Note: Descriptions are shown in the official language in which they were submitted.
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
1
PROCESS FOR THE PREPARATION OF
NEUTROPHIL INHIBITORY FACTOR
FIEZD OF THE INVENTION
The Application is a continuation-in-part of United
States Serial No. 09/644,942,, filed August 23, 2000.
The disclosure of which is incorporated herein by
reference.
The present invention relates to a method for the
preparation of a Neutrophil Inhibitory Factor (NIF)
comprising the cultivation of mammalian cells in an
animal component-free growth medium. The present
invention may be employed in large-scale preparation of
NIF. In addition, the present invention provides a
general method for the preparation of recombinant
proteins comprising the cultivation in an animal
component-free medium of mammalian cells, in particular
CHO cells, expressing an exogenous recombinant protein.
BACKGROUND AND INTRODUCTION TO THE INVENTION
NIFs are proteins that are specific inhibitors of
the activity of neutrophil cells. Neutrophils are a
member of the group of cell types known as granulocytes,
a subclass of the leukocyte family of cells.
Neutrophils are an important component of the
defense system in a host against microbial attack. In
response to soluble inflammatory mediators released at
the site of injury by cells, neutrophils enter .into ~tne
area of the injured tissue from the bloodstream and when
activated, kill foreign cells by phagocytosis and/or the
release of cytotoxic compounds, such as oxidants,
proteases and cytokines. Although the activity of
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
2
neutrophils is important to fight infection, they also
are known to damage the host tissue. Neutrophils may
give rise to an abnormal inflammatory response whereby
significant tissue damage may be caused by the release
of toxic substances at the vascular wall or in uninjured
tissue. Alternatively, neutrophils which adhere to a
capillary wall or aggregate in venules can produce
ischemic tissue damage.
Abnormal inflammatory response is implicated in the
pathogenesis of a variety of clinical disorders
including adult respiratory distress syndrome (ARDS);
ischemia-reperfusion injury following myocardial
infarction, shock, stroke, and organ transplantation;
acute and chronic allograft rejection; vasculitis;
sepsis; rheumatoid arthritis; head trauma; and
inflammatory skin diseases. Harlan et al., Immunol.
Rev., 114:5 (1990).
One of the specific activities that NIFs have been
reported to inhibit is adhesion of neutrophils to
vascular endothelial cells.
Certain NIFs have been isolated from hookworms and
related species, in particular the canine hookworm
(Ancylostoma caninum), Moyle et al., J. Biol. Chem.,
269:10008-15 (1994), and have been made by recombinant
methods. When isolated from parasitic worms, the NIF is
a glycoprotein. Recombinant NIFs produced by certain
expression systems have been reported to exhibit post-
translational glycosylation and sialylation.
NIFs have been reported to inhibit other aspects of
neutrophil activity, including the release of hydrogen
peroxide, release of superoxide anion, release of
myeloperoxidase, release of elastase, homotypic
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
3
neutrophil aggregation, adhesion to plastic surfaces,
adhesion to vascular endothelial cells, chemotaxis,
transmigration across a monolayer of endothelial cells
and phagocytosis. In particular NIF has been shown to
be effective in reducing infarct size in a rat re-
perfusion model of stroke. Jiang et al., Ann.
Neurology, 38:935-942 (1995); Jiang et al., Brain Res.,
788:25-34 (1998).
Certain Neutrophil Inhibitory Factors are described
in greater detail, along with methods of isolating them
from natural sources and of cloning them by recombinant
methods, in U.S. Patent No. 5,919,900, issued July 6,
1999, U.S. Patent No. 5,747,296, issued May 5, 1998, and
U.S. Patent No. 5,789,178, issued August 4, 1998. These
patent documents are incorporated herein by reference in
their entirety.
Heretofore, the cultivation of cells expressing NIF
has been carried out in growth media containing bovine
serum albumin. See, e.g., U.S. Patent No. 5,919,900.
As a general matter, the cultivation of cells
expressing recombinant proteins is most often conducted
in media which contain either animal-derived serum or
animal-extracted proteins. However, in view of the
increasing concerns in general over the use of animal
components and in particular over the contamination of
bovine products by pathogens, including contamination by
the organism giving rise to outbreaks of bovine
spongiform encephalopathy (BSE), there is a need for
growth medium which is free of animal components, e.g.,
serums and proteins. In U.S. Patent No. 5,122,469,
serum-free media are recited. However, serum-free media
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
4
have often not been optimized for cell growth, protein
production, and post-translational modification.
The present invention provides a animal serum-free
and animal protein-free medium as well as a method of
preparation of NIF using said serum- and protein-free
medium to provide NIF in high yields.
SUMMARY OF THE INVENTION
The present invention is directed to a process for
the preparation of Neutrophil Inhibitory Factor (NIF)
comprising the step of incubating a cell line expressing
NIF in an animal component-free growth medium.
Preferably, the NIF produced via the present invention
is a 257-amino acid protein, mature NIF-1FL (also termed
"NIF1") (SEQ. ID. N0. 3). The NIF so produced is
glycosylated and has a relative molecular weight of
about 38.3 to about 64.1 kDa. The glycan structures are
typically branched and may be capped by sialic acid
residues. The degree of glycosylation may vary, but
preferably, the NIF produced has a distribution of mono,
di, tri, and tetra-antennary glycan structures. More
preferably, the NIF produced is about 5 to about 25o
mono-sialylated, about 10 to about 30o di-sialylated,
about 15 to about 35o tri-sialylated, about 15 to about
45o tetra-sialylated and about 1 to about 20o non-
sialylated.
According to a preferred aspect of the invention,
the cell line expressing NIF is a Chinese Hamster Ovary
("CHO")cell line comprising the NIF gene, more
preferably a cell line which is not anchorage-dependent.
According to a most preferred aspect of the present
invention, the cell line is the CHO-K1 cell line (ATCC
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
CCL-61) modified by transfection with the glutamine
synthetase/methionine sulfoximine co-amplification
vector pEEl4 expressing the NIF1 gene. WO 87/04462 and
89110404 describe recombinant DNA sequences, vectors and
5 use of the glutamine synthetase system in expression
systems.
The most preferred cell line for use according to
the processes and methods of the present invention is
the cell line PFG01 (ATCC PTA-2503).
The preparation and cultivation of the most
preferred cell line expressing NIF is described in the
Examples 1 and 2 below.
A preferred embodiment of the invention is wherein
the animal component-free production growth medium
comprises:
(i) a CHO-III-PFM/glucose solution;
(ii) sodium hypoxanthine;
(iii) thymidine; and
(iv) yeast extract.
"CHO-III-PFM" refers to a protein-free medium
optimized for suspension culture of CHO cells which is
made without hypoxanthine and thymidine and which is
available from Life Technologies (Grand Island, NY).
"CHO-III-PFM glucose solution" refers to a CHO-III-PFM
medium made with added glucose, a preparation also
available, from Life Technologies. A preferred
CHO-III-PFM/glucose solution is custom formula No. 98-
0289 (Life Technologies, Rockville, MD, Grand Island,
'NY, a division of Invitrogen Corp., Carlsbad, CA) which
is a CHO-III-PFM/glucose solution having additional
glucose (3.45 g/L D-glucose) and which does not contain
hypoxanthine, thymidine or L-glutamine.
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
6
The CHO-III-PFM/glucose solution is itself animal
component-free (free of animal serum and animal "r
protein). It should be noted, however, that other
commercially available CHO cell cultivation media which
are animal component-free and which incorporate the
above-noted attributes and components of the CHO-III-PFM
media may also be used within the scope of the
invention.
A preferred yeast extract is that purchased under
the trade name Bacto (Difco/Becton-Dickinson). Other
commercially available yeast extracts also may be used..
A solution of phenol red, preferably a solution of
about 0.5o w/v thereof, may be added to the media for
use as a visual pH indicator. Such a phenol red
solution is more preferably used in the amount of about
0 to about 3.0 ml per liter of media.
A more preferred embodiment of the invention is
wherein the animal component-free production growth
medium comprises:
(i) CHO-III-PFM/glucose solution;
(ii) about 50 to about 100 ~,mol sodium
hypoxanthine per liter (i);
(iii) about 8 to about 32 ~mol thymidine
per liter (i); and
(iv) about 0.5 to about 5.0 grams per
liter (i) yeast extract.
According to a preferred aspect, sodium hypoxanthine and
thymidine are added as a lOmM sodium hypoxanthine/l.6mM
thymidine solution. Preferably about 5 to about 20 ml
30 of the sodium hypoxanthine/thymidine solution per liter
(i) are added.
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
7
A most preferred embodiment of the invention is
wherein the animal component-free production growth
medium comprises:
(i) CHO-III-PFM/glucose solution;
(ii) about 10.0 ml per liter (i) of a
lOmM sodium hypoxanthine/l.6mM thymidine solution; and
(iii) about 1.5 grams per liter (i) yeast
extract.
Optionally, about 0. 5 ml per liter (i) of a 0.5o w/v
solution of phenol ed may be added to the medium.
r
The present inv ention is also directed to a process
for the preparation of Neutrophil Inhibitory Factor
(NIF) comprising the steps of:
(i) providing an inoculum prepared by
incubating a cell li ne expressing NIF in an animal
component-free inoculum
growth medium; and
(ii) transferring said inoculum to a
vessel containing an
animal component-free
production
growth medium.
According to a preferred embodiment of this aspect
of the invention is the inoculum growth medium
comprises:
(i) a CHO-III-PFM/glucose solution;
(ii) sodium hypoxanthine;
(iii) thymidine;
(iv) an amino acid solution comprising
acids selected from the group consisting of L-aspartic
acid, L-glutamic acid,
L-asparagine, L-proline,
L-serine, and L-methionine;
(v) optionally, L-methionine
sulphoximine ("MSX" ): and
(vi) L-cysteine.
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
8
Optionally, a solution containing phenol red,
preferably a solution of about 0.5o w/v thereof, may be
added to this inoculum medium for use as a visual pH
indicator: preferably the solution is added in the
amount of about 0 to about 3.0 ml per liter of the
medium.
A more preferred embodiment of this aspect of the
invention is wherein the inoculum growth medium
comprises:
(i) CHO-III-PFM/glucose solution;
(ii) about 50 to about 100 ~,mol sodium
hypoxanthine per liter (i);
(iii) about 8 to about 32 ~mol thymidine
per liter (i);
(iv) addition of the following amino
acids in the noted amounts per liter (i): L-aspartic
acid (about 15 to about 90 mg); L-glutamic acid (about
12 to about 75 mg), L-asparagine (about 50 to about 300
mg), L-proline (about 6 to about 38 mg), L-serine (about
15 to about 90 mg) and L-methionine (about 7 to about 45
mg ) ;
(v) about 0 to about 75 ~mol
L-methionine sulpho~imine (MSX) per liter (i); and
(vi) about 10 to about 40 mg cysteine per
liter (i) .
The amino acids of (iv) may be conveniently added as
about 5 to about 30 ml per liter (i) of an amino acid
solution comprising L-aspartic acid (about 3.0 g/1), L-
glutamic acid (about 2.50 g/1), L-asparagine (about
10.00 g/1), L-proline (about 1.25 g/1), L-serine (about
3.0 g/1), and L-methionine (about 1.50 g/1). MSX may be
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
9
optionally added as about 0.5 to about 3 ml per liter
(i) of a 25mM L-methionine sulphoximine (MSX) solution
to give about 12.5 to about 75 mole MSX per liter.
Sodium hypoxanthine and thymidine may be conveniently
added as about 5 to about 20 ml of a lOmM sodium
hypoxanthine/l.6mM thymidine solution.
A most preferred embodiment of the invention is
wherein the inoculum growth medium comprises:
(i) CHO-III-PFM/glucose solution;
(ii) about 10.0 ml per liter (i) of a
lOmM sodium hypoxanthine/l.6mM thymidine solution;
(iii) about 20.0 ml per liter (i) of an
amino acid solution comprising L-aspartic acid (about
3.0 g/1), L-glutamic acid (about 2.5 g/1), L-asparagine
(about 10.0 g/1), L-proline (about 1.25 g/1), L-serine
(about 3.0 g/1), and L-methionine (about 1.5 g/1);
(iv) optionally about 1.0 ml per liter
(i) of an 25mM L-methionine sulphoximine (MSX) solution;
and
(v) about 25.0 mg per liter (i) of L-
cysteine. Optionally, about 0.5 ml per liter (i) of a
solution of about 0.5o w/v phenol red may be added to
the inoculum medium.
The present invention further relates to an animal
component-free growth medium, as described above. In
addition, the present invention relates to an animal
component-free inoculum growth medium.
Further, the present invention also relates to a
method for the preparation of a recombinant protein
comprising the cultivation of mammalian cells expressing
an exogenous recombinant protein in an animal component-
free growth medium of the present invention. In a
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
preferred embodiment, the mammalian cells are Chinese
Hamster Ovary cells transfected with a glutamine
synthetase plasmid vector comprising a nucleic molecule
having the DNA coding region for the recombinant
5 protein. Preferred vectors are a glutamine
synthetase/methionine sulfoximine co-amplification
vector, such as pEEl4 or pEE14.1 (Lonza Biologics,
Slough, UK).
Definitions
10 "Neutrophil Inhibitory Factor" or "NIF" refers to a
protein which may be isolated from natural sources or
made by recombinant methods. Neutrophil Inhibitory
Factor is a protein which is neither an antibody, a
member of the integrin or selectin families, nor a
member of the immunoglobulin superfamily of adhesive
proteins and which, when isolated from a parasitic worm,
is glycosylated. Recombinant NTF may or may not be
glycosylated or may be glycosylated to a variable
degrees this may be affected by the expression system
and/or culture conditions used in producing recombinant
NIF.
NIF1 or mature NIF-1FL refers to a protein which is
expressed in a proform, NIF-1FL (SEQ. ID. NO. 2), and
then, after synthesis, is cleaved (while within the
cell) to give mature NIF-1FL or NIF1 (SEQ. ID. N0. 3).
"NIFlcr" refers to the coding sequence for NIF1.
The term "NIF gene" refers to a nucleic acid
molecule which encodes a Neutrophil Inhibitory Factor.
Certain nucleic acid molecules which encode a NIF are
described in United States Patent No. 5,919,900.
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
11
The term "cell line expressing NIF" refers to a
cell line which has been transformed with a nucleic acid
molecule encoding a NIF so as to express a Neutrophil
Inhibitory Factor.
The cell line PFG01 is a CHO-K1 (ATCC-CCL-61) cell
line which has been transfected with the glutamine
synthetase/methionine sulfoximine co-amplication vector
pEEl4 expressing the NIF1 gene. Pfizer Inc. a Delaware
corporation, doing business at 235 East 42nd Street, New
York, New York made a deposit with the American Type
Culture Collection of cell line PFG01 (ATCC PTA-2503) on
September 27, 2000.
The term "CHO-III-PFM/glucose solution" refers to a
growth medium manufactured by Life Technologies (Grand
Island, NY; PFM = protein-free medium) with added
glucose developed specifically for the cultivation of
CHO cells.
The term "yeast extract" refers to a complex
supplement containing peptides which is extracted from
yeast cells and is free of animal-derived compounds.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 depicts the coding sequence for NIF1 (SEQ.
ID. NO. 1), the corresponding amino acid translation,
(SEQ. ID. NO. 2) and the amino acid sequence of mature
NIF1 (SEQ. ID. N0. 3). Nucleotides are numbered from
the 5'-end, and amino acids are numbered from the start
of the mature polypeptide (SEQ. ID. N0. 3.) (The
N-terminal Asn is indicated.) Numbers along the left-
hand margin denote the nucleotide number of the nucleic
acid sequence or amino acid number (bold) of the mature
NIF1 sequence of the first entry on each line. Peptides
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
12
identified by amino acid sequencing are underlined. The
peptides T-20 (SEQ. ID. N0. 4), T-22 (SEQ. ID. N0. 5),
D-96 (SEQ. ID. N0. 6), and D-102 (SEQ. ID. NO. 7) were
used to design forward and reverse primers for initial
and subsequent cloning purposes. Nucleotide sequences
in lower case represent the nucleotides added by the PCR
primers during rescue of the coding region for cloning
into the BSII shuttle vector (SEQ. ID. N0. 8). This
figure represents the sequence determined from both
strands of DNA using a {BSII/}{pEEl4/}{pSG5}NIFlcr
construct.
Figure 2 depicts the sequence for the full length
cDNA (SEQ. ID. N0. 9), as obtained from Ancylostoma mRNA
preparations, after cloning of the cDNA into 7~gt10/EcoRI
vectors, and subcloning into the BSII rescue vector.
The nucleotide sequence of NIF1 was determined using the
Sanger dideoxynucleotide sequencing method. Numbers
along the left margin indicate the number of nucleotides
fr~m the 5'-end of the sequence. The nucleotides
highlighted in bold type (313 through 1137) (SEQ. ID.
NO. 1) represent the coding region of NIF1.
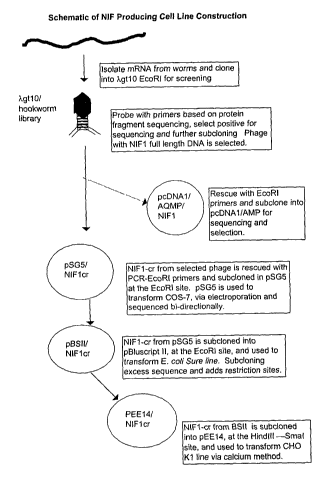
Figure 3 is a schematic of NIF producing cell line
construction and depicts a schematic representation of
the pathway from NIF1 cDNA to the pEEl4 vector which was
used to transfect CHO-K1 cells.
Figure 4 depicts the pEEl4 expression vector
construct employed in the construction of an NIF-
expressing cell line. As indicated by its designation,
the pEEl4/NIFlcx expression plasmid was derived from the
widely used 9.4kb pEEl4 expression vector (Lonza
Biologics, Slough, UK). The pEEl4 vector contains:
(1) a human CMV major immediate early promoter (hCMV-
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
13
MIE), (2) a multiple cloning site (MCS), (3) a SV40
early poly A site (pA), (4) a Col E1 origin of
replication (Col E1), (5) an ampicillin resistance gene
(Amp), and (6) the SV40 late promoter (SV40L) which
drives the glutamine synthetase minigene (GS-minigene).
The restriction endonuclease sites present in the
multiple cloning site are noted in this diagram. The 5'
HindIII insert site is slightly 5' to the MCS.
Figure 5 depicts additional non-coding sequences
(lower case) incorporated into the insert at both ends
of the coding sequence (upper case flanking "NIFIcr")
during the cloning process (SEQ. ID. NOS. 10 and 11).
The 5'-end of the insert sequences is shown to start at
the HindIII site in the pEEl4 expression vector (site
not shown on Figure 4), which are joined to the
complementary sequences from the 5'-HindIII site from
pBluescriptII shuttle vector ("BSII") polylinker. The
5' HindIII site is followed by an EcoRI site, provided
by the 5'-PCR NIFlcr rescue primer, used to clone the
NIFlcr sequences into BSII. The NIF1 coding region
sequence of NIF1, beginning at this EcoRI site extends
for approximately 850 nucleotides.
DETAILED DESCRIPTION OF THE INVENTION
The process of the present invention may be carried
out as described below. One of the advantages of the
present invention is that it does not involve the use of
animal components in any of the media, including the
inoculum growth medium, the production growth medium and
the nutrient feeds. This advantage is a significant in
view of increasing concerns over the use of animal-
derived substances in the production of medicinal drugs
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
14
(e. g., fear of transmission of BSE (Bovine Spongiform
Encephalopathy)). In addition, in contrast to
previously-used processes using media containing animal-
derived components, the process of the present invention
has a processing period which is several days shorter
and typically achieves appropriately glycosylated NIF
titers which are 3 to 4 times greater.
Preferred Cell Lines and NIFs
The present invention is preferably practiced with
mammalian cell lines, more preferably a recombinant
Chinese Hamster Ovary cell line derived from CHO-K1
(ATCC CCL-61), which has been transformed with a NIF-
expressing plasmid vector, preferably the pEEl4 vector
(Lonza Biologics; a glutamine synthetase/methionine
sulfoximine co-amplification vector containing HindIII,
XbaI, SmaI, SbaI, EcoRI, and BclI cloning site, wherein
the vector expresses glutamine synthetase and the cloned
gene) comprising NIF1 DNA (Example 1).
Construction of the NIF-producing cell line follows
procedures for the establishment of cell cultures
producing recombinant proteins which are known in the
art and are disclosed in U.S. Patent Nos. 5,919,900
5,747,296; 5,789,178; 5,591,639; 5,658,759; 5,849,522;
5,122,464; 5,770,359; and 5,827,739 International
Patent publication Nos. WO 87/04462; WO 89101036 WO
86/05807 and WO 89/10404; Bebbington, et al.,
Bio/Technology, 10:169-175 (1992), which are all hereby
incorporated by reference in their entirety.
Preferably, the cell line should be selected and
adapted prior to use, such that it easily forms a
suspension culture, hence is not anchorage-dependent and
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
is weaned over several generations from animal serum and
animal protein-containing media. In general, a
procedure for effectuating such an adaptation may be
performed by culturing the cell line analogously to that
5 set forth in Example 2 below.
The cell line designated PFG01 (ATCC PTA-2503) is
preferred for the process of the invention. The PFG01
cell line was derived from the CHO-K1 cell line (ATCC
CCL-61), as set forth below in Examples 1 and 2. The
10 PFG01 cell line was created via the transfection of the
CHO-K1 cell line (ATCC CCL-61) with the pEEl4 plasmid
vector containing the NIF1 gene. The PFG01 cell line
development was completed by generating a suspension
culture from the anchorage-dependent line and weaning
15 the recombinant cell from bovine serum.
Any of the NIFs produced via cells transformed by
the above-referenced methods may be produced according
to the process of the present invention. Preferably,
the NIF produced by the process of the present invention
is a 257-amino acid protein, mature NIF-1FL (NIF1) (SEQ.
ID. NO. 3) which is depicted in Figure 1. NIF1 is
produced by the transformed cells as a glycosylated and
sialylated protein with a relative molecular weight of
about 38.3 to about 64.1 kDa. According to a preferred
aspect, NIF1 is expressed as a 4lkD glycoprotein,
wherein about 30o to about 500 of its molecular weight
is made up of sugar moieties (glycans) oligosaccharides,
which may be branched and capped with sialic acid
residues. This particular NIF is described in detail in
Moyle et al., supra: see also, R. Webster et al.,
Xenobiotica, 29:1141-1155 (1999) and references cited
therein.
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
16
Preparation of NIF
The process for preparing NIF according to the
present invention involves the preparation of an
inoculum via the use of an animal component-free
inoculum growth medium, suspending the inoculum in a
vessel containing a production growth medium,
maintaining the culture of viable cells and harvesting
the NIF product. In one embodiment of the invention,
the generation of the inoculum culture is conducted by
growing a culture of PFG01 cells, which is then used to
"inoculate" the production reactor. This inoculum
culture is generated in shake flasks or in vessels,
ordinarily of a size smaller than the actual production
vessel.
The starting seed cells are initially suspended in
a pre-warmed inoculum growth medium. If the seed cells
are frozen, the seed cells expressing NIF, preferably
those of the PFG01 cell line (which expires NIF1), are
thawed in a bath, at a temperature of between about 30°C
and about 38°C, until the ice pellet has almost
completely melted. The thawed vial is ordinarily then
transferred to a bio-safe containment unit or cabinet
and the exterior of the vial is decontaminated by
standard means, e.g., wiping with alcohol pads, etc.
The cells are then suspended in a pre-warmed
inoculum growth medium comprising:
(i) a CH0-III-PFM/glucose solution;
(ii) sodium hypoxanthine, preferably from
about 50 to 100 ~,mol per liter (i);
(iii) thymidine, preferably from about 8
to about 32 ~,mol per liter (i):
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
17
(iv) an amino acid solution comprising
amino acids selected from the group consisting of
L-aspartic acid, L-glutamic acid, L-asparagine,
L-proline, L-serine and L-methionine;
(v) optionally L-methionine
sulphoximine; and
(vi) L-cysteine.
According to a preferred aspect, the inoculum
growth medium comprises:
(i) a CHO-III-PFM/glucose solution,
preferably Life Technologies, Custom Formula 98-0289;
with 3.45 g/1 D-glucose added without hypoxanthine,
thymidine, L-glutamine;
(ii) a sodium hypoxanthine/thymidine
solution, preferably HT supplement (100X) (Life
Technologies, Catalog No. 11067-030);
(iii) an amino acid solution, preferably
composed of acids selected from the group consisting of
L-aspartic acid, L-glutamic acid, L-asparagine, L-
proline, L-serine, and L-methionine;
(iv) optionally an L-methionine
sulphoximine (MSX) solution; and
(v) an L-cysteine solution.
Optionally, a solution containing phenol red, preferably
a solution of about 0.5% w/v thereof, may be added to
the media for use as a visual pH indicator. More
preferably it is added in the amount of about 0 to about
3.0 ml of a 0.5o w/v solution per liter medium, most
preferably about 0.5 ml per liter medium is added.
The amino acid solution, noted above, may be
conveniently prepared by dissolving the amino acids in
deionized water, adjusting the pH to approximately 8.0
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
18
with an aqueous base, preferably sodium hydroxide in
water, followed by sterile filtering.
The MSS solution may be prepared by dissolving the
MSX in deionized water and filtering the solution using
a 0.2 micron filter. Aliquots of the.MSX solution may
be placed into sterile tubes and may be kept for up to
three months or longer at temperatures, preferably below
5°C.
More preferably the inoculum growth medium
comprises:
(i) CHO-III-PFM/glucose solution (Life
Technologies, Custom Formula 98-0289; with 3.45 g/1 D-
glucose; without hypoxanthine, thymidine, L-glutamine);
(ii) about 5 to about 20 ml per liter (i)
of a IOmM sodium hypoxanthine/l.6mM thymidine solution;
(iii) amino acids in the noted amounts per
liter (i): L-aspartic acid (about 15 to 90 mg),
L-glutamic acid (about 12 to about 75 mg), L-asparagine
(about 50 to about 300 mg), L-proline (about 6 to about
38 mg), L-serine (about 15 to about 90 mg) and
L-methionine (about 7 to about 45 mg); more preferably
the amino acids are added by adding about 5 to about 30
ml per liter (i) of an amino acid solution comprising L-
aspartic acid (3.0 g/1; 22.5mM), L-glutamic acid (2.50
g/1; l7.OmM), L-asparagine (10.00 g/1; 75.7mM), L-
proline (1.25 g/1; 10.9mM), L-serine (3.0 g/1; 28.5mM),
and L-methionine (1.50 g/1; 10.1mM);
(iv) optionally about 12.5 to about 25
~.mol per liter (i) L-methionine sulphoximine, if
included, preferably as about 0.5 to about 3.0 ml per
liter (i) of an 25mM L-methionine sulphoximine (MSX)
solution; and
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
19
(v) about 10 to about 40 mg per liter
(i) of L-cysteine.
Most preferably the inoculum growth medium
comprises:
(i) CHO-III-PFM/glucose solution (Life
Technologies, Custom Formula 98-0289; with 3.45 g/1 D-
glucose; without hypoxanthine, thymidine, L-glutamine)~
(ii) about 10.0 ml per liter (i) of a
lOmM sodium hypoxanthine/l.6mM thymidine solution;
(iii) about 20.0 ml per liter (i) of an
amino acid solution comprising L-aspartic acid (about
3.0 g/1), L-glutamic acid (about 2.5 g/1), L-asparagine
(about 10.0 g/1), L-proline (about 1.25 g/1), L-serine
(about 3.0 g/1), and L-methionine (about 1.5 g/1)~
(iv) optionally about 1.0 ml per liter
(i) of an 25mM L-methionine sulphoximine (MSX) solution;
and
(v) about 25.0 mg per liter (i) of
L-cysteine.
The resultant inoculum growth medium is then
transferred into a shake flask, or other vessel, for use
in creating the inoculum, and the seed cells are
suspended in it.
At initiation, the inoculum culture may be sampled
and counted using, e.g., the Trypan Blue Dye Exclusion
method, to determine cell concentration and viability as
set forth in Cell and Tissue Culture: Laborator
Procedures in Biotechnology, A. Doyle and J.B.
Griffiths, eds. (John Wiley & Sons, Ltd., 1998). If the
cell concentration is greater than approximately 7.0 x
105 viable cells per ml ("vc/ml"), more pre-warmed growth
medium may be added to achieve a final concentration in
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
the range of about 2.0 x 105 vc/ml to about 6.0 x 105
vc/ml, but a concentration of about 5.0 x 105 vc/ml is
preferred.
The shake flask or vessel may then be incubated
5 with stirring at a temperature in the range of about 30
to about 38°C, preferably about 36.5 ~ 1°C; at a C02
concentration of about 2 to about 100, preferably, 5 ~
10; at a relative humidity of about 40 to about 900,
preferably 70 ~ 50; and a stirring rate of about 50 to
10 about 200 rpm, preferably 150 ~ 20 rpm (throw = 3/8 inch
in diameter). The flask may be sampled daily for cell
concentration and viability. More pre-warmed growth
medium may be added daily to maintain a concentration of
about 2.0 x 105 vc/ml to about 6.0 x 105 vc/ml,
15 preferably about 5.0 x 105 vc/m1. Tf the volume in the
vessel is exceeded by further additions of medium or the
cell density reaches about 1.0 x 106 vc/ml, the culture
may be split into two or more cultures which can be
diluted to about 5.0 x 105 vc/ml in new vessels.
20 Subsequently, each time the cell density reaches
about 1.0 x 106 vc/ml, the culture should be split up to
about 2.0 x 105 vc/ml in further vessels. This step
should be repeated in order to expand the seed train
until a sufficient volume is achieved to obtain a
seeding density of approximately 1.5 x 105 vc/m1 to 4.0 x
105 vc/ml, preferably about 2.0 x 105 vc/ml for a
bioreactor vessel. The inoculum ratio (volume of
inoculum culture/reactor liquid volume after
inoculation) is about 10 to about 200. The cell density
in the inoculum culture should be between about 1.0 x 106
vc/ml and about 2.5 x 106 vc/ml, preferably between about
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
21
1.0 x 106 vc/ml and about 1.5 x 106 vc/ml. The age of
the inoculum culture is approximately 3 to 4 days prior
to use in the actual production phase.
The medium used in the actual production
(production growth medium) stage for NIF differs from
that used for inoculum generation. The production
reactor is preferably operated under fed-batch
conditions, i.e., whereby nutrient solutions are
continuously fed into the reactor during the production
period.
The medium for the NIF production stage (production
growth medium) comprises:
(i) a CHO-III-PFM/glucose solution
(ii) a sodium hypoxanthine;
(iii) thymidine~ and
(iv) yeast extract.
Sodium hypoxanthine and thymidine may be conveniently
added as a sodium hypoxanthine/thymidine solution,
preferably HT supplement (100X) (Life Technologies,
Catalog No. 11067-030).
Preferably the production growth medium comprises:
(i) a CHO-III-PFM/glucose solution
(ii) sodium hypoxanthine, preferably from
about 50 to 100 ~.mol per liter (i):
(iii) thymidine, preferably from about 8
to about 32 ~mol per liter (i): and
(iv) about 0.5 to about 5 grams per liter
(i) yeast extract. The CHO-III-PFM/glucose solution is
preferably Life Technologies, Custom Formula 98-0289;
with 3.45 g/1 D-glucose; without hypoxanthine,
thymidine, L-glutamine.
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
22
Optionally, phenol red, preferably a solution of
about 0.5o w/v thereof, may be added for purposes of
facilitating pH measurement; more preferably in the
amount of about 0 to about 3.0 ml of that solution per
liter of medium, most preferably, in an amount of about
0.5 ml of that solution per liter of medium.
More preferably the production growth medium
comprises:
(i) CHO-III-PFM/glucose solution made by
Life Technologies, Custom Formula 98-0289; with 3.45 g/1
D-glucose; without hypoxanthine, thymidine, L-glutamine~
(ii) about 5 to about 20 ml per liter (i)
of a lOmM sodium hyp,oxanthine/l.6mM thymidine solution;
and
(iii) about 0.5 to about 5.0 grams per
liter (i) yeast extract.
Most preferably the production growth medium
comprises:
(i) CHO-III-PFM/glucose solution made by
Life Technologies, Custom Formula 98-0289; with 3.45 g/1
D-glucose; without hypoxanthine, thymidine, L-glutamine~
(ii) about 10.0 ml per liter (i) of a
lOmM sodium hypoxanthine/l.6mM thymidine solution; and
(iii) about 1.5 grams per liter (i) yeast
extract.
Preferably, two nutrient feeds are used over the
course of the production stage to supply the culture
with material needed for an advantageous growth rate.
One of the nutrient feeds is a glucose feed (Nutrient
Feed 1) at a concentration of from about 100 to about
500 g/1. This glucose feed is used to maintain the
glucose concentration in the reactor at approximately
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
23
0.1 to about 5.0 g/1, preferably about 2.0 g/1. This
feed is usually added at a rate of about 0.0 to about
6.0 grams of glucose per liter medium per day using a
suitable pump or other means for adding the glucose
spread out over time.
The second nutrient feed (Nutrient Feed 2)
comprises (i) CHO-III-PFM (5-fold concentration or "5X")
solution (made by Life Technologies, Custom Formula 99-
0180; with only 1X (one-fold for solubility reasons)
L-cystine, 3X (three-fold for solubility reasons)
L-tyrosine; without glucose, hypoxanthine, thymidine,
L-glutamine, sodium bicarbonate, or sodium chloride);
(ii) 25 to 100 ml per liter (i) of a lOmM sodium
hypoxanthine/l.6mM thymidine solution; and (iii) 5 to 20
grams per liter (i) yeast extract. More preferably, the
nutrient feed comprises (i) CHO-III-PFM (5X) solution
made by Life Technologies, Custom Formula 99-0180 (5X)
with 1X L-cystine, 3X L-tyrosine; without glucose,
hypoxanthine, Thymidine, L-glutamine, sodium
bicarbonate, sodium chloride; (ii) 50 ml per liter (i)
of a lOmM sodium hypoxanthine/l.6mM thymidine solution;
and (iii) 7.5 grams per liter (i) yeast extract. This
second feed is prepared by adding the lOmM
hypoxanthinell.6mM thymidine solution, preferably HT
supplement 100X (Life Technology) and the yeast extract
to the CH0-III-PFM (5X) solution, dissolving and mixing
together the components, adjusting the pH to about 6.8
to about 7.6 using sodium hydroxide, and then sterile
filtering the final solution. This second feed solution
is fed to the reactor continuously at a rate of
approximately 5 to about 50 ml per liter of culture at
inoculation per day starting at about 48 hours. This
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
24
addition is essential for achieving high productivity of
NIF with acceptable product quality.
The process of the present invention has been
performed in a 2-liter Wheaton bioreactor (B. Braun
Biotech Inc., Allentown, PA), controlled via a Foxboro
IA (Intelligence Application) computer system (The
Foxboro Company, Foxboro, MA), however any sterilizable
vessel may be used as the bioreactor so long as it has
an adequate mixing capability, sufficient feed inlets,
two for the nutrient feeds and one for pH control, and
one sampling port, is outfitted with gas inlet and
purging capabilities may be used. The vessel should
permit sufficient online process control. Preferably
the vessel is light-impermeable or of such a nature that
it may be covered to avoid direct exposure to ambient
light. After sterilization of the vessel, a sterile
conditioning solution may be employed to rinse out the
vessel, preferably either glutamine-free DMEM (Life
Technologies/ GibcoBRL: Catalog No. 11960-044) or
Dulbecco's Phosphate Buffered Saline (Life
Technologies/GibcoBRL, Catalog I~o. 14190-136). After an
adequate time period, depending on the size of the
vessel, the rinse medium is replaced with fresh, sterile
production medium. The temperature of the medium is
allowed to stabilize at a temperature in the range of
about 30 to about 38°C, preferably about 36.5 ~ 1°C, and
if necessary the pH should be adjusted to pH about 6.8
to about 7.6, preferably a pH of about 7.4, prior to
inoculation. The volume of the inoculum culture added
preferably creates an initial target inoculum density in
the reactor of about 1.0 x 105 viable cells/ml to about
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
5.0 x 105 viable cells/ml, preferably about 2.0 x 105
viable cells/ml.
The'contents of the vessel should be stirred at a
rate in the range of about 50 to about 200 rpm,
5 depending on the size and geometry of the vessel and the
impeller used, sufficient for thorough mixing of the
vessel contents. .Otherwise, the contents of the vessel
should be agitated in a manner which would be
commensurate to achieve the same degree of mixing. The
10 pH of the production culture should be maintained in the
range of about 6.8 to about 7.6, preferably about 7.40 ~
0.05, via appropriate control agents which do not
interfere with the viability and vitality of the cell
culture. In the case of the present invent-ion, C02 gas
15 and a solution of about 7.5o (w/v) NaHC03 is preferred as
the pH control agent. However, other common alkaline
solutions such as mixtures of NaHC03 and Na2C03, or
dilute NaOH may also be used successfully.
The dissolved oxygen concentration should be
20 maintained in the range of about 10 to about 1000 of air
saturation, preferably about 600 ~ 50 of air saturation
via appropriate control agent. The temperature of the
production culture should be maintained in the range of
about 30°C to about 38°C, preferably about 36.5°C +
1°C.
25 The glucose concentration of the production medium
preferably is maintained in the range of about 0.1 to
about 5.0 g/1; preferably about 2.0 g/1 ~ 0.5 g/1, by
means of a glucose feed solution (Nutrient Feed 1) which
is added in small amounts at intervals to maintain the
desired level.
Carbon dioxide gas and/or oxygen and/or air and/or
nitrogen gas, may be sparged into the culture on demand
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
26
to control the pH and dissolved oxygen. Nitrogen gas or
air may be directed to the headspace to assist dissolved
oxygen control and /or reduce foam generation.
The Nutrient Feed 2 is fed continuously at a rate
of approximately 5 to about 50 ml per liter of culture
at inoculation per day, preferably at a rate of about 25
ml per liter of culture at inoculation per day. This
feed should be started simultaneously with the glucose
feed, usually at about the 48 hour point.
The production culture should be sampled
immediately after inoculation. The following parameters
are usually measured immediately: the initial cell
density and viability; the off-line pH; the initial
glucose concentration; the initial lactate
concentration; the initial ammonia concentration; and
the initial osmolality. The on-line pH should be
adjusted if necessary. The bioreactor vessel should
preferably either be light-impermeable, or covered by an
opaque light-blocking covering to protect the production
medium from light. The production culture is usually
sampled daily for the following parameters: cell
density: culture viability; off-line pH; glucose
concentration; lactate concentration; ammonia
concentration; osmolality; and NIF concentration,
purification or characterization.
The glucose concentration should be maintained
between about 0.1 and about 5.0 g/liter, preferably
between about 1.5 and about 2.5 g/liter using Nutrient
Feed 1. Typically, the feed begins after about 48 hours
with an initial feed rate of approximately 2.0 g/(liter-
day), or approximately 2.0 to about 3.0 grams glucose
per 109 viable cells per day, using a calibrated pump
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
27
connected to an on/off timer using a 30 minute cycle.
The glucose feed rate should be adjusted each day if
necessary. The glucose consumption rate often changes
with culture age, but the required feed rate usually
remains within the range from about 0.0 to about 6.0
g/liter-day. The Nutrient Feed 2 is usually started at
about 48 hours.
The process of the present invention has been
carried out successfully in 2-liter stirred tank
bioreactors as well as in 10-liter, 50-liter and 100-
liter stirred tanks, and thus may be carried out on
virtually any scale. In stirred tank reactors and using
the PFG01 cell line, a NIF titer of approximately 4.0
Units/ml was reproducibly achieved in approximately
eleven days. Product quality of the NIF1 produced is
high based upon comparisons of post-translational
sialylation/glycosylation and rat pharmacokinetic (PK)
studies. PK studies of the NIF obtained by the methods
of the invention may be carried out according to the
protocols and techniques set forth in Webster et al.,
supra .
The process data for an actual 2-liter stirred tank
experiment is set forth in the Examples 3 to 8. In over
twenty similar reactor experiments carried out according
the process of the invention, the average concentration
of NIF1 produced in eleven days was approximately 4.2
Units/ml ~ 0.4 Units/ml, as measured by the assay set
forth above. Samples purified from these experiments
showed reproducible post-translational modification
(glycosylation/sialylation).
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
28
Determination of NIF Titer
The assay for NIF in a given sample may be
conducted by HPLC chromatography, or any other means by
which the concentration of NIF in a given sample may be
measured.
A preferred HPLC method utilizes an HPLC column
(Atlantis C5 2.0 x 50 mm, Phenomenex, Torrence CA)
outfitted with a Rheodyne SS column inlet filter (0.5
~,m) in line before the analytical column. Ancillary to
the column are a gradient pump, a variable wavelength uv
detector, an automatic sample injector with
heater/cooler, a column heater, and a data collection
integration system. Two mobile phases A and B are used:
typically phase A is 90/10/0.05 mixture of water (J. T.
Baker, HPLC grade), acetonitrile (HPLC grade) and
trifluoroacetic acid (Sigma, protein sequencing grade,
anhydrous) respectively; and phase B is a 90/1010.04
mixture of acetonitrile/water/trifluoroacetic acid.
These phases are prepared by stirring 900 ml and 100 ml
of the 90 to 10 components, followed by filtering,
degassing with stirring for several minutes,
transferring to reservoir, and finally adding the
trifluoroacetic acid (0.5 or 0.4 ml) with stirring for
approximately 10 seconds.
Typical HPLC conditions used are: injection volume
20 ~1 (samples in vials in an autosampler maintained at
20°C); uv detector at 210 nm; initial flow at 0.4 m1/min;
the initial A to B ratio of 75:25; column heater set at
30°C. The typical sample injection run time is about 44
minutes under such conditions.
The pump is ordinarily set on a gradient program.
A typical gradient program is as follows (Table I),
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
29
although this may be adjusted according to need and
setup:
TABLEI
time %A oB flow ~psi
0 75 25 0.4
20 48 52 0.4 600
25 0 100 0.8
30 0 100 0.8
35 75 25 0.8 1400
42 75 25 0.8
43 75 25 0.4
A standard sample of NIF is prepared from
concentrate and diluted to a known concentration in PBS
buffer (Dulbecco's phosphate buffered saline). Aliquots
of 0.5 ml of the dilute working standard may be kept
frozen. The aliquot is transferred to two autosample
vials and each is injected. The peak areas of NIF are
averaged. (standard concentration/average peak area =
response factor). The areas of the NIF peak in assay
samples are multiplied by the response factor to give
the NIF concentration in the sample.
Isolation/Purification from Culture
When the NIF concentration in the production vessel
has achieved a level in the range of about 1.0 to about
8.0 Units/ml NIF (or the production phase has run
between about 5 and about 20 days), the NIF may then be
recovered from the culture. The clarified culture fluid
is obtained by centrifugation to remove cells followed
by sterile filtration through an appropriate membrane,
preferably a 0.22 ~.m filter polyethersulfone (PES)
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
membrane. Once the filtration has been completed, the
clarified culture fluid is subjected to a number of
purification steps:
A. Chromatography Step 1: Q Sepharose Fast Flow
5 Anion Exchange Chromatography
The clarified fluid containing NIF is passed
through a Q Sepharose fast flow anion exchange
chromatographic column whereby the NIF becomes bound to
the column and is then eluted at a higher concentration
10 salt solution. A Q Sepharose column is conditioned with
1N sodium hydroxide, followed by equilibration with 50mM
Na2HP04\100mM NaCl solution (pH 7.0). The 0.22 ~m
filtered culture fluid is loaded onto the column,
followed by a washing with 50mM Na~HPO~\100mM NaCl
15 solution (pH 7.0), and elution with 50mM Na~HP04\250mM
NaCl solution (pH 7.0).
B. Concentration/Diafiltration Step 1
The purified eluate from the Q Sepharose column is
concentrated using a Pall 10000 MWCO Macrosep unit in a
20 centrifuge (Sorvall RCSC Plus, HS-4 rotor, 4000 rpm, 40
minutes). During the diafiltration, the concentrated
sample buffer is exchanged to 20mM Na~HP04, pH 6.0 by
performing 3 cycles of buffer addition followed by
centrifugation.
25 C. Chromatography Step 2: Phenyl Sepharose Fast
Flow Hydrophobic Interaction Chromatography
A Phenyl Sepharose column is conditioned with 1N
sodium hydroxide, followed by equilibration with a 20mM
Na~HP04/1. OM (NHQ) 2SOQ solution at pH 6. 0. An equal
30 volume of 20mM Na2HP04/2. OM (NH4) X504 solution (pH 6. 0) is
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
31
added to the concentrated and diafiltered Q sepharose
eluate prior to loading so that the sample is loaded in
20mM Na2HP04, 1. OM (NHQ) 2S04 solution (pH 6. 0) . The
diluted diafiltrate is loaded onto the column. NIF does
not bind to the column and is washed through with 20mM
Na~HP04/1.OM (NH4)2504 solution (pH 6.0) .
D. Concentration/Diafiltration Step 2
The purified effluent from the Phenyl Sepharose
column is concentrated and then diafiltered using a Pall
10000 MWCO Macrosep unit in a centrifuge (Sorvall RCSC
Plus, HS-4 rotor, 4000 rpm, 40 minutes). During the
diafiltration, the concentrated sample buffer is
exchanged to 25mM CH3C02Na, pH 4.1, by performing 3
cycles of buffer addition followed by centrifugation.
E. Virus Inactivation
The pH of the flow through post diafiltration is
adjusted to 3.7 with acetic acid. The sample is allowed
to remain at pH 3.7 for 30 to 45 minutes with stirring,
and re-adjusted to a pH of 4.1, then filtered through a
Millipore 0.22 ~m Steriflip filter.
F. Chromatography Step 3: DEAE Sepharose Fast
Flow Anion Exchange Chromatography
In this step, NIF is bound to the column and then
eluted using a solution with a higher salt
concentration. The DEAE Sepharose Fast Flow Anion
Exchange column is conditioned with 1N sodium hydroxide,
then equilibrated with a 25mM CH3COZNa solution (pH 4,1).
The sterile filtered (or DV50-filtered) material is then
loaded onto the column. The column is then washed with
a 25mM CH3C02Na solution (pH 4.1), and then washed with
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
32
either a 25mM CH3COZNa\30mM NaCl solution (pH 4.1) or a
25mM CH3C02Na\50mM NaCl solution (pH 4.1), followed by
elution with a 25mM CH3COZNa\300mM NaCl solution (pH
4.1). The eluate contains the NIF product.
G. Concentration/Diafiltration Step 3
The purified eluate from the DEAE Sepharose column
is concentrated and then diafiltered using a Pall 10000
MWCO Macrosep unit ,in a centrifuge (Sorvall RC5C Plus,
HS-4 rotor, 4000 rpm, 40 minutes). During the
diafiltration, the concentrated sample buffer is
exchanged to 25mM Na~HP04, pH 7.0, by performing 3 cycles
of buffer addition followed by centrifugation.
Measurement of Glycosylation
A protocol for the determination of the percentage
of zero-, mono-, di-, tri- and tetra sialylation is
described in Webster et al., Xenobiotica, 29(11):1141-
1155 (1999), which is hereby incorporated by reference.
A protocol for a determination of the total degree
of sialylation of NIF is an HPLC method using PA-10
columns (Dionex Ion Pac ATC-1 mobile phase conditioner,
Dionex CarboPac 4.6 x 50 mm PA-10 guard column and
Dionex CarboPac 4.6 x 250 mm PA-10 analytical column)
outfitted with a Dionex GP40 gradient pump, a Dionex
ED40 (EC detector used in pulsed amperometric detection
mode), a Dionex AS3500 autosampler and a Dionex PeakNet
5.1 software (for data acquisition and processing). The
assay employs two mobile phases A (0.2M NaOH
(Fisher)\50mM sodium acetate (Sigma ACS grade) and B
(0.2M NaOH\300mM sodium acetate). Typical running
conditions for the HPLC are: injection volume: 20 ~.1,
PAD detection (optimized carbohydrate waveform), flow
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
33
rate: 0.7 ml/min, initial mobile phase. A: 1000 and run
time: 45 minutes
The pump is ordinarily set on a gradient program.
A typical gradient program (Table II) is as follows,
although this may be adjusted according to need and
setup:
mTnT n -r z'
time oA oB flow
0 100 0 0.7
13 100 0 0
.
7
13.1 0 100 0.7
0 100 0.7
15.1 100 0 0.7
45 100 0 0.7
The purified NIF samples and a reference sialic
acid standard are prepared to a concentration of about
1.0 X 10-3 Units/ml. To 200 ~.1 aliquots of both the NIF
and reference samples is added 200 ~,1 of 0.2N HC1. The
aliquots are vortexed and centrifuged briefly, then
heated at 80°C for 1 hour. The samples are then cooled
in an ice bath for about 10 minutes, followed by further
vortexing and centrifuging, prior to allow them to
return to room temperature. A 20 ~,l sample is injected
for analysis. Results are then reported as a percentage
of the reference standard.
Determination of Neutrophil Inhibitory Activity
Assays for the determination of neutrophil
inhibitory activity which may be useful in verification
of the quality and biological activity of the NIF
produced by the cultured cell lines are the plastic
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
34
adherence assay, the calcein assay, the hydrogen
peroxide release assay and ELISA set forth below.
A. The Plastic Adherence Assa
i. Isolation of Neutrophils
Neutrophils are isolated from heparinized venous
blood using a one-step Ficoll-Hypaque gradient (Mono-
poly, ICN~ Biomedicals, Irvine, CA). Briefly, 5 ml whole
blood is layered onto 3 ml of Mono-poly resolving media
in a 16 x 100 mm glass tube. Separation of leukocytes
is achieved by centrifuging at 300 x g for 60 minutes at
20°C. The layer of cells containing neutrophils was
collected using a Pasteur pipette and cells were
suspended in 10 volumes of cold Delbeccos' modified
Eagle's medium (DMEM, Life Technologies, Gaithersburg
MD). Neutrophils were pelleted at 200 x g for 10
minutes at 4°C. The cell pellet was resuspended in 5 ml
cold ACK buffer (155mM NHQCl/lOmM KHC03, pH 7.4) and
incubated for 5 minutes at room temperature to lyse
contaminating red blood cells. Neutrophils were then
washed once by centrifugation and resuspended in HBSS
(1.33mM CaCl~, 0.5mM MgCl2, 0.04mM MgS04, 140mM NaCl, 5mM
KC1, 0.3mM KH~PO~, 0.3mM Na2HP04 with 5.6mM D-glucose and,
mg/1 phenol red) at an approximate concentration of
107 cellslml. Cell viability was determined by Tryptan
25 blue exclusion. These preparations were consistently
greater than 95o neutrophils as determined by automated
differential counting.
ii. The Plastic Adherence Assay
Stimulated human neutrophils will adhere to plastic
30 tissue culture ware and can be visualized by standard
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
phase contrast light microscopy. Neutrophils, isolated
as in step A above, are washed once and resuspended in
cold HSA buffer (RPMI without sodium phosphate (Life
Technologies), 1o human serum albumin (Calbiochem, San
5 Diego, CA), l.2mM CaCl2, l.OmM MgCl~, lOmM HEPES, pH 7.3)
at a concentration of 6.6 x 106 cell/ml. Neutrophils (20
~,l) are placed in a sterile microfuge tube and
stimulated with PMA (5 ~1 of a 800nM solution or 160nM
final concentration) for 5 minutes at 37°C. The sample
10 to be tested is added (20 ~.l) to tube containing the
stimulated cells, mixed gently, and 10 ~.~1 of the mixture
is immediately transferred to each well of a Terasaki-
style culture plate (Nalge Nunc International,
Naperville, IL). After an additional 5 minutes at 37°C,
15 the entire plate is immersed in Hanks' balanced salt
solution (JRH Biosciences, Lenexa, KS) and tapped to
dislodge non-adherent cells. The tap/rinse step is
repeated a total of six times. Cells adhered to the
plastic wells are visualized using a phase contrast
20 light microscope. Control wells with stimulated cells
and no test sample are scored "++++", control wells with
stimulated cells and the monoclonal antibody CLB-54
(directed against the integrin CDllb/CD18) axe scored
'-
25 B. Neutrophil-Huvec Adherence (Calcein) Assay
The adherence of human neutrophils to HUVEC
monolayers are monitored by using cells which are
preloaded with the fluorescent dye calcein-AM
(acetoxymethyl ester; Molecular Probes, Eugene, OR).
30 Human neutrophils are labeled with calcein as follows.
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
36
Neutrophils are pelleted~and resuspended in HBSS
containing 10 ~.g/ml calcein-AM at a final cell
concentration of approximately 107 cells/ml. The working
HBSS/calcein solution is prepared immediately before use
from a stock solution of calcein in dimethylsulfoxide
(10 mg/ml, stored at -20°C). Neutrophils are incubated
with calcein-AM for 30 minutes at 37°C with intermittent
mixing every 10 minutes. Labeled neutrophils are washed
once and resuspended in cold HSA buffer (RPMI without
sodium phosphate (Life Technologies), 1o human serum
albumin (Calbiochem, San Diego, CA), l.2mM CaCl2, l.OmM
MgCl2, lOmM HEPES, pH 7.3) at a concentration of 1.32 x
107 cell/ml. Cells are kept at 4°C until used.
Calcein-labeled neutrophils (175 ~1) are incubated
for 10 minutes at 20°C with a 175 ~.l test fraction in the
presence of 100 ng/ml PMA (Sigma, St. Louis, MO). A
stock solution of 1 mg/ml PMA was prepared in dimethyl
sulfoxide and routinely stored at -70°C. One hundred ~,l
of the test fraction/PMA-treated cells (6.6 x 105
neutrophils) are added to a confluent monolayer of
primary HUVECs (Clonetics, San Diego, CA) grown in a 96-
well microtiter plate (Costar, Cambridge, MA). After 30
minutes at 37°C, non-adherent cells are removed by
centrifuging inverted, sealed plates for 3 minutes at 75
x g. Adherent neutrophils were lysed by adding 100 ~.l
0.1 Triton X-100 (in 50mM Tris-HC1, pH 7.4) and the
fluorescent emission of calcein at 530 nm from 485 nm
excitation is reading using a Cytofluor fluorometric
plate reader (Millipore, Bedford, MA). Each data point
is performed in triplicate. In these experiments, 400
of the total input neutrophils, or approximately 2.6 x
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
37
105 cells, bind to the HUVEC monolayer in the absence of
inhibitor.
C. Hydrogen Peroxide Release Assay
Hydrogen peroxide release from stimulated human
neutrophils is determined by a modification of the
method described by Pick et al., J. Immun. Methods,
_38:161-170 (1980). Human neutrophils (6.6 x 106 cell/ml)
are resuspended in HBSS containing 10% fetal bovine
serum. Phenol red and Type IV horseradish peroxidase
(Sigma) are added to the cell suspension at final
concentrations of 83 ~g/ml and 0.01 Units/ml,
respectively. Five hundred microliters of this cell
suspension are added to 200 ~,l of test sample (in HBSS
containing 10o fetal. bovine serum). Cells are activated
with fMLP (Sigma) at a final concentration of 275~.M. A
stock solution of fMLP (500nM) is prepared in dimethyl
sulfoxide and stored at -20°C. The release assay is
performed in 1.5 ml plastic tubes (Eppendorf, Madison,
WI) that are precoated with fetal bovine serum for 60
minutes at 37°C; coated tubes are washed twice with
0.15N NaCl before use. The release action is allowed to
proceed for 90 minutes at 37°C, after which time the
cells are pelleted at 2000 x g for 3 minutes in an
Eppendorf Microfuge. Two hundred microliters of
supernatant fluid are transferred to a 96-well
microtiter plate and the reaction is stopped by the
addition of 10 ~.1 of 1N NaOH. Each data point is
performed in duplicate. Samples are quantitated at 610
nm with a ThermoMax plate reader (Molecular Devices,
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
38
Sunnyvale, CA). Hydrogen peroxide concentration was
calculated from an internal standard curve.
D. ELISA For NIF1
A polyclonal antibody directed against NIF1 is
prepared in rabbits using standard techniques. The
antibody is immunoaffinity-purified using resin composed
of rNIF1 coupled to uniform glass beads (Bioprocessing
Ltd., Consett, UK). A monoclonal antibody directed
against NIF1 is also prepared in mice using standard
techniques. The monoclonal antibody is purified from
mouse ascites fluid by protein A chromatography and
conjugated to horseradish peroxidase ("HRP") (Boehringer
Mannheim, Indianapolis, IN) following standard
protocols. The immunoaffinity-purified polyclonal
antibody is adsorbed to the wells of Immulon 2
polystryrene immunoassay plates (Dynatech Labs,
Chantilly, VA) and then blocked with bovine serum
albumin. Test samples containing NIF1 (100 ~l/well) are
added to the wells of the immunoassay plate, mixed using
a plate shaker, and incubated at 37°C for 3 hours. The
contents of the wells are removed and the wells washed
with phosphate buffered saline containing 0.020 Tween
20. Monoclonal antibody-HRP conjugate (100 ~ljwell) is
added to the wells, mixed as before, and incubated at
37°C for 2 hours. Unadsorbed monoclonal antibody-HRP
conjugate is rinsed away with phosphate buffered saline
containing 0.020 Tween 20 and HRP substrate (10 ml of
0.1M sodium acetate, pH 4.5, 0.0120 hydrogen peroxide,
plus 0.4 ml trimethylbenzidine, 3 mg/ml in 0.1M HCl) was
added to the wells. Color is allowed to develop for 10
minutes at room temperature when the reaction is stopped
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
39
with 1M sulfuric acid. The optical density at 450 nm is
determined using a Molecular Devices 96-well plate
reader. Standard curves are generated with samples
containing known concentrations.
Formulations
Pharmaceutical compositions of NIF may be
formulated and used as tablets, capsules or elixirs for
oral administration; suppositories for rectal
administration; sterile solutions, suspensions for
injectable administration; and the like. The dose and
method of administration can be tailored to achieve
optimal efficacy but will depend on such factors as
weight, diet, concurrent medication and other factors
which those skilled in the medical arts will recognize.
Generally, an amount between 0.01 mg/kg to 100 mg/kg
body weight/day is administered dependent upon the
potency of the composition used. Preferred embodiments
encompass pharmaceutical compositions prepared for
storage and subsequent administration which comprise a
therapeutically effective amount of NIF or an enriched
composition of NIF, as described herein in a
pharmaceutically acceptable carrier or diluent.
Acceptable carriers or diluents for therapeutic use are
well known in the pharmaceutical art, and are described,
for example, in Remington's Pharmaceutical Sciences,
Mack Publishing Co. (A. R. Gennaro, Ed. 1905).
Preservatives, stabilizers, dyes and even flavoring
agents may be provided in the pharmaceutical
composition. For example, sodium benzoate, sorbic acid
and esters of p-hydroxybenzoic acid may be added as
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
preservatives. In addition, antioxidants and suspending
agents may be used.
Injectables can be prepared in conventional forms,
either as liquid solutions or suspensions, solid forms
5 suitable for solution or suspension in liquid prior to
injection, or as emulsions. Suitable excipients are,
for example, water, saline, dextrose, mannitol, lactose,
lecithin, albumin, sodium glutamate, cysteine
hydrochloride or the like. In addition, if desired, the
10 injectable pharmaceutical compositions may contain minor
amounts of nontoxic auxiliary substances, such as
wetting agents, pH buffering agents, and the like. If
desired, absorption enhancing preparations (e. g.,
liposomes) may be utilized.
15 Utility
The NIF produced in the present invention may be
used in methods of treating in a mammal an inflammatory
condition characterized by abnormal neutrophil
activation or abnormal eosinophil activation comprising
20 administering to said mammal a therapeutically effective
amount of a NIF or their pharmaceutical compositions.
In practicing the preferred methods, NIFs or their
pharmaceutical compositions can be used alone or in
combination with one another, or in combination with
25 other therapeutic or diagnostic agents. These
compositions can be utilized in vivo, ordinarily in a
mammal, preferably in a human, or in vitro.
In employing NIFs or their pharmaceutical
compositions in vivo, the compositions can be
30 administered to the mammal in a variety of ways,
including parenterally, intravenously, subcutaneously,
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
41
intramuscularly, colonically, rectally, nasally or
intraperitoneally, employing a variety of dosage forms.
As will be readily apparent to one skilled in the art,
the useful in vivo dosage to be administered and the
particular mode of administration will vary depending
upon the mammalian species treated, the particular
composition employed, and the specific use for which
these compositions are employed. The determination of
effective dosage levels, that is the dosage levels
necessary to achieve the desired result, will be within
the ambit of one skilled in the art. Typically,
applications of compositions are commenced at lower
dosage levels, with dosage level being increased until
the desired effect is achieved.
1$ The dosage for a NIF or its pharmaceutical
compositions can range broadly depending upon the
desired effects and the therapeutic indication.
Typically, suitable dosages will be betweeri~about 0.01
mg and about 100 mg/kg, preferably between about 0.01
and about 10 mg/kg, body weight. Administration is
preferably parenteral, such as intravenous on a daily or
as-needed basis.
The NIF produced by the methods of the present
invention has potent neutrophil inhibitory activity and,
thus, may be used as an inhibitor of neutrophil
activity, including neutrophil activation in vitro, as
well as for preventing or treating in a mammal
inflammatory conditions characterized by abnormal
neutrophil activation. Thus, NIF will be useful in the
treatment of inflammation in which the abnormal
activation of neutrophils plays a significant role.
While applicants do not wish to be bound to any theory
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
42
or mode of activity, it is believed that this compound
will interfere with the inflammatory response which is
set into action by neutrophil-endothelial cell
interactions. Thus, where adhesion of neutrophils to
the endothelium is prevented, the neutrophils will be
unable to transmigrate to tissue to elicit a pro
inflammatory response with consequent tissue damage.
Inhibition of neutrophil-neutrophil adhesion and/or
aggregation by these NIFs should also prevent
microvascular occlusion. Thus, these NIFs will be
useful in treating a variety of clinical disorders,
including shock, stroke, acute and chronic allograft
rejection, vasculitis, autoimmune diabetes, rheumatoid
arthritis, head trauma, inflammatory skin diseases,
inflammatory bowel disease, adult respiratory distress
syndrome CARDS), ischemia-reperfusion injury following
myocardial infarction, in which neutrophil infiltration
and activation has been implicated and acute
inflammation caused by bacterial infection, such as
sepsis or bacterial meningitis.
The ability of the NIF produced by the present
invention to inhibit neutrophil activity makes it useful
in inhibiting the physiological processes of
inflammation, ischemia, and other neutrophil mediated
tissue damage. The specific activities of NIFs in
carrying out these related functions makes it
particularly useful as therapeutic and/or diagnostic
agents.
Antibodies, both monoclonal and polyclonal,
directed to the NTF produced by the present invention
are useful for diagnostic purposes and for the
identification of concentration levels of the subject
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
43
peptides in various biological fluids. Immunoassays
utilizing these antibodies may be used as a diagnostic
test, such as to detect infection of a mammalian host by
a parasitic worm or to detect NIF from a parasitic worm
in a tissue of the mammalian host. Also such
immunoassays may be used in the detection and isolation
of NIF from tissue homogenates, cloned cells and the
like. In another aspect of the present invention, NIFs
can be used in a test method to screen other compounds
to detect NIF mimics or to detect NIF antagonists for
their ability to affect NIF binding to the CDllb/CD18
receptor.
In yet another aspect of the present invention, the
NIF produced by the present invention with suitable
adjuvants can be used as a vaccine against parasitic
worm infections in mammals. Immunization with NIF
vaccine may be used in both the prophylaxis and therapy
of parasitic infections. NIF fragments and synthetic
polypeptides having the amino acid sequence of NIF may
also be used as vaccines. Disease conditions caused by
parasitic worms may be treated by administering to an
animal infested with these parasites substances which
antagonize NIF (such as NIF antagonists). Compounds may
be screened for their anti-NIF effect according to the
screening method described herein above. Examples of
such antihelminic agents include antibodies to NIF, both
naturally occurring antibodies isolated from serum and
polyclonal and monoclonal antibodies described above.
Chemically synthesized compounds which act as inhibitors
of NIF also are suitable antihelminic agents.
The following examples serve to illustrate the
process of the invention. The actual allowed ranges for
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
44
process control parameters and process scale may be
significantly broader.
EXAMPLES
L~VTn/fDT.~' l
Cell Line Expressing NIF
A. The Nucleic Acid Encoding NIF
The coding sequence for recombinant NIF was derived
from a canine hookworm (Ancylostoma) cDNA library to
which standard expression regulatory sequences were
added during plasmid construction. The nucleotide
sequence of NIF-1FL, mature NIF-1FL (NIF1) and the
corresponding full-length cDNA are presented in Figures
1 and 2, respectively. The nucleotide sequence in
Figure 2 has an open reading frame of 822 nucleotides
encoding a 274 amino acid polypeptide (nucleotides 313
through 1134).
B. Construction of the Expression Vector
The NIF1 cDNA described above was cloned into a
series of shuttle vectors and hosts, and finally into
the pEEl4 vector, as follows. Figure 3 is a schematic
representation of the pathway from NIF1 cDNA to pEEl4
vector which was used for transfecting CHO-K1 cells.
Some of the biochemicals used in the cell line
construction process and their respective suppliers are
as follows (Table III)
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
TTDT L~ TTT
Vector Supplier
Lambda gtl0/EcoRI Stratagene, La Jolla,
California
pcDNA1/Amp (sequencing Invitrogen, Carlsbad,
vector, not used in California
cloning )
pBluescript II KS+ Stratagene, La Jolla,
California
pSG5 Stratagene, La Jolla,
California
E. coli SLJRETM Stratagene, La Jolla,
California
The NIF1 coding region (~~NIFlcr") (SEQ. ID. N0. 1)
was rescued into the pSG5 vector and the nucleotide
5 sequence determined. The coding region itself
(disregarding the sequence immediately upstream from the
ATG start which was altered to promote expression)
corresponds exactly with bases 313 through 1137 of the
original NIF1 cDNA clone as illustrated in Figure 2.
10 COS-7 cells transfected with pSGS/NIFlcr produced
active NIF1. The NIF1 produced inhibited H202 production
by human neutrophils in a concentration dependent manner
as does hookworm-derived NIF. Neither control
transfection (cells transfected with a plasmid harboring
15 chloramphenicol acetyl transferase or mock transfected
cells) produced a NIF-like activity.
The limited multiple cloning site of pSG5
necessitated the passage of NIFlcr through a plasmid
capable of supplying different restriction sites on
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
46
opposite ends of the coding region. The pBluescript II
KS+ was selected due to the presence of an EcoRI site in
the middle of its extensive multiple cloning site and
its ease of manipulation. This was followed by cloning
into the pEEl4 expression plasmid. The expression
construct pEEl4/NIFlcr was used to transfect CHO K1
cells.
Proper isolation and sequence of final and
intermediate constructions was verified at critical
steps by restriction mapping or sequencing. The full
length NIF sequence was first verified by sequencing
when cloned into the pcDNAl/Amp sequencing vector (see,
Figure 2 for sequence). The coding region sequenced was
verified after cloning into the PSG5 shuttle vector by
bi-directional sequencing.
C. The Expression Vector
As indicated by its designation, the pEEl4/NIFlcr
expression plasmid was derived from the widely used
9.4kb pEEl4 expression vector (Zonza Biologics) shown at
Figure 4. The pEEl4 vector contains: (1) a human CMV
major immediate early promoter (hCMV-MIE), (2) a
multiple cloning site (MCS), (3) a SV40 early poly A
site (pA), (4) a Col E1 origin of replication (Col E1),
(5) an ampicillin resistance gene (Amp), and (6) the
SV40 late promoter (SV40Z) which drives the glutamine
synthetase minigene (GS-minigene). The restriction
endonuclease sites present in the multiple cloning site
are noted in this diagram. The 5' HindIII insert site
is slightly 5' to the MCS. Nucleotide sequences for
portions of the vector can be obtained from Bebbington
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
47
et al., Bio/Technology, 10:169-175 (1992) and Stephens
and Cockett, Nucleic Acids Research, 17:7110 (1989).
The pEEI4INIFIcr insert contains 825bp of NIF1
coding sequence (SEQ. TD. N0. 1), which codes for the
274 amino acids indicated in Figure 1 (SEQ. ID. N0. 2).
The mature NIF-1FL (NIF1) protein contains the 257 amino
acids coded by the sequences starting with codon 18, as
indicated in Figure 1 (SEQ. ID. N0. 3). Additional non-
coding sequences (SEQ. ID. NOS. 10 and 11) were
incorporated into the insert, at both ends of the coding
sequence during the cloning process, as shown in Figure
5.
The principal modification to the pEEl4 vector is
the insertion of the NIF1 coding sequence into the
vector's insert expression region between the pEEl4
HindIII (bp9292) and SmaI sites (bp9334}. This
construction allows for high level NIF1 expression under
the control of pEEl4's human CMV major intermediate
early (~~hCMV-MIE") promoter. The construction of NIFlcr
(cr = coding region) insert is depicted in Figure 5.
As shown in Figure 5, the 5'-end of the insert
sequences start at the HindIII site in the pEEl4
expression vector (site not shown on Figure 4), which
are joined to the complementary sequences from the 5'-
HindIII site from pBluescriptIl shuttle vector (~~BSII")
polylinker. The 5' HindIII site is followed by an EcoRI
site, provided by the 5'-PCR NIFlcr rescue primer, used
to clone the NIFlcr sequences into BSII. The NIF1
coding region sequence of NIF1, beginning at this EcoRI
site extends for approximately 850 nucleotides.
The NIFlcr is followed by the EcoRI site created by
the 3'-PCR NIF1 rescue primer used to create 3' end
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
48
needed fox cloning NIFIcr into the BSII. The 3'-end of
the coding region is followed by a PstI site from
pBluescriptII shuttle vector, and finally a SmaT site
and other sequence from pEEl4 (Figures 4 and 5). The
NIF1 protein coding sequences are shown in capitals, and
bars indicate the cleavage points for the indicated
restriction enzymes. The pEEl4 sequences between the
HindIII and SmaI sites are removed during NIFlcr
cloning.
D. Transfection of the Expression Vector
The pEEl4/NIFlcr vector was introduced in CHO-K1
cells (ATCC CCL-61) using a standard calcium method as
follows. For transformation, the CHO-K1 cells were
propagated in DMEM (Life Technologies/Gibco) in T-75
flasks at 37°C in a 7.5-10% C02 atmosphere. To each
500 ml DMEM was added: standard nutrients and 50 ml
fetal bovine serum (FBS). Prior to transfection, the
cells were removed from the flasks using porcine trypsin
as described above and washed with DMEM-S (DMEM prepared
as above but with dialyzed FBS) and seeded onto 10 cm
diameter tissue culture plates (Costar) at 1 x 106 cells
per plate. The cells were incubated at 37°C overnight.
Just before the cells were to be transformed, they were
rinsed once with DMEM without FBS. The DNA-calcium
phosphate precipitate was prepared in two steps as
follows. First 62 ~.l 2M calcium chloride was mixed with
10 ~g pEEl4/NIFlcr DNA and brought up to 500 ~,L with
sterile water. Next this mixture was added dropwise to
500 ~.l 2X HEPES buffered saline with constant gentle
agitation using a bubble stream. Once all of the DNA
mix was added the tube containing the DNA-calcium
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
49
phosphate precipitate was vortexed. The DNA-calcium
phosphate precipitate was diluted with 2 ml DMEM without
FBS and added to the 10 cm diameter dish containing the
CHO K1 cells. The plates were placed at 37°C, 7.5o COz
with gentle rocking for 4 hours. The medium and DNA-
calcium phosphate precipitate was removed from the cells
and replaced with 3 ml 15o glycerol in HEPES buffered
saline. After 90 seconds at 37°C, 10 ml DMEM without FBS
was added and immediately removed by aspiration. The
cells were then covered with 10 ml DMEM-S and incubated
for 24 hours at 37°C, 7.5o C02. The medium was replaced
with fresh DMEM-S containing 25 ~,M methionine
sulfoximine (MSX). The plates were incubated for an
additional 7 days when the C02 was raised to loo to lower
the pH of the medium. At this time the plates contained
many colonies of various sizes. The cells were removed
from the dishes by treatment with porcine trypsin as
before, collected by centrifuging as before, and
resuspended in 50 ml of an equal volume mixture of
conditioned medium and fresh DMEM-S supplemented with
20o dialyzed FBS and 25~.M MSX. The resuspended cells
were transferred into 96-well culture plates (100 ~,l per
well) and incubated at 37°C, 10o COZ to obtain individual
colonies. Seven days after plating, 100 ~.l cloning
medium (50o CHO K1 conditioned DMEM-S with 20o dialyzed
FBS, 50% fresh DMEM-S with 20o dialyzed FBS, 25~M MSX)
was added to replace medium lost to evaporation. Ten
days after plating, 201 wells contained individual
colonies and 3 wells contained 2 or 3 colonies.
Twenty days after plating, 15 wells exhibited
confluent growth and the cell-free supernatant fluids
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
were assayed using the plastic adhesion assay (above)
for NIF (and, thus, NIF1) activity. The positive clones
were expanded into 24-well culture plates as follows.
The cells were detached from the 96-well plates by
5 treatment with porcine trypsin and the digestion with
trypsin stopped with trypsin inhibitor. One ml of
cloning medium was added to each well and the plate was
incubated at 37°C, 10% C02. Three days later 500 ~l
cloning medium containing 25~M MSX was added. Seven
10 days after expansion, cells were assayed for NIF1
activity using the plastic adhesion and calcein assays.
Positive clones were expanded into 10 cm diameter tissue
culture dishes. One clone expressing the highest level
of NIF1 activity was frozen at approximately 1 X 106
15 cell/ml in cloning medium containing 20o dialyzed FBS,
25~,M MSX and 10o dimethyl sulfoxide.
This clone was subjected to two rounds of cloning
by limited dilution to ensure the final cell line
originated from a single transfected cell. The clone,
20 grown to confluence in the DMEM-S containing 100
dialyzed FBS and 25~.M MSX, was removed from a culture
dish by trypsin treatment, diluted with cloning medium
to 25 cells per ml, and plated in 96-well plates at 2.5
cells per well. The plates were incubated at 37°C, 100
25 COz. After 17 days in culture, 33 of the wells (those
exhibiting growth) were assayed for NIF activity using
the calcein assay. On the basis of growth rate and
expression of NIF activity, several of the clones were
grown to confluence in DMEM-S containing 10o dialyzed
30 FBS and 25~M MSX and frozen at approximately 1 X 106
cell/ml in cloning medium. All cultures were confirmed
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
51
to be producing NIF1 by ELISA (see Detailed Description
of the Invention).
EXAMPLE 2
Adaptation to Suspension Culture And Serum-Free Medium
A. Subculturing of Cell Line In T-flask Cultures
One of the cultures as prepared in Example 1 was
further grown in a medium consisting of DMEM:RPMI1640
50:50 (glutamine free) (50:50 mix of DMEM (Dulbecco's
Modified Eagle Medium, Gibco Catalog No. 11960) and
RPMI1640 (Roswell Park Memorial Institute, Gibco Catalog
No. 21870); 10o Certified Heat Inactivated Fetal Bovine
Serum (Gibco); with 1 ml per liter medium of a 25mM
(1000X) L-methionine sulfoximine stock solution (Sigma).
The medium was decanted off. The monolayer was
rinsed twice with 10 ml of Dulbecco's PBS (calcium and
magnesium free); the Dulbecco's PBS was decanted and 2
ml of versene was added to the monolayer. The culture
with versene was incubated at 37°C for 5 minutes. The
flask was rapped several times to dislodge the cells and
resuspended in an additional 18 ml of fresh medium and
split 1:5 to new T-flasks. The culture was incubatea az
37°C in 5o COZ and 70o humidity and designated as passage
X+1. A solution of 0.250 trypsin EDTA was used in the
place of versene for all subsequent subcultures in T-
flasks. Cultures were typically split 1:10 to 1:25 as
necessary twice per week. Cells were not allowed to
reach 1000 confluence if possible.
B. Adaptation of Culture to Suspension Growth
The culture was adapted to suspension growth in
shake flasks in CHO III PFM medium supplemented with
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
52
serum. The suspension culture was inoculated with cells
from T-flasks at passage X+4. The suspension medium
formulation consisted of CHO III PFM (Gibco Formula #
96-03345A); 10o Certified Heat Inactivated Fetal Bovine
Serum (Gibco 10082); 1 m1/1 of a 25mM (1000X)
L-methionine sulfoximine solution; and 10 m1/1 100x HT
supplement (Gibco 11067).
The suspension culture was inoculated at a density
of 1.7 x 105 cells/ml. The medium volume was 50 ml in a
250 ml Corning disposable shake flask. The culture was
incubated at 37°C with 5o COZ and 70o humidity on a
shaker at 130 rpm. The culture was split 1:3 to 1:5 as
needed when the cell density approached 1 x 106 cells/m1
and was never split to a density below 2 x 105 cells/ml.
The first passage of the cells in suspension shake
flask culture was designated passage X (X+5 from T-
flasks). The culture was continued out to passage X+7
with 10o fetal bovine serum. The adaptation to
suspension growth in serum supplemented medium took
approximately 20 days.
C. Adaptation to Serum-Free Suspension Growth
The suspension culture was adapted to serum free
growth by gradually decreasing the concentration of
serum in the medium. All other components of the medium
formulation were unchanged during the weaning process.
As with the suspension growth adaptation, cells were
maintained between 2.5 x 105 and 1 x 106 cells/ml by
splitting 1:3 to 1:5 as necessary. The culture was
incubated at 37°C with 5o COZ and 70o humidity on a
shaker at 130 rpm. At passage 8, the serum was reduced
to 50; at passage 9, to 20; at passage 10, to 10; at
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
53
passage 11, the cells were centrifuged and resuspended
in 40 ml fresh medium/10 ml conditioned medium, serum
concentration was maintained at 10 (large clumps and
cell debris were allowed to settle from the culture and
removed) at passages 12-15, serum was maintained at 10;
at passage 15, the medium was supplemented with 60 mg/1
L-aspartic acid, 120 mg/1 L-serine, 200 mg/1
L-asparagine and 60 mg/1 L-methionine added as a 50X
stock solution adjusted to pH 7.5 and filter sterilized;
at passages 16-21, the serum was reduced to Oo and the
amino acid supplements were maintained; and at passage
21, a pre-seed stock (PSS) frozen vial bank was prepared
at 1 x 107 cells/vial. Adaptation to serum free medium
took approximately 55 days. The serum-free suspension
culture as produced herein was designated PGF01.
D. Freezing the Culture
The cells as prepared above were prepared for
freezing and storage by centrifuging the cells at 10
minutes at 500 rpm in a Beckman GPR centrifuge. The
cells were resuspended in freezing medium consisting of
50% complete medium (CHO-III-PFM with 1 m1/1 25mM
(1000X) methionine sulfoximine stock solution, 10 m1/1
HT supplement, 20 m1/1 50X amino acid stock solution,
3 g/1 L-aspartic acid, 6 g/1 L-serine, 10 g/1
L-asparagine, 3 g/1 L-methionine); 50o conditioned
medium. Reconditioned" medium is one in which the cells
have been grown for a few days, the cells centrifuged
and separated out, and then filter sterilized; 10 g/1
bovine serum albumin as a protectant; and 75 m1/1 DMSO
(Sigma Cell Culture Tested D2650) to a density of 1 x 107
cells/ml and dispensed in cryovials. The cells were
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
54
frozen in a controlled rate freezer to -75°C at
1°C/minute and then transferred to liquid nitrogen for
storage.
E. Preparation of Serum-Free Suspension Master
Cell Bank (MCB)
A master cell bank was prepared from the Passage
X+21 cell culture removing one vial of the passage X+21
culture from liquid nitrogen storage and quickly thawed
in a 37°C water bath. The contents of the vial was
transferred to a 15 ml conical tube with 9 ml of fresh
medium consisting of CHO III PFM (Gibco Formula # 96-
03345A); 1 m1/1 of a 25mM (1000X) L-methionine
sulfoximine stock solution; 10 m1/1 100x HT supplement
(Gibco 11067); 20 m1/1 50X amino acid stock solution
adjusted to pH 7.5 and filter sterilized (stock
solution: 3 g/1 Z-aspartic acid, 6 g/1 L-serine; 10 g/1
L-asparagine and 3 g/1 L-methionine
The tube was centrifuged at 500 rpm for 10 minutes
in a Beckman GPR centrifuge. The supernatant was
decanted and the cells were dislodged from the bottom of
the tube. Ten ml of fresh medium was added to the tube
and the suspended cells were transferred to a Corning
250 ml shake flask. The medium volume was adjusted to
50 ml making the initial cell density in the culture 2 x
105 cells/ml. The culture was incubated overnight at
37°C in 5o COZ with 70o humidity on a rotary shaker at
100 rpm. After the first day of incubation, the rotary
shaker speed was increased to 130 rpm. After 5 passages
in serum free/animal protein free suspension growth, an
MCB was prepared at 1 x 107 cells/vial.
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
nvTnrtnT n
A. Medium for the Generation of the Inoculum
Culture
The culture medium for the inoculum culture was
5 prepared from the following components:
1.0 liter CHO-III-PFM solution with glucose (Life
Technologies, Custom Formula 98-0289 ; with 3.45 g/1 D-
glucose; without hypoxanthine, thymidine, L-glutamine);
10.00 m1/1 HT supplement (Life Technologies,
10 Catalog No. 11067-030: 100x = lOmM sodium hypoxanthine,
l.6mM thymidine);
20.00 m1/1 amino acid stock (as prepared in 3B
below)
1.00 m1/1 25mM L-methionine sulphoximine stock (as
15 prepared in 3C below);
25.00 mg/1 L-cysteine (Sigma) and
0.50 m1/1 phenol red (Sigma, 0.50 (w/v) solution).
B. Amino Acid Stock
The amino acid stock used in the inoculum culture
20 medium above was prepared by dissolving: 3.00 g/1
L-aspartic acid (Sigma), 2.50 g/1 L-glutamic acid
(Sigma), 10.00 g/1 L-asparagine (Sigma), 1.25 g/1
L-proline (Sigma), 3.00 g/1 L-serine (Sigma), and 1.50
g/1 L-methionine (Sigma) in deionized water to make a
25 one liter solution, adjusting the pH to 8.0 with aqueous
5N sodium hydroxide and then sterile filtering the
resultant solution.
C. L-Methionine Sulphoximine Stock
L-methionine sulfoximine (25 mmol, FW 180.2, Sigma)
30 was dissolved in one liter of deionized water. The
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
56
resultant solution was filtered using a 0.2 micron
filter. This solution may be kept at 4°C for up to 3
months, or can be stored frozen at -20°C or lower for
longer periods of time.
EXAMPLE 4
Inoculum Generation
A vial of frozen PFG01 seed cells were thawed in a
water bath at 36.5 ~ 1°C until only a small ice pellet
remained. The vial was transferred to the biosafety
cabinet and the exterior decontaminated with a sterile,
70o isopropanol wipe. The cells were resuspended in 25
ml of pre-warmed growth medium as prepared in Example 3A
and transferred into a 125 ml shake flask. The culture
was sampled using the Trypan Blue Dye Exclusion method
(Cell and Tissue Culture: Laboratory Procedures in
Biotechnology, A. Doyle and J.B. Griffiths, eds. (John
Wiley & Sons, Ltd., 1998)). If necessary, more pre-
warmed growth medium was added to adjust the final cell
concentration to approximately 5.0 x 105 vc/ml. The
flask was incubated with stirring at 36.5 ~ 1°C, C02
concentration of 5 ~ 10, a relative humidity of 70 ~ 50,
and a stirring rate of 170 ~ 5 rpm. The flask was
sampled daily to check cell concentration and viability.
Sufficient pre-warmed growth medium was added daily
to maintain a concentration of 5.0 x 105 vc/ml in the
flask. When the volume of the shaker flask reached 50
ml and the cell density reached 1.0 x 106 vc/ml, the
culture was transferred to a 250 ml shake flask and
diluted to 5.0 x 105 vc/ml in 100 ml. When the cell
density again reached 1.0 x 106 vc/ml, the culture split
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
57
and half transferred to another 250 ml shake flask and
diluted to 2.0 x 105 vc/ml. These steps of permitting
the seed train to expand was continued until a
sufficient volume was achieved to obtain a seeding
density of 2.0 x 105 vc/ml in the bioreactor. The ideal
inoculum ratio (volume of inoculum culture/reactor
liquid volume after inoculation) was judged to be about
to 200. Accordingly, the cell density in the
inoculum culture was adjudged to be ideally between 1.0
10 x 106 vc/ml and 2.0 x 106 vc/ml. The age of the inoculum
culture was approximately 3 days old.
wTrrtr~T ~ G
Medium For Use In The Production Bioreactor
A. Batch Medium
The batch medium for NIF1 production was prepared
by combining
1.00 liter CHO-III-PFM with glucose (Life
Technologies, Custom Formula 98-0289; with 3.45 g/1 D-
glucose; without hypoxanthine, thymidine, L-glutamine);
10.00 m1/1 HT supplement (Life Technologies);
1.50 g/1 yeast extract (Bacto, Difco/Becton
Dickinson); and
0.50 m1/1 phenol red (0.50 (w/v) solution, Sigma).
B. Nutrient Feed 1
For use as nutrient feed 1, 200 grams glucose (from
cerelose, Corn Products International) was dissolved in
deionized water to make one liter of solution. This
glucose feed is used to control the glucose
concentration in the reactor at a concentration of
approximately 1.5 to 2.5 g/1.
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
58
C. Nutrient Feed 2
For use as nutrient feed 2, the following
components are combined:
1.0 liter CHO-III-PFM 5X (adjust to pH 7.4) (Life
S Technologies, Custom Formula 99-0180; 5X with 1X
L-cystine, 3X L-tyrosine; without glucose, hypoxanthine,
thymidine, L-glutamine, sodium bicarbonate, and sodium
chloride).
50 m1/1 HT supplement (Life Technologies, Catalog
No. 11067-030 and 100x = lOmM sodium hypoxanthine,
l.6mM thymidine).
7.50 grams yeast extract (Difco, Bacto/Becton-
Dickinson).
The HT supplement and yeast extract was added to
the CHO-III-PFM 5X. The components were mixed until
completely dissolved, the pH was then adjusted to 7.4
using 5N sodium hydroxide, then the resulting solution
was sterile filtered.
n~rTnrtr~r rn P_'
Operation Of 2-Liter Stirred Tank Bioreactors
The production of NIF1 was performed in a 2-liter
Wheaton bioreactor (B. Braun Biotech Inc., Allentown,
PA), controlled via a Foxboro IA (Intelligence
Application) computer system (Foxboro Company, Foxboro,
MA). After sterilization of the bioreactor, one liter
of sterile conditioning solution, glutamine-free DMEM
(Life Technologies/GibcoBRL) was added. After four
hours, the rinse medium was replaced with one liter of
fresh, sterile production batch medium, as prepared in
Example 5A. The temperature of the medium was allowed
to stabilize at 36.5 ~ 1°C, and the pH was adjusted to pH
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
59
7.4. To obtain a target density in the reactor of
approximately 2.0 x 105 viable cells / ml, a volume of
about 200 ml inoculum culture was added to obtain a 1.2
liter initial liquid volume.
A. Operational Setpoints
The following parameters for operation of the
bioreactor were put in place.
Agitation: 100 rpm (4-inch diameter single plastic
vertical blade).
pH: 7.40 ~ 0.15 (Control agents: CO2 gas and a
solution of 7.5% (w/v) NaHC03).
Dissolved oxygen concentration: 600 ~ 5o air
saturation (Control agents: OZ and N2) .
Temperature: 36.5°C.
Nutrient Feed 1: 200 g/1 glucose solution - fed at
about 0.0 to about 6.0 g/(liter-day).
Gas flow: 200-300 ml/min constant air flow to the
headspace. C02 and 02 (for safety purposes 60o OZ in N2
was employed) were sparged into the culture on demand to
control pH and dissolved oxygen. N2 was directed to the
headspace at moments where the dissolved oxygen
concentration exceeded its upper limit (650).
Nutrient Feed 2 was fed continuously at a constant
rate of 30 ml/day (i.e., at a rate of 25 ml per liter of
culture at inoculation per day). This feed was started
simultaneously with the glucose feed at 48 hours and
maintained at this level over the course of the
production run.
B. Sampling and Maintenance
The bioreactor was sampled immediately after
inoculation. The following measurements were taken: the
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
initial cell density and viability; the off-line pH; the
initial glucose concentration; the initial lactate
concentration; the initial ammonia concentration; and
the initial osmolality. The on-line pH was adjusted
5 when necessary. The bioreactor was covered in black
plastic to protect the medium from light.
The bioreactor was sampled daily for the following
parameters: cell density; culture viability; off-line
pH; glucose concentration; lactate concentration;
10 ammonia concentration; osmolality; and mature NIF1
concentration (starting at 4 days). The glucose
concentration was maintained between 0.1 and 3.0 g/liter
using Nutrient Feed 1. The feed with Nutrient Feed 1
began after 48 hours with an initial feed rate of
15 approximately 2.0 g/(liter-day), or approximately 2.0 to
3.0 g glucose per 109 viable cells per day) using a
calibrated pump connected to an on/off timer using a 30
minute cycle. The glucose feed rate was adjusted each
morning as necessary. The required feed rate usually
20 remained within the range from 0.0 to 6.0 g/1-day. The
constant Nutrient Feed 2 was started simultaneously with
the glucose feed.
C. NIF1 Production Profile Using PFG01
The following measurements set forth in Table IV
25 below were observed for the run of the production of
NIF1 using PFG01 cells.
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
61
mT nT n rcT
DaypH Cell ViabilityNIF1 GlucoseLactateAmmonia Osmolality
Count (~) (Units/ml)(g/1) (mmol/1)immol/1)(mOsm/kg)
(vc/ml)
0 7.552.75E+05100.0 2.79
1 7.531.60E+0599.1 2.76 6 0.584 315
2 7.434.25E+0598.8 1.89
3 7.381.11E+06100.0 0.24 33 0.828 326
9 7.262.17E+0696.6 0.5 1.30 54 0.626 350
7.352.66E+0695.0 0.9 1.36 65 0.410 378
6 7.313.36E+0696.0 1.6 1.98 70 0.536 391
7 7.323.12E+0693.9 2.1 2.69 77 0.566 403
8 7.392.50E+0686.8 2.7 2.34 78 0.414 405
9 7.241.50E+0694.9 3.3 2.40
7.381.50E+0686.9 3.9 2.38
11 7.391.88E+0676.4 4.5 2.93 80 0.458 421
12 7.352.20E+0668.8 5.0 2.86 81 0.652 426
The cell count and viability measurements were
measured in accordance with the Trypan Blue Dye
5 Exclusion method as set forth in Cell and Tissue
Culture: Laboratory Procedures in Biotechnology, A.
Doyle and J.B. Griffiths, eds. (John Wiley & Sons, Ltd.,
1998). The NIF1 titer was measured by the protocol set
forth above in the Detailed Description of the
10 Invention. The glucose, lactate and ammonia
measurements were conducted using a Kodak Biolyzer Rapid
Analysis System. The osmolality was measured using an
Advanced Micro Osmometer, (Model 3330, Advanced
Instruments, Inc., Norwood, MA).
EXAMPLE 7
NIF1 Sialylation/Glycosylation Profile
The NIF1 harvested from a number of different
bioreactor runs were tested for degree of
sialylation/glycosylation, and compared with the results
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
62
for a standard sample. The standard NIF1 (STD) was
obtained from the cells of Example 1 that were adapted
to suspension growth as set forth in Example 2, but
nourished on media containing bovine serum albumin.
Sialylation/glycosylation profiles were examined via the
methods set forth in Webster et al., Xenobiotica,
29(11), pp. 1141-55 (1999) as follows.
A. Desialylation of NIF1
The procedure for desialylation of NIF1 uses acid
hydrolysis to release the sialic acid. NIF1 samples (2
mg/ml) were desialylated by the addition of 0.2N HCl
(1:1 V/V) and heated at 80°C for 1 hour. The sialic acid
liberated by the acid hydrolysis reactions is determined
using the thiobarbituric acid method developed by
Warren, J. Biol. Chem., 234, pp. 1971-5 (1959). The
incubation was terminated when no further increase in
free sialic acid was observed.
B. Total sialic acid determination
The sialic acid residues were released from NIF1
using acid hydrolysis (part A immediately above) and the
predominant sialic acid associated with the glycans of
the NIF protein, 5-acetylneuramic acid (neu5ac), was
analyzed by ion chromatography with pulsed amperometric
detection. Purified NIF1 samples were diluted to a
concentration of 0.1 mg/nil. The sample (200 ~l) was
then mixed with 200 ~.~1 of 0.2N HC1 and heated at 80°C for
1 hour. The sample was then pooled in an ice bath for
10 minutes and centrifuged. The supernatant (20 ~.1),
containing 5-acetylneuramic acid, was analyzed using a
Dionex Carbopac (PA-10 4.6 x 250 mm column (guard column
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
63
4.6 x 50 mm Carbopac (PA-10) with a 0.2M NaOH and 0.05M
C2H30~Na mobile phase (flow rate = 0.7 ml/min, mobile
phase conditioned with a Dionex Ion Pac ATC-1 mobile
phase conditioner) and detection was carried out using a
Dionex ED40 detector using the optimized carbohydrate
waveform setting. The 5-acetylneuramic acid standard
has a retention time of 10 minutes under the above HPLC
conditions. A wash step was performed after the elution
of 5-acetylneuramic acid (0.2 M NaOH and 0.3 M C~H302Na
for 5 minutes) and the column was re-equilibrated for 30
minutes before the next injection. The data are
presented as a percentage of a control batch (i.e.,
standard sample of NIF1 (STD) prepared from Example 1
cells adapted to suspension but made in the presence of
bovine serum albumin) and an increase in the value
presented for sialylation represent an increase in the
amount of sialic acid on the NIF1 molecule. The total
sialylation data is presented in Table V.
C. Oligosaccharide charge profile assay
N-linked oligosaccharides are released from~NIF1
using the enzyme peptide-N-Glycosidase F (PNGase-F).
The released oligosaccharides are labeled with 2-
aminobenzamide (2-AB) and separated on anion exchange
chromatography. Purified NIF1 was first diluted to a
concentration of about 0.05 mg/ml; 50 ~1 of the diluted
NIF was denatured by the addition of 4 ~l of 50 (w/v)
SDS and 6 ~,l of 1.44M (3-meracaptoethanol. After setting
the sample aside for about 5 minutes, 20 ~,l of 7.5o NP-
40 and 30 ~tl of (100mM Na~HP04, lOmM EDTA-disodium, pH
7.6) buffer were added to the sample. Following this,
10 ~l of 1mU/ml PNGase-F was added and the sample was
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
64
stored in an incubator for 18-24 hours at 37°C. Released
oligosaccharides were separated from deglycosylated
protein by ethanol precipitation, the supernatant was
removed and dried. This dried sample containing
oligosacharides were labeled with 2-AB using a labeling
kit (5 ~,l of labeling reagent used to re-suspend sample,
Signal 2-AB labeling kit (Oxford Glycoscience, product
number K-404). At the end of the incubation excess
labeling reagent was removed by chromatography on a
hydrophilic membrane (supplied with the kit). The
reagent mixture was loaded onto the disk in acetonitrile
and excess reagent removed with sequential acetonitrile
and acetonitrile/water washes. 2-AB labeled
oligosaccharides were eluted from the disk with water
and dried. The oligosaccharides labeled with 2-AB were
then analyzed by anion exchange HPLC on a Glycosep C
HPLC column (100 x 4.6 mm, Oxford Glycosystems).
Oligosaccharides were eluted in a gradient from 800
water/20o acetonitrile to 800 250mM ammonium acetate pH
4.5/200 acetonitrile over 35 minutes, flow rate was 0.3
ml/min. Fluorescence detection was performed with an
excitation wavelength of 330 nm and an emission
wavelength of 420 nm. A typical chromatogram consists
of peak clusters with uncharged species eluting first
followed by mono-, di-, tri- and tetra- sialylated
cluster.
A NIF1 batch (STD) was designated for profiling
control. The sialylation profile data is presented in
Table V.
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
nvTnrtmT n o
NIF1 Pharmacokinetic Profile
The NIFs harvested from a number of different
bioreactor runs were tested for pharmacokinetic
5 clearance and half-life data, and compared with the
results for a standard sample (STD) of NIF1 obtained
from the cells of Example 1 that were adapted to
suspension growth as set forth in Example 2, but made in
the presence of animal protein (BSA). The results are
10 listed in the following table as compared with a
standard sample of NIF1.
A. Preparation of Animals
Jugular vein catheterized Fischer 344 rats were
prepared by inserting a cannula (0.58mm I.D., 0.9mm
15 O.D., polythene tubing, Portex Ztd.) into the jugular
vein and the cannula was exteriorized at the back of the
neck using classical veterinary techniques. During the
surgery rats were anaesthetized with 70 mg/kg ketamine
HC1 (Vetalar, Parke-Davis Veterinary; 100 mg/ml) /10
20 mg/kg xylazine (Rompun injection 20, Bayer) administered
as an intra peritoneal injection. Following surgery,
reversal of the xylazine was carried out using a 1 ml/kg
injection of 1 in 5 diluted Antisedan (Pfizer Animal
Health, atipamezole; 5 mg/ml) administered as a
25 subcutaneous injection. Analgesia was provided for the
duration of the experiment (Buprenorphine HCl, 0.1 ml of
a 1 in 4 dilution of Vetergesic (Reckitt and Colman; 0.3
mg/ml)) administered subcutaneously. Following a
recovery period of two days, rats were dosed into the
30 tail vein (bolus) with NIF1 at a dose level of 2 mg/kg
(doses were made up at a concentration of 2 mg/ml and
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
66
administered on a 1 ml/kg basis). The use of jugular
vein catheterized rats allowed for serial sampling and
two rats were used to determine the pharmacokinetic of
each batch of NIF1. Blood samples (50 ~l) were removed
using the indwelling cannula into heparinized tubes at
the following time points: Pre Dose, 0.25, 1, 2, 4, 8,
12, 24 and 48 hours. The blood samples were
centrifuged, the plasma removed and stored frozen for
subsequent analysis. Plasma samples were analyzed for
NIF1 using a Delfia immunoassay (Example 8C).
B. Isolated perfused rat liver preparations
The isolated perfused rat liver (IPRL) preparation
was carried out using the methodology detailed in
Gardner et al., Xenobiotica, 25, pp. 185-187 (1995).
Male rats selected at a weight of approximately 250 g
were anaesthetized (Intraval) and surgery performed to
cannulate the bile duct, hepatic portal vein and
superior vena cava.
A perfusate consisting of a pH 7.4 Krebs high
bicarbonate buffer (610) containing washed human red
cell (l30) and l00 (w/v) bovine serum albumin (260) was
perfused through the liver at a flow rate of 15 ml/min.
The perfusate was oxygenated using 950 oxygen/5o carbon
dioxide and enters the liver via the hepatic portal
vein, exiting via the vena cava. The IPRL was run in
recirculating mode using a total perfusate volume of
150 ml. A solution containing taurocholic acid (24
mg/ml) was infused at a rate of 1.33 ml/h for the
duration of the experiment to maintain bile flow.
Asialo NIF1 (0.25 mg, Example 7A) was administered to
the reservoir and perfusate samples were withdrawn from
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
67
the reservoir after 2, 5, 10, 15, 20, 30, 45, 60, 75 and
90 minutes. Bile was collected for the duration of the
experiment (0-90 minutes). For the competition studies
asialo NIF (0.25 mg) was co-administered with asialo
fetuin (10 mg, Sigma A1908). The perfusate samples were
centrifuged and the supernatant removed and stored
frozen for subsequent analysis. Bile samples were also
stored frozen for analysis. The NIF1 concentration in
IPRh perfusate and bile samples was determined using the
Delfia immunoassay detailed for plasma (Example 8C).
C. Analysis of plasma samples for NIF1
Plasma samples were analyzed using a Dissociation
Enhanced Lanthanide Fluorescence Immunoassay (Delfia).
The assay is a "sandwich non-competitive immunoassay"
using europium-labeled monoclonal anti-NIF antibody as
detection reagent. Polyclonal rabbit anti-NIF
antibodies are bound to plates coated with anti-rabbit
antibody. NIF in samples or standards then binds to the
polyclonal antibodies and finally europium-labeled
monoclonal antibody binds to another epitope on the
bound NIF. Europium is determined after addition of
"enhancement" solution. In order to perform this assay
5 ~,1 of plasma was required.
The assay range in plasma was 0.1-40 ~,g/ml. The
accuracy of this assay was evaluated and the cumulative
variations were 10.5, 3.4, 6.3o at 0.1, 3 and 40 ~g/ml,
respectively. The day to day performance of the assay
was monitored using quality control samples. Delfia
immunoassays are well characterized and have been
previously used to determine the plasma concentrations
of many protein containing molecules including;
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
68
interferons (Ronnblom et al., APMIS, 105, pp. 531-536
(1997)), apolipoprotein D (Knipping et al., J.
Immunological Methods, 202, pp. 85-95 (1997)),
thyroglobulin (Dai et al., Clinical Biochemistry, 29,
pp. 461-465 (1996) and lipoprotein lipase (Wicker et
al., J. Immunological Methods, 192, pp. 1-11 (1996)).
D. Pharmacokinetic analysis
The pharmacokinetics were determined using standard
algorithms. The elimination rate constant (Kel) was
determined from the plot of concentrations in plasma
verses time using linear regression of the log (plasma
concentration) versus time. The half-life determined
using the following equation: Half-life = (Ln2)/Kel-area
under the plasma concentration time curve (AUC) was
calculated from time zero to the last data point using
the linear trapezoidal rule. The AUC was extrapolated
to infinity using the elimination rate constant.
Clearance was calculated using the relationship: Dose
divided by AUC ~o_~~. Volume of distribution was
calculated by the relationship: Clearance divided by the
elimination rate constant.
The hepatic extraction value in the IPRL was
calculated by dividing the clearance value obtained in
the IPRL by the IPRL flow rate (15 mllmin) and
multiplying this value by 100. The results of the
clearance and half-life determination are set forth in
Table V below. The NIFl titer was measured by the
method set forth above in the description.
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
69
TABLE V
Sialylation ClearanceHalf-
Profiles Life
Run Day TiterTotalZero Mono -Di Tri Tetra Tri
+ (ml/min/(h)
(Units(~) kg)
/ml) Tetra
STD 1.0 100 5.8 14.3 21.1 26.3 32.5 58.8 0.08 11.5
A 10 3.8 85.6 5.3 18.5 26.9 29.1 20.3 49.4 0.08 9.7
B 12 4.9 81.2 7.9 20.2 27.0 27.5 17.4 44.9 0.08 9.4
C 10 3.4 85.7 6.4 17.4 25.8 29.3 21.1 50.4 0.07 9.5
D 10 4.2 81.3 8.5 19.5 25.3 28.8 17.9 46.7 0.09 9.2
E 11 4.0 85.4 7.2 18.5 24.5 28.8 21.0 49.8 0.12 11.9
F 11 4.0 94.3 3.2 14.8 24.5 32.7 24.8 57.5 0.09 14.3
X
. Average 4.0 85.6 6.4 18.2 25.7 29.4 20.4 49.8 0.09 10.7
(Runs
A-
F)
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
1
266/066 SEQUENCE LISTING
<110> Pluschkell, Stefanie B.
Geldart, Roderick W.
Ho, Lewis
Koehler, Mark A.
Okediadi, Centy A.
Pias, Steven J.
Zhu, Marie M.
<120> PROCESS FOR THE PREPARATION OF NEUTROPHIL TNHIBITORY
FACTOR
<130> SUZANNE L. BIGGS: Corvas 266/066
<140>
<141>
<l60> 11
<170> PatentIn Ver. 2.0
<210> 1
<211> 825
<212> DNA
<213> Ancylostoma caninum
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
2
<220>
<221> CDS
<222> ( 1 ) . . ( 822 )
<400> 1
atg~ gag gcc tat ctt gtg gtc tta att gcc att get ggc ata get cat 48
Met Glu Ala Tyr Leu Val Val Leu Ile Ala Ile Ala Gly Ile Ala His
1 5 10 15
tcc aat gaa cac aac ctg agg tgc ccg cag aat gga aca gaa atg ccc 96
Ser Asn Glu His Asn Leu Arg Cys Pro Gln Asn Gly Thr Glu Met Pro
20 25 30
ggt ttc aac gac tog att agg ctt caa ttt tta gca atg cac aat ggt 144
Gly Phe Asn Asp Ser Ile Arg Leu Gln Phe Leu A1a Met His Asn Gly
35 40 45
tac aga tca aaa ctt gcg cta ggt cac atc agc ata act gaa gaa tcc 192
Tyr Arg Ser Lys Leu Ala Leu Gly His Ile Ser Ile Thr Glu Glu Ser
50 55 60
gaa agt gac gat gat gac gat ttc ggt ttt tta ccc gat ttc get cca 240
Glu Ser Asp Asp Asp Asp Asp Phe Gly Phe Leu Pro Asp Phe A1a Pro
65 70 75 80
agg gca tcg aaa atg aga tat ctg gaa tat gac tgt gaa get gaa aaa 288
Arg Ala Ser Lys Met Arg Tyr Leu Glu Tyr Asp Cys Glu Ala Glu Lys
85 90 95
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
3
agc gcc tac atg tcg get aga aat tgc tcg gac agt tct tct cca cca 336
Ser Ala Tyr Met Ser Ala Arg Asn Cys Ser Asp Ser Ser Ser Pro Pro
100 105 l10
gag ggc tac gat gaa aac aag tat att ttc gaa aac tca aac aat atc 384
Glu Gly Tyr Asp Glu Asn Lys Tyr Ile Phe Glu Asn Ser Asn Asn Ile
115 120 125
agt gaa get get ctg aag gcc atg atc tcg tgg gca aaa gag get ttc 432
Ser Glu Ala Ala Leu Lys Ala Met Ile Ser Trp Ala Lys Glu Ala Phe
130 135 140
aac cta aat aaa aca aaa gaa gga gaa gga gtt ctg tac cgg tcg aac 480
Asn Leu Asn Lys Thr Lys Glu Gly Glu G1y Val Leu Tyr Arg Ser Asn
145 150 155 160
cac gac ata tca aac ttc get aat ctg get tgg gac gcg cgt gaa aag 528
His Asp Ile Ser Asn Phe Ala Asn Leu Ala Trp Asp Ala Arg Glu Lys
165 170 175
ttt ggt tgc gca gtt gtt aac tgc cct ttg gga gaa atc gat gat gaa 576
Phe Gly Cys Ala Val Val Asn Cys Pro Leu G1y G1u Ile Asp Asp Glu
180 185 190
acc aac cat gat gga gaa acc tat gca aca acc atc cat gta gtc tgc 624
Thr Asn His Asp Gly Glu Thr Tyr Ala Thr Thr I1e His Val Val Cys
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
4
195 200 205
cac tac ccg aaa ata aac aaa act gaa gga cag ccg att tac aag gta 672
His Tyr Pro Lys Ile Asn Lys Thr G1u Gly Gln Pro Ile Tyr Lys Val
210 215 220
ggg aca cca tgc gac gat tgc agt gaa tac aca aaa aaa gca gac aat 720
Gly Thr Pro Cys Asp Asp Cys Ser Glu Tyr Thr Lys Lys Ala Asp Asn
225 230 235 240
acc acg tct gcg gat ccg gtg tgt att ccg gat gac gga gtc tgc ttt 768
Thr Thr Sex Ala Asp Pro Val Cys Ile Pro Asp Asp Gly Val Cys Phe
245 250 255
att ggc tcg aaa gcc gat tac gat agc aag gag ttt tat cga ttc cga 816
Ile Gly Ser Lys Ala Asp Tyr Asp Ser Lys Glu Phe Tyr Arg Phe Arg
260 265 270
825
gag tta tga
G1u Leu
<210> 2
<211> 274
<212> PRT
<213> Ancylostoma caninum
<400> 2
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
Met Glu Ala Tyr Leu Val Val Leu Ile Ala Ile Ala Gly Ile Ala His
1 5 10 15
Ser Asn Glu His Asn Leu Arg Cys Pro Gln Asn Gly Thr Glu Met Pro
20 25 30
Gly Phe Asn Asp Ser Ile Arg Leu Gln Phe Leu Ala Met His Asn Gly
35 40 45
Tyr Arg Ser Lys Leu Ala Leu Gly His Ile Ser Tle Thr Glu Glu Ser
50 55 60
Glu Ser Asp Asp Asp Asp Asp Phe Gly Phe Leu Pro Asp Phe A1a Pro
65 70 75 80
Arg Ala Ser Lys Met Arg Tyr Leu Glu Tyr Asp Cys Glu Ala Glu Lys
85 90 95
Ser Ala Tyr Met Ser Ala Arg Asn Cys Ser Asp Ser Ser Ser Pro Pro
100 105 110
Glu Gly Tyr Asp Glu Asn Lys Tyr I1e Phe Glu Asn Ser Asn Asn Ile
115 120 125
Ser Glu Ala Ala Leu Lys Ala Met Ile Ser Trp Ala Lys Glu A1a Phe
130 135 140
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
6
Asn Leu Asn Lys Thr Lys Glu Gly Glu Gly Val Leu Tyr Arg Ser Asn
145 150 155 160
His Asp I1e Ser Asn Phe Ala Asn Leu Ala Trp Asp Ala Arg Glu Lys
165 170 175
Phe Gly Cys Ala Val Val Asn Cys Pro Leu Gly Glu Ile Asp Asp Glu
180 185 190
Thr Asn His Asp Gly Glu Thr Tyr Ala Thr Thr Ile His Val Val Cys
195 200 205
His Tyr Pro Lys Ile Asn Lys Thr Glu G1y Gln Pro Ile Tyr Lys Val
210 215 220
Gly Thr Pro Cys Asp Asp Cys Ser Glu Tyr Thr Lys Lys Ala Asp Asn
225 230 235 240
Thr Thr Ser Ala Asp Pro Val Cys Ile Pro Asp Asp Gly Val Cys Phe
245 250 255
Ile Gly Ser Lys Ala Asp Tyr Asp Ser Lys Glu Phe Tyr Arg Phe Arg
260 265 270
Glu Leu
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
7
<210> 3
<211> 257
<212> PRT
<213> Ancylostoma caninum
<220>
<221> PEPTIDE
<222> (1)..(257)
<400> 3
Asn Glu His Asn Leu Arg Cys Pro Gln Asn Gly Thr Glu Met Pro Gly
1 5 10 15
Phe Asn Asp Ser Ile Arg Leu Gln Phe Leu Ala Met His Asn Gly Tyr
20 25 30
Arg Ser Lys Leu Ala Leu Gly His Ile Ser Ile Thr Glu Glu Ser Glu
35 40 45
Ser Asp Asp Asp Asp Asp Phe Gly Phe Leu Pro Asp Phe Ala Pro Arg
50 55 60
Ala Ser Lys Met Arg Tyr Leu Glu Tyr Asp Cys Glu Ala G1u Lys Ser
65 70 75 80
Ala Tyr Met Ser Ala Arg Asn Cys Ser Asp Ser Ser Ser Pro Pro Glu
85 90 95
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
8
Gly Tyr Asp Glu Asn Lys Tyr Ile Phe Glu Asn Ser Asn Asn Ile Ser
100 105 110
Glu Ala Ala Leu Lys Ala Met Ile Ser Trp Ala Lys Glu Ala Phe Asn
115 120 125
Leu Asn Lys Thr Lys Glu Gly Glu Gly Val Leu Tyr Arg Ser Asn His
130 135 140
Asp Ile Ser Asn Phe Ala Asn Leu Ala Trp Asp A1a Arg Glu Lys Phe
l45 150 155 160
Gly Cys Ala Val Val Asn Cys Pro Leu Gly Glu Ile Asp Asp Glu Thr
165 170 l75
Asn His Asp Gly Glu Thr Tyr Ala Thr Thr Ile His Val Val Cys His
180 185 190
Tyr Pro Lys Ile Asn Lys Thr Glu Gly Gln Pro Ile Tyr Lys Val Gly
195 200 205
Thr Pro Cys Asp Asp Cys Ser Glu Tyr Thr Lys Lys Ala Asp Asn Thr
210 215 220
Thr Ser Ala Asp Pro Val Cys I1e Pro Asp Asp Gly Val Cys Phe Ile
225 230 235 240
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
9
Gly Ser Lys Ala Asp Tyr Asp Ser Lys Glu Phe Tyr Arg Phe Arg Glu
245 250 255
Leu
<210> 4
<211> 8
<212> PRT
<213> Ancylostoma caninum
<400> 4
Lys Ala Met Ile Ser Trp A1a Lys
1 5
<210> 5
<211> 8
<212> PRT
<213> Ancylostoma caninum
<400> 5
Glu Phe Tyr Arg Phe Arg Glu Leu
1 5
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
<2l0> 6
<211> 11
<212> PRT
<213> Ancylostoma caninum
<400> 6
Asp Ile Ser Asn Phe A1a Asn Leu Ala Trp Asp
1 5 10
<210> 7
<211> 30
<212> PRT
<213> Ancylostoma caninum
<400> 7
Asp Glu Asn Lys Tyr Ile Phe Glu Asn Ser Asn Asn Ile Ser Glu Ala
1 5 10 15
Ala Leu Lys Ala Met I1e Ser Trp Ala Lys Glu A1a Phe Asn
25 30
<210> $
<211> 885
<212> DNA
<213> Partially Ancylostoma caninum
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
11
<400> 8
ggcgaattca ccatggaggc ctatcttgtg gtcttaattg ccattgctgg catagctcat 60
tccaatgaac acaacctgag gtgcccgcag aatggaacag aaatgcccgg tttcaacgac 120
tcgattaggc ttcaattttt agcaatgcac aatggttaca gatcaaaact tgcgctaggt l80
cacatcagca taactgaaga atccgaaagt gacgatgatg acgatttcgg ttttttaccc 240
gatttcgctc caagggcatc gaaaatgaga tatctggaat atgactgtga agctgaaaaa 300
agcgcctaca tgtcggctag aaattgctcg gacagttctt ctccaccaga gggctacgat 360
gaaaacaagt atattttcga aaactcaaac aatatcagtg aagctgctct gaaggccatg 420
atctcgtggg caaaagaggc tttcaaccta aataaaacaa aagaaggaga aggagttctg 480
taccggtcga accacgacat atcaaacttc gctaatctgg cttgggacgc gcgtgaaaag 540
tttggttgcg cagttgttaa ctgccctttg ggagaaatcg atgatgaaac caaccatgat 600
ggagaaacct atgcaacaac catccatgta gtctgccact acccgaaaat aaacaaaact 660
gaaggacagc cgatttacaa ggtagggaca ccatgcgacg attgcagtga atacacaaaa 720
aaagcagaca ataccacgtc tgcggatccg gtgtgtattc cggatgacgg agtctgcttt 780
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
12
attggctcga aagccgatta cgatagcaag gagttttatc gattccgaga gttatgaata 840
agtcgagacg tataaagaag ccaaggcaac gtaagcgaga atttc 885
<210> 9
<211> 1845
<212> DNA
<213> Ancylo toma caninum
<400> 9
agttctcaga tagtcacagt agcccttctt ttcattgtac acaagtgaag atgggcactt 60
catggtagtc gcgactcctt cattacagta aacatagtcg gatgtgcatc ccaacgaata 120
gtagccattc tgctttgtct tgcagtcaac ggtcttcgca atttgtggta cagcagcagg 180
agccggaggc tgcatcgctg gagctgctgg tggagctggc acaacagaag ccggaggtgg 240
agcaaccagt tcaggcgtgc agttctcagg atagtcgoag tagcccttct tctcatggta 300
tacaagtgaa gaatggaggc ctatcttgtg gtcttaattg ccattgctgg catagctcat 360
tccaatgaac acaacctgag gtgcccgcag aatggaacag aaatgcccgg tttcaacgac 420
tcgattaggc ttcaattttt agcaatgcac aatggttaca gatcaaaact tgcgctaggt 480
cacatcagca taactgaaga atccgaaagt gacgatgatg acgatttcgg ttttttaccc 540
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
13
gatttcgctc caagggcatc gaaaatgaga tatctggaat atgactgtga agctgaaaaa 600
agcgcctaca tgtcggctag aaattgctcg gacagttctt ctccaccaga gggctacgat 660
gaaaacaagt atattttcga aaactcaaac aatatcagtg aagctgctct gaaggccatg 720
atctcgtggg caaaagaggc tttcaaccta aataaaacaa aagaaggaga aggagttctg 780
taccggtcga accacgacat atcaaacttc gctaatctgg cttgggacgc gcgtgaaaag 840
tttggttgtc gcagttgtta actgcccttt gggagaaatc gatgatgaaa ccaaccatga 900
tggagaaacc tatgcaacaa ccatccatgt agtctgccac tacccgaaaa taaacaaaac 960
tgaaggacag ccgatttaca aggtagggac accatgcgac gattgcagtg atacacaaaa 1020
aaagcagaca ataccacgtc tgcggatccg gtgtgtattc cggatgacgg agtctgcttt 1080
attggctcga aagccgatta cgatagcaag gagttttatc gattccgaga gttatgaata 1140
agtcgagacg tataaagaag ccaaggcaac gtaagcgagc aagtctcgaa gacgatggag 1200
tcagcgaaag aggcggctgc caaagttggc gagcaggtgt cagatttttt ccaagggaac 1260
ccattttcca cgcctgtggg ccgcaagata gaacttgcca cgaacgcttc gattcttgca 1320
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
14
ctgagaattg gggtttgaac atggaaatct gtgatttcgt caataacact gaggacggtg 1380
ccaaagatgc tgtacgggct attcgcaaac gtctgcacac aaatatgtgt aagaataacg 1440
caatcgtcat gtacacatta acggtgctgg agacgtgcgt gaagaactgt ggccataatt 1500
tccacgtgct cgtatgttcc aaggactttg tgcaggattt ggtgaagttg atcggctcga 1560
agttcgatac gcctcagatt attcacgagc gtgtattgtc acttattcag gcttgggcag 1620
atgcattccg caatcaacca gatcttcagg gagtcgtaca ggtctatgaa gaacttgtta 1680
gtaagggggt tacattccct gcaactgatc tagacgctat ggcacctata ctaacaccaa 1740
aacaaacagt cttcactgag ccaaaggcat caacggctgt tccttcgcag tcaggtggag 1800
gacctagtta cgaggtggtc agccaaccag atggtccaat ttact 1845
<210> 10
<211> 43
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
sequences from cloning vectors
CA 02420071 2003-02-19
WO 02/16584 PCT/USO1/25733
<400> 10
ctgcagtcac cgtccttgac acaagcttga tatcgaattc acc 43
<210> 11
<211> 59
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Synthetic
sequences from cloning vectors
<400> 1l
ataagtcgag acgtataaag aagccaaggc aacgtaagcg agaattcctg cagcccggg 59
1