Note: Descriptions are shown in the official language in which they were submitted.
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
_1-
13-Alkvl Eoothilone Derivatives
The invention relates to 13-lower alkyl epothilones; intermediates and a
process for the
preparation of epothilones and 13-lower alkyl epothilones; a pharmaceutical
composition
comprising 13-lower alkyl epothilones; the use of such compounds for the
treatment of
tumor diseases; a method of treatment of warm-blooded animals; catalytic
antibodies
having aldolase activity; a process for enantioselectively resolving a racemic
mixture of aldol
synthons by means of antibody catalyzed retro-aldol reaction.
A number of chemical reactions have been catalyzed by monoclonal antibody
catalysts.
Most of those transformations were catalyzed with very high enantio- and
regioselectivity.
Several reactions were rerouted from their normal pathway and disfavored
reactions were
catalyzed. Recently, two aldolase monoclonal antibodies 38C2 and 33F12 were
generated
against a p-diketone hapten, 6-(4-glutaramidophenyl)-hexane-2,4-dione, using
reactive
immunization (J. Wagner, R.A. Lerner, C.F. Barbas III, Science 1995, 270,
1797). In
reactive immunization highly reactive chemicals are used as immunogens so that
a
chemical reaction occurs in the binding site of an antibody during its
induction. An analogue
of this chemical reaction later becomes part of a catalytic event.
Epothilones A and B represent a new class of microtubul e-stabilising
cytotoxic agents (see
Gerth, K. et al., J. Antibiot. 1996, 49, 560-3) of the formula:
ox
HC
N
wherein RX means hydrogen (epothilone A) or methyl (epothilone B).
These compounds have advantages over Taxol ~, a branded product already
introduced for
the treatment of tumours, that has the same mechanism of action but has
however a series
v OH v
W~ 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-2-
of disadvantages, such as very poor water solubility, making the preparation
of pharma -
ceutical formulations very difficult (at present, such formulations are
normally characterised
by the toxic side effects of the carrier materials), and inefficacy on a
series of tumours. The
advantages are as follows:
a) They have better water-solubility and are thus more readily accessible for
formulations.
b) It has been reported that, in cell culture experiments, they are also
active against the
proliferation of cells, which, owing to the overexpression of the P-
glycoprotein efflux pump
are "multidrug resistant", i. e., show resistance to treatment with other
chemotherapy agents
including Taxolo (see Bolag, D. M., et al., "Epothilones, a new class of
microtubule-
stabilizing agents with a Taxol-like mechanism of actions, Cancer Research
1995, 55, 2325-
33). And
c) it could be shown that they are still very effective in vitro against a
Taxol~-resistant
ovarian carcinoma cell line with modified ~-tubulin (see Kowalski, R. J., et
al., J. Biol. Chem.
1997, 272(4), 2534-2541 ).
Pharmaceutical application of the epothilones, for example for tumour
treatment, is possible
in an analogous manner to that described for Taxol, see, for example, US
5.641.803; US
5.496.804; US 5.565.478. One disadvantage of the epothilones is the relatively
low
therapeutic index, i.e. the dosage range between the necessary dose and the
maximum
tolerable dose is very small.
Surprisingly it has now been found that compounds of formula I
YR1
HO N
in which compounds
O OH O
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-3-
R, is methyl, hydroxymethyl, halomethyl, methylthio or methoxy;
R2 is hydrogen or methyl,
R3 is hydrogen or lower alkyl, and
Z is O or a bond;
can be obtained from chemical intermediates II obtained by a process of
enantioselectively
resolving a racemic mixture of an aldol synthon (_+)-II
OH
Ri ~g O
((~)-I I)
using catalytic antibodies.
Therefore, the invention relates to a process for enantioselectively resolving
a racemic
mixture of an aldol synthon, the racemic mixture including a first and a
second enantiomer
of the aldol synthon, said process comprising the following steps:
Step A: Catalyzing a retro-aldol reaction for enantioselectively converting
the first
enantiomer of the aldol synthon to form an aldehyde product while leaving the
second
enantiomer of the aldol synthon unmodified, said catalyst employing a
catalytic antibody;
and
Step B: Separating the aldehyde product from the unmodified second aldol
synthon.
In this process the racemic mixture need not to be a 50/50 mixture of the two
enantiomers.
Especially, the invention relates to a process wherein the racemic mixture of
the aldol
synthon of formula (~)-II is resolved to provide an unmodified enantiomer of
formula II* and
an aldehyde of formula III
W~ 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-4-
H H
~ \ +
R~ S O R~ S O R~ S
R R
(~)-I I I I* I I I
Preferably, in this process a catalytic antibody selected from the group
consisting of 8463,
85H6 and 93F3 is employed.
Preferably, in formula (~)-II R, represents methyl, hydroxymethyl, halomethyl,
methylthio or
methoxy, and R represents C,_5 alkyl, n-but-1-en-4-yl or halomethyl. More
preferably, R ~
represents methyl, hydroxymethyl, fluoromethyl, methylthio or methoxy, and R
represents
methyl, ethyl, n-propyl, n-butyl, n-pentyl, n-but-1-en-4-yl or fluoromethyl.
Most preferably, R ,
represents methyl, hydroxymethyl, methylthio or methoxy, and R represents
methyl, ethyl,
n-propyl, n-butyl, n-pentyl or n-but-1-en-4-yl.
Catalytic antibodies 8463, 85H6 and 93F3 were generated through immunization
of mice
against the hapten of formula IV
(IV).
coupled to the carrier protein keyhole limpet hemocyanine (KLH) according to
the
procedures described by J. Wagner et al, Science 1995, 270, 1798 and G. Kohler
et al,
Nature 1975, 256, 495. The antibodies were purified according to the methods
described by
V.E. Gouvernour et al, Science 1993, 262, 204, and deposited with the American
Type
Culture Collection (ATCC), 10801 University Blvd, Manassas, VA 20110-2209, USA
on
October 07, 1999 by The Scripps Research Institute under the deposit
designations PTA-
823, PTA-824 and PTA-825, respectively. 8463 and 93F3 operate with a catalytic
WO 01/27308 CA 02425620 2003-04-07 pCT/EP00/09817
-5-
proficiency of (k~plK"~lk"" > 10'3 M-'. These new catalysts present
significant advantages
with regard to the synthesis of epothilone and their analogs.
It is disclosed herein that catalytic antibodies 8463, 85H6 and 93F3 have
antipodal
reactivity with regard to catalytic antibody 38C2. These three antibodies are
especially
effective with regard to the catalytic resolution of compounds ( ~)-II. E.g.,
the antibodies
8463 is used in such resolution in a multi-gram scale in a quantity of between
0.00001 and
0.5 mol%.especially 0.0004, 0.003 or 0.005 mol%.
Deposits for hybridoma 8463, 85H6 and 93F3 were made in compliance with the
Budapest
Treaty requirements that the duration of the deposits should be for 30 years
from the date
of deposit at the depository or for the enforceable life of a patent that
matures from this
application, whichever is longer. The assignee of the present application has
agreed that if
the hybridoma deposit should die or be lost or destroyed when cultivated under
suitable
conditions, it will be promptly replaced on notification with a viable
specimen of the same
hybridoma.
Furthermore, the present invention relates to a process for the preparation of
a compound
of formula I, in which
R, is methyl, hydroxymethyl, halomethyl, methylthio or methoxy;
R2 is hydrogen, methyl,
R3 is hydrogen or lower alkyl, and
Z is O or a bond;
wherein a compound of formula V,
S~R~
PO
O
O - O
PO (V)
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-6-
in which P is terf-butyl-dimethylsilyl or another suitable protecting group
for a hydroxy group,
R, is methyl, hydroxymethyl which is protected by tart-butyl-dimethylsilyl or
another suitable
hydroxy protecting group, halomethyl, methylthio or methoxy, and the other
radicals have
the meaning as given above for formula I, is transformed into a compound of
formula I by
an olefine metathesis reaction in the presence of the catalyst of formula VI
Me~N NlMes
Ph
CI ~,~. ~~ ~
" Ru=.i
CI~ I
PCy3
in which compound of formula V Mes represents mesityl and Ph phenyl, followed
by the
detaching of the protecting group by a suitable reagent, and in which process
directly
before or after detaching the protecting groups present a compound of formula
I wherein Z
represents a bond can optionally be transformed by epoxidation in analogy to
the procedure
described in Example 26 of W099/43653 into a compound of formula I wherein Z
represents O, and, after carrying out the above process, if necessary for the
preparation of
a salt, converting a resulting free compound of the formula I into a salt or,
if necessary for
preparation of a free compound, converting a resulting salt of a compound of
the formula I
into the free compound.
It is a further objective of the invention to provide epothilone derivatives,
which, through
their advantageous biological and pharmacological properties, enable the
armamentarium
for the control of, in particular, proliferative diseases such as tumours to
be expanded. Also,
compounds have to be found, which have an improved therapeutic index compared
with
epothilones A and B.
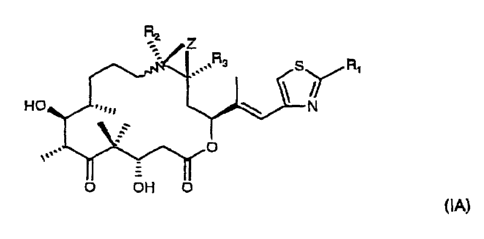
Surprisingly, 13-lower alkyl epothilone are pharmacologically highly effective
for the
indications mentioned herein. Hence, the invention relates also to 13-lower
alkyl epothifone
of formula (A wherein
R, is methyl, hydroxymethyl, halomethyl, methylthio or methoxy;
RZ is hydrogen, methyl,
WD 01/27308 CA 02425620 2003-04-07 pCT/EP00/09817
7_
R3 is lower alkyl, and
Z is O or a bond;
If Z represents a bond, a double bond is established between the both carbon
atoms
wearing the radical Z.
The compounds of formula IA and their pharmaceutically acceptable salts have
advantageous pharmaceutical properties. For example, they are active against
multidrug-
resistant cell lines and tumours and/or they have an improved therapeutic
index over natural
epothilones.
The general terms used hereinbefore and hereinafter preferably have within the
context of
this disclosure the following meanings, unless otherwise indicated:
The prefix "lower" denotes a radical having up to and including a maximum of
7, especially
up to and including a maximum of 5 carbon atoms, the radicals in question
being either
branched with single or multiple branching or unbranched.
Where the plural form is used for compounds, salts, and the like, this is
taken to mean also
a single compound, salt, or the like ("a" as an indefinite article or as a
numeral meaning
"one").
Asymmetric carbon atoms that are optionally present in the substituents may
exist in the
(R), (S) or (R,S) configuration, preferably in the (R) or (S) configuration.
Substituents on a
double bond or on a ring may be present in cis- (=Z-) or trans- (=E-) form.
The present
compounds may thus exist as mixtures of isomers or as pure isomers, preferably
as pure
diastereoisomers.
Lower alkyl has mono- or multiple-branching or, preferably, is unbranched or
and is in
particular methyl, ethyl, propyl, especially n-propyl, or butyl, especially n-
butyl. Very
preferably lower alkyl is methyl or ethyl.
Halogen is especially fluorine, chlorine, bromine, or iodine, in particular
fluorine or chlorine.
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
_g-
Halomethyl is fluoromethyl or chloromethyl, in particular fluoromethyl.
Preferably, in the compound of formula IA
R, is methyl, hydroxymethyl, halomethyl, methylthio or methoxy;
R2 is hydrogen or methyl,
R3 is lower alkyl, and
Z is O or a bond.
More preferably, in the compound of formula IA
R, is methylthio;
R2 is hydrogen;
R3 is lower alkyl, especially methyl or ethyl and
Z is O or a bond, prefeably a bond.
Furthermore, the present invention provides compounds of formula II*, which
are useful
intermediates for the preparation of epothilone derivatives of formula I.
In such compound of formula II*, preferably R , represents methyl,
hydroxymethyl,
halomethyl, lower alkoxy lower alkylenoxy methyl, preferably methoxy
methylenoxy methyl,
methylthio or methoxy, and R represents lower alkyl, n-but-1-en-4-yl or
halomethyl. Very
preferably, R, represents methylthio, and R represents methyl or ethyl.
Salts of compounds of formula I with a salt-forming group may be prepared in a
manner
known per se. Acid addition salts of compounds of formula I may thus be
obtained e.g. by
treatment with an acid or with a suitable anion exchange reagent.
Salts can usually be converted to free compounds, e.g. by treating with
suitable basic
agents, for example with alkali metal carbonates, -hydrogencarbonates, or -
hydroxides,
typically potassium carbonate or sodium hydroxide.
The macrocyclisation of a diene of formula V wherein P is tert-butyl-
dimethylsilyl or another
suitable hydroxy protecting group, R, is methyl, hydroxymethyl which is
protected by tert-
butyl-dimethylsilyl or another suitable hydroxy protecting group, halomethyl,
methylthio or
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09g17
_9_
methoxy; R2 is hydrogen or methyl, and R3 is hydrogen or lower alkyl, to a
compound of
formula I, wherein the radicals P, R,, R2 and R3 have the same meaning as in
the
compound of formula V and Z is a bond, takes place, preferably under the
following
conditions: The compound V is solved in a suitable, dry solvent or mixture of
solvents with a
boiling point between 30 and 60 °C, e.8., dichloro methane. The
catalyst of formula VI is
added, preferably, in an amount of 5 mol % to 30 mol %, very preferably
between 15 and
25 mol %, e.8., 20 mol %, and the cyclisation is carried out, e.8., at the
reflux temperature
of the solvent for a duration of 12 to 120 h, preferably 20 to 96 h. The
reaction can be
controlled by NMR analysis of the reaction mixture.
Starting materials
The starting materials are known, may be produced by known processes or are
commer -
cially available, or they may be produced as described in the following:
In the following preparation processes for intermediates, functional groups
which are to be
in protected form can be protected if necessary at suitable stages, whereby
selective pro -
tection or deprotection is also possible. The protecting groups and the
methods of introdu -
cing and/or removing them correspond to those named above under process a),
especial 1y
those named in the above-mentioned standard reference works or, in particular,
in the ex -
amples. As a rule, protecting groups are not mentioned in the following; the
following ex -
amples show where the usage of the protecting groups is appropriate or
necessary and can
therefore be regarded as a preferred instruction as to when protecting groups
should be
used and if compounds should be produced with other radicals. In the
following, protecting
groups are not mentioned at all the points where they are appropriately used.
The person
skilled in the art is clear as to where this usage ought to or must occur.
A compound of formula V wherein P is tert-butyl-dimethylsilyl or another
suitable hydroxy
protecting group, R, is methyl, hydroxymethyl which is protected by tert-butyl-
dimethylsilyl or
another suitable hydroxy protecting group, halomethyl, methylthio or methoxy;
R 2 is
hydrogen or methyl, and R3 is hydrogen or lower alkyl, can be obtained by the
esterification
of an alcohol of formula VII
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-10-
N
OH
(VII)
wherein the radicals R, and R3 have the same meaning as in the compound of
formula V,
with an acid of formula VIII
PO
H
O - O
PO
(VIII)
wherein P is tert-butyl-dimethylsilyl or another suitable hydroxy protecting
group and R 2 has
the same meaning as in the compound of formula V. The esterification can be
carried out
under conditions known per se, especially in the presence of a dehydrating
agent like DCC
(dicyclohexylcarbodiimide). For example, the acid can be solved in a suitable,
inert and dry
solvent, e.g. tetrahydrofuran or, preferably, dichloromethane, together with
the alcohol at a
temperature between -10 °C and room temperature, e.g., 0 °C.
Optionally, EDC and DMAP
(4-(dimethylamino)-pyridine) can be added and the mixture is stirred for 12 to
24 h, e.g.
16 h, of the same temperature.
The preparation of an acid of formula VIII wherein R 2 is hydrogen or methyl
and P is tert-
butyl dimethylsilyl, is, e.g., disclosed in WO 98/08849, especially in the
Example on page
30, the subject-matter of which is hereby incorporated into the present
application by
reference to this publication.
An alcohol of formula VII wherein the radicals R, and R3 have the same meaning
as in the
compound of formula V, can be obtained by the treatment a compound of formula
IX
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-11
R S~R~
N
O P'
(IX)
wherein the radicals R, and R3 have the same meaning as in the compound of
formula VII
and P' is a protecting group, with a reagent suitable to detach the protecting
group. E.g., if
P' is tent-butyl dimethylsilyl, treatment of IX with TBAF in a suitable
solvent, like tetrahydro-
furan, at a temperature between 0°C and room temperature, provides the
unprotected
alcohol VII. If R, comprises a hydroxy group protected by the same protecting
group P', the
treatment mentioned before also detaches the protecting group in R ,. The
protecting group
tent-butyl dimethylsilyl can be reintroduced by reacting VII with TBSCI ( tent-
butyl dimethylsilyl
chloride) in the presence of diisoproylethylamine in a suitable solvent, like,
e.g.,
dichloromethane, at a temperature between 0 °C and room temperature for
6 to 12 h, e.g.,
8 h.
A compound of formula IX is obtainable by a Wittig reaction of a compound of
formula X
R O S~R~
N
OP'
(X)
wherein the radicals R1, R3 and P' have the same meaning as in the compound of
formula
IX using, e.g., the Wittig reagent methyl-triphenylphosphonium iodide. The
Wittig reaction is
known as such. In the present case, the reaction can be carried out in dry
tetrahydrofuran
or another suitable solvent at temperatures between -10 °C and + 30
°C, preferably
between 10 °C and room temperature. After adding buthyl lithium to the
reagent methyl-
triphenylphosphonium iodide at 0 °C, the solution is stirred for about
30 minutes at room
temperature. Then the compound of formula X is added and the solution is
stirred for further
15 to 90 minutes at the same temperature.
WO ~l/273~g CA 02425620 2003-04-07 PCT/EP~O/09g17
-12-
A compound of formula X can be prepared by reacting a compound of formula II
wherein
the radicals R, and R3 have the same meaning as in the compound of formula X,
with a
reagent suitable to introduce a protecting group P', e.g. with TBSCI, in a
suitable solvent,
especially dimethylformamide, preferably in the presence of imidazole, at room
temperature
Compounds of formula (~)-II can be prepared by the reaction of a compound of
formula XI
S~R~
\ \ N
OHC
(XI)
wherein the radical R, has the same meaning as in the compound of formula (~)-
II, with a
compound of formula XII
R~O
(X11)
wherein the radical R3 has the same meaning as in the compound of formula L)-
II, in which
reaction the compound of formula XII is first solved in dry tetrahydrofuran
and a solution of
LDA is added at -78 °C. After stirring for between 1 and 4 h, the
solution is cooled further to
a temperature of about -100 °C and the compound of formula XI is added
at the same
temperature. The reaction mixture is stirred for further 30 to 120 minutes and
then
quenched by the addition of a suitable acidic reagent, e.g. an aqueous
solution of NH 4C1.
General process conditions
Stereoisomeric mixtures, e.g. mixtures of diastereoisomers, can be separated
into their cor -
responding isomers in a manner known per se by means of suitable separation
methods.
Diastereoisomeric mixtures may thus be separated into their individual
diastereoisomers by
means of fractionated crystallization, chromatography, solvent distribution,
and similar pro -
cedures, preferably according to the purification procedures described in the
Examples.
This separation may take place either at the stage of one of the starting com
pounds.
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-13-
Enantiomers may be separated through the for mation of diastereoisomeric
salts, for
example by salt formation with an enantiomer-pure chiral acid, or by means of
chromatography, for example by HPLC, using chromatographic substrates with
chiral
ligands. (Enantiomer separation is normally effected at the interme diate
stage).
All process steps described here can be carried out under known reaction
conditions, pre
ferably under those specifically mentioned, in the absence of or usually in
the presence of
solvents or diluents, preferably those that are inert to the reagents used and
able to dissol -
ve them, in the absence or presence of catalysts, condensing agents or
neutralisi ng agents,
for example ion exchangers, typically cation exchangers, for example in the H
+ form,
depending on the type of reaction and/or reactants at reduced, normal, or
elevated
temperature, for example in the range from -100°C to about
190°C; preferably from about -
80°C to about 150°C, for example at -80 to 60°C, at room
temperature, at - 20 to 40°C or at
the boiling point of the solvent used, under atmospheric pressure or in a
closed vessel, if
required under pressure, and/or in an inert, for example an argon or nitrogen,
atmosphere.
Salts may be present in all starting compounds and intermediates, if these
contain salt-
forming groups. Salts may also be present during the reaction of such
compounds, provided
that the reaction is not thereby disturbed.
At all reaction stages, isomeric mixtures that occur can be separated into
their individual
isomers, e.g. diastereoisomers or enantiomers, or into any mixtures of
isomers, e.g.
racemates or diastereoisomeric mixtures, for example analogously to methods
described
under "Additional process steps".
In certain cases, typically in dehydrogenation or aldol reactions, it is
possible to achieve
stereoselective reactions, allowing for example easier recovery of individual
isomers.
The solvents from which those can be selected which are suitable for the
reaction in
question include for example water, esters, such as lower alkyl-lower
alkanoate, e.g ethyl
acetate, ethers, such as aliphatic ethers, e.g. diethylether, or cyclic
ethers, e.g. tetra-
hydrofuran, liquid aromatic hydrocarbons, such as benzene or toluene,
alcohols, such as
methanol, ethanol or 1- or 2-propanol, nitrites, such as acetonitrile,
halogenated
hydrocarbons, such as methylene chloride, acid amides, such as
dimethylformamide,
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-14-
bases, such as heterocyclic nitrogen bases, e.g. pyridine, carboxylic acids,
such as lower
alkanecarboxylic acids, e.g. acetic acid, carboxylic acid anhydrides, such as
lower alkane
acid anhydrides, e.g. acetic anhydride, cyclic, linear, or branched
hydrocarbons, such as
cyclohexane, hexane, or isopentane, or mixtures of these solvents, e.g.
aqueous solutions,
unless otherwise stated in the description of the process. Such solvent
mixtures may also
be used in working up, for example by chromatography or partitioning.
The invention relates also to those embodiments of the process in which one
starts from a
compound obtainable at any stage as an intermediate and carries out the
missing steps, or
breaks off the process at any stage, or forms a starting material under the
reaction condi -
tions, or uses said starting material in the form of a reactive derivative or
salt, or produces a
compound obtainable by means of the process according to the invention under
the pro -
cess conditions therein, and further processes the said compound in situ. In
the preferred
embodiment, one starts from those starting materials which lead to the
compounds descri -
bed hereinabove as preferred, particularly as especially preferred, primarily
preferred, and/
or preferred above all.
In the preferred embodiment, compounds of formula I are prepared analogously
to the
processes and process steps defined in the examples.
The compounds of formula I, including their salts, are also obtainable in the
form of
hydrates, or their crystals may include for example the solvent used for
crystallisation
(present as solvates).
The invention relates also to a compound of formula I obtained by the process
of
preparation disclosed herein.
Salts are primarily the pharmaceutically acceptable salts of compounds of
formula I.
Such salts are formed, for example, as acid addition salts, preferably with
organic or inor -
ganic acids, from compounds of formula I with a basic nitrogen atom,
especially the
pharmaceutically acceptable salts. Suitable inorganic acids are, for example,
hydrohalic
acids, such as hydrochloric acid, sulphuric acid, or phosphoric acid. Suitable
organic acids
are, for example, carboxylic, phosphonic, sulphonic or sulphamic acids, for
example acetic
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-15-
acid, propionic acid, octanoic acid, decanoic acid, dodecanoic acid, glycolic
acid, lactic acid,
2-hydroxybutyric acid, gluconic acid, glucosemonocarboxylic acid, fumaric
acid, succinic
acid, adipic acid, pimelic acid, suberic acid, azelaic acid, malic acid,
tartaric acid, citric acid,
glucaric acid, galactaric acid, amino acids, such as glutamic acid, aspartic
acid, N-methyl-
glycine, acetylaminoacetic acid, N-acetylasparagine or N-acetylcysteine,
pyruvic acid,
acetoacetic acid, phosphoserine, 2- or 3-glycerophosphoric acid, malefic acid,
hydroxy-
maleic acid, methylmaleic acid, cyclohexanecarboxylic acid, benzoic acid,
salicylic acid, f-
or 3-hydroxynaphthyl-2-carboxylic acid, 3,4,5-trimethoxybenzoic acid, 2-
phenoxybenzoic
acid, 2-acetoxybenzoic acid, 4-aminosalicylic acid, phthalic acid,
phenylacetic acid,
glucuronic acid, galacturonic acid, methane- or ethane-sulphonic acid, 2-
hydroxy-
ethanesulfonic acid, etha ne-1,2-disulphonic acid, benzene sulphonic acid, 2-
naphthalene-
sulphonic acid, 1,5-naphthalene-disulphonic acid, N-cyclohexylsulphamic acid,
N-methyl-,
N-ethyl- or N-propyl-sulphamic acid, or other organic protonic acids, such as
ascorbic acid.
For isolation or purification purposes it is also possible to use
pharmaceutically unaccept -
able salts, for example picrates or perchlorates. Only the pharmaceutically
acceptable salts
or free compounds (if the occasion arises, in the form of pharmaceutical
preparations) attain
therapeutic use, and these are therefore preferred.
In view of the close relationship between the novel compounds in free form and
in the form
of their salts, including those salts that can be used as intermediates, for
example in the
purification or identification of the novel compounds, hereinbefore and
hereinafter any re
ference to the free compounds is to be understood as referring also to the
corresponding
salts, as appropriate and expedient.
The compounds of formula IA have valuable pharmacological properties, as
described
hereinbefore and hereinafter.
The efficacy of the compounds of formula IA as enhancers of micro tubule
polymerisation
may be proved as follows:
Stock solutions of the test compounds (10 mM) are prepared in DMSO and stored
at -20°C.
Microtubule protein is extracted from pigs' brain by two cycles of temperature-
dependent
depolymerisation/polymerisation, as known (see Weingarten et al., Biochemistry
1974; 13,
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-16-
5529-37). Working stock solutions of micro tubule protein (i.e. tubulin plus
microtubule-
associated proteins) are stored at -70°C. The degree of test-compound-
induced poly -
merisation of microtubule protein is determined basically as already known
(see Lin et al.,
Cancer Chem. Pharm. 1996, 38, 136-140). Drug or vehicle (DMSO, final
concentration 5%)
are diluted in 1x MEM buffer (100 mM MES, 1 mM EGTA, 1 mM MgCl2, pH 6.7) and
placed
in an eppendorf tube on ice. Following additon of microtubule protein (final
concentration
1 mg/ml), the polymerization reaction is started by transferring the
incubation mixtures to a
room-temperature water bath for 5 min. Then, the reaction mixtures are placed
in an
Eppendorf microcentrifuge and incubated for a further 15 minutes at room
temperature. The
samples are then centrifuged for 15 minutes at 14,000 rpm at room temperature,
in order to
separate polymerised from unpolymerised micro tubule protein. As an indirect
measure of
tubulin polymerisation, the protein concentration of the su pernatant (which
contains the
remaining non-polymerised, soluble microtubule protein) is determined by the
Lowry method
(DC Assay Kit, Bio-Rad Laboratories, Hercules, CA, USA), and the optical
density (OD) of
the colour reaction is measured at 750 nm using a spectrometer (Spectra Max
340,
Molecular Devices, Sunnyvale, CA, USA). The difference in OD's between samples
treated
with a test compound and vehicle-treated controls is compared with that
obtained with
incubations containing 25 pM epothilon B (positive control). The degree of
polymerisation
induced by a test compound is expressed relatively to the positive control
(100%). In this
test compounds of formula I exhibit a degree of polymerisation of 2 to 78 %,
in particular
39% to 78% for compounds of formula IA wherein R 1 is methylthio.
The efficacy against tumour cells may be demonstrated in the following way:
Stock solutions of the test compounds (10 mM) are prepared in DMSO and stored
at -20°C.
Human KB-31 and (multidrug-resistant, P-gp170 expressing) KB-8511 epidermoid
carcino -
ma cells originate from Dr. M. Baker, Roswell Park Memorial Institute
(Buffalo, NY, USA)
(description: see also Akiyama et al., Somat. Cell. Mol. Genetics 1985, 11,
117-126 and
Fojo A., et al., Cancer Res. 1985, 45, 3002-3007 - KB-31 and KB-8511 are both
derivati ves
of the KB cell line (ATCC) and are human epidermoid carcinoma cells. The cells
are
cultured as previously described (I. Utz, S. Hofer, U. Regenass, W. Hilbe, J.
Thaler, H.
Grunicke, and J. Hofmann, Int. J. Cancer 1984, 57, 104). Anti-proliferative
assays are
performed as previously described (T. Meyer, et al., Int. J. Cancer 1989, 43,
851 ). Briefly,
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
17-
cells are seeded at 1.5 x 103 cells/well into 96-well microtiter plates and
incubated
overnight. Compounds are added in serial dilutions on day 1. The plates are
than incubated
for an additional 4 days, after which the cells are fixed with 3.3 % v/v
glutaraldehyde,
washed with water and stained with 0.05% w/v methylene blue. After washing,
the dye is
eluted with 3 % HCI and the optical density measured at 665 nm with a
SpectraMax 340
(Molecular Devices, Sunnyvale, CA, USA). 1C50 values are determined by
mathematical
curve-fitting using the Softmax program currently version 2.6 ( Molecular
Devices, Sunny-
vale, CA, USA) using the formula (OD treated - OD start) / (OD control - OD
start) x 100.
The IC50 is defined as the drug concentration which leads to 50 % of cells per
well
compared to control cultures at the end of the incubation period.
Compounds of formula IA show for the KB-31 cell line an IC50 in the range of
0.05 and
250 nM, preferably between 1 and 50 nM.
Tests on other tumour cell lines, e.g., A549 (lung; ATCC CCL 185), NCIH460
(lung), HCT-
15 (colon; ATCC CCL 225), HCT-116 (colon), MCF-7 (breast; ATCC HTB 22), or Du-
145
(prostate; ATCC No. HTB 81 ) can be carried out in a comparable manner.
The in vivo efficacy may be demonstrated as follows: The models used are xeno -
trans-
plants of tumours, such as KB-31 or KB-8511 epidermoid tumours, in mice. The
anti-tumour
efficacy of the test compounds may be measured in female BLB/c nu/nu mice for
example
against the corresponding subcutaneously transplanted cell line. To this end,
tumour frag -
ments of about 25 mg are implanted into the left side of each of the mice (for
example 6
animals per dose). The test compound is administered for example on day 11
after trans -
plantation in different dosages (for example 0.1; 0.5; 1; 5 and 10 mg/kg), if
desired repea-
ting the administration, if required several times, after between two days and
two weeks.
The volumes of the tumours are determined for example after about 2 to 4 weeks
(e.g. two
weeks after the start of treatment). The tumour volumes are calculated by
measuring the
tumour diameter along two vertically arranged axes and according to published
methods
(see Evans et al., Brit. J. Cancer 1982, 45, 466-8). The anti-tumour efficacy
is determined
as the mean increase in tumour volume of the treated animals divided by the
mean increase
in tumour volume of the untreated animals (controls) and, after multiplication
by 100, is ex -
pressed as T/C%. Tumour regression (given in %) is calculated as the smallest
mean to -
mour volume (Vt) in relation to the mean tumour volume at the start of
treatment (Vo) ac -
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-18-
cording to the formula
regression = [1-(VtNo)] x 100.
In this case also, other cell lines can be used, for example those named above
in the
demonstration of efficacy against tumour cells.
Owing to these properties, the compounds of formula IA are suitable for the
treatment of
proliferative diseases, especially tumour diseases, including metastases; for
example solid
tumours such as lung tumours, breast tumours, colorectal tumours, prostate
tumours,
melanomas, brain tumours, pancreas tumours, neck tumours, bladder tumours,
neuro-
blastomas, throat tumours, but also proliferative diseases of blood cells,
such as leukaemia;
also for the treatment of other diseases which respond to treatment with micro
tubule
depolymerisation inhibitors, such as psoriasis. In one preferred embodiment of
the
invention, the compounds of formula IA are used for the treatment of prostate
tumours.
Especially preferred are the compounds named in the examples, or salts thereof
(especially
pharmaceutically acceptable salts), provided that a salt-forming group is
present.
Pharmaceutical preparations, methods, and uses
A compound of formula IA can be administered alone or in combination with one
or more
other therapeutic agents, possible combination therapy taking the form of
fixed combina -
tions or the administration of a compound of the invention and one or more
other therapeu -
tic agents being staggered or given independently of one another, or the
combined admini -
stration of fixed combinations and one or more other therapeutic agents. A
compound of
formula IA can besides or in addition be administered for tumour therapy in
combination
with chemotherapy, radiotherapy, immunotherapy, surgical intervention, or a
combination of
these. Long-term therapy is equally possible as is adjuvant therapy in the
context of other
treatment strategies, as described above. Other possible treatments are
therapy to maintain
the patient's status after tumour regression, or even chemopreventive therapy,
for example
in patients at risk.
WO 01/27308 CA 02425620 2003-04-07 pCT/EP00/09817
-19-
Therapeutic agents for possible combination are especially one or more
antiproliferative,
cytostatic or cytotoxic compounds, for example one or more chemotherapeutic
agent (s)
selected from the group comprising the classical chemotherapeutic agents, an
inhibitor of
polyamine biosynthesis, an inhibitor of protein kinase, especially of
serine/threo nine protein
kinase, such as protein kinase C, or of tyrosine protein kinase, such as
epidermal growth
factor receptor protein tyrosine kinase, a cytokine, a negative growth
regulator, such as
TGF-f3 or IFN-f3, an aromatase inhibitor, and a classical cytostatic.
The present invention relates also to pharmaceutical compositions that contain
a compound
of formula IA as active ingredient and that can be used especially in the
treatment of the
diseases mentioned above. Compositions for enteral administration, such as
nasal, buccal,
rectal or, especially, oral administration, and for parenteral administration,
such as
intravenous, intramuscular or subcutaneous administration, to warm-blooded
animals,
especially humans, are especially preferred. The compositions contain the
active ingredient
alone or, preferably, together with a pharmaceutically acceptable carrier. The
dosage of the
active ingredient depends upon the disease to be treated and upon the species,
its age,
weight, and individual condition, the individual pharmacokinetic data, and the
mode of
administration.
The invention relates also to pharmaceutical compositions for use in a method
for the pro -
phylactic or especially therapeutic treatment of warm-blooded animals,
including human,
especially suffering from a tumour disease, in particular breast cancer or
prostate cancer, to
a process for the preparation thereof (especially in the form of compositions
for the
treatment of tumours) and to a method of treating the above-mentioned
diseases, primarily
neoplastic diseases, especially those mentioned above.
In one preferred embodiment of the invention the tumour disease that is
treated is prostate
cancer or breast cancer.
The invention relates also to the use of compounds of formula IA for the
preparation of
pharmaceutical preparations which contain compounds of formula IA as active
component.
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-20-
Furthermore, the invention relates to a compound of formula IA for use in a
process for the
diagnostic or therapeutic treatment of humans and to the use of a compound of
formula IA
for the treatment of a tumour disease.
Preference is given to a pharmaceutical composition that is suitable for
administration to a
warm-blooded animal, especially a human or commercially useful mammal,
suffering from a
disease that is responsive to the enhancement of micro tubule polymerisation,
for example
psoriasis or especially a neoplastic disease, comprising a correspondingly
effective amount
of a compound of formula IA, or a pharmaceutically acceptable salt thereof
when salt-
forming groups are present, together with at least one pharmaceutically
acceptable carrier.
A pharmaceutical composition for the prophylactic or especially therapeutic
treatment of
neoplastic and other proliferative diseases of a warm-blooded animal,
especially a human
or a commercially useful mammal requiring such treatment, especially suffering
from such a
disease, comprising a new compound of formula IA, or a pharmaceutically
acceptable salt
thereof, as active ingredient in a quantity that is prophylactically or
especially therapeutically
active against said diseases, is likewise preferred.
Pharmaceutical preparations contain from about 0.000001 % to 95 % of the
active ingredi -
ent, whereby single-dose forms of administration preferably have from
approximately
0.00001 % to 90 % and multiple-dose forms of administration preferably have
from approxi -
mately 0.0001 to 0.5 % in the case of preparations for parenteral
administration or 1 % to
20 % active ingredient in the case of preparations for enteral administration.
Unit dosage
forms are, for example, coated and uncoated tablets, ampoules, vials,
suppositories or
capsules. Further dosage forms are, for example, ointments, creams, pastes,
foams, tinc -
tures, lipsticks, drops, sprays, dispersions, etc. Dosage unit forms, such as
coated tablets,
tablets or capsules, contain about 0.01 g to about 2 g, preferably about 0.02
g to about
1.0 g, of the active ingredient, in particular 0.02 to 0.6 g.
The pharmaceutical preparations of the present invention are prepared in a
manner known
per se, for example by means of conventional mixing, granulating, coating,
dissolving or
lyophilising processes.
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-21 -
Preference is given to the use of solutions of the active ingredient, and also
suspensions or
dispersions, which, for example in the case of lyophilised preparations which
contain the
active ingredient on its own or together with a carrier, for example mannitol,
can be made
up before use. The pharmaceutical preparations may be sterilised and/or may
contain
excipients, for example preservatives, stabilisers, wetting agents and/or
emulsifiers,
solubilisers, salts for regulating the osmotic pressure and/or buffers and are
prepared in a
manner known per se.
Suspensions in oil contain as the oil component the vegetable, synthetic, or
semi-synthetic
oils customary for injection purposes. In respect of such, special mention may
be made of
liquid fatty acid esters that contain as the acid component a long-chained
fatty acid having
from 8 to 22, carbon atoms, for example lauric acid, tridecylic acid, myristic
acid, penta-
decylic acid, palmitic acid, margaric acid, stearic acid, arachidic acid, be
henic acid or
corresponding unsaturated acids, for example oleic acid, elaidic acid, erucic
acid, brassidic
acid or linoleic acid, if desired with the addition of antioxidants, for
example vitamin E, ~i-
carotene or 3,5-di-tert-butyl-4-hydroxytoluene. The alcohol component of these
fatty acid
esters has a maximum of 6 carbon atoms and is a mono- or polyhydric, for exam
ple a
mono-, di- or trihydric, alcohol, or the isomers thereof, but especially
glycol and glycerol. As
fatty acid esters, the refore, the following are mentioned: ethyl oleate,
isopropyl myristate,
isopropyl palmitate, "Labrafil M 2375" (polyoxyethylene glycerol trioleate
from Gattefosse,
Paris), "Labrafil M 1944 CS" (unsaturated polyglycolised glycerides prepared
by alcoholysis
of apricot seed oil and consisting of glycerides and polyethylene glycol
ester; Gattefosse,
France), "Labrasol" (saturated polyglycolised glycerides prepared by
alcoholysis of TCM
and consisting of glycerides and polyethylene glycol ester; Gattefosse,
France), and/or
"Miglyol 812" (triglyceride of saturated fatty acids of chain length C $ to
C,2 from Huls AG,
Germany), but especially vegetable oils such as olive oil, cottonseed oil,
almond oil, castor
oil, sesame oil, soybean oil and more especially groundnut oil.
The manufacture of injectable preparations is usually carried out under
sterile conditions, as
is the filling, for example, into ampoules or vials, and the sealing of the
containers.
Pharmaceutical compositions for oral administration can be obtained, for
example, by com -
bining the active ingredient with one or more solid carriers, if need be
granulating a resulting
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-22-
mixture, and processing the mixture or granules, if desired, to form tablets
or tablet cores, if
need be by the inclusion of additional excipients.
Suitable carriers are especially fillers, such as sugars, cellulose
preparations, and/or
calcium phosphates, for example trical cium phosphate or calcium hydrogen
phosphate, and
also binders, such as starches, for example corn, wheat, rice or potato
starch, and/or
polyvinylpyrrolidone, and/or, if desired, disintegra tors, such as the above-
mentioned
starches, also carboxymethyl starch, crosslinked polyvinylpyrrolidone, alginic
acid or a salt
thereof, such as sodium alginate. Additional excipients are especially flow
conditioners and
lubricants, for example silicic acid, talc, stearic acid or salts thereof,
and/or polyethylene
glycol, or derivatives thereof.
Tablet cores may be provided with suitable, if need be enteric, coatings,
using inter alia con-
centrated sugar solutions which may comprise gum arabic, talc,
polyvinylpyrrolidone, poly -
ethylene glycol and/or titanium dioxide, or coating solutions in suitable
organic solvents or
solvent mixtures, or, for the preparation of enteric coatings, solutions of
suitable cellulose
preparations, such as acetylcellulose phthalate or
hydroxypropylmethylcellulose phthalate.
Dyes or pigments may be added to the tablets or tablet coatings.
Orally administrable pharmaceutical compositions also include hard capsules
consisting of
gelatin, and also soft, sealed capsules consisting of gelatin and a
plasticiser, such as gly -
cerol or sorbitol. The hard capsules may contain the active ingredient in the
form of granu -
les, for example in admixture with fillers, such as corn starch, binders,
and/or glidants, such
as talc or magnesium stearate, and if need be stabilisers. In soft capsules,
the active ingre-
dient is preferably dissolved or suspended in suitable liquid excipients, such
as fatty oils,
paraffin oil or liquid polyethylene glycols or fatty acid esters of ethylene
or propylene glycol,
to which stabilisers and detergents, for example of the polyoxyethylene
sorbitan fatty acid
ester type, may also be added.
The formulations suitable for parenteral administration are primarily aqueous
solutions of an
active ingredient in water-soluble form, e.g. a water-soluble salt, or aqueous
injectable
suspensions containing viscosity-increasing agents, e.g. sodium carboxymethyl
cellulose,
sorbitol and/or dextran, and where appropriate stabilisers. The active
ingredient, if need be
WO ~l/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-23-
together with excipients, can also be in the form of a lyophilisate and can be
made into a
solution before parenteral administration by the ad dition of suitable
solvents.
Solutions such as those used, for example, for parenteral administration can
also be emplo -
yed as infusion solutions.
The invention similarly relates to a process or a method for the treatment of
one of the abo -
ve-mentioned pathological conditions, especially a disease which responds to
an enhance-
ment of microtubule polymerisation, especially a corresponding neoplastic
disease. A com-
pound of formula IA can be administered as such or in the form of
pharmaceutical composi -
tions, prophylactically or therapeutically, preferably in an amount effective
against the said
diseases, to a warm-blooded animal, for example a human, requiring such
treatment, the
compounds especially being used in the form of pharmaceutical compositions. In
the case
of an individual having a bodyweight of about 70 kg the dose administered is
from approxi -
mately 0.1 mg to approximately 1 g, preferably from approximately 0.5 mg to
approximately
200 mg, of a compound of the present invention. Administration is preferably
effected e.g.
every 1 to 4 weeks.
The present invention also relates in particular to the use of a compound of
formula IA, or a
pharmaceutically acceptable salt thereof, especially a compound of formula IA
named as a
preferred compound, or a pharmaceutically acceptable salt thereof, as such or
in the form
of a pharmaceutical formulation containing at least one pharmaceutically
employable car -
rier, for the therapeutical and also prophylactic treatment of one or more of
the above disea -
ses.
The present invention also relates in particular to the use of a compound of
formula IA, or a
pharmaceutically acceptable salt thereof, especially a compound of formula IA
named as a
preferred compound, or a pharmaceutically acceptable salt thereof, for the
preparation of a
pharmaceutical formulation for the therapeutical and also prophylactic
treatment of one or
more of the above diseases.
The following examples illustrate the invention, but are not intended to
restrict their scope in
any way.
WO 01/27308 CA 02425620 2003-04-07 pCT/EP00/09817
-24-
General: 'H and'3C NMR spectra were measured in CDC13, respectively. Positive
ion
mass spectra, using the fast ion bombardment (FIB) technique, were obtained on
a VG
ZAB-VSE double focusing, high-resolution mass spectrometer equipped with
either a
cesium or sodium ion gun. Optical rotations were measured in a one-decimeter
(1.3 mL)
cell using an Autopol III automatic polarimeter. TLC was performed on glass
sheets
precoated with silica gel (Merck, Kieselgel 60, F254, Art. 5715). Column
chromato-
graphic separations were performed on silica gel (Merck, Kieselgel 60, 230-400
mesh,
Art. 9385) under pressure. THF was dried and distilled over sodium ketyl. All
antibody
reactions were degassed by passing a slow stream of Ar gas into the reaction
mixture
and carried out in Ar atmosphere. Temperatures are measured in degrees
Celsius. Unless
otherwise indicated, the reactions take place at room temperature.
Abbreviations used:
aqu. aqueous
BuLi butyl lithium
CC column chromatography
DBU 1,8-diazabicyclo[5.4.0]undec-7-a
ne
DMF N,N-dimethylformamide
DIBAL-H diisobutyl aluminum hydride
DMAP dimethylaminopyridine
DMSO dimethyl sulphoxide
EA ethyl acetate
equiv equivalents)
ESI-MS Electro-Spray Ionisation Mass Spectroscopy
EtOH ethanol
EtOAc acetic acid ethyl ester
HPLC high pressure liquid chromatography
HRMS high resolution mass spectrometry
Hunig's ethyl diisopropylamine
base
IC50 concentration leading to
50 % inhibition
LDA lithium diisopropylamide
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
- 25 -
Me methyl
NaHMDS sodium hexamethyl disilazide
NMR nuclear magnetic resonance
Ph phenyl
PBS phosphate buffered saline
Pr propyl
PTLC preparative thin layer chromatography
rt room temperature
tent tertiary
TBAF tetrabutyl ammonium fluoride
TBS or TBDMStent-butyl-dimethylsilyl
TFA trifluoroacetic acid
TFAA trifluoroacetic acid anhydride
THF tetrahydrofuran
TLC thin layer chromatography
TMS tetramethylsilane
Starting Materials - Synthesis of racemic aldols for the Examples 1 to 5.
General method: Ketone (1.1 equiv; 2.2 equiv for compounds ( ~)-11 of Example
5 and
Example 4) is added to a solution of LDA (0.5 M, 1.2 equiv; 2,4 equiv for
compounds ( ~)-11
of Example 5 and Example 4), freshly prepared from BuLi and i-Pr 2NH in THF)
at -78 °C.
The solution is stirred for 2 h at the same temperature, and then cooled to -
100 °C. The
corresponding aldehyde (1.0 equiv) in THF is added. The mixture is stirred at -
100 - -80 °C
for 0.5 to 1 h, and then quenched with a saturated solution of NH 4C1 and
allowed to warm to
rt. The mixture is diluted with water and extracted with EtOAc. The combined
organic layers
are washed with brine and dried over MgS04. Solvents are removed under vacuum
and the
residue is purified over silica gel (hexane-EtOAc) to afford the pure racemic
aldol product.
Compound 12 of Example 5: 'H NMR (400 MHz, CDC13): 8 7.08 (s, 1 H), 6.59 (s, 1
H), 4.85
(s, 2H), 4.76 (s, 2H), 4.61 (m, 1 H), 3.42 (s, 3H), 3.12 (br s, 1 H), 2.72 (m,
2H), 2.20 (s, 3H),
2.04 (s, 3H). '3C NMR (100.6 MHz, CDCI3): 8 210.3, 168.0, 154.0, 141.5, 119.3,
117.4,
96.8, 73.2, 66.6, 56.0, 49.0, 31.1, 14.8 ppm.
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-26-
Compound 13 of Example 5: 'H NMR (600 MHz, CDC13): 8 7.15 (s, 1 H), 6.58 (s, 1
H), 5.57
(d, J= 47.0 Hz, 2H), 4.59 (m, 1 H), 3.43 (d, J = 2.9 Hz, 1 H), 2.70 (m, 2H),
2.19 ( s, 3H), 2.01
s, 3H).'3C NMR (150.9 MHz, CDC13): 8209.0, 163.3, 153.4, 141.2, 118.0, 117.7,
81.1, 80.0,
72.5, 48.6, 30.8, 14.8 ppm.
Resolution of Thiazole Aldols (Examples 1 -5)
Example 1:
0
1
Antibody 8463 (65 mg, 0.000434 mmol) is added to a sterilized solution of 6.43
g
(26.9 mmol) of the racemic thiazole aldol (~)-1 in degassed CH3CN (10 - 20 mUg
aldol)
and a degassed buffer (PBS, pH 7.4, 200 mUg aldol) in a plastic bottle. The
mixture is
incubated at 37 °C for 5 days. At more than 98% consumption of the ent-
enantiomer as
judged by HPLC analysis, the mixture is filtered using Amicon to recover the
antibody and
the filtrate is passed through a reverse phase column (C-18) to elute first
water and then the
organic compounds using methanol as eluants. Solvents are removed under vacuum
and
the residue is purified by CC (silica gel, hexanes - EtOAc (3:1 )) to afford
the optically pure
aldol compounds 1 (99% ee) and the corresponding aldehyde 1a. Data for 1: [a]p-
33.1 (c
= 1.28, CHCI3); 'H NMR (400 MHz): b 6.89 (s, 1 H), 6.55 (s, 1 H), 4.58 (d, J =
8.6 Hz, 1 H),
3.57 (br s, 1 H), 2.70 (dd, J = 16.7, 9.4 Hz, 1 H), 2.66 (s, 3H), 2.64 (dd, J
= 16.7, 3.0 Hz, 1 H),
2.47 (q, J = 7.3 Hz, 2H), 2.00 (s, 3H), 1.04 (t, J = 7.3 Hz, 3H); MS: 240 (MH
+), 262 (MNa+).
HPLC (see Ex. 5): Solvent system A, Rt of 1, 16.20 min and ent-1, 18.12 min.
In analogy to the procedure described in Example 1, compounds 2 to 4 are
resolved.
Example 2:
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-27-
OH
\ ~ \
S' \S O
2
3.3 g, 12.8 mmol (t)-2 is resolved by 8463 (20 mg, 0.000133 mmol) in 7 days to
afford 2
with 96% ee; purified by CC (silica gel, hexanes - EtOAc (4:1 )). [ aJp
-35.2 (c = 2.05, CHCI3). 'H NMR (400 MHz): 8 6.93 (s, 1 H), 6.50 (s, 1 H),
4.58 (m, 1 H), 3.14
(d, J = 3.0 Hz, 1 H), 2.70 (d, J = 6.1 Hz, 2H), 2.67 (s, 3H), 2.20 (s, 3H),
2.06 (d, J = 1.2 Hz,
3H); MS: 258 (MH+), 280 (MNa+).
Example 3:
0
3
8.4 g (30.8 mmol) of (t)-3 is resolved by 8463 (20 mg, 0.000133 mmol) in 10
days to afford
3 with 99% ee; purified by CC (silica gel, hexanes - EtOAc (4:1 )). [ a]p -
35.6 (c = 0.92,
CHCI3); 'H NMR (400 MHz, CDCI3): 8 6.94 (s, 1 H), 6.57 (s, 1 H), 4.60 (t, J =
6.2 Hz, 1 H), 3.32
(br s, 1 H), 2.68 (s, 3H), 2.68 (m, 2H), 2.48 (q, J = 7.3 Hz, 2H), 2.07 (s,
3H), 1.07 (t, J = 7.3
Hz, 3H); MS: 272 (MH+), 294 (MNa+).
Example 4:
4
8.80 g (34.5 mmol) of (t)-4 is resolved by 8463 (250 mg, 0.00167 mmol) in 5
days to afford
4 in >99% ee; purified by CC (silica gel, hexanes - EtOAc (1:2)). [a]p
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
- 28 -
-23.4 (c = 1.08, CHCI3).' H NMR (500 MHz): 8 6.94(s, 1 H), 6.44 (s, 1 H), 5.21
(br s, 1 H), 4.76
(s, 2H), 4.50 (d, J = 9.3 Hz, 1 H), 3.32 (br, 1 H), 2.64 (dd, J = 16.3, 6.8
Hz, 1 H), 2.51 (dd, J =
16.3, 3.02 Hz, 1 H), 2.42 (q, J = 7.3 Hz, 2H), 1.86 (s, 3H), 0.96 (t, J = 7.3
Hz, 3H); MS: 278
(MNa+). HPLC (see Ex. 5): Solvent system B, R, of 4, 17.10 min and ent-4 18.42
min.
Example 5:
In analogy to the procedure described in Example 1, compounds of formula ( ~)-
II are
resolved by antibody 8463, 85H6 and 93F3 catalyzed retro-aldol reactions.
R~
n ((~)-I I)
Table 1
compounds of 8463 85H6 93F3
formula + -II ee % ee % ee
5: R = R, = Me 98 50 94 50 98 50
6: R = Pr, R, = Me 99 50 99 50 99 50
7: R = Bu, R, = Me 99 50 99 50 99 50
8: R = Pen, R, = Me 97 52 97 55 97 53
9: R = But-1-ene, R~ 98 52 98 50 99 50
= Me
10: R = CH2F, R1 = Me 96 54 99 55 98 52
11: R = Me, R, = CH20H 99 50 99 50 99 50
12: R = Me, R, = CH20MOM> 99 99 50 > 99
50 51
13: R = Me, R, = CH2F > 99 NT'a NTa
50
14: R = Me, R~ = OMe >95b >95b >95b
51 52 52
Numbers in parentheses represent the percent conversion.
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-29-
a: not tested
b: Peaks of the two enantiomers on HPLC trace not base-line separable.
HPLC conditions: For compounds of Examples 2 and 3 and compounds 5 to 10 and
14 of
Example 5, see Sinha et al, Org. Lett. 1999, 1, 1623 and supporting
information; ~,m~ = 254
nm; reverse phase ODR column, Daicel Chemical Industries.
Solvent systems: (A) acetonitrile-water (3:17) and 0.1 % TFA, (B) acetonitrile-
water (1:4) and
0.1 % TFA, (C) acetonitrile-water (3:7) and 0.1 % TFA, at a flow rate of 0.4
mUmin.
Compound 11: Solvent system B, R~ of 11, 12.56 min and ent-11 14.38 min.
Compound 12:
Solvent system C R~ of 12, 17.10 min and ent-12 18.42 min. Compound 13:
Solvent system
B, Rt of 13, 44.27 min and ent-13 48.72 min.
Example 6:
Y H Y
H( N N
O OH O OH O
A B
TFAA (4 equiv.) is added dropwise to a solution of the 2.8:1 mixture (70 mg,
0.096 mmol)
of stage 6.5 in dry CH2C12 (0.1 M solution) at 0 °C and the mixture is
stirred at the same
temperature for 4 h and then concentrated under vacuum. The residue is
dissolved in
EtOAc, washed with brine and dried over MgS04. Solvents are evaporated and the
resulting residue is purified by preparative TLC (PTLC) (silica gel, hexane -
EtOAc 2:1 )
to afford the compounds of the above formula (60 % A, 21 % B)
Physical data of A: [ aJp -69:5° (c = 0.23, CHC13); 'H NMR (600 MHz): b
6.95 (s, 1 H), 6.59 (s,
1 H), 5.33 (d, J = 10.5 Hz, 1 H), 5.20 (d, J = 7.0 Hz, 1 H), 4.15 (d, J = 10.9
Hz, 1 H), 3.77 (s,
1 H), 3.39 (d, J = 4.9 Hz, 1 H), 3.11 (qd, J = 7.0, 2.6 Hz, 1 H), 2.90 (br s,
1 H), 2.84 (dd, J =
14.5, 11.0 Hz, 1 H), 2.69 (s, 3H), 2.47 (dd, J = 14.9, 11.0 Hz, 1 H), 2.38
(dd, J = 15.4, 2.6 Hz,
1 H), 2.21 (m, 1 H), 2.08 (s, 3H), 2.03 (m, 3H), 1.93 (m, 1 H), 1.75 (m, 1 H),
1.73 (s, 3H), 1.59
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-30-
(m, 1 H), 1.39-1.24 (m, 3H), 1.33 (s, 3H), 1.17 (d, J = 6.6 Hz, 3H), 1.10 (s,
3H), 1.00 (d, J =
7.4 Hz, 3H), 0.99 (t, J = 7.4 Hz, 3H); HRMS: (C2SH43N05SNa = 528.2754) found
528.2735
(MNa+).
Physical data of B: [a]p -22.0° (c = 0.10, CHC13); 'H NMR (600 MHz): 8
6.96 (s, 1 H), 6.56 (s,
1 H), 5.46 (d, J = 11.0 Hz, 1 H), 5.25 (t, J = 7.0 Hz, 1 H), 4.01 (d, J = 10.6
Hz, 1 H), 3.76 (m,
1 H), 3.21 (quintet, J = 6.6 Hz, 1 H), 3.10 (d, J = 2.6 Hz, 1 H), 2.70 (s,
3H), 2.54 (dd, J = 15.4,
11.0 Hz, 1 H), 2.45 (m, 2H), 2.36 (dd, J = 14.9, 11.0 Hz, 1 H), 2.19 (m, 1 H),
2.13 (m, 1 H),
1.96 (m, 1 H), 1.89 (m, 1 H), 1.66 (m, 1 H), 1.60 (s, 3H), 1.57 (m, 1 H), 1.49
(m, 1 H), 1.29 (s,
3H), 1.28-1.20 (m, 2H), 1.18 (d, J = 6.5 Hz, 3H), 1.06 (s, 3H), 0.96 (d, J =
7.0 Hz, 3H), 0.94
(t, J = 7.4 Hz, 3H); HRMS: (C28H~NOSS = 506.2935) found 506.2926 (MN +).
Stage 6.1
TBSCI (1.5 equiv) is added to a solution of the compound of Example 1 (3.0 g,
12.6 mmol) and imidazole (3.0 equiv) in DMF (2M solution). The reaction
mixture is
stirred at rt for 24 h and worked up with ether and water. The organic layer
is separated
and the water phase is extracted with ether. The combined organic layer is
washed with
brine, dried over MgS04. Solvents are evaporated and the residue is purified
by CC
(silica gel, hexanes - EtOAc (10:1 )) to afford the pure silyl ether; [a]p -
44.4 (c = 0.98,
CHCI3); 'H NMR (400 MHz): 8 6.89 (s, 1 H), 6.50 (s, 1 H), 4.63 (dd, J = 9.1,
3.2 Hz, 1 H),
2.77 (dd, J = 14.4, 9.4 Hz, 1 H), 2.67 (s, 3H), 2.45 (m, 2H), 2.37 (dd, J =
14.4, 3.5 Hz, 1 H),
2.00 (d, J = 1.2 Hz, 3H), 1.01 (t, J = 7.0 Hz, 3H), 0.83 (s, 9H), 0.00 (s,
3H), -0.02 (s, 3H);
MS: 354 (MH+).
Stage 6.2
BuLi (1.1 equiv) is added to a heterogeneous mixture of MePPh31 (1.2 equiv) in
dry THF
(0.5 M solution) at 0 °C. After the mixture is stirred for 0.5 h at rt,
a solution of the
compound of stage 6.1 (570 mg, 1.61 mmol) in THF (2 M solution) is added. The
reaction mixture is stirred for an additional 0.5 h, and then quenched with a
saturated
solution of NH4C1 and extracted with ether. The combined organic layer is
washed with
brine and dried over MgS04. Solvents are removed under vacuum and the
resultant
residue is purified by CC (silica gel, hexanes - EtOAc (20:1 )) to afford the
pure
methylenated product. [a]p +4.6 (c = 1.00, CHCI3); 'H NMR (500 MHz): 8 6.90
(s, 1 H), 6.45
(s, 1 H), 4.75 (m, 2H), 4.23 (dd, J = 7.7, 5.2 Hz, 1 H), 2.69 (s, 3H), 2.28
(dd, J =13.2, 7.7 Hz,
WO 01/27308 CA 02425620 2003-04-07 pCT/EP00/09817
-31 -
1 H), 2.22 (dd, J = 13.2, 5.5 Hz, 1 H), 2.05 (q, J = 7.3 Hz, 2H), 1.99 (s,
3H), 1.01 (t, J = 7.3
Hz, 3H), 0.86 (s, 9H), 0.03 (s, 3H), -0.02 (s, 3H); MS: 352 (MH+).
Stage 6.3
TBAF (1.2 equiv) is added to a solution of the 292 mg (0.83 mmol) of the TBS
ether of
stage 6.2 in dry THF (0.3 M solution) at 0 °C. After stirred for 1 h at
this temperature,
the reaction mixture is diluted with water and extracted with EtOAc. The
combined
organic layer is washed with brine, dried over MgS04 and solvents are removed.
The
residue is purified by CC (silica gel, hexanes - EtOAc (4:1 )) to afford the
pure
deprotected product. [a]o -24.0 (c = 0.73, CHCI3); 'H NMR (400 MHz): 8 6.89
(s, 1 H), 6.55
(s, 1 H), 4.84 (d, J = 1.8 Hz, 1 H), 4.83 (s, 1 H), 4.24 (dd, J = 8.8, 4.1 Hz,
1 H), 2.66 (s, 3H),
2.48 (br s, 1 H), 2.36 (dd, J = 13.8, 4.1 Hz, 1 H), 2.26 (dd, J = 14.1, 9.1
Hz, 1 H), 2.05 (q, J =
7.3 Hz, 2H), 2.00 ( d, J = 0.9 Hz, 3H), 1.02 (t, J = 7.3 Hz, 3H); '3C NMR
(100.6 MHz): 8
164.4, 152.7, 147.8, 141.6, 118.7, 115.3, 111.2, 74.7, 42.9, 28.3, 19.0, 14.2,
12.1; MS: 238
(MH+), 260 MNa+).
Stage 6.4
EDC (2.0 equiv) and DMAP (0.1 equiv) are added to a solution of the acid of
formula
VIII (1.2 equiv) and 43 mg (0.18 mmol) of the thiazole alcohol of stage 6.3
(36 - 41, 1.0
equiv) in dry CH2CI2 (0.2 M solution) at 0 °C. After the reaction
mixture is stirred for 16 h
at 0 °C to rt, the solvent is evaporated under vacuum and the residue
is purified by CC
(silica gel, hexanes - EtOAc (10:1 )) to afford the corresponding pure ester.
[a]p -43.0 (c
= 1.43, CHCI3); 'H NMR (400 MHz): 8 6.92 (s, 1 H), 6.48 (s, 1 H), 5.77 (m, 1
H), 5.38 (dd, J =
7.9, 6.2 Hz, 1 H), 4.97 (dd, J = 17.3, 1.5 Hz, 1 H), 4.91 (br d, J = 10.2 Hz,
1 H), 4.76 (s, 2H),
4.31 (dd, J = 5.9, 4.1 Hz, 1 H), 3.71 (dd, J = 6.7, 2.0 Hz, 1 H), 3.14
(quintet, J = 6.7 Hz, 1 H),
2.67 (s, 3H), 2.51 (dd, J = 17.3, 3.8 Hz, 1 H), 2.46 (dd, J = 14.1, 7.9 Hz, 1
H), 2.37 (dd, J =
13.8, 5.9 Hz, 1 H), 2.24 (dd, J = 17.3, 5.9 Hz, 1 H), 2.06 (d, J = 1.2 Hz,
3H), 2.01 (m, 4H),
1.46 - 1.04 (m, 5H), 1.21 (s, 3H), 1.01 (d, J = 6.4 Hz, 3H), 1.01 (s, 3H),
1.00 (t, J = 7.4 Hz,
3H), 0.87 (d, J = 7.0 Hz, 3H), 0.87 (s, 9H), 0.85 (s, 9H), 0.08 (s, 3H), 0.02
(s, 3H), 0.01 (s,
6H); MS: 784 (MNa+).
Stage 6.5
0.2 equiv. of Grubbs' catalyst of formula VI
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-32-
Mes~N N~Mes
Ph
CI ~~"
/" Ru
C
PCy3
is added to a solution of the diene of stage 6.4 (116 mg, 0.15 mmol) in dry
CH2CIZ
(0.002 M solution) and the solution is stirred at reflux for 20 - 96 h. After
the reaction is
completed, as judged by'H NMR analysis, solvents are evaporated and the
residue is
purified by CC (silica gel, hexanes - EtOAc (12:1 )) to afford a mixture of
the
metathesized products which is taken to the next step without separation.
Example 7:
\ \ S~S~ ~Sw
HO ~~"~ \ N //N
.,,,, O
O OH O OH O
A g
The 1.5:1 mixture (77 mg, 0.10 mmol) of stage 7.5 is deprotected according to
the method
described in the final stage of Example 6 to give pure compounds A (50%) and B
(34%);
PTLC conditions: silica gel, hexanes - EtOAc (2:1 ).
Physical data of A: [a]p -80.6° (c = 0.50, CHCI3); 'H NMR (500 MHz): 8
6.98 (s, 1 H), 6.52 (s,
1 H), 5.39 (d, J = 10.3 Hz, 1 H), 5.21 (d, J = 8.5 Hz, 1 H), 4.11 (m, 1 H),
3.77 (br s, 1 H), 3.11
(qd, J = 6.6, 3.0 Hz, 1 H), 2.95 (d, J = 5.5 Hz, 1 H), 2.91 (dd, J =14.7,
11.4, 1 H), 2.86 (br s,
1 H), 2.69 (s, 3H), 2.49 (dd, J = 15.4, 11.0 Hz, 1 H), 2.40 (dd, J = 15.4, 3.0
Hz, 1 H), 2.21-
2.16 (m, 1 H), 2.13 (d, J = 1.5 Hz, 3H), 1.95 (br s, 1 H), 1.92 (br s, 1 H),
1.78-1.74 (m, 1 H),
1.72 (s, 3H), 1.62-1.55 (m, 1 H), 1.39-1.24 (m, 3H), 1.32 (s, 3H), 1.17 (d, J
= 7.0 Hz, 3H),
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-33-
1.10 (s, 3H), 1.00 (d, J = 7.0 Hz, 3H); HRMS: (C2,H42NOSSz = 524.2499) found
524.2522
(MH+).
Physical data of B: [a]o -33.3° (c = 0.12, CHCI3); 'H NMR (500 MHz): 8
6.98 (s, 1 H), 6.49 (s,
1 H), 5.50 (d, J = 9.6 Hz, 1 H), 5.31 (m, 1 H), 4.00 (d, J = 10.7 Hz, 1 H),
3.76 (m, 1 H), 3.20
(quintet, J = 6.3 Hz, 1 H), 3.02 (m, 1 H), 2.69 (s, 3H), 2.56-2.40 (m, 3H),
2.18 (m, 1 H), 2.14
(s, 3H), 1.98-1.84 (m, 2H), 1.66 (m, 1 H), 1.64 (s, 3H), 1.48 (m, 1 H), 1.33
(s, 3H), 1.24 (m,
3H), 1.17 (d, J= 7.0 Hz, 3H), 1.06 (s, 3H), 0.97 (d, J = 7.0 Hz, 3H); HRMS: (C
Z~H42NOSS2 =
524.2499) found 524.2515 (MH +).
Stage 7.1
Protection of the compound of Example 2 (1.58 g, 6.1 mmol) is protected
according to
the method described in stage 6.1 and affords the corresponding silyl ether;
CC
conditions: silica gel, hexanes - EtOAc (10:1 ). [a]o -47.1 (c = 0.95, CHCI3);
'H NMR
(400 MHz): b 6.92 (s, 1 H), 6.45 (s, 1 H), 4.62 (dd, J = 9.1, 3.5 Hz, 1 H),
2.79 (dd, J = 14.7,
9.1 Hz, 1 H), 2.69 (s, 3H), 2.44 (dd, J = 14.7, 3.5 Hz, 1 H), 2.17 (s, 3H),
2.05 (d, J = 1.5 Hz,
3H), 0.86 (s, 9H), 0.04 (s, 3H), 0.00 (s, 3H); MS: 372 (MH+).
Stage 7.2
The compound of stage 7.1 (620 mg, 1.67 mmol) is used according to the method
described in stage 6.2 to afford the methylenated reaction product; CC
conditions: silica
gel, hexanes - EtOAc (22:1 ); [a]p -9.7 (c = 0.90, CHCI3);'H NMR (500 MHz): 8
6.92 (s,
1 H), 6.40 (s, 1 H), 4.73 (m, 2H), 4.24 (dd, J = 7.3, 5.5 Hz, 1 H), 2.69 (s,
3H), 2.29 (dd, J =
13.2, 7.7 Hz, 1 H), 2.23 (dd, J = 13.2, 4.8 Hz, 1 H), 2.04 (d, J = 1.1 Hz,
3H), 1.75 (s, 3H),
0.88 (s, 9H), 0.04 (s, 3H), -0.01 (s, 3H); MS: 370 (MH+).
Stage 7.3
According to the method described in stage 6.3, the compound of stage 7.2 (498
mg, 1.35
mmol) is deprotected; CC conditions: silica gel, hexanes - EtOAc (5:1 ); [a]o -
44.8 (c =
1.23, CHCI3); 'H NMR (400 MHz): 8 6.92 (s, 1 H), 6.51 (s, 1 H), 4.87 (br s, 1
H), 4.82 (d, J =
0.9 Hz, 1 H), 4.25 (m, 1 H), 2.67 (s, 3H), 2.35 (dd, J = 13.5, 4.1 Hz, 1 H),
2.25 (dd, J = 14.1,
9.4 Hz, 1 H), 2.06 (d, J = 1.2 Hz, 3H), 1.78 (s, 3H); MS: 256 (MN +).
Stage 7.4
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-34-
According to the method described in stage 6.4, the compound of stage 7.3 (46
mg,
0.18 mmol) is used to afford the corresponding product which is purified by CC
(silica
gel, hexanes - EtOAc (12:1 )); [a]p -44.4 (c = 1.62, CHCI3); 'H NMR (400 MHz):
8 6.94 (s,
1 H), 6.42 (s, 1 H), 5.78 (m, 1 H), 5.39 (dd, J = 7.6, 4.6 Hz, 1 H), 4.97 (dg,
J = 17.0, 1.5 Hz,
1 H), 4.91 (br d, J = 10.0 Hz, 1 H), 4.76 (br s, 1 H), 4.74 (br s, 1 H), 4.31
(dd, J = 5.9, 3.8 Hz,
1 H), 3.71 (dd, J = 6.7, 2.0 Hz, 1 H), 3.14 (quintet, J = 6.8 Hz, 1 H), 2.67
(s, 3H), 2.53 (dd, J =
17.0, 3.8 Hz, 1 H), 2.44 (dd, J = 13.8, 7.9 Hz, 1 H), 2.35 (dd, J = 13.8, 5.9
Hz, 1 H), 2.25 (dd,
J = 17.3, 5.9 Hz, 1 H), 2.10 (d, J = 1.2 Hz, 3H), 2.01 (m, 2H), 1.74 (s, 3H),
1.47 -1.26 (m,
3H), 1.22 (s, 3H), 1.19 - 1.06 (m, 2H), 1.02 (d, J = 5.9 Hz, 3H), 1.01 (s,
3H), 0.87 (d, J = 6.8
Hz, 3H), 0.87 (s, 9H), 0.86 (s, 9H), 0.08 (s, 3H), 0.03 (s, 3H), 0.013 (s,
3H), 0.01 (s, 3H);
MS: 802 (MNa~).
Stage 7.5
According to the method described in stage 6.5, the diene of stage 7.4 (104
mg,
0.13 mmol) is metathesized to afford a mixture of the metathesized products
which is
taken to the next step without separation; CC conditions: silica gel, hexanes -
EtOAc
(14:1 ).
Example 8:
S~ ~S~
H( N
O OH O OH O
A B
The 2.4:1 mixture (60 mg, 0.078 mmol) of stage 8.5 is deprotected according to
the method
described in the final stage of Example 6 to yield pure compounds A (59%) and
B (24%);
PTLC conditions: silica gel, hexanes - EtOAc (3:1 ).
Physical data of A: [a]p -64.9° (c =1.46, CHCI3); 'H NMR (500 MHz): b
6.98 (s, 1 H), 6.51 (s,
1 H), 5.34 (d, J = 10.3 Hz, 1 H), 5.20 (dd, J =10.7, 3.0 Hz, 1 H), 4.10 (m, 1
H), 3.78 (m, 1 H),
3.11 (qd, J = 7.0, 3.3 Hz, 1 H), 2.86 (m, 2H), 2.79 (br s, 1 H), 2.69 (s, 3H),
2.48 (dd, J =15.4,
WO 01/27308 CA 02425620 2003-04-07 pCT~P00/09817
-35-
11.0 Hz, 1 H), 2.42 (dd, J = 15.4, 3.0 Hz, 1 H), 2.21 (m, 1 H), 2.14 (s, 3H),
2.04 (m, 3H), 1.93
(m, 1 H), 1.74 (m, 1 H), 1.38-1.25 (m, 3H), 1.33 (s, 3H), 1.18 (d, J = 6.6 Hz,
3H), 1.11 (s, 3H),
1.00 (d, J = 7.0 Hz, 3H), 0.99 (t, J = 7.4 Hz, 3H); HRMS: (C 28H~NO5S2 =
538.2655) found
538.2646 (MH+).
Physical data of B: [ajp -8.6° (c = 0.40, CHCI3); 'H NMR (600 MHz): b
6.98 (s, 1 H), 6.49 (s,
1 H), 5.46 (d, J = 10.5 Hz, 1 H), 5.26 (t, J = 7.0 Hz, 1 H), 3.99 (d, J = 10.5
Hz, 1 H), 3.75 (m,
1 H), 3.20 (quintet, J = 6.6 Hz, 1 H), 3.04 (d, J = 2.6 Hz, 1 H), 2.70 (s,
3H), 2.53 (dd, J = 14.9,
7.5 Hz, 1 H), 2.46 (dd, J = 15.4, 2.3 Hz, 1 H), 2.42 (br s, 1 H), 2.37 (dd, J
= 14.8, 10.9 Hz,
1 H), 2.21 (m, 1 H), 2.14 (d, J = 0.8 Hz, 3H), 2.12 (m, 2H), 1.95 (dd, J =
14.0, 7.4 Hz, 1 H),
1.89 (m, 1 H), 1.65 (m, 1 H), 1.52-1.45 (m, 1 H), 1.29 (s, 3H), 1.27-1.21 (m,
3H), 1.18 (d, J =
7.0 Hz, 3H), 1.06 (s, 3H), 0.96 (d, J = 7.0 Hz, 3H), 0.95 (t, J = 7.2 Hz, 3H);
HRMS:
(C28H~NOSS2 = 538.2655) found 538.2651 (MN +).
Stage 8.1
Protection of the compound of Example 3 (2.36 g, 8.7 mmol) is protected
according to
the method described in stage 6.1 and affords the corresponding silyl ether;
CC
conditions: silica gel, hexanes - EtOAc (12:1 ). [g]o -54.0 (c = 1.97, CHCI3);
' H NMR
(500 MHz): b 6.86 (s, 1 H), 6.39 (s, 1 H), 4.58 (dd, J = 9.1, 3.2 Hz, 1 H),
2.73 (dd, J = 14.5,
9.2 Hz, 1 H), 2.61 (s, 3H), 2.40 (m, 2H), 2.33 (dd, J = 14.5, 3.3 Hz, 1 H),
2.01 (s, 3H), 0.96 (t,
J = 7.3 Hz, 3H), 0.79 (s, 9H), -0.04 (s, 3H), -0.06 (s, 3H); MS: 408 (MNa+).
Stage 8.2
The compound of stage 8.1 (250 mg, 0.65 mmol) is used according to the method
described in stage 6.2 to afford the methylenated reaction product; CC
conditions: silica
gel, hexanes - EtOAc (20:1 ). [ajp +0.1 (c =1.55, CHCI3);'H NMR (400 MHz): S
6.91 (s,
1 H), 6.39 (s, 1 H), 4.75 (m, 2H), 4.21 (dd, J = 7.3, 5.6 Hz, 1 H), 2.69 (s,
3H), 2.26 (m, 2H),
2.05 (q, J = 7.6 Hz, 2H), 2.04 (d, J = 1.1 Hz, 3H), 1.01 (t, J = 7.3 Hz, 3H),
0.87 (s, 9H), 0.02
(s, 3H), -0.02 (s, 3H); MS: 384 (MH+).
Stage 8.3
According to the method described in stage 6.3, the compound of stage 8.2 (498
mg,
1.35 mmol) is deprotected; CC conditions: silica gel, hexanes - EtOAc (5:1 ).
[ajp -30.5
(c =1.13, CHCI3); 'H NMR (600 MHz): 8 6.92 (s, 1 H), 6.50 (s, 1 H), 4.88 (m, 1
H), 4.85 (br s,
1 H), 4:23 (dd, J = 9.7, 4.0 Hz, 1 H), 2.67 (s, 3H), 2.38 (dd, J =14.0, 3.9
Hz, 1 H), 2.24 (dd, J
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-36-
= 14.0, 9.2 Hz, 1 H), 2.10 (q, J = 7.5 Hz, 2H), 2.06 (d, J = 1.3 Hz, 3H), 1.03
(t, J = 7.5 Hz,
3H); MS: 270 (MH+).
Stage 8.4
According to the method described in stage 6.4, the compound of stage 8.3 (81
mg,
0.3 mmol) is used to afford the corresponding product which is purified by CC
(silica
gel, hexanes - EtOAc (12:1 )); [a]p - 39.2 (c = 1.10, CHC13); 'H NMR (400
MHz): 8 6.93 (s,
1 H), 6.41 (s, 1 H), 5.76 (m, 1 H), 5.37 (dd, J = 7.9, 6.2 Hz, 1 H), 4.96 (dq,
J = 17.3, 1.5 Hz,
1 H), 4.90 (br d, J = 10.2 Hz, 1 H), 4.76 (s, 2H), 4.31 (dd, J = 5.9, 4.1 Hz,
1 H), 3.70 (dd, J =
6.7, 2.0 Hz, 1 H), 3.13 (quintet, J = 7.0 Hz, 1 H), 2.67 (s, 3H), 2.50 (dd, J
= 17.3, 3.8 Hz, 1 H),
2.47 (dd, J = 14.1, 7.9 Hz, 1 H), 2.37 (dd, J = 14.1, 5.9 Hz, 1 H), 2.24 (dd,
J = 17.3, 5.6 Hz,
1 H), 2.09 (s, 3H), 2.07 - 1.96 (m, 4H), 1.48 - 1.26 (m, 3H), 1.21 (s, 3H),
1.20 - 1.04 (m,
2H), 1.02 (d, J = 6.4 Hz, 3H), 1.01 (s, 3H), 1.00 (t, J = 7.4 Hz, 3H), 0.88
(d, J = 6.8 Hz, 3H),
0.87 (s, 9H), 0.85 (s, 9H), 0.08 (s, 3H), 0.02 (s, 3H), 0.008 (s, 3H), 0.006
(s, 3H); 828 (MCI -)
Stagie 8.5
According to the method described in stage 6.5, the diene of stage 8.4 (110
mg, 0.14
mmol) is metathesized to afford a mixture of the metathesized products which
is taken
to the next step without separation; CC conditions: silica gel, hexanes -
EtOAc (14:1 ).
Examiple 9:
OH ~OH
HC N
OH O
A B
The 2.3:1 mixture (70 mg, 0.08 mmol) of stage 9.6 is deprotected according to
the method
described in the final stage of Example 6 to yield pure compounds A (63%) and
B (27%);
PTLC conditions: silica gel, hexanes - EtOAc (1:2).
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-37-
Physical data of A: [aJo -46.1 ° (c = 0.30, CHCI3); 'H NMR (600 MHz): S
7.10 (s, 1 H), 6.60 (s,
1 H), 5.35 (d, J = 10.9 Hz, 1 H), 5.20 (d, J = 7.4 Hz, 1 H), 4.91 (s, 2H),
4.13 (d, J = 10.9 Hz,
1 H), 3.77 (t, J = 3.3 Hz, 1 H), 3.28 (br m, 1 H), 3.11 (qd, J = 6.8, 3.0 Hz,
1 H), 2.85 (dd, J =
14.5, 11.2 Hz, 1 H), 2.62 (s, 3H), 2.48 (dd, J = 15.2, 11.2 Hz, 1 H), 2.39
(dd, J = 15.3, 2.7 Hz,
1 H), 2.20 (m, 1 H), 2.09 (s, 3H), 2.07-2.00 (m, 3H), 1.94 (m, 1 H), 1.75 (m,
1 H), 1.58 (m, 1 H),
1.35 (s, 3H), 1.30-1.22 (m, 3H), 1.17 (d, J = 6.8 Hz, 3H), 1.09 (s, 3H), 1.00
(d, J = 7.2 Hz,
3H), 0.99 (t, J = 7.3 Hz, 3H); HRMS: (C28H~NO6S = 522.2884) found 522.2889 (MN
+).
Physical data of B: [aJp -18.1 ° (c = 0.16, CHCI3); 'H NMR (600 MHz): 8
7.11 (s, 1 H), 6.58 (s,
1 H), 5.45 (d, J = 10.2 Hz, 1 H), 5.24 (t, J = 6.6 Hz, 1 H), 4.93 (s, 2H),
4.05 (d, J = 10.4 Hz,
1 H), 3.77 (s, 1 H), 3.75 (d, J = 4.8 Hz, 1 H), 3.20 (quintet, J = 6.9 Hz, 1
H), 3.05 (br s, 1 H),
2.60 (br s, 1 H), 2.54 (dd, J = 15.0, 10.6 Hz, 1 H), 2.46 (m, 2H), 2.37 (dd, J
= 14.4, 10.5 Hz,
1 H), 2.25-2.18 (m, 1 H), 2.10 (s, 3H), 2.10-2.04 (m, 1 H), 1.97 (m, 1 H),
1.90 (m, 1 H), 1.50 (m,
1 H), 1.28 (s, 3H), 1.28-1.20 (m, 4H), 1.18 (d, J = 6.7 Hz, 3H), 1.05 (s, 3H),
0.98 (d, J = 7.0
Hz, 3H), 0.95 (t, J = 6.5 Hz, 3H); HRMS: (C28H~NO6S = 522.2884) found 522.2888
(MH +).
Stage 9.1
Protection of the compound of Example 4 (3.2 g, 12.5 mmol) is protected
according to
the method described in stage 6.1 and affords the corresponding silyl ether;
CC
conditions: silica gel, hexanes - EtOAc (20:1). [aJo -32.1 (c = 1.21,
CHC13);'H NMR
(400 MHz): b 7.00 (s, 1 H), 6.50 (s, 1 H), 4.93 (s, 2H), 4.63 (dd, J = 9.4,
3.2 Hz, 1 H), 2.77
(dd, J = 14.4, 9.4 Hz, 1 H), 2.46 (m, 2H), 2.37 (dd, J = 14.3, 3.2 Hz, 1 H),
2.00 (s, 3H), 1.01
(t, J = 7.0 Hz, 3H), 0.93 (s, 9H), 0.83 (s, 9H), 0.11 (s, 6H), 0.01 (s, 3H), -
0.02 (s, 3H); MS:
484 (MH+), 506 (MNa+).
Stage 9.2
The compound of stage 9.1 (547 mg, 1.13 mmol) is used according to the method
described in stage 6.2 to afford the methylenated reaction product; CC
conditions: silica
gel, hexanes - EtOAc (40:1 ). [a]o +1.5 (c = 2.53, CHCI3); 'H NMR (400 MHz): S
7.01 (s,
1 H), 6.44 (s, 1 H), 4.95 (s, 2H), 4.75 (m, 2H), 4.22 (dd, J = 7.6, 5.6 Hz, 1
H), 2.29 (dd, J =
13.6, 7.6 Hz, 1 H), 2.24 (dd, J = 13.6, 5.6 Hz, 1 H), 2.05 (q, J = 7.3 Hz,
2H), 1.99 (s, 3H),
1.01 (t, J = 7.3 Hz, 3H), 0.95 (s, 9H), 0.87 (s, 9H), 0.12 (s, 6H), 0.02 (s,
3H), -0.03 (s, 3H);
MS: 482 (MH+), 504 (MNa+).
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-38-
Stage 9.3
According to the method described in stage 6.3 but using 2.4 equiv. of TBAF
instead of 1.2
equiv., the compound of stage 9.2 (180 mg, 0.37 mmol) is deprotected; CC
conditions:
silica gel, hexanes - EtOAc (1:2). [a]p -23.3 (c = 1.33, CHCI3); 'H NMR (400
MHz): b 6.99
(s, 1 H), 6.49 (s, 1 H), 4.82 (m, 5H), 4.22 (dd, J = 8.5, 4.4 Hz, 1 H), 3.08
(br s, 1 H), 2.34 (dd, J
= 14.1, 4.4 Hz, 1 H), 2.24 (dd, J = 14.1, 8.8 Hz, 1 H), 2.04 (q, J = 7.3 Hz,
2H), 1.93 (s, 3H),
1.01 (t, J = 7.3 Hz, 3H); MS: 254 (MH+), 276 (Mna+).
Stage 9.4
In a manner as described under stage 11.4 the compound of stage 9.3 (80 mg,
0.32
mmol) is allowed to react with TBSCI (60 mg, 0.38 mmol) and i-Pr2NEt (0.11 mL,
0.64
mmol) in dry CH2C12 (3 mL) at 0 °C to afford the protected derivative
which is purified by
CC (silica gel, hexanes - EtOAc, (3:1 )); [a]p -23.2 (c = 0.50, CHCI3); 'H NMR
(500 MHz): 8
7.03 (s, 1 H), 6.57 (s, 1 H), 4.94 (s, 2H), 4.89 (d, J = 1.5 Hz, 1 H), 4.86
(s, 1 H), 4.25 (dd, J =
9.2, 3.7 Hz, 1 H), 2.39 (dd, J = 14.0, 3.7 Hz, 1 H), 2.27 (dd, J = 14.0, 9.2
Hz, 1 H), 2.09 (q, J =
7.4 Hz, 2H), 2.04 (d, J = 1.1 Hz, 3H), 1.05 (t, J = 7.5 Hz, 3H), 0.94 (s, 9H),
0.12 (s, 6H);
'3CNMR (125.75 MHz): 8 171.9, 153.1, 147.9, 141.5, 118.8, 115.6, 111.4, 74.7,
63.2, 43.1,
28.4, 25.7, 18.2, 14.4, 12.2, -5.5; MS: 390 (MNa +).
Stage 9.5
According to the method described in stage 6.4, the compound of stage 9.4 (46
mg, 0.13
mmol) is used to afford the corresponding product which is purified by CC
(silica gel,
hexanes - EtOAc (15:1 ). [a]p -35.4 (c = 1.00, CHCI3); 'H NMR (400 MHz): 8
7.03 (s, 1 H),
6.48 (s, 1 H), 5.78 (m, 1 H), 5.39 (dd, J = 7.6, 6.2 Hz, 1 H), 4.97 (dg, J
=17.0, 1.4 Hz, 1 H),
4.94 (s, 2H), 4.92 (br d, J = 10.2 Hz, 1 H), 4.77 (s, 2H), 4.31 (dd, J = 5.6,
3.8 Hz, 1 H), 3.71
(dd, J = 7.0, 2.4 Hz, 1 H), 3.14 (quintet, J = 6.7 Hz, 1 H), 2.51 (dd, J =
17.0, 3.8 Hz, 1 H), 2.47
(dd, J = 15.0, 8.2 Hz, 1 H), 2.36 (dd, J = 15.0, 5.8 Hz, 1 H), 2.25 (dd, J =
17.0, 5.9 Hz, 1 H),
2.05 (s, 3H), 2.00 (m, 4H), 1.48 -1.25 (m, 5H), 1.22 (s, 3H), 1.02 (d, J = 6.8
Hz, 3H), 1.01
(s, 3H), 1.00 (t, J = 7.3 Hz, 3H), 0.94 (s, 9H), 0.88 (s, 9H), 0.87 (d, J =
6.7 Hz, 3H), 0.86 (s,
9H), 0.12 (s, 6H), 0.09 (s, 3H), 0.02 (s, 3H), 0.01 (s, 6H); MS: 892 (MH+).
Stage 9.6
WO 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-39-
According to the method described in stage 6.5, the diene of stage 9.5 (90 mg,
0.1 mmol)
is metathesized to afford a mixture of the metathesized products which is
taken to the
next step without separation; CC conditions: silica gel, hexanes - EtOAc (20:1
).
Example 10:
SY
HO ~-", ~ \ N
,,,,. O
O OH O OH O
A B
HF-pyridine (0.5 mL) is added to a solution of the 1.2:1 mixture (70 mg, 0.097
mmol) of
stage 10.5 in dry THF (0.05 M solution) at rt. The solution is stirred at the
same
temperature for 6 h. The completion of the reaction is judged by TLC. The
reaction
mixture is slowly poured into a cold aqueous solution of NaHC03 and extracted
with
EtOAc. The organic layer is washed with brine and dried over MgS04. Solvents
are
evaporated under reduced pressure. The residue is purified by PTLC (silica
gel,
hexanes - EtOAC (2:1 )) to yield the pure epothilones A (54%) and B (45%).
Physical data of A: [a]p -71.6 (c = 0.50, CHCI3); 'H NMR (600 MHz): 8 6.95 (s,
1 H), 6.60 (s,
1 H), 5.38 (d, J = 11.1 Hz, 1 H), 5.20 (d, J = 7.0 Hz, 1 H), 4.17 (d, J = 10.9
Hz, 1 H), 3.76 (br s,
1 H), 3.47 (m, 1 H), 3.11 (m, 1 H), 2.96 (br s, 1 H), 2.89 (dd, J = 14.3, 11.5
Hz, 1 H), 2.68 (s,
3H), 2.48 (dd, J = 15.0, 11.4 Hz, 1 H), 2.37 (dd, J = 15.0, 2.3 Hz, 1 H), 2.17
(m, 1 H), 2.08 (s,
3H), 1.97 -1.86 (m, 4H), 1.74 (m, 1 H), 1.72 (s, 3H), 1.57 (m, 1 H), 1.35 -
1.20 (m, 3H), 1.33
(s, 3H), 1.17 (d, J = 6.9 Hz, 3H), 1.09 (s, 3H), 0.99 (d, J = 7.1 Hz, 3H);
HRMS: (C ~H42N05S
= 492.2784; C2~H41N05SNa = 514.2603;) found 492.2760 (MH +), 514.2589 (MNa+).
Physical data of B: [a]p -30.2 (c = 0.5, CHCI3);'H NMR (600 MHz): 8 6.96 (s, 1
H), 6.56 (s,
1 H), 5.49 (dd, J = 10.8, 1.1 Hz, 1 H), 5.29 (m, 1 H), 4.01 (dt, J = 10.7, 2.5
Hz, 1 H), 3.75 (br s,
1 H), 3.19 (quintet, J = 6.1 Hz, 1 H), 3.13 (d, J = 3.2 Hz, 1 H), 2.69 (s,
3H), 2.53 (dd, J =14.9,
10.7 Hz, 1 H), 2.45 (dd, J = 14.7, 2.4 Hz, 1 H), 2.44 (t, J = 11.0 Hz, 1 H),
2.32 (m, 2H), 2.20
(m, 1 H), 2.08 (d, J = 0.9 Hz, 3H), 1.87 (m, 1 H), 1.82 (br s; 1 H), 1.63 (m,
1 H), 1.62 (s, 3H),
CA 02425620 2003-04-07
WO 01/27308 PCT/EP00/09817
-40-
1.55 (m, 1 H), 1.47 (m, 1 H), 1.28 (s, 3H), 1.22 (m, 1 H), 1.17 (d, J = 6.8
Hz, 3H), 1.10 (m,
1 H), 1.05 (s, 3H), 0.96 (d, J = 7.0 Hz, 3H); HRMS: (C2,H42N05S = 492.2784;
CZ,H4,NOSSNa
= 514.2603); found 492.2798 (MH +), 514.2640 (MNa+).
Stage 10.1
Protection of the compound 5 of Example 5 is protected according to the method
described in stage 6.1 and affords the corresponding silyl ether.
Stag a 10.2
The compound of stage 10.1 (500 mg, 1.48 mmol) is used according to the method
described in stage 6.2 to afford the methylenated reaction product; CC
conditions: silica
gel, hexanes - EtOAc (20:1 ). [a]p +1.0 (c = 0.50, CHCI3); 'H NMR (500 MHz): b
6.90 (s,
1 H), 6.45 (s, 1 H), 4.75 (m, 1 H), 4.70 (m, 1 H), 4.25 (dd, J = 7.7, 5.9 Hz,
1 H), 2.68 (s, 3H),
2.29 (dd, J = 13.6, 7.7 Hz, 1 H), 2.22 (dd, J = 13.6, 5.5 Hz, 1 H), 1.99 (s,
3H), 1.74 (s, 3H),
0.87 (s, 9H), 0.04 (s, 3H), -0.02 (s, 3H); MS: 338 (MH+).
Stage 10.3
According to the method described in stage 6.3, the compound of stage 10.2
(400 mg,
1.19 mmol) is deprotected; CC conditions: silica gel, hexanes - EtOAc (4:1 ).
[a]o -28.8
(c = 1.87, CHCI3); 'H NMR (500 MHz): 8 6.88 (s, 1 H), 6.54 (s, 1 H), 4.83 (s,
1 H), 4.79 (s,
1 H), 4.25 (dd, J = 8.8, 4.4 Hz, 1 H), 2.65 (s, 3H), 2.49 (br s, 1 H), 2.32
(dd, J = 13.6, 4.4 Hz,
1 H), 2.26 (dd, J = 14.0, 8.8 Hz, 1 H), 1.99 (s, 3H), 1.75 (s, 3H); MS: 224
(MN +).
Sta eq 10.4
According to the method described in stage 6.4, the compound of stage 10.3 (35
mg,
0.16 mmol) is used to afford the corresponding product which is purified by CC
(silica
gel, hexanes - EtOAc, (10:1 )); [a]o -34.7 (c = 1.95, CHCI3); 'H NMR (400
MHz): 8 6.92 (s,
1 H), 6.48 (s, 1 H), 5.76 (m, 1 H), 5.40 (dd, J = 7.6, 5.9 Hz, 1 H), 4.98 (dd,
J = 17.3, 2.1 Hz,
1 H), 4.90 (br d, J = 10.3 Hz, 1 H), 4.75 (s, 1 H), 4.73 (s, 1 H), 4.31 (dd, J
= 5.6, 3.8 Hz, 1 H),
3.71 (dd, J = 7.0, 2.4 Hz, 1 H), 3.13 (quintet, J = 6.7 Hz, 1 H), 2.67 (s,
3H), 2.50 (dd, J = 17.0,
3.8 Hz, 1 H), 2.45 (dd, J = 13.8, 7.9 Hz, 1 H), 2.34 (dd, J = 13.8, 5.6 Hz, 1
H), 2.25 (dd, J =
17.0, 5.6 Hz, 1 H), 2.05 (d, J = 1.2 Hz, 3H), 1.99 (m, 2H), 1.73 (s, 3H), 1.46-
1.05 (m, 5H),
1.21 (s, 3H), 1.01 (d, J = 5.9 Hz, 3H), 1.01 (s, 3H), 0.87 (s, 9H), 0.87 (d, J
= 6.5 Hz, 3H),
0.85 (s, 9H), 0.08 (s, 3H), 0.02 (s, 3H), 0.01 (s, 6H); '3C NMR (100.6 MHz):
8217.6, 171.1,
CA 02425620 2003-04-07
WO 01/27308 PCT/EP00/09817
-41 -
164.4, 152.5, 140.9, 138.8, 137.2, 121.0, 116.3, 114.3, 113.7, 77.61, 77.56,
74.0, 53.3,
45.2, 41.7, 40.3, 38.7, 34.3, 34.3, 31.5, 30.4, 27.0, 26.1, 26.0, 23.0, 22.6,
22.4, 20.3, 19.2,
18.4, 18.1, 17.6, 15.4, 14.4, 14.1, -3.7, -3.8, -4.4, -4.8; MS: 770 (MNa +).
Stage 10.5
According to the method described in stage 6.5, the diene of stage 10.4 (94
mg, 0.13
mmol) is metathesized to afford a mixture of the metathesized products which
is taken
to the next step without separation; CC conditions: silica gel, hexanes -
EtOAc (12:1 ).
Example 11:
\ S~OH t~OH
HO ~~." \ \ N
,~~ O
O OH O OH O
A B
The 1.5:1 mixture (100 mg, 0.12 mmol) of stage 11.6 is deprotected according
to the
method described in the final stage of Example 10 to yield pure compounds A
(55%) and B
(36%); PTLC conditions: silica gel, hexanes - EtOAc (1:2).
Physical data of A: [ a]p -70.0° (c = 0.29, CHCI3); 'H NMR (600 MHz): 8
7.10 (s, 1 H), 6.60 (s,
1 H), 5.39 (d, J = 10.7 Hz, 1 H), 5.21 (d, J = 9.0 Hz, 1 H), 4.91 (s, 2H),
4.16 (d, J = 9.8 Hz,
1 H), 3.75 (m, 1 H), 3.37 (br s, 1 H), 3.11 (qd, J = 6.9, 2.7 Hz, 1 H), 2.93
(br s, 1 H), 2.89 (dd, J
= 14.6, 11.3 Hz, 1 H), 2.62 (s, 1 H), 2.48 (dd, J = 15.3, 11.4 Hz, 1 H), 2.37
(dd, J = 15.1, 2.6
Hz, 1 H), 2.16 (d, J = 3.1 Hz, 3H), 1.94 (m, 2H), 1.77-1.73 (m, 1 H), 1.72 (s,
3H), 1.58 (m,
1 H), 1.34 (s, 3H), 1.24 (br s, 4H), 1.17 (d, J = 6.9 Hz, 3H), 1.09 (s, 3H),
0.97 (d, J = Hz, 3H);
HRMS: (CZ,H41NO6SNa = 530.2547) found 530.2544 (MNa +). Physical data of B:
[a]p -36.6°
(c = 0.24, CHCI3); 'H NMR (600 MHz): 8 7.11 (s, 1 H), 6.59 (s, 1 H), 5.49 (d,
J = 10.5 Hz, 1 H),
5.29 (t, J = 6.8 Hz, 1 H), 4.92 (s, 2H), 4.06 (d, J = 10.6 Hz, 1 H), 3.78 (br
s, 1 H), 3.75 (m, 1 H),
3.20 (quintet, J = 6.6 Hz, 1 H), 3.08 (br s, 1 H), 2.62 (s, 1 H), 2.54 (dd, J
= 15.0, 10.7 Hz, 1 H),
2.47 (m, 2H), 2.35 (m, 2H), 2.20 (m, 1 H), 2.16 (s, 3H), 1.89 (m, 1 H), 1.62
(s, 3H), 1.60-1.44
CA 02425620 2003-04-07
WO 01/27308 PCT/EP00/09817
- 42 -
(m, 2H), 1.28 (s, 3H), 1.24 (br s, 4H), 1.18 (d, J = 6.8 Hz, 3H), 1.05 (s,
3H), 0.97 (d, J = 7.0
Hz, 3H); HRMS: (C2~H42NO6S = 508.2727) found 508.2730 (MH +).
Sta eq 11.1
Protection of the compound 11 of Example 5 is protected according to the
method
described in stage 6.1 and affords the corresponding silyl ether.
Stage 11.2
The compound of stage 11.1 (715 mg, 1.52 mmol) is used according to the method
described in stage 6.2 to afford the methylenated reaction product; CC
conditions: silica
gel, hexanes - EtOAc (40:1 ). [g]o -1.3 (c = 1.33, CHCI3); 'H NMR (400 MHz): 8
7.01 (s,
1 H), 6.44 (s, 1 H), 4.95 (s, 2H), 4.75 (m, 1 H), 4.71 (br s, 1 H), 4.24 (dd,
J = 7.3, 5.3 Hz, 1 H),
2.29 (dd, J = 13.6, 7.3 Hz, 1 H), 2.21 (dd, J = 13.6, 5.3 Hz, 1 H), 1.98 (s,
3H), 1.74 (s, 3H),
0.94 (s, 9H), 0.86 (s, 9H), 0.12 (s, 6H), 0.03 (s, 3H), -0.02 (s, 3H); '3C NMR
(100.6 MHz): b
171.8, 153.3, 142.4, 142.3, 118.9, 115.1, 113.1, 77.6, 63.2, 45.5, 25.8, 25.7,
22.8, 18.2,
13.7, -4.7, -5.1, -5.5; MS: 468 (MH+), 490 (MNa+).
Stage 11.3
According to the method described in stage 6.3 but using 2.4 equiv. of TBAF
instead of 1.2
equiv., the compound of stage 11.2 (492 mg, 1.05 mmol) is deprotected; CC
conditions:
silica gel, hexanes - EtOAc (1:2). [g]o -35.0 (c = 0.48, CHCI3); 'H NMR (400
MHz): 8 7.01
(s, 1 H), 6.53 (s, 1 H), 4.85 (br s, 3H), 4.80 (s, 1 H), 4.25 (dd, J = 8.2,
4.1 Hz, 1 H), 2.32 (dd, J
= 13.8, 4.4 Hz, 1 H), 2.26 (dd, J = 13.5, 8.8 Hz, 1 H), 1.95 (s, 3H), 1.76 (s,
3H); MS: 240
(MH+), 262 (MNa+).
Sta eq 11.4
TBSCI (140 mg, 0.93 mmol) is added to a solution of the compound of stage 11.3
(180
mg, 0.75 mmol) and i-Pr2NEt (0.26 mL, 1.50 mmol) in dry CH2C12 (5 mL) at 0
°C and the
mixture is stirred at 0 °C to rt for 8h. The reaction mixture is worked
up with water and
CH2CI2. The combined organic layer is washed with water and dried over MgS04.
Solvents are evaporated and the protected derivative is purified by CC (silica
gel,
hexanes - EtOAc; 3:1 ); [g]o -26.3 (c =1.54, CHCI3); 'H NMR (500 MHz): b 7.03
(s, 1 H),
6.57 (s, 1 H), 4.94 (s, 2H), 4.88 (s, 1 H), 4.83 (s, 1 H), 4.27 (dd, J = 9.2,
4.4 Hz, 1 H), 2.35 (dd,
W~ 01/27308 CA 02425620 2003-04-07 PCT/EP00/09817
-43-
J = 14.0, 4.4 Hz, 1 H), 2.28 (dd, J = 14.0, 9.2 Hz, 1 H), 2.04 (s, 3H), 1.79
(s, 3H), 0.94 (s,
9H), 0.12 (s, 6H); MS: 354 (MH+).
Stage 11.5
According to the method described in stage 6.4, the compound of stagell .4 (65
mg, 0.18
mmol) is used to afford the corresponding product which is purified by CC
(silica gel,
hexanes - EtOAc (15:1 )); [a]o -38.2 (c = 1.03, CHCI3);'H NMR (500 MHz): 8
7.03 (s, 1 H),
6.49 (s, 1 H), 5.77 (m, 1 H), 5.41 (dd, J = 7.7, 5.9 Hz, 1 H), 4.97 (dd, J =
17.3, 1.5 Hz, 1 H),
4.93 (s, 2H), 4.92 (br d, J = 10.3 Hz, 1 H), 4.76 (s, 1 H), 4.74 (s, 1 H),
4.30 (dd, J = 5.6, 3.8
Hz, 1 H), 3.72 (dd, J = 7.0, 2.2 Hz, 1 H), 3.14 (quintet, J = 6.6 Hz, 1 H),
2.53 (dd, J = 17.2, 4.0
Hz, 1 H), 2.45 (dd, J = 14.0, 8.1 Hz, 1 H), 2.36 (dd, J = 14.0, 5.9 Hz, 1 H),
2.26 (dd, J = 16.9,
5.5 Hz, 1 H), 2.05 (s, 3H), 2.00 (m, 2H), 1.74 (s, 3H), 1.48 -1.27 (m, 3H),
1.22 (s, 3H), 1.15
-1.04 (m, 2H), 1.02 (d, J = 6.3 Hz, 3H), 1.01 (s, 3H), 0.94 (s, 9H), 0.88 (s,
9H), 0.87 (d, J =
6.3 Hz, 3H), 0.86 (s, 9H), 0.12 (s, 6H), 0.09 (s, 3H), 0.03 (s, 3H), 0.02 (s,
3H), 0.016 (s, 3H);
MS: 878 (MH+).
Stacie 11.6
According to the method described in stage 6.5, the diene of stage 11.5 (125
mg, 0.14
mmol) is metathesized to afford a mixture of the metathesized products which
is taken
to the next step without separation; CC conditions: silica gel, hexanes -
EtOAc (20:1 ).
Examgle 12
The efficacy of compounds of formula I as inhibitors of micro tubule
depolymerisation and
the efficacy against tumour cells was tested in the test procedures as
described herein-
above.
Table 2
Example Tubulin Polymerization KB-31 Cell Growth Inhibition
(%) (1C50 [nM])a
6A 11 150
6B 33 78.9
7A 39 24.8
7B 78 4.03
WO 01/27308 CA 02425620 2003-04-07 pCT/EP00/09817
-44-
8A 54 47.9
8B 64 17.5
10A 2 245
B 19 61.6
(a) Drug concentration required for half-maximal inhibition of KB-31 human
epidermoid
cancer cell growth was assessed after a 72h drug exposure by quantification of
cell mass
using a protein dye method .
Example 13: Dry capsules
3000 capsules, each of which contain 0.25 g of one of the compounds of the
formula IA
mentioned in the preceding Examples as active ingredient, are prepared as
follows:
Composition
Active ingredient 75.00 g
Lactose 750.00 g
Avicel PH 102 300.00 g
(microcrystalline cellulose)
Polyplasdone XL 30.00 g
(polyvinylpyrrolidone)
Magnesium stearate 9.00 g
Preparation process: The active ingredient is passed through a No. 30 hand
screen. The
active ingredient, lactose, Avicel PH 102 and Polyplasdone XL are blended for
15 minutes
in a mixer. The blend is granulated with sufficient water (about 500 mL),
dried in an oven at
35°C overnight, and passed through a No. 20 screen.
Magnesium stearate is passed through a No. 20 screen, added to the granulation
mixture,
and the mixture is blended for 5 minutes in a mixer. The blend is encapsulated
in No. 0 hard
gelatin capsules each containing an amount of the blend equivalent to 25 mg of
the active
ingredient.