Note: Descriptions are shown in the official language in which they were submitted.
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
METHOD FOR TREATMENT OF TUMORS USING
NORDIHYDROGUAIARETIC ACID DERIVATIVES
The invention described and claimed herein was made in part with funds
from Grant No. AI 32301 from the National Institutes of Health and U.S. Army
Medical Research Grant DAMD 17-93-C3122. The U.S. Government has certain
rights in the invention.
BACKGROUND OF THE INVENTION
1. Field of the Invention
The invention relates to the use of nordihydroguaiaretic acid
derivatives, in particular derivatives containing substituents of naturally
occuring amino acids, for the treatment of tumors and viral infections.
2. Background Information .
Carcinogenesis is a multistage event affected by a vaxiety of genetic
and epigenetic factors and is typified by the outbreak of wcontrolled cell
growth originated from different tissues. A universal goal for anticancer
research lies in the development of a clinical treatment that is highly
effective
in curtailment of tumor growth, non-toxic to the host, and is affordable for
most patients. Drugs that focus on the inhibition of targets that are unique
to
dividing cells should be effective chemotherapeutic agents without the risk of
substantial side effects.
Cells pass through many checkpoints as they proceed through the cell
cycle. Certain criteria must be met in order to pass each of these
checkpoints. In the G2/M transition, the most essential regulator is the
cyclin-
dependent l~inase CDC2. This lcinase binds tightly to the regulatory protein
cyclin B, and this complex, also called the maturation promoting factor (MPF),
is responsible for stimulating a myriad of events that lead to the cell's
entry
into early prophase (1): Not surprisingly, the loss or deactivation of either
component of the MPF will block cellular progression out of G2.
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
The expression and activity of the MPF is regulated at different levels.
Cyclin B protein levels slowly rise through the Gl and S phases of the cell
cycle, peak during the G2 to M phase transition, and drop sharply during
mitosis (2). The CDC2 protein, on the other hand, is always present during
the cell cycle, although levels rise slightly in the last stages of the G2
phase
(3). The activity of the protein is dependent on the association with the
appropriate cyclin, as well as on the dephosphorylation of its inhibitory
sites
by the phosphatase CDC25C (4,5). It has been shown that the failure of this
dephosphorylation initiates G2 arrest in response to DNA damage by radiation
or
chemical action. Recent evidence also suggests that any remaining active CDC2
may be transported outside the nucleus following DNA damage (6).
A number of naturally occurring derivatives of the plant lignan
nordihydroguaiaretic acid (NDGA) have been shown to block viral replication
through the inhibition of viral transcription. This earlier worle has shown
that
NDGA derivates, originally isolated from La~y~ea Tridehtata and subsequently
synthesized chemically, can inhibit the production of HIV (7,8), HSV (9), and
HPV transcripts (10) by the deactivation of their Spl-dependent promoters.
Unexpectedly, one of these derivatives, tetra-O-methyl NDGA, appears to
also induce cell cycle arrest in mammalian cell lines. The evidence presented
hereinbelow demonstrates that M4N is capable of inducing G2 arrest in
mammalian cells without detected toxicity, and supports the view that this
arrest is due to the inhibition of the cyclin-dependent kinase CDC2.
Human papillomavirus (HPV) infection causes unregulated cell growth
in many types of squamous epithelial cells, resulting in afflictions ranging
from
benign pallilomae (warts) to cervical, penile and mouth cancer. The strong
association of these cancers with HPV and the widespread occurrence of
infection
denotes the importance of developing an anti HPV therapy.
Most, if not all, viruses, including those replicatively active mutants, are
host dependent. They require the participation of certain cellular factors for
supporting viral growth. Host cellular factors, unlike viral proteins, are not
under mutational pressure and are in general, structurally invariable. Thus,
compounds that block the usage of these cellular factors at different stages
of
2
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
the viral life cycle are likely to be good candidates as mutation insensitive
antiviral drugs. Several studies using cellular factors as alternative targets
for
the inhibition of HIV-1 have been reviewed (11).
Applicants reported earlier that 3'-0 -methylated NDGA (i.e. Mal .4),
isolated from Creosote bush (La~~ea t~idehtata) can specifically block basal
HIV transcription, Tat-regulated transactivation, and HIV replication in human
cell culture(8, 12, 13). Mal.4 exerts its effects by interfering with the
binding
of transcription factor Sp 1 to the promoter of the HIV proviral template. The
target of Mal.4 is mapped to nucleotides -87 to -40, the Spl binding sites of
the HIV long terminal repeat (LTR). The unmodified NDGA, iya vitro, does not
inhibit HIV transcription and has no effect on Spl binding (8).
Isolation and purification of plant lignans, however, is labor intensive
and costly. In anticipation of the possible clinical use of plant lignans in
controlling Spl-regulated viral and tumor growth in humans, nine different
methylated NDGA activities were synthesized chemically using unmethylated
NDGA as the parent substrate in large quantities with low cost (7). At drug
concentrations below 30 ~.M, tetra-O-methyl NDGA was found to be most
effective in the control of replication HIV via inhibition of Sp 1 regulated
proviral transcription and transactivation (7). This study has since been
extended to the control of the growth of Herpes simplex virus (HSV-1 and
HSV-2) (9). Herpes simplex immediate early (IE) ICP4 gene is essential for
HSV replication (14). Its promoter region possesses eight Spl consensus
binding sites (15), five of which are required for ICP4 gene expression. It
thus
makes the ICP4 gene a good candidate for such testing. Applicants have found
that both 3-O-methyl NDGA (Mal. 4) and tetra-O-methyl NDGA (M4N)
are effective transcriptional inhibitors for HSV ICP4 gene expression in Vero
cells via the blocking of Sp 1 protein binding to the ICP4 promoter as shown
by the electrophoretic mobility shift assay (9).
When the anti-HSV activities of M4N and Mal. 4 were tested and
compared to that of acycloguanosine (acyclovir, ACV) in infected Vero cells,
Applicants observed that the ICSO for M4N varied between 11.7 ~,M to 4 p,M for
10 passages of HSV-1 and 4 passages of HSV-2 without obvious uprising
3
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
trend for requirement of higher drug concentration. However, the ICso for
ACV increased from 7 ~,M for the first viral passage to 444 ~.M for the tenth
passage of HSV-1 and to >88 ~M for the fourth passage of HSV-2 indicating
their rapid build-up of drug resistance against ACV in Vero cells.
S Consequently, while the selective index, S.I. (TCSO/ICSO) remained
relatively
stable for M4N, the S.I. for ACV dropped 60 fold following the viral passages
in
Vero cells (9). Thus M4N is a mutation insensitive drug. It can inhibit ACV
resistant HSV effectively (9).
Due to the fact that Spl is an important cellular transcription factor
(16), the possible inhibitory effect of this class of compounds on the
expression of Spl-regulated cellular genes should be addressed. Mal.4
cannot displace Spl once it is stably bound to its binding sites (8). It
therefore seemed likely that NDGA derivatives would have a greater effect on
Spl-regulated genes in proliferating cells than on the expression of
SpI-regulated housekeeping genes in stationary cells. In the former case,
the drug will be able to compete with Spl protein for the Spl sites in gene
promoters during DNA synthesis, while in the latter case, the drug may have
little effect on the transcribing chromatin of housekeeping genes with Spl
protein already stably bound at their promoters. This, in fact, has been shown
to be the case. As will be demonstrated below, by using gene array studies
with 9600 expressed genes, Applicants found products of most Spl regulated
genes remained at similar levels, and not affected by the drug treatment of
cervical cancer cells C3 in culture (Figure S). Even so, the relatively low
selective index of M4N certainly limits its use to the lowest effective
concentration if the drug must be used systemically. On the other hand,
human papilloma virus induces solid cervical and oral tumors initially through
the Spl regulated expression of HPV E6E7 genes (17). Applicants reasoned
that if drug can be delivered ih situ, and be kept only in the tumor area, the
drugs of high concentration may be used to effectively destroy the tumor with
little damage to the patients.
Survivin is an inhibitor of apoptosis that is abundantly expressed in many
human cancers (35), but not in normal adult human tissue, and is considered a
4
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
possible modulator of the terminal effector phase of cell death/survival.
(36).
Survivin is expressed in G2-M in a cell cycle-dependent manner, binding
directly
to mitotic spindle microtubules. It appears that survivin phosphorylation on
Thr34
may be required to maintain cell viability at cell division (37), and
expression of a
phosphorylation-defective survivin mutant has been shown to trigger apoptosis
in
several human melanoma cell lines (38). Phosphorylated survivin acts on the
caspase pathway to suppress the formation of caspase-3 and caspase-9, thereby
inhibiting apoptosis. (Ref. 39, page 10 presents an outline of apoptosis
signalling
pathways.) Thus, compounds that reduce the expression of survivin will be
expected to increase the rate of apoptosis and cell death. CDC-2 has been
shown
to be necessary for survivin phosphorylation (37).
SUMMARY OF THE INVENTION
Accordingly, it is one object of the invention to provide compounds and
compositions for use in the treatment of cancerous and noncancerous tumors
in animals, particularly in mammals, and most particularly in humans.
According to this aspect of the invention, novel nordihydroguaiaretic acid
derivatives are provided that inhibit tumor growth.
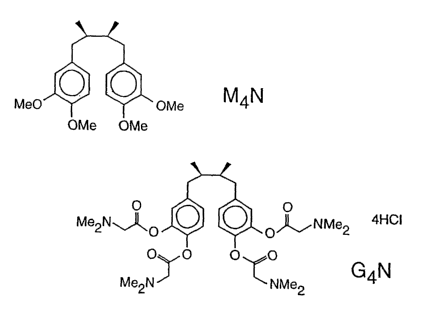
By nordihydroguaiaxetic acid derivatives is meant compounds of the
structure
. CH3 / ~ .R3
Rl /' ( \ .R4
RZ \ G'~g
(I)
wherein R1, RZ, R3 and R4 independently represent -OH, -OCH3,
-O(C=O)CH3, or an amino acid residue, but are not each -OH
simultaneously. Amino acid substituents are intended to include, inter alia,
alanine, arginine, asparagine, aspartate, cysteine, glutamate, gluamine,
glycine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine,
5
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
proline, serine, threonine, tryptophan, tyrosine, valine, 5-hydroxylysine, 4-
hydroxyproline, thyroxine, 3-methyllustidine, s-N-methyllysine, E-N,N,N-
trimethyllysine, aminoadipic acid, y-carboxyglutamic acid, phosphoserine,
phosphothreonine, phosphotyrosine, N-methylarginine, and N-acetyllysine.
Particularly preferred compounds for use according to the invention are
M4N and G4N, which are shown in Figure 1.
It is a further object of the invention to provide a method for treating
cancerous and noncancerous tumors by the use of these novel derivatives,
and by similar derivatives that are known in the art, but have not heretofore
been used for the treatment of tumors. The method should be especially
effective against rapidly proliferating cell types containing the cyclin
dependent l~inase CDC2. It is a further obj ect of the invention to provide a
method of inlubiting CDC2 in a eukaryotic cell cycle, particularly in an
animal
cell, more particularly in a mammalian cell, and most particularly in a human
cell.
Tumors to be treated include any tumor that is sensitive to the above-
mentioned compounds used according to the methods of the invention. In
particular, this includes rapidly dividing cancerous and benign tumors that
are
sensitive to inhibition of the cyclin-dependent kinase CDC2 cycle.
The term "cancerous tumor" is intended to include any malignant tumor
that may or may not have undergone metastasis. The term "noncancerous
tumor" is intended to include any benign tumor. These terms are used as
customarily understood by persons of skill in the art.
Examples of benign and malignant tumors which may be treated by the
compositions and methods of the invention can be found in Table 1-1 of Cancer
Biology (Raymond W. Ruddon, Cancer Biology, 3rd Ed., Oxford Univ.
Press, 1995, incorporated herein by reference). Tumors to be treated include
those that are known to be of viral origin, as well as those that are not of
viral
origin. The compositions and methods of the invention are expected to be
particularly useful in the treatment of solid tumors.
It is yet another object of the invention to provide a method of inhibiting
the cyclin-dependent kinase CDC2 cycle. This method will be useful in
6
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
inhibiting cell proliferation, particularly in rapidly dividing cell types.
In a preferred embodiment, the compounds and compositions
described herein are used in the treatment of HPV-induced tumors. HPV
induced tumors include in particular, but are not limited to, cervical, oral,
S penile and head and necl~ cancers that are associated with HPV infection.
The method comprises local application of nordihydroguaiaretic acid
derivatives, in particular tetra-O-methyinordihydroguaiaretic acid (M4N) and
tetraglycinal nordihydroguaiaretic acid (G4N), to cancerous and non-
cancerous HPV-induced tumors.
It is yet another object of the invention to provide a method of inhibiting
viral replication and growth by the administration of the compounds of formula
I containing amino acid substituents. Preferred for use in this method are
compounds in which the amino acid substituents Rl, R2 R3 and R4 are identical.
It is a further object of the invention to provide a method of inhibiting
1 S survivin production in a eukaryotic cell cycle in a cell that expresses
survivin,
particularly in a cancer cell. The inventors have found that the
nordihydroguaiaretic acid derivatives of the invention downregulate survivin
mRNA and protein levels and activate both CDC-2 and the caspase pathway,
thereby increasing the level of apoptosis in cell populations where survivin
is
expressed. This method should provide a treatment for cancers where survivin
is
expressed by suppressing or eliminating survivin expression, thereby
increasing
the rate of apoptosis.
It is contemplated that M4N, G4N and other derivatives will be
administered by local inj ection into the tumors, generally along with
pharmaceutically acceptable diluents, excipients and Garners. In preferred
embodiments, M4N is injected into tumors in the form of a DMSO solution,
and G4N is administered in PBS solution. The use of G4N will complement
the use of M4N, particularly in larger tumors (> 2 cm3), due to its water
solubility, which allows it to spread to a larger region of the tumor. Other
water-soluble and water-insoluble nordihydroguaiaretic acid derivatives can
be similarly employed, according to the invention. These may also be
employed in lipid based formulations for systemic delivery, as l~nown and
7
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
used in the art.
By pharmaceutically acceptable diluents, excipients and Garners is
meant such compounds as will be known to persons of skill in the art as being
compatible with M4N, G4N and other similar derivatives and suitable for local
administration to a human or other mammal according to the invention.
Although the examples hereinbelow describe administration by means of local
injection, other means of local administration, such as topical application or
targeted delivery to the tumor site, may also be used.
The amount of compound administered to obtain the desired treatment
effect will vary but can be readily determined by persons of skill in the art.
The amount of dosage, frequency of administration, and length of treatment
are dependent on the circumstances, primarily on the size and type of tumor.
However, dosages of from 10 mg to 20 mg of either M4N alone or with similar
amounts of G4N per gram tumor weight at intervals from daily to weelcly or
less frequently may be mentioned for purposes of illustration. Administration
of 50 ~,1 to 100 p.1 of M4N dissolved in DMSO at a concentration of 200 mg/ml,
either alone or in combination with G4N, is expected to be effective in many
cases for tumors of 1-1.5 cm3.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1. Structures of M4N and G4N
Figure 2A. HPV-16 LCR showing region of E6/E7 promoter (pPV16P97) and the
binding site for Spl protein.
Figure 2B. The effect of M4N on the E6/E7 promoter activity in C-33A cells.
(Inhibition of E6/E7 promoter driven luciferase gene transcription by
different
concentration of M4N)
Figures 3A-3C. Inhibition of Viral E6 and E7 RNA Transcripts by 40 ~.M
M4N. Total RNA isolated from C3 cells treated with either 40 ~,M M4N or
8
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
DMSO alone in growth media for 71 hours was subjected to relative RTPCR.
The RTPCR samples were removed after increasing cycles of amplification
and resolved on an agarose gel. The gel photographs (3A and 3B) indicate
these cycles, the presence of (+) or absence (-) of M4N in the growth media,
and two digests of a pGMT vector used as size markers. The amplification
map (2C) indicates the two expected size products of the amplification,
resulting from the alternate splicing of the early viral RNA transcript.
Figure 4A. Inhibition of C3 Cell Growth by M4N.
Figure 4B. Inhibition of C3 Cell Growth Following the Removal of M4N.
Figures 5A-5B. Effect of M4N on gene expression in C3 cells as examined by
the GENE Assay analysis. 5A. GENE expressed in C3 cells after > 2 hours of
DMSO treatment (C3 DMSO). 5B. GENE expressed in C3 cells after > 2 hours of
M4N treatment using DMSO as solvent (C3 M4N).
Figures 6A-6B. Visual observations of tumor-bearing mice following M4N
treatment. 6A. Mice bearing single tumors were treated with in situ injection
of DMSO (#3) or M4N (#7). Ih situ injection of M4N was also made to one of
the two tumors grown in mouse #9. 6B. M4N treated tumor (white scar) with
untreated tumor from the same mouse, #9 as described in Table 2.
Figure 7. Histopathology Effect of M4N and M4N/G4N on Tumor Growth in
Mice. First column from the panel presents the large size of tumors from
mouse #4, 10, 12, following DMSO treatment (CON) as compared to the
relatively small drug treated (M4N or M4N/G4N) lesions from mouse #12, 10,
27 and 20 (M4N). The subsequent photographs are examples of these tumors
examined at 100X magnification (A, B, C, DMSO treated, D untreated, E, F,
G, H, M4N or M4N/G4N treated) mice (Table 1 and Table 2).
Figure 8. HSV-1 replication in the absence of drugs (HSV-C, HSV-SC), in the
9
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
presence of ineffective drugs (ABDS1 ["HSV-ABDS1"], ABDSZ ["HSV-
ABDS2"])and in the presence of effective drugs (M4N ["HSV-4N"] and ACV
["HSV-ACV"]).
Figure 9. M4N Causes Growth Arrest in Mammalian Cells. (a-d) C3,
CEM-T4, C33a, and TC-1 cells were treated with different concentrations of
M4N. The number of cells present at the initiation of the experiment is
indicated as Day 0. After three days the number of viable cells were counted
and plotted versus the M4N concentration. (e) C3 cells were split into T-25
flaslcs with 5 X 103 cells per flask and given either M4N in I% DMSO in media
or 1% DMSO in media alone (first media change). After 3 days, one-half of
the M4N treated cells were given fresh media containing only 1% DMSO (1VI-
D), while the rest of the cells were given fresh media with the same
conditions
(second media change). The cells were counted daily and plotted versus the
time of treatment.
Figure 10. Cells Treated With M4N Arrest in G2/M. C3 cells (a), C33a
cells (b), CEM-T4 cells (c), and TC1 cells (d) were grown for three days in
media containing either 1% DMSO or 1% DMSO with M4N (M4N). The cells
were trypsinized, fixed with ethanol, stained with propidium iodide, and were
subsequently analyzed by flow cytomrnetry. The data is displayed as number
of cells (3-5 X 104 total cells) versus propidium iodide stain intensity. The
indicated stages of the cell cycle are labeled and correspond to the relative
cellular DNA compliment as determined by staining intensity.
Figure 11. C3 Cells Treated With 40~.M M4N Demonstrate G2 Cell Structures. C3
cells were grown on coverslips for three days in media containing either 1
DMSO (Control) or I% DMSO with 40q.M M4N (M4N). Samples were fixed with
ethanol and incubated with antibodies against a (green) and y (orange) tubulin
(a)
or with the DAPI DNA stain (b). Cells were examined by fluorescence
microscopy.
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
Figure 12. CDC2 and Viral Oncogenes are Reduced by M4N. C3 cells
were grown for different amounts of time (numbers are in hours) in media
containing either 1 % DMSO (D) or 1 % DMSO with 40 ~cM M4N (M). After the
specified times, total protein or total RNA was isolated from the cells.
S Western blots (a - top two panels) were performed using antibodies against
CDC2 or cyclin B with the same nitrocellulose filter. Kinase assays (a -
bottom two panels) were performed, following immunoprecipitation with
antibodies to cyclin B, by incubation with y-32P ATP and histone H1. The
coomassie stain of the PAGE gel is included as control for loading. Kinase
assays for 24 and 72 hour drug treatments were performed separately.
Northern blots (b) were performed on total RNA extracts. Filters were
incubated overnight with random-primed 32P-labeled DNA for CDC2 or GAPDH,
washed, and exposed to film for three days. The same filter was used to test
CDC2
and GAPDH RNA. rtPCR analysis (c) was performed on total RNA extracts with
primers hybridizing to regions within either HPV-16 E7 or GAPDH. Both primer
pairs were used in the same reactions, and the products were analyzed by
agarose
gel electrophoresis.
Figure 13. Electrophoretic mobility shift assay (EMSA) of G4N interaction with
the HIV Spl-binding sites (-87 to -49). (A) G4N inhibition of Sp1-167D binding
to 32P labeled HIV Spl DNA template. Lane 1, template alone; lane 2,
template plus 0.1 ~,g Sp1-167D; lanes 3-9, template incubated with increasing
concentrations of G4N (0.25 to 1.75 mM prior to the addition of 0.1 ~,g Sp1-
167D. (B) G4N displacement of Spl-167D bound to HIV template. Lane 1,
template alone; lane 2, template plus 0.1 ~,g Spl-167D plus 100-fold excess of
unlabeled template; lane 3, template plus 0.1 ~,g Spl-167D; lanes 4-10,
Spl/DNA complex challenged with increasing concentrations of G4N (0.25 to
1.75 mM); lane 11, template incubated in reaction buffer containing 1.75 mM
G4N. (C) Spl-167D displacement of G4N bound to template. Lane 1, template
alone; lanes 2-4, template plus increasing amounts of Spl-167D (0.075,
0.150, 0.300 ~.g); lanes 5-8, template incubated in reaction buffer containing
11
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
1.2 mM G4N followed by challenge with increasing amounts of Spl-167D
(0.075, 0.150, 0.300 ~.g), lane 8 received no Spl-167D. (D) Plot of
diminishing Spl-167D/DNA complex band intensities in response to
increasing concentrations of G4N used in (A) ---~--- and (B) -~-. The gels
used were 5% non-denaturing polyacrylamide with each lane receiving 5 p,1 of
each reaction volume as described in experimental section and Ref. [1].
Figure 14. Inhibition of HIV Tat-regulated transactivation in Cos cells by
G4N.
Figure 15. SIV production with presence of G4N. 107 174 x cells, a human T-
cell
lymphoma cell line, were mixed with a 24 hrs. harvest stock of SIV mac 239 (4
ng
of p27) for two hours at 37. Cells were resuspended and 1 x 105 cells in 100
~.1
medium were added to each well of three 96-well plates. Various concentrations
of
G4N from a freshly made stoclc were prepared and added to each of the six
designed well. Culture supernatants were collected after four and eight days
for
viral production analysis. Viral production was assayed by a modified p27
capsid
protein antigen capture ELISA as described in experimental section.
Figure 16. Inhibition of HIV p24 antigen production in H9 cells by G4N.
Inhibition in percentage was calculated by comparing p24 level from an
average of two duplicate cultures of G4N treated and not treated H9 cells 9
days following viral infection with a AZT resistant HIV strain, HIV-1RTMF.
Figure 17. RT-PCR Analysis of Survivin Gene Expression. (a) Top: Survivin
gene expression in C3 cells treated with 40 ,uM M4N for 24 hours and 72 hours,
respectively (lanes 3 and 4) and in the untreated controls (lanes 1 and 2).
Bottom:
the corresponding GAPDH controls. Band intensities were quantitated with Scion
Image. (b) Survivin RT-PCR product signals were normalized to those of the
GAPDH controls and plotted.
Figure 18. Drug concentration-dependent down-regulation of survivin protein.
(a)
C3 cells were incubated with various concentrations of M4N for 72 hours and
the
12
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
total cell lysate was immunoblotted against survivin. (b) Relative band
intensities
were quantitated by Scion Image and plotted against M4N concentration.
Figure 19. Iminunoblot analysis of caspase-3 cleavage in C3 cells treated with
M4N for 72 hours. (a) Western blot of caspase-3 showed cleavage of the 32 KD
procaspase-3 and the formation of the active 20 KD cleaved product. (b) Band
intensities were quantitated and plotted against M4N concentration.
DETAILED DESCRIPTION OF THE INVENTION
Experimental Methods
NDGA derivatives were synthesized chemically (7). Cell line C3 is a
HPV16E + L plus activated Ras transformed cell line of C57 BL/61~h origin
provided by W. Martin Kast of Loyola University Medical Center, Chicago,
Illinois, U.S.A. It is maintained and cultivated as described by Greenstone et
Al. (18) and Feltl~amp et al. (19, 20).
Synthesis of G4N:
Standard Procedure for the Preparation of ~Zeso-1,4-Bis [3,4 -
(dimethylaminoacetoxy) phenyl]- (2R,3S)- dimethylbutane Hydrochloride Salt
Tetraglycinyl NDGA, G4N. To a dichloromethane (250 ml) solution containing
NDGA (12.8 g, 42.3 mmol, 1.0 equiv) and N,N, -dimethylglycine (26.2 g, 254
mmol, 6.0 equiv) were added DCC (52.4 g, 254 mmol, 6.0 equiv) and DMAP
(2.32 g, 18.9 mmol, 1.0 equiv). The reaction mixture was stirred for 24 h
under nitrogen at room temperature. After the reaction mixture was filtered,
the solution was concentrated under reduced pressure. Acetone (250 ml)
was then added into the reaction flaslc and the solution was bubbled with
excess HCl(g). The water-soluble precipitate was dissolved in H20 and re-
precipitated twice at room temperature from acetone to give (1) (29, 2 g, 36.8
mmol) as a white solid in 87% yield. Proton NMR spectra were obtained on a
Varian Unity-400 (400 MHz) spectrometer by use of D20 solvent and TSP as
Internal standard. Carbon-13 NMR spectra were obtained on a Varian Unity-
13
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
400 (400 Mhz) spectometer by use of D20 as solvent. Carbon-13 chemical shifts
are referenced to the TSP singlet (80.0 ppm).
The synthesis is depicted in Scheme 1.
14
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
m
N
O °'
° °
O
U
x
z
N
p
N _
~O
U
x U t~
O
ON~
x
x
N
U .. ~ U
a
A ~ U
U
U
r; ~i A A
x
x
x
0
x
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
General Procedure. All reactions were carried out in oven-dried glassware
(120°C) under an atmosphere of nitrogen, unless as indicated otherwise.
Acetone, dichloromethane, 1,4-dioxane, ethyl acetate, hexanes, and
tetrahydrofuran were purchased from Mallinckrodt Chemical Co. Acetone was
dried with 4A molecular sieves and distilled. Dichloromethane, ethyl acetate,
and hexanes were dried and distilled from CaHz. 1,4 -Dioxane and
tetrahydrofuxan were dried by distillation from sodium and benzophenone
under an atmosphere of nitrogen. Nordihydroguaiaretic acid was purchased
from Fluka Chemical Co. N,N' -Dicyclohexylcarbodiimide (DCC),
4-dimethylaminopyridine (DMAP), morpholine, triethylamine, and potassium
carbonate were purchased from Merck Inc. 1 -Bromo-3-chloropropane, N,N
-dimethylglycine, and methylphosphorodichloridate were purchased from
Aldrich Chemical Co.
Analytical thin layer chromatography (TLC) was performed on precoated
plates (silica gel 60 F-254), purchased from Mercl~ Inc. Gas chromatographic
analyses were performed on a Hewlett-Packard 5890 Series II instrument
equipped with a 25-m cross-linked methyl silicone gum capillary column (0.32
mm i.d.). Nitrogen gas was used as a carrier gas and the flow rate was lcept
constant at 14.0 ml/min. The retention time (tR) was measured under the
following conditions: injector temperature 260°C, isothermal column
temperature 280°C. Gas chromatography and low resolution mass spectral
analyses were performed on a Hewlett-Packard 5890 Series II instrument
equipped with a Hewlett-Packard 5971A Mass Selective Detector and a
capillary HP-1 column. Separations by medium-pressure liquid
chromatography (MPLC) were performed at a flow rate of 120 ml/h by use of
a Jasco Model 880-PU intelligent HPLC pump. The MPLC packing material,
Reversed Phase Silica Gel C18 (particle size 0.035-0.070 mm), was purchased
from Knauer Co. Purification by gravity column chromatography was carned out
by use of Merely Reagents Silica Gel 60 (particle size 0.063-0.200 mm, 70-230
mesh ASTM).
Infrared (JR) spectra were measured on a Bomem Michelson Series
16
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
FT-IR spectrometer. The wave numbers reported are referenced to the
polystyrene 1601 cm' absorption. Absorption intensities are recorded by the
following abbreviations: s, strong; m, medium; w, weak. Proton NMR spectra
were obtained on a Varian Unity-400 (400 MHz) spectrometer by use of D20
as solvent and 3-(trimethylsilyl)propionic acid, sodium salt as internal
standard. Carbon-13 NMR spectra were obtained on a Varian Unity-400 (100
MHz) spectrometer by use of DZO as solvent. Carbon-13 chemical shifts are
referenced to the center of the 3-(trimethylsilyl)propionic acid, sodium salt
singlet (6 0.0 ppm). Multiplicities are recorded by the following
abbreviations:
s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; J. coupling
constant
(hertz). High-resolution mass spectra were obtained by means of a JEOL
JMS-HX110 mass spectrometer.
meso-l,4Bis[3,4(dimethyleminoacetoxy)phe 3S-dimethylbutane
Hydrochloride Salt (2). To a solution of NDGA (1, 12.81 g, 42.37 mmol, 1.0
equiv) and N,N-dimethylglycine (26.21 g, 254.2 mmol, 6.0 equiv) in
dichloromethane (250 ml) was added DCC (52.45 g, 254.2 mmol, 6.0 equiv)
and DMAP (5.176 g, 42.37 mmol, 1.0 equiv). The reaction mixture was stirred
for 24 h under nitrogen at room temperature. After dicyclohexylurea in the
reaction mixture was filtered off, the resultant solution was concentrated
under reduced pressure. Acetone (2S0 ml) was then added into the residue
and the resultant solution was bubbled with excess HCl (g). The precipitate
was dissolved in water and re-precipitated twice by use of acetone at room
temperature to give 2 (28.97 g, 36.86 mmol) as a white solid in 87% yield: 1H
NMR (D20, 400 MHz)8 0.78 (d, J = 6.0 Hz, 6 H. 2 x CH3), 1.73 (m, 2 H. 2 x
CH), 2.38 (dd, J=13.2, 9.6 Hz, 2 H. 2 x ArCH), 2.78 (dd, J = 13.2, 4.4 Hz, 2
H. 2 x ArCH), 3.03 (s, 24 H. 8 x CH3N), 4.53 (s, 8 H, 4 x CH2N), 7.22
(m, 4 H. 4 x ArH), 7.29 (d, J= 8.4 Hz, 2 H. 2 x ArH); 13C NMR(D20, 100 MHz)
8 18.11, 40.82, 41.73, 46.75, 59.59, 125.79, 126.58, 131.63, 140.66, 142.47,
146. 1 l, 167.84; IR (KBr) 3461 (br), 2963 (m), 1777 (s, C=O), 1620 (m), 1478
(m), 1377 (m), 1210(m), 1106 (m), 961 (w), 852 (w) cm 1; MS (FAB) of (2-4
HCl) m/z (relative intensity) 643 (M+, 30), 600 (20), 558 (43), 515 (20), 473
(42), 430 (13), 388 (26), 185 (18), 93 (38),58 (100), 44 (22); HRMS (FAB) of
17
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
(2 - 4 HCI) calcd for C34HsoNa.Os 642.3628, found 642.3614; Anal. Calcd for
C34Hs4N4O8C14: C, 51.78; H. 6.90; N. 7.10; O. 16.23. Found: C, 51.70; H. 6.85;
N.
7.05; O. 16.21.
It will be appreciated that by suitable substitution of other N,N-dimethyl-
substituted amino acids, additional amino acid substituted compounds of the
invention can be synthesized.
Example 1
Effect of M4N and several other NDGA derivatives of SP1-regulated HPV E6/E7
promoter activity.
The effect of M4N and several other NDGA derivatives of
SP1-regulated HPV E6E7 promoter activity was examined using luciferase as
a reporter. The assay depends upon DNA transfection of the HPV 16 LCR
(P97 promoter) fused to the luciferase reporter gene into C33A cells by
calcium phosphate methods. C33A is a cervical tumor cell line (ATCC
accession no. HTB-31) that does not contain any integrated HPV DNA, but
has transcription factors necessary for a robust expression of the HPV early
gene promoter. One day following DNA transfection various drug
concentrations dissolved with the help of dimethyl sulfoxide (DMSO) were
added to the cells. Thirty hours after drug treatment (so that the assay is
complete within the standard forty-eight hours for transient transfection
experiments), the cells were lysed and specific luciferase activity was
determined (Luciferase Assay Systems, Promega, U.S. Pat. No. 5,283,179).
As the M4N drug concentration was increased the specific luciferase activity
decreased.
The results (shown in Figure 2) demonstrate that M4N dramatically
reduces Spl regulated transcription initiation at the HPV E6/E7 promoter in
luciferase assay.
Example 2
Inhibition of E6/E7 mRNA synthesis following M4N treatment
Inhibition of E6/E7 mRNA synthesis following M4N treatment was
18
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
measured by RT-PCR in cervical cell line C3~ Relative RT-PCR was
performed with quantities of total cellular RNA standardized to the cell
numbers counted. The RT-PCR product was analyzed on a 2% agarose gel.
The results are shown in Fig. 3. The RT-PCR results indicated that the
amplified cDNAs of the expected size for E7 (321 bp) and E6 (204 bp) were
detected in the DMSO treated cells as early as cycle 22 of amplification.
These same products were barely detectable in the drug treated RNA
extracts following 30 cycles of amplification. No amplified products were
detected for the no template PCR control or from total RNA extracts of the
HPV16-negative C33a cell line.
Example 3 ,
Inhibition of cervical C3 cell growth by M4N treatment
HPV-16 transformed immortal mouse epithelial cells (C3 cells) were
plated at a density of 105 cells per vial. After 24 hours, %a of the vials
were
given growth media containing 40 ~,M M4N dissolved in 1% DMSO while the
other half were given growth media containing only 1 % DMSO. The results
are shown in Figure 4A. Within 24 hrs a difference in cell morphology
between drug treated and control C3 cells was observed. The growth and
division of the drug treated cells was markedly reduced in comparison to the
untreated control, while the fraction of viable cells compared to the total
cell
count remained constant for both drug treated and DMSO only control cells.
This indicates that M4N dramatically reduces cell division.
The effect on C3 growth following removal of M4N from the medium
was also examined. C3 cells were plated at a density of 104 cells per vial. At
time=0, 2/3 of the vials were given growth media supplemented with 40 ,uM
M4N in 1% DMSO. The remaining vials were given growth media containing
only 1% DMSO. After 73 hours, %2 of the vials that had received M4N in their
growth media were washed and media containing only 1% DMSO was added.
The othex 2/3 of the cell vials were washed and replaced with the same media
administered before. The results, shown in Figure 4B, indicate that the rate
of cell growth was not notably increased in M4N treated sample following the
19
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
change to drug-free media, indicating that M4N continues to significantly
reduce cell division even after its removal from the extracellular
environment.
Example 4
Analysis of cellular gene expression in C3 cells before and after 72 hrs of
drug treatment.
Gene expression with 9600 gene arrays was studied (Fig. S). Five
micrograms each of poly A+ RNA from 72 hrs. M4N (40 ,um) treated (C3 M4N)
and non-treated (C3 DMSO) was used in a pair of human 9600 gene array
hybridization study according to the procedure described in Genomics S 1,
313-324 1998. The hybridization image was captured by a color video
camera with a Nikon S5 mm AF micro Nilco lens and digitized by a Macintosh
LC630 computer. Such detection via enzyme substrate reaction of
color-forming enzymes in either single or dual-color mode is reproducible and
1 S extremely sensitive (can detect <S copies of transcript per cell with RNAs
from 107 cells).
The computer print outs showing differentially expressed genes (C3
M4N/C3 DMSO >10 and C3 DMSO/C3 M4N >10) were listed for examination.
Image files in TIFF format and data files in MS excel format are kept on ZIP
diskette. Gene names and clone m numbers are available for obtaining Image
clones for future northern blot confirmation.
Among a group of genes that are either up-regulated or
down-regulated 72 hrs after M4N treatment, the following are those
specifically related to cell division and apoptosis. Several other cell cycle
2S related genes axe also greatly upregulated in response to M4N. In addition
to
cyclin-dependent kinase CDC2 (Example 11), for example:
Increase
Cyclin-dependent kinase inhibitor (100X)
Apoptosis (APO-1) antigen (100X)
Death Domain Three DR3 (100X)
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
Ras-related protein R.AP-1 (60X)
Human Map I~inase (40X)
The following cell cycle related genes are greatly downregulated in response
to M4N:
Treated Untreated
Cyclin-dependent kinase 7 (5%) 100%
Human cytokine receptor (2%) 100%
Proliferating cell nuclear antigen,(1%) 100%
PCNA
Human TNF-related Apoptosis APOZ(3%) 100%
Cysteine protease (7%) 100%
At earlier time points, such as after one hour drug treatment, E6/E7
level was found to be similar with those in control cells while after 4.5 hrs,
E6/E7 were no longer detectable by RT-PCR (10). Gene expressions with
9600 gene arrays can be repeated with RNA isolated from these short-time
treated cells (1 hour and 5 hours) in order to further pin down the initial
cellular effects of the drug.
Example 5
Targeting C3 tumor growth in mice by local injection of M4N
Thirty six C57b1-16 NCR mice were injected with 5 x 105 C3 cells
between the shoulders on the backs of the mice. Twenty four of the mice
developed tumors within 20 days. Daily injection (50 ,u1-100 ~,l of M4N or
M4N/G4N) (200 mg/ml M4N in DMSQ, 200mg/ml G4N in PBS) showed
profound effect in tumor growth in animals, as shown in Tables 1 and 2, Fig. 6
and 7.
21
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
Table 1. M4N and G4N Effect on Growth of Single Tumors Developed
in Mice
Mouse Treatment Lesion Size (mm) Wt. Body
# Period Of wt.
Days 1-16 Day 1 Oay 7 Day Excised ay (g)
21 Lesion 1
(g) Oay
Day 16
16 Day
Oay 24
24
1 DMSO' 3x8x3.3 - 5x7x4 - 0_3 18.8- 20.2
2 OMSO 4.4x6x3.5 10x12x8 1.56 - 19.620.5 -
-
3 OMSO 0.8x0.8x1 - 10.5x11- 1.14 18.2- 16.1
x9
4 OMSO 2.8x3.8x2.5 - 18x11x9- 2.9 17.6- 202
Days 1-16
6 MaN - 9x8x5 - 0.2 - 19 19.2 -
7 MaN - 6x7x7 - - 0.1 18.2- 20.4
11 MaN 1 x1.3x1 9.5x10x9 - 0 19.5- 20.2
-
14 MaN 3.8x3.8x3.5 8x9x6 0.4 - 17 17.6 -
-
15 MaN - 5x4x4 - 0.1 - 18.920.0 -
16 MaN 2.8x2.8x2.8 9x6x4 0 - 17.217_6 -
-
17 MaN 2.3x2.3x2.3 6x6x4 0.2"x - 17.3- -
-
22
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
Days Oays
1-10 9-17
18 M4N GQN 3x2.8x3 8x7x5 - - 1 _0"' 18.8 - 21.1
19 MQN G,N - 5x5x5 - 0.2 - 18.2 19.9 -
21 MaN G4N 1.8x1.8x1.8 9x10x5 0.2 - 17.3 19.2 -
-
22 M4N G4N - 7x7x5 - - 0 17.9 - 19.5
27 MQN GIN 2.5x5x2.5 9x6x6 - - 1.8't' 20 - 20.7
28 MdN G,N 2.8x2.3x2.8 5x5x4 0.17 .- 18.1 19.8 -
-
29 M4N G4N 2.8x2.5x2.8 5x6x4 - 0.2 18.8 - 19.6
-
*DMSO = Vehicle for Drug
** Taken on Day 15
*** Lesion contained mostly necrotic cells as also found in lesions from
mouse 6, 7, 1 I, I4, I5, I7, 19, 21, 28, 19 (Fig. 6,7). There were no lesions
25 left in mouse #11 and #22 following drug treatments. Tumors found in
control
mouse #1, 2, 3, 4 contained growing cells (Fig. 2).
Experimental Procedures:
36 C57b1-16NCR mice were injected with 5 X 105 C3 cellslmouse.
30 Injections were 100 p.L made subcutaneously between the shoulders on the
backs of the mice. Cells were suspended in low-salt HBSS and suspension
uniformity was maintained by gentle vortexing.
24 mice developed tumors. Their lesion sizes were measured by dial
caliper. These mice were shaved, weight and treatment begun (Day 1). Four
35 mice were sequestered as controls. Control mice received 50 ~l DMSO
injected intratumorally daily. Experimental mice (10) received 50 p1 M4N
dissolved in DMSO(200mg/mL). An additional 10 mice received M4N
treatments for 8 days followed by G4N treatments (50 p1, 200mg/ml in PBS)
daily for 8 days. Injections were made to several regions of tumor. Mice
23
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
anesthetized
with
ether
or
metaphane
prior
to
injection.
Table
2.
M4N
and
G4N
Effect
on
Growth
of
Treated
Lesions
in
Mice
Carrying Multiple
Tumors
Mouse Treatment Body wt.
Period Lesion (g)
Size (mm)
Wt. Of Excised
Lesion (g)
Days 1-16 Day Day
Day 1 Day 1 24
7 Treated'
Not Treated'*
9 M4N 1.3x5x0.75 7x9x80.25 0.6 20.2 17.9
M4N 2.3x2.5x2.3 0.1 2.9 17.5 22.1
9.5x10x9
12 M4N 2.5x2.5x2.5 0.11 1.82 17.8 20.0
8x9x6
Oays Oays
1-9 10-18
M4N G4N 1.8x1.8x1.8 17 20.2
9x10x5 0.1
0.2
24 M4N G4N - 17.2 20.8
7x9x6 0 1.7
26 M4N G4N 5x3.3x2.5 19.3 20.6
7x7x7 0.2
1.9
*Drug
in
DMSO
was
injected
directly
to
the
tumor
regions
20 ** From adjacent
tumors deprived
of drug
Table
3.
Toxicity
Studies
of
G4N
in
Mice
Group # of Route Treatment Days of Morlalif~y
Mice per day Injection
9 '187.5mglkg 3 Subcutaneous 2X 6 0/3
2 375mglkg 3 Subcutaneous 1 X 6 0!3
3 750mglkg 4 Subcutaneous 1X 6 '(/4
4 375mglkg 2 IV 2X 6 0/2
CS'7BL-16NCR female mice from NCI were used in this experiment.
35 Tetraglycinal NDGA (G4N) was freshly made everyday in PBS in
concentrations of 75mg/ml. Injections of 0.05 ml for group 1, 0.1 ml for
groups
24
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
2 and 4, and 0.2 ml for group 3 per treatment were made for a period of 6
days. Experiments lasted seven days. Body weights were determined before
and after six days of injection. No significant weight changes were observed
during the experimental period.
All treated mice, controls (mouse numbers 1-4) and experimental mice
(mouse numbers 6, 7, 9, 10, 11, 12, 14, 15, 16, 17 M4N numbers 18-22, 24, 26-
29
M4N/G4N) exhibited swelling. Measurements of lesion sizes were made by dial
caliper. Some mice experienced mild bleeding due to injection.
The treatment regimen and results were as follows:
Day 10: Mice weighed again. All mice exhibited growth up to two grams.
Day 12: No treatments made.
Day 13: All mice have raised shin but to very different degrees. The slein
of one M4N treated mouse (#7) has split open through which the "dried-out
tumor" fell out.
Day 14: . Inj ection volume raised to 100 ~,1.
Day 15: One M4N treated mouse (#17) died due to overdose of
anesthesia/handling. The slcin at the lesion site of #17 cracked with the
"dried-out tumor" showing. It was dissected, and lesion excised and weighed.
Day
16: Four more MAN treated mice (#6, 14, 15, 16), three M4N/G4N treated mice
(#19,21,28) and one control mouse (#2) were euthanized, dissected and weighed.
Remaining control mice (#l, 3, 4) were examined non-invasively and were
carrying tumors.
Day 21: Tumor sizes from control mice were measured by dial caliper.
Observation: The skin at the lesion sites of mouse #10 and #12 (M4N treated
regions) cracked with the "dried-out tumor" showing.
Day 24: Mouse #7 skin recovered completely. The experiment was
terminated on this date. All remaining mice, M4N treated (#7, 9, 10, 11, 12)
and
M4N/G4N treated (#18, 20, 24, 26, 29) were euthanized, dissected, examined and
weighed.
The effects of M4N and M4N/G4N on C3 tiunor growth in mice are
summarized in Tables l and 2 and Figures 5 and 6. Table 1 shows the drug -
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
effect on C3 cell growth in mice carrying single tumors. The average weight
of four excised tumors of the control group was 1.48 g while weights of
lesions from M4N treated and M4N/G4N treated were 0.142 and 0.51 g
respectively. Drug treated lesions consisted mainly of dried out necrotic
cells
(Fig. 6). Tumors from the control group appeared homogenous and
contained actively growing cells. Table 2 shows the drug effect on C3 tumor
growth in mice carrying multiple tumors. In this study, drug was injected into
one of the tumors. The average weight of untreated tumors was 1.77 g while
that of M4N treated lesions was 0.15 g. Similar results were obtained
following M4N/G4N inj ection-the average weight of untreated tumors was 1.27
g, while that of the drug treated lesions was only 0.103 g.
The body weight changes of all mice during the entire experimental
period appeared insigW ficant (Table 1 and 2).
Example 6
Drug treated (M4N) and DMSO vehicle-treated or untreated tumors
(CON) from two groups of mice were prepared for histopathology
examination. The excised tumors were immediately fixed and then stored in
4% formaldehyde in phosphate buffered saline. The fixed tissue was then
dehydrated through a series of graded alcohols and xylene and embedded in
paraffin. The paraffin tissue blocks were thin sectioned and stained for
microscopy with hematoxylin and eosin. Histopathology studies showed that
the control tumors were unaffected by DMSO treatment and continued to
grow. They show the high nuclear/cytoplasmic ratio, pleomorphic nuclear
changes, high mitotic figures, spindle like sarcoma shape, and infiltration
into
the surrounding tissue characteristic of cancer cells. In contrast, those
tumors
receiving M4N treatment discontinued growth shortly after treatment began.
They
demonstrate sigiuficant necrosis and areno longer viable. There is a small
amount
of drug precipitate visible at higher magnification, and focal areas show
chronic
inflammation and fibrosis. This healing effect leads to the shedding of these
deceased tumor cells from the area. The same results are seen with M4N/G4N
treatment as with the M4N treatment alone. However, since G4N is water-
soluble, it
26
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
can spread to a larger area of the tumor than M4N. It is expected that G4N
when
used with M4N synergistically may be more effective in treating tumors of
large
sizes (i.e. greater than 2 cm3).
S Example 7
Effect of M4N on HSV-1 Shin Infection in Guinea pig
The drug M4N was also tested in inhibition of HSV-1 replication in skin
infections in guinea pigs. Guinea pig skin was pinched with needles and
HSV-1 suppression was applied topically to infect each pricked area. M4N
was then applied to the pricked infected area following infection daily for 6
days.
Six areas of bared baclc skin of a guinea pig were punched sterilely
with a 5=DIN needle. Two areas were infected with HSV-1 (HSV-C, culture
supernatant, or isolated HSV in saline, HSV-SC). The other four areas were
infected with HSV-SC. Fifteen minutes after infection, 30 ~ul of test
compounds (ABDS1, ABDS2, ACV and M4N (4N) in 60 mg/ml of DMSO were
applied to each punched infected region of an area, five times per day for six
days. ABDS1 and ABDS2 were included as negative controls. The
photograph in Figure 8 was talcen at day 6 and shows the extent of HSV-1
replication in the absence of drugs (HSV-C, HSV-SC), in the presence of
ineffective drugs (HSV-ABDS1, HSV-ABDS2) and in the presence of effective
drugs (HSV-M4N and HSV-ACV). It can be seen that six large confluent
blisters were developed in areas treated by HSV-C, HSV-SC, HSV-ABDS1,
HSV-ABDSZ, while no blisters were observed in infected areas following M4N
(4N) and ACV treatments. Clearcut results that M4N can block HSV replication
were obtained in this model system as shown by the disappearance of the skin
lesions and by no shedding of the virus 4 days after the drug treatment.
Initial
animal studies also showed M4N to be non-toxic to mice at concentrations as
high
as 300mg/kg when given intraperitoneally, and as high as 375 mg/kg when given
either subcutaneously or by IV (Table 3) (6).
27
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
Example 8
M4N for Clinical Treatment using ih situ Inj ection
Administration of M4N directly into tumors as a drug delivery route
provides several distinctive advantages. 1) M4N is a hydrophobic compound
and is exceedingly soluble in DMSO (200 mg/ml). Therefore only a small
volume of the drug solution is needed for injection in order to aclieve
effective dosage of the drug. In the mouse study described in Example 5,
above, daily injection of 50 ~,1 to 100 ~,1 for several days was sufficient to
completely stop tumor growth in mice. There have been several previous
studies on the use of large dosages (30 ml IV per treatment) of DMSO for
treating diseases (21). The results were not conclusive (22). However, since
tens of millions of people have been safely tested with large amounts of
DMSO worldwide in the past, it appears that DMSO should be safe as a
velicle for drug delivery when only small volume of it will be used (23).
2) By injection in situ, a majority of the drug residue remains insoluble and
concentrated in the tumor areas, and does not enter the circulatory system,
thus whole body toxicity is avoided. In addition, since enough drug remains
within the tumor to suppress its growth, continued injection of drug is
unnecessary after relatively few treatments. In the mouse study of Example
5, tumor cells continued to die even after discontinuation of M4N injections.
Thus when drug is directly targeted, tumor size becomes the determining
factor for the required amount of drug to be administered. The difference
between whole body weights of a human vs. a mouse becomes irrelevant. In
the mouse tumor studies, 20 mg/day for 10 days were more than sufficient to
eliminate tumors. There should be no reason to use a higher dosage than
this for treating a human tumor of comparable size (1-1.5 cm3). This should
reduce the risk considerably in human trials.
Example 9
M4N Treatment of Cells Blocks Cellular Proliferation
Our previous research on M4N indicated that it could inhibit viral
transcription by deactivation of Sp1-dependent promoters. Many mammalian
28
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
cell cycle genes also contain essential Sp 1 promoters and M4N may therefore
block their transcription. Tlus hypothesis was tested by examining the
antiproliferative effect of M4N on a munber of different cell lines. Low
concentrations (10 ~.M) of the parent compound, NDGA, have previously been
shown to induce apoptosis in mammalian cells (24). This effect, however,
can be circumvented by blocking one of the catechol oxygens or the addition
of a hydrophilic group to NDGA (25). Increasing amounts of the NDGA
derivative M4N were tested on cultures of the HPV-16/ras transformed C3 cell
line (26) to determine the optimal concentration required to inhibit
proliferation (Figure 9a). The cells respond well to M4N, ceasing division
after 72 hours over the range of concentration from 40 to 60 ACM. After three
days at these concentrations the number of cells remained equal to the count
at the initiation of treatment (day 0, Fig 9). A more modest reduction in cell
growth was observed at lower concentrations of the drug and some cell death
was seen at concentrations greater than 60 ~,M.
The antiproliferative effect of M4N on the C3 cell line is not solely due
to the drug's ability to deactivate the Spl-dependent HPV-16 E6/E7
oncogene promoter, as similar growth inhibition was observed in the HPV-16
transformed TC-1 cell line whose E6/E7 oncogenes are under control of a
non-Spl dependent retroviral promoter(27) (Figure 9d). In addition, growth
of the C33a cell line (Figure 9c), an HPV-negative human cervical cancer cell
line, and the CEM-T4 line (Figure 9b), a human leukemia cell line (28), was
also bloclced by treatment with M4N. In the four cell lines that were treated
with the drug, nearly all (>95%) of the arrested cells were viable until the
concentration of M4N exceeded a "threshold" value (60 ~.M for C3 cells, 40 ~.M
for TC-1 cells, etc.). Above these concentrations the percentage of viable
cells decreases precipitously. Interestingly, arrested cells maintained >95%
viability even after prolonged exposure to the drug. The C3 cells exhibited no
increase in cell death after eight days of treatment with 40 ~,M M4N (Figure
9e).
29
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
Example IO
Cells Treated With M4N Arrest in G2 Phase
Once it was established that cells treated with M4N cease proliferation
yet remain viable, analysis of cellular DNA content and fluorescence
examination of cell structures were used to determine the point in the cell
cycle where the cells arrest. Cells exposed to M4N for 72 hours demonstrated
increased G2/M DNA content relative to the controls (Figure l0a-d). The most
extreme responses were seen from the C3 and CEMT4 cell lines, in which >90% of
the cells show G2lM DNA content.
In order to distinguish between an arrest in G2 or a mitotic block,
antibodies against a tubulin (green) and 'y tubulin (red) were used to
determine the status of the centrosomes in the C3 cell line following 72 hours
M4N treatment. As shown in Figure l la, the centrosomes ofM4N treated
cells are duplicated but still located next to each other in the nucleus of
the
cell. Since centrosomes separate during early prophase, it can be concluded
that these cells have not begun mitosis. In contrast, the gamma tubulin
staining of the control cells has the diffuse pattern characteristic of G1 or
S
phase (29). A lack of chromatin condensation in the M4N treated cells was
also observed with DAPI staining (Figure 12b), additional evidence that the
cells have not moved forward out of G2 phase (30).
Example 11
Production of CDC2 is Inhibited by 40 ~,M M4N
Since progression of cells out of G2 is dependent on the production of
the MPF, the status of its protein components was examined in C3 cells
treated with 40 ,uM M4N. Asynchronous cells were grown for 24 or 72 hours in
media containing either M4N in 1% DMSO, or 1% DMSO alone. The cells
were harvested, and equal amounts of total cellular protein were analyzed by
western blotting. A marked reduction in the amount of CDC2 was observed
after 72 hours treatment with M4N (Figure 12a). However, levels of cyclin B,
detected by stripping and reprobing the same membrane, were found to be
unchanged. These results indicate that, under these conditions, the arrest is
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
not likely a response to p53 since it has been shown that overexpression of
p53 leads to a decrease in cyclin B (31, 32). Consistent with the results of
the
western analysis, CDC2 kinase activity was eliminated by 72 hours of M4N
treatment (Figure 12a). These experiments support the view that the drug
acts by inhibiting the production of the CDC2 protein, resulting in a loss of
activity of the MPF.
Our previous studies demonstrating the ability of M4N to block Spl-
dependent viral transcription suggest reduction of CDC2 mRNA levels as a
possible mechanism for the decrease of CDC2 protein. This is consistent
with the finding that the cyclin B protein, whose gene does not require Spl
for
its expression, is produced at nonnal levels while the CDC2 protein, whose
gene has two essential Spl sites in its promoter, is substantially reduced in
quantity. To test this hypothesis, northern blot analysis was performed on
RNA harvested from C3 cells treated with 40 ~.M M4N for 5 to 72 hours. As
shown in Figure 12b, the amount of CDC2 mRNA is reduced after only 24
hours treatment with M4N and nearly eliminated after 72 hours. Production of
the non-Spl regulated housekeeping gene GAPDH was used as an RNA loading
control, and its levels were not effected by 40 ~,1VI M4N.
The use of the C3 cell line allows us an additional control for analysis
of the mechanism of M4N mediated cell cycle arrest since other Spl-
dependent gene promoters are also likely to be inhibited by M4N treatment.
This possibility was examined in C3 cells by analyzing the effect of M4N on
transcription from the Spl dependent HPV-16 E6/E7 promoter. rtPCR
analysis of RNA isolated from C3 cells treated with 40 ~.M M4N for 5 to 72
hours demonstrated a clear reduction in the levels of the E7 transcript
(Figure
12c). GAPDH was again used as an internal control in this experiment, and
its levels were unaffected by drug treatment. These results provide additional
evidence that M4N reduces the transcripts of Spl regulated promoters.
Example 12
Inhibition of Spl-Binding Activity by G4N in a Gel Mobility-Shift Analysis.
Spl family proteins induce bends toward the major groove of DNA upon
31
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
binding (33). The zinc finger domain of the Sp1 protein is responsible for the
binding of the GC Box sequence 5'-GGGGCGGGG-3'. From computational
analysis, it was determined that G4N, the aminoester derivative of NDGA,
could form a stable complex with such a sequence in the major groove. To
determine whether G4N can serve as an Spl blocker as well as an Spl
displacer, we performed Spl/enhancer interaction studies in the presence or
absence of G4N by the gel mobility-shift analysis using only the DNB binding
domain of Spl for testing. In the blocking experiment, different
concentrations
of G4N were first incubated with 32P-labelled DNA in the binding buffer for 30
min at 25°C. DNA binding domain of recombinant Spl protein (Spl-167D)
was next added and incubated for additional 30 min in the presence of a large
excess of BSA protein. In the displacement study, the recombinant SPl-167D
was first allowed to bind DNA, G4N was then added at the second step of the
incubation. The G4N and Spl-167D concentrations and, the incubation and
gel electrophoresis conditions were identical in both studies (experimental
section). As shown in Figure 13, in either case, G4N was found to be able
keep DNA from interacting with Spl-167D protein. When only the DNA
binding domain of Spl alone was tested, G4N appeared to be more efficient
in displacement of the bound SpI than blocking Spl from binding to the
enhancer, as shown by the gel mobility-shift analysis (Figure 13, A,B,D). We
have also examined whether the bound G4N can be replaced by Spl-167D.
In this study, the inhibition of Spl-167D binding by G4N was first established
by the mobility-shift analysis (Fig 13C, lanes 2 and 5). When the G4N bound
template was challenged with additional Spl-167D, we observed a dosage
dependent increase of the band intensities of the Spl-I67D/DNA complex
(Fig 6C, Lanes 6,7) indicating the displacement of G4N by Spl-167D from the
template.
Example 13
Inhibition of Spl Regulated Tat-Transactivation of HIV Promoter Activity
by G4N.
As reported previously, methylated NDGA derivatives can block Spl
32
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
binding to the enhancer sites of a variety of viral promoters including HIV,
ICP4 of HSV, E6/E7 gene of HPV (8, 9, 10). We further tested the G4N
effect on the Tat-transactivation of HIV promoter activity in Cos cells by the
SEAP assay as previously described. Basal level of the HIV LTR driven SEAP.
expression was previously found to be barely detectable in Cos cells. There
were 60-fold or more increase in SEAP expression when Cos cells were
cotransfected with the CMV promoter driven Tat gene (8). Such Tat-driven
transactivation of the HIV LTR promoter activity was previously shown to be
Spl regulated (7, 8). In the presence of G4N, we observed inhibition of HIV
transactivation in a dose-dependent fashion (Figure 14). An average value
ICSO value of 36 ~,M for G4N was comparable to that of 3-O-methyl NDGA,
Mal .4 (ICSO 25 ~,M) and somewhat higher than that of tetra-methyl NDGA,
M4N (ICso 11 ~,M). The differences perhaps are due to the chemical nature of
the test compounds affecting the drug uptake to the cells.
33
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
Example 14
Inhibition of SIV-1 and HIV-1 Production in Cell Cultures by G4N.
Both HIV-1 and SIV are retroviruses that require integration into the
host genome to complete their replication. Both rely on host transcription
factors
for their proviral transcriptions. Spl plays a central role for such
expression in
these two viruses sharing an almost identical mode of transcription
regulation. In
anticipation of using SIV infected rhesus monkeys as animal model for testing
the antiviral effect of G4N, we have studied and compared the G4N effect in
inhibition of S1V in 174 x CEM cells with that of HIV in H9 cells. Cellular
toxicities of G4N in these two cell lines were also examined. For SIV
inhibition
study, 107 174 x CEM cells were mixed with high titer stock of SIVmac 239 at
37°C for two hours and then washed twice with cold PBS buffer to remove
the
unabsorbed virus. Cell suspension was aliquoted into each well of three 96
well plates. Various concentrations of the G4N solutions were made from
freshly prepared stoclc and aliquoted separately and each to six wells in a
column of one 96 well plate. Culture supemantants were collected every four
days post infection. (P.L) and fresh medium containing appropriate
concentrations of the drug were added to the culture following supernatant
collections. Viral production was assayed by a modified p27 core antigen
capture ELISA as shown (Figure 15). There was no SIV production detected
using G4N in concentrations above 5 CIM. At G4N concentrations below 2.5
CIM, SIV production was detected (Fig. 15) in culture supernatants from 4th
and 8th days post infected cultures as compared to viral production in the
absence of the drug. G4N (250 ~,M or less) showed no toxic effect on
uninfected
174 x CEM cells, as determined by the MTT assay (34).
A similar experiment was also carried out for the study of inhibition of
HIV-1
by G4N in H9 cells. The H9 cells were subcultured at 1 x 105/m1 and were
infected with an AZT resistant strain of HIV-1(HIV-1RTMF). G4N in different
concentrations was added two hours after infection. Fresh medium change
was made every four days. Cell growth in the presence of G4N was monitored
carefully during the nine-day experimental period. Viral production was
34
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
assayed by a p24 core antigen capture ELISA. As shown (Figure 16), G4N
concentration of 80 CIM completely inlubited HIV replication in H9 cells. An
ICSO of 12 ~,M CIIVI G4N for the inhibition of HIV-1 RTMF was found. Again,
there
was no detectable toxicity to uninfected H9 cells within the range of the
assay
(and below 250 ~,M).
Example 15
Effect of M4N Treatment on Survivin Gene Expression in C3 Cells
Materials and methods
Cell Culture. C3 cells were grown as monolayers in Iscove's Modified
Dulbecco's Medium (GIBCO BRL) supplemented with 5% fetal bovine serum
(GIBCO BRL) and maintained in a humid incubator at 37°C in a 5% COZ
environment.
M4N Treatment. C3 cells (5x106) were seeded in 150-mm plates and
allowed to attach to the plates. Twenty-four hours after seeding, cultures
were
washed twice with PBS and treated with M4N dissolved in 1% DMSO mixed with
the growth medium.
Cell extracts and immunoblotting. Cells were lysed in lysis buffer
containing 50 mM HEPES pH 7,250 mM NaCI, 0.1% (v/v) Nonidet P-40, 10%
glycerol, 1mM DTT, and 50 ~,1/ml protease inhibitor coclctail (Sigma). Protein
concentration of the extract was determined by the Bradford assay (Bio-Rad
Laboratories), and then 50 ,ug of protein was separated by SDS-PAGE and
electrotransferred to nitrocellulose membrane (ECL). Membranes were incubated
with primary antibodies against Survivin (Santa Cruz Biotechnology) and
caspase-
3 (Santa Cruz Biotechnology). Blots were then incubated with anti-rabbit
biotin-
conjugated secondary antibody and then with Avidx-APTM streptavidin-alkaline
phosphatase, and detected with CSPD~ substrate (Tropix).
RT-PCR analysis. rnRNA was isolated by the guanidinium thiocyanate and
phenol method from the cultured cells as described in Molecular Cloning (40).
A
343-base pair RT-PCR product was generated by using the survivin-sense
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
oligonucleotide primer 5'-GCATCGCCACCTTCAAGAACTGGCCC-3' and the
survivin-antisense oligonucleotide primer 5'-
CGGGTAGTCTTTGCAGTCTCTTCAAACTC-3'. GAPDH sense and anti-sense
primers 5'-GAATCTACTGGCGTCTTCACC-3' and 5'-
GTCATGAGCCCTTCCACGATGC-3' were used to generate a 238-base pair RT-
PCR product as a control. mRNA aliquots were incubated in 20 ,u1 reaction
buffer
containing 1 U or rRNAsin and DNAse at 75°C for 5 minutes followed by
reverse
transcription reaction with MMLV (Promega). The c-DNA products obtained
were amplified under the PCR conditions: 55°C for 55 seconds,
60°C for 55
seconds, and 72°C for 1 minute for 30 cycles. The PCR products were
separated
by electrophoresis on a 1.8% agarose gel containing ethidium bromide and
photographed under UV. The bands were quantitated by Scion Image and the
signal intensities of the survivin PCR reaction products were normalized to
those
of the GAPDH PCR products to generate a survivin gene down-regulation graph.
To determine whether the Spl-regulated survivin gene expression in C3
cells is reduced by M4N treatment, we treated the cells with 40 ~,M M4N for 24
hours and 72 hours. As shown in Figure 1, treatment of cells with M4N resulted
in
a significant decrease in survivin gene expression in a time-dependent manner.
Treatment with 40 ACM M4N for 24 hours and 72 hours resulted in 65% and 80%
reduction in survivin expression, respectively. Untreated cells did not show
any
reduction in survivin gene expression.
Survivin protein was also shown by immunoblotting to be downregulated
by 72 hours of M4N treatment. This downregulation was dosage-dependent
(Figure 2).
Example 16
Induction of Apoptosis with M4N treatment
Because our data showed that M4N resulted in survivin mRNA and protein
reduction, we investigated whether this reduction induces apoptosis since
survivin
has anti-apoptosis function. As shown by inununoblotting of caspase-3 (Figure
3),
treatment of M4N for 72 hours resulted in caspase-3 activation. This
activation
36
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
would be expected to result in an increase in an increase in apoptosis in
cells
treated with M4N.
37
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
References cited herein are listed below for convenience and are hereby
S incorporated by reference.
1. Nurse, P., Universal Control Mechanism Regulating Onset of M-Phase.
Nature, 344, 503-508 (1990).
2. Fang, F. and J.W. Newport, Evidence That the Gl-S and G2-M
Transitions Are Controlled by Different cdc2 Proteins in Higher
Eukaryotes. Cell, 66, 731-742(1991).
3. Dalton, S., Cell Cycle Regulation of the Human cdc2 Gene. The EMBO
Journal, 11, 1797-1804.(1992.)
4. Morgan, D.O., Principles of CDK Regulation. Nature, 374, 131-
134.(1995.)
5. Murray, A.VV., Creative Blocks: Cell-cycle Checlcpoints and Feedbaclc
Controls. Nature, 359, 599-604 (1992).
6. Kao, G.D., M. W.G., and R.J. Muschel, p34(Cdc2) Kinase Activity Is
Excluded From the Nucleus During the Radiation-induced G(2) Arrest
in HeLa Cells. J. Biol. Chem., 274, 34779-34784 (1999).
7. Hwu, J. R., Tseng, W. N., Gnabre, J., Giza, P. and Huang, R. C. C.
Antiviral
Activities of Methylated Nordihyd roguaiaxetic Acids (I) Synthesis, Structure
Identification and W hibition of Tat Regulated HIV Transactivation. J. Med.
Chem. 41:2994-3000(1998).
8. Gnabre, J. N., Brady, J. N., Clanton, D. J., Ito, Y., Dittmer, J., Bates,
R.
B. and Huang, R. C. Inhibition of Hurnan T_rmmunodeficiency
Virus Type 1 Transcription and Replication by DNA
Sequence-Selected Plant Lignans. Proc. Natl. Acad Sci U.S.A. 92,
11239 (1995).
9. Chen, H., et al., Antiviral Activities of Methylated Nordihydroguaiaretic
Acids. 2. Targeting Herpes Simplex Virus Replication by the Mutation
Insensitive Transcription Inhibitor Tetra-O-Methyl-NDGA. Journal of
Medicinal Chemistry, 41, 3001-3007 (1998).
38
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
10. Craigo, J., et al., Inhibition of Human Papillomavirus Type 16 Gene
Expression by Nordihydroguaiaretic Acid Plant Lignan Derivatives.
Antiviral Research, 47, 19-28 (2000).
11. Baba, M. Mini Review. Cellular Factors as Alternative Targets
S for inhibition of HIV- 1. Antiviral Res. 33, 144 i -1452 (1997).
I2. Gnabre, J. N., Ito, Y., Ma. Y. and Huang, R. C. (1996) Isolation of
Anti-HIV-1 Lignans from Larrea Tridentata by Counter-Current
Chromatography. J. Chromatogr. A 7I9, 353.
13. Gnabre, J. N., Huang, R. C., Bates, R. B., Burns, J. J., Calder, S.,
Malcomson, M. E. and McClure, K. J. (1995) Characterization of
Anti-HIV Lignans from Larrea Tridentata Tetrahedron S 1, 12203.
14. Honess, R. W., and Roizman, B. (1988) Regulation of Herpes Virus
Macromolecular Synthesis. 1. Cascade Regulation of Synthesis of Three
Groups of Viral Proteins. J. Virol. 1974. 14, 8.
1S 1S. Courey, A. J., and Tjian. R. (1988) Analysis of Spl in vivo Reveals
Multiple Transcription Domains. Including a Novel Glutamine-rich
Activation Motif. Cell SS, 887.
16. Some of the Spl-regulated cellular genes: Sartorelli, V.; Webster, K.
A.; Kedes, L. Muscle-specif c expresison of the cardiac alpha-actin
gene requires myoD l, Care-hox binding factor and Spl. Gene Dev.
1990, 4, 1811. Dailey, i., Roberts, S. B.; Heintz, N. Purification of the
histone H4 gene-specific transcription factors, H4TF-I and H4TF-2.
Gene Dev. 1988, 2, 1700. Means, A. L.; Farnham, P. J. Transcription
initiation form the dihydrogolate reductase promoter is positioned by
2S HIP-1 binding at the initiation site. Mel. Cell Biol. 1990, 10, 653.
Abravaya, K.; Phillips, B.; Morimoto, R. I. Heat shock-induced
interaction so heat shock transcription factor and human hsp70
promoter examined by in vive footprinting. Mel. Cell Biol. 1991, 11,
586. Leask, A.; Rosenberg, M.; Vassar, R.; Fuchs, E. Regulation fo a
human epidermal keratin gene: Sequences and nucleax factors involve
din keratinocyte-specific transcription. Gene Dev. 1990, 4, 1985.
Desjardins, E.; Hay, N. Repeated CT elements bound by zinc finger
39
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
proteins control the absolute and relative activities of the two principal
huyman cmyc promoter. Mel. Cell Biol. 1993, 13, 5710. Sanchez, H.
B.; Yieh, i., Osborne, T. F. Cooperation by sterol regulatory
element-binding proteins and Spl in sterol regulation of low-density
S lipoprotein receptor gene. J. Biol. Chem. 1995, 270, 1161. Lemaigre,
F. P.; Lafontaine, D. A.; Courtois, S. J.; Durviaux, S. M.; Rousseau, G.
G. Spl can displace GHF-1 from its distal binding site and stimulate
transcription form growth hormone gene promoter. Moll Cell. Biol.
1990, 10, 1811.
17. Phelps, W. C., Yee, C. L., Munger, K. and Howley, P. M. (1988) The
Human Papilloma Virus Type 16 E7 Gene Encodes Transactivation
and Transformation Functions Similar to Those of Adenovirus E/A. Cell
S3, S39-547.
18. Greenstone, H.L. Nieland, J.D., DeVisser, K.E., DeBruijn, M.L.,
1 S Kimbauer, R., Roden, R.B., Lowy, D.R., Kast, W.M. and Schiller, J.T.
(1998) Chimeric Papillomavirus Virus-Like particles Elicit Antitumor
T_mmullity Against the E7 Oncoprotein in an HPV16 Tumor Model.
PNAS 9S, 1800-1805.
19. Feltkamp, M.C., Vreugdenhil, G.R., Vierboom, M.P., Ras, E., Van der
Burg, S.H., Schegget, J. Ter, Melief, C.J.M. and Kast, W.M. (1995)CTL
Raised Against a Subdominant Epitope Offered as a Synthetic Peptide
Eradicate Human Papillonavirus Type 16-induced Tumors. European
Journal of Itnrnunology 2S, 2638-2642.
20. Feltkamp, M.C., Smits, H.L., Vierboom, M.P., Minaar, R.P., B.M.
2S Drijfhout, J.W., Schegget, J., Melief, C. and Kast, W.M. (1993)
Vaccination with Cytotoxic T Lymphocyte Epitope-Containing Peptide
Protects Against a Tumor Induced by Human Papillomavirus Type 16-
Transformed Cells. Eur. J. hnmunol. 23, 2242-2249.
21. Jacob, S. W. and Herschler (1986) Pharmacology of DMSO. Academic
Press, Inc.
22. Jack, C., and Torre, de la (1983) Biological Actions and Medical
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
Applications of Dimethyl Sulfoxide. New York Academy of Sciences, New
York, N.Y.
23. Spruance, S. L., McKeough. M. B. and Cardinal, J. R. (1983) Dimethyl
Sulfoxide as a vehicle for topical antiviral chemotherapy. Ann. N. Y.
Acad. Sci 411,28-33.
24. Biswal, S.S., et al., Glutathione Oxidation and Mitochondrial
Depolarization as Mechanisms of Nordihydroguaiaretic Acid-induced
Apoptosis in Lipoxygenase-def cient F15.12 Cells. Toxicol. Sci., 53, 77-
83.(2000.)
25. Schegg, K. M. and W.J. Welch, The Effect of Nordihydroguaiaretic Acid
and Related Lignans on Formyltetrahydxofolate Synthetase and
Carbodylesterase. Biochim. Biophys. Acta, 788, 167-180 (1984).
29. Feltkamp, M.C.W., et al., Vaccination With Cytotoxic T Lymphocyte
Epitope-containing Peptide Protects Against a Tumor Induced by Human
Papillomavirus Type 16-Transfromed Cells. Eur. J. Tmmunol., 23, 2242-2249
(1993).
27. Lin, K., et al., Treatment of Established Tumors With a Novel Vaccine
That Enhances Major Histocompatiblity Class II Presentation of Tumor
Antigen. Cancer Res, 56, 21-26.(1996.)
28. Foley, G.E., et al., Continuous Culture of Human Lyrnphoblasts From
Peripheral Blood of a Child With Acute Leukemia. Cancer, 18, 522-
529(1965).
29. Shiebel, E., Gamma-Tubulin Complexes: Binding to the Centrosome,
Regulation and Microtubule Nucleation. Current Opinion Cellular Biology,
12, 113-118(2000).
30. Marsden, M.P.F., Laemmli, U.K., Metaphase Chromosome Structure:
Evidence for a Radial Loop Model. Cell, 17, 849-858 (1979).
31. Taylor, W.R., et al., Mechanisms of 02 Arrest in Response to
Overexpression of p53. Molecular Biology of the Cell, 10, 3607-3622 (1999).
32. Innocente, S.A., et al., p53 Regulates a G2 Checkpoint Through Cyclin
B1. Proc. Natl. Acad. Sci USA, 96, 2147-2152(I999).
33. Sjottem, E.; Anderson, C.; Johansen, T. Structural and Functional
41
CA 02447045 2003-11-10
WO 02/089795 PCT/US02/14374
Analyses of DNA Bending Induced by Spl Family Transcription Factors. J.
Mol. Bio., 267, 490-504 (1997).
37.
34. Weislow, O.S.; Kiser, R.; Fine, D.L.; Bader, J.; Shoemaker, R. H.;
Boyd, M.R. New Soluble-Formazan Assay for HIV-1 Cytopathic
Effects: Application to High-Flux Screening of Synthetic and Natural
Products for AIDS-Antiviral Activity. J. Natl. Cancer Inst., 81(8),
557-586 (1989).
35. Li, F. and Altieri, D.C., Transcriptional analysis of human survivin gene
expression. Biochem. J. 344, 305-311(1999).
36. Li, F. and Altieri, D.C., The Cancer Apoptosis SurvivifZ gene:
Characterization of Locus and Transcriptional Requirements of Basal and
Cell Cycle-dependent Expression. Cancer Res. 59,3143-3151 (1999).
37. O'Connor, D.S. et al., Proc. Natl. Acad. Sci. (US) 97,13103-13107 (2000).
38. Grossman, D. et al., Proc. Natl. Acad. Sci. (US) 98,635-640(2001).
39. Studzinski, G.P. (ed.), Apoptosis, A Practical Approach (1999), page 10.
40. Sambrook and Russell, Molecular Cloning, 3rd ed.(2001).
42