Note: Descriptions are shown in the official language in which they were submitted.
CA 02444210 2003-10-09
wo 9sr3assa rc.-r~s9sroz~a9
Compounds and Method for Preparing Substituted 4-Phenyl
4-cyanocyclohexanoic Acids
Scope of the Invention
This invention covers intermediates and a synthetic route for making 4-cyano-4-
(3-cyclopentyloxy-4-methoxyphenyl)cyclohexanoic acid and its analogs. This
acid and
its named analogs are selective for inhibiting the catalytic site in the
phosphodiesterase
isoenzyme denominated IV (PDE IV hereafter) and as such the acids are useful
in
treating a number of diseases which can be moderated by affecting the PDE IV
enzyme
and its subtypes.
l0 Area of the Invention
Bronchial asthma is a complex, multifactorial disease characterized by
reversible narrowing of the airway and hyper-reactivity of the respiratory
tract to
external stimuli.
Identification of novel therapeutic agents for asthma is made difficult by the
fact
15 that multiple mediators are responsible for the development of the disease.
Thus, it
seems unlikely that eliminating the effects of a single mediator will have a
substantial
effect on all three components of chronic asthma. An alternative to the
"mediator
approach" is to regulate the activity of the cells responsible for the
pathophysiology of
the disease.
20 One such way is by elevating levels of cAMP (adenosine cyclic 3',5'-
monophosphate). Cyclic AMP has been shown to be a second messenger mediating
the
biologic responses to a wide range of hormones, neurotransmitters and drugs;
[Krebs
Endocrinology Proceedings of the 4th International Congress Excerpta Medica,
17-29,
1973]. When the appropriate agonist binds to specific cell surface receptors,
adenylate
25 cyclase is activated, which converts Mg~2-ATP to cAMP at an accelerated
rate.
Cyclic AMP modulates the activity of most, if not all, of the cells that
contribute
to the pathophysiology of extrinsic (allergic) asthma. As such, an elevation
of cAMP
would produce beneficial effects including: 1 ) airway smooth muscle
relaxation, 2)
inhibition of mast cell mediator release, 3) suppression of neutrophil
degranulation, 4)
30 inhibition of basophil degranulation, and 5) inhibition of monocyte and
macrophage
activation. Hence, compounds that activate adenylate cyclase or inhibit
phosphodiesterase should be effective in suppressing the inappropriate
activation of
airway smooth muscle and a wide variety of inflammatory cells. The principal
cellular -_
mechanism for the inactivation of cAMP is hydrolysis of the 3'-phosphodiester
bond by
CA 02444210 2003-10-09
WO 98134584 - PGTIUS98I02749
one or more of a family of isozymes referred to as cyclic nucleotide
phosphodiesterases
(PDEs).
It has now been shown that a distinct cyclic nucleotide phosphodiesterase
(PDE)
isozyme, PDE IV, is responsible for cAMP breakdown in airway smooth muscle and
inflammatory cells. [Torphy, "Phosphodiesterase Isozymes: Potential Targets
for
Novel Anti-asthmatic Agents" in New Drugs for Asthma, Barnes, ed. IBC
Technical
Services Ltd., 1989]. Research indicates that inhibition of this enzyme not
only
produces airway smooth muscle relaxation, but also suppresses degranulation of
mast
cells, basophils and neutrophils along with inhibiting the activation of
monocytes and
neutrophils. Moreover, the beneficial effects of PDE IV inhibitors are
markedly
potentiated when adenylate cyclase activity of target cells is elevated by
appropriate
hormones or autocoids, as would be the case in vivo. Thus PDE IV inhibitors
would be
effective in the asthmatic lung, where levels of prostaglandin E2 and
prostacyclin
(activators of adenylate cyclase) are elevated. Such compounds would offer a
unique
approach toward the pharmacotherapy of bronchial asthma and possess
significant
therapeutic advantages over agents currently on the market.
The process and intermediates of this invention provide a means for making
certain 4-substituted-4-(3,4-disubstitutedphenyl)cyclohexanoic acids which are
useful
for treating asthma, and other diseases which can be moderated by affecting
the PDE IV
enzyme and its subtypes. The final products of particular interest are fully
described in
U.S. patent 5,552,438 issues 03 September 1996.
Summary of the Invention
This invention relates a method for making a compound of formula I
X ~ R.
W Y ~ R"
RiX2
Rs (I)
R1 is -(CR4R5)nC(~)~(CR4R5)mR6, -(CR4R5)nC(~)NR4(CR4R5)mR6, _
(CR4R5)n0(CR4R5)mR6~ or -(CR4R5)rR6 wherein the alkyl moieties may be
optionally substituted with one or more halogens;
misOto2;
n is 1 to 4;
risOto6;
CA 02444210 2003-10-09
WO 98/34584 PCT/US98I02749
Rq. and RS are independently selected from hydrogen or a C 1 _2 alkyl;
R6 is hydrogen, methyl, hydroxyl, aryl, halo substituted aryl, aryloxyCl_3
alkyl,
halo substituted aryloxyCl-3 alkyl, indanyl, indenyl, C7-1 I polycycloalkyl,
tetrahydrofuranyl, furanyl, tetrahydropyranyl, pyranyl, tetrahydrothienyl,
thienyl,
tetrahydrothiopyranyl, thiopyranyl, C3-6 cycloalkyl, or a C4_6 cycloalkyl
containing
one or two unsaturated bonds, wherein the cycloalkyl and heterocyclic moieties
may be
optionally substituted by 1 to 3 methyl groups or one ethyl group;
provided that:
a) when R6 is hydroxyl, then m is 2; or
b) when R6 is hydroxyl, then r is 2 to 6; or
c) when R6 is 2-tetrahydropyranyl, 2-tetrahydrothiopyranyl,
2-tetrahydrofura.nyl, or 2-tetrahydrothienyl, then m is I or 2; or
d) when R6 is 2-tetrahydropyranyl, 2-tetrahydrothiopyranyl,
2-tetrahydrofuranyl, or 2-tetrahydrothienyl, then r is 1 to 6;
I S e) when n is 1 and m is 0, then R6 is other than H in
-(CR4R5)n0(CR4R5)mR6;
X is YR2, halogen, nitro, NH~, or formyl amine;
X2 is O or NRg;
Y is O or S(O)m~;
m' is 0, l, or 2;
R2 is independently selected from -CH3 or -CH2CH3 optionally substituted by
1 or more halogens;
R3 is hydrogen, halogen, CI-4 alkyl, CH2NHC(O)C(O)NH2, halo-substituted
C I-4 alkyl, -CH=CRg~Rg~, cyclopropyl optionally substituted by Rg~, CN, ORg,
CH20Rg, NRgR I0, CH2NRgR l0, C(Z')H, C(O)ORg, C(O)NRgR 10, or C=CRg~;
Rg is hydrogen or C I ~ alkyl optionally substituted by one to three
fluorines;
Rg~ is Rg or fluorine;
RIO isORgorRIl:
R1 I is hydrogen, or CIA alkyl optionally substituted by one to three
fluorines;
Z' is O, NR9, NORg, NCN, C(-CN)2, CRgCN, CRgN02, CRgC(O)ORg,
CRgC(O)NRgRg, C(-CN)N02, C(-CN)C(O)OR9, or C(-CN)C(O)NRgRg;
R' and R" are independently hydrogen or -C(O)OH;
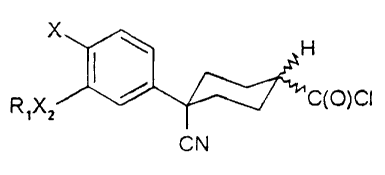
which method comprises treating a compound of formula II(a) or II(b)
X / H CN
X
R ~ ~ O ~ O
,Xz v R ~ H
R3 CN
R3
CA 02444210 2003-10-09
WO 98/34584 - PCT/L1S98/0.2749
II(a) II(b)
where R,, R3, X2 and X are the same as for formula (I), with lithium bromide
or
magnesium bromide in a polar solvent at a temperature between about 60°
and 100° C,
optionally under an inert atmosphere for a time sufficient for the reaction to
go to
completion.
This invention also relates to compounds of formula II per se.
In another aspect this invention relates to a one-pot method for making the
ketone of formula III starting with isovanillin,
X
O
R~X2
R3 (I~
where R 1, R3, X2 and X are the same as for formula (I), as more fully
described
herein below.
In yet a third aspect this invention relates to a process for preparing a
compound
of formula I which process comprises treating a compound of formula (IV) using
an
alkali metal cyanide, for example LiCN, in a compatible solvent such as
dimethylformamide which contains a small proportion of water
X
Br
R,X2 CHO
CN (N)
where, in formula III, R1, X and X2 are the same as in formula I.
In a further embodiment this invention relates to a process for making a
compound of formula I comprising treating an acyl nitrile of formula V with
water.
X
CtoaCN
I
RtX2
CN (V)
The X, X2 and R 1 groups in formula V are the same as those in formula I.
In yet a further embodiment this invention relates to compounds of
fomnula II
4
CA 02444210 2003-10-09
WO 98134584 PCT/ITS98/02749
T
O
R~XZ
R3 (B)
Rl is -(CR4R5)nC(O)O(CR4R5)mR6, -(CR4R5)nC(O)NR4(CR4R5)mR6, _
(CR4R5)n0(CR4R5)mR(, or -(CR4R5)rR6 wherein the alkyl moieties may be
optionally substituted with one or more halogens;
misOto2;
n is 1 to 4;
risOto6;
R4 and R5 are independently selected from hydrogen or a C 1 _2 alkyl;
R6 is hydrogen, methyl, hydroxyl, aryl, halo substituted aryl, aryloxyCl-3
alkyl,
halo substituted aryloxyC 1-3 alkyl, indanyl, indenyl, C'7_ 11 polycycloalkyl,
tetrahydrofuranyl, furanyl, tetrahydropyranyl, pyranyl, tetrahydrothienyl,
thienyl,
tetrahydrothiopyranyl, thiopyranyl, C3_6 cycloalkyl, or a C4_6 cycloalkyl
containing
one or two unsaturated bonds, wherein the cycloaIkyl and heterocyclic moieties
may be
optionally substituted by 1 to 3 methyl groups or one ethyl group;
provided that:
a) when R6 is hydroxyl, then m is 2; or
b) when R6 is hydroxyl, then r is 2 to 6; or
c) when R6 is 2-tetrahydropyranyl, 2-tetrahydrothiopyranyl,
2-tetrahydrofuranyl, or 2-tetrahydrothienyl, then m is 1 or 2; or
d) when R6 is 2-tetrahydropyranyl, 2-tetrahydrothiopyranyl,
2-tetrahydrofuranyl, or 2-tetrahydrothienyl, then r is 1 to 6;
e) when n is 1 and m is 0, then R( is other than H in
-(CR4R5)n0(CR4.R5)mR6;
X is YR2, halogen, nitro, NHZ, or formyl amine;
X2 is O or NRg;
Y is O or S(O)m~;
m' is 0, 1, or 2;
R2 is independently selected from -CH3 or -CH2CH3 optionally substituted by
1 or more halogens;
R3 is hydrogen, halogen, C 1-4 alkyl, CH2NHC(O)C(O)NH2, halo-substituted
Cl-4 alkyl, -CH=CRg~Rg~, cyciopropyl optionally substituted by Rg~, CN, ORg,
CH20Rg, NRgRIp, CH2NRgRlp, C(Z')H, C(O)ORg, C(O)NRgRlp, or C=CRg
Rg is hydrogen or C 1 ~ alkyl optionally substituted by one to three
fluorines;
Rg~ is Rg or fluorine;
R 10 is ORg or R 11;
5
CA 02444210 2003-10-09
WO 98134584 PGT/US98IOZ949
R 11 is hydrogen, or C 1 ~ alkyl optionally substituted by one to three
fluorines;
Z' is O, NR9> NORg, NCN, C(-CN)2, CRBCN, CR8N02, CRBC(O)ORg,
CRBC(O)NRBRg, C(-CN)N02, C(-CN)C(O)OR9, or C(-CN)C(O)NRBRg, and
T is CN or S02R where R is C1_6aikyl or CO_3alkylphenyl.
Specific Embodiments of the Invention
This process involves a nine step synthesis for preparing certain 4-
substituted-4-
(3,4-disubstitutedphenyI)cyclohexanoic acids. The starting material is
isovanillin, 3-
hydroxy-4-methoxybenzaldehyde, or an analog thereof. "Analog" means another 3
and/or 4 position substituent conforming to the definitions of R,, R,, X: and
X in the
definition of formula (I).
The compounds which are made by this process are PDE IV inhibitors. They
are useful for treating a number of diseases as described in U.S. patent
5,552,438 issued
3 September 1996.
The preferred compounds which can be made by this process are as follows:
Preferred Rl substitutents for the compounds of all named formulas are CH2-
cyclopropyl, CH2-CS_6 cycloalkyl, C4_6 cycloalkyl unsubstituted or substituted
with
OHC~-11 polycycioalkyl, (3- or 4-cycIopentenyl), phenyl, tetrahydrofuran-3-yl,
benzyl
or C 1-2 alkyl unsubstituted or substituted by 1 or more fluorines,
-(CH2)1-3C(O)O(CH2)0-2CH3, -(CH2)1-30(CH2)0-2CH3, and -(CH2)2-40H.
Preferred X groups for Formula (I), (II) or (III) are those wherein X is YR2
and
Y is oxygen. The preferred X2 group for Formula (I) is that wherein X2 is
oxygen.
Preferred R2 groups are a C 1_2 alkyl unsubstituted or substituted by 1 or
more
halogens. The halogen atoms are preferably fluorine and chlorine, more
preferably
fluorine. More preferred R2 groups are those wherein R? is methyl, or the
fluoro-
substituted alkyls, specifically a C 1 _2 alkyl, such as a -CF3, -CHF2, or -
CH2CHF~
moiety. Most preferred are the -CHF2 and -CH3 moieties.
Most preferred are those compounds wherein R 1 is -CH2-cyclopropyl,
cyclopentyl, 3-hydroxycyclopentyl, methyl or CF2H; X is YR2; Y is oxygen; X2
is
oxygen; and R2 is CF2H or methyl; and R3 is CN.
A representative schematic of this process is set out in Scheme I. This
graphical
representation uses specific examples to illustrate the general methodology
used in this
mvenuon.
6
CA 02444210 2003-10-09
wo 9sr3assa
Scheme I
PCT/fJS98/02'7.49
OH ~''~
H3C0 ~CI O
H CO
--~ NaBH4 3
O DMF, 125 °C DMF/MeOH
K2C03 w
H
tsovanillin 1-1
H OH
1-2
"' O
O
H3C0
Conc. HCI H CO / NaCN , ~ COOCH3
Toluene. 20 °C ~ ~ DMF, 55°C
W ~ CH3CN, Triton-B
CI CN
1_3 1_4
~O
H~CO
daOMe NaCI. HZO
~er~. as xH --. i °
DMSO.tso°c
cN
,~
,_>
N ~O
H~CO
CICH~CN Ligr , H
THFIaq. K01- DMFICH3CN
0 °C
Catalyst CN H20 COZH
i-8 95° C CN
Formula I
H C4 ~O
3 H
CN H1C0 Br
~ MgBrZ Or Liar
CN
O
---i ,
CN Ether H
1-8 1-9b N OM(H3
MgBr2 or Liar
Diglyme, 95 C
7
CA 02444210 2003-10-09
WO 98/34584 PCTIUS98/02'Z49
1-8 . 1-9b
t
I
II
a
O
H B
H3C0 ~ CN CN
OM(H)
OM(H
CN
CN
i -9a
Enolate A
(M = Li or Mgt
O O
H3C0 \ H H3CO ~ COCN
I t
COCN ~ H
1-10a CN CN 1-10b
H O H O 1
2 2
'O
H3C0 H H3C0 ~ COOH
I (
r
COOH H
CN CN
1-11a 1-11b
Referring to Scheme I, isovanillin, 3-hydroxy-4-methoxybenzaldehyde, is a
readily available starting material. It can be alkylated with an R,X moiety (X
= Cl, Br,
and I) as represented by cyclopentyl chloride. The reaction vessel is first
flushed with
an inert gas, for example nitrogen. A polar solvent such as DMF is then added
to the
8
CA 02444210 2003-10-09
WO 98134584 PCT/ITS98102749
vessel, then the isovanillin, then the R,X adduct, and some base. About 2
equivalents
of the R,X adduct versus the isovanillin are used. Likewise about 2
equivalents of base
are used, again relative to the isovanillin. The base can be any inorganic
base or a
carbonate. Here it is illustrated by potassium carbonate. The vessel contents
are heated
to about 125° C for about 90 to 120 minutes in which time the reaction
will have gone
to completion. The vessel contents are cooled to ambient temperature, filtered
to
remove the inorganic salts, and washed with an alcohol such as methanol. This
filtrate
contains the aldehyde, labeled 1-1.
The aldehyde is then reduced to the alcohol using an inorganic reducing agent.
To do this the filtrate from the foregoing reaction is treated with sodium
borohydride
and after workup affords the desired alcohol, 1-2 in 97% overall yield from
isovanillin.
This is achieved by cooling the filtrate to about 0° C after which a
reducing agent, here
sodium borohydride, is added. About 0.25 to 0.5 equivalents of this reducing
agent is
used. The temperature is keep at about 0° C during the addition of the
reducing agent
and for about 30 to 40 minutes thereafter. Then the temperature is allowed to
rise to
about room temperature after which about one-half an equivalent of HCI is
added to the
reaction vessel. The alcohol is then extracted into an organic solvent,
toluene is
illustrated, and washed with dilute sodium bicarbonate.
The top organic layer containing the alcohol is then treated with excess
concentrated hydrochloric acid at ambient temperature to afford, after workup,
the
desired benzyl chloride I-3. The chloride is isolated as a w/w solution in an
amide
solvent, DMF is illustrated, and treated with about a SO% molar excess of
sodium
cyanide at a mildly elevated temperature, here illustrated as 55° C.
This affords the
desired nitrilel-4.. The nitrile is isolated as a w/w solution in an
appropriate solvent
such as anhydrous acetonitrile and used directly in the next step.
The nitrite solution is charged with methyl acrylate. It is cooled to about -
10°
C, and slowly treated with a catalytic amount of Triton-B in the same solvent
as used to
dissolve the nitrite. The methyl acrylate is added in a 3 to 4-fold excess.
The reaction
is complete within 30 to 45 minutes after which the acrylate addition, the
pimelate
product, 1-5, is isolated as a w/w solution in toluene and treated with about
2
equivalents of sodium methoxide at about 75° C to give the ~-keto-ester
product, 1-6 .
The reaction solution is cooled and_neutralized to pH 7 with mineral acid such
as 6N
9
CA 02444210 2003-10-09
WO 98/34584 PCT/US98102749
hydrochloric acid. The solution is charged with dimethyl sulfoxide, sodium
chloride,
water, and heated, for example to about 150° C, to effect the
decarboxylation to give 1-
7. The ketone, 1-7, is isolated from the solvent system as an off white solid.
The dicarbonitrile 1-8 is prepared from the ketone by treating the ketone with
chloroacetonitrile in the presence of an inorganic base and a catalytic amount
of
benzyltriethylammonium chloride (BTEAC). The ketone is charged into a mixture
of
strong base (aqueous potassium hydroxide) and a water miscible solvent such as
tetrahydrofuran. A slight excess of chloroacetonitrile is added at reduced
temperature,
about 0° C or thereabouts. The reaction is maintained at about that
temperature for the
duration of the reaction, usually about 1 hour. The product is isolated and
usually it is
crystalline.
The dicarbonitrile is convened to the cyclohexanecarboxylic acid using a Lewis
acid catalyst; water is also needed to drive the reaction to the acid. Without
water
intermediates I-10a and I-10b may dimerize. This reaction is carried out by
charging a
vessel with solvents, in this instance exemplified by DMF, acetonitrile and
water, and
the Lewis acid (about 1.5 equivalents), Liar is illustrated, sweeping the
vessel with an
inert gas, adding the dicarbonitriie IIa or IIb, or a mixture of IIa and IIb
and heating the
vessel and its contents to about 100° C for a number of hours, 8 hours
being an
example. The acid is isolated by conventional means.
It should be noted that this reaction, that is the conversion of the epoxide
to the
acid, involves several intermediates which do not need to be isolated. It has
been found
that treating the epoxide with Liar yields intermediates 1-9a and 1-9b.
Intermediate 1-
9a is formed when Liar is added to the reaction pot. But intermediate i-9a
converts
back to the epoxide under the recited reaction conditions. Intermediate 1-9b
is also
formed but apparently reacts rapidly to form intermediates such as enolate A,
I-l0a and
1-lOb etc leading to product. So it appears 1-9a and 1-9b are formed, but that
1-9a
converts back to the epoxide which ultimately forms 1-9b which is then
converted to
other intermediates enroute to forming the acids of 1-l la and 1-l 1b.
Parenthetically
the designation "OM(H)" in I-9a and 1-9b means the metal salt of the alcohol
or the
alcohol per se, depending on the reaction conditions. Intermediate I-9b is
believed to
convert to the acyl nitriles of formulas 1-IQa and 1-lOb via the proposed
bracketed
intermediate. The existence of the_proposed bracketed intermediate (enolate)
has not
CA 02444210 2003-10-09
WO 98/34584 PCT/US98/02749
been fully confirmed. And while the acyl nitrites of 1-IOa and I-lOb have not
been
directly observed, indirect evidence exists for these compounds by virtue of
the fact the
bis-condensation product dimer B was isolated and is analogous to reported
compounds
where a similar bis-condensate is the product of an acyl nitrite.
Scheme 2
C(O)CN NC O O NC O O
NC
CN- -HCN
Ar ~' CN
CN Ar CN Ar CN Ar CN Ar
1-10a/b Dimer A Dimer B
Dimers such as dimer A are known to form from the likes of acyl nitrites 1-
l0a/b in the presence of HCN (Thesing, J.; Witzet, D.; Brehm, A. Angew Chem..
1956,
68, 425; and Hunig, S.; Schaller, R. Angew. Chem. Int. Ed. Engl.. 1982,
21,36).
And in addition, authentic samples of intermediates 1-l0a and 1-IOb were
prepared and found to convert to acids 1-1 la and 1-1 Ib when exposed to
water. The
equitorial isomer 1-IOa converted to the acid in an equitorial/axial ratio of
about 98:2
IS while the axial isomer I-l0a isomerized to a perponderance of the
equitorial isomer 1-
l la (77:23). It is believed the axial acyl nitrite converts to the equitorial
acyl nitrite via
the proposed bracketed enolate intermediate.
The second, following reaction scheme illustrates preparing the acids of
formula
(I) from the bromoaldehyde of formula (IV).
Scheme 3
11
CA 02444210 2003-10-09
WO 98/34584 PGT/US98/027.49
X
X ~ O S02 M~r2 I ~ Br
i
R~X2 ~ H ' Ether R,Xz CHO
CN 1 / CN
3-1 ._3-2
LiCN, DMF x ~ COCN
i
95° C R X ~ Y ''~H --'~ i-llalb
,z
CN
(1-l0a/b)
The following examples are provided to illustrate specifics of the invention,
not
to limit it. What is reserved to the inventors is set forth in the claims
appended hereto.
Specific Examples
Example 1
Preparation of 3-c~pentyloxy-4-methoxybenzaldeh~de.
A 12 liter round bottom flask equipped with an overhead stirrer, internal
thermometer, and a reflux condensor equipped with a nitrogen inlet was flushed
with
nitrogen. The flask was charged with dimethylformamide (2.4 L), isovanillin
(350 g,
2.3 mol, I equivalent), cyclopentyl chloride (481 g, 4.6 mol, 2.0 equivalent)
and
potassium carbonate (634 g, 4.6 mol, 2.0 equivalents). The vigorously stirred
suspension was heated to 125 °C for two hours or until the
disappearance of isovanillin.
The reaction was cooled to 20 -30 °C and filtered to remove the
inorganic salts. The
filter cake was rinsed with methanol ( I .0 L).
The clear, light-brown filtrate (DMF and methanol) containing the product, 3-
cyclopentyloxy-4-methoxybenzaldehyde, was used directly in the next step ( 100
solution yield).
Example 2
Preparation of 3-Cyclopent rLy-4-meth~xybenzyl cohol.
A 12 liter round bottom flask equipped with an overhead stirrer, internal
thermometer, and a reflux condensor equipped with a nitrogen inlet was flushed
with
nitrogen. The flask was charged with dimethylformamide (2.4 L), methanol ( I
.0 L),
and 3-cyclopentyloxy-4-methoxybenzaldehyde (506 g, 2.3 mol, I equivalent). The
contents of the flask were cooled to 0 to 5 °C followed by the addition
of sodium
borohydride (32.2 g, 0.85 mol, 0.37 equivalents). The reaction was maintained
at 0 to 5
°C for 30 minutes, and warmed to 20 to 25 °C for an additional 2
hours or until the
12
CA 02444210 2003-10-09
WO 98134584 PCT/LTS98/02749
disappearance of the aldehyde. A solution of 6N hydrochloric acid ( 195 mL,
1.17 mol,
0.51 equivalents) was added over 20 minutes. The reaction was concentrated
under
reduced pressure, and cooled to 20 to 25 °C.
The flask was charged with deionized water ( 1.9 L) and toluene ( 1.9 L). The
layers were separated, the organic layer was isolated, and washed twice with
deionized
water {2 x 800 mL). The product, 3-cyclopentyloxy-4-methoxybenzyl alcohol was
collected as a solution in toluene (97% solution yield) and used directly in
the next step.
Example 3
Preparation of 4-Chloromethyl-2-cvclo~entyloxy-1-methoxybenzene.
A 12 liter round bottom flask equipped with an overhead stirrer, internal
thermometer, and a reflux condensor equipped with a nitrogen inlet was flushed
with
nitrogen. The flask was charged with 3-cyclopentyloxy-4-methoxybenzyl alcohol
(495
g, 2.2 mol, 1 equivalent) in a solution of toluene. To the vigorously stirred
reaction at
22 °C was added concentrated hydrochloric acid (600 g, 2.75
equivalents}. The
reaction was maintained at 20 to 25 °C for 30 minutes. The top organic
layer was
isolated and the bottom acidic layer was discarded. To the top organic layer
was
charged a solution of 10% sodium bicarbonate (550 g, 0.6~ mo, 0.36
equivalents) and
t-butyl methyl ether (814 g). The contents of the flask were vigorously
stirred, and
allowed to settle. The product, 4-chloromethyl-2-cyclopentyloxy-1-
methoxybenzene,
was isolated as a solution in toluene and t-butyl methyl ether (96.8% solution
yield}.
This was used directly in the next step.
Example 4
Preparation of 4-Cyanomethyl-2-cvclo~entyloxy-1-methoxybenzene.
A 12 liter round bottom flask equipped with an overhead stirrer, and a
distillation apparatus was flushed with nitrogen. The flask was charged with 4-
chloromethyl-2-cyclopentyloxy-1-methoxybenzene {519 g, 2.15 mol, 1.0
equivalents)
in a solution of toluene and t-butyl methyl ether. The reaction was
concentrated under
reduce pressure to a residue. To the 12 liter flask was charged DMF (1.44 kg)
and
sodium cyanide (142 g, 2.9 mol, 1.35 equivalents). While sodium cyanide is
used
in this example, it is for illustrative purposes only and any other alkalin
metal
cyanide is suitable as well. ~e reactior. was heated to SS °C
for 6 hours or until deemed complete by the disappearance of the benzyl
chloride. The
reaction was concentrated under reduced pressure to a residue. To the flask
was
charged t-butyl methyl ether (2.30 kg) and deionized water (800 mL). The
contents of
the flask were vigorously stirred, and allowed to settle. The top organic
layer was
isolated, washed three times with deionized water (3 x 800 mL), and
concentrated
under atmospheric pressure to a residue. To the flask was added acetonitrile (
1.26 kg)
and the distillation was continued until an additional 400 mL of solvent was
collected.
The product, 4-cyanomethyl-2-cyclopentyloxy-1-methoxybenzene, was isolated as
a
solution in acetonitrile (92.2% yield). This was used directly in the next
step.
13
CA 02444210 2003-10-09
WO 98/34584 PCTIUS98/02749
Example 5
Prenaration of Dimethyl-4-cyano-4-(3-cycloQentyloxy-4-methoxy,_p
en~l,)p~melate
A 12 liter round bottom flask equipped with an overhead stirrer, internal
thermometer, and a reflux condensor equipped with a nitrogen inlet was flushed
with
nitrogen. The flask was charged with a solution of 4-cyanomethyl-2-
cyclopentyloxy-1-
methoxybenzene (460 g, 1.99 mol, 1.0 equivalent) in acetonitrile, and methyl
acrylate
(520 g, 6.0 mol, 3.0 equivalents). The contents of the flask was cooled to -10
°C. A
pressure equalizing addition fiinnel was charged with acetonitrile ( 1.1 L)
and
benzyltrimethyl ammonium hydroxide (a 40% w/w solution in methanol, 25 g, 0.06
mol, 0.03 equivalents). The contents of the addition funnel was added to the
flask. An
exotherm was observed, and after stirring for 30 minutes the contents of the
flask were
cooled to 20 °C. The reaction was concentrated under reduced pressure
to a residue.
To the residue was added toluene (2.6 L). This solution of dimethyl-4-cyano-4-
(3-
cyclopentyloxy-4-methoxy-phenyl)pimelate ( 90% solution yield) was used
directly in
1 S the next step.
Exam In a 6
Preparation of 4-Cyano-4-(3-cyclopentyloxy-4-methoxyphenyllcvclohexan-1-
A 12 liter round bottom flask equipped with an overhead stirrer, internal
thermometer, and a reflux condensor equipped with a nitrogen inlet was flushed
with
nitrogen. The flask was charged with a solution of dimethyl-4-cyano-4-(3-
cyclopentyloxy-4-methoxy-phenyl)pimelate ( 720 g, 1.78 mol, 1 equivalent) in
toluene,
and sodium methoxide (25 wt% in methanol, 545 g, 2.67 mol, 1.5 equivalents).
The
reaction was heated to 70 to 75 °C for 2 hours or until deemed complete
by the
disappearance of the pimelate. The reaction was cooled to 25 °C. A
solution of 6N
hydrochloric acid was added in order to adjust the pH to 6.8 - 7.2. The
reaction was
concentrated under vacuum to a residue. The flask was charged with
dimethylsulfoxide
(3.3 L), deionized water (250 mL) and sodium chloride (250 g).
The contents of the flask were heated to 145-155 °C, and held at
this
temperature for 2 hours. The reaction was cooled and concentrated under vacuum
to a
residue. To the residue was added water (1.9 L), ethyl acetate (1.25 L), and t-
butyl
methyl ether (620 mL). The solution was stirred and allowed to settle. The
layers were
separated, and the aqueous layer was re-extracted with ethyl acetate ( 1.25
L). The
combined organic layers were washed twice with deionized water (2 X 2.5 L).
The
organic layer was isolated and concentrated under reduced pressure to a
residue. To
this residue was added isopropanol ( 1.66 L) and heated to produce a solution
followed
by the slow addition of hexanes ( 1.66 L). The suspension was cooled to 5
°C over 30
minutes, and held at 0 to 5 °C for two hours. The product was filtered
and washed with
14
CA 02444210 2003-10-09
WO 98/34584 PCT/L1S98/02749
50-50 isopropanol-hexanes (840 mL) mixture at 0 °C. The product was
dried to afford
4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)cyclohexan-1-one (3I5 g, 56 % from
the pimelate).
Example 7
Preparation of cis-(+1-)-6-f3-(cyclo~entyloxX)-4-methoxyphen, l~~l-1-
oxobic clo(2.5]octane-2.6-dicarbonitrile.
A 5 liter round bottom flask equipped with an overhead stirrer, internal
thermometer, and a nitrogen inlet was flushed with nitrogen. The flask was
charged
with SO% potassium hydroxide (220 g) and tetrahydrofuran (550 mL). While
stirring at
room temperature, benzyltriethylammonium chloride (8.1 g, 0.035 mol, 0.05
equivalent) was added. The solution was cooled to 0 °C. To a pressure
equalizing
addition funnel was charged a solution containing tetrahydrofuran (550 mL), 4-
cyano-
4-{3-cyclopentyloxy-4-methoxyphenyl) cyclohexan-1-one (230 g, 0.73 mol, 1.0
equivalent), and chloroacetonitrile (59 g, 0.78 mol, I.07 equivalent) at room
temperature. While stirring the flasks contents at 0 °C, the solution
in the pressure
addition funnel was added over 15 minutes. The temperature was maintained
between
0 and 5 °C, and stirred for one hour. The reaction was warmed to 25
°C, diluted with
water (900 mL), and ethyl acetate (900 mL). The solution was stirred and
allowed to
settle for 30 minutes. The layers were separated, the organic layer was
isolated, and
concentrated by vacuum distillation to a residue. Methanol (540 mL) was added
and
the solution was heated to 40 °C. While cooling to 20 ° C over
90 minutes, hexanes
(540 mL) was added. Cooling was continued, and the product began to
crystallize at 10
°C. The suspension was then cooled to -5 °C and held at -5-0
°C for two hours. The
product was filtered and washed with a SO-50 methanol-hexanes mixture (300 mL)
at 0
°C. The product was dried to afford cis-(+/-)-6-[3-(cyclopentyloxy)-4-
methoxyphenyl]-
I-oxobicyclo[2.5]octane-2,6-dicarbonitrile (190 g, 73 %) as a white
crystalline solid.
Exam 1p a 8
Preparation of 1-9a
In a stoppered 12 dram screw-top vial was added diglyme (5.92 g) and the
epoxy nitrite (0.70 g, leq) of Example 7. This mixture was stirred with
heating in an
oil bath for 5 min. Then MgBr2~6H20 (0.906 g, 1.55 eq) was added. After 3 hr
no
starting material was detected. The reaction mixture was cooled, then mixed
with 5%
aqueous citric acid/ethyl acetate and the layers shaken and separated. The
second
extraction with ether/ethyl acetate gave some color extracted into organic
layer; but the
next extraction had no color. The organic fractions were combined and washed
with
water and brine and dried with MgS04. The product was crystallized from
hexane; mp
151-152 °C.
CA 02444210 2003-10-09
WO 98/34584 PGT/US98102749
Elemental analysis: C - 58.20, H - 5,82, Br - 18.44, N - 6.46; found C -
58.32, H
- 5.73, Br - 18.48 , N - 6.34. The structure was confirmed by X-ray structure
determination of a crystalline sample obtained from methyl alcohol.
EXample 9
Preparation of I-9b
To Mg (0.189 g, 2.02 eq) (polished with mortar and pestle) in ether was added
1,2-dibromoethane (1.55 g, 2.06 eq) in a small volume of ether to initiate the
Grignard.
When most of the magnesium was consumed and no more evolution of ethane was
observed, the reaction was stirred for an additional 0.5 hr at room
temperature after
which was added the epoxide of Example 7 (1.418; I eq) in a minimal amount of
dry
tetrahydrofuran at ambient temperature. After about 70 hr at room temperature
there
was obtained both the bromo cyanoalcohol ( 1-9b) and bromo cyanohydrin ( 1-9a)
in a
ratio of 6:1. The 1-9b product was isolated as an oil by prepartive HPLC. The
structure was confirmed by carbon and proton NMR.
Example 10
Preparation of Compound 3-1 - Epoxysulfone
To a 25m1 round-bottom flask equipped with a magnetic stir bar and a rubber
septum was charged 1.00 g of 4-cyano-4-(3-cyclopentyloxy-4-
methoxyphenyl)cyclohexan-1-one, 0.70 g of chloromethyl p-tolylsulfome, and 7
ml of
tetrahydrofuran. This was stirred, then 3 ml of 50% w/w aqueous NaOH and
benzyltrimeth ch (O.OSg) was added. This suspension was vigorously stirred for
2 hours
at room temperature. The reaction solution was transferred to a separatory
funnel to
which was added 50 ml of ethyl acetate and acidified with 6N HCI. The organic
layer
was retained, washed ZX with deionized water, dried with MgS04 and filtered to
2~ remove the salts.
Example 1 I
Preparation of Compound 3-2 - Bromoalde~de
To magnesium (0.048g, 1.03 eq, 0.021 mol; polished with mortar and pestle) in
ether was added 1,2-dibromoethane (0.40 g, 1.06 eq, 0.02 mol) under nitrogen.
Two
drops of iodine were added in ether to start the reaction, after which the
reaction was
heated gently. Once the Grignard had formed, the reaction flask was cooled to
about ~
°C and the epoxysulfone of Example 10 (0.93 g, 1 eq, 0.002 mol) was
added in
ether/methylene chloride. The reaction was followed via TLC (conditions:
silica gel
with cyclohexaneaoluene:acetonitcile:acetic acid 40:40:20:4). The reaction was
stirred
at 5° for 2 hours. The product was isolated by adding water and
ether/TBME to the
reaction mixture, and seperating the organic layers. These were washed with
water and
brine and dried over MgS04. Evaporation gave an oil which was flash _
chromatographed over 40 g of silica gel using a mixture of hexane and ethyl
acetate (5-
16
CA 02444210 2003-10-09
WO 98/34584 PCT/US98~2749
40% ethyl acetate). This gave a clear oil (0.49 g) containing about equal
proportions of
the equitorial and axial isomers as determined by proton NMR. Mass specrum
showed
a molecular ion at m/e 405 containing 1 bromine atom [C2pH24BrN03].
Example 12
Preparation of c-4-cyano-4-(3-cyclopentylox~4-methoxyphenyl)-r-
~clohexanecarboxylic acid.
A 5 liter round bottom flask equipped with an overhead stirrer, internal
thermometer> and a reflux condensor equipped with a nitrogen inlet was flushed
with
nitrogen. The flask was charged with dimethylformamide (580 g), acetonitrile
(480 g),
lithium bromide (72 g, 0.83 mol, 1.62 equivalents) and deionized water (20 g,
1.1 mol,
2.2 equivalents). The solution was stirred under nitrogen at 25 - 30 °C
followed by the
addition of cis-(+/-)-6-[3-(cyclopentyloxy)-4-methoxyphenyl]-1-
oxobicyclo[2.5]octane-
2,6-dicarbonitrile ( 180 g, 0.51 mol, I .0 equivalent). The reactor was heated
to 90 - 95
°C for 8 hours or until deemed complete by the disappearance of epoxy
nitrite. The
1 ~ contents of the flask was cooled to 20 °C , followed by the
addition of a sodium
hydroxide solution (92 g of sodium hydroxide, 2.3 mot, 4.5 equivalents,
dissolved in
200 mL of deionized water). The suspension was stirred at 20 °C for 30
minutes
followed by the addition of sodium hypochlorite (600 mL, 0.46 mot, 0.9
equivalents).
The contents of the flask were stirred for 90 minutes followed by the addition
of t-butyl
methyl ether ( 2.27 kg) and 6N HCl (644 mL, 3.86 mot, 7.5 equivalents). The
bottom
aqueous layer was back-extracted with t-butyl methyl ether (454 g), and the
combined
organic layer was washed four times with deionized water (4 X 800 mL). The
organic
layer was concentrated to a residue. To the flask was charged ethyl acetate
(900 g) and
heated to reflux. The contents of the flask was cooled to 50 °C
followed by the
addition of hexanes (672 g). The contents of the flask were cooled to 0
°C and held for
1 hour. The product was filtered and washed with cold ethyl acetate / hexanes
( 1/9, 175
g). The product was dried to afford c-4-cyano-4-(3-cyclopentyloxy-4-
methoxyphenyl)-
r-cyclohexanecarboxylic acid (125 g, 69 %) as an off white powder.
Example 13
Preparation of c-4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)-r-
c"r~clohexanecarbox,~c acid chloride
In a 1 neck flask equipped for nitrogen flow was combined c-4-cyano-4-(3-
cyclopentyloxy-4-methoxyphenyl)-r-cyclohexanecarboxylic acid (1.3728, I eq,
0.004
mot) and oxalyl chloride (4.5688, 9eq, 0.036 mot). One drop of dimethyl
formamide
was then added. This mixture was stirred at ambient temperature overnight.
After
evaporation under high vacuum this yielded the captioned product.
Example 14
17
CA 02444210 2003-10-09
VVO 98134584 PGT/US98I02749
Preparation of Form 1-l0a - 4-cyano-4-(3-cyclo~tyloxv-4-methox~ohenyl)-r-
cyclohexane acvl nitrite
In a flask a sample of the acid chloride (0.217 g, 0.006 mot, 1 eq) prepared
in
Example l3 was dissolved in CDC13 (2.34 mL). To this was solution (cooled to
5°C)
S was added trimethylsilyl cyanide (0.07g, 1.3 eq, 0.008 mot.) and a catalytic
amount of
ZnI2 (0.004g). This solution was refluxed overnight. This yielded 0.211g of
the
captioned product. IR: COCN, v 2220 cm-t ; C=O, v 1720cm- l . The isomeric
purity of
1-l0a was determined by hydrolyzing the acyl nitrite in warm water, the
product being
essentially pure compound 1-l la.
Example 15
Preparation of Form 1-lOb of 4-cyano-4-(3-cvcl~entyloxy-4-methoxYphenyl)-
r-cyclohexane acvl nitrite
In an experiment analogous to Example 14, the axial carboxylic acid was
convened to the acid chloride using oxalyl chloride and catalytic amount of
dimethyl
formamide. This acid chloride was convened directly to the cozresponding acyl
nitrite,
1-tOb, the isomer of the compound prepared in Example 14. The isomeric purity
was
assayed by hydrolyzing the acyl nitrite by stirring it in warm water for 20
hours.
Analytical HPLC determination showed that > 96% of the product had the form of
1-
l Ob.
18