Note: Descriptions are shown in the official language in which they were submitted.
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
DEVICES AND METHODS FOR B10CHIP MULTIPLEXING
This application claims the benefit of the priority date U.S.S.N. 60/145,840,
filed November 3, 2000.
This application is a continuation of U.S.S.N. 09/904,175, filed July 11, 2001
which is a continuation of
U.S.S.N. 09/760,384, filed 1/11/2001, which claims the benefit of the priority
date of U.S.S.N.
60/175,539, filed January 11, 2000 and PCT application US01/01150, filed
January 11, 2001.
FIELD OF THE INVENTION
The invention is directed to devices that allow for simultaneous multiple
biochip analysis. In particular,
the devices are configured to hold multiple cartridges comprising biochips
comprising arrays such as
nucleic acid arrays, and allow for high throughput analysis of samples.
BACKGROUND OF THE INVENTION
There are a number of assays and sensors for the detection of the presence
and/or concentration of
specific substances in fluids and gases. Many of these rely on specific
ligand/antiligand reactions as
the mechanism of detection. That is, pairs of substances (i.e. the binding
pairs or ligandlantiligands)
are known to bind to each other, while binding little or not at all to other
substances. This has been
the focus of a number of techniques that utilize these binding pairs for the
detection of the complexes.
These generally are done by labeling one component of the complex in some way,
so as to make the
entire complex detectable, using, for example, radioisotopes, fluorescent and
other optically active
molecules, enzymes, etc.
Other assays rely on electronic signals for detection. Of particular interest
are biosensors. At least
two types of biosensors are known; enzyme-based or metabolic biosensors and
binding or bioaffinity
sensors. See for example U.S. Patent No. 4,713,347; 5,192,507; 4,920,047;
3,873,267; and
references disclosed therein. While some of these known sensors use
alternating current (AC)
techniques, these techniques are generally limited to the detection of
differences in bulk (or dielectric)
impedance.
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
There are a variety of nucleic acid biosensors currently known. These include
nucleic acid biochips
based on fluorescent detection; see for example materials developed by
Affymetrix (including, but not
limited to, 5,800,992, 5,445,934, 5,744,305, and related patents and
materials), Nanogen (including,
but not limited to, 5,532,129, 5,605,662, 5,565,322 and 5,632,957 and related
patents and materials),
Southern (EP 0 373 023 B1) and Synteni/lncyte (WO 95/35505 and related patents
and materials).
Similarly, electronic detection of nucleic acids using electrodes is also
known; see, for example U.S.
Patent Nos. 5,591,578; 5,824,473; 5,705,348; 5,780,234 and 5,770,369;
U.S.S.N.s 08/873,598
08/911,589; and WO 98/20162; PCT/US98/12430; PCT/US98/12082; PCT/US99/10104;
PCT/US99/01705, and PCT/US99/01703 and related materials.
However, to date none of these methods have been used in highly parallel
systems to allow biochip
multiplexing. Accordingly, it is an object of the present invention to provide
devices and methods for
multiplex analysis of biochips, particularly nucleic acid biochips.
SUMMARY OF THE INVENTION
In accordance with the objects outlined above, the present invention provides
biochip cartridges
comprising one or more reaction chambers, such as a nucleic acid amplification
chamber. The
chambers are configured to include inlet and outlet ports, valves to control
the movement of fluid into
and out of the chamber and pumps.
In an additional aspect, the biochip cartridge comprises a detection chamber
with an array of
electrodes.
In an additional aspect, the biochip cartridge comprises one or more thermal
heaters.
DETAILED DESCRIPTION OF THE DRAWINGS
Figures 1A-1J depict a number of different detection chamber embodiments.
Figures 1A-1 F depict
alternative detection chamber geometries in which the inlet port 100 is
positioned at the top of the
chamber. In contrast, the outlet port 101 can have several configurations. For
example, in Figure 1A
the outlet port is located at the top of the chamber but does not vent to the
outside. In Figures 1 B and
1 D, the outlet port 101 is positioned at the top and vents outside. In Figure
1 C, the outlet port 101 is
located to the side of the chamber. In Figure 1 F, the outlet port 101 comes
off of the inlet port 100. In
Figures 1A through 1 E, the electrode array 103 is positioned within a
reaction chamber 102, which
may be formed from a gasket, a recess in the underlying printed circuit board
or from the housing 104.
In Figures 1A and 1 C, the reaction chamber is shaped like an inclined
diamond; in Figure 1 B, the
-2-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
reaction chamber is shaped like a diamond; in Figure 1 D, the reaction chamber
is circular in shape; in
Figure 1 E, the reaction chamber is triangular in shape; and, in Figure 1 F,
the reaction chamber is
shaped like a square. Figure 1 F depicts an embodiment in which reference
electrodes.106 are
located in the inlet and/or the outlet channels. The reference electrodes are
preferably AgCI. The
reference electrodes may be coated with AgCI before placing in the cartridge.
Alternatively, a coating
of AgCI may be applied to the reference electrodes while in the cartridge by
applying a voltage of
sufficient strength to an Ag electrode such that the silver is oxidized to
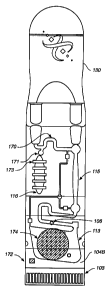
form Ag+. Figure 1 G depicts a
biochip 105 comprising a reaction chamber 102 with an electrode array 103, a
PCR chamber 115, a
buffer chamber 170, an air pimp chamber or other mechanism for moving fluid
116, one or more
valves for controlling the movement of fluid 171, temperature sensors 172,
heating elements
integrated into the device 173, a mixing element 174, reference electrodes
106, inlet 100 and outlet
101 ports, a microchannel 110, a silicon gasket 104B with a cutout for the
detection chamber 113 and
a cap 130. Figure 1 H depicts the top surtace of an electronic biochip. The
electrode array 103 is
configured such that each electrode is connected via a wire lead 109 to a
contact pad or interconnect
108 on the edge of the biochip. These metal contact pads can be used to make
contact between the
electronic biochip reader and the biochip using a standard computer edge card
connector.
Alternatively, a s depicted in Figure 11 and 1J, the electrical connections
can be made by transversing
through the board to the opposite side of the substrate. the opposite side of
the substrate can be
arranged in the mirror image configuration to the front side, or it can be
arranged in an alternative
fashion. Figure 1 K shows a biochip 105 with connects that transverse the
board making contact with
a pogo pin connector 176. The connector has an array of compliant pins 177, a
circuit board housing
177, and potentially an electronic multiplexer 178. The pin grid connector
ultimately plugs into an
instrument through some interface like an edge card connector via metal
fingers 179. In order to
ensure a good connection between the pogo pin connectors and the chip, it is
common to use some
type of fastener.
Figure 2 depicts the various components that can comprise a cartridge. In the
cartridge embodiment
depicted in Figure 2A, the detection chamber 102 contains an electrode array
103 connected via wires
109 to interconnects 108. The array is attached to a solid surface 105 which
can be made from any
number of materials as described below. In addition, registration pins 107 can
be attached to the
biochip to enable the addition of other components. Figure 2B depicts a rubber
gasket 104 with a cut
out for the detection chamber 113 and registration holes 112 for attaching the
gasket to the biochip
depicted in Figure 2A. Figure 2C depicts a housing 114, which can be made from
plastic and is
attached to the biochip via registration holes 112. The cartridge 114, may
optionally contain a cutout
113A for the detection chamber 103 and a recessed microchannel 110 running
from the inlet port 100
to the detection chamber 103.
-3-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
Figures 3A and 3B depict two views, top view (Figure 3A) and an angled front
view (Figure 3B) of the
cartridge holders 129 used to hold the cartridges during sample loading. As
shown in Figures 3A and
3B, the cartridge holder 129, can hold several cartridges 114 with attached
biochips 105. Also shown
is a cartridge cap 130, which can be taken on and off via a "snap-in lock".
Figures 3C and 3D depict different views of a cartridge with a cap. In Figure
3C, the cap 130, is
configured to include a snap-in Pock 132, that locks into a slot 133 in the
cartridge 114. In the
embodiment shown in Figure 3C, the cap has been configured to include a seal
134 for the sample
introduction module 136. Preferably, the seal 134 comprises a plastic plug
surrounded by cellulose or
another hydrophobic material that allows air to pass but not liquid. The
cartridge 114 has been
configured to include sample introduction module 136, and a PCR chamber 115.
The cartridge 114 is
attached to a biochip comprising a reaction chamber 102, an electrode array
103, connected via wires
109 to interconnects 108, a microchannel 110, and an outlet port 101. Figure
3D illustrates a side vies
of the cap 130, the cartridge 114 configured to contain a PCR chamber 115, and
a chip 105.
Figure 3E depicts a side view of several cartridge assemblies lined up as they
would appear in the
cartridge holder. The cap 130, is attached to the cartridge 114, which has
been configured to
comprise a sample introduction module 136 and a PCR chamber 115. Attached to
the cartridge is a
biochip 105.
Figures 4A and 4B depict different views of a multiplexing device 137. In
Figure 4A, a side and top
view of the multiplexing device 137 illustrates the cartridge/station pairs
139 and a drawer 138. In
Figure 4B, the multiplexing device 137 is illustrated with the stations 141
for holding the cartridges and
an open drawer 138.
Figure 5 illustrates a schematic of an electronic circuit designed to monitor
the sample temperature
with a thermal sensor inside the cartridge. This version of the design uses a
resistive temperature
device composed of a copper trace. The circuitry can be used in feedback
system for thermal control
of the cartridge temperature.
Figure 6 illustrates the thermal control logic. This feedback mechanism
employs a Proportional
Integral Derivative (PID) algorithm.
Figure 7 illustrates the layout of a multiplexing device. The device has eight
independent modules. In
this particular figure, each module has six card-edge connectors 141 and a
signal processing printed
circuit board 140. Directly underneath the modules is the power supply 145.
Adjacent to the power
-4-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
supply is a bus bar 150 for power distribution. Figure 7 depicts the stations
into which the cartridges
are inserted. A cartridge 114 with a cap 130 is shown inserted into an card
edge connector 141.
Figure 8 is a block diagram for the software application used to control the
multiplexing device.
Figures 9A-F depict a variety of different valves that may be used in the
present invention. Figure 9A
depicts a duck bill valve 144 within a microchannel 110 that can be used to
control the flow of liquid in
one direction, but not the other. Figure 9B depicts a cantilever valve 146. In
this embodiment,
voltage, applied via electrodes 147 is used to open and close the cantilever
valve 146. Figure 9C
depicts a plunger type valve mechanism. In this embodiment, a plunger valve
148 can be opened and
closed via the use of a shape memory wire 149. Figures 9D and 9E depict rotary
valves. In the
embodiment shown in Figure 9D, an external force must be applied to rotate the
rotary valve 151. In
Figure 9E, a shape memory wire 149 is used to rotate the rotary valve 151.
Figure 9F depicts a
thermally actuated valve that comprises a portion of the microchannel 110 with
a flexible membrane
152 that can be filled with liquid or air 153 for use in conjunction with a
heater 154.
Figure 9G depicts a generic pump design in which a chamber 156 through which
air and/or liquid can
flow is attached to an inlet port 100 with a valve, such as a cantilever valve
146, to control movement
into the chamber and an outlet port 101 with a valve 146A, to control movement
out of the chamber.
An external device 181, such as a PZT, can be used to compress chamber 156.
Alternatively, a
heater 182, can be actuated expanding the volume of the gas or liquid in
chamber 156.
Figure 10A is a side view of a biochip 105 depicting an embodiment in which a
thermal heater is
incorporated into the chip. In Figure 10 A, the resistive heaters 157 are
overlaid with a layer of copper
158. The copper layer is overlaid with printed circuit board 125, which is
covered with solder mask
159.
Figures 10B illustrates one means of creating thermal zones in a biochip 105.
In the embodiment
shown in Figure 10B,successive thermal zones 161, 162, 163 comprising several
rows of resistive
heaters 157 are overlaid with a serpentine microchannel 164. Discrete
temperature zones are
maintained by controlling the minimum separation distance between resistive
heater 161,162, and
163 as well as varying the thermal properties of the separating materials. the
device illustrated in
Figure 10B can be used in conjunction with a pumping device to transport a
fluid between temperature
zones and perform biological reactions that require heat cycling like PCR.
Figure 10C depicts a biochip analogous to Figure 10B, but made out of ceramic
165 with imbedded
heaters 166 and corresponding edge connections 108. The temperature difference
between the
-5-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
thermal zones is maintained by creating air pocket cut-outs 167 that have a
lower thermally
conductivity than the surrounding ceramic.
Figure 11 depicts a bar code reader 142 reading a bar code 143.
Figures 12 describes a bar code usage scenario.
Figure 13 highlights the benefits of using a bar code in combination with the
devices of the present
invention.
Figures 14 A and B depict a preferred embodiment of a biochip 105 with
registration holes 112 for the
attachment of a cartridge 114. In Figure 14A, the top surface of a biochip 105
is depicted showing the
registration holes 112, an electrode array 103 and interconnects 108. In
Figure 14B, a cartridge 114 is
shown overlaying the biochip 105 illustrated in Figure 14A. In the embodiment
shown, the cartridge
114 includes registration pins 107 for attaching the cartridge to the biochip.
Preferably, the registration
pins are plastic. Also shown are inlet 100 and outlet 101 ports, microchannels
110, a PCR chamber
115, a reaction chamber 102 and a sample introduction chamber 136. The cap
gasket 134 is depicted
as an insert within the sample introduction chamber 136.
Figure 15A depicts sample loading using a pipet tip 144 into a cartridge 114
inserted into a station 141
of a multiplexing device 137.
Figures 15B and C depict an alternative embodiment for attaching a cartridge
to a biochip. In Figure
15A, the biochip 105 is designed to have alignment slots 118. In Figure 15C,
the cartridge is
configured to have registration pins 107 that fit into the registration holes
on the sides of the biochip.
Also depicted in Figure 15C is the use of a pipet tip 144 for loading a sample
into a sample
introduction chamber 136.
Figure 16 depicts a sine wave and its corresponding vector notation.
Figure 17 depicts the visualization of the sine wave shown in Figure 16 using
vector notation. The two
values can be R and A, but as shown in the Figure 17 they can also be an (X,Y)
pair separated by one
quarter of an oscillation, i.e. by 90°.
Figures 18 and 19 are examples of R and 8 traces for the fourth harmonic ACE
voltammetry.
-6-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
Figures 20 and 21 depict that the R space signal distorts as the signal
shrinks relative to the size of
the background.
Figures 22 through 25 illustrate how the use of Cartesian coordinates
simplifies the dependence of of
D's parameters on those of S and B . This simplicity is exhibited when
graphing the same examples
shown previously, but now as (X,Y), as depicted in Figure 22 and 23 (medium
sized signal), and
Figure 24 and 25 (smaller signal).
Figure 26 depicts an AC voltage-4 trace that has a large signal relative to
the background. Figure 26
is obtained If, in a two-dimensional graph, the tip of the data vector as a
function of voltage (one point
is plotted every 10 mV) is plotted.
Figures 27and 28 depict the result when a frame of reference is chosen such
that the X and Y axes
straddles the signal. in this case, the signal contributes strongly to both X
and Y.
Figures 29 and 30 depicts the signal that is observed when an axis pair that
is roughly parallel and
perpendicular to the signal (rotated 45° with respect to the axes drawn
in Figure 26) is chosen. In this
case, very little of the signal contributes to the perpendicular vector.
Figures 31 through 35 illustrate the vectoral sum method. For signal
recognition based on the AC
voltage 4 trace model, a new pair of axes need to be chosen to straddle any
existing electrochemical
signal. fn order to choose such axes, a way is needed to measure the signal's
direction. One such
way is using a vectoral sum. Consider the grouping of three points shown in
Figure 31. If we consider
these points as vectors, we can add them by summing their coordinates. This
summation of the
vectors provides a reasonable angle for the best line through the data that
passes through (0,0). This
angle is called the "optimal phase." For our example, the summation is drawn
in Figure 32. Figure 33
shows how the three sample data points cluster around the line. An advantage
to this method is that
the results are weighted by the length of the vectors of the original data
points. For example, if we add
a small data point to the sample grouping, the results are shown in Figure 34
and 35.
Figures 36 through 41 illustrate the complications that must be considered
when using the vectoral
sum to calculate the optimal phase for fitting a signal. For example, if the
electrochemical signal is
shaped such that portions of it cancel each other out when completing the
calculation described
above, the first one half of the data must be rotated 180°. Taking the
data shown in Figure 26, we
calculate the optimal phase using the data as shown in Figure 36. The
resulting line is overlaid on the
original data, at Figure 37. The angle of the line drawn in Figures 36 and 37
(101 °) is what was used
to choose the X and Y axes (at ~45°) for this file. However, if the
signal is oriented differently relative
to the dividing line between rotated and unrotated segments, the stated
manipulation may not yield the
-7-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
proper angle. For example, if I take the above signal and rotate it 101
degrees clockwise, its optimal
phase should be 0°. However, the calculated value actually ends up as -
48° as shown in Figure 38.
To prevent this, a rotation boundary that is more perpendicular to the signal
than it is parallel is
chosen. Taking the vectoral sum of the absolute value of the coordinates of a
signal that's closer to
90, the resulting angle will be greater than 45°. Thus, for the above
case we find an angle of 10
degrees (see Figure 39), less than 45, and conclude that the signal is more
along 0 degrees.
Therefore, we rotate the half of the signal from the far side of the 90 degree
axis (see Figure 40).
Calculating the vectoral sum now yields a reasonable value for the optimal
phase: 1 °, similar to the
expected 0°. When the scan is examined in two dimensions, we can see
that the phase of the entire
scan is mostly along 120° (Figure 41).
Figures 42 through 46 illustrates the results obtained if the rapid
calculations necessary to fit
polynomials to the entire scan (one each along the 0 and 90° axes) are
pertormed. For example, the
background is approximated as shown in Figure 42 and 43. The approximation to
the background can
be subtracted, converting the scan into something that is much more purely
signal, as shown in Figure
44 and 45. Figure 46 depicts this as a two dimensional plot, from which an
optimal phase of
approximately 70° can be calculated.
Figures 47 through 52 illustrate how behaviors not modeled are detected. To
reduce total processing
time, the first thing to do is to check if a scan has any gross deviations
from the model that would
make fitting it meaningless. One such feature encountered in AC voltammetry
(fourth harmonic) has
been the sharp peak caused by the stripping of a metallic contaminant. Figure
47 shows an example
of one displayed in R-space. In X and Y (at ~ 45° from the optimal
phase), the sharp spike feature
remains clear, as shown in Figure 48 and 49.
The symmetry of this feature distinguishes it from our normal signal. One
method of monitoring this
symmetry is to separate out an approximate background and compare the
distribution of points above
the baseline with the distribution below. For example, if we subtract a
polynomial from the Y trace
above, we get the results shown in Figures 50 and 51. If we now examine the
distribution of data
above and below the approximated background, the presence of the spike causes
a larger range of
values to exist below the background line than above it, as shown in Figure
52.
Figures 53 through 58 illustrate the initial guess process required as a
starting point for iterative fitting
procedures. To guess parameters of signal position and signal height, the
known AC voltage-4
symmetry is used combined with knowledge of the characteristic width. Since
the average separation
between the two larger center lobes is known, the signal and shift are
duplicated, the two copies in
opposite directions for half of that separation. Subtracting one from the
other, the center lobes
_g_
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
interfere constructively. The absolute value of this resulting wave provides a
good estimation of the
height and position of the signal. This process is shown in Figures 53, 54 and
55 for a signal 11.9 tall
at a position of 0.20 with a center lobe separation of 0.072. The trace in
Figure 55 has its largest
value, 23.25, at a position of 0.20. The position matches well with the true
data value. (Both are
0.20. )
In Figure 56, the same signal is considered, but this time with an unusual
peak off to one side that's
slightly taller than the signal itself. In a simple maxima/minima search, this
would be likely to intertere
with the initial guess. However, using the initial guess, the signal remains
11.6 tall at a position of
0.20, as shown in Figure 57. Figure 58 is the overlay of a real data trace and
the corresponding initial
guess.
Figures 59 through 61 depict that for systems that are less well-behaved, the
boundary conditions may
be enforced during the fitting procedure. This can be done using various
equations described below.
lin Figure 59 the shape when the added term has 2n = 16 is compared with the
shape when 2n = 2. In
the case where 2n = 16, a values within ~ 7 of the expected are all equally
acceptable, with little added
penalty. However, with 2n = 2, there's an increasingly harsh penalty the
further a moves from the
expected value. Sharper constraints a result in the shapes depicted in Figure
60. More complicated
shapes may be used, as shown in Figure 61.
Figure 62 depicts the results when the fit is not reliable because the
difference between the fit and
date is too large.
Figures 63 through 65 depict the results obtained using procedures to refit
scans having no
observable signals.
Figures 66 and 67 depict the results obtained using the procedures described
herein. Figure 66
depicts the original data. Figure 67 depicts the data with the background
subtracted.
Figure 68 depicts the effect of various plasma treatments on the surtace
density of SAMs comprising
capture probes.
Figure 69 depicts the spectra of contaminants on a gold surtace after various
plasma treatment
procedures. The major contaminant peaks of carbon and oxygen are marked out
with gold peaks for
the oxygen plasma treatment alone, or for the oxygen plasma followed by
hydrogen plasma treatment.
_g_
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
Figure 70A-F depict the effect of different mixing techniques on hybridization
kinetics in an eSensorT"'
chamber. Figure 70A depicts effect of chip orientation (i.e., diffusion based
kinetics) on hybridization
kinetics and increased volume/z-dimension. Increased volume was obtained by
increasing the
thickness of the chamber using one (single tape), two (double tape) or three
(triple tape) layers of
tape. Chips were incubated either horizontally (H) or vertically (V). Figure
70B compares diffusion
based kinetics using chips oriented either vertically (vdiff) or horizontally
(hdiff) to mixing using a
recirculation pump (vpump). Figure 70C compares diffusion based
kinetics/vertical orientation (vdiff)
to bubble assisted PZT mixing using either a square wave (vpztsquare) or a
sine wave excitation
(vpztsine) waveform. Figure 70D compares diffusion based kineticslvertical
orientation (vdif~ to
thermal gradient based mixing (TG). Figure 70E compares diffusion based
kinetics/vertical orientation
to diffusion based kinetics in a biochannel/vertical orientation to diffusion
based kinetics in a
biochannel/horizontal orientation to bubble mixing in a biochannellhorizontal
orientation. Figure 70F
depicts acoustic based mixing (treat-H) to diffusion based kinetics using
either vertical (ctrl-V) or
horizontal (ctrl-H) chip orientation.
In Figure 71, there is illustrated a schematic block diagram of an exemplary
signal processing
approach. A digital to analog converter (DAC) receives a digital signal from a
signal source (such as
signal generating circuitry on the signal processing printed circuit board or
received from a connected
personal computer) and converts that signal into an analog signal which is
received by filter. The
characteristics of filter may be modified to provide frequency low-pass, high-
pass, or single or multiple
band-pass characteristics according to tailored the signal applied to the
electrodes of the E-Chem Cell.
In this embodiment, the filtered signal is passed through resistor R9 (110
Kohm) before passing
through a first auxiliary amplifier (AUX AMP). To reduce signal complexity and
cost, the signal is
desirably multiplexed through multiplexes (MUX) and distributed to a plurality
of auxiliary electrodes on
the E-Chem cell cartridge.
A set of reference electrodes is also disposed within the E-Chem Cell
cartridge, the outputs of which
are coupled to through a second multiplexes (MUX) and reference amplifier (REF
AMP) and resistor
R13 (110 Kohm) back to the input of first auxiliary amplifier.
Finally, a set of active electrodes (36 active electrodes in this embodiment)
are coupled via printed
circuit board traces to a third mutiplexer. The output of this active
electrode multiplexes is amplified by
an input signal amplifier (INPUT AMP), and after further optional signal
conditioning (such as filtering,
gain control and/or selection) is processed through a buffer amplifier (BUFFER
AMP) and converted
from analog to digital (ADC) form, so that it may be communicated, processed,
analyzed, stored or the
like in digital form.
-10-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
In~Figure 72 there is illustrated an embodiment of a Thermal Control Block
Diagram. A
microprocessor communicates with an external signal source or sink via a
serial communication
channel or link. Advantageously, microprocessor generates a control signal to
provide an indication of
an ON, OFF, or FLASH status to an LED logic circuit which is coupled with and
causes LED Drivers to
send signals to each of six slots causing each of the slots red or green
lights to be on, off, or flash.
Microprocessor also generates a signal to DAC. This signal is amplified to
power a heat sink blower
to control the temperature of the heat sink. A heat sink temperature sensor is
associated with the heat
sink and this sensor generates a temperature signal which is fed back to the
microprocessor in
feedback manner to control operation or non-operation of the heat sink blower
motor.
The microprocessor also generates a plurality of signals which are received by
a plurality of DAC and
driver amplifiers to a Pettier thermal block for each slot. A temperature
sensor is also associated with
each Pettier thermal block to provide a sensed temperature indication back to
the microprocessor for
controlling the Pettier thermal block drive signal in feedback manner.
In Figure 73 there is illustrated an exemplary layout for a signal processing
printed circuit board. Each
board includes an edge connector for coupling with a communication bus,
motherboard, or other
interconnect as are known in the art. Each board in this particular embodiment
further includes pad
selector circuitry, memory, a CPU/lock-in amplifier, buffers, serial
communication circuitry, waveform
signal generators, analog-to-digital converter (ADC), filters or filter
circuits, master gain circuit, current-
to-voltage converter, power regulators, and chip selector (reflmux).
DETAILED DESCRIPTION
The present invention is directed to devices designed to receive and analyze a
plurality of biochips,
each comprising an array of biological moieties, such as nucleic acids or
proteins, to allow high
throughput analysis and detection of target analytes in samples. Thus for
example a number of
samples (particularly patient samples) can be simultaneously analyzed, or
multiple assays can be run
on a single sample. The devices comprise a number of cartridge stations that
are configured to
receive the biochips, with different types of biochips allowing different
types of components. The
stations can include a wide variety of different components, including
thermocontrollers, signaling
systems, sensors for leak detection, alphanumeric displays, and detectors.
Preferred embodiments
include the use of biochips comprising electrodes that rely on electrochemical
detection, and thus the
devices and/or stations can comprise device boards and processors.
The biochip cartridges include substrates comprising the arrays of
biomolecules, and can be
configured in a variety of ways. For example, the chips can include reaction
chambers with inlet and
-11-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
outlet ports for the introduction and removal of reagents. In addition, the
cartridges can include caps
or lids that have microfluidic components, such that the sample can be
introduced, reagents added,
reactions done, and then the sample is added to the reaction chamber
comprising the array for
detection.
Accordingly, the present invention provides compositions and methods for
detecting the presence or
absence of target analytes in samples. As wilt be appreciated by those in the
art, the sample solution
may comprise any number of things, including, but not limited to, bodily
fluids (including, but not limited
to, blood, urine, serum, lymph, saliva, anal and vaginal secretions,
perspiration and semen, of virtually
any organism, with mammalian samples being preferred and human samples being
particularly
preferred); environmental samples (including, but not limited to, air,
agricultural, water and soil
samples); biological warfare agent samples; research sampies (i.e. in the case
of nucleic acids, the
sample may be the products of an amplification reaction, including both target
and signal amplification
as is generally described in PCTlUS99101705, such as PCR amplification
reaction); purified samples,
such as purified genomic DNA, RNA, proteins, etc.; raw samples (bacteria,
virus, genomic DNA, etc.};
as will be appreciated by those in the art, virtually any experimental
manipulation may have been done
on the sample.
The methods are directed to the detection of target anaiytes. By "target
anafyte" or "analyte" or
grammatical equivalents herein is meant any 'molecule or compound to be
detected and that can bind
to a binding species, defined below. Suitable analytes include, but are not
limited to, small chemical
molecules such as environmental or clinical chemical or pollutant~or
biomolecule,. including, but not
limited to, pesticides, insecticides, toxins, therapeutic and abused drugs,
hormones, antibiotics,
antibodies, organic materials, etc. Suitable biomolecules include, but are not
limited to, proteins
(including enzymes, immunoglobulins and glycoproteins), nucleic acids, lipids,
lectins, carbohydrates,
hormones, whole cells (including procaryotic (such as pathogenic bacteria) and
eucaryotic cells,
including mammalian tumor cells), viruses, spores, etc. Particularly preferred
analytes are proteins
including enzymes; drugs, cells; antibodies; antigens; cellular membrane
antigens and receptors
(neural, hormonal, nutrient, and cell surface receptors) or their ligands.
In a preferred embodiment, the target analyte is a protein. As will be
appreciated by those in the art,
there are a large number of possible proteinaceous target analytes that may be
detected using the
present invention. By "proteins" or grammatical equivalents herein is meant
proteins, oligopeptides
and peptides, derivatives and analogs, including proteins containing non-
naturally occurring amino
acids and amino acid analogs, and peptidomimetic structures. The side chains
may be in either the (R) .
or the (S) configuration. In a preferred embodiment, the amino acids are in
the (S) or L-configuration.
-12-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
As discussed below, when the protein is used as a binding ligand, it may be
desirable to utilize protein
analogs to retard degradation by sample contaminants.
Suitable protein target analytes include, but are not limited to, (1)
immunoglobulins, particularly IgEs,
IgGs and IgMs, and particularly therapeutically or diagnostically relevant
antibodies, including but not
limited to, for example, antibodies to human albumin, apolipoproteins
(including apolipoprotein E),
human chorionic gonadotropin, cortisol, a-fetoprotein, thyroxin, thyroid
stimulating hormone (TSH),
antithrombin, antibodies to pharmaceuticals (including antieptileptic drugs
(phenytoin, primidone,
carbariezepin, ethosuximide, valproic acid, and phenobarbitol), cardioactive
drugs (digoxin, lidocaine,
procainamide, and disopyramide), bronchodilators ( theophylline), antibiotics
(chloramphenicol,
sulfonamides), antidepressants, immunosuppresants, abused drugs (amphetamine,
methamphetamine, cannabinoids, cocaine and opiates) and antibodies to any
number of viruses or
bacteria outlined below.
As will be appreciated by those in the art, a large number of analytes may be
detected using the
present methods; basically, any target analyte for which a binding ligand,
described below, may be
made may be detected using the methods of the invention.
In a preferred embodiment, the target analytes are nucleic acids. By "nucleic
acid" or "oligonucleotide"
or grammatical equivalents herein means at least two nucleotides covalently
4inked together. A
nucleic acid of the present invention will generally contain phosphodiester
bonds, although in some
cases, as outlined below, nucleic acid analogs are included that may have
alternate backbones,
comprising, for example, phosphoramide (Beaucage et al., Tetrahedron
49(10):1925 (1993) and
references therein; Letsinger, J. Org. Chem. 35:3800 (1970); Sprinzl et al.,
Eur. J. Biochem. 81:579
(1977); Letsinger et al., Nucl. Acids Res. 14:3487 (1986); Sawai et al, Chem.
Lett. 805 {1984),
Letsinger et al., J. Am. Chem. Soc. 110:4470 (1988); and Pauwels et al.,
Chemica Scripts 26:141
91986)), phosphorothioate (Mag et al., Nucleic Acids Res. 19:1437 (1991 ); and
U.S. Patent No.
5,644,048), phosphorodithioate (Briu et al., J. Am. Chem. Soc. 111:2321
(1989), O-
methylphophoroamidite linkages (see Eckstein, Oligonucleotides and Analogues:
A Practical
Approach, Oxford University Press), and peptide nucleic acid backbones and
linkages (see Egholm, J.
Am. Chem. Soc. 114:1895 (1992); Meier et al., Chem. Int. Ed. Engl. 31:1008
(1992); Nielsen, Nature,
365:566 (1993); Carlsson et al., Nature 380:207 (1996), all of which are
incorporated by reference).
Other analog nucleic acids include those with bicyclic structures including
locked nucleic acids,
Koshkin et al., J. Am. Chem. Soc. 120:13252-3 (1998); positive backbones
(Denpcy et al., Proc. Natl.
Acad. Sci. USA 92:6097 (1995); non-ionic backbones (U.S. Patent Nos.
5,386,023, 5,637,684,
5,602,240, 5,216,141 and 4,469,863; Kiedrowshi et aL, Angew. Chem. Intl. Ed.
English 30:423 (1991);
Letsinger et al., J. Am. Chem. Soc. 110:4470 (1988); Letsinger et al.,
Nucleoside & Nucleotide
-13-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
13:1597 (1994); Chapters 2 and 3, ASC Symposium Series 580, "Carbohydrate
Modifications in
Antisense Research", Ed. Y.S. Sanghui and P. Dan Cook; Mesmaeker et al.,
Bioorganic & Medicinal
Chem. Lett. 4:395 (1994); Jeffs et al., J. Biomolecular NMR 34:17 (1994);
Tetrahedron Lett. 37:743
(1996)) and non-ribose backbones, including those described in U.S. Patent
Nos. 5,235,033 and
5,034,506, and Chapters 6 and 7, ASC Symposium Series 580, "Carbohydrate
Modifications in
Antisense Research", Ed. Y.S. Sanghui and P. Dan Cook. Nucleic acids
containing one or more
carbocyclic sugars are also included within the definition of nucleic acids
(see Jenkins et al., Chem.
Soc. Rev. (1995) pp169-176). Several nucleic acid analogs are described in
Rawls, C & E News
June 2, 1997 page 35. All of these references are hereby expressly
incorporated by reference. These
modifications of the ribose-phosphate backbone may be done to facilitate the
addition of ETMs, or to
increase the stability and half life of such molecules in physiological
environments.
As will be appreciated by those in the art, all of these nucleic acid analogs
may find use in the present
invention. In addition, mixtures of naturally occurring nucleic acids and
analogs can be made; for
example, at the site of conductive oligomer or ETM attachment, an analog
structure may be used.
Alternatively, mixtures of different nucleic acid analogs, and mixtures of
naturally occuring nucleic
acids and analogs may be made.
Particularly preferred are peptide nucleic acids (PNA) which includes peptide
nucleic acid analogs.
These backbones are substantially non-ionic under neutral conditions, in
contrast to the highly
charged phosphodiester backbone of naturally occurring nucleic acids. This
results in two
advantages. First, the PNA backbone exhibits improved hybridization kinetics.
PNAs have larger
changes in the melting temperature (Tm) for mismatched versus perfectly
matched basepairs. DNA
and RNA typically exhibit a 2-4°C drop in Tm for an internal mismatch.
With the non-ionic PNA
backbone, the drop is closer to 7-9°C. Similarly, due to their non-
ionic nature, hybridization of the
bases attached to these backbones is relatively insensitive to salt
concentration.
The nucleic acids may be single stranded or double stranded, as specified, or
contain portions of both
double stranded or single stranded sequence. The nucleic acid may be DNA, both
genomic and
cDNA, RNA or a hybrid, where the nucleic acid contains any combination of
deoxyribo- and ribo-
nucleotides, and any combination of bases, including uracil, adenine, thymine,
cytosine, guanine,
inosine, xathanine hypoxathanine, isocytosine, isoguanine, etc. A preferred
embodiment utilizes
isocytosine and isoguanine in nucleic acids designed to be complementary to
other probes, rather
than target sequences, as this reduces non-specific hybridization, as is
generally described in U.S.
Patent No. 5,681,702. As used herein, the term "nucleoside" includes
nucleotides as well as
nucleoside and nucleotide analogs, and modified nucleosides such as amino
modified nucleosides. In
addition, "nucleoside" includes non-naturally occurring analog structures.
Thus for example the
-14-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
individual units of a peptide nucleic acid, each containing a base, are
referred to herein as a
nucleoside.
Thus, in a preferred embodiment, the target analyte is a target sequence. The
term "target sequence"
or "target nucleic acid" or grammatical equivalents herein means a nucleic
acid sequence on a single
strand of nucleic acid. The target sequence may be a portion of a gene, a
regulatory sequence,
genomic DNA, cDNA, RNA including mRNA and rRNA, or others. As is outlined
herein, the target
sequence may be a target sequence from a sample, or a secondary target such as
a product of an
amplification reaction, etc. It may be any length, with the understanding that
longer sequences are
more specific. As will be appreciated by those in the art, the complementary
target sequence may
take many forms. For example, it may be contained within a larger nucleic acid
sequence, i.e. all or
part of a gene or mRNA, a restriction fragment of a plasmid or genomic DNA,
among others. As is
outlined more fully below, probes are made to hybridize to target sequences to
determine the
presence or absence of the target sequence in a sample. Generally speaking,
this term will be
understood by those skilled in the art. The target sequence may also be
comprised of different target
domains; for example, a first target domain of the sample target sequence may
hybridize to a capture
probe or a portion of capture extender probe, a second target domain may
hybridize to a portion of an
amplifier probe, a label probe, or a different capture or capture extender
probe, etc. The target
domains may be adjacent or separated as indicated. Unless specified, the terms
"first" and "second"
are not meant to confer an orientation of the sequences with respect to the 5'-
3' orientation of the
target sequence. For example, assuming a 5'-3' orientation of the
complementary target sequence,
the first target domain may be located either 5' to the second domain, or 3'
to the second domain.
Suitable target analytes include biomolecules associated with: (1) viruses,
including but not limited to,
orthomyxoviruses, (e.g. influenza virus), paramyxoviruses (e.g respiratory
syncytial virus, mumps
virus, measles virus), adenoviruses, rhinoviruses, coronaviruses, reoviruses,
togaviruses (e.g. rubella
virus), parvoviruses, poxviruses (e.g. variola virus, vaccinia virus),
enteroviruses (e.g. poliovirus,
coxsackievirus), hepatitis viruses (including A, B and C), herpesviruses (e.g.
Herpes simplex virus,
varicella-zoster virus, cytomegalovirus, Epstein-Barr virus), rotaviruses,
Norwalk viruses, hantavirus,
arenavirus, rhabdovirus (e.g. rabies virus), retroviruses (including HIV, HTLV-
I and -II), papovaviruses
(e.g. papillomavirus), polyomaviruses, and picornaviruses, and the like; and
(2) bacteria, including but
not limited to, a wide variety of pathogenic and non-pathogenic prokaryotes of
interest including
Bacillus; Vibrio, e.g. V. cholerae; Escherichia, e.g. Enterotoxigenic E. coli,
Shigella, e.g. S.
dysenteriae; Salmonella, e.g. S. typhi; Mycobacterium e.g. M. tuberculosis, M.
leprae; Clostridium, e.g.
C. botulinum, C. tetani, C. difficile, C.perfringens; Cornyebacterium, e.g. C.
diphtheriae;
Streptococcus, S. pyogenes, S. pneumoniae; Staphylococcus, e.g. S. aureus;
Haemophilus, e.g. H.
influenzae; Neisseria, e.g. N. meningitidis, N. gonorrhoeae; Yersinia, e.g. G.
IambIiaY, pestis,
-15-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
Pseudomonas, e.g. P. aeruginosa, P. putida; Chlamydia, e.g. C. frachomatis;
l3ordetella, e.g. 8.
pertussis; Treponema, e.g. T. palladium; and the like.
Other suitable target analytes include, but are not limited to, (1) enzymes
(and other proteins),
including but not limited to, enzymes used as indicators of or treatment for
heart disease, including
creatine kinase, lactate dehydrogenase, aspartate amino transferase, troponin
T, myoglobin,
fibrinogen, cholesterol, triglycerides, thrombin, tissue plasminogen activator
(tPA); pancreatic disease
indicators including amylase, lipase, chymotrypsin and trypsin; liver function
enzymes and proteins
including cholinesterase, bilirubin, and alkaline phosphotase; aldolase,
prostatic acid phosphatase,
terminal deoxynucleotidyl transferase, and bacterial and viral enzymes such as
HIV protease; (2)
hormones and cytokines (many of which serve as ligands for cellular receptors)
such as erythropoietin
(EPO), thrombopoietin (TPO), the interleukins (including IL-1 through IL-17),
insulin, insulin-like growth
factors (including IGF-1 and -2), epidermal growth factor (EGF), transforming.
growth factors (including
TGF-a and TGF-(3), human growth hormone, transferrin, epidermal growth factor
(EGF), low density
lipoprotein, high density lipoprotein, leptin, VEGF, PDGF, ciliary
neurotrophic factor, prolactin,
adrenocorticotropic hormone (ACTH), calcitonin, human chorionic gonadotropin,
cotrisol, estradiol,
follicle stimulating hormone (FSH), thyroid-stimulating hormone (TSH),
leutinzing hormone (LH),
progeterone and testosterone; and (3) other proteins (including a-fetoprotein,
carcinoembryonic
antigen CEA, cancer markers, etc.).
Suitable target analytes include carbohydrates, including but not limited to,
markers for breast cancer
(CA15-3, CA 549, CA 27.29), mucin-like carcinoma associated antigen (MCA),
ovarian cancer
(CA125), pancreatic cancer (DE-PAN-2), prostate cancer (PSA), CEA, and
colorectal and pancreatic
cancer (CA 19, CA 50, CA242).
Other suitable target analytes include metal ions, particularly heavy and/or
toxic metals, including but
not limited to, aluminum, arsenic, cadmium, selenium, cobalt, copper,
chromium, lead, silver and
nickel.
In a preferred embodiment, the methods of the invention are used to detect
pathogens such as
bacteria. In this embodiment, preferred target sequences include rRNA, as is
generally described in
U.S. Patent Nos. 4,851,330; 5,288,611; 5,723,597; 6,641,632; 5,738,987;
5,830,654; 5,763,163;
5,738,989; 5,738,988; 5,723,597; 5,714,324; 5,582,975; 5,747,252; 5,567,587;
5,558,990; 5,622,827;
5,514,551; 5,501,951; 5,656,427; 5.352.579; 5,683,870; 5,374,718; 5,292,874;
5,780,219; 5,030,557;
and 5,541,308, all of which are expressly incorporated by reference.
-16-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
As will be appreciated by those in the art, a large number of analytes may be
detected using the
present methods; basically, any target analyte for which a binding ligand,
described below, may be
made may be detected using the methods of the invention. While many of the
techniques described
below exemplify nucleic acids as the target analyte, those of skill in the art
will recognize that other
target analytes can be detected using the same systems.
If required, the target analyte is prepared using known techniques. For
example, the sample may be
treated to lyse the cells, using known lysis buffers, electroporation, etc.,
with purification and/or
amplification as needed, as will be appreciated by those in the art. When the
target analyte is a
nucleic acid, the target sequence may be amplified as required; suitable
amplification techniques are
outlined in PCT US99/01705, hereby expressly incorporated by reference. In
addition, techniques to
increase the amount or rate of hybridization can also be used; see for example
WO 99/67425 and
U.S.S.N.s 09/440,371 and 60/171,981, all of which are hereby incorporated by
reference.
The samples comprising the target analytes can be added to cartridges
comprising the biochips as is
outlined in greater detail below. By "cartridge" herein is meant a casing or
housing for the biochip. As
outlined herein, and as will be appreciated by those in the art, the cartridge
can take on a number of
configurations and can be made of a variety of materials. Suitable materials
include, but are not
limited to, fiberglass, teflon, ceramics, glass, silicon, mica, plastic
(including acrylics, polystyrene and
copolymers of styrene and other materials, polypropylene, polyethylene,
polybutylene, polycarbonate,
polyurethanes, TefIonT"', and derivatives thereof, etc.), etc. Particularly
preferred cartridge materials
are plastic (including polycarbonate and polyproplylene) and glass.
As will be appreciated by those in the art, the cartridge can comprise a
number of components,
including reaction chambers, inlet and outlet ports, heating elements
including thermoelectric
components, RF antennae, electromagnetic components, memory chips, sealing
components such as
gaskets, electronic components including interconnects, multiplexers,
processors, etc.
In a preferred embodiment, the cartridge comprises a reaction chamber.
Generally, the reaction
chamber comprises a space or volume that allows the contacting of the sample
to the biochip array.
The volume of the reaction chamber can vary depending on the size of the array
and the assay being
done. In general, reaction chamber ranges from 1 nl- to about 1 mL, with from
about 1 to about 250 p1
being preferred and from about 10 to about 100 u1 being especially preferred.
In some embodiments,
to avoid the introduction of air bubbles into the reaction chamber (which can
be disruptive to
detection), the reaction chamber is less than the size of the sample to be
introduced, to allow a slight
overflow and thus ensure that the reaction chamber contains little or no air.
-17-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
in a preferred embodiment, the biochip cartridge can be configured to include
additional chambers that
can used for any number of different reactions, such as sample preparation,
cell lysis, rare target
capturelconcentration, sample clean-up, nucleic acid amplification, including
PCR, post-amplification
clean-up, sample concentration, reagent storage, mixing baffles/devices, etc.
In other embodiments,
the reaction chamber may be configured for other types of reactions as
generally described below.
In a preferred embodiment, the biochip cartridge reaction chamber is
configured to include at least one
nucleic acid amplification chamber. However, multiple amplification chambers
may be used. That is,
a cartridge may comprise from about 1 to about 10 or more chambers, with 2, 3,
4, 5, 6, 7, 8 or 9 also
being preferred.
In a preferred embodiment, the biochip cartridge reaction chamber is
configured to include at least one
PCR chamber. However, multiple PCR chambers may be used. That is, a cartridge
may comprise
from about 1 to about 10 or more chambers, with 2, 3, 4, 5, 6, 7, 8 or 9 also
being preferred.
In a preferred embodiment, the chamber of the cartridge should be made from
biocompatible
materials. In particular, materials that provide a surface that retards the
non-specific binding of
biomolecules, e.g. a "non sticky" surface, are preferred. For example, when
the reaction chamber is
used for PCR or amplification reactions a "non sticky" surface prevents
enzymatic components of the
reaction mixture from sticking to the surface and being unavailable in the
reaction. In addition, the
biocompatible properties of the chamber may be improved by minimizing the
surface area.
Biocompatible materials include, but are not limited to, plastic (including
acrylics, polystyrene and
copolymers of styrene and other materials, polypropylene, polyethylene,
polybutylene, polyimide,
polycarbonate, polyurethanes, TeflonT"", and derivatives thereof, etc.) Other
configurations include
combinations of plastic and printed circuit board (PCB; defined below). For
example at least one side
of the chamber is printed circuit board, while one or more sides of the
chamber are made from plastic.
In a preferred embodiment, three sides of the chamber are made from plastic
and one side is made
from printed circuit board. In addition, the chambers, channels, valves,
pumps, etc. of the systems
described herein may be coated with a variety of materials to reduce non-
specific binding. These
include proteins such as caseins and albumins (bovine serum albumin, human
serum albumin, etc.),
parylene, other polymers, etc.
The reaction chamber of the cartridge comprises an inlet port for the
introduction of the sample to be
analyzed. Depending on the reaction being run, multiple inlet ports may be
used, that may feed from a
variety of storage chambers or from the outside of the chamber. The inlet port
may optionally
comprise a seal to prevent or reduce the evaporation of the sample or reagents
from the reaction
_18_
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
chamber. In a prefierred embodiment (as depicted in Figure 3C and 14B), the
seal comprises a
gasket,'or valve through which a pipette or syringe can be pushed. The gasket
or valve can be rubber
or silicone or other suitable materials, such as materials containing
cellulose.
The reaction chamber can be configured in a variety of ways. In a preferred
embodiment, the reaction
chamber is configured to minimize the introduction or retention of air bubbles
or other sample
impurities. Thus for example, as depicted in Figure 1, assuming that the
cartridge is held in an upright
angle, the inlet port allows the flow of fluid sample into the "bottom" of the
reaction chamber, to allow
the escape of air or fluid through the "top" of the reaction chamber, for
example through an outlet port.
Thus the fluid sample flows up into the reaction chamber and contacts the
array. Thus, in a preferred
embodiment, the reaction chamber further comprises an outlet port to allow air
or excess sample to
exit the reaction chamber. In some embodiments, the outlet port vents to
either a waste storage well,
as is further described below, to an external surface of the chip or
cartridge, or, in a preferred
embodiment, back into the inlet port. Thus for example a preferred embodiment
utilizes a system
wherein the exit port vents to the inlet port, preferably above the point of
loading. For example, when
a pipette is used to load the cartridge, the tip of the pipette extends below
the exit port, such that air
from the exit port is not introduced into the reaction chamber. In addition,
the materials of the cartridge
housing and biochip can be chosen to be similar in hydrophobicity or
hydrophilicity, to avoid the
creation of air bubbles.
In addition, in a preferred embodiment, the reaction chamber/inlet and/or
outlet ports optionally include
the use of valves. For example, a semi-permeable membrane or filter may be
used, that preferentially
allows the escape of gas but retains the sample fluid in the chamber. For
example, porous teflons
such as GortexT"" allow air but not fluids to penetrate.
In a preferred embodiment, a reaction chamber in the biochip cartridge (such
as a PCR chamber) has
one or more valves controlling the flow of fluids into and out of the chamber.
The number of valves in
the cartridge depends on the number of channels and chambers. Alternatively,
the biochip cartridge is
designed to include one or more loading ports or valves that can be closed off
or sealed after the
sample is loaded. It is also possible to have multiple loading ports into a
single chamber; for example,
a first port is used to load sample and a second port is used to add reagents.
In these embodiments,
the biochip cartridge may have a vent. The vent can be configured in a variety
of ways. In some
embodiments, as generally depicted in the Figures 1A-1G , the vent can be a
separate port, optionally
with a valve, that leads out of the reaction chamber. Alternatively, the vent
may be a loop structure
that vents liquid and/or air back into the inlet port, as generally depicted
in Figure 1 F.
-19-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
As will be appreciated by those in the art, a variety of different valves may
be used. Valves can be
multi cycle or single cycle valves. By "multicycle" valves is meant that the
valve can be opened and
closed more than once. By "single cycle valves" or "burst valves" or "one time
valves" herein is meant
a valve that is closed and then opened or opened and then closed but lacks a
mechanism for restoring
the valve to its original position. Valves may also be check valves, which
allow fluid flow in only one
direction, or bi-directional valves.
In a preferred embodiment, check valves are used to prevent fluid from going
in and out of the reaction
chamber during reactions. Generally check valves are used when in embodiments
in which it is
desirable to have fluids and/or air flow in one direction, but not the other.
For example, when the
chamber is filled and thus compressed, air and liquid flow out. Conversely,
valves can be used to
empty the chamber as well. Types of check valves that can be used include, but
are not limited to,
duck bill valves (Vernay, www.vernay.com), cantilevers, bubble valves, etc.
In a preferred embodiment, the valve is a duck bill valve as generally
depicted in Figure 9A. These
valves are "one way" valves, in that fluid can flow through in one direction
but not the other.
In a preferred embodiment, the valve is a cantilever valve. As will be
appreciated by those in the art,
there are a variety of different types of cantilever valves known in the art.
Cantilever valves can also
be configured for use in pumping systems as described below. In a preferred
embodiment, a
cantilever valve comprising a metal is used. In this embodiment, the
application of a voltage can
either open or close a valve. See Figure 9B.
In a preferred embodiment, a heat pump is incorporated into the system for
opening and closing the
cantilever valve. In this embodiment, the check valves are made out of metals
such as gold and
copper such that the check valve functions as a cantilever when heat is
applied. In other
embodiments, an actuating force is not used to pull down the valve, rather
they have a restraining
force that prevents them from going in the other direction.
Similarly, a thermally actuated" valve that comprises a portion of the
microchannel with a flexible
membrane filled with air or liquid can be used in conjunction with a heater.
The application of heat
causes the fluid to expand, blocking the channel. See Figure 9F.
In other embodiments, piezoelectric (PZT) mixers are used as valves. These can
be built out of silicon
(obtained from Frauhoffer), plastic (obtained from IMM) or PCB.
-20-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
Other materials can be used in combination with check valves include materials
that can be used to
block an inlet or an outlet port. Such materials include wax or other
polymeric materials, such as
polyethylene oxide)-polypropylene oxide)-polyethylene oxide) triblock
copolymers (PEO-PPO-PEO)
known commercially as Pluronics ( BASF; Pluronic F-127, Sigma) or Synperonic
(ICI), that melt for
use as membranes or plugs. These materials share the common feature that they
can go from a solid
to a liquid at a given temperature. These types of systems are used in
conjunction with heaters,
described below., For example, heat is applied to melt the material, thus
"opening" the valve.
In a preferred embodiment, the burst valve is a film of metal or polymer. In a
preferred embodiment, a
free standing gold film is used, that is constructed using standard techniques
as outlined herein, by
etching away a support surface. The gold membrane dissolves upon application
of a voltage and CI-
ions. See for example www.mchips.com: Santini, J.T., et al., 1999, Nature,
397:335-338; both of
which are incorporated by reference in their entirety.
In a preferred embodiment, a combination of check valves and wax plugs are
used. In other
embodiments, a combination of check valves and gold membranes are used.
Other means of making a valve include mechanical means. These can frequently
be bi-directional
valves. For example, a shape memory wire can be attached to a plunger blocking
a channel. By
applying a current to the wire, the wire contracts and moves the plunger out
of the way, thereby
opening the channel. Conversely, the plunger can be drawn into the channel to
block the channel.
See Figure 9C.
Other mechanical valves include rotary valves. Rotary valves can be configured
in a variety of ways,
as depicted in Figures 9D and 9E. In one embodiment, an external force must be
applied for rotation
(i.e., a screw driver or stepper motor). Alternatively, a shape memory wire
can be used, such that the
application of heat or current will shrink the wire to rotate the valve. See
Figure 9E.
In addition, commercially available valves may be used in to control the flow
of liquids from into and
out of the various chambers of the present invention. Examples of commercially
available valves
include, MEMS (micro-electro-mechanical systems) micro valves
(http://www.redwoodmicro.com).
TiNi liquid microvafve (TiNi Alloy Company, San Leandro, CA), TiNi pneumatic
microvalves (TiNi Alloy
Company, San Leandro, CA), silicon micro valves (Bosch, D., et al., Sensors
and Actuators A, 37-38
(1993) 684-692). Commercial/conventional valves also are available from
Measurement Specialities,
Inc., IC Sensors Division, Milpitas, CA (http:I/www.msiusa.com/icsensors);
Plast-O-Matic Valves, Inc.
(http:/Iwww_plastomatic.coml), Barworth Inc. (http://www.banuorthinc.com),
Mobile Electronics Solution
-21 -
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
(htto:Ilwww.mobifeelectronics.netll: Specrum Chromatograph
(http://www.lplc.cam); all of which are
hereby incorporated by reference in their entirety.
Other sources for obtaining valves and pumps include research foundations such
as Chronos
(http:l/www.memsrus.com): Institute for Microtechnology - Mainz
(http:/lwww.imm-mainz.del);
Microsystems integration group - Swiss Fed. Inst. of technology
(http://dmtwww.epfl.ch); University of
Washington (http://lettuce.me.washinaton.edu): The Berkeley Sensor and
Actuator Center (BSAC)
(htt-~/lwww.otl.berkeley.edulmems.html); University of Michigan, Microsystems
R&D Laboratory
(httyl/www.eecs.umich.edu/MEMSlfacilities.html); Caltech, MEMS Research
(http:/ltouch.caltech.edu/home/research/files/html/researchframe.html); all of
which are hereby
incorporated by reference in their entirety. .
In a preferred embodiment, either an "on chip" or "off chip" pump is used to
move fluids from one area
or chamber of the cartridge to another. A general design for a pump includes a
chamber through
which air and/or liquid can flow; an inlet and outlet port, and valves. Fluid
is moved through the pump
via the application of some force, such as heat, pressure, to the chamber. In
addition, pumps may be
designed for single use or be reusable. Generally, reusable pumps have valves,
i.e., check valves,
that bias the flow of fluid in one direction. Single use pumps lack valves.
Thus, almost any type of
pump can be built as long as a mechanism for changing the volume in the
chamber and restricting
back flow is included. For example, upon contraction of the chamber via a PZT
or other pressure
force, the fluid or gas is displaced out of the chamber through the check
valve. Upon removal of the
contractive force, the chamber expands and draws liquid in through the pump
inlet. Alternatively, a
heater can be placed inside the chamber and the temperature of the gas or
Liquid can be raised,
causing it to expand. Upon expansion, the liquid is forced out of the chamber
through the outlet.
Upon cooling the liquid, the fluid is draw in through the inlet. See Figure
9G.
There are two primary means by which fluid can be moved in the biochip
cartridge. These are: (1)
through the use of a pump that pushes the fluid in or out; or, (2) by suction
that pulls fluid in or out of
the chamber.
Generally, a device such as a moving piston is used to create suction, however
cooling of gases,
vacuum chambers and gas consuming reactions can be used. . When suction is
used to move liquid
in or out of the chamber, a vacuum may be created elsewhere in the system.
fn a preferred embodiment, an "air pump" is used to move the liquid out of the
PCR chamber. In this
embodiment, a chamber of air is incorporated in the chip with an "on chip"
heater. When the heater is
turned on, the air in the chamber expands according to PV = nRT.
-22-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
Preferably, heaters (as are also described below) are incorporated into the
middle of the chip. In
some embodiments, more than one heater is incorporated in a chip to create
"heater zones". Air
chambers or pockets are located over the heater zones. The air chambers are
connected to the
reaction chamber via a channel that runs up to the top of the reaction chamber
with a valve or a plug
blocking it off. When the air is heated, it expands. The resulting build up in
pressure forces the valve
or plug to move out of the way, thereby forcing the liquid out of the chamber
via an outlet port.
Other ways of moving liquid out of the reaction chamber or reaction chamber
include using a low
boiling liquid in place of air. In this embodiment, the low boiling liquid
expands when heated and
displaces the liquid contained in the reaction chamber. Alternatively, a
chemical reaction may be used
to move liquid out of the reaction chamber. For example, the chemical reaction
used to expand car air
bags may be used to move liquid out of the reaction chamber, or other
reactions in which gases are
generated.
Other types of pumps that can be used include syringe driven pumps. These
pumps can be actuated
either by expanding air behind the syringe or by mechanical means. For
example, TiNi alloys, nitinol
wire, or "shape memory metals" can be used to mechanically actuate a syringe
driven pump. By "TiNi
alloys", "nitinol wire" or "shape memory metals" herein is meant materials
that when heated above a
certain transition temperature contract (i.e., usually up to 3 to 5% over the
original length of the metal),
thereby changing shape. Other materials that change shape upon heating include
shape memory
plastics.
Pumps also may be created using spring loaded pistons. In this embodiment, a
spring that can be
released is compressed or restrained within the body of the cartridge. For
example, wax may be used
to hold a spring in its compressed state. Upon heating, the wax is melted, and
the spring is released,
thereby generating sufficient force to move a piston and displace liquid.
Other versions include
incorporating materials that change from solids to liquids at a given
transition temperature, or moving a
mechanical blockade from the spring's pathway.
Pumps that utilize PZT driven actuations are also known and may be
incorporated int this invention.
By "PZT" herein is meant a material comprised of lead, zirconium and titanium
which upon application
of a voltage undergoes a rearrangement of the crystal lattice and generates a
force and a
displacement. This so called piezoelectric effect can be used to constrict and
expand a pump
chamber and result in a net movement of liquid. Other materials like shape
memory alloys that under
a change in shape upon application of a current such that the temperature of
the metal is raised above
a certain transition temperature can also be used.
-23-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
In addition, commercially available micro pumps may be used in to move liquid
from one location to
another in the cartridge. Examples of commercially available pumps include,
moulded plastic micro
pumps available from IMM (see IiqanewsCcr~.imm.uni-mainz.de), thin film shape
alloy microactuators
(TiNi Alloy Company, San Leandro, CA), silicon micro pumps (see M. Richter &
J. Kruckow,
aktorik/paper/2000Jahresbericht/ Paper2, 16.11.00), .
In addition, based on the geometry of the chamber, air can be used to push
liquid out of the reaction
chamber or mix liquids within the reaction chamber. Whether the air pumps the
fluid or bubbles
through to generate a mixing effect is determine by the relative size of the
bubble, the geometry of the
chamber/channel and the surface tension of the liquid. Larger air-liquid
interfaces tend to favor mixing
over pumping. Mixing of liquid within the biochip cartridge can occur by
pumping the liquid back and
forth in the biochip cartridge.
In a preferred embodiment, mixing is used to enhance hybridization rates. In a
preferred embodiment,
mixing is accomplished by inducing acoustic streaming using a piezo-electric
transducer glued onto
the back of the cartridge and excited with 5 Khz a.c. waveform at 10 V~p.
In a preferred embodiment, mixing is accomplished by creating a thermal
gradient across a chip. For
example, a thermal gradient may be created by heating the bottom of the chip
to 65°C and cooling the
top of the cartridge cover to 10°C. This can be accomplished by placing
the chip between two pettier
heaters, or by using an imbedded heater and a single pettier or other
thermoelectric cooling devices.
In a preferred embodiment, mixing is accomplished by recirculating liquid in a
given chamber using an
on chip or "off chip" pump attached to a chip.
In other embodiments, biochannel based mixing can be used to enhance
hybridization rates. In this
embodiment, a bubble is intentionally introduced into one corner of the chip.
By alternately expanding
and contracting the bubble volume via the application of heat from either an
in chip or off chip heat
source, mixing occurs as a result of the pressure flow created by changing the
volume of the bubble
within the chip. Alternatively, resonance induced mixing of bubbles can be
done using PZT devices as
well.
In some embodiments, mixing may be accomplished using non-contact mixing
technologies like that
describe by Covaris, Inc.
In a preferred embodiment, heaters are incorporated onto or into the chip, to
allow "on chip" heating (in
addition, as described below, "off chip" thermocontrollers within the device
may also be used). In this
-24-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
embodiment, the reaction chamber is designed to maximize thermal conductivity
between the
chamber and the heater or thermocontroller. Generally, designs that minimize
thermal mass (i.e.,
making the surface of the chamber in contact with the heat source as thin as
possible), impose certain
geometric constraints to ensure the complete removal of liquid from the
chamber, incorporate
materials that are good thermal conductors (i.e., metals), and thermally
isolate the chamber from the
rest of the chip are preferred. Often one makes a trade off between minimizing
surface to volume
ratios to reduce surface area for the non-specific binding of biological
components and maximizing
surface-to-volume ratis in order to obtain rapid heat transfer rates for
heating and cooling.
1n a preferred embodiment, air pockets or vents are used to thermally isolate
the amplificationchamber
from the rest of the chip. That is, the there is a break in the continuity of
the cartridge around the
amplification chamber.
In a preferred embodiment, thermally conductive materials are incorporated
into or below the reaction
chamber, forming hybrid chambers. For example, by using "layers" of different
materials, effective
heaters are constructed. Thus for example, a preferred embodiment utilizes one
or more resistive
heaters in the form of resistive metallic inks can be applied to a first layer
of PC board. These heaters
are powered by interconnects. In a preferred embodiment, a thin sheet of a
thermally conductive
material, preferably a metal such as copper, is applied, to allow even heat
distribution. In a preferred
embodiment, the copper layer is then coated with a thin layer of biocompatible
material, such as
plastic. See Figure 10A.
The total thickness of the hybrid chamber may vary from a few microns to
millimeter dimension. A
preferred thickness is approximately 200 microns.
In a preferred embodiment, multiple thermal heaters are incorporated into the
device to allow for the
creation of multiple thermal zones. The temperature in the respective zones is
maintained via either
active or passive control. Frequently, thethermal connectivity of the
cartridge materials are taken into
account during the design. In one embodiment, a chip may contain a thermal
heater in the detection
chamber of the cartridge in order to maintain the temperature of the detection
chamber as well as
constructing unique temperature zones in another part of device. In one
embodiments, these
temperature zones may be maintained to allow an enzymatic reaction to run
efficiently. In another
embodiment, multiple temperature zones may be maintained to simulate the
temperatures normally
used during PCR heat cycling. In order to effect the necessary temperature,
the liquid can be
maintained stationary and the temperature of the amplification chamber cycled
(i.e. 95-55-72),
alternatively, the liquid can be pumped over different temperature zones in
order to obtain heat cycling
(figure 10 B). This embodiment can be realized in different material
substrates such as glass, plastic,
-25-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
ceramic and PCB.
Similarly, there may be portions of the substrate that require heating, and
those that do not. Thus
more than one heater may be incorporated into the substrate. Similarly, these
thermal zones may or
may not be thermally isolated firom other parts of the substrate. For example,
PC board is significantly
thermally insulative, and thus just putting distance between the heaters and
thermal zones and the
areas of the substrate that do not require heating may be sufficient. In other
embodiments, thermally
insulative materials may be incorporated. For example, when the substrate is a
ceramic material,
thermal isolation may be accomplished by cutting out sections of the ceramic
substrate such that solid
regions of ceramic are separated from one another by a "cut out" as shown in
Figure 10C.
Other embodiments include the incorporation of temperature sensors into the
substrate such that the
temperature throughout the board can be monitored. In a preferred embodiment,
temperature sensors
are created using resistive devices, including silicon diodes. Other
embodiments include the use of
capillary thermostats and limiters
(http~//www.thermodisc.comIBuIbAndCapillary.html).
As will be appreciated by those in the art, there are a variety of reaction
chamber geometries which
can be used in this way. Generally having the intersection of the inlet port
and the reaction chamber
be at the "bottom" of the cartridge, with a small aperture, with the reaction
chamber widening, is
preferred. fn addition, the "top" of the reaction chamber may narrow, as well.
Several embodiments
are depicted in Figure 2. Thus, preferred embodiments for the size and shape
of the reaction chamber
allow for smooth loading of the reaction chamber. Preferred embodiments
utilize reaction chamber
geometries that avoid the use of sharp corners or other components that serve
as points for bubble
formation.
In addition, in some embodiments, the reaction chamber can be configured to
allow mixing of the
sample. For example, when a sample and a reagent are introduced simultaneously
or separately into
the chamber, the inlet port and/or the reaction chamber can comprise weirs,
channels or other
components to maximize the mixing of the sample and reagent. In addition, as
is outlined below, the
reaction may utilize magnetic beads for mixing and/or separation.
In a preferred embodiment, the cartridge comprises a sealing and/or venting
mechanism to prevent
the cartridge from exploding due to a build up in pressure during a reaction,
or to prevent leakage of
the sample or reagents onto other parts of the substrate, particularly (in the
case of electronic
detection) onto electronic interconnects. As will be appreciated by those in
the art,.this may take on a
variety of different forms. In one embodiment, there is a gasket between the
biochip substrate
comprising the array and the cartridge, comprising sheets, tubes or strips.
Alternatively, there may be
-26-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
a rubber or silicone strip or tube used; for example, the housing may comprise
an indentation or
channel into which the gasket fits, and then the housing, gasket and chip are
clamped together.
Furthermore, adhesives can be used to attach the gasket to the cartridge, for
example, a double sided
adhesive can be used; for example, silicone, acryiic and combination adhesives
can be used to attach
the gasket to the biochip, which is then clamped into the cartridge as
described herein.
In some embodiments, the reaction chamber and biochip substrate are configured
such that a
separate sealing mechanism is not required. For example, the biochip substrate
can serve as one
"half' of the reaction chamber, with the array on the inside, and the reaction
chamber housing can
serve as the other "half'. Depending on the materials used, there may be an
optional adhesive to
attach the two. Alternatively, when there are arrays on both sides of the
substrate, the housing may
encompass the substrate.
Optional adhesives that may be used to seal the cartridge include, but are not
limited to, pressure
sensitive adhesives, thermal adhesives, etc. Other means of sealing the
cartridges include sonic
welding, laser bonding, and epoxys.
In a preferred embodiment, the reaction chamber is made entirely of plastic.
In another embodiment,
a PCB underlies all or a significant portion of the cartridge.. The cartridge
may be attached directly to
the PCB. Alternatively, the device can be built wholly in the PCB, ceramic, or
glass material with all of
the necessary or a large majority of the necessary functions integrated into
the device during the
manufacturing process.
Thus, in these embodiments, the volume of the reaction chamber can be set
either by forming a well in
the cartridge, such that the addition of the biochip substrate forms a
reaction chamber around the
array, or by using a flat cartridge and using a gasket or adhesive of a
defined depth, or by
combinations of the three.
In a preferred embodiment, the cartridge comprises a cap or lid. The cap may
be functional, as
outlined below when it comprises microfluidic components. In addition, the cap
may be designed for
safety purposes, to prevent the leakage of biological materials or cross-
contamination. Additionally,
the cap can be designed to be removable. As will be appreciated by those in
the art, the cap can take
on a wide variety of configurations. For example, in one embodiment, the cap
merely seals the inlet
port to prevent evaporation of the sample during the assay. In a preferred
embodiment, the cap may
comprise a number of additional elements for use in sample handling and
reagent storage, to allow for
a variety of different sample reactions. For example, a variety of
microfluidic components can be built
into the cap to effect a number of manipulations on a sample to ultimately
result in target analyte
-27-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
detection or quantitation. See generally PCT US00/10903, and references
outlined therein, all of which
are expressly incorporated by reference. These manipulations can include cell
handling (cell
concentration, cell lysis, cell removal, cell separation, etc.), separation of
the desired target analyte
from other sample components, chemical or enzymatic reactions on the target
analyte, detection of the
target analyte, etc. The devices of the invention can include one or more
wells for sample
manipulation, waste or reagents; microchannels (sometimes referred to as flow
channels) to and
between these wells, including microchannels containing electrophoretic
separation matrices; valves
to control fluid movement; on-chip pumps such as electroosmotic,
electrohydrodynamic, or
electrokinetic pumps. In addition, as outlined herein, portions of the
internal surtaces of the device
may be coated with a variety of coatings as needed, to reduce non-specific
binding, to allow the
attachment of binding ligands, for biocompatibility, for flow resistance, etc.
These microfluidic caps
can be made in a variety of ways, as will be appreciated by those in the art.
See for example
references described in PCT US00/10903, and references outlined therein, a14
of which are expressly
incorporated by reference.
When the cap of the cartridge is used as part of the assay, it may be
configured to include one or more
of a variety of components, herein referred to as "modules", that will be
present on any given device
depending on its use, and are connected as required by microchannels. These
modules include, but
are not limited to: sample inlet ports; sample introduction or collection
modules; cell handling modules
(for example, for cell lysis, cell removal, cell concentration, cell
separation or capture, cell growth,
etc.); separation modules, for example, for electrophoresis,
diefectrophoresis, gel filtration, ion
exchange/affinity chromatography (capture and release) etc.; reaction modules
for chemical or
biological alteration of the sample, including amplification of the target
analyte (for example, when the
target analyte is nucleic acid, amplification techniques are useful,
including, but not limited to
polymerase chain reaction (PCR), oligonucleotide ligation assay (OLA); strand
displacement
amplification (SDA), and nucleic acid sequence based amplification (NASBA) and
other techniques
outlined in WO 99/37819, PCT US00/19889, and US00/20476, all of which are
hereby incorporated by
reference in their entirety; chemical, physical or enzymatic cleavage or
alteration of the target analyte,
or chemical modification of the target; fluid pumps (including, but not
limited to, electroosmotic,
electrohydrodynamic, or electrokinetic pumps; fluid valves; thermal modules
for heating and cooling;
storage modules for assay reagents; mixing chambers; and detection modules.
In addition, while these microfluidic components are described herein as being
associated with the cap
of the cartridge, as wiN be appreciated by those in the art, these modules and
channels (as well as
other components outlined herein) may be located anywhere in the cartridge or
device. In addition,
some components may be in the device; for example, "off chip" pumps may be
located within one or
more stations of the device.
-28-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
The cartridge comprises at least one biochip, with some embodiments utilizing
one or more biochips
per cartridge. By "biochip" or equivalents herein is meant a substrate
comprising an array of distinct
biomolecules, particularly nucleic acids and proteins. There are a wide
variety of suitable nucleic acid
biochips, including those made using photolithographic techniques (such as the
Affymetrix
GeneChipT"'), spotting techniques (e.g. Synteni and Incyte), prining
techniques (Agilent and Rosetta),
three dimensional "gel pad" arrays, and those including electronic components
(e.g. Nanogen). A
preferred embodiment is described below and in U.S. Patent Nos. 5,591,578;
5,824,473; 5,705,348;
5,780,234 and 5,770,369; U.S.S.N.s 08/873,598 08/911,589; WO 98/20162;
W098/12430; '
W098/57158; WO 00/16089) W099/57317; W099/67425; WO00/24941; PCT US00/10903;
W000/38836; W099/37819; W099/57319 and PCTUS00/20476; and related materials,
all of which
are expressly incorporated by reference in their entirety.
It should be noted that one distinct advantage of the use of the electronic
detection methods outlined
herein is that real time monitoring of reactions and hybridization can occur.
That is, while systems
based on fluorescence require the removal of excess (e.g. unbound) signaling
probes (or target
sequences when the target sequence itself has been fluorescently labeled
during an amplification
reaction, for example), the electronic methods outlined herein do not. That
is, unless the probes
comprising the ETMs are bound to the surface, little or no signal is seen even
if unbound probes have
not been removed. This allows the monitoring of real-time reactions, as well
as multiple
measurements on the same array. Accordingly, while the discussion below is
directed mainly to the
use of biochips comprising an array of electrodes, other array technologies
are included in the present
invention.
In a preferred embodiment, the biochips comprise substrates with a plurality
of array locations. By
"substrate" or "solid support" or other grammatical equivalents herein is
meant any material that can
be modified to contain discrete individual sites appropriate of the attachment
or association of capture
ligands. Suitable substrates include metal surfaces such as gold, electrodes
as defined below, glass
and modified or functionalized glass, fiberglass, teflon, ceramics, mica,
plastic (including acrylics,
polystyrene and copolymers of styrene and other materials, polypropylene,
polyethylene,
polybutylene, polyimide, polycarbonate, polyurethanes, TeflonT"', and
derivatives thereof, etc.),
GETEK (a blend of polypropylene oxide and fiberglass), etc, polysaccharides,
nylon or nitrocellulose,
resins, silica or silica-based materials including silicon and modified
silicon, carbon, metals, inorganic
glasses and a variety of other polymers, with printed circuit board (PCB)
materials being particularly
preferred.
The present system finds particular utility in array formats, i.e. wherein
there is a matrix of addressable
detection electrodes (herein generally referred to "pads", "addresses" or
"'micro-locations"). By "array"
-29-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
herein is meant a plurality of capture ligands in an array format; the size of
the array will depend on the
composition and end use of the array. Arrays containing from about 2 different
capture ligands to
many thousands can be made. Generally, the array will comprise from two to as
many as 100,000 or
more, depending on the size of the electrodes, as well as the end use of the
array. Preferred ranges
are from about 2 to about 10,000, with from about 5 to about 1000 being
preferred, and from about 10
to about 100 being particularly preferred. In some embodiments, the
compositions of the invention
may not be in array format; that is, for some embodiments, compositions
comprising a single capture
ligand may be made as well. In addition, in some arrays, multiple substrates
may be used, either of
different or identical compositions. Thus for example, large arrays may
comprise a plurality of smaller
substrates.
In a preferred embodiment, the biochip comprises a substrate with at least one
surface comprising an
array, and in a preferred embodiment, an array of electrodes. By "electrode"
herein is meant a
composition, which, when connected to an electronic device, is able to sense a
current or charge and
convert it to a signal. Alternatively an electrode can be defined as a
composition which can apply a
potential to and/or pass electrons to or from species in the solution. Thus,
an electrode is an ETM as
described herein. Preferred electodes are known in the art and include, but
are not limited to, certain
metals and their oxides, including gold; platinum; palladium; silicon;
aluminum; metal oxide electrodes
including platinum oxide, titanium oxide, tin oxide, indium tin oxide,
palladium oxide, silicon oxide,
aluminum oxide, molybdenum oxide (Mo2O6), tungsten oxide (W03) and ruthenium
oxides; and
carbon (including glassy carbon electrodes, graphite and carbon paste).
Preferred electrodes include
gold, silicon, carbon and metal oxide electrodes, with gold being particularly
preferred.
The electrodes described herein are depicted as a flat surface, which is only
one of the possible
conformations of the electrode and is for schematic purposes only. The
conformation of the electrode
will vary with the detection method used and the configuration of the
cartridge. For example, flat
planar electrodes may be preferred for optical detection methods, or when
arrays of nucleic acids are
made, thus requiring addressable locations for both synthesis and detection.
Alternatively, for single
or low density analysis, the electrode may be in the form of a tube; this
allows a maximum of surface
area containing the nucleic acids to be exposed to a small volume of sample.
In a preferred embodiment, the detection electrodes are formed on a substrate.
In addition, the
discussion herein is generally directed to the formation of gold electrodes,
but as will be appreciated
by those in the art, other electrodes can be used as well. The substrate can
comprise a wide variety
of materials, as outlined above.
1n general, preferred materials include printed circuit board materials.
Circuit board materials ace
-30-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
those that comprise an insulating substrate that is coated with a conducting
layer and processed using
lithography techniques, particularly photolithography techniques, to form the
patterns of electrodes and
interconnects (sometimes referred to.in the art as interconnections or leads).
The insulating substrate
is generally, but not always, a polymer. As is known in the art, one or a
plurality of layers may be used,
to make either "two dimensional" (e.g. all electrodes and interconnections in
a plane) or "three
dimensionaP' (wherein the electrodes are on one surface and the interconnects
may go through the
board to the other side or wherein electrodes are on a plurality of surfaces)
boards. Three
dimensional systems frequently rely on the use of drilling or etching,
followed by electroplating with a
metal such as copper, such that the "through board" interconnections are made.
Circuit board
materials are often provided with a foil already attached to the substrate,
such as a copper foil, with
additional copper added as needed (for example for interconnections), for
example by electroplating.
The copper surface may then need to be roughened, for example through etching,
to allow attachment
of the adhesion layer.
Accordingly, in a preferred embodiment, the present invention provides
biochips (sometimes referred
to herein "chips") that comprise substrates comprising a plurality of
electrodes, preferably gold
electrodes. The number of electrodes is as outlined for arrays. Each electrode
preferably comprises
a self assembled monolayer as outlined herein. In a preferred embodiment, one
of the monolayer-
forming species comprises a capture ligand as outlined herein. In addition,
each electrode has an
interconnection, that is attached to the electrode at one end and is
ultimately attached to a device that
can control the electrode. That is, each electrode is independently
addressable.
In a preferred embodiment, the connections from the electrodes are made by
passing through the
substrate to produce a so called land grid array that can interface to a pogo
pin or like connector to
make connections from the chip to the instrument. In this embodiment, pogo pin
connectors are used
in place of edge card connectors. An example of a chip containing electrodes
arranged in a land grid
array is shown in Figure 1 H??. In this embodiment, rather than contain longer
interconnects, the
electrode array is one surface of the substrate, such as a PCR board or
ceramic substrate, and there
are "through board" or "through substrate" interconnects ending in pads. See
Figure 1 I??. When the
cartridge is placed in the device, these pads contact "pogo pin" type
connectors, thus saving space on
the chip and allowing for higher density arrays, if desired. See Figure 1J. In
some embodiments,
switching circuitry (multiplexers) can be built into the pogo pin connector.
Detection electrodes on circuit board material (or other substrates) are
generally prepared in a wide
variety of ways. In general, high purity gold is used, and it may be deposited
on a surface via vacuum
deposition processes (sputtering and evaporation) or solution deposition
(electroplating or electroless
processes). When electroplating is done, the substrate must initially comprise
a conductive material;
-31 -
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
fiberglass circuit boards are frequently provided with copper foil.
Frequently, depending on the
substrate, an adhesion layer between the substrate and the gold in order to
insure good mechanical
stability is used. Thus, preferred embodiments utilize a deposition layer of
an adhesion metal such as
chromium, titanium, titanium/tungsten, tantalum, nickel or palladium, which
can be deposited as above
for the gold. When electroplated metal (either the adhesion metal or the
electrode metal) is used,
grain refining additives, frequently referred to in the trade as brighteners,
can optionally be added to
alter surface deposition properties. Preferred brighteners are mixtures of
organic and inorganic
species, with cobalt and nickel being preferred.
In general, the adhesion layer is from about 100 A thick to about 25 microns
(1000 microinches). The
If the adhesion metal is electrochemically active, the electrode metal must be
coated at a thickness
that prevents "bleed-through"; if the adhesion metal is not electrochemically
active, the electrode metal
may be thinner. Generally, the electrode metal (preferably gold) is
deposited.at thicknesses ranging
from about 500 A to about 5 microns (200 microinches), with from about 30
microinches to about 50
microinches being preferred. In general, the gold is deposited to make
electrodes ranging in size from
about 5 microns to about 5 mm in diameter, with about 100 to 250 microns being
preferred. The
detection electrodes thus formed are then preferably cleaned and SAMs added,
as is discussed
below.
Thus, the present invention provides methods of making a substrate comprising
a plurality of gold
electrodes. The methods first comprise coating an adhesion metal, such as
nickel or palladium
(optionally with brightener), onto the substrate. Electroplating is preferred.
The electrode metal,
preferably gold, is then coated (again, with electroplating preferred) onto
the adhesion metal. Then
the patterns of the device, comprising the electrodes and their associated
interconnections are made
using lithographic techniques, particularly photolithographic techniques as
are known in the art, and
wet chemical etching. Frequently, a non-conductive chemically resistive
insulating material such as
solder mask or plastic is laid down using these photolithographic techniques,
leaving only the
electrodes and a connection point to the leads exposed; the leads themselves
are generally coated.
In one embodiment of the inventive structure, the solder mask is desirably
made of a solvent soluble
material rather than a water soluble material. Water soluble solder masks have
become standard in
the industry because of the environmental advantages of water soluble
materials generally.
Unfortunately, for a detector chip that is to be exposed to aqueous solutions,
water soluble materials
such as for example acetonitrile can dissolve when exposed to aqueous
solution.
The methods continue with the addition of SAMs, described below. In a
preferred embodiment, drop
deposition techniques are used to add the required chemistry, i.e. the
monolayer forming species, one
-32-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
of which is preferably a capture ligand comprising species. Drop deposition
techniques are well
known for making "spot" arrays. This is done to add a different composition to
each electrode, i.e. to
make an array comprising different capture ligands. Alternatively, the SAM
species may be identical
for each electrode, and this may be accomplished using a drop deposition
technique or the immersion
of the entire substrate or a surface of the substrate into the solution.
In a preferred embodiment, plasma treatments are used to generate a surface
free of major
contaminants prior to the deposition of SAMs comprising capture probes. This
method is particularly
useful for the activation of gold surfaces for the formation of SAMs
comprising capture probes with
packing densities close to the theoretical limit (see Figure 68). Plasma
methods can also be used for
the deposition of different capture probes on neighboring pads.
Plasma treatment in a barrel type machine (minimal ion bombardment) with
oxygen plasma is
commonly used in semiconductor processing to remove trace residues of organic
contaminants,
including photoresistant contaminants. Although treatment with oxygen plasma
can be used in the
methods of the present invention for generating a clean surface, this
treatment makes the insulator
layer separating the gold pads hydrophilic. Thus, it is difficult to spot
arrays without contamination_
In a preferred embodiment, a hydrogen plasma treatment is used. This procedure
recovers
hydrophobicity on organic surfaces because it converts hydrophilic C-O bonds
to C-H bonds. tn
addition, this treatment does not add contaminants to the gold surface.
Hydrogen plasma treatment
can be used alone, or in combination with an oxygen plasma treatment. Figure
69 illustrates the
effectiveness of an oxygen plasma treatment followed by a hydrogen plasma
treatment for removing
major contaminants from chips.
Combining an oxygen plasma treatment with a hydrogen plasma treatment can be
used on micro-
patterned photoresistant materials to generate hydrophobic surfaces next to
hydrophilic surfaces. The
hydrophobic and hydrophilic surfaces can either be adjacent to each other or
separated by a few
microns.
In a preferred embodiment, CF4 plasma can be used after an oxygen plasma
treatment to make
teflonT""-like compounds, i.e. C-F bonds, on the insulator, resulting in a
hydrophobic insulator surface.
In other embodiments, a chemical treatment can be combined with a plasma
treatment. For example,
chemical treatment with silanes can be used to make the oxide insulator
surface hydrophobic or
hydrophilic, while leaving non-oxide surfaces, such as gold, uncontaminated.
Whether the insulator
surface is hydrophobic or hydrophilic depends on the type of silane that
replaces the O-H bonds.
-33-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
Additionally, this procedure can be used in conjunctions with plasma cleaning
to enable selective
wetting of chemical and biological fluids on the chip surface or to prevent or
allow sticking of a given
species of solution to the surtace of the chip.
When the biochips comprise electrodes, there are a variety of additional
components in addition to the
chemistry outlined below, which may be present on the chip, including, but not
limited to,
interconnects, multiplexers, relay devices, filters, RF antennae, heating
elements, electromagnetic
components, etc.
Each electrode comprises an independent lead (interconnect) to transmit input
and electronic
response signals for each electrode of the array. In contrast to previous
systems which require the
ability to independently alter only input signals to each electrode but not
electronic response signals, it
is important in the present invention that both input and electronic response
signals be independently
monitorable for each electrode.
For a relatively small number of electrode pads andlor depending on the
desired size of the array,
providing direct connections using parallel circuits may be appropriate.
In a preferred embodiment, each electrode is individually connected to a
corresponding input of a
multiplexer via a corresponding interconnector. One problem presented in
conventional systems and
methods is the difficulty in providing electrical connections (inputs and/or
outputs) to a large number of
electrodes, particularly if the electrodes form a dense or close packed array.
Several solutions to this
problem have been identified, and include the use of circuitry that allows
signal processing either
simultaneously as sets of parallel circuits and connections, line-sample array
addressing, serially in a
time-domain multiplexed manner, or in parallel or serially using frequency
domain and/or time-domain
based separation techniques, among other available techniques, as are outlined
herein.
For example, a preferred method to connect a first multiplicity of circuits or
lines on the chip to a
smaller plurality of lines at a connector leading from the chip are to use a
switching device such as a
multiplexer (MUX) or relays to selectively couple circuits on the chip or
board with circuits off the
board.
The number of multiplexers will depend on the number of electrodes in the
array. In one embodiment,
a single MUX is utilized. In a preferred embodiment, a plurality of MUXs are
used. This can be done in
a variety of ways, as will be appreciated by those in the art; in one
embodiment, "sectors" of
electrodes are assigned to a particular MUX; thus for example, rows or columns
of the array may each
have their own MUX. Alternatively, submultiplexers are used; for example, a
column or row is
-34-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
connected to a respective sub-multiplexes, with the sub-multiplexes outputs
going to another
submultiplexer.
In a preferred embodiment, the multiplexes includes a binary counter which
receives the control signal
via the connector pad. The control signal is preferably a pulsed signal such
as a clock signal and
generates a sequential count to drive the decoders.
In a preferred embodiment, another way to connect a multiplicity of electrodes
on the substrate to a
smaller number of connector pads leading "off chip" is to use row-column
select signals to allow the
selection of individual electrodes.
Unfortunately, for structures and methods that access different electrodes or
groups of electrodes in a
time sequential manner, some correction or adjustment of the sensed
results.may be required when
the difference in time is sufficiently large to alter the results, in order to
maintain a calibration between
earlier sensed and read-out data and later sensed and read-out data. The need
for such adjustment
will depend upon the assay, reaction kinetics and the time separation which
may also be a function of
the number of electrodes to be sensed or read-out. For example, in some
embodiments it may be
entirely. reasonable to measure each of the 25 electrodes in a 5 X 5 array of
electrodes a few seconds
apart (e.g. 10 seconds apart); however, in other embodiments, the 4 minute
separation between the
first and last measurement may be unacceptable or difficult to compensate.
It is also desirable to consider the kinetics of reaction when the reaction
takes place on or near a
planar surface, such as the surface of the electrode. Diffusion rates may play
a more important role
than when the reaction occurs in solution. It is important to understand when
or over what period of
time the reaction takes place so that the measurements are taken at the
appropriate time. This may
be particularly important if an intermediate reaction product is to be sensed,
or if a series of
measurements are desirable, for example to do reaction kinetics.
Reaction kinetics are also an important consideration for the driving signals.
Biosensors are limited by
the chemical kinetics. For the class of molecules of interest here (DNA, DNA
fragments, proteins,
antibodies, and the like), each molecule has a maximum speed or velocity in
the medium. For
example, the molecules may typically be actively driven or moved in solution
at frequencies between
about 1 Hz and 10kHz, more typically between about 5 Hz and 5 kHz. At higher
frequencies, the
molecules only vibrate, while at lower frequencies the movement is not
particularly useful.
In addition, there is an assay volume, that is the accessible volume of the
assay, associated with each
driving signal frequency. As the frequency increases, the assay volume shrinks
in size and volume.
-35-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
This has implication for the distribution of electrodes and the driving signal
frequency.
One additional consideration for sensing or measuring a reaction is the
possible effect that the
reaction medium (such as solution components, sample components, reaction
components, etc.) may
have on the electrodes. Sometimes the electrodes will degrade, become
passivated, or otherwise
change over time thereby affecting the accuracy and uniformity of
measurements. Under such
conditions it is desirable to perform the sensing, measurement, or analysis
quickly, or at least
according to predetermined timings so that the data collected will be properly
interpreted.
In a preferred embodiment, one or more preamplifiers are used. As will be
appreciated by those in the
art, the preamplifier can be on the surface of the substrate, e.g. "on board"
or "on chip", or may be
provided in circuitry external to the array chip. It is preferable, however,
that the preamplifier be
included on the substrate to increase the signal-to-noise ratio of the signal
provided to the external
circuitry.
In a preferred embodiment, each individual electrode has an associated
preamplifier.
In a preferred embodiment, the array is divided into "sectors", wherein a
subset of the electrodes in the
array have an associated MUX and preamplifier. Similarly, other components of
the invention may be
associated with sectors.
In a preferred embodiment, impedance matching is done.
In a preferred embodiment, filters are used, including, but not limited to,
time domain filters and
frequency domain filters, and combinations.
In addition to electronic components, the electrodes of the invention in
preferred embodiments
comprise self-assembled monolayers (SAMs). The compositions of these SAMs will
vary with the
detection method used. In general, there are two basic detection mechanisms.
In a preferred
embodiment, detection of an ETM is based on electron transfer through the
stacked rr-orbitals of
double stranded nucleic acid. This basic mechanism is described in U.S. Patent
Nos. 5,591,578,
5,770,369, 5,705,348, and PCT US97/20014 and is termed "mechanism-1" herein.
Briefly, previous
work has shown that electron transfer can proceed rapidly through the stacked
rr-orbitals of double
stranded nucleic acid, and significantly more slowly through single-stranded
nucleic acid. Accordingly,
this can serve as the basis of an assay. Thus, by adding ETMs (either
covalently to one of the strands
or non-covalently to the hybridization complex through the use of
hybridization indicators, described
below) to a nucleic acid that is attached to a detection electrode via a
conductive oligomer, electron
-36-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
transfer between the ETM and the electrode, through the nucleic acid and
conductive oligomer, may
be detected.
Alternatively, the ETM can be detected, not necessarily via electron transfer
through nucleic acid, but
rather can be directly detected on an electrode comprising a SAM; that is, the
electrons from the ETMs
need not travel through the stacked rr orbitals in order to generate a signal.
As above, in this
embodiment, the detection electrode preferably comprises a self-assembled
monolayer (SAM) that
serves to shield the electrode from redox-active species in the sample. In
this embodiment, the
presence of ETMs on the surtace of a SAM, that has been formulated to comprise
slight "defects"
(sometimes referred to herein as "microconduits", "nanoconduits" or
"electroconduits") can be directly
detected. This basic idea is termed "mechanism-2" herein. Essentially, the
electroconduits allow
particular ETMs access to the surface. Without being bound by theory, it
should be noted that the
configuration of the electroconduit depends in part on the ETM chosen. For
example, the use of
relatively hydrophobic ETMs allows the use of hydrophobic electroconduit
forming species, which
effectively exclude hydrophilic or charged ETMs. Similarly, the use of more
hydrophilic or charged
species in the SAM may serve to exclude hydrophobic ETMs.
It should be noted that these defects are to be distinguished from "holes"
that allow direct contact of
sample components with the detection electrode. As is more fully outlined
below, the electroconduits
can be generated in several general ways, including but not limited to the use
of rough electrode
surfaces, such as gold electrodes formulated on PC circuit boards; or the
inclusion of at least fwo
different species in the monolayer, i.e. using a "mixed monolayer", at least
one of which is a
electroconduit-forming species (EFS). Thus, upon binding of a target analyte,
a soluble binding ligand
comprising an ETM is brought to the surface, and detection of the ETM can
proceed, putatively
through the "electroconduits" to the electrode. Essentially, the role of the
SAM comprising the defects
is to allow electronic contact of the ETM with the electronic surface of the
electrode, while still
providing the benefits of shielding the electrode from solution components and
reducing the amount of
non-specific binding to the electrodes. Viewed differently, the role of the
binding ligand is to provide
specificity for a recruitment of ETMs to the surface, where they can be
directly detected.
Thus, in either embodiment, as is more fully outlined below, an assay complex
is formed that contains
an ETM, which is then detected using the detection electrode.
Thus, in a preferred embodiment, the electrode comprises a monolayer,
comprising electroconduit
forming species (EFS). As outlined herein, the efficiency of target analyte
binding (for example,
oligonucleotide hybridization) may increase when the analyte is at a distance
from the electrode.
Similarly, non-specific binding of biomolecules, including the target
analytes, to an electrode is
-37-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
generally reduced when a monolayer is present. Thus, a monolayer facilitates
the maintenance of the
analyte away from the electrode surface. In addition, a monolayer serves to
keep charged species
away from the surtace of the electrode. Thus, this layer helps to prevent
electrical contact between
the electrodes and the ETMs, or between the electrode and charged species
within the solvent. Such
contact can result in a direct "short circuit" or an indirect short circuit
via charged species which may
be present in the sample. Accordingly, the monolayer is preferably tightly
packed in a uniform layer on
the electrode surface, such that a minimum of "holes" exist. The monolayer
thus serves as a physical
barrier to block solvent accesibility to the electrode.
By "monolayer" or "self-assembled monolayer" or "SAM" herein is meant a
relatively ordered assembly
of molecules spontaneously chemisorbed on a surface, in which the molecules
are oriented
approximately parallel to each other and roughly perpendicular to the surface.
A majority of the
molecules include a functional group that adheres to the surface, and a
portion that interacts with
neighboring molecules in the monolayer to form the relatively ordered array. A
"mixed" monolayer
comprises a heterogeneous monolayer, that is, where at least two different
molecules make up the
monolayer.
In general, the SAMs of the invention can be generated in a number of ways and
comprise a number
of different components, depending on the electrode surface and the system
used. For "mechanism-
1" embodiments, preferred embodiments utilize two monolayer forming species: a
monolayer forming
species (including insulators or conductive oligomers) and a conductive
oligomer species comprising
the capture binding ligand, although as will be appreciated by those in the
art, additional monolayer
forming species can be included as well. For "mechanism-2" systems, the
composition of the SAM
depends on the detection electrode surface. In general, two basic "mechanism-
2" systems are
described; detection electrodes comprising "smooth" surfaces, such as gold
ball electrodes, and those
comprising "rough" surfaces, such as those that are made using commercial
processes on PC circuit
boards. In general, without being bound by theory, it appears that monolayers
made on imperfect
surfaces, i.e. "rough" surfaces, spontaneously form monolayers containing
enough electroconduits
even in the absence of EFS, probably due to the fact that the formation of a
uniform monolayer on a
rough surface is difficult. "Smoother" surfaces, however, may require the
inclusion of sufficient
numbers of EFS to generate the electroconduits, as the uniform surfaces allow
a more uniform
monolayer to form. Again, without being bound by theory, the inclusion of
species that disturb the
uniformity of the monolayer, for example by including a rigid molecule in a
background of more flexible
ones, causes electroconduits. Thus "smooth" surfaces comprise monolayers
comprising three
components: an insulator species, a EFS, and a species comprising the capture
ligand, although in
some circumstances, far example when the capture ligand species is included at
high density, the
capture ligand species can serve as the EFS. "Smoothness" in this context is
not measured physically
-38-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
but rather as a function of an increase in the measured signal when EFS are
included. That is, the
signal from a detection electrode coated with monolayer forming species is
compared to a signal from
a detection electrode coated with monolayer forming species including a EFS.
An increase indicates
that the surface is relatively smooth, since the inclusion of a EFS served to
facilitate the access of the
ETM to the electrode. It should also be noted that while the discussion herein
is mainly directed to
gold electrodes and thiol-containing monolayer forming species, other types of
electrodes and
monolayer-forming species can be used.
It should be noted that the "electroconduits" of mechanism-2 systems do not
result in direct contact of
sample components with the electrode surface; that is, the electroconduits are
not large pores or holes
that allow physical access to the electrode. Rather, without being bound by
theory, it appears that the
electroconduits allow certain types of ETMs, particularly hydrophobic ETMs, to
penetrate sufficiently
into the monolayer to allow detection. However, other types of redox active
species, including some
hydrophilic species, do not penentrate into the monolayer, even with
electroconduits present. Thus, in
general, redox active species that may be present in the sample do not give
substantial signals as a
result of the electroconduits. While the exact system will vary with the
composition of the SAM and the
choice of the ETM, in general, the test for a suitable SAM to reduce non-
specific binding that also has
sufficient electroconduits for ETM detection is to add either ferrocene or
ferrocyanide to the SAM; the
former should give a signal and the latter should not.
Accordingly, in mechanism-1 systems, the monolayer comprises a first species
comprising a
conductive oligomer comprising the capture binding ligand, as is more fully
outlined below, and a
second species comprising a monolayer forming species, including either or
both insulators or
conductive oligomers.
In a preferred embodiment, the monolayer comprises electroconduit-forming
species. By
"electroconduit-forming species" or "EFS" herein is meant a molecule that is
capable of generating
su~cient electroconduits in a monolayer, generally of insulators such as alkyl
groups, to allow
detection of ETMs at the surface. In general, EFSs have one or more of the
following qualities: they
may be relatively rigid molecules, for example as compared to an alkyl chain;
they may attach to the
electrode surface with a geometry different from the other monolayer forming
species (for example,
alkyl chains attached to gold surfaces with thiol groups are thought to attach
at roughly 45° angles,
and phenyl-acetylene chains attached to gold via thiols are thought to go down
at 90° angles); they
may have a structure that sterically interferes or interrupts the formation of
a tightly packed monolayer,
for example through the inclusion of branching groups such as alkyl groups, or
the inclusion of highly
flexible species, such as polyethylene glycol units; or they may be capable of
being activated to form
electroconduits; for example, photoactivatible species that can be selectively
removed from the
-39-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
surface upon photoactivation, leaving electroconduits.
Preferred EFSs include conductive oligomers, as defined below, and phenyl-
acetylene-polyethylene
glycol species, as well as asymmetrical SAM-forming disulfide species such as
described in U.S.S.N.
09/847,113, filed May 1, 2001, hereby expressly incorporated by reference.
However, in some
embodiments, the EFS is not a conductive oligomer.
In a preferred embodiment, the monolayer comprises conductive oligomers. By
"conductive oligomer"
herein is meant a substantially conducting oligomer, preferably linear, some
embodiments of which are
referred to in the literature as "molecular wires". By "substantially
conducting" herein is meant that the
oligomer is capable of transferring electrons at 100 Hz. Generally, the
conductive oligomer has
substantially overlapping rr-orbitals, i.e. conjugated rr-orbitals, as between
the monomeric units of the
conductive oligomer, although the conductive oligomer may also contain one or
more sigma (a) bonds.
Additionally, a conductive oligomer may be defined functionally by its ability
to inject or receive
electrons into or from an associated ETM. Furthermore, the conductive oligomer
is more conductive
than the insulators as defined herein. Additionally, the conductive oligomers
of the invention are to be
distinguished from electroactive polymers, that themselves may donate or
accept electrons.
In a preferred embodiment, the conductive oligomers have a conductivity, S, of
from between about
10~ to about 104 S2-'cm-', with from about 10-5 to about 103 W'cm' being
preferred, with these S
values being calculated for molecules ranging from about 20A to about 200A. As
described below,
insulators have a conductivity S of about 10-' W'cm-' or lower, with less than
about 10~ O-'cm' being
preferred. See generally Gardner et al., Sensors and Actuators A 51 (1995) 57-
66, incorporated
herein by reference.
Desired characteristics of a conductive oligomer include high conductivity,
sufficient solubility in
organic solvents and/or water for synthesis and use of the compositions of the
invention, and
preferably chemical resistance to reactions that occur i) during nucleic acid
synthesis (such that
nucleosides containing the conductive oligomers may be added to a nucleic acid
synthesizer during
the synthesis of the compositions of the invention), ii) during the attachment
of the conductive oligomer
to an electrode, or iii) during hybridization assays. In addition, conductive
oligomers that will promote
the formation of self-assembled monolayers are preferred.
The oligomers of the invention comprise at least two monomeric subunits, as
described herein. As is
described more fully below, oligomers include homo- and hetero-oligomers, and
include polymers.
In a preferred embodiment, the conductive oligomer has the structure depicted
in Structure 1:
-40-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
Structure 1
--/---Y-1-f B ~ D Y
a
n m
As will be understood by those in the art, all of the structures depicted
herein may have additional
atoms or structures; e.g. the conductive oligomer of Structure 1 may be
attached to ETMs, such as
electrodes, transition metal complexes, organic ETMs, and metallocenes, and to
nucleic acids, or to
several of these. Unless otherwise noted, the conductive oligomers depicted
herein will be attached at
the left side to an electrode; that is, as depicted in Structure 1, the left
"Y" is connected to the electrode
as described herein. If the conductive oligomer is to be attached to a nucleic
acid, the right "Y", if
present, is attached to the nucleic acid, either directly or through the use
of a Linker, as is described
herein.
In this embodiment, Y is an aromatic group, n is an integer from 1 to 50, g is
either 1 or zero, a is an
integer from zero to 10, and m is zero or 1. When g is 1, B-D is a bond able
to conjugate with
neighboring bonds (herein referred to as a "conjugated bond"), preferably
selected from acetylene, B-
D is a conjugated bond, preferably selected from acetylene, alkene,
substituted alkene, amide, azo, -
C=N- (including -N=C-, -CR=N- and -N=CR-), -Si=Si-, and -Si=C- (including -
C=Si-, -Si=CR- and -
CR=Si-). When g is zero, a is preferably 1, D is preferably carbonyl, or a
heteroatom moiety, wherein
the heteroatom is selected from oxygen, sulfur, nitrogen, silicon or
phosphorus. Thus, suitable
heteroatom moieties include, but are not limited to, -NH and -NR, wherein R is
as defined herein;
substituted sulfur; sulfonyl (-SOZ-) sulfoxide (-SO-); phosphine oxide (-PO-
and -RPO-); and
thiophosphine (-PS- and -RPS-). However, when the conductive oligomer is to be
attached to a gold
electrode, as outlined below, sulfur derivatives are not preferred.
By "aromatic group" or grammatical equivalents herein is meant an aromatic
monocyclic or polycyclic
hydrocarbon moiety generally containing 5 to 14 carbon atoms (although larger
polycyclic rings
structures may be made) and any carbocylic ketone or thioketone derivative
thereof, wherein the
carbon atom with the free valence is a member of an aromatic ring. Aromatic
groups include arylene
groups and aromatic groups with more than two atoms removed. For the purposes
of this application
aromatic includes heterocycle. "Heterocycle" or "heteroaryl" means an aromatic
group wherein 1 to 5
of the indicated carbon atoms are replaced by a heteroatom chosen from
nitrogen, oxygen, sulfur,
phosphorus, boron and silicon wherein the atom with the free valence is a
member of an aromatic ring,
and any heterocyclic ketone and thioketone derivative thereof. Thus,
heterocycle includes thienyl,
furyl, pyrrolyl, pyrimidinyl, oxalyl, indolyl, purinyl, quinolyl, isoquinolyl,
thiazolyl, imidozyl, etc.
Importantly, the Y aromatic groups of the conductive oligomer may be
different, i.e. the conductive
-41 -
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
oligomer may be a heterooligomer. That is, a conductive oligomer may comprise
a oligomer of a
single type of Y groups, or of multiple types of Y groups.
The aromatic group may be substituted with a substitution group, generally
depicted herein as R. R
groups may be added as necessary to affect the packing of the conductive
oligomers, i.e. R groups
may be used to alter the association of the oligomers in the monolayer. R
groups may also be added
to 1 ) alter the solubility of the oligomer or of compositions containing the
oligomers; 2) alter the
conjugation or electrochemical potential of the system; and 3) alter the
charge or characteristics at the
surtace of the monolayer.
In a preferred embodiment, when the conductive oligomer is greater than three
subunits, R groups are
preferred to increase solubility when solution synthesis is done. However, the
R groups, and their
positions, are chosen to minimally effect the packing of the conductive
ofigomers on a surface,
particularly within a monolayer, as described below. In general, only small R
groups are used within
the monolayer, with larger R groups generally above the surFace of the
monolayer. Thus for example
the attachment of methyl groups to the portion of the conductive oligomer
within the monolayer to
increase solubility is preferred, with attachment of longer alkoxy groups, for
example, C3 to C10, is
preferably done above the monolayer surface. In general, for the systems
described herein, this
generally means that attachment of sterically significant R groups is not done
on any of the first two or
three oligomer subunits, depending on the average length of the molecules
making up the monolayer.
Suitable R groups include, but are not limited to, hydrogen, alkyl, alcohol,
aromatic, amino, amido,
nitro, ethers, esters, aldehydes, sulfonyl, silicon moieties, halogens, sulfur
containing moieties,
phosphorus containing moieties, and ethylene glycols. In the structures
depicted herein, R is
hydrogen when the position is unsubstituted. It should be noted that some
positions may allow two
substitution groups, R and R', in which case the R and R' groups may be either
the same or different.
By "alkyl group" or grammatical equivalents herein is meant a straight or
branched chain alkyl group,
with straight chain alkyl groups being preferred. If branched, it may be
branched at one or more
positions, and unless specified, at any position. The alkyl group may range
from about 1 to about 30
carbon atoms (C1 -C30), with a preferred embodiment utilizing from about 1 to
about 20 carbon atoms
(C1 -C20), with about C1 through about C12 to about C15 being preferred, and
C1 to C5 being
particularly preferred, although in some embodiments the alkyl group may be
much larger. Also
included within the definition of an alkyl group are cycloalkyl groups such as
C5 and C6 rings, and
heterocyclic rings with nitrogen, oxygen, sulfur or phosphorus. Alkyl also
includes heteroalkyl, with
heteroatoms of sulfur, oxygen, nitrogen, and silicone being preferred. Alkyl
includes substituted alkyl
groups. By "substituted alkyl group" herein is meant an alkyl group further
comprising one or more
-42-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
substitution moieties "R", as defined above.
By "amino groups" or grammatical equivalents herein is meant -NH2, -NHR and -
NRZ groups, with R
being as defined herein.
By "nitro group" herein is meant an -NOZ group.
By "sulfur containing moieties" herein is meant compounds containing sulfur
atoms, including but not
limited to, this-, thio- and sulfo- compounds, thiols (-SH and -SR), and
sulfides (-RSR-). By
"phosphorus containing moieties" herein is meant compounds containing
phosphorus, including, but
not limited to, phosphines and phosphates. By "silicon containing moieties"
herein is meant
compounds containing silicon.
By "ether" herein is meant an -O-R group. Preferred ethers include alkoxy
groups, with -O-(CHZ)ZCH3
and -O-(CHz)4CH3 being preferred.
By "ester" herein is meant a -COOR group.
By "halogen" herein is meant bromine, iodine, chlorine, or fluorine. Preferred
substituted alkyls are
partially or fully halogenated alkyls such as CF3, etc.
By "aldehyde" herein is meant -RCHO groups.
By "alcohol" herein is meant -OH groups, and alkyl alcohols -ROH.
By "amido" herein is meant -RCONH- or RCONR- groups.
By "ethylene glycol" or "(poly)ethylene glycol" herein is meant a -(O-CH2-
CHZ)n group, although each
carbon atom of the ethylene group may also be singly or doubly substituted,
e.g. -(O-CRZ-CRZ)~ , with
R as described above. Ethylene glycol derivatives with other heteroatoms in
place of oxygen (i.e. -(N-
CH~ CHZ)~ or -(S-CHa-CH~)~ , or with substitution groups) are also preferred.
Preferred substitution groups include, but are not limited to, methyl, ethyl,
propyl, alkoxy groups such
as -O-(CHZ)2CH3 and -O-(CHz)4CH3 and ethylene glycol and derivatives thereof.
Preferred aromatic groups include, but are not limited to, phenyl, naphthyl,
naphthalene, anthracene,
phenanthroline, pyrole, pyridine, thiophene, porphyrins, and substituted
derivatives of each of these,
-43-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
included fused ring derivatives.
In the conductive oligomers depicted herein, when g is 1, B-D is a bond
linking two atoms or chemical
moieties. In a preferred embodiment, B-D,is a conjugated bond, containing
overlapping or conjugated
rr-orbitals.
Preferred B-D bonds are selected from acetylene (-C=C-, also called alkyne or
ethyne), alkene (-
CH=CH-, also called ethylene), substituted alkene (-CR=CR-, -CH=CR- and -CR=CH-
), amide (-NH-
CO- and -NR-CO- or -CO-NH- and -CO-NR-), azo (-N=N-), esters and thioesters (-
CO-O-, -O-CO-, -
CS-O- and -O-CS-) and other conjugated bonds such as (-CH=N-, -CR=N-, -N=CH-
and -N=CR-), (-
SiH=SiH-, -SiR=SiH-, -SiR=SiH-, and -SiR=SiR-), (-SiH=CH-, -SiR=CH-, -SiH=CR-,
-SiR=CR-, -
CH=SiH-, -CR=SiH-, -CH=SiR-, and -CR=SiR-). Particularly preferred B-D bonds
are acetylene,
alkene, amide, and substituted derivatives of these three, and azo. Especially
preferred B-D bonds
are acetylene, alkene and amide. The oligomer components attached to double
bonds may be in the
trans or cis conformation, or mixtures. Thus, either B or D may include
carbon, nitrogen or silicon.
The substitution groups are as defined as above for R.
When g=0 in the Structure 1 conductive oligomer, a is preferably 1 and the D
moiety may be carbonyl
or a heteroatom moiety as defined above.
As above for the Y rings, within any single conductive oligomer, the B-D bonds
(or D moieties, when
g=0) may be all the same, or at least one may be different. For example, when
m is zero, the terminal
B-D bond may be an amide bond, and the rest of the B-D bonds may be acetylene
bonds. Generally,
when amide bonds are present, as few amide bonds as possible are preferable,
but in some
embodiments all the B-D bonds are amide bonds. Thus, as outlined above for the
Y rings, one type of
B-D bond may be present in the conductive oligoiner within a monolayer as
described below, and
another type above the monolayer level, for example to give greater
flexibility for nucleic acid
hybridization when the nucleic acid is attached via a conductive oligomer.
In the structures depicted herein, n is an integer from 1 to 50, although
longer oligomers may also be
used (see for example Schumm et al., Angew. Chem. Int. Ed. Engl. 1994
33(13):1360). Without
being bound by theory, it appears that for efficient hybridization of nucleic
acids on a surtace, the
hybridization should occur at a distance from the surface, i.e. the kinetics
of hybridization increase as
a function of the distance from the surface, particularly for long
oligonucleotides of 200 to 300
basepairs. Accordingly, when a nucleic acid is attached via a conductive
oligomer, as is more fully
described below, the length of the conductive oligomer is such that the
closest nucleotide of the
nucleic acid is positioned from about 6A to about 100A (although distances of
up to 500A may be
-44-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
used) from the electrode surface, with from about 15A to about.60A being
preferred and from about
25A to about 60A also being preferred. Accordingly, n will depend on the size
of the aromatic group,
but generally will be from about 1 to about 20, with from about 2 to about 15
being preferred and from
about 3 to about 10 being especially preferred.
In the structures depicted herein, m is either 0 or 1. That is, when m is 0,
the conductive oligomer may
terminate in the B-D bond or D moiety, i.e. the D atom is attached to the
nucleic acid either directly or
via a linker. In some embodiments, for example when the conductive oligomer is
attached to a
phosphate of the ribose-phosphate backbone of a nucleic acid, there may be
additional atoms, such
as a linker, attached between the conductive oligomer and the nucleic acid.
Additionally, as outlined
below, the D atom may be the nitrogen atom of the amino-modified ribose.
Alternatively, when m is 1,
the conductive oligomer may terminate in Y, an aromatic group, i.e. the
aromatic group is attached to
the nucleic acid or linker.
As will be appreciated by those in the art, a large number of possible
conductive oligomers may be
utilized. These include conductive oligomers falling within the Structure 1
and Structure 8 formulas, as
well as other conductive oligomers, as are generally known in the art,
including for example,
compounds comprising fused aromatic rings or Teflon-like oligomers, such as -
(CFZ)~ , -(CHF)" and
-(CFR)~ . See for example, Schumm et al., Angew. Chem. Intl. Ed. Engl. 33:1361
(1994); Grosshenny
et al., Platinum Metals Rev. 40(1 ):26-35 (1996); Tour, Chem. Rev. 96:537-553
(1996); Hsung et al.,
Organometallics 14:4808-4815 (1995); and references cited therein, all of
which are expressly
incorporated by reference.
Particularly preferred conductive oligomers of this embodiment are depicted
below:
Structure 2
-f-Y-/-B-D Y
a
n m
Structure 2 is Structure 1 when g is 1. Preferred embodiments of Structure 2
include: a is zero, Y is
pyrrole or substituted pyrrole; a is zero, Y is thiophene or substituted
thiophene; a is zero, Y is furan or
substituted furan; a is zero, Y is phenyl or substituted phenyl; a is zero, Y
is pyridine or substituted
pyridine; a is 1, B-D is acetylene and Y is phenyl or substituted phenyl (see
Structure 4 below). A
preferred embodiment of Structure 2 is also when a is one, depicted as
Structure 3 below:
Structure 3
~y-g-p Y
n m
-45-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
Preferred embodiments of Structure 3 are: Y is phenyl or substituted phenyl
and B-D is azo; Y is
phenyl or substituted phenyl and B-D is acetylene; Y is phenyl or substituted
phenyl and B-D is alkene;
Y is pyridine or substituted pyridine and B-D is acetylene; Y is thiophene or
substituted thiophene and
B-D is acetylene; Y is furan or substituted furan and B-D is acetylene; Y is
thiophene or furan (or
substituted thiophene or furan) and B-D are alternating alkene and acetylene
bonds.
Most of the structures depicted herein utilize a Structure 3 conductive
oligomer. However, any
Structure 3 oligomers may be substituted with any of the other structures
depicted herein, i.e.
Structure 1 or 8 oligomer, or other conducting oligomer, and the use of such
Structure 3 depiction is
not meant to limit the scope of the invention.
Particularly preferred embodiments of Structure 3 include Structures 4, 5, 6
and 7, depicted below:
Structure 4
R R R R
p R h R R m
Particularly preferred embodiments of Structure 4 include: n is two, m is one,
and R is hydrogen; n is
three, m is zero, and R is hydrogen; and the use of R groups to increase
solubility.
Structure 5
R R R R
O
H
H
R R ~ R R m
When the B-D bond is an amide bond, as in Structure 5, the conductive
oligomers are pseudopeptide
oligomers. Although the amide bond in Structure 5 is depicted with the
carbonyl to the left, i.e. -
CONH-, the reverse may also be used, i.e. -NHCO-. Particularly preferred
embodiments of Structure
include: n is two, m is one, and R is hydrogen; n is three, m is zero, and R
is hydrogen (in this
embodiment, the terminal nitrogen (the D atom) may be the nitrogen of the
amino-modified ribose);
and the use of R groups to increase solubility.
Structure 6
R R R R R ~ R
O
N
R~R R ~ R~R n \ R R
Preferred embodiments of Structure 6 include the first n is two, second n is
one, m is zero, and all R
groups are hydrogen, or the use of R groups to increase solubility.
Structure 7
-46-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
R R R R
- S V
R~R / ~ n
Preferred embodiments of Structure 7 include: the first n is three, the second
n is from 1-3, with m
being either 0 or 1, and the use of R groups to increase solubility.
In a preferred embodiment, the conductive oligomer has the structure depicted
in Structure 8:
Structure 8
~C-G-C J
n m
In this embodiment, C are carbon atoms, n is an integer from 1 to 50, m is 0
or 1, J is a heteroatom
selected from the group consisting of oxygen, nitrogen, silicon, phosphorus,
sulfur, carbonyl or
sulfoxide, and G is a bond selected from alkane, alkene or acetylene, such
that together with the two
carbon atoms the C-G-C group is an alkene (-CH=CH-), substituted alkene (-
CR=CR-) or mixtures
thereof (-CH=CR- or -CR=CH-), acetylene (-C=C-), or alkane (-CRS CRZ , with R
being either
hydrogen or a substitution group as described herein). The G bond of each
subunit may be the same
or different than the G bonds of other subunits; that is, alternating
oligomers of alkene and acetylene
bonds could be used, etc. However, when G is an alkane bond, the number of
alkane bonds in the
oligomer should be kept to a minimum, with about six or less sigma bonds per
conductive oligomer
being preferred. Alkene bonds are preferred, and are generally depicted
herein, although alkane and
acetylene bonds may be substituted in any structure or embodiment described
herein as will be
appreciated by those in the art.
In some embodiments, for example when ETMs are not present, if m=0 then at
least one of the G
bonds is not an alkane bond.
In a preferred embodiment, the m of Structure 8 is zero. In a particularly
preferred
embodiment, m is zero and G is an alkene bond, as is depicted in Structure 9:
Structure 9
R
Y-t-
n ~m
R
The alkene oligomer of structure 9, and others depicted herein, are generally
depicted in the preferred
-47-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
trans configuration, although oligomers of cis or mixtures of trans and cis
may also be used. As
above, R groups may be added to alter the packing of the compositions on an
electrode, the
hydrophilicity or hydrophobicity of the oligomer, and the flexibility, i.e.
the rotational, torsional or
longitudinal flexibility of the oligomer. n is as defined above.
In a preferred embodiment, R is hydrogen, although R may be also alkyl groups
and polyethylene
glycols or derivatives.
In an alternative embodiment, the conductive oligomer may be a mixture of
different types of
oligomers, for example of structures 1 and 8.
In addition, in some embodiments, the terminus of at least some of the
conductive oligomers in the
monolayer are electronically exposed. By "electronically exposed" herein is
meant that upon the
placement of an ETM in close proximity to the terminus, and after initiation
with the appropriate signal,
a signal dependent on the presence of the ETM may be detected. The conductive
oligomers may or
may not have terminal groups. Thus, in a preferred embodiment, there is no
additional terminal group,
and the conductive oligomer terminates with one of the groups depicted in
Structures 1 to 9; for
example, a B-D bond such as an acetylene bond. Alternatively, in a preferred
embodiment, a terminal
group is added, sometimes depicted herein as "Q". A terminal group may be used
for several
reasons; for example, to contribute to the electronic availability of the
conductive oligomer for detection
of ETMs, or to alter the surface of the SAM for other reasons, for example to
prevent non-specific
binding. For example, there may be negatively charged groups on the terminus
to form a negatively
charged surface such that when the nucleic acid is DNA or RNA the nucleic acid
is repelled or
prevented from lying down on the surtace, to facilitate hybridization.
Preferred terminal groups include
-NHS, -OH, -COOH, and alkyl groups such as -CH3, and (poly)alkyloxides such as
(poly)ethylene
glycol, with -OCH~CHzOH, -(OCH2CH~0)~H, -(OCHZCH~O)3H, and -(OCHZCH20)~H being
preferred.
In one embodiment, it is possible to use mixtures of conductive oligomers with
different types of
terminal groups. Thus, for example, some of the terminal groups may facilitate
detection, and some
may prevent non-specific binding.
It will be appreciated that the monolayer may comprise different conductive
oligomer species, although
preferably the different species are chosen such that a reasonably uniform SAM
can be formed. Thus,
for example, when nucleic acids are covalently attached to the electrode using
conductive oligomers, it
is possible to have one type of conductive oligomer used to attach the nucleic
acid, and another type
functioning to detect the ETM. Similarly, it may be desirable to have mixtures
of different lengths of
conductive oligomers in the monolayer, to help reduce non-specific signals.
Thus, for example,
-48-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
preferred embodiments utilize conductive oligomers that terminate below the
surface of the rest of the
monolayer, i.e. below the insulator layer, if used, or below some fraction of
the other conductive
oligomers. Similarly, the use of different conductive oligomers may be done to
facilitate monolayer
formation, or to make monolayers with altered properties.
In a preferred embodiment, the monolayer forming species are "interrupted"
conductive oligomers,
containing an alkyl portion in the middle of the conductive oligomer.
In a preferred embodiment, the monolayer comprises photoactivatable species as
EFSs. This general
scheme is depicted in Figure 11 of 09!626,096, incorporated by reference.
Photoactivatable species
are known in the art, and include 4,5-dimethoxy-2-nitrobenzyl ester, which can
be photolyzed at 365
nm for 2 hours.
In a preferred embodiment, the monolayer may further comprise insulator
moieties. By "insulator"
herein is meant a substantially nonconducting oligomer, preferably linear. By
"substantially
nonconducting" herein is meant that the insulator wiff not transfer electrons
at 100 Hz. The rate of
electron transfer through the insulator is preferably slower than the rate
through the conductive
oligomers described herein.
In a preferred embodiment, the insulators have a conductivity, S, of about
10'' S2-'cm' or lower, with
less than about 10-8 SZ-'cm-' being preferred. See generally Gardner et al.,
supra.
Generally, insulators are alkyl or heteroalkyl oligomers or moieties with
sigma bonds, although any
particular insulator molecule may contain aromatic groups or one or more
conjugated bonds. By
"heteroalkyl" herein is meant an alkyl group that has at least one heteroatom,
i.e. nitrogen, oxygen,
sulfur, phosphorus, silicon or boron included in the chain. Alternatively, the
insulator may be quite
similar to a conductive oligomer with the addition of one or more heteroatoms
or bonds that serve to
inhibit or slow, preferably substantially, electron transfer.
Suitable insulators are known in the art, and include, but are not limited to,
-(CH~)~ , -(CRH)~ , and -
(CRZ)~ , ethylene glycol or derivatives using other heteroatoms in place of
oxygen, i.e. nitrogen or
sulfur (sulfur derivatives are not preferred when the electrode is gold).
As for the conductive oligomers, the insulators may be substituted with R
groups as defined herein to
alter the packing of the moieties or conductive oligomers on an electrode, the
hydrophilicity or
hydrophobicity of the insulator, and the flexibility, i.e. the rotational,
torsional or longitudinal flexibility of
the insulator. For example, branched alkyl groups may be used. Similarly, the
insulators may contain
_ q.9 _
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
terminal groups, as outlined above, particularly to influence the surface of
the monolayer.
In a preferred embodiment, the insulator species included in the SAM utilizes
novel methods and
compositions comprising asymmetric disulfides. As outlined herein, the signals
generated from label
probes can be dependent on the behavior or properties of the SAM. SAMs
comprising "nanoconduits"
or "electroconduits", as outlined in U.S.S.N. 60/145,912 hereby expressly
incorporated herein by
reference in its entirety, give good signals. Thus, the present invention
provides asymmetric insulators
based on disulfides, wherein one of the arms being a longer alkyl chain (or
other SAM forming
species) and the other arm comprising either a shorter alkyl chain or a bulky
group, such as a
branched alkyl group, that can be polar or nonpolar) for creating the
nanoconduits. Exemplary
species and methods of making are described in U.S.S.N. 09/847,113. See also
Mukaiyama
Tetrahedron Lett. 1968, 5907; Boustany Tetrahedron Lett. 1970 3547; Harpp
Tetrahedron Lett. 1970
3551; and Oae, J. Chem. Soc. Chem. Commun, 1977, 407, all of which are
expressly incorporated
herein by reference.
The length of the species making up the monolayer will vary as needed. As
outlined above, it appears
that hybridization is more efficient at a distance from the surface. The
species to which nucleic acids
are attached (as outlined below, these can be either insulators or conductive
oligomers) may be
basically the same length as the monolayer forming species or longer than
them, resulting in the
nucleic acids being more accessible to the solvent for hybridization. In some
embodiments, the
conductive oligomers to which the nucleic acids are attached may be shorter
than the monolayer.
As will be appreciated by those in the art, the actual combinations and ratios
of the different species
making up the monolayer can vary widely, and will depend on whether mechanism-
1 or -2 is used.
Generally, either two or three component systems are preferred for mechanism-2
systems. Three
component systems utilize a first species comprising a capture probe
containing species, attached to
the electrode via either an insulator or a conductive oligomer. The second
species are conductive
oligomers, and the third species are insulators. In this embodiment, the first
species can comprise
from about 90% to about 1 %, with from about 20% to about 40% being preferred.
For nucleic acids,
from about 30% to about 40% is especially preferred for short oligonucleotide
targets and from about
10% to about 20% is preferred for longer targets. The second species can
comprise from about 1 % to
about 90%, with from about 20% to about 90% being preferred, and from about
40% to about 60%
being especially preferred. The third species can comprise from about 1 % to
about 90%, with from
about 20% to about 40% being preferred, and from about 15% to about 30% being
especially
preferred. To achieve these approximate proportions, preferred ratios of
firstaecondahird species in
SAM formation solvents are 2:2:1 for short targets, 1:3:1 for longer targets,
with total thiol
concentration (when used to attach these species, as is more fully outlined
below) in the 500 uM to 1
-50-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
mM range, and 833 uM being preferred.
Alternatively, two component systems can be used. In one embodiment, for use
in either mechanism-
1 or mechanism-2 systems, the two components are the first and second species.
In this embodiment,
the first species can comprise from about 1 % to about 90%, with from about 1
% to about 40% being
preferred, and from about 10% to about 40% being especially preferred. The
second species can
comprise from about 1 % to about 90%, with from about 10% to about 60% being
preferred, and from
about 20% to about 40% being especially preferred. Alternatively, for
mechanism-1 or mechanism-2
systems, the two components are the first and the third species. In this
embodiment, the first species
can comprise from about 1 % to about 90%, with from about 1 % to about 40%
being preferred, and
from about 10% to about 40% being especially preferred. The second species can
comprise from
about 1 % to about 90%, with from about 10% to about 60% being preferred, and
from about 20% to
about 40% being especially preferred.
In a preferred embodiment, the deposition of the SAM is done using aqueous
solvents. As is generally
described in Steel et al., Anal. Chem. 70:4670 (1998), Herne et al., J. Am.
Chem. Soc. 119:8916
(1997), and Finklea, Electrochemistry of Organized Monolayers of Thiols and
Related Molecules on~
Electrodes, from A.J. Bard, Electroanalytical Chemistry: A Series of Advances,
Vol. 20, Dekker N.Y.
1966-, all of which are expressly incorporated by reference, the deposition of
the SAM-forming species
can be done out of aqueous solutions, frequently comprising salt.
The covalent attachment of the conductive oligomers and insulators may be
accomplished in a variety
of ways, depending on the electrode and the composition of the insulators and
conductive oligomers
used. In a preferred embodiment, the attachment tinkers with covalently
attached nucleosides or
nucleic acids as depicted herein are covalently attached to an electrode.
Thus, one end or terminus of
the attachment linker is attached to the nucleoside or nucleic acid, and the
other is attached to an
electrode. In some embodiments it may be desirable to have the attachment
Pinker attached at a
position other than a terminus, or even to have a branched attachment linker
that is attached to an
electrode at one terminus and to two or more nucleosides at other termini,
although this is not
preferred. Similarly, the attachment linker may be attached at two sites to
the electrode, as is generally
depicted in Structures 11-13. Generally, some type of linker is used, as
depicted below as "A" in
Structure 10, where "X" is the conductive oligomer, "I" is an insulator and
the hatched surface is the
electrode:
Structure 10
A -X
A I
I
-51-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
In this embodiment, A is a linker or atom. The choice of "A" will depend in
part on the characteristics
of the electrode. Thus, for example, A may be a sulfur moiety when a gold
electrode is used.
Alternatively, vnhen metal oxide electrodes are used, A may be a silicon
(silane) moiety attached to the
oxygen of the oxide (see for example Chen et al., Langmuir 10:3332-3337
(1994); Lenhard et al., J.
Electroanal. Chem. 78:195-201 (1977), both of which are expressly incorporated
by reference). When
carbon based electrodes are used, A may be an amino moiety (preferably a
primary amine; see for
example Deinhammer et al., Langmuir 10:1306-1313 (1994)). Thus, preferred A
moieties include, but
are not limited to, silane moieties, sulfur moieties (including alkyl sulfur
moieties), and amino moieties.
In a preferred embodiment, epoxide type linkages with redox polymers such as
are known in the art
are not used.
Although depicted herein as a single moiety, the insulators and conductive
oligomers may be attached
to the electrode with more than one''A" moiety; the "A" moieties may be the
same or different. Thus,
for example, when the electrode is a gold electrode, and "A" is a sulfur atom
or moiety, multiple sulfur
atoms may be used to attach the conductive oligomer to the electrode, such as
is generally depicted
below in Structures 11, 12 and 13. As will be appreciated by those in the art,
other such structures
can be made. In Structures 11, 12 and 13, the A moiety is just a sulfur atom,
but substituted sulfur
moieties may also be used.
Structure 11
s
--s ~ X or ~
Structure 12
S R
-$ x or I
Structure 13
S R
g~Xorl
-52-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
It should also be noted that similar to Structure 13, it may be possible to
have a a conductive oligomer
terminating in a single carbon atom with three sulfur moities attached to the
electrode. Additionally,
although not always depicted herein, the conductive oligomers and insulators
may also comprise a "Q"
terminal group.
In a preferred embodiment, the electrode is a gold electrode, and attachment
is via a sulfur linkage as
is well known in the art, i.e. the A moiety is a sulfur atom or moiety.
Although the exact characteristics
of the gold-sulfur attachment are not known, this linkage is considered
covalent for the purposes of
this invention. A representative structure is depicted in Structure 14, using
the Structure 3 conductive
oligomer, although as for all the structures depicted herein, any of the
conductive oligomers, or
combinations of conductive oligomers, may be used. Similarly, any of the
conductive oligomers or
insulators may also comprise terminal groups as described herein. Structure 14
depicts the "A" linker
as comprising just a sulfur atom, although additional atoms may be present
(i.e. linkers from the sulfur
to the conductive oligomer or substitution groups). In addition, Structure 14
shows the sulfur atom
attached to the Y aromatic group, but as will be appreciated by those in the
art, it may be attached to
the B-D group (i.e. an acetylene) as well.
Structure 14
S~Y-'B-D~Y
n m
In a preferred embodiment, the electrode is a carbon electrode, i.e. a glassy
carbon electrode, and
attachment is via a nitrogen of an amine group. A representative structure is
depicted in Structure 15.
Again, additional atoms may be present, i.e. Z type linkers andlor terminal
groups.
Structure 15
H-y-Y B D Y
n m
Structure 16
O-Si-r-Y-B-D-h-Y-1-
~n ~ ~m
-53-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
In Structure 16, the oxygen atom is from the oxide of the metal oxide
electrode. The Si atom may be
combined with other atoms, i.e. be a silicon moiety containing substitution
groups. Other attachments
for SAMs to other electrodes are known in the art; see for example Napier et
al., Langmuir, 1997, for
attachment to indium tin oxide electrodes, and also the chemisorption of
phosphates to an indium tin
oxide electrode (talk by H. Holden Thorpe, CHI conference, May 4-5, 1998).
The SAMs of the invention can be made in a variety of ways, including
deposition out of organic
solutions and deposition out of aqueous solutions. The methods outlined herein
use a gold electrode
as the example, although as will be appreciated by those in the art, other
metals and methods may be
used as well. In one preferred embodiment, indium-tin-oxide (1T0) is used as
the electrode.
In a preferred embodiment, a gold surface is first cleaned. A variety of
cleaning procedures may be
employed, including, but not limited to, chemical cleaning or etchants
including Piranha solution
(hydrogen peroxide/sulfuric acid) or aqua regia (hydrochloric acid/nitric
acid), electrochemical
methods, flame treatment, plasma treatment or combinations thereof.
Following cleaning, the gold substrate is exposed to the SAM species. When the
electrode is ITO, the
SAM species are phosphonate-containing species. This can also be done in a
variety of ways,
including, but not limited to, solution deposition, gas phase deposition,
microcontact printing, spray
deposition, deposition using neat components, etc. A preferred embodiment
utilizes a deposition
solution comprising a mixture of various SAM species in solution, generally
thiol-containing species.
Mixed monolayers that contain nucleic acids are usually prepared using a two
step procedure. The
thiolated nucleic acid is deposited during the first deposition step
(generally in the presence of at feast
one other monolayer-forming species) and the mixed monolayer formation is
completed during the
second step in which a second thiol solution minus nucleic acid is added.
Optionally, a second step
utilizing mild heating to promote monolayer reorganization.
In a preferred embodiment, the deposition solution is an organic deposition
solution. In this
embodiment, a clean gold surface is placed into a clean vial. A binding ligand
deposition solution in
organic solvent is prepared in which the total thiol concentration is, between
micromolar to saturation;
preferred ranges include from about 1 pM to 10 mM, with from about 400 uM to
about 1.0 mM being
especially preferred. In a preferred embodiment, the deposition solution
contains thiol modified DNA
(i.e. nucleic acid attached to an attachment linker) and thiol diluent
molecules (either conductive
oligomers or insulators, with the latter being preferred). The ratio of
nucleic acid to diluent (if present)
is usually between 1000:1 to 1:1000, with from about 10:1 to about 1:10 being
preferred and 1:1 being
especially preferred. The preferred solvents are tetrahydrofuran (THF),
acetonitrile, dimethylforamide
(DMF), ethanol, or mixtures thereof; generally any solvent of sufficient
polarity to dissolve the capture
-54-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
ligand can be used, as long as the solvent is devoid of functional groups that
will react with the
surface. Sufficient nucleic acid deposition solution is added to the vial so
as to completely cover the
electrode surface. The gold substrate is allowed to incubate at ambient
temperature or slightly above
ambient temperature for a period of time ranging from seconds to hours, with 5-
30 minutes being
preferred. After the initial incubation, the deposition solution is removed
and a solution of diluent
molecule only (from about 1 pM to 10 mM, with from about 100 uM to about 1.0
mM being preferred)
in organic solvent is added. The gold substrate is allowed to incubate at room
temperature or above
room temperature for a period of time (seconds to days, with from about 10
minutes to about 24 hours
being preferred). The gold sample is removed from the solution, rinsed in
clean solvent and used.
In a preferred embodiment, an aqueous deposition solution is used. As above, a
clean gold surface is
placed into a clean vial. A nucleic acid deposition solution in water is
prepared in which the total thiol
concentration is between about 1 uM and 10 mM, with from about 1 pM to about
200 uM being
preferred. The aqueous solution frequently has salt present (up to saturation,
with approximately 1 M
being preferred), however pure water can be used. The deposition solution
contains thiol modified
nucleic acid and often a thiol diluent molecule. The ratio of nucleic acid to
diluent is usually between
between 1000:1 to 1:1000, with from about 10:1 to about 1:10 being preferred
and 1:1 being especially
preferred. The nucleic acid deposition solution is added to the vial in such a
volume so as to
completely cover the electrode surface. The gold substrate is allowed to
incubate at ambient
temperature or slightly above ambient temperature for 1-30 minutes with 5
minutes usually being
sufficient. After the initial incubation, the deposition solution is removed
and a solution of diluent
molecule only (10 uM -1.0 mM) in either water or organic solvent is added. The
gold substrate is
allowed to incubate at room temperature or above room temperature until a
complete monolayer is
formed (10 minutes-24 hours). The gold sample is removed from the solution,
rinsed in clean solvent
and used.
In a preferred embodiment, the deposition solution comprises a zwitterionic
compound, preferably
betaine. Preferred embodiments utilize betaine and Tris-HCI buffers.
In a preferred embodiment, as outlined herein, a circuit board is used as the
substrate for the gold
electrodes. Formation of the SAMs on the gold surface is generally done by
first cleaning the boards,
for example in a 10% sulfuric acid solution for 30 seconds, detergent
solutions, aqua regia, plasma,
etc., as outlined herein. Following the sulfuric acid treatment, the boards
are washed, for example via
immersion in two Milli-Q water baths for 1 minute each. The boards are then
dried, for example under
a stream of nitrogen. Spotting of the deposition solution onto the boards is
done using any number of
known spotting systems, generally by placing the boards on an X-Y table,
preferably in a humidity
chamber. The size of the spotting drop will vary with the size of the
electrodes on the boards and the
-55-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
equipment used for delivery of the solution; for example, for 250 NM size
electrodes, a 30 nanoliter
drop is used. The volume should be sufficient to cover the electrode surface
completely. The drop is
incubated at room temperature for a period of time (sec to overnight, with 5
minutes preferred) and
then the drop is removed by rinsing in a Milli-Q water bath. The boards are
then optionally treated with
a second deposition solution, generally comprising insulator in organic
solvent, preferably acetonitrile,
by immersion in a 45°C bath. After 30 minutes, the boards are removed
and immersed in an
acetonitrile bath for 30 seconds followed by a milli-Q water bath for 30
seconds. The boards are dried
under a stream of nitrogen. Preferrably, only the water rinse is employed.
In a preferred embodiment, the detection electrode comprising the SAM (or the
sites on the array, for
non-electrode embodiments) further comprises capture binding ligands,
preferably covalently
attached. By "binding ligand" or "binding species" herein is meant a compound
that is used to probe
for the presence of the target analyte, that will bind to the target analyte.
In general, for most of the
embodiments described herein, there are at least two binding ligands used per
target analyte
molecule; a "capture" or "anchor" binding ligand (also referred to herein as a
"capture probe",
particularly in reference to a nucleic acid binding ligand) that is attached
to the detection electrode as
described herein, and a soluble binding ligand (frequently referred to herein
as a "signaling probe" or a
"label probe"), that binds independently to the target analyte, and either
directly or indirectly comprises
at least one ETM. However, it should be noted that for fluorescence-based
nucleic acid detection
systems, the target sequence is generally amplified, and during amplification,
a fluorescent label is
added; thus these systems generally comprise only two elements, the capture
probe and the labeled
target. Again, the discussion below is directed to the use of electrodes and
electrochemical detection,
but as will be appreciated by those in the art, fluorescent based systems can
be used as well.
Generally, the capture binding ligand allows the attachment of a target
analyte to the detection
electrode, for the purposes of detection. As is more fully outlined below,
attachment of the target
analyte to the capture binding ligand may be direct (i:e. the target analyte
binds to the capture binding
ligand) or indirect (one or more capture extender ligands may be used).
In a preferred embodiment, the binding is specific, and the binding ligand is
part of a binding pair. By
"specifically bind" herein is meant that the ligand binds the analyte, with
specificity sufficient to
differentiate between the analyte and other components or contaminants of the
test sample. However,
as will be appreciated by those in the art, it will be possible to detect
analytes using binding that is not
highly specific; for example, the systems may use different binding ligands,
for example an array of
different ligands, and detection of any particular analyte is via its
"signature" of binding to a panel of
binding figands, similar to the manner in which "electronic noses" work. The
binding should be
sufficient to allow the analyte to remain bound under the conditions of the
assay, including wash steps
-56-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
to remove non-specific binding. In some embodiments, for example in the
detection of certain
biomolecules, the binding constants of the analyte to the binding ligand will
be at least about 10~' to 10-
9 M-', with at least about 10-5 to 10'9 being preferred and at least about 10-
'to 10'9 M-' being particularly
preferred.
As will be appreciated by those in the art, the composition of the binding
ligand will depend on the
composition of the target analyte. Binding ligands to a wide variety of
analytes are known or can be
readily found using known techniques. For example, when the analyte is a
single-stranded nucleic
acid, the binding ligand is generally a substantially complementary nucleic
acid. Alternatively, as is
generally described in U.S. Patents 5,270,163, 5,475,096, 5,567,588,
5,595,877, 5,637,459,
5,683,867, 5,705,337, and related patents, hereby incorporated by reference,
nucleic acid "aptamersn
can be developed for binding to virtually any target analyte. Similarly the
analyte may be a nucleic
acid binding protein and the capture binding ligand is either a single-
stranded or double-stranded
nucleic acid; alternatively, the binding ligand may be a nucleic acid binding
protein when the analyte is
a single or double-stranded nucleic acid. When the analyte is a protein, the
binding ligands include
proteins (particularly including antibodies or fragments thereof (FAbs,
etc.)), small molecules, or
aptamers, described above. Preferred binding ligand proteins include peptides.
For example, when
the analyte is an enzyme, suitable binding ligands include substrates,
inhibitors, and other proteins
that bind the enzyme, i.e. components of a multi-enzyme (or protein) complex.
As will be appreciated
by those in the art, any two molecules that will associate, preferably
specifically, may be used, either
as the analyte or the binding ligand. Suitable analyte/binding ligand pairs
include, but are not limited
to, antibodieslantigens, receptors/ligand, proteins/nucleic acids; nucleic
acids/nucleic acids,
enzymes/substrates and/or inhibitors, carbohydrates (including glycoproteins
and glycolipids)/lectins,
carbohydrates and other binding partners, proteins/proteins; and protein/small
molecules. These may
be wild-type or derivative sequences. In a preferred embodiment, the binding
ligands are portions
(particularly the extracellular portions) of cell surface receptors that are
known to multimerize, such as
the growth hormone receptor, glucose transporters (particularly GLUT4
receptor), transferrin receptor,
epidermal growth factor receptor, low density lipoprotein receptor, high
density lipoprotein receptor,
leptin receptor, interleukin receptors including IL-1, IL-2, IL-3, IL-4, IL-5,
IL-6, IL-7, IL-8, IL-9, IL-11, IL-
12, IL-13, IL-15 and IL-17 receptors, VEGF receptor, PDGF receptor, EPO
receptor, TPO receptor,
ciliary neurotrophic factor receptor, prolactin receptor, and T-cell
receptors. Similarly, there is a wide
body of literature relating to the development of binding partners based on
combinatorial chemistry
methods.
In this embodiment, when the binding ligand is a nucleic acid, preferred
compositions and techniques
are outlined in U.S. Patent Nos. 5,591,578; 5,824,473; 5,705,348; 5,780,234
and 5,770,369; U.S.S.N.s
081873,598 08/911,589; WO 98/20162; W098/12430; W098/57158; WO 00/16089)
W099/57317;
W099/67425; WO00/24941; PCT US00/10903; WO00/38836; W099/37819; W099/57319 and
-57-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
PCTUS00/20476; and related materials, all of which are expressly incorporated
by reference in their
entirety.
The method of attachment of the capture binding ligands to the attachment
linker (either an insulator
or conductive oligomer) will generally be done as is known in the art, and
will depend on both the
composition of the attachment linker and the capture binding ligand. In
general, the capture binding
ligands are attached to the attachment linker through the use of functional
groups on each that can
then be used for attachment. Preferred functional groups for attachment are
amino groups, carboxy
groups, oxo groups and thiol groups. These functional groups can then be
attached, either directly or
indirectly through the use of a linker, sometimes depicted herein as "Z".
Linkers are well known in the
art; for example, homo-or hetero-bifunctional linkers as are well known (see
1994 Pierce Chemical
Company catalog, technical section on cross-linkers, pages 155-200,
incorporated herein by
reference). Preferred Z linkers include, but are not limited to, alkyl groups
(including substituted alkyl
groups and alkyl groups containing heteroatom moieties), with short alkyl
groups, esters, amide,
amine, epoxy groups and ethylene glycol and derivatives being preferred, with
propyl, acetylene, and
Cz alkene being especially preferred. Z may also be a sulfone group, forming
sulfonamide linkages.
In this way, capture binding ligands comprising proteins, lectins, nucleic
acids, small organic
molecules, carbohydrates, etc. can be added.
A preferred embodiment utilizes proteinaceous capture binding ligands. As is
known in the art, any
number of techniques may be used to attach a proteinaceous capture binding
ligand to an attachment
linker. A wide variety of techniques are known to add moieties to proteins.
A preferred embodiment utilizes nucleic acids as the capture binding ligand.
While most of the
following discussion focuses on nucleic acids, as will be appreciated by those
in the art, many of the
techniques outlined below apply in a similar manner to non-nucleic acid
systems as well, and to
systems that rely on attachment to surfaces other than metal electrodes.
The capture probe nucleic acid is covalently attached to the electrode, via an
"attachment linker", that
can be either a conductive oligomer (required for mechanism-1 systems) or an
insulator. By
"covalently attached" herein is meant that two moieties are attached by at
least one bond, including
sigma bonds, pi bonds and coordination bonds.
Thus, one end of the attachment linker is attached to a nucleic acid (or other
binding ligand), and the
other end (although as will be appreciated by those in the art, it need not be
the exact terminus for
either) is attached to the electrode. Thus, any of the structures depicted
herein may further comprise
-58-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
a nucleic acid effectively as a terminal group. Thus, the present invention
provides compositions
comprising nucleic acids covalently attached to electrodes as is generally
depicted below in Structure
17:
Structure 17
F,-(X or l) -F2-nucleic acid
fn Structure 17, the hatched marks on the left represent an electrode. X is a
conductive oligomer and I
is an insulator as defined herein. F, is a linkage that allows the covalent
attachment of the electrode
and the conductive oligomer or insulator, including bonds, atoms or linkers
such as is described
herein, for example as "A", defined below. F2 is a linkage that allows the
covalent attachment of the
conductive oligomer or insulator to the nucleic acid, and may be a bond, an
atom or a linkage as is
herein described. F~ may be part of the conductive oligomer, part of the
insulator, part of the nucleic
acid, or exogeneous to both, for example, as defined herein for "Z".
In a preferred embodiment, the capture probe nucleic acid is covalently
attached to the electrode via
an attachment linker. The covalent attachment of the nucleic acid and the
attachment linker may be
accomplished in several ways. In a preferred embodiment, the attachment is via
attachment to the
base of the nucleoside, via attachment to the backbone of the nucleic acid
(either the ribose, the
phosphate, or to an analogous group of a nucleic acid analog backbone), or via
a transition metal
ligand, as described below. The techniques outlined below are generally
described for naturally
occurring nucleic acids, although as will be appreciated by those in the art,
similar techniques may be
used with nucleic acid analogs, and in some cases with other binding ligands.
Similarly, most of the
structures herein depict conductive oligomers as the attachment linkers, but
insulators such as alkyl
chains are preferred in many embodiments.
In a preferred embodiment, the attachment linker is attached to the base of a
nucleoside of the nucleic
acid. This may be done in several ways, depending on the linker, as is
described below. In one
embodiment, the linker is attached to a terminal nucleoside, i.e. either the
3' or 5' nucleoside of the
nucleic acid. Alternatively, the linker is attached to an internal nucleoside.
The point of attachment to the base will vary with the base. Generally,
attachment at any position is
possible. In some embodiments, for example when the probe containing the ETMs
may be used for
hybridization (i.e. mechanism-1 systems) , it is preferred to attach at
positions not involved in
-59-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
Structure 18
hydrogen bonding to the complementary base. Thus, for example, generally
attachment is to the 5 or
6 position of pyrimidines such as uridine, cytosine and thymine. For purines
such as adenine and
guanine, the linkage is preferably via the 8 position. Attachment to non-
standard bases is preferably
done at the comparable positions.
In one embodiment, the attachment is direct; that is, there are no intervening
atoms between the
attachment linker and the base. In this embodiment, for example, attachment
linkers comprising
conductive oligomers with terminal acetylene bonds are attached directly to
the base. Structure 18 is
an example of this linkage, using a Structure 3 conductive oligomer and
uridine as the base, although
other bases and attachment linkers can be used as will be appreciated by those
in the art:
0
--[-Y-B-D~ Y
~NH
N' \'O
O
It should be noted that the pentose structures depicted herein may have
hydrogen, hydroxy,
phosphates or other groups such as amino groups attached. In addition, the
pentose and nucleoside
structures depicted herein are depicted non-conventionally, as mirror images
of the normal rendering.
In addition, the pentose and nucleoside structures may also contain additional
groups, such as
protecting groups, at any position, for example as needed during synthesis.
In addition, the base may contain additional modifications as needed, i.e. the
carbonyl or amine groups
may be altered or protected.
In an alternative embodiment, the attachment is any number of different Z
linkers, including amide and
amine linkages, as is generally depicted in Structure 19 using uridine as the
base and a Structure 3
oligomer as the attachment linker: ,
Structure 19:
NHz
Y D Y
n m
N O
O
-60-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
In this embodiment, Z is a linker. Preferably, Z is a short linker of about 1
to about 10 atoms, with from
1 to 5 atoms being preferred, that may or may not contain alkene, alkynyl,
amine, amide, azo, imine,
etc., bonds. Linkers are known in the art; for example, homo-or hetero-
bifunctional linkers as are well
known (see 1994 Pierce Chemical Company catalog, technical section on cross-
linkers, pages
155-200, incorporated herein by reference). Preferred Z linkers include, but
are not limited to, alkyl
groups (including substituted alkyl groups and alkyl groups containing
heteroatom moieties), with short
alkyl groups, esters, amide, amine, epoxy groups and ethylene glycol and
derivatives being preferred,
with propyl, acetylene, and CZ alkene being especially preferred. Z may also
be a sulfone group,
forming sulfonamide linkages as discussed below.
In a preferred embodiment, the attachment of the nucleic acid and the
attachment tinker is done via
attachment to the backbone of the nucleic acid. This may be done in a number
of ways, including
attachment to a ribose of the ribose-phosphate backbone, or to the phosphate
of the backbone, or
other groups of analogous backbones.
As a preliminary matter, it should be understood that the site of attachment
in this embodiment may be
to a 3' or 5' terminal nucleotide, or to an internal nucleotide, as is more
fully described below.
In a preferred embodiment, the attachment linker is attached to the ribose of
the ribose-phosphate
backbone. This may be done in several ways. As is known in the art,
nucleosides that are modified at
either the 2' or 3' position of the ribose with amino groups, sulfur groups,
silicone groups, phosphorus
groups, or oxo groups can be made (Imazawa et al., J. Org. Chem., 44:2039
(1979); Hobbs et al., J.
Org. Chem. 42(4):714 (1977); Verheyden et al., J. Orrg. Chem. 36(2):250
(1971); McGee et al., J.
Org. Chem. 61:781-785 (1996); Mikhailopulo et al., Liebigs. Ann. Chem. 513-519
(1993); McGee et
al., Nucleosides & Nucleotides 14(6):1329 (1995), all of which are
incorporated by reference). These
modified nucleosides are then used to add the attachment linkers.
A preferred embodiment utilizes amino-modified nucleosides. These amino-
modified riboses can then
be used to form either amide or amine linkages to the conductive oligomers. In
a preferred
embodiment, the amino group is attached directly to the ribose, although as
will be appreciated by
those in the art, short linkers such as those described herein for "Z" may be
present between the
amino group and the ribose.
In a preferred embodiment, an amide linkage is used for attachment to the
ribose. Preferably, if the
conductive oligomer of Structures 1-3 is used, m is zero and thus the
conductive oligomer terminates
in the amide bond. In this embodiment, the nitrogen of the amino group of the
amino-modified ribose
is the "D" atom of the conductive oligomer. Thus, a preferred attachment of
this embodiment is
-61 -
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
depicted in Structure 20 (using the Structure 3 conductive oligomer as the
attachment linker):
Structure 20
0
~Y B ~Y'-C'-H'-"bas
n
As will be appreciated by those in the art, Structure 20 has the terminal bond
fixed as an amide bond.
In a preferred embodiment, a heteroatom linkage is used, i.e. oxo, amine,
sulfur, etc. A preferred
embodiment utilizes an amine 4inkage. Again, as outlined above for the amide
linkages, for amine
linkages, the nitrogen of the amino-modified ribose may be the "D" atom of the
conductive oligomer
when the Structure 3 conductive oligomer is used. Thus, for example,
Structures 21 and 22 depict
nucleosides with the Structures 3 and 9 conductive oligomers, respectively, as
the attachment linkers,
using the nitrogen as the heteroatom, athough other heteroatoms can be used:
Structure 21
~ ~ 0
--I-Y-B-f~Y~Z~ N bas
~n ~ t H
In Structure 21, preferably both m and t are not zero. A preferred Z here is a
methylene group, or
other aliphatic alkyl linkers. One, two or three carbons in this position are
particularly useful for
synthetic reasons.
Structure 22
R
~ ~ \O
-~ Y-j--f-Z~ N base
H
R
In Structure 22, Z is as defined above. Suitable linkers include methylene and
ethylene.
In an alternative embodiment, the attachment linker is covalently attached to
the nucleic acid via the
phosphate of the ribose-phosphate backbone (or analog) of a nucleic acid. In
this embodiment, the
attachment is direct, utilizes a linker or via an amide bond. Structure 23
depicts a direct linkage, and
Structure 24 depicts linkage via an amide bond (both utilize the Structure 3
conductive oligomer,
although Structure 8 conductive oligomers are also possible as well as any
number of other
attachment linkers). Structures 23 and 24 depict the conductive oligomer in
the 3' position, although
the 5' position is also possible. Furthermore, both Structures 23 and 24
depict naturally occurring
phosphodiester bonds, although as those in the art will appreciate, non-
standard analogs of
-62-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
phosphodiester bonds may also be used.
Structure 23
base
O
O
--rY-B-~Y~Z~ ~=O ors
~n ~ ~ m~ ~t
O
In Structure 23, if the terminal Y is present (i.e. m=1 ), then preferably Z
is not present (i.e. t=0). If the
terminal Y is not present, then Z is preferably present.
Structure 24 depicts a preferred embodiment, wherein the terminal B-D bond.is
an amide bond, the
terminal Y is not present, and Z is a linker, as defined herein.
Structure 24
0
~Y-B-D-r-Y-I I
~n
In a preferred embodiment, the attachment linker is covalently attached to the
nucleic acid via a
transition metal ligand. In this embodiment, the attachment linker is
covalently attached to a iigand
which provides one or more of the coordination atoms for a transition metal.
In one embodiment, the
ligand to which the attachment linker is attached also has the nucleic acid
attached, as is generally
depicted below in Structure 25. Alternatively, the attachment linker is
attached to one ligand, and the
nucleic acid is attached to another ligand, as is generally depicted below in
Structure 26. Thus, in the
presence of the transition metal, the attachment linker is covalently attached
to the nucleic acid. Both
of these structures depict Structure 3 conductive oligomers, although other
attachment linkers may be
utilized. Structures 25 and 26 depict two representative structures:
Structure 25
nucleic acid
-/-Y-B-D~Y~Z~L
~n ~ ~m t ~~~M
Lr
Structure 26
-63-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
nucleic acid
~Y-B-D~Y~Z~L., .L-
~n ~ ~m ~ ~t 'O
M
Lr
In the structures depicted herein, M is a metal atom, with transition metals
being preferred. Suitable
transition metals for use in the invention include, but are not limited to,
cadmium (Cd), copper (Cu),
cobalt (Go), palladium (Pd), zinc (Zn), iron (Fe), ruthenium (Ru), rhodium
(Rh), osmium (Os), rhenium
(Re), platinium (Pt), scandium (Sc), titanium (Ti), Vanadium (V), chromium
(Cr), manganese (Mn),
nickel (Ni), Molybdenum (Mo), technetium (Tc), tungsten (W), and iridium (1r).
That is, the first series
of transition metals, the platinum metals (Ru, Rh, Pd, Os, Ir and Pt), along
with Fe, Re, W, Mo and Tc,
are preferred. Particularly preferred are ruthenium, rhenium, osmium,
platinium, cobalt and iron.
L are the co-ligands, that provide the coordination atoms for the binding of
the metal ion. As will be
appreciated by those in the art, the number and nature of the co-ligands will
depend on the
coordination number of the metal ion. Mono-, di- or polydentate co-ligands may
be used at any
position. Thus, for example, when the metal has a coordination number of six,
the L from the terminus
of the conductive oligomer, the L contributed from the nucleic acid, and r,
add up to six. Thus, when
the metal has a coordination number of six, r may range from zero (when all
coordination atoms are
provided by the other two ligands) to four, when all the co-ligands are
monodentate. Thus generally, r
will be from 0 to 8, depending on the coordination number of the metal ion and
the choice of the other
ligands.
In one embodiment, the metal ion has a coordination number of six and both the
ligand attached to the
conductive oligomer and the ligand attached to the nucleic acid are at least
bidentate; that is, r is
preferably zero, one (i.e. the remaining co-ligand is bidentate) or two (two
monodentate co-ligands are
used).
As will be appreciated in the art, the co-ligands can be the same or
different. Suitable iigands fall into
two categories: ligands which use nitrogen, oxygen, sulfur, carbon or
phosphorus atoms (depending
on the metal ion) as the coordination atoms (generally referred to in the
literature as sigma (e) donors)
and organometallic ligands such as metallocene ligands (generally referred to
in the literature as pi (rr)
donors, and depicted herein as Lm). Suitable nitrogen donating ligands are
well known in the art and
include, but are not limited to, NHS; NHR; NRR'; pyridine; pyrazine;
isonicotinamide; imidazole;
bipyridine and substituted derivatives of bipyridine; terpyridine and
substituted derivatives;
phenanthrolines, particularly 1,10-phenanthroline (abbreviated phen) and
substituted derivatives of
phenanthrolines such as 4,7-dimethylphenanthroline and dipyridol[3,2-a:2',3'-
c]phenazine (abbreviated
dppz); dipyridophenazine; 1,4,5,8,9,12-hexaazatriphenylene (abbreviated hat);
9,10-
-64-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
phenanthrenequinone diimine (abbreviated phi); 1,4,5,8-tetraazaphenanthrene
(abbreviated tap);
1,4,8,11-tetra-azacyclotetradecane (abbreviated cyclam), EDTA, EGTA and
isocyanide. Substituted
derivatives, including fused derivatives, may also be used. In some
embodiments, porphyrins and
substituted derivatives of the porphyrin family may be used. See for example,
Comprehensive
Coordination Chemistry, Ed. Wilkinson et al., Pergammon Press, 1987, Chapters
13.2 (pp73-98), 21.1
(pp. 813-898) and 21.3 (pp 915-957), all of which are hereby expressly
incorporated by reference.
Suitable sigma donating ligands using carbon, oxygen, sulfur and phosphorus
are known in the art.
For example, suitable sigma carbon donors are found in Cotton and Wilkenson,
Advanced Organic
Chemistry, 5th Edition, John Wiley & Sons, 1988, hereby incorporated by
reference; see page 38, for
example. Similarly, suitable oxygen ligands include crown ethers, water and
others known in the art.
Phosphines and substituted phosphines are also suitable; see page 38 of Cotton
and Wilkenson.
The oxygen, sulfur, phosphorus and nitrogen-donating ligands are attached in
such a manner as to
allow the heteroatoms to serve as coordination atoms.
In a preferred embodiment, organometallic ligands are used. In addition to
purely organic compounds
for use as redox moieties, and various transition metal coordination complexes
with a-bonded organic
ligand with donor atoms as heterocyclic or exocyclic substituents, there is
available a wide variety of
transition metal organometalVic compounds with rr-bonded organic ligands (see
Advanced tnorganic~
Chemistry, 5th Ed., Cotton & Wilkinson, John Wiley & Sons, 1988, chapter 26;
Organometallics, A
Concise Introduction, Elschenbroich et al., 2nd Ed., 1992, VCH; and
Comprehensive Organometallic
Chemistry II, A Review of the Literature 1982-1994, Abel et al. Ed., Vol. 7,
chapters 7, 8, 10 & 11,
Pergamon Press, hereby expressly incorporated by reference). Such
organometallic ligands include
cyclic aromatic compounds such as the cyclopentadienide ion [C5H5(-1)] and
various ring substituted
and ring fused derivatives, such as the indenylide (-1 ) ion, that yield a
class of bis(cyclopentadieyl)
metal compounds, (i.e. the metallocenes); see for example Robins et al., J.
Am. Chem. Soc.
104:1882-1893 (1982); and Gassman et al., J. Am. Chem. Soc. 108:4228-4229
(1986),
incorporated by reference. Of these, ferrocene [(C5H5)2Fe] and its derivatives
are prototypical
examples which have been used in a wide variety of chemical (Connelly et al.,
Chem. Rev. 96:877-
910 (1996), incorporated by reference) and electrochemical (Geiger et al.,
Advances in
Organometallic Chemistry 23:1-93; and Geiger et al., Advances in
Organometallic Chemistry 24:87,
incorporated by reference) electron transfer or "redox" reactions. Metallocene
derivatives of a variety
of the first, second and third row transition metals are potential candidates
as redox moieties that are
covalently attached to either the ribose ring or the nucleoside base of
nucleic acid. Other potentially
suitable organometallic ligands include cyclic arenes such as benzene, to
yield bis(arene)metal
compounds and their ring substituted and ring fused derivatives, of which
bis(benzene)chromium is a
-65-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
prototypical example, Other acyclic rr-bonded ligands such as the allyl{-1)
ion, or butadiene yield
potentially suitable organometallic compounds, and all such ligands, in
conjuction with other rr-bonded
and 6-bonded ligands constitute the general class of organometallic compounds
in which there is a
metal to carbon bond. Electrochemical studies of various dimers and oligomers
of such compounds
with bridging organic ligands, and additional non-bridging ligands, as well as
with and without metal-
metal bonds are potential candidate redox moieties in nucleic acid analysis.
When one or more of the co-ligands is an organometallic ligand, the ligand is
generally attached via
one of the carbon atoms of the organometallic ligand, although attachment may
be via other atoms for
heterocyclic ligands. Preferred organometallic ligands include metallocene
ligands, including
substituted derivatives and the metalloceneophanes (see page 1174 of Cotton
and Wlkenson, supra).
For example, derivatives of metallocene ligands such as
methylcyclopentadienyl, with multiple methyl
groups being preferred, such as pentamethylcyclopentadienyl, can be used to
increase the stability of
the metallocene. In a preferred embodiment, only one of the two metallocene
ligands of a metallocene
are derivatized.
As described herein, any combination of ligands may be used. Preferred
combinations include: a) all
ligands are nitrogen donating ligands; b) all ligands are organometallic
ligands; and c) the ligand at the
terminus of the attachment linker is a metallocene ligand and the ligand
provided by the nucleic acid is
a nitrogen donating ligand, with the other ligands, if needed, are either
nitrogen donating ligands or
metallocene ligands, or a mixture. These combinations, using the conductive
oligomer of Structure 3,
are depicted in Structures 27 (using phenanthroline and amino as
representative ligands), 28 (using
ferrocene as the metal-ligand combination) and 29 (using cyclopentadienyl and
amino as
representative ligands).
Structure 27
~Y-~B-D~Y~Z
n m
-N N-
w.,e
.M,
H~~base
Structure 28
-(-Y-B-D-H-Y-/-[-Z t O
4..",mIGJ_..
base
-66-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
Structure 29
~Y-B--D~~Z t
w.:M.,,, ~'O
~~bas
Again, other attachment linkers such as alkyl groups may also be utilized.
In a preferred embodiment, the ligands used in the invention show altered
fluorescent properties
depending on the redox state of the chelated metal ion. As described below,
this thus serves as an
additional mode of detection of electron transfer between the ETM and the
electrode.
In addition, similar methods can be used to attach proteins to the detection
electrode; see for example
U.S. Patent No. 5,620,850, hereby incorporated by reference.
In a preferred embodiment, as is described more fully below, the ligand
attached to the nucleic acid is
an amino group attached to the 2' or 3' position of a ribose of the ribose-
phosphate backbone. This
ligand may contain a multiplicity of amino groups so as to form a polydentate
ligand which binds the
metal ion. Other preferred ligands include cyclopentadiene and phenanthroline.
In a preferred embodiment, the capture probe nucleic acids (or other binding
ligands) are covalently
attached to the electrode via an insulator (i.e. the attachment linker is an
insulator). The attachment of
nucleic acids (and other binding ligands) to insulators such as alkyl groups
is well known, and can be
done to the base or the backbone, including the ribose or phosphate for
backbones containing these
moieties, or to alternate backbones for nucleic acid analogs.
In a preferred embodiment, there may be one or more different capture probe
species on the surface.
In some embodiments, there may be one type of capture probe, or one type of
capture probe
extender, as is more fully described below. Alternatively, different capture
probes, or one capture
probe with a multiplicity of different capture extender probes can be used.
Similarly, it may be
desirable (particularly in the case of nucleic acid analytes and binding
ligands in mechanism-2
systems) to use auxiliary capture probes that comprise relatively short probe
sequences, that can be
used to "tack down" components of the system, for example the recruitment
linkers, to increase the
concentration of ETMs at the surface.
In a preferred embodiment, a number of capture probes are designed and used
for each target
sequence. That is, a single electrode pad of the array may have 1 probe to the
target analyte, or a
-67-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
plurality of probes to the same target sequence, preferably (but not required
to be) non-overlapping.
This is particularly preferred for long target sequences. In this embodiment,
at least two different
capture probes are used, with at least 3, 4, 5, 6, 7, 8, 9 or 10 being
preferred, and 8 being particularly
preferred.
Thus the present invention provides substrates comprising at least one
detection electrode comprising
monolayers and capture binding ligands, useful in target analyte detection
systems.
In a preferred embodiment, the compositions further comprise a solution or
soluble binding ligand,
although as is more fully described below, for mechanism-1 systems, the ETMs
may be added in the
form of non-covalently attached hybridization indicators. Solution binding
ligands are similar to
capture binding ligands, in that they bind, preferably specifically, to target
analytes. The solution
binding ligand (generally referred to herein as label probes when the target
analytes are nucleic acids)
may be the same or different from the capture binding ligand. Generally, the
solution binding ligands
are not directly attached to the surface. The solution binding ligand either
directly comprises a
recruitment linker that comprises at least one ETM (Figure 4A from
601190,259), or the recruitment
linker binds, either directly (Figure 4A) or indirectly (Figure 4E), to the
solution binding ligand.
Thus, "solution binding ligands" or "soluble binding ligands" or "signal
carriers" or "label probes" or
"label binding ligands" with recruitment tinkers comprising covalently
attached ETMs are provided.
That is, one portion of the label probe or solution binding ligand directly or
indirectly binds to the target
analyte, and one portion comprises a recruitment linker comprising covalently
attached ETMs. In
some systems, for example in mechanism-1 nucleic acid systems, these may be
the same. Similarly,
for mechanism-1 systems, the recruitment linker comprises nucleic acid that
will. hybridize to detection
probes. The terms "electron donor moiety", "electron acceptor moiety", and
"ETMs" (ETMs) or
grammatical equivalents herein refers to molecules capable of electron
transfer under certain
conditions. It is to be understood that electron donor and acceptor
capabilities are relative; that is, a
molecule which can lose an electron under certain experimental conditions will
be able to accept an
electron under different experimental conditions. It is to be understood that
the number of possible
electron donor moieties and electron acceptor moieties is very large, and that
one skilled in the art of
electron transfer compounds will be able to utilize a number of compounds in
the present invention.
Preferred ETMs include, but are not limited to, transition metal complexes,
organic ETMs, and
electrodes.
In a preferred embodiment, the ETMs are transition metal complexes. Transition
metals are those
whose atoms have a partial or complete d shell of electrons. Suitable
transition metals for use in the
invention are listed above.
- 68 -
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
The transition metals are complexed with a variety of ligands, L, defined
above, to form suitable
transition metal complexes, as is well known in the art.
Preferred ETMs comprise metallocenes, particularly ferrocene.
In addition to transition metal complexes, other organic electron donors and
acceptors may be
covalently attached to the nucleic acid for use in the invention. These
organic molecules include, but
are not limited to, riboflavin, xanthene dyes, azine dyes, acridine orange,
N,N'-dimethyl-2,7-
diazapyrenium dichloride (DAP2*), methylviologen, ethidium bromide, quinones
such as N,N'-
dimethylanthra(2,1,9-det6,5,10-d'e'f~diisoquinofine dichloride (ADIQZ+);
porphyries ([meso-tetrakis(N-
methyl-x-pyridinium)porphyrin tetrachloride], varlamine blue B hydrochloride,
Bindschedler's green;
2,6-dichloroindophenol, 2,6-dibromophenolindophenol; Brilliant crest blue (3-
amino-9-dimethyl-amino-
10-methylphenoxyazine chloride), methylene blue; Nile blue A
(aminoaphthodiethylaminophenoxazine
sulfate), indigo-5,5',7,7'-tetrasulfonic acid, indigo-5,5',7-trisulfonic acid;
phenosafranine, indigo-5-
monosulfonic acid; safranine T; bis(dimethylglyoximato)-iron(II) chloride;
induline scarlet, neutral red,
anthracene, coronene, pyrene, 9-phenylanthracene, rubrene, binaphthyl, DPA,
phenothiazene,
fluoranthene, phenanthrene, chrysene, 1,8-diphenyl-1,3,5,7-octatetracene,
naphthalene,
acenaphthalene, perylene, TMPD and analogs and subsitituted derivatives of
these compounds.
In one embodiment, the electron donors and acceptors are redox proteins as are
known in the art.
However, redox proteins in many embodiments are not preferred.
The choice of the specific ETMs will be influenced by the type of electron
transfer detection used, as is
generally outlined below. Preferred ETMs are metallocenes, with ferrocene
being particularly
preferred.
Accordingly, the present invention provides methods and compositions useful in
the detection of
nucleic acids and other target analytes. As will be appreciated by those in
the art, the compositions of
the invention can take on a wide variety of configurations. As is more fully
outlined below, preferred
systems of the invention work as follows. A target nucleic acid sequence is
attached (via
hybridization) to an electrode comprising a monolayer, generally including
conductive oligomers. This
attachment can be either directly to a capture probe on the surface, or
indirectly, using capture
extender probes. In some embodiments, the target sequence itself comprises the
ETMs.
Alternatively, a label probe is then added, forming an assay complex. The
attachment of the label
probe may be direct (i.e. hybridization to a portion of the target sequence),
or indirect (i.e. hybridization
to an amplifier probe that hybridizes to the target sequence), with all the
required nucleic acids forming
an assay complex. As a result of the hybridization of the first portion of the
label probe, the second
-69-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
portion of the label probe, the "recruitment linker", containing the ETMs is
brought into spatial proximity
to the SAM surtace on the electrode, and the presence of the ETM can then be
detected electronically.
Thus, in a preferred embodiment, the present invention provides electrodes
comprising monolayers
comprising SAM forming species and capture probes, useful in nucleic acid (or
other target analyte)
detection systems. In a preferred embodiment, the compositions further
comprise a label probe. The
label probe is nucleic acid, generally single stranded, although as more fully
outlined below, it may
contain double-stranded portions. In mechanism-2 systems, the label probe
comprises a first portion
that is capable of hybridizing to a component of the assay complex, defined
below, and a second
portion that does not hybridize to a component of an assay complex and
comprises at least one
covalently attached ETM.
Without being bound by theory, it appears that in "mechanism-2" systems,
electron transfer is
facilitated when the ETM is able to penetrate ("snuggle") into the monolayer
to some degree. That is,
in general, it appears that hydrophobic ETMs used with hydrophobic SAMs give
rise to better (greater)
signals than ETMs that are charged or more hydrophilic. Thus, for example,
ferrocene in solution can
penetrate the monolayers of the examples and give a signal when
electroconduits are present, while
ferrocyanide in solution gives little or no signal. Thus, in general,
hydrophobic ETMs are preferred iri
mechanism-2 systems; however, transition metal complexes, although charged,
with one or more
hydrophobic ligands, such as Ru and Os complexes, also give rise to good
signals. Similarly, electron
transfer between the ETM and the electrode is facilitated by the use of
linkers or spacers that allow the
ETM some flexibility to penetrate into the monolayer; thus the N6 compositions
of the invention have a
four carbon tinker attaching the ETM to the nucleic acid.
In a preferred embodiment, a plurality of ETMs are used. The use of multiple
ETMs provides signal
amplification and thus allows more sensitive detection limits. As discussed
below, while the use of
multiple ETMs on nucleic acids that hybridize to complementary strands can
cause decreases in Tms
of the hybridization complexes depending on the number, site of attachment and
spacing between the
multiple ETMs, this is not a factor when the ETMs are on the recruitment
linker, since this does not
hybridize to a complementary sequence. Accordingly, pluralities of ETMs are
preferred, with at least
about 2 ETMs per recruitment linker being preferred, and at least about 10
being particularly preferred,
and at least about 20 to 50 being especially preferred. In some instances,
very large numbers of
ETMs (100 to 1000) can be used.
As will be appreciated by those in the art, the portion of the label probe (or
target, in some
embodiments) that comprises the ETMs (termed herein a "recruitment linker" or
"signal carrier") can
be nucleic acid, or it can be a non-nucleic acid linker that links the first
hybridizable portion of the label
-70-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
probe to the ETMs. That is, since this portion of the label probe is not
required for hybridization, it
need not be nucleic acid, although this may be done for ease of synthesis. In
some embodiments, as
is more fully outlined below, the recruitment linker may comprise double-
stranded portions. Thus, as
will be appreciated by those in the art, there are a variety of configurations
that can be used. In a
preferred embodiment, the recruitment linker is nucleic acid (including
analogs), and attachment of the
ETMs can be via (1) a base; (2) the backbone, including the ribose, the
phosphate, or comparable
structures in nucleic acid analogs; (3) nucleoside replacement, described
below; or (4) metallocene
polymers, as described below. In a preferred embodiment, the recruitment
linker is non-nucleic acid,
and can be either a metallocene polymer or an alkyl-type polymer (including
heteroalkyl, as is more
fully described below) containing ETM substitution groups. These options are
generally depicted in
the Figures.
In a preferred embodiment, the recruitment linker is a nucleic acid, and
comprises covalently attached
ETMs. The ETMs may be attached to nucleosides within the nucleic acid in a
variety of positions.
Preferred embodiments include, but are not limited to, (1) attachment to the
base of the nucleoside, (2)
attachment of the ETM as a base replacement, (3) attachment to the backbone of
the nucleic acid,
including either to a ribose of the ribose-phosphate backbone or to a
phosphate moiety, or to
analogous structures in nucleic acid analogs, and (4) attachment via
metallocene polymers, with the
fatter being preferred.
In addition, as is described below, when the recruitment linker is nucleic
acid, it may be desirable to
use secondary label probes, that have a first portion that will hybridize to a
portion of the primary label
probes and a second portion comprising a recruitment linker as is defined
herein. This is generally
depicted in Figure 16H of U.S.S.N. 60/190,259.
In a preferred embodiment, the ETM is attached to the base of a nucleoside as
is generally outlined
above for attachment of the attachment linkers. Attachment can be to an
internal nucleoside or a
terminal nucleoside.
The covalent attachment to the base will depend in part on the ETM chosen, but
in general is similar to
the attachment of conductive oligomers to bases, as outlined above. Attachment
may generally be
done to any position of the base. In a preferred embodiment, the ETM is a
transition metal complex,
and thus attachment of a suitable metal ligand to the base leads to the
covalent attachment of the
ETM. Alternatively, similar types of linkages may be used for the attachment
of organic ETMs, as will
be appreciated by those in the art.
In one embodiment, the C4 attached amino group of cytosine, the C6 attached
amino group of
-71 -
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
adenine, or the C2 attached amino group of guanine may be used as a transition
metal ligand.
Ligands containing aromatic groups can be attached via acetylene linkages as
is known in the art (see
Comprehensive Organic Synthesis, Trost et al., Ed., Pergamon Press, Chapter
2.4: Coupling
Reactions Between sp2 and sp Carbon Centers, Sonogashira, pp521-549, and pp950-
953, hereby
incorporated by reference). Structure 30 depicts a representative structure in
the presence of the
metal ion and any other necessary Iigands;.Structure 30 depicts uridine,
although as for all the
structures herein, any other base may also be used.
La is a ligand, which may include nitrogen, oxygen, sulfur or phosphorus
donating ligands or
organometallic ligands such as metallocene ligands. Suitable La ligands
include, but are not limited to,
phenanthroline, imidazole, bpy and terpy. L~ and M are as defined above.
Again, it will be appreciated
by those in the art, that a linker ("Z") may be included between the
nucleoside and the ETM.
Similarly, as for the attachment linkers, the linkage may be done using a
linker, which may utilize an
amide linkage (see generally Telser et al., J. Am. Chem. Soc. 111:7221-7226
(1989); Telser et a(., J.
Am. Chem. Soc. 111:7226-7232 (1989), both of which are expressly incorporated
by reference).
These structures are generally depicted below in Structure 31, which again
uses uridine as the base,
although as above, the other bases may also be used:
Structure 31
L
:M
~~4
In this embodiment, L is a ligand as defined above, with L~ and M as defined
above as well.
Preferably, L is amino, phen, byp and terpy.
-72-
Structure 30
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
In a preferred embodiment, the ETM attached to a nucleoside is a metallocene;
i.e. the L and L~ of
Structure 31 are both metallocene ligands, Lm, as described above. Structure
32 depicts a preferred
embodiment wherein the metallocene is ferrocene, and the base is uridine,
although other bases may
be used:
Preliminary data suggest that Structure 32 may cyclize, with the second
acetylene carbon atom
attacking the carbonyl oxygen, forming a furan-like structure. Preferred
metallocenes include
ferrocene, cobaltocene and osmiumocene.
In a preferred embodiment, the ETM is attached to a ribose at any position of
the ribose-phosphate
backbone of the nucleic acid, i.e. either the 5' or 3' terminus or any
internal nucleoside. Ribose in this
case can include ribose analogs. As is known in the art, nucleosides that are
modified at either the 2'
or 3' position of the ribose can be made, with nitrogen, oxygen, sulfur and
phosphorus-containing
modifications possible. Amino-modified and oxygen-modified ribose is
preferred. See generally PCT
publication WO 95/15971, incorporated herein by reference. These modification
groups may be used
as a transition metal ligand, or as a chemically functional moiety for
attachment of other transition
metal ligands and organometallic ligands, or organic electron donor moieties
as will be appreciated by
those in the art. In this embodiment, a linker such as depicted herein for "Z"
may be used as well, or a
conductive oligomer between the ribose and the ETM. Preferred embodiments
utilize attachment at
the 2' or 3' position of the ribose, with the 2' position being preferred.
Thus for example, the
conductive oligomers depicted in Structure 13, 14 and 15 may be replaced by
ETMs; alternatively, the
ETMs may be added to the free terminus of the conductive oligomer.
In a preferred embodiment, a metallocene serves as the ETM, and is attached
via an amide bond as
depicted below in Structure 33. The examples outline the synthesis of a
preferred compound when
-73-
Structure 32
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
the metallocene is ferrocene.
Structure 33
base
O
NH
'~M
In a preferred embodiment, amine linkages are used, as is generally depicted
in Structure 34.
Structure 34
BASE
O
NH
()
ETM
Z is a linker, as defined herein, with 1-16 atoms being preferred, and 2-4
atoms being particularly
preferred, and t is either one or zero.
In a preferred embodiment, oxo linkages are used, as is generally depicted in
Structure 35.
Structure 35
BASE
0
0
ETM
In Structure 35, Z is a linker, as defined herein, and t is either one or
zero. Preferred Z linkers include
alkyl groups including heteroalkyl groups such as (CH~)n and (CH2CHz0)n, with
n from 1 to 10 being
preferred, and n = 1 to 4 being especially preferred, and n=4 being
particularly preferred.
Linkages utilizing other heteroatoms are also possible.
In a preferred embodiment, an ETM is attached to a phosphate at any position
of the ribose-phosphate
backbone of the nucleic acid. This may be done in a variety of ways. In one
embodiment,
phosphodiester bond analogs such as phosphoramide or phosphoramidite linkages
may be
-74-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
incorporated into a nucleic acid, where the heteroatom (i.e. nitrogen) serves
as a transition metal
ligand (see PCT publication WO 95/15971, incorporated by reference).
Alternatively, the conductive
oligomers depicted in Structures 23 and 24 may be replaced by ETMs. In a
preferred embodiment,
the composition has the structure shown in Structure 36.
Structure 36
BASE
O
O
O = ~ -O -lZ) t-ETM
O
In Structure 36, the ETM is attached via a phosphate linkage, generally
through the use of a linker, Z.
Preferred Z linkers include alkyl groups, including heteroalkyl groups such as
(CH2)~, (CH~CH~O)~, with
n from 1 to 10 being preferred, and n = 1 to 4 being especially preferred, and
n=4 being particularly
preferred.
When the ETM is attached to the base or the backbone of the nucleoside, it is
possible to attach the
ETMs via "dendrimer" structures, as is more fully outlined below. As is
generally depicted in the
Figures, alkyl-based linkers can be used to create multiple branching
structures comprising one or
more ETMs at the terminus of each branch (although internal ETMs can be used
as well). Generally,
this is done by creating branch points containing multiple hydroxy groups,
which optionally can then be
used to add additional branch points. The terminal hydroxy groups can then be
used in
phosphoramidite reactions to add ETMs, as is generally done below for the
nucleoside replacement
and metallocene polymer reactions. The branch point can be an internal one or
a terminal one, and
can be a chemical branch point or a nucleoside branch point.
In a preferred embodiment, an ETM such as a metallocene is used as a
"nucleoside replacement",
serving as an ETM. For example, the distance between the two cyclopentadiene
rings of ferrocene is
similar to the orthongonal distance between two bases in a double stranded
nucleic acid. Other
metallocenes in addition to ferrocene may be used, for example, air stable
metallocenes such as those
containing cobalt or ruthenium. Thus, metallocene moieties may be incorporated
into the backbone of
a nucleic acid, as is generally depicted in Structure 37 (nucleic acid with a
ribose-phosphate
backbone) and Structure 38 (peptide nucleic acid backbone). Structures 37 and
38 depict ferrocene,
although as will be appreciated by those in the art, other metallocenes may be
used as well. In
general, air stable metallocenes are preferred, including metallocenes
utilizing ruthenium and cobalt as
the metal.
-75-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
Structure 37
BASE
O
O
0-P=O
-Z
Fe
--Z
..-n
In Structure 37, Z is a linker as defined above, with generally short, alkyl
groups, including
heteroatoms such as oxygen being preferred. Generally, what is important is
the length of the linker,
such that minimal perturbations of a double stranded nucleic acid is effected,
as is more fully ,
described below. Thus, methylene, ethylene, ethylene glycols, propylene and
butylene ace all
preferred, with ethylene and ethylene glycol being particularly preferred. In
addition, each Z linker may
be the same or different. Structure 37 depicts a ribose-phosphate backbone,
although as wilt be
appreciated by those in the art, nucleic acid analogs may also be used,
including ribose analogs and
phosphate bond analogs.
Structure 38
HN
OI
~ DEBASE
.N
Cs\C-O
HN/ Z-
Fe
CiZ
/ ~O
HN
O
DEBASE
/N
~C=O
HN
-76-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
In Structure 38, preferred Z groups are as listed above, and again, each Z
linker can be the same or
different. As above, other nucleic acid analogs may be used as well.
In addition, although the structures and discussion above depict metallocenes,
and particularly
ferrocene, this same general idea can be used to add ETMs in addition to
metallocenes, as nucleoside
replacements or in polymer embodiments, described below. Thus, for example,
when the ETM is a
transition metal complex other than a metallocene, comprising one, two or
three (or more) ligands, the
ligands can be functionalized as depicted for the ferrocene to allow the
addition of phosphoramidite
groups. Particularly preferred in this embodiment are complexes comprising at
least two ring (for
example, aryl and substituted aryl) ligands, where each of the ligands
comprises functional groups for
attachment via phosphoramidite chemistry. As will be appreciated by those in
the art, this type of
reaction, creating polymers of ETMs either as a portion of the backbone of the
nucleic acid or as "side
groups" of the nucleic acids, to allow amplification of the signals generated
herein, can be done with
virtually any ETM that can be functionalized to contain the correct chemical
groups.
Thus, by inserting a metallocene such as ferrocene (or other ETMs) into the
backbone of a nucleic
acid, nucleic acid analogs are made; that is, the invention provides nucleic
acids having a backbone
comprising at least one metallocene. This is distinguished from nucleic acids
having metallocenes
attached to the backbone, i.e. via a ribose, a phosphate, etc. That is, two
nucleic acids each made up
of a traditional nucleic acid or analog (nucleic acids in this case including
a single nucleoside), may be
covafently attached to each other via a metallocene. Viewed differently, a
metallocene derivative or
substituted metallocene is provided, wherein each of the two aromatic rings of
the metallocene has a
nucleic acid substitutent group.
In addition, as is more fully outlined below, it is possible to incorporate
more than one metallocene into
the backbone, either with nucleotides in between and/or with adjacent
metallocenes. When adjacent
metallocenes are added to the backbone, this is similar to the process
described below as
"metallocene polymers"; that is, there are areas of metallocene polymers
within the backbone.
In addition to the nucleic acid substitutent groups, it is also desirable in
some instances to add
additional substituent groups to one or both of the aromatic rings of the
metallocene (or ETM). For
example, as these nucleoside replacements are generally part of probe
sequences to be hybridized
with a substantially complementary nucleic acid, for example a target sequence
or another probe
sequence, it is possible to add substitutent groups to the metallocene rings
to facilitate hydrogen
bonding to the base or bases on the opposite strand. These may be added to any
position on the
metallocene rings. Suitable substitutent groups include, but are not limited
to, amide groups, amine
groups, carboxylic acids, and alcohols, including substituted alcohols. In
addition, these substitutent
77 -
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
groups can be attached via linkers as well, although in general this is not
preferred.
In addition, substituent groups on an ETM, particularly metallo~enes such as
ferrocene, may be added
to alter the redox properties of the ETM. Thus, for example, in some
embodiments, as is more fully
described below, it may be desirable to have different ETMs attached in
different ways (i.e. base or
ribose attachment), on different probes, or for different purposes (for
example, calibration or as an
internal standard). Thus, the addition of substituent groups on the
metallocene may allow two different
ETMs to be distinguished.
In order to generate these metallocene-backbone nucleic acid analogs, the
intermediate components
are also provided. Thus, in a preferred embodiment, the invention provides
phosphoramidite
metallocenes, as generally depicted in Structure 39:
Structure 39
PG-O
Z -AROMATIC
RING
IM
Z -AROMATIC
RING
O
NCH2CHZC- -N
~ wCH
~CH3
I H \CH
3
~
\
CH3
H3C
In Structure 39, PG is a protecting group, generally suitable for use in
nucleic acid synthesis, with
DMT, MMT and TMT all being preferred. The aromatic rings can either be the
rings of the
metallocene, or aromatic rings of ligands for transition metal complexes or
other organic ETMs. The
aromatic rings may be the same or different, and may be substituted as
discussed herein.
-78-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
Structure 40 depicts the ferrocene derivative:
Structure 40
PG-O
Z-
Fe
Z
O
3
NCHaCH2C-P- ~ ~C \ CH
CH CH3
H3C/ \CH3
These phosphoramidite analogs can be added to standard oligonucleotide
syntheses as is known in
the art.
Structure 41 depicts the ferrocene peptide nucleic acid (PNA) monomer, that
can be added to PNA
synthesis as is known in the art:
Structure 41
PG--NH
Fe
Z
O=C\
\0H
In Structure 41, the PG protecting group is suitable for use in peptide
nucleic acid synthesis, with
MMT, boc and Fmoc being preferred.
These same intermediate compounds can be used to form ETM or metallocene
polymers, which are
added to the nucleic acids, rather than as backbone replacements, as is more
fully described below.
In a preferred embodiment, the ETMs are attached as polymers, for example as
metallocene
polymers, in a "branched" configuration similar to the "branched DNA"
embodiments herein and as
outlined in U.S. Patent No. 5,124,246, using modified functionalized
nucleotides. The general idea is
as follows. A modified phosphoramidite nucleotide is generated that can
ultimately contain a free
hydroxy group that can be used in the attachment of phosphoramidite ETMs such
as metallocenes.
-79-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
This free hydroxy group could be on the base or the backbone, such as the
ribose or the phosphate
(although as will be appreciated by those in the art, nucleic acid analogs
containing other structures
can also be used). The modified nucleotide is incorporated into a nucleic
acid, and any hydroxy
protecting groups are removed, thus leaving the free hydroxyl. Upon the
addition of a
phosphoramidite ETM such as a metallocene, as described above in structures 39
and 40, ETMs,
such as metallocene ETMs, are added. Additional phosphoramidite ETMs such as
metallocenes can
be added, to form "ETM polymers", including "metallocene polymers" as depicted
herein, particularly
for ferrocene. In addition, in some embodiments, it is desirable to increase
the solubility of the
polymers by adding a "capping" group to the terminal ETM in the polymer, for
example a final
phosphate group to the metallocene as is generally depicted in Figure 12.
Other suitable solubility
enhancing "capping" groups will be appreciated by those in the art. It should
be noted that these
solubility enhancing groups can be added to the polymers in other places,
including to the ligand rings,
for example on the metallocenes as discussed herein
In a preferred embodiment, (as depicted in the figures of U.S.S.N. 09/626,096)
the 2' position of a
ribose of a phosphoramidite nucleotide is first functionalized to contain a
protected hydroxy group, in
this case via an oxo-linkage, although any number of linkers can be used, as
is generally described
herein for Z linkers. The protected modified nucleotide is then incorporated
via standard
phosphoramidite chemistry into a growing nucleic acid. The protecting group is
removed, and the free
hydroxy group is used, again using standard phosphoramidite chemistry to add a
phosphoramidite
metallocene such as ferrocene. A similar reaction is possible for nucleic acid
analogs. For example,
using peptide nucleic acids and the metallocene monomer shown in Structure 41,
peptide nucleic acid
structures containing metallocene polymers could be generated.
Thus, the present invention provides recruitment linkers of nucleic acids
comprising "branches" of
metallocene polymers as is generally depicted in Figures 12 and 13. Preferred
embodiments also
utilize metallocene polymers from one to about 50 metallocenes in length, with
from about 5 to about
20 being preferred and from about 5 to about 10 being especially preferred.
In addition, when the recruitment linker is nucleic acid, any combination of
ETM attachments may be
done.
In a preferred embodiment, the recruitment linker is not nucleic acid, and
instead may be any sort of
linker or polymer. As will be appreciated by those in the art, generally any
linker or polymer that can be
modified to contain ETMs can be used. In general, the polymers or linkers
should be reasonably
soluble and contain suitable functional groups for the addition of ETMs.
-80-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
As used herein, a "recruitment polymer" comprises at least two or three
subunits, which are covalently
attached. At least some portion of the monomeric subunits contain functional
groups for the covalent
attachment of ETMs. In some embodiments coupling moieties are used to
covalently link the subunits
with the ATMs. Preferred functional groups for attachment are amino groups,
carboxy groups, oxo
groups and thiol groups, with amino groups being particularly preferred. As
will be appreciated by
those in the art, a wide variety of recruitment polymers are possible.
Suitable linkers include, but are not limited to, alkyl linkers (including
heteroalkyl (including
(poly)ethylene glycol-type structures), substituted alkyl, aryalkyl linkers,
etc. As above for the
polymers, the linkers will comprise one or more functional groups for the
attachment of ETMs, which
will be done as will be appreciated by those in the art, for example through
the use homo-or hetero-
bifunctional linkers as are well known (see 1994 Pierce Chemical Company
catalog, technical section
on cross-linkers, pages 155-200, incorporated herein by reference).
Suitable recruitment polymers include, but are not limited to, functionalized
styrenes, such as amino
styrene, functionalized dextrans, and polyamino acids. Preferred polymers are
polyamino acids (both
poly-D-amino acids and poly-L-amino acids), such as polylysine, and polymers
containing lysine and
other amino acids being particularly preferred. Other suitable polyamino acids
are polyglutamic acid,
polyaspartic acid, co-polymers of lysine and glutamic or aspartic acid, ca-
polymers of lysine with
alanine, tyrosine, phenylalanine, serine, tryptophan, and/or proline.
In a preferred embodiment, the recruitment linker comprises a metallocene
polymer, as is described
above.
The attachment of the recruitment linkers to the first portion of the label
probe will depend on the
composition of the recruitment linker, as will be appreciated by those in the
art. When the recruitment
linker is nucleic acid, it is generally formed during the synthesis of the
fsrst portion of the label probe,
with incorporation of nucleosides containing ETMs as required. Alternatively,
the first portion of the
label probe and the recruitment linker may be made separately, and then
attached. For example,
there may be an overlapping section of complementarity, forming a section of
double stranded nucleic
acid that can then be chemically crosslinked, for example by using psoralen as
is known in the art.
When non-nucleic acid recruitment linkers are used, attachment of the
linker/polymer of the
recruitment linker will be done generally using standard chemical techniques,
such as will be
appreciated by those in the art. For example, when alkyl-based linkers are
used, attachment can be
similar to the attachment of insulators to nucleic acids.
-81 -
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
In addition, it is possible to have recruitment linkers that are mixtures of
nucleic acids and non-nucleic
acids, either in a linear form (e.g. nucleic acid segments linked together
with alkyl linkers) or in
branched forms (nucleic acids with alkyl "branches" that may contain ETMs and
may be additionally
branched).
In a preferred embodiment, it is the target sequence itself that carries the
ETMs, rather than the
recruitment linker of a label probe. For example, as is more fully described
below, it is possible to
enzymatically add triphosphate nucleotides comprising the ETMs of the
invention to a growing nucleic
acid, for example during a polymerase chain reaction (PCR). As will be
recognized by those in the art,
while several enzymes have been shown to generally tolerate modified
nucleotides, some of the
modified nucleotides of the invention, for example the "nucleoside
replacement" embodiments and
putatively some of the phosphate attachments, may or may not be recognized by
the enzymes to
allow incorporation into a growing nucleic acid. Therefore, preferred
attachments in this embodiment
are to the base or ribose of the nucleotide.
Thus, for example, PCR amplification of a target sequence, as is well known in
the art, will result in
target sequences comprising ETMs, generally randomly incorporated into the
sequence. The system
of the invention can then be configured to allow detection using these ETMs,
as is generally depicted
in Figures 16A, 16B and 16D of U.S.S.N. 60/190,259. ,
Alternatively, as outlined more fully below, it is possible to enzymatically
add nucleotides comprising
ETMs to the terminus of a nucleic acid, for example a target nucleic acid. In
this embodiment, an
effective "recruitment linker" is added to the terminus of the target
sequence, that can then be used for
detection. Thus the invention provides compositions utilizing electrodes
comprising monolayers of
conductive oligomers and capture probes, and target sequences that comprise a
first portion that is
capable of hybridizing to a component of an assay complex, and a second
portion that does not
hybridize to a component of an assay complex and comprises at least one
covalently attached
electron transfer moiety. Similarly, methods utilizing these compositions are
also provided.
It is also possible to have ETMs connected to probe sequences, i.e. sequences
designed to hybridize
to complementary sequences. Thus, ETMs may be added to non-recruitment linkers
as well. For
example, there may be ETMs added to sections of label probes that do hybridize
to components of the
assay complex, for example the first portion, or to the target sequence as
outlined above. These ETMs
may be used for electron transfer detection in some embodiments, or they may
not, depending on the
location and system. For example, in some embodiments, when for example the
target sequence
containing randomly incorporated ETMs is hybridized directly to the capture
probe, as is depicted in
Figure 16A of U.S.S.N. 60/190,259, there may be ETMs in the portion
hybridizing to the capture probe.
-82-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
If the capture probe is attached to the electrode using a conductive oligomer,
these ETMs can be used
to detect electron transfer as has been previously described. Alternatively,
these ETMs may not be
specifically detected.
Similarly, in some embodiments, when the recruitment linker is nucleic acid,
it may be desirable in
some instances to have some or all of the recruitment linker be double
stranded. In one embodiment,
there may be a second recruitment linker, substantially complementary to the
first recruitment linker,
that can hybridize to the first recruitment linker. In a preferred embodiment,
the first recruitment linker
comprises the covalently attached ETMs. In an alternative embodiment, the
second recruitment linker
contains the ETMs, and the first recruitment linker does not, and the ETMs are
recruited to the surtace
by hybridization of the second recruitment linker to the first. In yet another
embodiment, both the first
and second recruitment linkers comprise ETMs. It should be noted, as discussed
above, that nucleic
acids comprising a large number of ETMs may not hybridize as well, i.e. the Tm
may be decreased,
depending on the site of attachment and the characteristics of the ETM. Thus,
in general, when
multiple ETMs are used on hybridizing strands, generally there are less than
about 5, with less than
about 3 being preferred, or alternatively the ETMs should be spaced
sufficiently far apart that the
intervening nucleotides can sufficiently hybridize to allow good kinetics.
In one embodiment, non-covafently attached ETMs may be used. In one
embodiment, the ETM is a
hybridization indicator. Hybridization indicators serve as ETMs that will
preferentially associate with
double stranded nucleic acid, usually reversibly, similar to the method of
Millan et al., Anal. Chem.
65:2317-2323 (1993); Millan et al., Anal. Chem. 662943-2948 (1994), both of
which are hereby
expressly incorporated by reference. In this embodiment, increases in the
local concentration of
ETMs, due to the association of the ETM hybridization indicator with double
stranded nucleic acid at
the surface, can be monitored using the monolayers comprising the conductive
oligomers.
Hybridization indicators include intercalators and minor and/or major groove
binding moieties. In a
preferred embodiment, intercalators may be used; since intercalation generally
only occurs in the
presence of double stranded nucleic acid, only in the presence of double
stranded nucleic acid will the
ETMs concentrate. Intercalating transition metal complex ETMs are known in the
art. Similarly, major
or minor groove binding moieties, such as methylene blue, may also be used in
this embodiment.
Similarly, the systems of the invention may utilize non-covalently attached
ETMs, as is generally
described in Napier et al., Bioconj. Chem. 8:906 (1997), hereby expressly
incorporated by reference.
In this embodiment, changes in the redox state of certain molecules as a
result of the presence of
DNA (i.e. guanine oxidation by ruthenium complexes) can be detected using SAMs
comprising
conductive oligomers.
-83-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
Thus, the present invention provides electrodes comprising monolayers
comprising conductive
oligomers, generally including capture probes, and either target sequences or
label probes comprising
recruitment linkers containing ETMs. Probes of the present invention are
designed to be
complementary to a target sequence (either the target sequence of the sample
or to other probe
sequences, as is described below), such that hybridization of the target
sequence and the probes of
the present invention occurs. As outlined below, this complementarity need not
be perfect; there may
be any number of base pair mismatches which will interfere with hybridization
between the target
sequence and the single stranded nucleic acids of the present invention.
However, if the number of
mutations is so great that no hybridization can occur under even the least
stringent of hybridization
conditions, the sequence is not a complementary target sequence. Thus, by
"substantially
complementary" herein is meant that the probes are sufficiently complementary
to the target
sequences to hybridize under normal reaction conditions.
Generally, the nucleic acid compositions of the invention are useful as
oligonucleotide probes. As is
appreciated by those in the art, the length of the probe will vary with the
length of the target sequence
and the hybridization and wash conditions. Generally, oligonucleotide probes
range from about 8 to
about 50 nucleotides, with from about 10 to about 30 being preferred and from
about 12 to about 25
being especially preferred. In some cases, very long probes may be used, e.g.
50 to 200-300
nucleotides in length. Thus, in the structures depicted herein, nucleosides
may be replaced with
nucleic acids.
A variety of hybridization conditions may be used in the present invention,
including high, moderate
and low stringency conditions; see for example Maniatis et al., Molecular
Cloning: A Laboratory
Manual, 2d Edition, 1989, and Short Protocols in Molecular Biology, ed.
Ausubel, et al, hereby
incorporated by referenece. The hybridization conditions may also vary when a
non-ionic backbone,
e.g. PNA is used, as is known in the art. In addition, cross-linking agents
may be added after target
binding to cross-link, i.e. covalently attach, the two strands of the
hybridization complex.
As will be appreciated by those in the art, the systems of the invention may
take on a large number of
different configurations, as is generally depicted in the Figures of U.S.S.N.
09/626,096 (the Figures in
the next paragraphs refer to the figures of U.S.S.N. 09/626,096). In general,
there are three types of
systems that can be used: (1 ) systems in which the target sequence itself is
labeled with ETMs (see
Figures 16A, 16B and 16D); (2) systems in which label probes directly
hybridize to the target
sequences (see Figures 16C and 16H); and (3) systems in which label probes are
indirectly hybridized
to the target sequences, for example through the use of amplifier probes (see
Figures 16E, 16F and
16G).
-84-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
In general, for all the systems outlined herein, both for nucleic acids and
other target analytes, the
invention provides assay complexes that minimally comprise a target analyte
and a capture binding
ligand. For nucleic acid target sequences, by "assay complex" herein is meant
the collection of
hybridization complexes comprising nucleic acids, including probes and
targets, that contains at least
one label (preferably an ETM in the electronic methods of the present
invention) and thus allows
detection. The composition of the assay complex depends on the use of the
different probe
components outlined herein. The assay complexes may also include label probes,
capture extender
probes, label extender probes, and amplifier probes, as outlined herein and in
U.S.S.N. 09/626,096,
depending on the configuration used.
The assays are generally run under stringency conditions which allow formation
of the label probe
hybridization complex only in the presence of target. Stringency can be
controlled by altering a step
parameter that is a thermodynamic variable, including, but not limited to,
temperature, formamide
concentration, salt concentration, chaotropic salt concentration pH, organic
solvent concentration, etc.
These parameters may also be used to control non-specific binding, as is
generally outlined in U.S.
Patent No. 5,681,697. Thus it may be desirable to perform certain steps at
higher stringency
conditions; for example, when an initial hybridization step is done between
the target sequence and
the label extender and capture extender probes. Running this step at
conditions which favor specific
binding can allow the reduction of non-specific binding.
The reactions outlined herein may be accomplished in a variety of ways, as
will be appreciated by
those in the art. Components of the reaction may be added simultaneously, or
sequentially, in any
order, with preferred embodiments outlined below. In addition, the reaction
may include a variety of
other reagents. These include reagents like salts, buffers, neutral proteins
(e.g. albumin), detergents,
etc which may be used to facilitate optimal hybridization and detection,
and/or reduce non-specific or
background interactions. Also reagents that otherwise improve the efficiency
of the assay, such as
protease inhibitors, nuclease inhibitors, anti-microbial agents, etc., -may be
used, depending on the
sample preparation methods and purity of the target.
Accordingly, the present invention provides biochips, with covalently attached
capture binding ligands
(e.g. capture probes). The biochips are incorporated into the cartridges of
the invention and then fitted
into the stations of the multiplexing devices of the invention for running
assays.
In a preferred embodiment, the biochips are attached to the rest of the
cartridge in a wide variety of
ways. In one embodiment, the biochip is made directly on a portion of the
cartridge and is thus
incorporated into the system. Alternatively, as outlined herein, when the
biochip is formulated on a
85 _
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
different substrate than the remainder of the cartridge, there are a variety
of attachment mechanisms
that can be used, depending on the composition and configuration of the two
substrates. For
example, when the biochip is formulated on printed circuit board
material,~there can be "pins" or "rods"
that are inserted into holes, with subsequent fusion (for example, using
solvents or heat). Similarly,
surtace-to-surtace heat or solvent fusion may be done. Alternatively,
adhesives can be used to glue
the two together. Similarly, these techniques can be used with additional
sealing components such as
gaskets. Alternatively, the biochip may "snap" into the cartridge, using
components such as molded
plastic snapping devices.
The present invention further provides for holders for the cartridges for
loading with samples, prior to
loading the cartridges into the stations of the device. In general, as will be
appreciated by those in the
art, the holders may be configured in a wide variety of ways, depending on the
configuration of the
cartridges and caps, if present. For example, holders that align cartridges
such that standard reagent
handling tools can be used are preferred. As shown in the Figures, holders
that allow the use of
mulltichannel pipettemen or robotic systems based on 96 well formats are
preferred. The holders may
also include the caps, positioned for easy use, or reagents and/or buffer
components. In general, the
holders are fabricated out of materials resistant to the chemicals and
reagents used in the assays.
The cartridges of the invention are designed to be inserted into stations in a
multiplexing device. As
will be appreciated by those in the art and described below, the devices of
the invention can take on a
wide variety of conformations, depending on the desired components, the end
use, the ultimate
desired size of the instrument, etc.
Each multiplexing device has a number of different stations into which the
cartridges are inserted. The
cartridge/station pair can be configured in a variety of ways to include the
use of "snap-in" locks,
asymmetry such that the cartridge only fits into the device in a particular
orientation, different size
stations for different size cartridges (for example, some rare amount of tests
may require special
handling and the machines may be designed with special stations for these
tests). This embodiment
may also utilize electronic sensors that detect the presence or absence of a
cartridge, or whether the
cartridge is correctly positioned.
In general, the number of stations per device will vary with the desired use.
Preferred embodiments
utilize at least two or three stations, with at least 5 -100 being preferred,
and from about 25-50 being
particularly preferred, with 48 being especially preferred. In general, the
devices are laid out as a
matrix, with columns and rows of stations.
As outlined herein, each station can have a number of different functional
components, including, but
-86-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
not limited to, interconnects to electronic components, thermocontrollers,
signaling systems, sensors
fior leak detection, alphanumeric displays, and detectors.
In a preferred embodiment, when the cartridge comprises a biochip that relies
on electrodes for
detection, the stations comprise matching interconnects for the biochip, to
allow electronic
communication between the chip and the device.
In a preferred embodiment, each station comprises an individual thermal
controller. "Thermal
controller" or "thermocontroller" in this context includes elements that can
both heat and cool the
cartridges and thus the samples in the cartridges as well. In general, given
the size and function of the
systems, it is desirable to utilize small, fast thermocontrollers. There are a
wide variety of known
suitable thermocontrollers, including Pettier systems.
In general, the thermocontroller should be able to heat/cool samples ranging
from 0 to about 100°C
and at a rate ranging from 0.01 °C/sec to 10°C/sec.
It should be noted that a thermocontroller can be used after an assay to
destroy the biological material
in the cartridge. That is, it is frequently desirable to minimize the exposure
ofi health care workers and
lab workers to potentially dangerous samples, and to facilitate the disposal
of these materials. The
thermocontroller can be used to heat the spent sample at extreme temperatures
for some period of
time in order to kill or destroy the sample. In addition, heating in
conjunction with the addition of other
generally harsh reagents (strong acid, strong base, etc.) can also be used.
Furthermore, in some
embodiments, an RF antennae is used to generate plasma that is pumped into the
chamber after fluid
evacuation to destroy all biological material.
In one embodiment, rather than each station comprising an individual thermal
controller, sets (for
example, rows or columns) of the stations share a thermal controller. In an
alternative embodiment,
the multiplexing device comprises a single thermal controller.
In a preferred embodiment, the devices of the invention include a "Stat Slot",
where a cartridge can be
put in and read right away at one station, rather than run as a sequence. In
general, the temperature
at this station may be preset.
In a preferred embodiment, the stations of the device include signaling
systems. For example, a
system of lights, particularly colored lights, at each station can be used to
indicate the status of the
cartridge or the assay: cartridge present or absent, assay in progress, error,
assay completed, etc. In
addition, the configuration of the lights may be the code (particularly for
color blind people); two lights
_g~_
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
for cartridge in, flashing lights for assay finished, etc. Again, these
signaling systems may be at each
station or at sets of stations.
In a preferred embodiment, the devices of the invention include an
alphanumeric display to allow the
display of data or other information. For example, this display may be used in
conjunction with a
barcode reader, described below, to show the operator which cartridge was
inserted (e.g. the HIV
panel, the HCV panel, the infectious disease panel, the breast cancer SNP
panel, etc.), or other data
about the cartridge (lot or batch number, etc.). In addition, the display can
be used to give the
operator the test results, etc. As for the signaling systems, a display can be
at each station, or there
may be displays for sets of stations or for the whole device.
In a preferred embodiment, each station of the device may be configured to
allow electrophoresis or
dielectrophoresis on the biochip. That is, as is generally described in
W099/67425 and U.S.S.N.
09!171,981, hereby incorporated by reference, there may be additional
electrodes or electronic
components to allow the concentration and/or movement of analytes to the
surFace of the array.
Similarly, as is described in W099/67425 and U.S.S.N. 09/171,981, the
electrophoresis or
dielectrophoresis electrodes may be contained on the biochip.
In a preferred embodiment, the device (or alternatively, each station)
comprises a barcode reader to
read a corresponding barcode on the cartridge. These barcodes may be used for
a wide variety of
purposes, including, but not limited to, identifying the sample (e.g. patient
number or code), the test
being done, the batch number of the chip, calibration information, assay
protocols including cycle time,
signal processing requirements, etc.
In addition, the barcode can be used to control the instrument. For example,
instrument control may
be through the use of a keyboard, a mouse or a barcode reader. Thus, for
example, there may be
barcodes on the cartridges to indicate the identity of the chip, but also on a
card to scan for starting the
assay, stopping the assay, downloading the data, etc. In a preferred
embodiment, the card of barcode
commands are found in a drawer or storage compartment of the device, outlined
herein.
In a preferred embodiment, each station comprises a memory chip reader. Again,
in this embodiment,
each cartridge comprises a memory chip, that can have sample information (e.g.
patient number or
code), the test being done, the batch number of the chip, calibration
information, assay protocols,
etc.), or what the user interface looks like (for example, not a number but
"HIV positive"), etc.
In a preferred embodiment, each station comprises a memory chip writer to add
information to the
cartridge, such as what test was done, the date, the results, etc.
_88_
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
In a preferred embodiment, each station has encryption components in
conjunction with the cartridge,
to encrypt patient information. There is a growing concern regarding the
confidentiality of patient
information, particularly with regard to employment and insurance issues. Thus
for example, in some
embodiments, the devices of the invention will not allow the operator to know
the results of the test.
Rather, the output will be a confirmation that the test was performed
correctly and a viable answer
received, but will say nothing about the actual test being done or the
results. The test results
themselves, in addition to the patient information, can be encrypted and sent
to a remote location as
outlined below for processing, decryption or storage.
In a preferred embodiment, the device may include drawers or storage
compartments to allow the
storage of reagents, cartridges, caps, holders, pipettemen, etc.
In a preferred embodiment, for example, when fluorescence dyes are used in the
assays, fluorescent
readers are used. In one embodiment, the device comprises a reader at each
station. Alternatively, in
a preferred embodiment, the device comprises a single reader that is moved,
either by moving the
reader or by moving the stations to a single reader within the device. Thus,
in some embodiments,
there are motors, pulleys, cords, etc. to allow the movement of stations,
cartridges or readers.
In a preferred embodiment, the devices of the invention comprise liquid
handling components,
including components for loading and unloading fluids at each station or sets
of stations. The liquid
handling systems can include robotic systems comprising any number of
components. In addition,
any or all of the steps outlined herein may be automated; thus, for example,
the systems may be
completely or partially automated.
As will be appreciated by those in the art, there are a wide variety of
components which can be used,
including, but not limited to, one or more robotic arms; plate handlers for
the positioning of microplates;
holders with cartridges and/or caps; automated lid or cap handlers to remove
and replace lids for wells
on non-cross contamination plates; tip assemblies for sample distribution with
disposable tips;
washable tip assemblies for sample distribution; 96 well loading blocks;
cooled reagent racks;
microtitler plate pipette positions (optionally cooled); stacking towers for
plates and tips; and computer
systems.
Fully robotic or microfluidic systems include automated liquid-, particle-,
cell- and organism-handling
including high throughput pipetting to perform ail steps of screening
applications. This includes liquid,
particle, cell, and organism manipulations such as aspiration, dispensing,
mixing, diluting, washing,
accurate volumetric transfers; retrieving, and discarding of pipet tips; and
repetitive pipetting of
identical volumes for multiple deliveries from a single sample aspiration.
These manipulations are
_89_
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
cross-contamination-free liquid, particle, cell, and organism transfers. This
instrument performs
automated replication of microplate samples to filters, membranes, and/or
daughter plates, high-
density transfers, full-plate serial dilutions, and high capacity operation.
In a preferred embodiment, chemically derivatized particles, plates,
cartridges, tubes, magnetic
particles, or other solid phase matrix with specificity to the assay
components are used. The binding
surfaces of microplates, tubes or any solid phase matrices include non-polar
surfaces, highly polar
surfaces, modified dextran coating to promote covalent binding, antibody
coating, affinity media to bind
fusion proteins or peptides, surface-fixed proteins such as recombinant
protein A or G, nucleotide
resins or coatings, and other affinity matrix are useful in this invention.
In a preferred embodiment, plattorms for multi-well plates, multi-tubes,
holders, cartridges, minitubes,
deep-well plates, microfuge tubes, cryovials, square well plates, filters,
chips,.optic fibers, beads, and
other solid-phase matrices or platform with various volumes are accommodated
on an upgradabl'e
modular plattorm for additional capacity. This modular platform includes a
variable speed orbital
shaker, and multi-position work decks for source samples, sample and reagent
dilution, assay plates,
sample and reagent reservoirs, pipette tips, and an active wash station.
In a preferred embodiment, thermocycler and thermoregulating systems are used
for stabilizing the
temperature of heat exchangers such as controlled blocks or platforms to
provide accurate
temperature control of incubating samples from O~C to 100~C; this is in
addition to or in place of the
station thermocontrollers.
In a preferred embodiment, interchangeable pipet heads (single or multi-
channel ) with single or
multiple magnetic probes, affinity probes, or pipetters robotically manipulate
the liquid, particles, cells,
and organisms. Multi-well or multi-tube magnetic separators or platforms
manipulate liquid, particles,
cells, and organisms in single or multiple sample formats.
In some embodiments, for example when electronic detection is not done, the
instrumentation will
include a detector, which can be a wide variety of different detectors,
depending on the labels and
assay. In a preferred embodiment, useful detectors include a microscopes) with
multiple channels of
fluorescence; plate readers to provide fluorescent, ultraviolet and visible
spectrophotometric detection
with single and dual wavelength endpoint and kinetics capability, fluroescence
resonance energy
transfer (FRET), luminescence, quenching, two-photon excitation, and intensity
redistribution; CCD
cameras to capture and transform data and images into quantifiable formats;
and a computer
workstation.
-90-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
These instruments can fit in a sterile laminar flow or fume hood, or are
enclosed, self-contained
systems, for cell culture growth and transformation in multi-well plates or
tubes and for hazardous
operations. The living cells may be grown under controlled growth conditions,
with controls for
temperature, humidity, and gas for time series of the live cell assays.
Automated transformation of
cells and automated colony pickers may facilitate rapid screening of desired
cells.
Flow cytometry or capillary electrophoresis formats can be used for individual
capture of magnetic and
other beads, particles, cells, and organisms.
The flexible hardware and software allow instrument adaptability for multiple
applications. The
software program modules allow creation, modification, and running of methods.
The system
diagnostic modules allow instrument alignment, correct connections, and motor
operations. The
customized tools, labware, and liquid, particle, cell and organism transfer
patterns allow difFerent
applications to be performed. The database allows method and parameter
storage. Robotic and
computer interfaces allow communication between instruments.
In a preferred.embodiment, the robotic apparatus includes a central processing
unit which ,
communicates with a memory and a set of input/output devices (e.g., keyboard,
mouse, monitor,
printer, etc.) through a bus. Again, as outlined below, this may be in
addition to or in place of the CPU
for the multiplexing devices of the invention. The general interaction between
a central processing
unit, a memory, input/output devices, and a bus is known in the art. Thus, a
variety of different
procedures, depending on the experiments to be run, are stored in the CPU
memory.
These robotic fluid handling systems can utilize any number of different
reagents, including buffers,
reagents, samples, washes, assay components such as label probes, etc.
In a preferred embodiment, the devices of the invention include sensors for
leak detection. These are
generally of two types; either electronic measurements of resistance or the
spiking of the assay with
optical or detectable tags. This may be particularly important in some
embodiments where
biohazardous materials or caustic chemicals are being tested.
In a preferred embodiment, the devices of the invention comprise a device
board that can be used to
do a variety of analyses, including signal processing, digital lock-in,
comprising logic circuits, etc., as
outlined herein.
In a preferred embodiment, the devices of the invention comprise a device
board that can be used to
do a variety of analyses, including signal processing, digital lock-in,
comprising logic circuits, etc., as
-91-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
outlined herein.
In a preferred embodiment, the systems of the invention comprise a processor
or central processing
unit (CPU) with an associated memory. The associated memory can be memory on-
board the
processor and optional memory coupled to the processor via an external memory
bus. This processor
(CPU) can be physically contained within the apparatus itself, can be
connected to the apparatus via a
cable, or can be connected using wireless technology. There can be one or more
per device or one
can be shared among devices.
For example, in one embodiment the systems of the invention provide a
motherboard on which are
mounted the CPU and associated memory. The motherboard may desirably provide
connectors for
mechanically and electrically connecting with one or a plurality of edge
connector mountable printed
circuit cards having the signal processing circuits formed thereon. The
edge.connectors provide
signal and power connections with the motherboard. In one embodiment, the CPU
and memory, in
conjunction with an operating system, support the menu or command driven
operation and analysis
described elsewhere herein. Software andlor firmware executing in the
processor and/or within the
signal processing printed circuit card components are used to control the
operation of the apparatus,
devices, and system.
In yet another embodiment, the system of the invention are configured and
operate in the manner of a
computer peripheral device coupled to an external personal computer. In this
type of implementation,
each signal processing printed circuit card may be connected to the personal
computer by a separate
communication channel or link (such as for example, by one or more serial,
parallel, SCSI, Fire-wire,
blue-tooth, or other wired or wireless communication channel or link), or
multiple signal processing
printed circuit cards may be multiplexed to share a smaller number of
communication channels or
links. Typically, the multiple signal processing printed circuit cards will
interconnect via a
communication bus, such as may be provided by a motherboard or other
interconnect structure.
Each signal processing board may have a unique address (locally or globally
unique) such that
communications directed between signal processing cards or between a signal
processing printed
circuit card to the processor may be identified with the signal processing
card and interpreted and/or
routed accordingly. The PC contains an application program that controls the
instrument and collects
data from the instrument. Those workers having ordinary skill in the art in
light of the description ,
provided here will appreciate that there are numerous ways of connecting
specialized instrumentation
using digital and/or analog circuits, such as the devices and apparatus
describe here, and therefore
not described in greater detail here.
In one particular embodiment, an apparatus having six sensor slots on each of
eight separate printed
-92-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
circuit based signal processing cards is coupled with a personal computer via
at least one serial
interface, such as an RS-232 or RS-485 serial link. Advantageously, a
plurality of such serial ports on
each of the personal computer and apparatus are provided to increase
bandwidth. In one
embodiment, three serial input/output interfaces are provided.
In still another embodiment, a processor or CPU with associated memory may be
provided directly
one each signal processing printed board.
In a preferred embodiment, the devices of the invention include a localization
device, such as a Global
Positioning System (GPS) as are known in the art. This may find particular use
in agriculture and
biowarfare uses, as well as remote diagnosis of problems.
In a preferred embodiment, the devices of the invention include components for
the communication of
data, assay results, patient information, etc. to an off device location.
Thus, for example, one or more
modems (including both telephone and cable modems) , Internet cards, infrared
ports, etc. may be
included in the devices to allow the transmission of data and other relevant
information (barcode
information, assay conditions and protocols, operator identification, time
stamps, etc.) to a remote
location such as a general information repository, hospitals, doctor's
offices, epidemiology centers,
pharmacies, government centers, insurance providers, etc.
In a preferred embodiment, the devices of the invention include components for
wireless
communication systems, to allow this transmission of data in the absence of
physical electronic or
communications connections. In addition, wireless receivers can be included.
Accordingly, the present invention provides methods and compositions for the
multiplex analysis of
samples and target analytes. Samples (either raw samples or treated samples
(e.g. amplified,
purified, etc.)) are loaded into the cartridges of the invention, optional
caps are put on, and the
cartridges loaded into a station of the device. Additional reagents are added
as necessary, and assay
complexes formed.
Once the assay complexes of the invention are made, that minimally comprise a
target sequence and
a label probe, detection proceeds with electronic initiation. Without being
limited by the mechanism or
theory, detection is based on the transfer of electrons from the ETM to the
electrode.
Detection of electron transfer, I.e. the presence of the ETMs, is generaffy
initiated electronically, with
voltage being preferred. A potential is applied to the assay complex. Precise
control and variations in
the applied potential can be via a potentiostat and either a three electrode
system (one reference, one
-93-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
sample (or working) and one counter electrode) or a two electrode system (one
sample and one
counter electrode). This allows matching of applied potential to peak
potential of the system which
depends in part on the choice of ETMs and in part on the other system
components, the composition
and integrity of the monolayer, and what type of reference electrode is used.
As described herein,
ferrocene is a preferred ETM.
In some embodiments, co-reductants or co-oxidants are used as is generally
described in
W000/16089, hereby expressly incorporated by reference.
The presence of the ETMs at the surface of the monofayer can be detected in a
variety of ways. A
variety of detection methods may be used, including, but not limited o,
optical detection (as a result of
spectral changes upon changes in redox states), which includes fluorescence,
phosphorescence,
luminiscence, chemiluminescence, electrochemiluminescence, and refractive.
index; and electronic
detection, including, but not limited to, amperommetry, voltammetry,
capacitance and impedence.
These methods include time or frequency dependent methods based on AC or DC
currents, pulsed
methods, lock-in techniques, filtering (high pass, low pass, band pass), and
time-resolved techniques
including time-resolved fluoroscence.
In one embodiment, the efficient transfer of electrons from the ETM to the
electrode results in
stereotyped changes in the redox state of the ETM. With many ETMs including
the complexes of
ruthenium containing bipyridine, pyridine and imidazole rings, these changes
in redox state are
associated with changes in spectral properties. Significant differences in
absorbance are observed
between reduced and oxidized states for these molecules. See for example
Fabbrizzi et al., Chem.
Soc. Rev. 1995 pp197-202). These differences can be monitored using a
spectrophotometer or
simple photomultiplier tube device.
In this embodiment, possible electron donors and acceptors include all the
derivatives listed above for
photoactivation or initiation. Preferred electron donors and acceptors have
characteristically large
spectral changes upon oxidation and reduction resulting in highly sensitive
monitoring of electron
transfer. Such examples include Ru(NH3)4py and Ru(bpy)Zim as preferred
examples. It should be
understood that only the donor or acceptor that is being monitored by
absorbance need have ideal
spectral characteristics.
In a preferred embodiment, the electron transfer is detected fluorometrically.
Numerous transition
metal complexes, including those of ruthenium, have distinct fluorescence
properties. Therefore, the
change in redox .state of the electron donors and electron acceptors attached
to the nucleic acid can
be monitored very sensitively using fluorescence, for example with Ru(4,7-
biphenyl2-phenanthroline)32.~
-94-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
. The production of this compound can be easily measured using standard
fluorescence assay
techniques. For example, laser induced fluorescence can be recorded in a
standard single cell
fluorimeter, a flow through "on-line" fluorimeter (such as those attached to a
chromatography system)
or a multi-sample "plate-reader" similar to those marketed for 96-well immuno
assays.
Alternatively, fluorescence can be measured using fiber optic sensors with
nucleic acid probes in
solution or attached to the fiiber optic. Fluorescence is monitored using a
photomuitiplier tube or other
light detection instrument attached to the fiber optic. The advantage of this
system is the extremely
small volumes of sample that can be assayed.
1n addition, scanning fluorescence detectors such as the Fluorlmager sold by
Molecular Dynamics are
ideally suited to monitoring the fluorescence of modified nucleic acid
molecules arrayed on solid
surfaces. The advantage of this system is the large number of electron
transfer probes that can be
scanned at once using chips covered with thousands of distinct nucleic acid
probes.
Many transition metal complexes display fluorescence with large Stokes shifts.
Suitable examples
include bis- and trisphenanthroline complexes and bis- and trisbipyridyl
complexes of transition metals
such as ruthenium (see Juris, A., Balzani, V., et. al. Coord. Chem. Rev., V.
84, p. 85-277, 1988).
Preferred examples display efficient fluorescence (reasonably high quantum
yields) as well as low
reorganization energies. These include Ru(4,7-biphenylz phenanthroline)3z+,
Ru(4,4'-Biphenyl-2,2'-
bipyridine)3z+ and platinum complexes (see Cummings et al., J. Am. Chem. Soc.
118:1949-1960
(1996), incorporated by reference). Alternatively, a reduction in fluorescence
associated with
hybridization can be measured using these systems.
In a further embodiment, electrochemiluminescence is used as the basis of the
electron transfer
detection. With some ETMs such as Ruz+(bpy)3, direct luminescence accompanies
excited state
decay. Changes in this property are associated with nucleic acid hybridization
and can be monitored
with a simple photomultiplier tube arrangement (see Blackburn, G. F. Chn.
Chem. 37: 1534-1539
(1991); and Juris et al., supra.
In a preferred embodiment, electronic detection is used, including
amperommetry, voltammetry,
capacitance, and impedence. Suitable techniques include, but are not limited
to, electrogravimetry;
coulometry (including controlled potential coulometry and constant current
coulometry); voltammetry
(cyclic voltammetry, pulse voltammetry (norii~al pulse voltammetry, square
wave voltammetry,
differential pulse voltammetry, Osteryoung square wave voltammetry, and
coulostatic pulse
techniques); stripping analysis (aniodic stripping analysis, cathiodic
stripping analysis, square wave
stripping voltammetry); conductance measurements (electrolytic conductance,
direct analysis); time-
-95-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
dependent electrochemical analyses (chronoamperometry, chronopotentiometry,
cyclic
chronopotentioinetry and amperometry, AC polography, chronogalvametry, and
chronocoulometry);
AC impedance measurement; capacitance measurement; AC voltammetry; and
photoelectrochemistry.
In a preferred embodiment, monitoring electron transfer is via amperometric
detection. This method of
detection involves applying a potential (as compared to a separate reference
electrode) between the
nucleic acid-conjugated electrode and a reference (counter) electrode in the
sample containing target
genes of interest. Electron transfer of differing efficiencies is induced in
samples in the presence or
absence of target nucleic acid; that is, the presence or absence of the target
nucleic acid, and thus the
label probe, can result in different currents.
The device for measuring electron transfer amperometrically involves sensitive
current detection and
includes a means of controlling the voltage potential, usually a potentiostat.
This voltage is optimized
with reference to the potential of the electron donating complex on the label
probe. Possible electron
donating complexes include those previously mentioned with complexes of iron,
osmium, platinum,
cobalt, rhenium and ruthenium being preferred and complexes of iron being most
preferred.
In a preferred embodiment, alternative electron detection modes are utilized.
For example,
potentiometric (or voltammetric) measurements involve non-faradaic (no net
current flow) processes
and are utilized traditionally in pH and other ion detectors. Similar sensors
are used to monitor
electron transfer between the ETM and the electrode. In addition, other
properties of insulators and of
conductors (such as resistance conductivity, impedance and capicitance) could
be used to monitor
electron transfer between ETM and the electrode. Finally, any system that
generates a current (such
as electron transfer) also generates a small magnetic field, which may be
monitored in some
embodiments.
It should be understood that one benefit of the fast rates of electron
transfer observed in the
compositions of the invention is that time resolution can greatly enhance the
signal-to-noise results of
monitors based on absorbance, fluorescence and electronic current. The fast
rates of electron
transfer of the present invention result both in high signals and stereotyped
delays between electron
transfer initiation and completion. By amplifying signals of particular
delays, such as through the use
of pulsed initiation of electron transfer and "lock-in" amplifiers of
detection, and Fourier transforms.
In a preferred embodiment, electron transfer is initiated using alternating
current (AC) methods.
Without being bound by theory, it appears that ETMs, bound to an electrode,
generally respond
similarly to an AC voltage across a circuit containing resistors and
capacitors.
-96-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
There are a variety of techniques that can be used to increase the signal,
decrease the noise, or make
the signal more obvious or detectable in a background of noise. That is, any
technique that can serve
to better identify a signal in the background noise may find use in the
present invention. These
techniques are generally classified in three ways: (1) variations in the type
or methods of applying the
initiation signals (i.e. varying the "input" to maximize or identify the
sample signal); (2) data processing,
i.e. techniques used on the "output" signals to maximize or identify the
sample signal; and (3)
variations in the assay itself, i.e. to the electrode surface or to the
components of the system, that
allow for better identification of the sample signal. Thus, for example,
suitable "input" AC methods
include, but are not limited to, using multiple frequencies; increasing the AC
amplitude; the use of
square wave ACV; the use of special or complicated waveforms; etc. Similarly,
suitable "output" AC
techniques include, but are not limited to, monitoring higher harmonic
frequencies; phase analysis or
filters; background subtration techniques (including but not limited to
impedance analysis and the use
of signal recognition or peak recognition techniques); digital filtering
techniques; bandwidth narrowing
techniques (including lock-in detection schemes particularly digital lock in);
Fast Fourier Transform
(FFT) methods; correlation and/or convolution techniques; signal averaging;
spectral analysis; etc.
Additionally, varying components of the assay can be done to result in the
sample signal and the noise
signal being altered in a non-parallel fashion; that is, the two signals
respond non-linearly with respect
to each other. These techniques are described in W000/16089 and O'Connor et
al., J. Electroanal.
Chem. 466(2):197-202 (1999), hereby expressly incorporated by reference.
In general, non-specifically bound label probes/ETMs show differences in
impedance (e.g. higher
impedances) than when the label probes containing the ETMs are specifically
bound in the correct
orientation. In a preferred embodiment, the non-specifically bound material is
washed away, resulting
in an effective impedance of infinity. Thus, AC detection gives several
advantages as is generally
discussed below, including an increase in sensitivity, and the ability to
"filter out" background noise. In
particular, changes in impedance (including, for example, bulk impedance) as
between non-specific
binding of ETM-containing probes and target-specific assay complex formation
may be monitored.
Accordingly, when using AC initiation and detection methods, the frequency
response of the system
changes as a result of the presence of the ETM. By "frequency response" herein
is meant a
modification of signals as a result of electron transfer between the electrode
and the ETM. This
modification is different depending on signal frequency. A frequency response
includes AC currents at
one or more frequencies, phase shifts, DC offset voltages, faradaic impedance,
etc.
Once the assay complex including the target sequence and label probe is made,
a first input electrical
signal is then applied to the system, preferably via at least the sample
electrode (containing the
complexes of the invention) and the counter electrode, to initiate electron
transfer between the
_97_
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
electrode and the ETM. Three electrode systems may also be used, with the
voltage applied to the
reference and working electrodes. The first input signal comprises at least an
AC component. The AC
component may be of variable amplitude and frequency. Generally, for use in
the present methods,
the AC amplitude ranges from about 1 mV to about 1.1 V, with from about 10 mV
to about 800 mV
being preferred, and from about 10 mV to about 500 mV being especially
preferred. The AC
frequency ranges from about 0.01 Hz to about 100 MHz, with from about 10 Hz to
about 10 MHz being
preferred, and from about 100 Hz to about 20 MHz being especially preferred.
The use of combinations of AC and DC signals gives a variety of advantages,
including surprising
sensitivity and signal maximization.
1n a preferred embodiment, the first input signal comprises a DC component and
an AC component.
That is, a DC offset voltage between the working and counter electrodes is
swept through the
electrochemical potential of the ETM (for example, when ferrocene is used, the
sweep is generally
from 0 to 500 mV) (or alternatively, the working electrode is grounded and the
counter electrode is
swept from 0 to -500 mV). The sweep is used to identify the DC voltage at
which the maximum
response of the system is seen. This is generally at or about the
electrochemical potential of the ETM.
Once this voltage is determined, either a sweep or one or more uniform DC
offset voltages may be
used. DC offset voltages of from about -1 V to about +1.1 V are preferred,
with from about -500 mV to
about +800 mV being especially preferred, and from about -300 mV to about 500
mV being particularly
preferred. In a preferred embodiment, the DC offset voltage is not zero. On
top of the DC offset
voltage, an AC signal component of variable amplitude and frequency is
applied. If the ETM is
present, and can respond to the AC perturbation, an AC current will be
produced due to electron
transfer between the electrode and the ETM. These voltages are meaningful
numbers for a Ag vs an
AgCI reference electrode.
Thus, the devices of the invention preferably provide voltage sources capable
of delivering both AC
and DC currents.
For defined systems, it may be sufficient to apply a single input signal to
differentiate between the
presence and absence of the ETM (i.e. the presence of the target sequence)
nucleic acid.
Alternatively, a plurality of input signals are applied. As outlined herein,
this may take a variety of
forms, including using multiple frequencies, multiple DC offset voltages, or
multiple AC amplitudes, or
combinations of any or all of these.
Thus, in a preferred embodiment, multiple DC offset voltages are used,
although as outlined above,
DC voltage sweeps are preferred. This may be done at a single frequency, or at
two or more
_98_
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
frequencies.
In a preferred embodiment, the AC frequency is varied. At different
frequencies, different molecules
respond in different ways. As will be appreciated by those in the art,
increasing the frequency
generally increases the output current. However, when the frequency is greater
than the rate at which
electrons may travel between the electrode and the ETM, higher frequencies
result in a loss or
decrease of output signal. At some point, the frequency will be greater than
the rate of electron
transfer between the ETM and the electrode, and then the output signal will
also drop.
In a preferred embodiment, multiple frequencies with a small AC voltage is
applied and the
fundamental of each is evaluated. Alternatively, a preferred embodiment
utilizes several frequencies
with a large AC voltage, and the harmonics of each are evaluated. Similarly,
preferred embodiments
utilize several frequencies with a large AC voltage where the effect of the
different frequencies on the
system can result in an output that is different from the sum of the outputs
at individual frequencies.
In one embodiment, detection utilizes a single measurement of output signal at
a single frequency.
That is, the frequency response of the system. in the absence of target
sequence, and thus the
absence of label probe containing ETMs, can be previously determined to be
very low at a particular
high frequency. Using this information, any response at a particular
frequency, will show the presence
of the assay complex. That is, any response at a particular frequency is
characteristic of the assay
complex. Thus, it may only be necessary to use a single input frequency, and
any changes in
frequency response is an indication that the ETM is present, and thus that the
target sequence is
present.
In a preferred embodiment, the input signals and data processing steps are
done to increase the non-
linearity of the system. That is, for example, the ferrocene response reacts
non-linearly, producing a
harmonic response in the signal above that in the background; this harmonic
signal from AC
voltammetry is most likely the result of a harmonic distortion due to the
nonlinear response of the
electrochemical cell; see Yap, J, of Electroanalytical Chem. 454:33 (1998);
hereby incorporated by
reference. Thus, any techniques that increase this non-linearity are
desirable. In a preferred
embodiment, techniques are used to increase the higher harmonic signals; thus,
frequency and
phase-sensitive lock-in detection is performed at both the fundamental
frequency of the applied
waveform and also at multiples of the fundamental frequency (i.e. the higher
harmonics) or just one.
Since the background capacitance responds relatively linearly to AC signals (a
sine wave input AC
voltage results in a relatively nondistorted sine wave output), very little
upper harmonic current is
produced in the background. This gives a dramatic increase in the signal to
noise ratio. Thus,
detection at the higher harmonic frequencies, particularly the third, fourth
and fifth harmonics
_99_
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
(although the harmonics from second to tenth or greater can also be used) is
shown to result in
dramatic suppression of the background currents associated with non-Faradaic
processes (like double
layer charging) that can overwhelm the signal from the target molecules. In
this way, tthe evaluation
of the system at higher harmonic frequencies and phases can lead to signficant
improvements in the
detection limits and clarity of signal. However, in some embodiments, the
analysis of higher
harmonics is not desired.
Thus, in a preferred embodiment, one method of increasing the non-linear
harmonic response is to
increase or vary the amplitude of the AC perturbation, although this may also
be used in monitoring
the fundamental frequency as well. Without being bound by theory, it appears
that increasing the
amplitude increases the driving force nonlinearly. Thus, generally, the same
system gives an
improved response (i.e. higher output signals) at any single frequency through
the use of higher
overpotentials at that frequency. Thus, the amplitude may be increased at high
frequencies to
increase the rate of electron transfer through the system, resulting in
greater sensitivity. In addition,
this may be used, for example, to induce responses in slower systems such as
those that do not
possess optimal spacing configurations.
In a preferred embodiment, measurements of the system are taken at at least
two separate amplitudes
or overpotentials, with measurements at a plurality of amplitudes being
preferred. As noted above,
changes in response as a result of changes in amplitude may form the basis of
identification,
calibration and quantification of the system. In addition, one or more AC
frequencies can be used as
well.
In a preferred embodiment, harmonic square wave AC voltage is used; see
Baranski et al., J.
Electroanal. Chem. 373:157 (1994), incorporated herein by reference, although
in some embodiments
this is not preferred. This gives several potential advantages. For example,
square waves are easier
to create digitally and the pulse shape of the square wave can allow for
better discrimination against
charging capacitance. In sinusoidal harmonic AC voltammetry, harmonic signals
provide better signal
to background since faradaic response can be more nonlinear than charging
capacitance. The same
concept applies to SW harmonic AC voltage. The key difference between the two
techniques is the
frequency spectrum of the AC waveform. A singular frequency sinusoidal
waveform contains just the
fundamental frequency whereas a singular square wave contains the fundamental
frequency as well
as all odd harmonics. The technique looks at the even harmonics where the
ratio of faradaic current
to capacitance current is enhanced. All the odd harmonics have single AC
voltage peaks while all the
even harmonics have double AC voltage peaks. This is opposite to the case of
sinusoidal harmonic
AC voltage of a system that has a non-reversible redox couple.
-100-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
In a preferred embodiment, multiple frequency AC voltage is used. The idea is
to create a waveform
consisting of multiple frequencies with the same amplitude or different
amplitudes to excite an
electrochemical cell in an AC voltage fashion. The method benefits from fast
Fourier transform or joint
time-frequency transform to analyze the cell response. A JTFT spectrogram of a
multiple frequencies
AC voltage provides information on the driven (or fundamental) frequencies as
well as their harmonic
components. Some possible data analyses are: 1 ) comparison of response of
fundamental
frequencies, 2) comparison of all harmonic frequencies, 3) comparison of the
response of one
particular harmonic frequency of all excited frequencies, and 4) all analyses
possible by standard
single frequency AC voltage.
Accordingly, in a preferred embodiment, a fast Fourier transform is done, as
is generally outlined in the
examples. Fourier transform analysis is a preferred method for improving
signal to noise and isolating
desired signals when sinusoidal electrochemistry is done. Typical AC
techniques rely on
measurements of the primary frequency only. With sinusoidal voltammetry (and
other inputs)
observation at higher harmonics allows discrimination of signals primarily
based on kinetics. For
example, both fast and slow redox events would give similar peaks (provided
the AC frequency was
not too high) at the primary frequency. However, at higher harmonics, some
redox molecules viiould
generate signals while others would not. Using FFT analysis, all the various
frequency components of
a response to a sinusoidal input can be observed at once.
Similarly, in a preferred embodiment, a joint time-frequency transform (JTFT)
is done.
In a preferred embodiment, digital lock-in techniques are used. In the past,
digitized raw data from the
electrochemical cell have been analyzed by either fast Fourier transform or
some complex form of joint
time-frequency transform analysis. The major draw back of these methods is the
enormous
computational time associated with frequency transformation techniques.
Digital lock-in, on the other
hand, is simple and fast. In principle, digital lock-in is identical to analog
lock-in. In the former case,
the bandwidth narrowing process is done mathematically by multiplying the cell
response by a
sinusoidal with the same frequency as the input voltage, but with 90°
phase shift. The technique has
the same limitation as its analog counterpart since only one frequency can be
analyzed at a time.
However, unlike analog lock-in, other frequencies can also be analyzed
sequentially (or in parallel with
a more powerful processor) since the raw data is archived. For an input
voltage of
~n Edc+.Y't-I-Lr'p~l~(~~ (1 )
the cell's response is essentially
I ~t~ - ~In ~V~lI~YICt)t (pn ~ - ~I ~n ~h~ZY~nfJJt~ - r ~n ~V~COS~YICOt)
-101 -
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
To find the voltage dependent coefficients h for the frequency (nQ w) we
multiply the response by 2
Sin(wno t) and -2 Cos(wno t) and apply a low pass filter to get the real and
imaginary components.
The low pass filtering used in this example is a simple moving average.
Mathematically, the process
is expressed as
1 ~1 ~l'" (v~S'i~(nc~t)-I"" (v)Cos(nwt) 2Si~(wn~t)dt-_
tl-t0 ~ n
I~n (v) t- St~r(2nOcot) ~ I'jz (v) , for t,-to »T (3)
tl-t0 ~ tl,t0
In a preferred embodiment, background subtraction of the current vector and
phase optimization is
done.
In a preferred embodiment, correlation and/or convolution techniques are used.
In this embodiment,
many scans of the same electrode. Rather than looking for a peak in a single
scan, many scans are
viewed and a common correlation between the scans. For instance, it is
possible that a bump in the
noise appears near 180 mV for a negative, even if no ferrocene is present.
However, it is unlikely that
the same bump will appear in the same place if the frequencies are scanned.
Thus, preferred
embodiments take scans at many frequencies and only count a positive if a peak
occurs in all of them.
This is a very simple correlation; more complex correlations may be done as
well.
In a preferred embodiment, signal recovery is done using signal recognition
and background
subtraction. In this embodiment, the idea is to fit the cell response to two
summed functions, one that
describes the signal and the other that models the background capacitive
current. Once the functions
are constructed, the signal is easily recovered from the response by
subtracting the fitted background
capacitive current. This signal recognition scheme is applicable to any system
where the signal has a
behavior and shape that is relatively well known. The following example
illustrates how such a
scheme can be applied to the systems of the invention.
The response from an electrochemical cell can be processed with a lock-in
amplifier or equivalent
bandwidth-narrowing technique. This is one of many methods of increasing
signal to background
using some form of bandwidth-narrowing technique.
In a preferred embodiment, spectral analysis of the signal is done. In this
embodiment, filtering
techniques in the frequency domain make use of means, variances, densities,
autocorrelation
-102-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
functions, and power spectral densities of the signal and apply it to the
present systems to enhance
the signal to noise ratio (see Schwartz et al., Signal Processing: Discrete
Spectral Analysis, Detection,
and Estimation, N.Y. McGraw HIII, 1975, hereby incorporated by reference).
In a preferred embodiment, digital filtering techniques are used. These
include, but are not limited to,
match filter, Weiner filtering, Kalman, Finite Impulse Response, infinite
impulse response, narrow band
filtering, etc.
In a preferred embodiment, a match filter is used; see Ziemer et al.,
"Principles of Communication
Systems, Modulation and Noise", 4th Ed. John Wiley & Sons Inc., New York, 465-
471, 1988; and
Helstrom, C. W., "Statistical Theory of Signal Detection", Pergamon Press,
Oxford, 112-115, 1968,
both of which are incorporated by reference. In its simplest form, a match
filter is a signal processing
technique that "weights" the measured response (signal plus noise) samples by
some corresponding
known signal amplitude and convolutes the two signals to enhance signal to
noise.
In a preferred embodiment, a Weiner filter is used (see Press, supra; and
Elliot et al., Fast Transforms:
Algorithm, Analysis, Applications N.Y. Academic Press (1982), both of which
are incorporated by
reference). Weiner filtering involves finding an optimal filter that removes
noise or background from
the "corrupted" signal. This signal processing method works in conjunction
with Fourier transform
techniques. The idea is as follows. Due to poor signal to noise or a large
background, the output from
the instrument is a "corrupted" signal
c(t) = s(t) + n(t)
where s(t) is the signal and n(t) is the noise. Note that s(t) is not the
signal we're after, it is composed
of the true uncorrupted signal u(t) convolved with some known response
function r(t) (In the case of
the CMS system with a redox couple, u(t) is the Nernstian). In other words,
s(t) _ ~ r(t -i )u(i )di
In frequency space, the relation is
S(w) = R(e~)U(w)
where S, R, and U are the Fourier transform of s, r, and u, respectively. The
uncorrupted signal can
be recovered by finding the optimal filter c~(t) or its Fourier counterpart
d~(w) which when applied to the
measured signal c(t) or C(c~), and then deconvolved by r(t) or R(c~), produces
a signal that
approximates the uncorrupted signal u(t) or U(w) with
U(~) = C'(~)~(w)
R(w )
-103-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
In general the optimal filter is defined as
S(~ ) Iz
~(~ ) - I S(~ ) Iz + I N(~ ).1z
In a preferred embodiment, a kalman filter is used, which is a recursive-
estimation filtering technique
that tracks the current value of a changing signal in the presence of noise.
See Kalman et al., A New
Approach to Linear Filtering and Prediction Problems, Trans. ASME J. Basic
Engineering, Seires D,
82, March 35, 1960; Elliot Ed. Handbook of Digital Signal Processing:
Engineering Applications" ,
Academic Press, San Diego, p908, 1987; Chui et al., Kalman Filtering: with
Real Time Applications",
Springer-Verlag, New York, 1987; all of which are expressly incorporated by
reference.
In a preferred embodiment, the non-linear harmonic response is increased by
inducing an
asymmetrical response. In a preferred embodiment, this is done by using a
system that has a non-
reversible redox couple. For example, ferrocene is a redox couple that is very
reversible. Thus, the
ferrocenes subtended by the ac voltage at a given point, get oxidized on the
upswing of the ac voltage
and reduced on the down swing. However, If a semi-reversible or non-reversible
redox couple is
used, for example, the molecule will get oxidized on the up swing and not
reduced (or a portion) on the
downswing; or vice versa. This will produce even greater non-linearities at
certain frequencies.
Three examples of ways to perform this are: use an ETM molecule that gets
degraded in the oxidized
form, like luminol, use co-reduction or redox mediation, and use enzyme
coupled mediation, as
generally described in W000116089.
In a preferred embodiment, electron transfer is initiated using alternating
current (AC) methods. In
addition, the use of AC techniques allows the significant reduction of
background signals at any single
frequency due to entities other than the ETMs, i.e. "locking out" or
"filtering" unwanted signals. That is,
the frequency response of a charge carrier or redox active molecule in
solution will be limited by its
diffusion coefficient and charge transfer coefficient. Accordingly, at high
frequencies, a charge carrier
may not diffuse rapidly enough to transfer its charge to the electrode, andlor
the charge transfer
kinetics may not be fast enough. This is particularly significant in
embodiments that do not have good
monolayers, i.e. have partial or insufficient monolayers, i.e. where the
solvent is accessible to the
electrode. As outlined above, in DC techniques, the presence of "holes" where
the electrode is
accessible to the solvent can result in solvent charge carriers "short
circuiting" the system, i.e. they
reach the electrode and generate background signal. However, using the present
AC techniques, one
or more frequencies can be chosen that prevent a frequency response of one or
more charge carriers
in solution, whether or not a monolayer is present. This is particularly
significant since many biological
fluids such as blood contain significant amounts of redox active molecules
which can interfere with
-104-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
amperometric detection methods.
In a preferred embodiment, measurements of the system are taken at at least
two separate
frequencies, with measurements at a plurality of frequencies being preferred.
A plurality of
frequencies includes a scan. For example, measuring the output signal, e.g.,
the AC current, at a low
input frequency such as 1 - 20 Hz, and comparing the response to the output
signal at high frequency
such as 10 - 100 kHz will show a frequency response difference between the
presence and absence
of the ETM. In a preferred embodiment, the frequency response is determined at
at least two,
preferably at least about five, and more preferably at least about ten
frequencies.
After transmitting the input signal to initiate electron transfer, an output
signal is received or detected.
The presence and magnitude of the output signal will depend on a number of
factors, including the
overpotential/amplitude of the input signal; the frequency of the input AC
signal; the composition of the
intervening medium; the DC offset; the environment of the system; the nature
of the ETM; the solvent;
and the type and concentration of salt. At a given input signal, the presence
and magnitude of the
output signal will depend in general on the presence or absence of the ETM,
the placement and
distance of the ETM from the surface of the monolayer and the character of the
input signal. In some
embodiments, it may be possible to distinguish between non-specific binding of
label probes and the
formation of target specific assay complexes containing label probes, on the
basis of impedance.
In a preferred embodiment, the output signal comprises an AC current. As
outlined above, the
magnitude of the output current will depend on a number of parameters. By
varying these parameters,
the system may be optimized in a number of ways.
In general, AC currents generated in the present invention range from about 1
femptoamp to about 1
milliamp, with currents from about 50 femptoamps to about 100 microamps being
preferred, and from
about 1 picoamp to about 1 microamp being especially preferred.
!n a preferred embodiment, the output signal is phase shifted in the AC
component relative to the input
signal. Without being bound by theory, it appears that the systems of the
present invention may be
sufficiently uniform to allow phase-shifting based detection. That is, the
complex biomolecules of the
invention through which electron transfer occurs react to the AC input in a
homogeneous manner,
similar to standard electronic components, such that a phase shift can be
determined. This may serve
as the basis of detection between the presence and absence of the ETM, and/or
differences between
the presence of target-specific assay complexes comprising label probes and
non-specific binding of
the label probes to the system components.
-105-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
The output signal is characteristic of the presence of the ETM; that is, the
output signal is
characteristic of the presence of the target-specific assay complex comprising
label probes and ETMs.
In a preferred embodiment, the basis of the detection is a difference in the
faradaic impedance of the
system as a result of the formation of the assay complex. Faradaic impedance
is the impedance of
the system between the electrode and the ETM. Faradaic impedance is quite
different from the bulk
or dielectric impedance, which is the impedance of the bulk solution between
the electrodes. Many
factors may change the faradaic impedance which may not effect the bulk
impedance, and vice versa.
Thus, the assay complexes comprising the nucleic acids in this system have a
certain faradaic
impedance, that will depend on the distance between the ETM and the electrode,
their electronic
properties, and the composition of the intervening medium, among other things.
Of importance in the
methods of the invention is that the faradaic impedance between the ETM and
the electrode is
signficantfy different depending on whether the label probes containing the
ETMs are specifically or
non-specifically bound to the electrode. .
Accordingly, the present invention further provides electronic devices or
apparatus for the detection of
analytes using the compositions of the invention. The apparatus includes a
test chamber for receiving
a sample olution which has at least a first measuring or sample electrode, and
a second measuring
or counter electrode. Three electrode systems are also useful. The first and
second measuring
electrodes are in contact with a test sample receiving region, such that in
the presence of a liquid test
sample, the two electrophoresis electrodes may be in electrical contact.
In a preferred embodiment, the apparatus also includes detection electrodes
comprising a single
stranded nucleic acid capture probe covalently attached via an attachment
linker, and a monolayer
comprising conductive oligomers, such as are described herein.
The apparatus further comprises an AC voltage source electrically connected to
the test chamber; that
is, to the measuring electrodes. Preferably, the AC voltage source is capable
of delivering DC offset
voltage as well.
In a preferred embodiment, the apparatus further comprises a processor capable
of comparing the
input signal and the output signal. The processor is coupled to the electrodes
and configured to
receive an output signal, and thus detect the presence of the target nucleic
acid.
Once made, the multiplexing devices and cartridges of the invention find use
in a wide variety of
applications. In particular, the compositions of the invention find use in
hybridization assays. As will
be appreciated by those in the art, electrodes can be made that have a single
species of nucleic acid,
i.e. a single nucleic acid sequence, or multiple nucleic acid species.
-106-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
Recent focus has been on the analysis of the relationsliip between genetic
variation and phenotype by
making use of polymorphic DNA markers. Previous work utilized short tandem
repeats (STRs) as
poiymorphic positional markers; however, recent focus is on the use of single
nucleotide
polymorphisms (SNPs), which occur at an average frequency of more than 1 per
kilobase in human
genomic DNA. Some SNPs, particularly those in and around coding sequences, are
likely to be the
direct cause of therapeutically relevant phenotypic variants andlor disease
predisposition. There are a
number of well known polymorphisms that cause clinically important phenotypes;
for example, the
apoE2/3/4 variants are associated with different relative risk of Alzheimer's
and other diseases (see
Cordor et al., Science 261(1993)). Multiplex PCR amplification of SNP loci
with subsequent
hybridization to oligonucleotide arrays has been shown to be an accurate and
reliable method of
simultaneously genotyping at least hundreds of SNPs
The present invention is directed to methods of determining the sequence of a
target nucleic acid at a
particular position, using electrochemical detection on an electrode. The
invention preferably includes
the detection (and optionally quantification) of differences or variations of
sequences (e.g. SNPs) using
electrode arrays for detection of the variation.
As is known in the art, there are a number of techniques that can be used to
detect or determine the
identity of a base at a particular location in a target nucleic acid,
including, but not limited to, the use of
temperature, competitive hybridization of perfect and imperfect probes to the
target sequence,
sequencing by synthesis, for example using single base extension techniques
(sometimes referred to
as "minisequencing"), the oligonucleotide ligase amplification (OLA) reaction,
rolling circle amplification
(RCA), allelic PCR, competitive hybridization and InvaderT"" technologies. In
addition, the present
invention is directed to a novel invention that capitalizes on novel
properties of surface-bound arrays,
and uses "competimers" to reduce non-specific binding.
Thus, the compositions of the present invention may be used in a variety of
research, clinical, quality
control, or field testing settings.
In a preferred embodiment, the probes are used in genetic diagnosis. For
example, probes can be
made using the techniques disclosed herein to detect target sequences such as
the gene for
nonpolyposis colon cancer, the BRCA1 breast cancer gene, P53, which is a gerie
associated with a
variety of cancers, the Apo E4 gene that indicates a greater risk of
Alzheimer's disease, allowing for
easy presymptomatic screening of patients, mutations in the cystic fibrosis
gene, or any of the others
well known in the art.
-107-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
In an additional embodiment, viral and bacterial detection is done. using the
complexes of the
invention. In this embodiment, probes are designed to detect target sequences
from a variety of
bacteria and viruses. For example, current blood-screening techniques rely on
the detection of anti-
HIV antibodies. The methods disclosed herein allow for direct screening of
clinical samples to detect
HIV nucleic acid sequences, particularly highly conserved HIV sequences. In
addition, this allows
direct monitoring of circulating virus within a patient as an improved method
of assessing the efficacy
of anti-viral therapies. Similarly, viruses associated with leukemia, HTLV-I
and HTLV-II, may be
detected in this way. Bacterial infections such as tuberculosis, clymidia and
other sexually transmitted
diseases, may also be detected, for example using ribosomal RNA (rRNA) as the
target sequences.
In a preferred embodiment, the nucleic acids of the invention find use as
probes for toxic bacteria in
the screening of water and food samples. For example, samples may be treated
to lyse the bacteria
to release its nucleic acid (particularly rRNA), and then probes designed to
recognize bacterial strains,
including, but not limited to, such pathogenic strains as, Salmonella,
Campylobacter, Vibrio cholerae,
Leishmania, enterotoxic strains of E. coli, and Legionnaire's disease
bacteria. Similarly,
bioremediation strategies may be evaluated using the compositions of the
invention.
In a further embodiment, the probes are used for forensic "DNA fingerprinting"
to match crime-scene
DNA against samples taken from victims and suspects.
In an additional embodiment, the probes in an array are used for sequencing by
hybridization.
Thus, the present invention provides for extremely specific and sensitive
probes, which may, in some
embodiments, detect target sequences without removal of unhybridized probe.
This will be useful in
the generation of automated gene probe assays.
Alternatively, the compositions of the invention are useful to detect
successful gene amplification in
PCR, thus allowing successful PCR reactions to be an indication of the
presence or absence of a
target sequence. PCR may be used in this manner in several ways. For example,
in one
embodiment, the PCR reaction is done as is known in the art, and then added to
a composition of the
invention comprising the target nucleic acid with a ETM, covalently attached
to an electrode via a
conductive oligomer with subsequent detection of the target sequence.
Alternatively, PCR is done
using nucleotides labelled with a ETM, either in the presence of, or with
subsequent addition to, an
electrode with a conductive oligomer and a target nucleic acid. Binding of the
PCR product containing
ETMs to the electrode composition will allow detection via electron transfer.
Finally, the nucleic acid
attached to the electrode via a conductive polymer may be one PCR primer, with
addition of a second
primer labelled with an ETM. Elongation results in double stranded nucleic
acid with a ETM
-108-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
and electrode covalently attached. In this way, the present invention is used
for PCR detection of
target sequences.
In a preferred embodiment, the arrays are used for mRNA detection. A preferred
embodiment utilizes
either capture probes or capture extender probes that hybridize close to the
3' polyadenylation tail of
the mRNAs. This allows the use of one species of target binding probe for
detection, i.e. the probe
contains a poly-T portion that will bind to the poly-A tail of the mRNA
target. Generally, the probe will
contain a second portion, preferably non-poly-T, that will bind to the
detection probe (or other probe).
This allows one target-binding probe to be made, and thus decreases the amount
of different probe
synthesis that is done.
In a preferred embodiment, the use of restriction enzymes and ligation methods
allows the creation of
"universal" arrays. In this embodiment, monolayers comprising capture probes
that comprise
restriction endonuclease ends, as is generally depicted in Figure 7 of PCT
US97/20014 . By utilizing
complementary portions of nucleic acid, while leaving "sticky ends", an array
comprising any number
of restriction endonuclease sites is made. Treating a target sample with one
or more of these
restriction endonucleases allows the targets to bind to the array. This can be
done without knowing
the sequence of the target. The target sequences can be ligated, as desired,
using standard methods
such as ligases, and the target sequence detected, using either standard
labels or the methods of the
invention.
The present invention provides methods which can result in sensitive detection
of nucleic acids. In a
preferred embodiment, less than about 10 X 106 molecules are detected, with
less than about 10 X 105
being preferred, less than 10 X 104 being particularly preferred, less than
about 10 X 103 being
especially preferred, and less than about 10 X 102 being most preferred. As
will be appreciated by
those in the art, this assumes a 1:1 correlation between target sequences and
reporter molecules; if
more than one reporter molecule (i.e. electron transfer moeity) is used for
each target sequence, the
sensitivity will go up.
All references cited herein, including all patent applications are
incorporated by reference in their
entirety.
EXAMPLES
Example 1
Signal Analysis
The present invention utilizes electrochemical techniques to detect various
biological and chemical
-109-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
targets. Generally these techniques yield signals with an informative or a
characteristic shape, size,
and location. By creating a computer program that recognizes a signal's
characteristic features, we
can distinguish signals from background phenomena and extract any relevant
information necessary
for accurate detection.
Fitting to a Model
It's generally possible to design a family of equations and a set of boundary
conditions that describe
the signals that can arise from a given measurement technique. This
mathematical description is
called a "model." Sometimes the model is based on underlying scientific
theory, but in many cases it
may simply be an approximation that matches the observed signal behavior. In
most cases the model
is "non-linear," comprising equations that are more complicated than basic
polynomials.
There are several ways to fit data to a non-linear model, but they commonly
involve the following
steps: 1 ) the rapid detection of any common behaviors not described by the
model, 2) an initial guess,
3) iterative improvement and evaluation, repeated as necessary, 4) the
detection and correction of
common erroneous fits, if any, and 5) a final evaluation to judge the quality
of the fit. Once a fit is
chosen, the values of important parameters can be extracted for use in further
data analysis.
Vector Notation for Describing AC Signals
As an example illustrating the signal recognition methods described in this
report, I will use what is
currently our most common electrochemical technique: Alternating Current (AC)
Voltammetry
monitored at the fourth harmonic. This technique yields an AC signal (a sine
wave) that varies its
amplitude (height, R) and phase (position, 8) as a function of the input DC
voltage. As long as we
monitor at a known frequency, it only takes two values to define such a wave.
Figure 16 depicts a
sine wave and its corresponding vector notation.
The two values can be R and 8, but as shown in the figure they can also be an
(X,Y) pair separated by
one quarter of an oscillation, i.e. by 90°. One way to simplify the
visualization of such a system is by
using what is called vector notation, demonstrated in four configurations in
Figure 17.
It's important to observe that the values (R,8) and (X,Y) are different but
interchangeable ways of
describing the same vector. The vector itself is what represents the sine wave
and, therefore, the
data. Furthermore, the difference between the primed and unprimed values
(those on the right side of
the diagram versus those on the left) is only a rotated frame of reference (as
indicated, for example,
by the relative positions of the dotted lines in the polar coordinate
diagrams). This rotation also does
-110 -
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
not alter the data, but can be useful as described in later sections.
One important attribute of vector notation is that the vectors add exactly
like the corresponding waves.
For example, if two vectors point in roughly opposite directions, when they
add they tend to cancel one
another, leaving only a small residual vector. This exactly models how it is
possible to add two waves
together in such a way as to have "destructive interference," where the
resulting amplitude is less than
each of the inputs. As long as all waves have the same frequency, vectors will
model their
interference with one another.
In AC Voltariimetry, we monitor the oscillations at a given frequency as a
function of an input voltage.
Since vector notation exactly models sinusoidal behavior at any single, known
frequency, in my
description of our fitting I will only describe the data as a vector.
Choosing to Fit in X and Y
During any experiment acquiring vectoral data, it is common for scientists to
only actively monitor the
value of R (even if both R and B are recorded). This is because, depending on
the system and
experimental setup, the frame of reference may change from one instrument to
another or from one
day to the next. R, however, does not change with the frame of reference.
(Remember that, in polar
coordinates, a frame of reference rotation only changes 8.)
However, in order for signal recognition to work we need a model, a
mathematical description of the
shapes we expect to observe. The simpler and less varied the shape, the easier
the description and
recognition. Figures 18 and 19 are examples of R and 6 traces for fourth
harmonic AC voltammetry
(AC voltage-4).
The four-lobed shape in R-space is characteristic of medium to large signals,
but as the signal shrinks
relative to the size of the background, the R-space signal distorts.
Furthermore, 8 traces of scans
with larger signals are quite different from those with smaller ones. Figures
20 and 21 depict
examples of a smaller signal.
This complex (R,8) behavior is a characteristic of vectoral traces that
comprise both signal and
background. If we have signal s (described by RS and 6S) with a background B
(described by RB
and AB), then the data is D = S + B . D is described by Ro and 60, which have
dependence on the
signal and background values:
-111 -
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
RD _ . JARS sine S + RB sin6 B ~2 + ~RS cose S + RB cos9 B ~~
RS sinA S + RB sinA B
a D = a~~t~n
Rs rose s + ~RB cos9 B
The complexity of R and 8 traces comes from the fact that Ro depends on all
four parameters (RS, AS,
RB, and 6B), as does 00. However, if we use Cartesian coordinates to describe
the data instead of
polar ones, then D is described by Xo and Yo, which are:
XD = Xs + Xa
and
YD = Ys + Ya
Using Cartesian coordinates simplifies the dependence of D's parameters on
those of S and B
This simplicity is exhibited when graphing the same examples shown previously,
but now as (X,Y), as
depicted in Figure 22 and 23 (medium sized signal), and Figure 24 and 25
(smaller signal).
The smaller signal is now qualitatively similar to the medium signal, and is
therefore more likely to be
described by the same mathematical model. Because of this, we chose to fit in
X and Y. (For
simplicity in conceiving a model and in computation during fitting, we chose
to fit independently in X
and Y instead of fitting both dimensions simultaneously.)
The Model Assumed for AC voltage-4
We have compared the characteristic shape exhibited by AC voltage-4 signals to
several different
mathematical expressions. The four-lobed profile immediately suggested we use
an equation related
to the third derivative of a peak shape, and after making many comparisons, we
concluded that the
third derivative of a Gaussian (G"') was a very good approximation to an AC
voltage-4 signal. As for
0 the background, a polynomial (P) should be sufficient to account for the
majority of the different
shapes that we observe. (The order of polynomial (and therefore the number of
parameters needed to
-112 -
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
describe its shape) will depend on the length of the scan. Longer scans will
require higher order
polynomials to account for the same scan features.) This translates to the
following equations, where
I (Y) is the data, I s (v) is the signal, and I a (Y) is the background:
I (Y) = I s (v) + I a (v) _ ~Gx ~. ~(~) + GY .. ~(v)~.+ f px (y) '+- Pr (v)l
_ f Gx ...(y) + Px (V ),+ [GY ...(y) + PY (V )]
This leads to the final equations we used for our AC voltage-4 model:
~(v) = Gx ~ ~ ~ (y) + Px (V )
and
Y(y) ! GY ... (y) + PY (v)
We have also created fitting procedures for systems with more than one label
(differentiated by their
position in voltage space). They use a model analogous to that described
above, but with the
underlying assumption of more than one signal, that is:
I (~) = I E (v) + ~ I S,n (~)
n
As for boundary conditions, note that the Gaussian derivatives G,~" and G,,"'
each have three
parameters: one for height, one for width, and one for location (in voltage).
The height has no
restrictions, corresponding only to the number of electrochemical labels that
are signaling. However,
in order to represent a true electrochemical signal, a fit's width must fall
within a reasonable range.
Furthermore, signals in the independent X and Y fits must be close to one
another (in voltage space)
to assure that they both correspond to the same electrochemical label. Later I
will discuss how these
boundary conditions may be "enforced" to assure a meaningful fit.
Optimal Phase
As mentioned previously, the choice of reference frame is arbitrary. As far as
the data is concerned,
one (X,Y) pair is just as good as another, rotated (X',Y') pair. However,
since the model is often an
approximation to reality instead of an exact theoretical description, the
model may impose a preferred
- 113-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
frame of reference. This is true of the AC voltage-4 system described above.
As an example, lets consider one AC voltage-4 trace that has a large signal
relative to the
background. If, in a two-dimensional graph, we plot the tip of the data vector
as a function of voltage
(one point is plotted every 10 mV), it results in Figure 26.
If we choose a frame of reference such that the X and Y axes straddle the
signal, as shown above,
then the signal contributes strongly to both X and Y, as shown in Figure 27
and 28.
However, if we choose an axis pair that is roughly parallel and perpendicular
to the signal (rotated 45°
with respect to the axes drawn in Figure 26), very little of the signal
contributes to the perpendicular
vector as shown in Figure 29 and 30.
Furthermore, we can see that the four-lobed shape we chose for the AC voltage-
4 model does not
describe the six lobes of the perpendicular trace. If we were to try to use
the model to fit the data
using the parallel and perpendicular (X,Y) pair, we would only be able to
extract the signal out of the
parallel component, thus losing one of the dimensions of our data.
Instruments generally assign the X and Y axes based on the phase of the AC
input driving force.
Because this choice does not take into account the electrochemical system,
it's possible that it may
lead to the parallellperpendicular trouble described above. Therefore, for
signal recognition based on
the above model, it's best to choose a new pair of axes assured to straddle
any existing
electrochemical signal.
In order to choose such axes, we need a way to measure the signal's direction.
We could fit a line to
the signal in polar coordinates, but we can't use basic linear fitting since
the X and Y signals are
independent of one another. For example, imagine a signal aligned along the Y
axis. if we attempt a
linear, least squares fit (the most common type of fitting), the resulting
line is not along the signal but
rather along the X axis, with equal number of points above and below the
fitting line. This is because
the data, when considered only as the values in the Y direction, has no X
dependence.
A non-linear fit would work, but would be an iterative procedure and so would
take more processing
time than we'd care to use. Instead, we'd rather use a faster way involving
simpler mathematical
operations. One such way is using a vectoral sum. Consider the grouping of
three points shown in
Figure 31.
If we consider these points as vectors, we can add them by summing their
coordinates. The vectoral
-114 -
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
sum will have coordinates
X=fix;
and
Y=~Y;
This summation of the vectors provides a reasonable angle for the best line
through the data that
passes through (0,0).
Y
Bop, = arctanC Y ~ = arctan
X ~ x;
We call this angle the "optimal phase." For our example, the summation is
drawn in Figure 32. Figure
33 shows how the three sample data points cluster around the line. For them,
the optimal phase is
64°.
An advantage to this method is that the results are weighted by the length of
the vectors of the original
data points. That is, if a data point has a small amplitude (as it will if it
represents a segment of a scan
where no signal exists), it has a smaller impact on the value of the optimal
phase. For example, if we
add a small data point to the sample grouping, the results are shown in Figure
34 and 35. The new
point changes the optimal phase by less than three degrees.
1f desired, it is possible to give the small values even less weight. A more
generic expression for the
optimal phase has its summations weighted by the lengths (r;) of the
individual data points' vectors.
Increasing the value of n places less and less emphasis on the small data
points. However, in all of
our current fitting programs, we use the equation as written above, equivalent
to the case where n
remains zero.
~ n ( 2 2 '/Z
LJ Y)Yi ~, Y; ~xr + Y;
a op, = arctan ' ~~ = arctan
2 2 /
~x'~~ ~x~~xj +Y~
r
-115 -
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
We can use this method to calculate the optimal phase for fitting a signal,
but there are complications
that must be considered. For many techniques (including AC voltage-4), the
electrochemical signal is
shaped such that portions of it may cancel each other out when completing the
calculation described
above. To avoid this, we first rotate one half of the data 180°. Taking
the data shown in Figure 26, we
calculate the optimal phase using the data as shown in Figure 36. The
resulting line is overlaid on the
original data, at Figure 37.
The angle of the line drawn in Figures 36 and 37 (101 °) is what was
used to choose the X and Y axes
(at ~45°) for this file. Unfortunately, however, there can be a further
complication. If the signal is
oriented differently relative to the dividing line between rotated and
unrotated segments, the stated
manipulation may not yield the proper angle. For example, if I take the above
signal and rotate it 101
degrees clockwise, its optimal phase should be 0°. However, the
calculated value actually ends up as
-48° as shown in Figure 38.
To prevent this, we need to choose a rotation boundary that is more
perpendicular to the signal than it
is parallel. We can do so by first determining whether the signal lies mostly
along 0 degrees or mostly
along 90. If we take the vectoraf sum of the absolute value of the coordinates
of a signal that's closer
to 90, the resulting angle
arctan ~~x,~
will be greater than 45°. On the other hand, for the above case we find
an angle of 10 degrees (see
Figure 39), less than 45, and conclude that the signal is more along 0
degrees. Therefore, we rotate
the half of the signal from the far side of the 90 degree axis (see Figure
40). Calculating the vectoral
sum now yields a reasonable value for the optimal phase: 1 °, similar
to the expected 0°.
One final complication is the fact that we don't want to find the optimal
phase for an entire scan, but
rather for any electrochemical signal present in a scan. We want to ignore any
background.
Unfortunately, if the signal is small, the optimal phase calculations outlined
above would be dominated
by the phase of the background. To avoid this, we perform an approximate
background subtraction
before calculating the vectoral sum.
For example, if we examine the scan represented by Figures 24 and 25, we see a
large contribution
by the background. If we examine the scan in two dimensions, we can see that
the phase of the entire
scan is mostly along 120° (Figure 41).
-116 -
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
However, if we perform the rapid calculations necessary to fit polynomials to
the entire scan (one each
along the 0 and 90° axes), we can approximate the background as shown
in Figure 42 and 43. In this
instance, we are using the symmetry of the AC voltage-4 to our advantage. If a
signal has equal .
contributions above and below the background (as is true of any even harmonic
AC voltage), then a
polynomial fit will tend to follow the centerline of that signal. This makes
the polynomial fit to the scan
an excellent estimation of the background. To calculate the polynomial, we
currently use a "general
polynomial singular value decomposition fit".
This approximation to the background can then be subtracted, converting the
scan into something that
is much more purely signal, as shown in Figure 44 and 45. Figure 46 depicts
this as a two
dimensional plot, from which we can see that now, using the techniques
outlined earlier in this section,
we could calculate an optimal phase of approximately 70°.
In summary, the basic procedure for choosing the X and Y axes is as follows:
1. Fit an approximate background along each of the 0 and 90°
directions. Subtract them from the
scan, leaving a residual trace dominated by signal.
2. Take the absolute value of all coordinates (along 0 and 90°).
Calculate a vectoral sum. Determine
if its angle (A abs) is closer to 0° or 90°.
3. If 8 abs is closer to 0°, select all data points to the left of the
90° axis. If closer to 90°, take all data
points below the 0° axis. Rotate these 180°. (For simplicity in
calculation, we have used the 0° and
90° axes as our boundaries. If a more accurate determination of 6oP, is
required, we can instead rotate
the set of points that falls outside the span of 8abs ~ 90°).
4. Calculate a vectoral sum. Its angle is the "optimal phase" (8 op,).
5. Choose the X and Y axes at ~45° from 8 opt.
Checking for Behaviors Not Modeled
To reduce total processing time, it's best to notice early if a scan has any
gross deviations from the
model that would make fitting it meaningless. One such feature we have
encountered in AC
voltammetry (fourth harmonic) has been the sharp peak caused by the stripping
of a metallic
contaminant. Figure 47 shows an example of one displayed in R-space.
- 117 -
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
In X and Y (at ~ 45° from the optimal phase), the sharp spike feature
remains clear, as shown in
Figure 48 and 49.
The symmetry of this feature distinguishes it from our normal signal: although
AC voltage-4 signals
have equal portions above and below the background, this spike does not.
One quick but rough method of monitoring this symmetry is to separate out an
approximate
background (as we had done to determine the optimal phase) and then compare
the distribution of
points above the baseline with the distribution below. For example, if we
subtract a polynomial from
the Y trace above, we get the results shown in Figures 50 and 51.
If we now examine the distribution of data above and below the approximated
background, we find
that the presence of the spike causes a larger range of values to exist below
the background line than
above it, as shown in Figure 52.
In this example, the standard deviation of the data below the line is about
2'/z times larger than the
standard deviation above. We can take this as an indication of symmetry
different from that expected
of an AC voltage-4 signal, since AC voltage-4 signals are distributed evenly
about the assumed
background and therefore have ratios closer to 1. By setting a range of
acceptable values, this rough
method allows for the rapid detection and rejection of scans with large
spikes. Currently, we consider
a ratio less than 2.25 and greater than 1/2.25 to be acceptable for further
fitting. However, it is
important to be aware that this value can depend on unusual parameter, such as
scan length.
The Initial Guess
Iterative fitting procedures all require a starting point, an initial guess
for the values of the model's
equations that would match the data. Iterations (discussed in the following
section) then improve upon
these guesses in gradual steps. For systems with simple models, there is often
only a need for a
single, predetermined initial guess. In that case, the guess is an adequate
starting point for all
possible data. However, for models that comprise complex shapes (such as the
model we use for AC
voltage-4), an accurate initial guess based on each individual scan can lead
to more rapid fitting and
can reduce or eliminate any tendency to create erroneous fits.
Again, we can use symmetry to our advantage. First, for a signal with symmetry
such that it's
distributed equally above and below the baseline, we can use a fast polynomial
fit to calculate an initial
guess for the background. As was true in the optimal phase calculation
outlined in a previous section,
this polynomial will tend to fit to the centerline of any signal, thus making
a good background
-118-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
estimation. An improvement to get better background estimation is down by
"nailing down" the edges,
thereby reducing edge effects.
When we subtract out the estimation of the background, what remains ("the
residual") should be
mostly signal (if any). To estimate that signal, we can begin with the fact
that a given measurement
technique and chemical system generally yields signals with characteristic
behaviors. For sensors
probed with AC voltage-4, all signals have similar widths. An initial guess
based on the most common
width is therefore appropriate.
To guess the remaining parameters of signal position and signal height, we can
again use the known
AC voltage-4 symmetry, this time combined with knowledge of the characteristic
width. Since we
know the average separation between the two larger center lobes, we can
duplicate the signal and
shift the two copies in opposite directions for half of that separation. If we
then subtract one from the
other, the center lobes interfere constructively. The absolute value of this
resulting wave provides a
good estimation of the height and position of the signal. This process is
shown in Figures 53, 54 and
55 for a signal 11.9 tall at a position of 0.20 with a center lobe separation
of 0.072.
The trace in Figure 55 has its largest value, 23.25, at a position of 0.20.
The position matches well
with the true data value. (Both are 0.20.) The constructive interference
height should be about twice
the center-to-peak signal height, which means the interference plot gives an
initial guess of %2 x 23.25
= 11.6. This is only 3% different from the input value. (This difference
occurs because the shift used
in the above process was 0.062, different from the actual 0.072 separation of
the sample signal.)
One important advantage of the above method (rather than if we were to simply
search for maxima
and minima in the raw data) is that it amplifies only those features with the
expected width and
symmetry of the signal. Consider the same signal as above, but this time with
an unusual peak off to
one side that's slightly taller than the signal itself. (See Figure 56.) In a
simple maxima/minima
search, this would be likely to interfere with the initial guess. However,
using the procedure outlined
above, the initial guess will remain 11.6 tall at a position of 0.20, as shown
in Figure 57.
We therefore have a method of calculating an initial guess for all parameters
(the signal height,
position, and width, plus the background) that involves only 1) calculating a
polynomial using a rapid
(linear) fitting method, 2) searching for the largest number in a set, and 3)
simple arithmetic. As an
example of the power of this technique, Figure 58 is the overlay of a real
data trace and the
corresponding initial guess.
Optimization and Dynamic Range
-119 -
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
Once an initial guess has been made, we can use any of a number of standard
non-linear regression
algorithms to optimize the fit. (We currently use a version based on the
Levenberg-Marquardt method.
This is discussed in Chapter 15 of Numerical Recipes in C: The Art of
Scientific Computing, second
edition, by W. H. Press, S. A. Teukolsky, W. T. Vetterling, and B. P.
Flannery, Cambridge University
Press, New York (1992). It was originally presented by D. W. Marquardt as the
"maximum
neighborhood" method in Journal of the Society, for Industrial and Applied
Mathematics, 11 (1963)
431-441.) While they differ in the details, the basic procedures are all the
same. The initial guess is
compared with the actual data, and the fit is altered based on this comparison
in an attempt to
minimize the "error". We iterate (repeat this process continuously) until the
successive reductions in
the error are smaller than the "precision" (a pre-set constant). In this case,
the fit is said to have
"converged". However, if the data doesn't match the entire shape described by
the model, it's possible
that the alterations to the fit won't reduce the error. (This will happen, for
example, if the scan contains
neither a signal nor noise that looks like a signal.) If the error doesn't
become smaller than the
precision within the "maximum number of iterations" (also a pre-set constant),
the fit is said to have
"diverged".
The error and precision are often defined such that their values have units.
For example, if the data is
a current measured in picoamps; then the error and precision are in picoamps
squared. This is most
useful when all expected signals are of similar size, because it considers
everything in absolute terms
and will not attempt to optimize fits to small features. However, quantitative
analytical techniques
generally require a wide dynamic range. For example, one may need to examine
signals that are two
picoamps tall with the same ease as one examines two billion picoamp signals.
To achieve such a
dynamic range, we normalize data to the initial guess for the signal height.
This allows the small
signals to be fit just as well as the large, with only the shape and the
background noise affecting the fit.
Boundary Conditions and Weighting
The question of boundary conditions arose earlier when discussing the choice
of a model. For some
systems, certain parameters may be reasonable only within a certain range. For
example, for AC
voltage-4 the width of the center two lobes of a sensor signal always falls
between 110 and 265 mV,
and is most commonly between 150 and 200 mV. There are several ways to enforce
these
boundaries, two of which are discussed here.
If the initial guess for a scan is fairly accurate and the scan's signal is
sizeable relative to the
background, the fitting procedure will generally lock into the signal
properly. There will be no need to
enforce the boundary conditions. However, if there's no signal or the signal
is obscured, it's possible
for parameters to drift outside of their acceptable ranges. Now, for a well-
behaved system, the only
-120-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
fits that exhibit this drift have no true signals. We can therefore discard
fits with unacceptable values
and consider the corresponding scans to be comprised only of background. (See
"Quantitation of
Negatives" below.) This is the procedure most widely used in our current
software.
However, for systems that are less well-behaved, we may prefer to enforce the
boundary conditions
during the fitting itself rather than after the fact. We can do this by adding
an additional term to the
equation that describes the error for each constrained parameter, penalizing
the fit as its parameters
deviate from the desired range. For example, if E is the error as defined by
the non-linear regression
technique, and if the parameter a is to be constrained to fall close to some
expected value a, then we
can replace E with E', where k is some constant and n is an integer:
E'=E+k~a-al2n
Remember that, during the iterative optimization, the goal is to minimize the
error. If we use the
equation above, then the farther a is from a the larger the error becomes and
therefore the less
favorable the fit. We can use the value of k to determine how important it is
to constrain the parameter
relative to the standard error E (and also relative to any other parameters'
constraints). The value of n
affects how unfavorable a certain range of values is. For example, in the
graph in Figure 59 we
compare the shape when the added term has 2n = 16 with when 2n = 2. In the
case where 2n = 16, a
values within ~ 7 of the expected are all equally acceptable, with little
added penalty. However, with
2n = 2, there's an increasingly harsh penalty the further a moves from the
expected value.
It's important to note that there's nothing critical about the form of the
added term, so long as it's
always positive. For example, if we wanted a sharper constraint on a near its
true value but not so
much dependence when far off, we could use an expression like:
z
E' = E + k1 arctan a - a
c
This equation leads to shapes like the ones depicted in Figure 60.
Even more complicated shapes may be used. For example, for the case of our AC
voltage-4 center
lobe pairs (described previously), we may wish to use the shapes shown in
Figure 61, which may be
defined by:
- 121 -
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
br f l + tanh~c; ~a - si ~~~+ t~ ba+tt ~1- t~~c.i+t~ ~a - s.i+t~ ~~
im W
where there are n steps up, m steps down, the s's are the locations of the
steps, the 6's are their
heights, and the c's control their steepness.
There is another way to use the error to control where various segments of a
fit lock into a scan. The
mathematical definition of the error E (often called the mean square error, or
MSE) is below:
E - 1 ~ ~Fi _ Di ~z
z
N ;m
In this equation, N is the number of data points, F; is the value of the fit
for data point number i, D; is
the corresponding data value, and 6's is the standard deviation in this data
value. This standard
deviation is a measure of the uncertainty in D;, and is generally known only
if several different
measurements were averaged to create D;. (For data that has not been averaged,
or whose
averaging information has been lost, a value of a's = 1 is assumed, saying
that al! values are known
with equal certainty.) By dividing by ds, we're saying that a larger fitting
error is acceptable for a given
point if that data value was uncertain in the first place.
Irrespective of any uncertainty in data values, we can use the same kind of
manipulation to force the fit
to match the data more closely in certain data ranges than in others by
introducing a weighting w; for
each point. Data points with larger w;'s (a heavier weighting) will be fit
more closely than those with
smaller w;'s:
z
_ _1 N z ~F'i - DJ J
Eweighted - ~ Wi 2
N ,., 6 ;
(Note that this is exactly the same as introducing an alternate standard
deviation equal to ~ J ~wi .)
Now, this does not directly force the signal portion of the fit to exist in a
region with large weighting. It
only forces the fit as a whole to be tighter in this region relative to the
other segments of the scan.
However, the equations that describe signal are more localized than those that
describe background,
and they generally can exhibit much more curvature. Because of this, putting
larger weightings w;
around the expected signal position tends to force the signal to lock in near
that value.
-122-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
Since the initial guess is such an important factor when calculating a good
fit, we need to use the
weighting in that step as well. To accomplish this, we can multiply the Shift
and Subtracts value
described in the "Initial Guess" section by some function, either based on the
weightings w; or on the
term added to create E', depending on the method.
Detecting and Correcting Flawed Fits
After completing the optimization and converging to a fit, it's still possible
for that fit to have locked in
improperly. For example, there can be edge effects. That is, since the
computer has no data outside
the scan range, it is completely free to assume any shape for the data outside
that range. Because of
this, the fitting procedure may conclude that an unusual background
oscillation at the scan's edge is
actually a signal. This kind of fit needs to be discarded, or perhaps avoided
by using the weightings
described above.
Other possible errors may be corrected. For example, in AC voltage-4 we fit in
X and Y
independently. Because of this, for small signals or for scans with wavy
backgrounds, it's possible for
the program to lock into incorrect lobes of the signal in either X or Y. That
is, the fit may name a
satellite and a central peak as the two central peaks. This will manifest
itself as a fit to signal positions
that are substantially different in X and Y. In these cases, we can take the
scan (X or Y) that locked in
too far from the expected position and refit. We base a new initial guess on
the incorrect fit (keeping
the background and the signal width, but correcting the signal position) and
restart the iterative
optimization. If this does not remedy the problem (or if the original
difference in position is very large),
then generally it means that we've locked into noise rather than a signal, so
we discard this fit.
Judgment of Fit Suitability
Once a fit has converged and those fits with common flaws have been discarded
or corrected, we
need to judge if the fit has too much error. That is, we need to make sure
that we've locked into a real
signal. For example, examine the graph shown in Figure 62.
Although the fit may closely follow the average path of the data, in the above
case the fit isn't reliable
because the difference between the fit and the data is too large. There is too
much noise in the scan.
We can judge this quantitatively by setting a threshold value for an
acceptable error E. In the case of
AC voltage-4, since we fit in X and Y independently, we set a threshold value
for maximum noise
allowable in R-space:
-123-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
ER = Ex z + EY z
ER must be less than some empirically-determined value in order for the fit to
be considered as having
locked into a true signal.
Quantitation of Negatives
Once all of the above procedures have been completed, many scans have had fits
diverge or be
discarded. These are classified as having no observable signal, and so are
refit using only
background as the model. For example, Figure 63 is the R composite, ~z + Yz .
fn Figure 64 is the
R composite with the background polynomials subtracted,
( _ z ( _ z
background ~ + \Y Ybockground
After the background has been fit, it's often desirable to extract a
quantitative estimation of the size of
the largest signal that may be hidden within the residual noise. Because the
software recognizes
shape, all we need to consider are oscillations in the noise that are similar
to the shape of the modeled
signals. (In AC voltage-4, signals have a characteristic period in voltage
space: the center lobes
commonly complete one cycle in about 0.16 volts.) By subtracting out the
background fit and
examining only the residual, we remove the low frequency background. To remove
the high frequency
noise, (thus considering only the signal-like oscillations), we push the
residual through a low pass
filter. To determine the appropriate filter parameters for AC voltage-4, we
averaged the power spectra
of the X and Y traces of several thousand files with signals. This yielded an
average frequency (V-')
profile which we used to choose the appropriate parameters to pass all
signals. We commonly use an
IFIR low pass filter. (The Interpolated FIR filter is described by Y. N. Neuvo
et al. in IEEE Trans.
Acoust., Speech, Signal Processing, vol. ASSP-32, pp. 563-570, June 1984.).
Any scan passed
through this filter now holds only those oscillations that might represent an
obscured signal. Figure 65
shows the filtered, signal-like noise is drawn on top of the residual (Raw)
from the previous example.
For AC voltage-4, since the vectoral signal is being fit independently in X
and Y, the filtered residual
can be quantified as follows:
f Zt2Y2dR center-to-peak = ~~.~lte~ed R,~ ~ _ ~ filteredX,~,,s z +
filteredY,~,,s z
-124-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
The RMS measurement was multiplied by sqrt[2] to convert it to a center-to-
peak measure. We
named this residual current value i~.
i~ gives an estimate of how much signal-like noise exists across the scan, but
does not account for 1 )
any attenuation due to the filter, 2) for the fact that our signals are
localized in a single region of the
scan rather than spread across its entire length, or 3) for any possible
limitations of the non-linear
optimization when extracting a signal from noise. Because of this, i~ alone
would underestimate the
largest possible hidden signal. Therefore we include a multiplicative factor
"C" such that C*i~ is equal
to the largest peak height (iP) that might be obscured. (To find the value of
C, we calculated iP/i~ for
several thousand files from many different experiments. C was set equal to the
value of iP/i~ at which
the signals disappear.) In the above example, the largest possible missed
signal is C*i~ = 7.57 x 10-'2.
Background Subtraction and Information Extraction
For scans with signals, once we have a fit, we're armed with all the
information necessary for data
analysis. Using the model as a guide, we can use the fit parameters to
calculate the equation for the
background alone and subtract this from the data. For example, in AC voltage-
4. we can subtract the
polynomial in X and in Y. Figure 66 is the original data, Figure 67 is the
data with the background
subtracted.
We can also calculate values of interest. For example, for AC voltage-4 we can
calculate the peak
height ip and the peak position Eo based on the values fit in X and Y:
1 P = Z PX 2 -~- Z PY 2
2 2 2 2
EoxiPx -I-EDY1PY Eaxipx '+-EoYIPY
Eo = z z - z
IPX + IPY IP
In such a fashion, we are able to reduce an entire scan to a simple subset of
experimentally
meaningful numbers.
Conclusion
We have devised an automated method of fitting that reduces a data scan to a
small number of
parameters from which all experimentally meaningful information is extracted.
Although we have
focused on two-dimensional, vectoral data, simplified versions of the methods
described here apply to
-125-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
1-D scans. The important steps in the outlined procedure include: 1 ) assuming
a model, 2) using
vectoral sums to calculate an optimal phase, 3) checking for behaviors that do
not conform to the
model, 4) making an initial guess, using the inherent properties of the signal
to minimize the effects of
aberrant noise, 5) iterate to optimize the fit, perhaps while implementing a
weighting scheme, 6)
enforcing boundary conditions, 7) detecting and correcting flawed fits, 8)
judging fit suitability, and 9)
extracting meaningful quantitative information. By programming a computer to
follow this process, we
have derived an automated method that extracts meaningful data from one scan
in less time than it
takes the instrumentation to measure the next one.
Example 2
Rapid Extraction of Phase and Amplitude from a Noisy, Digitized Sine Wave of
Known Frequency
In extremely clean systems that create pure sine waves, one can determine a
wave's phase by finding
its zero-crossings and can measure its amplitude by finding local maxima and
minima. If finding these
extrema is too difficult, one can instead calculate the RMS of the wave, in
which case the amplitude
will be the square root of 2) times the extracted RMS value. However, pure
sine waves are rare in real
world systems such as the systems described herein. Noise in the signal, makes
finding maxima,
minima, and zero-crossings more difficult as methods to extract values.
Furthermore, the RMS
method measures power irrespective of frequency, so as the target signal
decreases in amplitude,
high frequency noise begins to dominate and swamp out the true signal.
Non-linear regression techniques can reliably extract values from noisy data,
but they are iterative ,
processes, and as such can take large amounts of computational time. This
example uses only sums
(and differences), and then renormalization and a basic coordinate
transformation. As such, it is
extremely rapid. Also, the sums which comprise all of the data reduction (from
some large number of
points per cycle to just two points per cycle) can easily be programmed into
an embedded device, thus
allowing more rapid data transfer from the acquisition device (e.g. the
instrument of the invention) to
the data manipulation and storage device (e.g. computer).
The following equations are used to extract phase and amplitude from a
sinusoidal signal of known
frequency that has been digitized such that it has 4n points per cycle. The
summations that create X
and Y are a means of averaging away any random noise, and are simple enough to
be programmed
into the firmware.
-126-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
If (in Cartesian coordinates) we plot y=Rsin(2~cft+~)ahd x=Rsin 2~~,ft+~~+~ ,
then we create a circle of amplitude R centered at the origin. If we plot only
one quarter of a
circle, then we are using the values from half of one cycle (the first quarter
for y and the second for x).
For this subset, we can calculate average values of y and x, called y 1/2cycle
and x 1/2cyclev
Yv2~ycle=1/4-pljRsin(2~z+~)dz= ~R(sin~+cos~)=R~~~sin~~+4J and
\0
xvzcy~le=1/Z11/4jJ Rsin(2~z+~)dz=~sin~-cos~)=R R~cos~~+4J
1/4
These values describe a point slightly within the arc of the quarter circle
whose angle bisects the arc's
sweep, and from them we can recover the original phase and amplitude:
2 2
x+
and R= Y
2 2l~
Note that, to include information from the entire cycle when calculating these
average values, we can
simply use the fact that sin(x) _ -sin(x + n/2) to find the same results:
1/4 3/4 1
y-1 1 f Rsin(2~z+~)dz-3/411 2 ~ Rsin(2~z+~)dz =R~~~sin~~+4J
2 1/4-0 0 / v2
x- 1 1 ~~ Rsin(2~z+~ )dz- I f Rsin(2nz+~ )dz = R~2~~os(~+~
2 1/2-1/4 ~/4 1-3/4 3/a 4
- 127 -
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
We can exploit this behavior as follows: First, acquire an AC signal and lock
into a desired frequency
component. Choose the acquisition rate such that the number of points per
cycle is evenly divisible by
four - it can be written as 4n where n is an integer. The first n points
correspond to the first quarter
cycle, the second n points the second quarter, etc.
If we add all n data points in the first quarter of the cycle and subtract
from them all n points in the third
quarter, then we will have an unnormalized value, call it Y---2n~ y ,
analogous to the y described
above. Similarly, adding together the points in the second quarter and
subtracting those in the fourth
will yield X---2n~x . To extract the phase and amplitude from the data, we
replace y and x by Yl2
and Xl2n in the equations for ~ and R above, but with a few alterations
necessitated by the
digitization:
~t~ '' - 4 (m n~ and R=
ZYl~ Cn
where
n ~~ cos( 2 nJJz+~~ sin( 2 nl,z
In the above equation for R, C~ has the following values:
X2+Y2
-128-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
n C" n C"
1 1.00000012 0.900959
2 0.92388013 0.900864
3 0.91068414 0.900789
4 0.90612715 0.900728
0.90402920 0.900548
6 0.90289325 0.900464
7 0.90220830 0.900419
8 0.90176450 0.900353
9 0.90146070 0.900335
0.90124390 0.900328
11 0.901082inf 0.900316
(As n becomes very large, these equations approach those described above for
the continuous case.
1/n goes to zero, and the value of 0.900316 listed for C",, is actually two
times the square root of two
over pi.)
The equations for ~ and C" the previous page describe a procedure by which we
can extract phase
and amplitude from a sinusoidal signal of known frequency that has been
digitized such that it has 4n
points per cycle. The summations that create X and Y are a means of averaging
away any random
noise, and are simple enough that they can be programmed into the firmware.
Example 3
DNA hybridization assays on an eSensorT"" chip typically require an incubation
time of 4 to 6 hours for
a 10 nM range target concentration. This time period was chosen based on
difFusion based
hybridization times in large volume (i.e., 0.5 ml) cartridges. However, for
smaller volume cartridges
(i.e., 100 ~I), a 4 to 6 hour incubation time is not sufficient to achieve
saturated hybridization signals.
Thus, a number of different connective mixing techniques were evaluated in an
eSensor T"' chip in
order to accelerate DNA hybridization.
Experimental Protocols and Materials:
HFE-H was chosen as the model assay system. All chips and reagents used were
from the same
-129-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
stock in order to eliminate sample or chip performance variations:
Chips used: DC 668
Capture probe: D2002
Target concentration: 10 nM (unless otherwise specified)
Signaling probes: D2005 and D2004 (300 nM each)
Hybridization buffer: prepared according to standard protocols
Heating systems: Depending on the type of mixing used, different methods of
heating were
employed. For example, chips were incubated in a connective oven or on chip
heating
devices were used.
Controls: Diffusion based hybridization was used as the control
Horizontal vs Vertical orientation of chip:
Method of heating chips: connective oven at 35°C.
Slow hybridization kinetics was observed in chips that were incubated in a
horizontal orientation for
100 NI volume cartridges. At 10 nM target concentration chips incubated
horizontally did not reach
saturation signal values by 5 hours,. However, if the concentration of the
target was increased to 100
nM, saturation signal values were observed within 5 to 6 hours.
Increasing the thickness of the chamber.also improved the performance of the
horizontally incubated
chip due to increased volume/z-dimension. See Figure 70A.
Chips that were incubated vertically performed better and reached saturation
signal values with 2 to 3
hours for a 10 nM target concentration.
Recirculation Pumping
Method of Heating: connective oven at 35°C.
Chips were attached to a mini-peristaltic pump using micro bore peek tubing
with a total dead volume
of nearly 7 NI (< 10% of chip volume of 8- to 100 NI). The fluid was
recirculated at approximately 100
pllmin. The entire pump, tubing and chip assembly was placed inside an oven at
35°C.
Measurements were made in real time by placing the eSensorT""-600board inside
the oven.
Saturation signals for the pumped chips were obtained within 1 hour. See
Figure 70B.
Bubble Assisted Piezo (PZT) Mixing
-130-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
Cartridge covers were drilled with 500 pm diameter holes that served as sites
for bubbles when the
hybridization buffer was added to the chip. A piezo-electric transducer was
glued on the back of the
cartridge and excited with 5 Khz a.c. waveform at 10Vp-p. This device was
operated in two modes, a
square wave or a sine wave excitation waveform. PZT based chips performed
better than diffusion
contols, requiring 2.5 hours to reach saturation. See Figure 70C.
Thermal Gradient Based Mixing
A thermal gradient was created across an assembled chip by heating the bottom
of the chip to 65°C
and cooling the top of the cartridge cover to 10°C. The thermal
gradient was created by jacketing the
chip between two pettier heaters. The diffusion control chip was heated using
a pettier heater rather
than the convective oven. Using pettier heating improved hybridization
kinetics in the vertical diffusion
control chip such that a saturation signal was observed within 2 hours. The
chip in which thermal
gradient mixing was used performed even better and reached saturation values
within one hour. See
Figure 70D.
Biochannel with Bubble Pump Based Mixing
A microchannel was place on top of the eSensorT"" chip by cutting adhesive
tape into approximately 1
mm wide thin strips and placing these strips on top of the eSensorT"" chip. A
bubble was intentionally
introduced into one corner of the chip. This bubble was then utilized to
enhance mixing by alternately
expanding and contracting the bubble volume by heating and cooling the bubble
area. This approach
allows mixing due to the pressure flow created by changing the volume of the
bubble within the chip.
However, the addition of microchannels decreased the chip volume to
approximately 20 NI, resulting in
slower kinetics in this system. Using the bubble pump accelerated the
kinetics, but the kinetics were
still not as good as observed with other mixing techniques. Typical results
are shown in Figure 70E.
Acoustic Streaming
The proprietary technology of Covaris, Inc. was used to generate mixing in the
chip. In this method, a
fluid jet surrounds the chip cartridge, through which acoustic waves are
transferred to the chip. A non-
standard cartridge of 20 NI was used and the assays were performed without
temperature control as
no temperature control is currently feasible with this system. Saturation
signal levels were lower using
this system regardless of whether mixing was used. Typical results are shown
in Figure 70F.
Conclusions
Mixing TechniqueSaturation SignalTime to SaturationRate Enhancement
(nA)
(hours) Over Diffusion
Control
Vertical Diffusion50 3 not applicable
(35C, oven)
-131-
CA 02427669 2003-05-02
WO 02/43864 PCT/USO1/44364
Vertical Diffusion60 2 not applicable
(35C, pettier)
Thermal Gradient60 1 2X
Recirculation 50 1 3X
Pumping
Bubble Assisted 50 2.5 1.2X
Acoustic Streaming
-132-