASPINAT, T132N, K194N3 10
pGC-60 pVL1392N-term:ANNTNYTNWT 3 P2: 14
pGC-61 pVL1392N-term: ATNITLNYTANTT 3 P2: 38
pGC-62 pVL1392N-term: AANSTGNITINGT 3 P2: 35
pGC-63 pVL1392N-term: AVNWTSNDTSNST 3 P2: 66
pGC-68 pVL1392AN N-term extension + R2T 1 37
Table 2: The plasmid column shows the number of the GCB polypeptide. The
vector column
shows the plasmid vector used for expression of the polypeptide. The mutation
column shows
the amino acid exchanges of the GCB polypeptide_ N-terminal extentions are
described as N-
term followed by the amino acid residues that makes up the extension. The
Activity column
gives the units per liter of GCB activity measured by the GCB Activity Assay
on the
supernatant from Sf9 insect cells infected with one single plaque and grown in
3 ml of media in
a 6-well plate. Those labelled with P2 are activity measured of supernatant
from virus infection
cells grown in 15 ml T75 flasks.
Table 3
GCB polypeptide Vmax Km
VJildtype 0.57 87.7
Cerezyme 0.52 91.9
pGC36 0.60 70.6
pGC38 0.48 44.0
pGC56 0.39 32.2
pGC60 0.57 79.1
pGC61 0.74 100.5
pGC62 0.86 110.8
pGC63 0.51 83.1
Table 3: Calculated Vmax and Km for uptake in the J774E macrophage cell line
of the
different GCB polypeptides. Vmax and Km was calculated from dosis response
curve (See Fig.
1). The uptake of selected GCB polypeptides are shown in Figure 1
As can be seen from table 3, an increase in Vm~ was observed for the N-
terminally
extended GCB polypeptides (pGC60, pGC6l, and pGC62).
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
68
EXAMPLE 3
Glycosylation of GCB polypeptides of the ifzvezztiozz expressed in izzsect
cells
MALDI-TOF mass spectrometry was used to investigate the amount of carbohydrate
attached to GCB polypeptides expressed in Sf9 cells.
The 6 GCB polypeptide variants investigated all contained additional potential
N-
glycosylation sites compared to wtGCB.
WtGCB contains 5 potential N-glycosylation sites of which only 4 are used.
The 6 GCB polypeptide variants were:
GC-36: ASPINATSPINAT-GCB,
GC-38: ASPINAT-GCB(K194N,K321N),
GC-60: ANNTNYTNWT-GCB,
GC-61: ATNITLNYTANTT-GCB,
GC-62: AANSTGNITINGT-GCB, and
GC-63: AVNWTSNDTSNST-GCB.
WtGCB:
The theoretical peptide mass of wtGCB is 55 591 Da. WtGCB has 5 potential N-
glycosylation
sites of which only 4 are used. As the two most common N-glycan structures on
recombinant
proteins expressed in Sf9 cells are Man3GlcNAc2Fuc and Man3GlcNAc2 having
masses of
1038.38 Da and 892.31 Da, respectively, the expected mass of wtGCB carrying 4
N-glycans is
between 59 159 Da and 59 743 Da.
MALDI-TOF mass spectrometry of wtGCB shows the broad peak typical of
glycoproteins with a peak mass of 59.3 kDa in accordance with the expected
mass of wtGCB
carrying 4 N-glycans.
GC-36 (ASPINATSPINAT-GCB):
The theoretical peptide mass of GC-36 is 56 829 Da. The N-terminal extension
contains two
additional potential glycosylation sites at N5 and N11 compared to wtGCB.
Assuming that the
wtGCB part of the variant is glycosylated like wtGCB, the variant has 6
potential N-
glycosylation sites.
As the two most common N-glycan structures on recombinant proteins expressed
in Sf9
cells are Man3GlcNAc2Fuc and Man3GlcNAc2 having masses of 1038.38 Da and
892.31 Da,
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
69
respectively, the expected mass of GC-36 carrying 4 N-glycans is between 60
397 Da and 60
981 Da, the expected mass of GC-36 carrying 5 N-glycans is between 61 289 Da
and 62 019
Da, and the expected mass of GC-36 carrying 6 N-glycans is between 62 181 Da
and 63 057
Da.
MALDI-TOF mass spectrometry of GC-36 shows a rather broad peak with a peak
mass
between 61.5 kDa and 62.9 kDa in accordance with the expected mass of GC-36
carrying
either 5 or 6 N-glycans.
N-terminal amino acid sequence analysis of GC-36 showed that N5 is completely
glycosylated while N11 is partially glycosylated in complete agreement with
the result
obtained using mass spectrometry.
GC-38 (ASPINAT-GCB(K194N,K321N)):
The theoretical peptide mass of GC-38 is 56 217 Da. The N-terminal extension
contains one
additional potential glycosylation sites at N5 compared to wtGCB. In addition,
the
substitutions of Lys194 and Lys321 with Asn-residues introduce two additional
potential N-
glycosylation sites. Assuming that the wtGCB part of the variant is
glycosylated like wtGCB,
the variant has 7 potential N-glycosylation sites.
Based on the same considerations as those used for GC-36, the expected mass of
GC-38
carrying 4 N-glycans is between 59 785 Da and 60 369 Da, the expected mass of
GC-38
carrying 5 N-glycans is between 60 677 Da and 61 407 Da, the expected mass of
GC-38
carrying 6 N-glycans is between 61 569 Da and 62 445 Da, and the expected mass
of GC-38
carrying 7 N-glycans is between 62 461 Da and 63 483 Da.
MALDI-TOF mass spectrometry of GC-38 shows a major peak with a peak mass of
63.1 kDa in accordance with the expected mass of GC-38 carrying 7 N-glycans.
In addition, a
minor peak with a peak mass of 62.3 kDa is seen which corresponds to GC-38
carrying 6 N-
glycans.
N-terminal amino acid sequence analysis of GC-38 showed that N5 is completely
glycosylated.
GC-60 (ANNTNYTNWT-GCB):
The theoretical peptide mass of GC-60 is 56 770 Da. The N-terminal extension
contains three
additional potential glycosylation sites at N2, N5 and N8 compared to wtGCB.
Assuming that
the wtGCB part of the variant is glycosylated like wtGCB, the variant has 7
potential N-
glycosylation sites.
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
Based on the same considerations as those used for GC-36 the expected mass of
GC-60
carrying 4 N-glycans is between 60 338 Da and 60 922 Da, the expected mass of
GC-60
carrying 5 N-glycans is between 61230 Da and 61 960 Da, the expected mass of
GC-60
carrying 6 N-glycans is between 62122 Da and 62 998 Da, and the expected mass
of GC-60
carrying 7 N-glycans is between 63 014 Da and 64 036 Da.
MALDI-TOF mass spectrometry of GC-60 shows two broad peaks with peak masses of
61.9 kDa and 62.8 kDa in accordance with the expected mass of GC-60 carrying
either 5 or 6
N-glycans.
N-terminal amino acid sequence analysis of GC-60 showed that N2 is mainly
glycosylated, N5 is completely glycosylated while N8 is only seldom
glycosylated in
acceptable agreement with the result obtained using mass spectrometry.
GC-61 (ATNITLNYTANTT-GCB):
The theoretical peptide mass of GC-61 is 56 970 Da. The N-terminal extension
contains three
additional potential glycosylation sites at N3, N7 and N11 compared to wtGCB.
Assuming that
the wtGCB part of the variant is glycosylated like wtGCB, the variant has 7
potential N-
glycosylation sites.
Based on the same considerations as used for GC-36, the expected mass of GC-61
carrying 4 N-glycans is between 60 538 Da and 61 122 Da, the expected mass of
GC-61
carrying 5 N-glycans is between 61430 Da and 62 160 Da, the expected mass of
GC-61
carrying 6 N-glycans is between 62 322 Da and 63 198 Da, and the expected mass
of GC-61
carrying 7 N-glycans is between 63 214 Da and 64 236 Da.
MALDI-TOF mass spectrometry of GC-61 shows a very broad peak with peak mass
between 61.5 kDa and 63.0 kDa in accordance with the expected mass of GC-61
carrying
either 5 or 6 N-glycans.
N-terminal amino acid sequence analysis of GC-61 showed that N3 is completely
glycosylated while N7 and N11 are partially glycosylated in acceptable
agreement with the
result obtained using mass spectrometry.
GC-62 (AANSTGNITINGT-GCB):
The theoretical peptide mass of GC-62 is 56 806 Da. The N-terminal extension
contains three
additional potential glycosylation sites at N3, N7 and Nl l compared to wtGCB.
Assuming that
the wtGCB part of the variant is glycosylated like wtGCB, the variant has 7
potential N-
glycosylation sites.
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
71
Based on the same considerations as those used for GC-36, the expected mass of
GC-62
carrying 4 N-glycans is between 60 374 Da and 60 958 Da, the expected mass of
GC-62
carrying 5 N-glycans is between 61266 Da and 61996 Da, the expected mass of GC-
62
carrying 6 N-glycans is between 62158 Da and 63 034 Da, and the expected mass
of GC-62
carrying 7 N-glycans is between 63 050 Da and 64 072 Da.
MALDI-TOF mass spectrometry of GC-62 shows two broad peaks with peak masses of
61.6 kDa and 62.7 kDa in accordance with the expected mass of GC-62 carrying
either 5 or 6
N-glycans.
N-terminal amino acid sequence analysis of GC-62 showed that N3 is completely
glycosylated while N7 and Nl 1 are partially glycosylated in acceptable
agreement with the
result obtained using mass spectrometry.
GC-63 (AVNWTSNDTSNST-GCB):
The theoretical peptide mass of GC-63 is 56 969 Da. The N-terminal extension
contains three
additional potential glycosylation sites at N3, N7 and N11 compared to wtGCB.
Assuming that
the wtGCB part of the variant is glycosylated like wtGCB, the variant has 7
potential N-
glycosylation sites.
Based on the same considerations as those used for GC-36, the expected mass of
GC-63
carrying 4 N-glycans is between 60 537 Da and 61 121 Da, the expected mass of
GC-63
carrying 5 N-glycans is between 61429 Da and 62 159 Da, the expected mass of
GC-63
carrying 6 N-glycans is between 62 321 Da and 63 197 Da, and the expected mass
of GC-63
carrying 7 N-glycans is between 63 213 Da and 64 235 Da:
MALDI-TOF mass spectrometry of GC-63 shows a major peak with a peak mass of
61.9 kDa in accordance with the expected mass of GC-63 carrying 5 N-glycans.
In addition, a
minor peak with a peak mass of 62.9 kDa is seen which corresponds to GC-63
carrying 6 N-
glycans.
N-terminal amino acid sequence analysis of GC-63 showed that N3 ans N7 are
partially
glycosylated. It was not possible to evaluate the glycosylation status of N11.
Furthermore, insect cell expressed N-terminally extended glycosylated
polypeptide
(GC-6 and GC-13) was subjected to N-terminal amino acid sequence analysis
(using Procize
from PE Biosystems, Foster City, CA). The sequencing cycle was blank for the
Asn residue in
both ANIT and ASP1NAT N-terminal peptide additions, demonstrating that the
introduced
glycosylation site is glycosylated.
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
72
When subjecting GC-13 to mass spectrophometry using the MALDI-TOF techniques
on the Voyager DERP instrument (from PE-Biosystems, Foster City, CA) the
following results
were obtained:
The wildtype and ASPINAT-extended wildtype expressed in insect cells gave
average
masses very close to the calculated mass of 59,727 Da and 61,421 Da,
respectively, assuming
that four glycosylation sites were occupied by the carbohydrates
FucGlcNAc2Man3.
EXAMPLE 4
Constructiof2~plasmids for ex~ressiof2 of FSH
A gene encoding the human FSH-alpha subunit was constructed by assembly of
synthetic oligonucleotides by PCR using methods similar to the ones described
in Stemmer et
al. (I995) GefZe I64, pp. 49-53. The native FSH-alpha signal sequence was
maintained in order
to allow secretion of the gene product. The codon usage of the gene was
optimised for high
expression in mammalian cells. Furthermore, in order to achieve high gene
expression, an
intron (from pCI-Neo (Promega)) was included in the 5' untranslated region of
the gene. The
synthetic gene was subcloned behind the CMV promoter in pcDNA3.l/Hygro
(Invitrogen).
The sequence of the resulting plasmid, termed pBvdH977, is given in SEQ ID
N0:3 (FSH-
alpha-coding sequence at position 1225 to 1570). Similarly, a synthetic gene
encoding the
wildtype human FSH-beta subunit was constructed. Also in this construct, the
native signal
sequence was maintained (except for a Lys to Glu mutation at position 2) in
order to allow
secretion, and the codon usage was optimised for high expression and an intron
was included
in the recipient vector (pcDNA3.1/Zeo (Invitrogen)). The sequence of the
resulting FSH-beta -
containing plasmid, termed pBvdH1022, is given in SEQ 117 N0:4 (FSH-beta-
coding sequence
at position 1231 to 1617). A plasmid containing both the FSH-alpha and the FSH-
beta
2o encoding synthetic genes was generated by subcloning the FSH-alpha
containing NruI-PvuII
fragment from pBvdH977 into pBvdH1022 linearized with NruI. The resulting
plasmid, in
which the FSH-alpha and FSH-beta-expression cassettes are in direct
orientation, was termed
pBvdH1100.
Expression of FSH ifa CHO cells
FSH was expressed in Chinese Hamster Ovary (CHO) Kl cells, obtained from the
American Type Culture Collection (ATCC, CCL-61).
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
73
For transient expression of FSH, cells were grown to 95% confluency in serum-
containing media (MEMa with ribonucleotides and deoxyribonucleotides (Life
Technologies
Cat # 32571-028) containing 1:10 FBS (BioWhittaker Cat # 02-701F) and 1:100
penicillin and
streptomycin (BioWhittaker Cat # 17-602E), or Dulbecco's MEM/Nut.-mix F-12
(Ham) L-
glutamine, 15 mM Hepes, pyridoxine-HCl (Life Technologies Cat # 31330-038)
with the same
additives. FSH-encoding plasmids were transfected into the cells using
Lipofectamine 2000
(Life Technologies) according to the manufacturer's specifications. 24-48 hrs
after
transfection, culture media were collected, centrifuged and filtered through
0.22 micrometer
filters to remove cells.
to Stable clones expressing FSH were generated by transfection of CHO Kl cells
with
FSH-encoding plasmids followed by incubation of the cells in selective media
(for instance one
of the above media containing 0.5 mg/ml zeocin for cells transfected with
plasmid
pBvdH1100). Stably transfected cells were isolated and sub-cloned by limited
dilution. Clones
that produced high levels of FSH were identified by ELISA.
More specifically, the concentration of FSH in samples was quantified by use
of a
commercial immunoassay (DRG FSH EIA, DRG Instruments GmbH, Marburg, Germany).
DRG FSH EIA is a solid phase immunosorbent assay (ELISA) based on the sandwich
principle. The microtiter wells are coated with a monoclonal antibody directed
towards a
unique antigenic site on the FSH-0 subunit. An aliquot of FSH-containing
sample (diluted in
2o H20 with 0.1% BSA) and an anti-FSH antiserum conjugated with horseradish
peroxidase are
added to the coated wells. After incubation, unbound conjugate is washed off
with water. The
amount of bound peroxidase is proportional to the concentration of FSH in the
sample. The
intensity of colour developed upon addition of substrate solution is
proportional to the
concentration of FSH in the sample.
Large-scale production of FSH in CHO cells
The cell line CHO Kl 1100 5, stably expressing human FSH, was passed 1:10 from
a
confluent culture and propagated as adherent cells in serum-containing medium
Dulbecco's
MEM/Nut.-mix F-12 (Ham) L-glutamine, 15 mM Hepes, pyridoxine-HCl (Life
Technologies
3o Cat # 31330-038), 1:10 FBS (BioWhittaker Cat # 02-701F), 1:100 penicillin
and streptomycin
(BioWhittaker Cat # 17-602E) until confluence in a 10 layer cell factory (NUNC
#165250).
The media was then changed to serum-free media: Dulbecco's MEM/Nut.-mix F-12
(Ham) L-
glutamine, pyridoxine-HCl (Life Technologies Cat # 21041-025) with the
addition of 1:500
ITS-A (Gibco/BRL # 51300-044), 1:500 EX-CYTE VLE (Serological Proteins Inc. #
81-129)
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
74
and 1:100 penicillin and streptomycin (BioWhittaker Cat # 17-602E).
Subsequently, every 24
h, culture media were collected and replaced with 1 fresh liter of the same
serum-free media.
The collected media was filtered through 0.22 ~ m filters to remove cells.
Growth in cell
factories was continued with daily harvests and replacements of the culture
media until FSH
yields dropped below one-fourth of the initial expression level (typically
after 10-15 days).
EXAMPLE 5
l0 Puri 'catiore of FSH wildtype ayad variaf~ts
Three chromatographic steps were employed to obtain highly purified FSH. First
an
anion exchanger step, then hydrophobic interaction chromatography (HIC) and
finally an
immunoaffinity step using an FSH-(3 specific monoclonal antibody.
Culture supernatants were prepared as described in Example 4. Filtered culture
supernatants were concentrated 10 to 20 times by ultrafiltration (10 kD cut-
off membrane), pH
was adjusted to 8.0 and conductivity to 10 - 15 mS/cm, before application on a
DEAE
Sepharose (Pharmacia) anion exchanger column, which had been equilibrated in
ammonium
acetate buffer (0.16 M, pH 8.0). Semipurified FSH was recovered both in the
unbound flow-
through fraction as well as in the wash fraction using 0.16 M ammonium
acetate, pH 8Ø The
flow through and wash fractions were pooled and ammonium sulfate was added
from a stock
solution (4.5 M) to obtain a final concentration of 1.5 M (NH4)2SO4. The pH
was adjusted to
7Ø
The partially purified FSH was subsequently applied on a 25 ml butyl Sepharose
(Pharmacia) HIC column. After application, the column was washed with at least
3 column
volumes of 1.5 M (NH4)2504, 20 mM ammonium acetate, pH 7 (until the absorbance
at 280
nm reached baseline level) and FSH was eluted with 4 column volumes of buffer
B (20 mM
ammonium acetate, pH 7). FSH enriched fractions from the HIC step were pooled,
concentrated and diafiltrated using Vivaspin 20 modules, 10 kD cut-off
membrane
(Vivascience), to a 50 mM sodium phosphate, 150 mM NaCl, pH 7.2.
3o For the third chromatographic step, an anti-FSH-(3 monoclonal antibody (RDI-
FSH909,
Research Diagnostics) was immobilized to CNBr-activated Sepharose (Pharmacia)
using a
standard procedure from the supplier. Approximately 1 mg antibody was coupled
per ml resin.
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
The immunoaffinity resin was packed in plastic columns and equilibrated with
50 mM sodium
phosphate, 150 mM NaCI, pH 7.2 before application.
The buffer exchanged eluate from the butyl HIC step was applied on the
antibody
column by use of gravity flow. This was followed by several washing steps in
50 mM sodium
5 phosphate solutions (0.5 M NaCl and 1 M NaCI, both pH 7.2). Elution was
performed using
either 1 M NH3 or 0.6 M NH3, 40% (v/v) isopropanol and the eluate was
immediately
neutralized with 1 M acetic acid to pH 6-8.
The purified FSH bulk product was concentrated and diafiltrated using Vivaspin
20
modules, 10 kD cut-off membrane (Vivascience), to a 50 mM sodium phosphate,
150 mM
to NaCI, pH 7.2. For subsequent storage, BSA was added to 0.1% (w/v) and the
purified FSH was
microfiltrated using a 0.22 ~m filter prior to storage at - 80°C.
SDS-PAGE, run under non-dissociating conditions (without boiling), showed
wildtype
FSH migrating as an apparant 42~3 kDa band, slightly diffuse due to
heterogeneity in the
attached carbohydrates. The purity was about 80-90%. N-terminal sequencing
showed that the
15 a-chain had the expected N-terminal sequence starting with residue 1 (SEQ
1D N0:5) and the
(3-chain starting with residue 3 (SEQ ID N0:6). These N-terminal sequences
have been found
previously for recombinant FSH produced in CHO cells (Olijve, W. et al. (1996)
Mol. Hufn.
Reprod. 2, 371-382).
2o EXAMPLE 6
FSH in vitro activi , assay
6.1 FSH assay Outline
It has previously been published that activation of the FSH receptor by FSH
Ieads to an
increase in the intracellular concentration of cAMP. Consequently,
transcription is activated at
25 promoters containing multiple copies of the cAMP response element (CRE). It
is thus possible
to measure FSH activity by use of a CRE luciferase reporter gene introduced
into CHO cells
expressing the FSH receptor.
6.2 Construction of a CHO FSH-R / CRE-luc cell line
30 Stable clones expressing the human FSH receptor were produced by
transfection of
CHO Kl cells with a plasmid containing the receptor cDNA inserted into pcDNA3
(Invitrogen) followed by selection in media containing 600 microg/ml 6418.
Using a
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
76
commercial CAMP-SPA RIA (Amersham), clones were screened for the ability to
respond to
FSH stimulation. On the basis of these results, an FSH receptor-expressing CHO
clone was
selected for further transfection with a CRE-luc reporter gene. A plasmid
containing the
reporter gene with 6 CRE elements in front of the Firefly luciferase gene was
co-transfected
with a plasmid conferring Hygromycin B resistance. Stable clones were selected
in the
presence of 600 microg/ml 6418 and 400 microg/ml Hygromycin B. A clone
yielding a robust
luciferase signal upon stimulation with FSH (ECSO ~ 0.01 ICT/ml) was obtained.
This CHO
FSH-R / CRE-luc cell line was used to measure the activity of samples
containing FSH.
6.3 FSH luciferase assay
To perform activity assays, CHO FSH-R / CRE-luc cells were seeded in white 96
well
culture plates at a density of about 15,000 cells/well. The cells were in 100
~1 DMEM/F-12
(without phenol red) with 1.25% FBS. After incubation overnight (at
37°C, 5% C02), 25 ~,1 of
sample or standard diluted in DMEM/F-12 (without phenol red) with 10% FBS was
added to
each well. The plates were further incubated for 3 hrs, followed by addition
of 125 ~,1 LucLite
substrate (Packard Bioscience). Subsequently, plates were sealed and
luminescence was
measured on a TopCount luminometer (Packard) in SPC (single photon counting)
mode.
EXAMPLE 7
Construction afzd afzal serf a varia>2t form of FSH coz2tai>2in~ two N=linked
,~lycosylatior2s,at
the N-termiszus of the alpha suburzit
A construct encoding a modified form of FSH-alpha, having two additional sites
for N-
linked glycosylation at its N-terminus was generated by site-directed
mutagenesis using
standard DNA techniques known in the art. A DNA fragment encoding the sequence
Ala-Asn-
lle-Thr-Val-Asn-Ile-Thr-Val was inserted immediately upstream of the mature
FSH-alpha
sequence in pBvdH977. The sequence of the resulting plasmid, termed pBvdH1163,
is given in
SEQ ID N0:7 (modified FSH-alpha-encoding sequence at position 1225 to 1599). A
plasmid
encoding both subunits was constructed by subcloning the FSH-containing NruI-
PvuII
3o fragment from pBvdHl 163 into pBvdH1022 (Example 4), which had been
linearized with
PvuII. The resulting plasmid was termed pBvdH1208.
For expression of the variant form of FSH containing two N-linked
glycosylations at
the N-terminus of the alpha subunit (termed FSH1208), CHO K1 cells were
transfected with
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
77
pBvdH1208 or co-transfected with a combination of pBvdH1163, encoding the
modified alpha
subunit and pBvdH1022, encoding the wildtype beta subunit. Transient
expressions, isolation
of stable expression clones, and large-scale production of FSH1208 were
performed as
described for wildtype FSH in Example 4.
The FSH content of samples was analysed by Western Blotting: Proteins were
separated by SDS-PAGE and a standard Western blot was performed using rabbit
anti human
FSH (AHP519, Serotec) or mouse anti human FSH-alpha (MCA338, Serotec) as
primary
antibody, and an ImmunoPure Ultra Sensitive ABC Peroxidase Staining Kit
(Pierce) for
detection. Western blotting showed that FSH1208 had a larger molecular mass
than wildtype
to FSH, indicating that the introduction of acceptor sites for N-linked
glycosylation at the N-
terminus of the alpha subunit indeed lead to hyperglycosylation of FSH. For
analysis of pI,
samples were separated on pH 3-7 IEF gels (NOVEX). After electrophoresis,
proteins were
blotted onto Immobilon-P (Millipore) membranes and a Western blot was
performed as
described above, using the same antibodies and detection kit. Isoelectric
focusing demonstrated
that the FSH forms in the FSH1208 samples were found in a lower pI range than
wildtype
FSH. Thus, the pH interval for FSH1208 isoforms was about 3.0-4.5 versus about
4.0-5.2 for
wildtype FSH. This indicated that FSHI208 molecules are on average more
negatively charged
than the wild type, which is attributed to the presence of additional sialic
acid residues.
FSH1208 was purified and characterized as described in Example 5. SDS-PAGE,
run
under non-dissociating conditions (without boiling), showed FSH1208 migrating
as an
apparent 55~5 kDa band, slightly diffuse due to heterogeneity in the attached
carbohydrates.
The purity was about 80-90%. N-terminal sequencing showed that while the (3-
chain had the
same N-terminal sequence as wildtype FSH, the sequence of a-chain was in
agreement with
this subunit carrying the expected N-terminal extension ANITVNITV, in which
both
asparagines residues are glycosylated.
The specific activity of FSH1208 was determined by measurement of the ih vitro
bioactivity (FSH luciferase assay, Example 6) and the FSH content of the
samples by ELISA.
The specific activity of FSH1208 was found to be about one-third of that of
the wildtype
reference.
3o A pharmacokinetic study performed as follows:
Immature 26-27 days old female Sprague-Dawley rats were injected i.v. with 3-4
microg FSH, produced, purified and analyzed as described above. Subsequently,
blood samples
were taken at various time-points after injection. FSH concentrations in serum
samples were
determined by ELISA, as described above.
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
78
Ih vivo bioactivity of wildtype recombinant FSH and variant forms may be
evaluated by
the ovarian weight augmentation assay (Steelman and Pohley (1953)
E>zdocrinology 53, 604-
616). Furthermore, the ability of FSH and variant forms to stimulate
maturation of follicles in
laboratory animals may be detected with e.g. ultrasound equipment. The
experiment showed
that 24 hours after injection of equal amounts of wildtype FSH and FSH1208,
the sera of
FSH1208-treated animals contained more than 10 fold more remaining
immunoreactive
material than the sera from animals treated with wildtype FSH.
EXAMPLE 8
Cozzstruction ayzd anal s~fotheY FSH variayzts containin,~ additional
~lycosylatiorz sites
Plasmids encoding variant forms of FSH-alpha and FSH-beta containing
additional
sites for N-linked glycosylation were generated by site-directed mutagenesis
using standard
DNA techniques known in the art. The following amino acid substitutions and/or
insertions
were generated:
FSH1147: Amino acid Tyr58 of mature FSH-beta altered to Asn
FSH1349: N-terminus of mature FSH-alpha altered from APD QDC... to: APNDTVNFT
QDC
FSH1354: N-terminus of mature FSH-beta altered from NS CEL ... to: NSNITVNITV
CEL ...
Plasmids encoding the variant forms were transiently expressed in CHO K1 cells
as
2o described in Example 4. Plasmids encoding FSH-alpha variants were co-
transfected with a
plasmid encoding wild-type FSH-beta and vice versa.
Western and isoelectric focusing were performed on culture media samples as
described above. The variant forms had higher molecular weights than the wild-
type, indicating
that the additional acceptor sites for N-linked glycosylation had indeed been
glycosylated.
Furthermore, isoelectric focusing showed that the different isoforms of the
three FSH variants
were spread over a lower pI range than the wildtype. This strongly suggests
that the variant
forms had a higher sialic acid content than the wildtype.
In vitro FSH activities of the resulting media samples were analysed as
described in
Example 6.3. All three variant forms were able to stimulate the CHO FSH-R /
CRE-luc cells,
indicating that these variant FSH forms have retained significant FSH
activity.
While the foregoing invention has been described in some detail for purposes
of clarity and
understanding, it will be clear to one skilled in the art from a reading of
this disclosure that
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
79
various changes in form and detail can be made without departing from the true
scope of the
invention. For example, all the techniques, methods, compositions, apparatus
and systems
described above may be used in various combinations. All publications,
patents, patent
applications, or other documents cited in this application are incorporated by
reference in their
entirety for all purposes to the same extent as if each individual
publication, patent, patent
application, or other document were individually indicated to be incorporated
by reference for
all purposes.
SUBSTITUTE SHEET (RULE 26)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
SEQUENCE LISTING
<110> MAXYGEN APS
<120> N-TERMINALLY EXTENDED POLYPEPTIDES
<130> 0217W0210
<170> PatentIn Ver. 2.1
<210> 1
<211> 497
<212> PRT
<213> Homo sapiens
<220>
<221> MOD_RES
<222> (495)
<223> R or H
<400> 1
Ala Arg Pro Cys Ile Pro Lys Ser Phe Gly Tyr Ser Ser Val Val Cys
1 5 10 15
Val Cys Asn Ala Thr Tyr Cys Asp Ser Phe Asp Pro Pro Thr Phe Pro
20 25 30
Ala Leu Gly Thr Phe Ser Arg Tyr Glu Ser Thr Arg Ser Gly Arg Arg
35 40 45
Met Glu Leu Ser Met Gly Pro Ile Gln Ala Asn His Thr Gly Thr Gly
50 55 60
Leu Leu Leu Thr Leu Gln Pro Glu Gln Lys Phe Gln Lys Val Lys Gly
65 70 75 80
Phe Gly Gly Ala Met Thr Asp Ala Ala Ala Leu Asn Ile Leu Ala Leu
85 90 95
Ser Pro Pro Ala Gln Asn Leu Leu Leu Lys Ser Tyr Phe Ser Glu Glu
100 105 110
Gly Ile Gly Tyr Asn Ile Ile Arg Val Pro Met Ala Ser Cys Asp Phe
115 120 125
Ser Ile Arg Thr Tyr Thr Tyr Ala Asp Thr Pro Asp Asp Phe Gln Leu
130 135 140
His Asn Phe Ser Leu Pro Glu Glu Asp Thr Lys Leu Lys Ile Pro Leu
145 150 155 160
Ile His Arg Ala Leu Gln Leu Ala Gln Arg Pro Val Ser Leu Leu Ala
165 17 0 17 5.
Ser Pro Trp Thr Ser Pro Thr Trp Leu Lys Thr Asn Gly Ala Val Asn
180 185 190
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
Gly Lys Gly Ser Leu Lys Gly Gln Pro Gly Asp Ile Tyr His Gln Thr
195 200 205
Trp Ala Arg Tyr Phe Val Lys Phe Leu Asp Ala Tyr Ala Glu His Lys
210 215 220
Leu Gln Phe Trp Ala Val Thr Ala Glu Asn Glu Pro Ser Ala Gly Leu
225 230 235 240
Leu Ser Gly Tyr Pro Phe Gln Cys Leu Gly Phe Thr Pro Glu His Gln
245 250 255
Arg Asp Phe Ile Ala Arg Asp Leu Gly Pro Thr Leu Ala Asn Ser Thr
260 265 270
His His Asn Val Arg Leu Leu Met Leu Asp Asp Gln Arg Leu Leu Leu
275 280 285
Pro His Trp Ala Lys Val Val Leu Thr Asp Pro Glu Ala Ala Lys Tyr
290 295 300
Val His Gly Ile Ala Val His Trp Tyr Leu Asp Phe Leu Ala Pro Ala
305 310 315 320
Lys Ala Thr Leu Gly Glu Thr His Arg Leu Phe Pro Asn Thr Met Leu
325 330 335
Phe Ala Ser Glu Ala Cys Val Gly Ser Lys Phe Trp Glu Gln Ser Val
340 345 350
Arg Leu Gly Ser Trp Asp Arg Gly Met Gln Tyr Ser His Ser Ile Ile
355 360 365
Thr Asn Leu Leu Tyr His Val Val Gly Trp Thr Asp Trp Asn Leu Ala
370 375 380
Leu Asn Pro Glu Gly Gly Pro Asn Trp Va1 Arg Asn Phe Val Asp Ser
385 390 395 400
Pro Ile Ile Val Asp Ile Thr Lys Asp Thr Phe Tyr Lys Gln Pro Met
405 410 415
Phe Tyr His Leu Gly His Phe Ser Lys Phe Ile Pro Glu Gly Ser Gln
420 425 430
Arg Val Gly Leu Val Ala Ser Gln Lys Asn Asp Leu Asp Ala Val Ala
435 440 445
Leu Met His Pro Asp Gly Ser Ala Val Val Val Val Leu Asn Arg Ser
450 455 460
Ser Lys Asp Val Pro Leu Thr Ile Lys Asp Pro Ala Val Gly Phe Leu
465 470 475 480
Glu Thr Ile Ser Pro Gly Tyr Ser Ile His Thr Tyr Leu Trp Xaa Arg
485 490 495
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
Gln
<210> 2
<211> 1551
<212> DNA
<213> Homo Sapiens
<400> 2
atggctggca gcctcacagg attgcttcta cttcaggcag tgtcgtgggc atcaggtgcc 60
cgcccctgca tccctaaaag cttcggctac agctcggtgg tgtgtgtctg caatgccaca 120
tactgtgact cctttgaccc cccgaccttt cctgcccttg gtaccttcag ccgctatgag 180
agtacacgca gtgggcgacg gatggagctg agtatggggc ccatccaggc taatcacacg 240
ggcacaggcc tgctactgac cctgcagcca gaacagaagt tccagaaagt gaagggattt 300
ggaggggcca tgacagatgc tgctgctctc aacatccttg ccctgtcacc ccctgcccaa 360
aatttgctac ttaaatcgta cttctctgaa gaaggaatcg gatataacat catccgggta 420
cccatggcca gctgtgactt ctccatccgc acctacacct atgcagacac ccctgatgat 480
ttccagttgc acaacttcag cctcccagag gaagatacca agctcaagat acccctgatt 540
caccgagcac tgcagttggc ccagcgtccc gtttcactcc ttgccagccc ctggacatca 600
cccacttggc tcaagaccaa tggagcggtg aatgggaagg ggtcactcaa gggacagccc 660
ggagacatct accaccagac ctgggccaga tactttgtga agttcctgga tgcctatgct 720
gagcacaagt tacagttctg ggcagtgaca gctgaaaatg agccttctgc tgggctgttg 780
agtggatacc ccttccagtg cctgggcttc acccctgaac atcagcgaga cttaattgcc 840
cgtgacctag gtcctaccct cgccaacagt actcaccaca atgtccgcct actcatgctg 900
gatgaccaac gcttgctgct gccccactgg gcaaaggtgg tgctgacaga cccagaagca 960
gctaaatatg ttcatggcat tgctgtacat tggtacctgg actttctggc tccagccaaa 1020
gccaccctag gggagacaca ccgcctgttc Cccaacacca tgctctttgc ctcagaggcc 1080
tgtgtgggct ccaagttctg ggagcagagt gtgcggctag gctcctggga tcgagggatg 1140
cagtacagcc acagcatcat cacgaacctc ctgtaccatg tggtcggctg gaccgactgg 1200
aaccttgccc tgaaccccga aggaggaccc aattgggtgc gtaactttgt cgacagtccc 1260
atcattgtag acatcaccaa ggacacgttt tacaaacagc ccatgttcta ccaccttggc 1320
catttcagca agttcattcc tgagggctcc cagagagtgg ggctggttgc cagtcagaag 1380
aacgacctgg acgcagtggc attgatgcat cccgatggct ctgctgttgt ggtcgtgcta 1440
aaccgctcct ctaaggatgt gcctcttacc atcaaggatc ctgctgtggg cttcctggag 1500
acaatctcac ctggctactc cattcacacc tacctgtggc gtcgccagtg a 1551
<210> 3
<211> 6186
<212> DNA
<213> Artificial sequence
<220>
<221> exon
<222> (1225)..(1572)
<223> Coding sequence for human FSH-alpha
<400> 3
gacggatcgg gagatctccc gatcccctat ggtcgactct cagtacaatc tgctctgatg 60
ccgcatagtt aagccagtat ctgctccctg cttgtgtgtt ggaggtcgct gagtagtgcg 120
cgagcaaaat ttaagctaca acaaggcaag gcttgaccga caattgcatg aagaatctgc 180
ttagggttag gcgttttgcg ctgcttcgcg atgtacgggc cagatatacg cgttgacatt 240
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
gattattgactagttattaa tagtaatcaa ttacggggtcattagttcatagcccatata300
tggagttccgcgttacataa cttacggtaa atggcccgcctggctgaccgcccaacgacc360
cccgcccattgacgtcaata atgacgtatg ttcccatagtaacgccaatagggactttcc420
attgacgtcaatgggtggac tatttacggt aaactgcccacttggcagtacatcaagtgt480
atcatatgccaagtacgccc cctattgacg tcaatgacggtaaatggcccgcctggcatt540
atgcccagtacatgacctta tgggactttc ctacttggcagtacatctacgtattagtca600
tcgctattaccatggtgatg cggttttggc agtacatcaatgggcgtggatagcggtttg660
actcacggggatttccaagt ctccacccca ttgacgtcaatgggagtttgttttggcacc720
aaaatcaacgggactttcca aaatgtcgta acaactccgccccattgacgcaaatgggcg780
gtaggcgtgtacggtgggag gtctatataa gcagagctctctggctaactagagaaccca840
ctgcttactggcttatcgaa attaatacga ctcactatagggagacccaagctggctagc900
ttattgcggtagtttatcac agttaaattg ctaacgcagtcagtgcttctgacacaacag960
tctcgaacttaagctgcagt gactctctta aggtagccttgcagaagttggtcgtgaggc1020
actgggcaggtaagtatcaa ggttacaaga caggtttaaggagaccaatagaaactgggc1080
ttgtcgagacagagaagact cttgcgtttc tgataggcacctattggtcttactgacatc1140
cactttgcctttctctccac aggtgtccac tcccagttcaattacagctcttaaaagctt1200
ggtaccgagctcggatccgc cacc atg gac gcc gcc 1251
tac tac egc aag tac
Met Asp Tyr Tyr Arg Lys Ala Ala
Tyr
1 5
atc ttc gtg acc ctg agc gtg ttc gtg ctg agc gcc 1299
ctg ctg cac cac
Ile Phe Val Thr Leu Ser Val Phe Val Leu Ser Ala
Leu Leu His His
15 20 25
ccc gac cag gac tgc ccc gag tgc cag gag ccc ttc 1347
gtg acc ctg aac
Pro Asp Gln Asp Cys Pro Glu Cys Gln Glu Pro Phe
Val Thr Leu Asn
30 35 40
ttc age ccc ggc gcc ccc atc ctg atg ggc tgc ttc 1395
cag cag tgc tgc
Phe Ser Pro Gly Ala Pro Ile Leu Met Gly Cys Phe
Gln Gln Cys Cys
45 50 55
agc cgc tac ccc acc ccc ctg cgc aag acc ctg gtg 1443
gcc agc aag atg
Ser Arg Tyr Pro Thr Pro Leu Arg Lys Thr Leu Val
Ala Ser Lys Met
60 65 70
cag aag gtg acc agc gag agc acc gtg gcc agc tac 1491
aac tgc tgc aag
Gln Lys Val Thr Ser Glu Ser Thr Val Ala Ser Tyr
Asn Cys Cys Lys
75 80 85
aac cgc acc gtg atg ggc ggc ttc gag aac acc gcc 1539
gtg aag gtg cac
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
Asn Arg Val Thr Val Met Gly Gly Phe Lys Val Glu Asn His Thr Ala
90 95 100 105
tgc cac tgc agc acc tgc tac tac cac aag agc taatctagag ggcccgttta 1592
Cys His Cys Ser Thr Cys Tyr Tyr His Lys Ser
110 115
aacccgctgatcagcctcgactgtgccttctagttgccagccatctgttgtttgcccctc1652
ccccgtgccttccttgaccctggaaggtgCcactcccactgtcctttcctaataaaatga1712
ggaaattgcatcgcattgtctgagtaggtgtcattctattctggggggtggggtggggca1772
ggacagcaagggggaggattgggaagacaatagcaggcatgctggggatgcggtgggctc1832
tatggcttctgaggcggaaagaaccagctggggctctagggggtatccccacgcgccctg1892
tagcggcgcattaagcgcggcgggtgtggtggttacgcgcagcgtgaccgctacacttgc1952
cagcgccctagcgcccgctcctttcgctttcttcccttcctttctcgccacgttcgccgg2012
ctttccccgtcaagctctaaatcggggcatccctttagggttccgatttagtgctttacg2072
gcacctcgaccccaaaaaacttgattagggtgatggttcacgtagtgggccatcgccctg2132
atagacggtttttcgccctttgacgttggagtccacgttctttaatagtggactcttgtt2192
ccaaactggaacaacactcaaccctatctcggtctattcttttgatttataagggatttt2252
ggggatttcggcctattggttaaaaaatgagctgatttaacaaaaatttaacgcgaatta2312
attctgtggaatgtgtgtcagttagggtgtggaaagtccccaggctccccaggcaggcag2372
aagtatgcaaagcatgcatctcaattagtcagcaaccaggtgtggaaagtccccaggctc2432
cccagcaggcagaagtatgcaaagcatgcatctcaattagtcagcaaccatagtcccgcc2492
cctaactccgcccatcccgcccctaactccgcccagttccgcccattctccgccccatgg2552
ctgactaattttttttatttatgcagaggccgaggccgcctctgcctctgagctattcca2612
gaagtagtgaggaggcttttttggaggcctaggcttttgcaaaaagctcccgggagcttg2672
tatatccattttcggatctgatcagcacgtgatgaaaaagcctgaactcaccgcgacgtc2732
tgtcgagaagtttctgatcgaaaagttcgacagcgtctccgacctgatgcagctctcgga2792
gggcgaagaatctcgtgctttcagcttcgatgtaggagggcgtggatatgtcctgcgggt2852
aaatagctgcgccgatggtttctacaaagatcgttatgtttatcggcactttgcatcggc2912
cgcgctcccgattccggaagtgcttgacattggggaattcagcgagagcctgacctattg2972
catctcccgc cgtgcacagg gtgtcacgtt gcaagacctg cctgaaaccg aactgcccgc 3032
tgttctgcag ccggtcgcgg aggccatgga tgcgatcgct gcggccgatc ttagccagac 3092
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
gagcgggttcggcccattcggaccgcaaggaatcggtcaatacactacatggcgtgattt3152
catatgcgcgattgctgatccccatgtgtatcactggcaaactgtgatggacgacaccgt3212
cagtgcgtccgtcgcgcaggctctcgatgagctgatgctttgggccgaggactgccccga3272
agtccggcacctcgtgcacgcggatttcggctccaacaatgtcctgacggacaatggccg3332
cataacagcggtcattgactggagcgaggcgatgttcggggattcccaatacgaggtcgc3392
caacatcttcttctggaggccgtggttggcttgtatggagcagcagacgcgctacttcga3452
gcggaggcatccggagcttgcaggatcgccgcggctccgggcgtatatgctccgcattgg3512
tcttgaccaactctatcagagcttggttgacggcaatttcgatgatgcagcttgggcgca3572
gggtcgatgcgacgcaatcgtccgatccggagccgggactgtcgggcgtacacaaatcgc3632
ccgcagaagcgcggccgtctggaccgatggctgtgtagaagtactcgccgatagtggaaa3692
ccgacgccccagcactcgtccgagggcaaaggaatagcacgtgctacgagatttcgattc3752
caccgccgccttctatgaaaggttgggcttcggaatcgttttccgggacgccggctggat3812
gatcctccagcgcggggatctcatgctggagttcttcgcccaccccaacttgtttattgc3872
agcttataatggttacaaataaagcaatagcatcacaaatttcacaaataaagcattttt3932
ttcactgcattctagttgtggtttgtccaaactcatcaatgtatcttatcatgtctgtat3992
accgtcgacctctagctagagcttggcgtaatcatggtcatagctgtttcctgtgtgaaa4052
ttgttatccgctcacaattccacacaacatacgagccggaagcataaagtgtaaagcctg4112
gggtgcctaatgagtgagctaactcacattaattgcgttgcgctcactgcccgctttcca4172
gtcgggaaacctgtcgtgccagctgcattaatgaatcggccaacgcgcggggagaggcgg4232
tttgcgtattgggcgctcttCCC,~'CttCCtCgctcactgactcgctgcgctcggtcgttcg4292
gctgcggcgagcggtatcagctcactcaaaggcggtaatacggttatccacagaatcagg4352
ggataacgcaggaaagaacatgtgagcaaaaggccagcaaaaggccaggaaccgtaaaaa4412
ggccgcgttgctggcgtttttccataggctccgcccccctgacgagcatcacaaaaatcg4472
acgctcaagtcagaggtggcgaaacccgacaggactataaagataccaggcgtttccccc4532
tggaagctccctcgtgcgctctcctgttccgaccctgccgcttaccggatacctgtccgc4592
ctttctcccttcgggaagcgtggcgctttctcaatgctcacgctgtaggtatctcagttc4652
ggtgtaggtcgttcgctccaagctgggctgtgtgcacgaaccccccgttcagcccgaccg4712
ctgcgccttatccggtaactatcgtcttgagtccaacccggtaagacacgacttatcgcc4772
actggcagcagccactggtaacaggattagcagagcgaggtatgtaggcggtgctacaga4832
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
gttcttgaagtggtggcctaactacggctacactagaaggacagtatttggtatctgcgc4892
tctgctgaagccagttaccttcggaaaaagagttggtagctcttgatccggcaaacaaac4952
caccgctggtagcggtggtttttttgtttgcaagcagcagattacgcgcagaaaaaaagg5012
atctcaagaagatcctttgatcttttctacggggtctgacgctcagtggaacgaaaactc5072
acgttaagggattttggtcatgagattatcaaaaaggatcttcacctagatccttttaaa5132
ttaaaaatgaagttttaaatcaatctaaagtatatatgagtaaacttggtctgacagtta5192
ccaatgcttaatcagtgaggcacctatctcagcgatctgtctatttcgttcatccatagt5252
tgcctgactccccgtcgtgtagataactacgatacgggagggcttaccatctggccccag5312
tgctgcaatgataccgcgagacccacgctcaccggctccagatttatcagcaataaacca5372
gccagccggaagggccgagcgcagaagtggtcctgcaactttatccgcctccatccagtc5432
tattaattgttgccgggaagctagagtaagtagttcgccagttaatagtttgcgcaacgt5492
tgttgccattgctacaggcatcgtggtgtcacgctcgtcgtttggtatggcttcattcag5552
ctccggttcccaacgatcaaggcgagttacatgatcccccatgttgtgcaaaaaagcggt5612
tagctccttcggtcctccgatcgttgtcagaagtaagttggccgcagtgttatcactcat5672
ggttatggcagcactgcataattctcttactgtcatgccatccgtaagatgcttttctgt5732
gactggtgagtactcaaccaagtcattctgagaatagtgtatgcggcgaccgagttgctc5792
ttgcccggcgtcaatacgggataataccgcgccacatagcagaactttaaaagtgctcat5852
cattggaaaacgttcttcggggcgaaaactctcaaggatcttaccgctgttgagatccag5912
ttcgatgtaacccactcgtgcacccaactgatcttcagcatcttttactttcaccagcgt5972
ttctgggtgagcaaaaacaggaaggcaaaatgccgcaaaaaagggaataagggcgacacg6032
gaaatgttgaatactcatactcttcctttttcaatattattgaagcatttatcagggtta6092
ttgtctcatgagcggatacatatttgaatgtatttagaaaaataaacaaataggggttcc6152
gcgcacattt ccccgaaaag tgccacctga cgtc 6186
<210> 4
<211> 5651
<212> DNA
<213> Artificial sequence
<220>
<221> exon
<222> (1231)..(1617)
<223> Coding sequence for human FSIi-beta
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<400>
4
gacggatcgggagatctcccgatcccctatggtcgactctcagtacaatctgctctgatg60
ccgcatagttaagccagtatctgctccctgcttgtgtgttggaggtcgctgagtagtgcg120
cgagcaaaatttaagctacaacaaggcaaggcttgaccgacaattgcatgaagaatctgc180
ttagggttaggcgttttgcgctgcttcgcgatgtacgggccagatatacgcgttgacatt240
gattattgactagttattaatagtaatcaattacggggtcattagttcatagcccatata300
tggagttccgcgttacataacttacggtaaatggcccgcctggctgaccgcccaacgacc360
cccgcccattgacgtcaataatgacgtatgttcccatagtaacgccaatagggactttcc420
attgacgtcaatgggtggactatttacggtaaactgcccacttggcagtacatcaagtgt480
atcatatgccaagtacgccccctattgacgtcaatgacggtaaatggcccgcctggcatt540
atgcccagtacatgaccttatgggactttcctacttggcagtacatctacgtattagtca600
tcgctattaccatggtgatgcggttttggcagtacatcaatgggcgtggatagcggtttg660
actcacggggatttccaagtctccaccccattgacgtcaatgggagtttgttttggcacc720
aaaatcaacgggactttccaaaatgtcgtaacaactccgccccattgacgcaaatgggcg780
gtaggcgtgtacggtgggaggtctatataagcagagctctctggctaactagagaaccca840
ctgcttactggcttatcgaaattaatacgactcactatagggagacccaagctggctagc900
ttattgcggtagtttatcacagttaaattgctaacgcagtcagtgcttctgacacaacag960
tctcgaacttaagctgcagtgactctcttaaggtagccttgcagaagttggtcgtgaggc1020
actgggcaggtaagtatcaaggttacaagacaggtttaaggagaccaatagaaactgggc1080
ttgtcgagacagagaagactcttgcgtttctgataggcacctattggtcttactgacatc1140
cactttgcctttctctccacaggtgtccactcccagttcaattacagctcttaaaagctt1200
ggtaccgagctcggatctatcgatgccaccatg gag ctg cag 1254
acc ttc ttc
ttc
Met Glu Leu Gln
Thr Phe Phe
Phe
1 5
ctg ttc tgc tgc tgg aag gcc atc tgc tgc aac agc tgc gag ctg acc 1302
Leu Phe Cys Cys Trp Lys Ala Ile Cys Cys Asn Ser Cys Glu Leu Thr
15 20
aac atc acc atc gcc atc gag aag gag gag tgc cgc ttc tgc atc agc 1350
Asn Ile Thr Ile Ala Ile Glu Lys Glu Glu Cys Arg Phe Cys Ile Ser
25 30 35 40
atc aac acc acc tgg tgc gcc ggc tac tgc tac acc cgc gac ctg gtg 1398
Ile Asn Thr Thr Trp Cys Ala Gly Tyr Cys Tyr Thr Arg Asp Leu Val
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
45 50 55
tacaag gaccccgcc cgccccaagatc cagaagacctgc accttcaag 1446
TyrLys AspProAla ArgProLysIle GlnLysThrCys ThrPheLys
60 65 70
gagctg gtgtacgag acggtccgggtg cccggctgcgcc caccacgcc 1494
GluLeu ValTyrGlu ThrValArgVal ProGlyCysAla HisHisAla
75 80 85
gacagc ctgtacacc taccccgtggcc acccagtgccac tgcggcaag 1542
AspSer LeuTyrThr TyrProValAla ThrGlnCysHis CysGlyLys
90 95 100
tgcgac agcgacagc accgactgcacc gtgcgcggcctg ggccccagc 1590
CysAsp SerAspSer ThrAspCysThr ValArgGlyLeu GlyProSer
105 110 115 120
tactgc agcttcggc gagatgaaggag taactcgaga ctagagggcc 1637
TyrCys SerPheG1y GluMetLysGlu
125
cgtttaaacc cgctgatcag cctcgactgt gccttctagt tgccagccat ctgttgtttg 1697
cccctccccc gtgccttcct tgaccctgga aggtgccact cccactgtcc tttcctaata 1757
aaatgaggaa attgcatcgc attgtctgag taggtgtcat tctattctgg ggggtggggt 1817
ggggcaggacagcaagggggaggattgggaagacaatagcaggcatgctggggatgcggt1877
gggctctatggcttctgaggcggaaagaaccagctggggctctagggggtatccccacgc1937
gccctgtagcggcgcattaagcgcggcgggtgtggtggttacgcgcagcgtgaccgctac1997
acttgccagcgccctagcgcccgctcctttcgctttcttcccttcctttctcgccacgtt2057
cgccggctttccccgtcaagctctaaatcggggcatccctttagggttccgatttagtgc2117
tttacggcacctcgaccccaaaaaacttgattagggtgatggttcacgtagtgggccatc2177
gccctgatagacggtttttcgccctttgacgttggagtccacgttctttaatagtggact2237
cttgttccaaactggaacaacactcaaccctatctcggtctattcttttgatttataagg2297
gattttggggatttcggcctattggttaaaaaatgagctgatttaacaaaaatttaacgc2357
gaattaattctgtggaatgtgtgtcagttagggtgtggaaagtccccaggctccccaggc2417
aggcagaagtatgcaaagcatgcatctcaattagtcagcaaccaggtgtggaaagtcccc2477
aggctccccagcaggcagaagtatgcaaagcatgcatctcaattagtcagcaaccatagt2537
CCCgCCCCtaactccgcccatCCCgCCCCtaactccgcccagttccgcccattctccgcc2597
ccatggctgactaattttttttatttatgcagaggccgaggccgcctctgcctctgagct2657
attccagaagtagtgaggaggcttttttggaggcctaggcttttgcaaaaagctcccggg2717
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
agcttgtatatccattttcggatctgatcagcacgtgttgacaattaatcatcggcatag2777
tatatcggcatagtataatacgacaaggtgaggaactaaaccatggccaagttgaccagt2837
gccgttccggtgctcaccgcgcgcgacgtcgccggagcggtcgagttctggaccgaccgg2897
ctcgggttctcccgggacttcgtggaggacgacttcgccggtgtggtccgggacgacgtg2957
accctgttcatcagcgcggtccaggaccaggtggtgccggacaacaccctggcctgggtg3017
tgggtgcgcggcctggacgagctgtacgccgagtggtcggaggtcgtgtccacgaacttc3077
cgggacgcctccgggccggccatgaccgagatcggcgagcagccgtgggggcgggagttc3137
gccctgcgcgacccggccggcaactgcgtgcacttcgtggccgaggagcaggactgacac3197
gtgctacgagatttcgattccaccgccgccttctatgaaaggttgggcttcggaatcgtt3257
ttccgggacgccggctggatgatcctccagcgcggggatctcatgctggagttcttcgcc3317
caccccaacttgtttattgcagcttataatggttacaaataaagcaatagcatcacaaat3377
ttcacaaataaagcatttttttcactgcattctagttgtggtttgtccaaactcatcaat3437
gtatcttatcatgtctgtataccgtcgacctctagctagagcttggcgtaatcatggtca3497
tagctgtttcctgtgtgaaattgttatccgctcacaattccacacaacatacgagccgga3557
agcataaagtgtaaagcctggggtgcctaatgagtgagctaactcacattaattgcgttg3617
cgctcactgcccgctttccagtcgggaaacctgtcgtgccagctgcattaatgaatcggc3677
caacgcgcggggagaggcggtttgcgtattgggcgctcttccgcttcctcgctcactgac3737
tcgctgcgctcggtcgttcggctgcggcgagcggtatcagctcactcaaaggcggtaata3797
cggttatccacagaatcaggggataacgcaggaaagaacatgtgagcaaaaggccagcaa3857
aaggccaggaaccgtaaaaaggccgcgttgctggcgtttttccataggctccgcccccct3917
gacgagcatcacaaaaatcgacgctcaagtcagaggtggcgaaacccgacaggactataa3977
agataccaggcgtttccccctggaagctccctcgtgcgctctcctgttccgaccctgccg4037
cttaccggatacctgtccgcctttctcccttcgggaagcgtggcgctttctcaatgctca4097
cgctgtaggtatctcagttcggtgtaggtcgttcgctccaagctgggctgtgtgcacgaa4157
ccccccgttcagcccgaccgctgcgccttatccggtaactatcgtcttgagtccaacccg4217
gtaagacacgacttatcgccactggcagcagccactggtaacaggattagcagagcgagg4277
tatgtaggcggtgctacagagttcttgaagtggtggcctaactacggctacactagaagg4337
acagtatttggtatctgcgctctgctgaagccagttaccttcggaaaaagagttggtagc4397
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
tcttgatccggcaaacaaaccaccgctggtagcggtggtttttttgtttgcaagcagcag4457
attacgcgcagaaaaaaaggatctcaagaagatcctttgatcttttctacggggtctgac4517
gctcagtggaacgaaaactcacgttaagggattttggtcatgagattatcaaaaaggatc4577
ttcacctagatccttttaaattaaaaatgaagttttaaatcaatctaaagtatatatgag4637
taaacttggtctgacagttaccaatgcttaatcagtgaggcacctatctcagcgatctgt4697
ctatttcgttcatccatagttgcctgactccccgtcgtgtagataactacgatacgggag4757
ggcttaccatctggccccagtgctgcaatgataccgcgagacccacgctcaccggctcca4817
gatttatcagcaataaaccagccagccggaagggccgagcgcagaagtggtcctgcaact4877
ttatccgcctccatccagtctattaattgttgccgggaagctagagtaagtagttcgcca4937
gttaatagtttgcgcaacgttgttgccattgctacaggcatcgtggtgtcacgctcgtcg4997
tttggtatggcttcattcagctccggttcccaacgatcaaggcgagttacatgatccccc5057
atgttgtgcaaaaaagcggttagctccttcggtcctccgatcgttgtcagaagtaagttg5117
gccgcagtgttatcactcatggttatggcagcactgcataattctcttactgtcatgcca5177
tccgtaagatgcttttctgtgactggtgagtactcaaccaagtcattctgagaatagtgt5237
atgcggcgaccgagttgctcttgcccggcgtcaatacgggataataccgcgccacatagc5297
agaactttaaaagtgctcatcattggaaaacgttcttcggggcgaaaactctcaaggatc5357
ttaccgctgttgagatccagttcgatgtaacccactcgtgcacccaactgatcttcagca5417
tcttttactttcaccagcgtttctgggtgagcaaaaacaggaaggcaaaatgccgcaaaa5477
aagggaataagggcgacacggaaatgttgaatactcatactcttcctttttcaatattat5537
tgaagcatttatcagggttattgtctcatgagcggatacatatttgaatgtatttagaaa5597
aataaacaaataggggttccgcgcacatttccccgaaaagtgccacctgacgtc 5651
<210> 5
<211> 92
<212> PRT
<213> Homo Sapiens
<400> 5
Ala Pro Asp Val Gln Asp Cys Pro Glu Cys Thr Leu Gln Glu Asn Pro
1 5 10 15
Phe Phe Ser Gln Pro Gly Ala Pro Ile Leu Gln Cys Met Gly Cys Cys
20 25 30
Phe Ser Arg Ala Tyr Pro Thr Pro Leu Arg Ser Lys Lys Thr Met Leu
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
35 40 45
Val Gln Lys Asn Val Thr Ser Glu Ser Thr Cys Cys Val Ala Lys Ser
50 55 60
Tyr Asn Arg Val Thr Val Met Gly Gly Phe Lys Val Glu Asn His Thr
65 70 75 80
Ala Cys His Cys Ser Thr Cys Tyr Tyr His Lys Ser
85 90
<210> 6
<211> 111
<212> PRT
<213> Homo Sapiens
<400> 6
Asn Ser Cys Glu Leu Thr Asn Ile Thr Ile Ala Ile Glu Lys Glu Glu
1 5 10 15
Cys Arg Phe Cys Ile Ser Ile Asn Thr Thr Trp Cys Ala Gly Tyr Cys
20 25 30
Tyr Thr Arg Asp Leu Val Tyr Lys Asp Pro AIa Arg Pro Lys Ile Gln
35 40 45
Lys Thr Cys Thr Phe Lys Glu Leu Val Tyr Glu Thr Val Arg Val Pro
50 55 60
Gly Cys Ala His His Ala Asp Ser Leu Tyr Thr Tyr Pro Val Ala Thr
65 70 75 80
Gln Cys His Cys Gly Lys Cys Asp Ser Asp Ser Thr Asp Cys Thr Val
85 90 95
Arg Gly Leu Gly Pro Ser Tyr Cys Ser Phe Gly Glu Met Lys Glu
100 105 110
<210> 7
<211> 6213
<212> DNA
<213> Artificial sequence
<220>
<221> exon
<222> (1225)..(1599)
<223> Coding sequence for modified FSH-alpha
<400> 7
gacggatcgg gagatctccc gatcccctat ggtcgactct cagtacaatc tgctctgatg 60
ccgcatagtt aagccagtat ctgctccctg cttgtgtgtt ggaggtcgct gagtagtgcg 120
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
cgagcaaaatttaagctacaacaaggcaaggcttgaccgacaattgcatgaagaatctgc180
ttagggttaggcgttttgcgctgcttcgcgatgtacgggccagatatacgcgttgacatt240
gattattgactagttattaatagtaatcaattacggggtcattagttcatagcccatata300
tggagttccgcgttacataacttacggtaaatggcccgcctggctgaccgcccaacgacc360
cccgcccattgacgtcaataatgacgtatgttcccatagtaacgccaatagggactttcc420
attgacgtcaatgggtggactatttacggtaaactgcccacttggcagtacatcaagtgt480
atcatatgccaagtacgccccctattgacgtcaatgacggtaaatggcccgcctggcatt540
atgcccagtacatgaccttatgggactttcctacttggcagtacatctacgtattagtca600
tcgctattaccatggtgatgcggttttggcagtacatcaatgggcgtggatagcggtttg660
actcacggggatttccaagtctccaccccattgacgtcaatgggagtttgttttggcacc720
aaaatcaacgggactttccaaaatgtcgtaacaactccgccccattgacgcaaatgggcg780
gtaggcgtgtacggtgggaggtctatataagcagagctctctggctaactagagaaccca840
ctgcttactggcttatcgaaattaatacgactcactatagggagacccaagctggctagc900
ttattgcggtagtttatcacagttaaattgctaacgcagtcagtgcttctgacacaacag960
tctcgaacttaagctgcagtgactctcttaaggtagccttgcagaagttggtcgtgaggc1020
actgggcaggtaagtatcaaggttacaagacaggtttaaggagaccaatagaaactgggc1080
ttgtcgagacagagaagactcttgcgtttctgataggcacctattggtcttactgacatc1140
cactttgcctttctctccacaggtgtccactcccagttcaattacagctcttaaaagctt1200
ggtaccgagctcggatccgccacc atg gcc gcc 1251
gac tac
tac cgc
aag tac
Met Asp Ala Ala
Tyr Tyr
Arg Lys
Tyr
1 5
atc ttc gtg acc gtg ctg agc gcc 1299
ctg ctg cac
agc
gtg
ttc
ctg
cac
Ile Phe Val Thr Val Leu Ser Ala
Leu Leu His
Ser
Val
Phe
Leu
His
15 20 25
aac atc gtt aac gtg cag tgc ccc 1347
acc atc gac
acc
gtg
gcc
ccc
gac
Asn Ile Val Asn e Thr Val Gln Cys Pro
Thr Il Val Ala Asp
Pro Asp
30 35 40
gag tgc acc ctg cag gag aac ccc ttc ttc agc cag ccc ggc gcc ccc 1395
Glu Cys Thr Leu Gln Glu Asn Pro Phe Phe Ser Gln Pro Gly Ala Pro
45 50 55
atc ctg cag tgc atg ggc tgc tgc ttc agc cgc gcc tac ccc acc ccc 1443
Ile Leu Gln Cys Met Gly Cys Cys Phe Ser Arg Ala Tyr Pro Thr Pro
60 65 70
ctg cgc agc aag aag acc atg ctg gtg cag aag aac gtg acc agc gag 1491
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
Leu Arg Ser Lys Lys Thr Met Leu Val Gln Lys Asn Val Thr Ser Glu
75 80 85
agc acc tgc tgc gtg gcc aag agc tac aac cgc gtg acc gtg atg ggc 1539
Ser Thr Cys Cys Val Ala Lys Ser Tyr Asn Arg Val Thr Val Met Gly
90 95 100 105
ggc ttc aag gtg gag aac cac acc gcc tgc cac tgc agc acc tgc tac 1587
Gly Phe Lys Val Glu Asn His Thr Ala Cys His Cys Ser Thr Cys Tyr
110 115 120
tac cac aag agc taatctagag ggcccgttta aacccgctga tcagcctcga 1639
Tyr His Lys Ser
125
ctgtgccttctagttgccagCCatCtgttgtttgCCCCtCCCCCgtgCCttCCttgaCCC1699
tggaaggtgccactcccactgtcctttcctaataaaatgaggaaattgcatcgcattgtc1759
tgagtaggtgtcattctattctggggggtggggtggggcaggacagcaagggggaggatt1819
gggaagacaatagcaggcatgctggggatgcggtgggctctatggcttctgaggcggaaa1879
gaaccagctggggctctagggggtatccccacgcgccctgtagcggcgcattaagcgcgg1939
cgggtgtggtggttacgcgcagcgtgaccgctacacttgccagcgccctagcgcccgctc1999
ctttcgctttcttcccttcctttctcgccacgttcgccggctttccccgtcaagctctaa2059
atcggggcatccctttagggttccgatttagtgctttacggcacctcgaccccaaaaaac2119
ttgattagggtgatggttcacgtagtgggccatcgccctgatagacggtttttcgccctt2179
tgacgttggagtccacgttctttaatagtggactcttgttccaaactggaacaacactca2239
accctatctcggtctattcttttgatttataagggattttggggatttcggcctattggt2299
taaaaaatgagctgatttaacaaaaatttaacgcgaattaattctgtggaatgtgtgtca2359
gttagggtgtggaaagtccccaggctccccaggcaggcagaagtatgcaaagcatgcatc2419
tcaattagtcagcaaccaggtgtggaaagtccccaggctccccagcaggcagaagtatgc2479
aaagcatgcatctcaattagtcagcaaccatagtcccgcccctaactccgcccatcccgc2539
ccctaactccgcccagttccgcccattctccgccccatggctgactaattttttttattt2599
atgcagaggccgaggccgcctctgcctctgagctattccagaagtagtgaggaggctttt2659
ttggaggcctaggcttttgcaaaaagctcccgggagcttgtatatccattttcggatctg2719
atcagcacgtgatgaaaaagcctgaactcaccgcgacgtctgtcgagaagtttctgatcg2779
aaaagttcgacagcgtctccgacctgatgcagctctcggagggcgaagaatctcgtgctt2839
tcagcttcgatgtaggagggcgtggatatgtcctgcgggtaaatagctgcgccgatggtt2899
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
tctacaaagatcgttatgtttatcggcactttgcatcggccgcgctcccgattccggaag2959
tgcttgacattggggaattcagcgagagcctgacctattgcatctcccgccgtgcacagg3019
gtgtcacgttgcaagacctgcctgaaaccgaactgcccgctgttctgcagccggtcgcgg3079
aggccatggatgcgatcgctgcggccgatcttagccagacgagcgggttcggcccattcg3139
gaccgcaaggaatcggtcaatacactacatggcgtgatttcatatgcgcgattgctgatc3199
cccatgtgtatcactggcaaactgtgatggacgacaccgtcagtgcgtccgtcgcgcagg3259
ctctcgatgagctgatgctttgggccgaggactgccccgaagtccggcacctcgtgcacg3319
cggatttcggctccaacaatgtcctgacggacaatggccgcataacagcggtcattgact3379
ggagcgaggcgatgttcggggattcccaatacgaggtcgccaacatcttcttctggaggc3439
cgtggttggcttgtatggagcagcagacgcgctacttcgagcggaggcatccggagcttg3499
caggatcgccgcggctccgggcgtatatgctccgcattggtcttgaccaactctatcaga3559
gcttggttgacggcaatttcgatgatgcagcttgggcgcagggtcgatgcgacgcaatcg3619
tccgatccggagccgggactgtcgggcgtacacaaatcgcccgcagaagcgcggccgtct3679
ggaccgatggctgtgtagaagtactcgccgatagtggaaaccgacgccccagcactcgtc3739
cgagggcaaaggaatagcacgtgctacgagatttcgattccaccgccgccttctatgaaa3799
ggttgggcttcggaatcgttttccgggacgccggctggatgatcctccagcgcggggatc3859
tcatgctggagttcttcgcccaccccaacttgtttattgcagcttataatggttacaaat3919
aaagcaatagcatcacaaatttcacaaataaagcatttttttcactgcattctagttgtg3979
gtttgtccaaactcatcaatgtatcttatcatgtctgtataccgtcgacctctagctaga4039
gcttggcgtaatcatggtcatagctgtttcctgtgtgaaattgttatccgctcacaattc4099
cacacaacatacgagccggaagcataaagtgtaaagcctggggtgcctaatgagtgagct4159
aactcacattaattgcgttgcgctcactgcccgctttccagtcgggaaacctgtcgtgcc4219
agctgcattaatgaatcggccaacgcgcggggagaggcggtttgcgtattgggcgctctt4279
ccgcttcctcgctcactgactcgctgcgctcggtcgttcggctgcggcgagcggtatcag4339
ctcactcaaaggcggtaatacggttatccacagaatcaggggataacgcaggaaagaaca4399
tgtgagcaaaaggccagcaaaaggccaggaaccgtaaaaaggccgcgttgctggcgtttt4459
tccataggctccgcccccctgacgagcatcacaaaaatcgacgctcaagtcagaggtggc4519
gaaacccgacaggactataaagataccaggcgtttccccctggaagctccctcgtgcgct4579
ctcctgttccgaccctgccgcttaccggatacctgtccgcctttctcccttcgggaagcg4639
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
tggcgctttctcaatgctcacgctgtaggtatctcagttcggtgtaggtcgttcgctcca4699
agctgggctgtgtgcacgaaccccccgttcagcccgaccgctgcgccttatccggtaact4759
atcgtcttgagtccaacccggtaagacacgacttatcgccactggcagcagccactggta4819
acaggattagcagagcgaggtatgtaggcggtgctacagagttcttgaagtggtggccta4879
actacggctacactagaaggacagtatttggtatctgcgctctgctgaagccagttacct4939
tcggaaaaagagttggtagctcttgatccggcaaacaaaccaccgctggtagcggtggtt4999
tttttgtttgcaagcagcagattacgcgcagaaaaaaaggatctcaagaagatcctttga5059
tcttttctacggggtctgacgctcagtggaacgaaaactcacgttaagggattttggtca5119
tgagattatcaaaaaggatcttcacctagatccttttaaattaaaaatgaagttttaaat5179
caatctaaagtatatatgagtaaacttggtctgacagttaccaatgcttaatcagtgagg5239
cacctatctcagcgatctgtctatttcgttcatccatagttgcctgactccccgtcgtgt5299
agataactacgatacgggagggcttaccatctggccccagtgctgcaatgataccgcgag5359
acccacgctcaccggctccagatttatcagcaataaaccagccagccggaagggccgagc5419
gcagaagtggtcctgcaactttatccgcctccatccagtctattaattgttgccgggaag5479
ctagagtaagtagttcgccagttaatagtttgcgcaacgttgttgccattgctacaggca5539
tcgtggtgtcacgctcgtcgtttggtatggcttcattcagctccggttcccaacgatcaa5599
ggcgagttacatgatcccccatgttgtgcaaaaaagcggttagctccttcggtcctccga5659
tcgttgtcagaagtaagttggccgcagtgttatcactcatggttatggcagcactgcata5719
attctcttactgtcatgccatccgtaagatgcttttctgtgactggtgagtactcaacca5779
agtcattctgagaatagtgtatgcggcgaccgagttgctcttgcccggcgtcaatacggg5839
ataataccgcgccacatagcagaactttaaaagtgctcatcattggaaaacgttcttcgg5899
ggcgaaaactctcaaggatcttaccgctgttgagatccagttcgatgtaacccactcgtg5959
cacccaactgatcttcagcatcttttactttcaccagcgtttctgggtgagcaaaaacag6019
gaaggcaaaatgccgcaaaaaagggaataagggcgacacggaaatgttgaatactcatac6079
tcttcctttttcaatattattgaagcatttatcagggttattgtctcatgagcggataca6139
tatttgaatgtatttagaaaaataaacaaataggggttccgcgcacatttccccgaaaag6199
tgccacctgacgtc 6213
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<210> 8
<211> 5
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD_RES
<222> (5)
<223> T or S
<400> 8
Ala Ser Asn Ile Xaa
1 5
<210> 9
<211> 6
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide ,
<220>
<221> MOD RES
<222> (6)
<223> T or S
<400> 9
Ser Pro Ile Asn Ala Xaa
1 5
<210> 10
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD RES
<222> (7)-
<223> T or S
<400> 10
Ala Ser Pro Ile Asn Ala Xaa
1 5
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<210> 11
<211> 11
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD RES
<222> (4)
<223> T or S
<220>
<221> MOD_RES
<222> (8)
<223> T or S
<400> 11
Ala Asn Ile Xaa Ala Asn Ile Xaa Ala Asn Ile
1 5 10
<210> 22
<211> 14
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD RES
<222> (4)
<223> T or S
<220>
<221> MOD RES
<222> (9)
<223> T or S
<220>
<221> MOD_RES
<222> (14)
<223> T or S
<400> 12
Ala Asn Ile Xaa Gly Ser Asn Ile Xaa Gly Ser Asn Ile Xaa
1 5 10
<210> 13
<211> 13
<212> PRT
<213> Artificial Sequence
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD RES
<222> (5)
<223> T or S
<220>
<221> MOD_RES
<222> (9)
<223> T or S
<220>
<221> MOD_RES
<222> (13)
<223> T or S
<400> 13
Ala Ser Asn Ser Xaa Asn Asn Gly Xaa Leu Asn Ala Xaa
1 5 10
<210> 14
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD RES
<222> (4)
<223> T or S
<220>
<221> MOD RES
<222> (7)-
<223> T or S
<220>
<221> MOD_RES
<222> (10)
<223> T or S
<400> 14
Ala Asn His Xaa Asn Glu Xaa Asn Ala Xaa
1 5 10
<210> 15
<211> 7
<212> PRT
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD_RES
<222> (7)
<223> T or S
<400> 15
Gly Ser Pro Ile Asn Ala Xaa
1 5
<210> 16
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD RES
<222> (7)-
<223> T or S
<220>
<221> MOD_RES
<222> (13)
<223> T or S
<400> 16
Ala Ser Pro Ile Asn Ala Xaa Ser Pro Ile Asn Ala Xaa
1 5 10
<210> 17
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD_RES
<222> (4)
<223> T or S
<220>
<221> MOD_RES
<222> (7)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<223> T or S
<220>
<221> MOD_RES
<222> (10)
<223> T or S
<400> 17
Ala Asn Asn Xaa Asn Tyr Xaa Asn Trp Xaa
1 5 10
<210> 18
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD RES
<222> (5)
<223> T or S
<220>
<221> MOD_RES
<222> (9)
<223> T or S
<220>
<221> MOD_RES
<222> (12)
<223> T or S
<400> 18
Ala Thr Asn Ile Xaa Leu Asn Tyr Xaa Ala Asn Xaa Thr
1 5 10
<210> 19
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD_RES
<222> (5)
<223> T or S
<220>
<221> MOD_RES
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<222> (9)
<223> T or S
<220>
<221> MOD_RES
<222> (13)
<223> T or S
<400> 19
Ala Ala Asn Ser Xaa Gly Asn Ile Xaa Ile Asn Gly Xaa
1 5 10
<210> 20
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD_RES
<222> (5)
<223> T or S
<220>
<221> MOD_RES
<222> (9)
<223> T or S
<220>
<221> MOD_RES
<222> (13)
<223> T or S
<400> 20
Ala Val Asn Trp Xaa Ser Asn Asp Xaa Ser Asn Ser Xaa
1 5 10
<210> 21
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD_RES
<222> (5)
<223> T or S
<220>
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<221> MOD_RES
<222> (9)
<223> T or S
<220>
<221> MOD_RES
<222> (13)
<223> T or S
<400> 21
Ala Val Asn Trp Xaa Ser Asn Asp Xaa Ser Asn Ser Xaa
1 5 10
<210> 22
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD_RES
<222> (4)
<223> T or S
<220>
<221> MOD_RES
<222> (7)
<223> T or S
<220>
<221> MOD_RES
<222> (10)
<223> T or S .
<400> 22
Ala Asn Asn Xaa Asn Tyr Xaa Asn Ser Xaa
1 5 10
<210> 23
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 23
Ala Asn Asn Thr Asn Tyr Thr Asn Trp Thr
1 5 10
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<210> 24
<211> 15
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Linker
<400> 24
Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser
1 5 10 15
<210> 25
<211> 35
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Primer
<400> 25
cgcagatctg atggctggca gcctcacagg attgc 35
<210> 26
<211> 37
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Primer
<400> 26
ccggaattcc catcactggc gacgccacag gtaggtg 37
<210> 27
<211> 35
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Primer
<400> 27
acgcgagctc gcccctgcat ccctaaaagc ttcgg 35
<210> 28
<211> 54
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Primer
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<400> 28
gcgttgacgg cagtcagagt tgacagaagg gccagccagc aaaggatagt catg 54
<210> 29
<211> 62
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Primer
<400> 29
ctagcatgac tatcctttgc tggctggccc ttctgtcaac tctgactgcc gtcaacgcag 60
ct 62
<210> 30
<211> 48
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Primer
<400> 30
cctgctactg ctcccagcag cagtgaaaga gtccaaagtg gcagcatg 48
<210> 31
<211> 56
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Primer
<400> 31
ctagcatgct gccactttgg actctttcac tgctgctggg agcagtagca ggagct 56
<210> 32
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Primer
<400> 32
cagctggcca tgggtacccg g 21
<210> 33
<211> 4
<212> PRT
<213> Artificial Sequence
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<220>
<223> Description of Artificial Sequence: N-terminal
peptide addition
<400> 33
Ala Asn Ile Thr
1
<210> 34
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: N-terminal
peptide addition
<400> 34
Ala Ser Pro Ile Asn Ala Thr
1 5
<210> 35
<211> 48
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Primer
<400> 35
tgggcatcag gtgccaacat tacagcccgc ccctgcatcc ctaaaagc 48
<210> 36
<211> 24
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Primer
<400> 36
tttactgttt tcgtaacagt tttg 24
<210> 37
<211> 48
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Primer
<400> 37
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
gcaggggcgg gctgtaatgt tggcacctga tgcccacgac actgcctg 48
<210> 38
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD_RES
<222> (1) .(13)
<223> "Xaa" represents a variable amino acid
<400> 38
Ala Xaa Asn Xaa Thr Xaa Asn Xaa Thr Xaa Asn Xaa Thr
1 5 10
<210> 39
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD_RES
<222> (1) .(10)
<223> "Xaa" represents a variable amino acid
<400> 39
Ala Asn Xaa Thr Asn Xaa Thr Asn Xaa Thr
1 5 10
<220> 40
<211> 81
<212> DNA
<213> Artificial Sequence
<220>
<221> modified_base
<222> (1)..(81)
<223> "n" represents a, t, c, g, other or unknown
<220>
<223> Description of Artificial Sequence: Primer
<400> 40
gtgtcgtggg catcaggtgc cnnsaaydns achdnsaayd nsachdnsaa ydnsachgcc 60
cgcccctgca tccctaaaag c 81
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<210> 41
<211> 27
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Primer
<400> 42
ggcacctgat gcccacgaca ctgcctg 27
<210> 43
<211> 68
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Primer
<220>
<221> modified_base
<222> (1)..(68)
<223> "nnn" is a mixture of trinucleotide colons for all
natural amino acid residues, except proline
<400> 43
cgtgggcatc aggtgccaac nnnachaayn nnachaaynn nachgcccgc ccctgcatcc 60
ctaaaagc 68
<210> 44
<211> 30
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Primer
<400> 44
gttggcacct gatgcccacg acactgcctg 30
<210> 45
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD_RES
<222> (4)
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<223> variable amino acid
<220>
<221> MOD_RES
<222> (12)
<223> F or L
<400> 45
Ala Phe Asn Xaa Thr Leu Asn Lys Thr Trp Asn Xaa Thr
1 5 10
<210> 46
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial. Sequence: Synthetic
peptide
<400> 46
Thr Met Asn Asn Thr Trp Asn Trp Thr Trp Asn Trp Thr
1 5 10
<210> 47
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 47
Ala Leu Asn Ser Thr Gly Asn Leu Thr Val Asp Gly Thr
1 5 10
<210> 48
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 48
Ala Ser Asn Ser Thr Phe Asn Leu Thr Glu Asn Leu Thr
1 5 10
<210> 49
<211> 12
<212> PRT
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 49
Thr Arg Asn Val Thr Ile Asn Cys Thr Asn Ser Thr
1 5 l0
<210> 50
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 50
Ala Leu Asn Trp Thr Tyr Asn Gly Thr Lys Asn Val Thr
1 5 10
<210> 51
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 51
Ala Ala Asn Trp Thr Val Asn Phe Thr Gly Asn Phe Thr
1 5 10
<210> 52
<211> 12
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD RES
<222> (2)
<223> variable amino acid
<220>
<221> MOD_RES
<222> (4)
<223> variable amino acid
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<400> 52
Ala Xaa Asn Xaa Thr Val Asn Ser Thr Asn Val Thr
1 5 10
<210> 53
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 53
Ala Asn Asn Phe Thr Phe Asn Gly Thr Leu Asn Leu Thr
1 5 l0
<210> 54
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 54
Ala Gly Asn Trp Thr Ala Asn Val Thr Val Asn Val Thr
1 5 10
<210> 55
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 55
Ala Gly Asn Ser Thr Ser Asn Val Thr Gly Asn Trp Thr
1 5 10
<210> 56
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<400> 56
Ala Val Asn Ser Thr Met Asn Ile His Ala Ile Pro Pro
1 5 10
<210> 57
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 57
Ala Gly Asn Gly Thr Val Asn Gly Thr Ile Asn Gly Thr
1 5 10
<210> 58
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD RES
<222> (8)-
<223> variable amino acid
<400> 58
Ala Val Asn Ser Thr Gly Asn Xaa Thr G1y Asn Trp Thr
1 5 10
<210> 59
<211> 12
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 59
Ala Gly Asn Gly Thr Asn Gly Thr Ser Asn Leu Thr
1 5 10
<210> 60
<211> 13
<212> PRT
<213> Artificial Sequence
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 60
Ala Met Asn Ser Thr Lys Asn Ser Thr Leu Asn Ile Thr
1 5 10
<210> 61
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 61
Ala Phe Asn Tyr Thr Ser Lys Asn Ser Thr
1 5 10
<210> 62
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 62
Ala Val Asn Ala Thr Met Asn Trp Thr Ala Asn Gly Thr
1 5 10
<210> 63
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 63
Ala Ser Asn Ser Thr Asn Asn Gly Thr Leu Asn Ala Thr
1 5 10
<210> 64
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 64
Ala Arg Asn Lys Thr Lys Asn Phe Thr I1e Asn Leu Thr
1 5 10
<210> 65
<211> 12
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 65
Ala Pro Asn Ile Thr Asn Asp Thr Val Asn Met Thr
1 5 10
<210> 66
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 66
Ala Gln Asn Lys Thr Phe Asn Phe Thr Met Asn Cys Thr
1 5 10
<210> 67
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 67
Ala Leu Asn Val Thr Trp Asn Cys Thr Leu Asn Leu Thr
1 5 10
<210> 68
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
peptide
<400> 68
Ala Leu Asn Thr Thr Trp Thr Asn Leu Thr
1 5 10
<210> 69
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 69
Ala Asn Thr Thr Asn Phe Thr Asn Glu Thr
1 5 10
<210> 70
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 70
Ala Asn Trp Thr Asn Arg Thr Asn Cys Thr
1 5 10
<210> 71
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 71
Ala Asn Trp Thr Asn Phe Thr Asn Trp Thr
1 5 10
<210> 72
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<400> 72
Pro Thr Gly Leu Ile Gly Thr Asn Phe Thr
1 5 10
<210> 73
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 73
Ala Asn Trp Thr Asn Lys Thr Asn Phe Thr
1 5 10
<210> 74
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 74
Ala Asn Asn Thr Asn Leu Thr Asn Ala Thr
1 5 10
<210> 75
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 75
Ala Asn Tyr Thr Asn Trp Thr Asn Phe Thr
1 5 10
<210> 76
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<400> 76
Ala Asn Thr Thr Asn Gln Thr Asn Asp Thr
1 5 10
<210> 77
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 77
Ala Asn Arg Thr Asn Trp Thr Asn Thr Thr
1 5 10
<210> 78
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 78
Pro Thr Ala Thr Asn His Thr Asn Ser Thr
1 5 10
<210> 79
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 79
Ala Asn Trp Thr Asn Gln Thr Asn Gln Thr
1 5 10
<210> 80
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 80
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
Ala Asn Trp Thr Asn Trp Thr Asn Ala Thr
1 5 10
<210> 81
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 82
Ala Asn Phe Thr Asn Lys Thr Asn Met Thr
1 5 10
<210> 83
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 83
Ala Asn His Thr Asn Glu Thr Asn Ala Thr
1 5 10
<210> 84
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD_RES
<222> (3)
<223> C or W
<400> 84
Ala Asn Xaa Thr Asn Phe Thr Asn Glu Thr
1 5 10
<210> 85
<211> 9
<212> PRT
<213> Artificial Sequence
<220>
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 85
Ala Asn Leu Asp Lys Leu His Lys His
1 5
<210> 86
<211> 11
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 86
Ala Asn Cys Phe Thr Asn G1n Thr Asn Phe Thr
1 5 10
<210> 87
<211> 11
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 87
Ala Asn Trp Thr Asn Trp Thr Asn Glu Trp Thr
1 5 10
<210> 88
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 88
Ala Asn Cys Thr Asn Trp Thr Asn Cys Thr
1 5 10
<210> 89
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
CA 02412882 2002-12-13
WO 02/02597 - PCT/DKO1/00459
peptide
<400> 89
Cys His Pro Tyr Asn Trp Thr Asn Trp Thr
1 5 10
<210> 90
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 90
Ala Asn Glu Thr Asn Tyr Thr Asn Glu Thr
1 5 10
<210> 91
<211> 7
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 91
Ala Asn Trp Thr Asn Trp Thr
1 5
<210> 92
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 92
Ala Lys Pro Tyr Lys Ser Tyr Lys Phe Tyr
1 5 10
<210> 93
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
CA 02412882 2002-12-13
WO 02/02597 - PCT/DKO1/00459
<400> 93
Ala Asn Ile Thr Asn Lys Thr Asn Trp Thr
1 5 10
<210> 94
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 94
Ala Asn Trp Thr Asn Met Thr Asn Ile Thr
1 5 10
<210> 95
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 95
Ala Asn Asn Thr Asn Arg Thr Asn Phe Thr
1 5 10
<210> 96
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 96
Ala Asn Trp Thr Asn Trp Thr Asn Trp Thr
1 5 10
<210> 97
<211> 11
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<400> 97
Ala Asn Trp Arg Thr Asn His Thr Asn Lys Thr
1 5 10
<210> 98
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 98
Ala Asn Gln Thr Asn 21e Thr Asn Trp Thr
1 5 10
<210> 99
<211> 11
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 99
Ala Asn Phe Thr Asn Val Ala Thr Asn Gln Thr
1 5 10
<210> 100
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD_RES
<222> (1)
<223> most probable amino acid
<220>
<221> MOD_RES
<222> (2)
<223> most probable amino acid
<220>
<221> MOD_RES
<222> (5)
<223> variable amino acid
CA 02412882 2002-12-13
WO 02/02597 - PCT/DKO1/00459
<220>
<221> MOD RES
<222> (9)
<223> most probable amino acid
<400> 100
A1a Asn Thr Thr Xaa Leu Thr Asn Lys Thr
1 5 10
<210> 101
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD_RES
<222> (6)
<223> S or C
<400> 101
A1a Asn Lys Thr Asn Xaa Thr Asn Ile Thr
1 5 10
<210> 102
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<220>
<221> MOD_RES
<222> (9)
<223> most probable amino acid
<400> 102
Ala Asn Trp Thr Asn Cys Thr Asn Ile Thr
1 5 10
<210> 103
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
CA 02412882 2002-12-13
WO 02/02597 ~ PCT/DKO1/00459
<220>
<221> MOD_RES
<222> (6)
<223> F or L
<400> 103
Ala Asn Trp Thr Asn Xaa Thr Asn Trp Thr
1 5 10
<210> 104
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 104
Cys Gln Leu Asp Arg Ser Thr Asn Glu Thr
1 5 10
<210> 105
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 105
Ala Asn Asn Thr Asn Tyr Thr Asn Trp Thr
1 5 10
<210> 106
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 106
Ala Asn Asn Thr Asn Tyr Thr Asn Trp Thr
1 5 10
<210> 107
<211> 12
<212> PRT
<213> Artificial Sequence
CA 02412882 2002-12-13
WO 02/02597 PCT/DKO1/00459
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 107
Ala Ala Asn Asp Thr Asn Trp Thr Val Asn Cys Thr
1 5 10
<210> 108
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 108
Ala Thr Asn Ile Thr Leu Asn Tyr Thr Ala Asn Thr Thr
1 5 10
<210> 109
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 109
Ala Ala Asn Ser Thr Gly Asn Ile Thr Ile Asn Gly Thr
1 5 10
<210> 110
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 110
Ala Val Asn Trp Thr Ser Asn Asp Thr Ser Asn Ser Thr
1 5 10
<210> 111
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
CA 02412882 2002-12-13
WO 02/02597 - PCT/DKO1/00459
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 111
Ala Ser Pro Ile Asn Ala Thr Ser Pro Ile Asn Ala Thr
1 5 10
<210> 112
<211> 4
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Linker
<400> 112
Gly Gly Gly Gly
1
<210> 113
<211> 4
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Linker
<400> 113
Gly Asn Ala Thr
<210> 114
<211> 8
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 114
Asn Ser Thr Gln Asn Ala Thr Ala
1 5
<210> 115
<211> 14
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
peptide
<400> 115
Ala Asn Leu Thr Val Arg Asn Leu Thr Arg Asn Val Thr Val
Image