Note: Descriptions are shown in the official language in which they were submitted.
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
MICROPARTICLES OF BIODEGRADABLE POLYMER
ENCAPSULATING A BIOLOGICALLY ACTIVE SUBSTANCE AND
SUSTAINED RELEASE PHARMACEUTICAL FORMULATIONS
s CONTAINING SAME.
The present invention relates to novel microparticles of biodegradable polymer
encapsulating a water-soluble or water-insoluble biologically active
substance, a
method for preparing same and sustained release pharmaceutical formulation
to comprising those microparticles.
Many different methods of preparation of microparticles are described in the
literature (Herrmann et al., European Journal of Pharmaceutics and
Biopharmaceutics 45 (1998) 75-82). The methods presently used for the
is preparation of microparticles from hydrophobic polymers generally are
organic
phase separation and solvent removal techniques.
The solvent removal techniques can be divided into solvent evaporation,
solvent
extraction, spray drying and supercritical fluid technology. In solvent
evaporation
20 or solvent extraction techniques, a drug containing organic polymer
solution is
emulsified into an aqueous or another organic solution. The drug is dissolved,
dispersed or emulsified in the inner organic polymer solution.
These solvent removal techniques for production of microspheres by evaporation
2s or extraction necessitate the step of preparing a stable emulsion of
organic
droplets before solvent removal. The size and characteristics of the final
microspheres depend on this step during which a stable emulsion in the
presence
of the solvent is a prerequisite. The proportions of organic solvent and
aqueous
phase in the solvent removal methods are carefully maintained so as to control
3o the solvent migration in the aqueous phase. Below a certain ratio organic
solvent/aqueous phase, the formation of droplets is not possible any more (see
H.
Sah, "Microencapsulation techniques using ethyl acetate as a dispersed solvent
1
CONFIRMATION COPY
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
effects of its extraction rate on the characteristics of PLGA microspheres,"
Journal
of controlled release, 47 (3) 1997, 233-245). In some methods, solvent is even
added to the aqueous phase in order to saturate it and to prevent the solvent
migration during the formation of the primary emulsion.
s
Several related patents and published applications describe various aspects of
these processes.
EP 0 052 105 B2 (Syntex) describes a microcapsule prepared by the phase
to separation technique using a coacervation agent such as mineral oils and
vegetable oils.
EP 0 145 240 B1 (Takeda) discloses a method for encapsulating a water-soluble
compound by thickening the inner phase of a W/O emulsion, building a W/O/W
is and subjecting the emulsion to an "in water drying" process. This method
brings
different drawbacks such as: the necessity of using a thickening agent to
retain
the drug, and the multi-step procedure including two emulsification steps and
the
"in water drying" step.
zo EP 0 190 833 B1 (Takeda) describes a method for encapsulating a water-
soluble
drug in microcapsules by increasing the viscosity of a primary W/0 emulsion to
150-5,000 cp (by the procedure of increasing the polymer concentration in the
organic phase or by adjusting the temperatures) prior to formation of a second
W/O/W emulsion which is then subjected to "in water drying". The drawbacks of
2s this procedure are the complexity of the necessary steps, including
formation of
two emulsions (W/O and W/O/W) one after the other, and the step of "in-water
drying".
US 5,407,609 (Tice/SRI) describes a microencapsulation process for highly
3o water-soluble agents. This process involves the distinct steps of forming a
primary
O/W emulsion, the external aqueous phase being preferably saturated with
polymer solvent. This O/W emulsion is then poured to a large volume of
extraction medium in order to extract immediately the solvent. The drawback of
2
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
this method is that the O/V1I emulsion is formed in the presence of the
organic
solvent in a small volume. The solvent is subsequently removed by extraction
in a
large aqueous volume. The polymeric droplets are prevented to harden in the
primary emulsion, allowing the migration of the drug into the external phase.
s
WO 95/11008 (Genentech) describes a method for the encapsulation of
adjuvants into microspheres. The process comprises the three distinct steps of
preparing a primary W/O emulsion, followed by the production of a W/O/W and
finally the hardening of the microspheres by extraction of the solvent. As
already
Io mentioned above, the drawback of such a method is the complication due to a
multi-step procedure separating droplet production from solvent elimination.
EP 0 779 072 A1 (Takeda) describes an "in-water drying" method used for the
removal of solvent after production of a W/O/W or a O/W emulsion. It is
is mentioned that the O/W method is preferable for active substances insoluble
or
sparingly soluble in water.
WO 00/62761 discloses a method for the preparation of microparticles
encapsulating water-soluble biologically active substances with an extremely
high
2o encapsulation rate thanks to the optimal reduction of diffusion for the
substance
to be encapsulated. That method comprises the steps of first incorporating a
biodegradable polymer in an organic liquid phase comprising at least one
organic
non-water miscible solvent, then pouring said organic phase being into an
aqueous liquid phase having a volume which is sufficient to dissolve said
organic
Zs solvent, said aqueous phase containing a surfactant, and homogenizing the
resulting organic/aqueous phase in order to perform in one single step the
microparticle formation and the organic solvent removal. The microparticles
are
collected at the end of the homogenization step by filtration and then vacuum
dried at room temperature (see Examples 1 to 6).
The applicant has now surprisingly found that performing the sequence of steps
of the method disclosed in WO 00/62761 with the difference that microparticles
3
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
collected at the end of the homogenization step are suspended without vacuum
drying thereof in a lyophilisation medium, yields novel microparticles of
compartmentalized structure: they are non porous microparticles of irregular
spheroidal shapes wherein pocket microparticles contain microparticles of
smaller
s size, the active substance being evenly distributed within the polymer
matrix.
Probably due to that compartmentalized structure and/or the low level
diffusion
external to the microparticles during the preparation process thereof, those
microparticles have the advantageous property of releasing the active
substance
io in a regular and slow manner. When the core loading of active substance is
below
a threshold value, those microparticles show no or a very low burst, probably
due
to a molecular dispersion of the active principle within the polymer matrix.
Other
novel microparticles of similar structure having those advantageous properties
are
obtained when performing the same sequence of steps with a water-insoluble
is biologically active substance.
The invention thus concerns microparticles of biodegradable polymer
encapsulating a water-soluble or water-insoluble biologically active
substance,
wherein pocket microparticles contain microparticles of smaller size, wherein
said
2o microparticles are obtainable by a method comprising
(a) pouring an organic liquid phase comprising, in a dissolved state the
biodegradable polymer and in a uniformly distributed state the biologically
active substance, in a non-water miscible organic solvent showing a low
solubility in water, into an aqueous liquid phase of sufficient volume to
2s dissolve said organic solvent, said aqueous phase containing a surfactant,
and homogenizing the resulting organic/aqueous phase, thereby forming a
suspension of microparticles, and
(b) filtering the suspension obtained in (a), optionally washing the
microparticles with water, suspending the microparticles without vacuum
3o drying thereof in a lyophilization medium and freeze-drying.
4
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
When the biologically active substance is water-soluble, the organic liquid
phase
may be prepared by dissolving or dispersing that substance in a volume of
water,
dissolving the biodegradable polymer in a 10 to 100 larger volume of non-water
miscible organic solvent showing a low solubility in water, and mixing under
s vigorous agitation the aqueous and the organic solutions obtained, e.g. by
pouring the aqueous solution into the organic solution and homogenizing the
mixture at high rotation speed, for example using a Polytron PT 6100 (PT-DA
3020/2TM shaft) at 10 000 to 30 000 rpm.
io When the biologically active substance is water-insoluble, the organic
liquid
phase may be prepared by dissolving that substance together with the
biodegradable polymer in a non-water miscible organic solvent showing a low
solubility in water.
is One of the specific features in the process for preparing the
microparticles is that
no stable emulsion comprising organic solvent droplets occurs in step (a) when
pouring the organic liquid phase into an aqueous liquid phase of sufficient
volume
to dissolve said organic solvent. Avoiding such a step results in a better
retention
of the biologically active substance and a direct harvesting of the
microparticles
2o after their formation.
Because the microparticle formation and the solvent removal are done together
in
one single step in this process, the water-soluble biologically active
substance is
quickly kept inside the microparticles which have an impermeable wall. Thereby
2s any diffusion of the active substance external to the microparticles is at
a low
level, the encapsulation rate is very high and the amount of the biologically
active
substance on the surface of the microparticles is minimal. Hence a release of
the
active substance in a regular and slow manner. When the core loading is low
enough for a very fine dispersion, probably a molecular dispersion, of the
active
3o principle within the polymer matrix, those microparticles show no or a very
low
burst.
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
Those microparticles with no or a very low burst may show an initial release
of the
active substance of less than 10 % during the first 24 hours, or even below 3%
during the first 48 hours.
s The organic solvents used in the process of the present invention are non-
water
miscible solvents showing a low solubility in water such as esters (e.g. ethyl
acetate, butyl acetate), halogenated hydrocarbons (e.g. dichloromethane,
chloroform, carbon tetrachloride, chloroethane, dichloroethane,
trichloroethane),
ethers (e.g. ethyl ether, isopropyl ether), aromatic hydrocarbons (e.g.
benzene,
io toluene, xylene), carbonates (e.g. diethyl carbonate), or the like.
Although these
solvents are generally classified by the person skilled in the art as non-
water
miscible solvent, they are actually sparingly miscible in water, having a low
solubility in water. For instance, for ethyl acetate and dichloromethane, the
solubility is respectively 8.70% and 1.32% (by weight) in water at 20-
25°C (see
is A.K. Doolittle Ed., Properties of individual solvents, in The technology of
solvents
and plasticizers, chpt. 12. Wiley, New York, 1954, pp. 492-742). One of the
preferred solvent is ethyl acetate.
The above-mentioned organic solvents can be used alone or in mixtures of two
or
2o more different solvents.
The volume of the aqueous liquid phase must be sufficient to dissolve, or
extract,
the total amount of organic solvent used. If this is not the case, the
microparticles
cannot be sufficiently hardened. Those "soft" microparticles may therefore
melt
2s among each other during the filtration process.
Accordingly, the amount of organic solvent is kept as low as possible to get a
viscous organic phase and to minimize the necessary volume of the aqueous
phase. In all of the following embodiments, the volume of the aqueous phase is
3o chosen to be capable of dissolving at least the complete amount of organic
solvent.
6
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
The maximal value of the ratio solvent/water (w/w) in the present invention
should
therefore preferably be 0.087 and 0.013 for ethyl acetate and dichloromethane
respectively. In the examples given below, the ratio ethyl acetate/aqueous
phase
ranges from 0.007 to 0.06. The encapsulating efficiency improves if the volume
of
s aqueous phase increases.
A surfactant is added to the aqueous phase in order to keep the precipitating
biodegradable polymer in fine independent particles. An ideal surfactant gives
a
viscosity to the aqueous phase that approaches the viscosity of the organic
io phase.
An electrolyte may also be optionally added to the aqueous solution to create
repulsion between the particles and preventing aggregation. As a preferred
electrolyte, sodium chloride is used in the aqueous phase and leads to a
higher
is encapsulating efficiency.
The aqueous solution can also be buffered to obtain good pH conditions for the
drug concerning stability and release.
2o When a solvent such as ethyl acetate is used, it has been surprisingly
found that
the encapsulation efficiency is increased when using cold solutions, by
optimizing
the solubility of the solvent in water, by reducing the aqueous solubility of
the
drug, and by slowing down its diffusion. In other words, the present invention
achieves the effect of further reducing the already small amount of diffusion
of
2s internal particle substances to the exterior.
A water-soluble biologically active substance is dispersed as such or as an
aqueous solution into one of the above-mentioned non-miscible organic solvent.
In some embodiments of the process, the biologically active substance is
present
3o in solid state in the organic phase during the entrapment procedure, thus
slowing
down the solubilisation into the aqueous liquid phase.
7
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
The thus obtained liquid organic phase containing the biologically active
substance is used to dissolve the biodegradable polymer.
The appropriate biodegradable polymers comprise poly(lactides),
poly(glycolides),
s copolymers thereof or other biodegradable polymers such as other aliphatic
polymers, polycitric acid, polymalic acid, polysuccinates, polyfumarates, poly
hydroxybutyrates, polycaprolactones, polycarbonates, polyesteramides, poly
anhydrides, poly(amino acids), polyorthoesters, polycyano-acrylates,
polyetheresters, poly(dioxanone)s, copolymers of polyethylene glycol (PEG),
1o polyorthoesters, biodegradable polyurethanes, polyphosphazenes.
Other biocompatible polymers are polyacrylic acid, polymethacrylic acid,
acrylic
acid-methacrylic acid copolymers, dextran stearate, ethylcellulose, acetyl
cellulose, nitrocellulose, etc. These polymers may be homopolymers or
is copolymers of two or more monomers, or mixtures of the polymers.
A particularly interesting biodegradable polymer is poly(D-L-lactide-co-
glycolide).
The biologically active substance and the polymer can also be incorporated in
2o separate organic phases. The polymer is dissolved in another above-
mentioned
organic non-water miscible solvent. Preferred solvents include ethyl acetate
or
dichloromethane. More preferred is when the solvent used to dissolve the
polymer is the same solvent as that use for incorporating the biologically
active
substance. The thus obtained separated organic phases are poured together to
2s form a homogenous organic phase before addition to the aqueous phase.
If the biologically active substance and/or the biodegradable polymer is not
or is
only slightly soluble in one of the above-mentioned solvent, for instance in
the
preferred solvent ethyl acetate, a sufficient amount of co-solvents such those
3o comprised among the family of benzyl alcohol, DMSO, DMF, ethyl alcohol,
methyl
alcohol, acetonitrile and the like, may optionally be used for that purpose.
8
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
A better encapsulating efficiency can be achieved by an appropriate setting of
the
physic chemical parameters such as surfactant capacity, viscosity,
temperature,
ionic strength, pH and buffering potential during the homogenization of the
organic inner phase into the aqueous phase. By carefully adjusting the
production
s parameters, the precipitating polymer can be surprisingly well formed into
homogeneously dispersed particles.
Preferably, the amount of solvent used to dissolve the biodegradable polymer
is
kept to a minimum in order to be soluble as quickly as possible (most
preferably
io at once) in the aqueous phase. If the amount of solvent is high, the amount
of
aqueous phase has to be too large on a practical point of view.
The concentration of polymer in the organic phase is adjusted to 5-90% (by
weight), preferably between about 10 and 50%, depending on the polymer and
is solvent used.
In the case that the concentration of polymer in the organic solvent is high,
the
viscosity of this phase, depending on the polymer used, may be increased.
2o The viscosity of the polymer solution may be comprised between 1000 and
40,000 centipoise (cp) (Brookfield viscosity), more preferably between 2,000
and
30,000 cp, even more preferably between 3,000 and 20,000 cp.
Using solvents like ethyl acetate for dissolving the polymer, the solubility
of the
2s solvent in the aqueous phase is increased by lowering the temperature of
both
the organic and the aqueous phases, accelerating the solvent migration and
therefore also the encapsulation rate.
In process of the present invention, the temperature of the organic phase
ranges
3o between about -10°C and 30°C, and preferably between about
0°C and 10°C.
For ethyl acetate, the temperature ranges preferably between about 2°C
and 5°C.
The temperature of the polymeric organic phase and the temperature of the
9
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
aqueous phase are the same or different and are adjusted in order to increase
the solubility of the solvent in the aqueous phase.
The obtained organic phase for use as the inner polymer and biologically
active
s substance containing phase is added to a aqueous outer phase under a
homogenization procedure to give microparticles.
For the homogenization procedure, a method of creating dispersion is used.
This
dispersion can be realized for example with any apparatus capable of shaking,
to mixing, stirring, homogenizing or ultrasonicating.
Different agents influencing the physico-chemical characteristics of the
resultant
medium may be added. For instance, surfactants, such as for example an anionic
surfactant (e.g. sodium oleate, sodium stearate, sodium lauryl sulfate), a
nonionic
~s surfactant (e.g. polyoxyethylene-sorbitan fatty acid ester (Tween 80, Tween
60,
products available from Atlas Powder Co, U.S.A.), a polyoxyethylene castor oil
derivative (HCO-60, HCO-50, products available from Nikko Chemicals, Japan)),
polyvinyl pyrrolidone, polyvinyl alcohol, carboxymethyl-cellulose, lecithin or
gelatine.
In specific embodiments of the present invention, a surfactant comprised among
the family of anionic, non-ionic agents or other agents capable of reducing
the
surface tension of the polymeric dispersion can be added. Suitably, therefore,
are
nonionic surfactants such as Tween (for example Tween 80), anionic
surfactants,
2s nonionic surfactant like polyvinyl alcohol or others. These surfactants
can, in
general, be used alone or in combination with other suitable surfactants. The
concentration of the surfactant is selected in order to disperse and stabilize
the
polymer particles, and possibly also to give a viscosity approaching the
viscosity
of the organic phase.
The preferred concentration of the surfactant in the aqueous phase ranges
therefore between about 0.01-50% (by weight), preferably between about 5 and
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
30%. The viscosity depending on the surfactant used and on its concentration
ranges between about 1,000-8,000 cp (Brookfield viscosity), preferably about
3,000-5,000 cp.
s Optionally salts comprised among the family of sodium chloride, potassium
chloride, carbonates, phosphates and the like can be added to the aqueous
phase to adjust ionic strength and to create a Zeta potential between the
polymer
particles, leading to particle repulsion.
1o Additional buffering agents may be added to the aqueous phase to maintain a
specific pH. So, the internal aqueous phase may be supplemented with a pH
regulator for retaining stability or solubility of the biologically active
substance,
such as carbonic acid, acetic acid, oxalic acid, citric acid, phosphoric acid,
hydrochloric acid, sodium hydroxide, arginine, lysine or a salt thereof. The
pH of
is the formulations of this invention is generally about 5 to 8, preferably
about 6.5 to
7.5.
The temperature of the aqueous phase can be adjusted to the temperature of the
inner organic phase. The temperature range is from about -10°C to
30°C, more
2o preferably between 0° and 10° C and even more preferably from
between 2°C
and 5°C.
The microparticles of the present invention can be prepared in any desired
size,
ranging from 1~m to about 500~~m, by varying the parameters such as polymer
2s type and concentration in the organic phase, volumes and temperature of the
organic and aqueous phase, surfactant type and concentration, homogenization
time and speed. The mean particle size of the microparticles ranges generally
from 10 to 200ym, more preferably from 20 to 200ym, even more preferably from
30 to 150~m.
A number of water-soluble active substances can be encapsulated by the process
of the present invention.
11
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
Preferably, the encapsulated soluble substance is a peptide, a polypeptide, a
protein and their related pharmaceutically acceptable salts. The salt of
peptide is
suitably a pharmacologically acceptable salt. Such salts include salts formed
with
s inorganic acids (e.g. hydrochloric acid, sulfuric acid, nitric acid),
organic acids
(e.g. carbonic acid, bicarbonic acid, succinic acid, acetic acid, propionic
acid,
trifluoroacetic acid) etc. More preferably, the salt of peptide is a salt
formed with
an organic acid (e.g. carbonic acid, bicarbonic acid, succinic acid, acetic
acid,
propionic acid, trifluoroacetic acid) with greater preference given to a salt
formed
io with acetic acid. These salts may be mono-, di- or tri-salts.
Examples of water-soluble active substances which can be encapsulated in the
microparticles of the present invention include, but are not limited to,
peptides,
polypeptides and proteins such as luteinizing hormone releasing hormone
is (LHRH) or derivatives of LHRH comprising agonists or antagonists,
melanocyte
stimulating hormone (MSH), thyrotropin releasing hormone (TRH), thyroid
stimulating hormone (TRH), follicule stimulating hormone (FSH), human
chorionic
gonadotropin (HCG), parathyroid hormone (PTH), human placental lactogen,
somatostatin and derivatives, gastrin, prolactin, adreno-corticotropic hormone
20 (ACTH), growth hormones (GH), growth hormone releasing hormone (GHRH),
growth hormone releasing peptide (GHRP), calcitonin, oxytocin, angiotensin,
enkephalins, endorphin, enkephalin, kyotorphine, interferons, interleukins,
tumor
necrosis factor (TNF), erythropoetin (EPO), colony stimulating factors (G-CSF,
GM-CSF, M-CSF), thrombopoietin (TPO), platelet derived growth factor,
fibroblast
25 growth factors (FGF), nerve growth factors (NGF), insulin like growth
factors
(IGF), amylin peptides, leptin, RGD peptides, bone morphogenic protein (BMP),
substance P, serotonin, GABA, tissue plasminogen activator (TPA), superoxide
dismutase (SOD), urokinase, kallikrein, glucagon, human serum albumin, bovine
serum albumin, gamma globulin, immunomodulators (EGF, LPS), blood
3o coagulating factor, lysozyme chloride, polymyxin B, colistin, gramicidin,
bacitracin
and the like.
12
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
A number of other unlimiting examples of water-soluble substances or
particularly
a water-soluble form of the following substances can be encapsulated by the
process of the present invention.
s These substances comprise for instance anticancer drugs such as actinomycin
D,
bleomycin, carboplatin, carmustine, chlorambucil, cisplatin, cladribine,
cyclophosphamide, cytarabine, dacarbazine, daunorubicin, doxorubicin,
estramustine, etoposide, floxuridine, fludarabine, fluorouracil,
hexamethylmelamine, hydroxyurea, idarubicin, ifosfamide, asparaginase,
io lomustine, mechlorethamine, melphalan, mercaptopurine, methotrexate,
mithramycin, mitomycin C, mitotane, mitozantrone, oxaliplatine, pentostatin,
procarbazine, streptozocin, teniposide, thioguanine, thiopeta, vinblastine,
vincristine, , an aromatase inhibitor such as Fradrazol or Anastrazol and the
like;
antibiotics such as tetracyclines, penicillins, sulfisoxazole, ampicillin,
is cephalosporins, erytromycin, clindamycin, isoniazid, amikacin,
chloramphenicol,
streptomycin, vancomycin, salvicin and the like.
Other examples of such substances comprise analgesics and antiinflammatory
agents include acetaminophen, acetylsalicylic acid, methylprodnisolone,
ibuprofen
2o diclofenac sodium, indomethacin sodium, flufenamate sodium, pethidine
hydrochloride, levorphanol tartrate, morphine hydrochloride, oxymorphone and
the like; anesthetics such as xylocaine and the like; antiulcer agents include
metoclopramide, ranitidine hydrochloride, cimetidine hydrochloride, histidine
hydrochloride, and the like anorexics such as dexedrine, phendimetrazine
2s tartrate, and the like; antitussives such as noscapine hydrochloride,
dihydrocodeine phosphate, ephedrine hydrochloride, terbutaline sulfate,
isopreterenol hydrochloride, salbutamol sulfate, and the like; antiepileptics
such
as acetazolamide sodium, ethosuximide, phenytoin sodium, diazepam and the
like; antidepressants such as isocarboxazide, phenelzine sulfate,
clomipramine,
3o noxiptilin, imipramine, and the like anticoagulants such as heparin or
warfarin,
and the like.
13
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
Other unlimiting examples comprise sedatives such as chlorpromazine
hydrochloride, scopolamine methylbromide, antihistaminics such as
diphenhydramine hydrochloride, ketotifen fumarate, chlorpheniramine maleate,
methoxy-phenamine hydrochloride and the like.
s
Other unlimiting examples comprise cardiotonics such as etilefrine
hydrochloride,
aminophylline and the like; antiasthmatics such as terbutaline sulfate,
theophylline, ephedrine, cetirizin and the like; antifungals such as
amphotericin B,
nystatin, ketoconazole, and the like; antiosteopotic agents such as
io bisphosphonates, e.g. alendronate, antiarrhytmic agents such as propranolol
hydrochloride, alprenolol hydrochloride, bufetolol hydrochloride, oxyprenolol
hydrochloride and the like; antitubercular agents such as isoniazid,
ethambutol,
and the like; hypotensive, diuretic agents such as captopril, ecarazine,
mecamylamine hydrochloride, clonidine hydrochloride, bunitrolol hydrochloride
is and the like; hormones such as prednisolone sodium sulfate, betamethasone
sodium phosphate, hexestrol phosphate, dexamethasone sodium sulfate and the
like; antigens from bacteria, viruses or cancers, antidiabetics such as
glipizide,
phenformin hydrochloride, buformin hydrochloride, glymidine sodium,
methformin,
and the like; cardiovascular agents such as propanolol hydrochloride,
2o nitroglycerin, hydralazine hydrochloride, prazosin hydrochloride and the
like;
diuretics such as spironolactone, furosemide and the like; and enzymes,
nucleic
acids, plant extracts, anti-malarials, psychotherapeutics, hemostatic agents,
etc.
Examples of water-insoluble biologically active substances which can be
2s encapsulated in the microparticles of the present invention include, but
are not
limited to, anesthetics such as lidocaine and the like, anorexics such as
phendimetrazine, antiarthritics such as methylprednisolone, ibuprofen and the
like, antiasmathics such as terbutaline and the like, antibiotics such as
sulfisoxazole, cephalosporins, tetracyclines, erythromycin, clindamycin and
the
30 like, antifungals such as amphotericin B, nystatin, ketoconazole and the
like,
antivirals such as acyclovir, amantadine and the like, anticancer agents such
as
methotrexate, etretinate, aromatase inhibitors such as Exemestane, Formestane,
14
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
Letrozole, Vorozole, Aminoglutetimide and the like, anticoagulants such as
warfarin and the like, anticonvulsants such as phenytoin and the like,
antidepressants such as amoxapine and the like, antihistamines such as
dephenydramine, chlorpheniramine and the like, hormones such as insulin,
s progestins, thyroxines, estrogens, corticoids, androgens and the like,
tranquilizers
such as chlorpromazine, reserpine, chlordiazepoxide and the like,
antispasmodics
such as Belladonna alkaloids, dicyclomine and the like, vitamins and minerals,
cardiovascular agents such as prazosin, nitroglycerin, propanolol,
hydralazine,
linsidomin, verapamil and the like, peptides and proteins such as LHRH,
io somatostatin, vasopressin and the like, prostaglandins, nucleic acids,
carbohydrates, fats, narcotics such as morphine, codeine and the like,
psychotherapeutics, anti-malarials, diuretics such as furosemide,
spironolactone
and the like, and antiulcer drugs such as ranitidine, cimetidine and the like.
is Preferred substances include Tamoxiphen, 4-OH Tamoxiphen, a derivative
thereof, a non-soluble LHRH derivative such as Triptorelin pamoate and a non-
soluble somatostatin derivative such as OctreotideTM, LanreotideT"' or
vapreotide
pamoate.
2o The invention also concerns a sustained release pharmaceutical formulation
which comprises a suspension of the above microparticles in a pharmaceutically
acceptable vehicle. Preferably the initial release of the active substance
during
the first 24 hours is less than 10 %. More preferably the initial release
during the
first 48 hours is less than 3 %.
2s
The invention also relates to a method of preparing the above microparticles,
which comprises
(a) pouring an organic liquid phase comprising, in a dissolved state the
biodegradable polymer and in a uniformly distributed state the biologically
3o active substance, in a non-water miscible organic solvent showing a low
solubility in water, into an aqueous liquid phase of sufficient volume to
dissolve said organic solvent, said aqueous phase containing a surfactant,
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
and homogenizing the resulting organic/aqueous phase, thereby forming a
suspension of microparticles, and
(b) filtering the suspension obtained in (a), optionally washing the
microparticles with water, suspending the microparticles without vacuum-
s drying thereof in a lyophilization medium and freeze-drying.
The examples that follow are set forth as an aid in understanding the present
invention, and provide some examples of the many embodiments that are
potentially available for the present invention. They are not intended to
limit the
to scope of the invention.
The following description will be better understood by referring to Figures
1A, 1B,
2A and 2B, 3, 4 and 5.
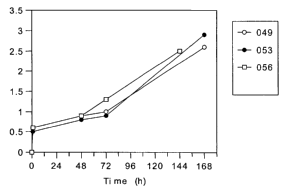
is Figures 1A, 1B, 3 and 4 are curves representing the in vitro release
profiles of
batches (49, 53 and 56) and (58, 59 and 60) of Triptorelin acetate
encapsulating
microparticles, batch 4 of Tamoxifen encapsulating microparticles, and batch 5
of
4-OH-Tamoxiphen, respectively.
2o Figure 2A represents the variation of the cum AUC (cumulated area under the
curve) of the Triptorelin acetate level for batches 56 and 69 of Triptorelin
acetate
as a function of time for rats, injected on day 1 with a suspension of
Triptorelin
acetate encapsulating microparticles of batches 56 and 69.
2s Figure 2B represents the variation of serum testosterone levels as a
function of
time for rats previously housed close to female rats, injected on day 1 with a
suspension of Triptorelin acetate encapsulating microparticles of batches 56
and
57 and non treated rats as a control.
3o Figures 5A and 5B represent scanning electron microscopy photographs of a
Triptorelin acetate encapsulating microparticle of batch 57. Those photographs
show a primary polymeric non porous pocket microparticle containing secondary
16
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
polymeric microparticles of smaller size, the active substance being evenly
distributed within the polymer matrix.
Figure 6 represents a transmission electron microscopy photograph of a
s Triptorelin acetate encapsulating microparticle of batch 57 cut in a thin
layer. That
photograph shows a polymeric non porous pocket microparticle containing
secondary microparticles of smaller size.
Example 1 Preparation of different batches of Triptorelin acetate
encapsulating
io microparticles
The following sequence of steps was performed for each batch:
1. About 940 mg of D-Trps-LHRH acetate (Triptorelin acetate) were dissolved in
is 9.4 g of sterile distilled water. This aqueous phase solution was cooled to
4 °C.
2. About 25.0 g of poly(D-L-lactide-co-glycolide) (PLGA) with a ratio of
lactide to
glycolide of 50/50 and a weight average molecular weight of 45,000 were
dissolved in 250 g of ethyl acetate at room temperature. This organic phase
2o solution was cooled to 4°C.
3 .The aqueous phase solution was poured into the organic phase solution and
the mixture was homogenized using a Polytron PT 6100 (PT-DA 3020/2TM shaft)
at 20 000 rpm during 2 minutes.
2s
4. This w/o preparation was poured into about 8500 g of aqueous phase
containing 20% (w/w) of polyoxyethylene sorbitan fatty acid ester (Tween 80)
and
possibly 84.4 g of sodium chloride, in a reactor kept at a temperature of
4°C.
30 5. The homogenization was performed using a Polytron PT 6100 (PT6020/2TM
shaft) at 3000-3500 rpm during 5 minutes, thereby forming a suspension of
microparticles
17
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
6. The microparticles ~ivere collected by filtration and washed with about 9 I
of
sterile distilled water yielding a bulk of microparticles.
s 7. The bulk was possibly frozen, kept overnight and thawed.
8. The microparticles were suspended in a lyophilization medium consisting of
mannitol, Tween 80 and sodium carboxy-methyl-cellullose using a magnetic
rod at 200 rpm, and possibly homogenized using an IKA-T25 homogenizes at
io 8000 rpm during 20 minutes or at 9500 rpm during 30 minutes (batches 58-
60). The suspension was freeze-dried.
The obtained microparticle lyphilisate showed less than 2 % residual water.
is The entrapment efficiency was measured by UV spectrometry on the bulk of
microparticles and by HPLC on the lypholisate and the particle size
distribution
was determined using a laser granulometer (Mastersizer~, Malvern Instruments).
Batch 49, obtained using a reactor with a conic lower part in steps 4 and 5,
no
2o sodium chloride in step 4, no step 7, and no homogenization in step 8,
showed a
mean particle size of 92.5 ~m and an entrapment efficiency of 81.8 % on the
lyophilisate.
Batch 53, obtained using a reactor with a conic lower part in steps 4 and 5,
84.4 g
zs sodium chloride in step 4, no step 7, and no homogenization in step 8,
showed a
mean particle size of 96.1 ~m and an entrapment efficiency of 75.4 % on the
lyophilisate.
Batch 56, obtained using a reactor with a conic lower part in steps 4 and 5,
no
3o sodium chloride in step 4, and step 7, and homogenization in step 8, showed
a
mean particle size of 70.0 ym and an entrapment efficiency of 87.7 % on the
bulk.
18
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
Batch 57, obtained using a reactor with a conic lower part in steps 4 and 5,
84.4 g
sodium chloride in step 4, no step 7 and homogenization in step 8, showed a
mean particle size of 84.3 ~m and an entrapment efficiency of 91.7 % on the
bulk.
s Batch 58, obtained using a reactor with a flat bottom in steps 4 and 5, no
sodium
chloride in step 4, no step 7, and homogenization in step 8, showed a mean
particle size of 39.2 pm.
Batch 59, obtained using a reactor with a conic bottom in steps 4 and 5, no
to sodium chloride in step 4, no step 7, and homogenization in step 8, showed
a
mean particle size of 59.3 Vim.
Batch 60, obtained as batch 59 but with a different PLGA 50/50 having a lower
average molecular weight, showed a mean particle size of 87.1 Vim.
Batch 69 obtained using a reactor with a flat bottom in steps 4 and 5, no
sodium
chloride in step 4, no step 7, showed a mean particle size of 56.5 ~m and an
entrapment efficiency of 70.6 %.
2o Example 2 In vitro release profile of Triptorelin acetate encapsulating
microparticles
The lyophilisate microparticles were put in a methanol/water mixture under
stirring
at 200 rpm at 37 °C in a test representative of the physiological
conditions in the
zs human body. Samples from this mixture were analyzed as a function of time
by
HPLC.
The in vitro release curves for the seven batches of Triptorelin acetate
encapsulating microparticles are represented in Figures 1A and 1B.
Those curves show for all batches a release of the therapeutically active
substance of less than 3 % during the first 48 hours.
19
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
Example 3A Effect of injection of Triptorelin acetate encapsulating
microparticles
in rats on the serum Triptorelin acetate levels
s Protocol: 6 adult male rats are given an intramuscular injection of a
suspension of
Triptorelin acetate encapsulating microparticles in sterile distilled water.
Blood
samples are then collected regularly from day 1 (day of the injection) through
day
35 for determining of Triptorelin acetate levels.
io Figure 2A represents the variation of the cum AUC (cumulated area under the
curve) of the Triptorelin acetate level for batches 56 and 69 of Triptorelin
acetate
as a function of time.
That curve shows that the cum AUC, i.e. the burst, is less than 10 % after 24
is hours and the variation of that parameter is linear until day 35.
Example 3B Effect of injection of Triptorelin acetate encapsulating
microparticles
in rats on the serum testosterone levels
20 6 adult male rats previously housed close to female rats (for testosterone
stimulation) are given an intramuscular injection of a suspension of
Triptorelin
acetate encapsulating microparticles in sterile distilled water. Blood samples
are
then collected regularly from day 1 (day of the injection) through day 42 for
determining of testosterone levels.
2s
The curves of mean testosterone levels as a function of time (expressed in
days)
for Triptorelin acetate encapsulating microparticles of batches 56 and 57 and
a
control, are represented in Figure 2C.
3o Those curves show that satisfying testosterone levels below 3.5 nmol/I
corresponding to a castration condition are obtained for all samples as from
day 5
to day 36.
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
Example 4 Preparation of Tamoxifen encapsulating microparticles
Batch 4 of Tamoxifen encapsulating microparticles was prepared using a
s sequence of steps similar to that described in Example 1 for batch 56, with
the
main difference that in the first step water-insoluble Tamoxifen is dissolved
in the
ethyl acetate solution together with the PLGA 50/50 having a average molecular
weight of 45,000, the following steps being very similar to steps 4 to 8.
io That batch showed a mean particle size of 49.6 ~m as determined by laser
granulometry and an encapsulation efficiency of 82.8 %.
Example 5 In vitro release profile of Tamoxifen encapsulating microparticles
is The lyophilisate microparticles were put in a methanol/water mixture under
stirring
at 200 rpm at 37 °C in a test representative of the physiological
conditions in the
human body. Samples from this mixture were analyzed as a function of time by
HPLC.
2o The in vitro release curve for batch 4 of Tamoxifen encapsulating
microparticles is
represented in Figure 3.
That curve shows a release of the therapeutically active substance of less
than
% during the first 48 hours, and a linear release up to 1 month.
2s
Example 6 Preparation of 4-OH-Tamoxifen encapsulating microparticles and in
vitro release profile thereof
Batch 5 of the Z isomer of 4-OH-Tamoxifen encapsulating microparticles was
3o prepared using a sequence of steps similar to that described in Example 1
for
batch 56, with the main difference that in the first step water-insoluble 4-OH-
Tamoxifen is dissolved in the ethyl acetate solution together with the PLGA
50/50
21
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
having a average molecular weight of 45,000, the following steps being very
similar to steps 4 to 8.
That batch showed a mean particle size of 53.98 ~m as determined by laser
s granulometry and an encapsulation efficiency of 66.92 % on the lyophilisate.
An in vitro release test similar to that described in Example 2 showed a
release of
the therapeutically active substance of about 9.2 %, i.e. a burst of less than
10
during the first 24 hours, and a linear release up to 500 hours (see Figure
4).
io
Example 7 Preparation of RC-160 encapsulating microparticles
The following sequences of steps was performed:
is 1. About 175.0 mg of Vapreotide acetate were dissolved in 2.0 g of sterile
distilled water. This aqueous phase solution was cooled to 4°C.
2. About 5.0 g of poly(D-L-lactide-co-glycolide) (PLGA) with a ratio of
lactide
to glycolide of 50/50 and a weight average molecular weight of 45,000 Dalton
2o were dissolved in 50.0 g of ethyl acetate at room temperature. This organic
solution was cooled to 4°C.
3. The aqueous phase solution was poured into the organic phase solution
and the mixture was homogenized using a Polytron PT 6100 (PT-DA
zs 3020/2TM shaft) at 20,000 rpm during 2 minutes.
4. This w/o preparation was poured into about 1687.5 g of aqueous phase
containing 20% (w/w) of polyoxyethylene sorbitan fatty acid ester (Tween 80)
in a reactor kept at a temperature of 4°C.
22
21
CA 02435415 2003-07-21
WO 02/058672 PCT/CH02/00048
5. The homogenization was performed using a Polytron PT 6100
(PT6060/2TM shaft) at 3,000 rpm during 5 minutes, thereby forming a
suspension microparticles.
s 6. The microparticles were collected by filtration and washed with bout 1.7
I of
sterile distilled water yielding a bulk of microparticles.
7. The bulk was possibly frozen, kept overnight and thawed.
l0 8. The microparticles were suspended in a lyophilization medium consisting
of mannitol and sodium carboxy-methyl-cellulose (and possibly Tween 80)
using an IKA T25 homogenizes at 9,500 rpm during 30 minutes. The
suspension was poured on a tray and freeze-dried.
is 9. The obtained freeze-dried microparticles were sieved on 106 Vim.
The obtained freeze-dried microparticles showed less than 2% residual water.
The entrapment efficiency was measured by HPLC on the freeze-dried
microparticles and the particles size distribution was determined using a
laser
2o granulometer (MastersizerTM, Malvern Instruments).
An in vitro release test similar to that described in Example 2 showed a
release of
the therapeutically active substance of less than 10 % during the first 24
hours,
and a linear release up to 500 hours.
23