Note: Descriptions are shown in the official language in which they were submitted.
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
INTERFERON GAMMA POLYPEPTIDE VARIANTS
FIELD OF THE INVENTION
The present invention relates to novel interferon gamma polypeptide variants
having
inteneron gamma (IFNG) activity, methods for their preparation, pharmaceutical
compositions comprising the polypeptide variants and their use in the
treatment of diseases,
in particular for the treatment of interstitial pulmonary diseases, such as
idiopathic pulmonary
fibrosis.
BACKGROUND OF THE INVENTION
Interferon gamma (IFNG) is a cytolune produced by T-lymphocytes and natural
filler
cells and exists as a homodimer of two noncovalently bound polypeptide
subunits. The
mature form of each dimer comprises 143 amino acid residues (shown in SEQ ID
N0:17),
the precursor form thereof includes 166 amino acid residues (shown in SEQ ID
N0:18).
Each subunit has two potential N-glycosylation sites (Aggarwal et al., Human
Cytokines, Blackwell Scientific Publications, 1992) at positions 25 and 97.
Depending on the
degree of glycosylation the molecular weight of IFNG in dimer form is 34-50
kDa (Farrar et
al., Ann. Rev. Immunol, 1993, 11:571-611).
The primary sequence of wild-type human IFNG (huIFNG) was reported by Gray et
al. (Nature 298:859-863, 1982), Taya et al. (EMBO J. 1:953-958, 1982), Devos
et al.
(Nucleic Acids Res. 10:2487-2501, 1982) and Rinderknecht et al. (J. Biol.
Chem. 259:6790-
6797, 1984), and in EP 0 077 670, EP 0 089 676 and EP 0 110 044. The 3D
structure of
huIFNG was reported by Ealick et al. (Science 252:698-702, 1991).
Various naturally-occurring or mutated forms of the IFNG subunit polypeptides
have
been reported, including one comprising a Cys-Tyr-Cys N-terminal amino acid
sequence
(positions (-3)-(-1) relative to SEQ ID N0:17), one comprising an N-terminal
methionine
(position -1 relative to SEQ ID N0:17), and various C-terminally truncated
forms
comprising 127-134 amino acid residues. It is known that 1-15 amino acid
residues may be
deleted from the C-terminus without abolishing IFNG activity of the molecule.
Furthermore,
heterogenecity of the huIFNG C-terminus was described by Pan et al. (Eur. J.
Biochem.
166:145-149, 1987).
HuIFNG muteins were reported by Slodowski et al. (Eur. J. Biochem. 202 :1133-
1140, 1991), Luk et al. (J. Biol. Chem. 265:13314-13319, 1990), Seelig et al.,
(Biochemistry
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
2
27:1981-1987, 1988), Trousdale et al. (Invest. Ophthalmol. Vis. Sci. 26:1244-
1251, 1985),
and in EP 146354. A natural huIFNG variant was reported by Nishi et al. (J.
Biochem.
97:153-159, 1985).
US 6,046,034 discloses thermostable recombinant huIFNG (rhulFNG) variants
having
incorporated up to 4 pairs of cysteine residues to enable disulphide bridge
formation and thus
stabilization of the IFNG variant in homodimer form.
WO 92/08737 discloses 1FNG variants comprising an added methionine in the N-
terminal end of the full (residues 1-143) or partial (residues 1-132) amino
acid sequence of
wild-type human IFNG. EP 0 219 781 discloses partial huIFNG sequences
comprising amino
acid residues 3-124 (of SEQ ID N0:17). US 4,832,959 discloses partial huIFNG
sequences
comprising residues 1-127, 5-146 and 5-127 of an amino acid sequence that
compared to
SEQ ID N0:17 has three additional N-terminal amino acid residues (Cys-Tyr-
Cys). US
5,004,689 discloses a DNA sequence encoding huIFNG without the 3 N-terminal
amino acid
residues (Cys-Tyr-Cys) and its expression in E. coli. EP 0 446 582 discloses
E. coli produced
rhuIFNG free of an N-terminal methionine. US 6,120,762 discloses a peptide
fragment of
huIFNG comprising residues 95-134 thereof (relative to SEQ ID N0:18).
High level expression of rhuIFNG was reported by Wang et al. (Sci. Sin. B
24:1076-
1084, 1994).
Glycosylation variation in rhuIFNG has been reported by Curling et al.
(Biochem. J.
272 :333-337, 1990) and Hooker et al., (J. of Interferon and Cytolcine
Research, 1998, 18:
287-295).
Polymer-modification of rhulFNG was reported by Kita et al. (Drug Des. Deliv.
6 :157-167, 1990), and in EP 236987 and US 5109120.
WO 92/22310 discloses asialoglycoprotein conjugate derivatives of interferons,
inter
alia huIFNG.
IFNG fusion proteins have been described. For instance, EP 0 237 019 discloses
a
single chain polypeptide having region exhibiting interferon (3 activity and
one region
exhibiting IFNG activity.
EP 0 158 198 discloses a single chain polypeptide having a region exhibiting
IFNG
activity and a region exhibiting IL-2 activity. Several references described
single chain
dimeric IFNG proteins, e.g. Landar et al. (J. Mol. Biol., 2000, 299:169-179).
WO 99/02710 discloses single chain polypeptides, one example among many being
IFNG.
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
WO 99/03887 discloses PEGylated variants of polypeptides belonging to the
growth
hormone superfamily, wherein a non-essential amino acid residue located in a
specified
region of the polypeptide has been replaced by a cysteine residue. IFNG is
mentioned as one
example of a member of the growth hormone super family, but modification
thereof is not
discussed in any detail.
1FNG has been suggested for treatment of interstitial lung diseases (also
known as
Interstitial Pulmonary Fibrosis (IPF) (Ziesche et al. (N. Engl. J. Med.
341:1264-1269, 1999
and Chest 110:Supp1:25S, 1996) and EP 0 795 332) for which purpose IFNG can be
used in
combination with prednisolone. In addition to IPF, granulomatous diseases
(Bolinger et al,
Clinical Pharmacy, 1992, 11:834-850), certain mycobacterial infections (N.
Engl. J. Med.
330:1348-1355, 1994), ludney cancer (J. Urol. 152:841-845, 1994),
osteopetrosis (N. Engl. J.
Med. 332:1594-1599, 1995), scleroderma (J. Rheumatol. 23:654-658, 1996),
hepatitis B
(Hepatogastroenterology 45:2282-2294, 1998), hepatitis C (Int. Hepatol.
Communic. 6:264-
273, 1997), septic shock (Nature Medicine 3:678-681, 1997), and rheumatoid
arthritis may be
treated with IFNG.
As a pharmaceutical compound rhuIFNG is used with a certain success, above
all,
against some viral infections and tumors. rhuIFNG is usually applicable via
parenteral,
preferably via subcutaneous, injection. Maximum serum concentrations have been
found after
seven hours. The half-life in plasma is 30 minutes after iv administration.
For this reason
efficient treatment with rhuIFNG involves frequent injections. The main
adverse effects
consist of fever, chills, sweating, headache, myalgia and drowsiness. These
effects are
associated with injecting rhuIFNG and are observed within the first hours
after injection.
Rare side effects are local pain and erythema, elevation of liver enzymes,
reversible granulo-
and thrombopenia and cardiotoxicity.
WO 01/36001 discloses novel IFNG conjugates comprising a non-polypeptide
moiety
attached to an IFNG polypeptide which have been modified by introduction
and/or deletion
of attachment sites for such non-polypeptide moieties, e.g. PEG and
glycosylation sites.
It is well known that when N-glycosylated molecules, such as IFNG, are
produced in
a glycosylating host not all potential N-glycosylation sites are fully
utilized. This means that
quite often a mixture of proteins having a varying degree of ih vivo N-
glycosylation is
obtained, which in turn has the consequence that subsequent purification is
necessary.
Furthermore, it is often time-consuming and cumbersome to separate identical
proteins
having a varying degree of glycosylation. It has now surprisingly been found
that by
substitution of one or more amino acid residues located close to an in vivo N-
glycosylation
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
4
site (independently of whether said iya vivo N-glycosylation site is naturally
occurnng in
IFNG or whether the in vivo N-glycosylation site has been introduced, such as
described in
WO 01/36001) it is possible to obtain an increased fraction of fully
glycosylated IFNG
molecules. In particular, it has been found that changing the naturally
occurring N-
glycosylation site N-Y-S at positions 97, 98 and 99 of hIFNG to N-Y-T gives
rise to a
dramatically increased fraction of fully glycosylated IE'NG molecules.
BRIEF DISCLOSURE OF THE INVENTION
Thus, in a first aspect the present invention relates to an interferon gamma
(IFNG)
polypeptide variant exhibiting IFNG activity and having the amino acid
sequence shown in
SEQ m N0:1 ([S99T]huIFNG), or a fragment thereof exhibiting IFNG activity.
In a further aspect the present invention relates to a variant of SEQ m N0:1,
including variant of fragments of SEQ m NO:1 (such as SEQ )D NOS:2-16),
wherein said
variant comprises at least one further modification and exhibits IFNG
activity.
In still further aspects the present invention relates to a nucleotide
sequence encoding
a polypeptide variant of the invention.
In even further aspects the present invention relates to an expression vector
comprising a nucleotide sequence of the invention, and to a glycosylating host
cell
comprising a nucleotide sequence of the invention or an expression vector of
the invention.
The present invention also relates to a pharmaceutical composition comprising
a
polypeptide variant of invention, to a polypeptide variant of the invention or
to a
pharmaceutical composition of the invention for use as a medicament.
Even further aspects of the present invention relates to the use of a
polypeptide variant
of the invention or to the use of a pharmaceutical composition of invention
for the
manufacture of a medicament for treatment of interstitial lung diseases.
Analogously, the present invention also relates to a method for treating or
preventing
interstitial lung diseases, said method comprising administering to a mammal,
in particular a
human being, in need thereof an effective amount of a polypeptide variant of
the invention or
a pharmaceutical composition of the invention.
A still further aspect of the invention relates to a population of IFNG
polypeptide
variants, or a composition comprising a population of IFNG polypeptide
variants, wherein
said population comprises at least 70% of the IFNG polypeptide variant of the
invention.
In another aspect the present invention relates to a method of increasing the
degree of
i~ vivo N-glycosylation of a parent IFNG polypeptide that comprises at least
one ifa vivo N-
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
glycosylation site with the amino acid sequence N-X-S, wherein X is any amino
acid residue
except proline, said method comprising substituting the serine residue in said
N-X-S amino
acid sequence with a threonine residue to obtain an IFNG variant.
In still another aspect the present invention relates to a method for
producing an IFNG
5 polypeptide variant of the invention, said method comprising
(a) culturing a glycosylating host cell comprising a nucleotide sequence which
encodes an IFNG polypeptide variant of the invention under conditions
conducive
for expression of the polypeptide variant;
(b) optionally reacting said polypeptide variant with a non-polypeptide moiety
zfa vitro
under conditions conducive for the conjugation to take place; and
(c) recovering the polypeptide variant.
Other aspects of the present invention will be apparent from the below
description.
BRIEF DESCRIPTION OF THE DRAWINGS
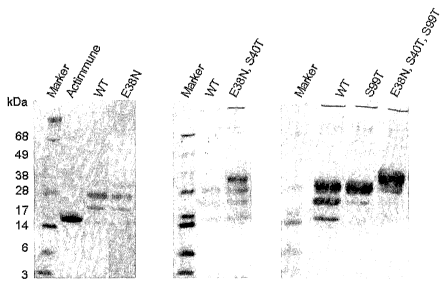
Fig. 1 is a Western blot of optimised glycosylation variants of rhuIFNG.
Left side Westena blot: Lane 1: standard, Lane 2: Actimmune~, Lane 3: rhuIFNG,
Lane 4:
[E38N]rhuIFNG. Middle Weste~a blot: Lane 1: standard, Lane 2: rhuIFNG, Lane 3:
[E38N+S40T]rhuIFNG. Right side Western blot: Lane 1: standard, Lane 2:
rhuIFNG, Lane 3:
[S99T]rhuIFNG, Lane 4: [E38N+S40T+S99T]rhuIFNG.
Fig. 2 shows the IFNG activity in serum-time curve after subcutaneous
administration
in rats. ~: Actimmune~, ~: rhuIFNG, ~: [E38N+S40T+S99T]rhuIFNG.
The same dose was administered for all compounds (1.15 x 107 AU/kg).
Fig. 3 shows the IFNG activity in serum-time curve after subcutaneous
administration
in rats. ~: [N16C+S99T]rhuIFNG (5 kDa mPEG attached), ~: [N16C+S99T]rhuIFNG
(10
kDamFEGattached), ~: [E38N+S40T+S99T]rhuIFNG.
The [E38N+S40T+S99T] variant was administered in a dose of 1.15 x 107 AU/kg,
whereas the two PEGylated variants were administered in a dose of 4.6 x 106
AU/lcg.
DETAILED DESCRIPTION OF THE INVENTION
Defiyaitzohs
In the context of the present application and invention the following
definitions apply:
The term "conjugate" (or interchangeably "conjugated polypeptide" or
"conjugated
variant") is intended to indicate a heterogeneous (in the sense of composite
or chimeric)
molecule formed by the covalent attachment of one or more polypeptide
variants) to one or
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
6
more non-polypeptide moieties. The term covalent attachment means that the
polypeptide
variant and the non-polypeptide moiety are either directly covalently joined
to one another, or
else are indirectly covalently joined to one another through an intervening
moiety or moieties,
such as a bridge, spacer, or linkage moiety or moieties. Preferably, a
conjugated polypeptide
variant is soluble at relevant concentrations and conditions, i.e. soluble in
physiological fluids
such as blood. Examples of conjugated polypeptide variants of the invention
include
glycosylated and/or PEGylated polypeptide variants. The term "non-conjugated
polypeptide
variant" may be used about the polypeptide part of the conjugated polypeptide
variant.
The term "non-polypeptide moiety" is intended to indicate a molecule that is
capable
of conjugating to an attachment group of the IFNG polypeptide variant.
Preferred examples
of such molecules include polymer molecules, lipophilic compounds, sugar
moieties or
organic derivatizing agents. It will be understood that the non-polypeptide
moiety is linked to
the polypeptide through an attachment group of the polypeptide variant. Except
where the
number of non-polypeptide moieties, such as polymer molecule(s), attached to
the IFNG
polypeptide variant is expressly indicated every reference to "a non-
polypeptide moiety"
attached to the 1FNG polypeptide variant or otherwise used in the present
invention shall be a
reference to one or more non-polypeptide moieties attached to the IFNG
polypeptide variant.
The term "polymer molecule" is defined as a molecule formed by covalent
linkage of
two or more monomers, wherein none of the monomers is an amino acid residue.
The term
"polymer" may be used interchangeably with the term "polymer molecule".
The term "sugar moiety" is intended to indicate a carbohydrate molecule
attached by
in vivo or iyz vitro glycosylation, such as N- or O-glycosylation.
An "N-glycosylation site" has the sequence N-X-SlT/C", wherein X is any amino
acid residue except proline, N is asparagine and S/T/C is either serine,
threonine or cysteine,
preferably serine or threonine, and most preferably threonine. An "O-
glycosylation site" is
the OH-group of a serine or threonine residue.
The term "attachment group" is intended to indicate an amino acid residue
group
capable of coupling to the relevant non-polypeptide moiety such as a polymer
molecule or a
sugar moiety. Useful attachment groups and their matching non-polypeptide
moieties are
apparent from the table below.
AttachmentAmino acid Examples of Conjugation Reference
non-
group polypeptide method/activated
moiety
PEG
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
-NH2 N-terminal, Polymer, e.g. mPEG-SPA Shearwater
Lys PEG Inc.
Tresylated Delgado et
mPEG al,
critical
reviews
in Therapeutic
Drug Carrier
Systems
9(3,4):249-304
(1992)
-COOH C-term, Asp, Polymer, e.g. mPEG-Hz Shearwater
PEG Inc
Glu
Sugar moiety In vitro coupling
-SH Cys Polymer, e.g. PEG- Shearwater
PEG, Inc
vinylsulphone Delgado et
al,
PEG-maleimide critical
reviews
in Therapeutic
Sugar moiety Iya vitro couplingDrug Carrier
Systems
9(3,4):249-304
(1992)
-OH Ser, Thr, Sugar moiety he vivo O-linked
OH-,
Lys glycosylation
-CONH2 Asn as part Sugar moiety Ih vivo
of
an N- glycosylation
glycosylation
site
Aromatic Phe, Tyr, Sugar moiety Ih vitro coupling
Trp
residue
-CONHa Gln Sugar moiety In vitro couplingYan and Wold,
Biochemistry,
1984, Jul
31;
23(16): 3759-65
Aldehyde Oxidized Polymer, e.g. PEGylation Andresz et
PEG, al.,
Ketone carbohydrate PEG-hydrazide 1978,
Makromol.
Chem. 179:301;
WO 92/16555,
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
8
WO 00/23114
GuanidinoArg Sugar moiety Ifz vitro couplingLundblad
and
Noyes,
Chimical
Reagents
for
Protein
Modification,
CRC Press
Inc.
Boca Raton,
FI
ImidazoleHis Sugar moiety Ifz vitro couplingAs for
ring guanidine
For irz vivo N-glycosylation, the term "attachment group" is used in an
unconventional
way to indicate the amino acid residues constituting an N-glycosylation site
(with the
sequence N-X-S/T/C, wherein X is any amino acid residue except proline, N is
asparagine
and S/T/C is either serine, threonine or cysteine, preferably serine or
threonine, and most
preferably threonine). Although the asparagine residue of the N-glycosylation
site is the one
to which the sugar moiety is attached during glycosylation, such attachment
cannot be
achieved unless the other amino acid residues of the N-glycosylation site is
present.
Accordingly, when the non-polypeptide moiety is a sugar moiety and the
conjugation is to be
achieved by N-glycosylation, the term "amino acid residue comprising an
attachment group
for the non-polypeptide moiety" as used in connection with alterations of the
amino acid
sequence of the IFNG polypeptide is to be understood as one, two or all of the
amino acid
residues constituting an N-glycosylation site is/are to be altered in such a
manner that either a
functional N-glycosylation site is introduced into the amino acid sequence,
removed from
said sequence or a functional N-glycosylation site is retained in the amino
acid sequence (e.g.
by substituting a serine residue, which already constitutes part of an N-
glycosylation site,
with a threonine residue and vice versa).
In the present application, amino acid names and atom names (e.g. CA, CB, CD,
CG,
SG, NZ, N, O, C, etc) are used as defined by the Protein DataBank (PDB)
(www.pdb~org)
which are based on the IITPAC nomenclature (IUPAC Nomenclature and Symbolism
for
Amino Acids and Peptides (residue names, atom names etc.), Eur. J. Bioclzem.,
138, 9-37
(1984) together with their corrections in Eur. J. Biochem., 152, 1 (1985). CA
is sometimes
referred to as Ca, CB as Chi. The term "amino acid residue" is intended to
indicate an amino
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
9
acid residue contained in the group consisting of alanine (Ala or A), cysteine
(Cys or C),
aspartic acid (Asp or D), glutamic acid (Glu or E), phenylalanine (Phe or F),
glycine (Gly or
G), histidine (His or H), isoleucine (lle or I), lysine (Lys or K), leucine
(Leu or L),
methionine (Met or M), asparagine (Asn or N), proline (Pro or P), glutamine
(Gln or Q),
arginine (Arg or R), serine (Ser or S), threonine (Thr or T), valine (Val or
V), tryptophan (Trp
or W), and tyrosine (Tyr or Y) residues.
Numbering of amino acid residues in this document is from the N-terminus of
[S99T]hulFNG without signal peptide (i.e. SEQ ID NO:l) or, where relevant,
from the N-
terminus of huIFNG without signal peptide (i.e. SEQ ID N0:17).
The terminology used for identifying amino acid positions/substitutions is
illustrated
as follows: G18 indicates position 18 occupied by glycine in the amino acid
sequence shown
in SEQ ID N0:1 or SEQ ID N0:17. G18N indicates that the Gly residue of
position 18 has
been replaced with an Asn. Multiple substitutions are indicated with a "+",
e.g. G18N+S20T
means an amino acid sequence which comprises a substitution of the Gly residue
in position
18 with an Asn and a substitution of the Ser residue in position 20 with Thr.
Alternative
substitutions are indicated with a "/". For example, G18S/T covers the
following individual
substitutions: G18S and G18T. Deletions are indicated by an asterix. For
example, G18*
indicates that the Gly residue in position 18 has been deleted. Insertions are
indicated the
following way: Insertion of an additional Ser residue after the Gly residue
located at position
18 is indicated as G18G5. Combined substitutions and insertions are indicated
in the
following way: Substitution of the Gly residue at position 18 with an Ser
residue and
insertion of an Ala residue after the position 18 amino acid residue is
indicated as G18SA.
The term "nucleotide sequence" is intended to indicate a consecutive stretch
of two or
more nucleotide molecules. The nucleotide sequence may be of genomic, cDNA,
RNA,
semisynthetic, synthetic origin, or any combinations thereof.
The term "polymerase chain reaction" or "PCR" generally refers to a method for
amplification of a desired nucleotide sequence in vitro, as described, for
example, in US
4,683,195. In general, the PCR method involves repeated cycles of primer
extension
synthesis, using oligonucleotide primers capable of hybridising preferentially
to a template
nucleic acid.
"Cell", "host cell", "cell line" and "cell culture" are used interchangeably
herein and
all such terms should be understood to include progeny resulting from growth
or culturing of
a cell.
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
"Transformation" and "transfection" are used interchangeably to refer to the
process
of introducing DNA into a cell.
"Operably linked" refers to the covalent joining of two or more nucleotide
sequences,
by means of enzymatic ligation or otherwise, in a configuration relative to
one another such
5 that the normal function of the sequences can be performed. For example, the
nucleotide
sequence encoding a presequence or secretory leader is operably linked to a
nucleotide
sequence for a polypeptide if it is expressed as a preprotein that
participates in the secretion
of the polypeptide: a promoter or enhancer is operably linlced to a coding
sequence if it
affects the transcription of the sequence; a ribosome binding site is operably
linked to a
10 coding sequence if it is positioned so as to facilitate translation.
Generally, "operably linked"
means that the nucleotide sequences being linked are contiguous and, in the
case of a
secretory leader, contiguous and in reading phase. Linking is accomplished by
ligation at
convenient restriction sites. If such sites do not exist, then synthetic
oligonucleotide adaptors
or linkers are used, in conjunction with standard recombinant DNA methods.
The term "modification", as used herein, covers substitution, insertion and
deletion.
The terms "mutation" and "substitution" are used interchangeably herein.
The term "introduce" is primarily intended to mean substitution of an existing
amino
acid residue, but may also mean insertion of an additional amino acid residue.
The term "remove" is primarily intended to mean substitution of the amino acid
residue to be removed for another amino acid residue, but may also mean
deletion (without
substitution) of the amino acid residue to be removed.
The term "amino acid residue comprising an attachment group for the non-
polypeptide moiety" is intended to indicate that the amino acid residue is one
to which the
non-polypeptide moiety binds (in the case of an introduced amino acid residue)
or would
have bound (in the case of a removed amino acid residue).
The term "one difference" or "differs from" as used in connection with
specific
modifications is intended to allow for additional differences being present
apart from the
specified amino acid difference. Thus, in addition to amino acid residue
alterations disclosed
herein aiming at optimising the utilization of glycosylation sites or removing
and/or
introducing amino acid residues comprising an attachment group for a non-
polypeptide
moiety, the IFNG polypeptide variant may, if desired, comprise other
modifications that are
not related to such alterations. These may, for example, include truncation of
the C-terminus
by one or more amino acid residues, addition of one or more extra residues at
the N- and/or
C-terminus, e.g. addition of a Met residue at the N-terminus, addition of the
amino acid
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
11
sequence Cys-Tyr-Cys at the N-terminus, as well as "conservative amino acid
substitutions",
i.e. substitutions performed within groups of amino acids with similar
characteristics, e.g.
small amino acids, acidic amino acids, polar amino acids, basic amino acids,
hydrophobic
amino acids and aromatic amino acids. Examples of conservative substitutions
in the present
invention may in particular be selected from the groups listed in the table
below.
1 Alanine (A) Glycine (G) Serine (S) Threonine (T)
2 Aspartic acid Glutamic acid
(D) (E)
3 Asparagine (N) Glutamine (Q)
4 Arginine (R) Histidine (H) Lysine (K)
5 Isoleucine (I) Leucine (L) Methionine (M) Valine (V)
6 Phenylalanine Tyrosine (Y) Tryptophan (W)
(F)
The term "at least one" as used about a non-polypeptide moiety, an amino acid
residue, a substitution, etc is intended to mean one or more.
The term "AITCS~" or "Area Under the Curve when administered subcutaneously"
is
used in its normal meaning, i.e. as the area under the IFNG activity in serum-
time curve,
where the 1FNG polypeptide variant has been administered subcutaneously, in
particular
when administered subcutaneously in rats. Once the experimental IFNG activity-
time points
have been determined, the AUCS~ may conveniently be calculated by a computer
program,
such as GraphPad Prism 3.01.
The term "functional in vivo half-life" is used in its normal meaning, i.e.
the time at
which 50% of the biological activity of the polypeptide is still present in
the bodyltarget
organ, or the time at which the activity of the p'olypeptide is 50% of the
initial value.
As an alternative to determining functional ifi vivo half-life, "serum half-
life" may be
determined, i.e. the time at which 50% of the polypeptide circulates in the
plasma or
bloodstream prior to being cleared. Determination of serum half-life is often
more simple
than determining the functional ifa vivo half-life and the magnitude of serum
half-life is
usually a good indication of the magnitude of functional in vivo half-life.
Alternatively terms
to serum half-life include "plasma half-life", "circulating half-life", "serum
clearance",
"plasma clearance" and "clearance half-life". The serum half-life may
conveniently by
determined in rats, cf. the Materials and Method section herein. It is
important to note that the
term "serum half-life", when used herein, for a given 1FNG polypeptide variant
must be
determined for a sample that has been administered intravenously (iv).
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
12
The term "serum" is used in its normal meaning, i.e. as blood plasma without
fibrinogen and other clotting factors.
The polypeptide is normally cleared by the action of one or more of the
reticuloendothelial systems (RES), kidney, spleen or liver, or by specific or
unspecific
proteolysis. The term "renal clearance" is used in its normal meaning to
indicate any
clearance taking place by the kidneys, e.g. by glomerular filtration, tubular
excretion or
tubular elimination. Normally, renal clearance depends on physical
characteristics of the
polypeptide, including molecular weight, size (relative to the cutoff for
glomerular filtration),
symmetry, shapelrigidity, charge, attached carbohydrate chains and the
presence of cellular
receptors for the polypeptide. A molecular weight of about 67 kDa is normally
considered to
be a cut-off-value for renal clearance. Renal clearance may be measured by any
suitable
assay, e.g. an established ira vivo assay. For instance, renal clearance may
be determined by
administering a labelled (e.g. radiolabelled or fluorescence labelled)
conjugated polypeptide
to a patient and measuring the label activity in urine collected from the
patient. Reduced renal
clearance is determined relative to the reference molecule, such as huIFNG,
[S99T]huIFNG
or Actimmune~. The functionality to be retained is normally selected from
antiviral,
antiproliferative, immunomodulatory or IFNG receptor binding activity.
The term "increased" as used about the functional in vivo half-life or serum
half life is
used to indicate that the relevant half-life of the IFNG variant is
statistically significantly
increased relative to that of a reference molecule, such as glycosylated
huIFNG (SEQ m
N0:17), glycosylated [S99T]huIFNG (SEQ )D NO:1) or Actimmune~ (SEQ m N0:34 -
produced in E. coli), when administered intravenously and when determined
under
comparable conditions. Thus, interesting IFNG polypeptide variants are such
variants, which,
has an increased functional in vivo half-life or an increased serum half-life
as compared to
any of the reference molecules mentioned above.
More particularly, interesting IFNG variants are such variants where the ratio
between
the serum half-life (or functional in vivo half-life) of said variant and the
serum half-life (or
functional in vivo half-life) of huIFNG or [S99T]hulFNG in their glycosylated
forms is at
least 1.25, more preferably at least 1.50, such as at least 1.75, e.g. at
least 2, even more
preferably at least 3, such as at least 4, e.g. at least 5, when administered
intravenously, in
particular when administered intravenously in rats.
Other examples of interesting IFNG variants are such variants where the ratio
between the serum half-life (or functional ira vivo half-life) of said variant
and the serum half-
life (or functional ifa vivo half-life) of Actixnmune~ (SEQ m N0:34 - produced
in E. coli) is
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
13
at least 2 more preferably at least 3, such as at least 4, e.g. at least 5,
even more preferably at
least 6, such as at least 7, e.g. at least 8, most preferably at least 9, such
as at least 10, when
administered intravenously, in particular when administered intravenously in
rats.
The term "increased" as used about the AUCS~ is used to indicate that the Area
Under
the Curve for an IFNG variant of the invention, when administered
subcutaneously, is
statistically significantly increased relative to that of a reference
molecule, such as
glycosylated huIFNG (SEQ m N0:17), glycosylated [S99T]huIFNG (SEQ ID NO:1) or
Actimmune~ (SEQ m N0:34 - produced in E. coli), determined under comparable
conditions. Thus, preferred IFNG variants are such variants, which have an
increased AUCS~,
as compared to any of the reference molecules mentioned above. Evidently, the
same amount
of IFNG activity should be administered for the IFNG variant of the invention
and the
reference molecule. Consequently, in order to make direct comparisons between
different
1FNG molecules, the AUCS~ values may be normalized, i.e. they may be expressed
as
AUCS~ldose administered.
Particular preferred IFNG variants are such variants where the ratio between
the
AUCS~ of said variant and the AUCS~ of glycosylated huIFNG or glycosylated
[S99T]huIFNG
is at least 1.25, such as at least 1.5, e.g. at least 2, more preferably at
least 3, such as at least
4, e.g. at least 5 or at least 6, even more preferably at least 7, such as at
least 8, e.g. at least 9
or at least 10, most preferably at least 12, such as at least 14, e.g. at
least 16, at least 18 or at
least 20, in particular when administered (subcutaneously) in rats.
Other examples of particular preferred IFNG variants are such variants where
the ratio
between the AUCS~ of said variant and the AUCS~ of Actimmune~ is at least 100,
more
preferably at least 150, such as at least 200, e.g. at least 250, even more
preferably at least
300, such as at least 400 e.g. at least 500, most preferably at least 750,
such as at least 1000,
e.g. at least 1500 or at least 2000, in particular when administered
(subcutaneously) in rats.
The term "Tmax,sc" is used about the time in the IFNG activity in serum-time
curve
when the highest IFNG activity in serum is observed. Preferred IFNG variants
of the
invention, are such variants which have an increased T,T,a,;.s~ as compared to
Actimmune~
and/or as compared to glycosylated huIFNG. More particularly, such preferred
variants have
a Tm~,sc (when determined after subcutaneous administration in rats) of at
least 200 min, such
as at least 250 min, e.g. at least 300 min, more preferably at least 350 min,
such as at least
400 min.
The term "reduced irnmunogenicity" is intended to indicate that the IFNG
polypeptide variant gives rise to a measurably lower immune response than a
reference
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
14
molecule, e.g. huIFNG or Actimmune~, as determined under comparable
conditions. The
immune response may be a cell or antibody mediated response (see, e.g., Roitt:
Essential
Immunology (8th Edition, Blackwell) for further definition of immunogenicity).
Normally,
reduced antibody reactivity is an indication of reduced immunogenicity.
Reduced
immunogenicity may be determined by use of any suitable method known in the
art, e.g. ifz.
vivo or ifa vitYO.
In the present context the terms "increased glycosylation", "increased degree
of in
vivo N-glycosylation" or "increased degree of N-glycosylation" are intended to
indicate
increased levels of attached carbohydrate molecules, normally obtained as a
consequence of
increased (or better) utilization of glycosylation site(s). It is well-known
(Hooker et al., 1998,
J. Interferon and Cytokine Res. 18, 287-295 and Sarenva et al., 1995, Biochem
J., 308, 9-14)
that when huIFNG is expressed in CHO cells only about 50% of the IFNG
molecules utilizes
both glycosylation sites, about 40% utilizes one glycosylation site (1N), and
about 10% is not
glycosylated (ON). The increased degree of iya vivo N-glycosylation may be
determined by
any suitable method known in the art, e.g. by SDS-PAGE. One convenient assay
for
determining increased glycosylation is the method described in the section
entitled
"Determination of Increased Glycosylation" in the Materials and Methods
section herein.
When used herein the term "population of IFNG polypeptide variants" or
"composition comprising a population of IFNG polypeptide variants" is intended
to cover a
composition comprising at least two IFNG polypeptides glycosylated to a
different extent. As
will be understood the present invention provides means for obtaining a
population of IFNG
polypeptides, wherein an increased amount of the INFG molecules present in the
population
is fully glycosylated.
Thus, the present invention also relates to a homogeneous population of IFNG
polypeptides of the invention (i.e. a population, wherein most of the IFNG
polypeptides are
fully glycosylated) or a composition comprising a homogenous population of
IFNG
polypeptides of the invention. For example, the population of IFNG
polypeptides may
contain at least 70% of the IFNG polypeptide of the invention, preferably at
least 75%, such
as at least 80%, e.g. at least 85%, more preferably at least 90%, such as at
least 95%, e.g. at
least 96%, even more preferably at least 97%, such as at least 98%, e.g. at
least 99%.
The term "exhibiting IFNG activity" is intended to indicate that the
polypeptide
variant has one or more of the functions of native huIFNG or rhuIFNG,
including the
capability to bind to an IFNG receptor and cause transduction of the signal
transduced upon
huIFNG-binding of its receptor as determined in vztro or in vivo (i.e. in
vzt~o or in vzvo
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
bioactivity). The IFNG receptor has been described by Aguet et al. (Cell
55:273-280, 1988)
and Calderon et al. (Proc. Natl. Acad. Sci. USA 85:4837-4841, 1988). A
suitable assay for
testing IFNG activity is the assay entitled "Primary Assay" disclosed herein.
When using the
"Primary Assay" described herein, polypeptide variants "exhibiting 1FNG
activity" have a
5 specific activity of at least 5% as compared to the rhuIFNG. It will be
understood, that
depending on which specific modifications are performed, for example whether
the variant is
PEGylated or not, may lead to activities over a wide range. Thus, examples of
specific
activities may range from as low as 5% to as high as 150% as compared to
rhuIFNG. For
example, the specific activity may be at least 10% (e.g. 10-125%), such as at
least 15% (e.g.
10 15-125%), e.g. at least 20% (such as 20-125%), at least 25% (e.g. 25-125%),
at least 30%
(e.g. 30-125%), at least 35% (e.g. 35-125%), at least 40% (e.g. 40-125%), at
least 45% (e.g.
45-125%), at least 50% (e.g. 50-125%), at least 55% (e.g. 55-125%), at least
60% (e.g. 60-
125%), at least 65% (e.g. 65-125%), at least 70% (e.g. 70-125%), at least 75%
(e.g. 75-
125%), at least 80% (e.g. 80-125%) or at least 90% (e.g. 90-110%) as compared
to the
15 specific activity of rhuIFNG.
An "IFNG polypeptide" is a polypeptide exhibiting IFNG activity, i.e. the term
"IFNG polypeptide" is used about any IFNG molecule (independently of whether
this
molecule is huIFNG, a truncated form thereof, or a variant thereof) as long as
said IFNG
molecule exhibits IFNG activity as defined herein. The term "IFNG polypeptide"
is used
herein about the polypeptide in monomer or dimeric form, as appropriate. For
instance, when
specific substitutions are indicated these are normally indicated relative to
the huIFNG
polypeptide monomer. When reference is made to the IFNG molecule of the
invention this is
normally in dimeric form (and thus, e.g., comprises two IFNG polypeptide
monomers
modified as described). The dimeric form of the IFNG polypeptides may be
provided by the
normal association of two monomers or be in the form of a single chain dimeric
IFNG
polypeptide.
The term "parent" is intended to indicate the 1FNG polypeptide to have the
glycosylation sites) improved in accordance with the present invention.
Although the parent
polypeptide to be modified by the present invention may be any polypeptide
with IFNG
activity, and thus be derived from any origin, e.g. a non-human mammalian
origin, it is
preferred that the parent polypeptide is huIFNG with the amino acid sequence
shown in SEQ
117 N0:17 or a fragment thereof.
A "fragment" is a part of the full-length IFNG polypeptide sequence (e.g. a
fragment
of the full-length huIFNG polypeptide shown in SEQ ID N0:17 or a fragment of
the full-
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
16
length [S99T]huIFNG polypeptide variant shown in SEQ )D N0:1) exhibiting IFNG
activity,
e.g. a C-terminally or N-terminally truncated version thereof. Specific
examples of IFNG
polypeptide variant fragments include [S99T]huIFNG C-terminally truncated with
1-15
amino acid residues, e.g. with 1 amino acid residue (SEQ ll~ N0:2), 2 amino
acid residues
(SEQ ID N0:3), 3 amino acid residues (SEQ ID N0:4), 4 amino acid residues (SEQ
JD
N0:5), 5 amino acid residues (SEQ ID N0:6), 6 amino acid residues (SEQ ID
N0:7), 7
amino acid residues (SEQ ID N0:8), 8 amino acid residues (SEQ ll~ N0:9), 9
amino acid
residues (SEQ ID NO:10), 10 amino acid residues (SEQ ll~ NO:11), 11 amino acid
residues
(SEQ ID N0:12), 12 amino acid residues (SEQ ID N0:13), 13 amino acid residues
(SEQ ID
N0:14), 14 amino acid residues (SEQ ID N0:15) or 15 amino acid residues (SEQ
ID N0:16)
andlor N-terminally truncated with 1-3 amino acid residues. Specific examples
of huIFNG
fragments include huIFNG, which is C-terminally truncated with 1-15 amino acid
residues,
e.g. with 1 amino acid residue (SEQ ID N0:19), 2 amino acid residues (SEQ ID
NO:10), 3
amino acid residues (SEQ ID NO:21), 4 amino acid residues (SEQ m N0:22), 5
amino acid
residues (SEQ ID N0:23), 6 amino acid residues (SEQ ID NO:24), 7 amino acid
residues
(SEQ ID N0:25), 8 amino acid residues (SEQ m N0:26), 9 amino acid residues
(SEQ ID
N0:27), 10 amino acid residues (SEQ ll~ N0:28), 11 amino acid residues (SEQ >D
N0:29),
12 amino acid residues (SEQ ID N0:30), 13 amino acid residues (SEQ ID N0:31),
14 amino
acid residues (SEQ >D N0:32) or 15 amino acid residues (SEQ ID N0:33) andlor N-
terminally truncated with 1-3 amino acid residues.
As indicated above, the IFNG polypeptide variant may comprise at least one
further
modification in addition to the S99T substitution as long as said variant
exhibits IFNG
activity, i.e. the variant may be a variant of [S99T]huIFNG, or a variant of a
fragment of
[S99T]huIFNG. Specific examples of such variants are variants having
introduced and/or
removed amino acid residues comprising an attachment group for a non-
polypeptide moiety.
Other examples of variants of [S99T]huIFNG (and fragments thereof] are the
variants
described in the "Background of the invention" section above and include, e.g.
[S99T]huIFNG with the N-terminal addition Cys-Tyr-Cys or with Met, and the
cysteine-
modified variants disclosed in US 6,046,034.
Normally, the variant according to the invention is encoded by a nucleotide
sequence,
which, compared to the nucleotide sequence encoding the parent IFNG
polypeptide, has been
modified in accordance with the present invention.
This may, however, not always be the case, since the variant polypeptide may
be
subjected to C- or N-terminal truncation during posttranslational processing,
e.g. by C- or N-
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
17
terminal cleavage by proteases in the cell, in the expression media, during
purification, etc.,
so that the resulting variant polypeptide is a truncated version of the
originally produced
variant polypeptide (for example, although a full-length variant is initially
produced, a C-
terminally truncated variant polypeptide may be obtained due to
posttranslational processing
of the full-length variant polypeptide). In this case, the term "parent"
should be construed as
the truncated form to be modified in accordance with the invention.
The term "variant" is intended to cover a polypeptide, which differs in one or
more
amino acid residues from its parent polypeptide (normally SEQ m N0:17 or any
of the
truncated forms thereof shown in SEQ ID NOS:19-33), typically in 1-15 amino
acid residues
(such as in 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 or 15 amino acid
residues), e.g. in 1-10
amino acid residues, in 1-5 amino acid residues or in 1-3 amino acid residues.
The term "functional site" is intended to indicate one or more amino acid
residues
which is/are essential for, or otherwise involved in, the function or
performance of IFNG.
Such amino acid residues are "located at" the functional site. The functional
site may be
determined by methods known in the art and is preferably identified by
analysis of a structure
of the polypeptide complexed to a relevant receptor, such as the IFNG
receptor.
The term "huIFNG" is intended to mean the mature form of wild-type human IFNG
having the amino sequence shown in SEQ ID N0:17.
The term "rhuIFNG" is intended to cover the mature form of wild-type human
IFNG
having the amino acid sequence shown in SEQ m N0:17, which has been produced
by
recombinant means.
The term "[S99T]huIFNG" is used to indicate the mature form of wild-type human
IFNG, wherein the serine residue in position 99 has been replaced with a
threonine residue
(disclosed in SEQ m N0:1).
When used herein the term "glycoyslated huIFNG" indicates that the huIFNG
polypeptide is produced in a cell capable of glycosylating the polypeptide
and, therefore, the
huIFNG polypeptide is glycosylated at its native N-glycosylation sites
(position 25 and 97 of
SEQ ID NO:17).
In a similar way, the term "glycoyslated huIFNG variant" indicates that the
IFNG
polypeptide variant is produced in a cell capable of glycosylating the
polypeptide variant.
When used herein the term "Actimmune~" refers to the 140 amino acid form
(Actimmune~ is C-terminally truncated with 4 amino acid residues and includes
one N-
terminal Met residue) of IFNG (disclosed in SEQ ID NO:34) achieved by
fermentation of a
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
18
genetically engineered E. coli bacterium. Further information of Actimmune~ is
available on
www.actimmune.com.
Interferon gamma polypeptide variants of the present invention
IFNG variants of the invention with optinzr.'sed irz vivo ~lycosylation sites
As indicated previously, it has surprisingly been found that glycosylation of
the
naturally occurring N-glycosylation site located in position 97 of huIFNG may
be increased,
i.e. an increased fraction of fully, or substantially fully, glycosylated IFNG
molecules may be
obtained, by substituting the serine residue located in position 99 of huIFNG
(or fragments
thereof) with a threonine residue. Inspection of Fig. 1 reveals that a
significant increase in the
degree of fully glycosylated IFNG polypeptide can be achieved. For the
[S99T]huIFNG
(SEQ ID N0:1) polypeptide variant it can be seen that about 90% of the
polypeptide variants
present in the harvested medium utilized both N-glycosylation site, whereas
only about 60%
of the rhuIFNG polypeptides present in the harvested medium was fully
glycosylated.
Accordingly, in a first aspect the present invention relates to an IFNG
polypeptide
variant exhibiting IFNG activity and having the amino acid sequence shown in
SEQ ID NO:l
(i.e. [S99T]huIFNG), or fragment thereof exhibiting IFNG activity.
As already discussed above, it is known that C-terminally truncated forms of
huIFNG
retain activity, and in some cases even have increased activity, compared to
hulFNG. Thus, in
an interesting embodiment of the invention the IFNG polypeptide variant of the
invention is a
fragment of SEQ ll~ NO:1, which is C-terminally truncated with 1-15 amino acid
residues,
typically C-terminally truncated with 1-10 amino acid residues. Specific
examples of such C-
terminally truncated forms of SEQ ID NO:l are disclosed in SEQ ID NOS:2-16.
The IFNG
polypeptide fragment according to this embodiment of the present invention
exhibits 1FNG
activity.
It will be understood that the glycosylated IFNG polypeptide variants
according to
this aspect should be expressed recombinantly in a glycosylating host cell,
preferably a
mammalian host cell, such as any of those mentioned in the section entitled
"Coupling to a
sugar moiety".
As explained above, only about 50-60% of the total population of expressed
IFNG
polypeptides are fully glycosylated when rhuIFNG is expressed in CHO cells.
Thus, one of
the main advantages of the IFNG polypeptide variants of the present invention
is the higher
utilization of the position 97 in vivo N-glycosylation site, which in turn has
the consequence
that a more homogenous population is obtained compared to huIFNG. Due to the
more
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
19
homogenous population, compositions (e.g. the harvested medium) comprising
such a
population of IFNG polypeptide variants do not require the same cumbersome and
time-
consuming purification as rhulFNG.
Thus, in a further aspect the present invention relates to a population of
1FNG
polypeptide variants, or to a composition comprising a population of IFNG
polypeptide
variants, wherein said population comprises at least about 70% of an 1FNG
polypeptide
variant of the invention. Preferably, the composition comprises at least 75%,
more preferably
at least 80%, even more preferably at least 85%, such as about 90% of an IFNG
polypeptide
variant of the invention.
More particularly, the invention relates to a population of IFNG polypeptide
variants,
or to a composition comprising a population of IFNG polypeptide variants,
wherein said
population comprises at least 70%, preferably at least 75%, more preferably at
least 80%,
even more preferably at least 85%, such as about 90% of the IFNG polypeptide
variant
having the amino acid sequence shown in SEQ ~ NO:1.
Analogously, the invention also relates to a population of IFNG polypeptide
variants,
or to a composition comprising a population of IFNG polypeptide variants,
wherein said
population comprises at least 70%, preferably at least 75%, more preferably at
least 80%,
even more preferably at least 85%, such as about 90% of an IFNG polypeptide
variant
fragment having an amino acid sequence selected from the group consisting of
SEQ m
N0:2, SEQ m N0:3, SEQ ~ N0:4, SEQ m NO:S, SEQ ID N0:6, SEQ m NO:7, SEQ m
N0:8, SEQ JD N0:9, SEQ ID NO:10, SEQ )D NO:11, SEQ m N0:12, SEQ m N0:13, SEQ
m N0:14, SEQ m N0:15 and SEQ m N0:16.
In addition to the already mentioned S99T mutation required for optimisation
of the in
vivo N-glycosylation site at position 97 in rhulFNG, other i~a vivo
glycosylation sites, which
have been introduced into SEQ m NO:1 or fragments thereof (e.g. in order to
increase the
serum half-life and/or to increase the AUCS~) may be optimised. Normally, the
ih vivo
glycosylation site is an N-glycosylation site, but also an O-glycosylation
site is contemplated
as relevant for the present invention. This optimisation may be achieved by
performing a
modification, preferably a substitution, in a position, which is located close
to a glycosylation
site, in particular close to an iTZ vivo N-glycosylation. Typically, such an
ioz vivo N-
glycoyslation site is an introduced ih vivo N-glycosylation site. Specific
examples of suitable
positions to introduce in vivo N-glycosylation sites are disclosed in WO
01/36001 and further
below.
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
An amino acid residue "located close to" a glycosylation site is usually
located in
position -4, -3, -2, -1, +1, +2, +3 or +4 relative to the amino acid residue
of the glycosylation
site to which the carbohydrate is attached, preferably in position -1, +1, or
+3, in particular in
position +1 or +3. Thus, the amino acid residue located close to an in vivo N-
glycosylation
5 site (having the sequence N-X-S/T/C) may be located in position -4, -3, -2, -
l, +1, +2, +3 or
+4 relative to the N-residue.
When position +2 relative to the N-residue is modified it will be understood
that only
a limited number of modifications are possible since in order to
maintain/introduce an in vivo
N-glycosylation site, the amino acid residue in said position must be either
Ser, Thr or Cys.
10 In a particular preferred embodiment of the invention, the modification of
the amino
acid residue in position +2 relative to the in vivo N-glycosylation site is a
substitution where
the amino acid residue in question is replaced with a Thr residue. If, on the
other hand, said
amino acid residue is already a Thr residue it is normally not preferred or
necessary to
perform any substitutions in that position. When X is modified, X should not
be Pro and
15 preferably not Trp, Asp, Glu and Leu. Further, the amino acid residue to be
introduced is
preferably selected form the group consisting of Phe, Asn, Gln, Tyr, Val, Ala,
Met, Ile, Lys,
Gly, Arg, Thr, His, Cys and Ser, more preferably Ala, Met, Ile, Lys, Gly, Arg,
Thr, His, Cys
and Ser, in particular Ala or Ser.
When position +3 relative to the N-residue is modified, the amino acid residue
to be
20 introduced is preferably selected from the group consisting of His, Asp,
Ala, Met, Asn, Thr,
Arg, Ser and Cys, more preferably Thr, Arg, Ser and Cys. Such modifications
are particular
relevant if the X residue is a Ser residue.
Thus, with respect to the naturally present ifz vivo N-glycosylation, it is
contemplated
that the N-glycosylation site at position 97 may be further optimised by
performing a
modification, such as a substitution, in a position selected from the group
consisting of E93,
K94, L95, T96, Y98, V 100 and T101 (i.e. at position -4, -3, -2, -1, +1, +3 or
+4 relative to
N97). Specific examples of substitutions performed in position 98 of SEQ ll~
NO:1 (or
fragments thereof) include Y98F, Y98N, Y98Q, Y98V, Y98A, Y98M, Y98I, Y98K,
Y98G,
Y98R, Y98T, Y98H, Y98C and Y98S, preferably Y98A, Y98M, Y98I, Y98K, Y98G,
Y98R,
Y98T, Y98H, Y98C and Y98S, in particular Y98S. Specific examples of
substitutions
performed in position 100 of SEQ ll~ NO:1 (or fragments thereof) include V
100H, V 100D,
V 100A, V 100M, V 100N, V 100T, V 1008, V 1005, or V 100C, in particular V
100T, V 1008,
V100S or V100C.
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
21
In a similar way, with respect to the irz vivo N-glycosylation site at
position 25 it is
contemplated that this site may be further optimised by performing a
modification, such as a
substitution, in a position selected from the group consisting of D21, V22,
A23, D24, G26,
L28 and F29 (i.e. at position -4, -3, -2, -1, +1, +3 or +4 relative to N25).
Specific examples of
substitutions performed in position 26 of SEQ ID NO:1 (or fragments thereof)
include G26F,
G26N, G26Y, G26Q, G26V, G26A, G26M, G26I, G26K, G26R, G26T, G26H, G26C and
G26S, preferably G26A, G26M, G26I, G26K, G26R, G26T, G26H, G26C and G26S, more
preferably G26A and G26S, in particular G26A. Specific examples of
substitutions
performed in position 28 of SEQ ID NO:1 (or fragments thereof) include G28H,
G28D,
G28A, G28M, G28N, G28T, G28R, G28S, or G28S, in particular G28A, G28T, G28R,
G28S
or G28C.
Obviously, any of the modifications mentioned in connection with optimisation
of
glycosylation at position 97 may be combined with any of the above-mentioned
modifica-
tions performed in connection with optimisation of glycosylation at position
25.
IFNG variants of the ihvefZtion with increased AUC~, and/or i~zcYeased serum
hal -li a
In a further aspect the present invention relates to a variant of the IFNG
polypeptide
having the amino acid sequence shown in SEQ ID NO:1, wherein said variant
exhibits 1FNG
activity.
Moreover, the present invention also relates to a variant of an IFNG
polypeptide
fragment having an amino acid sequence selected from the group consisting of
SEQ ~
N0:2, SEQ ID N0:3, SEQ ID N0:4, SEQ ID NO:S, SEQ ID N0:6, SEQ ID N0:7, SEQ ID
N0:8, SEQ 117 N0:9, SEQ ID NO:10, SEQ ID N0:11, SEQ ~ N0:12, SEQ ID N0:13, SEQ
ID N0:14, SEQ m N0:15 and SEQ ID NO:16, wherein said variant exhibits IFNG
activity.
Thus, such a variant comprises at least one further modification compared to
SEQ ID
NOS:1-16.
In order to avoid too much disruption of the structure and function of the
[S99T]huIFNG polypeptide variant (or fragments thereof) the total number of
amino acid
residues to be modified in accordance with the present invention typically
does not exceed
15. Usually, the 1FNG polypeptide variant comprises 1-10 modifications
relative to the amino
acid sequence shown in SEQ ID NO:l, such as 1-8, 2-8, 1-5, 1-3 or 2-5
modifications relative
to the amino acid sequence shown in SEQ ID NO:l. Preferably, the
modifications) is/are a
substitution(s). It will be understood that similar considerations hold true
for variants of
fragments of the IFNG polypeptide variant having the amino acid sequence shown
in SEQ ID
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
22
N0:1. Thus, when the variant is a variant of any of the sequences disclosed in
SEQ m
NOS:2-16, such a variant usually comprises less than 15 modifications,
typically 1-10
modifications, relative to the relevant amino acid sequence shown in SEQ )D
NOS:2-16, such
as 1-8, 2-8, 1-5, 1-3 or 2-5 modifications relative to the relevant the amino
acid sequence
shown in SEQ )D NOS:2-16. Preferably, the modifications) is/are a
substitution(s).
Thus, normally such an IFNG polypeptide variant (i.e. a variant which
comprises at
least one further modification in addition to the S99T substitution) comprises
an amino acid
sequence which differs from the amino acid sequence shown in SEQ >D NO: l (or
fragments
thereof) in l, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 or 15 amino acid
residues.
In a preferred embodiment of the invention, the IFNG variant (further)
comprises at
least one introduced and/or at least one removed amino acid residue comprising
an
attachment group for a non-polypeptide moiety.
By removing or introducing an amino acid residue comprising an attachment
group
for the non-polypeptide moiety it is possible to specifically adapt the
polypeptide so as to
make the molecule more susceptible to conjugation to the non-polypeptide
moiety of choice,
to optimse the conjugation pattern (e.g. to ensure an optimal distribution of
non-polypeptide
moieties on the surface of the IFNG polypeptide variant) and thereby obtain a
new conjugate
molecule, which exhibits IFNG activity and in addition one or more improved
properties as
compared to huIFNG- or rhuIFNG-based molecules available today. For instance,
by
introduction of attachment groups, the IFNG polypeptide variant is boosted or
otherwise
altered in the content of the specific amino acid residues to which the
relevant non-
polypeptide moiety binds, whereby a more efficient, specific and/or extensive
conjugation is
achieved. By removal of one or more attachment groups it is possible to avoid
conjugation to
the non-polypeptide moiety in parts of the polypeptide in which such
conjugation is
disadvantageous, e.g. to an amino acid residue located at or near a functional
site of the
polypeptide (since conjugation at such a site may result in inactivation or
reduced IFNG
activity of the resulting conjugated polypeptide due to impaired receptor
recognition).
Further, it may be advantageous to remove an attachment group located closely
to another
attachment group in order to avoid heterogeneous conjugation to such groups.
In interesting
embodiments more than one amino acid residue of the IFNG polypeptide is
altered, e.g. the
alteration embraces removal as well as introduction of amino acid residues
comprising
attachment sites for the non-polypeptide moiety of choice. This embodiment is
considered of
particular interest in that it is possible to specifically design the IFNG
polypeptide variant so
as to obtain an optimal conjugation to the non-polypeptide moiety.
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
23
In addition to the removal and/or introduction of amino acid residues the
polypeptide
variant may comprise other modifications, e.g. substitutions, that are not
related to
introduction and/or removal of amino acid residues comprising an attachment
group for the
non-polypeptide moiety. Examples of such modifications include conservative
amino acid
substitutions and/or introduction of Cys-Tyr-Cys or Met at the N-terminus.
The exact number of attachment groups available for conjugation and present in
the
IFNG polypeptide variant in dimeric form is dependent on the effect desired to
be achieved
by the conjugation. The effect to be obtained is, e.g. dependent on the nature
and degree of
conjugation (e.g. the identity of the non-polypeptide moiety, the number of
non-polypeptide
moieties desirable or possible to conjugate to the polypeptide, where they
should be
conjugated or where conjugation should be avoided, etc.).
It will be understood that the amino acid residue comprising an attachment
group for a
non-polypeptide moiety, either it be removed or introduced, is selected on the
basis of the
nature of the non-polypeptide moiety part of choice and, in most instances, on
the basis of the
conjugation method to be used. For instance, when the non-polypeptide moiety
is a polymer
molecule such as a polyethylene glycol- or polyalkylene oxide-derived molecule
amino acid
residues capable of functioning as an attachment group may be selected from
the group
consisting of cysteine, lysine, aspartic acid, glutamic acid and arginine. In
particular, cysteine
is preferred. When the non-polypeptide moiety is a sugar moiety the attachment
group is, e.g.
an in vivo glycosylation site, preferably an N-glycosylation site.
Whenever an attachment group for a non-polypeptide moiety is to be introduced
into
or removed from the IFNG polypeptide having the amino acid sequence shown as
SEQ ID
NO: l (or fragments thereof), the position of the polypeptide to be modified
is conveniently
selected as follows:
The position is preferably located at the surface of the IFNG polypeptide, and
more
preferably occupied by an amino acid residue that has more than 25% of its
side chain
exposed to the solvent, preferably more than 50% of its side chain exposed to
the solvent, as
determined on the basis of a 3D structure or model of IFNG in its dimeric
form, the structure
or model optionally further comprising one or two IFNG receptor molecules.
Such positions
are listed in Example 1 herein.
Also of interest is to modify any of the 23 C-terminal amino acid residues of
the
parent IFNG polypeptide (in particular by introduction of amino acid residues
comprising an
attachment group for the non-polypeptide moiety, such as Cys residues) since
such residues
are believed to be located at the surface of the IFNG polypeptide.
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
24
In addition, it may be of interest to modify one or more amino acid residues
located in
the loop regions of the IFNG polypeptide since most amino acid residues within
these loop
regions are exposed to the surface and located sufficiently far away from
functional sites so
that non-polypeptide moieties, such as polymer molecules, in particular PEG
molecules,
and/or N-glycosylation sites, may be introduced without impairing the function
of the
molecule. Such loops regions may be identified by inspection of the three-
dimensional
structure of huIFNG. The amino acid residues constituting said loop regions
are residues
N16-K37 (the "A-B loop"), F60-S65 (the "B-C loop"), N83-S84 (the "C-D loop")
and Y98-
L103 (the "D-E loop").
The amino acid residues constituting the IFNG receptor binding site are Q1,
D2, Y4,
V5, E9, K12, G18, H19, 520, D21, V22, A23, D24, N25, G26, T27, L30, K34, K37,
K108,
H111, E112, I114, Q115, A118, E119 (see also Example 2 herein). In general, it
is preferred
that attachment groups for a non-polypeptide moiety (such as additional N-
glycosylation sites
and/or cysteine residues) are not introduced into this site of the molecule.
In order to determine an optimal distribution of attachment groups, the
distance
between amino acid residues located at the surface of the IFNG polypeptide is
calculated on
the basis of a 3D structure of the IFNG dimeric polypeptide. More
specifically, the distance
between the CB's of the amino acid residues comprising such attachment groups,
or the
distance between the functional group (NZ for lysine, CG for aspartic acid, CD
for glutamic
acid, SG for cysteine) of one and the CB of another amino acid residue
comprising an
attachment group are determined. In case of glycine, CA is used instead of CB.
In the IFNG
polypeptide part of the invention any of said distances is preferably more
than 8 A, in
particular more than 10A in order to avoid or reduce heterogeneous
conjugation.
Also, the amino acid sequence of the 1FNG polypeptide variant may differ from
that
of SEQ ll~ NO:1 (or fragments thereof) in that one or more amino acid residues
constituting
part of an epitope has been removed, preferably by substitution to an amino
acid residue
comprising an attachment group for the non-polypeptide moiety, so as to
destroy or inactivate
the epitope. Epitopes of [S99T]huIFNG, huIFNG or rhulFNG may be identified by
use of
methods known in the art, also known as epitope mapping, see, e.g. Romagnoli
et al., Biol
Chem, 1999, 380(5):553-9, DeLisser HM, Methods Mol Biol, 1999, 96:11-20, Van
de Water
et al., Clin Immunol Immunopathol, 1997, 85(3):229-35, Saint-Remy JM,
Toxicology, 1997,
119(1):77-81, and Lane DP and Stephen CW, Curr Opin Irnrr~unol, 1993, 5(2):268-
71. One
method is to establish a phage display library expressing random oligopeptides
of e.g. 9
amino acid residues. IgGl antibodies from specific antisera towards
[S99T]huIFNG, huIFNG
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
or rhuIFNG are purified by immunoprecipitation and the reactive phages are
identified by
immunoblotting. By sequencing the DNA of the purified reactive phages, the
sequence of the
oligopeptide can be determined followed by localization of the sequence on the
3D-structure
of the IFNG. The thereby identified region on the structure constitutes an
epitope that then
5 can be selected as a target region for introduction of an attachment group
for the non-
polypeptide moiety.
Functional in vivo half-life and serum half-life is e.g. dependent on the
molecular
weight of the polypeptide variant and the number of attachment groups needed
for providing
increased half-life may depend on the molecular weight of the non-polypeptide
moiety in
10 question. In one embodiment, the IFNG polypeptide variant of the invention
has a molecular
weight of at least 67 kDa, in particular at least 70 kDa as measured by SDS-
PAGE according
to Laemmli, U.K., Nature Vol 227 (1970), p680-85. IFNG has a Mw in the range
of about
34-50 kDa, and therefore additional about 20-40kDa is required to obtain the
desired effect.
This may, e.g., be provided by 2-4 lOkDa PEG molecules or by a combination of
additional
15 ih vivo glycosylation sites and additional PEG molecules, or as otherwise
described herein.
Preferably, a conjugated IFNG polypeptide variant according to the invention
comprises 1-10 (additional) non-polypeptide moieties, such as 1-8, 2-8, 1-5, 1-
3 or 2-5
(additional) non-polypeptide moieties. Typically, a conjugated variant
comprises 1-3
(additional) non-polypeptide moieties, such as 1, 2 or 3 (additional) non-
polypeptide
20 moieties.
As mentioned above, under physiological conditions huIFNG exists as a dimeric
polypeptide. The polypeptide is normally in homodimeric form (e.g. prepared by
association
of two IFNG polypeptide molecules prepared as described herein). However, if
desired the
IFNG polypeptide variant may be provided in single chain form, wherein two
IFNG
25 polypeptide monomers are linked via a peptide bond or a peptide linker.
Providing the IFNG
polypeptide variant in single chain form has the advantage that the two
constituent IFNG
polypeptides may be different which can be advantageous, e.g., to enable
asymmetric
mutagenesis of the polypeptides. For instance, PEGylation sites can be removed
from the
receptor-binding site from one of the monomers, but retained in the other.
Thereby, after
PEGylation one monomer has an intact receptor-binding site, whereas the other
may be fully
PEGylated (and thus provide significantly increased molecular weight).
IFNG variants of the ircveratiora wherein tlae nora polypeptide moiety is a
sugar moiety
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
26
In a preferred embodiment of the invention the IFNG variant of SEQ ID NO: l
(or
fragments thereof) comprises at least one introduced glycosylation site and/or
at least one
removed glycosylation site. Preferably, the glycosylation site is an irz vzvo
N-glycosylation
site, i.e. the non-polypeptide moiety is a sugar moiety, e.g. an O-linked or N-
Iinlced sugar
moiety, preferably an N-linked sugar moiety.
In one interesting embodiment of the invention said variant comprises at least
one
introduced glycosylation site, in particular an introduced if2 vivo N-
glycosylation site.
Preferably, the introduced glycosylation site is introduced by a substitution.
For instance, an izz vivo N-glycosylation site may be introduced into a
position of the
IFNG polypeptide of SEQ ID NO:1 (of fragments thereof) comprising an amino
acid residue
exposed to the surface. Preferably said surface-exposed amino acid residue has
at least 25%
of the side chain exposed to the surface, in particular at least 50% of its
side chain exposed to
the surface. Details regarding determination of such positions can be found in
Example 1
herein.
The N-glycosylation site is introduced in such a way that the N-residue of
said site is
located in said position. Analogously, an O-glycosylation site is introduced
so that the S or T
residue making up such site is located in said position. It should be
understood that when the
term "at least 25% (or 50%) of its side chain exposed to the surface" is used
in connection
with introduction of an iy2 vivo N-glycosylation site this term refers to the
surface accessibility
of the amino acid side chain in the position where the sugar moiety is
actually attached. In
many cases it will be necessary to introduce a serine or a threonine residue
in position +2
relative to the asparagine residue to which the sugar moiety is actually
attached and these
positions, where the serine or threonine residues are introduced, are allowed
to be buried, i.e.
to have less than 25% (or 50%) of their side chains exposed to the surface of
the molecule.
Furthermore, in order to ensure efficient glycosylation it is preferred that
the in vivo
glycosylation site, in particular the N residue of the N-glycosylation site or
the S or T residue
of the O-glycosylation site, is located within the 118 N-terminal amino acid
residues of the
IFNG polypeptide, more preferably within the 97 N-terminal amino acid
residues. Still more
preferably, the in vivo glycosylation site is introduced into a position
wherein only one
mutation is required to create the site (i.e. where any other amino acid
residues required for
creating a functional glycosylation site is already present in the molecule).
For instance, substitutions that lead to introduction of an additional N-
glycosylation
site at positions exposed at the surface of the IFNG polypeptide and occupied
by amino acid
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
27
residues having at least 25% of the side chain exposed to the surface (in a
structure with
receptor molecule) include:
Q1N + P3S/T, P3N+V5S/T, K6N+A8S/T, E9N+L11S/T, K12S/T, K13N+F15S/T,
Y14N+N16S/T, G18S/T, G18N, G18N+S20T, H19N+D21S/T, D21N+A23S/T,
G26N+L28S/T, G31N+L33S/T, K34N+W36S/T, K37S/T, K37N+E39S/T, E38N,
E38N+S40T, E39N+D41S/T, S40N+R42S/T, K55N+F57S/T, K58N+F60S/T, K61S/T,
K61N+D63S/T, D62N+Q64S/T, D63N, D63N+S65T, Q64N+I66S/T, S65N+Q67S/T, Q67N,
Q67N+S69T, K68N+V70S/T, E71N+I73S/T, T72N+K74S/T, K74N+D76S/T,
E75N+M77S/T, K80S/T, V79N+F81S/T, K80N+F82S/T, N85S/T, S84N+K86S/T, K87S/T,
K86N+K88S/T, K87N+R89S/T, D90N+F92S/T, E93N+L95S/T, K94N, K94N+T96S,
T101N+L103S/T, D102N+N104S/T, L103N+V105S/T, Q106S/T, E119N, E119N+S121T,
P122N+A124S/T, A123N+K125S/T, A124N, A124N+T126S, K125N+G127S/T,
T126N+K128S/T, G127N+R129S/T, K128N+K130S/T, R129N+R131S/T, K130N,
K130N+S132T, R131N+Q133S/T, S132N+M134S/T, Q133N+L135S/T, M134N+F136S/T,
L135N+R137S/T, F136N+G138S/T, R137N+R139S/T, G138N+R140S/T, R139N+A141S/T,
R140N and R140N+S142T, the substitution being indicated relative to
[S99T]huIFNG with
the amino acid sequence shown in SEQ ID NO 1 (or relative to the relevant
fragment thereof
having the amino acid sequence shown in SEQ ID NOS:2-16). S/T indicates a
substitution to
a serine or threonine residue, preferably a threonine residue.
Substitutions that lead to introduction of an additional N-glycosylation site
at
positions exposed at the surface of the IFNG polypeptide having at least 50%
of the side
chain exposed to the surface (in a structure with receptor molecule) include:
P3N+V5S/T, K6N+A8S/T, K12S/T, K13N+F15S/T, G18S/T, D21N+A23S/T,
G26N+L28S/T, G31N+L33S/T, K34N+W36S/T, K37N+E39S/T, E38N, E38N+S40S/T,
E39N+D41S/T, K55N+F57S/T, K58N+F60S/T, K61S/T, D62N+Q64S/T, Q64N+I66S/T,
S65N+Q67S/T, K68N+V70S/T, E71N+I73S/T, E75N+M77S/T, N85S/T, S84N+K86S/T,
K86N+K88S/T, K87N+R89S/T, K94N, K94N+T96S, TlOlN+L103S/T, D102N+N104S/T,
L103N+V105S/T, Q106S/T, P122N+A124S/T, A123N+K125S/T, A124N, A124N+T126S,
K125N+G127S/T, T126N+K128S/T, G127N+R129S/T, K128N+K130S/T,
R129N+R131S/T, K130N, K130N+S132T, R131N+Q133S/T, S132N+M134S/T,
Q133N+L135S/T, M134N+F136S/T, L135N+R137S/T, F136N+G138S/T, R137N+R139S/T,
G138N+R140S/T, R139N+A141S/T, R140N and R140N+S142T, the substitution being
indicated relative to [S99T]huIFNG with the amino acid sequence shown in SEQ
ID NO 1
(or relative to the relevant fragment thereof having the amino acid sequence
shown in SEQ
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
28
~ NOS:2-16). S/T indicates a substitution to a serine or threonine residue,
preferably a
threonine residue.
Substitutions where only one amino acid substitution is required to introduce
an N
glycosylation site include K12S/T, G18S/T, G18N, K37S/T, E38N, M45N, I49N,
K61S/T,
D63N, Q67N, V70N, K80S/T, F82N, N85S/T, K87S/T, K94N, Q106S/T, E119N, A124N,
K130N and R140N, in particular K12S/T, G18N, G18S/T, K37S/T, E38N, K61S/T,
D63N,
Q67N, K80S/T, N85S/T, K94N, Q106S/T, A124N, K130N, and R140N (positions with
more
than 25% of its site chain exposed to the surface (in a structure without
receptor molecule)),
or more preferably G18N, E38N, D63N, Q67N, K94N, S99N, A124N, K130N and R140N
(positions with more than 50% of its side chain exposed to the surface in a
structure without
receptor molecule).
Usually, it is not preferred to introduce N-glycosylation sites in the region
constituting
the receptor binding site (except in special cases, cf. the section entitled
"Variants with a
reduced receptor affinity"). Accordingly, the mutations Q1N+P3S/T, E9N+L11S/T,
G18N,
G18N+S20T, H19N+D21S/T, D21N+A23S/T, G26N+L28S/T, K34N+W36S/T,
K37N+E39S/T, E119N and E119N+S 121T should normally not be performed, unless a
reduced receptor affinity is desired.
Particular preferred variants of the present invention include a variant of
SEQ m
NO:1 (or fragments thereof having the amino acid sequence shown in SEQ 1D
NOS:2-16),
wherein said variant exhibits IFNG activity and which comprises at least one
substitution
selected from the group consisting of K12S, K12T, G18S, G18T, E38N, E38N+S40T,
K61S,
K61T, N85S, N85T, K94N, Q106S and Q106T, more preferably selected from the
group
consisting of K12T, G18T, E38N+S40T, K61T, N85T, K94N and Q106T, even more
preferably selected from the group consisting of K12T, G18T, E38N+S40T, K61T
and N85T,
in particular E38N+S40T.
In another interesting embodiment of the invention, the variant of SEQ )D NO:1
(or
fragments thereof having the amino acid sequence shown in SEQ m NOS:2-16)
comprises at
least two introduced glycosylation sites, in particular at least two
introduced N-glycosylation
sites. The at least two modifications, in particular substitutions, leading to
the introduction of
the at least two introduced N-glycosylation sites may preferably be selected
from the group
consisting of K12S, K12T, G18S, G18T, E38N, E38N+S40T, K61S, K61T, N85S, N85T,
K94N, Q106S and Q106T, more preferably selected from the group consisting of
K12T,
G18T, E38N+S40T, K61T, N85T, K94N and Q106T, even more preferably selected
from the
group consisting of K12T, G18T, E38N+S40T, K61T and N85T. Specific examples of
such
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
29
substitutions giving rise to a variant comprising at least two additional N-
glycosylation sites
include: K12T+G18T, K12T+E38N+S40T, K12T+K61T, K12T+N85T, G18T+E38N+S40T,
G18T+K61T, G18T+N85T, E38N+S40T+K61T, E38N+S40T+N85T and K61T+N85T.
From the above lists of substitutions, it is preferable to select
substitutions located
within the 118 N-terminal amino acid residues, in particular within the 97 N-
terminal amino
acid residues.
The 1FNG polypeptide variant of the invention may contain a single additional
in vivo
glycosylation site per monomer (as compared to SEQ ID NO: l or fragments
thereof).
However, in order to become of a sufficient size to increase the serum half
life it is often
desirable that the polypeptide comprises more than one additional iYZ vivo N-
glycosylation
site, in particular 2-7 or 2-5 additional zfz vivo N-glycosylation sites, such
as 2, 3, 4 or 5 zrz
vivo N-glycosylation sites. Such in vivo N-glycosylation sites are preferably
introduced by
one or more substitutions described in any of the above lists.
Furthermore, it will be understood that any of the above-mentioned
modifications
may be combined with any of the modifications disclosed in the section
entitled "IFNG
varzant of the inveyztion witla optimises' in vivo glycasylatioyz sites", in
particular with the
substitution G26A.
IFNG variants of the ifzvention wherein the hoyz polypeptide moiety is a
molecule, which has
cystei>ze as afz attaclameyzt group
In another preferred embodiment of the invention the IFNG variant of SEQ ID
NO:1
(or fragments thereof) comprises at least one introduced cysteine residue. For
instance, a
cysteine residue may be introduced into a position of the IFNG polypeptide of
SEQ ID NO: l
(or fragments thereof) comprising an amino acid residue exposed to the
surface. Preferably
said surface-exposed amino acid residue has at least 25% of the side chain
exposed to the
surface, in particular at least 50% of its side chain exposed to the surface.
Details regarding
determination of such positions can be found in Example 1 herein.
For instance, substitutions that lead to introduction of a cysteine residue at
positions
exposed at the surface of the IFNG polypeptide and occupied by amino acid
residue having at
least 25% of the side chain exposed to the surface (in a structure with
receptor molecule)
include: Q1C, D2C, P3C, K6C, E9C, N10C, K13C, Y14C, N16C, G18C, H19C, D21C,
N25C, G26C, G31C, K34C, N35C, K37C, E38C, E39C, S40C, K55C, K58C, N59C, K61C,
D62C, D63C, Q64C, S65C, Q67C, K68C, E71C, T72C, K74C, E75C, N78C, V79C, K80C,
N83C, S84C, N85C, K86C, K87C, D90C, E93C, K94C, T101C, D102C, L103C, N104C and
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
El 19C, the substitution being indicated relative to [S99T]huIFNG with the
amino acid
sequence shown in SEQ ID NO 1 (or relative to the relevant fragment thereof
having the
amino acid sequence shown in SEQ m NOS:2-16).
Substitutions that lead to introduction of a cysteine residue at positions
exposed at the
5 surface of the IFNG polypeptide and occupied by amino acid residue having at
least 50% of
the side chain exposed to the surface (in a structure with receptor molecule)
include: P3C,
K6C, N10C, K13C, N16C, D21C, N25C, G26C, G31C, K34C, K37C, E38C, E39C, K55C,
K58C, N59C, D62C, Q64C, S65C, K68C, E71C, E75C, N83C, S84C, K86C, K87C, K94C,
TlOlC, D102C, L103C and N104C, the substitution being indicated relative to
10 [S99T]huIFNG with the amino acid sequence shown in SEQ m NO 1 (or relative
to the
relevant fragment thereof having the amino acid sequence shown in SEQ m NOS:2-
16).
Usually, it is not preferred to introduce a cysteine residue (and subsequently
attaching
the cysteine residue to a non-polypeptide moiety) in the region constituting
the receptor
binding site (except in special cases, cf. the section entitled "Variaf2ts
with a reduced Yeceptor
15 aff-chity"). Accordingly, the mutations Q1C, E9C, G18C, H19C, D21C, G26C,
K34C, K37C
and E119C should normally not be performed, unless a reduced receptor affinity
is desired.
More preferably, said cysteine residue is introduced by a substitution
selected from
the group consisting of NlOC, N16C, E38C, N59C, N83C, K94C, N104C and A124C.
In another interesting embodiment of the invention, the variant of SEQ ~ NO:1
(or
20 fragments thereof having the amino acid sequence shown in SEQ m NOS:2-16)
comprises at
least two introduced cysteine residues. The at least two modifications, in
particular
substitutions, leading to the introduction of the at least two cysteine
residues may preferably
be selected from the group consisting of NlOC, N16C, E38C, N59C, N83C, K94C,
N104C
and A124C. Specific examples of such substitutions giving rise to a variant
comprising at
25 least two introduced cysteine residues include: N10C+N16C, N10C+E38C,
N10C+N59C,
N10C+N83C, N10C+K94C, NlOC+N104C, N10C+A124C, N16C+E38C, N16C+N59C,
N16C+N83C, N16C+K94C, N16C+N104C, N16C+A124C, E38C+N59C, E38C+N83C,
E38C+K94C, E38C+N104C, E38N+A124C, N59C+N83C, N59C+K94C, N59C+N104C,
N59C+A124C, N83C+K94C, N83C+K94C, N83C+N104C, N83C+A124C, K94C+N104C,
30 K94C+A124C and N104C+A124C.
As will be understood the introduced cysteine residues) may preferably be
conjugated to a non-polypeptide moiety, such as PEG or more preferably mPEG.
The
conjugation between the cysteine-containing polypeptide variant and the
polymer molecule
may be achieved in any suitable manner, e.g. as described in the section
entitled
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
31
"Coz2jugatioyz to a polyrrzer molecule", e.g. in using a one step method or in
the stepwise
manner referred to in said section. The preferred method for PEGylating the
IFNG
polypeptide variant is to covalently attach PEG to cysteine residues using
cysteine-reactive
PEGS. A number of highly specific, cysteine-reactive PEGs with different
groups (e.g.
orthopyridyl-disulfide, maleimide and vinylsulfone) and different size PEGS (2-
20 l~Da, such
as 5 kDa, 10 kDa, 12 l~Da or 15 kDa) are commercially available, e.g. from
Shearwater
Polymers Inc., Huntsville, AL, USA).
It will be understood that any of the above-mentioned modifications may be
combined
with any of the modifications disclosed in the section entitled "IFNG variants
of the iyzventio>z
with optimised in vivo glycosylation sites", in particular with the
substitution G26A.
IFNG variants of the irzveyztion whereifz the first >zon polypeptide moiety is
a sugar moiety
afzd the second norz polypeptide moiety is a molecule, which has cysteirze as
au attachmefzt
group
In a further preferred embodiment of the invention the IFNG variant of SEQ ID
NO:1
(or fragments thereof) comprises at least one introduced N-glycosylation site
and at least one
introduced cysteine residue. Such variants may be prepared by selecting the
residues
described in the two preceding sections describing suitable positions for
introducing N-
glycosylation sites and cysteine residues, respectively. However, in a
preferred embodiment
of the invention said variant comprises substitutions selected from the group
consisting of
K12T+N16C, K12T+E38C, K12T+N59C, K12T+N83C, K12T+K94C, K12T+N104C,
K12T+A124C, G18T+N10C, G18T+E38C, G18T+N59C, G18T+N83C, G18T+K94C,
G18T+N104C, G18T+A124C, E38N+S40T+N10C, E38N+S40T+N16C,
E38N+S40T+N59C, E38N+S40T+N83C, E38N+S40T+K94C, E38N+S40T+N104C,
E38N+S40T+A124C, K61T+N10C, K61T+N16C, K61T+E38C, K61T+N83C, K61T+K94C,
K61T+N104C, K61T+A124C, N85T+N10C, N85T+N16C, N85T+E38C, N85T+N59C,
N85T+K94C, N85T+N104C, N85T+A124C, K94N+N10C, K94N+N16C, K94N+E38C,
K94N+N59C, K94N+N83C, K94N+N104C, K94N+A124C, Q106T+N10C, Q106T+N16C,
Q106T+E38C, Q106T+N59C, Q106T+N83C, Q106T+K94C and Q106T+A124C, more
preferably from the group consisting of E38N+S40T+NlOC, E38N+S40T+N16C,
E38N+S40T+N59C, E38N+S40T+N83C, E38N+S40T+K94C and E38N+S40T+N104C.
As will be understood the introduced cysteine residue may preferably be
conjugated
to a non-polypeptide moiety, such as PEG or more preferably mPEG. The
conjugation
between the cysteine-containing polypeptide variant and the polymer may be
achieved in any
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
32
suitable manner, e.g. as described in the section entitled "Conjugation to a
polyfzzer
molecule", e.g. in using a one step method or in the stepwise manner referred
to in said
section. A suitable polymer is VS-mPEG or OPSS-mPEG.
Furthermore, it will be understood that any of the above-mentioned
modifications
may be combined with any of the modifications disclosed in the section
entitled "IFNG
variant of tlae invention with optr.'mised in vivo glycosylation sites", in
particular with the
substitution G26A.
IFNG variants with a reduced receptor affinity
One way to increase the serum half-life of an IFNG polypeptide would be to
decrease
the receptor-mediated internalisation and thereby decrease the receptor-
mediated clearance.
The receptor-mediated internalisation is dependent upon the affinity of the
IFNG
dimer for the IFNG receptor complex and, accordingly, an IFNG variant with a
decreased
affinity to the 1FNG receptor complex is expected to be internalised, and
hence cleared, to a
lesser extent.
The affinity of the IFNG dimmer to its receptor complex may be decreased by
performing one or more modifications, in particular substitutions, in the
receptor binding site
of the IFNG polypeptide. The amino acid residues which constitute the receptor
binding site
is defined in Example 2 herein. One class of substitutions that may be
performed is
conservative amino acid substitutions. In another embodiment, the modification
performed
gives rise to the introduction of an N-glycosylation site.
Thus, in a further particular preferred embodiment of the invention the IFNG
variant
of SEQ ID NO:1 (or fragments thereof) comprises at least one modification,
such as a
substitution, in the receptor binding site (as defined herein). More
particularly, the IFNG
polypeptide comprises at least one modification, preferably a substitution,
which creates an irz
vivo N-glycosylation site, in said receptor binding site. For instance, such
substitutions may
be selected from the group consisting of Q1N+P3S/T, D2N+Y4S/T, Y4N+K6S/T,
V5N+E7S/T, E9N+L11S/T, K12N+Y14S/T, G18N, G18N+S20T, H19N+D21S/T,
S20N+V22S/T, D21N+A23S/T, V22N+D24S/T, D24N+G26S/T, G26N+L28S/T,
L30N+I32S/T, K34N+W36S/T, K37N+E39S/T, K108N+I110S/T, H111N+L113S/T,
E112N+I114S/T, I114N+V116S/T, Q115N+M117S/T, A118N+L120S/T, E119N and
E119N+S121T, preferably from the group consisting of Q1N+P3S/T, D2N+Y4S/T,
E9N+L11S/T, K12N+Y14S/T, G18N, G18N+S20T, H19N+D21S/T, S20N+V22S/T,
D21N+A23S/T, K34N+W36S/T, K37N+E39S/T, H111N+L113S/T, Q115N+M117S/T,
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
33
A118N+L120S/T, E119N and E119N+S121T (introduction of N-glycosylation sites in
positions comprising an amino acid residue having at least 25% of its side
chain exposed to
the surface), more preferably from the group consisting of Q1N+P3S/T,
D2N+Y4S/T,
E9N+L11S/T, G18N, G18N+S20T, H19N+D21S/T, S20N+V22S/T, D21N+A23S/T,
K34N+W36S/T, K37N+E39S/T, Q115N+M117S/T, A118N+L120S/T, E119N and
E119N+S121T (introduction of N-glycosylation sites in positions comprising an
amino acid
residue having at least 50% of its side chain exposed to the surface), even
more preferably
from the group consisting of Q1N+P3T, D2N+Y4T, E9N+L11T, G18N+S20T, H19N+D21T,
S20N+V22T, D21N+A23T, K34N+W36T, K37N+E39T, Q115N+M117T, A118N+L120T
and E119N+S 121T, most preferably from the group consisting of G18N+S20T,
H19N+D21T, D21N+A23T and E119N+S 121T, in particular D21N+A23T.
Such variants are contemplated to exhibit a reduced receptor affinity as
compared to
huIFNG or Actimmune~. The receptor affinity may be measured by any suitable
assay and
will be known to the person skilled in the art. One example of a suitable
assay for
determining the receptor binding affinity is the BIAcore~ assay described in
Michiels et al.
Int. J. Biochem. Cell Biol. 30:505-516 (1998). Using the above-identified
assay, IFNG
variants considered useful for the purposes described herein are such IFNG
variants, wherein
the binding affinity (Kd) is 1-95% of the Kd-value of glycosylated
[S99T]huIFNG or
Actimmune0. For example the Kd-value of the IFNG polypeptide may be 1-75% or 1-
50%,
such as 1-25%, e.g. 1-20% or even as low as 1-15%, 1-10% or 1-5% of the Kd-
value of
glycosylated [S99T]huIFNG or Actimmune~.
Typically, such IFNG variants having reduced receptor affinity will exhibit a
reduced
IFNG activity, e.g. when tested in the "Primary Assay" described herein. For
example, the
IFNG polypeptide variant may exhibit 1-95% of the specific activity of
Actimunne~ or
rhuIFNG, e.g. 1-75%, such as 1-50%, e.g. 1-20% or 1-10% of the specific
activity of
Actimunne~ or rhuIFNG.
As mentioned above, such IFNG variants are contemplated to possess an
increased
serum-half due to the reduced receptor-mediated clearance. Therefore, the IFNG
polypeptide
variants according to the aspect of the invention are contemplated to fulfil
the requirements
with respect to increased serum-half described previously herein in connection
with the
definition of increased serum half-life.
Evidently, any of the above-mentioned modifications giving rise to a reduced
receptor
binding affinity may be combined with any of the other modifications disclosed
herein, in
particular the modifications mentioned in the sections entitled "IFNG variants
of the
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
34
invention with optimised N-glycosylation sites", "IFNG variants of the
invention wherein the
non polypeptide moiety is a sugar nzoiety", "IFNG variants of the invention
wherein the non-
polypeptide moiety is a molecule, which has cysteirze as an attachment group"
and "IFNG
variants of the invention wherein the first rzorz polypeptide moiety is a
sugar moiety and the
second non-polypeptide moiety is a molecule, which lzas cysteine as an
attachment group",
such as G26A, E38N+S40T and combinations thereof.
Conjugation Methods
The non polypeptide moiety
As indicated further above the non-polypeptide moiety is preferably selected
from the group
consisting of a polymer molecule, a lipophilic compound, a sugar moiety (e.g.
by way of in
vivo N-glycosylation) and an organic derivatizing agent. All of these agents
may confer
desirable properties to the IFNG polypeptide variant, in particular increased
AUCS~, increased
serum half-life and/or reduced immunogenicity. The polypeptide variant is
normally
conjugated to only one type of non-polypeptide moiety, but may also be
conjugated to two or
more different types of non-polypeptide moieties, e.g. to a polymer molecule
and a sugar
moiety, to a lipophilic group and a sugar moiety, to an organic derivating
agent and a sugar
moiety, to a lipophilic group and a polymer molecule, etc. When conjugated to
two different
types of non-polypeptide moieties these are preferably a sugar moiety and a
polymer moiety.
The conjugation to two or more different non-polypeptide moieties may be done
simultaneous or sequentially. In the following sections "Conjugation to a
lipophilic
compound", "Conjugation to a polymer molecule", "Conjugation to a sugar
moiety" and
"Conjugation to an organic derivatizing agent" conjugation to specific types
of non-
polypeptide moieties is described.
Conjugation to a lipoplzilic compound
The polypeptide variant and the lipophilic compound may be conjugated to each
other, either directly or by use of a linker. The lipophilic compound may be a
natural
compound such as a saturated or unsaturated fatty acid, a fatty acid diketone,
a terpene, a
prostaglandin, a vitamine, a carotenoide or steroide, or a synthetic compound
such as a
carbon acid, an alcohol, an amine and sulphonic acid with one or more alkyl-,
aryl-, alkenyl-
or other multiple unsaturated compounds. The conjugation between the
polypeptide variant
and the lipophilic compound, optionally through a linker may be done according
to methods
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
known in the art, e.g. as described by Bodanszky in Peptide Synthesis, John
Wiley, New
York, 1976 and in WO 96/12505.
Conjugatzora to a polymer molecule
5 The polymer molecule to be coupled to the polypeptide variant may be any
suitable
polymer molecule, such as a natural or synthetic homo-polymer or
heteropolymer, typically with
a molecular weight in the range of 300-100,000 Da or 1000-50,000 Da, such as
in the range of
2000-40,000 Da or 2000-30,000 Da, e.g. in the range of 2000-20,000 Da, 2000-
10,000 or 1000-
5000 Da. More particularly, the polymer molecule, such as PEG, in particular
mPEG, will
10 typically have a molecular weight of about 2, 5, 10, 12, 15, 20, 30, 40 or
50 kDa, in particular a
molecular weight of about 5 kDa, about 10 kDa, about 12 kDa, about 15 kDa or
20 about kDa.
When used about polymer molecules herein, the word "about" indicates an
approximate average molecular weight and reflects the fact that there will
normally be a
certain molecular weight distribution in a given polymer preparation.
15 Examples of homo-polymers include a polyol (i.e. poly-OIL, a polyamine
(i.e. poly-
NH2) and a polycarboxylic acid (i.e. poly-COOIT). A hetero-polymer is a
polymer, which
comprises one or more different coupling groups, such as, e.g., a hydroxyl
group and an amine
group.
Examples of suitable polymer molecules include polymer molecules selected from
the
20 group consisting of polyalkylene oxide (PAO), including polyalkylene glycol
(PAG), such as
polyethylene glycol (PEG) and polypropylene glycol (PPG), branched PEGS, poly-
vinyl alcohol
(PVA), poly-carboxylate, poly-(vinylpyrolidone), polyethylene-co-malefic acid
anhydride,
polystyrene-co-malic acid anhydride, dextran including carboxymethyl-dextran,
or any other
biopolymer suitable for reducing immunogenicity and/or increasing functional
in vivo half-life
25 and/or serum half-life. Another example of a polymer molecule is human
albumin or another
abundant plasma protein. Generally, polyalkylene glycol-derived polymers are
biocompatible,
non-toxic, non-antigenic, non-immunogenic, have various water solubility
properties, and are
easily excreted from living organisms.
PEG is the preferred polymer molecule to be used, since it has only few
reactive groups
30 capable of cross-linking compared, e.g., to polysaccharides such as
dextran, and the like. In
particular, monofunctional PEG, such as, monomethoxypolyethylene glycol
(mPEG), is of
interest since its coupling chemistry is relatively simple (only one reactive
group is available for
conjugating with attachment groups on the polypeptide). Consequently, the risk
of cross-linking
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
36
is eliminated, the resulting conjugated polypeptide variantss are more
homogeneous and the
reaction of the polymer molecules with the polypeptide is easier to control.
To effect covalent attachment of the polymer molecules) to the polypeptide
variant,
the hydroxyl end groups of the polymer molecule must be provided in activated
form, i.e.
with reactive functional groups (examples of which include primary amino
groups, hydrazide
(HZ), thiol, succinate (SUC), succinimidyl succinate (SS), succinimidyl
succinamide (SSA),
succinimidyl proprionate (SPA), succinimidy carboxymethylate (SCM),
benzotriazole
carbonate (BTC), N-hydroxysuccinimide (NHS), aldehyde, nitrophenylcarbonate
(NPC), and
tresylate (TRES)). Suitably activated polymer molecules are commercially
available, e.g.
from Shearwater Polymers, Inc., Huntsville, AL, USA. Alternatively, the
polymer molecules
can be activated by conventional methods known in the art, e.g. as disclosed
in WO
90/13540. Specific examples of activated linear or branched polymer molecules
for use in
the present invention are described in the Shearwater Polymers, Inc. 1997 and
2000
Catalogue (Functionalized Biocompatible Polymers for Research and
pharmaceuticals,
Polyethylene Glycol and Derivatives, incorporated herein by reference).
Specific examples of
activated PEG polymers include the following linear PEGS: NHS-PEG (e.g. SPA-
PEG,
SSPA-PEG, SBA-PEG, SS-PEG, SSA-PEG, SC-PEG, SG-PEG, and SCM-PEG), and NOR-
PEG), BTC-PEG, EPOX-PEG, NCO-PEG, NPC-PEG, CDI-PEG, ALD-PEG, TRES-PEG, VS-
PEG, IODO-PEG, and MAL-PEG, and branched PEGs such as PEG2-NHS and those
disclosed
in US 5,932,462 and US 5,643,575, both of which references are incorporated
herein by
reference. Furthermore, the following publications, incorporated herein by
reference, disclose
useful polymer molecules and/or PEGylation chemistries: US 5,824,778, US
5,476,653, WO
97/32607, EP 229,108, EP 402,378, US 4,902,502, US 5,281,698, US 5,122,614, US
5,219,564, WO 92/16555, WO 94/04193, WO 94/14758, WO 94/17039, WO 94/18247, WO
94/28024, WO 95/00162, WO 95/11924, W095113090, WO 95/33490, WO 96/00080, WO
97/18832, WO 98/41562, WO 98/48837, WO 99/32134, WO 99/32139, WO 99/32140, WO
96/40791, WO 98/32466, WO 95/06058, EP 439 508, WO 97/03106, WO 96/21469, WO
95/13312, EP 921 131, US 5,736,625, WO 98/05363, EP 809 996, US 5,629,384, WO
96/41813, WO 96/07670, US 5,473,034, US 5,516,673, EP 605 963, US 5,382,657,
EP 510
356, EP 400 472, EP 183 503 and EP 154 316.
Specific examples of activated PEG polymers particularly preferred for
coupling to
cysteine residues, include the following linear PEGS: vinylsulfone-PEG (VS-
PEG),
preferably vinylsulfone-mPEG (VS-mPEG); maleimide-PEG (MAL-PEG), preferably
maleimide-mPEG (MAL-mPEG) and orthopyridyl-disulfide-PEG (OPSS-PEG),
preferably
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
37
orthopyridyl-disulfide-mPEG (OPSS-mPEG). Typically, such PEG or mPEG polymers
will
have a size of about 5 kDa, about 10 kD, about 12 kDa or about 20 lcDa.
The conjugation of the polypeptide variant and the activated polymer molecules
is
conducted by use of any conventional method, e.g. as described in the
following references
(which also describe suitable methods for activation of polymer molecules):
Harris and
Zalipsky, eds., Polyethylene glycol) Chemistry and Biological Applications,
AZC, Washington;
R.F. Taylor, (1991), "Protein immobilisation. Fundamental and applications",
Marcel Del~lcer,
N.Y.; S.S. along, (1992), "Chemistry of Protein Conjugation and Crosslinking",
CRC Press,
Boca Raton; G.T. Hermanson et al., (1993), "Immobilized Affinity Ligand
Techniques",
Academic Press, N.Y.). For PEGylation to cysteine residues (see above) the
IFNG variant is
usually treated with a reducing agent, such as dithiothreitol (DDT) prior to
PEGylation. The
reducing agent is subsequently removed by any conventional method, such as by
desalting.
Conjugation of PEG to a cysteine residue typically takes place in a suitable
buffer at pH 6-9 at
temperatures varying from 4°C to 25°C for periods up to 16
hours.
The skilled person will be aware that the activation method andlor conjugation
chemistry
to be used depends on the attachment groups) of the polypeptide variant as
well as the
functional groups of the polymer (e.g. being amino, hydroxyl, carboxyl,
aldehyde or sulfydryl).
The PEGylation may be directed towards conjugation to all available attachment
groups on
the polypeptide variant (i.e. such attachment groups that are exposed at the
surface of the
polypeptide) or may be directed towards specific attachment groups, e.g. the N-
terminal
amino group (US 5,985,265). Furthermore, the conjugation may be achieved in
one step or in
a stepwise manner (e.g. as described in WO 99/55377).
It will be understood that the PEGylation is designed so as to produce the
optimal
molecule with respect to the number of PEG molecules attached, the size and
form (e.g.
whether they are linear or branched) of such molecules, and where in the
polypeptide variant
such molecules are attached. For instance, the molecular weight of the polymer
to be used may
be chosen on the basis of the desired effect to be achieved. For instance, if
the primary purpose
of the conjugation is to achieve a conjugate having a high molecular weight
(e.g. to reduce renal
clearance) it is usually desirable to conjugate as few high molecular weight-
polymer molecules
as possible to obtain the desired molecular weight. When a high degree of
epitope shielding is
desirable this may be obtained by use of a sufficiently high number of low
molecular weight
polymer (e.g. with a molecular weight of about 5,000 Da) to effectively shield
all or most
epitopes of the polypeptide. For instance, 2-~, such as 3-6, of such polymers
may be used.
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
38
In connection with conjugation to only a single attachment group on the
protein (as
described in US 5,985,265), it may be advantageous that the polymer molecule,
which may
be linear or branched, has a high molecular weight, e.g. about 20 lcDa.
Normally, the polymer conjugation is performed under conditions aiming at
reacting all
available polymer attachment groups with polymer molecules. Typically, the
molar ratio of
activated polymer molecules to polypeptide variant is 1000-1, in particular
200-1, e.g. 100-1,
such as 10-1 or 5-1 in order to obtain optimal reaction. However, also
equimolar ratios may be
used.
It is also contemplated according to the invention to couple the polymer
molecules to the
polypeptide variant through a linker. Suitable linkers are well known to the
skilled person. A
preferred example is cyanuric chloride (Abuchowski et al., (1977), J. Biol.
Chem., 252,
3578-3581; US 4,179,337; Shafer et al., (1986), J. Polym. Sci. Polym. Chem.
Ed., 24, 375-378.
Subsequent to the conjugation residual activated polymer molecules are blocked
according to methods known in the art, e.g. by addition of primary amine to
the reaction
mixture, and the resulting inactivated polymer molecules removed by a suitable
method.
Coupling to a sugaY ruoiety
The coupling of a sugar moiety may take place in vivo or in vitYO. In order to
achieve
iyz vivo glycosylation of a polypeptide with IFNG activity, which have been
modified so as to
introduce one or more iyz vivo glycosylation sites (see the section "IFNG
variants of the
inveyztion wherezyz the zzon polypeptide moiety is a sugar moiety), the
nucleotide sequence
encoding the polypeptide must be inserted in a glycosylating, eukaryotic
expression host. The
expression host cell may be selected from fungal (filamentous fungal or
yeast), insect or
animal cells or from transgenic plant cells. Furthermore, the glycosylation
may be achieved
in the human body when using a nucleotide sequence encoding the polypeptide
variant of the
invention in gene therapy. In one embodiment the host cell is a marnrnalian
cell, such as an
CHO cell, BHK or HEK cell, e.g. HEI~293, or an insect cell, such as an SF9
cell, or a yeast
cell, e.g. Saccharorzzyces cerevisiae, Pichia pastoris or any other suitable
glycosylating host,
e.g. as described further below. Optionally, sugar moieties attached to the
1FNG polypeptide
variant by z~z vivo glycosylation are further modified by use of
glycosyltransferases, e.g. using
the glycoAdvance~ technology marketed by Neose, Horsham, PA, USA. Thereby, it
is
possible to, e.g., increase the sialyation of the glycosylated IFNG
polypeptide variant
following expression and in vivo glycosylation by CHO cells.
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
39
Covalent ira vitro coupling of glycosides to amino acid residues of IFNG
polypepeptides may be used to modify or increase the number or profile of
carbohydrate
substituents. Depending on the coupling mode used, the sugars) may be attached
to a)
arginine and histidine, b) free carboxyl groups, c) free sulfhydryl groups
such as those of
cysteine, d) free hydroxyl groups such as those of serine, threonine, tyrosine
or
hydroxyproline, e) aromatic residues such as those of phenylalanine or
tryptophan or f) the
amide group of glutamine. These amino acid residues constitute examples of
attachment
groups for a sugar moiety, which may be introduced and/or removed in the IFNG
polypeptide. Suitable methods of iyi vitro coupling are described, for
example, in WO
87/05330 and in Aplin et al., CRC Crit Rev. Biochem., pp. 259-306, 1981. The
iya vitro
coupling of sugar moieties or PEG to protein- and peptide-bound Gln-residues
can also be
carried out by transglutaminases (TGases), e.g. as described by Sato et al.,
1996 Biochemistry
35, 13072-13080 or in EP 725145.
Coupling to ayz organic derr.'vatizifag agent
Covalent modification of the IFNG polypeptide variant may be performed by
reacting
(an) attachment groups) of the polypeptide variant with an organic
derivatizing agent.
Suitable derivatizing agents and methods are well known in the art. For
example, cysteinyl
residues most commonly are reacted with a-haloacetates (and corresponding
amines), such as
chloroacetic acid or chloroacetamide, to give carboxymethyl or
carboxyamidomethyl
derivatives. Cysteinyl residues also are derivatized by reaction with
bromotrifluoroacetone,
a-bromo-(3-(4-imidozoyl)propionic acid, chloroacetyl phosphate, N-
alkylmaleimides, 3-nitro-
2-pyridyl disulfide, methyl 2-pyridyl disulfide, p-chloromercuribenzoate, 2-
chloromercuri-4-
nitrophenol, or chloro-7-nitrobenzo-2-oxa-1,3-diazole. Histidyl residues are
derivatized by
reaction with diethylpyrocarbonateat pH 5.5-7.0 because this agent is
relatively specific for
the histidyl side chain. Para-bromophenacyl bromide also is useful; the
reaction is preferably
performed in 0.1 M sodium cacodylate at pH 6Ø Lysinyl and amino terminal
residues are
reacted with succinic or other carboxylic acid anhydrides. Derivatization with
these agents
has the effect of reversing the charge of the lysinyl residues. Other suitable
reagents for
derivatizing a-amino-containing residues include imidoesters such as methyl
picolinimidate;
pyridoxal phosphate; pyridoxal; chloroborohydride; trinitrobenzenesulfonic
acid; O-
methylisourea; 2,4-pentanedione; and transaminase-catalyzed reaction with
glyoxylate.
Arginyl residues are modified by reaction with one or several conventional
reagents, among
them phenylglyoxal, 2,3-butanedione, 1,2-cyclohexanedione, and ninhydrin.
Derivatization
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
of arginine residues requires that the reaction be performed in alkaline
conditions because of
the high pKa of the guanidine functional group. Furthermore, these reagents
may react with
the groups of lysine as well as the arginine guanidino group. Carboxyl side
groups (aspartyl
or glutamyl) are selectively modified by reaction with carbodiimides (R-N=C=N-
R'), where
5 R and R' are different allcyl groups, such as 1-cyclohexyl-3-(2-morpholinyl-
4-ethyl)
carbodiimide or 1-ethyl-3-(4-azonia-4,4-dimethylpentyl) carbodiimide.
Furthermore, aspartyl
and glutaxnyl residues are converted to asparaginyl and glutaminyl residues by
reaction with
ammonium ions.
10 Blocking of fufzctioyzal site
It has been reported that excessive polymer conjugation can lead to a loss of
activity
of the polypeptide to which the polymer is conjugated. This problem can be
eliminated, e.g.,
by removal of attachment groups located at the functional site or by blocking
the functional
site prior to conjugation. These latter strategies constitute further
embodiments of the
15 invention, the first strategy being exemplified further above, e.g. by
removal of lysine
residues which may be located close to the functional site and/or by
introducing cysteine
residues and/or ih vivo glycosylation sites at positions not interfering with
functional sites.
More specifically, according to the second strategy the conjugation between
the
polypeptide variant and the non-polypeptide moiety is conducted under
conditions where the
20 functional site of the IFNG polypeptide variant is blocked by a helper
molecule capable of
binding to the functional site of the polypeptide variant. Preferably, the
helper molecule is
one, which specifically recognizes a functional site of the polypeptide
variant, such as a
receptor. Alternatively, the helper molecule may be an antibody, in particular
a monoclonal
antibody recognizing the polypeptide variant. In particular, the helper
molecule may be a
25 neutralizing monoclonal antibody.
The polypeptide variant is then allowed to interact with the helper molecule
before
effecting conjugation. This ensures that the functional site of the
polypeptide variant is
shielded or protected and consequently unavailable for derivatization by the
non-polypeptide
moiety such, as a polymer. Following its elution from the helper molecule, the
conjugate
30 between the non-polypeptide moiety and the polypeptide variant can be
recovered with at
least a partially preserved functional site.
The subsequent conjugation of the polypeptide variant having a blocked
functional
site to a polymer, a lipophilic compound, a sugar moiety, an organic
derivatizing agent or any
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
41
other compound is conducted in the normal way, e.g. as described in the
sections above
entitled "Conjugation to ....".
In a further embodiment the helper molecule is first covalently linlced to a
solid phase
such as column paclcing materials, for instance Sephadex or agarose beads, or
a sui~ace, e.g.
reaction vessel. Subsequently, the polypeptide variant is loaded onto the
column material
carrying the helper molecule and conjugation carned out according to methods
known in the
art, e.g. as described in the sections above entitled "Conjugatiofa to ....".
This procedure
allows the conjugated polypeptide variant to be separated from the helper
molecule by
elution. The conjugated polypeptide variant is eluted by conventional
techniques under
physico-chemical conditions that do not lead to a substantive degradation of
the conjugated
polypeptide variant. The fluid phase containing the conjugated polypeptide
variant is
separated from the solid phase to which the helper molecule remains covalently
linked. The
separation can be achieved in other ways: For instance, the helper molecule
may be
derivatized with a second molecule (e.g. biotin) that can be recognized by a
specific binder
(e.g. streptavidin). The specific binder may be linked to a solid phase
thereby allowing the
separation of the conjugated polypeptide variant from the helper molecule-
second molecule
complex through passage over a second helper-solid phase column which will
retain, upon
subsequent elution, the helper molecule-second molecule complex, but not the
conjugated
polypeptide variant. The conjugated polypeptide variant may be released from
the helper
molecule in any appropriate fashion. De-protection may be achieved by
providing conditions
in which the helper molecule dissociates from the functional site of the
polypeptide variant to
which it is bound. For instance, a complex between an antibody to which a
polymer is
conjugated and an anti-idiotypic antibody can be dissociated by adjusting the
pH to an acid or
alkaline pH.
Conjugation of a tagged polypeptide variaT2t
In an alternative embodiment the 1FNG polypeptide variant is expressed, as a
fusion
protein, with a tag, i.e. an amino acid sequence or peptide stretch made up of
typically 1-30,
such as 1-20 amino acid residues. Besides allowing for fast and easy
purification, the tag is a
convenient tool for achieving conjugation between the tagged 1FNG polypeptide
variant and
the non-polypeptide moiety. In particular, the tag may be used for achieving
conjugation in
microtiter plates or other carriers, such as paramagnetic beads, to which the
tagged
polypeptide can be immobilised via the tag. The conjugation to the tagged IFNG
polypeptide
variantin, e.g., microtiter plates has the advantage that the tagged
polypeptide variant can be
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
42
immobilised in the microtiter plates directly from the culture broth (in
principle without any
purification) and subjected to conjugation. Thereby, the total number of
process steps (from
expression to conjugation) can be reduced. Furthermore, the tag may function
as a spacer
molecule ensuring an improved accessibility to the immobilised polypeptide
variant to be
conjugated. The conjugation using a tagged polypeptide variant may be to any
of the non-
polypeptide moieties disclosed herein, e.g. to a polymer molecule such as PEG.
The identity of the specific tag to be used is not critical as long as the tag
is capable of
being expressed with the polypeptide variant and is capable of being
immobilised on a
suitable surface or carrier material. A number of suitable tags are
commercially available, e.g.
from Unizyme Laboratories, Denmark. For instance, the tag may any of the
following
sequences:
His-His-His-His-His-His (SEQ m N0:35),
Met-Lys-His-His-His-His-His-His (SEQ 1D N0:36),
Met-Lys-His-His-Ala-His-His-Gln-His-His (SEQ )D N0:37),
Met-Lys-His-Gln-His-Gln-His-Gln-His-Gln-His-Gln-His-Gln (SEQ ID N0:38,
(all available from Unizyme Laboratories, Denmark)
or any of the following:
EQKLT SEEDL (SEQ ID NO:39) (a C-terminal tag described in Mol. Cell. Biol.
5:3610-16,
1985),
DYKDDDDK (SEQ m N0:40) (a C- or N-terminal tag),
YPYDVPDYA (SEQ ~ NO:41)
Antibodies against the above tags are commercially available, e. g. from ADI,
Aves
Lab and Research Diagnostics.
The subsequent cleavage of the tag from the polypeptide may be achieved by use
of
commercially available enzymes.
Methods of preparing an IFNG polypeptide variant of the invention
The IFNG polypeptide variant may be produced by any suitable method known in
the
art. Such methods include constructing a nucleotide sequence encoding the
polypeptide
variant and expressing the sequence in a suitable transformed or transfected
host. However,
polypeptide variants of the invention may be produced, albeit less
efficiently, by chemical
synthesis or a combination of chemical synthesis or a combination of chemical
synthesis and
recombinant DNA technology.
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
43
The nucleotide sequence of the invention encoding an IFNG polypeptide variant
(in
monomer or single chain form) may be constructed by isolating or synthesizing
a nucleotide
sequence encoding the parent IFNG, such as hulFNG with the amino acid sequence
SEQ ID
N0:17 or a fragment thereof, and then changing the nucleotide sequence so as
to effect
introduction (i.e. insertion or substitution) or deletion (i.e. removal or
substitution) of the
relevant amino acid residue(s).
The nucleotide sequence is conveniently modified by site-directed mutagenesis
in
accordance with well-known methods, see, e.g., Mark et al., "Site-specific
Mutagenesis of the
Human Fibroblast Interferon Gene", Proc. Natl. Acad. Sci. USA, 81, pp. 5662-66
(1984); and
US 4,588,585.
Alternatively, the nucleotide sequence is prepared by chemical synthesis, e.g.
by
using an oligonucleotide synthesizer, wherein oligonucleotides are designed
based on the
amino acid sequence of the desired polypeptide variant, and preferably
selecting those codons
that are favored in the host cell in which the recombinant polypeptide variant
will be
produced. For example, several small oligonucleotides coding for portions of
the desired
polypeptide variant may be synthesized and assembled by PCR, ligation or
ligation chain
reaction (LCR). The individual oligonucleotides typically contain 5' or 3'
overhangs for
complementary assembly.
Once assembled (by synthesis, site-directed mutagenesis or another method),
the
nucleotide sequence encoding the polypeptide variant is inserted into a
recombinant vector
and operably linked to control sequences necessary for expression of the IFNG
polypeptide
variant in the desired transformed host cell.
d It should of course be understood that not all vectors and expression
control
sequences function equally well to express the nucleotide sequence encoding an
IFNG
polypeptide variant described herein. Neither will all hosts function equally
well with the
same expression system. However, one of shill in the art may make a selection
among these
vectors, expression control sequences and hosts without undue experimentation.
For
example, in selecting a vector, the host must be considered because the vector
must replicate
in it or be able to integrate into the chromosome. The vector's copy number,
the ability to
control that copy number, and the expression of any other proteins encoded by
the vector,
such as antibiotic markers, should also be considered. In selecting an
expression control
sequence, a variety of factors should also be considered. These include, for
example, the
relative strength of the sequence, its controllability, and its compatibility
with the nucleotide
sequence encoding the polypeptide variant, particularly as regards potential
secondary
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
44
structures. Hosts should be selected by consideration of their compatibility
with the chosen
vector, the toxicity of the product coded for by the nucleotide sequence,
their secretion
characteristics, their ability to fold the polypeptide correctly, their
fermentation or culture
requirements, and the ease of purification of the products coded for by the
nucleotide
sequence.
The recombinant vector may be an autonomously replicating vector, i.e. a
vector,
which exists as an extrachromosomal entity, the replication of which is
independent of
chromosomal replication, e.g. a plasmid. Alternatively, the vector is one
which, when
introduced into a host cell, is integrated into the host cell genome and
replicated together with
the chromosomes) into which it has been integrated.
The vector is preferably an expression vector, in which the nucleotide
sequence
encoding the IFNG polypeptide variant is operably linked to additional
segments required for
transcription of the nucleotide sequence. The vector is typically derived from
plasmid or viral
DNA. A number of suitable expression vectors for expression in the host cells
mentioned
herein are commercially available or described in the literature. Useful
expression vectors for
eukaryotic hosts, include, for example, vectors comprising expression control
sequences from
SV40, bovine papilloma virus, adenovirus and cytomegalovirus. Specific vectors
are, e.g.,
pCDNA3.1(+)~IIyg (Invitrogen, Carlsbad, CA, USA) and pCI-neo (Stratagene, La
Jola, CA,
USA). Useful expression vectors for bacterial hosts include known bacterial
plasmids, such
as plasmids from E. coli, including pBR322, pET3a and pETl2a (both from
Novagen Inc.,
WI, USA), wider host range plasmids, such as RP4, phage DNAs, e.g., the
numerous
derivatives of phage lambda, e.g. , NM989, and other DNA phages, such as M13
and
filamentous single stranded DNA phages. Useful expression vectors for yeast
cells include
the 2~. plasmid and derivatives thereof, the POT1 vector (US 4,931,373), the
pJS037 vector
described in (Okkels, Ann. New York Acad. Sci. 782, 202-207, 1996) and pPICZ
A, B or C
(Invitrogen). Useful vectors for insect cells include pVL941, pBG311 (Cate et
al., "Isolation
of the Bovine and Human Genes for Mullerian Inhibiting Substance And
Expression of the
Human Gene In Animal Cells", Cell, 45, pp. 685-98 (1986), pBluebac 4.5 and
pMelbac
(both available from Invitrogen).
Other vectors for use in this invention include those that allow the
nucleotide
sequence encoding the IFNG polypeptide variant to be amplified in copy number.
Such
amplifiable vectors are well known in the art. They include, for example,
vectors able to be
amplified by DHFR amplification (see, e.g., Kaufman, U.S. Pat. No. 4,470,461,
Kaufman and
Sharp, "Construction Of A Modular Dihydrafolate Reductase cDNA Gene: Analysis
Of
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
Signals Utilized For Efficient Expression", Mol. Cell. BioL, 2, pp. 1304-19
(1982)) and
glutamine synthetase ("GS") amplification (see, e.g., US 5,122,464 and EP
338,841).
The recombinant vector may further comprise a DNA sequence enabling the vector
to
replicate in the host cell in question. An example of such a sequence (when
the host cell is a
5 mammalian cell) is the SV40 origin of replication. When the host cell is a
yeast cell, suitable
sequences enabling the vector to replicate are the yeast plasmid 2~
replication genes REP 1-3
and origin of replication.
The vector may also comprise a selectable marker, e.g. a gene the product of
which
complements a defect in the host cell, such as the gene coding for
dihydrofolate reductase
10 (DHFR) or the Schizosaccharomyces pombe TPI gene (described by P.R.
Russell, Gene 40,
1985, pp. 125-130), or one which confers resistance to a drug, e.g.
ampicillin, kanamycin,
tetracyclin, chloramphenicol, neomycin, hygromycin or methotrexate. For
filamentous fungi,
selectable markers include amdS, pyre, arcB, niaD, sC.
The term "control sequences" is defined herein to include all components,
which are
15 necessary or advantageous for the expression of the IFNG polypeptide
variant. Each control
sequence may be native or foreign to the nucleic acid sequence encoding the
polypeptide
variant. Such control sequences include, but are not limited to, a leader,
polyadenylation
sequence, propeptide sequence, promoter, enhancer or upstream activating
sequence, signal
peptide sequence, and transcription terminator. At a minimum, the control
sequences include
20 a promoter.
A wide variety of expression control sequences may be used in the present
invention.
Such useful expression control sequences include the expression control
sequences associated
with structural genes of the foregoing expression vectors as well as any
sequence known to
control the expression of genes of prokaryotic or eukaryotic cells or their
viruses, and various
25 combinations thereof.
Examples of suitable control sequences for directing transcription in
mammalian cells
include the early and late promoters of SV40 and adenovirus, e.g. the
adenovirus 2 major late
promoter, the MT-1 (metallothionein gene) promoter, the human cytomegalovirus
immediate-early gene promoter (CMV), the human elongation factor 1oc (EF-la)
promoter,
30 the Drosophila minimal heat shock protein 70 promoter, the Rous Sarcoma
Virus (RSV)
promoter, the human ubiquitin C (UbC) promoter, the human growth hormone
terminator,
SV40 or adenovirus Elb region polyadenylation signals and the Kozak consensus
sequence
(Kozak, M. J Mol Biol 1987 Aug 20;196(4):947-50).
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
46
In order to improve expression in mammalian cells a synthetic intron may be
inserted
in the 5' untranslated region of the nucleotide sequence encoding the IFNG
polypeptide
variant. An example of a synthetic intron is the synthetic intron from the
plasmid pCI-Neo
(available from Promega Corporation, WI, USA).
Examples of suitable control sequences for directing transcription in insect
cells
include the polyhedrin promoter, the P10 promoter, the Autographa califonaica
polyhedrosis
virus basic protein promoter, the baculovirus immediate early gene 1 promoter
and the
baculovirus 39K delayed-early gene promoter, and the SV40 polyadenylation
sequence.
Examples of suitable control sequences for use in yeast host cells include the
promoters of the yeast a-mating system, the yeast triose phosphate isomerase
(TPI) promoter,
promoters from yeast glycolytic genes or alcohol dehydogenase genes, the ADH2-
4c
promoter and the inducible GAL promoter.
Examples of suitable control sequences for use in filamentous fungal host
cells
include the ADH3 promoter and terminator, a promoter derived from the genes
encoding
Aspergillus oryzae TAKA amylase triose phosphate isomerase or alkaline
protease, an A.
niger oc-amylase, A. uiger or A. yaidulans glucoamylase, A. hidulans
acetamidase,
Rhizomucor miehei aspartic proteinase or lipase, the TPIl terminator and the
ADH3
terminator.
Examples of suitable control sequences for use in bacterial host cells include
promoters of the lac system, the trp system, the TAC or TRC system and the
major promoter
regions of phage lambda.
The nucleotide sequence of the invention, whether prepared by site-directed
mutagenesis, synthesis or other methods, may or may not also include a
nucleotide sequence
that encode a signal peptide. The signal peptide is present when the
polypeptide is to be
secreted from the cells in which it is expressed. Such signal peptide, if
present, should be one
recognized by the cell chosen for expression of the polypeptide variant. The
signal peptide
may be homologous (e.g. be that normally associated with huIFNG) or
heterologous (i.e.
originating from another source than huIFNG) to the polypeptide or may be
homologous or
heterologous to the host cell, i.e. be a signal peptide normally expressed
from the host cell or
one which is not normally expressed from the host cell. Accordingly, the
signal peptide may
be prokaryotic, e.g. derived from a bacterium such as E. coli, or eulcaryotic,
e.g. derived from
a mammalian, or insect or yeast cell.
The presence or absence of a signal peptide will, e.g., depend on the
expression host
cell used for the production of the polypeptide variant, the protein to be
expressed (whether it
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
47
is an intracellular or intracellular protein) and whether it is desirable to
obtain secretion. For
use in filamentous fungi, the signal peptide may conveniently be derived from
a gene
encoding an Aspergillus sp. amylase or glucoamylase, a gene encoding a
Rhizomucor miehei
lipase or protease or a Humicola lanuginosa lipase. The signal peptide is
preferably derived
from a gene encoding A. oryzae TAKA amylase, A. niger neutral a-amylase, A.
niger acid-
stable amylase, or A. niger glucoamylase. For use in insect cells, the signal
peptide may
conveniently be derived from an insect gene (cf. WO 90/05783), such as the
lepidopteran
Manduca sexta adipokinetic hormone precursor, (cf. US 5,023,328), the honeybee
melittin
(Invitrogen), ecdysteroid UDPglucosyltransferase (egt) (Murphy et al., Protein
Expression
and Purification 4, 349-357 (1993) or human pancreatic lipase (hpl) (Methods
in Enzymology
284, pp. 262-272, 1997).
A preferred signal peptide for use in mammalian cells is that of huIFNG or the
murine
Ig kappa light chain signal peptide (Coloma, M (1992) J. Imm. Methods 152:89-
104). For use
in yeast cells suitable signal peptides have been found to be the a-factor
signal peptide from
S. cereviciae. (cf. US 4,870,008), the signal peptide of mouse salivary
amylase (cf. O.
Hagenbuchle et al., Nature 289, 1981, pp. 643-646), a modified
carboxypeptidase signal
peptide (cf. L.A. Valls et al., Cell 48, 1987, pp. 887-897), the yeast BAR1
signal peptide (cf.
WO 87/02670), and the yeast aspartic protease 3 (YAP3) signal peptide (cf. M.
Egel-Mitani
et al., Yeast 6, 1990, pp. 127-137).
Any suitable host may be used to produce the IFNG polypeptide variant,
including
bacteria, fungi (including yeasts), plant, insect, mammal, or other
appropriate animal cells or
cell lines, as well as transgenic animals or plants. Examples of bacterial
host cells include
grampositive bacteria such as strains of Bacillus, e.g. B. brevis or B.
subtilis, Pseudomorzas or
Streptomyces, or gramnegative bacteria, such as strains of E. coli. The
introduction of a
vector into a bacterial host cell may, for instance, be effected by protoplast
transformation
(see, e.g., Chang and Cohen, 1979, Molecular General Genetics 168: 111-115),
using
competent cells (see, e.g., Young and Spizizen, 1961, Journal of Bacteriology
81: 823-829,
or Dubnau and Davidoff-Abelson, 1971, Joun2al of Molecular Biology 56: 209-
221),
electroporation (see, e.g., Shigekawa and Dower, 1988, BioteclzfZiques 6: 742-
751), or
conjugation (see, e.g., Koehler and Thorne, 1987, Journal of Bacteriology 169:
5771-5278).
Examples of suitable filamentous fungal host cells include strains of
Aspergillus, e.g.
A. oryzae, A. rziger, or A. rzidulans, Fusarium or Trichodenna. Fungal cells
may be
transformed by a process involving protoplast formation, transformation of the
protoplasts,
and regeneration of the cell wall in a manner known per se. Suitable
procedures for
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
48
transformation of Aspergillus host cells are described in EP 238 023 and US
5,679,543.
Suitable methods for transforming Fusarium species are described by Malardier
et al., 1989,
Gene 78: 14.7-156 and CVO 96/00787. Yeast may be transformed using the
procedures
described by Becleer and Guarente, In Abelson, J.N. and Simon, M.L, editors,
Guide to Yeast
Genetics and Molecular Biology, Methods in Ezzzymology, Volume 194, pp 182-
187,
Academic Press, Inc., New York; Ito et al., 1983, Jour~zal of Bacteriology
153: 163; and
Hinnen et al., 1978, Proceedings of the National Academy of Sciences USA 75:
1920.
Examples of suitable yeast host cells include strains of Saccharonzyces, e.g.
S.
cerevisiae, Sclzizosacclzaromyces, Kluyverotzzyces, Pichia, such as P.
pastoris or P.
metlzanolica, Hansenula, such as H. Polymorpha or Yarrowia. Methods for
transforming
yeast cells with heterologous DNA and producing heterologous polypeptides
therefrom are
disclosed by Clontech Laboratories, Inc, Palo Alto, CA, USA (in the product
protocol for the
Yeastmaker~ Yeast Tranformation System Kit), and by Reeves et al., FEMS
Microbiology
Letters 99 (1992) 193-198, Manivasakam and Schiestl, Nucleic Acids Research,
1993, Vol.
21, No. 18, pp. 4414-4415 and Ganeva et al., FEMS Microbiology Letters 121
(1994) 159-
164.
Examples of suitable insect host cells include a Lepidoptora cell line, such
as
Spodoptera frugiperda (Sf9 or Sf21) or Trichoplusioa ni cells (High Five) (US
5,077,214).
Transformation of insect cells and production of heterologous polypeptides
therein may be
performed as described by Invitrogen.
Examples of suitable mammalian host cells include Chinese hamster ovary (CHO)
cell lines, (e.g. CHO-Kl; ATCC CCL-61), Green Monkey cell lines (COS) (e.g.
COS 1
(ATCC CRL-1650), COS 7 (ATCC CRL-I65I)); mouse cells (e.g. NS/O), Baby Hamster
Kidney (BHK) cell lines (e.g. ATCC CRL-1632 or ATCC CCL-10), and human cells
(e.g.
HEK 293 (ATCC CRL-1573)), as well as plant cells in tissue culture. Additional
suitable cell
lines are known in the art and available from public depositories such as the
American Type
Culture Collection, Roclcville, Maryland. Also, the mammalian cell, such as a
CHO cell, may
be modified to express sialyltransferase, e.g. 1,6-sialyltransferase, e.g. as
described in US
5,047,335, in order to provide improved glycosylation of the IFNG polypeptide
variant.
Methods for introducing exogenous DNA into mammalian host cells include
calcium
phosphate-mediated transfection, electroporation, DEAF-dextran mediated
transfection,
liposome-mediated transfection, viral vectors and the transfection method
described by Life
Technologies Ltd, Paisley,°UK using Lipofectamin 2000. These methods
are well known in
the art and e.g. described by Ausbel et al. (eds.), 1996, Current Protocols in
Molecular
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
49
Biology, John Wiley & Sons, New York, USA. The cultivation of mammalian cells
are
conducted according to established methods, e.g. as disclosed in (Animal Cell
Biotechnology,
Methods and Protocols, Edited by Nigel Jenkins, 1999, Human Press Inc, Totowa,
New
Jersey, USA and Harrison MA and Rae IF, General Techniques of Cell Culture,
Cambridge
University Press 1997).
In order to produce a glycosylated polypeptide a eulcaryotic host cell, e.g.
of the type
mentioned above, is used.
In the production methods of the present invention, the cells are cultivated
in a
nutrient medium suitable for production of the polypeptide variant using
methods known in
the art. For example, the cell may be cultivated by shake flask cultivation,
small-scale or
large-scale fermentation (including continuous, batch, fed-batch, or solid
state fermentations)
in laboratory or industrial fermenters performed in a suitable medium and
under conditions
allowing the polypeptide variant to be expressed and/or isolated. The
cultivation takes place
in a suitable nutrient medium comprising carbon and nitrogen sources and
inorganic salts,
using procedures known in the art. Suitable media are available from
commercial suppliers
or may be prepared according to published compositions (e.g., in catalogues of
the American
Type Culture Collection). If the polypeptide variant is secreted into the
nutrient medium, the
polypeptide variant can be recovered directly from the medium. If the
polypeptide variant is
not secreted, it can be recovered from cell lysates.
The resulting polypeptide variant may be recovered by methods known in the
art. For
example, the polypeptide variant may be recovered from the nutrient medium by
conventional procedures including, but not limited to, centrifugation,
filtration, extraction,
spray drying, evaporation, or precipitation.
The polypeptide variants may be purified by a variety of procedures known in
the art
including, but not limited to, chromatography (e.g., ion exchange, affinity,
hydrophobic,
chromatofocusing, and size exclusion), electrophoretic procedures (e.g.,
preparative
isoelectric focusing), differential solubility (e.g., ammonium sulfate
precipitation), HPLC, or
extraction (see, e.g., Protein Purification, J.-C. Janson and Lars Ryden,
editors, VCH
Publishers, New York, 1989). Specific methods for purifying polypeptides
exhibiting IFNG
activity are disclosed in EP 0 110 044 and unexamined Japanese patent
application No.
186995/84.
The biological activity of the IFNG polypeptide variant can be assayed by any
suitable method known in the art. Such assays include antibody neutralization
of antiviral
activity, induction of protein kinase, oligoadenylate 2,5-A synthetase or
phosphodiesterase
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
activities, as described in EP 0 041 313 B 1. Such assays also include
immunomodulatory
assays (see, e.g., US 4,753,795), growth inhibition assays, and measurement of
binding to
cells that express interferon receptors. Specific assays are described in the
Materials and
Methods section herein.
5
Pharmaceutical compositions and uses thereof
Furthermore, the present invention relates to improved methods of treating or
preventing, in particular, inflammatory diseases, e.g. interstitial lung
diseases, such as
idiopathic pulmonary fibrosis, but also granulomatous diseases; cancer, in
particular ovarian
10 cancer; infections such as pulmonary atypical mycobacterial infections;
bone disorders (e.g. a
bone metabolism disorder so as malignant osteopetrosis); autoimmune diseases
such as
rheumatoid arthritis; as well as other diseases such as multiresistent
tuberculosis;
cryptococcal meningitis; cystic fibrosis and liver fibrosis, in particular
liver fibrosis
secondary to hepatitis C, said method comprising administering to a mammal, in
particular a
15 human being, in need thereof an effective amount of a polypeptide variant
of the invention or
a composition of the invention; the key advantages being less frequent and/or
less intrusive
administration of more efficient therapy, and optionally a lower rislc of
immune reactions
with the therapeutically active compound(s).
The molecule of the invention is preferably administered in a composition
including a
20 pharmaceutically acceptable carrier or excipient. "Pharmaceutically
acceptable" means a
carrier or excipient that does not cause any untoward effects in patients to
whom it is
administered. Such pharmaceutically acceptable carriers and excipients are
well known in the
art (Remington's Pharmaceutical Sciences, lath edition, A. R. Gennaro, Ed.,
Maclc
Publishing Company [1990]; Pharmaceutical Formulation Development of Peptides
and
25 Proteins, S. Frokjaer and L. Hovgaard, Eds., Taylor & Francis [2000] ; and
Handbook of
Pharmaceutical Excipients, 3rd edition, A. Kibbe, Ed., Pharmaceutical Press
[2000]).
The molecules of the invention can be used "as is" and/or in a salt form
thereof.
Suitable salts include, but are not limited to, salts with alkali metals or
alkaline earth metals,
such as sodium, potassium, calcium and magnesium, as well as e.g. zinc salts.
These salts or
30 complexes may by present as a crystalline andlor amorphous structure.
The variant of the invention is administered at a dose approximately
paralleling that
employed in therapy with known commercial preparations of IFNG such as
Actimmune~ or
as specified in EP 0 795 332. The exact dose to be administered depends on the
circumstances. Normally, the dose should be capable of preventing or lessening
the severity
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
51
or spread of the condition or indication being treated. It will be apparent to
those of skill in
the art that an effective amount of variant or composition of the invention
depends, inter alia,
upon the disease, the dose, the administration schedule, whether the
polypeptide variant or
composition is administered alone or in conjunction with other therapeutic
agents, the serum
half-life of the compositions, and the general health of the patient.
The present invention also relates to an IFNG polypeptide variant according to
the
present invention, or a pharmaceutical composition according to the present
invention, for use
as a medicament.
Furthermore, the invention also relates to the use of z) an IFNG variant
according to
the present invention, or ii) a pharmaceutical composition of the invention,
for the
manufacture of a medicament, a pharmaceutical composition or a lit-of-parts
for the
treatment of diseases selected from the group consisting of inflammatory
diseases, such as
interstitial lung diseases, in particular idiopathic pulmonary fibrosis;
cancer, in particular
ovarian cancer; infections, such as pulmonary atypical mycobacterial
infections; bone
disorders (e.g. a bone metabolism disorder so as malignant osteopetrosis);
granulomatous
diseases; autoimmune diseases such as rheumatoid arthritis; multiresistent
tuberculosis;
cryptococcal meningitis; cystic fibrosis and liver fibrosis, in particular
liver fibrosis
secondary to hepatitis C. Most preferably the disease is an interstitial lung
disease, in
particular idiopathic pulmonary fibrosis.
Also disclosed are improved means of delivering the molecules or preparations,
optionally additionally comprising glucocorticoids.
The preferred dosing is 1-4, more preferably 2-3, micrograms/kg patient weight
of the
polypeptide component per dose. The preferred dosing is 100-350, more
preferably 100-150
micrograms glucocorticoidlkg patient weight per dose.
The invention also relates to a kit of parts suitable for the treatment of
interstitial lung
diseases comprising a first pharmaceutical composition comprising the active
components a)
or ii) mentioned above and a second pharmaceutical composition comprising at
least one
glucocorticoid, each optionally together with a pharmaceutically acceptable
carrier and/or
excipient.
The variant of the invention can be formulated into pharmaceutical
compositions by
well-known methods. Suitable formulations are described by Remington's
Pharmaceutical
Sciences by E.W.Martin and TJS 5,183,746.
The pharmaceutical composition may be formulated in a variety of foams,
including
liquid, gel, lyophilized, powder, compressed solid, or any other suitable
form. The preferred
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
52
form will depend upon the particular indication being treated and will be
apparent to one of
skill in the art.
The pharmaceutical composition may be administered orally, subcutaneously,
intravenously, intracerebrally, intranasally, transdermally,
intraperitoneally, intramuscularly,
intrapulmonary, vaginally, rectally, intraocularly, or in any other acceptable
manner, e.g.
using PowderJect or Protease technology. The formulations can be administered
continuously by infusion, although bolus injection is acceptable, using
techniques well
known in the art, such as pumps or implantation. In some instances the
formulations may be
directly applied as a solution or spray. The preferred mode of administration
will depend
upon the particular indication being treated and will be apparent to one of
shill in the art.
The pharmaceutical composition of the invention may be administered in
conjunction
with other therapeutic agents. These agents may be incorporated as part of the
same
pharmaceutical composition or may be administered separately from the
polypeptide variant
of the invention, either concurrently or in accordance with any other
acceptable treatment
schedule. In addition, the polypeptide variant or pharmaceutical composition
of the invention
may be used as an adjunct to other therapies. In particular, combinations with
glucocorticoids
as described in EP 0 795 332 are considered.
In a further aspect the invention relates to a method of treating a mammal
having
circulating antibodies against huIFNG or rhuIFNG, which method comprises
administering
an IFNG variant which has the bioactivity of IFNG and which does not react
with said
antibodies. The compound is preferably a variant as described herein and the
mammal is
preferably a human being. The mammals to be treated may suffer from any of the
diseases
listed above for which IFNG is a useful treatment. Furthermore, the invention
relates to a
method of making a pharmaceutical product for use in treatment of mammals
having
circulating antibodies against huIFNG or rhuIFNG, wherein an 1FNG variant
which has the
bioactivity of IFNG and which does not react with such is formulated into an
injectable or
otherwise suitable formulation. The term "circulating antibodies" is intended
to indicate
autoantibodies formed in a mammal in response to having been treated with any
of the
commercially available IFNG preparations.
Also contemplated is use of a nucleotide sequence encoding an IFNG polypeptide
variant of the invention in gene therapy applications. In particular, it may
be of interest to use
a nucleotide sequence encoding an IFNG polypeptide variant described in the
section above
entitled "IFNG variants of tlae invention wherein the non polypeptzde moiety
is a sugar
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
53
»ioiety". The glycosylation of the polypeptide variant is thus achieved during
the course of
the gene therapy, i.e. after expression of the nucleotide sequence in the
human body.
Gene therapy applications contemplated include treatment of those diseases in
which
the polypeptide variant is expected to provide an effective therapy.
Local delivery of IFNG variant using gene therapy may provide the therapeutic
agent
to the target area while avoiding potential toxicity problems associated with
non-specific
administration.
Both iya vitro and iya vivo gene therapy methodologies are contemplated.
Several methods for transferring potentially therapeutic genes to defined cell
populations are known. For further reference see, e.g., Mulligan, "The Basic
Science Of
Gene Therapy", Science, 260, pp. 926-31 (1993). These methods include:
Direct gene transfer, e.g., as disclosed by Wolff et al., "Direct Gene
transfer Into
Mouse Muscle In vivo", Science 247, pp. 1465-68 (1990);
Liposome-mediated DNA transfer, e.g., as disclosed by Caplen et al., "Liposome-
mediated CFTR Gene Transfer to the Nasal Epithelium Of Patients With Cystic
Fibrosis"
Nature Med., 3, pp. 39-46 (1995); Crystal, "The Gene As A Drug", Nature Med.,
1, pp.- 15-
17 (1995); Gao and Huang, "A Novel Cationic Liposome Reagent For Efficient
Transfection
of Mammalian Cells", Biochem.Biophys Res. Comm., 179, pp. 280-85 (1991);
Retrovirus-mediated DNA transfer, e.g., as disclosed by Kay et al., "In vivo
Gene
Therapy of Hemophilia B: Sustained Partial Correction In Factor IX-Deficient
Dogs",
Science, 262, pp. 117-19 (1993); Anderson, "Human Gene Therapy", Science, 256,
pp.808-
13(1992);
DNA Virus-mediated DNA transfer. Such DNA viruses include adenoviruses
(preferably Ad-2 or Ad-5 based vectors), herpes viruses (preferably herpes
simplex virus
based vectors), and parvoviruses (preferably "defective" or non-autonomous
parvovirus
based vectors, more preferably adeno-associated virus based vectors, most
preferably AAV-2
based vectors). See, e.g., Ali et al., "The Use Of DNA Viruses as Vectors for
Gene Therapy"
Gene Therapy, 1, pp. 367-84 (1994); US 4,797,368, and US 5,139,941.
Parehterals
An example of a pharmaceutical composition is a solution designed for
parenteral
administration. Although in many cases pharmaceutical solution formulations
are provided in
liquid form, appropriate for immediate use, such parenteral formulations may
also be
provided in frozen or in lyophilized form. In the former case, the composition
must be
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
54
thawed prior to use. The latter form is often used to enhance the stability of
the active
compound contained in the composition under a wider variety of storage
conditions, as it is
recognized by those skilled in the art that lyophilized preparations are
generally more stable
than their liquid counterparts. Such lyophilized preparations are
reconstituted prior to use by
the addition of one or more suitable pharmaceutically acceptable diluents such
as sterile
water for injection or sterile physiological saline solution.
In case of parenterals, they are prepared fox storage as lyophilized
formulations or
aqueous solutions by mixing, as appropriate, the polypeptide variant having
the desired
degree of purity with one or more pharmaceutically acceptable carriers,
excipients or
stabilizers typically employed in the art (all of which are termed
"excipients"), for example
buffering agents, stabilizing agents, preservatives, isotonifiers, non-ionic
detergents,
antioxidants and/or other miscellaneous additives.
Buffering agents help to maintain the pH in the range which approximates
physiological conditions. They are typically present at a concentration
ranging from about 2
mM to about 50 mM. Suitable buffering agents for use with the present
invention include
both organic and inorganic acids and salts thereof such as citrate buffers
(e.g., monosodium
citrate-disodium citrate mixture, citric acid-trisodium citrate mixture,
citric acid-monosodium
citrate mixture, etc.), succinate buffers (e.g., succinic acid-monosodium
succinate mixture,
succinic acid-sodium hydroxide mixture, succinic acid-disodium succinate
mixture, etc.),
tartrate buffers (e.g., tartaric acid-sodium tartrate mixture, tartaric acid-
potassium tartrate
mixture, tartaric acid-sodium hydroxide mixture, etc.), fumarate buffers
(e.g., fumaric acid-
monosodium fumarate mixture, fumaric acid-disodium fumarate mixture,
monosodium
fumarate-disodium fumarate mixture, etc.), gluconate buffers (e.g., gluconic
acid-sodium
glyconate mixture, gluconic acid-sodium hydroxide mixture, gluconic acid-
potassium
glyuconate mixture, etc.), oxalate buffer (e.g., oxalic acid-sodium oxalate
mixture, oxalic
acid-sodium hydroxide mixture, oxalic acid-potassium oxalate mixture, etc.),
lactate buffers
(e.g., lactic acid-sodium lactate mixture, lactic acid-sodium hydroxide
mixture, lactic acid-
potassium lactate mixture, etc.) and acetate buffers (e.g., acetic acid-sodium
acetate mixture,
acetic acid-sodium hydroxide mixture, etc.). Additional possibilities are
phosphate buffers,
histidine buffers and trimethylamine salts such as Tris.
Preservatives may be added to retard microbial growth, and are typically added
in
amounts of about 0.2%-1% (w/v). Suitable preservatives for use with the
present invention
include phenol, benzyl alcohol, meta-cresol, methyl paraben, propyl paraben,
octadecyldimethylbenzyl ammonium chloride, benzalkonium halides (e.g.
benzallconium
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
chloride, bromide or iodide), hexamethonium chloride, alkyl parabens such as
methyl or
propyl paraben, catechol, resorcinol, cyclohexanol and 3-pentanol.
Isotonicifiers may be added to ensure isotonicity of liquid compositions and
include
polyhydric sugar alcohols, preferably trihydric or higher sugar alcohols, such
as glycerin,
5 erythritol, arabitol, xylitol, sorbitol and mannitol. Polyhydric alcohols
can be present in an
amount between 0.1% and 25% by weight, typically 1% to 5%, talung into account
the
relative amounts of the other ingredients.
Stabilizers refer to a broad category of excipients, which can range in
function from a
bullring agent to an additive which solubilizes the therapeutic agent or helps
to prevent
10 denaturation or adherence to the container wall. Typical stabilizers can be
polyhydric sugar
alcohols (enumerated above); amino acids such as arginine, lysine, glycine,
glutamine,
asparagine, histidine, alanine, omithine, L-leucine, 2-phenylalanine, glutamic
acid, threonine,
etc., organic sugars or sugar alcohols, such as lactose, trehalose, stachyose,
mannitol, sorbitol,
xylitol, ribitol, myoinisitol, galactitol, glycerol and the like, including
cyclitols such as
15 inositol; polyethylene glycol; amino acid polymers; sulfur-containing
reducing agents, such
as urea, glutathione, thioctic acid, sodium thioglycolate, thioglycerol, a-
monothioglycerol
and sodium thiosulfate; low molecular weight polypeptides (i.e. <10 residues);
proteins such
as human serum albumin, bovine serum albumin, gelatin or immunoglobulins;
hydrophilic
polymers such as polyvinylpyrrolidone; monosaccharides such as xylose,
mannose, fructose
20 and glucose; disaccharides such as lactose, maltose and sucrose;
trisaccharides such as
raffinose, and polysaccharides such as dextran. Stabilizers are typically
present in the range
of from 0.1 to 10,000 parts by weight based on the active protein weight.
Non-ionic surfactants or detergents (also known as "wetting agents ") may be
present to
help solubilize the therapeutic agent as well as to protect the therapeutic
polypeptide against
25 agitation-induced aggregation, which also permits the formulation to be
exposed to shear
surface stress without causing denaturation of the polypeptide. Suitable non-
ionic surfactants
include polysorbates (20, 80, etc.), polyoxamers (184, 188 etc.), Pluronic~
polyols,
polyoxyethylene sorbitan monoethers (Tween~-20, Tween~-80, etc.).
Additional miscellaneous excipients include bulking agents or fillers (e.g.
starch),
30 chelating agents (e.g. EDTA), antioxidants (e.g., ascorbic acid,
methionine, vitamin E) and
cosolvents.
The active ingredient may also be entrapped in microcapsules prepared, for
example,
by coascervation techniques or by interfacial polymerization, for example
hydroxymethylcellulose, gelatin or poly-(rnethylmethacylate) microcapsules, in
colloidal
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
56
drug delivery systems (for example liposomes, albumin microspheres,
microemulsions, nano-
particles and nanocapsules) or in macroemulsions. Such techniques are
disclosed in
Remington's Pharmaceutical Sciences, supra.
Parenteral formulations to be used for in vivo administration must be sterile.
This is
readily accomplished, for example, by filtration through sterile filtration
membranes.
In a preferred embodiment of the invention said pharmaceutical composition
comprises the i) IFNG variant of the invention, ii) a buffering agent, in
particular a salt of an
organic acid, capable of maintaining the pH between 5.0-6.5, iii) a
stabilizer, in particular an
organic sugar or sugar alcohol, iv) a non-ionic surfactant, and v) sterile
water. More
particularly, the buffering agent is selected from the group consisting of
acetate, succinate
and citrate, the stabilizer is mannitol or sorbitol, the non-ionic surfactant
is Tween~-20 or
Tween~-80. Preferably, the pharmaceutical composition does not include any
preservatives.
Sustained release preparations
Suitable examples of sustained-release preparations include semi-permeable
matrices
of solid hydrophobic polymers containing the variant, the matrices having a
suitable form
such as a film or microcapsules. Examples of sustained-release matrices
include polyesters,
hydrogels (for example, poly(2-hydroxyethyl-methacrylate) or
poly(vinylalcohol)),
polylactides, copolymers of L-glutamic acid and ethyl-L-glutamate, non-
degradable ethylene-
vinyl acetate, degradable lactic acid-glycolic acid copolymers such as the
ProLease~
technology or Lupron Depot~ (injectable microspheres composed of lactic acid-
glycolic acid
copolymer and leuprolide acetate), and poly-D-(-)-3-hydroxybutyric acid. While
polymers
such as ethylene-vinyl acetate and lactic acid-glycolic acid enable release of
molecules for
long periods such as up to or over 100 days, certain hydrogels release
proteins for shorter
time periods. When encapsulated polypeptides remain in the body for a long
time, they may
denature or aggregate as a result of exposure to moisture at 37°C,
resulting in a loss of
biological activity and possible changes in immunogenicity. Rational
strategies can be
devised for stabilization depending on the mechanism involved. For example, if
the
aggregation mechanism is discovered to be intermolecular S-S bond formation
through thio-
disulfide interchange, stabilization may be achieved by modifying sulfhydryl
residues,
lyophilizing from acidic solutions, controlling moisture content, using
appropriate additives,
and developing specific polymer matrix compositions.
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
57
Oral adnaifZistration
For oral administration, the pharmaceutical composition may be in solid or
liquid
form, e.g. in the form of a capsule, tablet, suspension, emulsion or solution.
The
pharmaceutical composition is preferably made in the form of a dosage unit
containing a
given amount of the active ingredient. A suitable daily dose for a human or
other mammal
may vary widely depending on the condition of the patient and other factors,
but can be
determined by persons skilled in the art using routine methods.
Solid dosage forms for oral administration may include capsules, tablets,
suppositories, powders and granules. In such solid dosage forms, the active
compound may
be admixed with at least one inert diluent such as sucrose, lactose, or
starch. Such dosage
forms may also comprise, as is normal practice, additional substances, e.g.
lubricating agents
such as magnesium stearate. In the case of capsules, tablets and pills, the
dosage forms may
also comprise buffering agents. Tablets and pills can additionally be prepared
with enteric
coatings.
The variants may be admixed with adjuvants such as lactose, sucrose, starch
powder,
cellulose esters of alkanoic acids, stearic acid, talc, magnesium stearate,
magnesium oxide,
sodium and calcium salts of phosphoric and sulphuric acids, acacia, gelatin,
sodium alginate,
polyvinyl-pyrrolidine, and/or polyvinyl alcohol, and tableted or encapsulated
for
conventional administration. Alternatively, they may be dissolved in saline,
water,
polyethylene glycol, propylene glycol, ethanol, oils (such as corn oil, peanut
oil, cottonseed
oil or sesame oil), tragacanth gum, and/or various buffers. Other adjuvants
and modes of
administration are well known in the pharmaceutical art. The carrier or
diluent may include
time delay material, such as glyceryl monostearate or glyceryl distearate
alone or with a wax,
or other materials well known in the art.
The pharmaceutical compositions may be subjected to conventional
pharmaceutical
operations such as sterilization and/or may contain conventional adjuvants
such as
preservatives, stabilizers, wetting agents, emulsifiers, buffers, fillers,
etc., e.g. as disclosed
elsewhere herein.
Liquid dosage forms for oral administration may include pharmaceutically
acceptable
emulsions, solutions, suspensions, syrups and elixirs containing inert
diluents commonly used
in the art, such as water. Such compositions may also comprise adjuvants such
as wetting
agents, sweeteners, flavoring agents and perfuming agents.
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
58
Topical administration
Formulations suitable for topical administration include liquid or semi-liquid
preparations suitable for penetration through the skin (e.g., liniments,
lotions, ointments,
creams, or pastes) and drops suitable for administration to the eye, ear, or
nose.
Pulmonary deliveYy
Formulations suitable for use with a nebulizer, either jet or ultrasonic, will
typically
comprise the polypeptide variant dissolved in water at a concentration of,
e.g., about 0.01 to
25 mg of variant per mL of solution, preferably about 0.1 to 10 mg/mL. The
formulation may
also include a buffer and a simple sugar (e.g., for protein stabilization and
regulation of
osmotic pressure), and/or human serum albumin ranging in concentration from
0.1 to 10
mg/ml. Examples of buffers that may be used are sodium acetate, citrate and
glycine.
Preferably, the buffer will have a composition and molarity suitable to adjust
the solution to a
pH in the range of 3 to 9. Generally, buffer molarities of from 1 mM to 50 mM
are suitable
for this purpose. Examples of sugars which can be utilized are lactose,
maltose, mannitol,
sorbitol, trehalose, and xylose, usually in amounts ranging from 1 % to 10% by
weight of the
formulation.
The nebulizer formulation may also contain a surfactant to reduce or prevent
surface
induced aggregation of the protein caused by atomization of the solution in
forming the
aerosol. Various conventional surfactants can be employed, such as
polyoxyethylene fatty
acid esters and alcohols, and polyoxyethylene sorbitan fatty acid.esters.
Amounts will
generally range between 0.001 % and 4% by weight of the formulation. An
especially
preferred surfactant for purposes of this invention is polyoxyethylene
sorbitan monooleate.
Specific formulations and methods of generating suitable dispersions of liquid
particles of the invention are described in WO 94/20069, US 5,915,378, US
5,960,792, US
5,957,124, US 5,934,272, US 5,915,378, US 5,855,564, US 5,826,570 and US
5,522,385
which are hereby incorporated by reference.
Formulations for use with a metered dose inhaler device will generally
comprise a
finely divided powder. This powder may be produced by lyophilizing and then
milling a
liquid variant formulation and may also contain a stabilizer such as human
serum albumin
(HSA). Typically, more than 0.5% (w/w) HSA is added. Additionally, one or more
sugars or
sugar alcohols may be added to the preparation if necessary. Examples include
lactose
maltose, mannitol, sorbitol, sorbitose, trehalose, xylitol, and xylose. The
amount added to the
formulation can range from about 0.01 to 200% (w/w), preferably from
approximately 1 to
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
59
50%, of the variant present. Such formulations are then lyophilized and milled
to the desired
particle size.
The properly sized particles are then suspended in a propellant with the aid
of a
surfactant. The propellant may be any conventional material employed for this
purpose, such
as a chlorofluorocarbon, a hydrochlorofluorocarbon, a hydrofluorocarbon, or a
hydrocarbon,
including trichlorofluoromethane, dichlorodifluoromethane,
dichlorotetrafluoroethanol, and
1,1,1,2-tetrafluoroethane, or combinations thereof. Suitable surfactants
include sorbitan
trioleate and Soya lecithin. Oleic acid may also be useful as a surfactant.
This mixture is then
loaded into the delivery device. An example of a commercially available
metered dose
inhaler suitable for use in the present invention is the Ventolin metered dose
inhaler,
manufactured by Glaxo Inc., Research Triangle Park, N.C.
Formulations for powder inhalers will comprise a finely divided dry powder
containing variant and may also include a bulking agent, such as lactose,
sorbitol, sucrose, or
mannitol in amounts which facilitate dispersal of the powder from the device,
e.g., 50% to
90% by weight of the formulation. The particles of the powder shall have
aerodynamic
properties in the lung corresponding to particles with a density of about 1
g/cm2 having a
median diameter less than 10 micrometers, preferably between 0.5 and 5
micrometers, most
preferably of between 1.5 and 3.5 micrometers. An example of a powder inhaler
suitable for
use in accordance with the teachings herein is the Spinhaler powder inhaler,
manufactured by
Fisons Corp., Bedford, Mass.
The powders for these devices may be generated and/or delivered by methods
disclosed in US 5,997,848, US 5,993,783, US 5,985,248, US 5,976574, US
5,922,354, US
5,785,049 and US 5,654,007.
Mechanical devices designed for pulmonary delivery of therapeutic products,
include
but are not limited to nebulizers, metered dose inhalers, and powder inhalers,
all of which are
familiar to those of skill in the art. Specific examples of commercially
available devices
suitable for the practice of this invention are the Ultravent nebulizer,
manufactured by
Mallinckrodt, Inc., St. Louis, Missouri; the Acorn lI nebulizer, manufactured
by Marquest
Medical Products, Englewood, Colorado; the Ventolin metered dose inhaler,
manufactured
by Glaxo Inc., Research Triangle Park, North Carolina; the Spinhaler powder
inhaler,
manufactured by Fisons Corp., Bedford, Massachusetts; the "standing cloud"
device of Inhale
Therapeutic Systems, Inc., San Carlos, California; the AIR inhaler
manufactured by
Alkermes, Cambridge, Massachusetts; and the AERx pulmonary drug delivery
system
manufactured by Aradigm Corporation, Hayward, California.
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
The invention is further described in the following examples. The examples
should
not, in any manner, be understood as limiting the generality of the present
specification and
claims.
5 MATERIALS AND METHODS
Materials
CHO-K1 cells (available from American Type Culture Collection (ATCC #CCL-61)).
HeLa cells (available from American Type Culture Collection (ATCC #CCL-2)).
10 ISRE-Luc was obtained fromStratagene, La Jolla USA.
pCDNA 3.1/hygro was obtained from Invitrogen, Carlsbad USA.
Restricion enzymes and polymerases were obtained from New England Biolabs
Inc., Beverly,
USA.
DMEM medium: Dulbecco's Modified Eagle Media (DMEM), 10% fetal bovine serum
and
15 Hygromycin B were obtained from Life Technologies A/S, Copenhagen, Denmark.
LucLite substrate was obtained from Packard Bioscience, Groningen, The
Netherlands.
TopCount luminometer was obtained from Packard Bioscience, Groningen, The
Netherlands.
Biotinylated polyclonal anti-human 1FNG antibody, BAF285, was obtained
available from
R&D Systems Inc., Minneapolis, USA.
20 Horse Radish Peroxidase-conjugated streptavidin, P0397, was obtained from
DAKO,
Copenhagen, Denmark.
TMB blotting reagent was obtained from KEM-EN-TEC, Copenhagen, Denmark.
Methods
25 Ihterf'ero~c Assay Outline
It has previously been published that IFNG interacts with and activates IFNG
receptors on HeLa cells. Consequently, transcription is activated at promoters
containing an
Interferon Stimulated Response Element (ISRE). It is thus possible to screen
for agonists of
interferon receptors by use of an ISRE coupled luciferase reporter gene (ISRE-
luc) placed in
30 HeLa cells.
PrifnaYy Assay
HeLa cells are co-txansfected with ISRE-Luc and pCDNA 3.1/hygro and foci (cell
clones) are created by selection in DMEM media containing Hygromycin B. Cell
clones are
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
61
screened for luciferase activity in the presence or absence of IFNG. Those
clones showing the
highest ratio of stimulated to unstimulated luciferase activity are used in
further assays.
To screen polypeptide variants, 15,000 cells/well are seeded in 96 well
culture plates
and incubated overnight in DMEM media. The next day the polypeptide variants
as well as a
known standard are added to the cells in various concentrations. Actimmune~
was used as
"known standard"; A vial of Actimmune~ was diluted to 300 IU/ml in DMEM, 5%
FBS and
stored at -80°C until use.
The plates are incubated for 6 hours at 37°C in a 5% COZ air atmosphere
LucLite
substrate (Packard Bioscience, Groningen, The Netherlands) is subsequently
added to each
well. Plates are sealed and luminescence measured on a TopCount luminometer
(Packard) in
SPC (single photon counting) mode.
Each individual plate contains wells incubated with IFNG as a stimulated
control and
other wells containing normal media as an unstimulated control. The ratio
between stimulated
and unstimulated luciferase activity serves as an internal standard for both
IFNG activity and
experiment-to-experiment variation. For each IFNG sample, the amount of units
were
calculated relative to the Actimmune~ standard and given as AU.
Determimatiom of increased degree of glycosylatiofa
To determine the various degrees of glycosylation of IFNG variant monomers, a
SDS-
PAGE gel is run under standard conditions and transferred to a nitrocellulose
membrane.
Western blotting is done according to standard procedures using a biotinylated
polyclonal
anti-human IFNG antibody (BAF285 from R & D Systems) as primary antibody and
Horse
Radish Peroxidase-conjugated streptavidin (P0397from DAKO) as secondary
antibody
followed by staining with TMB blotting reagent (KEM-EN-TEC, Copenhagen,
Denmarlc).
The distribution of IFNG variant monomers having varying degrees of
glycosylation is made
by visual inspection of the stained membrane.
Determination of AUCs
The AUCS° is determined by one 200 ~1 bolus subcutaneous administration
of equal
amount (on an activity basis) of the IFNG polypeptide variant of the invention
in rats.
For these experiments, female Sprag-Dawley rats, weiging between 220-260
grams,
are used. The IFNG polypeptide is formulated in sodium succinate (720 mg/1),
mannitol 40
g/1), polysorbat 20 (100 mg/1) at pH 6Ø
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
62
Before subcutaneous administration, one blood sample is drawn in the tail-vein
to
ensure that no background IFNG activity can be detected. After administration,
blood
samples are withdrawn from the tail vein after 10 min, 20 min, 40 min, 60 min,
120 min, 240
min, 480 min, 720 min, 1440 min, 1620 min, 1920 min and 2880 min (sometimes
also 3600
min). Serum is prepared by letting the blood sample coagulate for 20 min at
room
temperature followed by centrifugation at 5000g, 20 min at room temperature.
The serum is
then isolated and stored at -80°C until determination of IFNG activity
using the "Primary
Assay" described above. It should be noted that when the amount of units were
determined in
serum samples from PK studies in rats, the Actimmune~ standard was diluted in
DMEM, 5%
FBS and 5 %rat serum.
The amount of units in serum (AU/ml) against time (min) is then plotted and
the
AUCS~ is calculated using GraphPad Prism 3.01.
Similar experiments are performed on huIFNG in its glycosylated form and/or
Actimmune~ in order to assess the increase in AUCS~ of the IFNG polypeptide
variant of the
invention as compared to huTFNG in its glycosylated form and/or Actimmune~.
SeYUm half life
The serum half-life is determined by one 200 ~1 bolus intravenous
administration of
equal amount (on an activity basis) of the IFNG polypeptide variant of the
invention in rats.
For these experiments, female Sprag-Dawley rats, weighing between 220-260
grams,
are used. The IFNG polypeptide variant is formulated in sodium succinate (720
mg/1),
mannitol 40 g/1), polysorbat 20 (100 mg/1) at pH 6Ø
Before intravenous administration, one blood sample is drawn in the tail-vein
to
ensure that no background IFNG activity can be detected. After administration
in one tail
vein, blood samples are withdrawn from the other tail vein after 5 min, 10
min, 20 min, 40
min, 60 min, 120 min, 240 min, 480 min, 720 min, 1440 min, 1620 min, 1920 min
and 2880
min. Serum is prepared by letting the blood sample coagulate for 20 min at
room temperature
followed by centrifugation at 5000g, 20 min at room temperature. The serum is
then isolated
and stored at -80 ° C until determination of IFNG activity using the
"Primary Assay"
described above. It should be noted that when the amount of units were
determined in serum
samples from PK studies in rats, the Actimmune~ standard was diluted in DMEM,
5% FBS
and 5 %rat serum.
The amount of units in serum (AU/ml) against time (min) is then plotted and
the
serum half-life is calculated using WinNonLin Pro 3.3.
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
63
Similar experiments are performed on huIFNG in its glycosylated form, and/or
Actimmune0 in order to assess the increase in serum half-life of the 1FNG
polypeptide
variant of the invention as compared to huIFNG in its glycosylated form and/or
Actimmune~.
Ide~ztificatzora of surface exposed amzsao acid residues
Structures
Experimental 3D structures of huIFNG determined by X-ray crystallography have
been reported by: Ealick et.al. Science 252:698-702 (1991) reporting on the C-
alpha trace of
an IFNG homodimer. Walter et.al. Nature 376:230-235 (1995) reporting on the
structure of
an IFNG homodimer in complex with two molecules of a soluble form of the IFNG
receptor.
The coordinates of this structure have never been made publicly available.
Thiel et. al.
Structure 8:927-936 (2000) reporting on the structure of an IFNG homodimer in
complex
with two molecules of a soluble form of the IFNG receptor having a third
molecule of the
receptor in the structure not making interactions with the 1FNG homodimer.
Accessible Surface Area (ASA)
The computer program Access (B. Lee and F.M.Richards, J. Mol.Biol. 55: 379-400
(1971)) version 2,(Copyright (c) 1983 Yale University) was used to compute the
accessible
surface area (ASA) of the individual atoms in the structure. This method
typically uses a
0
probe-size of 1.4A and defines the Accessible Surface Area (ASA) as the area
formed by the
centre of the probe. Prior to this calculation all water molecules, hydrogen
atoms and other
atoms not directly related to the protein are removed from the coordinate set.
Fractional ASA of side chain
The fractional ASA of the side chain atoms is computed by division of the sum
of the
ASA of the atoms in the side chain with a value representing the ASA of the
side chain atoms
of that residue type in an extended ALA-x-ALA tripeptide. See Hubbard,
Campbell &
Thornton (1991) J.Mol.Biol.: 220,507-530. For this example the CA atom is
regarded as a
part of the side chain of Glycine residues but not for the remaining residues.
The following
table are used as standard 100% ASA for the side chain:
Ala 69.23 AZ Leu 140.76 A2
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
64
Arg 200.35 A2 Lys 162.50
Asn 106.25 A2 Met 156.08
Asp 102.06 A2 Phe 163.90 A2
Cys 96.69 AZ Pro 119.65 A2
Gln 140.58 A2 Ser 78.16
Glu 134.61 AZ Thr 101.67
Gly 32.28 A2 Trp 210.89 AZ
His 147.00 AZ Tyr 176.61 A2
Ile 137.91 AZ Val 114.14 A2
Residues not detected in the structure are defined as having 100% exposure as
they
are thought to reside in flexible regions.
DetermifZizzg distafaces betweezz atoms:
The distance between atoms was determined using molecular graphics software
e.g.
Insightll v. 98.0, MSI INC.
Deten~zzzzatiozz of receptor bifzdizzg site:
The receptor-binding site is defined as comprising of all residues having
their
accessible surface area changed upon receptor binding. This is determined by
at least two
ASA calculations; one on the isolated ligand(s) in the ligand(s)/receptor(s)
complex and one
on the complete ligand(s)/receptor(s) complex.
EXAMPLES
Example 1- Determination of surface-exposed amino acid residues
The X-ray structure used was of an IFNG homo-dimer in complex with two
molecules of a soluble form of the 1FNG receptor having a third molecule of
the IFNG
receptor in the structure not making interactions with the IFNG homodimer
reported by Thiel
et.al. Structure 8:927-936 (2000). The structure consists of the IFNG
homodimer wherein the
two molecules are labeled A and B. For construction purposes there is an
additional
methionine placed before the 1FNG sequence labeled MO and the sequence is C-
terminally
truancuted with ten residues (Q133 being the last residue in the constructed
molecules). The
MO is removed from the structure in all the calculations of this example. The
structure of the
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
two IFNG monomers has very weak electron density after residue 120 and
residues were only
modeled until residue T126. Therefore, residues 5121-T126 were removed from
the structure
prior to the calculations in this example. The two receptor fragments labeled
C and D make
direct interactions with the IFNG homodimer and a third receptor molecule
labeled E makes
5 no contact with the IFNG homodimer and are not included in these
calculations.
Surface exposure:
Performing fractional ASA calculations on the homodimer of molecules A and B
excluding MO and S 121-T126 in both molecules resulted in the following
residues having
10 more than 25% of their side chain exposed to the surface in at least one of
the monomers: Q1,
D2, P3, K6, E9, N10, K12, K13, Y14, N16, G18, H19, 520, D21, A23, D24, N25,
G26, T27,
G31, K34, N35, K37, E38, E39, 540, K55, K58, N59, K61, D62, D63, Q64, S65,
Q67, K68,
E71, T72, K74, E75, N78, V79, K80, N83, 584, N85, K86, K87, D90, E93, K94,
N97, 599,
T101, D102, L103, N104, Hl l l, Q115, A118 and E119.
15 The following residues had more than 50% of their side chain exposed to the
surface
in at least one of the monomers: Q1, D2, P3, K6, E9, N10, K13, N16, G18, H19,
520, D21,
A23, D24, N25, G26, T27, G31, K34, K37, E38, E39, K55, K58, N59, D62, Q64,
565, K68,
E71, E75, N83, 584, K86, K87, K94, N97, 599, T101, D102, L103, N104, Q115,
A118,
E119.
20 Performing fractional ASA calculations on the homodimer of molecules A and
B
excluding MO and 5121-T126 in both molecules and including the receptor
molecules C and
D resulted in the following residues had more than 25% of their side chain
exposed to the
surface in at least one of the monomers: Q1, D2, P3, K6, E9, N10, K13, Y14,
N16, G18,
H19, D21, N25, G26, G31, K34, N35, K37, E38, E39, 540, K55, K58, N59, K61,
D62, D63,
25 Q64, 565, Q67, K68, E71, T72, K74, E75, N78, V79, K80, N83, 584, N85, K86,
K87, D90,
E93, K94, N97, 599, T101, D102, L103, N104, E119.
The following residues had more than 50% of their side chain exposed to the
surface
in at least one of the monomers: P3, K6, N10, K13, N16, D21, N25, G26, G31,
K34, K37,
E38, E39, K55, K58, N59, D62, Q64, 565, K68, E71, E75, N83, 584, K86, K87,
K94, N97,
30 599, T101, D102, L103 and N104.
All of the above positions are targets for modification in accordance with the
present
invention.
Comparing the two lists, results in K12, 520, A23, D24, T27, Hl l l, Q115 and
A118
being removed from the more than 25% side chain ASA list upon receptor
binding, and Q1,
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
66
D2, E9, G18, H19, 520, A23, D24, T27, Q115, A118 and E119 being removed from
the more
than 50% side chain ASA list upon receptor binding.
Residues not determined in the structure are treated as fully surface exposed,
i.e.
residues 5121, P122, A123, A124, K125, T126, 6127, K128, 8129, K130, 8131,
5132,
Q133, M134, L135, F136, 8137, 6138, 8139, 8140, A141, 5142, Q143. These
residues also
constitute separate targets for introduction of attachment groups in
accordance with the
present invention (or may be viewed as belonging to the group of surface
exposed amino acid
residues, e.g. having more than 25% or more than 50% exposed side chains).
Example 2 - Determination of the receptor binding site:
Performing ASA calculations as described above results in the following
residues of
the IFNG molecule having reduced ASA in at least one of the monomers in the
complex as
compared to the calculation on the isolated dimer: Ql, D2, Y4, V5, E9, K12,
G18, H19, 520,
D21, V22, A23, D24, N25, G26, T27, L30, K34, K37, K108, H111, E112, I114,
Q115, A118,
E119.
Example 3 - Design of a cassette for expression of IFNG with optimised eodon
usage
The DNA sequence, GenBank accession number X13274, encompassing a full-length
cDNA encoding mature hulFNG with its native signal peptide, was modified in
order to
facilitate high expression in CHO cells. Codons of the huIF'NG nucleotide
sequence were
modified by making a bias in the codon usage towards the codons frequently
used in 7zomo
sapie~s. Subsequently, certain nucleotides in the sequence were substituted
with others in
order to introduce recognition sites for DNA restriction endonucleases.
Primers were
designed such that the gene could be synthesised.
The primers were assembled to the synthetic gene by one-step PCR using the
platinum Pfx-polymerase kit (Life Technologies) and standard three-step PCR
cycling
parameters. The assembled gene was amplified by PCR using the same conditions
and has
the sequence shown in SEQ ID N0:42. The synthesised gene was cloned into
pcDNA3.1/hygro (InVitrogen) between the BamHI and the XbaI sites, resulting in
pIGY-22.
pIGY-22 was transfected into CHO Kl cells by use of Lipofectaim2000 (Life
Technologies) as transfection agent. 24 hours later the culture medium was
harvested and
assayed for IFNG activity and concentration by Elisa. Using the Primary assay
described
herein, an activity of 1.4 x 107 AU/ml was obtained.
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
67
Exam-ple 4 - Site directed muta e~'nesis
Generation of glycosylation variants
To introduce mutations in IFNG, oligonucleotides were designed in such a way
that
PCR-generated changes could be introduced in the expression plasmid (pIGY-22)
by
classical two-step PCR.
Two vector primers were used together with specific mutation primers:
ADJ013: 5'-GATGGCTGGCAACTAGAAG-3' (antisense downstream vector primer) (SEQ
ID N0:43) and ADJ014: 5'-TGTACGGTGGGAGGTCTAT-3' (SEQ ID N0:44) (sense
upstream vector primer)
The S99T variant was generated by classical two-step PCR, using ADJ013 and
ADJ014 as vector primers, ADJ093 (5'-GTTCAGGTCTGTCACGGTGTAATTGGTCAG-
~~~1
CTT-3') (SEQ ID N0:45) and ADJ094 (5'-AAGCTGACCAATTACACCGTGACAGA-
CCTGAAC-3' ) (SEQ 1D N0:46) as mutation primers, and pIGY-22 as template. The
447 by
PCR product was subcloned into pcDNA3.1/Hygro (InVitrogen) using BamHI and
XbaI,
leading to plasmid pIGY-48.
pIGY-48 was transfected into CHO K1 cells by use of Lipofectaim2000 (Life
Technologies) as transfection agent. 24 hours later the culture medium was
harvested and
assayed for IFNG activity. Using the Primary assay described herein, the
following activity
was obtained: 5.1 x 106 AUlml.
The E38N+S40T+S99T variant was generated by classical two-step PCR, using
ADJ013 and ADJ014 as vector primers, ADJ091 (5'-CATGATCTTCCGATCGGTCTC-
GTTCTTCCAATT-3') (SEQ ll~ N0:47) and ADJ092 (5'-AATTGGAAGAACGAGACC-
GATCGGAAGATCATG-3' ) (SEQ ID NO:48) as mutation primers, and pIGY-48 as
template. The 447 by PCR product was subcloned into pcDNA3.l/Hygro
(InVitrogen) using
BamHI and XbaI, leading to plasmid pIGY-54.
pIGY-54 was transfected into CHO K1 cells by use of Lipofectaim2000 (Life
Technologies) as transfection agent. 24 hours later the culture medium was
harvested and
assayed for IFNG activity. Using the Primary assay described herein, an
activity of 1.3 x 107
AU/ml was obtained.
Using similar standard techniques as described above, a number of full-length
IFNG
glycosylation variants were prepared. These variants are compiled in Table 1
below.
Generation of C-terminally truncated IFNG variants
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
68
C-terminally truncated 1NFG variants, containing a stop codon immediately
downstream of the codon for Leu135, were generated by one-step PCR using pIGY-
22,
pIGY-48 and pIGY-54 as templates, followed by subcloning of the PCR products
into
pcDNA3.1/Hygro (InVitrogen) using BamHI and XbaI. The primers used for
construction of
these variants were: ADJ014 (see above, upstream) and: 5'-GAGTCTAGATTACAGCAT-
CTGGCTTCTCTT-3' (SEQ ID N0:49) (downstream). The resulting plasmids were
termed
pIGY-72 (wild-type 1FNG truncated after Leul35), pIGY-73 (S99T variant
truncated after
Leu135) and pIGY-74 (E38N+S40T+ S99T truncated after Leu135).
Geyzeratior~ of cysteizze-cot2taizziz2g IFNG varia3zts
INFG variants containing cysteine residues were generated using Stratagene's
QuikChange~'XL site-directed mutagenesis kit, according to the manufacturer's
specifications. Seven IFNG variants, each containing one introduced cysteine,
were generated
using pIGY-48 as template: N10C+S99T, N16C+S99T, E38C+S99T, N59C+S99T,
N83C+S99T, K94C+S99T and S99T+N104C. Similarly, six IFNG variants, each
containing
one introduced cysteine, were generated using pIGY-54 as template:
N10C+E38N+S40T+S99T, N16C+E38N+S40T+S99T, E38N+S40T+ N59C+S99T,
E38N+S40T+ N83C+S99T, E38N+S40T+ K94C+S99T and E38N+S40T+S99T+N104C.
Example 5 - PEGylation of cysteine-containing variants
All buffers were de-oxidized prior to use. Protein concentrations were
estimated by
measuring A280.
PEGylatiozz usizzg the OPSS coupli>zg chezzzzstry
7.2 ml of 1.3 mg/ml of the IFNG variant N16C+S99T (full-length) in 5 mM sodium
succinate, 4% mannitol, 0.01 % Tween 20, pH 6.0, was reduced by incubation
with 300 x.10.5
M DTT for 30 minutes at room temperature. The IFNG variant was desalted by
running 3
aliquots of 2.5 ml on a NAP25 gel filtration column (Pharmacia) in buffer A
(50 mM sodium
phosphate, 1 mM EDTA, pH 8.1). Each aliquote eluted in 3.5 ml.
mPEG-OPSS (I0 KDa) was dissolved in buffer A to a concentration of 2 mg/ml and
added in equal volume to the reduced and desalted IFNG variant and incubated
for 60 min
with gentle shaking at room temperature.
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
69
11 ml of the reaction mixture was concentrated to 1-6 ml using a Vivaspin20
column
(VivaScience) and remaining mPEG was removed by gel filtration using a
Sephacryl S-100
column (Pharmacia) equilibrated in buffer A.
The PEGylated IFNG variant was diafiltered into 5 mM sodium succinate, 4%
mannitol, pH 6.0 using a Vivaspin 6 column (VivaScience) and Tween 20 was
added to
0.01%. The purified FEGylated 1FNG variant had a specific activity of 1.3 x
106 AU/mg as
measured in the Primary Assay described herein (15% of the specific activity
of the
corresponding non-PEGylated IFNG variant).
PEGylatioh using the MAL couplif2g chemistYy
1.6 ml of 1.5 mglml of the IFNG variant N59C+S99T (full-length) in 5 mM sodium
succinate, 4% mannitol, 0.01% Tween 20, pH 6.0 was reduced by incubation with
64 x,10.5
M DTT for 30 minutes at room temperature. The IFNG variant was desalted on a
NAP25 gel
filtration column (Pharmacia) in buffer A (50 mM sodium phosphate, 1mM EDTA,
pH 8.1).
The INFG variant eluted in 3.5 ml.
mPEG-MAL (5 kDa) was dissolved in buffer A to a concentration of 0.5 mg/ml and
added in equal volume to reduced and desalted IFNG variant and incubated for
120 minutes
with gentle shaking at room temperature.
Ammonium sulphate was added to a concentration of 0.9 M and the PEGylated IFNG
variant was applied onto a 1 ml ResourceT"" phenyl column (Pharmacia)
equilibrated in buffer
B (20 mM sodium phosphate, 0.9 M ammonium sulphate, pH 6.6). The column was
washed
with 5 column volumes of buffer B before elution of the bound PEGylated IFNG
variant in a
linear gradient from 0-50% buffer C (20 mM sodium phosphate, pH 6.6) over 30
column
volumes. The PEGylated IFNG variant eluted around 0.6 M ammonium sulphate.
Fractions containing PEGylated IFNG variant were pooled and diafiltered into 5
mM
sodium succinate, 4% mannitol, pH 6.0 using a Vivaspin 6 column (VivaScience)
and Tween
20 was added to 0.01%. The purified PEGylated IFNG variant had a specific
activity of 2.4 x
106 AU/mg as measured in the Primary Assay described herein (15% of the
specific activity
of the corresponding non-PEGylated IFNG variant).
Example 6 - Expression of IFNG and IFNG variants in mammalian cells
For transient expression of IFNG, cells were grown to 95% confluency in media
(Dulbecco's MEM/Nut.-mix F-12 (Ham) L-glutamine, 15 mM Hepes, pyridoxine-HCl
(Life
Technologies Cat # 31330-038)) containing 1:10 fetal bovine serum
(BioWhittalcer Cat # 02-
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
701F) and 1:100 penicillin and streptomycin (BioWhittaker Cat # 17-602E).1FNG-
encoding
plasmids were transfected into the cells using Lipofectamine 2000 (Life
Technologies)
according to the manufacturer's specifications. 24 hrs after transfection,
culture media were
collected and assayed for IFNG activity. Furthermore, in order to quantify the
relative
5 number of glycosylation sites utilized, Western blotting was performed using
harvested
culture medium.
Stable clones expressing IFNG were generated by transfection of CHO K1 cells
with
IFNG-encoding plasmids followed by incubation of the cells in media containing
0.36 mg/ml
hygromycin. Stably transfected cells were isolated and sub-cloned by limited
dilution. Clones
10 producing high levels of IFNG were identified by ELISA.
Example 7 - Lame-scale production
Stable cell lines expressing 1FNG or variants were grown in Dulbecco's
MEM/Nut.-
rnix F-12 (Ham) L-glutamine, 15 mM Hepes, pyridoxine-HCl (Life Technologies
Cat #
15 31330-038), 1:10 fetal bovine serum (BioWhittaker Cat # 02-701F), 1:100
penicillin and
streptomycin (BioWhittaker Cat # 17-602E) in 1700 cm2 roller bottles (Corning,
# 431200)
until confluence. The media was then changed to 300 ml UltraCHO with L-
glutamine
(BioWhittaker Cat # 12-724Q) with the addition of 1:500 EX-CYTE VLE
(Serological
Proteins Inc. # 81-129) and 1:100 penicillin and streptomycin (BioWhittaleer
Cat # 17-602E).
20 After 48 hours of growth, the media was replaced with fresh UltraCHO with
the same
additives. After another 48 hours of growth, the media was replaced with
Dulbecco's
MEM/Nut.-mix F-12 (Ham) L-glutamine, pyridoxine-HCl (Life Technologies Cat #
21041-
025) with the addition of 1:100 TTS-A (GibcoBRL # 51300-044), 1:500 EX-CYTE
VLE
(Serological Proteins Inc. # 81-129) and 1:100 penicillin and streptomycin
(BioWhittalcer Cat
25 # 17-602E). Subsequently, every 24 h, culture media were harvested and
replaced with 300
ml of fresh serum-free media with the same additives. The collected media were
filtered
through 0.22 ~m filters to remove cells.
Example 8 - Purification
30 The filtrate was microfiltrated (0.22 pm) before ultrafiltration to
approximately 1/15
volume using a Millipore TFF system. On the same system the concentrate was
diafiltrated
using 10 mM Tris, pH 7.6. Ammonium sulphate was added to a concentration of
1.7 M and
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
71
after stirring the precipitate was removed by centrifugation at 8000 rpm for
25 minutes in a
Sorvall centrifuge using a GS3 rotor.
The supernatant was applied onto a 25 ml Phenyl High Performance (Pharmacia)
column previously equilibrated in 10 mM Tris, 1.7 M ammonium sulphate, pH 7.6.
After
application the column was washed with 3 column volumes of 10 mM Tris, 1.7 M
ammonium sulphate, pH 7.6 and the bound IFNG variant was then eluted in a
linear gradient
over 10 column volumes to 100% 10 mM Tris, pH 7.6. The flow-through as well as
the
eluted IFNG variant was fractionated. Fractions enriched in the IFNG variant
were pooled
and buffer exchanged by diafiltration into 10 mM Tris, pH 9.0, using a
Vivaf1ow200 system
(VivaScience) with a molecular weight cut-off of 10,000 Da.
The 1FNG variant was then applied onto a 18 ml Q-sepharose Fast Flow
(Pharmacia)
column previously equilibrated in 10 mM Tris, pH 9Ø After application the
column was
washed with 3 column volumes of 10 mM Tris, pH 9,0 before eluting the bound
IFNG
variant in a gradient from 0-100% 10 mM Tris, 0.5 M NaCI, pH 9.0, over 15
column
volumes. The flow-through as well as the eluted IFNG variant was fractionated.
Fractions
enriched in the IFNG variant were pooled and buffer exchanged intol0 mM sodium
phosphate, pH 7.0, by diafiltration using a Vivaspin20 (VivaScience) column
with a
molecular weight cut-off of 10,000 Da.
Then, the IFNG variant was applied onto an 8 ml CHT ceramic hydroxyapatite
column (Biorad) previously equilibrated in 10 mM sodium phosphate, pH 7Ø
After
application the column was washed with 5 column volumes of 10 mM sodium
phosphate, pH
7.0, before elution of the bound IFNG variant in a gradient from 0-60% 500 mM
sodium
phosphate, pH 7.0, over 30 column volumes. The flow-through as well as the
eluted IFNG
variant was fractionated. Fractions containing the IFNG variant were pooled
and buffer
exchanged into 5 mM sodium succinate, 4% mannitol, pH 6.0, using a VivaSpin20
column
(VivaScience) and Tween 20 was subsequently added to a concentration of 0.01%.
The IFNG
variant was sterile filtered and stored at -80°C.
Alternatively, the IFNG variants may be purified according to the below
purification
scheme:
The filtrate is microfiltrated (0.22 pm) before ultrafiltration to
approximately 1/15
volume using a Millipore TFF system. On the same system the concentrate is
diafitrated
using 10 mM Tris, pH 7.6, after which pH is adjusted to 9.0 and precipitate is
removed by
microfiltration.
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
72
The sample is applied onto a Q-sepharose Fast Flow (Pharmacia) column
previously
equilibrated in 10 mM Tris, pH 9Ø After application the column is washed
with 3 column
volumes of 10 mM Tris, pH 9.0 before eluting the bound IFNG variant in a
gradient from 0-
100% 10 mM Tris, 0.5 M NaCI, pH 9.0 over 15 column volumes. The flow-through
as well
as the eluted IFNG variant is fractionated. Fractions enriched in the INFG
variant are pooled,
and pH is adjusted to 7.6. Ammonium sulphate is added to 1.5 M and after
stirring the
precipitate is removed by centrifugation.
The IFNG variant is then applied onto a Phenyl Sepharose High Performance
(Pharmacia) previously equilibrated in 10 mM Tris, 1.5 M ammonium sulphate, pH
7.6.
After application the column is washed with 3 column volumes of 10 mM Tris,
1.5 M
ammonium sulphate, pH 7.6, and the bound IFNG variant is then eluted in a
linear gradient
over 10 column volumes to 100% 10 mM Tris, pH 7.6. The flow-through as well as
the
eluted 1FNG variant is fractionated. Fractions enriched in the INFG variant
are pooled and
ammonium sulphate is adjusted to 1.7 M.
Then, the IFNG variant is applied onto a Butyl Sepharose column previously
equilibrated in 10 mM sodium phosphate, 1.7 M ammonium sulphate, pH 7.6. After
application the column is washed with 10 mM sodium phosphate, 1.7 M ammonium
sulphate,
pH 7.6, before eluting the bound 1FNG variant in a step using 10 mM sodium
phosphate, pH
6.5. The flow-through as well as the eluted IFNG variant is fractionated.
Fractions enriched in the IFNG variant are then pooled and applied onto a
hydroxy-
apatite column previously equilibrated in 10 mM sodium phosphate, pH 6.5.
After
application the column is washed with 5 column volumes of 10 mM sodium
phosphate, pH
6.5, before eluting the bound IFNG variant in a linear gradient from 0-100%
500 rnM sodium
phosphate, pH 6.5, over 30 column volumes. The flow-through as well as the
eluted IFNG
variant is fractionated.
Fractions containing the 1NFG variant are pooled and buffer exchanged into a
buffer
containing 5 mM sodium succinate, 4% mannitol, pH 6Ø Tween 20 is
subsequently added to
a concentration of 0.01%. The IFNG variant is sterile filtered and stored at -
~0°C.
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
' 73
Example 9 - Activity of variants and PEGylated variants
Using the "primary Assay" described above, the following activity data (after
transient transfection) were obtained:
Mutations Activity (AU/ml)% activity
of
full-length
wt
Wild-type (full-length) 1.4 x 107 -
S99T (full-length) 5.1 x 106 36%
E38N (full-length) 1.4 x 107 100%
E38N +S40T (full-length) 9.9 x 106 71%
E38N+S40T+S99T (full-length) 1.3 x 107 93%
E38N+K61T (full-length) 1.2 x 107 86%
E38N+K61T+S99T (full-length) 1.4 x 106 10%
N10C+S99T (full-length) 2.5 x 106 18%
N16C+S99T (full-length) 1.1 x 107 79%
E38C+S99T (full-length) 1.0 x 107 71%
S99T+N104C (full-length) 5.0 x 106 36%
N10C+E38N+S40T+S99T (full-length)1.5 x 106 11%
N16C+E38N+S40T+S99T (full-length)3.7 x 106 26%
E38N+S40T+N59C+S99T (full-length)1.1 x 107 79%
E38N+S40T+N83C+S99T (full-length)7.2 x 105 5%
E38N+S40T+K94C+S99T (full-length)9.4 x 105 7%
E38N+S40T+S99T+N104C (full-length)2.3 x 106 16%
Wild-type (truncated) 6.3 x 106 45%
S99T (truncated) 3.5 x 106 25%
E38N+S40T+S99T (truncated) 4.0 x 106 29%
Table 1: Activity of variants of full-length and truncated rhuIFNG
polypeptides after transient transfection
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
74
Using the "primary Assay" described above, the following specific activity
data (after
purification) were obtained:
Mutations Specific activity % activity of
(AU/mg) full-length wt
Wild-type (full-length) 2.1 x 10' -
S99T (full-length) 2.2 x 107 105%
E38N+S40T+S99T (full-length) 1.4 x 107 67%
NlOC+S99T (full-length) 3.8 x 106 18%
N16C+S99T (full-length) 2.3 x 106 11%
N16C+S99T (full-length+10 kDa mPEG) 1.3 x 106 6%
E38C+S99T (full-length) 3.4 x 106 16%
N59C+S99T (full-length) 6.3 x 106 30%
N59C+S99T (full-length+5 kDa mPEG)2.4 x 106 11%
S99T+N104C lfull-len~thl 3.5 x 106 17%
Table 2: Activity of variants of full-length rhuIFNG polypeptides after
purification
The activity of a number of the PEGylated variants were measured by comparing
the
activity of the PEGylation products by samples which have been subjected to
the same
PEGylation procedure (see Example 5 above), but without actually adding PEG to
the
reaction medium. The results are compiled in Table 3 below:
Activity relative to non-PEGylated
Mutations product subjected to "PEGylation
procedure"
(%)
N10C+S99T (full-length+5 kDa mPEG) 23
N16C+S99T (full-length+5 lcDa mPEG) 59
E38C+S99T (full-length+10 kDa mPEG)1~ 41
S99T+N104C (full-length+5 kDa mPEGI 42
Table 3: Activity of PEGylated variants of full-length rhuIFNG polypeptides
1) ODSS coupling chemistry employed. MAL coupling chemistry employed for all
other PEGylated variants
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
It should be emphasized that the activity data shown in Table 1, reflect a
combination
of specific activity and expression level in CHO-Kl cells. It can therefore be
concluded that
all variants show a comparable expression level/specific activity to relative
to rhuIFNG.
As can be seen in Table 2, all cysteine variants show a decreased specific
activity
5 compared to rhuIFNG while variants containing the E38N, S40T and/or S99T
mutations
retained a specific activity comparable to rhuIFNG. When the cysteine variants
were
PEGyIated with either 5 or 10 kDa a further 2-3 fold drop in specific activity
was observed
(Table 3). Without being limited to any specific theory, it could be
speculated that the
decrease in specific activity could be due to decreased receptor binding
caused by steric
10 hindrance of the conjugated PEG group.
Example 10 - Assessment of utilization of N-glycosylation sites
In order to quantify the relative number of glycosylation sites utilized,
western
blotting was performed using harvested culture medium (see Fig. 1). For the
wild-type
15 rhuIFNG (full-length), it was estimated that about 50% utilized both
glycosylation sites (2N),
about 40% utilized one glycosylation site (1N), and about 10% was not
glycosylated (ON).
These data are in agreement with previously published data by Hooker et al.,
1998, J.
Interferon and Cytol~ine Res. 18, 287-295 and Sarenva et al., 1995, Biochem
J., 308, 9-14.
As it appears from Fig. 1, the S99T variant (full-length) utilizes its two
glycosylation
20 sites significantly more efficiently than the corresponding wild-type. For
the S99T variant, it
was estimated that about 90% utilized both glycosylation sites (2N), about 7%
utilized one
glycosylation site (1N), and about 3% was not glycosylated (ON).
Moreover, it is apparent from Fig. 1 that the introduced glycosylation site at
position
38 is significantly better utilized for the variant E38N+S40T (full-length)
compared to the
25 non-optimised variant E38N (full-length).
These data clearly demonstrate that better utilization of glycosylation sites,
independently of whether these sites are naturally occurring or introduced,
can be achieved
by introducing a threonine residue rather than a serine residue in position +2
relative to the
asparagine residue.
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
76
Example 11- Pharmacolcinetic studies
The AUC for subcutaneous administration (AUCS~) in rats was determined as
described hereinbefore for a number of IFNG variants. The results are compiled
in Table 4
and in Figs. 2 and 3.
AUCS~/dose AUC c variant AUCtc,variant Tmax,sc
Variant (min X g)/ml AUCsc,Actlmmunen AUCsc,full-length, wt (fin)
Actimmune~' 0.3-0.4 - 0.013-0.027 43
rhulFNG (full-length)15-24 37-80 - 362-446
E38N+S40T+S99T1~ 111-192 277-640 4.6-13 308-374
N16C+S99T2~ 37 92-123 1.5 2.5 247
N16C+S99T3~ 114 285-380 4.8-7.6 249
Table 4: Pharmacokinetic data for subtunaceous administration in rats
1) full-length
2) full-length, 5 kDa mPEG attached to the introduced cysteine residue
3) full-length, 10 lcDa mPEG attached to the introduced cysteine residue
Referring to Figs. 2 and 3 and Table 4, it is evident that the variants
(including
PEGylated variants) have a significantly higher AUC, when administered
subcutaneously, as
compared to rhuIFNG and, in particular, when compared to the commercially
available
Actimmune~. Referring to Fig. 3 it should be noted that the administered dose
of the two
PEGylated variants were reduced 2.5 fold compared to the administered dose of
the
[E38N+S40T+S99T] variant.
Evidently, this opens up the possibility of administering lower doses, thereby
obtaining fewer side effects, andlor administering the active principle less
frequently than
today thereby obtaining an improved patient compliance.
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
SEQUENCE LISTING
<110> Maxygen Holdings
<120> Interferon gamma polypeptide variants
<130> 231wo410 - INFG variants
<140>
<141>
<160> 49
<170> PatentIn Ver. 2.1
<210> 1
<211> 143
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: [S99T]huIFNG
<400> 1
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Thr Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg Ser Gln Met Leu Phe Arg Gly Arg Arg Ala Ser Gln
130 135 140
<210> 2
<211> 142
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated [S99T]huIFNG
1
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
<400> 2
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Thr Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg Ser Gln Met Leu Phe Arg Gly Arg Arg Ala Ser
130 135 140
<210> 3
<211> 141
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated [S99T]huIFNG
<400> 3
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Thr Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
2
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg Ser Gln Met Leu Phe Arg Gly Arg Arg Ala
130 135 140
<210> 4
<211> 140
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated [S99T]huIFNG
<400> 4
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
A1a Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Thr Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr G1y Lys
115 120 125
Arg Lys Arg Ser Gln Met Leu Phe Arg Gly Arg Arg
130 135 140
<210> 5
<211> 139
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated [S99T]huIFNG
<400> 5
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
AIa Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
3
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Thr Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg Ser Gln Met Leu Phe Arg Gly Arg
130 135
<210> 6
<211> 138
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated [S99T]huIFNG
<400> 6
Gln Asp Pro Tyr Val Lys G1u Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe GIu Lys Leu Thr
85 90 95
Asn Tyr Thr Val Thr Asp Leu Asn Val Gln Arg Lys A1a Ile His Glu
100 105 110
Leu IIe Gln Val Met Ala GIu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg Ser Gln Met Leu Phe Arg Gly
130 135
<210> 7
4
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
<211> 137
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated [S99T]huIFNG
<400> 7
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Thr Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg Ser Gln Met Leu Phe Arg
130 135
<210> 8
<211> 136
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated [S99T]huIFNG
<400> 8
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Thr Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg Ser Gln Met Leu Phe
130 135
<210> 9
<211> 135
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated [S99T]huIFNG
<400> 9
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Thr Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg Ser Gln Met Leu
130 135
<210> 10
<211> 134
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated [S99T]huIFNG
6
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
<400> 10
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys G1u Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Thr Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg Ser G1n Met
130
<210> 11
<211> 133
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated [S99T]huIFNG
<400> 11
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Va1 Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Va1 Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Thr Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile G1n Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
7
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
115 120 125
Arg Lys Arg Ser Gln
130
<210> 12
<211> 132
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated [S99T]huIFNG
<400> 12
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
A1a Gly His Ser Asp Val Ala Asp Asn G1y Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Thr Val Thr Asp Leu Asn Val Gln Arg Lys A1a Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg Ser
130
<210> 13
<211> 131
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated [S99T]huIFNG
<400> 13
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
8
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
35 40 45
Ile Va1 Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Va1 Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Thr Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro A1a Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg
130
<210> 14
<211> l30
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated [S99T]huIFNG
<400> 14
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Tle Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Thr Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pra Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys
130
<210> 15
<211> 129
9
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated [S99T]huIFNG
<400> 15
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Thr Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg
<210> 16
<211> 128
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated [S99T]huIFNG
<400> 16
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala G1y His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Thr Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
<210> 17
<211> 143
<212> PRT
<213> Homo Sapiens
<400> 17
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Ser Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg Ser Gln Met Leu Phe Arg Gly Arg Arg Ala Ser Gln
130 135 140
<210> 18
<211> 166
<212> PRT
<213> Homo Sapiens
<400> 18
Met Lys Tyr Thr Ser Tyr Ile Leu Ala Phe Gln Leu Cys Ile Val Leu
1 5 10 15
Gly Ser Leu Gly Cys Tyr Cys Gln Asp Pro Tyr Val Lys Glu A1a Glu
20 25 30
Asn Leu Lys Lys Tyr Phe Asn Ala Gly His Ser Asp Val Ala Asp Asn
35 40 45
Gly Thr Leu Phe Leu Gly Ile Leu Lys Asn Trp Lys Glu Glu Ser Asp
50 55 60
11
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
Arg Lys Ile Met Gln Ser Gln Ile Val Ser Phe Tyr Phe Lys Leu Phe
65 70 75 80
Lys Asn Phe Lys Asp Asp Gln Ser Ile Gln Lys Ser Val Glu Thr Ile
85 90 95
Lys Glu Asp Met Asn Val Lys Phe Phe Asn Ser Asn Lys Lys Lys Arg
100 105 110
Asp Asp Phe Glu Lys Leu Thr Asn Tyr Ser Val Thr Asp Leu Asn Val
115 120 125
Gln Arg Lys Ala Ile His Glu Leu Ile Gln Val Met Ala Glu Leu Ser
130 135 140
Pro Ala Ala Lys Thr Gly Lys Arg Lys Arg Ser Gln Met Leu Phe Arg
145 150 155 160
Gly Arg Arg Ala Ser Gln
165
<210> 19
<211> 142
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated huIFNG
<400> 19
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Ser Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg Ser Gln Met Leu Phe Arg Gly Arg Arg Ala Ser
130 135 140
12
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
<210> 20
<211> 141
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated huIFNG
<400> 20
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Va1 Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Ser Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu I1e Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg Ser Gln Met Leu Phe Arg Gly Arg Arg Ala
130 135 140
<210> 21
<211> 140
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated huIFNG
<400> 21
Gln Asp Pro Tyr Val Lys Glu Ala G1u Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Va1 Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met G1n Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
13
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Ser Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Va1 Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg Ser Gln Met Leu Phe Arg Gly Arg Arg
130 135 140
<210> 22
<211> 139
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated huIFNG
<400> 22
G1n Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Ser Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg Ser Gln Met Leu Phe Arg Gly Arg
130 135
<210> 23
<211> 138
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated huIFNG
14
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
<400> 23
Gln Asp Pro Tyr Val Lys Glu A1a Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Ser Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg Ser Gln Met Leu Phe Arg Gly
130 135
<210> 24
<211> 137
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated huIFNG
<400> 24
Gln Asp Pro Tyr Val Lys G1u Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser I1e Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Ser Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg Ser Gln Met Leu Phe Arg
130 135
<210> 25
<211> 136
<212> PRT
<223> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated huIFNG
<400> 25
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Ser Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg Ser Gln Met Leu Phe
130 135
<210> 26
<211> 135
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated huIFNG
<400> 26
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
16
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Ser Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg Ser Gln Met Leu
230 135
<210> 27
<211> 134
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated huIFNG
<400> 27
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Ser Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg Ser Gln Met
130
<210> 28
17
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
<211> 133
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated huIFNG
<400> 28
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Ser Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 210
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg Ser Gln
130
<210> 29
<211> 132
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated huIFNG
<400> 29
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
18
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Ser Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg Ser
130
<210> 30
<211> 131
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated huIFNG
<400> 30
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Ser Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys Arg
130
<210> 31
<211> 130
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated huIFNG
19
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
<400> 31
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val A1a Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Ser Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
Arg Lys
130
<210> 32
<211> 129
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: C-terminally
truncated huIFNG
<400> 32
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Ser Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala A1a Lys Thr Gly Lys
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
115 120 125
Arg
<210> 33
<211> 128
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:.C-terminally
truncated huIFNG
<400> 33
Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe Asn
1 5 10 15
Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly Ile
20 25 30
Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys Ile Met Gln Ser Gln
35 40 45
Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp Gln
50 ' 55 60
Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val Lys
65 70 75 80
Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu Thr
85 90 95
Asn Tyr Ser Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His Glu
100 105 110
Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly Lys
115 120 125
<210> 34
<211> 140
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Actimmune(r)
<400> 34
Met Gln Asp Pro Tyr Val Lys Glu Ala Glu Asn Leu Lys Lys Tyr Phe
1 5 10 15
Asn Ala Gly His Ser Asp Val Ala Asp Asn Gly Thr Leu Phe Leu Gly
20 25 30
Ile Leu Lys Asn Trp Lys Glu Glu Ser Asp Arg Lys I1e Met Gln Ser
35 40 45
Gln Ile Val Ser Phe Tyr Phe Lys Leu Phe Lys Asn Phe Lys Asp Asp
50 55 60
21
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
Gln Ser Ile Gln Lys Ser Val Glu Thr Ile Lys Glu Asp Met Asn Val
65 70 75 80
Lys Phe Phe Asn Ser Asn Lys Lys Lys Arg Asp Asp Phe Glu Lys Leu
85 90 95
Thr Asn Tyr Ser Val Thr Asp Leu Asn Val Gln Arg Lys Ala Ile His
100 105 110
Glu Leu Ile Gln Val Met Ala Glu Leu Ser Pro Ala Ala Lys Thr Gly
115 120 125
Lys Arg Lys Arg Ser Gln Met Leu Phe Arg Gly Arg
130 135 140
<210> 35
<211> 6
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Tag
<400> 35
His His His His His His
1 5
<210> 36
<211> 8
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Tag
<400> 36
Met Lys His His His His His His
1 5
<210> 37
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Tag
<400> 37
Met Lys His His Ala His His Gln His His
1 5 10
<210> 38
<211> 14
<212> PRT
<213> Artificial Sequence
22
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
<220>
<223> Description of Artificial Sequence: Tag
<400> 38
Met Lys His G1n His Gln His Gln His Gln His Gln His Gln
1 5 10
<210> 39
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Tag
<400> 39
G1u Gln Lys Leu Ile Sex Glu Glu Asp Leu
1 5 10
<210> 40
<211> 8
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Tag
<400> 40
Asp Tyr Lys Asp Asp Asp Asp Lys
1 5
<210> 41
<211> 9
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Tag
<400> 41
Tyr Pro Tyr Asp Val Pro Asp Tyr Ala
1 5
<210> 42
<211> 498
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Expression
casette optimised for expression of interferon
gamma in CHO cells
<400> 42
atgaagtaca caagctatat cctggccttt cagctgtgca tcgtgctggg ctccctgggc 60
tgctattgcc aggaccctta cgtgaaggag gccgagaacc tgaagaagta ctttaacgcc 120
ggccacagcg atgtggccga caatggcaca ctgtttctgg gcatcctgaa gaattggaag 180
23
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
gaggagagcg atcggaagat catgcagtcc cagatcgtgt ccttctattt caagctgttt 240
aagaatttca aggacgatca gtccatccag aagtccgtgg agaccatcaa ggaggacatg 300
aacgtgaagt ttttcaatag caataagaag aagagagacg atttcgagaa gctgaccaat 360
tactccgtga cagacctgaa cgtgcagaga aaggccatcc acgagctgat ccaggtgatg 420
gccgagctgt cccccgccgc caagaccggc aagagaaaga gaagccagat gctgttcaga 480
ggcagacggg ccagccag 498
<210> 43
<211> 19
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Primer
<400> 43
gatggctggc aactagaag 19
<210> 44
<211> 19
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Primer
<400> 44
tgtacggtgg gaggtctat 19
<210> 45
<211> 33
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Primer
<400> 45
gttcaggtct gtcacgctgt aattggtcag ctt 33
<210> 46
<211> 33
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Primer
<400> 46
aagctgacca attacaccgt gacagacctg aac 33
<210> 47
<211> 33
<212> DNA
<213> Artificial Sequence
24
CA 02443277 2003-10-03
WO 02/081507 PCT/DK02/00226
<220>
<223> Description of Artificial Sequence: Primer
<400> 47
catgatcttc cgatcggtct cgttcttcca att 33
<210> 48
<211> 33
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Primer
<400> 48
aattggaaga acgagaccga tcggaagatc atg 33
<210> 49
<211> 30
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Primer
<400> 49
gagtctagat tacagcatct ggcttctctt 30