Note: Descriptions are shown in the official language in which they were submitted.
CA 02419503 2003-02-14
MONITORING PROTEINS FOR THE ACTIVITIES OF
LOW-MOLECULAR-WEIGHT GTP-BINDING PROTEINS
BACKGROUND OF THE INVENTION
Field of the Invention
The present invention relates to monitoring proteins for the
activity of low-molecular-weight GTP-binding proteins, genes
encoding the proteins, expression vectors encoding the genes, cells
and transgenic animals carrying the expression vectors, methods for
the activity of low-molecular-weight GTP-binding proteins which use
the proteins, and screening procedures for the substances which
regulate the activity of low-molecular-weight GTP-binding proteins.
Description of the Related Art
There are many intracellular signaling molecules. Among them,
low-molecular-weight GTP-binding proteins, often called as GTP-
binding proteins hereafter, have been extensively studied, because
there are many proteins belonging to this group and because they
play critical roles as molecular switches of various signal
transduction cascades. The low-molecular-weight GTP-binding
proteins consist of Ras-family, Rho-family, Rab-family, Ran-family,
etc (ref.1). These low-molecular-weight GTP-binding proteins
function as critical molecular switches of cell growth, cytoskeleton,
intracellular trafficking, and nuclear transport. The low-molecular-
weight GTP-binding proteins cycle between GTP-bound inactive and
GTP-bound active forms (Fig.1). The GTP-bound form binds to and
1
CA 02419503 2003-02-14
activates specific target proteins. The conversion of the inactive
GDP-bound form to the active GTP-bound form is catalyzed by
guanine nucleotide exchange factors (GEFs) and the reverse reaction
is catalyzed by GTPase activating proteins (GAPS). The GTPase
activating protein stimulates the GTP hydrolysis on the low-
molecular-weight GTP-binding protein, cleaving GTP to phosphate
and GDP.
A number of low-molecular-weight GTP-binding proteins have
been already isolated, which have aroused a question as to their
functional difference in the context of cells and tissues. To study this
question, the activities of low-molecular-weight GTP-binding
proteins have to be monitored in the living cells and tissues.
To know the activities of the low-molecular-weight GTP-
binding proteins, the ratio of GTP-bound to GDP-bound forms of the
low-molecular-weight GTP-binding proteins has to be determined.
Currently, the following two methods are used routinely.
(1) 32Pi-labeling method: The low-molecular-weight GTP-binding
proteins are purified from cells labeled with 3zPi. GTP and GDP
bound to them are separated and quantified by thin layer
chromatography (ref. 2).
(2) Pull-down method: Target-proteins that bind to the low-
molecular-weight GTP-binding proteins are pre-bound to
agarose beads and incubated with cell Iysates. Since the GTP-
bound form, but not GDP-bound form, binds to the target
proteins with high affinity, only the GTP-bound form can be
2
CA 02419503 2003-02-14
collected by this method. Then, the amount of GTP-bound
forms is quantified by SDS-PAGE and immunoblotting (ref. 3).
However, both methods are applicable only to the cell lysates;
therefore, no method have been applicable for the measurement of
the activity of low-molecular-weight GTP binding proteins in living
cells.
It has been revealed that different biochemical reactions are
processed not only at various intracellular organelles but also at
various cytoplasmic Iocalizations. Furthermore, the importance of
low-molecular-weight GTP-binding proteins has been shown also in
the higher brain function and the organ development. Thus, to
monitor the activity of low-molecular-weight GTP-binding proteins
in living cells and tissues are essential not only to understand the Life,
but also to develop a new drug. However, the biochemical methods
described previously require cell lysates; therefore, it has been
impossible to know where in the living cells or tissues the low-
molecular-weight GTP-binding proteins are activated.
Meanwhile, green fluorescent protein (GFP) has been
successfully used to visualize the localization of proteins in living
cells (ref. 4). GFP is a group of proteins isolated from various animals
such as Aequorea Victoria and emanates mostly green fluorescence
and is extensively used to determine the intracellular localization of
proteins. Groups of GFP include cyan-emitting mutant of GFP (CFP),
yellow-emitting mutant of GFP (YFP), enhanced CFP (ECFP),
enhanced YFP (EYFP), and enhanced blue-emitting mutant of GFP
3
CA 02419503 2003-02-14
(EBFP), which are collectively called GFP hereafter. These GFPs are
excited with lights of different wavelengths and emanated lights of
longer wavelengths.
GFPs can be applicable to fluorescence resonance energy
transfer (FRET) (ref. 5). FRET is a phenomenon as described below.
Assuming two fluorescent proteins A and B, which emanate lights of
emission wavelengths of Aaem and Abem at excitation wavelengths of
Aaex and Abex, respectively. If molecule A is in close proximity of
molecule B and if ~aem overlaps Abex, excited energy of molecule A
is transferred to molecule B, and the latter emanates a light of Abem.
This phenomenon is called FRET and can be applicable to measure
the distance between two fluorescent molecules. In this situation,
molecule A and B are called as donor and acceptor, respectively.
Application of FRET includes detection of conformational
change of proteins that are labeled with two fluorescent substances.
Two sets of GFP-derived proteins, "EBFP and EGFP" and "ECFP and
EYFP," are known to provide such FRET pairs. For example, calcium
concentration has been measured by a fusion protein consisting of
EBFP, EGFP, and calmodulin. However, this single-molecule
monitoring protein based on the technology of GFP and FRET is
currently known only for the measurement of calcium and cAMP.
SUMMARY OF THE INVENTION
The present invention aims at providing monitoring proteins
which measure the activity of low-molecular-weight GTP-binding
4
CA 02419503 2003-02-14
proteins in non-destructive manners, genes encoding said monitoring
proteins, expression vectors containing said genes, cells and
transgenic animals carrying said expression vectors, methods for
measurement of the activity of low-molecular-weight GTP-binding
proteins which use said monitoring proteins, particularly methods
for the determination of the ratio of GTP-bound to GDP-bound low-
molecular-weight GTP-binding proteins in living cells, and screening
procedures for the regulatory substances of low-molecular-weight
GTP-binding proteins.
In summary, the present invention relates to:
<1> Monitoring proteins for low-molecular-weight GTP-binding
proteins consisting of: fused proteins, wherein the fused proteins
include at Ieast the Iow-molecular-weight GTP-binding protein, a
target protein of said Iow-molecular-weight GTP-binding proteins, a
GFP donor protein, and a GFP acceptor protein, whole ox part of
which are directly or indirectly connected each other, in a state
wherein each of the protein retains its function,
<2> genes encoding said monitoring proteins for Iow-molecular-
weight GTP-binding proteins,
<3> expression vectors which contain the genes described in <2>,
<4> cells Transformed by the expression vectors described in <3>,
<5> transgenic animals which contain the expression vectors
described in <3>,
<6> a method for measurement of the activity of the low-molecular-
weight GTP-binding proteins comprising: the step of detecting FRET
CA 02419503 2003-02-14
of the monitoring proteins for the low-molecular-weight GTP-
binding proteins described in <1 >,
<7> a method for measurement of the activity of the low-molecular-
weight GTP-binding proteins comprising: the step of detecting FRET
of the monitoring proteins for the low-molecular-weight GTP-
binding proteins in the cells described in <4> or transgenic animals
described in <5>,
<8> a screening method for the regulator of the activity of low-
molecular-weight GTP-binding proteins comprising: (a) the step of
culturing cells described in <4> in the presence of the specimens and
(b) the step of measuring the activity change of low-molecular-weight
GTP-binding proteins.
BRIEF DESCRIPTION OF THE DRAWINGS
Fig.1 shows an example of the regulation of the low-molecular-
weight GTP-binding proteins. In the present figure, adducing Ras as
an example of low-molecular-weight GTP-binding proteins, the
regulation of low-molecular-weight GTP-binding proteins is
schematically presented. The low-molecular-weight GTP-binding
protein is inactive when it is bound to GDP. Guanine nucleotide
exchange factor (GEF) promotes exchange of GDP with GTP, thereby
activating the Iow-molecular-weight GTP-binding protein. The
activated GTP-bound low-molecular-weight GTP-binding protein
changes its conformation, thereby binding to and activating the target
proteins. The activated Iow-molecular-weight GTP-binding protein
6
CA 02419503 2003-02-14
hydrolyses GTP to GDP and Pi in the presence of GTPase activating
protein (GAP), thereby returning to the inactive GDP-bound state.
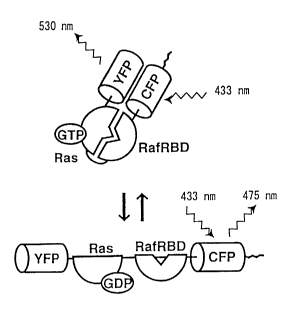
Fig. 2 shows an example of the principle of the measurement of
the activity of Iow-molecular-weight GTP-binding protein based on
FRET technology. In this figure, Ras and Raf are adduced as
examples of low-molecular-weight GTP-binding proteins and their
target proteins, respectively. Cyan-emitting mutant of GFP (CFP),
which is adduced as an example of the GFP donor protein, emanates
fluorescence of 475 nm by excitation at a wavelength of 433 nm.
Meanwhile, yellow-emitting mutant of GFP (YFP), which is adduced
as an example of the GFP acceptor protein, emanates fluorescence of
530 nm by excitation at a wavelength of 505 nm. In the present
invention, CFP and YFP are used as the GFP donor and GFP acceptor
proteins, respectively. As shown in the lower part of the Fig. 2, the
energy of excited CFP is not effectively transferred to YFP before Ras
activation, because YFP and CFP are positioned remotely. However,
upon stimulation (for example, addition of epidermal growth factor
(EGF)), activated Ras is induced to bind to the Ras-binding domain
(RBD) of Raf, which brings YFP in close proximity of CFP, thereby
causing the energy transfer from CFP to YFP, followed by the
emission of 530-nm wavelength. Thus, by measuring the FRET
efficiency before and after the stimulation (namely, before and after
the Ras activation), the activity of Ras can be measured.
Fig. 3 shows an example of the structure of pRafras1722.
pCAGGS, an expression vector used to express Rafras1722, has been
7
CA 02419503 2003-02-14
reported previously. A cDNA encoding a fusion protein consisting of
EYFP-Ras-RafRBD (Ras-binding domain)-ECFP from the amino-
terminus is inserted downstream of CAG promoter as shown in the
figure.
Fig. 4 shows an example of the nucleotide sequence and the
amino-acid sequence decoded from the nucleotide sequence of the
coding region of the plasmid pRafras2722.
Fig. 5 shows an example of the nucleotide sequence and the
amino-acid sequence decoded from the nucleotide sequence of the
coding region of the plasmid pRafras1722 (continued).
Fig. 6 shows an example of the nucleotide sequence and the
amino-acid sequence decoded from the nucleotide sequence of the
coding region of the plasmid pRafras1722 (continued).
Fig. 7 shows an example of the fluorescent profile of the
expressed protein Rafras1722. HEK293T cells were transfected with
pRafras1722 and an expression vector for guanine nucleotide
exchange factor Sos (pCAGGS-mSos) or GTPase activating protein
for Gaplm (pEF-Bos-Gaplm) by calcium phosphate coprecipitation
method. Forty-eight hours after transfection, cells were lysed and
cleared by centrifugation. Fluorescent intensity of the supernatant
was examined with a fluorescent spectrometer from 450 to 550 nm
wavelength range at an excitation wavelength of 433 nm. The right
panel shows the fluorescent profiles of cells transfected with
pRafras2722 and pCAGGS-mSos or pEF-Bos-Gaplm.
8
CA 02419503 2003-02-14
Fig. 8 shows an example of the correlation of the GTP/GDP
ratio (GTP/ (GDP + GTP)) bound to the GTP-binding protein of the
expressed Rafras2722 with the ratio of fluorescent intensity at 530 nm
to fluorescent intensity at 475 nm (Em A5so/ Em 1~~). HEK293T cells
were transfected with pRafras1722 and various amounts of an
expression vector for Sos (pCAGGS-mSos) or GTPase activating
protein for Gaplm (pEF-Bos-Gaplm). Forty-eight hours after
transfection, cells were labeled with 32Pr, and Rafras2722 was
immunoprecipitated with anti-GFP antibody, followed by separation
and quantitation of guanine nucleotides bound to Rafras1722 by thin
layer chromatography. In parallel, cell lysates were analyzed for the
fluorescent profiles to obtain the ratio of fluorescent intensity at 530
run to 475 nm (Em A53a/ Em A4~5) at an excitation wavelength of 433
nm. Note that the ratio of fluorescent intensity increases with the
increasing amount of GTP on Rafras1722.
Fig. 9 shows an example of the establishment of cell lines
expressing Rafras1722. NIH3T3 cells were transfected with
pRafras1722 to obtain a cell Iine, named 3T3-Rafras. Cells were Iysed
and analyzed by immunoblotting with anti-GFP antibody.
Molecular-weight size-markers are shown at the Ieft.
Fig.10 shows an example of analysis of Ras activation using
3T3-Rafras cell's. 3T3-Rafras cells were stimulated with EGF (1
lZg/ml) and fluorescent spectra (wavelength range from 450 nm to
550 nm) were obtained before and after stimulation.
9
CA 02419503 2003-02-14
Fig.11 shows an example of the structure of pRai-chu311. The
expression vector is as same as Fig. 3.
Fig.12 shows an example of the nucleotide sequence and the
amino-acid sequence decoded from the nucleotide sequence of the
coding region of the plasmid pRai-chu311.
Fig.13 shows an example of the nucleotide sequence and the
amino-acid sequence decoded from the nucleotide sequence of the
coding region of the plasmid pRai-chu311 (continued).
Fig.14 shows an example of the nucleotide sequence and the
amino-acid sequence decoded from the nucleotide sequence of the
coding region of the plasmid pRai-chu311 (continued).
Fig.15 shows an example of the fluorescent profile of expressed
protein Rai-chu311. HEK293T cells were transfected with pRai-
chu311 and an expression vector for guanine nucleotide exchange
factor C3G (pCAGGS-C3G, described in ref. 9) or GTPase activating
protein for rapIGAPII (pCAGGS-rapIGAPII, described in ref. 9) by
calcium phosphate coprecipitation method. Forty-eight hours after
transfection, cells were Iysed and cleared by centrifugation.
Fluorescent intensity of the supernatant was scanned with a
fluorescent spectrometer from 450 to 550 nm wavelength range at an
excitation wavelength of 433 run. The right panel shows the
fluorescent profile of cells transfected with pRai-chu311 and
pCAGGS-C3G or pCAGGS-rapIGAPII.
Fig.16 shows an example of the structure of pRai-chu158. The
expression vector is same as Fig. 3.
CA 02419503 2003-02-14
Fig.17 shows an example of the nucleotide sequence and the
amino-acid sequence decoded from the nucleotide sequence of the
coding region of the plasmid pRai-chu158.
Fig.18 shows an example of the nucleotide sequence and the
amino-acid sequence decoded from the nucleotide sequence of the
coding region of the plasmid pRai-chu158 (continued).
Fig.19 shows an example of the nucleotide sequence and the
amino-acid sequence decoded from the nucleotide sequence of the
coding region of the plasmid pRai-chu158 (continued).
Fig. 20 shows an example of the fluorescent profile of expressed
protein Rai-chu158. HEK293T cells were transfected with pRai-
chu158 and an expression vector for guanitne nucleotide exchange
factor CaIDAG-GEFIII (pCAGGS-CaIDAG-GEFIII, described in ref.
10) or GTPase activating protein for GAPImI (pEF-Bos-GAPlm) by
calcium phosphate coprecipitation method. Forty-eight hours after
transfection, cells were lysed and cleared by centrifugation.
Fluorescent intensity of the supernatant was scanned with a
fluorescent spectrometer from 450 to 550 nm wavelength range at an
excitation wavelength of 433 nm. The right panel shows the
fluorescent profile of cells transfected with pRai-chu258 and
pCAGGS-CaIDAG-GEFIII or pEF-Bos-GAPlm.
Fig. 21 shows an example of the nucleotide sequence and the
amino-acid sequence decoded from the nucleotide sequence of the
coding region of the plasmid pRai-chu119.
11
CA 02419503 2003-02-14
Fig. 22 shows an example of the nucleotide sequence and the
amino-acid sequence decoded from the nucleotide sequence of the
coding region of the plasmid pRai-chu119 (continued).
Fig. 23 shows an example of the nucleotide sequence and the
amino-acid sequence decoded from the nucleotide sequence of the
coding region of the plasmid pRai-chu119 (continued).
Fig. 24 shows an example of the fluorescent profile of expressed
protein Rai-chu119. HEK293T cells were transfected with pRai-
chu119 or pRafras1722 and an expression vector for guanine
nucleotide exchange factor Sos (pCAGGS-mSos) by calcium
phosphate coprecipitation method. Twenty-four hours after
transfection, temperature of the cell culture was changed to 33 ~C or
40 ~C. After further 24 hrs incubation, cells were lysed and cleared by
centrifugation. Fluorescent intensity of the supernatant was scanned
with a fluorescent spectrometer from 450 to 550 nm wavelength
range at an excitation wavelength of 433 nm. The right panel shows
the fluorescent profile of cells transfected with pRafras1722 or pRai-
chu119 and pCAGGS-mSos. Rai-chu119 responded more efficiently
to the guanine nucleotide exchange factor than did the wild-type
(Rafras1722).
Fig. 25 shows an example of the time course of fluorescent
intensities of ECFP and EYFP after the addition of epidermal growth
factor (EGF). Cells were illuminated at a wavelength of 430 nm to
obtain time-lapse fluorescence images at a wavelength of 475 nm and
12
CA 02419503 2003-02-14
530 nm, which were then used to determine the fluorescent
intensities of ECFP and EYFP, respectively.
Fig. 26 shows an example of the change in the fluorescent
intensities of ECFP and EYFP of Rafras1722 by the expression of
various kinds of guanine nucleotide exchange factors and GTPase
activating proteins. HEK293T cells were transfected with pRafras1722
and expression vectors for guanine nucleotide exchange factors or
GTPase activating proteins by the calcium coprecipitation method.
Twenty-four hours later, cells were lysed and cleared by
centrifugation. By using the supernatant, fluorescent intensities at
475 nm and 530 nm were determined at an excitation wavelength of
433 nm with a fluorescent spectrometer. The ratio of the latter to the
former (fluorescence ratio) is shown in the graph.
Fig. 27 shows an example of the change in the fluorescent
intensities of ECFP and EYFP of Rai-chu404 by the expression of
various kinds of guanine nucleotide exchange factors and GTPase
activating proteins. HEK293T cells were transfected with pRai-
chu404 and expression vectors for guanine nucleotide exchange
factors or GTPase activating proteins by calcium coprecipitation
method. Twenty-four hours later, cells were lysed and cleared by
centrifugation. By using the supernatant, fluorescent intensities at
475 nm and 530 nm were determined at an excitation wavelength of
433 nm with a fluorescent spectrometer. The ratio of the latter to the
former (fluorescence ratio) is shown in the graph.
13
CA 02419503 2003-02-14
Fig. 28 shows an example of the time course and intracellular
distribution of the fluorescence ratio of EYFP to ECFP in COS1 cells
transfected with pRai-chu101X or pRai-chu404X and stimulated with
EGF. COS1 cells transfected with pRai-chu101X or pRai-chu404X
were cultured for 24 hrs. The medium was changed to MEM without
phenol-red and serum before imaging. Cell images were obtained
with an imaging system consisting of Metamorph image analyzing
software (Roper Scientific Japan) and inverted fluorescent microscope
Axiovert 100 (Carl Zeiss) equipped with Xenon lamp, revolving filter
changers for excitation filters and emission filters (LUDL electronic),
and high sensitivity cooled CCD camera Micromax 450 (Photometrix).
Cells were illuminated with an excitation wavelength of 430 nm and
fluorescent images of ECFP donor protein at 475 nm and EYFP
acceptor protein at 530 nm were obtained every 30 sec. After data
acquisition, from blue to red colors was assigned to each pixel of the
digital images, depending on the levels of EYFP/ECFP fluorescence
ratios. Meanwhile, the intensity of ECFP is assigned to the intensity
of each pixel. From time-lapse images, only the images at the
indicated time point are shown. By the simulation of EGF, the
fluorescence ratio, which reflects the FRET efficiency, gradually
increases from the periphery to the center of the cells expressing Rai-
chu101X. In contrast, the activity of Rap1 increases from the center to
the periphery of the cells expressing Rai-chu404X. Thus, the invented
monitoring proteins can monitor the spatio-temporal change in the
activity of Ras-family G proteins.
14
CA 02419503 2003-02-14
Fig. 29 shows an example of the time course and intracellular
distribution of the fluorescence ratio of EYFP to ECFP in subconfluent
COS1 cells transfected with pRai-chu101X and stimulated with EGF.
Experiments were performed similarly to Fig. 28 except that
subconfluent COS1 cells were used. Upon stimulation with EGF, the
fluorescence ratio, which reflects the FRET efficiency, increases from
the periphery where cells are not in contact with the neighboring cells.
In contrast, at the region where the COS cells are in contact with the
neighboring cells, the increase in FRET efficiency is suppressed.
Fig. 30 shows an example of the time course and intracellular
distribution of the fluorescence ratio of EYFP to ECFP in PC12 cells
transfected with pRai-chu101X or pRai-chu404X and stimulated with
nerve growth factor. PC12 cells transfected with pRai-chu101X or
pRai-chu404X were cultured more than 24 hrs. After changing the
medium to MEM without serum and phenol-red, cells were
stimulated with nerve growth factor and observed as in Fig. 29. Only
the figures at the indicated time points are shown. Upon stimulation
of Rai-chu101X expressing cells with nerve growth factor, the
fluorescence ratio, which reflects the FRET efficiency, increases from
the periphery to the center. Then, after 180 min, when the neuronal
extension is visible, the increase in FRET efficiency is limited mostly
at these extended neurites. In contrast, in the cells expressing Rai-
chu404X, the activity increases from the center to the periphery and is
suppressed at the differentiated extended neurites. This observation
indicates that Ras is activated from the periphery and Rap1 from the
CA 02419503 2003-02-14
center during the neuronal differentiation, and that high Ras activity
is maintained at the extended neurites. This observation further
indicates that each Ras-family G protein is activated at different
intracellular localization and demonstrates the usefulness of the
invented monitoring proteins to obtain the spatio-temporal
information of the activity of Ras=family G proteins.
Fig. 31 shows an example of the structure of pRai-chu1011X.
The basal vector is as same as Fig. 3.
Fig. 32 shows an example of the structure of pRai-chu2054X.
The basal vector is as same as Fig. 3.
Fig. 33 shows an example of the structure of pRai-chu2212X.
The basal vector is as same as Fig. 3.
Fig. 34 shows an example of the fluorescence profile of Rai-
chu1011X (wild type), Rai-chu1012X (activated form), and Rai-
chu1013X (inactive form). HEK293T cells were transfected with pRai-
chu1011X, pRai-chu1012X, or pRai-chu1013X by the calcium
phosphate method. Forty-eight hours Later, cells were Iysed and
centrifuged to obtain supernatant, which was analyzed with a
fluorescent spectrometer to obtain the fluorescent profiles from 450
nm to 550 nm.
Fig. 35 shows an example of the fluorescence profile of Rai-
chu2054X (wild type) and Rai-chu1052X (activated form). HEK293T
cells were transfected with pRai-chu1054X or pRai-chu1052X by
calcium phosphate method. Foray-eight hours Later, cells were lysed
16
CA 02419503 2003-02-14
and centrifuged to obtain supernatant, which was then analyzed by
spectrometer to obtain the fluorescent profiles from 450 nm to 550 nm.
Fig. 36 shows an example of the fluorescence profile of Rai-
chu1212X (wild type) and Rai-chu2220X (activated form). HEK293T
cells were transfected with pRai-chu1212X or pRai-chu1220X by
calcium phosphate method. Forty-eight hours later, cells were lysed
and centrifuged to obtain supernatant, which was then analyzed with
a fluorescent spectrometer to obtain the fluorescent profiles from 450
nm to 550 nm.
Fig. 3~ shows an example of the time course and intracellular
distribution of the fluorescence ratio of EYFP to ECFP in COS1 cells
transfected with pRai-chu1011x and stimulated with EGF.
Experiments were performed similarly to Fig. 28. Upon stimulation
with EGF, the fluorescence ratio, which reflects the FRET efficiency,
increases diffusely within one minute, followed by increase at the
membrane ruffles and decrease in the central region. This spatio-
temporal distribufiion of Rac activity is remarkably different from
those of Ras and Rap1 examined by Ra-chu101X or Rai-chu404X,
respectively. This observation indicates that the invented monitoring
proteins can obtain the spatio-temporal information of the activities
of Rho-family G proteins.
DESCRIPTION OF THE PREFERRED EMBODIMENTS
The invented monitoring proteins for the activity of low
molecular-weight GTP-binding proteins, called monitoring proteins
17
CA 02419503 2003-02-14
hereafter, utilize the GTP-dependent binding to the target proteins by
the low-molecular-weight GTP-binding proteins and provide
extremely useful tools which can measure the activity low-molecular-
weight GTP-binding proteins in living cells. The invented
monitoring proteins consist of low-molecular-weight GTP-binding
protein, its target protein, GFP donor protein, and GFP acceptor
protein, which are ligated directly or indirectly so that each
component functions properly. Therefore, these fusion proteins have
structures that amino acid sequences of said proteins are ligated
directly or indirectly. Notably, each component does not need to
consist of the full-length protein if it retains its function.
In this disclosure, where it should be exactly expressed as "a
part of protein," it is simply called as "a protein'; Adducing target
protein as an example, when it should be called as "a part of target
protein,' it is simply expressed as "a target protein."
In the monitoring protein described in this disclosure, the
binding of GTP to the low-molecular-weight GTP-binding protein, or
exchange of GDP with GTP, induces intrarnolecular binding of the
low-molecular-weight GTP-binding protein to the target protein,
changing the level of FRET efficiency. Fig. 2 shows the schematic
representation of the principle of the measurement of the activity of
low-molecular-weight GTP-binding proteins with the invented
monitoring proteins. In this invention, the FRET efficiency means the
ratio of the intensity of acceptor fluorophore to the intensity of the
18
CA 02419503 2003-02-14
acceptor fluorophore. This will be described in detail in the following
paragraphs.
For the FRET measurement, the following three factors demand
consideration. I) Overlap of the emission wavelength of the GFP
donor and the excitation wavelength of the GFP acceptor. II)
Distance between the donor and acceptor. III) Moment of the
emission from donor and the moment of the excitation of the acceptor.
Furthermore, by the structural tension caused by the fused proteins,
GFPs may not form chromophore efficiently. Therefore, a probe
wherein the energy is transferred efficiently from the GFP donor to
the GFP acceptor by FRET can be constructed only after fulfilling
many strict conditions. However, the conditions wherein FRET is
always observed has not been reported and usually, construction of
such probes require enormous efforts. In other words, a FRET-based
probe cannot be easily constructed on the known techniques and can
be prepared only after many try-and-error experiments and many
sophisticated experiments. The invented monitoring proteins are
generated after such efforts to obtain the desired effects. In this probe
the specific binding of GTP-bound low-molecular-weight GTP-
binding protein to the target protein is designed to greatly change the
level of FRET efficiency; therefore it is extremely useful in many
applications.
The order of the components of the probes can be selected by
the change in the level of FRET efficiency after the activation of the
low-molecular-weight GTP-binding proteins, which will be simply
19
CA 02419503 2003-02-14
called the change in FRET efficiency. Larger the change in FRET
efficiency, more sensitive is the detection of the activation of the low-
molecular-weight GTP-binding proteins. The most desirable aspects
of the probes include that the carboxyl-terminus of the low-
molecular-weight GTP-binding proteins is bound directly or
indirectly to the amino-terminus of the target protein (1) and that the
carboxyl-terminus of the target protein is bound directly or indirectly
to the amino-terminus of low-molecular-weight GTP-binding
proteins (2). Particularly, when the low-molecular-weight GTP-
binding proteins belong to the Ras-family, the aspect (1) is preferable;
when the low-molecular-weight GTP-binding proteins belong to the
Rho-family, the aspect (2) is preferable. GFP acceptor and GFP donor
proteins are ligated directly or indirectly to either the amino-terminus
or the carboxyl-terminus of the G protein-target protein complex. In
particularly preferable aspects, the G protein-target protein complex
binds to GFP acceptor at its amino-terminus and to GFP donor at its
carboxyl-terminus. Therefore, the invented monitoring protein
preferably consists of, from the amino-terminus, GFP acceptor
protein, low-molecular-weight GTP-binding protein, its target
protein, and GFP donor protein, which are bound to each other
directly or indirectly. When it is called indirectly, it means that the
proteins are linked each other with a peptide spacer as will be
described later.
There is no restriction in the kind of the low-molecular-weight
GTP-binding proteins in the invented monitoring protein; however,
CA 02419503 2003-02-14
from the view of its usefulness, preferably it should belong to Ras-
superfamily G proteins, particularly to Ras-family or Rho-family.
More preferably, low-molecular-weight GTP-binding protein should
be chosen among H-Ras, K-Ras, N-Ras, R-Ras, RaplA, RaplB, Rap2A,
and Rap2B that belong to the Ras-family, or RhoA, RhoB, RhoC, Racl,
Rac2, and Cdc42 that belong to the Rho-family.
There is no restriction in the species of target proteins, if they
bind to the low-molecular-weight GTP-binding proteins in a GTP-
dependent manner. For the viewpoint of usefulness, they are Raf and
RaIGDS for Ras-family and Pak or mDia for the Rho-family.
Furthermore, as pairs of low-molecular-weight GTP-binding
protein and its target protein, from the viewpoint of usefulness and
specificity, the following are preferable: the low-molecular-weight
GTP-binding protein is H-Ras and the target protein is Raf, the low-
molecular-weight GTP-binding protein is RaplA and the target
protein is RaIGDS, the low-molecular-weight GTP-binding protein is
Rac1 and the target protein is Pak, the low-molecular-weight GTP-
binding protein is Cdc42 and the target protein is Pak, and the low-
molecular-weight GTP-binding protein is RhoA and the target
protein is mDia.
Any of the GFP-related proteins can be used as the GFP
acceptor protein. From the functional viewpoint, EGFP and EYFP are
preferable. Similarly, any of the GFP-related proteins can be used as
GFP donor protein and from the functional viewpoint, ECFP and
EBFP are preferable.
21
CA 02419503 2003-02-14
From the viewpoints of usefulness, specificity, and sensitivity,
most preferable combinations of the constituents of the probes are as
following: (1) The low-molecular-weight GTP-binding protein is H-
Ras, the target protein is Raf, GFP donor protein is ECFP, and GFP
acceptor protein is EYFP. (2) The low-molecular-weight GTP-binding
protein is RaplA, the target protein is RaIGDS, GFP donor protein is
ECFP, and GFP acceptor protein is EYFP. (3) The low-molecular-
weight GTP-binding protein is Racl, the target protein is Pak, GFP
donor protein is ECFP, and GFP acceptor protein is EYFP. (4) The
low-molecular-weight GTP-binding protein is Cdc42, the target
protein is Pak, GFP donor protein is ECFP, and GFP acceptor protein
is EYFP. (5) The low-molecular-weight GTP-binding protein is RhoA,
the target protein is mDia, GFP donor protein is ECFP, and GFP
acceptor protein is EYFP.
From the viewpoint of the change in FRET efficiency, the
preferable orders of the low-molecular-weight GTP-binding protein,
target protein, GFP donor protein, and GFP acceptor protein in the
invented probes are as following: EYFP-H-Ras-Raf-ECFP, EYFP-
RaplA-RaIGDS-ECFP, EYFP-Pak-Rac1-ECFP, EYFP-Pak-Cdc42-ECFP,
and EYFP-mDia-RhoA-ECFP. Notably, the orders of EYFP and ECFP
can be changeable.
The low-molecular-weight GTP-binding protein does not
necessarily consist of full-length peptide and can be a part of low-
molecular-weight GTP-binding protein, if it can bind to the target
protein. This property of a part of low-molecular-weight GTP-
22
CA 02419503 2003-02-14
binding protein can be tested, for example, by examining its binding
to target proteins after in vitro loading of GTP by any known
methods. The binding of a part of low-molecular-weight GTP-
binding protein to the target protein can be detected, for example, by
immunoprecipitating the target protein and detecting the part of low-
molecular-weight GTP binding protein by immunoblotting.
Followings are the examples of the parts of low-molecular-weight
GTP-binding protein: amino-acids 1 to 180, or preferably 1 to 172, of
H-Ras and Rapl; amino-acids 1 to 204, or preferably 28 to 204, of R-
Ras; amino-acids 1 to 277 of Racl; amino-acids 1 to 176 of Cdc42 and
RhoA.
Sometimes, the change in FRET efficiency can be increased by
trimming the amino- and/ or carboxyl-terminal regions of the low-
molecular-weight GTP-binding protein. Therefore, "a part of low-
molecular-weight GTP-binding protein" includes those with at least
one amino-acid deletion, preferably 1 to 28, more preferably 17 to 28
amino-acid deletions. For example, the change in FRET efficiency is
larger in the probe with the carboxyl-terminal deletion to amino acid
170 than one to amino acid 180. Therefore, the carboxyl-terminus of
low-molecular-weight GTP-binding protein should be trimmed for,
at least one, preferably 9 to 20, more preferably 17 amino acids.
The amino-terminal and carboxyl-terminal regions generally
indicate up to 30 amino-acid regions from either amino-terminus or
carboxyl-terminus of the low-molecular-weight GTP-binding protein.
23
CA 02419503 2003-02-14
Similarly, the target protein does not necessarily consist of full-
length peptide and can be a part of the target protein, if it can bind to
low-molecular-weight GTP-binding protein. Notably, the nature of a
part of target protein can be examined similarly as described for Iow-
molecular-weight GTP-binding protein. Followings are such
examples: in case of Raf (Genbank/EMBL accession number: X03484),
preferably the Ras-binding region (amino acid 51 to 204), more
preferably 51 to 131; in case of RaIGDS (Genbank/EMBL accession
number: U24417), preferably amino acid 202 to 309, more preferably
amino acid 211 to 297; in case of Pak1 (Genbank/EMBL accession
number: NM002576), amino acid 68 to 150; in case of rnDia1
(Genbank/EMBL accession number: E17362), amino acid 68 to 240,
more preferably 68 to 180.
Meanwhile, GFP donor and/or GFP acceptor protein does not
necessarily constitute of full-length peptide and can be a parf of the
target protein, only if it can be used as FRET pairs. Sometimes,
trimming the carboxyl-terminal regions of these proteins increases
the change in FRET efficiency. For example, GFP donor and/or GFP
acceptor protein preferably possesses at least one, more preferably
one to eleven amino acids deletion. In case of EYFP, its carboxyl
region may have preferably at least one, more preferably one to
eleven amino-acids deletion: In case of ECFP, its carboxyl region may
have preferably at least one, more preferably one to eleven amino
acids deletion. Here the carboxyl-terminal region is defined as the
amino acid region of 1 to 20, preferably up to 11 amino acid from the
z4
CA 02419503 2003-02-14
carboxyl terminus of GFP-related proteins. Whether the trimmed
GFP proteins function as FRET pairs can be examined as follows: The
GFP proteins are expressed in E. coli and then the fluorescent
spectrum of the cell lysates are obtained by using the cell lysates.
Furthermore, the GFP donor and jor acceptor proteins can
possess mutations. These mutations can be introduced to any amino
acid regions as far as they do not inhibit FRET. One aspect of such
mutation is a GFP mutant (Phe64Leu, Va168Leu, Ser72Ala, Ile67Thr).
Introduction of these mutations are preferable because they may
increase the efficiency of fluorophore maturation or change in FRET
efficiency.
'The mutation can also be introduced into the low-molecular-
weight GTP-binding protein or target proteins. For example, by
introducing a point mutation, the sensitivity to guanine nucleotide
exchange factors or GTPase activating proteins can be increased.
These mutations can be introduced to at any amino acid regions as
far as they do not inhibit the intramolecular binding of low-
molecular-weight GTP-binding protein and target proteins. Aspects
of such mutations include amino-acid substitution, insertion, and/ or
deletion. For example, Ile36Leu mutation in H-Ras amino-acid
sequence increases the sensitivity of the probe to the GTPase
activating protein. As a result, the dynamic range of the probe can be
increased. These mutant H-Ras proteins are preferably used in the
invented monitoring proteins. These mutations can be easily
introduced by using either restriction enzymes or by PCR.
CA 02419503 2003-02-14
In the invented monitoring protein, the spatial arrangement of
each component protein affects its function. By changing the spatial
arrangement, the change in FRET efficiency can be remarkably
increased. For example, by the insertion of a spacer peptide between
the protein components, the change in FRET efficiency can be
modulated. To increase the change in FRET efficiency, such spacers
are preferably inserted between the low-molecular-weight GTP-
binding protein and the target protein. The length of spacer peptide,
which can consist of any amino acids, is preferably between 1 to 30,
more preferably 1 to 10. By inserting these peptides, the change in
FRET efficiency and/or the folding of GFP can be enhanced. For the
proper conformational arrangement, the spacer peptides may
preferably consist of many glycine residues.
Another preferable aspect of the monitoring proteins is that the
monitoring protein is fused to other peptides or proteins at either the
amino-terminus or the carboxyl-terminus. Particularly, by fusing
intracellular localization signals such as endoplasmic reticulum-
localization signal or membrane localization signal, the monitoring
proteins can measure the local activity of Iow-molecular-weight GTP-
binding proteins. Furthermore, as will be described later, the monitor
can measure the local ratio of GTP-bound to GDP-bound low-
molecular-weight GTP-binding proteins.
In the invented monitoring proteins, the activation of low-
molecular-weight GTP-binding protein by GTP loading will cause the
intramolecular binding of the low-molecular-weight GTP-binding
26
CA 02419503 2003-02-14
protein to the target protein, thereby inducing the conformational
change of the probe, thereby changing the relative direction and
distance between the GFP donor and the GFP acceptor proteins.
Therefore, by the excitation at predetermined wavelength, the
increase in FRET efficiency from the donor to acceptor proteins can
be monitored. Such change in FRET efficiency is affected by the
positioning of GFP donor and GFP acceptor proteins after the
conformational change of the probe. Fox example, shortening of the
distance between GFP donor and GFP acceptor proteins will increase
in FRET efficiency, and the lengthening of it will decrease the FRET
efficiency. Dynamic range of the FRET efficiency, in the other words,
the difference between the maximum and the minimum FRET
efficiency, can be tuned to the desired Ievel by inserting spacer
peptides, depending on the property of constituent proteins.
This invention also provides the genes encoding the invented
monitoring proteins. Such genes can be constructed by obtaining the
sequence information from Genbank etc., by PCR amplification, or by
using restriction enzymes and ligase.
Followings are names and Genbank jEMBL accession numbers
of proteins used preferably as constituents of monitoring proteins.
Accession numbers are shown in the parenthesis.
(1) low-molecular-weight GTP-binding proteins
H-Ras (V00574), K-Ras (L00045 - L00049), N-Ras (L00040 -
L00043), R-Ras (M24948, M14949), RaplA (X12533), RaplB
(X08004), Rap2A (X12534),Rap2B (X52987), RhoA (L25080),
27
CA 02419503 2003-02-14
RhoB (X06820), RhoC (X06821), Rac1(M29870), Rac2
(NM002872), Rac3 (NM005052), Cdc42 (M57298)
(2) target protein
Raf (X03484), RaIGDS (U14417), Pak2 (NM002576), mDia1
(E27362)
(3) GFP donor and acceptor proteins
EGFP (U76561), EYFP (U73901), ECFP (AB041904)
EBFP described in ref. 6 carries the following three mutations:
Phe64Leu, Tyr66His, Tyr145Phe.
The present invention also provides the expression vectors
encoding the genes. Such vectors are obtained by inserting the genes
of the monitoring proteins into any known prokaryotic expression
vectors including pGEX-2T (Amersham), eukaryotic expression
vectors including pCAGGS (ref. 7), or viral vectors including pShuttle
(Clontech). As an expression vector, expression plasmids are used
preferably.
The present invention also provides cells or transgenic animals
carrying the expression vectors. Such cells can be obtained by
introducing the expression vectors into the cells. There is no
restriction in the method of introducing the genes into the cells,
including calcium phosphate coprecipitation method, Iipofection, or
electroporation. Any prokaryotic or eukaryotic cells can be used as
the host. Followings are some examples of eukaryotic cells; human
embryonic kidney cell HEK293T, monkey kidney cell COS, human
28
CA 02419503 2003-02-14
umbilical venous endothelial cell; and prokaryotic cells, including E.
coli. Meanwhile, by microinjecting the expression vector into mouse
fertilized eggs, transgenic mouse can be obtained.
The present invention also provides the method for the
measurement of the activify of low-molecular-weight GTP-binding
proteins. In this method, by measuring the FRET efficiency of said
monitoring protein, the activity of low-molecular-weight GTP-
binding proteins can be measured. Moreover, by measuring the FRET
of said transformed cells or transgenic animals, the activity of Iow-
molecular-weight GTP-binding proteins in these cells and animals
can be measured. By preparing a calibration curve of GTP/GDP ratio
of the low-molecular-weight GTP-binding proteins against FRET
efficiency of the probe, the data on the FRET efficiency can be
correlated with the data of GTP/GDP on the low-molecular-weight
GTP-binding proteins.
Followings are such examples.
(1) A method using spectrometer
Cells that can express monitoring proteins are cultured in the
condition wherein the monitoring proteins are expressed. Cells can
be lysed by any methods, preferably by using buffer containing
Triton X-100. The cell lysates are illuminated at an excitation
wavelength of GFP donor protein (ex. 433 nm) and spectrogram is
obtained with any known spectrometers. Based on the spectrogram,
for example, the ratio of fluorescent intensity of donor protein at a
29
CA 02419503 2003-02-14
wavelength of 475 nm vs fluorescent intensity of acceptor protein at a
wavelength of 530 nm ((fluorescent intensity at an wavelength of 530
nm)/ (fluorescent intensity at an wavelength of 475 nm)) is calculated
to estimate the FRET efficiency. Because the FRET efficiency after
GTP loading to the low-molecular-weight GTP-binding proteins
(namely, activation of low-molecular-weight GTP-binding proteins)
is higher than that before GTP binding, the FRET efficiency can be
used to measure the activation of low-molecular-weight GTP-binding
proteins. The activation of low-molecular-weight GTP-binding
proteins can be induced by co-expressing guanine nucleotide
exchange factor expression vector such as pCAGGS-Sos (ref. 9) or by
stimulating the cells with growth factors such as EGF. Similarly,
inactivation of low-molecular-weight GTP-binding proteins can be
induced by co-expressing expression vectors for GTPase activating
proteins such as pEF-Bos-GAPlm (ref. 9). Meanwhile, since the FRET
efficiency is influenced by the distance and direction of the GFP
donor and the GFP acceptor, the change in protein conformation can
also be detected by the change in FRET efficiency.
(2) A method using fluorescence microscope
The change in FRET efficiency before and after the activation of
low-molecular-weight GTP-binding proteins can be directly
examined by observing the invented cells or transgenic animals
expressing monitoring proteins with any fluorescence microscope.
CA 02419503 2003-02-14
The activation and inactivation of low-molecular-weight GTP-
binding proteins can be induced similarly to (1).
Any microscope can be used; however, inverted fluorescence
microscope (Carl Zeiss, Axiovert 100) equipped with revolving filter
changers containing excitation and emission filters and high
sensitivity cooled CCD camera. More preferably, the filter changers
and CCD camera are controlled by Metamorph imaging software
(Roper Scientific Japan).
The cells or animals are illuminated at the excitation
wavelength of GFP donor protein and the image is obtained at the
emission wavelength of the donor protein. Then, the image is
obtained at the wavelength of the fluorescence of the acceptor protein.
By calculating the ratio of the intensities of the both images, FRET
efficiency at each pixel can be obtained. The calibration of FRET data
with GTP/GDP ratio can be performed as following. First, various
activation levels of low-molecular-weight GTP-binding protein is
achieved by expressing various amounts of guanine nucleotide
exchange factor such as Sos. Then, FRET efficiency in these cells is
examined with a fluorescent microscope by the method. In parallel,
similarly-prepared cells are lysed and used to measure the ratio of
GTP-bound to GDP-bound low-molecular-weight GTP-binding
proteins as described (ref. 2). Lastly, the data of FRET efficiency are
plotted against the GTP/ GDP ratio of the monitoring proteins. In
other words, both FRET efficiency and GTP/GDP ratio of the
monitoring proteins are measured in various conditions, which data
31
CA 02419503 2003-02-14
are used to prepare calibration curve. By this method, by simply
observing the cells or animals with a fluorescent microscope and
measuring the FILET efficiency, the GTP/GDP level at each time
point and each place can be determined. Therefore, this method
allows us to know the activation status of low-molecular-weight
GTP-binding protein very easily in living cells, and furthermore,
these data can be correlated with the GTP/ GDP ratio. Similar method
for preparing the calibration curve can be also applicable to the
method (1).
The present invention provides the monitoring proteins that
envision non-destructive measurement of the activation status of
low-molecular-weight GTP-binding proteins, and also its genes etc.
The present invention also provides cells and transgenic animals that
express and encode the useful monitoring protein, and also the
method to measure the activity of low-molecular-weight GTP-
binding proteins. Therefore, this invention enables us to know the
activation status of low-molecular-weight GTP-binding proteins by
non-destructive methods. These features will have a great benefit not
only in the field of bioscience but also in the development of drugs,
for example, therapeutic and prophylactic drugs for cancer,
autoimmune disease, and allergic disease.
Another aspect of this invention is the screening methods for
the substances which regulate the activity of Iow-molecular-weight
GTP-binding proteins. Namely,
32
CA 02419503 2003-02-14
(a) procedure wherein substances are incubated with cells carrying
the expression vector for and expressing the monitoring protein of
low-molecular-weight GTP-binding protein, and
(b)procedure wherein the activity change of low-molecular-weight
GTP-binding protein is detected.
According to this screening method, substances or their salts that
can change the activity of low-molecular-weight GTP-binding protein
{namely, the regulatory substances of low-molecular-weight GTP-
binding protein) can be effectively screened by preparing cells that
express the monitoring proteins for fhe low-molecular-weight GTP-
binding protein and constructing bioassay system. The subject can be
any materials, but is preferably peptide, protein, non-peptide
materials, synthetic materials, and fermented materials.
The invented screening procedure can be performed {i) in the
presence of the activators of low-molecular-weight GTP-binding
proteins, ox (ii) in the absence of the activator of low-molecular-
weight GTP-binding proteins. Here, the activator of low-molecular-
weight GTP-binding proteins means substances that active low-
molecular-weight GTP-binding proteins, for example, cell gxowth
factors such as epidermal growth factor, or cytokines such as
interleukin; however, the activator is not limited to these materials.
The regulatory substances of low-molecular-weight GTP-binding
proteins can be detected as materials that either increase or decrease
the activity of low-molecular-weight GTP-binding proteins in case of
33
CA 02419503 2003-02-14
method (i), and as materials that increase the activity of low-
molecular-weight GTP-binding proteins in case of method (ii).
Detailed description of the invented screening procedure is as
follows. In the presence or absence of the activator, the cells
expressing the invented monitoring proteins are incubated with the
substances in case of procedure (a) (aspect 1). There is no limitation in
the method of incubation, for example, the cells can be cultured in the
presence of the substances. In parallel, as a control, the cells are kept
in the same condition without incubating with the substances (aspect
2). Then, in procedure (b), the activity of low-molecular-weight GTP-
binding protein is measured. By comparing the activity measured in
aspect 1 with that in aspect 2, the regulatory substances of low-
molecular-weight GTP-binding protein can be screened. The activity
of low-molecular-weight GTP-binding protein is measured by
quantitating the FRET efficiency.
In conclusion, the substances that enhance the activity of low-
molecular-weight GTP-binding proteins in case (i) is the regulatory
substances that increase the activity of low-molecular-weight GTP-
binding proteins, and, in contrast, the materials that suppress the
activity of low-molecular-weight GTP-binding proteins in case (i) is
the regulatory substances that decrease the activity of low-molecular-
weight GTP-binding proteins. Furthermore, the substances that
enhance the activity of low-molecular-weight GTP-binding proteins
in case (ii) are the regulatory substances that increase the activity of
low-molecular-weight GTP-binding proteins.
34
CA 02419503 2003-02-14
References
Followings are the list of references described in this disclosure.
1. Bos, J. L.1997. Ras-like GTPases. Biochim. Biophys. Acta
1333:M19-M31.
2. Satoh, T. and Y. Kaziro.1995. Measurement of Ras-bound guanine
nucleotide in stimulated hematopoietic cells. Method. Enzymol.
255:149-155.
3. Franke, B., J. W. N. Akkerman, and J. L. Bos.1997. Rapid Ca2+-
mediated activation of Rap1 in human platelets. EMBO J.15:252-
259.
4. Tsien, R. Y. and A. Miyawaki.1998. Seeing the machinery of live
cells. Science 280:1954-1955.
5. Pollok, B. A. and R. Heim.1999. Using GFP in FRET-based
applications. Trends Cell Biol. 9:57-60.
6. Miyawaki, A., J. Llopis, R. Heim, J. M. McCaffery, J. A. Adams, M.
Ikura, and R. Y. Tsien.1997. Fluorescent indicators for Ca2+ based
on green fluorescent proteins and calmodulin. Nature 388:882-887.
7. Niwa, H., K. Yamamura, and J. Miyazaki.1991. Efficient selection
for high-expression transfectants with a novel eukaryotic vector.
Gene 108:193-200.
8. DeClue, J. E., J. C. Stone, R. A. Blanchard, A. G. Papageorge, P.
Martin, K. Zhang, and D. R. Lowy. A ras effector domain mutant
which is temperature sensitive for cellular transformation:
CA 02419503 2003-02-14
interactions with GTPase-activating protein and NF-1. Mol.Cell
Biol.11:3132-3138,1991.
9. Ohba, Y., N. Mochizuki, S. Yamashita, A. M. Chan, J. W. Schrader,
S. Hattori, K. Nagashima, and M. Matsuda. Regulatory proteins of
R-Ras, TC-21/R-Ras2, and M-Ras/R-Ras3. J. Biol. Chem.
275:20020-20026, 2000.
lO.Yamashita, S., N. Mochizuki, Y. Ohba, M. Tobiume, Y. Okada, H.
Sawa, K. Nagashima, and M. Matsuda. GaIDAG-GEFIII activation
of Ras, R-Ras, and Rapl. J. Biol. Chem. 275:25488-25493, 2000.
11.T. Gotoh, S. Hattori, S. Nakamura, H. Kitayama, M. Noda, Y. Takai,
K. Kaibuchi, H. Matsui, O. Hatase, H. Takahashi, T. Kurata, and M.
Matsuda. Identification of Rap1 as a target for Crk SH3 domain-
binding guanine nucleotide-releasing factor, C3G. Mol.Cell.Biol.
15:6746-6753,1995.
36
CA 02419503 2003-02-14
EXAMPLES
Hereinafter, this invention will be described with examples;
however, the content of this invention shall not be limited to these
examples. Note that human H-Ras, human c-Rafl, human RaplA,
human RaIGDS, human R-Ras, human Racl, human Cdc42, human
RhoA, human Pakl, and human mDia1 will be called simply as Ras,
Raf, RaplA, RaIGDS, R-Ras, Racl, Cdc42, RhoA, Pakl, and mDial,
respectively.
Eample 1 Measurement of Ras activity by the use of Rafras1722.
(1) Construction of a gene encoding a chimera of Ras and Raf
(i) Amplification of Ras gene
Using Ras cDNA (Genbank/EMBL accession number: V00574) as a
template, sense primer hRasXh (5'-
CTCGAGATGACGGAATATAAGCTGGTGGTG-3") (sequence
number:1), anti-sense primer Ras172Raf (5'-
AGTGTTGCTTGTCTTAGAAGGGGTACCACCTCCGGAGCCGTTC
AGCTTCCGCAGCTTGTG-3') (sequence number: 2), and heat stable
DNA polymerase Pfx (Gibco-BRL, Bethesda, U.S.A.), DNA fragment
corresponding to amino acid 1 to 172 of Ras was amplified by
polymerase chain reaction (PCR).
Sense primer hRasXh consists of the underlined recognition
sequence of restriction enzyme XhoI and a DNA sequence
corresponding to the amino acid 1 to 8 of Ras. Meanwhile, anti-sense
primer Ras172Raf consists of a complementary DNA sequence of Raf
37
CA 02419503 2003-02-14
corresponding to amino-terminal region of the Ras binding domain
(amino acid 61 to fi7), spacer sequence (underlined), and DNA
sequence of Ras corresponding to amino acid166 to 172.
(ii) Amplification of Raf gene
Using Raf cDNA (Genbank/EMBL accession number: X03484)
as a template, sense primer RafRBD-F1 (5'-
GGTACCCCTTCTAAGACAAGCAACACT -3")(sequence number: 3),
anti-sense primer RafRBDn2 (5'-
GCGGCCGCCCAGGAAATCTACTTGAAGTTC -3') (sequence
number: 4), and said Pfx, DNA corresponding to amino acid 51 to 131
of Raf were amplified by PCR.
Sense primer RafRBD-F1 consists of the underlined recognition
sequence of restriction enzyme KpnI and a DNA sequence
corresponding to the amino acid 51 to 57 of Raf. Anti-sense primer
RafRBDn2 consists of a complementary DNA sequence of Raf
corresponding to the carboxyl-terminal region of the Ras binding
domain (amino acid 125 to 131).
Using a mixture of the amplified DNA fragments described in
(i) and (ii) as templates, sense primer hRasXh, anti-sense primer
RafRBDn2, and said Pfx, DNA of a chimera of Ras and Raf was
amplified by PCR. Then, the obtained DNA fragment was cloned into
pCR-bluntII-TOPO (Invitrogen), followed by transformation of E. coli.
Then, the E. coli was cultured and plasmids were prepared by the
SDS-alkaline method.
38
CA 02419503 2003-02-14
(2) Construction of pFret2, an expression vector encoding EYFP and
ECFP
(i) Construction of pCAGGS-P7
The multiple cloning site of pBluescript-SKII (+) (Stratagene) is
PCR-amplified with primer P7 (5'-
CGCCAGGGTTTTCCCAGTCACGAC-3')(sequence number: 5) and
primer P8 (5'-AGCGGATAACAATTTCACACAGGAAAC-
3')(sequence number: 6) as described. pCAGGS (ref. ~ was cleaved
with EcoRI and blunt-ended with Klenow enzyme, and ligated with
the PCR-amplified fragment, generating pCAGGS-P7.
(ii) Amplification of EYFP gene
In this example, EYFP was obtained from EGFP
(Genbank/EMBL accessionnumber: U76561) by introducing six
amino acid substitution
(Leu65Phe;Thr66Gly;Va169Leu;G1n70Lys;Ser73Ala;Thr204Tyr) by use
of PCR-mediated mutagenesis. Then, full length cDNA of EYFP was
obtained by using the EYFP gene as template, sense primer GFP-N2
(5'-GGATCCGGCATGGTGAGCAAGGGCGAGGAG-3') (sequence
number: 7), anti-sense primer GFP-N3 (5'-
GGATCCGGTACCTCGAGCTTGTACAGCTCGTCCATG-3')
(sequence number: 8), and said Pfx.
Sense primer GFP-N2 consists of the underlined recognition
sequence of BamHI, three-bases spacer, and the nucleotide sequence
39
CA 02419503 2003-02-14
corresponding to amino acid 1 to 7 of EYFP. Antisense primer GFP-
N3 consists of the underlined restriction sequences of BamHI, KpnI,
and XhoI, and the complementary sequence corresponding to the
carboxyl-terminus of ECFP (amino acid 233 to 239).
(iii) Amplification of ECFP gene
In this example, ECFP was obtained from EGFP
(Genbank/EMBL accession number: U76561} by introducing six
amino acid substitution (Tyr6TTrp; Asn147I1e; Met154Thr; Va1164A1a)
by use of PCR-mediated mutagenesis. Then, full length DNA of ECFP
was obtained by using the EYFP gene as a template, sense primer
XFPNot2 (5'- GCGGCCGCATGGTGAGCAAGGGCGAGGAGC -3')
(sequence number: 9), anti-sense primer XFP-Bgl (5'-
AGATCTACAGCTCGTCCATGCCGAGAG -3') (sequence number:
10), and said Pfx.
Sense primer XFPNot2 consists of the underlined recognition
sequence of NotI and the nucleotide sequence corresponding to
amino acid 1 to 8 of ECFP. Antisense primer XFP-Bgl consists of the
underlined restriction site of BgIII and the complementary sequence
corresponding to the carboxyl-terminus of ECFP (amino acid 231 to
23~.
(iv) Construction of pFret2
pCAGGS-P7 obtained in (i) was cleaved with XhoI and partially
filled in with Klenow enzyme in the presence of dTTP and dCTP.
CA 02419503 2003-02-14
EYFP obtained in (ii) was cleaved with BamHI and partially filled in
with Klenow enzyme in the presence of dATP and dGTP. These two
fragments were ligated by T4 DNA ligase. Then, the plasmid was
cleaved with NotI and BgIII and ligated with ECFP which was
obtained in (iii) and cleaved with the same restriction enzymes. The
obtained plasmid was named as pFret2.
(3) Construction of pRafras1722, an expression plasmid for the
monitoring protein of Ras.
pFret2 as described in (2)-(iv) was cleaved with XhoI and NotI
and ligated by using T4 DNA ligase with the chimeric gene described
in (1)-(iii) cleaved with the same restriction enzymes, generating
pRaf ras1722.
The structure and the nucleotide sequence of the coding region
(sequence number:11) and predicted amino acid sequence (sequence
number:12) are shown in Fig. 3 and Fig. 4 to 6, respectively. Detailed
explanation is as follows.
nt 1- 717 : Aequorea EYFP
nt 718 - 723 : Linker
nt 724 -1239 : Ras
nt 1240 -1257: Linker
nt 1258 -1500: Raf
nt 1501-1509 : Linker
nt 1510 - : Aequorea ECFP
2220
41
CA 02419503 2003-02-14
(4) Expression of Ras monitoring protein Rafras1722 in mammalian
cells and its spectrum analysis
HEIC293T cells derived from human embryonic kidney cells
were cultured in DMEM (Nissui) containing 10% fetal calf serum.
pRafras1722 described in (3) and an expression vector of guanine
nucleotide exchange factor Sos (pCAGGS-mSos) or an expression
vector of GTPase activating protein Gaplm (pCAGGS-mSos) were
transfected into HEIC293T cells by calcium coprecipitation method.
After transfection, HEK293T cells were further cultured in DMEM
containing 10 % FBS to allow the Ras monitoring protein expressed.
Forty-eight hours later, cells were washed with phosphate-buffered
saline and lysed in lysis buffer (20 mM Tris-HCl, pH 7.5,150 mM
NaCI, 5 mM MgCl2, 0.1 % Triton X-100). The cell lysates were
centrifuged at 10,000 x g and the supernatant was collected.
The supernatant was transferred into 1 ml cuvette of
fluorescent spectrometer (Nippon Bunko, FP-750) and fluorescent
intensity was analyzed from 450 nm to 550 nm at an excitation
wavelength of 433 nm. The obtained fluorescent profile is shown in
Fig. 7.
The transfected HEK293T cells were also labeled with 32P1 and
lysed. Ras monitoring protein was immunoprecipitated with anti-
GFP antibody and bound GTP and GDP were separated by thin layer
chromatography. By this method, the FRET efficiency ((fluorescent
intensity at 530 nm) / (fluorescent intensity at 475 nm) at an
excitation wavelength of 433 nm) can be correlated with the actual
42
CA 02419503 2003-02-14
GTP/GDP ratio (Fig. 8). In the Fig. 8, FRET efficiency and GTP-
binding are shown as (fluorescent intensity (530 nm/475 nm)) and
(GTP/(GDP + GTP) (%)), respectively.
(5) Expression of Ras monitoring protein in mammalian cells and
analysis with time-lapse fluorescent microscope.
COS7 cells derived from monkey kidney cells were cultured in
phenol-red-free MEM (Nissui) containing 10 % fetal calf serum.
pRafras1722 described in (3) was transfected into COS7 cells by
calcium phosphate method. After transfection, COS7 cells were
cultured in phenol-red-free MEM (Nissui) containing 10% fetal calf
serum to allow the expression of Ras monitoring protein. Forty-eight
hours after transfection, cells were observed with time-lapse
fluorescent microscope.
Cell images were obtained with an imaging system consisting
of Metamorph image analyzing software (Roper Scientific Japan) and
inverted fluorescent microscope Axiovert 100 (Carl Zeiss) equipped
with Xenon lamp, revolving filter changers for excitation filters and
emission filters (LUDL electronic), and high sensitivity cooled CCD
camera Micromax 450 (Photometrix). Excitation filters, emission
filters and dichroic mirrors were obtained from Omega.
Cells were illuminated with an excitation wavelength of 430 nm
and fluorescent images of ECFP donor protein at 4~5 nm and
fluorescent images of EYFP acceptor protein at 530 nm were acquired.
After data acquisition, each pixel of the digital images was assigned
43
CA 02419503 2003-02-14
from blue to red colors, depending on the levels of EYFP/ECFP
fluorescent ratios.
Example 2 Establishment of cell lines for the measurement of Ras
activation
Mouse fibroblast NIH3T3 cells were cultured in DMEM
(Nissui) containing 10% fetal calf serum. pRafras1722 and pSV2neo
(Genbank/EMBL: U02434) were co-transfected into NIH3T3 cells
with FuGene6 (Roche). Forty-eight hours after transfection, cells were
replated at 1:10 and cultured in said DMEM containing 0.5 mg/ml
6418 (Gibco-BRL). Medium was replaced every three days. After 2
weeks, cells of well-isolated colonies were cloned and named as 3T3-
Rafras cells.
The 3T3-Rafras cells were cultured in DMEM containing 10%
fetal calf serum and 0.5 mg/ml 6418 to allow the expression of
monitoring protein for Ras activity. Then, the expression of said
protein was examined by anti-Ras antibody (Transduction Lab) by
immunoblotting. Expression of expected ca. 80 kDa protein was
confirmed (Fig. 9).
Furthermore, the cells were stimulated with epidermal growth
factor (EGF) (Sigma), and the FRET efficiency before and after EGF
stimulation was analyzed as described in example 1. The fluorescence
profile before and after EGF stimulation is shown in Fig.10.
Example 3 Measurement of RaplA activity by using Rai-chu311
44
CA 02419503 2003-02-14
(1) Construction of a gene encoding a chimera of RaplA and RaIGDS
(i) Amplification of RaplA gene
Using RaplA cDNA (Genbank/EMBL accession number:
X12533) as a template, sense primer hRaplXh (5'-
GGCTCGAGATGCGTGAGTACAAGCTAGTGG--3") (sequence
number:13), anti-sense primer Rap172RalGDS (5'-
GCGGATGATACAGCAGTCGCCACCTCCGGATCCGCCGGTACC
TCCACCACCGGTTCCACCTCCGGAGCCATTGATCTTTGACTTTG
CAGAAG -3') (sequence number:14), and heat stable DNA
polymerase Pfx (Gibco-BRL, Bethesda, U.S.A.), DNA corresponding
to the amino acid 1 to 172 of RaplA was amplified by polymerase
chain reaction (PCR).
Sense primer hRaplXh consists of the underlined recognition
sequence of restriction enzyme XhoI and the DNA sequence
corresponding to the amino acid 1 to 8 of RaplA. Meanwhile, anti-
sense primer Rap172RalGDS consists of a complementary DNA
sequence of RaIGDS (Genbank/EMBL accession number : U14417)
corresponding to the amino-terminal region of the Rap1 binding
domain (amino acid 211 to 217), spacer sequence (underlined), and
DNA sequence of RaplA corresponding to amino acid166 to 172.
(ii) Amplification of RaIGDS gene
Using RaIGDS cDNA (Genbank/ EMBL accession number:
U14417) as a template, sense primer RaIGDS-F (5'-
GGCGACTGCTGTATCATCCGC -3")(sequence number:15), anti-
sense primer RaIGDSR (5'-
CA 02419503 2003-02-14
CGCGGCCGCCCCGCTTCTTGAGGACAAAGTC -3') (sequence
number:16), and said Pfx, DNA corresponding to amino acid 51 to
131 of Raf were amplified by polymerase chain reaction (PCR).
Sense primer RaIGDS-F consists of a DNA sequence
corresponding to the amino acid 211 to 217 of RaIGDS. Meanwhile,
anti-sense primer RaIGDSR consists of the underlined Not restriction
sequence and the complementary DNA sequence of RaIGDS
corresponding to carboxyl-terminal region of the Rap1 binding
domain (amino acid 291 to 29~.
Using a mixture of the amplified DNAs described in (i) and (ii)
as templates, sense primer hRaplXh, anti-sense primer RaIGDSR, and
said Pfx, DNA of a chimera of Rap1 and RaIGDS was amplified by
PCR. Then, the obtained DNA fragment was cloned into pCR-bluntII-
TOPO (Invitrogen), followed by transformation of E. coli. Then, the E.
coli was cultured and plasmids were prepared by SDS-alkaline
method.
(2) Construction of pRai-chu311, an expression plasmid for the
monitoring protein of Rapl.
In example 1-(2)-(ii), antisense primer GFP-dllR (5'-
GGATCCGGTACCTCGAGGGCGGCGGTCACGAACTCCAGCAG-
3')(sequence number:17} was used instead of the primer GFP-N3 to
obtain a cDNA of EYFP that lacks eleven amino acids of the carboxyl
terminus. This truncated EYFP cDNA was replaced with the
corresponding region of pFRET2. This vector was cleaved with XhoI
46
CA 02419503 2003-02-14
and NotI, and ligated with the chimeric gene described in (1)-(ii)
cleaved with the same restriction enzymes by using T4 DNA ligase,
generating pRai-chu311.
The structure and the nucleotide sequence of the coding region
(sequence number:18) and predicted amino acid sequence (sequence
number:19) are shown in Fig.11 and Figs.12 to 14, respectively.
Detailed explanation is as follows.
nt 1- 684 : Aequorea EYFP
nt 685 - 690 : Linker
nt 691-1206 : Rap1
nt 120 -1257 : Linker
nt 1258 -1515 : RaIGDS
nt 1516 -1521 : Linker
nt 1522 - 2235 : Aequorea ECFP
(3) Expression of RaplA monitoring protein Rai-chu311 in
mammalian cells and its spectra analysis
Analysis was performed as in example 1-(4). The fluorescent
profile is shown in Fig.15.
Example 4 Measurement of R-Ras activity by using Rai-chu158
(1) Construction of pRai-chu158
(i) Amplification of R-Ras gene
Using R-Ras cDNA (Genbank/ EMBL accession number:
M14948, M14949) as a template, sense primer RRas28F (5'-
47
CA 02419503 2003-02-14
CCCCTCGAGACACACAAGCTGGTGGTC -3")(sequence number:
20), anti-sense primer RRas204R (5'-
GCCGGTACCGCCACTGGGAGGGCTCGGTGGGAG -3') (sequence
number: 21), and heat stable DNA polymerase Pfx (Gibco-BRL,
Bethesda, U.S.A.), DNA corresponding to amino acid 28 to 204 of R-
Ras was amplified by polymerase chain reaction (PCR).
Sense primer RRas28F consists of the underlined XhoI restriction
sequence and the DNA sequence corresponding to the amino acid 28
to 33 of R-Ras. Anti-sense primer RRas204R consists of the
underlined KpnI restriction sequence and the complementary DNA
sequence corresponding to the carboxyl-terminal region of R-Ras
(amino acid 198 to 204).
(ii) Preparation of the restriction fragment.
The PCR product obtained in (i) was cleaved with XhoI and
KpnI.
(iii) Construction of pRai-chu158, an expression plasmid for R-Ras
activity monitoring protein.
pRafras1722 obtained in example 1 was cleaved with XhoI and
KpnI to obtain a DNA fragment that lacks the Ras gene. This plasmid
was ligated with the restriction fragment obtained in (ii), generating
pRai-chu158.
The structure and the nucleotide sequence of the coding region
(sequence number: 22) and predicted amino acid sequence (sequence
48
CA 02419503 2003-02-14
number: 23) are shown in Fig.16 and Figs.17 to 19, respectively.
Detailed explanation is as follows.
nt 1- 717 : Aequorea EYFP
nt 718 - 723 : Linker
nt 724 -1251 : R-Ras
nt 1252 -1257: Linker
nt 1258 -1500: Raf
nt 1501-1509 : Linker
nt 1510 - : Aequorea ECFP
2220
(3) Expression of R-RasA monitoring protein Rai-chu158 in
mammalian cells and its spectra analysis
Analysis was performed as in the example 1 (4). The fluorescent
profile is shown in Fig. 20.
Example 5 Construction of a gene encoding a monitoring protein
which carries a temperature-sensitive mutation in the effector-
binding domain of Ras.
(1) Construction of pRai-chu119
(i) Amplification of a mutated Ras gene
Using the cDNA used in example 1, sense primer hRasXh
(described in example 1), anti-sense primer RasI38LR (5'-
GGAATCCTCTAGAGTGGGGTCG -3') (sequence number: 24), and
the DNA polymerase Pfx, DNA corresponding to amino acid 1 to 39
of Ras was amplified by polymerase chain reaction (PCR).
49
CA 02419503 2003-02-14
Antisense primer RasI38LR consists of a DNA sequence
corresponding to the amino acid 35 to 42 of Ras wherein a codon for
Ile is substituted for Leu as indicated by underline. This point
mutation is known to generate a temperature sensitive Ras mutant
(ref. 8).
Similarly, using the cDNA used in example 1, sense primer
RasI36LF (5'-CGACCCCACTCTAGAGGATTCC-3')(sequence
number 25), anti-sense primer Ras172Raf (described in example 1),
and the DNA polymerase Pfx, DNA corresponding to amino acid 32
to 172 of Ras was amplified by polymerase chain reaction (PCR). By
using the mixture of these two amplified DNA fragments as a
template, sense primer hRasXh, and anti-sense primer Ras172Raf, a
DNA fragment corresponding to amino acid 1 to 172 of Ras that
contains a point mutation of Ile36Leu was amplified by PCR.
(ii) Preparation of restriction fragment.
The PCR product obtained in (i) was cleaved with XhoI and
KpnI.
(iii) Construction of pRai-chu119, an expression plasmid for R-Ras
activity monitoring protein.
pRafras1722 obtained in example 1 was cleaved with XhoI and
KpnI to obtain a DNA fragment that lacks the Ras gene. This plasmid
was ligated with the restriction fragment obtained in (ii), generating
pRai-chu119.
CA 02419503 2003-02-14
The structure and the nucleotide sequence of the coding region
(sequence number: 26) and predicted amino acid sequence (sequence
number: 27) are shown in Figs. 21 to 23.
(2) Expression of Ras monitoring protein Rai-chu119 in mammalian
cells and its spectrum analysis
HEK293T cells derived from human embryonic kidney cells
were cultured in DMEM (Nissui) containing 10 % fetal calf serum.
pRafras1722 described in (3) or pRai-chu119 with an expression
vector of guanine nucleotide exchange factor Sos (pCAGGS-mSos)
were transfected into HEK293T cells by calcium coprecipitation
method. After transfection, HEK293T cells were further cultured in
DMEM containing 10 % FBS to allow the Ras monitoring protein
being expressed. Twenty-four hours after transfection, temperature of
the incubator was changed to 33 or 40 °C. After further 24 hrs, cells
were washed with phosphate-buffered saline and lysed in lysis buffer
(20 mM Tris-HCI, pH 7.5,150 mM NaCI, 5 mM MgClz 0.1 % Triton ~-
100). The cell lysates were centrifuged at 10,000 x g and the
supernatant was collected.
The supernatant was transferred into 1 ml cuvette of
fluorescent spectrometer (Nippon Bunko, FP-750) and fluorescent
intensity was analyzed from 450 nm to 550 nm at an excitation
wavelength of 433 nm. The obtained fluorescent profile is shown in
Fig. 24.
51
CA 02419503 2003-02-14
Example 6 Generation of transgenic mice expressing Rafras1722 and
the measurement of Ras activation using cardiac myocytes.
(1) pRafras1722 described in example 1 was cleaved with restriction
enzyme SpeI and BamHI, and subjected to agarose electrophoresis to
obtain 4.5 kb DNA fragment containing promoter, intron, coding
region, and poly A signal. The DNA fragment was electro-eluted
from the gel and purified with Qiagen-tip 20 (Qiagen). The DNA was
injected to mouse fertilized egg (DBF1, SLC Co.) and transferred to
oviduct of ICR mouse. From the offspring, tail DNA was obtained by
Proteinase K treatment, phenol-chloroform extraction, and
isopropanol precipitation. DNA was dissolved in double distilled
water at 37 ~C.
(2) By using the mouse cDNAs as templates, sense primer RafRBDx
(5'-CTCGAGCCTTCTAAGACAAGCAACACT-3') (sequence number:
28), and anti-sense primer XFPNseq (5'-
CGTCGCCGTCCAGCTCGACCAG -3') (sequence number: 29), PCR
was performed. With these primers, a DNA fragment corresponding
to the junction of Raf and ECFP in Rafras1722 gene was amplified.
Appearance of the expected 314 by fragment indicates the integration
the Rafras1722 gene into the mouse genome. Among 35 newborn
mice, 7 were positive.
(3) The PCR-positive F1 mice were mated with C57/Black mice (SLC,
Hamamatsu, Japan). From the newborn F2 mice, cardiac ventricle
52
CA 02419503 2003-02-14
was excised and cut into pieces. By the addition of PBS containing
0.05 % trypsine and 0.5 mM EDTA for 10 min at 37 °C, cardiac
myocytes were harvested. By repeating this procedure for 6 times,
myocytes were collected. Then, after the addition of DMEM
containing 10% FBS and low-speed centrifugation, myocytes were
plated onto dishes.
{4) The obtained myocytes were replated on glass base dishes.
After 6 hours incubation in serum-free DMEM, EGF was added and
cells were observed as described in example 1- (5). The time-course
of fluorescent intensities of ECFP and EYFP upon EGF stimulation is
shown in Fig. 25. The EGF-induced activation of Ras was observed in
primary cardiac myocytes derived from transgenic mice.
Example 7 Specificity of Rafras1722 to guanine nucleotide exchange
factors and GTPase activating proteins.
By the same procedure shown in example 1- {4), specificity of
Rafras1722 to guanine nucleotide exchange factors and GTPase
activating proteins was examined. Expression vectors for GAPlm, R-
RasGAP, rapGAPII, mSosl, RasGRF, CaIDAG-GEFI, C3G, PDZ-GEF1,
and KIAA0351 were used for GTPase activating protein or guanine
nucleotide exchange factors. As shown in Fig. 26, the FRET efficiency
was decreased by GAPlm, a Ras GAP, but not by R-RasGAP or
rapIGAPII, GAPs for R-Ras or Rapl. In contrast, the FRET efficiency
was increased by GEFs for Ras including mSosl, RasGRF, and
53
CA 02419503 2003-02-14
CaIDAG-GEFII, but not by GEFs for the other Ras-family G proteins
including CaIDAG-GEFI, C3G, PDZ-GEF1, and KIAA0351. These
observations indicate that the FRET efficiency of Rafras1722 is
regulated by the guanine nucleotide exchange factors and GTPase
activating proteins for the authentic Ras.
Example 8 Construction of a Rap monitoring protein Rai-chu404 and
its specificity to guanine nucleotide exchange factors and GTPase
activating proteins.
(1) Construction of a gene encoding Rai-chu404, a chimera of RaplA
and Raf
By PCR mutagenesis, seven amino-acid substitutions
(Thr66Gly; Va169Leu; Ser73Ala; Met154Thr; Va1164A1a; Ser176G1y;
Thr204Tyr) were introduced into EGFP (Genbank/EMBL accession
number: U76561), which was substituted fox EYFP of Rai-chu311
described in example 3. Then, the KpnI/NotI fragment encoding
RaIGDS in this modified Rai-chu311 was replaced with the
KpnI/NotI fragment of Rafras1722 encoding Raf, generating pRai-
chu4Q4. The nucleotide sequence and predicted amino-acid
sequence of the coding region are shown in sequence number 30 and
31, respectively.
(2) The effect of guanine nucleotide exchange factors on the FRET
efficiency of Rai-chu404 was examined as described in example 7.
54
CA 02419503 2003-02-14
PDZ-GEF1, C3G, CaIDAG-GEFI, CaIDAG-GEFIII were used as
guanine nucleotide exchange factors for Rapl. As controls, guanine
nucleotide exchange factors for Ras including CaIDAG-GEFII,
mSosland RasGRF and a guanine nucleotide exchange factor for Ral,
KIAA0351, was used as described in reference 9. As shown in Fig. 27,
only the guanine nucleotide exchange factors for Rap1 could increase
the FRET efficiency of Rai-chu404.
Example 9 Construction of Rai-chu101X and Rai-chu404X,
monitoring proteins that contain the CAAX box of K-Ras.
In the experimental protocol shown in example 1-(1)-(i), a
primer with XbaI restriction site (sequence number 32) was used
instead of the forward primer XFP-Bgl. T'he amplified ECFP gene was
ligated to the CAAX region of K-Ras gene as described in reference 11.
The fused ECFP-CAAX gene was substituted for the ECFP gene of
Rafras1722 or Rai-chu404. The resulting vectors were designated as
pRai-chu101X and pRai-chu404X. The nucleotide sequences of the
coding regions (sequence number: 33 and 35) and predicted amino
acid sequences (sequence number: 34 and 36) are shown.
Example 10 Visualization of the activity of Ras and Rap1 in
COS1 cells expressing Rai-chu101X and Rai-chu404X.
COS1 cells transfected with pRai-chu101X or pRai-chu404X
described above were cultured for 24 hrs. Cell images were obtained
with an imaging system described in the example 1-(5). The time-
CA 02419503 2003-02-14
course of fluorescence intensities of ECFP and EYFP and fluorescence
ratio (EYFP/ ECFP} in the cells stimulated with EGF are shown in Fig.
28. This figure is displayed by the IMD mode wherein the regions of
high fluorescence ratio are shown in red, those of low fluorescence
ratio are shown in blue, and the intensity reflects that of ECFP. By the
stimulation of EGF, the fluorescence ratio, which reflects the FRET
efficiency, gradually increases from the periphery to the center in the
cells expressing Rai-chu101X. Meanwhile, the activity increases from
the center to the periphery in the cells expressing Rai-chu404X. When
similar experiments were performed with semiconfluent cells, it
became clear that Ras could not be activated where cells were in
contact with neighboring cells and that Ras was activated only from
the free edges (Fig. 29}. Thus, the invented monitoring proteins
enables us to obtain the spatio-temporal information on the activity
of Ras-family G proteins.
Example 11 Visualization of the activation of Ras and Rap1 in PC12
cells expressing Rai-chu101X and Rai-chu404X.
PC12 cells txansfected with pRai-chu101X or pRai-chu404X
described above were cultured for 24 hrs. Cell images were obtained
with an imaging system described in the exampole 1 (5). The time-
course of fluorescence intentisities of ECFP and EYFP and
fluorescence ratio (EYFP/ECFP) in the cells stimulated with nerve
growth factor are shown in Fig. 30. This figure is displayed by IMD
mode wherein the regions of high fluorescence ratio are shown in red,
56
CA 02419503 2003-02-14
those of low fluorescence ration are shown in blue, and the intensity
reflects that of ECFP. During the induction of neuronal differentiation
of PC12 cells, Ras was activated from the periphery of the cell body in
the induction phase, whereas after differentiation the Ras activity,
which is known to be essential for the survival of the cells, were
maintained only at the extended neurites. Namely, it became evident
that the activation of Ras occurs at different intracellular regions
during the stages of differentiation. In contrast to Ras, Rap1 is
activated from the perinuclear region and suppressed at the neurites.
This observation indicates that the Ras-family G proteins are
regulated differently at the different intracellular localization.
Example 12 Construction of a monitoring protein for Racl, Raichu-
1011X
(1) Construction of a chimeric gene between Rac1 and Pakl.
By using the PCR-based procedure described in the examples 1
and 9 pRai-chu1011X was obtained with the cDNAs of Rac1
(Genbank/EMBL accession number M29870) and Pak1
(Genbank/ EMBL accession number NM002576) used as templates.
The structure (Fig. 31) and the nucleotide sequence of the coding
region (sequence number: 37) and predicted amino acid sequence
(sequence number: 38) are shown.
Detailed explanation is as follows.
nt 1- 684: Aequorea EYFP
57
CA 02419503 2003-02-14
nt 685 - 690: linker
nt 691- 939: Pak1
nt 940 - 969: linker
nt 970 -1497: Rac1
nt 1498 -1506:linker
nt 1507 - 2217:Aequorea ECFP
nt 2218 - 2229:linker
nt 2230 - 2289:carboxyl-terminal region of K-Ras (CAAX
box)
(2) Construction of mutants of the chimeric gene
By the PCR-mediated mutagenesis, thymine was substituted for
guanine at position 1004 of pRai-chu1011X (sequence number 37),
whereby Val was substituted for Gly in the predicted amino acid
sequence, generating pRai-chu1012X. Similarly, adenine was
substituted for cytosine at position 1019, whereby Asn was
substituted for Thr in the predicted amino acid sequence, generating
pRai-chu1013X. In the mutant protein Rai-chu1012X, the GTPase
activity is decreased, rendering this mutant constitutively active.
Meanwhile, in the mutant protein Rai-chu1013X, the binding of Rac1
to GTP is decreased, rendering this protein inactive.
(3) Expression of Rac1 monitoring proteins in mammalian cells and
their analysis by spectrometer.
Rai-chu1011X, Rai-chu1012X, and Rai-chu1013X were expressed
in the cells and analyzed by the method described in the example 1-
58
CA 02419503 2003-02-14
(4). The obtained profiles of spectrum are shown in Fig. 34. The FRET
efficiency of Rai-chu1013X is lower than those of the wild-type Rai-
chu1011X and the active form Rai-chu1012X.
Example 13 Construction of a monitoring protein for Cdc42, Raichu-
1054X
(1) Construction of a chimeric gene between Cdc42 and Pakl.
By using PCR-mediated method described in the examples 1
and 9 with cDNAs of Cdc42 (Genbank/ EMBL accession number
M57298) and Pak1 (Genbank/EMBL accession number NM002576) as
templates, pRai-chu1054X was obtained. The structure (Fig. 32 and
the nucleotide sequence of the coding region (sequence number: 39
and predicted amino acid sequence (sequence number: 40) are shown.
Detailed explanation is as follows.
nt 1 - 684: Aequorea EYFP
nt 685 - 690: Linker
nt 691- 939: Pak1
nt 940 - 969: Linker
nt 970 -1494: Cdc42
nt 1495 -1503: Linker
nt 1504 - 2214: Aequorea ECFP
nt 2215 - 2226: Linker
nt 2227 - 2286: Carboxyl-terminal region of K-Ras (CAAX box)
59
CA 02419503 2003-02-14~
(2) construction of mutants of the chimeric gene
By the PCR-mediated mutagenesis, nucleotide thymine was
substituted for guanine at position 1001 of pRai-chu1054X (sequence
number 39), by which Val was substituted for Gly in the predicted
amino acid sequence, generating pRai-chu1052X. In the mutant
protein Rai-chu1052X, the GTPase activity is decreased, rendering
this mutant constitutively active.
(3) Expression of Cdc42 monitoring protein Rai-chu1054X in
mammalian cells and their analysis by spectrometer.
Rai-chu1054X and Rai-chu1052X were expressed in the cells
and analyzed by the method described in the example 1-(4). The
obtained spectral profiles are shown in Fig. 35. The FRET efficiency of
the wild-type Rai-chu1054X is lower than that of the active form Rai-
chu1052X.
Example 14 Construction of a monitoring protein for RhoA, Raichu-
1214X
(1) Construction of a chimeric gene between RhoA and mDial.
By using PCR method described in the examples 1 and 9, pRai-
chu1214X was obtained with cDNAs of RhoA (Genbank/EMBL
accession number L25080) and mDia1 (Genbank/EMBL accession
number E17361) used as templates. The structure (Fig. 33 and the
nucleotide sequence of the coding region (sequence number: 41 and
predicted amino acid sequence (sequence number: 42) are shown.
CA 02419503 2003-02-14
Detailed explanation is as follows.
nt 1- 684: Aequorea EYFP
nt 685 - 696:Linker
nt 697 -1092:mDia1
nt 1093 -1110:Linker
nt 1111 -1677:RhoA
nt 1678 -1686:Linker
nt 1687 - Aequorea ECFP
2397:
nt 2398 - 2409: Linker
nt 2410 - 2469: Carboxyl-terminal region of K-Ras (CAAX box)
(2) construction of mutants of the chimeric gene
By the PCR-mediated mutagenesis, nucleotide thymine and
cytosine were substituted for adenine and guanine at positiosns1298
and 1299 of pRai-chu1214X (sequence number 41), by which Leu was
substituted for Gln in the predicted amino acid sequence, generating
pRai-chu1220X. In the mutant protein Rai-chu1220X, the GTPase
activity is decreased, rendering this mutant constitutively active.
(3) Expression of RhoA monitoring protein Rai-chu1054X in
mammalian cells and their analysis by spectrometer.
Rai-chu1214X and Rai-chu1220X were expressed in the cells
and analyzed by the method described in the example 1 (4). The
obtained profiles of spectrum are shown in Fig. 36. The FRET
61
CA 02419503 2003-02-14
efficiency of the wild-type Rai-chu1214X is lower than that of the
active form Rai-chu1220X.
Example 15 Visualization of Rac1 activation in the COS1 cells
expressing Rai-chu1011X.
COS1 cells were replated to glass-base dishes. pRai-
chu1011X described in example 12 was txansfected into COS1 cells.
After 24 hours, cells were imaged by the fluorescent microscope
system described in example 1-(5). The time-course of fluorescence
intensities of ECFP and EYFP and fluorescence ratio (EYFP/ECFP) in
the cells stimulated with nerve growth factor are shown in Fig. 37.
Within one minute after EGF stimulation, Rac1 is activated diffusely
in the cells and then the activation was localized to the membrane
ruffles, where cell membrane moves dynamically. Thus, the invented
monitoring proteins enables us to obtain the spatio-temporal
information on the activity of Rho-family G proteins. The pattern of
the Rac1 activation was different from those of Ras or Rapl, further
supporting the specificity of the monitoring proteins.
Sequences:
Sequence number 1: Nucleotide sequence of a primer based on the
XhoT restriction sequence and the nucleotide sequence of human H-
Ras.
62
CA 02419503 2003-02-14
Sequence number 2: Nucleotide sequence of a primer based on the
nucleotide sequences of human c-Raf1 and human H-Ras.
Sequence number 3: Nucleotide sequence of a primer based on the
KpnI restriction sequence and the nucleotide sequence of human c-
Rafl.
Sequence number 4: Nucleotide sequence of a primer based on the
NotI restriction sequence and the nucleotide sequence of human c-
Rafl.
Sequence number 5: Nucleotide sequence of a primer based on the 5'
sequence of the multiple cloning site of pBluescript-SKII (+).
Sequence number 6: Nucleotide sequence of a primer based on the 3'
sequence of the multiple cloning site of pBluescript-SKII (+).
Sequence number 7: Nucleotide sequence of a primer based on the
BamHI restriction sequence and the nucleotide sequence of EYFP.
Sequence number 8: Nucleotide sequence of a primer based on the
restriction sequences of BamHI, KpnI, and XhoI and the nucleotide
sequence of ECFP.
63
CA 02419503 2003-02-14
Sequence number 9: Nucleotide sequence of a primer based on the
NotI restriction sequence and the nucleotide sequence of ECFP.
Sequence number 10: Nucleotide sequence of a primer based on the
BgIII restriction sequence and the nucleotide sequence of ECFP.
Sequence number 11: Nucleotide sequence of a plasmid based on the
nucleotide sequences of human H-Ras, human c-Rafl, EYFP, and
ECFP.
Sequence number 12: Amino acid sequence predicted from the
nucleotide sequence of the plasmid shown in the sequence number 11.
Sequence number 13: Nucleotide sequence of a primer based on the
XhoI restriction sequence and the nucleotide sequence of human
RaplA.
Sequence number 14: Nucleotide sequence of a primer based on the
nucleotide sequences of human human RaIGDS and human RaplA.
Sequence number 15: Nucleotide sequence of a primer based on the
nucleotide sequence of human RaIGDS.
64
CA 02419503 2003-02-14
Sequence number 16: Nucleotide sequence of a primer based on the
NotI restriction sequence and the nucleotide sequence of human
RaIGDS.
Sequence number 17: Nucleotide sequence of a primer based on the
restriction sequences of BamHI, KpnI, and XhoI and the nucleotide
sequence of ECFP.
Sequence number 18: Nucleotide sequence of a plasmid based on the
nucleotide sequences of human RaplA, human RaIGDS, EYFP, and
ECFP.
Sequence number 19: Amino acid sequence predicted from the
nucleotide sequence of the plasmid shown in sequence number 18.
Sequence number 20: Nucleotide sequence of a primer based on the
XhoI restriction sequence and the nucleotide sequence of human R-
Ras.
Sequence number 21: Nucleotide sequence of a primer based on the
KpnI restriction sequence and the nucleotide sequence of human R-
Ras.
CA 02419503 2003-02-14
Sequence number 22: Nucleotide sequence of a plasmid based on the
nucleotide sequences of human R-Ras, human c-Rafl, EYFP, and
ECFP.
Sequence number 23: Amino acid sequence predicted from the
nucleotide sequence of the plasmid shown in sequence number 22.
Sequence number 24: Nucleotide sequence of a primer based on the
nucleotide sequence of human H-Ras.
Sequence number 25: Nucleotide sequence of a primer based on the
nucleotide sequence of human H-Ras.
Sequence number 26: Nucleotide sequence of a plasmid based on the
nucleotide sequences of human H-Ras, human c-Rafl, EYFP, and
ECFP.
Sequence number 27: Amino acid sequence predicted from the
nucleotide sequence of the plasmid shown in sequence number 29.
Sequence number 28: Nucleotide sequence of a primer based on the
H-Ras binding region of human c-Rafl.
Sequence number 29: Nucleotide sequence of a primer based on the
nucleotide sequence of ECFP.
66
CA 02419503 2003-02-14
Sequence number 30: Nucleotide sequence of a plasmid based on the
nucleotide sequences of human RaplA, human c-Rafl, EYFP, and
ECFP.
Sequence number 31: Amino acid sequence predicted from the
nucleotide sequence of the plasmid shown in sequence number 30.
Sequence number 32: Nucleotide sequence of a primer based on the
nucleotide sequence of ECFP.
Sequence number 33: Nucleotide sequence of a plasmid based on the
nucleotide sequences of human H-Ras, human c-Rafl, EYFP, ECFP,
and human K-Ras.
Sequence number 34: Amino acid sequence predicted from the
nucleotide sequence of the plasmid shown in sequence number 33.
Sequence number 35: Nucleotide sequence of a plasmid based on the
nucleotide sequences of human RaplA, human c-Rafl, EYFP, ECFP,
and human K-Ras.
Sequence number 36: Amino acid sequence predicted from the
nucleotide sequence of the plasmid shown in sequence number 33.
67
CA 02419503 2003-02-14
Sequence number 37: Nucleotide sequence of a plasmid based on the
nucleotide sequences of human Racl, human Pakl, EYFP, ECFP, and
human K-Ras.
Sequence number 38: Amino acid sequence predicted from the
nucleotide sequence of the plasmid shown in sequence number 37.
Sequence number 39: Nucleotide sequence of a plasmid based on the
nucleotide sequences of human Cdc42, human Pakl, EYFP, ECFP,
and human K-Ras.
Sequence number 40: Amino acid sequence predicted from the
nucleotide sequence of the plasmid shown in sequence number 39.
Sequence number 41: Nucleotide sequence of a plasmid based on the
nucleotide sequences of human RhoA, human mDial, EYFP, ECFP,
and human K-Ras.
Sequence number 42: Amino acid sequence predicted from the
nucleotide sequence of the plasmid shown in sequence number 41.
The value of this invention for the industry
The present invention provides monitoring proteins for the
activity of low-molecular-weight GTP-binding proteins, cells and
transgenic animals expressing the monitoring proteins useful for the
68
CA 02419503 2003-02-14
measurement of the activity of low-molecular-weight GTP-binding
proteins in non-destructive manners, methods for measurement of
the activity of low-molecular-weight GTP-binding proteins which
use the proteins, more in detail, methods that measure the ratio of
GTP-bound to GDP-bound forms of the low-molecular weight GTP-
binding proteins that are applicable with living cells, and screening
procedures for the regulatory substances of low-molecular-weight
GTP-binding proteins.
69