Note: Descriptions are shown in the official language in which they were submitted.
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
SYSTEMS AND PROCESSES FOR REMOVAL
OF POLLUTANTS FROM A GAS STREAM
Field of the Invention
The invention relates to the systems and processes for removal of pollutants,
such as
oxides of sulfur, oxides of nitrogen, oxides of carbon, totally reduced
sulfides, fly ash,
mercury compounds, and elemental mercury from gases generated from the burning
of fossil
fuels and other process gases with electronic control of operational
parameters such as,
differential pressure across the system, gas temperature, and removal
efficiency. The systems
and processes of the invention employ oxides of manganese as the primary
sorbent to effect
removal of pollutants, such as oxides of sulfur and/or oxides of nitrogen, and
may further
employ other sorbent materials and chemical additives separately and in
conjunction with
oxides of manganese to effect the removal of other target pollutants, e.g.,
using alumina to
remove mercury.
Background of the Invention
During combustion of fuels that contain sulfur compounds, oxides of sulfur
(SOx),
such as sulfur dioxide (S02), and sulfur trioxide (S03) are produced as a
result of oxidation of
the sulfur. Some fuels may contain nitrogen compounds that contribute to the
formation of
oxides of nitrogen (NOx), which are primarily formed at high temperatures by
the reaction of
nitrogen and oxygen from the air used for the reaction with the fuel. These
reaction
compounds, SOx and NOx, are reported to form acids that may contribute to
"acid rain."
Federal and state regulations dictate the amount of these and other
pollutants, which may be
emitted. The regulations are becoming more stringent and plant operators are
facing greater
difficulties in meeting the regulatory requirements. Many technologies have
been developed
for reduction of SOx and NOx, but few can remove both types of pollutants
simultaneously in
a dry process or reliably achieve cost effective levels of reduction.
In the past to meet the regulatory requirements, coal-burning power plants
have often
employed a scrubbing process, which commonly uses calcium compounds to react
with SOx
to form gypsum. This waste product is normally discarded as a voluminous
liquid slurry in
an impoundment and ultimately is capped with a clay barrier, which is then
covered with
topsoil once the slurry is de-watered over time. Alternatively, some power-
plant operators
have chosen to burn coal that contains much lower amounts of sulfur to reduce
the quantities
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
2
of SOx emitted to the atmosphere. In the case of NOx, operators often choose
to decrease the
temperature at which the coal is burned. This in turn decreases the amount of
NOx produced
and therefore emitted; however, low temperature combustion does not utilize
the full heating
value of the coal, resulting in loss of efficiency.
Turbine plants normally use natural gas, wluch contains little or no sulfur
compounds,
to power the turbines, and therefore virtually no SOx is emitted. On the other
hand at the
temperature that the turbines are commonly operated, substantial NOx is
produced. In
addition to Selective Catalytic Reduction (SCR) processes for conversion of
NOx to nitrogen,
water vapor, and oxygen, which can be safely discharged, some operators choose
to reduce
the temperature at which the turbines are operated and thereby reduce the
amount of NOx
emitted. With lower temperatures the full combustion/heating value of natural
gas is not
realized, resulting in loss of efficiency. Unfortunately for these operators,
newer
environmental regulation will require even greater reduction of SOx and NOx
emissions
necessitating newer or more effective removal technologies andlor further
reductions in
efficiency.
Operators of older coal-burning power plants are often running out of space to
dispose
of solid wastes associated with use of scrubbers that use calcium compounds to
form gypsum.
Operators of newer plants would choose to eliminate the problem from the
outset if the
technology were available. Additionally, all power plants, new and old, are
faced with
upcoming technology requirements of further reducing emissions of NOx and will
have to
address this issue in the near future. Thus, plants that currently meet the
requirements for
SOx emissions are facing stricter requirements for reduction of NOx for which
there has been
little or no economically feasible technology available.
The nitrogen oxides, which are pollutants, are nitric oxide (NO) and nitrogen
dioxide
(NOa) or its dimer (N2O4). The relatively inert nitric oxide is often only
removed with great
difficulty relative to NOZ. The lower oxide of nitrogen, nitrous oxide (N20),
is not considered
a pollutant at the levels usually found in ambient air, or as usually
discharged from air
emission sources. Nitric oxide (NO) does however; oxidize in the atmosphere to
produce
nitrogen dioxide (N02). The sulft~r oxides considered being pollutants are
sulfur dioxide
(S02) and sulfur trioxide (S03).
Typical sources of iutrogen and sulfur oxide pollutants are power plant stack
gases,
automobile exhaust gases, heating-plant stack gases, and emissions from
various industrial
process, such as smelting operations and nitric and sulfuric acid plants.
Power plant
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
emissions represent an especially formidable source of nitrogen oxides and
sulfur oxides, by
virtue of the very large tonnage of these pollutants and such emissions
discharged into the
atmosphere annually. Moreover, because of the low concentration of the
pollutants in such
emissions, typically 500 ppm or less for nitrogen oxides and 3,000 ppm or less
for sulfur
dioxide, their removal is difficult because very large volumes of gas must be
treated.
Of the few practical systems, which have hitherto been proposed for the
removal of
nitrogen oxides from power plant flue gases, all have certain disadvantages.
Various
methods have been proposed for the removal of sulfur dioxide from power plant
flue gases,
but they too have disadvantages. For example, wet scrubbing systems based on
aqueous
allcaline materials, such as solutions of sodium carbonate or sodium sulfite,
or slurries of
magnesia, lime or limestone, usually necessitate cooling the flue gas to about
SS° C in order
to establish a water phase. At these temperatures, the treated gas requires
repeating in order
to develop enough buoyancy to obtain an adequate plume rise from the stack.
U.S. Pat. No.
4,369,167 teaches removing pollutant gases and trace metals with a lime
slurry. A wet
scrubbing method using a limestone solution is described in U.S. Pat. No.
5,199,263.
Considerable work has also been done in an attempt to reduce NOx pollutants by
the
addition of combustion catalysts, usually organo-metallic compounds, to the
fuel during
combustion. However, the results of such attempts have been less successful
than staged
combustion. NOx oxidation to N2 is facilitated by asnmoiua, methane, et al.
which is not
effected by SOx is described in U.S. Pat. No. 4,112,053. U.S. Pat. No.
4,500,281 teaches the
limitations of organo-metallic catalysts for NOx removal versus staged
combustion. Heavy
metal sulfide with ammonia is described for reducing NOx in stack gases in
U.S. Pat. No.
3,981,971.
Many fuels, and particularly those normally solid fuels such as coal, lignite,
etc., also
contain substantial amounts of bound or fuel sulfur with the result that
conventional
combustion produces substantial amounts of SOx pollutants which are also
subject to
pollution control. It has generally been the opinion of workers in the art
that those conditions
employed in staged combustion, particularly two-stage rich-lean combustion for
NOx
reduction, will likewise lower the level of SOx emissions. However, it has
been found that
little or no reduction in SOx emissions can be obtained in a two-stage, rich-
lean combustion
process. Indeed, it has been found that the presence of substantial amounts of
sulfur in a fuel
also has a detrimental effect on NOx reduction in a two-stage, rich-lean
process.
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
4
Considerable effort has been expended to remove sulfur from normally solid
fuels,
such as coal, lignite, etc. Such processes include wet scrubbing of stack
gases from coal-fired
burners. However, such systems are capital intensive and the disposal of wet
sulfite sludge,
which is produced as a result of such scrubbing techniques, is also a problem.
Cost
inefficiencies result from the often-large differential pressures across a wet
scrubber removal
system; differential pressures in excess of 30 inches of water column (WC) are
not unusual.
Also, the flue gases must be reheated after scrubbing in order to send them up
the stack, thus
reducing the efficiency of the system. Both U.S. Pat. Nos. 4,102,982 and
5,366,710 describe
the wet scrubbing of SOx and NOx.
In accordance with other techniques, sulfur scavengers are utilized, usually
in
fluidized bed burners, to act as scavengers fox the sulfur and convert the
same to solid
compounds which are removed with the ash. The usual scavengers in this type of
operation
include limestone (calcium carbonate) and dolomite (magnesium-calcium
carbonate) because
of availability and cost. However, the burning techniques are complex and
expensive to
operate and control; and the burner equipment is comparatively expensive.
Dissolving coal
or like material in a molten salt compound is described in U.S. Pat. No.
4,033,113. U.S. Pat.
No. 4,843,980 teaches using alkali metal salt during the combustion of coal or
other
carbonaceous material with further efficiency by adding a metal oxide. A
sulfur scavenger
added upstream to a combustion zone is described in U.S. Pat. No. 4,500,281.
The combustion gas stream from a coal-burning power plant is also a major
source of
airborne acid gases, fly ash, mercury compounds, and elemental mercury in
vapor form. Coal
contains various sulfides, including mercury sulftde. Mercury sulfide reacts
to form elemental
mercury and SOx in the combustion boiler. At the same time other sulfides are
oxidized to
SOx and the nitrogen in the combustion air is oxidized to NOx. Downstream of
the boiler, in
the ducts and stack of the combustion system, and then in the atmosphere, part
of the
elemental mercury is re-oxidized, primarily to mercuric chloride (HgCl2). This
occurs by
reactions with chloride ions or the like normally present in combustion
reaction gases flowing
through the combustion system of a coal-burning power plant.
Many power plants emit daily amounts of up to a pound of mercury, as elemental
mercury and mercury compounds. The concentration of mercury in the stream of
combustion
gas is about 4.7 parts per billion (ppb) or 0.0047 parts per million (ppm).
Past efforts to
remove mercury from the stream of combustion gas, before it leaves the stack
of a power
plant, include: (a) injection, into the combustion gas stream, of activated
carbon particles or
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
S
particulate sodium sulfide or activated alumina without sulfur; and (b)
flowing the
combustion gas stream through a bed of activated particles. When activated
carbon particle
injection is employed, the mercuric chloride in the gas stream is removed from
the gas stream
in a bag house and collected as part of a powder containing other pollutants
in particulate
form. Mercuric chloride and other particulate mercury compounds that may be in
the gas
stream can be more readily removed from the gas stream at a bag house than can
elemental
mercury. Activated carbon injection for mercury removal along with an
activated particle
bed is described in U.S. Pat. No. 5,672,323.
When the gas stream flows through a bed of activated carbon particles, mercury
compounds are adsorbed on the surface of the activated carbon particles and
remain there.
Elemental mercury, usually present in vapor form in combustion gases, is not
adsorbed on the
activated carbon to any substantial extent without first being oxidized into a
compound of
mercury. U.S. Pat. No. 5,607, 496 teaches the oxidation of mercury and
subsequent
absorption to particles and utilization of alumina are described therein.
Sodium sulfide particle injection can be utilized to form mercuric sulfide
(HgS),
which is more readily removable from the gas stream at a bag house than is
elemental
mercury. The conversion of mercury to a sulfide compound with subsequent
capture in a
dust separator is detailed in U.S. Pat. No. 6,214,304.
Essentially, all of the above techniques create solid waste disposal problems.
The
solids or particulates, including fly ash, collected at the bag house and the
spent activated
carbon removed from the bed of activated carbon, all contain mercury compounds
and thus
pose special problems with respect to burial at landfills where strictly
localized containment
of the mercury compounds is imperative. The concentration of mercury compounds
in
particulates or solids collected from a bag house is relatively minute;
therefore, a very small
quantity of mercury would be dispersed throughout relatively massive volumes
of a landfill,
wherever the bag house solids or paxticulates are dumped. Moreover, with
respect to
activated carbon, that material is relatively expensive, and once spent
activated carbon
particles are removed from an adsorbent bed, they cannot be easily regenerated
and used
again.
In the activated alumina process, mercury compounds in the gas stream can be
adsorbed and retained on the surface of activated particles, but much of the
elemental
mercury will not be so affected. Thus elemental mercury in the combustion gas
stream is
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
6
oxidized to form mercury compounds (e.g. mercuric chloride), and catalysts are
employed to
promote the oxidation process. However, such processes do not capture SOX and
NOX.
The use of oxides of manganese to remove sulfur compounds from gas streams is
known in the art. Oxides of manganese are known to form sulfates of manganese
from SOX
and nitrates of manganese from NOX when contacted with a gas containing these
pollutants.
U.S. Pat. No. 1,851,312 describes an early use of oxides of manganese to
remove sulfur
compounds from a combustible gas stream. U.S. Pat. No. 3,150,923 describes a
dry bed of
oxides of manganese to remove SOX. A wet method to remove SOX with oxides of
manganese is described in U.S. Pat. No. 2,984,545. A special filter
impregnated with
manganese oxide to remove totally reduced sulfur compounds is described in
U.S. Pat. No.
5,112,796. Another method in U.S. Pat. No. 4,164,545 describes using an ion
exchange resin
to trap the products of manganese oxide and SOX and NOX. The use of certain
types of
oxides of manganese to remove SOx is disclosed U.S. Pat. Nos. 3,723,598 and
3,898,320.
Some of the known methods of bringing oxides of manganese in contact with a
gas stream,
i.e., sprayed slurries, beds of manganese ore or special filters, have been
cumbersome.
Although the prior art teaches the use of oxides of manganese to remove SOX
and/or NOX,
they do not teach an adaptable system or process that can capture SOX and/or
NOX and other
pollutants with oxides of manganese and monitor and adjust system operational
parameters,
such as differential pressure, to provide real-time system control.
Bag houses have traditionally been used as filters to remove particulates from
high
volume gas streams. U.S. Pat. No. 4,954,324 describes a bag house used as a
collector of
products generated through the use of ammonia and sodium bicarbonate to remove
SOX and
NOx from a gas stream. U.S. Pat. No. 4,925,633 describes a bag house as a site
of reaction for
SOx and NOXwith the reagents, ammonia and alkali. U.S. Pat. No. 4,581,219
describes a bag
house as a reactor for highly efficient removal of SOX only with a calcium-
based reagent and
alkaline metal salt. Although these prior art discloses and teach the use of
bag houses for
removal of particulates and as a reaction chamber, they do not teach the use
of bag houses in
an adaptable system capable of monitoring and adjusting system operational
parameters, such
as differential pressure, to capture SOX and/or NOX and other pollutants with
oxides of
manganese.
In view of the aforementioned problems of known processes for removal of SOX,
NOx, mercury compounds, and elemental mercuxy as well as other pollutants from
combustion gases, process gases, and other industrial waste gases, it would be
desirable to
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
7
provide a dry process for removal of SOX and NOX as well as other pollutants
from a gas
stream. It is further desirable to have a dry removal process that eliminates
the environmental
impacts of the disposal of large volumes of mercury containing solids and
particulates and
significant amounts of gypsum generated during SOX wet removal processes.
Wet removal processes can result in significant differential pressures across
a removal
system. Differential pressures above 30 inches of water column have been
observed in wet
removal processes. Such large differential pressures are costly because
significant energy
must be expended to counter the differential pressure and provide a waste gas
stream with
sufficient energy to flow up and out of a stack. A system and process that can
accomplish
pollutant removal with minimal or controlled differential pressure across the
system therefore
would be desirable and cost effective for most industry sectors processing or
emitting
significant amounts of combustion gases, process gases, and other industrial
gases.
The calcium compounds utilized in SOX wet scrubbing methods form gypsum in the
process. They are purchased and consumed in significant quantities and once
gypsum is
formed the calcium compounds cannot be recovered, at least not cost-
effectively. Thus, it
would be desirable to have a removal method employing a sorbent that not only
can remove
pollutants from a gas stream but that can be regenerated, recovered, and then
recycled or
reused for removal of additional pollutants from a gas stream.
To realize such a system and process, it would need to incorporate process
controls
and software that can monitor and adjust operational parameters from computer
stations
onsite or at remote locations through interface with a sophisticated
electronics network
incorporating an industrial processor. This would allow a technician to
monitor and adjust
operational parameters in real-time providing controls of such operational
parameters as
system differential pressure and pollutant capture rates or removal
efficiencies. Such a
network would be desirable for its real-time control and off site
accessibility.
In light of increased energy demand and recent energy shortages, it would be
desirable to be able to return to operational utility idled power plants that
have been
decormnissioned because their gypsum impoundments have reached capacity. This
could be
accomplished with retrofits of a system employing a regenerable sorbent in a
dry removal
process that does not require the use of calcium compounds. Such a system
would also be
readily adapted and incorporated into new power plants that may be coming on
line. Utility
plants and independent power plants currently in operation could readily be
retrofitted with
such a system. Further, such a system could be of significant value in
enabling emissions
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
8
sources to comply with emission standards or air quality permit conditions.
With the
reductions in emissions of pollutants such as NOx and SOx, marketable
emissions trading
credits could be made available or non-attainment areas for state or national
ambient air
quality standards may be able to achieve attainment status. Such scenarios
would allow for
development in areas where regulatory requirements previously prohibited
industrial
development or expansion.
The systems and processes of the present invention in their various
embodiments can
achieve and realize the aforementioned advantages, objectives, and desirable
benefits.
Summary of the Invention
The invention is directed to an adaptable system for wet removal and
combination
wet and dry removal of SOx and/or NOx andlor other pollutants from gases and
to processes
employing the system.
In an embodiment of the invention the adaptable system for wet removal of
target
pollutants from gases with minimal differential pressure across the system is
comprised of at
least one reaction zone which is a wet scrubber. The wet scrubber is supplied
with an acidic
aqueous slurry of a sorbent of regenerable oxides of manganese and is
configured far
introduction of a gas containing at least one target pollutant at a
temperature below the
boiling point of the slurry. The gas is contacted with the sorbent for a time
sufficient to effect
capture of the target pollutant at a targeted capture rate set point for the
target pollutant. The
gas is substantially stripped of the target pollutant through the formation of
a reaction product
of the target pollutant and the oxides of manganese. The reaction zone is
further configured
to allow the gas to be vented from the reaction zone. Differential pressure
across the system
is regulated so that any differential pressure across the system is no greater
than a
predetermined level.
The system may have a single wet scrubber, or multiple wet scrubber in series
for
removal of target pollutants. In a dual stage removal system, the two reaction
zones of the
system may be both wet scrubbers, a wet scrubber followed by a dry scrubber,
or a dry
scrubber followed by a wet scrubber.
In another embodiment of the invention, the system is utilized in processes
the
removal of target pollutants from a gas stream. Gas containing a target
pollutant is
introduced into the reaction zone of the system. The gas is contacted with the
sorbent in the
sorbent slurry of the system for a time sufficient to effect the capture of
the target pollutant at
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
9
a targeted capture rate set point for the target pollutant through the
formation of a reaction
product of the target pollutant and oxides of manganese to substantially strip
the gas of the
target pollutant;. The gas is vented gas from the reaction zone. These
processes can be
carried in single reaction zone or in multiple reaction zones of the system.
In another embodiment of the invention a process for the regeneration of
oxides of
manganese from a solution containing sulfate and nitrate anions and manganese
cations
formed when the reaction product of the removal of SOx and NOx from a gas
stream with a
sorbent of oxides of manganese, comprising the steps of
A. providing first and second anion exchangers having an anion exchange resin
loaded therein, the anion exchange resin having chloride in the exchange
position on the
resin;
B. passing a solution containing sulfate and nitrate anions through the first
anion
exchanger to elute the chloride to form manganese chloride while capturing the
sulfate anion
on the resin;
C. passing the solution containing nitrate anions through the second anion
exchanger to elute the chloride to form manganese chloride while capturing the
nitrate anion
on the resin;
D. adding a soluble carbonate or hydroxide compound to the solution to
precipitate manganese carbonate or manganese hydroxide;
D. separating the manganese carbonate or manganese hydroxide from the
solution; and
E. heating the manganese carbonate or manganese hydroxide to form regenerated
oxides of manganese.
Brief Description of the Drawings
Figure 1 is a schematic block diagram showing a system according to the
invention.
Figure 2 is a schematic block diagram showing a system according to the
invention.
Figure 3 is a schematic block diagram showing a system according to the
invention.
Figure 4 is a block diagram showing a system according to the invention.
Figure 5 is a block diagram showing a system according to the invention.
Figure 6 is a perspective view of a commercially available bag house.
Figure 7 is an end elevation view of a commercially available bag house.
Figure ~ is a top plan view of a commercially available bag house.
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
Figure 9 is a side elevation view of a commercially available bag house.
Figure 10 is a sectional view of an inverted bag house according to the
invention.
Figure 11 is a top plan view of an inverted bag house according to the
invention.
Figure 12 is a flow diagram of a bag house reactor according to the invention.
5 Figure 13 is a block diagram of a system according to the invention.
Figure 14 is a block diagram of a system according to the invention.
Figure 15 is a block diagram of a system according to the invention.
Figure 16 is a flow diagram an electrouc control system useful in the
invention.
Figure 17 is electronic control panel display.
10 Figure 18 is electronic control panel display.
Figure 19 is electronic control panel display.
Figure 20 is a block diagram of a control sub-element according to the
invention for
regulating differential pressure.
Figure 21 is a control sub-element according to the invention for control of
SOX or
NOX capture rate or sorbent feed rate.
Figure 22 is a control sub-element according to the invention for control of
bag house
gas inlet temperature.
Figure 23 is a control sub-element according to the invention for control of
variable
venturi position(s).
Figure 24 is a control sub-element according to the invention for control of
SOX or
NOX capture rate, differential pressure, and sorbent feed rate.
Figure 25 is a control sub-element according to the invention for control of
SOX or
NOX capture rate, differential pressure, sorbent feed rate, and variable
venturi position.
Figure 26 is a block diagram of a system and process according to the
invention.
Figure 27 is a block diagram of a system and process according to the
invention.
Figure 28 is a block diagram of system according to the invention.
Figure 29 is a graph plotting NOX values over time.
Figure 30 is a graph plotting SOX values over time.
Detailed Description of the Invention
The invention relates to systems and processes for removal of SOX and/or NOX
as
well as other pollutants, from a gas stream. In the invention, gas containing
SOX ald/or NOX
is introduced into a first reaction zone where the gas is contacted with a
sorbent of
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
11
regenerable oxides of manganese and/or regenerated oxides of manganese. The
sorbent may
interact with the pollutants in a gas stream as a catalyst, a reactant, an
absorbent or an
adsorbent. The oxides of manganese react with the SOx and the NOx to form,
respectively,
sulfates of manganese and nitrates of manganese.
"Nitrates of manganese" is used herein to refer to and include the various
forms of
manganese nitrate, regardless of chemical formula, that may be formed through
the chemical
reaction between NOx and the sorbent and includes hydrated forms as well.
Similarly, "sulfates of manganese" is used herein to refer to and include the
various
forms of manganese sulfate, regardless of chemical formula that may be formed
through the
chemical reaction between SOx and the sorbent and includes hydrated forms as
well.
"Target pollutant(s)" means the pollutant or pollutants that are targeted for
removal in
the system.
"Substantially stripped" means that a pollutant has been removed from a gas at
about
a targeted capture rate whether by interaction with a sorbent or physical
removal in a solid-
gas separator. With respect to pollutants removed by interaction with a
sorbent, it further
contemplates that removal up to a targeted capture rate for that pollutant may
be commenced
in a first reaction zone and completed in a subsequent reaction.
"Reacted sorbent" means sorbent that has interacted with one or more
pollutants in a
gas whether by chemical reaction, adsorption or absorption. The term does not
mean that all
reactive or active sites on the sorbent have been utilized since all such
sites may not actually
be utilized.
"Unreacted sorbent" means virgin sorbent that has not intereacted with
pollutants in a
gas.
Some of the reaction zones may also serve as solid-gas separators rendering
the gas
free of solids and particulates, such as sorbent, whether reacted or
unreacted, fly ash, and
mercury compounds, so as to allow the gas that is substantially stripped of
SOx and/or NOx
or other pollutants to be vented from the reaction zone and passed to another
reaction zone or
routed up a stack to be vented into the atmosphere. The solids and
particulates which include
the reacted and unreacted sorbent, fly ash, and the like, are retained within
reaction zones that
are solid-gas separators and may be subsequently removed for further
processing.
Reaction zones may be mufti-stage removal systems which would incorporate
additional reaction zones. The reaction zones utilized in single stage, dual
stage, or multi-
stage removal may be a fluidized bed, a pseudo-fluidized bed, a reaction
column, a fixed bed,
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
12
a pipe/duct reactor, a moving bed, a bag house, an inverted bag house, bag
house reactor,
serpentine reactor, and a cyclone/multiclone.
The gases that may be processed in the invention are most gases containing SOx
and/or NOx. Such gases may be generated by the combustion of fossil fuels in
power plants,
heating plants and various industrial processes, such as the production of
taconite pellets by
taconite plants, refineries and oil production facilities, gas turbines, and
paper mills.
Combustion for heating and other process steps at such facilities generate
waste or flue gases
that contain SOx and NOx in various concentrations, typically but not limited
to 500 ppm or
less for NOx and 3000 ppm or less for SOx. Further, the gases may contain
other removable
pollutants, such as fly ash, and mercury (Hg), as elemental Hg in vapor form
or mercury
compounds in particulate form, in small concentration, e.g., 0.0047 ppm (4.7
ppb). The gases
may further contain hydrogen sulfide and other totally reduced sulfides (TRS)
and other
pollutants. These gases may typically have temperatures typically ranging from
ambient
temperature to below the thermal decomposition temperatures) of nitrates of
manganese and
to below the thermal decomposition temperatures) of sulfates of manganese.
Gases
generally within this temperature range can be processed in the system of the
invention.
The primary sorbent useful in the invention are oxides of manganese, which may
be
found in manganese ore deposits or derived synthetically. Manganese compounds
of interest
occur in three different oxidation states of +2, +3, and +4; this gives rise
to a range of
multivalent phases, which provide oxides of manganese with a great diversity
of atomic
structures and thus mineral forms. Examples of these mineral forms include,
but are not
limited to, pyrolusite (Mn02), ramsdellite (Mn02), manganite (MnOOH or
MnZO3'HZO),
groutite (MnOOH), and vernadite (MnO2'nH20) to name a few. This is reported by
Jerry E.
Post in his article "Manganese Oxide Minerals: Crystal structures and economic
and
environmental significance," Proc. Nat'1. Acad. Sci, IJ.S.A., Vol. 96, pp.
3447-3454, March
1999, the disclosure of which is incorporated herein by this reference.
One of the most common of the various forms of oxides of manganese is
manganese
dioxide, Mn02. The pyrolusite form of this mineral is often the primary
mineral form in
manganese deposits. Pyrolusite is composed predominantly of the compound Mn02.
This
oxide of manganese exhibits at least two crystalline forms. One is the gamma
form, which is
nearly amorphous. The other is a beta form that exhibits pronounced
crystalline structure.
The term "oxides of manganese" as used herein is intended to refer and include
the various
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
13
forms of manganese oxide, their hydrated forms, and crystalline forms, as well
as manganese
hydroxide (e.g. Mn(OH)2), etc.
With reference to the removal of SOx and/or NOx, the relative capture or
removal
efficiencies of oxides of manganese may be understood by the below
calculations) of
loading rates. In order to assess the economics of the system and processes of
the invention,
it is necessary to determine the gas removal efficiencies of the sorbent. Gas
capture efficiency
based upon test results may be calculated by dividing weight of gas removed by
weight of
sorbent. This provides an approximate picture of system operations, but does
not account for
stoichiometry of the reactions or interference between reactive gases in a
multiple-gas
system. The stoichiometric gas capture ratio is described below.
For the purpose of this assessment the overall reactions believed to occur
between the
sorbent, oxides of manganese, and sulfur dioxide (S02) and nitric oxide (NO)
are shown
below, with molecular weights shown above each species.
87 64 151
Mn02 ~SOha~ + SO2 (gas) _> MnS04 ~S°ha~ (1 mole Mn02 captures 1
mole S02)
87 60 32 179
MnO2 ~S°~,a> + 2N0 (gash + 02 (gas) _> Mn(N03)z ~S°ha~ (1 mole
Mn02 captures 2 moles NO)
These reactions may occur in multiple steps. Molecular weights are shown above
each species. Based on these reactions, the theoretical maximum stoichiometric
gas capture
by weight of MnO2 sorbent is the ratio of the molecular weights of the
products versus the
reactants which is 73% for S02 or 69% for NO, for systems containing only one
reactive gas.
For a system containing two reactive gases, depending on reaction
characteristics, the
maximum stoichiometric gas capture will be lower for both gases. If reaction
speeds are
assumed to be equal for both reactive gases, maximum stoichiometric gas
capture for each
gas should be proportional to the percentage of each gas present.
For example, during a 48-hour test, two reactive gases, SOZ and NO were
present at
approximately 430 ppm and 300 ppm, respectively. Total weights of reactive
inlet gases
treated were:
SOa = 98.45 1b. NO = 47.02 1b. total =145.47 1b.
Therefore, SOZ and NO represented 67.7% and 32.3% respectively, ofreactive
gases present.
If the theoretical maximum stoichiometric gas capture for a single-gas system
is corrected to
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
14
these reactive gas weight proportions, the theoretical maximum percentage
capture for each
gas by Mn02 weight is:
502: (0.73 single-gas) x (0.67 for the 48-hr. test) = 0.489 = 48.9%
NO: (0.69 single-gas) x (0.323 for the 48-hr. test) = 0.223 = 22.3%
Therefore, the theoretical maximum weights of gases captured by 289 1b., for
example, of
sorbent for the 48-hour test would be:
502: (289 1b. Sorbent) x (0.489) =141.4 1b. S02
NO: (289 1b. Sorbent) x (0.323) = 98.35 1b. NO
Actual gas capture experienced in the 48-hour test was 23.94 1b. of S02 and
4.31 1b. of NO.
For the 2-gas system, stoichiometric gas capture was:
502: (23.94 1b. captured) / (141.4 1b. S02 possible) =16.9% (of theoretical
maximum)
NO: (4.31 1b. captured) / (64.41 1b. possible) = 6.69% (of theoretical
maximum)
Oxides of manganese, once reacted with SOx and NOx to form sulfates of
manganese
and nitrates of manganese respectively, can be regenerated. There are
essentially two general
methods of regeneration, thermal decomposition and chemical decomposition.
In thermal decomposition, the sulfates of manganese and/or nitrates of
manganese are
heated in an oxidizing atmosphere whereupon manganese oxide is formed and
nitrogen
dioxide and/or sulfur dioxide are desorbed and captured. The captured nitrogen
dioxide or
sulfur dioxide can be reacted with other chemicals to produce marketable
products.
In the chemical decomposition or regeneration of manganese oxide, the sulfates
of
manganese and/or nitrates of manganese are dissolved from the used sorbent in
a dilute acidic
aqueous slurry to which, after separation and recovery of the washed sorbent,
other
compounds such as alkali or hydroxides or carbonates may be added and
manganese oxide is
precipitated out of solution and removed. The solution, now free of oxides of
manganese,
can be routed on for further processing or production of marketable products
such as alkali or
ammonium sulfates and nitrates. The regeneration of manganese oxide and
production of
useful or marketable products through thermal or chemical decomposition is
further
discussed below.
In the process of regeneration, the regenerated oxides of manganese are in
particle
fonn and are defined by the chemical formula MnOx, where X is about 1.5 to
2Ø The
regeneration process may be engineered to yield oxides of manganese having a
particle size
ranging from 0.1 to 500 microns. Oxides of manganese in this range are useful
in the
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
invention. Preferably, the oxides of manganese will have a particle size of
less than 300
microns, and more preferably of less than 100 microns. The regenerable oxides
of
manganese and/or regenerated oxides of manganese are typically fine, powdery,
particulate
compounds.
Reactivity of dry sorbents may generally be related to its particle surface
area.
Particles or particulates all have weight, size, and shape, and in most cases
they are of
inconsistent and irregular shape. In the case of fine powders it is often
desirable to know how
much surface area a given quantity of powder exhibits, especially for
particles that are
chemically reactive on particle surfaces, or are used as sorbents, thickeners
or fillers.
10 (Usually measurements of surface area properties are done to compare
several powders for
performance reasons.) Particles may also have microscopic pores, cracks and
other features
that contribute to surface area.
The BET (Brunauer-Emmett-Teller) method is a widely accepted means for
measuring the surface area of powders. A powder sample is exposed to an inert
test gas, such
15 as nitrogen, at given temperature and pressures, and because the size of
the gas molecules are
known at those conditions, the BET method determines how much test gas covers
all of the
exterior surfaces, exposed pores and cracks with essentially one layer of gas
molecules over
all of the particles in the powder sample. Optionally, the analyst can use
other test gases such
as helium, argon or krypton; and can vary from 1 to 3 relative test pressures,
or more, for
better accuracy. From this, a measure of total surface area is calculated and
usually reported
in units of square meters of particle surface area per gram of powder sample
(m2/g).
Generally, coarse and smooth powders often range in magnitude from 0.001 to
0.1 m2/g of
surface area, and fine and irregular powders range from 1 to 1000 m2/g. Since
the
interactions between a sorbent and the pollutant occurs primarily at the
surface of sorbent
particle, surface area correlates with removal efficiency. The oxides of
manganese useful in
the invention are fine and irregular powders and thus may have a surface area
ranging from 1
to 1000 m2/g. Preferably the sorbent will have a surface area of greater than
15 m2/g, and
more preferably of greater than 20 m2/g.
With reference to Figure 1, a system according to the invention is illustrated
in block
diagram form. The system 10 may be seen as comprised of a feeder 20 and a
first reaction
zone 30 and a second reaction zone 3~. The feeder 20 would contain a supply of
sorbent of
regenerable oxides of manganese and/or regenerated oxides of manganese. The
feeder 20 is
configured to handle and feed oxides of manganese, which, upon regeneration,
are in particle
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
16
form and defined by the chemical formula MnOX where X is about 1.5 to 2Ø The
first
reaction zone 30 is configured for introduction of the sorbent in a gas
containing SOX and
NOX. In one embodiment, the first reaction zone 30 may be a section of
pipe/duct, possibly
configured as a fluidized bed, a pseudo-fluidized bed, a reaction column, a
fixed bed, a
pipe/duct reactor, a moving bed, a bag house, an inverted bag house, bag house
reactor,
serpentine reactor, and a cyclone/multiclone. The second reaction zone 38 a
fluidized bed, a
pseudo-fluidized bed, a reaction column, a fixed bed, a pipelduct reactor, a
moving bed, a bag
house, an inverted bag house, bag house reactor, serpentine reactor, and a
cyclone/multiclone. Preferably, the second reaction zone is a bag house, such
as
commercially available bag house, an inverted bag house according to the
invention, or a bag
house reactor according to the invention.
The gas containing SOx and NOx, or other pollutants, comes from a gas source
15
external to the system. The gas is introduced into the first reaction zone 30
and is contacted
with sorbent introduced into the first reaction zone 30 from the feeder 20 and
is contacted
with the sorbent for a time sufficient to primarily effect SOX capture at a
targeted SOX
capture rate. For purpose of discussion, and not wishing to be held to a
strict interpretation,
with respect to effecting a certain capture, it has been observed that oxides
of manganese can
more readily capture SOZ in a gas stream absent of NO, and also can more
readily capture NO
in a gas stream absent of SOZ, than when the gas stream contains both SO2 and
NO. SOX
capture tends to proceed at a much faster rate than NOX capture when the two
pollutants are
present in a gas stream.
The gas and sorbent may be introduced separately or commingled before
introduction
into a reaction zone. Once the gas and sorbent have been contacted for
sufficient time, the
SOx is captured by reacting with the sorbent to form sulfates of manganese to
substantially
strip the gas of SOx. The gas substantially stripped of SOX passes from the
first reaction zone
into the second reaction zone 38. The second reaction zone 38 is configured
for
introduction of sorbent and the gas substantially stripped of SOX. In the
second reaction zone
38, the gas is further contacted with sorbent for a time sufficient to
primarily effect NOX
capture at a targeted NOX capture rate. The NOX is captured by reacting with
the sorbent to
30 form nitrates of manganese to substantially strip the gas of NOX. The
second reaction zone 38
is further configured so that the gas which has been substantially stripped of
both SOX and
NOX is rendered free of reacted and unreacted sorbent. The gas may then be
vented from the
second reaction zone 38 to a stack 40 where the gas is released to the
atmosphere.
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
17
Differential pressure across the reactor system is regulated by a control sub-
element
(not shown in Figure 1) so that any differential pressure across the system is
no greater than a
pre-determined level. As is later described, the control sub-element may
control other system
parameters such as feeder rate, SOx and/or NOx capture rate, and the inlet gas
temperature
into the reaction zones. Thus, the system of the invention is highly adaptable
and, in another
embodiment, is generally comprised of a feeder 20, a first reaction zone 30, a
second reaction
zone 3~, and at least one control sub-element for regulating process
parameters.
In another embodiment of the invention, the system is comprised of a feeder 20
as
previously described and a modular reaction unit 60 comprised of at least
three
interconnected reaction zones. With reference to Figure 2, where the reaction
zones are three
intercomlected bag houses 62, 64, 66, the modular reaction unit may be
understood. The bag
houses 62, 64, 66 are connected so that a gas containing SOx and/or NOx can be
routed
through any one of the bag houses, any of the two bag houses in series, or all
of the at least
three bag houses in series or in parallel or any combination of series or
parallel. Each bag
house is separately connected to the feeder 20 and to the external gas source
15. Through
these connections, sorbent and gas can be introduced into each bag house where
SOx and
NOx capture can occur when the gas is contacted with sorbent for a time
sufficient to allow
formation of sulfates of manganese, nitrates of manganese, or both. The system
in this
embodiment may also include control sub-elements 50 (not shown) for regulating
various
process parameters. The reaction zones of the modular unit 60 are not limited
to bag houses
and may be any combination of reaction zones useful in the inventory. If the
bag houses are
operated independently of each other, then the section of pipe or duct
(pipe/duct) preceding
the bag house and that which is connected to an inlet of each bag house
conveys gas into each
bag house and is also configured as a first reaction zone 30, a pipe/duct
reactor, into which
gas containing SOx and NOx flows along with the sorbent. The gas is mixed with
the sorbent
in the pipe/duct reactor for a sufficient time to achieve SOx capture at a
targeted capture rate.
In this mode, the system operates as illustrated in Figure 1 with each bag
house 62, 64, 66
being a second reaction zone 3~ into which the gas that has been substantially
stripped of
SOx passes from the first reaction zone 30, pipe/duct reactor.
With reference to Figure 3, another embodiment of the invention is shown. In
this
embodiment, the system 10 is comprised of a feeder 20, and three bag houses
70, 76, and 7S,
a common conduit 73 and a diverter valve 74. Gas and sorbent are introduced
into the first
bag house 70 which serves as a first reaction zone of a two-staged SOx/NOx
removal system
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
18
where primarily SOX capture occurs. The gas substantially stripped of SOx then
passes from
the first bag house 70 into the common conduit 73. As shown in Figure 3, the
common
conduit 73 is Y-shaped, but may be of any shape that allows gas to flow from
the first bag
house 72 and to be directed to the second and third bag houses 76, 78 which
each function as
the second reaction zone of a two-staged SOX/NOX removal system.
In the Y-shaped common conduit 73 can be seen a diverter valve 74 illustrated
as a
dotted line at the fork of the "Y". The diverter valve 74 is positioned in the
cormnon conduit
73 so as to direct the flow of gas from the first bag house 70 to the second
bag house 76
and/or the third bag house 78. The diverter valve 74 has variable positions,
in the first
position gas from the first bag house 70 is directed to the second bag house
76, in the second
(variable) position gas from the first bag house 70 is directed to both the
second and third bag
houses 76,78, and in the third position, as illustrated in Figure 3, the gas
from the first bag
house 70 is directed to the third bag house 78. Gas exiting the second and
third bag houses
76 and 78 may be vented and directed for further processing or handling (e.g.
directed to
stack 40 or directed to a subsequent reactor for Hg removal). The system of
this embodiment
may incorporate any combination of the reaction zones useful in the invention
and is not
intended to be limited to bag houses.
However, when the reaction zones are bag houses, the system illustrated in
Figure 3
may further comprise an off line loading circuit 42. The off line loading
circuit 42 is brought
into use after the filter bags have been pulsed to clean them of filter cake
so reacted sorbent
can be removed for recycling or regeneration. There may be more than one off
line loading
circuit 42, as shown in Figure 3, each separately connected to a bag house 76
and 78. The
off line loading circuit is connected to a sorbent feeder and a bag house via
an off line
loading circuit conduit and incorporates a fan for blowing air commingled with
sorbent into
the bag houses 76 and 78 in order to pre-load the fabric filter bags in the
bag houses by
building a filter cake thereon. The air passing through the bags and cake
thereon is vented
from the bag house. When the bag house is ready to come back on line, the off
line loading
circuit can be closed or switched off and the diverter valve 74 moved to a
position to permit
the flow of process gas through the bag house that is being brought back on
line.
When NOX is captured by the sorbent, the sorbent may not be completely loaded
or
spent thus having remaining reactive sites. Even though it may no longer be
effective as an
efficient sorbent for NOX at this point, the sorbent may have reactive sites
that could be
utilized efficiently for SOX capture. Thus, the partially loaded reacted
sorbent or NOX-
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
19
reacted sorbent in a second reaction zone of a two-stage SOX/NOX removal
system could be
removed from the second reaction zone and fed into the first reaction zone to
allow additional
SOX capture with, or loading onto, the sorbent. This would decrease the
frequency at which
sorbent regeneration is needed and reduce the amount of virgin or unreacted
sorbent that
would need to be introduced into the first reaction zone.
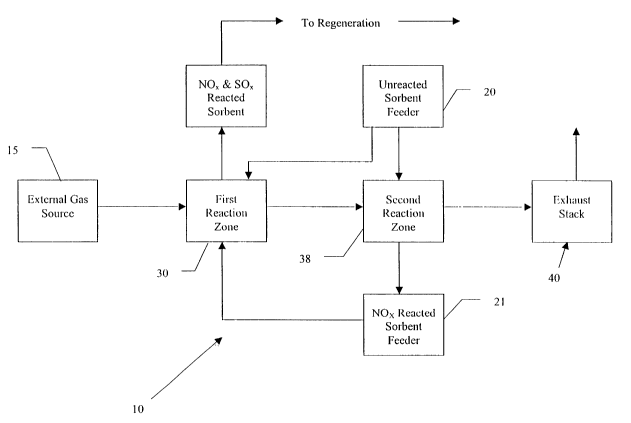
With reference to Figure 4 a system according to the invention utilizing
counter-flow
feed of NOX-reacted sorbent is illustrated in a block flow diagram. The system
10 is
comprised of a first reaction zone 30, a second reaction zone 38, a feeder 20
containing virgin
or unreacted sorbent, and a NOx-reacted sorbent feeder 21. The first reaction
zone 30 of
system 10 is connected to external gas source 15 and gas flows from the
external gas source
to the first reaction zone 30, from the first reaction zone 30 to the second
reaction zone 38,
and from the second reaction zone 38 is either vented to stack 40 or directed
on to another
system unit such as a mercury-sorbent reactor (not shown). The feeder 20 can
feed virgin or
unreacted sorbent into the first reaction zone 30 and the second reaction zone
38. NOX-
15 reacted sorbent is removed from the second reaction zone and is conveyed
from the second
reaction zone to the first reaction zone via NOX-reacted sorbent feeder 21
where the NOX-
reacted sorbent with available reaction sites is further contacted with a gas
containing both
SOX and NOx to remove and capture SOX.
Using reacted sorbent feeders allows sorbent to be recycled to a reaction zone
where
unreacted sites on the surface of the sorbent can be utilized. Through the
mechanical
operations of removing reacted sorbent from a reaction zone and returning it
to the same or
another reaction zone, the amount of virgin or unreacted sorbent that has to
be introduced into
the system is reduced. A sorbent may be recycled this way several times before
regeneration
is necessary due to the reduction in available reaction sites on the surface
of sorbent particles.
This represents significant cost savings and more economical and complete use
of the
sorbent.
During operation, the surfaces of sorbent particles may become obstructed, for
example, by compaction or agglomeration. The physical manipulation and
handling of the
reacted sorbent re-orients the particles making unexposed surfaces available
to capture
targeted pollutants.
The recycling of reacted sorbent in this way may proceed as shown in Figure 4
in a
counter-flow manner as discussed above. Recycling may also proceed by removing
reacted
sorbent from a reaction zone conveying it to a reacted sorbent feeder and
introducing or re-
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
introducing the reacted sorbent into the same reaction zone. This is shown in
Figure 28,
where reacted sorbent feeder 21A receives reacted sorbent conveyed from the
first reaction
zone 30 and reacted sorbent from reacted sorbent feeder 21A is re-introduced
into the first
reaction zone 30. Further, reacted sorbent from second reaction zone 38 is
conveyed to
reacted sorbent feeder 21B and re-introduced into the second reaction zone 38.
This may be
desirable where a first targeted pollutant is being captured in the first
reaction zone and a
second targeted pollutant is being captured in the second reaction zone. If,
for example, SOx
is being captured in the first reaction zone 30, the SOx reacted sorbent when
it is spent or
ceases to be effective for SOx removal, can then be routed for regeneration
and recovery of
10 sulfates as alkali or ammonium sulfate, useful commercial product.
Similarly, if NOx is the
pollutant being captured in the second reaction zone 38, the NOx reacted
sorbent can be
removed when it ceases to be effective for NOx removal and directed for
regeneration and
recovery to produce alkali or ammonium nitrates, again, useful commercial by-
products.
Capture rates may be affected by the gas inlet temperature as it enters a
reaction zone
15 and may need to be adjusted, cooled or heated to achieve a desired capture
rate for SOx
and/or NOx. This can be accomplished with a heat exchanger. As is illustrated
in Figure 5,
the system may further include a heat exchanger preceding each reaction zone
of a system of
the invention. In Figure 5, the system of the invention as illustrated is
substantially the same
as the illustration of Figure 1, depicting first and second reaction zones 30
and 38, feeder 20,
20 external gas source 15, and stack 40. In Figure 5, heat exchangers 72A, 72B
have been
introduced into the system before each reaction zone. The heat exchangers 72A,
72B may be
utilized to heat or cool the gas stream prior to entry into each reaction
zone. As the gas enters
into the system, if the gas temperature is above the thermal decomposition
temperatures) of
either sulfates of manganese or nitrates of manganese, the heat exchangers
72A, 72B will
operate to cool the gas to a desired temperature based upon whether SOx
capture or NOx
capture is the primary pollutant captured in the reaction zone. Similarly, if
the gas were
below a desired temperature set point, the heat exchangers 72A, 72B will
operate to heat the
gas to the desired temperature. The heat exchangers 72A, 72B may be a gas-to-
gas cooler or
a heater unit, or other suitable means for accomplishing heating and cooling
of gases to
assure that the gas inlet temperature at a targeted temperature or within an
acceptable range.
As previously mentioned above, the gases entering the system from external gas
source 15 may be any of a variety of process or industrial gases. These gases
when generated
encompass a range of temperatures. Due to simple economics and the design of
various
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
21
plants and facilities for efficient use of waste heat which is captured or
transferred to provide
heat for various processes at a facility, these process gases will typically
have a temperature
ranging from 250° F. to 350° F or 120° C to 180°
C. In less typical situations, these gases
may have temperatures upwards of 1000° F, or 540° C. Gases at
these temperatures are
readily processed in the systems of the invention and the heat exchangers 72A,
72B can be
utilized to maintain the gas within these temperature ranges if desired. The
system can also
process gases at much higher temperatures such as 1000° F. For purposes
of SOx and NOx
capture, the gas temperature should not exceed, respectively, the thermal
decomposition
temperatures) of sulfates of manganese and nitrates of manganese. Given that
different
forms or species of these sulfates and nitrates, the thermal decomposition
temperature would
depend upon the species formed during capture. It has been reported that that
sulfates of
manganese may thermally decompose at temperatures approximating 950°C.
Similarly,
nitrates of manganese are believed to thermally decompose at temperatures
ranging up to
260 °C. The system of the invention can process gases approaching these
thermal
decomposition temperatures. But, more typically, the system in practice will
be operated in
temperature ranges approximating those of process gases from industrial
sources.
Heat or waste heat from the process gases of a facility may be utilized in the
regeneration and recovery processes discussed herein below. Further, the waste
heat may be
utilized for purposes of sorbent preheating which serves to "activate" sorbent
prior to
introduction into a reaction zone. Although the exact mechanism of activation
is not known,
it is generally known that oxides of manganese can be "activated" with heat.
Thus, as can be
seen in Figure 28, a system according to the invention may further include a
sorbent preheater
22 which may actually be part of or separate from sorbent feeder 20. The
source of heat for
the sorbent preheater may be any heat source, but waste heat from facility
processes can be
economically efficiently utilized for this purpose.
The SOx and/or NOx capture rate may be regulated by the amount of sorbent fed
into
the reaction zones. In order to regulate capture rate, gas measuring devices,
such as
continuous emission monitors (CEMS), are utilized to measure the composition
of the gas at
the inlet to the reaction zone and at the outlet of the reaction zone. With
reference to Figure
14, the gas flows from the external gas source 15 and past GEMS 80A where the
gas
composition is measured prior to entry into first reaction zone 30. Another
CEMS 80B is
provided after the first reaction zone 30 to measure the concentration of the
gas substantially
stripped of SOx and/or NOx as it passes from the first reaction zone 30. As in
Figure 1, the
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
22
gas may be vented to a stack 40, passed to a second reaction zone 38, or
another system unit
for further processing.
In the system of the invention, a bag house may serve as a reaction zone
and/or as a
solid gas separator, since bag houses are solid-gas separators. A
conventional, commercially
available bag house 82 is depicted in Figures 6 through 9. Figure 6 is a
perspective view of a
bag house 82. Figure 7 is an end elevation view showing a bag house 82. Figure
8 is a top
plan view of a bag house 82. Figure 9 is a side elevation view of a bag house
82. Within the
bag house 82 are a plurality of bags 88 also referred to as filter fabric bags
shown in Figures
7 through 9. As can be seen in figure 7 through 9, the bag house 82 has a
plurality of filter
fabric bags 88 suspended therein. Typically, they are suspended from a frame
or support
structure at the top of the bag house 82. The filter bags 88 may be of various
shapes, e.g.,
conical or pyramidal, and include an internal frame and suitable fabric
filter. Those skilled
in the art would be able to select suitable filter fabric materials from those
commercially
available. Gas and entrained sorbent enters the bag house 82 through the bag
house inlet 92,
shown in Figures 7 through 9, and by virtue of an applied differential
pressure, gases are
forced through the fabric of the bags 88 and the entrained sorbents are
separated from the gas
by forming a filter cake on the surface of the bags 88. The filter cake thus
formed is a
reaction medium where pollutants are contacted with and removed by the
sorbent. The
commingled gases and sorbents move vertically upward and contact the fabric
and/or the
filter cake formed thereon. The bags 88 are configured to permit the gases to
be directed
from the outside to the inside of the bags to a conduit at the top of the bag
house 82 and then
to the bag house outlet 98, shown in Figures 6 through 9.
While the bag house 82 is in operation, the filter bags 88 may be periodically
pulsed
or otherwise agitated in order to adjust differential pressure across the bag
house 82, which
frees some or all of the filter cake and allows gas to flow more freely
through the filter cake
and the fabric filter bags. If the filter cake is allowed to get too thick,
excess differential
differential pressure across the bag house or the system of the invention may
result. Thus, the
pulse intensity or frequency can be utilized to regulate or adjust
differential pressure. When
the bag house 82 is taken off line, the bags 88 may be pulsed to free the bags
88 of virtually
all reacted and unreacted sorbent not otherwise removed during normal
operations. The
reacted and unreacted sorbent or filter cake fall from the bags 88 by gravity
into a hopper 112
(seen in Figures 7 and 9) at the bottom of the bag house 82 for subsequent
removal from the
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
23
bag house hopper 112. Removal from the hopper 112 may be accomplished with a
screw
conveyor or by other appropriate means, even manually.
A thicker filter cake will lead to increased removal efficiency, but at the
price of extra
power required to force the external gas source through the reaction zone. In
one example,
more power is required for an induction fan to pull exhaust gases through the
bag house when
the filter cake thickness is greater. The differential pressure may thus be
maintained at an
optimal level, trading off increased power requirements against the increased
pollutant
removal. In addition, the thicker the filter cake the longer the residence
time of the sorbent
material in the system. Longer residence time of the gas in the filter cake
results in better
removal efficiencies. Higher sorbent loading rates results in less material
that will have to be
regenerated. This may also be taken into consideration in setting the
differential pressure set
point.
In Figures 7 and 9, the plurality of filter bags is shown in position within
the bag
house. Also shown near the top of the bag house 82 is a pulse valve 124
utilized to pulse the
fabric bags 88 in order to reduce filter cake thickness or to free the filter
cake from the bags
88. The bag house may be provided with a number of pulse valves 124. During
operation,
these pulse valves 124 may be activated sequentially or randomly in order to
pulse the bags
88 in order to regulate and control differential pressure across the bag house
82 or the system
as a whole. When the bag house is taken off line, the bags may be pulsed to
free the bags of
virtually all filter cake so that reacted and unreacted sorbent may be
removed.
The bag house illustrated in Figure 6 through 9 is of a conventional design.
In
Figures 10 and 11, a novel bag house according to the invention is
illustrated. This bag
house, which can be utilized in the system of the invention, is referred to as
an inverted bag
house 140. The inverted bag house 140 eliminates the need for high can
velocities, and
permits downward, vertical flow of gases and reacted and unreacted sorbent.
The inverted
bag house 140 is comprised of a bag house housing 142, at least one inlet 145,
a plurality of
fabric filter bags 88, a support structure 149 for the filter bags, a hopper
152 to receive and
collect reacted and unreacted sorbent, am outlet 154, and a conduit 158. The
bag house
housing permits the introduction of gases and reacted and unreacted sorbent
entrained in the
gases, has a top and a bottom and is configured for gases to flow vertically
downward from
the top to the bottom of the bag house. The inlet 145 is located near the top
of the bag house
housing and is configured for the introduction of gases and reacted and
unreacted sorbent
entrained in the gases into the bag house. The plurality of fabric filter bags
88 are configured
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
24
to allow gas to flow from the outside of the bags 88 to the inside of the bags
88 under an
applied differential pressure and to prevent the passage of reacted and
unreacted sorbent from
the outside to the inside of the bags 88, thereby separating reacted and
unreacted sorbent from
the gas and forming a filter cake on the bags 88. The support structure 149 is
configured to
receive and support the fabric filter bags 88 and to provide openings through
which reacted
and unreacted sorbent may be freely passed downward into the hopper 152 by
gravity. The
hopper 152 is configured to receive the reacted and unreacted sorbent and to
permit the
removal of the reacted and unreacted sorbent. The inverted bag house 140 also
has an outlet
154 located near the bottom of the housing 142 below the bags 88 and above the
hopper 152.
The outlet 154 is connected to a conduit 158 located below the fabric filter
bags 88 and
positioned to receive gas passing through the fabric filter bags. Conduit 158
conveys gas to
the outlet so that the gas may be vented or passed from the inverted bag house
140.
In Figure 12, a bag house reactor 150 of the invention is illustrated. This
bag house
reactor 150 can also be utilized in the system in place of a conventional bag
house. The bag
house reactor 150 has interior surface 154 and exterior surface 152. It may be
viewed as
having an upper section 156, central section 157 and lower section 158.
Generally located in
the central and/or lower sections 157, 158 is a variable venturi 160. The
purpose of the
variable venturi 160 is to adjust the velocity of gas flowing through the
venturi opening
within the bag house reactor 150. The variable venturi 160 is configured to
adjust the
position of the variable venturi by varying the space or distance between the
variable venturi
160 and the interior surface 152 of the bag house reactor 150. W order to'
vary position a
variable venturi position detector 367 shown in Figure 23) for determining the
position of the
variable venturi 160 and a variable venturi positioner 368 (shown in Figure
23) for adjusting
the position of the variable venturi 160 are provided.
With the variable venturi 160 contacting the interior surface 154 of the bag
house
reactor 150, gas cannot flow from the lower section 158 to the central and
upper sections 156,
157 of the bag house. By opening the space between the variable venturi 160
and the interior
surface 154, gas is allowed to flow through the reactor 150. Gas introduced
through gas
distribution conduit 164 and the gas distribution port 162 flows from the
lower section 158 to
above the variable venturi 160 and into the central and upper sections 156,
157, and to the
filter bags 88. When the space between the variable venturi 160 and the
interior surface 154
is wide, the gas flows at lower velocities which allows some of the sorbent
suspended above
the variable venturi 160 to fall into the hopper 112.
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
There is also a sorbent distribution port 166 connected to a sorbent feed
conduit 168.
The sorbent distribution port 166 is positioned above the variable venturi 160
to allow the
introduction of sorbent into the upper section 156 of the bag house reactor
150. The sorbent
distribution port 166 is configured to allow introduction of sorbent into the
bag house. Port
5 162 is configured to allow introduction of gas into the bag house reactor.
The bag house reactor 150 has a plurality of fabric filter bags 88 secured
therein. The
fabric filter bags are mounted in the upper section 156 of the bag house
reactor 150 and
extend downward into the central section 157. At the bottom of the bag house
reactor in the
lower section 158, is a sorbent hopper 112 where reacted and unreacted sorbent
is collected.
10 The sorbent hopper is connected to outlet 172. Outlet 172 has an outlet
valve 176 which in
the open position allows for the removal of sorbent from the hopper 112. A
vent 180 is
located in the top section 156 of the bag house reactor 150. Gases flowing
through the bag
house reactor 150 pass from the bag house reactor 150 through the vent 180 and
may be
directed on for further processing or venting to the atmosphere.
15 Sorbent entrained in gases containing pollutants such as SOx and NOx can
begin
reacting with the sorbent during transport in the sorbent feeder conduit 168.
Since SOx is
more reactive than NOX, the more reactive SOx is primarily captured while it
is being
transported to the bag house reactor 150 in the first sorbent feeder conduit
164. At lower gas
velocities the larger solids will abrade into finer solids and re-fluidize.
The finer solids will
20 travel upward through the opening between the variable venturi 160 and the
interior surface
154 where the sorbent is suspended to create a pseudo fluidized-bed above the
variable
venturi 160 and the finest particules will travel upwards to form a filter
cake on the surface of
the fabric filter bags 88. By adjusting the position of the variable venturi
160 increasing or
decreasing the space between the variable venturi 160 and the interior surface
154 of the bag
25 house reactor 150 gas velocity is correspondingly decreased or increased.
In operation, the
variable venturi may be positioned to achieve a gas velocity sufficient to
suspend a selected
coarse fraction sorbent just above the orifice to create a pseudo-fluidized
bed which may
primarily or preferentially capture SOx, since SOx is more reactive than NOx.
Partially
stripped gas flows upward from the pseudo-fluidized bed carrying the finer
fraction sorbent
onto the filter bags. The resulting filter cake provides a reaction medium
where "slower"
reactions, such as NOx removal may occur. The variable venturi 160 position
may be
adjusted to achieve the desired thickness of filter cake on the fabric bags 88
thereby
increasing or decreasing the differential pressure across the system also to
balance overall
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
26
differential pressure by changing the venturi restriction. The fabric filter
bags 88 may also be
pulsed to partially remove filter cake and thus regulate differential
pressure. The gas flow
rate entering port 162 can be adjusted to regulate upward gas velocity so that
the bags 88 may
be pulsed to allow some of the loaded sorbent to fall into the hopper 112
without being
reentrained in the gas or redeposited on the bags 88.
Using the variable venturi 160, one can operate the system so that sorbent
suspended
above the venturi, loaded with the faster reacting gases, can primarily be
captured by falling
to the hopper before being carried up to the filter bags 88. The fraction of
sorbent loaded
with faster reacting gases can then be removed from the hopper 112 by opening
the outlet
valve 176 so that that fraction may be removed from the hopper 112 through the
outlet 172.
Later the fabric filter bags 88 can be pulsed to release the sorbent loaded
with slower reacting
gases which would then fall through the variable venturi 160 into the hopper
112. The
sorbent loaded with slower reacting gases could then be removed from the
hopper through
loaded sorbent outlet 172 after the outlet valve 176 has been opened. This
could allow for the
separate processing of the different loaded sorbent fractions to regenerate
the sorbent and
produce useful by-products.
Differential pressure, which represents sorbent filter cake thiclmess, is only
one of
several process parameters that can be controlled in the system in order to
achieve desired
levels of SOx and NOx removal efficiencies and cost advantages of the system.
NOx and
SOx removal efficiency may be regulated by various processes, including
sorbent feeder rate
and temperature control at the inlet to the reaction zones of the system.
These controls are
achieved by the control sub-elements or electronics, which include hardware
and software
and also are referred to herein below as control loops.
Referring now to Figure 13, a differential pressure control loop 300 is
illustrated.
External gas source 15 is illustrated feeding first reaction zone 30, which in
turn feeds
generally an output gas stream 316, which can feed either stack 40 or second
reaction zone
38. The differential pressure across first reaction zone 30 may be measured as
illustrated as
difference in pressure between the inlet pressure 306 and the outlet pressure
304. In the
example illustrated, inlet pressure 306 and outlet pressure 304 feed a
differential pressure cell
308, which sends a differential pressure signal 310 to a differential pressure
controller 302.
Differential pressure controller 302 can be any appropriate controller,
including a
proportional integral derivative (PZD) controller. As used herein, Pm
controllers may be
understood to operate using any combination of the proportional, integral, and
derivative
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
27
components. Differential pressure controller 302 can accept a set point 312,
indicating the
desired differential pressure across first reaction zone 30. Set point 312 can
be human or
computer generated. As discussed below, differential pressure controller 302,
and other
controllers, may be implemented as a stand-alone controller, distributed
control system, as a
PID block in a programmable logic controller (PLC), or as a set of discrete
calculations
within a PLC. Differential pressure controller 302 generates an output signal
314 to control
the differential pressure across first reaction zone 30. In embodiments where
first reaction
zone 30 includes a bag house or uses solids-filtering media, differential
pressure controller
302 output signal 314 may control the shaking, pulsing, or other removal of
sorbent which
has formed a filter cake on the filter medium.
In one embodiment, first reaction zone 30 includes numerous filter bags which
can
have an exterior containing sorbent material and an interior having a lower
pressure, acting to
pull the sorbent material against the bag filter media. In one example of the
invention, a
compressed air jet, pulse valve 124, is periodically discharged within the
interior of the filter.
In one embodiment, the compressed air pulse is sufficiently strong to dislodge
a portion of
caked sorbent material from the filter material even during normal operation
of the bag
house, not requiring the shut down of the bag house. In one embodiment, the
individual bags
are sequentially pulsed to dislodge a portion of caked sorbent material. The
frequency of the
pulsing may be increased in order to maintain a thinner filter cake thickness.
Thus,
increasing the frequency of the periodic pulsing of each filter bag will
maintain a smaller
filter cake thickness, and thus result in a smaller differential pressure
across the bag house as
a whole. In one embodiment, filter bags are grouped by row, with each row
periodically
pulsed at the same instant. In some embodiments, output 314 from differential
pressure
controller 302 includes a frequency for pulsing filters within a bag house
reaction zone.
Differential pressure controller 302, in response to a higher differential
pressure than set
point, may increase the frequency of filter pulsing through output 314.
Conversely, in
response to a lower differential pressure than set point, differential
pressure controller 302
may decrease the frequency of filter pulsing through output 314.
In one embodiment, the individual filter bags are formed of cylindrical filter
media
disposed about a rigid cylindrical cage, with the compressed air jet, pulse
valve 124, disposed
within the cylindrical rigid cage. After a period of time, the sorbent
material filter cake
builds up on the outside of the filter media, forming a thick filter cake. The
pulsed air jet can
force the filter media momentarily away from the cylindrical rigid cage,
thereby cracking the
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
28
caked sorbent material and dislodging it, thereby allowing the sorbent
material to fall under
gravity to be collected and removed from the reaction zone.
A thicker filter cake can lead to increased pollutant removal efficiency, but
at the
price of extra power required to force the external gas source through the
reaction zone. In
one example, more power is required for an induction fan to pull exhaust gases
through the
bag house when the filter cake thickness is greater. The differential pressure
may thus be
maintained at an optimal level, trading off increased power requirements
against the
increased pollutant removal. In addition, as the filter cake thickness
increases the contact or
residence time of the gas with sorbent material in the system increases,
resulting in more
complete reaction. Therefore less material will have to be regenerated. This
may also be
taken into consideration in defining the differential pressure set point.
Referring now to Figure 14, an emissions control loop 320 is illustrated. A
gas stream
may be seen to flow from gas source 15, through a first continuous emission
monitor system
(CEM) 80A, then to first reaction zone 30, then to a second CEM 80B. A sorbent
feeder 20
may be seen to feed material to first reaction zone 30. Feeder 20 may be a
screw feeder
having a variable speed screw, auger, pneumatic conveyor, or other method to
move sorbent,
within.
CEM 80A and CEM SOB can represent a NOx analyzer and or a SOx analyzer. Iii
one
embodiment, CEM 80A is a chemiluminescent monitor, for example, Thermo
Electron model
42H. In one embodiment, CEM 80A includes a SOxmonitor such as Bovar Western
Research model 921NMP, utilizing a spectrophotometric method. In some
embodiments,
CEM 80A and CEM SOB include both NOx and SOx analyzers. A feed controller 322
may
be seen to accept a first input 328 from an outlet CEM signal 325. Controller
input 328 may
be used as a feedback signal to control the feeder rate. In some embodiments,
a feeder
controller 322 also has a second input 330 accepting an inlet measurement
signal 324, also
including pollutant concentration data. Second input 330 may be used to
display the
incoming gas concentrations and/or to calculate percentage removal set points
in the system.
Feeder controller 322 also accepts a set point signal 326, indicating the
desired feed rate
and/or the desired NOx or SOx concentration exiting first reaction zone 30.
Feeder controller
output 332 can be a variable frequency drive signal, among other available
signals, to control
the speed of feeder 20.
Feeder controller 322 may be any suitable controller, including a Pm
controller
utilizing any combination of its individual modes. In one embodiment, set
point 326 is set at
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
29
a desired concentration for either NOx or SOx, depending on the embodiment.
The gas
concentration signal 325 from CEM 80B can be used by feeder controller 322 to
calculate
output signal 332. When the gas concentration is higher than indicated as
desirable by set
point 326, output 332 can be increased to increase the speed of feeder 20,
which will put
more sorbent into first reaction zone 30, thereby dropping the pollutant
concentration.
Conversely, when pollutant gas concentration 325 is lower than required,
feeder controller
output 332 can be decreased to decrease the rate of sorbent addition from
feeder 20 into first
reaction zone 30.
Refernng now to Figure 15, the gas to be cleaned may be seen to flow from
extenzal
gas source 15, through a first heat exchanger 72A, through first reaction zone
30, through
second heat exchanger 72B, through a second reaction zone 38, and to stack 40.
Figure 15
illustrates a system having two reaction zones and two heat exchangers. The
temperature to
the first reaction zone 30 may be seen to be controlled by a first temperature
controller 340,
which accepts a set point 344 and a temperature input 342, and generates an
output 346 to
first heat exchanger 72A. As previously discussed, the maximum desired
temperature in the
reaction zone may depend on the thermal decomposition temperatures) of the
sulfates of
manganese or nitrates of manganese, depending on whether NOx and/or SOx are
being
removed. Lower temperature set points will be above the dew point of the
system and
adjusted automatically or manually as needed. In one embodiment, the
temperature to be
controlled is measured at the reaction zone itself, rather than at the outlet
from the heat
exchanger, in order to more directly measure the temperature in the reaction
zone. In one
embodiment, temperature controller 340 output 346 may be a variable analog
signal or other
variable signals used to control a variable speed blower to control the outlet
temperature from
heat exchanger 72A. Temperature controller 340 may increase/decrease the
cooling air
passing through heat exchanger 72A when the temperature in first reaction zone
30 is
greater/less than set point 344.
A second temperature controller 350 may be seen to accept a temperature input
352
from second reaction zone 38 and a set point 354, and to generate an output
356 for heat
exchanger 72B. Second temperature controller 350 may be similar to first
temperature
controller 340. In one embodiment, heat exchanger 72B is used to cool the
incoming gas,
using ambient air as the cooling medium. As discussed previously with respect
to
temperature controller 340, second temperature controller 350 may
increase/decrease the
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
output to a variable speed drive coupled to a blower when the temperature of
second reaction
zone 38 is greater/less than set point 354.
Figure 15 also illustrates how a first feeder 20A may feed material to first
reaction
zone 30. A second feeder 20B may be used to feed sorbent material to second
reaction zone
5 38. First feeder 20A and second feeder 20B may be controlled as previously
described with
respect to feeder 20 in Figure 14.
Refernng now to Figure 16, a control and data acquisition system 400 for
controlling
and monitoring the previously described processes is illustrated. System 400
may be seen to
include generally a programmable logic controller (PLC) 402 and a local on-
site computer
10 440. Both PLC 402 and local computer 440 may be coupled to the World Wide
Web 424.
PLC 402 and local computer 440 may be accessed over World Wide Web 424 by a
user PC
428, a hand-held computer such as a Pahn Pilot 430, and other devices 426
which can access
World Wide Web 424.
PLC 402 may be seen to include a PLC rack 403. In one example, PLC 402 is an
15 Allen Bradley PLC. In one example, the Allen Bradley PLC is a PLC 5. PLC
rack 403 may
be seen to include a PLC processor module 408, and Ethernet module 410, and a
DC power
supply 412. PLC 402 may be seen to include an output bus 406, for example a
Control net
bus 406. Bus 406, in the present example, may be seen to be coupled to
numerous
input/output cards 404. Input/output cards 404 may be seen to include a
discrete I/O cards
20 404A, mixed discrete and analog I/O cards 404B, discrete I/O cards 404C,
discrete and
analog I/O cards 404D, more discrete and analog cards I/O 404E, a variable
frequency drive
card 404F, and a second variable frequency drive card 4046. The discrete I/O
may be
commonly used to accept inputs from discrete switches such as limit switches,
and the output
used to open and shut valves and to start and stop motors. The analog I/O may
be used to
25 accept input analog measurements from sensors and to control variable
position output
devices. The variable frequency drive outputs may be used to control variable
speed motors,
for example, variable speed motors used to control airflow pass the heat
exchangers.
PLC 402 may be seen to be coupled to an Ethernet hub 420 via an Ethernet cable
418.
In one embodiment, a DSL modem 422 enables Ethernet hub 420 to be accessed
from World
30 Wide Web 424. Local computer 440 may also be seen to be coupled to Ethernet
hub 420 via
an Ethernet cable 444. Ethernet cable 444 can be coupled to an Ethernet card
446. Similarly,
local computer phone line 442 may be coupled to a PC modem card 450. The PC
modem
card can provide access to World Wide Web 424 when a DSL modem line is not
available or
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
31
is not fiuictioning. Local computer 440 may be seen to include software 448
which can
include, for example, Microsoft Windows 2000 as an operating system that is
providing both
server and terminal functionality. Software component 448 can include a~i
Allen Bradley
OLE Process Control (OPC) module 452, as well as an Intellution ~ OPC server
component
454. The IFIX process monitoring and control package by Intellution is used in
one
embodiment. An Intellution process database component 456 may also be
included. Allen
Bradley OPC server 452 can provide communication between local on-site
computer and
Allen Bradley PLC 402.
Intellution OPC server 454 can provide communication between the Allen Bradley
inputs and outputs and the Intellution process monitoring and control system
residing within
local computer 440. Intellution process database 456 may be used to monitor
and control the
entire process. Intellution Work Space 458 may be used to allow access to
monitor, display,
and change current data, and a historical data area 460 may be used to trend
historical process
data. An Access/Oracle RDB component 462 may also be included to provide
database
reporting. In one embodiment, a report module, for example, a Microsoft Excel
or Crystal
report component 464 may also be provided. In some embodiments, an Intellution
web
server component 466 is provided, as is a Microsoft Internet Information
Server (IIS) module
468. In some embodiments, local on-site computer 440 has a local terminal or
CRT as well
to display, monitor, and change data residing in the Intellution Work Space
458.
In some embodiments, most or all of the controls discussed below in the
present
application are implemented within control system 400. In one embodiment, most
or all
controls are implemented within Allen Bradley PLC 402. For example, PID
control blocks
can be implemented using provided Allen Bradley PID blocks, or the blocks can
be created
from primitive mathematical operations using ladder logic. Control blocks such
as the table
blocks and selector blocks of Figures 24 and 25 may be implemented within
Allen Bradley
PLC 402 using standard blocks. Local on-site computer 440 may be used to store
and output
values such as PID set points and selector switch values from local computer
440 to registers
or control blocks within PLC 402. For example, the set points to heat
exchanger, differential
pressure, and feed rate control blocks may reside within local computer 440
and be
downloaded to PLC 402. The set points may be obtained by local computer 440
from a local
terminal and/or from World Wide Web 424 from devices 426, 428, and/or 430,
protected by
appropriate security. Local computer 440 can be used to provide historical
trending, operator
interface, alarming, and reporting.
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
32
Refernng now to Figure 17, a process graphic 450, as displayed on a human-
machine
interface is displayed. Process graphic 450 may be displayed, for example, on
an Intellution
IFIX system. Process graphic 450 can be updated in real time and can reside on
a personal
computer, for example. Process graphic 450 includes a manual switch 458 and an
automatic
switch 459 for controlling the control mode of the differential pressure
across the bag house.
Process graphic 450 also includes a table of values 460 including the
differential pressure set
point, the actual differential pressure and the inlet temperature to the bag
house. An output
table 462 is also illustrated, including the bag house outlet temperature, the
flue gas flow rate,
the inlet pressure to the bag house and the outlet pressure from the bag
house. A bag house
452 is shown diagrarnrnatically including an inlet 454 and an outlet 456. An
outlet emission
table 464 is also illustrated, including the 502, the NOx level, and the 02
level. Process
graphic 450 may be used to monitor and control the bag house differential
pressure, as
previously discussed.
Referring now to Figure 18, a process graphic 470 is illustrated as may be
displayed
on an Intellution IFIX process graphic. Process graphic 470 can monitor and
control the
absorbent feeder speed, including an increase button 471 and a decrease button
472. The
actual feeder speed in pounds of sorbent per hour is illustrated at feeder
speed 483. A
scrubber inlet table 473 is illustrated, including a S02 level, a NO level, a
N02 level, a NOx
level, a CO level, and an 02 level. A scrubber outlet table 474 includes the
same levels as the
inlet, but at the scrubber outlet. A NOx control section 475 on the process
graphic includes a
manual button 476 and an auto button 477, as well as a set point 478. In
automatic mode, set
point 478 may be used to control the feeder speed using the NOX set point.
Similarly, an S02
control section 479 includes a manual control button 480 and an auto control
button 481, as
well as a set point 482. In automatic mode, set point 479 may be used to
control the feeder
speed using the S02 set point.
Referring now to Figure 19, a process graphic 490 is illustrated, as may be
found on a
process control and monitoring station. A cooler 491 is illustrated, having an
inlet 492 and an
outlet 493, with the inlet and outlet temperatures being displayed in real
time. Cooler 491
may be a heat exchanger as previously discussed. Process graphic 490 includes
a manual
button 494 and an auto button 495. The bag house inlet temperature is
displayed at 498 as is
the cooler set point 497. When in the automatic mode, the fan speed may be
controlled by a
Pm controller using set point 497. Process graphic 490 also includes an outlet
emission table
496, including the S02 level, the NOx level, and the 02 level.
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
33
Referring now to Figure 20, differential pressure control loop 300 is
illustrated in
block diagram form. Differential pressure controller 302 may be seen to accept
set point 312
and actual differential pressure 310, and to generate output signal 314 to
control the
differential pressure across bag house 30. As previously discussed,
differential pressure set
point 312 may be set taking into accoumt the desired pollutant removal target
of the system,
the power required to force gas through the filters, and the desired rate of
sorbent
replenislunent.
Referring now to Figure 21, sorbent feeder control loop 320 is illustrated in
block
diagram form. As previously discussed, feeder control loop 320 can include a
reaction zone
CEM unit 80B that generates an output signal from the NOX and/or SOX emission
analyzers.
Emissions/Feeder controller 322 can accept the NOX or SOX measured emission
level through
controller input 328, and accepts a set point 326 indicating the desired NOX
and/or SOX
concentration. Controller 322 may also send a controller output 332 to sorbent
feeder 20. As
previously discussed, sorbent feeder 20 may be a variable speed screw feeder,
accepting a
variable analog drive signal among others as its input from feeder controller
322. The
process trade-offs in setting set point 326 are as previously described.
Figure 22 illustrates a control loop 341 for controlling the temperature of
bag house
82. Temperature controller 340 is as previously described with respect to
Figure 15.
Temperature controller 340 accepts a bag house temperature input 342 and
desired bag house
input temperature set point 344, generating controller output 346 which can be
fed as a fan
speed control to heat exchanger 72A. The control scheme rationale is as
previously described
with respect to Figure 15.
Referring now to Figure 23, a variable venturi control loop 361 is
illustrated. Figure
23 illustrates a venturi position controller 360, which accepts a venturi
position set point 362
and an actual venturi position input 364, generating a controller output 366
which can be
accepted by a variable venturi positioner at 368. The actual position of the
variable venturi
position may be measured by a position detector 367. In one embodiment, the
variable
venturi position may be measured in units of 0 to 100%. Venturi set point 362
may be set as
a function of one of several desired process parameters.
The variable venturi position may be set to control the space between the
variable
venturi 160 and interior surface 154, the cross-sectional flow area, available
for the bag house
inlet gas to flow around the flow occluding devise, variable venturi 160,
thereby controlling
the fluidization velocity of the gas. When the flow cross-sectional axea is
decreased, the gas
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
34
flow velocity increases, which can be used to support a deeper fluidized bed
depth of sorbent
material. If the gas flow velocity is made very high, only the densest sorbent
particles will be
able to descend against the swiftly rising gas and be collected from the
system. If the fluid
velocity is set very low, even the lightest particles will be able to settle
out of the system
quickly, thereby increasing the need for regeneration or recycling of material
back to the
reaction zone for more loading. A higher gas flow velocity will, in effect,
create a fluidized
bed reactor, having a fluidized bed of sorbent material held in place by the
upwardly rising
gas stream. A rapidly moving gas stream will also carry more sorbent particles
to the fabric
bags 88 filter to form a filter cake. Conversely, a slowly moving gas flow
around the variable
venturi 160 will allow many sorbent particles to fall and be collected prior
to becoming caked
upon the bags 88. A deeper fluidized bed will create higher differential
pressures and a
shallow fluidized bed will create lower differential pressures. Removal
efficiencies may be
taken into consideration when setting SOx and/or NOx fluidized bed depth.
Variable venturi
controller 360 may be any suitable controller, including a PID controller,
utilizing any
combination of its modes.
Referring now to Figure 24, a control scheme 370 is illustrated for
controlling sorbent
feeder 20 using one set of inputs selected from the group including NOx
concentration, SOx
concentration, and reactor zone differential pressure. The control of sorbent
feeder 20 may
be accomplished by selecting one of the aforementioned control inputs, where
the selection
may be based on the greatest deviation from set point or error.
An error generator 373 may be seen to accept several actual measurement
signals 384,
as well as several set points 385. The actual signals and set points may be
used to generate
corresponding errors, for example, using subtraction. Error generator 373 may
be seen in this
example to output a NOx error 373A, a SOx error 373B, and a differential
pressure error
373C. The outputs from error generator 373 may be accepted by an error
selector gate 374,
with one of the input errors selected and output as the error to a controller
error input 382.
Error selector gate 374 may be operated manually to accept one of the several
input errors in
some embodiments. In other embodiments, error selector gate 374 may
automatically select
the largest error or deviation, to control based on the process variable or
parameter most
requiring attention. For example, sorbent feeder 20 may be controlled based
upon the NOx
concentration, the SOx concentration, or the differential pressure across the
reaction zone.
Error selector gate 374 may select the highest deviation, or the highest
percent of
deviation, of these three error inputs. Error selector gate 374 can generate a
selector output
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
386 which can be used to select which of the inputs a gain selector 372 is to
select. Similarly,
error selector gate 374 may output a selector output 383 which can be accepted
by a set point
selector gate 376 to select from various set points provided to the selector
gate.
A gain table 371 may be implemented as a table in a fixed database, for
example, a
5 series of registers in a PLC. Gain table 371 may be seen to include a NOx
gain 371A, a SOx
gain 371B, and a differential pressure gain 371C. The gains from gain table
371 may be seen
to feed gain selector block 372. A gain selector output 377 may be sent to a
controller gain
input 379.
A set point table 375 may be seen to include a NOx set point 375A, a SOx set
point
10 375B, and a differential pressure set point 375C. The set points may be
used as inputs to
selector gate 376, with selector output 383 being used to select one of the
input set points.
Selector gate 376 may be seen to output one of the selected set points to
controller set point
input 380.
Control scheme 370 thus provides a system and method for controlling the
sorbent
15 feeder rate based upon any one of the NOx concentrations, the SOx
concentration or the
differential pressure across the reaction zone. This can be accomplished using
the selector
blocks previously discussed while only requiring a single controller.
Controller 378 can be,
for example, a Pm controller, using any combination of its individual modes.
Referring now to Figure 25, a control scheme 390 is illustrated, similar in
some
20 respects to control scheme 370 of Figure 24. Control scheme 390 includes
similar control
blocks, tables, and outputs as previously described in Figure 24. Control
scheme 390 further
includes the variable venturi control as one of the possible sets of inputs,
gains, and set points
to be used to control sorbent feeder 20. Gain table 371 may be seen to include
a variable
venturi gain 371D. Error generator 373 may be seen to generate a variable
venturi error
25 373D. Set point table 375 may be seen to include a variable venturi set
point 375D. Control
scheme 390 may thus operate in a manner similar to control scheme 370 of
Figure 24, but
allowing for control based on the venturi position.
Various components of the system of the invention have been discussed above.
Many
of the components of the system are commercially available from various
original equipment
30 manufacturers and are known to those of ordinary skill in the art. Further,
one skilled in the
art will recognize and understand that the reaction zones and other units of
the system of the
invention may be connected by pipes, ducts, and lines , etc. which allow gas
and/or sorbent to
flow through and within the system and that reaction zones are in flow through
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
36
communication in dual and multi stage embodiments of the invention. In
addition to the
aforementioned system components, the system may further include various
hoppers,
conveyors, separators, recirculation equipment, horizontal and vertical
conveyors, eductors.
Further, there may be modulating diverter valves, vibrators associated with
feeders,
compressors to provide instrument air to pulse filter fabric bags, as well as
various meters and
sampling ports.
In addition to removing SOx and NOx, the system and processes of the invention
can
be utilized to remove mercury (Hg) and fly ash. Gases emanating from
combustion of fuels,
which contain mercury and sulfides, include mercury compounds, mercury vapor,
ash, SOx
and NOx. These gases and solids are commingled with oxides of manganese and
are
transported at a sufficient velocity as a gas-solids mixture to a reactor,
which may be a bag
house or other reactor/separating device. During transport and during
residence in the reactor,
oxidation-reduction reactions occur. These reactions cause the conversion of
mercury vapor
to mercury compound(s), and sorbent and/or alumina adsorb the mercury
compound(s). As
disclosed above, SOx and NOx are removed through reaction with oxides of
manganese to
form sulfate and nitrate compounds of manganese. These reaction products,
unreacted
sorbent (if any) alumina, adsorbed mercury, and ash are trapped and collected
in the bag
house and clean, substantially stripped gases are vented to the stack. Thus,
during the
processing of gases with the system of the invention, mercury and mercury
compounds may
also be removed. The reacted and unreacted sorbent when removed from the
reaction zones
of the system may be further processed to generate useful products and to
regenerate the
sorbent as described herein below.
The system of the invention in its various embodiments may be utilized in a
process
for removal of oxides of sulfur and/or oxides of nitrogen, mercury (compounds
and vapor),
and other pollutants from a gas stream. The processes generally involve
providing a system
according to the invention, whether single stage, dual-stage, or mufti-stage.
Gas and sorbent
are introduced into a reaction zone and contacted for a time sufficient to
effect capture of the
targeted pollutants) thereby substantially stripping the gas of the targeted
pollutant(s). In a
single-stage removal process, the reaction zone would need to be a solid-gas
separator
operating as a reaction zone or else followed by a solid-gas separator in
order to render the
gas that has been substantially stripped of a target pollutant free of solids
so that the gas may
either be vented or directed for further processing. In a dual-stage removal
process, the
second reaction would preferably be a solid-gas separator operating as a
reaction zone. And,
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
37
in a mufti-stage removal process the last reaction zone in the series of
reaction zones through
which the process gas is directed would need to be a solid-gas separator
operating as a
reaction zone or else followed by a solid-gas separator in order to render the
gas that has been
substantially stripped of a target pollutant free of solids so that the gas
may either be vented
or directed for further processing. Generally, configuring the systems and
processes of the
invention to incorporate a solid-gas separator as the last reaction zone in a
sequence of
removal steps would be most economical and efficient.
A process according to the invention is described below using single-stage and
dual-
stage systems of the invention for purposes of illustration. It should be
readily understood by
those skilled in the art that the processes as described can be adapted to
mufti-stage removals
and to removal of various targeted pollutants with or without the addition of
other sorbent
materials or chemical additives, as appropriate.
Removal of SOX and/or NOX can be accomplished in a single single-stage removal
system. Sorbent and gas containing SOX and/or NOX are introduced into a
reaction zone 30
where the gas and sorbent are contacted for a time sufficient to substantially
strip the gas of
SOX and/or NOX. If SOX is the primary target pollutant, the gas may be
introduced at
temperatures typically ranging from about ambient temperature to below the
thermal
decomposition temperatures) of sulfates of manganese. If NOX is the primary
target
pollutant, the gas would be introduced at temperatures typically ranging from
about ambient
temperature to below the thermal decomposition temperatures) of nitrates of
manganese. If
both pollutants are present, NOX will not be captured if the temperature of
the gas is above
the thermal decomposition temperature of nitrates of manganese. In the
reaction zone, the
gas would be contacted with the sorbent for a time sufficient to effect
capture of the pollutant
at a targeted capture rate. If both pollutants are to be captured, the capture
rate for the
primary targeted pollutant would control or utilize a control sub-element,
such as control loop
320 of Figure 14 or control loop 390 of Figure 25. The capture rate for the
targeted
pollutants can be monitored and adjusted. The reaction zone would preferably
be a solid-gas
separator that renders the gas free of solids, such as reacted and unreacted
sorbent and any
other particulate matter in the gas so that the gas may be vented from the
reaction zone or
directed for further processing, after contacting the gas with sorbent for a
sufficient time.
In a dual-stage removal process, a system of the invention having at least two
reaction
zones, first and second reaction zone 30, 38 as in Figure 1, is provided. It
should be
understood that the system could be a system of the invention such as the
modular reaction
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
38
units illustrated in Figures 2 and 3. With reference to Figure 2, an.y of the
bag houses 62, 64,
66 could serve as first and second reaction zones 30, 38 depending upon how
the gas is
directed through the system. Further, with reference to Figure 3, the first
bag house 70 would
correspond to first reaction zone 30 and either or both of the second and
third bag houses 76,
78 would correspond to second reaction zone 38. Additionally, it is understood
that other
reaction zones may be substituted for the bag houses of Figures 2 and 3 and
the process as
described could be carried out.
However, for purposes of illustration, the dual-stage removal process is
discussed
with reference to Figure 1. In this process of the invention, gas and sorbent
are introduced
into first reaction zone 30 . The gas is contacted with the sorbent for
sufficient time to
primarily effect SOx capture at a targeted capture rate. The gas is rendered
free of solids and
then vented from the first reaction zone 30. Sorbent and the gas that has been
substantially
stripped of SOx are then introduced into second reaction zone 38. In the
second reaction
zone, the gas is contacted with the sorbent for a sufficient time to primarily
effect NOx
capture at a targeted capture rate. The gas is rendered free of solids and
then vented from the
second reaction zone 38. The vented gas may be directed to stack 40 to be
vented or emitted
into the atmosphere or directed on for further processing.
With the processes of the invention, other pollutants that can be captured
with oxides
of manganese can be removed. For example, without being limited or bound by
theory,
Applicants believe that mercury compounds adsorb onto oxides of manganese.
Applicants
further believe that, in the system and processes of the invention, elemental
mercury is
oxidized to form oxides of mercury which also adsorb onto oxides of manganese.
Additionally, hydrogen sulfide (HZS) and other totally reduced sulfides (TRS)
can be
removed utilizing oxides of manganese. More specifically, Applicants postulate
that the
sulfur in TRS may be oxidized to form SOZ which is known to react with oxides
of
manganese to form sulfates of manganese. Further still, Applicants believe
that CO is
oxidized to COZ which in turn reacts with the sorbent to form carbonated of
manganese
(MnC03) from which useful products can be recovered and oxides of manganese
regenerated.
It is known that mercury compounds may be removed from gases by adsorption on
fly
ash and/or alumina. Thus, alumina may be introduced with the sorbent in a
reaction zone for
purposes of removing mercury compounds and elemental mercury that has be
oxidized to
form oxides of mercury. Thus, elemental mercury that is not oxidized and
therefore not
captured by the sorbent in a first or second reaction zone may be captured in
a third reaction
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
39
zone, which may be referred to as a mercury-alumina reactor or an alumina
reactor. With
respect to single-stage removal, the mercury compounds may be removed in a
reaction zone
by contacting the gas with sorbent for a time sufficient for the mercury
compounds to adsorb
on to the sorbent, and alumina if mixed with the sorbent to thereby
substantially strip the gas
of mercury. Further, if the reaction zone is a solid-gas separator, mercury
compounds
adsorbed to fly ash would also be removed, thereby substantially stripping the
gas of mercury
compounds. In a dual-stage, the mercury compounds would similarly be removed,
but
depending upon which reaction zone is also a solid gas separator.
Thus, the system and process of the invention are readily understood to
include and
contemplate the removal of not only SOx and/or NOx but other pollutants, such
as mercury
compounds, elemental mercury, CO, CO2, TRS, and H2S.
The system and process of the invention has been tested at several power
plants
utilizing a SOx and/or NOx removal demonstration unit embodying a system
according to the
invention. The demonstration unit utilized a bag house as the second reaction
zone and a
pipe/duct as a first reaction zone in a dual stage removal system. The test
runs and results are
summarized in the following examples.
Example 1
NOx concentrations were determined using EPA method 7E, chemiluxninesent
analysis method, and analyzed with a model 42H chemiluminescent instrument
manufactured
by Thermo Electron Inc. Sulfur dioxide (SO2) concentrations were measured
utilizing, a
spectrophotometric analysis method employing a Bovar Western Research
Spectrophotometric model 921NMP instrument. In order to obtain accurate and
reliable
emission concentrations, sampling and reporting was conducted in accordance
with US EPA
Reference CFR 40, Part 60, Appendix A, Method 6C. Gas flow rates in standard
cubic feet
per minute (scfin) were measured using AGA method #3, utilizing a standard
orifice plate
meter run. The demonstration was conducted utilizing a series of test runs on
live gas
streams from a power plant. Said power plant operates steam boilers which are
fired on high
sulfur coal. During test runs, NOx and SOZ concentration readings were taken
continuously
alternating from the inlet and the outlet of the demonstration unit. Gas flow
rates were
measured continuously. The demonstration tests were performed utilizing two
different
forms of sorbent. The tests conducted utilized various forms of oxides of
manganese as
sorbent. The tests were performed with and without bag house filter pulsing.
The following
table summarizes the results and operational parameters:
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
Range of Operation Parameters
Range of NO~c Concentrations Processed by the 14.14 to 320
Demonstration Unit ppm
Range of SOZ Concentrations Processed by the Demonstration300 to 1800
Unit ppm
Range of Gas Flow through the Demonstration Unit 250 to 2000
scfm
Range of Pressure Across the Bag House 0.5" to 10.0"
of Ha0
Range of Bag House Temperatures 60 F to 246
F
Maximum NOx steady state Removal Rate 96.0%
Maximum SOz steady state Removal Rate 99.8%
Example 2
A test using the demonstration unit according to the invention, utilizing
oxides of
manganese as the sorbent was conducted on a simulated gas stream containing
varying levels
5 of NOx. Oxides of manganese powders that were used during this test
described generally by
60% of particles less than 45 microns in size and having a BET surface area of
approximately
30 m2/g. IW owing that there is a competition for reaction sites between S02
and NOx, a
series of tests was conducted to gather data on the efficiency of NOX capture
in the absence
of 502. Synthetic NOx gas was made on site by use of high-concentration bottle
gas which
10 was diluted into the inlet gas stream and processed by the demonstration
unit. The bag house
was pre-loaded with oxides of manganese prior to introduction of test gas by
operating the
demonstration unit's blower at high speed (typically about 1200 scfm), and
feeding the
oxides of manganese into the gas stream at a high rate (between 40% and 90% of
feeder
capacity) in order to form a suitable filter cake on the fabric bags in the
bag house. Gas from
15 cylinders containing NOx, 20% NO, and 20% N02, (20,000 ppm) was metered
into the bag
house inlet through a rotameter-type flow gage. NOx concentrations were
measured at the
bag house inlet and outlet on an alternating basis throughout the testing with
the
demonstration unit's continuous emissions monitoring system (CEMS), utilizing
a Thermo
Electron model 42H Chemilmninescent instrument. In order to obtain accurate
and reliable
20 emission concentrations, sampling and reporting was conducted in accordance
with US EPA
Reference CFR 40, Part 60, Appendix A, Method 6C.
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
41
Tests were performed at varying levels of bag house differential pressure
(measured
in inches of water column) and flow rates (measured in scfin). The NOx inlet
concentrations
ranged from 18.3-376.5 ppm with flow rates ranging from 260-1000. It has been
determined
that varying levels of filter cake thickness affect the NOx and SOz removal. A
thicker filter
cake increases the quantity of sorbent exposed to the gas, thus increasing the
micro-reaction
zone within the filter cake. As a representation of the sorbent filter cake
depth, the
differential pressure across the bag house (referred to as ~P) was measured
between 2.00"-
9.67" of WC (expressed in inches of water column). NOx concentrations were
recorded once
the system was in steady state and the readings were stable for up to 20
minutes. The
following table illustrates the level of NOx removal achieved as a function of
inlet
concentration, gas flow rate, and bag house differential pressure:
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
42
Summary of Bottle Gas NOx Reduction Test
R Inlet Outlet % QP Flow
un NOx NOx ate
No. ( ) ( ) Reduction(in. H2O)(scfm)
m m
1 25.5 3.3 87.1 2.00 60
2 140.1 8.5 94.0 3.86 500
3 102.0 10.5 89.7 7.71 1000
4 324.9 17.4 94.7 7.78 1000
195.0 15.1 92.3 7.85 1000
6 46.7 8.4 81.9 7.85 1000
7 200.3 32.5 83.8 3.0 to 1000
4.0
8 28.2 6.2 78.0 7.80 500
9 57.8 11.4 80.3 2.10 500
84.9 8.9 89.5 3.80 500
11 86.0 8.9 89.7 3.80 500
12 194.5 11.5 94.1 3.80 500
13 317.5 12.7 96.0 3.80 500
14 376.5 26.7 92.9 2.10 500
376.5 26.7 92.9 2.10 500
16 18.3 4.0 78.1 4.45 509
17 83.5 8.7 89.6 4.45 509
18 40.1 5.9 85.3 .45 509
19 83.5 8.7 89.6 4.45 509
21.5 4.5 79.2 4.74 500
21 45.7 6.5 85.8 4.75 500
22 92.1 8.6 90.7 .75 500
23 201.1 11.5 94.3 .76 500
24 317.5 14.0 95.6 .79 500
52.1 10.0 80.9 9.67 1000
26 82.4 12.0 85.5 9.67 1000
27 105.4 13.2 87.5 9.65 1000
28 224.0 18.5 91.8 9.67 1000
29 328.4 23.1 93.0 9.67 1000
100.2 15.0 85.0 9.67 1000
Example 3
A further test of the demonstration unit according to the invention utilizing
oxides of
5 . manganese as the sorbent, was conducted on a live exhaust gas slipstream
from a 170 MW
coal fired boiler. The boiler was operating on high sulfur coal of
approximately 4-6% sulfur,
resulting in emission concentrations of SOZ in the range of 1200-2000 ppm and
NOx
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
43
concentrations in the range of 280-320 ppm. A slipstream averaging 1000 scfm
was diverted
from the main staclc exhaust and routed to the demonstration unit for reaction
and sorption by
the sorbent oxides of manganese. SOZ and NOx concentrations were measured at
the scrubber
inlet and outlet of the bag house on an alternating basis throughout the
testing with the
demonstration unit's continuous emissions monitoring system (CEMS). S02
concentrations
were measured utilizing a Bovar Western Research model 921NMP
spectrophotometric
analyzer and NOx concentrations were measured utilizing a Thermo Electron
model 42H
chemiluminescent instrument. In order to obtain accurate and reliable emission
concentrations, sampling and reporting was conducted in accordance with US EPA
Reference
CFR 40, Part 60, Appendix A, Method 6C.
S02 removal efficiencies of 99.8% and NOx removal efficiencies of 75.3% were
achieved while processing on average 1000 scfin of exhaust gas at temperatures
typically
ranging from 150° F to 250° F. Test runs were conducted with
varying levels of bag house
differential pressures ranging from 0.5" to 8.6" of WC, which represents
various levels of
filter cake thickness. Tests were also conducted with different rates of bag
house filter bag
pulsing and varying levels of oxides of manganese feed rates. Oxides of
manganese powders
that were used during this test described generally by 60% of particles less
than 45 microns in
size and having a BET surface area of approximately 30 m2/g. The following
table gives an
example of SO2 and NOx data collected during a test in which 1000 scfm was
processed by
the dry scrubber at an inlet temperature of 250 °F, and a differential
pressure of 5.75" of WC.
Data was collected once the demonstration unit was in a steady state of NOx
and S02
removal for a period of 30 minutes. The results are summarized in the below
table:
Pollutant Inlet Outlet ppm % Removal
ppm ppm
Oxides of Nitrogen 285.9 70.5 75.3%
(NOx)
Sulfur Dioxide (S02)1703 3.9 99.8%
Example 4
An additional series of demonstration tests of the demonstration unit,
utilizing oxides
of manganese as the sorbent, was conducted on a live exhaust gas slipstream
from a 75 MW
coal fired boiler. This boiler was operating on Powder River Basin (PRB) coal,
resulting in
emission concentrations of SOZ in the range of 340-500 ppm with NOx
concentrations in the
range of 250-330 ppm. A slipstream ranging from 500-1000 scfin was diverted
from the
main stack exhaust and routed to the demonstration unit for reaction and
sorption by the
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
44
oxides of manganese. Oxides of manganese powder that were used during this
test described
generally by 60% of particles less than 45 microns in size and having a BET
surface area of
approximately 30 ma/g. S02 and NOx concentrations were measured at the bag
house inlet
and outlet on an alternating basis throughout the test with the demonstration
unit's continuous
emissions monitoring system (CEMS). S02 concentrations were measured utilizing
a Bovar
Western Research model 921NMP spectrophotometric instrument and NOx
concentrations
were measured utilizing a Thermo Electron model 42H chemiluminescent
instrument. In
order to obtain accurate and reliable emission concentrations, sampling and
reporting was
conducted in accordance with LTS EPA Reference CFR 40, Part 60, Appendix A,
Method 6C.
SOZ and NOx reduction efficiencies were measured at 99.9% and 91.6%
respectively.
Testing was conducted with varying degrees of differential pressure (4P)
across the bag
house to affect the residence time of the targeted pollutants. Reaction
chamber temperatures
ranged from 150° F to 280° F. It was determined that longer
residence times resulted in
improved capture rates for NOx. However, the fact that the S02 reaction occurs
so rapidly
and completely, the SOZ reduction efficiency remains nearly complete (99.9%)
at even the
lowest of residence times. While operating the scrubber at 0.5"-1.0" of WC
across the bag
house, a pollutant concentration reduction efficiency of 99.8% for S02 and
40.0% for NOx
was achieved. It is from these results that the concept for a two stage
reaction chamber
system develops, whereby the first reaction chamber captures the majority of
S02 and a small
fraction of NOx, while the second "polishing" stage completes the NOx removal
to desired
levels of efficiency, predetermined and controlled by the system operator.
Data was collected
once the dry scrubber was in a steady state of NOx and SOZ removal for a
period of 30
minutes. The following table gives an example of SO2 and NOx data collected
during a
testing in which 500 scfm was processed by the demonstration unit at an inlet
temperature of
250 °F, and a differential pressure of 8.7" of WC:
ollutant Inlet Outlet pm % Removal
ppm ppm
Oxides of Nitrogen268.1 22.4 91.6%
(NOx)
Sulfur Dioxide 434.3 0.5 99.9%
(S02)
Example 5
In an attempt to determine the effectiveness of S02 and NOx removal, a series
of lab-
scale tests were conducted utilizing a glass reactor. The reactor was designed
to mimic the
gas-solid interactions known to be present in the aforementioned demonstration
unit. The
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
glass reactor had a diameter of 2 inches with a length of approximately 24
inches. 50.0
grams of oxides of manganese were suspended in the reactor using a fritted
glass filter
allowing for flow of the gas stream, while keeping the oxides of manganese
suspended.
Approximately 3 inches above the fluidized bed of oxides of manganese, a
sintered stainless
5 steel filter was arranged to simulate a bag house filter bag. The reactor
was heated during the
testing to 250°F.and the gas flow rate was metered at a constant 6
liters per minute (lpm).
Simulated exhaust gas was produced by use of a calibration gas standaxd having
the
following composition: COZ= 17.35%, NOx=391 ppm, S02=407 ppm, CO=395 ppm, and
balance N2 . The simulated flue gas stream passed through the fluidized bed of
oxides of
10 manganese, where the flow carried a portion of the sorbent up onto the
filter, thus creating a
filter cake, which mimics a bag house reactor chamber.
S02 and NOx concentrations were measured continuously from the reactor outlet
utilizing a continuous emissions monitoring system (CEMS). S02 concentrations
were
measured utilizing a Bovar Western Research model 921NMP spectrophotometric
instrument
15 and NOx concentrations were measured utilizing a Thermo Electron model 42H
chemiluminescent instrument. In order to obtain accurate and reliable emission
concentrations, sampling and reporting was conducted in accordance with US EPA
Reference
CFR 40, Part 60, Appendix A, Method 6C. Removal efficiencies of 99.9% for SOz
as well as
99.9% for NOx were measured and duplicated for several test runs. Inlet
temperature was
20 250° F, with a differential pressure of 2.00" of WC. The following
table gives an example of
SOZ and NOx data collected during testing in which 6 lpm of gas was processed
by a glass
reactor:
PollutantInletOutletSorbent % Flow ~P Temp.Time with
(ppm)(ppm) Weight Removal rate (in H20)( >94%
(g) (lpm) F) Removal
Oxides
of
Manganese
Type
A
NOX 391 17.21 50 95.6% 6 2.00 250 29 min
SOZ 407 0.1 50 99.9% 6 2.00 250 >54 min
Oxides
of
Manganese
Type
B
NOX 391 0.1 50 99.9% 6 2.00 250 60 min
SOZ 407 0.1 50 99.9% 6 2.00 250 >90
Oxides
on
Manganese
Type
C
NOX 391 0.2 50 99.9% 6 2.00 250 34 min
SOz 407 0.1 50 99.9% 6 2.00 250 >68 min
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
46
The tests of this Example 5 were conducted with three different lots of
manganese
oxide sorbent. Figures 29 and 30 are, respectively, graphs plotting NOx and
SOx
concentrations at the outlet of the glass reactor versus time. The three
different oxides of
manganese are represented by the symbols "O" for type A sorbent, "D" for type
B sorbent,
and "~" for type C sorbent in Figure 29 and 30. Type A sorbent is an oxide of
manganese
powder generally at 60% of particles less than 45 microns in size and having a
BET surface
area of approximately 30 mz/g. Type B sorbent is an oxide of manganese powder
generally
at 100% of particles less than 45 microns in size and having a BET surface
area of
approximately 200 m2/g. Type C sorbent is an oxide of manganese powder
generally at 80%
of particles less than 45 microns in size and having a BET surface area of
approximately 90
m2/g. The graph of Figure 30, confinns the above statements regarding near
immediate and
complete SOx capture upon contact with the sorbent. The graph of Figure 29
shows a range
of capture efficiency over time for NOx and that different forms of oxide
manganese may be
able to provide more efficient capture of NOx. The type B sorbent performed
the best before
break-through, followed by type C. Useful captures were observed for all three
types. With
the process controls of the invention a wide variety of oxides of manganese
can be utilized to
effect removal at targeted capture rates. Further, the graphs of Figures 29
and 30 show that
high removal or capture rates can be achieved and sustained over time. The
operational
parameters of the systems of the invention can be monitored and adjusted to
attain and
maintain removal or capture rates at these lugh levels.
As mentioned above, the reacted or loaded sorbent can be recycled and/or
regenerated
after being removed from a reaction zone. For recycling purposes the reacted
sorbent may
simply be reintroduced into another reaction zone. For example with reference
to Figure 4,
the system has first and second reaction zones 30, 38 which are connected to
feeder 20 which
contains unreacted or virgin sorbent. Gas from external gas source 15 is
introduced into first
reaction zone 30 along with sorbent fed from feeder 20. The gas is contacted
with sorbent for
a time sufficient to remove a target pollutant, such as SOx, and after being
rendered free of
solids is vented from the first reaction zone 30. The gas is then introduced
in the second
reaction zone 38 along with sorbent from feeder 20. In the second reaction
zone 38, the gas
is contacted with gas for a time sufficient to remove another target
pollutant, here NOx.
During operation, the level of NOx loading on the reacted sorbent in second
reaction zone 38
reaches the point where the sorbent no longer efficiently removes NOx. When
the point is
reached, the NOx. reacted sorbent is removed from the second reaction zone 38
and conveyed
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
47
or transported to NOx reacted sorbent feeder 21. The NOx reacted sorbent,
which has unused
reactive sites available for further SOx capture, is fed or introduced into
the first reaction
zone 30 for additional loading or reaction with SOx in the gas introduced from
external gas
source 15. When the recycled NOx reacted sorbent reaches the point where SOx
capture can
no longer be achieved at a targeted rate of removal, the now NOx and SOx
reacted (or
loaded) sorbent is removed from the first reaction zone axed routed for
regeneration. In this
way, the amount of virgin or unreacted sorbent that is utilized in the first
reaction zone can be
reduced and the additional load or reactive sites available on the NOx reacted
sorbent can be
utilized.
During a wet regeneration process the reacted surfaces of the sorbent may be
removed
and the remaining sorbent may be refreshed. This will be understood with
reference to Figure
26. In a wet regeneration, reacted sorbent is removed from a reaction zone, a
reaction
chamber in Figure 26, and washed in an aqueous dilute acid rinse. Since the
interaction
between pollutants and the sorbent is believed to be a surface-controlled
phenomenon, only a
small fraction of the oxides of manganese is reacted with the pollutant. It is
this small
fraction of the sorbent that can be removed by washing or rinsing which
thereby "activates"
the sorbent by making unreacted surface area available. The solubility in
water of nitrates of
manganese is greater than the solubility of sulfates of manganese by at least
an order of
magnitude in cold water and by at least several orders of magnitude in warm to
hot water.
This differential in solubility can be advantageously utilized in the
regeneration process.
The sulfates and nitrates of manganese on the surface of the sorbent particles
dissolve
off into solution in the dilute acid bath, leaving clean sorbent that can be
readily separated
from the rinse or bath by known means, such as settling and decanting,
filtering, centrifuging
or other suitable techniques. As is further discussed below, the clear
filtrate or solution
containing dissolved sulfates and/or nitrates of manganese are directed to a
regeneration
vessel for regeneration of sorbent and production of useful by-products. The
clean sorbent is
then dried in, for example, a kiln to remove excess moisture. The heat for
this drying step
may be waste heat generated by combustion which is transferred or exchanged
from
combustion or process gases at an industrial or utility plant. After drying,
the clean sorbent
may be pulverized as necessary to reduce the clean sorbent to particle sizes
useful in the
system of the invention. The cleaned or "activated" sorbent is then conveyed
or otherwise
transported to the mireacted sorbent feeders) and thus, recycled.
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
48
Again with reference to Figure 26, the regeneration of sorbent and production
of
useful by-products can be understood. The solution or filtrate containing the
dissolved
sulfates and nitrates of manganese is passed from the acidic bath to a
regeneration vessel to
which alkali hydroxides such as potassium hydroxide (KOH) or sodium hydroxide
(NaOH),
or ammonium hydroxide (NH40H) is added. The addition of these hydroxides,
yield
respectively, a solution containing nitrates and/or sulfates of potassium,
sodium, or
ammonium and a precipitate of manganese hydroxide (Mn(OH)2). These solutions
can be
made into fertilizer products or other products such as explosives. Air or
oxygen may be
bubbled into or otherwise introduced into the reaction vessel to further the
regeneration of the
sorbent. The precipitate may be removed with or without the prior introduction
of air or
f
oxygen and then dried and heated to form oxides of manganese, MnOx where X is
between
about 1.5 to 2Ø
Instead of hydroxide compounds, soluble carbonate compounds, e.g., alkali
carbonates, such as potassium carbonate (K2C03), sodium carbonate (NaZC03), or
ammonium carbonate ( (NH4)ZC03) may be added to the solution or filtrate in an
regeneration
vessel. The addition of carbonates will yield a manganese carbonate
precipitate and a
solution containing nitrates and/or sulfates of potassium, sodium, or
ammonium. The
precipitate is separated from the solution, dried and heated to thermally
decompose the
manganese carbonate to form oxides of manganese and C02 gas which may be
vented or
captured and containerized as a marketable product. The oxides of manganese
may be
further heated in an oxidizing atmosphere to complete the sorbent
regeneration, to form
oxides of manganese, MnOx where X is between about 1.5 to 2.O.The oxides of
manganese
are separated from the solution, much as the cleaned or reactivated sorbent
after the acid
wash step, and are then dried and pulverized before being conveyed to a virgin
or unreacted
sorbent feeder. The filtrate from the separation containing useful sulfates
and nitrates that can
then be further processed into marketable products.
Oxides of manganese may also be regenerated in a dry or thermal regeneration
process, taking advantage of the thermal decomposition temperatures) of
nitrates of
manganese. This regeneration process may be understood with reference to
Figure 27. The
process illustrated and discussed herein is based upon a removal process where
NOx is the
target pollutant with nitrates of manganese being formed in the removal step
in the reaction
zone, a reaction chamber in Figure 27. The NOx reacted sorbent is removed from
the
reaction chamber and conveyed to a first kiln. In the first kiln, the reacted
sorbent is heated
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
49
to a temperature at or above the thermal decomposition temperatures) of
nitrates of
manganese and N02 desorbs or is otherwise driven off. Oxides of manganese,
MnOx where
X ranges from about 1.5 to 2.0 are formed in the first kiln which may be
heated with waste
process heat from the local plant. The regenerated oxides of manganese from
the first lciln
may be conveyed to a second kiln heated with waste process heat. Air or oxygen
are
introduced into the second kiln to more completely oxidize the regenerated
sorbent so that the
X of MnOx ranges from about 1.5 to 2Ø
If the sorbent was SOx-reacted the thermal regeneration would proceed much as
described for NOx, except the first kiln would be heated to a temperature at
or above the
thermal decomposition temperature of sulfates of manganese and S02 would
desorb or
otherwise driven off. With out being bound by theory, Applicants believe that
nitrates of
manganese thermally decompose at temperatures between about 130° C to
about 260° C,
while sulfates of manganese tend to liquefy at the temperatures over which
nitrates of
manganese thermally decompose. Applicants further believe that sulfates of
manganese
heated to these temperatures in the presence of a reducing agent, e.g., CO,
H2, etc., will
decompose to S02 and MnO. Thus, if the sorbent were reacted with both SOx and
NOx,
NOZ could be driven off first by heating reacted sorbent in a kiln to a first
temperature at
which nitrates of manganese thermally decompose so that N02 can be generated
and directed
for further processing. A reducing agent could then be introduced and the
reacted sorbent
further heated to desorb SOZ. Alternatively, the reacted sorbent could be
heated to a second
temperature, the thermal decomposition temperature of sulfates of manganese
with SOZ being
desorbed and directed for further processing. The desorbed SO2 can be directed
to a wet
scrubber containing water and an optional oxidant to form sulfuric acid. This
acid liquor can
then be marketed as is or further processed. This further processing would
involve the
addition of an ammonium or alkali hydroxide solution to form useful sulfates.
In either case,
the regenerated sorbent is further heated in an oxidizing atmosphere to more
completely
oxidize the regenerated sorbent so that the X of MnOx ranges from about 1.5 to
2.O.Referring
back to Figure 27, the desorbed N02 can be directed to a wet scrubber
containing water and
an oxidant to form nitric acid. This acid liquor can then be marketed as is or
further
processed. This further processing would involve the addition of an ammonium
or alkali
hydroxide solution to form useful nitrates, such as I~OH as illustrated in
figure 27.
In addition to regeneration of sorbent and production of useful by-products
from the
sulfates and nitrates of manganese, elemental mercury can be recovered from
NOx, SOx
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
reacted sorbent that further has mercury compounds adsorbed thereon can be
processed to
generate and recover elemental mercury. The reacted sorbent is removed from a
reaction
zone of a system according to the invention and conveyed to a first kiln, the
reacted sorbent is
heated to a first temperature to desorb NOZ which is routed for further
processing into
5 marketable products. The reacted sorbent is then heated a second temperature
to desorb
elemental mercury which is routed to a condenser for recovery as a marketable
product. The
sorbent is then rinsed to wash away any ash and to dissolve sulfates of
manganese into
solution to form a liquor. Any ash in the liquor is separated out and the ash
routed for further
handling. Alkali or armnonium hydroxide is added to the liquor to form an
unreacted sorbent
10 precipitate of oxides of manganese and a liquor containing alkali or
ammonium sulfates. The
liquor contains rinsed sorbent. The rinsed sorbent and unreacted sorbent
precipitate and are
separated from the liquor and the liquor is routed for further processing into
marketable
products or for distribution andlor sale as a useful by-product. The rinsed
sorbent and sorbent
precipitate are dried to form unreacted sorbent which can then be pulverized
to de-
15 agglomerate the unreacted sorbent.
Ion exchange can also be utilized as a mechanism for the separation and
recovery of
useful sulfate and nitrates. The dissolved sulfates and nitrates of manganese
in the filtrate or
solution left after washing SOx and/or NOx reacted sorbent can be processed in
anion
exchangers, permitting the recovery manganese cations and separation of the
sulfate and
20 nitrate aeons. To accomplish this separation, the aqueous solution
containing dissolved
sulfates and nitrates is passed across or through a bed or column of an anion
exchange resin
that has an affinity for one of the two anions to remove one of the two
anions. The resin with
absorb the anion, for instance the sulfate, while permitting the nitrate to
pass through the bed
or column. Additionally, the solution stripped of sulfate can then be passed
across or through
25 a second bed or coluiml of yet a second anion exchange resin to remove the
nitrate. After the
resin is loaded, the vessel containing the resin can be taken off line and the
resin therein
stripped of the captured anion and recovered for reuse.
Suitable anion exchange resins and vessels are known to and readily identified
by
those skilled in the art. For purposes of illustration, the anion exchange
resin has a chloride
30 in the exchange position on the resin. The chloride is eluted while
capturing the sulfate
and/or nitrate anions. The solution, after passing through the anion exchanger
or exchangers
in series, will contain manganese chloride from which manganese carbonate or
manganese
hydroxide is precipitated with the addition of a soluble carbonate or
hydroxide compound;
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
51
and oxides of manganese can be regenerated from the precipitate as discussed
above. The
sulfates and/or nitrates loaded on the resin can in turn be eluted with a
solution containing
chlorides of potassium, sodium or ammonium in order to generate useful
sulfates and nitrate
by-products for marketing or further processing. The filtrate or solution left
over after
precipitate formation can be utilized for this purpose.
Liquid mercury can also be recovered from mercury adsorbed to alumina in an
alumina reactor. The mercury-reacted alumina from the reactor is heated to
drive off or
desorb mercury. The mercury vapor is then directed to a condenser where it is
condensed to
form liquid mercury which is a marketable product.
With the system and processes of the invention, CO and C02 in a gas stream can
also
be captured. Applicants believe that CO in a gas stream is oxidized to form
C02 when
contacted with the sorbent. The C02 in turn reacts with oxides of manganese to
form
MnC03. Thus, in the processes of the invention, manganese carbonate may be
formed either
during the capture of CO and CO2 with the sorbent or during a regeneration
step in which
soluble carbonate compounds are utilized. Manganese carbonate is insoluble in
water. Thus,
sorbent that has been utilized to capture CO and C02 must be thermally
regenerated. Sorbent
loaded with manganese carbonate must be removed from the system of the
invention and
heated to thermally regenerate oxides of manganese, releasing COa which may be
compressed and containerized for sale or other marketable purposes. The
heating of the
loaded sorbent may be carried out in either two stages or in two separate
heating units or
kilns. In the first stage, the sorbent would be heated to thermally decompose
the manganese
carbonate, driving off COZ after which the sorbent would be further heated to
complete the
sorbent regeneration. In the second stage, the heating would continue either
in the same or a
second hearting unit or kiln. The second heating stage preferably would be in
an oxidizing
atmosphere being carried out with the introduction of air or oxygen in order
to complete the
regeneration of the sorbent to form oxides of manganese, MnOx where X is
between about
1.5 to 2Ø
The above examples of regeneration processes are provided by way of example
and
are not intended to limit the processes, both known and unknown, for
regeneration of oxides
of manganese and for recovery of useful and marketable by-products that may be
incorporated into the processes of the invention.
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
52
The combustion of fossil fuels (e.g., coal, oil, and natural gas) liberates
three major air
pollutants: (1) particulates (2) sulfur dioxide (SOa) and (3) oxides of
nitrogen (NOx). Wet
scrubbing, electrostatic precipitators and bag houses can remove particulates
such as fly ash.
Using mechanical filters or electrostatic precipitators does not remove SOZ,
503, NOZ, N204,
NO, or NZO3. Prior technologies have used wet scrubbing for the process as a
means of
sorbing SOx and NOx, Water is effective as a scrubbing medium for the removal
of SOZ;
removal efficiencies can be improved by the addition of chemical absorbents
such as calcimn,
magnesium and sodium. However, nitrogen oxide (NO) is essentially insoluble in
water,
even with the use of sorbtion chemicals. Residence times required and liquid-
to-gas surface
areas have proven to be impractical where high gas flow rates are encountered
such as boiler
flue gas.
Some of the economics involved in the wet scrubbing process involve high-
energy
consumption; on the average 4-5% of a plant gross power generation is consumed
in the
process. For example: (1) high differential pressure of a venturi / absorber
tower requires 30"
of WC or a bag house and scrubber combination requires even higher static
pressures. (2)
Large volumes of high pressure scrubbing liquor injected through nozzles into
the scrubbing
apparatus. (3) Slurry tanks requiring continual vigorous agitation. (4) High
horsepower
required to force water-saturated non-buoyant flue gas up the stack.
Environmental drawbacks of existing systems include large quantities of
minerals
used as sorbents and the insoluble sulfites or sulfate formed from the
scrubbing reaction. The
precipitate is then taken to landfills or holding ponds. Some other
disadvantages of existing
systems are fouling of the internal scrubber components with hard scale,
increasing
operational labor and maintenance costs. Some complex regenerative systems use
large
quantities of chemicals required to react with the millions of gallons of
slurry used every day.
The dry scrubbing process described in this patent is effective in removing
nearly all
NOx and SOx . Differential pressure requirements through the scrubber should
typically not
exceed 10 inches of water column and residence times within the sorbent cake
are typically
less than 1 second. Volumes of sorbent used in this invention in comparison to
the wet slurry
volumes are miniscule and recharging of reaction zones are done periodically.
While stack
gases remain dry and hot, some waste heat will be used in the drying of washed
and re-
generated sorbent. Operational costs of the reaction zones) are similar to
operating an ash
bag house; also capital expenditures are estimated to be reasonable requiring
standard off the-
shelf equipment and instrumentation.
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
53
As a summary, the equipment is used in the dry scrubbing process is much less
complex than the wet scrubber process thus requiring lower operational
maintenance costs
and a reduced operating staff. Chemical and raw material costs are expected to
be similar
with less waste effluent produced. The major cost savings will be in the
reduced power
consumption expected to be significantly less than that of a wet scrubbing
system, with fan
horsepower reduction making up the majority of the savings.
Although economics favor the use of dry scrubbing processes, removal of target
pollutants with the sorbent can be accomplished with wet methods or
combinations of wet
and dry methods. In the system of the invention, wet scrubbers can serve as
reaction zones in
the place of or in combination with the reaction zones previously described
for dry removal.
Wet scrubbers useful in the systems and processes of the invention may be of
several types,
including but not limited to slurry, sprayer, cascading, and others known to
those skilled in
the art. Whether the wet scrubber is a slurry, sprayer, cascading or other
know type of
scrubber, an acidic slurry of oxides of manganese is utilized. The acidity
serves to enhance
the effective removal of the target pollutants. For SOx and/or NOx removal,
the pH of the
slurry is preferably 2.0 or less and more preferably between about 1.5 and
about 1.75
The gas should be introduced into the wet scrubber at a temperature below the
boiling
point of the solution or slurry to prevent excess evaporation of the sorbent
slurry. Since gases
processed in the system of the invention typically are at elevated
temperatures, the gas may
be cooled to below the boiling point utilizing a heat exchangers 72A, 72B
preceding the
reaction zone as is shown in Figure 5. The gas containing taxget pollutants is
introduced into
the wet scrubber and contacted with the slurry for a time sufficient to effect
capture of a
target pollutant at a target capture rate set point for pollutants such as SOx
and/or NOx, CO
and/or COZ or TRS, forming respectively, sulfates and/or nitrates of
manganese, manganese
carbonate, or sulfates of manganese. While the sorbent itself is not soluble
in the slurry,
reactions products such as sulfates and nitrates of manganese are and dissolve
immediately
into solution. Manganese carbonate, being insoluble in aqueous solution, does
not dissolve.
With respect to the removal of CO and/or C02 to manganese carbonate, when the
sorbent is no longer effective for pollutant removal at a target capture rate
set point, the
reacted sorbent must be separated from the slurry for regeneration of the
sorbent and recovery
of useful by-products. This is accomplished through the thermal decomposition
of the
manganese carbonate as previously described.
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
54
With respect to the sulfates and/or nitrates of manganese formed in wet
removal, the
sorbent must be periodically separated from the solution. The sorbent, which
is by virtue of
being in solution, is essentially clean or "activated" and can be returned to
the scrubber in a
slurry or added to a slurry requiring additional sorbent. The point at which
periodic
separation would need to be carried out generally depends upon the capacity of
the slurry to
retain additional solute in solution, the saturation point of the solution.
The frequency at
which separation must be carried may be affected through temperature
adjustment, since
generally a saturated solution can dissolve additional solute at increased
temperatures.
However, as previously noted, the temperature should not be increased to the
boiling point of
the solution. Further, simply increasing the volume of the slurry with the
addition of acidic
aqueous solution can decrease the separation frequency, as long as the wet
scrubber has
sufficient capacity for the increased volume of the slurry. Further still, the
periodic
separation can be minimized by bleeding of the aqueous solution containing
solute from the
scrubber and with simultaneous feeding of additional, fresh aqueous solution
into the
scrubber to maintain the slurry in an unsaturated state. The solution that has
been bled from
the wet scrubber can be retained in a holding tank, vessel or other suitable
container until a
sufficient volume has be accumulated and then processed to regenerate oxides
of manganese
and to recover useful and marketable by-products.
hi a single stage wet removal process a single reaction zone, a wet scrubber,
is
utilized to remove the target pollutants.. The rate of reaction is related to
the solubility of the
reaction product of target pollutant and the sorbent. For example the
solubility of SOx is
greater than the solubility of NOx in an aqueous solution; and therefore, a
longer residence
time is required for NOx removal than for SOx removal. The gas once
substantially stripped
of the target pollutant is vented from the scrubber either to a stack or for
further processing.
A single wet scrubber can be utilized to remove one or more target pollutants;
however, the
residence time of the gas in the wet scrubber will be driven by a combination
of the solubility
of the less soluble of the target pollutants and the target capture rate of
that target pollutant.
In a single-stage wet removal system the gas is introduced into a reaction
zone configured for
the introduction of the gas and contacted with the sorbent containing slurry
for a time
sufficient to effect capture of the target pollutants) at the target capture
rate set point of the
target pollutant(s). The target pollutant is captured by reaction with the
sorbent to form
reaction products. The reaction products may be soluble in the aqueous
solution, as with
nitrates and sulfates of manganese. Or they may be insoluble as with
carbonates of
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
manganese formed during the removal of CO and/or COa. Wet removal methods can
be
utilized in either case; but are better suited for removal of target
pollutants that form soluble
reaction products.
Wet removal can also be accomplished in multiple stages with at least two
reaction
5 zones in series of which at least one of the reaction zones is a wet
scrubbers. This can be
illustrated with reference to dual-stage removal of SOx and NOx. In dual-stage
removal, first
and second reaction zones are provided. With both reactions zones being wet
scrubber s, gas
is introduced into the first reaction zone which is configured for the
introduction of a gas
containing target pollutants, in this case SOx and NOx. In the first reaction
zone, the gas is
10 contacted with a sorbent containing slurry for a time sufficient to effect
SOx capture at a
target SOx capture rate set point. The SOx is captured by reacting with the
sorbent to form
sulfates of manganese to substantially strip the gas of SOx. The gas which is
substantially
stripped of SOx is vented from the first reaction zone and passed to a second
reaction zone,
also a wet scrubber, configured for the introduction of the gas substantially
stripped of SOx.
15 In the second wet scrubber the gas is contacted with the sorbent contaiung
slurry for a time
sufficient to effect NOx capture at a target NOx capture rate set point. The
NOx is captured
by reacting with the sorbent to form nitrates of manganese to substantially
strip the gas of
NOx. The gas that has been substantially stripped of SOx and NOx is vented
from the second
reaction zone. It is readily understood by those skilled in the art that more
than two wet
20 scrubbers could be utilized in series to effect capture of multiple target
pollutants and that the
sequence of pollutant removal, in a mufti-stage removal process, would be
determined by the
relative solubilities of the reaction products generated from target
pollutants with the sorbent.
Dual stage removal may also be carried out with one of the reaction zones
being a wet
scrubber and the other reaction zone being selected from the group consisting
of a fluidized
25 bed, a pseudo-fluidized bed, a reaction column, a fixed bed, a pipe/duct
reactor, a moving
bed, a bag house, an inverted bag house, bag house reactor, serpentine
reactor, and a
cyclone/multiclone. Again using SOx and NOx for illustrative purposes, the
removal can
proceed by first wet SOx removal and dry NOx removal or by first dry SOx
removal and wet
NOx removal. Regardless of the sequence, wet removal and dry removal would
proceed as
30 previously described, with the gas substantially stripped of SOx being
directed from the first
reaction zone to the second reaction where NOx removal would occur: In a wet-
dry removal
system, the first reaction zone would be a wet scrubber; and in dry-wet
removal system the
second reaction zone would be a wet scrubber. For the dry removal stage, the
dry reaction
CA 02424120 2003-04-O1
WO 02/28513 PCT/USO1/28473
56
zone or scrubber , whether the first or second in sequence, is selected from
the
aforementioned group.
Where the reaction product of the target pollutant is soluble in aqueous
solution, the
surface area of the oxide of manganese sorbent is not as critical in a wet
removal system, i.e.,
a scrubber, as opposed to a dry removal system. Further, particle size may not
be as critical
with a liquid medium as opposed to a gas medium; however, particles must be
small enough
so that the sorbent remains sufficiently mixed in the slurry. Agitators can be
used to keep the
sorbent sufficiently mixed in the slurry. Generally, oxides of manganese
useful as a sorbent
for dry removal methods are similarly useful for wet removal methods.
The systems of the invention including those that incorporate wet scrubbers
are
adaptable; and process parameters, such as differential pressure, inlet gas
temperature, and
removal efficiency, are monitored and controlled in wet removal systems of the
invention
with electronic controls just as in dry removal systems according to the
invention.
While exemplary embodiments of this invention and methods of practicing the
same
have been illustrated and described, it should be understood that various
changes,
adaptations, and modifications might be made therein without departing from
the spirit of the
invention and the scope of the appended claims.