Note: Descriptions are shown in the official language in which they were submitted.
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
PRION-BINDING ACTIVITY IN SERUM AND PLASMA DETERMINED AS PLASMINOGEN AND
FIBRINOGEN
Field of the Invention
The present invention concerns a method and
agents to detect transmissible spongiform encephalopathies
as well as agents for the prevention and treatment of
respective infections.
Background Art
According to all available evidence, the agents
causing transmissible spongiform encephalopathies, termed
prions, are devoid of informational nucleic acids and
consist of an "infectious" protein (termed PrPSc) capable
of converting a normal host protein called PrPC into a
likeness of themselves. The only organ system in which
histopathological damage and its clinical sequelae can be
demonstrated as a consequence of infection with prions is
the nervous system (Brandner et al., 1996). This
consideration applies to both the human transmissible
spongiform encephalopathies, such as Creutzfeldt-Jakob
disease, Gerstmann-Straussler-Scheinker Syndrome, Kuru and
fatal familial insomnia, and all known prion
encephalopathies of animals (Weber and Aguzzi, 1997). The
latter comprise scrapie in sheep, bovine spongiform
encephalopathy, and chronic wasting diseases of mule, deer
and exotic ungulates (GVeissmann and Aguzzi, 1997).
However, there is no doubt that prions,
herewith operationally defined as the infectious agents
causing transmissible spongiform encephalopathies, can
colonize organs other than the central and peripheral
nervous system, and can be demonstrated in extracerebral
compartments (Aguzzi et al., 1997). The problem of which
organ systems can harbour infectivity is further
complicated by the existence of prion strains. Just like
strains of conventional viruses, prions can come in various
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
2
different flavors, each one of which has its specific
preferences with regard to the host range which is
infectible and also to the type of cells in which it
replicates (Aguzzi, 1998). One paradoxical situation, which
is of immediate relevance to the question of blood safety,
is exemplified by the radically different organ tropism of
the BSE agent in cows and in humans. BSE prions seem to be
largely confined to the neural compartment of cows, even
after oral exposure (Wells et al., 1998). A very accurate
study of the pathogenesis of experimental BSE in cows upon
feeding 100 grams of infected brain has disclosed that
there is only a short and transient period during which
infectivity can be demonstrated in the terminal ileum
(Wells et al., 1998). At later time points, BSE prions can
only be shown in brain, spinal cord, and dorsal root
ganglia. The exact localization of BSE in the terminal
ileum is not known. It is being discussed whether
infectivity resides in Pet'er's patches or in the neural
compartment which comprises the Plexus submucosus Meissner
and the Plexus myentericus Auerbach. There is a great body
of circumstantial evidence that BSE prions can provoke new
variant Creutzfeldt-Jakob Disease (nvCJD) (Bruce et al.,
1997; Chazot et al., 1996; Hill et al., 1997; Will et al.,
1996), but no absolutely final evidence has been produced.
For the purpose of the following discussion we will regard
the evidence that BSE and new variant Creutzfeldt-Jakob
Disease caused by the same agent as sufficiently verified
(Aguzzi and Weissmann, 1996). Upon passage into humans, and
consecutive progression to manifest nvCJD, prions
experience a dramatic shift in their organotropism. Instead
of remaining confined mainly to neural structures, they can
be detected in many organs belonging to the immune system,
including most notably tonsils, spleen, and as recently
demonstrated, the appendix (Hilton et al., 1998). It is,
therefore, unavoidable to conclude that the tropism of the
infectious agent for various structures depends on both the
strains of prions in question (and therefore it is in part
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
3
autonomous to its carrier) and on the species in which
prion disease manifests itself (Aguzzi and V7eissmann,
1998).
These considerations are not only of academic
interest. In fact, the transmissibility of the agent by
iatrogenic manipulations (i.e. blood transfusions, organ
transplants, etc.) is crucially affected by such
parameters.
Horizontal Transmissibility of Human Prions:
Prion diseases of humans are undoubtedly
transmissible. However, transmission is achieved only under
particular circumstances. One could say that in this
respect prion diseases fulfill the characteristics of
transmissibility delineated by Semmelweiss for puerperal
fever: these affections are infectious but not contagious.
Direct transmissions of brain-derived material from a
patient suffering from Creutzfeldt-Jakob disease to other
persons have documentedly resulted in transmission of
disease. A particularly tragic case occurred in the early
seventies in Zurich, when electrodes used for cortical
recordings from Creutzfeldt-Jakob patients were sterilized
(formaldehyde and alcohol) and used in additional patients.
Disease was transmitted to the very young recipients
(Bernoulli et al., 1977). Also, transplantation of cornea
has most likely resulted in transmission of disease (Duffy
et al., 1974).
Despite these tragic dimensions, cases of
iatrogenic transmission of CJD via neurosurgical procedures
have remained rather rare. This is not totally understood,
given that the frequency of subclinical CJD must be much
higher than that of manifest disease, and that most
neurosurgical instruments are not sterilized in a way that
would reliably inactivate prions. Therefore, the quite rare
nature of iatrogenic transmission is likely to indicate
that host factors, in addition to the virulence of prions,
may affect the probability that infection takes place. This
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
4
notion is strengthened by the epidemiology of iatrogenic
CJD (iCJD) upon transmission of contaminated dura mater. It
has been estimated that several thousands patients,
predominantly in Japan, may have been exposed to the CJD
agent via preparations of cadaveric dura mater which had
been contaminated with prions. However, it appears that
less then 2% of those exposed have developed disease so
far. While we can rejoice about this low efficiency in the
"take" of infectivity, we do not fully understand the
biological basis for the apparent protection enjoyed by
most subjects exposed to CJD prions. The largest problem
with iatrogenic transmission has occurred as result of
administration of pituitary hormones of cadaveric origin
(Gibbs et al., 1985). Preparations of growth hormone and of
gonadotropins contaminated with human prions have caused
the death of more then 80 persons, predominantly children.
Due to the long latency that can be expected when the agent
is introduced into extracerebral sites, such as via
intramuscular injection, it must be assumed that further
cases from this procedure, which has been stopped more than
a decade ago, will arise in the future.
Besides its tragic human dimension and the harm
that it has cost to the patients and to their physicians,
the pituitary hormone disaster needs to be understood in
detail, because the anterior lobe of the pituitary gland is
not a part of the central nervous system. Therefore, these
events may serve as a paradigm for transmission of prions
via contaminated extracerebral tissue that does not belong
to the canonical sites of replication of prions. The
observation that latency after intracerebral contamination
is much shorter than latency after peripheral infection is
in good agreement with experimental data from various
animal models, and suggest that a rather lengthy phase of
extracerebral events (which may include replication of the
agent, and invasion of specific extraneuronal systems) may
be a precondition to prion neuroinvasion (Aguzzi, 1997).
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
Factors influencing the neurotropism of prions:
There is good reason to suspect that
neuroinvasive processes in the course of prion infections
are very tightly controlled. Perhaps the best argument in
this respect derives from the observation that the
incubation times of experimental animals inoculated
intraperitoneally with scrapie prions are extremely
reproducible. Upon inoculation with a known amount of
standard inoculum, the experience in various laboratories
has been that latencies between inoculation and first
clinical symptoms display standard variations in the order
of only a few percent points (Klein et al., 1997). If prion
neuroinvasion were a totally random process, one would
expect a large variability in the incubation times, which
would depend on processes governed by chance. However, if
some rate-limiting processes control neuroinvasion, these
may be responsible for the remarkable precision of the
incubation times. Indeed, we very much hope that this
interpretation is correct because if such processes exist
they might be amenable to manipulation, which in turn may
represent a post-exposure strategy to prevent overt prion
disease. Indeed, various mechanisms have been explored by
which neuroinvasions may be accomplished.
A first phase or neuroinvasion seems to be
widespread colonization of the immune system. This
colonization can be visualized by homogenizing spleen,
lymph nodes, tonsils, and also appendix, and injecting the
homogenates into suitable experimental animals. The
dilution of the homogenates at which 500 of the
experimental animals become sick, contains one ID50 of the
infectious agent in each inoculum.
The second phase of neuroinvasion seems to be
dependent upon a compartment which cannot be replaced by
adoptive bone marrow transfer (Blattler et al., 1997) and
which may be represented by the peripheral nervous system
and/or the follicular dendritic cells resistant to germinal
center of secondary lymphatic organs. It appears that this
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
6
second compartment necessitates the expression of normal
prion protein in order to support neuroinvasion (Blattler
et al., 1997).
Neuroinvasion is dependent on a functional
immune system, and immunodeficient mice do not develop
disease after inoculation with a moderate dose of the agent
(Fraser et al., 1996; Kitamoto et al., 1991; Lasmezas et
al., 1996; 0'Rourke et al., 1994). One crucial component of
the immune system necessary for neuroinvasion has been
traced to the physical presence of terminally mature B-
lymphocytes . To date, it is not clear whether B cells are
required because they bind physically prions and carry them
to sites of neuroinvasion, or whether B cells produce
factors, or induce processes, which are indirectly
responsible for facilitating neuroinvasion (Klein et al.,
1997). Given the requirement for B-lymphocytes secreting
lymphotoxin for the maturation of follicular dendritic
cells, and the fact that follicular dendritic cells
accumulate large amounts of scxapie prions in experimental
situations, it is tempting to speculate that the main
function of B-lymphocytes in the aforementioned process
consists in allowing FDCs to mature.
The cellular and molecular basis of prion
neuroinvasion:
Following experimental inoculation of mice with
prions at peripheral sites, there is typically a prolonged,
clinically silent replication phase of the infectious agent
within the lymphoreticular system (LRS). This occurs prior
to detectable neuroinvasion by prions and the subsequent
occurrence of neurological symptoms. During this
preclinical latency period, prions may replicate to high
titers within lymphoreticular tissues. Elucidating the cell
types in which prions replicate within the peripheral
lymphoid tissue and - crucially - how prions are
transported to the central nervous system (CNS) is of great
interest and clinical importance. Despite considerable
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
7
evidence implicating the role of the immune system in
peripheral prion pathogenesis, there have been few studies
on the identity of the cells involved in this process. It
has been shown many years ago that whole-body irradiation
of mice with gamma rays fails to influence prion
pathogenesis or incubation time of scrapie. This has been
taken as an argument against significant involvement of
proliferating cells in the lymphoreticular phase of prion
propagation. Instead, follicular dendritic cells (FDC) have
been considered as the prime cell type for prion
replication within lymphoid tissue since PrPSc accumulates
in the follicular dendritic network of scrapie infected
wild-type and nude mice (Kitamoto et al., 1991). In
addition, severe combined immuno deficient mice (SCID),
which lack mature B- and T-cells, and which do not appear
to have functional FDCs, are highly resistant to scrapie
after intraperitoneal inoculation and fail to replicate
prions in the spleen (Fraser et al., 1996; Kitamoto et al.,
1991; Zasmezas et al., 1996; 0'Rourke et al., 1994).
Interestingly, bone-marrow reconstitution of SLID mice with
wild-type spleen cells restores full susceptibility to
scrapie after peripheral infection (Eraser et al., 1996;
Klein et al., 1998). These findings suggest that an intact,
or at least partially functional, immune system comprising
lymphocytes and FDC is required for efficient transfer of
prions from the site of peripheral infection to the CNS.
The time course for the development of scrapie
disease following intracerebral or intraperitoneal
inoculation is highly reproducible and is primarily
dependent on the dose of the inoculum. Therefore,
neuroinvasion by prions migrating from peripheral lymphoid
tissue may depend on tightly controlled, rate-limiting
reactions. In order to identify such rate-limiting steps
during prion neuroinvasion, PrPC deficient mice bearing
PrP-overexpressing cerebral neurografts were infected
intraperitoneally (i.p.). No disease was observed in the
grafts, suggesting that neuroinvasion depends on PrP
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
8
expression in extracerebral sites. This was further
underlined by reconstitution of the lymphoid system with
PrPC expressing cells, which restores infectivity in the
lymphoid tissue, but still fails to transport prions to the
nervous system.
As prions can be detected in lymphoreticular
tissues, an understanding of the peripheral pathogenesis is
of immediate importance in assessing risks of iatrogenic
transmission of human BSE via exposure to blood or tissues
from preclinical cases, and possibly from contaminated
surgical instruments, or even blood and blood products.
Additionally, such advances might pave the way for the
development of sensitive diagnostic tests and the means to
block prion neuroinvasion. Why is contamination of the
blood supply with prions an important issue? The main
problem is new variant CJD. For one thing, we by far do not
know as much about the epidemiology and iatrogenic
transmissibility of this new disease as we do for sporadic
CJD (sCJD). What is most unsettling, the distribution of
preclinical disease in Great Britain and possibly in other
countries is very obscure, and the little knowledge that is
being gathered is far from reassuring (Will et al., 1999).
Moreover, there is all reason to believe that nvCJD may be
much more "lymphoinvasive" than its sporadic counterpart.
In particular, nvCJD prions can be easily detected in
lymphatic organs such as tonsils and appendix (Hill et al.,
1999; Hill et al., 1997; Hilton et al., 1998), a fact that
was previously demonstrated to be true for scrapie
(Schreuder et al., 1997; Schreuder et al., 1998; Vankeulen
et al., 1996), but not for sCJD prions. While all available
evidence points to follicular dendritic cells as the prion
reservoir in lymphatic organs, splenic lymphocytes of
experimentally inoculated mice can be infected with prions
(Raeber et al., 1999). Although prion infectivity of
circulating lymphocytes appear to be at least two logs
lower than that detected in splenic lymphocytes (Raeber et
al., 1999), the possibility that circulating lymphocytes
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
9
may be in equilibrium with their splenic siblings call for
cautionary measures. The nature of the latter is still
matter of controversy and debate: leukodepletion has been
advocated, but at present there is no certainty about its
efficacy, and even whether the presently available
technologies for leukoreduction are necessary and/or
sufficient for decreasing the threat to blood supply that
derives from nvCJD. In addition, it has to be taken into
account that, even if blood prion infectivity were to be
originally contained in lymphocytes in vivo, lysis of cells
may lead to contamination of non-particulate fractions and,
in the absence of appropriate measures of removal, of
stable blood products.
The second consideration applies so secondary
prophylaxis. Given the very large numbers of infectious BSE
material that has entered the human food chain, it is
possible that many individuals harbor preclinical nvCJD. It
is imperative and urgent to develop strategies that will
help control spread of the agent and that will hopefully
prevent the clinical outbreak of symptoms in these persons.
Possible targets for the interference with neuroinvasion
are rate-limiting processes that control prion replication
within the infected individual. In light of the knowledge
discussed above, treatments that target the neuro-immune
interface of prion replication and neuroinvasion (Aguzzi
and Collinge, 1997) seem a promising area for research
aimed at post-exposure prophylaxis.
Methods to detect prions and their limitation:
In the age of real-time kinetic polymerase
chain reaction (PCR), we have become very spoiled with
respect to the detection thresholds which we demand from
assays geared at detecting viral contaminants in blood.
Consider the case of HIV: here the introduction of
quantitative PCR technologies has pushed the limit of
detection in blood and blood products down to quasi-
perfection. Even when PCR techniques have not proved that
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
useful, or have not yet met with such widespread
acceptance, ultrasensitive immunochemical methods, such as
time-resolved fluorescent ELISA, have progressed to a
degree of sophistication that is highly satisfactory for
most screening application. So why do we still have a
problem with prion detection in blood?
The most formidable problem derives from the
unique biology of the prion. According to more-or-less
accepted wisdom, infectious prions are likely to consist
solely of the PrPSc protein, which has exactly the same
amino acid structure as the normal cellular protein PrPC. A
more noncommittal way of wording this fact would be to
state that PrPSc is the only known surrogate marker for
prion infectivity: this latter statement is likely to be
agreeable upon by both the proponents of the protein-only
hypothesis and by those who still believe that the
infectious agent is a virus.
The consequence of the fact mentioned above for
prion detection is obvious: if prion-specific nucleic acids
do not exist, any PCR-based screening assay to detect said
nucleic acid will not be an option. Therefore, we are left
with immunochemical assays. Besides being less sensitive
than PCR by several orders of magnitude, these are also
fraught with a series of prion-specific problems. The
biggest trouble, again, derives directly from the peculiar
biology of TSE agents. As explained above, PrPSc possesses
the same chemical composition as PrPC, and the latter is a
membrane-bound protein that is normally found in many cell
types of healthy individuals including white blood cells
(Aguz~i and Weissmann, 1997). Although PrPC and PrPSc
differ in a number of physical properties, it appears to be
extremely difficult to develop immunological reagents which
reliably differentiate between these two isoforms. Only one
monoclonal antibody has been described to react with PrPSc
but not with PrPC, and its practical usefulness remains to
be demonstrated since fourteen months after its publication
no follow-up studies have appeared and even the company
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
11
which developed this reagent in the first place does not
appear to use it in its in-house screening assay for BSE
prions.
The hitherto best method for the detection of
prions is by performing Western blot analysis with
homogenized brain tissue that has been digested with
proteinase K (PK). The digestion is necessary since for
Western blot analysis the secondary structure is broken up
so that no difference is found any more between cellular
prions (PrPC) and pathological prions (PrPSc), however,
while PrPC is readily digested by PK under specified
conditions, PrPSC is only degraded to relatively large
fragments called PrP~~-30.
It is also already known to concentrate
proteins by adsorbing them to so called magnetic beads (MB)
to which a specific antibody is bound. However, the
application of such a concentration method to PrP has been
assumed to be impossible due to the specific features of
prions.
Thus, still a great need exists to have a
sensitive method or test to detect small amounts of prions
of the PrPSc confirmation not only for diagnosis but also
for further investigating the disease, as well as agents to
perform such tests.
Brief Description of the Invention
Hence it is one object of the present invention
to provide a method for the detection of the pathological
prion protein as PrPSc or PrP2~-30, respectively.
Tt is another object of the present invention
to provide a method for searching for prion in particular
PrPSc interacting agents.
Still another object of the present invention
are agents specifically recognizing PrPSc and/or PrP2~-30.
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
12
Still another object of the present invention
are solid phase materials such as e.g. magnetic beads
carrying such agents and composition comprising same.
Still another object of the present invention
are compositions comprising such agents for purifying body
fluids and sterilization of surgical and diagnostic
instruments.
Still another object is to provide an improved
method for diagnosing transmissible spongiform
encephalopathies (TSE) and means therefor.
PrPSc is also termed "infectious protein".
PrPSc means a priors protein with a confirmation which
differs from the "normal" confirmation of PrPc in healthy
organisms which do not show or develop any signs of TSE.
A "priors binding site which selectively
interacts with PrPSc but not with PrPc" means a molecule or
part of a molecule which can bind to PrPSc but fails to
bind to PrPc. Such a binding site can be provided e.g. by
a low molecular organic compound, a peptide or protein as
well as by antigen binding sites of antibodies, wherein
said term "antibodies" inter'alia, comprises conventional
antibodies, scFv-fragments (Fab) and (Fab2) fragments. The
term "selectively" in this context means that a compound
having the said binding site reacts at least twofold,
preferably at least fivefold, preferably at least tenfold,
stronger with the priors protein in the PrPSc confirmation
than with the priors protein in the PrPc confirmation. Most
preferably, the binding site shows at least the selectivity
for PrPSc as shown by plasminogen.
"A factor with priors binding activity" means a
compound which can bind to a priors protein and carries the
selective priors binding site of the invention. The factor
can be a low molecular compound but preferably is a peptide
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
13
of at least 10 amino acids length or a protein, which,
however, can carry further nonprotein residues such as
carbohydrate residues or lipid residues. Said factor can
be e.g. of animal or human origin or synthetic.
"Selective prion binding site as contained in
plasminogen" means a binding site as provided by a peptide
or protein having all or part of the amino acid sequence as
contained in a plasminogen of animal or human origin which
selectively interacts with PrPSc but not with PrPc.
"Derivative of plasminogen plasminogen" means a
peptide or protein which carries at least one amino acid
addition, substitution or deletion compared to the
naturally occurring plasminogen or fragment thereof but
still capable of selectively interacting with PrPSc and not
with PrPc. Such derivatives can easily be prepared, e.g.
by site directed mutagenesis of the nucleic acid encoding
the naturally occurring plasminogen or by peptide
synthesis.
"Fragment of plasminogen" means a part of a
naturally occurring plasminogen which part is capable of
selectively interacting with PrPSc and not with PrPc.
"Carboxy terminus of PrPSc'~ means the first ...
amino acids from the carboxy terminus of a prion protein.
"Ligand" means a compound which selectively
binds to the complex of PrPSc with the binding factor
according to the invention but does not interact with free
PrPSc or free factor alone. Such a ligand can be e.g. an
antibody or another receptor type protein.
The present invention interalia relates to a
method for the concentration of PrPSc or digestion products
thereof, wherein a body fluid or fluidized organ is treated
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
14
with solid phase material, the material at least partly
carries a prion binding site which selectively interacts
with PrPSc but not with PrPc. The selectivity of a given
binding site can easily be determined as further described
herein below. The sufficient selectivity is shown, e.g.,
by plasminogen.
In this context, "fluidized organ" means tissue
derived from an organ and solubilized by mechanical
procedures, sonication, or other procedures in order to
bring into solution or suspension a significant proportion
of its constituents.
In a further preferred embodiment the fluidized
organ is a homogenized tissue preferably of the central
nervous system, preferably homogenized brain tissue.
In a further preferred method, the body fluid,
such as blood, plasma, serum, urine, or lymph is treated
with a proteinase, preferably with proteinase K (PK). In a
further preferred method the selective prion binding site
is contained in a factor with prion binding activity
(PrPB). Such factor is preferably a peptide or protein.
In case of the peptide or protein, it is of sufficient
length to allow stable binding to PrPSc or the digestion
product thereof. A sufficient binding stability (or
affinity) is shown, e.g. by plasminogen.
In a further preferred method the solid phase
material carries a PrPB as can be found in blood serum or
blood plasma, preferably as contained in fraction II of the
ammonium sulfate precipitation of serum or plasma.
In a further preferred method the solid phase
material carries a PrPB as can be found in plasminogen,
fibrinogen or in plasma fraction I or ammonium sulfate
precipitation.
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
In a further preferred method the PrPB is
selected from plasminogen, fibrinogen, sPrPBII and PPrPBII
or fragments thereof which fragments show the same
selectivity as the complete protein.
The present invention further relates to a
method for the detection and optionally quantification of
PrPSc or digestion products thereof which method comprises
the step of selectively binding PrPSc or the digestion
product thereof to a prion binding site as defined above.
In a preferred embodiment the prion binding
site is contained in a factor with prion binding activity.
In a preferred embodiment, the factor is
selected from factors as contained in blood serum, blood
plasma, serum or plasma fractions II of ammonium sulfate
precipitation, plasma fraction I of ammonium sulfate
precipitation or as provided by plasminogen, fibrinogen,
sPrPBII and PPrPBII or fragments thereof which show a
similar selectivity as the complete protein.
The detection of the produced complex between
PrPSc and the prion binding site can be done by any method
currently applicable to the detection of such protein
complexes, e.g., by any type of affinity assays or in
particular immunoassays as, e.g., described in "The
Immunoassay Handbook" of David Wild, Second Edition ISPNO-
33-72306-6, Nature Publishing Group. In order to achieve
high sensitivity, a fluorescence detection method is
preferred.
The present invention further provides for the
first time a composition comprising a PrPSc or digestion
product thereof and a material carrying a prion binding
site which selectively interacts with PrPSc but not with
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
16
PrPc. According to a preferred embodiment, the prion
binding site is contained in a factor with prion binding
activity, preferably selected from a factor as contained in
blood serum, blood plasma, serum or plasma fraction II of
ammonium sulfate precipitation, plasma fraction I of
ammonium sulfate precipitation or as contained in
plasminogen, fibrinogen, sPrPBII and PPrPBII.
The present invention further provides a solid
phase material which carries a prion binding site of the
invention, which binding site is preferably contained in a
factor with prion binding activity which factor is
preferably a factor selected from a factor as contained in
plasma serum, blood plasma, serum or plasma fraction II of
ammonium sulfate precipitation, plasma fraction I of
ammonium sulfate precipitation or as contained in
plasminogen, fibrinogen, sPrPBII and PPrPBII.
The present invention further provides a
protein complex which comprises a PrPSc and a factor with a
prion binding site which selectively interacts with PrPSc
but not PrPc, preferably selective PrPSc a binding site as
contained in plasminogen.
The present invention further provides a test
kit for the detection of pathological prion protein such as
PrPSc, in body fluids or organs such as blood, urine,
cerebrospinal fluid, brain tissue, lymph nodes, tonsils
which kit comprises a factor with a prion binding site
according to the invention and the factor is preferably
selected from a factor as contained in blood serum, blood
plasma, serum or plasma fraction II of ammonium sulfate
precipitation, plasma fraction I of ammonium sulfate
precipitation or as contained in plasminogen, fibrinogen,
sPrPBII and PPrPBII.
The present invention further provides an assay
for the diagnosis of human transmissible human spongiform
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
17
encephalopathies or prion encephalopathies of animals
comprising the step of contacting the sample to be tested
with a prion binding site which selectively interacts with
PrPSc but not with PrPc. In a preferred embodiment the
sample is derived from blood. The tested animal or human
is diagnosed positive, i.e. for running risk of developing
a transmissible spongiform encephalopathy if PrPSc can be
detected in the tested sample.
The present invention for the first time allows
the detection of PrPSc in a sample which may contain also
normal PrPc by using a binding factor for PrPc which
contains the selective prion binding site according to the
invention.
In a further aspect of the invention, the
carboxy terminal part of PrPSc is the target for the
selective binding site. It has surprisingly been found
that the carboxy terminus of PrPSc provides a binding site
which allows discrimination between PrPSc and PrPc, which
binding site is a preferred target for the binding sites
according to the present invention.
It has further surprisingly been found that the
binding sites according to the present invention strongly
discriminate between PrPSc and PrPc of different species
although it is known that sequence variations exist between
prion proteins from different species. Therefore, PrPSc of
different species can be detected by the use of a single
prion binding site according to the invention. A very much
preferred binding site is the binding site as contained in
human plasminogen. It was surprisingly found that the
plasminogen binding site interacts with disease associated
prion protein from species. The findings according to the
invention further suggest that a property common to PrP~C
of various species rather than the peculiarities private to
the specific protein structure of individual PrPSc
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
18
molecules are responsible for binding to the binding sites
in plasminogen.
As a carrier for use in accordance with the
present invention, essentially all materials can be
considered that are currently available to perform
biological assays for detecting a given compound in a
biological sample. Such a carrier includes magnetic beads,
filter stripes, walls of microtiter plates.
According to one aspect of the present
invention, the detection of PrPSc could be done
"indirectly" in that first a complex is formed between
PrPSc and the binding site according to the present
invention and the so-formed complex is then selectively
detected by a ligand, such as an antibody. Such a ligand
which selectively interacts with an epitope formed by the
interaction of PrPSc and its binding factor is also highly
selective and detects such a complex only but neither
detects free PrPSc nor free factor according to the
invention only.
The products according to the invention are
useful to provide new diagnostic kits which contain all
ingredients for performing assays in order to detect PrPSc.
Such kits may contain in addition to the products according
to the invention buffers, reagents for detecting a product
being the result of the presence of PrPSc in a sample, the
working instructions on how to reliably perform an assay.
Since PrPSc as such is an essential element in
the development of TSE reducing the availability of free
PrPSc can be a useful means to avoid and or reduce the
speed of BSE development. A means for reducing free PrPSc
is the binding of PrPSc to a binding site according to the
invention, which binding site hence can form part of the
pharmaceutical composition.
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
19
Alternatively, the binding site according to
the invention can be used for removing PrPSc from a
biological bacteria, e.g., in the form of a dialysis
wherein blood of the animal to be tested is continuously
brought into contact with a carrier containing the binding
site of the invention.
The present invention hence allows for the
first time a reliable method for diagnosing human
transmissible spongiform encephalopathies and priors
encephalopathies of animals in which method the material of
the animal to be tested is brought into contact with the
binding site according to the invention.
In a preferred method of the present invention
for the concentration of PrPSc or digestion products
thereof, a body fluid, such as e.g. blood, urine,
cerebrospinal fluid etc., or fluidized organ, such as brain
tissue, lymph nodes, tonsils etc., is treated with a solid
phase material such as magnetic beads(MB) whereby at least
part of said material or beads, respectively, carries a
priors binding site. A preferred priors binding site is a
factor with priors binding activity (PrPB).
The method works very well with a fluidized
organ, in particular homogenized tissue of central nervous
system, preferably homogenized brain tissue.
In cases where the priors binding site can only
distinguish PrPC and PrPSc in digested form, it is
necessary to digest the fluid or fluidized organ prior to
the actual concentration step. A suitable digestion is
obtained by digestion with proteinase K (PK), whereby it is
important to inactivate the proteinase K prior to the
addition of the solid phase, e.g. MBs.
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
Methods are, however, preferred which do not
require the digestion of the starting material in order to
achieve selectivity for the PrPSc.
Preferred solid phase materials carrying PrPB
can be prepared by coupling such materials with blood
serum, or blood plasma, such as fresh frozen plasma of
mammals, whereby an excess of protein is present during the
coupling procedure. Such factors are designated spPrPB (s=
serum and p= plasma) (see below). Even more preferred solid
materials carrying PrPB are prepared by coupling purified
plasminogen or fibrinogen or fragments thereof to solid
phase such as the beads(see below).
Very suitable solid materials are magnetic
beads since they can easily be treated with specific
components of interests and easily be collected by applying
a magnetic field.
A further preferred method of the present
invention concerns the detection (and optionally
quantification) of PrPSC or digestion products thereof,
wherein PrPSc is first concentrated as described above,
optionally also with previous digestion of the fluid or
fluidized organ, and then detected and optionally compared
with a standard. A suitable detection method is Western
blot analysis. Such test may furthermore be embodied by
other detection methods such as a microtiter plate format
immunoassay (e. g. ELISA assay), an immunoprecipitation
assay, a BIACORE assay, immunocytochemical assay, histoblot
assay etc.
Besides of the above mentioned methods, the
present invention also concerns factors with prion binding
activities such as sPrPBII, which is a prion binding
activity in fraction II of ammonium sulfate precipitation
of serum or pPrPBII which is a factor with prion binding
activity in fraction II of ammonium sulfate precipitation
of normal or fresh frozen plasma. Said factors are of
course subject matter of the present invention in any form,
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
2l
such as in isolated form, or as ingredient in a
composition, e.g. in a fraction of ammonium sulfate
precipitation.
Said factors can be obtained by concentration
and/or isolation of PrPBs whereby serum or plasma is
subjected to fractionated ammonium sulfate precipitation
thus that a PrPB of interest is precipitated, preferably in
only one fraction. A further purification can be obtained
by the application of further protein isolation methods.
The factors of the present invention are not
only suitable for the detection of prions, in particular
PrPSC~ but they have further applications in methods for
the purification and removal of pathological prion protein
from body fluids and organs, such as blood, urine,
cerebrospinal fluid, brain tissue, lymph nodes, tonsils
etc., or for the sterilization of surgical and/or
diagnostic tools, basing on the affinity of PrPB for the
pathological prion protein. They are furthermore tools for
a therapy regimen based on the modulation of production of
PrPB for preventing the spread of prions in the body.
Especially suitable in this respect is plasminogen, that is
also especially suitable for the purification of body
fluids, e.g. blood units. Such purification may e.g. be
performed by treating fluids with PrPBIp, in particular
with immobilized plasminogen or plasma fractions containing
same.
Also part of the present invention is a test
for the detection of pathological prion protein such as
PrPSc in body fluids or organs such as blood, urine,
cerebrospinal fluid, brain tissue, lymph nodes, tonsils
etc, that utilizes the specific binding properties of PrPB
to pathological prion protein. Such test can be embodied as
a microtiter plate format immunoassay, e.g. EZISA assay, an
immunoprecipitation assay, a BIACORE assay,
immunogytochemical assay, histoblot assay etc.
Also the DNA sequences specific for
biosynthesis of PrPB are comprised by the present invention
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
22
as well as vectors able to express such DNA sequences in
suitable hosts.
Furthermore comprised by the present invention
are: a method for purification of PrPB by using PrP~~-30 as
bait; monoclonal and polyclonal antibodies produced in
animals such as mice, rabbits, chicken etc., and directed
against PrPB; single-chain Fv fragments and other types of
fragments of antibodies produced in recombinant phages or
in other recombinant systems, and directed against PrPB; a
test predictive of susceptibility to prion diseases based
on polymorphisms of PrPB, or on variations in the strength
and pattern of production of PrPB; a transgenic animal,
e.g. mouse that overproduces PrPB in brain, lymph nodes, or
other organs, to be used in a bioassay for prions; a
knockout animal, in particular a mouse, which is devoid of
PrPB, to be used in a bioassay for prions; a production
method of PrPB by expressing a DNA sequence specific for
the biosynthesis of PrPB in a suitable host cell, such as
bacteria, yeast, fungi, or eukaryotic cells, and by
purification of PrPB from the aforementioned organisms; a
use of natural or synthetic, preferably purified PrPB as a
medicament for therapeutical applications in humans and
animals; a vaccination of organisms with natural or
synthetic PrPB, in particular plasminogen; a diagnostic
assay for human and/or animal diseases resulting from
abnormal production and/or metabolism of PrPB.
Brief Description of the Drawings
Figure 1 is a scheme showing the IAP method.
Figure 2 shows GVestern Blots and IAP
experiments of dilution experiments, whereby lanes 1 to 6
and 10 represent usual Western Blots and lanes 7 to 9 and
11 to 13 represent immuno affinity purification (IAP).
Figure 3 is a scheme showing the prion affinity
assay (PAA) method.
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
23
Figure 4 represents Western Blots showing
positive and begative controls of the PAA.
Figure 5 shows the observation that beads
coated with sheep anti mouse IgG Abs by DYNAL bind PrPSc
but not PrP27-30. Upon preincubation with normal mouse
serum PrP27-30 is also bound.
Figure 6 represents Western Blots showing the
results with serum proteins that are coupled to beads. The
* means that the coupling was performed in the presence of
an excess of proteins.
Figure 7 shows the effect of the addition of
PK-treated brain homogenate to the assay.
Figure 8 represents Western Blots showing the
results with PrP-deficient material.
Figure 9 represents Western Blots showing PAA
of ammonium sulfate precipitates.
Figure 10 represents Western Blots showing PAA
of ammonium sulfate precipitates that are not covalently
crosslinked to the beads.
Figure 11 shows the result of the PAA of the 58
fractions of human plasma that were obtained by
chromatography and differential precipitation and tested
for binding activity.
Figure 12 represents Western Blots showing the
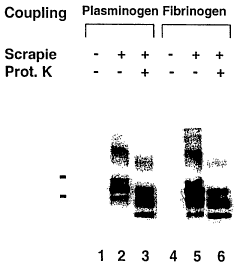
results with purified plasminogen and fibrinogen.
Figure 13 represents Western Blots showing the
calcium dependency of the binding activity of plasminogen
and fibrinogen.
Figure 14 represents Western Blots showing the
dependency of the binding activity of plasminogen on the
native state of the proteins.
Figure 15 represents Western Blots showing PAA
of plasminogen that is not covalently crosslinked to the
beads.
Figure 16 shows the concepts of the bioassay.
Figure 17 shows the results of the bioassay.
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
24
Figure 18 shows PrPSc binding activity of
plasminogen to different species.
Figure 19 shows precipitation of human PrP~'~D by
plasminogen.
Detailed Description of the Invention
As already mentioned above, there is a great
need for a detection method for low concentrations of PrPSc
that can be used as a diagnostic test for transmissible
spongiform encephalopathies (TSEs).
There are basically three diagnostic principles
for TSEs: histopathological detection of the typical
spongiform changes in the CNS, detection of the scrapie-
specific isoform of the prion protein, and the bioassay
that detects infectivity. All these methods have
limitations: histopathology is not useful for preclinical
diagnosis since the structural changes appear late in the
incubation period. Detection of the scrapie the Western
specific is form of prion protein is more sensitive but
still much less sensitive than the bioassay. The bioassay
can, in principle, detect as little as 1 infectious unit
but can last months or even years.
The hitherto used Western blot technique is
based on the partial protease resistance of PrPSc that
allows to distinguish between PrPC and PrPSc. After
protease treatment, PrP~~-3S - the protease resistant core
of PrPSc - can be detected but not PrPC which is completely
digested.
Although due to the "stickiness" of prions it
was generally assumed that immuno affinity purification
(IAP) cannot be applied, it has now been found that
concentration can be achieved by applying magnetic beads
(MB) carrying a prion binding site, preferably a factor
with prion protein binding activity (PrPB).
Thus, because the sensitivity of detection of
absolute amounts of PrP~~-30 is a function of antibody
affinity, and cannot be easily increased for each given
antibody, in the scope of the present invention, despite of
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
the hitherto assumed problems, first an "immuno affinity
purification" (IAP) assay has been developed, using
antibodies covalently Crosslinked to solid phase material,
e.g. magnetic beads. Because the monoclonal antibody (6H4
purchased at Prionics, Zurich, Switzerland, described in
Korth et al., 1997), originally used for the development of
the IAP, is not able to distinguish between PrPC and PrPSC
(it binds both undigested forms as well as digested PrPSC,
i.e. PrP27-30), it is necessary to perform Proteinase K
digestion prior to the IAP (see Fig.1).
For the development of the present IAP method,
the following model system was used: Two tests were
performed to determine the efficiency of the method. On the
one hand, small amounts of a scrapie-infected mouse brain
homogenate were diluted with water and then subjected to
the PrPSc concentration method. On the other hand, small
amounts of a scrapie-infected brain homogenate was diluted
with brain homogenate of non-infected mice in order to
simulate a real situation in which a brain homogenate
contains low amounts of PrPSc (see Fig.2).
In Figure 2, lanes 1 to 6 and 10 represent
usual Western Blots and lanes 7 to 9 and 11 to 13 represent
immuno affinity purification (IAP). PrnPo is material from
PrP deficient mice. MB are of course only used for IAP
whereby 6H4 refers to MB coupled with 6H4 antibodies and -
refers to uncoupled MBs. PRPC refers to brain homogenate of
non-infected mice and PrPSc refers to brain homogenate of
scrapie-infected mice. PK refers to Proteinase K digestion
whereby - refers to non digestion and + to digested
homogenate. The same abbreviations are used for the
following figures.
For prion analysis in homogenate, in particular
of brain tissue, it is important to use in a first
homogenation step low concentration of ionic detergent,
followed by low speed centrifugation, preferably 500 g 30
minutes, 4°C applied twice. For following steps high
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
26
concentration of non-ionic detergent is used and a protein
concentration of the homogenate of at most 5 mg/ml.
Conditions for the proteinase K digestion are
preferably 50 ~,g/ml PK, 37°C and at least half an hour.
Suitable incubation conditions for the beads
with homogenate are e.g. about 1.5 hours at room
temperature, whereby for low concentrations longer
incubation times might be preferable.
The concentration step in said first attempt
was carried out by adding to digested homogenate magnetic
beads (MB) carrying said 6H4.
If a digestion step is needed, it has to be
performed prior to the concentration step, whereby the
digestion, usually by proteinase K, has to be stopped prior
to the concentration step by deactivating the proteinase
e.g. with phenyl methyl sulfonyl fluoride or another agent
known to the skilled person.
By applying the method of the present invention
for e.g. brain tissue homogenate, PrP27-30 can be
concentrated up to amounts detectable by Western blot
analysis from tissue comprising much less pathological
prion protein than needed for the hitherto known tests.
Using largely the same procedure, the above
described method can also be applied as prion affinity
assay (PAA) by exchanging the monoclonal antibody 6H4 by
other substances to be examined, for example in order to
find a binding partner for PrPSc (see Fig.3).
As~a positive control of this assay 6H4 (see
Figure 4, lanes 1-3) is used and as a negative control
mouse IgG or mouse albumin (see Figure 4, lanes 4-9).
In order to investigate whether a given mouse
serum container IgG that specifically recognize PrPs°
magnetic beads that are already coated by the company DYNAZ
with sheep antibodies directed against mouse IgGs were used
after preincubation with mouse serum. These beads - used
without preincubation - were the first negative control
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
27
(see Figure 5, lanes l-2). As a second negative control
these beads preincubated with normal mouse serum were used
in order to show that IgGs from normal mouse serum do not
bind to any form of PrP (see Figure 5, lanes 3-4).
Surprisingly the beads alone showed an affinity to PrPs° but
not to PrP~'-3°. Upon preincubation with normal mouse serum
also PrP2'-3° is bound. Therefore it was hypothised that the
sheep antibodies from DYNAL recognize a molecule that is
associated with PrPs° but digested away after PK-treatment.
As PrP~'-3° is bound upon preincubation with normal mouse
serum, this serum might contain the molecule with affinity
to prps°.
The beads coupled to total mouse serum proteins
did not show any affinity to any form of PrP. However, if
the coupling of the total serum was performed in the
presence of an excess of protein the beads showed the same
binding to PrP~'-3° as the monoclonal antibody 6H4 (see
Figure 6, lanes 4-6) whereas the beads that were coupled in
the presence of an excess of albumine still did not show
any affinity to any form of PrP (see Figure 6, lanes 1-3).
Though it was not possible to measure any difference of the
coupling efficency of the two conditions it might be that
offering an excess of proteins causes a sponge on the
surface of the beads that binds PrP2'-ao. We also checkled
whether PK-treated brain homogenate might enhance the
binding as in the case of bound PrP~'-3° total PK-digested
brain homogenate is present: the addition of PK-digested
brain homogenate from wild-type C57BL/6 mice or Prnp°~° mice
allowed to bind PrPs° in addition to PrPz'-3° ( see Figure 7,
lanes 1-3); the addition of inactive PK had no influence on
the binding activity (see Figure 7, lanes 7-9). If coupled
in the presence of an excess the activity of binding prP2'-3o
was also found in the serum of man, sheep, cow and in the
serum of terminally scrapie-sick C57BL/6 mice (data not
shown).
Apart from an artefact it might well be that
serum of several species contains activities (collectively
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
28
termed PrPB) that interact specifically with the pathogenic
isoform of the prion protein and that are kinetically
favoured in binding to the beads. The affinity to PrPz'-so
could then be understood assuming that native PrPs° present
in sick mice is saturated with PrP$ which might be released
upon proteolytic digest. Alternatively, partial proteolysis
may expose PrPB binding sites on PrPs°. However, the fact
that the addition of PK-treated brain homogenate allows to
bind PrPs° indicates that there might be several different
interactions leading to our observations.
The template-directed refolding hypothesis
predicts that PrP~ and PrPs° form heterodimers during the
conversion process. Therefore we investigated whether PrPB
is identical with PrPC. However, when coupling in excess
PrPB activity was present in the serum of Prnp°~° mice at
levels similar to those of wild-type mice, implying that
PrP~ does not contribute to the binding activity (see
Figure 8).
If PrPg activity is not only caused by the
special coupling conditions, it should be possible to
"purify" it by fractionating mouse serum by differential
ammonium sulfate precipitation. Indeed, it was possible to
precipitate PrPB at an ammonium sulfate saturation below
50% whereby coupling of each fraction was performed in the
presence of an excess of protein (see Figure 9). While
purified rabbit immunoglobulins against total mouse serum
did not contain PrPB (data not shown), they efficiently
bound PrP2'-3° upon preincubation with full mouse serum (see
Figure 10, lanes 1-3) or with proteins precipitating
between 25o and 50o ammonium sulfate saturation (see Figure
10, lanes 4-6). Preincubation with proteins precipitating
between 75o and 1000 ammonium sulfate saturation did not
lead to PrPB activity (see Figure 10, lanes 7-9). This
finding is important as it shows that the PrPB activity is
a property of one or more serum proteins independent of the
covalent crosslink to the surface of the beads.
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
29
As the ammonium sulfate fractionation worked
with human serum as well (data not shown), 58 fractions of
human plasma were obtained by chromatography and
differential precipitation and tested for binding activity
to form an idea of the identity of PrPs. All fractions were
not coupled in the presence of an excess of proteins.
Therefore the results can directly be compared with 6H4 or
mouse IgG. 20 fractions tested positive: Plasminogen,
fibrinogen, antithrombin III, antithrombin III heparin
complex, C1 esterase inhibitor, factor IX and several
fractions containing protein mixtures (see Figure 11).
Purified plasminogen and also purified fibrinogen bound
PrPs° in addition to PrP2'-3° ( see Figure 12 ) . Out of
the 38
fractions that tested negative, 6 contained purified
proteins: Prothrombin complex concentrate, albumin,
activated prothrombin complex concentrate, factor XIII and
thrombin.
As mentioned, there are some hints that the
binding of PrP~'-3° is caused by different effects. The
activity that binds PrP2'-3° is termed spPrPB (s=serum and
p=plasma) as it is present in serum and in plasma. Said
activity is comparable to the activity found for
plasminogen and fibrinogen. Plasminogen and fibrinogen were
furthermore characterised as they both bind also prPs°.
As calcium is an important cofactor in the
coagulation cascade it was investigated whether PrPB
activity is still intact if coagulation is inhibited by
complexing calcium. In the presence of 10 mM EDTA the
pathogenic PrPs° and PrP~'-3° were still bound by plasminogen
(see Figure 13, lanes 1-3) but only PrP2'-3° by fibrinogen
(see Figure 13, lanes 4-6). At least in the case of
plasminogen this finding speaks against the possibility
that the PrPB activity is due to unspecific coagulation.
Because PrPB selectively interacts with the pathogenic PrP
but not with PrP~, interaction may be conformation-
specific. When the assay was carried out in the presence of
6M urea the fraction containing purified plasminogen didn't
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
bind PrPs° nor PrP~~-3° ( see Figure 14, lanes 8-9 ) under
these conditions PrPs° becomes protease-sensitive (see
Figure 14, lanes 14-15). As the conformation of PrPs° is
thought to be responsible for the PK resistancy we conclude
from this experiment that the interaction of plasminogen
and PrPs° is conformation-dependant.
Furthermore it could be shown that PrPB
activity of plasminogen is not dependent on the covalent
crosslink to the beads by using magnetic beads coated with
antibodies directed against plasminogen and preincubated
with plasminogen (see Figure 15, lanes 3-4). There are two
negative controls: 1. If beads coated with antibodies
against plasminogen are not at all preincubated (see Figure
15, lanes 1-2) or preicubated with albumin (see Figure 15,
lanes 5-6), the pathogenic isoform of PrP is not bound. 2.
If beads coated with albumin are preincubated with
plasminogen there is also no binding to the pathogenic
isoform of PrP (see Figure 15, lanes 7-8).
Furthermore it could be shown that at least
spPrPB does not only bind the pathogenic PrP but also
infectivity. For this purpose we inoculated indicator tga20
mice i.c. with 0.20 of the paramagnetic beads before
eluting the other 990 of the beads and performing a western
blot (see Figure 16). The animals that were inoculated with
beads that bind the pathogenic PrP did all develop the
disease (see Figure 17, lanes 4,5 and 7).
It was also determined whether the interaction
between plasminogen and disease-associated prion protein
represents a universal feature of spongiform
encephalopathies. Human plasminogen (100~g) was linked to
tosyl-activated paramagnetic Dynabeads M-280(Dynal, Oslo, 1
ml). Brain tissues from a healthy mouse (Fig. 18, lane 1),
a scrapie-affected mouse (lanes 2-3), pooled brains of
Swiss non-affected cows (lane 4) and brains of BSE-affected
cows of various breeds (lanes 5-10) were homogenized as
described and tested for the presence of PrPSc . For this,
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
31
50~.g (mouse) or 1mg(cow) homogenate were incubated with
paramagnetic beads coupled to anti-PrP monoclonal antibody
6H4 (data not shown), BSA (negative control; data not
shown), or plasminogen. Bead eluates (24u1) were run on
SDS-PAGE (5% stacking - 12% resolving) and blotted on
nitrocellulose membranes (Schleicher & Schuell, Dassel).
For detection of disease-associated PrP, membranes were
incubated with 6H4(Prionics, Zurich)as primary antibody and
rabbit-a-mouse IgGl-HRP (Zymed, San Francisco) as secondary
antibody. Membranes were then developed using ECL
detection reagents. Signals were recorded on film and/or
quantified using a Kodak ImageStation. In all cases,
plasminogen immobilized to magnetic beads captured PrPSc
from each species when subjected to the precipitation
assay. It has been reported that various breeds of sheep
are variably susceptible to scrapie. Susceptibility was
mapped to polymorphisms at codons 136, 154, 171 within the
sheep Prnp gene. Because these polymorphisms occur at the
carboxy terminus of the protein and affect basic amino
acids, and indirect evidence implies that the carboxy
terminus of PrPSc may participate to the binding to
plasminogen, we have investigated whether genetic
susceptibility to scrapie in sheep might correlate with the
ability of PrPSc to bind plasminogen. Brain tissue from
non-affected and scrapie-affected sheep with the Prnp
genotypes at codons 136, 154 and 171 of VHQ/ARQ (Fig. 18,
lanes 11-13), VRQ/ARQ (lanes 14-16), and VRQ/ARR (lane 17-
19) were homogenized and subjected to the prion affinity
assay. Plasminogen precipitated PrPSc from all sheep
genotypes investigated. Fig. 18 eluates from plasminogen
beads incubated with brain homogenates were subjected to
Western blot analysis. Species and breeds are indicated
over the respective lanes. Infection with scrapie or with
BSE, and digestion of samples with proteinase K, are marked
with "+" and "-" signs. Numbers listed underneath each
lane indicate individual cows and sheep of various breeds
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
32
and Prnp genotypes. Plasminogen beads immobilized PrPSc in
all samples tested.
In addition, we tested brain tissues (500~g)
from several patients who died of sporadic Creutzfeldt-
Jakob disease, Alzheimer's disease (Fig. 19) and
Binswanger's disease (data not shown) with the prion
affinity assay. In all assays performed with homogenates
of CJD patients, plasminogen was able to precipitate PrPSc,
while no signal was detectable with homogenates of non-CJD
patients. Unambiguous positive signals were obtained from
cases with plaque-like, patchy-perivacuolar and synaptic
pattern of PrP depositions. The intensity of the prp~J~
signals in the precipitation assays correlated closely with
histopathological findings (Fig. 19). In Fig. 19
plasminogen precipitated PrP~'~D from brain homogenate of
three Swiss sCJD patients (a, b, c) exhibiting extensive
plaque-like (a) or scant synaptic accumulation (b,c) of
PrPcJD. For control we used brain homogenate from a patient
suffering from Alzheimer's disease (d). Proteinase IC
digestion was carried out as indicated with "+" signs over
the corresponding lanes. Corresponding brain sections
immunostained with antibody 3F4 (available from Dr. Richard
Kascksak, Albert Einstein College, The Bronx, New York, USA
or Draco, Denmark, Botrup) to PrP are displaced on the
right side. In each case, the plasminogen-based assay and
the ~nlestern blot show congruent results. In Alzheimer's
disease, PrPc was detectable (-), but not PrPSc (+).
Scale bars are 50 Vim.
Examples:
Example 1: IAP method
The IAP protocol is the following: Bring the
brain tissue in a 15 ml FALCON tube, put it on ice and
leave it there for all steps. Add Homogenate Buffer (0.50
DOC / 0.5o NP-40 in PBS) to get 10 0 (w/v) homogenate. Pass
the tissue through a 18 gauge needle and a 22 gauge needle
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
33
by sucking up and down for 15 times each. Centrifuge the
homogenate for 30 minutes at 500 g and 4°C. Keep the
supernatant. Determine the protein concentration.
Centrifuge the homogenate for 30 minutes at 500 g and 4°C.
Keep the supernatant. If the protein concentration is
higher than 10 mg/ml then bring the homogenate to a protein
concentration of 10 mg/ml using the homogenate buffer.
Bring the homogenate to a protein concentration of 5 mg/ml
and 3o Tween 20 / 3o NP-40 all in PBS. Add to the tissue
homogenate Proteinase K to get a final concentration of 50
~g/ml. Incubate for 60 minutes at 37°C. Add PMSF to get a
final concentration of 5 mM. Add 0.25 volumes of IAP buffer
(3o Tween 20 /3 o NP-40 in PBS). Resuspend the magnetic
beads (covered with 6H4) according to the protocol
described below) thoroughly. Pipette out 100 ~,1. Remove
buffer. Add the homogenate to the beads and incubate the
bead-sample mixture with continous mixing for 1.5 hours at
room temperature. Collect the beads using the MPC (strong
magnet). Wash three times with 1 ml Washing Buffer (2%
Tween 20 / 2% NP-40 in PBS) and once with 1 ml PBS by
vortexing for 15 seconds at room temperature and by using
the MPC. Spin down the beads, discard the remaining
supernatant using again the MPC. Add 24 ~,1 x Loading Buffer
(50 mM Tris pH 6,8; 2o SDS; 0.010 bromphenol blue; l00
glycerol). Heat to 95~C for 5 minutes. If the samples are
stored at -20~C then heat them again for 30 seconds at 95~C
before performing SDS-PAGE followed by western Blot:
Assemble the glass plates according to the manufacturer's
instructions. Prepare in a Falcon tube the appropriate
volume of the Resolving Gel (2.1 ml H20, 1.5 ml 40
Acrylamid, 1.3 ml 1.5 M Tris pH 8.8, 50 ~,1 10 o SDS, 50 ~,1
% Ammoniumpersulfat, 2 ~,1 TEMED). Mix the components in
the order shown. Polymerization will begin as soon as the
TEMED has been added. Pour the acrylamide solution into the
gap between the glass plates. Leave sufficient space for
the stacking gel (the length of the comb plus 1 cm). Using
a pasteur pipette carefully overlay the acrylamide with
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
34
water. Place the gel in a vertical position at room
temperature. After polymerization is complete (30 minutes),
pour off the overlay and wash the top of the gel several
times with deionized water to remove any unpolymerized
acrylamide. Prepare in a Falcon tube the appropriate volume
of the Stacking Gel (1.48 ml H20, 0.25 ml 40 % Acrylamid,
0.25 ml 1.0 M Tris pH 6.8, 20 ~,l 10 % SDS, 20 ~,l 10 0
Ammoniumpersulfat, 2 ~,I TEMED). Mix the components in the
order shown. Polymerization will begin as soon as the TEMED
has been added. Pour the stacking gel solution directly
onto the surface of the polymerized resolving gel.
Immediately insert a clean Teflon comb into the stacking
gel solution, being careful to avoid trapping air bubbles.
Place the gel in a vertical position at room temperature.
After polymerization is complete (30 minutes), remove the
Teflon comb carefully. Mount the gel in the electrophoresis
apparatus. Add Running buffer to the top and bottom
reservoirs. Remove any (25 mM Tris, 250 mM glycine, 0.1 0
SDS) bubbles that become trapped at the bottom of the gel
between the glass plates. Load 24 ~,1 of each of the samples
in a predetermined order into the bottom of the wells (1.
well: Zow -range marker). hoad an equal volume of lx Gel-
loading Buffer into any wells that are unused. Attach the
electrophoresis apparatus to an electric power supply (the
positive electrode should be connected to the bottom
reservoir). Apply 10 V/cm to the gel. After the dye front
has moved into the resolving gel (30 minutes), increase the
voltage to 14 V(cm and run the gel until the bromophenol
blue reaches the bottom of the resolving gel (1 hour). Then
turn off the power supply. Cut six sheets of absorbent
paper (Whatman 3MM or equivalent) and one sheet of
nitrocellulose to the size of the gel (6cm x 8 cm). If the
paper overlaps the edge of the gel, the current will short-
circuit the transfer and bypass the gel, preventing
efficient transfer. Wet the absorbent paper, the
nitrocellulose and the gel by soaking in Transfer (39 mM
glycine, 48 mM Tris, 0.037 o SDS, 20 % methanol) Buffer. On
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
the bottom plate of the apparatus (the anode), assemble the
gel, nitrocellulose, and paper in this order:
bottom electrode,
three layers absorbent paper soaked in transfer
buffer,
one nitrocellulose membrane soaked in transfer
buffer,
polyacrylamide gel slightly wetted with
transfer buffer,
three layers absorbent paper soaked in transfer
buffer.
Check carefully for air bubbles and gently
remove them either by using a gloved hand or by rolling a
pipet over the sandwich. Dry any buffer that may surround
the gel-paper sandwich. Carefully place the upper electrode
(the cathode) on top of the stack. Put a weight on it.
Connect the electrodes and commence transfer. Running time
is 1 hour with a current of 1 mA/cm2. After transfer,
disconnect the power source. Carefully disassemble the
apparatus. Mark membrane to follow orientation (usually by
snipping off lower left-hand corner, the number one lane).
Rinse the membrane three times with TBS-T. Add Blocking
Buffer (5 0 (w/v) nonfat dry milk in TBS-T). Incubate at
room temperature with agitation for 30 minutes. Rinse the
membrane three times with TBS-T. Add to 2.5 ~,1 of mAB 6H4
(2 mg/ml) 12.5 ml of 10 (w/v) nonfat dry milk in TBS-T.
Incubate at room temperature with agitation for 1 hour or
overnight at 4°C. Remove the membrane from the antibody
solution. and wash three times for 10 minutes each in TBS-T.
Add to 1.25 ~,l of relativ anti mouse IgGl-HRP 12.5 ml of 1%
(w/v) nonfat dry milk in TBS-T. Incubate at room
temperature with agitation for 1 hour. Remove the membrane
from the antibody solution and wash three times for 15
minutes each in TBS-T. Mix 1 ml of detection solution 1
with 1 ml of detection solution 2 from the ECZ Western
blotting detection reagents (Amersham Pharmacia Biotech).
Incubate for precisely 1 minute at room temperature without
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
36
agitation. Drain off excess detection reagent by putting
the membrane on a absorbent paper. Gently place the
membrane, protein side down, on a SaranWrap. Close
Saranrnlrap to form a envelope avoiding pressure on the
membrane. Place the membrane, protein side up, in the film
cassette. Work as quickly as possible. Switch off the
lights and carefully place a sheet of autoradiography film
such as (Hyperfilm ECL) on top of the membrane, close the
cassette and expose for some seconds (15", 30").
Example 2: PAA method
Couple the protein of interest to magnetic
beads: Bring 100 ~.g of protein into approx. 1m1 of Coupling
Buffer (0.1 M borate buffer pH 9.5; dissolve 6.183 g H3B03
in 800 ml distilled water, Adjust pH to 9.5 using 5 M NaOH
and adjust volume to 1000 ml with distilled water; if
necessary, change buffer by dialysis). (If coupling was
performed in the presence of an excess, 1 mg was used for 1
ml of coupling buffer.) Make a homogeneous suspension of
the Dynabeads M-280 Tosylactivated by Dynal using a pipette
and by vortexing for approximately 1 min. Pipette out 1 ml
of Dynabeads and wash as follows: Place the tube in the
DYNAL MPC. Leave to separate for 2 minutes. Remove the
supernatant taking care not to disturb the Dynabeads.
Remove the tube from the Dynal MPC and resuspend the
Dynabeads in PBS. Repeat these steps and resuspend the
Dynabeads in the coupling buffer containing the antibodies.
Incubate for 24~h at 37°C with tilt rotation. Place the
tube in the magnet for 3 minutes and remove the
supernatant. Wash the coated Dynabeads six times: 2 x in
PBS/BSA (add 0.1 % (w/v) bovine serum albumin (final
concentration) to PBS), pH 7.4 for 5 minutes at room
temperature; 1 x in Blocking Buffer (0.2 M Tris pH 8.5 with
0.1 0 (w/v) BSA: dissolve 2.42 g Tris in 80 ml distilled
water. Adjust pH to 8.5 using 1 M HC1, add 0.1 o BSA and
adjust volume to 100 ml with distilled water) for 4 h at
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
37
37°C~ 1 x in PBS/BSA, pH 7.4 for 5 minutes at room
temperature; 1 x in 1o Tween 20 for 10 minutes;l x in
PBS/BSA, pH 7.4 for 5 minutes at room temperature. Store
the coated Dynabeads in PBS/BSA pH 7.4, 0.02% sodium aide.
Then prepare Sample I: Add 1 ml of PAA Buffer (3 % NP-40 /
3 o Tween 20 in PBS) to 10 ~,1 of not infected brain
homogenate (Protein concentration 5 mg/ml; 0.5% DOC / 0.5
NP-40). Then prepare Sample II and III: Add 1 ml of PAA
Buffer (3 o NP-40 / 3 o Tween 20 in PBS) to 10~ ~.l of
infected brain homogenate (Protein concentration 5 mg/ml;
0.5% DOC / 0.5 NP-40). Incubate Sample I and Sample II for
30 minutes at 37°C without PK. Incubate Sample III for 30
minutes at 37°C with PK at final concentration of 50 ~g/ml
(add 50 ~,l of PK 1mg/ml). Add PMSF to all samples to get a
final concentration of 5 mM (add 50 ~,1 of 100 mM PMSF).
Resuspend the Magnetic Beads thoroughly. Pipette out 100
~,1. Add the beads to the Samples and incubate the bead-
sample mixture with continuous mixing for 1.5 hours at room
temperature. Collect the beads using the MPC. Wash three
times with 1 ml Washing Buffer and once with 1 ml PBS by
vortexing for 15 seconds at room temperature and by using
the MPC. Spin down the beads, discard the remaining
supernatant using again the MPC. Add 24 ~,l 1 x Zoading
Buffer. Heat to 95°C for 5 minutes. If the samples are
stored at -20°C then heat them again for 30 seconds at 95°C
before loading on the gel.
As a positive control of this assay 6H4 is used
and as a negative control mouse IgG or mouse albumin (see
Figure 4).
Example 3
In order to investigate whether a given mouse
serum containes IgG that specifically recognize prps°
magnetic beads that are already coated by the company DYNAL
with sheep antibodies directed against mouse IgGs were used
after preincubation with mouse serum. These beads were the
first negative control. As a second negative control these
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
38
beads preincubated with normal mouse serum were used in
order to show that IgGs from normal mouse serum do not bind
to any form of PrP. Surprisingly the beads alone showed an
affinity to PrPs° but not to PrP2'-3°. Upon preincubation
with normal mouse serum also PrP2'-3° is bound (see Figure
5). Therefore it was hypothised that the sheep antibodies
from DYNAL recognize a molecule that is associated with
PrPs° but digested away after PK-treatment. As PrP2'-3° is
bound upon preincubation with normal mouse serum, this
serum might contain the molecule with affinity to prPs°.
Example 4
The beads coupled to total mouse serum proteins
did not show any affinity to any form of PrP. However, if
the coupling of the total serum was performed in the
presence of an excess of protein the beads showed the same
binding to PrP2'-3° as the monoclonal antibody 6H4 whereas
the beads that were coupled in the presence of an excess of
albumine still did not show any affinity to any form of PrP
(see Figure 6). Though it was not possible to measure any
difference of the coupling efficency of the two conditions
it might be that offering an excess of proteins causes a
sponge on the surface of the beads that binds PrP2'-so.
Example 5
We also checkled whether PK-treated brain
homogenate might enhance the binding as in the case of
bound PrP2'-so total PK-digested brain homogenate is present:
the addition of PK-digested brain homogenate from wild-type
C57BL/6 mice or Prnp°~° mice allowed to bind PrPs°
in
addition to prPz'-3o~ the addition of inactive PK had no
influence on the binding activity (see Figure 7).
Example 6
If coupled in the presence of an excess the
activity of binding PrP~'-3° was also found in the serum of
man, sheep, cow and in the serum of terminally scrapie-sick
C57BL/6 mice (data not shown).
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
39
Example 7
The template-directed refolding hypothesis
predicts that PrP~ and PrPs° form heterodimers during the
conversion process. Therefore we investigated whether PrPB
is identical with PrP~. However, when coupling in excess
PrPB activity was present in the serum of Prnp°~° mice at
levels similar to those of wild-type mice, implying that
PrP~ does not contribute to the binding activity (se Figure
8) .
Example 8
If PrPB activity is not only caused by the
special couspling conditions, it should be possible to
"purify" it by fractionating mouse serum by differential
ammonium sulfate precipitation. Indeed, it was possible to
precipitate PrPB at an ammonium sulfate saturation below
50% whereby coupling of each fraction was performed in the
presence of an excess of protein (see Figure 9). While
purified rabbit immunoglobulins against total mouse serum
did not contain PrPB (data not shown), they efficiently
bound PrP~'-3° upon preincubation with full mouse serum or
with proteins precipitating between 25o and 50% ammonium
sulfate saturation. Preincubation with proteins
precipitating between 75o and 1000 ammonium sulfate
saturation did not lead to PrPB activity (see Figure 10).
This finding is important as it shows that the PrPB
activity is a property of one or more serum proteins
independent of the covalent crosslink to the surface of the
beads.
Example 9
As the ammonium sulfate fractionation worked
with human serum as well (data not shown), 58 fractions of
human plasma were obtained by chromatography and
differential precipitation and tested for binding activity
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
to form an idea of the identity of PrPB. All fractions were
not coupled in the presence of an excess of proteins.
Therefore the results can directly be compared with 6H4 or
mouse IgG. 20 fractions tested positive: Plasminogen,
fibrinogen, antithrombin III, antithrombin III heparin
complex, C1 esterase inhibitor, factor IX and several
fractions containing protein mixtures (see Figure 11).
Purified plasminogen and also purified fibrinogen bound
PrPs° in addition to PrP2'-so ( see Figure 12 ) . Out of the 38
fractions that tested negative, 6 contained purified
proteins: Prothrombin complex concentrate, albumin,
activated prothrombin complex concentrate, factor XIII and
thrombin.
Example 10
As calcium is an important cofactor in the
coagulation cascade it was investigated whether PrPB
activity is still intact if coagulation is inhibited by
complexing calcium. In the presence of 10 mM EDTA the
pathogenic PrPs° and PrPz'-so were still bound by plasminogen
but only PrP2'-3° by fibrinogen (Fig. 13). At least in the
case of plasminogen this finding speaks against the
possibility that the PrPB activity is due to unspecific
coagulation.
Example 11
Because PrPB selectively interacts with the
pathogenic PrP but not with PrPC, interaction may be
conformation-specific. When the assay was carried out in
the presence of 6M urea the fraction containing purified
plasminogen didn' t bind PrPs° nor PrPz'-3°; under these
conditions PrPs° becomes protease-sensitive (Fig. 14). As
the conformation of PrPs° is thought to be responsible for
the PK resistancy we conclude from this experiment that the
interaction of plasminogen and PrPs° is conformation-
dependant.
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
4l
Example 12
Furthermore it could be shown that PrPB
activity of plasminogen is not dependent on the covalent
crosslink to the beads by using magnetic beads coated with
antibodies directed against plasminogen and preincubated
with plasminogen (Fig, 15).
Example 13
Furthermore it could be shown that at least
spPrPH does not only bind the pathogenic PrP but also
infectivity. For this purpose we inoculated indicator tga20
mice i.c. with 0.20 of the paramagnetic beads before
eluting and performing a western blot. The animals that
were inoculated with beads that bind the pathogenic PrP did
all develop the disease (Fig. 16, Fig. 17).
Example 14
The prior art offers a large number of options
for determining and characterizing the binding
characteristics of a given peptide or protein to a certain
target.. Binding assay for determining the selectivity of a
PrPSc specific binding partner using solid state-bound
technologies includes e.g. microtiter plate formats,
paramagnetic beads, non-magnetic beads, plasmon surface
resonance, interferometry, coincidence detection, mass
spectrometry/mass spectroscopy, electrospray analysis, and
combinations thereof. For use in the present invention the
following two approaches are preferred.
1. The peptides or protein or fragments
thereof to be tested are coupled to a solid phase material:
a. Use micro particles such as magnetic beads
as solid phase and perform immunopreciptiation.
Couple the peptides to magnetic beads. Incubate
the beads with PrPSc, PrP27-30 or PrPC. Detect whether the
prion protein has bound to the peptides either by western
blot analysis or by microparticle immunoassay.
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
42
b. Use surface of a micro titer plate as solid
phase and perform ELISA.
Coat the surface of the wells of a micro titer
plate with the peptides. Add PrPSc, PrP27-30 or PrPC to the
wells. Detect whether the prion protein has bound to the
peptides by ELISA.
2. PrPSc (or PrP27-30) and PrPC are coupled to
a solid phase material:
a. Use micro particles such as magnetic beads
as solid phase and perform immunopreciptiation.
Couple PrPSc (or PrP27-30) and PrPC,
respectively, to magnetic beads. Incubate the beads with
the peptides or protein fragments to be tested. Detect
whether the peptides have bound to the PrPSc but not to
PrPC either by Western blot analysis or by microparticle
immunoassay.
b. Use surface of a micro titer plate as solid
phase and perform ELISA.
Coat the surface of the wells of a micro titer
plate with PrPSc (or PrP27-30) and PrPC, respectively. Add
peptides or protein fragments to be tested to the wells.
Detect whether the peptides have bound to PrPSc but not to
PrPC by ELISA.
Example 15
The prior art offers many possibilities to
determine and detect certain parts of a protein which are
involved in specific binding of the protein to a certain
target.
Method to identify suitable fragments of
plasminogen as PrPSc specific binding partners includes
e.g. forward genetic selection using phage display,
ribosomal display, bacterial protein fragment affinity
assay, and combinations or derivations thereof.
Accordingly, those parts of plasminogen that are involved
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
43
in the specific binding to PrPSc can be determined as
follows:
I. Produce peptide libraries of plasminogen
fragments and/or mutants displayed on phage, expose them to
a solid phase coated with the pathological prion protein
and select for the clones with the maximal binding affinity
to PrPSc but minimal affinity to PrPC.
2. Express fragments and/or mutants of
plasminogen in a host cell such as bacteria, yeast, fungi
or eukaryotic cells, purify the peptides, label them and
test them for binding activity.
3. Express fusion proteins with plasminogen
fragments and/or mutants in a host cell and test them for
PrPSc affinity in a binding assay.
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
44
References
Aguzzi, A. (1997). Neuro-immune connection in
spread of prions in the body? The Lancet 349, 742-743.
Aguzzi, A. (1998). Protein conformation
dictates prion strain. Nat Med 4, 1125-6.
Aguzzi, A., Blattler, T., Klein, M. A., Raber,
A. J., Hegyi, I., Frigg, R., Brandner, S., and Weissmann,
C. (1997). Tracking prions: the neurografting approach.
Cell Mol Life Sci 53, 485-95.
Aguzzi, A., and Collinge, J. (1997). Post-
exposure prophylaxis after accidental prion inoculation.
Lancet 350, 1519-20.
Aguzzi, A., and Weissmann, C. (1997). Prion
research: the next frontiers. Nature 389, 795-798.
Aguzzi, A., and Weissmann, C. (1998).
Spongiform encephalopathies. The prion's perplexing
persistence. Nature 392, 763-4.
Aguzzi, A., and Weissmann, C. (1996).
Spongiform encephalopathies: a suspicious signature. Nature
383, 666-7.
Bernoulli, C., Siegfried, J., Baumgartner, G.,
Regli, F., Rabinowicz, T,, Gajdusek, D. C., and Gibbs, C.
J. (1977). Danger of accidental person-to-person
transmission of Creutzfeldt-Jakob disease by surgery
[letter]. Lancet 1, 478-479.
Blattler, T., Brandner, S., Raeber, A. J.,
Klein, M. A., Voigtlander, T., Weissmann, C., and Aguzzi,
A. (1997). PrP-expressing tissue required for transfer of
scrapie infectivity from spleen to brain. Nature 389, 69-
73.
Brandner, S., Isenmann, S., Raeber, A.,
Fischer, M., Sailer, A., Kobayashi, Y., Marino, S.,
Weissmann, C., and Aguzzi, A. (1996). Normal host prion
protein necessary for scrapie-induced neurotoxicity. Nature
379, 339-43.
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
Bruce, M. E., Will, R. G., Ironside, J. W.,
McConnell, I., Drummond, D., Suttie, A., McCardle, L.,
Chree, A., Hope, J., Birkett, C., Cousens, S., Eraser, H.,
and Bostock, C. J. (1997). Transmissions to mice indicate
that 'new variant' CJD is caused by the BSE agent [see
comments]. Nature 389, 498-501.
Chazot, G., Broussolle, E., Lapras, C.,
Blattler, T., Aguzzi, A., and Kopp, N. (1996). New variant
of Creutzfeldt-Jakob disease in a 26-year-old French man
[letter]. Lancet 347, 1181.
Duffy, P. , Wolf, J. , Collins, G. , DeVoe, A. G. ,
Streeten, B., and Cowen, D. (1974). Possible person-to-
person transmission of Creutzfeldt-Jakob disease. N Engl J
Med 290, 692-3.
Eraser, H., Brown, K. L., Stewart, K.,
McConnell, I., McBride, P., and Williams, A. (1996).
Replication of Scrapie in Spleens of Scid Mice Follows
Reconstitution With Wild-Type Mouse Bone Marrow. Journal of
General Virology 77, 1935-1940.
Gibbs, C. J., Jr., Joy, A., Heffner, R.,
Franko, M., Miyazaki, M., Asher, D. M., Parisi, J. E.,
Brown, P. W., and Gajdusek, D. C. (1985). Clinical and
pathological features and laboratory confirmation of
Creutzfeldt-Jakob disease in a recipient of pituitary-
derived human growth hormone. N Engl J Med 313, 734-8.
Hill, A. F., Butterworth, R. J., Joiner, S.,
Jackson, G., Rossor, M. N., Thomas, D. J., Frosh, A.,
Tolley, N., Bell, J. E., Spencer, M., King, A., A1-Sarraj,
S., Ironside, J. W., Lantos, P. L., and Collinge, J.
(1999). Investigation of variant Creutzfeldt-Jakob disease
and other human prion diseases with tonsil biopsy samples.
Lancet 353, 183-9.
Hill, A. F., Desbruslais, M., Joiner, S.,
Sidle, K. C. , Lowland, I . , Collinge, J. , Doey, L. J. , and
Lantos, P. (1997). The same prion strain causes vCJD and
BSE [letter] [see comments]. Nature 389, 448-50.
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
46
Hill, A. F., Zeidler, M., Ironside, J., and
Collinge, J. (1997). Diagnosis of new variant Creutzfeldt-
Jakob disease by tonsil biopsy. Lancet 349, 99.
Hilton, D. A., Fathers, E., Edwards, P.,
Ironside, J. W., and Zajicek, J. (1998). Prion
immunoreactivity in appendix before clinical onset of
variant Creutzfeldt-Jakob disease [letter]. Lancet 352,
703-4.
Kitamoto, T., Muramoto, T., Mohri, S., Doh ura,
K., and Tateishi, J. (1991), Abnormal isoform of prion
protein accumulates in follicular dendritiC cells in mice
with Creutzfeldt-Jakob disease. J.Virol. 65, 6292-6295.
Klein, M. A., Frigg, R., Flechsig, E., Raeber,
A. J., Kalinke, U., Bluethmann, H., Bootz, F., Suter, M.,
Zinkernagel, R. M., and Aguzzi, A. (1997). A crucial role
for B cells in neuroinvasive scrapie. Nature 390, 687-90.
Klein, M. A., Frigg, R., Raeber, A. J.,
Flechsig, E., Hegyi, I., Zinkernagel, R. M., Weissmann, C.,
and Aguzzi, A. (1998). PrP expression in B lymphocytes is
not required for prion neuroinvasion. Nat Med 4, 1429-33.
Korth, C., Stierli, B., Streit, P., Moser, M.,
Schaller, 0., Fischer, R., SChulz-SChaeffer, W.,
Kretzschmar, H., Raeber, A., Braun, U., Ehrensperger, F.,
Hornemann, S., Glockshuber, R., Riek, R., Billeter, M.,
Wuthrich, K., and Oesch, B. (1997). Prion (PrPs°)-specific
epitope defined by a monoclonal antibody. Nature 390, 74-7.
Lasmezas, C. I., Cesbron, J. Y,, Deslys, J. P.,
Demaimay, R. , Adj ou, K. T. , Rioux, R. , Lemaire, C . , Locht,
C., and Dormont, D. (1996). Immune system-dependent and -
independent replication of the scrapie agent. J Virol 70,
1292-5.
0'Rourke, K. I., Huff, T. P., Leathers, C. W.,
Robinson, M. M., and Gorham, J. R. (1994). SLID mouse
spleen does not support scrapie agent replication. J. Gen.
Virol. 75, 1511 - 1514.
Raeber, A. J., Klein, M. A., Frigg, R.,
Flechsig, E., Aguzzi, A., and Weissmann, C, (1999). PrP-
CA 02413742 2002-12-20
WO 02/00713 PCT/EPO1/03481
47
dependent association of prions with splenic but not
circulating lymphocytes of scrapie-infected mice. EMBO J
18, 2702-2706.
Schreuder, B. E., van Keulen, L. J., Smits, M.
A., Langeveld, J. P., and Stegeman, J. A. (1997). Control
of scrapie eventually possible? Vet Q 19, 105-13.
Schreuder, B. E., van Keulen, L. J., Vromans,
M, E., Langeveld, J. P., and Smits, M. A. (1998). Tonsillar
biopsy and PrPSc detection in the preclinical diagnosis of
scrapie. Vet Rec 142, 564-8.
Vankeulen, L. J. M., Schreuder, B. E. C.,
Meloen, R. H., Mooijharkes, G., Vromans, M. E. W., and
Langeveld, J. P. M, (1996). Immunohistochemical Detection
of Prion Protein in Lymphoid Tissues of Sheep With Natural
Scrapie. Journal of Clinical Microbiology 34, 1228-1231.
Weber, T., and Aguzzi, A. (1997). The spectrum
of transmissible spongiform encephalopathies. Intervirology
40, 198-212.
Weissmann, C., and Aguzzi, A. (1997). Bovine
spongiform encephalopathy and early onset variant
Creutzfeldt- Jakob disease. Curr Opin Neurobiol 7, 695-700.
Wells, G. A., Hawkins, S. A., Green, R. B.,
Austin, A. R., Dexter, I., Spencer, Y. I., Chaplin, M. J.,
Stack, M. J., and Dawson, M. (1998), Preliminary
observations on the pathogenesis of experimental bovine
spongiform encephalopathy (BSE): an update. Vet Rec 142,
103-6.
Will, R., Cousens, S., Farrington, C., Smith,
P., Knight, R., and Ironside, J. (1999). Deaths from
variant Creutzfeldt-Jakob disease. Lancet 353, 9157-9158.
Will, R., Ironside JW, Zeidler M, Cousens SN,
Estibeiro K, Alperovitch A, Poser S, Pocchiari M, Hofman A,
and Smith (1996). A new variant of Creutzfeldt-Jakob
disease in the UK. Lancet 347, 921-925.