Note: Descriptions are shown in the official language in which they were submitted.
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
SYNTHESIS AND ANTIMICROBIAL ACTIVITY OF
NOVEL DICATIONIC "REVERSED AMIDINES"
[0001] CROSS-REFERENCE TO RELATED APPLICATIONS
[0002] The present application claims priority to United States Provisional
Application
No. 60/246,244 filed November 6, 2000, the disclosure of which is incorporated
herein by
reference in its entirety.
[0003] STATEMENT OF GOVERNMENT SUPPORT
[0004] This invention was made with government support under grant numbers
ROlAI 46365-OlA2 and RO1GN61587 from the National Institutes of Health. The
United
States government has certain rights in this invention.
[0005] FIELD OF THE INVENTION
[0006] This invention relates to the treatment of microbial infections caused
by
Mycobacterium tuberculosis, Tzyparzosoma spp., Candida albicans, Aspergillus
spp.,
Cryptosporidium parvum, Giardia lamblia, Plasmodium spp., Pneunzocystis
carinii,
Toxoplasma gondii, Fusariunz solani, and Cryptococcus neoformans.
[0007] BACKGROUND OF THE INVENTION
[0008] The incidence of microbial infections (e.g., mycobacterial, fungal and
protozoal infections) in the immunocompromised population has significantly
increased over
the past several years. In particular, Candida species, especially Candida
albicans, are often
significant pathogens in patients infected with human immunodeficiency virus
(HIV).
Another pathogen, Pzzeunzocystis carinii, causes a form of pneumonia (PCP)
that is believed
to be one of the leading causes of death in patients suffering from AIDS.
[0009] Human African trypanosomiasis (HAT) has reemerged as a threat to over
60
million people. Current estimates are that between 350,000 and 450,000 people
are infected.
[0010] Other severe and life-threatening microbial infections are caused by
Mycobacterium tuberculosis, Aspergillus spp., Cryptosporidium parvum, Giardia
larnblia,
Plasmodium spp., Toxoplasma gondii, Fusarium solani, and Cryptococcus
neofornzans.
[0011] The antimicrobial properties of dicationic molecules have been studied
since
1
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
the 1930's. Compounds of this type have typically utilized amidine groups as
the cationic
moieties, and their activities against a number of pathogens including
Cryptosporidiurn
parvurn, Giar~dia lamblia, Leishmania spp., Plasrnodium spp., Pneumocystis
carinii,
Toxoplasma gondii, Trypanosoma spp., Candida albicans, Aspergillus spp., and
Cryptococcus neofor~rnans have been reported. See e.g., King, H. et al., Ann.
Trop. Med.
Parasitol. 1938, 32,177-192; Blagburn, B. L. et al., Antimicrob. Agents
Chemother. 1991, 35,
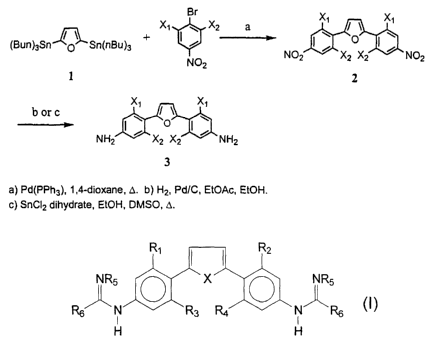
1520- 1523; Bell, C. A. et al., Antinzicr~ob. Agents Chemother~. 1991, 35,
1099-1107; Bell, et
al., Aratirnicrob. Agents Chemother~. 1990, 34, 1381-1386; Kirk, R.et al.,
Ann. Trop. Med.
Parastiol. 1940, 34, 181=197; Fulton, J. D. Ann. Trop. Med. Parasitol. 1940,
34, 53-66;
Ivady, V. G. et al., Monatschr~. Kinder~heilkd. 1958, 106, 10-14; Boykin, D.
W. et al., J. Med.
Chem. 1995, 38, 912-916; Boykin, D. W. et al., J. Med. Chem. 1998, 41, 124-
129;
Francesconi et al., J. Med. Chem.,1999, 42, 2260-2265; Lindsay, D. S. et al.,
Antimicr~ob.
Agents Chemother. 1991, 35, 1914-1916; Lourie, E. M; et al., Ann. Trop. Med.
Parasitol.
1939, 33, 289-304; Lourie, E. M. et al., Ann. Trop. Med. Pa>~asitol. 1939, 33,
305-312; Das,
B. P. et al., JMed. Chem. 1976, 20, 531-536; Del Poeta, M. et al., J.
Antimicrob. Chemother.
1999, 44, 223-228; Del Poeta, M. et al., Antimicr~ob. Agents Chemother~. 1998,
42, 2495-
2502; Del Poeta, M. et al., Antimicrob. Agents Clzemother. 1998, 42, 2503-
2510.
[0012] Despite the broad range of activity exhibited by diamidines, only one
compound of this chemical type, pentamidine, has seen significant clinical
use. Pentamidine
has been used clinically against African trypanosomiasis, antimony-resistant
leishmaniasis
and P. carinii pneumonia. See e.g., Apted, F. I. C., Pharmacol. There. 1980,
Il, 391-413;
Bryceson, A. D. M.et al., Ti~ans. Roy. Soc. T>"op. Med. Hyg. 1985, 79, 705-
714; Hughes, W.
T.; et al., Antimicrob. Agents Chemother~. 1974, 5, 289-293.
[0013] A number of compounds in this class of dicationic molecules have been
shown
to bind to the minor-groove of DNA at AT-rich sites and the details of their
interaction with
the minor-groove have been elucidated from biophysical studies and from
several crystal
structures. It is hypothesized that these types of molecules exert their
biological activity by
first binding to DNA and then by inhibiting one or more of several DNA
dependent enzymes
(i.e., topoisomerases, nucleases, etc.) or possibly by direct inhibition of
transcription. See,
Tanious, F. A. et al., J. Biomol. Struct. & Dyn. 1994, 1l, 1063-1083.; Wilson,
W. D. et al.,
J. Am. Chern. Soc. 1998, 120, 1'0310-10321; Bailly, C.et al., Anti-Cancer Drug
Design, 1999,
14, 47-60; Mazur, et al., J. Molecular Biology 2000, 300, 321-337; Trent, J.
O.; et al., J.
Med. Chem. 1996, 36, 4554-4562; Guerri, A. et al., Nucleic Acids Res. 1998,
26, 2873-2878;
Laughton, C. A. et al., Biochemistry 1996, 35, 5655 -5661; Beerman, T. A. et
al., Biochim.
2
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
Biophys. Acta 1992, 1131, 52-61; Bell, C. A.; et al., Antimicr~ob. Agents
Chenaother°. 1993,
37, 2668-2673; Dykstra, C. C. et al., Antimicrob. Agents Chenaother. 1994, 38,
1890-1898;
Hildebrandt, E. et al., J. Euk. Microbial. 1998, 45, 112-121; Henderson, D. et
al., Nature
Medicine 1995, l, 525-527; Fitzgerald, D. J.; et al., J. Biol. Chena 1999,
274, 27128-27138.
[0014] 2,5-Diphenylfuran and 2,4-diphenylfuran diamidines have been found to
be
highly effective treatments in animal models for Pneumocystis carinii and
Cryptospor~idiurn
parvum. See Blagburn, B.L. et al., Antirnicf~ob. Agents Chenaother". 1991; 35,
1520- 1523;
Boykin, D. W. et al., J. Med. Chem. 1995, 38, 912-916; Boykin, D. W. et al.,
J. Med. Chern.
1998, 41, 124-129; Francesconi, I. et al., J. Med. Chern. 1999, 42, 2260-2265;
Tidwell, R. R.
J . Par~asitol. 1998, 84, 851-856. Furthermore, these molecules have shown
antifimgal
activity in vitro against Candida albicans and Cryptococcus neoformans. See,
Del Poeta, M.
et al., J. Antimicrob. Chemother. 1999, 44, 223-228; Del Poeta, M. et al.,
Antimicrob. Agents
Chemother. 1998, 42, 2495-2502; Del Poeta, M. et al., Antimicrob. Agents
Chenaother. 1998,
42, 2503-2510.
[0015] Although there are reports of antimicrobial activity of guanidino
compounds,
this class of cationic compounds has not been studied as extensively as their
amidino analogs.
See Lourie et al., Ann. Ti~op. Med. Par"asitol. 1937, 31, 435-445.
[0016] SUMMARY OF THE INVENTION ,
[0017] The synthesis, DNA-binding affinities and antimicrobial properties of
2,5-bis
~ [alkyl (or aryl) imino] aminophenyl~ furans and thiophenes are described.
These compounds
have the imino group of the amidine attached to an "anilino" nitrogen (in
contrast to the
known amidino furans in which the imino group is directly attached to the aryl
ring). These
compounds, hereafter, are referred to as "reversed" amidines. The various
effects of placing
substituents on the central phenyl rings of the 2,5-diphenylfuran framework of
this class of
compounds are also described.
[0018] One aspect of the invention relates to novel compounds that are useful
in
treating microbial infections caused by Mycobacterium tuberculosis,
Tryparaosoma spp.,
Candida albicans, Asper~gillus spp., Cr yptospor~idium parvurn, Giardia
lamblia, Plasmodium
spp., Pneumocystis carinii, Toxoplasma gondii, Fusar~ium solani, and
Cryptococcus
heofor~maras. Compounds of the present invention have a structure according to
Formula I:
3
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
NR5 ~ ~X~ ~ NR5
R ~ N R N"
6 I 3 R4 I R6
[0019] H H
[0020] wherein:
[0021] Rl, R2, R3 and R4 are each independently selected from the group
consisting of
H, alkyl, alkoxy, halide, and alkylhalide groups;
[0022] RS is H, alkyl or aryl;
[0023] R6 is H, alkyl, aryl, or NR~RB, wherein R~ and R8 are each
independently
selected from the group consisting of H, alkyl and aryl; and
[0024] X is O, S or NR9, wherein R~ is H or alkyl.
[0025] Additional aspects of the invention include pharmaceutical compositions
comprising a compound having a structure according to Formula I, or a
pharmaceutical salt
thereof (i.e., an "active compound"), in a pharmaceutically acceptable
carrier.
Pharmaceutical compositions of the present invention are useful in the
treatment of microbial
infections caused by Mycobacterium tuberculosis, Trypanosoma spp., Caudida
albicans,
Aspergillus spp., C~yptosporidium parvum, Giardia lamblia, Plasmodium spp.,
Pneumocystis
carir~ii, Toxoplasma gondii, Fusarium solarci, and Cryptococcus r~eoformans.
[0026] Certain aspects of the invention relate to methods of treating
microbial
infections caused by Mycobacterium tuberculosis, Trypanosoma spp., Candida
albicans,
Aspergillus spp., Cryptosporidium parvum, Giardia lamblia, Plasmodium spp.,
Prceumocystis
carinii, Toxoplasma gor~dii, Fusarium sola~i, and Cryptococcus neoformans, in
a subject in
need of such treatment. The method comprises administering to the subject a
compound
according to Formulas (I), or a pharmaceutically acceptable salt thereof, in
an amount
effective to treat the microbial infection.
[0027] A further aspect of the present invention is the use of the active
compounds
described herein for the manufacture of a medicament for the treatment of
microbial
infections caused by Mycobacterium tuberculosis, Tyypanosoma spp., Candida
albicar~s,
Aspergillus spp., Cryptosporidium parvum, Giardia larnblia, Plasrnodiuna spp.,
Prceumocystis
carinii, Toxoplasma gorrdii, Fusarium solani, and Cryptococcus neoformans in a
subject in
need of such treatment.
[0028] The foregoing and other aspects of the present invention are explained
in detail
in the specification set forth below.
4
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
[0029] BRIEF DESCRIPTION OF THE DRAWINGS
[0030] FIG. 1 illustrates various chemical schemes that may be useful in the
synthesis
of compounds of the present invention.
[0031] DETAILED DESCRIPTION OF THE INVENTION
[0032] The present invention now will be described more fully hereinafter with
reference to the accompanying specification and drawings, in which preferred
embodiments
of the invention are shown. This invention may, however, be embodied in many
different
forms and should not be construed as limited to the embodiments set forth
herein. Rather,
these embodiments are provided so that this disclosure will be thorough and
complete, and
' will fully convey the scope of the invention to those skilled in the art.
[0033] The terminology used in the description of the invention herein is for
the
purpose of describing particular embodiments only and is not intended to be
limiting of the
' invention. As used in the description of the invention and the appended
claims, the singular
forms "a", "an" and "the" are intended to include the plural forms as well,
unless the context
clearly indicates otherwise.
[0034] Unless otherwise defined, all technical and scientific terms used
herein have
the same meaning as commonly understood by one of ordinary skill in the art to
which this
invention belongs. All publications, patent applications, patents, and other
references
mentioned herein are incorporated by reference in their entirety.
[0035] With respect to the compounds of the formulas as used herein, the term
"alkyl"
refers to C1-10 inclusive, linear, branched, or cyclic, saturated or
unsaturated (i.e., alkenyl
and alkynyl) hydrocarbon chains, including for example, methyl, ethyl, propyl,
isopropyl,
butyl, isobutyl, tert-butyl, pentyl, hexyl, octyl, ethenyl, propenyl, butenyl,
pentenyl, hexenyl,
octenyl, butadienyl, propynyl, butynyl, pentynyl, hexynyl, heptynyl, and
allenyl groups. The
term "alkyl" specifically includes cycloakyl hydrocarbon chains, which as used
herein refers
to C3 to C6 cyclic alkyl, such as cyclopropyl, cyclobutyl, cyclopentyl, and
cyclohexyl. In the
present invention, preferred alkyls are the lower alkyls. The term "lower
alkyl" refers to Cl
to C4 linear or branched alkyl, such as methyl, ethyl, propyl, butyl,
isopropyl, sec-butyl, and
tent-butyl.
[0036] The term "alkyl" also encompasses substituted alkyls, which include
aminoalkyls, hydroalkyls, oxygen-substituted alkyls (i. e., alkoxy groups),
and halogen-
substituted alkyls (i. e., alkyl halides, polyhaloalkyls). The term
"aminoalkyl," as used herein,
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
refers to C 1 to C4 linear or branched amino-substituted alkyl, wherein the
term "amino"
refers to the group NR'R", and wherein R' and R" are independently selected
from H or lower
alkyl as defined above, i.e., -NH2, -NHCH3, -N(CH3)2, etc. The term
"hydroxyallcyl" as used
herein refers to C 1 to C4 linear or branched hydroxy-substituted alkyl, i.
e., -CHZOH, -
(CH2)ZOH, etc. The term "alkoxy" as used herein refers to C 1 to C4 oxygen-
substituted alkyl,
i. e., -OCH3. The term "loweralkoxy," as used herein, refers to C 1 to C4
linear or branched
alkoxy, such as methoxy, ethoxy, propyloxy, butyloxy, isopropyloxy, and t-
butyloxy.
[0037] The terms "halide" has its conventional meaning and refers to fluoro,
chloro,
bromo, and iodo groups. Preferred halide groups include chloro groups, and
preferred alkyl
halides of the present invention include CF3.
[0038] The term "aryl" as used herein refers to C3 to C10 cyclic aromatic
groups such
as phenyl, naphthyl, and the like, and specifically includes substituted aryl
groups including
but not limited to tolyl, substituted phenyl, and substituted naphthyl. Aryl
groups may be
substituted with halide, amino, nitro, and the like. Heterocyclic aromatic
rings and polycyclic
aromatic groups are also included in this definition of "aryl." Specific
examples of aryl
groups encompassed by the present invention include but are not limited to
substituted and
unsubstituted pyridine, quinoline, cyclopentadienyl, phenyl, furan, thiophene,
pyrrole, pyran,
imidazole, isothiazole, isoxazole, pyrazole, pyrazine, pyrimidine, and the
like. Preferred aryl
groups include pyridine, substituted pyridine (e.g., 5-methylpyridine), and
quinoline.
[0039] The compounds of the present invention are also useful in the form of
their
pharmaceutically acceptable salt forms. Such salts may include, but are not
limited to, the
gluconate, lactate, acetate, tartarate, citrate, phosphate, borate, nitrate,
sulfate, hydrobromide
and hydrochloric salts of the compounds. Compomds of Formula (I) and their
pharmaceutically acceptable salts are referred to herein as "active compounds"
or "active
agents."
[0040] The compounds represented by Formula (I) may be formed by synthesis
procedures that are described in the Examples below, as well as by certain
methods known in
the art. Some of these known methods are set forth below in the Examples by
description or
by reference (the disclosures of which are all incorporated herein by
reference in their
entirety).
[0041] As noted above, the compounds, methods and compositions of the present
invention are useful for treating infections caused by Mycobacterium
tuberculosis,
Trypa~osoma spp., Cahdida albicans, Aspergillus spp., Cryptosporidiurn parvum,
Giardia
lamblia, Plasmodium spp., Pt~eumocystis carirZii, Toxoplasma go~dii,
Fusaf°ium solani, and
6
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
Cryptococcus neoformarZS. In a preferred embodiment, the methods and
compositions of the
present invention are used to treat Mycobacter-iurrz tuberculosis infections.
In another
preferred embodiment, the methods and compositions of the present invention
are used to
treat Candida albicans infections. In another preferred embodiment, the
methods and
compositions of the present invention axe used to treat infections caused by
Aspergillus spp.
In another preferred embodiment, the methods and compositions of the present
invention are
used to treat infections caused by Ti~ypanosoma spp. The present invention is
useful for
treating all known species of Trypanosoma, with Trypanosoma brucei rhodesiense
and
Tr ypanosoma cruzi being particularly preferred.
Table I. Examples of inventive compounds
R1 ~ ~ R~
w I \~ I ~
Y R2 Rz Y
Compound X R1, Y
Number R2
'
5a O Rl R2 = H NHAm
=
6a O Rl = = H NHC(=NH)-2-Pyr
R2
6b O Rl = = H NHC(=NH)Ph
R2
6c O Rl = = H NHC(=NH)Ph-4-CH3
Ra
6d O Rl R2 = H NHC(=NH)-c-hexane
=
6e O Rl = = H NHC(=NH)CH3
R2
6f O Rl = RZ = CH3 NHC(=NH)CH3
H,
5b O Rl = R2 = CH3 NHAm
H,
5c O Rl = Ra = OCH3 NHAm
H,
5d O Rl = Ra = Cl NHAm
H,
5e O Rl = R2 = CF3 NHAm
H,
5f O Rl = = CH3 NHAm
R2
6 O Rl = RZ = CH3 NHC(=NH)Ph
H,
6h O Rl = R2 = CH3 NHC( NH)-2-Pyr
H,
6i O Rl = R2 = CH3 NHC(=NH)-2-Qu
H,
6' O Rl = R2 = CH3 NHC(=NH)-2-Pyr-5-CH3
H,
6k O Rl = R2 = OCH3 NHC(=NH)-2-Pyr
H,
6I O Rl = R2 = CI NHC(=NH)-2-Pyr
H,
6m O Rl = = CH3 NHC(=NH)-2-Pyr
R2
DB _686_ S Rl = NHAm
R2
=
H
DB 653 S RI = = H NHC(--NH)Ph
R2
[0042] Examples of compounds of the present invention are set forth in Table
1, in
which Pyr means pyridine, c-hexane means cyclohexane, Ph means phenyl, Am
means
amidine, and Qu means quinoline.
7
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
[0043] In one embodiment of the invention, a subject afflicted with a
microbial
infection described herein is administered a therapeutically-effective amount
of the
compound of Formula (I), or a pharmaceutically acceptable salt thereof. A
"therapeutically-
effective" amount as used herein is an amount of a compound of Formula (I)
that is sufficient
to alleviate (e.g., mitigate, decrease, reduce) at least one of the symptoms
associated with the
microbial infection. It is not necessary that the administration of the
compound eliminate the
symptoms of the infection, as long as the benefits of administration of
compound outweigh
the detriments. Likewise, the terms "treat" and "treating", as used herein,
are not intended to
mean that the subject is necessarily cured of the microbial infection, or that
all clinical signs
thereof axe eliminated, only that some alleviation or improvement in the
condition of the
subject is effected by administration of the compound of Formula (I).
[0044] Suitable subjects of the present invention include humans and animals.
When
the subject is an animal, mammals are preferred, with livestock and primates
being
particularly preferred. Humans axe the most preferred subjects. Subjects may
be adult,
adolescent, juvenile, infant, or neonatal.
[0045] Subjects may be administered the compounds and compositions of the
present
invention by any suitable means. Exemplary means are oral administration
(e.g., in the form
of a liquid or solid), intramusculax injection, subcutaneous injection, and
intravenous
injection. Pharmaceutical formulations of the present invention comprise
active compounds
of the invention in a pharmaceutically acceptable carrier. Suitable
pharmaceutical
formulations include those suitable for inhalation, oral, rectal, topical,
(including buccal,
sublingual, dermal, vaginal and intraocular), parenteral (including
subcutaneous, intradermal,
intramuscular, intravenous and intraarticular) and transdermal administration.
The most
suitable route of administration in any given case may depend upon the
anatomic location of
the condition being treated in the subject, the nature and severity of the
condition being
treated, and the particular active compound which is being used. The
formulations may
conveniently be presented in unit dosage form and may be prepared by any of
the methods
well known in the art.
[0046] In the manufacture of a medicament according to the invention (the
"formulation"), active compounds or the pharmaceutically acceptable salts
thereof (the
"active compounds") axe typically admixed with, inter nlia, an acceptable
carrier. The carrier
must, of course, be acceptable in the sense of being compatible with any other
ingredients in
the formulation and must not be deleterious to the patient. The carrier may be
a solid or a
liquid, or both, and is preferably formulated with the compound as a unit-dose
formulation,
8
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
for example, a tablet, which may contain from 0.5% to 99% by weight of the
active
compound. One or more active compounds may be incorporated in the formulations
of the
invention, which formulations may be prepared by any of the well known
techniques of
pharmacy consisting essentially of admixing the components, optionally
including one or
more accessory therapeutic ingredients.
[0047] Formulations suitable for oral administration may be presented in
discrete
units, such as capsules, cachets, lozenges, or tablets, each containing a
predetermined amount
of the active compound; as a powder or granules; as a solution or a suspension
in an aqueous
or non-aqueous liquid; or as an oil-in-water or water-in-oil emulsion. Such
formulations may
be prepared by any suitable method of pharmacy which includes the step of
bringing into
association the active compound and a suitable carrier (which may contain one
or more
accessory ingredients as noted above). In general, the formulations of the
invention are
prepaxed by uniformly and intimately admixing the active compound with a
liquid or finely
divided solid carrier, or both, and then, if necessary, shaping the resulting
mixture. For
example, a tablet may be prepared by compressing or molding a powder or
granules
containing the active compound, or optionally with one or more accessory
ingredients.
Compressed tablets may be prepared by compressing, in a suitable machine, the
compound in
a free-flowing form, such as a powder or granules optionally mixed with a
binder, lubricant,
inert diluent, and/or surface active/dispersing agent(s). Molded tablets may
be made by
molding, in a suitable machine, the powdered compound moistened with an inert
liquid
binder. Formulations for oral administration may optionally include enteric
coatings known
in the art to prevent degradation of the formulation in the stomach and
provide release of the
drug in the small intestine.
[004] Formulations of the present invention suitable for parenteral
administration
comprise sterile aqueous and non-aqueous injection solutions of the active
compound, which
preparations are preferably isotonic with the blood of the intended recipient.
These
preparations may contain anti-oxidants, buffers, bacteriostats and solutes
which render the
formulation isotonic with the blood of the intended recipient. Aqueous and non-
aqueous
sterile suspensions may include suspending agents and thickening agents. The
formulations
may be presented in unit\dose or multi-dose containers, for example sealed
ampoules and
vials, and may be stored in a freeze-dried (lyophilized) condition requiring
only the addition
of the sterile liquid cannier, for example, saline or water-for-injection
immediately prior to
use. Extemporaneous injection solutions and suspensions may be prepared from
sterile
powders, granules and tablets of the kind previously described. For example,
in one aspect of
9
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
the present invention, there is provided an injectable, stable, sterile
composition comprising a
compound of Formula (I) , or a salt thereof, in a unit dosage form in a sealed
container. The
compound or salt is provided in the form of a lyophilizate which is capable of
being
reconstituted with a suitable pharmaceutically acceptable carrier to form a
liquid composition
suitable for injection thereof into a subject. The unit dosage form typically
comprises from
about 10 mg to about 10 grams of the compound or salt. When the compound or
salt is
substantially water-insoluble, a sufficient amount of emulsifying agent which
is
physiologically acceptable, may be employed in sufficient quantity to emulsify
the compound
or salt in an aqueous carrier.
[0049] Further, the present invention provides . liposomal formulations of the
compounds disclosed herein and salts thereof. The technology for forming
liposomal
suspensions is well known in the art. When the compound or salt thereof is an
aqueous-
soluble salt, using conventional liposome technology, the same may be
incorporated into lipid
vesicles. In such an instance, due to the water solubility of the compound or
salt, the
compound or salt will be substantially entrained within the hydrophilic center
or core of the
liposomes. The lipid layer employed may be of any conventional composition and
may
either contain cholesterol or may be cholesterol-free. When the compound or
salt of interest
is water-insoluble, again employing conventional liposome formation
technology, the salt
may be substantially entrained within the hydrophobic lipid bilayer which
forms the structure
of the liposome. In either instance, the liposomes produced may be reduced in
size through
the use of standard sonication and homogenization techniques or other
techniques known in
the art.
[0050] Of course, the liposomal formulations containing the pharmaceutically
active
compounds identified with the methods described herein may be lyophilized to
produce a
lyophilizate which may be reconstituted with a pharmaceutically acceptable
carrier, such as
water, to regenerate a liposomal suspension.
[0051] In addition to the active compounds, the pharmaceutical formulations
may
contain other additives, such as pH-adjusting additives. In particular, useful
pH-adjusting
agents include acids, such as hydrochloric acid, bases or buffers, such as
sodium lactate,
sodium acetate, sodium phosphate, sodium citrate, sodium borate, or sodium
gluconate.
Further, the compositions may contain microbial preservatives. Useful
microbial
preservatives include methylparaben, propylparaben, and benzyl alcohol. The
microbial
preservative is typically employed when the formulation is placed in a vial
designed for
multidose use.
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
[0052] Pharmaceutical formulations of the present invention may comprise
compounds of the present invention in lyophilized form. Alternatively,
pharmaceutical
formulations of the present invention may comprise compounds of the present
invention in a
pharmaceutically acceptable carrier. Such pharmaceutical formulations are
generally made
by admixing the compounds described herein with a pharmaceutically acceptable
carrier.
Pharmaceutically acceptable carriers are preferably liquid, particularly
aqueous, carriers, the
selection of which are known in the art. For the purpose of preparing such
formulations, the
compound may be mixed in a buffered saline (e.g., pH 6 to 8) or conventional
culture media.
The formulation may be stored in a sterile glass container sealed with a
rubber stopper
through which liquids may be injected and formulation withdrawn by syringe.
[0053] With respect to all the methods described herein, a therapeutically
effective
dosage of any specific compound, the use of which is in the scope of present
invention, may
vary somewhat from compound to compound and subject to subject, and will
depend upon
the condition of the subject and the route of delivery. A dosage from about
0.5 mg/kg to
about 15 mg/kg of subject body weight, or about 20 mg/kg of subject body
weight, or even
about 25 mglkg of subject body weight may be employed for intravenous
injection or oral
administration.
[0054] The concentration of the compound of the present invention or a
pharmaceutically acceptable salt thereof in a formulation of the present
invention may be
determined by the skilled artisan and will vary according to certain
conditions, including the
characteristics of subject being treated (e.g., species, age, weight), the
severity and type of the
infecting virus or the strain that the subject is being vaccinated against,
the dosage form being
used, and the like.
[0055] The compounds of the present invention may be administered in
conjunction
with other antiviral compounds, as may be determined by the skilled artisan.
[0056] The present invention is explained in greater detail in the Examples
which follow.
These examples are intended as illustrative of the invention, and are not to
be taken as limiting
thereof.
[0057] EXAMPLE 1
[0058] General Methodology: Chemical Synthesis and Analysis
[0059] Melting points were determined with a MEL-TEMP° 3.0 capillary
melting
point apparatus and are uncorrected. 1H nuclear magnetic resonance spectra
were recorded
on a Varian Unity+300 or a Varian VRX 400 instrument, with peak assignments
relative to
11
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
residual DMSO (2.49ppm) or CHCl3 (7.24ppm). Mass spectra were recorded on a VG
Instruments 70-SE spectrometer at the Georgia Institute of Technology,
Atlanta, GA.
Elemental analyses were performed by Atlantic Microlab, Norcross, GA. All
final
compounds were dried i~ vacuo (oil pump) at 50-60 °C for at least 36
hours before elemental
analysis. Unless otherwise stated, all reagent chemicals and solvents
(including anhydrous
solvents) were purchased from Aldrich Chemical Co., Fisher Scientific, or
Lancaster
Synthesis and used as received. Acetonitrile (CaH2), triethylamine (CaH2), and
ethanol
(Mg/I2) were distilled from the indicated. drying agent. 2,6-Dimethyl-4-
nitrobromobenzene
and S-(2-Naphthylmethyl) thioacetimidate were prepared according to the
literature. See
B.M. Wepster, Rec. Trav. Chim. 73, 809-818 (1954); D.N. Kravtsov, J.
Organometal. Chem.
36, 227-237 (1972); B.G. Shearer et al., Tetrahedron Lett. 38, 179-182 (1997).
[0060] FIG. 1 sets forth representative schemes for the synthesis of compounds
of the
present invention. Although the schemes set forth in the figure relate to
furan compounds,
the methods illustrated therein may also be carried out with analogous
compounds such as
thiophene compounds. Referring to FIG. l, the synthesis for target compounds
required the
corresponding diamino compounds (Scheme 1). The synthesis of the diamino
compounds
begins with Stille coupling between a 2,5-distannyl furan or thiophene and
substituted
bromonitroarenes to form the corresponding 2,5-bis[nitrophenyl] furans and
thiophenes.
Reduction of the 2,5-bis-nitrophenyl heterocycles either by catalytic
hydrogenation or by
stannous chloride produced the desired diamino compounds (Scheme 1). The
required
diamino analogs were obtained by the two step conversion of 2,5-
bis[nitrophenyl] furans and
thiophenes to amines involving Pd(0) coupling of benzophenone imine to form
the
corresponding arylimino compounds. The diguanidinium analogs were prepared by
the
reaction of the aryl diamines with Boc-protected S-methylthiourea in the
presence of
mercuric chloride (Scheme 2). Reaction of the aryl diamines with two
equivalents of S-(2-
naphthylmethyl)thiobenzimidate produced the "reversed" amidines in good yields
(Scheme
4).
[0061] The 2,5-bis(4-aminophenyl)furans and -thiophenes (Scheme 1) were
prepared
in good yield by reduction of the corresponding bis-nitro derivatives using
either catalytic
hydrogenation (Pd/C), stannous chloride, or iron/AcOH. The bis-nitro
derivatives, in tum,
were prepared by the palladium catalyzed coupling of 2,5-bis(tri-n-
butylstannyl)furan or 2,5-
bis(trimethylstannyl)thiophene with the appropriate halonitrobenzene.
[0062] In the following examples, compound numbers refer to the corresponding
compounds in FIG. 1.
12
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
[0063] EXAMPLE 2
[0064] Preparation of 2,5-bis(4-nitrophenyl)furans
[0065] The following representative procedures are variations of a general
procedure
previously described in A. Kumar et al., Heterocyclic Conam. 5, 301-304
(1999).
[0066] 2,5-Bis(4-nitr~ophenyl)furan (Compound 2a). Yield: 88%; orange fluffy
solid;
mp 269-270 °C (not recrystallized), lit. mp 270-272 °C, Ling, C.
et al., J. Am. Chern. Soc.
1994,116, 8784-8792.
[0067] 2, 5-Bis(2-methyl-4-hitr~ophenyl)furar~ (Compound 2b). To a solution of
2-
bromo-5-nitrotoluene (4.32 g, 20 mmol) and tetrakis (triphenylphospine)
palladium (0) (0.40
g) in anhydrous 1,4-dioxane (50 ml) was added 2,5-bis (tri-n-butylstannyl)
furan (6.46 g, 10
mmol) and the mixture was heated overnight under nitrogen at 95-100 °C.
The resulting
orange suspension was diluted with hexanes (15 ml), cooled to room-
temperature, and
filtered to give, after rinsing with hexanes, an orange solid (3.10 g), mp 241-
243 °C. The
product was recrystallized from DMF (100 ml) to give a bright orange fluffy
solid (2.87 g,
85%), mp 242-243 °C. 1H NMR (DMSO-d6): 2.69 (s, 6H), 7.31 (s, 2H), 8.12
(m, 4H), 8.23
(s, 2H). Anal. Calcd. for C18H14NZO5 (338.31): C, H, N.
[0068] 2, 5-Bis(2-methoxy-4-nitr~ophenyl)furan (Compound 2c). Yield: 77%;
bright
orange granular solid; mp 308-310 °C (DMF). tH NMR (DMSO-d6): 4.10 (s,
6H), 7.37 (s,
2H), 7.90 (s, 2H), 7.94 (d, 2H), 8.22 (d, 2H). Anal. Calcd. for
C18H14N20~~O.1H20 (372.11):
C, H, N.
[0069] 2,5-Bis(2-chloro-4-nitrophenyl)furan (Compound 2d). Yield: 71%; fluffy
orange solid; mp 247-247.5 °C (DMF/MeOH). 1H NMR (DMSO-d6): 7.70 (s,
2H), 8.29 (dd,
J = 8.8, 2.2 Hz, 2H), 8.36 (d, J = 8.8 Hz, 2H), 8.43 (d, J = 2.2 Hz, 2H).
Anal. Calcd. for
C16H8C12N205 (379.15): C, H, N.
[0070] 2,5-Bis(4-vitro-2-trifluorornethylphenyl)furan (Compound 2e). Yield:
74%;
fluffy golden needles; mp 158.5-159 °C (EtOH). 1H NMR (DMSO-d6): 7.38
(s, 2H), 8.24 (d,
J = 8.7 Hz, 2H), 8.57 (d, J = 2:4 Hz, 2H), 8.62 (dd, J = 8.6, 2.4 Hz, 2H).
Anal. Calcd. for
C18H8F6N205 (446.26): C, H, N.
13
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
[0071] 2,5-Bis(2,6-dimethyl-4-nit~ophenyl)fur°ah (Compound 2f). Yield:
65%; yellow
needles; mp 156.5-157.5 °C (DMF/EtOH/H20). 1H NMR (DMSO-d6): 2.34 (s,
12H), 6.85
(s, 2H), 8.04 (s, 4H). Atzal. Calcd. for C2oH1gN2O5 (366.36): C, H, N.
[0072] EXAMPLE 3
[0073) Preparation of 2,5-bis(4-aminophenyl)furans
[0074] (The following procedures are representative)
[0075] 2, S-Bis(4-aminophe~cyl)furan (Compound 3a). Yield: 94%; pale
green/tari
solid; mp 218-221 °C,1it46 mp 213-216 °C. MS (EI): m/z 250 (M+).
[0076] 2, 5-Bis(4-amino-2-methylphenyl)fur an (Compound 3b). To a suspension
of
the bis-nitro derivative 2b (2.87 g) in EtOAc (90 ml) and dry EtOH (10 ml) was
added Pd/C
(10%) (0.40 g) and the mixture was hydrogenated on a Parr apparatus at an
initial pressure of
~SOpsi. After the uptake of hydrogen subsided (generally 3-6 hours), the
resulting solution
was filtered over Celite and the pale yellow to colorless filtrate was
concentrated ire vacuo to
near dryness to give, after dilution with hexanes, the pure diamine as a pale
yellow/green
solid (2.17 g, 91%), mp 174-176 °C, which required no purification. 1H
NMR (DMSO-d6):
2.33 (s, 6H), 5.15 (br s, 4H), 6.42 (s, 2H), 6.46 (m, 4H), 7.35 (d, 2H). MS
(EI): m/z 278
(M+).
[0077] 2,5-Bis(4-amino-~-methoxyphenyl)furah (Compound 3c). The original oiI
was
reconcentrated with benzene to give a yellow/tan solid which was triturated
with ether.
Yield: 79%; mp 201-202.5 °C. 1H NMR (DMSO-d6): 3.80 (s, 6H), 5.25 (br
s, 4H), 6.24 (dd,
J = 8.3, 2.0 Hz, 2H), 6.30 (d, J = 1.9 Hz 2H), 6.56 (s, 2H), 7.48 (d, J = 8.4
Hz, 2H). MS (EI):
m/z 310 (M+).
[0078] 2, 5-Bis(4-amino-2-chlorophenyl)fu~an (Compound 3d). To a suspension of
the
corresponding bis-nitro derivative 2d (1.22 g, 3.2 mmol) in dry EtOH (100 ml)
and DMSO
(20 ml) was added SnCl2~2H20 (5.80 g, 25.7 mmol) and the mixture was heated
under
nitrogen at 80 °C. After 4-5 hours, TLC showed that starting material
had been consumed,
and thus the mixture was cooled, neutralized with NaOH (aq), and extracted
with EtOAc.
The extract was washed with water, brine, then dried (Na2SO4) and
concentrated. The
resulting oil was crystallized from benzenelhexane with partial concentration
to give a light
74
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
brown solid (0.74 g, 71%), mp 191.5-193 °C. Catalytic hydrogenation was
not explored. 1H
NMR (DMSO-d6): 5.60 (br s, 4H), 6.61 (dd, J = 8.6, 2.2 Hz, 2H), 6.68 (d, J =
2.2 Hz 2H),
6.82 (s, 2H), 7.56 (d, J = 8.6 Hz, 2H). MS (EI): rnlz 318 (M+).
[0079] 2,5-Bis(4-amino-~-trifluof~omethylphenyl)furan (Compound 3e). Original
red
oil crystallized from EtOAclhexanes in two crops as a red/orange solid.
Combined yield:
81%; mp (first/major crop) 89.5-91 °C; mp (second crop) 91.5-92
°C. 1H NMR (DMSO-d6):
5.79 (br s, 4H), 6.52 (s, 2H), 6.82 (dd, J = 8.4, 2.4 Hz, 2H), 6.98 (d, J =
2.2 Hz, 2H), 7.43 (d,
J = 8.4 Hz, 2H). MS (EI): m/z 386 (M+).
[0080] 2, S-Bis(4-amino-2, 6-dimethylphenyl)furan (Compound 3f). Yield: 99%;
white
fluffy solid; mp 144.5-146 °C. 1H NMR (DMSO-d6): 2.01 (s, 6H), 5.06 (br
s, 4H), 6.24 (s,
2H), 6.29 (s, 4H). MS (EI): rnlz 306 (M+).
[0081] EXAMPLE 4
[0082] Preparation of 2,5-bis(4 N,N'-di-BOC-guanidinophenyl)furan derivatives
[0083] (The following procedures are representative) (See Scheme 2).
[0084] 2, 5-Bis(4-N,N'-di-BOCguanidinophenyl)furan (Compound 4a). To a room-
temperature solution of 2,5-bis(4-aminophenl)furan (0.626 g, 2.5 mmol) and 1,3-
bis(tert-
butoxycarbonyl)-2-methyl-2-thiopseudourea (1.56 g, 5.3 mmol) in anhydrous DMF
was
added triethylamine (1.59 g, 15.7 mmol) followed by mercury(II) chloride (1.57
g, 5.8 mmol)
and the resulting suspension was stirred at room-temperature for 22 hours.
After diluting
with CH2Cl2 and sodium carbonate solution, the suspension was filtered over
Celite and the
filtrate was washed well with water (3X) and finally with brine. After drying
(NaZS04), the
solvent was removed in vacuo and the residue was diluted with MeOH to give the
BOC-
protected bis-guanidine as a pale yellow solid. The collected product was
purified by
reprecipitation from CH2C12/MeOH to give a fluffy yellow solid (1.25 g, 68%),
mp >400 °C
dec. 1H NMR (CDC13): 1.50 and 1.53 (2s, 36H), 6.65 (s, 2H), 7.66 (s, 8H),
10.38 (br s, 2H),
11.61 (br s, 2H).
[0085] 2,5-Bis(2-methyl-4-N,N'-di-BOCguanidinophenyl)furan (Compound 4b).
Yellow solid, mp >250 °C dec. Yield: 62%. 1H NMR (CDC13): 1.51 and 1.52
(2s, 36H),
2.53 (s, 6H), 6.60 (s, 2H), 7.40 (s, 2H), 7.62 (d, 2H), 7.74 (d, 2H), 10.34
(s, 2H), 11.62 (br s,
2H).
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
[0086] 2,5-Bis(2-methoicy-4-N,N'-di-BOCguauidinopherzyl)furan (Compound 4c).
Yellow solid, mp >300 °C dec. Yield: 79%. 1H NMR (CDCl3): 1.50 and 1.53
(2s, 36H),
3.95 (s, 6H), 6.95 (s, 2H), 7.13 (d, 2H), 7.59 (s, 2H), 7.86 (d, 2H), 10.36
(s, 2H), 11.55 (br s,
2H).
[0087] 2,5-Bis(2-chloro-4-N,N'-di-BOCguarcia'ir~ophercyl)furah (Compound 4d).
Pale
yellow/tan solid, mp >400 °C dec. Yield: 63%. 1H NMR (CDC13): 1.52 (s,
36H), 7.17 (s,
2H), 7.63 (dd, 2H), 7.79 (d, 2H), 7.88 (d, 2H), 10.43 (s, 2H), 11.59 (br s,
2H).
[0088] 2,5-Bis(2-trifluoromethyl-~-N,N'-di-BOC guanidinophenyl)furan (Compound
4e). Bright orange solid. Yield: 88%. 1H NMR (CDC13): 1.51 and 1.53 (2s, 36H),
6.77 (s,
2H), 7.82 (d, 2H), 7.94 (s, 2H), 8.00 (d, 2H), 10.52 (s, 2H), 11.59 (br s,
2H).
[0089] 2, 5-Bis(2, 6-dimethyl-4-N,N'-di-BOC guanidihophenyl)fura~ (Compound
4f).
Pale yellow/off white solid, mp >300 °C dec. Yield: 89%. 1H NMR
(CDCl3): 1.51 and 1.53
(2s, 36H), 2.23 (s, 12H), 6.31 (s, 2H), 7.33 (s, 4H), 10.27 (s, 2H), 11.63 (br
s, 2H).
[0090] EXAMPLE 5
[0091] Deprotection of N,N'-di-BOC guanidines
[0092] (The following procedures are representative).
[0093] 2, 5-Bis(4-guanidircophenyl)furan dihydrochloride (Compound Sa). A
solution
of the corresponding N,N'-di-BOCguanidine (1.19 g, 1.62 mmol) in CH2C12 (15
ml) was
diluted with dry EtOH (10 ml) and saturated at ice-water bath temperature with
anhydrous
HCI. The solution was then stirred at room-temperature for 2-3 days (drying
tube), with the
product slowly precipitating (shorter reaction times generally gave incomplete
deprotection).
The resulting suspension was concentrated to near dryness, with the solid then
taken up in hot
EtOH. After filtering to clarify, the solution was concentrated to near
dryness to give a
suspension, which was diluted with ether and collected to yield, after drying
in vacuo at 50-
60 °C for 2 days, the bis-guanidine dihydrochloride as an off white/tan
solid (0.66 g,
quantitative), mp >300 °C dec. 1H NMR (DMSO-d6): 7.12 (s, 2H), 7.31 (d,
4H), 7.58 (br s,
8H), 7.86 (d, 4H), 10.09 (br s, 2H). MS (FAB, thioglycerol): mlz 335.3 (MH+,
100). Anal.
Calcd. for C18H18N60~2HC1~0.25EtOH (407.30): C, H, N.
16
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
[0094] 2,S-Bis(4-guanidino-2-methylphenyl)furan dihydrochloride (Compound Sb).
Tan solid, mp 26S-271 °C dec. 1H NMR (DMSO-d6): 2.53 (s, 6H), 6.93 (s,
2H), 7.17 (m,
4H), 7.56 (br s, 8H), 7.82 (d, 2H), 10.06 (br s, 2H). MS (FAB, thioglycerol):
m/z 363.3
(MH+, 100). Anal. Calcd. for C2oHZZN60~2HCl~1.SH2O~0.66EtOH (496.93): C, H, N.
[0095] 2, 5-Bis(4-guanidino-2-methoxyphenyl)furan dihydr~ochloride (Compound
Sc).
Light brown solid. 1H NMR (DMSO-d6): 3.95 (s, 6H), 6.92 (dd, 2H), 6.99 (d,
2H), 7.02 (s,
2H), 7.58 (br s, 8H), 7.95 (d, 2H), 10.08 (br s, 2H). MS (EI): m/z 3S2 (M+ -
NH2CN, 38.0),
310 (100), 267 (38.9), 251 (8.8), 1SS (18.7). Anal. Calcd. for
CaoHaaN60s~2HCl~1.OH20~0.33EtOH (SOO.S7): C, H, N.
[0096] ~, 5-Bis(2-chlor o-4-guanidinophenyl)fuf°an dihydrochloride
(Compound Sd).
Tan solid, mp 300-304 °C dec. 1H NMR (DMSO-d6): 7.31 (s, 2H), 7.33 (d,
2H), 7.47 (s, 2H),
7.72 (br s, 8H), 8.04 (d, 2H). MS (DCI, ammonia): m/z 365, 363, 361 (MH+ -
NH2CN, 8, S2,
78), 323, 321, 319 (11, 66, 100). Anal. Calcd. for C18H16C12N60~2HC1~O.SHZO
(485.21): C,
H, N, Cl.
[0097] 2,5 - Bis (4-guanidino-2-tr~~uoromethylphenyl) furan dilzyd~ochloride
(Compound Se). Orange/red solid. 1H NMR (DMSO-d6): 6.99 (s, 2H), 7.63 (d, 2H),
7.69 (s,
2H), 7.79 (br s, 8H), 7.91 (d, 2H), 10.37 (br s, 2H). MS (CI, isobutane): m/z
471 (MH+, 14),
429 (100), 387 (19). Anal. Calcd. for C2oH16F6N6O ~2HCl~0.67H20~0.67EtOH
(586.24): C,
H, N.
[0098] 2, 5-Bis(~-guanidino-2, 6-dimethylphenyl)fur an dihydrochloride
(Compound
Sf). ,Off white solid. 1H NMR (DMSO-d6): 2.20 (s, 12H), 6.56 (s, 2H), 7.01 (s,
4H), 7.57
(br s, 8H), 10.09 (br s, 2H). MS (FAB, thioglycerol): mlz 391.2 (MH+, 100).
Anal. Calcd. for
C22H26N60~2HC1~O.SH2O (472.41): C, H, N.
[0099] 2,S-Bis~4-(benzimidoyl)aminophenylJthiophene Free base: yellow
crystalline
solid, mp 284-286 °C dec (DMF/MeOH/H20). Yield: 3S%. 1H NMR (DMSO-d6):
6.41 (br s,
4NH), 6.91 (d, 4H), 7.40 (s, 2H), 7.44 (d, 6H), 7.62 (d, 4H), 7.97 (d, 4H).
Hydrochloride:
yellow/orange solid, mp 304-306 °C dec. .1H NMR (DMSO-d6): 7.56 (d,
4H), 7.67 (t, 4H),
7.70 (s, 2H), 7.77 (t, 2H), 7.90 (d, 4H), 7.95 (d, 4H), 9.13 (br s, 2H), 9.94
(br s, 2H), 11.71 (br
s, 2H). MS (EI): rnlz 472 (M+, 35.1), 369 (76.8), 266 (100), 103 (49.5), 76
(14.8). Anal.
Calcd. for C3oH24N4S~2HCl~O.SHaO (S54.S2): C, 64.97; H, 4.91; N, 10.10. Found:
C, 64.95;
17
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
H, 4.89; N, IO.I4.
[00100] ~,5-Bisj~-methyl-4-(2 pyy~idylimi~o)amifaophenylJthiophene Free base:
yellow
crystals, mp 152-153 °C (EtOH/H20). Yield: 20%. 1H NMR (DMSO-d6): 2.46
(s, 6H), 6.60
(br s, 4NH), 6.82 (d, 2H), 6.90 (s, 2H), 7.16 (s, 2H), 7.42 (d, 2H), 7.55 (m,
2H), 7.95 (t, 2H),
8.30 (d, 2H), 8.63 (dd, 2H). Hydrochloride: Yellow powder, mp xxx °C
dec. 'H NMR
(DMSO-d6): 2.54 (s, 6H), 7.36 (s, 2H), 7.39 (d, 2H), 7.48 (s, 2H), 7.66 (d,
2H), 7.85 (m, 2H),
8.22 (t, 2H), 8.47 (d, 2H), 8.89 (d, 2H), 9.37 (br s, 2H), IO.I2 (br s, 2H),
11.87 (br s, 2H).
MS (EI):. Ahal. Calcd. for C3oH26N6S~2.SHC1~1.25H20 (616.30): C, 58.46; H,
5.07; N,
13.64; Cl, 14.38. Found: C, 58.83; H, 4.92; N, 13.68; Cl, 14.02.
[00101] EXAMPLE 6
[00102] Preparation of Compounds 6b-6c (Scheme 3)
[00103] ~ The original route to the reversed amidines (which was used to
prepare 6b-c) is
as follows.
[00I 04] 2, 5-Bis j4-(be~zzimidoylamino)phenylJfur~an Dihydrochloride
(Compound 6b)
To a chilled solution of 2,5-bis(4-aminophenyl)furan (0.25 g, 1.0 mmol) in dry
acetonitrile
(10 ml) was added triethylamine (0.22 g, 2.1 mmol) followed dropwise by
benzoyl chloride
(0.30 g, 2.1 mmol) and the resulting suspension was stirred at room-
temperature for 3 hours.
Water (10 ml) was then added and the precipitate was collected, rinsed with
water, followed
by MeOH, and finally dried in vacuo to give 2,5-bis(4-benzamidophenyl)furan as
a tan solid
(0.44 g, 96%), mp 312-314.5 °C. 1H NMR (DMSO-d6): 6.98 (s, 2H), 7.52-
7.62 (m, 6H), 7.80
(d, 4H), 7.89 (d, 4H), 7.97 (d, 4H), 10.33 (br s, 2H).
[00105] The intermediate bis(benzamide) (0.44 g, 0.96 mmol) was suspended in
anhydrous dichloromethane (40 ml) and treated with freshly distilled thionyl
chloride (0.68 g,
5.7 mmol) along with 2 drops of DMF and the mixture was refluxed with vigorous
stirring
until a solution was obtained (20 hours). The solution was then concentrated
in vacuo to give
a yellow solid, which was co-evaporated with dry benzene. The obtained imidoyl
chloride
was dissolved in anhydrous dichloromethane (40 ml) and the solution was
saturated at
ice/water-bath temperature with anhydrous ammonia and sealed. After stirring
overnight at
room-temperature, the turbid mixture was concentrated to give a yellow solid,
which was
triturated with O.SN NaOH, collected, and air dried. This free-base (0.44 g,
100%) was
dissolved in boiling EtOH (50 ml), filtered, and at ice-bath temperature was
treated with dry
HCI. After failed attempts at inducing precipitation with the addition of
ether, the solution
18
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
was concentrated (high vacuum) to give the dihydrochloride as an orange
hygroscopic solid,
mp 242-248 °C. 1H NMR (DMSO-d6): 7.26 (s, 2H), 7.58 (d, 4H), 7.67 (t,
4H), 7.78 (t, 2H),
7.95 (d, 4H), 8.03 (d, 4H), 9.12 (br s, 2H), 9.94 (br s, 2H), 11.66 (br s,
2H). MS (EI): m/z
456 (M+, 100), 353 (63), 250 (62), 221 (16), 130 (15), 103 (41), 76 (14), 44
(22). Anal.
Calcd. for C3oH24N4O~2HCI~O.H~0.1 (C2H5)(545.87):C, H, N.
[00I06] 2,5 - Bis ~4 - ~(4-methylbenzimidoyl) amino) phenyl) furan
Dihydrochloride
(Compound 6c). Following the above procedure, 2,5-bis[(4-
methylbenzamido)phenyl]furan
was first obtained as a pale yellow solid by reaction of 2,5-bis(4-
aminophenyl)furan (0.50 g,
2.0 mmol) with 4-methylbenzoyl chloride (0.65 g, 4.2 mmol). Yield: 0.96 g,
99%; mp 348-
350.5 °C. 1H NMR (DMSO-d6): 2.39 (s, 6H), 6.98 (s, 2H), 7.34 (d, 4H),
7.79 (d, 4H), 7.89
(dd, 8H), 10.25 (br s, 2H).
[00107] Subsequent conversion of the bis(benzamide) to the amidine was
accomplished
as above, with the exception that the precipitated product following reaction
with ammonia
was collected by filtration and rinsed with EtOH to give the free base
directly (Yield: 34%).
The dihydrochloride was obtained as an orange oily solid which crystallized in
vacuo, mp
227-240 °C (hygroscopic). ~H NMR (DMSO-d6): 2.44 (s, 6H), 7.24 (s, 2H),
7.46 (d, 4H),
7.56 (d, 4H), 7.87 (d, 4H), 8.01 (d, 4H), 9.02 (br s, 2H), 9.89 (br s, 2H),
11.66 (br s, 2H). MS
(EI): m/z 484 (M+, 59), 367 (86), 250 (100), 221 (21), 130 (20), 117 (69), 90
(19), 44 (36).
Anal. Calcd. for C32H28N40~2HCl~0.5H20 (566.51): C, H, N.
[00108] EXAMPLE 7
[00109] Alternative preparation of bis-~[alkyl(or aryl)imino]aminophenyl}furan
derivatives (Scheme 4)
[00110] The following experimental is representative. In some cases, the
product was
purified by recrystallization.
[00111 ] 2, 5-Bis~2-methyl-4-(2 pyr~idylimino)amihophenylJfurah ((Compound
6h). To a
solution of 2,5-bis(4-amino-2-methylphenyl)furan (0.30 g, 1.08 mmol) in dry
MeCN (5 ml)
was added dry EtOH (15 ml) and the solution was chilled briefly on an
ice/water bath. S-(2-
Naphthylmethyl)thiobenzimidate hydrobromide (0.815 g, 2.27 mmol) was then
added and the
mixture was stirred overnight at room-temperature. The resulting solution was
concentrated
to an oil, which was triturated with ether to give a yellow solid. The solid
was collected,
dissolved in EtOH and basified with NaOH (11~, and the free base was extracted
into EtOAc.
19
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
After drying (Na2S04) and removing the most of the solvent, the resulting
suspension was
diluted with excess ether to give a fluffy yellow solid (0.36 g, 69%), mp 188-
189 °C, which
required no purification. 1H NMR (DMSO-d6): 2.51 (s, 6H), 6.60 (br s, 4NH),
6.77 (s, 2H),
6.87 (m, 4H), 7.55 (dd, 2H), 7.74 (d, 2H), 7.95 (m, 2H), 8.31 (d, 2H), 8.63
(d, 2H).
[00112] To prepare the hydrochloride salt, the free base was suspended in EtOH
(40m1)
and treated with dry HCl gas for 5-10 min at ice-bath temperature. Continued
stirring of the
resulting solution for 15-20 minutes gave an orange suspension which was
diluted with ether
(40 ml) and filtered to yield an orange powder (0.40 g), mp >180 °C
dec. 1H NMR (DMSO-
d6): 2.62 (s, 6H), 7.08 (s, 2H), 7.44 (d, 2H), 7.47 (s, 2H), 7.85 (dd, 2H),
7.99 (d, 2H), 8.22 (t,
2H), 8.49 (d, 2H), 8.89 (d, 2H), 9.36 (br s, 2H), 10.13 (br s, 2H), 11.88 (br
s, 2H). MS (EI):
mlz 486 (M+, 100), 382 (77.9), 278 (12.8), 104 (20.0), 78 (8.8), 43 (28.9).
Ahal. Calcd. for
C3oHa6N60~3.5HC1~0.5Hz0 (623.20): C, H, N, Cl.
[00113] 2,5-Bis~4-(~ py~idylimino)aminophenylJfuran (Compound 6a) Free base:
yellow crystalline solid, mp 221-223 °C (DMF/EtOH/Ha0). Yield: 65% 1H
NMR (DMSO-
d6): 6.80 (br s, 4NH), 6.94 (s, 2H), 7.03 (d, 4H), 7.56 (m, 2H), 7.77 (d, 4H),
7.96 (m, 2H),
8.32 (d, 2H), 8.64 (m, 2H). Hydrochloride: Orange/red powder, mp >175
°C dec. 1H NMR
(DMSO-d6): 7.26 (s, 2H), 7.58 (d, 4H), 7.85 (dd, 2H), 8.03 (d, 4H), 8.22 (t,
2H), 8.52 (d,
2H), 8.89 (d, 2H), 9.39 (br s, 2H), 10.16 (br s, 2H), 11.91 (br s, 2H). MS
(EI): m/z 458 (M+,
100), 354 (49.1), 250 (27.6), 221 (8.9), 130 (9.4), 105 (13.6), 78 (8.6).
Ahal. Calcd. for
C28Hz2N60~3.5HC1 (586.12): C, H, N, Cl.
[00114] 2,5-Bis~4-(cyclohexylimiho)aminophenylJfu~an (Compound 6d) Free base:
pale yellow needles, mp 242-243 °C dec (EtOAc). Yield: 17%. 1H NMR
(DMSO-d6): 1.18-
1.90 (m, 20H), 2.14 (m, 2H), 5.71 (br s, 4NH), 6.82 (s, ZH), 7.63 (d, 4H). [A
41 % yield of
the mono-amidine/mono-amine (free base, yellow solid, mp 195-196 °C)
was islolated by
chromatography on silica (EtOAc-MeOH, 9:1). The insoluble nature of the
reaction medium
was the likely cause of the incomplete reaction.] Dihydrochloride: tan/peach
solid mp 244-
248 °C dec. 1H~NMR (DMSO-d6): 1.27 (m, 6H), 1.63-1.96 (m, 14H), 2.72
(m, 2H), 7.22 (s,
2H), 7.40 (d, 4H), 7.96 (d, 2H), 8.60 (br s, 2H), 9.34 (br s, 2H), 11.39 (br
s, 2H). MS (FAB,
thioglycerol): nalz 469.4 (MH+, 100). Anal. Calcd. for
CsoHsaN40~2HCl~0.75EtOH~0.25H2O (580.60): C, H, N.
[00115] 2,5-Bis~4-(benzimidoyl)amino-2-methylphe~tylJfunah (Compound 6g) Free
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
base: yellow crystalline solid. Yield: 60%. 'H NMR (DMSO-d6): 2.48 (s, 6H),
6.50 (br s,
4NH), 6.75 (s, 2H), 6.84 (s, 4H), 7.44 (m, 6H), 7.71 (d, 2H), 7.95 (d, 4H).
Hydrochloride:
orange/yellow hygroscopic solid. 1H NMR (DMSO-d6): 2.61 (s, 6H), 7.03 (s, 2H),
7.38-7.44
(m, 4H), 7.63-7.68 (m, 4H), 7.75-7.80 (m, 2H), 7.94 (d, 6H). MS (EI): m/z 484
(M+, 100),
381 (87.2), 278 (37.9), 235 (5.4), 218 (3.1), 190 (5.5), 144 (11.1), 103
(32.8), 76 (9.3). Anal.
Calcd. for C32H28N~0~2HC1~O.SH20~569.39): C, H, N.
[00116] 2,S-Bis(2-methyl-4-(2-quinolylinaino)arninophenylJfuran (Compound 6i).
Free
base: orange powdery crystals, mp 168-169 °C (EtOH). Yield: 52%. 1H NMR
(DMSO-d6):
2.54 (s, 6H), 6.80 (s, 2H), 6.95 (m, 4H), 7.69 (m, 2H), 7.78 (d, 2H), 7.84 (m,
2H), 8.07 (d,
2H), 8.12 (d, 2H), 8.44 (d, 2H), 8.50 (d, 2H). Dihydrochloride: orange solid,
mp >185 °C
dec. 1H NMR (DMSO-d6): 2.65 (s, 6H), 7.10 (s, 2H), 7.50 (m, 4H), 7.85 (m, 2H),
8.01 (m,
2H), 8.20 (d, 2H), 8.26 (d, 2H), 8.46 (d, 2H), 8.80 (d, 2H), 9.44 (br s, 2H),
10.21 (br s, 2H),
11.98 (br s, 2H). MS (FAB, thioglycerol): rralz 587.2 (MH+, 100). Anal. Calcd.
for
C38H3oN60~2.OHCl~1.75HZO (691.13): C, H, N, Cl.
[00117] 2,5-Bis~2-methyl-4-(5-methyl-2 pyridylimino)aminophenylJfuf~an
(Compound
6j). Free base: yellow crystalline solid, mp 156-158 °C (Et20/hexanes).
Yield: 74%. 1H
NMR (DMSO-d6): 2.37 (s, 6H), 2.50 (s, 6H), 6.55 (br s, 4NH), 6.75 (s, 2H),
6.85 (m, 4H),
7.70-7.76 (m, 4H), 8.18 (d, 2H), 8.45 (s, 2H). Hydrochloride: orange solid, mp
>175 °C dec.
1H NMR (DMSO-d6): 2.49 (s, 6H), 2.62 (s, 6H), 7.08 (s, 2H), 7.43 (d, 2H), 7.47
(s, 2H), 7.85
(dd, 2H), 7.98 (d, 2H), 8.03 (d, 2H), 8.42 (d, 2H), 8.74 (s, 2H), 9.29 (br s,
2H), 10.07 (br s,
2H), 11.83 (br s, 2H). MS (EI): m/z 514 (M~, 19.2), 396 (100), 278 (34.5), 144
(8.0), 118
(33.6), 91 (13.6), 43 (22.8). Anal. Calcd. for C32H3oN6O~3.25HC1~0.75HZO
(646.62): C, H,
N, Cl.
[00118] 2,5-Bis~~-nZethoxy-4-(~ pyridylimino)aminophenylJfi~r~an (Compound
6k).
Free base: Bright yellow crystalline solid, mp 196-197 °C (EtOAc/Et20).
Yield: 75%. IH
NMR (DMSO-d6): 3.92 (s, 6H), 6.64 and 6.67 (d, 2H and s, 2H, overlapping a
broad NH
signal), 6.89 (s, 2H), 7.55 (dd, 2H), 7.86 (d, 2H), 7.95 (m, 2H), 8.32 (d,
2H), 8.63 (d, 2H).
Dihydrochloride: brick orange solid, mp >180 °C dec. 1H NMR (DMSO-d6):
4.00 (s, 6H),
7.16 (s, 2H), 7.18 (d, 2H), 7.34 (s, 2H), 7.85 (dd, 2H), 8.13 (d, 2H), 8.22
(t, 2H), 8.49 (d, 2H),
8.89 (d, 2H), 9.39 (br s, 2H), 10.15 (br s, 2H), 11.89 (br s, 2H). MS (EI):
m/z 518 (M+, 100),
414 (90.0), 371 (13.3), 310 (12.7), 267 (9.7), 155 (6.02), 104 (25.9), 77
(9.6), 43 (13.6).
21
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
;y,.:. ,~w" ~: :.~ ~w.: :. . .....,. _._._ _
a
x
Anal. Calcd. for CsoHzsNsO~2.OHC1~2.OHz0 (627.51): C, H, N, Cl.
2,5-Bisj2-chloro-4-(2 pyridylimino)aminophenylJfur~an (Compound 61). Free
[00119]
a cr stalline solid, mp 189-190 °C (EtOH). Yield: 25%. 1H NMR {DMSO-
ds):
base: orang y
4NH 7.02 {dd, 2H), 7.08 (d, 2H), 7.17 (s, 2H), 7.56 (m, 2H), 7.93-7.98 (m,
4H),
6.85 {br s, ),
2H . Dihydrochloride: yellow/orange solid, mp >180 °C dec. ~H
8.29 (d, 2H), 8.64 (m,
SO-d : 7.45 (s, 2H), 7.60 (d, 2H), 7.80 (s, 2H), 7.86 (dd, 2H), 8.23 (m, 4H),
8.50
NMR (DM s)
0 d 2H), 9.54 (br s, 2H), 10.23 (br s, 2H), 11.98 (br s, 2H). MS (EI): rnlz
530,
(d, 2H), 8.9 ( ,
+ 13,2, 69.8, 100), 426, 424, 422 (7.9, 48, 72.4), 322, 320, 318 (2.6, 17.4,
26.6).
528, 526 (M ,
Anal. Calcd. for CzsHzoClzNsO~2.OHCl~l.SHzO (627.35): C, H, N, Cl.
2 5-Bisj2,6-dimethyl-~-(2 py~'idylimino)aminophenylJfuran (Compound 6m).
[00120]
ale ellow crystals, mp 206-207 °C (EtOH). Yield: 80°l0. ~H NMR
(DMSO-ds):
Free base: p Y
I-I 6.47 (s, 2H), 6.55 (br s, 4NH), 6.69 (s, 4H), 7.54 (m, 2H), 7.94 (m, 2H),
8.29
2.19 (s, 12 ),
2 d 2H). Hydrochloride: fluffy yellow solid, mp >x~ °C dec. 1H NMR
(d, 2H), 8.6 ( ,
2.28 (s, 12H), 6.68 (s, 2H), 7.28 (s, 4H), 7.84 (m, 2H), 8.21 (t, 2H), 8.48
(d,
(DMSO-ds):
.37 br s, 2H), 10.12 {br s, 2H), 11.87 (br s, 2H). MS (EI): m/z 514 (M+,
2H), 8.88 (d, 2H), 9 W
3g,7 306 {100), 291 (16.0), 148 (45.4), 104 (56.3), 77 (31.0). Anal. Calcd.
for
8.5), 410 ( ),
C3zHsoNsO~3.75HC1~0.5Hz0 (660.35): C, H, N, Cl.
[00121] EXAMPLE 8
[00122] Purification of acetamidines
The following acetamidines were purified anal characterized as the HBr salt
[00123]
without conversion to the free base.
2 5-Bisj4-(acetimidoyl)aminophenylJ.~uYan DihydYObromide {Compo~d 6e).
[00124]
an a solid, mp 307-309.5 °C dec (MeOH/EtOAc). Yield: 57%. 1H NMR
Fluffy tanlor g
70 °C : 2.37 (s, 6H), 7.17 (s, 2H), 7.40 (d, 4H), 7.94 (d, 4H), 8.52
(br s, 2H), 9.43
{DMSO-ds, )
11.13 br s, 2H). MS (FAB, thioglycerol): mlz 333.2 (MH~,100). Anal. Calcd. for
(br s, 2H), (
CzoHzoN40~2.OHBr (494.23): C, H, N.
~ 5 - Bis j4 - (acetimidoyl) amino - 2 - methylphenYlJ.fur~an Dihydrobr~omide
[00125]
d 6 . Fluffy tanlyellow solid, mp 282.5-284 °C dec (MeOH/EtOAc). Yield:
64%.
(Compoun
DMSO-ds, 70 °C): 237 (s, 6H), 2.57 (s, 6H),, 6.99 (s, 2H), 7.27 (d,
2H), 7.28 (s,
H NMR
8 d 2H , 8.52 (br s, 2H), 9.42 (br s, 2H), 11.12 (br s, 2H). MS (FAB,
thioglycerol):
2H), 7.8 ( , )
22
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
mlz 361.2 (MH+, 100). Anal. Calcd. for C2oHaoN40~2.OHBr~0.3MeOH (531.89): C,
H, N.
[00126] EXAMPLE 9
[00127] Preparation of pyridine-2-thiocarboxamide
[00128] Adapting the general method of Taylor, a mixture of 2-cyanopyridine
(7.28 g,
7.0 mmol) and thioacetamide (10.52 g, 14.0 mmol) was treated with 60 ml of HCl-
saturated
DMF, and the solution was stirred vigorously in an open flask on an oil bath
set initially at 80
°C (the temperature gradually rising to 95 °C over the coarse of
the reaction). Taylor, E. C. et
al., J. Aria. Chem. Soc. 1960, ~2, 2656-2657. After 80 minutes (TLC
monitoring), the
resulting orange suspension was cooled, neutralized with con. NaOHlice, and
extracted with
EtOAc. The extract was washed with water (3x) and then concentrated to a light
brown solid
which was triturated with warm water and collected. The dried product was
passed over a
silica gel column eluting with EtOAc:hexanes (2:1) to give, after removal of
most of the
solvent and dilution with hexanes, a yellow crystalline solid (6.36 g, 66%),
mp 136-137 °C;
litmp 137 °C.
[00129] S-Methylpyridihe-2-thioca~boxamide. Prepared as above from 2-cyano-5-
methylpyridine with a reaction time of 30 minutes. See Moynehan, T. M. et al.,
J. Chem. Soc.
1962, 2637-2658. Yield: 59%. Gold crystals, mp 172.5-173 °C. 1H NMR
(CDC13): 2.39 (s,
3H), 7.60 (br s, NH), 7.61 (dd, 1H), 8.31 (d, 1H), 8.57 (d, 1H), 9.42 (br s,
NH).
[00130] EXAMPLE 10
[00131] Preparation of S-(2-naphthylmethyl)thioimidates
[00132] The following new S-(2-naphthylmethyl)thioimidates were prepared
according
to the literature by reaction of the appropriate thioamide with (2-
bromomethyl)naphthalene in
refluxing CHC13 (EtOH-free) for 1.5 hr. See Shearer, B. G.; et al.,
Tetrahedron Lett. 1997,
38, 179-182. After dilution with ether, the precipitated product was
collected, rinsed with
ether, and dried in vacuo.
[00133] S-(2-Naphthylmethyl)cyclohexanethioimidate~HB~. Yield: 91%. White
solid,
mp 192-192.5 °C. 1H NMR (DMSO-d6): 1.14-1.32 (m, 3H), 1.45-1.53 (m,
2H), 1.63 (d, 1H),
1.76 (d, 2H), 1.87 (d, 2H), 2.84 (t, 1H), 4.73 (s, 2H), 7.53-7.56 (m, 3H),
7.89-7.97 (m, 3H),
8.01 (s, 1 H).
23
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
[00134] S-(2-Naphthylmethyl)thiobenzimidate~HBr. Yield: 94%. White solid, mp
210-212 °C dec. 1H NMR (DMSO-d6): 4.90 (s, 2H), 7.54-7.62 (m, 2H), 7.62-
7.66 (m, 3H),
7.78-7.82 (m, 1H), 7.88-7.99 (m, SH), 8.06 (s, 1H).
[00I35] S-(2-Naphthylmethyl)-2-pyridylthioimidate~HBr. Yield: 58%. White
fluffy
solid, mp 192 °C dec. 1H NMR (DMSO-d6): 4.80 (s, 2H), 7.53-7.57 (m,
2H), 7.59-7.62 (dd,
1 H), 7.76-7.79 (m, 1 H), 7.90-7.97 (m, 3 H), 8.05 (s, 1 H), 8.10-8 .14 (m, 1
H), 8.26 (d, 1 H),
8.78-8.80 (m, 1 H).
[00136] S-(2-Naphthylmethyl)-5-methyl-2-pyridylthioimidate~HBr. Yield: 65%.
White
fluffy solid, mp 190-191 °C dec. 1H NMR (DMSO-d6): 2.42 (s, 3H), 4.79
(s, 2H), 7.53-7.57
(m; 2H), 7.59-7.62 (dd, 1 H), 7.90-7.97 (m, 4H), 8.05 (s, 1 H), 8.17 (d, 1 H),
8.64 (s, 1 H).
[00137] S-(2-Naphthylmethyl)-2-quinolylthioimidate~HBr. Yield: 23%. Light tan
fluffy solid, mp 184-186 °C dec. 1H NMR (DMSO-d6): 4.73 (s, 2H), 7.53-
7.55 (m, 2H),
7.62-7.64 (dd, 1 H), 7.78 (t, 1 H), 7.87-7.97 (m, 4H), 8.07 (s, 1 H), 8.12 (d,
2H), 8.28 (d, 1 H),
8.66 (d, 1 H).
[00138] EXAMPLE 11
[OOI39j Biological Testing Of Inventive Compounds: Materials and Methods
[00140] Differences in thermal melting values (OTm) were determined and DNA
samples were prepared as previously described in Boykin, D. W. et al., J. Med
Chem. 1998,
41, 124-129 and Francesconi, I. et al., J. tiled. Chem. 1999, ~2, 2260-2265.
[00141] Mycobacterium tuberculosis susceptibility testing. The compounds of
the
present invention were tested against M. tuberculosis H37Rv in BACTEC 12B
medium using
a fluorometric broth microdilution assay, the Microplate Alamar Blue Assay
(MABA),
according to Collins, L. et al., Autimic~ob. Agents Chemother. 1997, 41, 1004-
1009.
Compounds were initially assessed at 6.25 ug/ml and those effecting a
reduction in
fluorescence of at least 90% relative to untreated cultures were further
evaluated for MIC by
testing at lower concentrations. The MIC was defined as the lowest
concentration of
compound effecting a reduction of > 90% of the relative fluorescence units
relative to a
control culture. Antimycobacterial data were provided by the Tuberculosis
Antimicrobial
Acquisition and Coordinating Facility (TAACF)) through a research and
development
contract with the U.S. National Institute of Allergy and Infectious Diseases.
24
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
[00142] Antifungal Test organisms. The fungi used in this study for all the
compounds in Table 1 included two reference strains C albicans A39 and
Aspergillus
fumigatus (strain 168.95). Expanded studies on 6j employed the fungi listed in
Table 3.
[00143] Medium. Antifungal susceptibility testing was performed with RPMI 1640
medium (Sigma Chemical Co., St. Louis, Mo.) with glutamine, but without sodium
bicarbonate and buffered at pH 7.0 with 0.165 M morpholinopropanesulfonic
acid.
[00144] Antifungal in vitro susceptibility testing. Experiments for
determination of
MICs of yeasts were performed by the broth macrodilution method according to
the
recommendations of the National Committee for Clinical Laboratory Standards.
See
Natio~tal Committee for Clinical Laboratory Standards. Refet~ehce method for
broth
elilutiof~ susceptibility testing of yeasts Document M27-T. Tentative
standard, National
Committee for Clinical Laboratory Standards, Wayne, Pa., 1995). The only
difference
compared to the standardized method was the choice of drug dilutions, which
ranged from
100 to 0.09 ~glml. Briefly, this method specifies the use of an inoculum grown
at 35°C and
adjusted to a concentration of 0.5 x 103 to 2.5 x 103 CFU/ml, incubation of
the culture at
35°C, and reading at 48 h for all yeasts except for C. neoformans, for
which the results are
interpreted at 72 h. The MIC was defined as the culture with the lowest drug
concentration in
which a visual turbidity less than or equal to 80% inhibition compared to that
produced by the
growth control tube was observed.
[00145] The minimum fungicidal concentration (MFC) was determined by plating
100
~,l aliquots from tubes showing complete inhibition of growth on Sabouraud
agar plates. The
lowest drug concentration that yielded three or fewer colonies was recorded as
the MFC.
[00146] Molds were tested by the same method but with the following
modifications.
Isolates were grown on Sabouraud dextrose agar at 30°C, after adequate
sporulation occurred
(4 to 14 days); conidia were harvested by flooding the colonies with a sterile
solution of
0.85% NaCI and 0.05% Tween 80 in sterile distilled water. Inocula were
prepared with a
hemocytometer for counting and were then diluted with RPM1 1640 medium to
obtain a final
inoculum size of approximately 0.5 x 103 to 2.5 x 103 CFU/ml. The inoculum
size was
verified by plating an aliquot of the inoculum. The cultures were incubated at
30°C for 48 to
72 h or until growth in the control tube was visible.
[00147] EXAMPLE 12
(00148] Results of Biological Testing
[00149] Melting temperatures were measured for the compounds 5 and 6 bound to
poly
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
dA~dT to obtain a qualitative evaluation of the DNA binding affinity of these
drug candidates
(Table 2). The difference in Tm values between the drug-DNA complexes and free
DNA in
solution (OTm) provides a useful tool to assess the interaction strength of
the molecules with
DNA. Since several of the compounds bound very strongly to poly dA~dT (Table
2), the
interaction of these compounds with the Dickerson-Drew dodecamer d
(CGCGAATTCGCG)2 (SEQ ID NO:1), a DNA with a different and shorter AT sequence
and
different groove characteristics, was also studied. The reduced binding of the
drugs to the
dodecamer reflected by the lower OTm values of the drug-dodecamer complexes
(Table 2),
allowed for a better relative comparison of the DNA binding affinity of these
putative minor-
groove binding compounds.
Table 2. Irz vitro Antimicrobial Activities and DNA binding results for
inventive compounds.
C. albicaus Asner~illus fumi~atus MTb DNA Affinitiecb
Comp. MIC MFC MIC MFC % MIC ~Tm ~Tm
No. ( ml) ( ml) ( /ml ( /ml) Inha ( ml) (AT) (oligo)
Sa 12.5 25 100 >100 3.13 21.6 10.8
6a Nt nt nt nt 1.56 19.6 7.5
6b 25 50 nt nt 1.56 28.6 15.0
6c >100 nt nt nt _<6.25 >28 15.4
~
6d >I00 nt I00 >100 _<6.25 26.7 12.8
6e >100 nt 100 >100 0 nd 15.9 6.0
6f >100 nt >100 nt 18 nd 14.5 2.4
Sb 1.04 2.08 33.4 33.4 _<1 17.8 6.9
Sc 10 100 100 100 16 15.2 2.8
Sd 10 100 100 100 4 26.1 4.7
Se 10 10 100 100 8 5.9 0
Sf Nt nt nt nt 2.7 1.4
6 3.12 6.25 nt nt 3.13 24.9 7.8
6h 10 10 10 100 < 1 22.6 8.9
6i 100 100 100 nt 7.5 1.1
6' _< 1 10 10 nt 2 24.3 10.8
6k _<1 _<1 _<1 _<1 _<1 19.0 7.8
61 100 >100 >100 nt 8 5.2 0
6m 10 >100 10 >100 5.7 1.2
DB 686 4.17 4.17 < 1
DB 653 >I00 >100 20.2 I2.8
(a) % Inhibition at 6.25 ~,g/ml
(b) AT = poly dA~dT; oligo -- d(CGCGAATTCGCG)2
(c) nt = not tested
(d) nd = not determined
[00150] The parent diguanidino compound Sa showed a strong affinity for DNA as
26
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
judged by the OTm values for both poly dA~dT (21.6) and the dodecamer (10.8).
These
values compare well to those (25 and I 1.7) for the parent amidine 2,5-bis [4-
amidinophenyl]
furan suggesting little difference in affinities in this core furan structure
for the amidine and
guanidine cationic centers. Based on the comparison of OTm values for ,binding
to the
dodecamer for the parent diguanidino Sa and for the various reversed amidine
congeners 6a-
6e, several interesting effects resulting from structural variation of the
terminal groups are
noted. First, reversed amidines bearing phenyl, substituted phenyl or
cyclohexyl terminal
groups (6b, 6c and 6d) showed an increase in affinity over that of the parent
diguanidino 5a.
In a related series of diamidines, such an increase in affinity with
increasing bulk of terminal
groups was attributed to increased van der Waals interactions of the terminal
groups with the
walls of the minor-groove and such is likely the case in this system.
Interestingly, the OTm
value for the compound with a terminal 2-pyridyl group 6a is significantly
lower than that for
its phenyl counterpart 6b. The lower, affinity of 6a may suggest a different
binding mode or
different base pair selectivity for the two structurally closely related
dicationic analogs.
However, introduction of a methyl group on each of he two phenyl rings of the
2,5-
diarylfuran system with a terminal phenyl group, 6g, resulted in lowering of
the OTm value to
one similar to that of its pyridyl counterpart, 6h. The use of small alkyl
terminal groups,
methyl groups as found in 6e and 6f, also Ied to a significant drop in binding
affinity.
[00151 ] The placement of a single substituent on each of the two phenyl rings
of the
2,5-diarylfuran framework produced striking differences in ~Tm values for both
the
diguanidine and reversed amidine series. Again, based on the comparison of ~Tm
values for
binding to the dodecamer for the diguanidine series, it is noted that
placement of single
substituent of roughly the same size but of differing electronic properties on
the phenyl rings
resulted in a lowering of the ~Tm values; compare the values of Sa with Sb-Se.
The most
dramatic effect was observed for the strongest electron-withdrawing group, the
CF3 of 5e,
which reduced the OTm value to zero.
[00152] The OTm values for a series of compounds with terminal 2-pyridyl
groups (6a,
6h-6m) showed a different sensitivity to substituents. In this case, the
introduction of a single
substituent on each of the two phenyl rings of the 2,5-diarylfuran framework
caused little
effect on the OTm values when the substituent was methyl (6h) or methoxy (6k).
However,
introduction of a chloro group (61) resulted in significant reduction of the
value, perhaps in
part due to a pK effect. The detrimental . effect of the chloro group on the
~Tm was much
greater for the pyridyl derivative 61 than for the analogous guanidine Sd,
possibly due to the
27
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
lower basicity of the reversed amidine in comparison to the guanidine. In
agreement with the
results from the diguanidine series, the introduction of two methyl groups on
each of the core
phenyl rings (6m) also dramatically reduced the DNA affinity. Interestingly,
replacement of
the terminal 2-pryidyl with a 2-quinoyl group (6I) also resulted in a
significant reduction of
the ~Tm suggesting definite limits on the dimensions of the terminal group. On
the other
hand, the introduction of a methyl group on the terminal pyridyl ring (6j)
slightly enhanced
the binding affinity.
[00153] The antimicrobial data for these compounds are also summarized in
Table 2.
The greatest activity amongst the diguanidino compounds was found for Sa and
5b, which
showed good in vitro activity against both Candida albicans and Mycobacterium
tuberculosis. These compounds gave MIC values between 1 to 3 ~,g/ml against
both
organisms. Both compounds were fungicidal against C. albicans. The 2,5-bis
[alkylimino]
aminophenyl] furans (6d, 6e, 6f), in general, did not show significant
antimicrobial activity,
although compound 6d, with the larger cyclohexyl group, did exhibit some
activity against
M. tuberculosis.
[00154] The 2,5-bis [arylimino] aminophenyl] fixrans can be divided into two
groups:
those in which the terminal group is phenyl or substituted phenyl and those in
which it is 2-
pyridyl or substituted 2-pyridyl. The terminal phenyl group class of compounds
did not
exhibit high antifungal activity; however, both 6b and 6g did show significant
activity against
M. tuberculosis with MIC values of 0.78 and 1.56 ~,g/ml, respectively. ' Four
of the
compounds in the terminal pyridyl group class (6a, 6h, 6j and 6k) showed
promising activity
against M. tuberculosis with MIC values ranging from 1.0 to 2.0 ~,g/ml. Most
of the 2-pyridyl
compounds exhibited only moderate antifungal activity. As exceptions, 6j and
6k showed
activity at the MIC level of < 1.0 ~g/ml against C. albicans, and 6k showed a
similar level of
activity against Aspergillus fumigatus.
[00155] In order to evaluate the spectrum of antifungal activity of these
compounds,
pyridyl-substituted amidines 6j and 6k were selected for studies against other
pathogenic
fungi (Table 3). Compound 6j was quite effective against C. albicans and
exhibited
fungicidal activity against several strains. Compound 6j was less effective
versus both
Asper~gillus species. Compound 6j showed good fungicidal activity versus
Rhizopus arrhizus;
however, it was not very effective against the mold-Fusay~iurn solani.
Compound 6k did not
show significant activity in the expanded fungus panel.
[00156] To summarize, dicationic 2,5-bis (4-guanidinophenyl) furans Sa-Sf, 2,5-
bis [4-
(arylimino) aminophenyl] furans 6a-6c, 6g-6m, and 2,5-bis [4-(alkylimino)
aminophenyl]
28
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
furans 6d-6f have been synthesized starting from 2,5-bis [tri-n-butylstannyl]
furan. Thermal
melting studies with poly dA~dT and the duplex oligomer d(CGCGAATTCGCG)z (SEQ
ID
NO: 1) demonstrated high DNA binding affinities for a number of the compounds.
Both the
guanidines and the reversed amidines in the 2,5-diarylfuran series have strong
DNA binding
properties.
[00157] Compounds in both these classes show both antifungal and anti-
mycobacterial
activity. In addition, they may be broad-spectrum anti-fungal agents. Of the
nineteen novel
dicationic compounds synthesized, six (6a, 6b, 5b, 6h, 6j, 6k) exhibited MICs
of 2 ~,g/ml or
less versus Mycobacterium tuberculosis. Of the nineteen screened against
Candida albicahs,
four gave MICs of 2 ~,g/ml or less (5a, 5b, 6j, 6k) and two (5a, 6k) were
fungicidal, unlike a
standard antifungal drug Fluconazole which was fungistatic. One of the tested
compounds
(6k) exhibited a MIC of 1 ~,g/ml and was also fungicidal for Aspergillus
fumigates. Some
compounds possessed inhibitory activity against Cryptococcus eeoformans but
all appeared
less potent for this pathogenic yeast compared with C. albicans and A.
fumigates.
[00158] Evaluation against Trypar~osoma brucei rhodesiehse in vitro showed
that the
(arylimino) aminophenyl furans, especially when aryl is 2-pyridyl, were
effective in the 0.02
to 0.1 ~.g/mL range; approximately 1/10 as effective as pentamidine and
furamidine. In
contrast, these new compounds are ten times more effective than pentamidine
and
furamidine, and comparable in activity to benznidazole, against T. cruzi.
Table 3. Evaluation of Compound 6j against an Expanded Fungus Panel
Genus, species, isolate numberMIC 80% MIC 100% MFC
Aspergillus flavus 194.99 3.12 6.25 >100
Aspergillus flavus 107.96 3.12 50 >100
Aspergillus flavus 141.88 3.12 50 >100
Aspergillus fumigates 168.95 50 50 >100
Aspergillus fumigates 182.99 3.12 50 >100
Aspergillus fumigates 119.00 3.12 50 >100
Aspergillus fumigates 165.86 50 50 >100
As ergillus fumigates 153.90 3.12 50 >100
Fusarium solani 152.89 3.12 50 50
Rlzizopus arrhizus 117.89 0.78 1.56 1.56
Ca~adida albicarcs 116.98 1.56 1.56 >100
Candida albicahs 159.95 0.79 1.58 3.12
Candida albicar~s 149.97 0.78 1.56 6.25
Candida albicans 156.97 1.56 3.15 >100
Candida albicarts 126.97 1.56 3.12 3.12
Cayzdida albicans 117.00 0.78 3.12 25
Candida albicans A39 1.56 1.56 25
29
CA 02425135 2003-04-03
WO 02/057224 PCT/USO1/47238
Cryptococcus heoformans H99- - 1.56 I 1.56 ~ >100
MIC and MFC values are ~,g/ml. MIC 80% = 80% of inoculum is inhibited. MIC
100% _
100% of inoculum is inhibited. MFC = minimum fungicidal concentration.
[00159] In the specification, and examples there have been disclosed typical
preferred
embodiments of the invention and, although specific terms are employed, they
are used in a
generic and descriptive sense only and not for the purposes of limitation, the
scope of the
invention being set forth in the following claims.