Note: Descriptions are shown in the official language in which they were submitted.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
NUCLEASE RESISTANT CHIMERIC OLIGONUCLEOTIDES
Cross Reference to Related Applications
This application claims the benefit of U.S. Application No. 10/013,295,
filed December 10, 2001; U.S. Application No. 09/996,292, filed November 2~,
2001; and U.S. Provisional Application No. 601302,682, filed July 3, 2001.
Field of the Invention
The present invention relates to novel nuclease-resistant oligomeric
compounds and to novel methods for increasing the nuclease resista~lce of
oligomeric compounds.
B_ ackground of the Invention
Efficacy and sequence specific behavior of antisense oligonucleotides
(ONs) in biological systems depend upon their resistance to enzymatic
degradation. It is therefore essential, when designing potent antisense drugs,
to
combine features such as high binding affinity and mismatch sensitivity with
nuclease resistance. Unmodified-phosphodiester antisense oligonucleotides are
degraded rapidly in biological fluids containing hydrolytic enzymes (Shaw,
J.P.;
Kent, K.; Bird, J.; Fishback, J.; Froehler, B. Nucleic Acids Res. 1991,19, 747-
750;Woolf, T.M.; Jennings, C.G.B.; Rebagliati, M; Melton, D.A. Nucleic Acids
Res. 1990, I8, 1763-1769), and the first generation of modified antisense
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-2-
oligonucleotide drugs, such as 2'-deoxyphosphorothioate oligonucleotides, were
also subj ect to enzymatic degradation (Maier, M.; Bleicher, K.; Kalthoff, H.;
Bayer, E. Biomed. Pept., Pi°oteins Nucleic Acids 1995, l, 235-241;
Agrawal, S.;
Temsamani, J.; Tang, J.Y. P~oc. Natl. Acad. Sci. 1991, 88, 7595-7599).
Extensive
stability against the various nucleases present in biological systems can best
be
achieved by modified oligonucleotides. Since 3' exonuclease activity is
predominantly responsible for enzymatic degradation in serum-containing
medium and in various eukaryotic cell lines, modifications located at the 3'-
terminus significantly contribute to the nuclease resistance of an
oligonucleotide
(Shaw, J.-P.; Kent, K.; Bird, J.; Fishback, J.; Froehler, B. Nucleic Acids
Res. 1991,
19, 747-750; Maier, M.; Bleicher, K.; Kalthoff, H.; Bayer, E. Biof~aed. Pept.,
Proteins Nucleic Acids 1995,1, 235-241).
Extensive modifications have been made to the phosphodiester linkages
and sugar moieties of oligonucleotides, while modifications to the
heterocyclic
base moieties have been relatively limited, due to a desire to maintain the
specific
hydrogen bonding motifs required for base pair specificity (For a review see,
Herdewijn, P. Ahtiseyase Nucleic Acids Drug Dev. 2000, 10, 297-310). The 2'-
position is attractive for derivatization because it offers the advantages of
enhancing both nuclease resistance and binding affinity (Manoharan, M.
Bioclzif~a.
Biophys. Acta 1999,1489, 117-130; Kawasaki, A. M.; Casper, M. D.; Prakash, T.
P.; Manalili, S.; Sasmor, H.; Manoharan, M.; Cook, P. D. Nucleosides
Nucleotides
1999,18, 1419-1420).
A large number of nucleobase modifications, which were designed to
enhance the binding affinity of antisense oligonucleotides to their
complementary
target strands, have recently been introduced (Beaucage, S. L.; Iyer, R. P.
Tet~ahedf°ooa 1993; 49, 6123-94; Cook, P. D. Asmu. Rep. Med. Claem.
1998, 33,
313-325; Goodchild, J. Bioco~ejugate Chemist~y,1990; 1, 165-87; TJhlmann, E.;
Peyman, A. Glaem. Rev. 1990, 90, 543-84. For reviews see: Uhlmami, E.; Peyman,
A. Claem. Rev. 1990, 90, 543-584; Milligan, J. F.; Matteucci, M. D.; Martin,
J. C.
J. Med. Claem. 1993, 36, 1923-37; Cook, P. D. Antisense medicinal chemistry.
In:
Antisense Research and Application, A Handbook of Expef°imehtal
Pharmacology
(ed. Crooke, S. T.), pp. 51-101. Springer-Verlag, New York, 1998). Some
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-3-
heterocyclic modifications have been shown to enhance the binding affinity of
nucleic acids through increased hydrogen bonding and/or base stacking
interactions. Examples of such heterocyclic modifications include 2,6-
diaminopurine, which allows fox a third hydrogen bond with thymidine and
replacement of the hydrogen atom at the C5 position of pyrimidine bases with a
propynyl group, resulting in increased stacking interactions (Chollet, A.;
Chollet-
Damerius, A.; I~awashima, E. H. Chezzz. Scripta 1986, 26, 37-40; Wagner, R.
W.;
Matteucci, M. D.; Lewis, J. G.; Guttierrez, A. J.; Moulds, C.; Froehler, B. C.
Science 1993, 260, 1510-1513).
More recently, several tricyclic cytosine analogs, such as phenoxazine,
phenothiazine (Lin, I~.-Y.; Jones, R. J.; Matteucci, M. J. Am. Chezzz. Soc.
1995,
117, 3873-3874) and tetrafluorophenoxazin (Wang, J.; Lin, K.-Y., Matteucci, M.
TetrahedYOn Lett. 1998, 39, 8385-8388), have been developed and have been
shown to hybridize to guanine and, in case of tetrafluorophenoxazin, also with
adenine. The tricyclic cytosine analogs have also been shown to enhance
helical
thermal stability by extended stacking interactions.
The helix-stabilizing properties of the tricyclic cytosine analogs are further
improved with G-clamp, a cytosine analog with an aminoethoxy moiety attached
to the rigid phenoxazine scaffold (Lin, I~.-Y.; Matteucci, M. J. Am. Chem.
Soc.
1998, 120, 8531-8532). Binding studies have demonstrated that a single G-clamp
enhances the binding affinity of a model oligonucleotide to its complementary
target DNA or RNA with a ~T", of up to 18° relative to 5-methyl
cytosine
(dC5"'~, the highest known affinity enhancement for a single modification. The
gain in helical stability does not compromise the binding specificity of the
oligonucleotides, as the Tm data indicate an even greater discrimination
between
the perfectly matched and mismatched sequences as compared to dCSme. The
tethered amino group may serve as an additional hydrogen bond donor that
interacts with the Hoogsteen face, namely the 06, of a complementary guanine.
The increased affinity of G-clamp is thus most likely mediated by the
combination
of extended base stacking and additional hydrogen bonding.
The enhanced binding affinity of the phenoxazine derivatives together
with their uncompromised sequence specificity makes them valuable nucleobase
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-4-
analogs for the development of more potent antisense-based drugs. Promising
data have been derived from ih vitro experiments demonstrating that
heptanucleotides containing phenoxazine substitutions axe capable of
activating
RNaseH, enhance cellular uptake, and exhibit an increased antisense activity
(Lin,
K.-Y.; Matteucci, M. J. Am. Ghefya. Soc. 1998, 1~0, 8531-8532). The activity
enhancement was even more pronounced in the case of G-clamp, as a single
substitution was shown to significantly improve the ifz vitro potency of a
20mer
2'-deoxyphosphorothioate oligonucleotide (Flanagan, W. M.; Wolf, J.J.; Olson,
P.; Grant, D.; Lin, K.-Y.; Wagner, R. W.; Matteucci, M. Proc. Natl. Acad. Sci.
USA, 1999, 96, 3513-3518).
The efficacy and sequence specificity of oligonucleotides in biological
systems is dependent, in part, upon their nuclease stability. Resistance to
the
many nucleases present in biological systems is best achieved by modified
oligonucleotides. It is therefore essential, when designing modified
nucleotides,
to evaluate and optimize their resistance to enzymatic degradation.
Summary of the Invention
The present invention relates to novel nuclease-resistant oligomeric
compounds and to novel methods for increasing the nuclease resistance of
oligomeric compounds.
In preferred embodiments, the compounds of the invention relate to
oligomeric compounds of formula V:
W~ Y
Bx
n
Y~ A~
W2
V
wherein:
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-5-
n is from 3 to about 50;
each Yl is, independently, an internucleoside linking group;
Yz is oxygen or an internucleoside linking group;
Y3 is oxygen or an internucleoside linking group;
each Bx is an optionally protected heterocyclic base moiety;
each A1 is, independently, hydrogen or a sugar substituent group;
WI is hydrogen, a hydroxyl protecting group or a modified nucleoside
selected from the group consisting of
HO Bx
~Vi
HO Vi Bx HO V' Bx HO ~ Bx
O~O~CH3
HO O Bx HO S Bx
HO Bx
~OH
OH
NHA4
HO O Bx H O Bx
and ,
Aa SA3
WZ is hydrogen, a hydroxyl protecting group or a modified nucleoside
selected from the group consisting of
Bx V1 Bx u1 Bx
OH
Bx ~ Bx N Bx
V1 i
p O~O~CH3 OH
O Bx S Bx O Bx
HO OH HO OH
H ~A4
O Bx O Bx
and '
AZ SA3
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-6-
each A~ is, independently, allcyl, alkenyl, allcynyl, aryl, alkaxyl, O-allcyl,
O-
aryl, amino, substituted amino, -SH, -SA3, thiolether, F, or morpholino;
each A3 is, independently, H, a sulfur protecting group, aryl, alkaryl,
substituted or unsubstituted Cl-Clo alkyl, substituted or wsubstituted Ca-Clo
alkenyl, substituted or unsubstituted CZ-Clo alkynyl, or alkaryl, wherein said
substitution is OAS or SAS;
each A4 is, independently, H, a nitrogen protecting group, substituted or
unsubstituted C1-Clo allcyl, substituted ox unsubstituted C2-Cio alkenyl,
substituted
or unsubstituted C2-Clo alkynyl, or alkaryl, wherein said substitution is OAS
or
SAS;
each AS is, independently, hydrogen, C1-Clo alkyl, cycloalkyl or aryl;
each Vl is, independently, O or S;
wherein at least one of Wl and W2 is not hydrogen or a hydroxyl
protecting group and at least one internucleoside linking group is not a
phosphodiester linking group.
In certain preferred embodiments, the internucleoside linking groups of the
compounds of formula V are phosphorus-containing internucleoside linking
groups. In still more preferred embodiments, at least one internucleoside
linking
group of the compounds of formula V is other than phosphodiester, and more
preferably, greater than 90% of the internucleoside linking groups of the
compounds of formula V are non-phosphorous containing internucleoside linking
groups. In even more preferred embodiments, greater than 90% of the
internucleoside linking group of the compounds of formula V are
phosphorothioate linking groups.
In certain other embodiments of the invention, the oligomeric compounds
of formula V comprise gapmers, hemimers or inverted gapmers. In more
preferred embodiments, the oligomexic compounds of formula V comprise at least
one 2'-O-CH2CH~-O-CH3 sugar substituent group in at least one region of the
gapmer, hemimer of inverted gapmer.
In other embodiments of the invention, the oligomeric compounds of
formula V comprise at least one nucleoside wherein Bx is a polycyclic
heterocyclic base moiety. In more preferred embodiments, the oligomeric
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
_7_
compounds of formula V comprise at least one nucleoside wherein Bx is,
independently, of the formula:
VI
wherein
A6 is O or S;
A~ is CH2, N-CH3, O or S;
each A$ and A9 is hydrogen or one of A$ and A9 is hydrogen and the other
of A8 and A9 is selected from the group consisting of:
-O-(CH~)pi G -O (CH2)p1 N Q
and Q2 p2
wherein:
G is -CN, -OAIO, -SAIO, -N(H)Alo, -ON(H)Alo or -
C(=NH)N(H)Aio~
Ql is H, -NHAIO, -C(=O)N(H)Alo, -C(-S)N(H)Alo or -
C(=NH)N(H)Aio~
each Q2 is, independently, H or Pg;
Alo is H, Pg, substituted or unsubstituted CI-Clo alkyl, acetyl,
benzyl,
-(CHZ)p3NHZ, -(CHZ)p3N(H)Pg, a D or L a-amino acid, or a peptide derived from
D, L or racemic a-amino acids;
Pg is a nitrogen, oxygen or thiol protecting group;
each p1 is, independently, from 2 to about 6;
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
_g_
p2 is from 1 to about 3; and
p3 is from 1 to about 4.
In another embodiment of the invention, Y3 of formula V is an
inten~ucleoside likning group and Wl of formula V is a modified nucleoside. In
another embodiment of the invention, Ya of formula V is an internucleoside
linking group and W2 of formula V is a modified nucleoside.
In certain preferred embodiments of the invention, each sugar substituent
group of formula V is, independently, -O-CHZCH20CH3, -O(CH2)20N(CH3)Z, -O-
(CH2)~-O-(CH2)2-N(CH3)Z, -O-GH3, -OCH2CH2CH2NHz, -CHa-CH=CH2, or
fluoro.
In another preferred embodiment, the invention relates to methods of
enhancing the nuclease resistance of an oligomeric compound comprising
providing at least one modified nucleoside at either the 3' or 5' terminus of
the
oligomeric compound to give a modified oligomeric compound of formula V,
such that at least one of Wl and WZ of formula V is not hydrogen or a hydorxyl
protecting group.
Brief Description of the Drawings
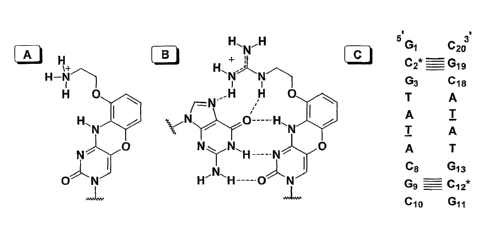
Figure 1 A depicts the structure of the tricyclic cytosine analog G-clamp,
Figure 1 B depicts guanidinyl G-clamp hybridized to complementary guanosine,
and Figure 1 C depicts a palindromic decamer duplex that was used for x-ray
crystallography. The five hydrogen bonds formed between C* and G are
indicated by horizontal lines. C* refers to guanidinyl G-clamp and T refers to
2'-
O-MOE-T.
Figure 2 depicts a Fourier (2Fo-F~) sum electron density map (contoured at
1.250') around a guanidinyl G-clamp nucleoside analog and guanosine that
confirms the formation of five hydrogen bonds, which are indicated by thin
solid
lines with distances shown in t~.
Figure 3 depicts the base stacking that occurs between a guanidinyl G-
clamp nucleobase analog and guanosine viewed approximately along the vertical
to the phenoxazine rings.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
_g_
Figure 4 depicts the degradation of oligonucleotides 157 (open triangles)
and 158 (closed circles) with SVPD as a function of incubation time and
compared to degradation of an unmodified control oligonucleotide 159 (closed
diamonds) as determined by CGE analysis.
Figure 5 depicts the velocity of the hydrolysis of oligonucleotide 159 with
BIPD as a function of the concentration of co-incubated oligonucleotides 157
(open triangles) and 158 (closed circles).
Figure 6A depicts the relative units of a full-length L/D clumeric
oligonucleotide before administration to BalbC mice; Figure 6 B depicts the
relative units of a full-length L/D chimeric oligonucleotide that was present
in the
liver one hour after administration of a 25 mg/kg dose by IV bolus into BalbC
mice; Figure 6 C depicts the relative units of a full-length L/D chimeric
oligonucleotide that was present in the kidney one hour after administration
of a
25 mg/kg dose by IV bolus into BalbC mice; Figure 6 D depicts the relative
units
of a full-length L/D chimeric oligonucleotide that was present in the spleen
one
hour after administration of a 25 mg/kg dose by IV bolus into BalbC mice; and
Figure 6 E depicts the relative units of a full-length L/D chimeric
oligonucleotide
that was present in the lung one hour after administration of a 25 mg/kg dose
by
IV bolus into BalbC mice.
Figure 7A depicts the relative units of a full-length L/D chimeric
oligonucleotide before administration to BalbC mice; Figure 7 B depicts the
relative units of a full-length L/D chimeric oligonucleotide that was present
in the
liver 24 housr after achninistration of a 25 mg/kg dose by IV bolus into BalbC
mice; Figure 7 C depicts the relative units of a full-length L/D chimeric
oligonucleotide that was present in the kidney 24 hours after administration
of a
25 mg/kg dose by IV bolus into BalbC mice; Figure 7 D depicts the relative
units
of a full-length L/D chimeric oligonucleotide that was present in the spleen
24
hours after administration of a 25 mg/kg dose by IV bolus into BalbC mice; and
Figure 7 E depicts the relative units of a full-length L/D chimeric
oligonucleotide
that was present in the lung 24 hours after administration of a 25 mglkg dose
by
IV bolus into BalbC mice.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-10-
Detailed Description of the Preferred Embodiments
In the context of this invention, the terms "oligomer" and "oligomeric
compound" refer to a plurality of naturally-occurnng or non-naturally-
occurring
nucleosides joined together in a specific sequence. The terms "oligomer" and
"oligomeric compound" include oligonucleotides, oligonucleotide analogs,
oligonucleosides and chimeric oligomeric compounds where there are more than
one type of internucleoside linkages dividing the oligomeric compound into
regions. Oligomeric compounds are typically structurally distinguishable from,
yet functionally interchangeable with, naturally-occurnng or synthetic wild-
type
oligonucleotides. Thus, oligomeric compounds include all such stt-uctures that
function effectively to mimic the structure and/or function of a desired RNA
or
DNA strand, for example, by hybridizing to a target.
In the context of this invention, the term "oligonucleotide" refers to an
oligomer or polymer of ribonucleic acid (RNA) or deoxyribonucleic acid (DNA)
or mimetics thereof. This term includes oligonucleotides composed of naturally-
occurring nucleobases, sugars and covalent internucleoside (backbone) linkages
as
well as oligonucleotides having non-naturally-occurnng portions that function
similarly. Such modified or substituted oligonucleotides are often preferred
over
native forms because of desirable properties such as, for example, enhanced
cellular uptake, enhanced affinity for nucleic acid target and increased
stability in
the presence of nucleases.
As is known in the art, a nucleoside is a base-sugar combination. The base
portion of the nucleoside is normally a heterocyclic base. The two most common
classes of such heterocyclic bases are the purines and the pyrimidines.
Nucleotides are nucleosides that further include a phosphate group covalently
linked to the sugar portion of the nucleoside. For those nucleosides that
include a
pentofuranosyl sugar, the phosphate group can be linked to either the 2', 3'
or 5'
hydroxyl moiety of the sugar. In forming oligonucleotides, the phosphate
groups
covalently link adjacent nucleosides to one another to form a linear polymeric
compound. In turn the respective ends of this linear polymeric structure can
be
further joined to form a circular structure. However, open linear structures
are
generally preferred. Within the oligonucleotide structure, the phosphate
groups
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-11-
are commonly referred to as forming the internucleoside backbone of the
oligonucleotide. The normal linkage or backbone of RNA and DNA is a 3' to 5'
phosphodiester linkage.
Specific examples of preferred oligomeric compounds useful in this
invention include those having modified backbones or non-naturally occurring
internucleoside linkages. As defined in this specification, modified backbones
include those having a phosphorus atom in the backbone and those that do not
have a phosphorus atom in the backbone. For the purposes of this
specification,
and as sometimes referenced in the art, modified oligonucleotides that do not
have
a phosphorus atom in their internucleoside backbone can also be considered to
be
oligonucleosides.
Preferred modified oligonucleotide backbones include, for example,
phosphorothioates, chiral phosphorothioates, phosphorodithioates,
phosphotriesters, aminoalkylphosphotriesters, methyl and other alkyl
phosphonates including 3'-alkylene phosphonates, 5'-alkylene phosphonates and
chiral phosphonates, phosphinates, phosphoramidates including 3'-amino
phosphoramidate and aminoalkylphosphoramidates, thionophosphoramidates,
thionoalkylphosphonates, thionoalkylphosphotriesters, selenophosphates and
boranophosphates having normal 3'-5' linkages, 2'-5' linked analogs of these,
and
those having inverted polarity wherein one or more internucleotide linkages is
a 3'
to 3', 5' to 5' or 2' to 2' linkage. Preferred oligonucleotides having
inverted
polarity comprise a single 3' to 3' linkage at the 3'-most internucleotide
linkage i.e.
a single inverted nucleoside residue which may be abasic (the nucleobase is
missing or has a hydroxyl group in place thereof). Various salts, mixed salts
and
free acid forms are also included.
reuresentative phosphorus containing linkages
phosphorodithioate (-O-P(S)(S)-O-);
phosphorothioate (-O-P(S)(O)-O-);
phosphoramidate (-O-P(O)(NJ2)-O-);
phosphonate (-O-P(J)(O)-O-);
phosphotriesters (-O-P(O J)(O)-O-);
phophosphoramidate (-O-P(O)(NJ)-S-);
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-12-
thionoalkylphosphonate (-O-P(S)(J)-O-);
thionoalkylphosphotriester (-O-P(O)(OJ)-S-);
phosphoramidate (-N(J)-P(O)(O)-O-);
boranophosphate (-R$-P(O)(O)-J-);
where J denotes a substituent group which is commonly hydrogen or an
alkyl group or a more complicated group that varies from one type of linkage
to
another.
Representative United States patents that teach the preparation of the
above-noted phosphorus-containing linkages include, but are not limited to,
U.S.:
3,687,808; 4,469,863; 4,476,301; 5,023,243; 5,177,196; 5,188,897; 5,264,423;
5,276,019; 5,278,302; 5,286,717; 5,321,131; 5,399,676; 5,405,939; 5,453,496;
5,455,233; 5,466,677; 5,476,925; 5,519,126; 5,536,821; 5,541,306; 5,550,111;
5,563,253; 5,571,799; 5,587,361; 5,194,599; 5,565,555; 5,527,899; 5,721,218;
5,672,697 and 5,625,050, certain of which are commonly owned with this
application, and each of which is herein incorporated by reference.
Preferred modified backbones that do not include a phosphorus atom
therein are those that are formed by short chain alkyl or cycloalkyl
internucleoside
linkages, mixed heteroatom and alkyl or cycloalkyl internucleoside linkages,
or
one or more short chain heteroatomic or heterocyclic internucleoside linkages.
These include those having morpholino linkages (formed in part from the sugar
portion of a nucleoside); siloxane backbones; sulfide, sulfoxide and sulfone
backbones; formacetyl and thioformacetyl backbones; methylene formacetyl and
thioformacetyl backbones; riboacetyl backbones; alkene containing backbones;
sulfamate backbones; methyleneimino and methylenehydrazino backbones;
sulfanate and sulfonamide backbones; amide backbones; and others having mixed
N, O, S and CH2 component parts.
representative non-nhosnhorus containing linkages
thiodiester (-O-C(O)-S-); _
thionocarbamate (-O-C(O)(NJ)-S-);
siloxane (-O-Si(J)Z-O-);
carbamate (-O-C(O)-NH- and -NH-C(O)-O-)
sulfamate (-O-S(O)(O)-N- and -N-S(O)(O)-N-;
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-13-
morpholino sulfamide (-O-S(O)(N(morpholino)-);
sulfonamide (-O-SOz-NH-);
sulfide (-CHz-S-CHz-);
sulfonate (-O-SOz-CHz-);
N,N'-dimethylhydrazine (-CHz-N(CH3)-N(CH3)-);
thioformacetal (-S-CHz-O-);
fonnacetal (-O-CHz-O-);
thioketal (-S-C(J)z-O-); and
ketal (-O-C(J)z-O-);
amine (-NH-CHz-CHz-);
hydroxylamine (-CHz-N(J)-O-);
hydroxylimine (-CH=N-O-); and
hydrazinyl (-CHz-N(H)-N(H)-).
where J denotes a substituent group which is commonly hydrogen or an
alkyl group or a more complicated group that varies from one type of linkage
to
another.
Representative United States patents that teach the preparation of the
above-noted oligonucleosides include, but are not limited to, U.S.: 5,034,506;
5,166,315; 5,185,444; 5,214,134; 5,216,141; 5,235,033; 5,264,562; 5,264,564;
5,405,938; 5,434,257; 5,466,677; 5,470,967; 5,489,677; 5,541,307; 5,561,225;
5,596,086; 5,602,240; 5,610,289; 5,602,240; 5,608,046; 5,610,289; 5,618,704;
5,623,070; 5,663,312; 5,633,360; 5,677,437; 5,792,608; 5,646,269 and
5,677,439,
certain of which are commonly owned with this application, and each of which
is
herein incorporated by reference.
In certain preferred oligonucleotide mimetics, both the sugar and the
internucleoside linkage, i.e., the backbone, of the nucleotide units are
replaced
with novel groups. The base units are maintained for hybridization with an
appropriate nucleic acid target compound. One such oligomeric compound, an
oligonucleotide mimetic that has been shown to have excellent hybridization
properties, is referred to as a peptide nucleic acid (PNA). In PNA compounds,
the
sugar-backbone of an oligonucleotide is replaced with an amide containing
backbone, in particular an aminoethylglycine backbone. The nucleobases are
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-14-
retained and are bound directly or indirectly to aza nitrogen atoms of the
amide
portion of the backbone. Representative United States patents that teach the
preparation of PNA compounds include, but are not limited to, U.S.: 5,539,082;
5,714,331; and 5,719,262, each ofwhich is herein incorporated by reference.
Further teaching of PNA compounds can be found in Nielsen et al.,
Scieface,1991,
254, 1497-1500.
Among the preferred compounds of this invention are oligonucleotides
with phosphorothioate backbones and oligonucleotides with heteroatom
backbones, and in particular -CHZ-NH-O-CH2-, -CH2-N(CH3)-O-CHZ- [known as
a methylene (methylimino), MMI backbone or more generally as
methyleneimino], -CH2-O-N(CH3)-CH2-, -CH2-N(CH3)-N(CH3)-CH2- and -O-
N(CH3)-CHZ-CH2- of the above referenced U.S. patent 5,489,677, and the amide
backbones of the above referenced U.S: patent 5,602,240. Also preferred are
oligonucleotides having morpholino backbone structures of the above-referenced
U.S. patent 5,034,506.
"Bx," as used herein, is intended to indicate a heterocyclic base moiety.
Heterocyclic base moieties (often referred to in the art simply as a "bases"
or a
"nucleobases") amenable to the present invention include naturally or non-
naturally occurring nucleobases. One or more functionalities of the base can
bear
a protecting group. As used herein, "unmodified" or "natural" nucleobases
include
the purine bases adenine (A) and guanine (G), and the pyrimidine bases thymine
(T), cytosine (C) and uracil (U). Modified nucleobases include other synthetic
and natural nucleobases such as 5-methylcytosine (5-me-C), 5-hydroxymethyl
cytosine, xanthine, hypoxanthine, 2-aminoadenine, 6-methyl and other alkyl
derivatives of adenine and guanine, 2-propyl and other alkyl derivatives of
adenine and guanine, 2-thiouracil, 2-thiothymine and 2-thiocytosine, 5-
halouracil
and cytosine, 5-propynyl (-C---C-CH3) uracil and cytosine and other alkynyl
derivatives of pyrimidine bases, 6-azo uracil, cytosine and thymine, 5-uracil
(pseudouracil), 4-thiouracil, 8-halo, 8-amino, 8-thiol, 8-thioalkyl, 8-
hydroxyl and
other 8-substituted adenines and guanines, 5-halo particularly 5-bromo, 5-
trifluoromethyl and other 5-substituted uracils and cytosines, 7-methylguanine
and
7-methyladenine, 2-F-adenine, 2-amino-adenine, 8-azaguanine and 8-azaadenine,
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-15-
7-deazaguanine and 7-deazaadenine and 3-deazaguanine and 3-deazaadenine.
Further modified nucleobases include tricyclic pyrimidines such as phenoxazine
cytidine(1H-pyrimido[5,4-b][1,4]benzoxazin-2(3H)-one), phenothiazine cytidine
(1H-pyrimido[5,4-b][1,4]benzothiazin-2(3H)-one), G-clamps such as a
substituted
phenoxazine cytidine (e.g. 9-(2-aminoethoxy)-H-pyrimido[5,4-b][1,4]benzoxazin-
2(3H)-one), carbazole cytidine (2H-pyrimido[4,5-b]indol-2-one), pyridoindole
cytidine (H-pyrido[3',2':4,5]pyrrolo[2,3-d]pyrimidin-2-one). Modified
nucleobases include those in which the purine or pyrimidine base is replaced
with
other heterocycles, for example 7-deaza-adenine, 7-deazaguanosine, 2-
aminopyridine and 2-pyridone.
Further nucleobases include those disclosed in United States Patent No.
3,687,808, those disclosed in The Concise Encyclopedia Of Polymer Science And
Engineering, pages 858-859, Kroschwitz, J.L, ed. John Wiley & Sons, 1990,
those
disclosed by Englisch et al., Angewandte Chemie, International Edition, 1991,
30,
613, and those disclosed by Sanghvi, Y.S., Chapter 15, Antisense Research and
Applications, pages 289-302, Crooke, S.T. and Lebleu, B. , ed., CRC Press,
1993.
Certain of these nucleobases are particularly useful for increasing the
binding
affinity of the oligomeric compounds of the invention. These include 5-
substituted pyrimidines, 6-azapyrimidines and N-2, N-6 and O-6 substituted
purines, including 2-aminopropyladenine, 5-propynyluracil and 5-
propynylcytosine. 5-Methylcytosine substitutions have been shown to increase
nucleic acid duplex stability by 0.6- 1.2°C (Sanghvi, Y.S., Crooke, S.T.
and
Lebleu, B., eds., Antisense Research and Applications, CRC Press, Boca Raton,
1993, pp. 276-278) and are presently preferred base substitutions, even more
particularly when combined with 2'-O-methoxyethyl sugar modifications.
Representative United States patents that teach the preparation of certain
of the above noted modified nucleobases as well as other modified nucleobases
include, but are not limited to, the above noted U.S. 3,687,808, as well as
U.S.:
4,845,205; 5,130,302; 5,134,066; 5,175,273; 5,367,066; 5,432,272; 5,457,187;
5,459,255; 5,484,908; 5,502,177; 5,525,711; 5,552,540; 5,587,469; 5,594,121,
5,596,091; 5,614,617; 5,645,985; 5,830,653; 5,763,588; 6,005,096; 5,681,941,
and
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-16-
5,750,692, certain of which are commonly owned with the instant application,
and
each of which is herein incorporated by reference.
In one aspect of the present invention oligomeric compounds are prepared
having one or more heterocyclic base moieties comprising a polycyclic
heterocyclic base moiety. As used herein the term polycyclic heterocyclic base
moiety is intended to include compounds comprising at least 3 or more fused
rings. A number of tricyclic and some tetracyclic heterocyclic compounds have
been prepared and substituted for naturally ocurring heterocyclic base
moieties in
oligomeric compounds. The resulting oligomeric compounds have been used in
antisense applications to increase the binding properties of for example a
modified
strand to a target strand. The more studied modifications have been targeted
to
guanosines and are commonly referred to as cytidine analogs.
In one aspect of the present invention a polycyclic heterocyclic base
moiety has the formula:
mz
Rm / Ris
NH Ri4
Rm
Representative cytosine analogs that make 3 hydrogen bonds with a
guanosine in a second strand or elsewhere in the same strand include 1,3-
diazaphenoxazine-2-one (Rlo- O, Rll - R14= H) [Kurchavov, et al., Nucleosides
and Nucleotides, 1997, 16, 1837-1846], 1,3-diazaphenothiazine-2-one (Rlo= S,
Ri m Rm= H), [Lin, K.-Y.; Jones, R. J.; Matteucci, M. J. Am. Chem. Soc. 1995,
117, 3873-3874] and 6,7,8,9-tetrafluoro-1,3-diazaphenoxazine-2-one (Rlo = O,
Rl i - Ri4 = F) [Wang, J.; Lin, K.-Y., Matteucci, M. Tetrahedron Lett. 1998,
39,
8385-8388]. Incorporated into oligonucleotides these base modifications were
shown to hybridize with complementary guanine and the latter was also shown to
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-17-
hybridize with adenine and to enhance helical thermal stability by extended
stacking interactions.
Further helix-stabilizing properties have been observed when a cytosine
analogs having an aminoethoxy moiety attached to the rigid 1,3-diazaphenoxa-
zine-2-one scaffold (Rlo =_ O, Rl i = -O-(CHz)z-NHz, Rlz-14=H, this analog has
been
given a particular name "G-clamp") [Lin, K.-Y.; Matteucci, M. J. Am. Chem.
Soc. 1998, 120, 8531-8532]. Binding studies demonstrated that a single
incorporation could enhance the binding affinity of a model oligonucleotide to
its
complementary target DNA or RNA with a OTm of up to 18 °C relative to 5-
methyl cytosine (dC5"'~, which is the highest known affinity enhancement for a
single modification, yet. On the other hand, the gain in helical stability
does not
compromise the specificity of the oligonucleotides. The Tm data indicate an
even
greater discrimination between the perfect match and mismatched sequences
compared to dCSme. It was suggested that the tethered amino group serves as an
additional hydrogen bond donor to interact with the Hoogsteen face, namely the
06, of a complementary guanine thereby forming 4 hydrogen bonds. This means
that the increased affinity of G-clamp is mediated by the combination of
extended
base stacking and additional specific hydrogen bonding.
Further polycyclic heterocyclic base moieties and methods of using them
that are amenable to the present invention are disclosed in United States
Patent
Serial Number 6,028,183, which issued on May 22, 2000, and United States
Patent Serial Number 6,007,992, which issued on December 28, 1999, the
contents of both are commonly assigned with this application and are
incorporated
herein in their entirety. Such compounds include those having the formula:
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-18-
Wherein Rl1 includes (CH3)2N-(CHa)2-O-; HZN-(CH2)3-; Ph-CHa-O-
C(=O)-N(H)-(CHZ)3-; H2N-; Fluorenyl-CHZ-O-C(=O)-N(H)-(CHa)s-;
Phthalimidyl-CH2-O-C(=O)-N(H)-(CH2)3-; Ph-CH2-O-C(=O)-N(H)-(CH2)Z-O-;
Ph-CHZ-O-C(=O)-N(H)-(CHZ)3-O-; (CH3)2N-N(H)-(CH2)Z-O-; Fluorenyl-CH2-O-
C(=O)-N(H)-(CH2)2-O-; Fluorenyl-CH2-O-C(=O)-N(H)-(CHZ)3-O-; H2N-(CHZ)a-
O-CH2-; N3-(CHa)2-O-CH2-; H2N-(CHa)2-O-, and NH2C(--NH)NH-.
Also disclosed are polycyclic heterocyclic compounds of the formula:
Wherein
Rloa is O, S or N-CH3;
Rm is A(Z)Xl,wherein A is a spacer and Z independently is a label
bonding group bonding group optionally bonded to a detectable label, but Rl is
is
not amine, protected amine, nitro or cyano;
Xl is 1, 2 or 3; and
Rb is independently -CH=, -N=, -C(C1_8 alkyl= or -C(halogen)=,
but no adjacent Rb are both -N=, or two adjacent Rb are taken together to form
a
ring having the structure:
I'°_R
RJR
where R~ is independently -CH=, -N=, -C(C1_8 alkyl= or -
C(halogen)=, but no adjacent Rb are both -N=.
The enhanced binding affinity of the phenoxazine derivatives together
with their uncompromised sequence specificity makes these polycyclic
heterocyclic base moieties valuable nucleobase analogs for the development of
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-19-
more potent antisense-based drugs. In fact, promising data have been derived
from in vitro experiments demonstrating that heptanucleotides containing
phenoxazine substitutions are capable to activate RNaseH, enhance cellular
uptal~e
and exhibit an increased antisense activity [Lin, K.-Y.; Matteucci, M. J. Am.
Chem. Soc. 1998, 120, 8531-8532]. The activity enhancement was even more
pronounced when the heterocyclic heterocyclic base moiety was the "G-clamp"
where a single substitution was shown to significantly improve the in vitro
potency of 20 mer 2'-deoxyphosphorothioate oligonucleotides [Flanagan, W. M.;
Wolf, J.J.; Olson, P.; Grant, D.; Lin, I~.-Y.; Wagner, R. W.; Matteucci, M.
Proc.
Natl. Acad. Sci. USA, 1999, 96, 3513-3518]. Nevertheless, to optimize
oligonucleotide design and to better understand the impact of these polycyclic
heterocyclic base modifications on biological activity, it is important to
evaluate
their effect on nuclease stability of the oligomers.
Further polycyclic heterocyclic base moieties comprising tricyclic and
tetracyclic heteroaryl compounds amenable to the present invention include
those
having the formulas:
R14
wherein R14 is N02 or both R14 and R1z are independently -CH3. The
synthesis of these compounds is dicslosed in United States Patent Serial
Number
5,434,257, which issued on July 18, 1995, United States Patent Serial Number
5,502,177, which issued on March 26, 1996, and United States Patent Serial
Number 5,646, 269, which issued on July 8, 1997, the contents of which are
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-20-
commonly assigned with this application and are incorporated herein in their
entirety.
Further polycyclic heterocyclic base moieties amenable to the present
invention also disclosed in the "257, 177 and 269" Patents include those
having
the formula:
A
(X) ~ (~')b
NH
~N\ /N
~O
a and b are independently 0 or 1 with the total of a and b being 0 or 1;
A is N, C or CH;
X is S, O, C=O, NH or NCH2, R~;
Y is C=O;
Z is taken together with A to form an aryl or heteroaryl ring structure
comprising 5 or 6 ring atoms wherein the heteroaryl ring comprises a single O
ring heteroatom, a single N ring heteroatom, a single S ring heteroatom, a
single O
and a single N ring heteroatom separated by a carbon atom, a single S and a
single
N ring heteroatom separated by a C atom, 2 N ring heteroatoms separated by a
carbon atom, or 3 N ring heteroatoms at least 2 of which are separated by a
carbon
atom, and wherein the aryl or heteroaryl ring carbon atoms are unsubstituted
with
other than H or at least 1 nonbridging ring carbon atom is fubstituted with
RZ° or
=O;
or Z is taken together with A to form an aryl ring structure comprising 6
ring atoms wherein the aryl ring carbon atoms are unsubstituted with other
than H
or at least 1 nonbridging ring carbon atom is substituted with R6 or =O;
R6 is independently H, C1_6 alkyl, C2_6 alkenyl, C~_6 alkynyl, NOa, N(R3)z,
CN or halo, or an R6 is taken together with an adjacent Z group R6 to complete
a
phenyl ring;
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-21-
RZ° is , independently, H, Cl_~ alkyl, C2_6 alkyl, CZ_6 alkenyl, C2_6
alkynyl,
N02, N(RZ1)Z, CN, or halo, or an RZ° is taken together with an adjacent
Ra° to
complete a ring containing 5 or 6 ring atoms, and tautomers, solvates and
salts
thereof;
R21 is, independently, H or a protecting group;
R3 is a protecting group or H; and tautomers, solvates and salts thereof.
More specific examples included in the "257, 177 and 269" Patents are
compounds of the formula:
~ RI6
16 O ~ ~ ~ R16
Rt6
or ~Rlz
N alz
~16
,N N
N
O
O~ N
6
R
R16 R16
.~~~1 R
i
HN
N, (O or S
O~N
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-22-
wherein each RI6, is, independently, selected from hydrogen and various
substituent groups.
The present invention provides oligomeric compounds comprising a
plurality of linked nucleosides wherein the preferred internucleoside linkage
is a
3',5'-linkage. Alternatively, 2',5'-linkages can be used (as described in U.S.
Application Serial No. 09/115,043, filed July 14, 1990. A 2',5'-linkage is one
that
covalently connects the 2'-position of the sugar portion of one nucleotide
subunit
with the 5'-position of the sugar portion of an adj acent nucleotide subunit.
The compounds described herein may have asymmetric centers. Unless
otherwise indicated, all chiral, diastereomeric, and racemic forms are
included in
the present invention. Geometric isomers may also be present in the compounds
described herein, and all such stable isomers are contemplated by the present
invention. It will be appreciated that compounds in accordance with the
present
invention that contain asymmetrically substituted carbon atoms may be isolated
in
optically active or racemic forms or by synthesis.
The present invention includes all isotopes of atoms occurring in the
intermediates or final compounds. Isotopes include those atoms having the same
atomic number but different mass numbers. By way of example, and without
limitation, isotopes of hydrogen include tritium and deuterium.
As used herein, the term "sugar substituent group" refers to optionally
protected groups that are attached to selected sugar moieties at the 2', 3',
or 5'-
position. Sugar substituent groups have also been attached to heterocyclic
base
moieties for example by attachment at amino functionalities.
A representative list of sugar substituent groups amenable to the present
invention include hydroxyl, C1-C2o alkyl, CZ-C2o alkenyl, CZ-C2o alkynyl, CS-
C2o
aryl, O-alkyl, O-alkenyl, O-alkynyl, O-alkylamino, O-alkylalkoxy, O-
alkylaminoalkyl, O-alkyl imidazole, S-alkyl, S-alkenyl, S-alkynyl, NH-alkyl,
NH-
alkenyl, NH-alkynyl, N-dialkyl, O-aryl, S-aryl, NH-aryl, O-aralkyl, S-aralkyl,
NH-aralkyl, N-phthalimido, halogen (particularly fluoro), amino, thiol, keto,
carboxyl, nitro, nitroso, nitrile, trifluoromethyl, trifluoromethoxy,
imidazole,
azido, hydrazino, hydroxylamino, isocyanato, sulfoxide, sulfone, sulfide,
disulfide, silyl, aryl, heterocycle, carbocycle, intercalators, reporter
groups,
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-23-
conjugates, polyamine, polyamide, polyalkylene glycol, and polyethers of the
formula (O-alkyl)",, where m is 1 to about 10. Preferred among these
polyethers
are linear and cyclic polyethylene glycols (PEGS), and (PEG)-containing
groups,
such as crown ethers and those which are disclosed by Ouchi et al. (Dr ug
Design
and Discovery 1992, 9, 93), Ravasio et al. (.I. Or~g. Chem. 1991, 56, 4329)
and
Delgardo et. al. (Critical Reviews in Therapeutic Drug Car~Yier~ Systems 1992,
9,
249), each of which is herein incorporated by reference in its entirety.
Further
sugar modifications are disclosed in Cook, P.D., Anti-Carzcer° Drug
Design, 1991,
6, 585-607. Fluoro, O-alkyl, O-alkylamino, O-alkyl imidazole, O-
alkylaminoalkyl, and alkyl amino substitution is described in United States
Patent
Application serial number 08/398,901, filed March 6, 1995, entitled Oligomeric
Compounds having Pyrimidine Nucleotides) with 2' and 5' Substitutions, hereby
incorporated by reference in its entirety.
Additional sugar substituent groups amenable to the present invention
include -SRl and -N(Rl)Z groups, wherein each Rl is, independently, hydrogen,
a
protecting group or substituted or unsubstituted alkyl, alkenyl, or alkynyl.
2'-S-Rl
nucleosides are disclosed in United States Patent No. 5,670,633, issued
September
23, 1997, hereby incorporated by reference in its entirety. The incorporation
of 2'-
S Rl monomer synthons are disclosed by Hamm et al., J. Or~g. Clzem., 1997, 62,
3415-3420. 2'-N(Rl)2 nucleosides are disclosed by Goettingen, M., J. Org.
Chern., 1996, 61, 6273-6281; and Polushin et al., Tetralzed>~on Lett., 1996,
37,
3227-3230.
Further representative sugar substituent groups can include groups having
the structure of one of formula I or II:
lzqZl~zS)q5
ZO (CHa)ql O q2 (CH2)q4 J E
~4
2$ q3
I II
wherein:
Zo is O, S or NH;
J is a single bond, O or C(=O);
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-24-
E is C1-Cio alkyl, N(Rs)(R6), N(Rs)(R~), N=C(Rsa)(Rsa), N=C(Rsa)(R~a) or
has formula III;
~N-R9
N C'
R8 N Rli
R12
III
each R8, R9, Rl l and Rla is, independently, hydrogen, C(O)R13, substituted
or unsubstituted C1-Clo alkyl, substituted or unsubstituted C2-Clo alkenyl,
substituted or unsubstituted CZ-Clo alkynyl, alkylsulfonyl, arylsulfonyl, a
chemical
functional group or a conjugate group, wherein the substituent groups are
selected
from hydroxyl, amino, alkoxy, carboxy, benzyl, phenyl, nitro, thiol,
thioalkoxy,
halogen, alkyl, aryl, alkenyl and alkyriyl;
or optionally, Rl l and R12, together form a phthalimido moiety with the
nitrogen atom to which they are attached;
each R13 is, independently, substituted or unsubstituted C1-Clo alkyl,
trifluoromethyl, cyanoethyloxy, methoxy, ethoxy, t-butoxy, allyloxy, 9-
fluorenylmethoxy, 2-(trimethylsilyl)-ethoxy, 2,2,2-trichloroethoxy, benzyloxy,
butyryl, iso-butyryl, phenyl or aryl;
RS is hydrogen, a iutrogen protecting group or -T-L,
R5a is hydrogen, a nitrogen protecting group or -T-L,
T is a bond or a linking moiety;
L is a chemical functional group, a conjugate group or a solid support
material;
each R~ and R~ is, independently, H, a nitrogen protecting group,
substituted or unsubstituted Cl-Clo alkyl, substituted or unsubstituted CZ-Cio
alkenyl, substituted or unsubstituted CZ-Clo alkynyl, wherein said
substitution is
hydroxyl, amino, alkoxy, carboxy, benzyl, phenyl, nitro, thiol, thioalkoxy,
halogen, alkyl, aryl, alkenyl, alkynyl; NH3+, N(R14)(Rls), guanidino or acyl
where
said acyl is an acid amide or an ester;
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-25-
or R~ and R~, together, are a nitrogen protecting group, are joined in a ring
structure that optionally includes an additional heteroatom selected from N
and O
or are a chemical functional group;
each R14 and Rls is, independently, H, C1-Clo alkyl, a nitrogen protecting
group, or R14 and Rls, together, are a nitrogen protecting group;
or R14 and R15 axe joined in a ring structure that optionally includes an
additional heteroatom selected from N and O;
Z4 is OX, SX, or N(X)2;
each X is, independently, H, Ci-C$ alkyl, C1-Cg haloalkyl,
C(=NH)N(H)R16, C(=O)N(H)R16 or OC(=O)N(H)R16;
RI6 is H or C1-C8 alkyl;
ZI, Z2 and Z3 comprise a ring system having from about 4 to about 7
carbon atoms or having from about 3 to about 6 carbon atoms and 1 or 2
heteroatoms wherein said heteroatoms are selected from oxygen, nitrogen and
sulfur and wherein said ring system is aliphatic, unsaturated aliphatic,
aromatic, or
saturated or unsaturated heterocyclic;
ZS is alkyl or haloalkyl having 1 to about 10 carbon atoms, alkenyl having
2 to about 10 caxbon atoms, alkynyl having 2 to about 10 carbon atoms, aryl
having 6 to about 14 carbon atoms, N(RS)(R6) ORS, halo, SRS or CN;
each q1 is, independently, an integer from 1 to 10;
each q2 is, independently, 0 or l;
q3 is 0 or an integer from 1 to 10;
q4 is an integer from 1 to 10;
q5 is from 0, 1 or 2; and
provided that when q3 is 0, q4 is greater than 1.
Representative sugar substituent groups of Formula I are disclosed in
United States Patent Application Serial No. 09/130,973, filed August 7, 1998,
entitled "Capped 2'-Oxyethoxy Oligonucleotides," hereby incorporated by
reference in its entirety.
Representative cyclic sugar substituent groups of Formula II are disclosed
in United States Patent Application Serial No. 09/123,108, filed July 27,
1998,
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-26-
entitled "RNA Targeted 2'-Modified Oligonucleotides that are Conformationally
Preorganized," hereby incorporated by reference in its entirety.
Particularly preferred sugar substituent groups include O[(CHZ)p10]p2CH3,
O(CH2)pl OCH3, O(CHZ) plNH2, O(CH2) plCH3, O(CH2) p1 ONH2, and O(CH2)
plON[(CHZ)plCH3)]2, where p1 and p2 are from 1 to about 10.
Some preferred oligomeric compounds of the invention contain at least
one nucleoside having one of the following sugar substituent groups: C1 to Clo
lower alkyl, substituted lower alkyl, alkaryl, aralkyl, O-alkaryl or O-
aralkyl, SH,
SCH3, OCN, Cl, Br, CN, CF3, OCF3, SOCH3, S02CH3, ONO2, NOz, N3, NH2,
heterocycloalkyl, heterocycloalkaryl, aminoalkylamino, polyalkylamino,
substituted silyl, an RNA cleaving group, a reporter group, an intercalator, a
group
for improving the pharmacokinetic properties of an oligomeric compound, or a
group for improving the pharmacodynamic properties of an oligomeric compound,
and other sugar substituent groups having similar properties. A preferred
modification includes 2'-methoxyethoxy [2'-O-CHZCH2OCH3, also known as 2'-
O-(2-methoxyethyl) or 2'-1V1OE] (Martin et al., Helv. Claifra. Acta, 1995, 78,
486),
i. e., an alkoxyalkoxy group. A further preferred modification is 2'-
dimethylaminooxyethoxy, i.e., a O(CH2)20N(CH3)Z group, also known as 2'-
DMAOE. Representative aminooxy sugar substituent groups are described in co-
owned United States Patent Application serial number 09/344,260, filed June
25,
1999, entitled "Aminooxy-Functionalized Oligomers"; and United States Patent
Application serial number 091370,541, filed August 9, 1999, entitled "Aminooxy-
Functionalized Oligomers and Methods for Making Same;" hereby incorporated
by reference in their entirety.
Other preferred modifications include 2'-methoxy (2'-O-CH3), 2'-
aminopropoxy (2'-OCH2CH2CHZNH2) and 2'-fluoro (2'-F). Similar modifications
may also be made at other positions on nucleosides and oligomers, particularly
the
3' position of the sugar on the 3' terminal nucleoside or at a 3'-position of
a
nucleoside that has a linkage from the 2'-position such as a 2'-5' linked
oligomer
and at the 5' position of a 5' terminal nucleoside. Oligomers may also have
sugar
mimetics such as cyclobutyl moieties in place of the pentofuranosyl sugar.
Representative United States patents that teach the preparation of such
modified
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-27-
sugars structures include, but are not limited to, U.S. Patents 4,981,957;
5,118,800; 5,319,080; 5,359,044; 5,393,878; 5,446,137; 5,466,786; 5,514,785;
5,519,134; 5,567,811; 5,576,427; 5,591,722; 5,597,909; 5,610,300; 5,627,0531
5,639,873; 5,646,265; 5,658,873; 5,670,633; and 5,700,920, certain of which
are
connnonly owned, and each of which is herein incorporated by reference, and
commonly owned United States patent application 08/468,037, filed on June 5,
1995, also herein incorporated by reference.
Representative guanidino sugar substituent groups that are shown in
formula III are disclosed in co-owned United States Patent Application
09/612,531, entitled "Guinidinium Functionalized Oligomers and Methods", filed
July 7, 2000, hereby incorporated by reference in its entirety.
Representative acetamido sugar substituent groups are disclosed in United
States Patent Application 09/378,568, entitled "2'-O-Acetamido Modified
Monomers and Oligomers", filed August 19, 1999, hereby incorporated by
reference in its entirety.
Representative dimethylaminoethyloxyethyl sugar substituent groups are
disclosed in International Patent Application PCT/LTS99/17895, entitled "2'-O-
Dimethylaminoethyloxyethyl-Modified Oligonucleotides", filed August 6, 1999,
hereby incorporated by reference in its entirety.
A further prefered modification includes Locked Nucleic Acids (LNAs) in
which the 2'-hydroxyl group is linked to the 3' or 4' carbon atom of the sugar
ring
thereby forming a bicyclic sugar moiety. The linkage is preferably a methelyne
(-
CH2-)n group bridging the 2' oxygen atom and the 4' carbon atom wherein n is 1
or 2. LNAs and preparation thereof are described in WO 98/39352 and WO
99/14226.
It is not necessary for all positions in a given compound to be uniformly
modified, and in fact more than one of the aforementioned modifications may be
incorporated in a single compound or even at a single nucleoside within an
oligonucleotide.
The present invention also includes oligomeric compounds that are
chimeric compounds. "Chimeric" oligomeric compounds or "chimeras," in the
context of this invention, are oligomeric compounds, particularly
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-28-
oligonucleotides, that contain two or more chemically distinct regions, each
made
up of at least one monomer unit, i.e., a nucleotide in the case of an
oligonucleotide
compound. Chimeric oligonucleotides typically contain at least one region
wherein the oligonucleotide is modified so as to confer increased resistance
to
nuclease degradation, increased cellular uptake, and/or increased binding
affinity
for the target nucleic acid upon the oligonucleotide. An additional region of
the
oligonucleotide may serve as a substrate for enzymes capable of cleaving
RNA:DNA or RNA:RNA hybrids. By way of example, RNase H is a cellular
endonuclease which cleaves the RNA strand of an RNA:DNA duplex. Activation
of RNase ,H, therefore, results in cleavage of the RNA target, thereby greatly
enhancing the efficiency of oligonucleotide inhibition of gene expression.
Consequently, comparable results can often be obtained with shorter
oligonucleotides when chimeric oligonucleotides are used, compared to
phosphorothioate deoxyoligonucleotides hybridizing to the same target region.
Cleavage of the RNA target can be routinely detected by gel electrophoresis
and,
if necessary, associated nucleic acid hybridization techniques known in the
art.
Chimeric oligomeric compounds of the invention may be formed as
composite structures of two or more oligonucleotides, modified
oligonucleotides,
oligonucleosides and/or oligonucleotide mimetics as described above. Such
compounds have also been referred to in the art as hybrids or gapmers.
Representative United States patents that teach the preparation of such hybrid
structures include, but are not limited to, U.S.: 5,013,830; 5,149,797;
5,220,007;
5,256,775; 5,366,878; 5,403,711; 5,491,133; 5,565,350; 5,623,065; 5,652,355;
5,652,356; and 5,700,922, certain of which are commonly owned with the instant
application, and each of which is herein incorporated by reference in its
entirety.
In certain embodiments, the oligomeric compounds of the invention can be
chimeric oligonucleotides, including " gapmers," "inverted gapmers," or
"hemimers." In a "hemimer," a single terminal (either 5' or 3') region of the
oligonucleotide contains modified nucleosides. When both termini of the
oligonucleotide contain modified nucleosides, the oligonucleotide is called a
"gapmer" and the modified 5'- and 3'-terminal regions are referred to as
"wings".
In a gapmer, the 5' and 3' wings can contain nucleosides modified in the same
or
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-29-
different manner. In an "inverted gapmer" a central region of the
oligonucleotide
contains modified nucleosides.
The present invention provides compounds and methods that are useful for
enhancing the nuclease resistance of oligomeric compounds. More specifically,
the present invention is directed to oligomeric compounds that exhibit
enhanced
nuclease resistance, and to methods for improving the nuclease stability of
oligomeric compounds. As noted above, resistance to enzymatic degradation is
an
important feature of antisense oligonucleotide therapeutics, and the efficacy
of
antisense oligonucleotide drugs has been hampered by the activity of nucleases
present in biological systems. Surprisingly, it has been discovered that
certain
modifications of oligomeric compounds enhance their nuclease stability. Novel
methods for increasing the nuclease stability of oligomeric compounds
involving
the incorporation of modified nucleosides have also been discovered.
The present invention is directed to nuclease-resistant oligomeric
compounds that may be useful as pharmaceuticals. Antisense oligonucleotides
can be designed to bind in predictable ways to certain nucleic acid target
sequences, which can cause selective inhibition of the expression of genes
whose
products lead to disease. Antisense oligonucleotides can bind to specific
complementary regions on mRNA, thereby inhibiting protein biosynthesis through
the disruption of processes such as splicing, polyadenylation, correct RNA
folding, translocation and initiation of translation of mRNA, or ribosome
movement along the mRNA. The oligomeric compounds of the invention
typically exhibit enhanced nuclease resistance and can be used as effective
antisense oligonucleotides in therapeutic applications for the treatment of
specific
diseases. The methods of the invention can also be used to increase the
efficacy
of antisense oligonucleotides as therapeutics through enhancement of the
nuclease
resistance of oligomeric compounds.
Preferred embodiments of the invention include nuclease resistant
oligomeric compounds that comprise at least one modified 5' or 3' terminal
nucleoside or nucleotide and at least one internucleoside linking group other
than
phosphodiester, and optionally comprise modified 2' substituent groups in the
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-30-
gapmer, hemimer, and inverted gapmer configuration and one or more modified
nucleobases.
The tricyclic cytosine analogs phenoxazine and 9-
(aminoethoxy)phenoxazine (G-clamp) have been shown to significantly enhance
the nuclease resistance of oligonucleotides. Phenoxazine and G-clamp were
incorporated into model oligomers with a natural phosphodiester backbone and
enzymatic degradation was monitored after treatment with snake venom
phosphodiesterase. A single incorporation of either phenoxazine or G-clamp at
the 3' terminus completely protected the oligonucleotides against 3'
exonuclease
attack. The nuclease resistance of oligonucleotides containing phenoxazine and
G-clamp is not believed to be caused by low binding affinity for the enzyme's
active site, as the modified oligonucleotides are capable of slowing down the
degradation of a natural DNA fragment by bovine intestinal mucosal
phosphodiesterase in a dose-dependent manner. No significant difference was
observed between phenoxazine and G-clamp in terms of their effects on nuclease
resistance and their capacity to inhibitmuclease activity.
A guanidinyl moiety can be added to an oligonucleotide by postsynthetic
guanidinylation of a primary amino group tethered to either the 2'-position or
to
the phenoxazine ring system of a tricyclic cytosine analog (G-clamp). The
former
amino group can be selectively deprotected and guanidinylated on the solid
support, while the aminoethoxy tether of G-clamp can be guanidinylated in
aqueous solution after deprotection and cleavage of the oligonucleotide from
the
support. Both methods have been successfully used to synthesize and
characterize
various guanidinyl-modified oligonucleotides. The conversion of a primary
amine
to a guanidinium moiety, which has a significantly higher pKa than a primary
amine, allows a positive charge to be introduced to the oligonucleotide, which
is
maintained over a wide pH range. The introduction of cationic residues at the
2'-
position greatly enhances the nuclease resistance of oligonucleotides
(Prakash, T.
P.; Kawasaki, A. M.; Vasquez, G.; Fraser, A. S.; Casper, M. D.; Cook, P. D.;
Manoharan, M. Nucleosides Nucleotides 1999,18, 1381-1382). X-ray
crystallography studies of a decamer duplex containing guanidinyl G-clamp
nucleotides revealed an additional Hoogsteen bond between the imino or amino
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-31-
nitrogens of the tethered guanidinium and N7 of a complementary guanine base,
which was the first observation of a single base pair within a nucleic acid
duplex
containing a total number of five hydrogen bonds.
The current method of choice for the preparation of oligomeric compounds
uses support media. Support media is used to attach a first nucleoside or
larger
nucleosidic synthon which is then iteratively elongated to give a final
oligomeric
compound. Support media can be selected to be insoluble or have variable
solubility in different solvents to allow the growing oligomer to be kept out
of or
in solution as desired. Traditional solid supports are insoluble and are
routinely
placed in a reaction vessel while reagents and solvents react and or wash the
growing chain until cleavage frees the final oligomer. More recent approaches
have introduced soluble supports including soluble polymer supports to allow
precipitating and dissolving the bound oligomer at desired points in the
synthesis
(Graven et al., Chem. Rev., 1997, 97, 489-510).
Representative support media that are amenable to the methods of the
present invention include without limitation: controlled pore glass (CPG);
oxalyl-
controlled pore glass (see, e.g., Alul, et al., Nucleic Acids Research 1991,
19,
1527); TENTAGEL Support, (see, e.g., Wright, et al., Tetrahedron Letters 1993,
34, 3373); or POROS, a copolymer of polystyrene/divinylbenzene available from
Perceptive Biosystems. The use of a soluble support media, polyethylene
glycol), with molecular weights between 5 and 20 kDa, for large-scale
synthesis
of phosphorothioate oligonucleotides is described in, Bonora et al., Organic
Process Research & Development, 2000, 4, 225-231. Equipment for such
synthesis is sold by several vendors including, for example, Applied
Biosystems
(Foster City, CA). Any other means for such synthesis known in the an may
additionally or alternatively be employed. It is well known to use similar
techniques to prepare oligonucleotides such as the phosphorothioates and
alkylated derivatives.
Activated phosphorus compositions (e.g. compounds having activated
phosphorus-containing substituent groups) may be used in coupling reactions
for
the synthesis of oligomeric compounds. As used herein, the term "activated
phosphorus composition" includes monomers and oligomers that have an
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-32-
activated phosphorus-containing substituent group that is reactive with a
hydroxyl
group of another monomeric or oligomeric compound to form a phosphorus-
containing internucleotide linkage. Such activated phosphorus groups contain
activated phosphorus atoms in pIii valence state. Such activated phosphorus
atoms
are known in the art and include, but are not limited to, phosphoramidite, H-
phosphonate, phosphate triesters and chiral auxiliaries. A preferred synthetic
solid
phase synthesis utilizes phosphoramidites as activated phosphates. The
phosphoramidites utilize Plji chemistry. The internzediate phosphite compounds
are subsequently oxidized to the Pv state using known methods to yield, in a
preferred embodiment, phosphodiester or phosphorothioate internucleotide
linkages. Additional activated phosphates and phosphites are disclosed in
Tetrahedron Report Number 309 (Beaucage and Iyer, Tetralzed~on, 1992, 48,
2223-2311).
A representative list of activated phosphorus containing monomers or
oligomers include those having the formula:
Bx
m
wherein
each Bx is, independently, a heterocyclic base moiety or a blocked
heterocyclic base moiety; and
each Rl~ is, independently, H, a blocked hydroxyl group, a sugar
substituent group, or a blocked substituent group;
W~ is an hydroxyl protecting group, a nucleoside, a nucleotide, an
oligonucleoside or an oligonucleotide;
Rl8 is N(Ll)L2;
each Ll and LZ is, independently, Cl_6 alkyl;
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-33-
or Ll and Lz are joined together to forth a 4- to 7-membered heterocyclic
ring system including the nitrogen atom to which LI and Lz are attached,
wherein
said ring system optionally includes at least one additional heteroatom
selected
from O, N and S; and
R19 is Xl;
Xl is Pg-O-, Pg-S-, C1-Clo straight or branched chain alkyl, CH3(CHz)ps-
O- or
RzoRz iN-;
p5 is from 0 to 10;
Pg is a protecting group;
each Rzo and Rzl is, independently, hydrogen, Cl-Clo alkyl, cycloalkyl or
aryl;
or optionally, Rzo and Rzl, together with the nitrogen atom to which they
are attached form a cyclic moiety that may include an additional heteroatom
selected from O, S and N; or
Rl8 and R19 together with the phosphorus atom to which Rl $ and Rl9 are
attached form a chiral auxiliary.
Groups that are attached to the phosphorus atom of internucleotide
linkages before and after oxidation (R18 and R19) can include nitrogen
containing
cyclic moieties such as morpholine. Such oxidized internucleoside linkages
include a phosphoromorpholidothioate linkage (Wilk et al., Nucleosides afid
nucleotides, 1991, 10, 319-322). Further cyclic moieties amenable to the
present
invention include mono-, bi- or tricyclic ring moieties which may be
substituted
with groups such as oxo, acyl, alkoxy, alkoxycarbonyl, alkyl, alkenyl,
alkynyl,
amino, amide, azido, aryl, heteroaryl, carboxylic acid, cyano, guanidine,
halo,
haloalkyl, haloalkoxy, hydrazine, ODMT, alkylsulfonyl, nitre, sulfide,
sulfone,
sulfonamide, thiol and thioalkoxy. A preferred bicyclic ring structure that
includes nitrogen is phthalimido.
In the context of this specification, alkyl (generally C1-C20), alkenyl
(generally C2-C20), and alkynyl (generally C2-C20) groups include but are not
limited to substituted and unsubstituted straight chain, branch chain, and
alicyclic
hydrocarbons, including methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl,
octyl,
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-34-
nonyl, decyl, undecyl, dodecyl, tridecyl, tetradecyl, pentadecyl, hexadecyl,
heptadecyl, octadecyl, nonadecyl, eicosyl and other higher carbon alkyl
groups.
Further examples include 2-methylpropyl, 2-methyl-4-ethylbutyl, 2,4-
diethylbutyl, 3-propylbutyl, 2,8-dibutyldecyl, 6,6-dimethyloctyl, 6-propyl-6-
butyloctyl, 2-methylbutyl, 2-methylpentyl, 3-methylpentyl, 2-ethylhexyl and
other
branched chain groups, allyl, crotyl, propargyl, 2-pentenyl and other
unsaturated
groups containing a pi bond, cyclohexane, cyclopentane, adamantane as well as
other alicyclic groups, 3-penten-2-one, 3-methyl-2-butanol, 2-cyanooctyl, 3-
methoxy-4-heptanal, 3-nitrobutyl, 4-isopropoxydodecyl, 4-azido-2-nitrodecyl, 5-
mercaptononyl, 4-amino-1-pentenyl as well as other substituted groups.
Representative alkyl substituents are disclosed in United States Patent No.
5,212,295, at column 12, lines 41-50, hereby incorporated by reference in its
entirety.
A number of chemical functional groups can be introduced into
compounds of the invention in a blocked form and subsequently deblocked to
form a final, desired compound. Such as groups directly or indirectly attached
at
the heterocyclic bases, the internucleoside linkages and the sugar substituent
groups at one or more or the 2', 3' and 5'-positions. Protecting groups can be
selected to block functional groups located in a growing oligomeric compound
during iterative oligonucleotide synthesis while other positions can be
selectively
deblocked as needed. In general, a blocking group renders a chemical
functionality of a larger molecule inert to specific reaction conditions and
can later
be removed from such functionality without substantially damaging the
remainder
of the molecule (Greene and Wuts, Protective Groups in Organic Synthesis, 3rd
ed, John Wiley & Sons, New York, 1999). For example, the nitrogen atom of
amino groups can be blocked as phthalimido groups, as 9-
fluorenylmethoxycarbonyl (F1VIOC) groups, and with triphenylmethylsulfenyl, t-
BOC or benzyl groups. Carboxyl groups can be blocked as acetyl groups.
Representative hydroxyl protecting groups are described by Beaucage et al.,
Tetrahedron 1992, 48, 2223. Preferred hydroxyl protecting groups are acid-
labile,
such as the trityl, monomethoxytrityl, dimethoxytrityl, trimethoxytrityl, 9-
phenylxanthine-9-yl (Pixyl) and 9-(p-methoxyphenyl)xanthine-9-yl (MOX).
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-35-
Chemical functional groups can also be "blocked" by including them in a
precursor form. Thus, an azido group can be used considered as a "blocl~ed"
form
of an amine since the azido group is easily converted to the amine. Further
representative protecting groups utilized in oligonucleotide synthesis are
discussed
in Agrawal, et al., Protocols for Oligonucleotide Conjugates, Eds, Humana
Press;
New Jersey, 1994; Vol. 26 pp. 1-72.
Examples of hydroxyl protecting groups include, but are not limited to, t-
butyl, t-butoxymethyl, methoxymethyl, tetrahydropyranyl, 1-ethoxyethyl, 1-(2-
chloroethoxy)ethyl, 2-trimethylsilylethyl, p-chlorophenyl, 2,4-dinitrophenyl,
benzyl, 2,6-dichlorobenzyl, diphenylmethyl, p,p=-dinitrobenzhydryl, p-
nitrobenzyl, triphenylmethyl, trimethylsilyl, triethylsilyl, t-
butyldimethylsilyl, t-
butyldiphenylsilyl, triphenylsilyl, benzoylformate, acetate, chloroacetate,
trichloroacetate, trifluoroacetate, pivaloate, benzoate, p-phenylbenzoate, 9-
fluorenylmethyl carbonate, mesylate and tosylate.
Examples of thiol (sulfur) protecting groups include, but axe not limited to,
benzyl, substituted benzyls, diphenylmethly, phenyl, t-butyl, methoxymethyl,
thiazolidines, acetyl and benzoyl. Further thiol protecting groups are
illustrated in
Greene and Wuts, ibid.
Additional amino-protecting groups include but are not limited to,
carbamate-protecting groups, such as 2-trimethylsilylethoxycarbonyl (Teoc), 1-
methyl-1-(4-biphenylyl)ethoxycarbonyl (Bpoc), t-butoxycarbonyl (BOC),
allyloxycarbonyl (Alloc), 9-fluorenylinethyloxycarbonyl (Fmoc), and benzyl-
oxycarbonyl (Cbz); aanide-protecting groups, such as formyl, acetyl,
trihaloacetyl,
benzoyl, and nitropherlylacetyl; sulfonamide-protecting groups, such as 2-
nitrobenzenesulfonyl; and imine- and cyclic imide-protecting groups, such as
phthalimido and dithiasuccinoyl. Equivalents of these amino-protecting groups
are also encompassed by the compounds and methods of the present invention.
Some preferred amino-protecting groups are stable to acid treatment and
can be selectively removed with base treatment which make reactive amino
groups selectively available for substitution. Examples of such groups are the
Fmoc (E. Atherton and R.C. Sheppard in The Peptides, S. Udenfriend, J.
Meienhofer, Eds., Academic Press, Orlando, 1987, volume 9, p.1), and various
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-36-
substituted sulfonylethyl carbamates exemplified by the Nsc group (Samukov et
al., Tetrahedron Lett, 1994, 35:7821; Verhart and Tesser, Rec. Trav. Chim.
Pays-
Bas, 1987, 107:621).
In some especially preferred embodiments, the nucleoside components of
the oligomeric compounds are connected to each other by optionally protected
phosphorothioate internucleoside linkages. Representative protecting groups
for
phosphorus containing internucleoside linkages such as phosphite,
phosphodiester
and phosphorothioate linages include ~-cyanoethyl, diphenylsilylethyl, 8-
cyanobutenyl, cyano p-xylyl (CPX), N-methyl-N-trifluoroacetyl ethyl (META),
acetoxy phenoxy ethyl (APE) and butene-4-yl groups. See for example U.S.
Patents Nos. 4,725,677 and Re. 34,069 (~3-cyanoethyl); Beaucage, S.L. and
Iyer,
R.P., Tetrahedron, 49 No. 10, pp. 1925-1963 (1993); Beaucage, S.L. and Iyer,
R.P., Tetrahedron, 49 No. 46, pp. 10441-10488 (1993); Beaucage, S.L. and Iyer,
R.P., Tetrahedron, 48 No. 12, pp.
The present invention also includes pharmaceutical compositions and
formulations that include the oligomeric compounds of the invention. The
pharmaceutical compositions of the present invention may be achninistered in a
number of ways depending upon whether local or systemic treatment is desired
and upon the area to be treated. Administration may be topical (including
ophthalmic and to mucous membranes including vaginal and rectal delivery),
pulmonary, e.g., by inhalation or insufflation of powders or aerosols,
including by
nebulizer; intratracheal, intranasal, epidermal and transdermal), oral or
parenteral.
Paxenteral administration includes intravenous, intraaxterial, subcutaneous,
intraperitoneal or intramuscular injection or infusion; or intracranial, e.g.,
intrathecal or intraventricular, administration. Oligonucleotides with at
least one
2'-O-methoxyethyl modification are believed to be particularly useful for oral
administration.
Pharmaceutical compositions and formulations for topical administration
may include transdermal patches, ointments, lotions, creams, gels, drops,
suppositories, sprays, liquids and powders. Conventional pharmaceutical
carriers,
aqueous, powder or oily bases, thickeners and the like may be necessary or
desirable. Coated condoms, gloves and the like may also be useful. Preferred
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-37-
topical formulations include those in which the oligomeric compounds of the
invention are in admixture with a topical delivery agent such as lipids,
liposomes,
fatty acids, fatty acid esters, steroids, chelating agents and surfactants.
Preferred
lipids and liposomes include neutral (e.g. dioleoylphosphatidyl DOPE
ethanolamine, dimyristoylphosphatidyl choline DMPC, distearolyphosphatidyl
choline) negative (e.g. dimyristoylphosphatidyl glycerol DMPG) and cationic
(e.g.
dioleoyltetramethylaminopropyl DOTAP and dioleoylphosphatidyl ethanolamine
DOTMA). Oligomeric compounds of the invention may be encapsulated within
liposomes or may form complexes thereto, in particular to cationic liposomes.
Alternatively, oligomeric compounds may be complexed to lipids, in particular
to
cationic lipids. Preferred fatty acids and esters include but are not limited
arachidonic acid, oleic acid, eicosanoic acid, lauric acid, caprylic acid,
capric acid,
myristic acid, palmitic acid, stearic acid, linoleic acid, linolenic acid,
dicaprate,
tricaprate, monoolein, dilaurin, glyceryl 1-monocaprate,
1-dodecylazacycloheptan-2-one, an acylcarnitine, an acylcholine, or a C1_10
alkyl
ester (e.g. isopropylmyristate IPM), monoglyceride, diglyceride or
pharmaceutically acceptable salt thereof. Topical formulations are described
in
detail in United States patent application 091315,29 filed on May 20, 1999
which
is incorporated herein by reference in its entirety.
Compositions and formulations for oral administration include powders or
granules, microparticulates, nanoparticulates, suspensions or solutions in
water or
non-aqueous media, capsules, gel capsules, sachets, tablets or minitablets.
Thickeners, flavoring agents, diluents, emulsifiers, dispersing aids or
binders may
be desirable. Preferred oral formulations axe those in which oligomeric
compounds of the invention are administered in conjunction with one or more
penetration enhancers surfactants and chelators. Preferred surfactants include
fatty acids and/or esters or salts thereof, bile acids and/or salts thereof.
Preferred
bile acids/salts include chenodeoxycholic acid (CDCA) and
ursodeoxychenodeoxycholic acid (LTDCA), cholic acid, dehydrocholic acid,
deoxycholic acid, glucholic acid, glycholic acid, glycodeoxycholic acid,
taurocholic acid, taurodeoxycholic acid, sodium tauro-24,25-dihydro-fusidate,
sodium glycodihydrofusidate. Preferred fatty acids include arachidonic acid,
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-38-
undecanoic acid, oleic acid, lauric acid, caprylic acid, capric acid, myristic
acid,
palmitic acid, stearic acid, linoleic acid, linolenic acid, dicaprate,
tricaprate,
monoolein, dilaurin, glyceryl 1-monocaprate, 1-dodecylazacycloheptan-2-one, an
acylcarnitine, an acylcholine, or a monoglyceride, a diglyceride or a
pharmaceutically acceptable salt thereof (e.g. sodium). Also preferred are
combinations of penetration enhancers, for example, fatty acids/salts in
combination with bile acids/salts. A particularly preferred combination is the
sodium salt of lauric acid, capric acid and UDCA. Further penetration
enhancers
include polyoxyethylene-9-lauryl ether, polyoxyethylene-20-cetyl ether.
Oligomeric compounds of the invention may be delivered orally in granular form
including sprayed dried particles, or complexed to form micro or
nanoparticles.
Oligonucleotide complexing agents include poly-amino acids; polyimines;
polyacrylates; polyalkylacrylates, polyoxethanes, polyalkylcyanoacrylates;
cationized gelatins, albumins, starches, acrylates, polyethyleneglycols (PEG)
and
starches; polyalkylcyanoacrylates; DEAF-derivatized polyimines, pollulans,
celluloses and starches. Particularly preferred complexing agents include
chitosan, N-trimethylchitosan, poly-L-lysine, polyhistidine, polyornithine,
polyspermines, protamine, polyvinylpyridine, polythiodiethylamino-
methylethylene P(TDAE), polyaminostyrene (e.g. p-amino),
poly(methylcyanoacrylate), poly(ethylcyanoacrylate), poly(butylcyanoacrylate),
poly(isobutylcyanoacrylate), poly(isohexylcynaoacrylate), DEAE-methacrylate,
DEAE-hexylacrylate, DEAF-acrylamide, DEAE-albumin and DEAE-dextran,
polymethylacrylate, polyhexylacrylate, poly(D,L-lactic acid), poly(DL-lactic-
co-
glycolic acid (PLGA), alginate, and polyethyleneglycol (PEG). Oral
formulations
for oligonucleotides and their preparation axe described in detail in United
States
applications 08/886,829 (filed July 1, 1997), 09/108,673 (filed July 1, 1998),
09/256,515 (filed February 23, 1999), 09/082,624 (filed May 21, 1998) and
09/315,298 (filed May 20, 1999) each of which is incorporated herein by
reference in their entirety.
Compositions and formulations for parenteral, intrathecal or
intraventricular administration may include sterile aqueous solutions which
may
also contain buffers, diluents and other suitable additives such as, but not
limited
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-39-
to, penetration enhancers, carrier compounds and other pharmaceutically
acceptable carriers or excipients.
Pharmaceutical compositions of the present invention include, but are not
limited to, solutions, emulsions, and liposome-containing formulations. These
compositions may be generated from a variety of components that include, but
are
not limited to, preformed liquids, self emulsifying solids and self
emulsifying
semisolids.
The pharmaceutical formulations of the present invention, which may
conveniently be presented in unit dosage form, may be prepared according to
conventional techniques well known in the pharmaceutical industry. Such
techniques include the step of bringing into association the active
ingredients with
the pharmaceutical carriers) or excipient(s). In general the formulations are
prepared by uniformly and intimately bringing into association the active
ingredients with liquid carriers or finely divided solid carriers or both, and
then, if
necessary, shaping the product.
The compositions of the present invention may be formulated into any of
many possible dosage forms such as, but not limited to, tablets, capsules, gel
capsules, liquid syrups, soft gels, suppositories, and enemas. The
compositions of
the present invention may also be formulated as suspensions in aqueous, non-
aqueous or mixed media. Aqueous suspensions may further contain substances
which increase the viscosity of the suspension including, for example, sodium
carboxymethylcellulose, sorbitol and/or dextran. The suspension may also
contain stabilizers.
In one embodiment of the present invention the pharmaceutical
compositions may be formulated and used as foams. Pharmaceutical foams
include formulations such as, but not limited to, emulsions, microemulsions,
creams, jellies and liposomes. While basically similar in nature these
formulations vary in the components and the consistency of the final product.
The
preparation of such compositions and formulations is generally known to those
skilled in the pharmaceutical and formulation arts and may be applied to the
formulation of the compositions of the present invention.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-40-
The compositions of the present invention may be prepared and formulated
as emulsions. Emulsions axe typically heterogenous systems of one liquid
dispersed in another in the form of droplets usually exceeding 0.1 ~,m in
diameter.
(Idson, in Pharmaceutical Dosage Forms, Liebennan, Rieger and Banker (Eds.),
1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 199; Rosoff, in
Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988,
Marcel Dekker, Inc., New York, N.Y., Volume 1, p. 245; Block in Pharmaceutical
Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc.,
New York, N.Y., volume 2, p. 335; Higuchi et al., in Remington's
Pharmaceutical
Sciences, Mack Publishing Co., Easton, PA, 1985, p. 301). Emulsions are often
biphasic systems comprising of two immiscible liquid phases intimately mixed
and dispersed with each other. W general, emulsions may be either water-in-oil
(w/o) or of the oil-in-water (o/w) variety. When an aqueous phase is finely
divided into and dispersed as minute droplets into a bulk oily phase the
resulting
composition is called a water-in-oil (w/o) emulsion. Alternatively, when an
oily
phase is finely divided into and dispersed as minute droplets into a bulk
aqueous
phase the resulting composition is called an oil-in-water (o/w) emulsion.
Emulsions may contain additional components in addition to the dispersed
phases
and the active drug which may be present as a solution in either the aqueous
phase, oily phase or itself as a separate phase. Pharmaceutical excipients
such as
emulsifiers, stabilizers, dyes, and anti-oxidants may also be present in
emulsions
as needed. Pharmaceutical emulsions may also be multiple emulsions that are
comprised of more than two phases such as, for example, in the case of oil-in-
water-in-oil (o/w/o) and water-in-oil-in-water (w/o/w) emulsions. Such complex
formulations often provide certain advantages that simple binary emulsions do
not. Multiple emulsions in which individual oil droplets of an o/w emulsion
enclose small water droplets constitute a w/o/w emulsion. Likewise a system of
oil droplets enclosed in globules of water stabilized in an oily continuous
provides
an o/w/o emulsion.
Emulsions are characterized by little or no thermodynamic stability.
Often, the dispersed or discontinuous phase of the emulsion is well dispersed
into
the external or continuous phase and maintained in this form through the means
of
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-41-
emulsifiers or the viscosity of the formulation. Either of the phases of the
emulsion may be a semisolid or a solid, as is the case of emulsion-style
ointment
bases and creams. Other means of stabilizing emulsions entail the use of
emulsifiers that may be incorporated into either phase of the emulsion.
Emulsifiers may broadly be classified into four categories: synthetic
surfactants,
naturally occurring emulsifiers, absozption bases, and finely dispersed solids
(Idson, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.),
1988, Marcel Dekker, Inc., New York, N.Y., volume l, p. 199).
Synthetic surfactants, also known as surface active agents, have found
wide applicability in the formulation of emulsions and have been reviewed in
the
literature (Rieger, in Pharmaceutical Dosage Forms, Lieberman, Rieger and
Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 285;
Idson, in Pharmaceutical Dosage Forms, Liebennan, Rieger and Banker (Eds.),
Marcel Dekker, Inc., New York, N.Y., 1988, volume 1, p. 199). Surfactants are
typically amphiphilic and comprise a hydrophilic and a hydrophobic portion.
The
ratio of the hydrophilic to the hydrophobic nature of the surfactant has been
termed the hydrophile/lipophile balance (HLB) and is a valuable tool in
categorizing and selecting surfactants in the preparation of formulations.
Surfactants may be classified into different classes based on the nature of
the
hydrophilic group: nonionic, anionic, cationic and amphoteric (Rieger, in
Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988,
Maxcel Dekker, Inc., New York, N.Y., volume 1, p. 285).
Naturally occurring emulsifiers used in emulsion formulations include
la~lolin, beeswax, phosphatides, lecithin and acacia. Absorption bases possess
hydrophilic properties such that they can soak up water to form w/o emulsions
yet
retain their semisolid consistencies, such as anhydrous lanolin and
hydrophilic
petrolatum. Finely divided solids have also been used as good emulsifiers
especially in combination with surfactants and in viscous preparations. These
include polar inorganic solids, such as heavy metal hydroxides, nonswelling
clays
such as bentonite, attapulgite, hectorite, kaolin, montmorillonite, colloidal
aluminum silicate and colloidal magnesium aluminum silicate, pigments and
nonpolar solids such as carbon or glyceryl tristearate.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-42-
A large variety of non-emulsifying materials are also included in emulsion
fonnulations and contribute to the properties of emulsions. These include
fats,
oils, waxes, fatty acids, fatty alcohols, fatty esters, humectants,
hydrophilic
colloids, preservatives and antioxidants (Block, in Pharmaceutical Dosage
Forms,
Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York,
N.Y., volume 1, p. 335; Idson, in Pharmaceutical Dosage Forms, Lieberman,
Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume l,
p. 199). Hydrophilic colloids or hydrocolloids include naturally occurring
gums
and synthetic polymers such as polysaccharides (for example, acacia, agar,
alginic
acid, carrageenan, guar gum, karaya gum, and tragacanth), cellulose
derivatives
(for example, carboxymethylcellulose and carboxypropylcellulose), and
synthetic
polymers (for example, carbomers, cellulose ethers, and carboxyvinyl
polymers).
These disperse or swell in water to form colloidal solutions that stabilize
emulsions by forming strong interfacial films around the dispersed-phase
droplets
and by increasing the viscosity of the external phase.
Since emulsions often contain a munber of ingredients such as
carbohydrates, proteins, sterols and phosphatides that may readily support the
growth of microbes, these formulations often incorporate preservatives.
Commonly used preservatives included in emulsion formulations include methyl
paraben, propyl paraben, quaternary ammonium salts, benzalkonium chloride,
esters of p-hydroxybenzoic acid, and boric acid. Antioxidants are also
commonly
added to emulsion formulations to prevent deterioration of the formulation.
Antioxidants used may be free radical scavengers such as tocopherols, alkyl
gallates, butylated hydroxyanisole, butylated hydroxytoluene, or reducing
agents
such as ascorbic acid and sodium metabisulfite, and antioxidant synergists
such as
citric acid, tartaric acid, and lecithin.
The application of emulsion formulations via dermatological, oral and
parenteral routes and methods for their manufacture have been reviewed in the
literature (Idson, in Pharmaceutical Dosage Forms, Lieberman, Rieger and
Banker
(Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 199). Emulsion
formulations for oral delivery have been very widely used because of reasons
of
ease of formulation, efficacy from an absorption and bioavailability
standpoint.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-43-
(Rosoff, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.),
1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 245; Idson, in
Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988,
Marcel Dekker, Inc., New York, N.Y., volume l, p. 199). Mineral-oil base
laxatives, oil-soluble vitamins and high fat nutritive preparations are among
the
materials that have commonly been administered orally as o/w emulsions.
In one embodiment of the present invention, the compositions of
oligomeric compounds and nucleic acids are formulated as microemulsions. A
microemulsion may be defined as a system of water, oil and amphiphile which is
a
single optically isotropic and thermodynamically stable liquid solution
(Rosoff, in
Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988,
Marcel Dekker, Inc., New York, N.Y., volume 1, p. 245). Typically
microemulsions are systems that are prepared by first dispersing an oil in an
aqueous surfactant solution and then adding a sufficient amount of a fourth
component, generally an intermediate chain-length alcohol to form a
transparent
system. Therefore, microemulsions have also been described as
thermodynamically stable, isotropically clear dispersions of two immiscible
liquids that are stabilized by interfacial films of surface-active molecules
(Leung
and Shah, in: Controlled Release of Drugs: Polymers and Aggregate Systems,
Rosoff, M., Ed., 1989, VCH Publishers, New York, pages 185-215).
Microemulsions commonly are prepared via a combination of three to five
components that include oil, water, surfactant, cosurfactant and electrolyte.
Whether the microemulsion is of the water-in-oil (w/o) or an oil-in-water
(o/w)
type is dependent on the properties of the oil and surfactant used and on the
structure and geometric packing of the polar heads and hydrocarbon tails of
the
surfactant molecules (Schott, in Remington's Pharmaceutical Sciences, Mack
Publishing Co., Easton, PA, 1985, p. 271).
I The phenomenological approach utilizing phase diagrams has been
extensively studied arid has yielded a comprehensive knowledge, to one skilled
in
the art, of how to formulate microemulsions (Rosoff, in Pharmaceutical Dosage
Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New
York, N.Y., volume 1, p. 245; Block, in Pharmaceutical Dosage Forms,
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-44-
Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York,
N.Y., volume 1, p. 335). Compared to conventional emulsions, microemulsions
offer the advantage of solubilizing water-insoluble drugs in a formulation of
thermodynamically stable droplets that are formed spontaneously.
Surfactants used in the preparation of microemulsions include, but are not
limited to, ionic surfactants, non-ionic surfactants, Brij 96, polyoxyethylene
oleyl
ethers, polyglycerol fatty acid esters, tetraglycerol monolaurate (ML310),
tetraglycerol monooleate (M0310), hexaglycerol monooleate (P0310),
hexaglycerol pentaoleate (P0500), decaglycerol monocaprate (MCA750),
decaglycerol monooleate (M0750), decaglycerol sequioleate (50750),
decaglycerol decaoleate (DA0750), alone or in combination with cosurfactants.
The cosurfactant, usually a short-chain alcohol such as ethanol, 1-propanol,
and 1-
butanol, serves to increase the interfacial fluidity by penetrating into the
surfactant
film and consequently creating a disordered film because of the void space
generated among surfactant molecules. Microemulsions may, however, be
prepared without the use of cosurfactants and alcohol-free self emulsifying
microemulsion systems are known in the art. The aqueous phase may typically
be, but is not limited to, water, an aqueous solution of the drug, glycerol,
PEG300,
PEG400, polyglycerols, propylene glycols, and derivatives of ethylene glycol.
The oil phase may include but is not limited to, materials such as Captex 300,
Captex 355, Capmul MCM, fatty acid esters, medium chain (C8-C12) mono, di,
and tri-glycerides, polyoxyethylated glyceryl fatty acid esters, fatty
alcohols,
polyglycolized glycerides, saturated polyglycolized C8-C10 glycerides,
vegetable
oils and silicone oil.
Microemulsions are particularly of interest from the standpoint of drug
solubilization and the enhanced absorption of drugs. Lipid based
microemulsions
(both o!w and w!o) have been proposed to enhance the oral bioavailability of
drugs, including peptides (Constantinides et al., Pharmaceutical Research,
1994,
11, 1385-1390; Ritschel, Meth. Find. Exp. Clin. Pharmacol., 1993, 13, 205).
Microemulsions afford advantages of improved drug solubilization, protection
of
drug from enzymatic hydrolysis, possible enhancement of drug absorption due to
surfactant-induced alterations in membrane fluidity and permeability, ease of
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-45-
preparation, ease of oral administration over solid dosage forms, improved
clinical
potency, and decreased toxicity (Constantinides et al., Pharmaceutical
Research,
1994, 11, 1385; Ho et al., J. Pharm. Sci., 1996, 85, 138-143). Often
microemulsions may form spontaneously when their components are brought
together at ambient temperature. This may be particularly advantageous when
formulating thermolabile drugs, peptides or oligonucleotides. Microemulsions
have also been effective in the transdermal delivery of active components in
both
cosmetic and pharmaceutical applications. It is expected that the
microemulsion
compositions and formulations of the present invention will facilitate the
increased systemic absorption of oligonucleotides and nucleic acids from the
gastrointestinal tract, as well as improve the local cellular uptake of
oligonucleotides and nucleic acids within the gastrointestinal tract, vagina,
buccal
cavity and other areas of administration.
Microemulsions of the present invention may also contain additional
components and additives such as sorbitan monostearate (Grill 3), Labrasol,
and
penetration enhancers to improve the properties of the formulation and to
enhance
the absorption of the oligomeric compounds and nucleic acids of the present
invention. Penetration enhancers used in the microemulsions of the present
invention may be classified as belonging to one of five broad categories -
surfactants, fatty acids, bile salts, chelating agents, and non-chelating non-
surfactants (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems,
1991, p. 92). Each of these classes has been discussed above.
There are many organized surfactant structures besides microemulsions
that have been studied and used for the formulation of drugs. These include
monolayers, micelles, bilayers and vesicles. Vesicles, such as liposomes, have
attracted great interest because of their specificity and the duration of
action they
offer from the standpoint of drug delivery. As used in the present invention,
the
term "liposome" means a vesicle composed of amphiphilic lipids arranged in a
spherical bilayer or bilayers.
Liposomes are unilamellar or multilamellar vesicles which have a
membrane formed from a lipophilic material and an aqueous interior. The
aqueous portion contains the composition to be delivered. Cationic liposomes
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-46-
possess the advantage of being able to fuse to the cell wall. Non-cationic
liposomes, although not able to fuse as efficiently with the cell wall, are
taken up
by macrophages in vivo. In order to cross intact mammalian slcin, lipid
vesicles
must pass through a series of fine pores, each with a diameter less than 50
nm,
under the influence of a suitable transdermal gradient. Therefore, it is
desirable to
use a liposome which is highly deformable and able to pass through such fine
pores.
Further advantages of liposomes include; liposomes obtained from natural
phospholipids are biocompatible and biodegradable; liposomes can incorporate a
wide range of water and lipid soluble drugs; liposomes can protect
encapsulated
drugs in their internal compartments from metabolism and degradation (Rosoff,
in
Pharmaceutical Dosage Forms, Liebernian, Rieger and Banker (Eds.),1988,
Marcel Dekker, Inc., New York, N.Y., volume 1, p. 245). Important
considerations in the preparation of liposome formulations are the lipid
surface
charge, vesicle size and the aqueous volume of the liposomes.
Liposomes are useful for the transfer and delivery of active ingredients to
the site of action. Because the liposomal membrane is structurally similar to
biological membranes, when liposomes are applied to a tissue, the liposomes
start
to merge with the cellular membranes. As the merging of the liposome and cell
progresses, the liposomal contents are emptied into the cell where the active
agent
may act.
Liposomal formulations have been the focus of extensive investigation as
the mode of delivery for many drugs. There is growing evidence that for
topical
administration, liposomes present several advantages over other formulations.
Such advantages include reduced side-effects related to high systemic
absorption
of the achninistered drug, increased accumulation of the administered drug at
the
desired target, and the ability to administer a wide variety of drugs, both
hydrophilic and hydrophobic, into the skin.
Several reports have detailed the ability of liposomes to deliver agents
including high-molecular weight DNA into the skin. Compounds including
analgesics, antibodies, hormones and high-molecular weight DNAs have been
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-47-
administered to the skin. The majority of applications resulted in the
targeting of
the upper epidermis.
Liposomes fall into two broad classes. Cationic liposomes are positively
charged liposomes which interact with the negatively charged DNA molecules to
form a stable complex. The positively charged DNA/liposome complex binds to
the negatively charged cell surface and is internalized in an endosome. Due to
the
acidic pH within the endosome, the liposomes are ruptured, releasing their
contents into the cell cytoplasm (Wang et al., Biochem. Biophys. Res. Commun.,
1987, 147, 980-985).
Liposomes which are pH-sensitive or negatively-charged, entrap DNA
rather than complex with it. Since both the DNA and the lipid are similarly
charged, repulsion rather than complex formation occurs. Nevertheless, some
DNA is entrapped within the aqueous interior of these liposomes. pH-sensitive
liposomes have been used to deliver DNA encoding the thymidine kinase gene to
cell monolayers in culture. Expression of the exogenous gene was detected in
the
target cells (Zhou et al., Journal of Controlled Release, 1992, 19, 269-274).
One major type of liposomal composition includes phospholipids other
than naturally-derived phosphatidylcholine. Neutral liposome compositions, for
example, can be formed from dimyristoyl phosphatidylcholine (DMPC) or
dipalinitoyl phosphatidylcholine (DPPC). Anionic liposome compositions
generally are formed from dimyristoyl phosphatidylglycerol, while anionic
fusogenic liposomes are formed primarily from dioleoyl
phosphatidylethanolamine (DOPE). Another type of liposomal composition is
formed from phosphatidylcholine (PC) such as, for example, soybean PC, and egg
PC. Another type is formed from mixtures of phospholipid and/or
phosphatidylcholine and/or cholesterol.
Several studies have assessed the topical delivery of liposomal drug
formulations to the shin. Application of liposomes containing interferon to
guinea
pig skin resulted in a reduction of skin herpes sores while delivery of
interferon
via other means (e.g. as a solution or as an emulsion) were ineffective
(Weiner et
al., Journal of Drug Targeting, 1992, 2, 405-410). Further, an additional
study
tested the efficacy of interferon administered as part of a liposomal
formulation to
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-48-
the administration of interferon using an aqueous system, and concluded that
the
liposomal formulation was superior to aqueous administration (du Plessis et
al.,
Antiviral Research, 1992, 18, 259-265).
Non-ionic liposomal systems have also been examined to determine their
utility in the delivery of drugs to the skin, in particular systems comprising
non-
ionic surfactant and cholesterol. Non-ionic liposomal formulations comprising
NovasomeTM I (glyceryl dilaurate/cholesterol/polyoxyethylene-10-stearyl ether)
and NovasomeTM II (glyceryl distearate/ cholesterol/polyoxyethylene-10-stearyl
ether) were used to deliver cyclosporin-A into the dermis of mouse skin.
Results
indicated that such non-ionic liposomal systems were effective in facilitating
the
deposition of cyclosporin-A into different layers of the skin (Hu et al.
S.T.P.Pharma. Sci., 1994, 4, 6, 466).
Liposomes also include "sterically stabilized" liposomes, a teen which, as
used herein, refers to liposomes comprising one or more specialized lipids
that,
when incozporated into liposomes, result in enhanced circulation lifetimes
relative
to liposomes lacking such specialized lipids. Examples of sterically
stabilized
liposomes are those in which part of the vesicle-forming lipid portion of the
liposome (A) comprises one or more glycolipids, such as monosialoganglioside
GMI, or (B) is derivatized with one or more hydrophilic polymers, such as a
polyethylene glycol (PEG) moiety. While not wishing to be bound by any
particular theory, it is thought in the art that, at least for sterically
stabilized
liposomes containing gangliosides, sphingomyelin, or PEG-derivatized lipids,
the
enhanced circulation half life of these sterically stabilized liposomes
derives from
a reduced uptake into cells of the reticuloendothelial system (RES) (Allen et
al.,
FEBS Letters, 1987, 223, 42; Wu et al., Cancer Research, 1993, 53, 3765).
Various liposomes comprising one or more glycolipids are known in the
art. Papahadjopoulos et al. (Ann. N.Y. Acad. Sci., 1987, 507, 64) reported the
ability of monosialoganglioside GMI, galactocerebroside sulfate and
phosphatidylinositol to improve blood half lives of liposomes. These findings
were expounded upon by Gabizon et al. (Proc. Natl. Acad. Sci. U.S.A., 1988,
85,
6949). U.S. Patent No. 4,837,028 and WO 88/04924, both to Allen et al.,
disclose
liposomes comprising (1) sphingomyelin and (2) the ganglioside GMi or a
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-49-
galactocerebroside sulfate ester. U.S. Patent No. 5,543,152 (Webb et al.)
discloses liposomes comprising sphingomyelin. Liposomes comprising 1,2-sn-
dimyristoylphosphatidylcholine are disclosed in WO 97/13499 (Lim et al.).
Many liposomes comprising lipids derivatized with one or more
hydrophilic polymers, and methods of preparation thereof, are known in the
art.
Sunamoto et al. (Bull. Chem. Soc. Jpn., 1980, 53, 2778) described liposomes
comprising a nonionic detergent, 2C1215G, that contains a PEG moiety. Illum et
al. (FEBS Lett., 1984, 167, 79) noted that hydrophilic coating of polystyrene
particles with polymeric glycols results in significantly enhanced blood half
lives.
Synthetic phospholipids modified by the attachment of carboxylic groups of
polyalkylene glycols (e.g., PEG) are described by Sears (U.S. Patent Nos.
4,426,330 and 4,534,899). I~libanov et al. (FEBS Lett., 1990, 268, 235)
described
experiments demonstrating that liposomes comprising phosphatidylethanolamine
(PE) derivatized with PEG or PEG stearate have significant increases in blood
circulation half lives. Blume et al. (Biochimica et Biophysica Acta, 1990,
1029,
91) extended such observations to other PEG-derivatized phospholipids, e.g.,
DSPE-PEG, formed from the combination of distearoylphosphatidylethanolamine
(DSPE) and PEG. Liposomes having covalently bound PEG moieties on their
external surface are described in European Patent No. EP 0 445 131 Bl and WO
90/04384 to Fisher. Liposome compositions containing 1-20 mole percent of PE
derivatized with PEG, and methods of use thereof, are described by Woodle et
al.
(U.S. Patent Nos. 5,013,556 and 5,356,633) and Martin et al. (U.S. Patent No.
5,213,804 and European Patent No. EP 0 496 813 Bl). Liposomes comprising a
number of other lipid-polymer conjugates are disclosed in WO 91/05545 and U.S.
Patent No. 5,225,212 (both to Martin et al.) and in WO 94/20073 (Zalipsky et
al.)
Liposomes comprising PEG-modified ceramide lipids are described in WO
96/10391 (Choi et al.). U.S. Patent Nos. 5,540,935 (Miyazaki et al.) and
5,556,948 (Tagawa et al.) describe PEG-containing liposomes that can be
further
derivatized with functional moieties on their surfaces.
A limited number of liposomes comprising nucleic acids are known in the
art. WO 96140062 to Thierry et al. discloses methods for encapsulating high
molecular weight nucleic acids in liposomes. U.S. Patent No. 5,264,221 to
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-50-
Tagawa et al. discloses protein-bonded liposomes and asserts that the contents
of
such liposomes may include an antisense RNA. U.S. Patent No. 5,665,710 to
Rahman et al. describes certain methods of encapsulating oligodeoxynucleotides
in liposomes. WO 97/04787 to Love et al. discloses liposomes comprising
antisense oligonucleotides targeted to the raf gene.
Transfersomes are yet another type of liposomes, and are highly
defonnable lipid aggregates which are attractive candidates for drug delivery
vehicles. Transfersomes may be described as lipid droplets which are so highly
deformable that they are easily able to penetrate through pores which are
smaller
than the droplet. Transfersomes are adaptable to the environment in which they
are used, e.g. they are self optimizing (adaptive to the shape of pores in the
skin),
self repairing, frequently reach their targets without fragmenting, and often
self
loading. To make transfersomes it is possible to add surface edge-activators,
usually surfactants, to a standard liposomal composition. Transfersomes have
been used to deliver serum albumin to the skin. The transfersome-mediated
delivery of serum albumin has been shown to be as effective as subcutaneous
inj ection of a solution containing serum albumin.
Surfactants find wide application in formulations such as emulsions
(including microemulsions) and liposomes. The most common way of classifying
and ranking the properties of the many different types of surfactants, both
natural
and synthetic, is by the use of the hydrophile/lipophile balance (HLB). The
nature
of the hydrophilic group (also known as the "head") provides the most useful
means for categorizing the different surfactants used in formulations (Rieger,
in
Pharmaceutical Dosage Forms, Marcel Dekker, Inc., New York, NY, 1988, p.
285).
If the surfactant molecule is not ionized, it is classified as a nonionic
surfactant. Nonionic surfactants find wide application in pharmaceutical and
cosmetic products and are usable over a wide range of pH values. In general
their
HLB values range from 2 to about 18 depending on their structure. Nonionic
surfactants include nonionic esters such as ethylene glycol esters, propylene
glycol esters, glyceryl esters, polyglyceryl esters, sorbitan esters, sucrose
esters,
and ethoxylated esters. Nonionic alkanolamides and ethers such as fatty
alcohol
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-51-
ethoxylates, propoxylated alcohols, and ethoxylated/propoxylated block
polymers
are also included in this class. The polyoxyethylene surfactants are the most
popular members of the nonionic surfactant class.
If the surfactant molecule carries a negative charge when it is dissolved or
dispersed in water, the surfactant is classified as anionic. Anionic
surfactants
include carboxylates such as soaps, acyl lactylates, acyl amides of amino
acids,
esters of sulfuric acid such as alkyl sulfates and ethoxylated alkyl sulfates,
sulfonates such as alkyl benzene sulfonates, aryl isethionates, acyl taurates
and
sulfosuccinates, and phosphates. The most important members of the anionic
surfactant class are the alkyl sulfates and the soaps.
If the surfactant molecule carries a positive charge when it is dissolved or
dispersed in water, the surfactant is classified as cationic. Cationic
surfactants
include quaternary ammonium salts and ethoxylated amines. The quaternary
ammonium salts are the most used members of this class.
If the surfactant molecule has the ability to carry either a positive or
negative charge, the surfactant is classified as amphoteric. Amphoteric
surfactants
include acrylic acid derivatives, substituted alkylamides, N-alkylbetaines and
phosphatides. The use of surfactants in drug products, formulations and in
emulsions has been reviewed (Rieger, in Pharmaceutical Dosage Forms, Marcel
Dekker, Inc., New York, NY, 1988, p. 285).
In one embodiment, the present invention employs various penetration
enhancers to effect the efficient delivery of nucleic acids, particularly
oligomeric
compounds, to the skin of animals. Most drugs are present in solution in both
ionized and nonionized forms. However, usually only lipid soluble or
lipophilic
drugs readily cross cell membranes. It has been discovered that even non-
lipophilic drugs may cross cell membranes if the membrane to be crossed is
treated with a penetration enhancer. In addition to aiding the diffusion of
non-
lipophilic drugs across cell membranes, penetration enhancers also enhance the
permeability of lipophilic drugs. Penetration enhancers may be classified as
belonging to one of five broad categories, i.e., surfactants, fatty acids,
bile salts,
chelating agents, and non-chelating non-surfactants (Lee et al., Critical
Reviews in
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-52-
Therapeutic Drug Carner Systems, 1991, p.92). Each of the above mentioned
classes of penetration enhancers are described below in greater detail.
hi connection with the present invention, surfactants (or "surface-active
agents") are chemical entities which, when dissolved in an aqueous solution,
reduce the surface tension of the solution or the interfacial tension between
the
aqueous solution and another liquid, with the result that absorption of
oligonucleotides through the mucosa is enhanced. In addition to bile salts and
fatty acids, these penetration enhancers include, for example, sodium lauryl
sulfate, polyoxyethylene-9-lauryl ether and polyoxyethylene-20-cetyl ether)
(Lee
et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, p.92); and
perfluorochemical emulsions, such as FC-43. Takahashi et al., J. Pharm.
Pharmacol., 1988, 40, 252).
Various fatty acids and their derivatives which act as penetration
enhancers include, for example, oleic acid, lauric acid, capric acid (n-
decanoic
acid), myristic acid, palmitic acid, stearic acid, linoleic acid, linolenic
acid,
dicaprate, tricaprate, monoolein (1-monooleoyl-rac-glycerol), dilaurin,
caprylic
acid, arachidonic acid, glycerol 1-monocaprate, 1-dodecylazacycloheptan-2-one;
acylcarnitines, acylcholines, Ci_io alkyl esters thereof (e.g., methyl,
isopropyl and
t-butyl), and mono- and di-glycerides thereof (i.e., oleate, laurate, caprate,
myristate, palmitate, stearate, linoleate, etc.) (Lee et al., Critical Reviews
in
Therapeutic Drug Carrier Systems, 1991, p.92; Muranishi, Critical Reviews in
Therapeutic Drug Carner Systems, 1990, 7, 1-33; El Hariri et al., J. Pharm.
Pharmacol., 1992, 44, 651-654).
The physiological role of bile includes the facilitation of dispersion and
absorption of lipids and fat-soluble vitamins (Bnmton, Chapter 38 in: Goodman
&
Gilman's The Pharmacological Basis of Therapeutics, 9th Ed., Hardman et al.
Eds., McGraw-Hill, New York, 1996, pp. 934-935). Various natural bile salts,
and
their synthetic derivatives, act as penetration enhancers. Thus the term "bile
salts"
includes any of the naturally occurnng components of bile as well as any of
their
synthetic derivatives. The bile salts of the invention include, for example,
cholic
acid (or its pharmaceutically acceptable sodium salt, sodium cholate),
dehydrocholic acid (sodium dehydrocholate), deoxycholic acid (sodium
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-53-
deoxycholate), glucholic acid (sodium glucholate), glycholic acid (sodium
glycocholate), glycodeoxycholic acid (sodium glycodeoxycholate), taurocholic
acid (sodium taurocholate), taurodeoxycholic acid (sodium taurodeoxycholate),
chenodeoxycholic acid (sodium chenodeoxycholate), ursodeoxycholic acid
(UDCA), sodium tauro-24,25-dihydro-fusidate (STDHF), sodium
glycodihydrofusidate and polyoxyethylene-9-lauryl ether (POE) (Lee et al.,
Critical Reviews in Therapeutic Drug Carrier Systems, 1991, page 92; Swinyard,
Chapter 39 In: Remington's Pharmaceutical Sciences, 18th Ed., Gennaro, ed.,
Mack Publishing Co., Easton, PA, 1990, pages 782-783; Muranishi, Critical
Reviews in Therapeutic Drug Carrier Systems, 1990, 7, 1-33; Yamamoto et al.,
J.
Pharm. Exp. Ther., 1992, 263, 25; Yamashita et al., J. Pharm. Sci., 1990, 79,
579-
583).
Chelating agents, as used in connection with the present invention, can be
defined as compounds that remove metallic ions from solution by forming
complexes therewith, with the result that absorption of oligonucleotides
through
the mucosa is eWanced. With regards to their use as penetration enhancers in
the
present invention, chelating agents have the added advantage of also serving
as
DNase inhibitors, as most characterized DNA nucleases require a divalent metal
ion for catalysis and are thus inhibited by chelating agents (Jarrett, J.
Chromatogr.,
1993, 618, 315-339). Chelating agents of the invention include but are not
limited
to disodium ethylenediaminetetraacetate (EDTA), citric acid, salicylates
(e.g.,
sodium salicylate, 5-methoxysalicylate and homovanilate), N-acyl derivatives
of
collagen, laureth-9 and N-amino acyl derivatives of beta-diketones
(enamines)(Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems,
1991, page 92; Muranishi, Critical Reviews in Therapeutic Drug Carrier
Systems,
1990, 7, 1-33; Buur et al., J. Control Rel., 1990, 14, 43-51).
As used herein, non-chelating non-surfactant penetration enhancing
compounds can be defined as compounds that demonstrate insignificant activity
as
chelating agents or as surfactants but that nonetheless enhance absorption of
oligonucleotides through the alimentary mucosa (Muranishi, Critical Reviews in
Therapeutic Drug Carner Systems, 1990, 7, 1-33). This class of penetration
enhancers include, for example, unsaturated cyclic ureas, 1-alkyl- and 1-
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-54-
alkenylazacyclo-alkanone derivatives (Lee et al., Critical Reviews in
Therapeutic
Drug Carrier Systems, 1991, page 92); and non-steroidal anti-inflammatory
agents
such as diclofenac sodium, indomethacin and phenylbutazone (Yamashita et al.,
J.
Pharm. Pharmacol., 1987, 39, 621-626).
Agents that enhance uptake of oligonucleotides at the cellular level may
also be added to the pharmaceutical and other compositions of the present
invention. For example, cationic lipids, such as lipofectin (Junichi et al,
U.S.
Patent No. 5,705,188), cationic glycerol derivatives, and polycationic
molecules,
such as polylysine (Lollo et al., PCT Application WO 97/30731), are also known
to enhance the cellular uptake of oligonucleotides.
Other agents may be utilized to enhance the penetration of the
administered nucleic acids, including glycols such as ethylene glycol and
propylene glycol, pyrrols such as 2-pyrrol, azones, and terpenes such as
limonene
and menthone.
Certain compositions of the present invention also incorporate Garner
compounds in the formulation. As used herein, "carrier compound" or "carrier"
can refer to a nucleic acid, or analog thereof, which is inert (i.e., does not
possess
biological activity per se) but is recognized as a nucleic acid by in vivo
processes
that reduce the bioavailability of a nucleic acid having biological activity
by, for
example, degrading the biologically active nucleic acid or promoting its
removal
from circulation. The coadministration of a nucleic acid and a Garner
compound,
typically with an excess of the latter substance, can result in a substantial
reduction of the amount of nucleic acid recovered in the liver, kidney or
other
extracirculatory reservoirs, presumably due to competition between the carrier
compound and the nucleic acid for a common receptor. For example, the recovery
of a partially phosphorothioate oligonucleotide in hepatic tissue can be
reduced
when it is coadministered with polyinosinic acid, dextran sulfate, polycytidic
acid
or 4-acetamido-4'isothiocyano-stilbene-2,2'-disulfonic acid (Miyao et al.,
Antisense Res. Dev., 1995, 5, 115-121; Takakura et al., Antisense & Nucl. Acid
Drug Dev., 1996, 6, 177-183).
In contrast to a carrier compound, a "pharmaceutical carrier" or
"excipient" is a pharmaceutically acceptable solvent, suspending agent or any
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-55-
other pharmacologically inert vehicle for delivering one or more nucleic acids
to
an animal. The excipient may be liquid or solid and is selected, with the
planned
manner of administration in mind, so as to provide for the desired bulk,
consistency, etc., when combined with a nucleic acid and the other components
of
a given pharmaceutical composition. Typical pharmaceutical Garners include,
but
are not limited to, binding agents (e.g., pregelatinized maize starch,
polyvinylpyrrolidone or hydroxypropyl methylcellulose, etc.); fillers (e.g.,
lactose
and other sugars, microcrystalline cellulose, pectin, gelatin, calcium
sulfate, ethyl
cellulose, polyacrylates or calcium hydrogen phosphate, etc.); lubricants
(e.g.,
magnesium stearate, talc, silica, colloidal silicon dioxide, stearic acid,
metallic
stearates, hydrogenated vegetable oils, corn starch, polyethylene glycols,
sodium
benzoate, sodium acetate, etc.); disintegrants (e.g., starch, sodium starch
glycolate,
etc.); and wetting agents (e.g., sodium lauryl sulphate, etc.).
Pharmaceutically acceptable organic or inorganic excipient suitable for
non-parenteral administration which do not deleteriously react with nucleic
acids
can also be used to formulate the compositions of the present invention.
Suitable
pharmaceutically acceptable carriers include, but are not limited to, water,
salt
solutions, alcohols, polyethylene glycols, gelatin, lactose, amylose,
magnesium
stearate, talc, silicic acid, viscous paraffin, hydroxymethylcellulose,
polyvinylpyrrolidone and the like.
Formulations for topical administration of nucleic acids may include
sterile and non-sterile aqueous solutions, non-aqueous solutions in common
solvents such as alcohols, or solutions of the nucleic acids in liquid or
solid oil
bases. The solutions may also contain buffers, diluents and other suitable
additives. Pharmaceutically acceptable organic or inorganic excipients
suitable
for non-parenteral administration which do not deleteriously react with
nucleic
acids can be used.
Suitable pharmaceutically acceptable excipients include, but are not
limited to, water, salt solutions, alcohol, polyethylene glycols, gelatin,
lactose,
amylose, magnesium stearate, talc, silicic acid, viscous paraffin,
hydroxymethylcellulose, polyvinylpyrrolidone and the like.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-56-
The compositions of the present invention may additionally contain other
adjunct components conventionally found in pharmaceutical compositions, at
their
art-established usage levels. Thus, for example, the compositions may contain
additional, compatible, pharmaceutically-active materials such as, for
example,
antipruritics, astringents, local anesthetics or anti-inflammatory agents, or
may
contain additional materials useful in physically formulating various dosage
forms
of the compositions of the present invention, such as dyes, flavoring agents,
preservatives, antioxidants, opacifiers, thickening agents and stabilizers.
However, such materials, when added, should not unduly interfere with the
biological activities of the components of the compositions of the present
invention. The formulations can be sterilized and, if desired, mixed with
auxiliary
agents, e.g., lubricants, preservatives, stabilizers, wetting agents,
emulsifiers, salts
for influencing osmotic pressure, buffers, colorings, flavorings and/or
aromatic
substances and the like which do not deleteriously interact with the nucleic
acids)
of the formulation.
Aqueous suspensions may contain substances which increase the viscosity
of the suspension including, for example, sodium carboxymethylcellulose,
sorbitol
and/or dextran. The suspension may also contain stabilizers.
The compounds of the invention may also be admixed, encapsulated,
conjugated or otherwise associated with other molecules, molecule structures
or
mixtures of compounds, as for example, liposomes, receptor targeted molecules,
oral, rectal, topical or other formulations, for assisting in uptake,
distribution
and/or absorption. Representative United States patents that teach the
preparation
of such uptake, distribution and/or absorption assisting formulations include,
but
are not limited to, U.S.: 5,108,921; 5,354,844; 5,416,016; 5,459,127;
5,521,291;
5,543,158; 5,547,932; 5,583,020; 5,591,721; 4,426,330; 4,534,899; 5,013,556;
5,108,921; 5,213,804; 5,227,170; 5,264,221; 5,356,633; 5,395,619; 5,416,016;
5,417,978; 5,462,854; 5,469,854; 5,512,295; 5,527,528; 5,534,259; 5,543,152;
5,556,948; 5,580,575; and 5,595,756, each of which is herein incorporated by
reference.
The oligomeric compounds of the invention encompass any
pharmaceutically acceptable salts, esters, or salts of such esters, or any
other
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-57-
compound which, upon administration to an animal including a human, is capable
of providing (directly or indirectly) the biologically active metabolite or
residue
thereof. Accordingly, for example, the disclosure is also drawn to prodrugs
and
pharmaceutically acceptable salts of the compounds of the invention,
pharmaceutically acceptable salts of such prodrugs, and other bioequivalents.
The term "prodrug" indicates a therapeutic agent that is prepared in an
inactive form that is converted to an active form (i.e., drug) within the body
or
cells thereof by the action of endogenous enzymes or other chemicals and/or
conditions. In particular, prodrug versions of the oligonucleotides of the
invention
are prepared as SATE [(S-acetyl-2-thioethyl) phosphate] derivatives according
to
the methods disclosed in WO 93124510 to Gosselin et al., published December 9,
1993 or in WO 94/26764 and U.S. 5,770,713 to Imbach et al.
The term "pharmaceutically acceptable salts" refers to physiologically and
pharmaceutically acceptable salts of the compounds of the invention: i.e.,
salts
that retain the desired biological activity of the parent compound and do not
impart undesired toxicological effects thereto.
Pharmaceutically acceptable base addition salts are formed with metals or
amines, such as alkali and alkaline earth metals or organic amines. Examples
of
metals used as cations are sodium, potassium, magnesium, calcium, and the
like.
Examples of suitable amines are N,N'-dibenzylethylenediamine, chloroprocaine,
choline, diethanolamine, dicyclohexylamine, ethylenediamine,
N-methylglucamine, and procaine (see, for example, Berge et al.,
"Pharmaceutical
Salts," J. of Pharma Sci., 1977, 66, 1-19). The base addition salts of said
acidic
compounds are prepared by contacting the free acid form with a sufficient
amount
of the desired base to produce the salt in the conventional manner. The free
acid
form may be regenerated by contacting the salt form with an acid and isolating
the
free acid in the conventional manner. The free acid forms differ from their
respective salt forms somewhat in certain physical properties such as
solubility in
polar solvents, but otherwise the salts are equivalent to their respective
free acid
for purposes of the present invention. As used herein, a "pharmaceutical
addition
salt" includes a pharmaceutically acceptable salt of an acid form of one of
the
components of the compositions of the invention. These include organic or
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-58-
inorganic acid salts of the amines. Preferred acid salts are the
hydrochlorides,
acetates, salicylates, nitrates and phosphates. Other suitable
pharmaceutically
acceptable salts are well known to those skilled in the art and include basic
salts of
a variety of inorganic and organic acids, such as, for example, with inorganic
acids, such as for example hydrochloric acid, hydrobromic acid, sulfuric acid
or
phosphoric acid; with organic carboxylic, sulfonic, sulfo or phospho acids or
N-substituted sulfamic acids, for example acetic acid, propionic acid,
glycolic
acid, succinic acid, malefic acid, hydroxymaleic acid, methylmaleic acid,
fumaric
acid, malic acid, tartaric acid, lactic acid, oxalic acid, gluconic acid,
glucaric acid,
glucuronic acid, citric acid, benzoic acid, cinnamic acid, mandelic acid,
salicylic
acid, 4-aminosalicylic acid, 2-phenoxybenzoic acid, 2-acetoxybenzoic acid,
embonic acid, nicotinic acid or isonicotinic acid; and with amino acids, such
as
the 20 alpha-amino acids involved in the synthesis of proteins in nature, for
example glutamic acid or aspartic acid, and also with phenylacetic acid,
methanesulfonic acid, ethanesulfonic acid, 2-hydroxyethanesulfonic acid,
ethane-1,2-disulfonic acid, benzenesulfonic acid, 4-methylbenzenesulfonic
acid,
naphthalene-2-sulfonic acid, naphthalene-1,5-disulfonic acid, 2- or
3-phosphoglycerate, glucose-6-phosphate, N-cyclohexylsulfamic acid (with the
formation of cyclamates), or with other acid organic compounds, such as
ascorbic
acid. Pharmaceutically acceptable salts of compounds may also be prepared with
a pharmaceutically acceptable canon. Suitable pharmaceutically acceptable
cations are well known to those skilled in the art and include alkaline,
alkaline
earth, ammonium and quaternary ammonium cations. Carbonates or hydrogen
carbonates are also possible.
For oligomeric compounds, preferred examples of pharmaceutically
acceptable salts include but are not limited to (a) salts formed with cations
such as
sodium, potassium, ammonium, magnesium, calcium, polyamines such as
spermine and spermidine, etc.; (b) acid addition salts formed with inorganic
acids,
for example hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric
acid,
nitric acid and the like; (c) salts formed with organic acids such as, for
example,
acetic acid, oxalic acid, tartaric acid, succinic acid, malefic acid, fumaric
acid,
gluconic acid, citric acid, malic acid, ascorbic acid, benzoic acid, tannic
acid,
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-59-
palmitic acid, alginic acid, polyglutamic acid, naphthalenesulfonic acid,
methanesulfonic acid, p-toluenesulfonic acid, naphthalenedisulfonic acid,
polygalacturonic acid, and the like; and (d) salts formed from elemental
anions
such as chlorine, bromine, and iodine.
The materials, methods, and examples presented herein are intended to be
illustrative, and are not intended to limit the scope of the invention. All
publications, patent applications, patents, and other references mentioned
herein
are incorporated by reference in their entirety. Unless otherwise defined, all
technical and scientific terms are intended to have their art-recognized
meanings.
EXAMPLE 1
5'-O-DMT-L-thymidine (2).
HO T DMTO T
OH DMTCI, Py. OH
1 D~ 2
Compound 1 (800 mg, 3.3 mmol, (prepared according to Smejkal, J. et. al.
Collect. Czecla. Chem. Comrnun. 1964, 29, 2809-2813 and Jung, M. E. et czl.
TetYahedron Lett. 1998, 39, 4615-4618) was dried over PZOS under high vacuum
overnight at 40°C. It was then co-evaporated with anhydrous pyridine
(10 mL).
The residue obtained was dissolved in pyridine (9 mL) under axz argon
atmosphere. 4-Dimethylaminopyridine (40 mg, 0.33 mmol), and DMT chloride
(DMT-Cl, 1.33 g, 3.93 mmol) were added to the mixture and the reaction mixture
was stirred at room temperature until all of the starting material disappeared
(12
h). Methanol (0.5 mL) was added and solvent was removed iya vacuo. The
residue was chromatographed and eluted with ethyl acetate: exane, 6:4. to give
2
(1.42 g, 79 %). Rf= 0.17 (with ethyl acetate : hexane, 6 : 4). MS (ES~) m/z
543
(M-H).
EXAMPLE 2
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-60-
5'-O-DMT-L-thymidine-3'-O-[(2-cyanoethyl)-N,1V diisopropylphosphor-
amidite] (3).
Compound 2 (1.00 g, 1.84 mmol) was co-evaporated with toluene (20 mL).
To the residue N,N diisopropylamine tetrazolide (0.16 g, 0.92 mural) was added
and dried over Pa05 under high vacuum overnight at 40°C. The dried
reaction
mixture was dissolved in anhydrous acetonitrile:CHZCIz (9:2 mL) and 2-
cyanoethyl-N,N,N~,N~-tetraisopropylphosphoramidite (1.2 mL, 3.68 mmol) was
added. The reaction mixture was stirred at ambient temperature for 4 h under
an
inert atmosphere. The progress of the reaction was monitored by TLC
(hexane:ethyl acetate 1:1). The solvent was evaporated, the residue was
dissolved
in ethyl acetate (70 mL) and washed with 5% aqueous NaHC03 (40 mL). The
ethyl acetate layer was dried over anhydrous NaaSO4 and concentrated. Residue
obtained was chromatographed (ethyl acetate:hexane, 3:2 as eluent) to give 3
as a
foam (0.82g, 60.3%). Rf= 0.47 (ethyl acetate:hexane, 3:2). 31P NMR (CDC13) ~
149.98, 149.57 ppm; MS (API-ES) r~z/z 743.3 (M-H)
EXAMPLE 3
5'-O-DMT-L-thymidine-3'-O-succinyl CPG (4).
Compound 2 (0.2 g, 0.37 mmol) was mixed with succinic anhydride
(0.074 g, 0.73 mmol) and DMAP (0.023 g, 0.19 rmnol). The mixture was dried
over PZOS overnight in vacuum. To this Cl-CHZ-CHz-Cl (1.1 mL) and
triethylamine (0.2 mL, 1.46 mmol) were added. The reaction mixture was heated
at 60 °C for 2 h. Diluted the reaction mixture with CH2C12 (20 mL),
washed with
5 % aqueous citric acid (20 mL), water (20 mL) and brine (20 mL). The organic
phase was dried over anhydrous NaZS04 and concentrated in vacuo (0.22 g, 93%)
as a foam. Rf= 0.23 (5% MeOH in CHaCl2: MeOH). The residue obtained was
used as such for the next reaction. 1H NMR (200 MHz, CDC13) 8 1.08 (t, 9H, J =
7.18 Hz), 1.4 (s, 3H), 1.92 (m, 2H), 2.54 (s, 6H), 2.62 (m, 10H), 2.91 (m,
1H),
3.37-3.74 (m, 5H), 3.61 (s, 6H), 4.29 (t, 2H, J= 4.52 Hz), 5.38 (t, 1H, J =
5.48 Hz),
6.05 (d, 1H, J = 4.5 Hz), 6.85 (d, 4H, J = 8.78 Hz), 7.26-7.42 (m, 9H), 7.62
(s,
1H); 13C NMR (50 MHz, CDC13) 811.33, 28.17, 28.63, 28.89, 37.70, 31.34,
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-61-
55.03, 63.48, 75.43, 83.62, 84.22, 86.94, 111.35, 113.12, 126.98,
127.81,127.92,
129.88, 134.98, 135.07, 135.68, 144.03, 150.61, 158.52, 164.54, 171.62,
175.98.
The succinyl derivative (0.19 g, 0.25 mrnol) was dried over PZOS ifz vacuo
at 40° C overnight. Anhydrous DMF (0.62 mL) was added followed by 2-(1H-
benzotriazole)-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (0.081 g,
0.25
mmol) and N-methylmorpholine (55 ~,L, 0.5 mmol). Vortexed to give a clear
solution. To this anhydrous DMF (2.4 mL) and activated CPG (1.08 g, 115.2
mmol/g, particle size 120/200, mean pore diameter 520 ~) were added. It was
then allowed to shake on a shaker for 18 h. Aliquot was withdrawn to estimate
the loading capacity. Filtered the functionalized CPG and washed thoroughly
with DMF, CH3CN and Et20. Dried in vacuo overnight. Suspended the
functionalized CPG (3) in capping solution (2 mL, Cap A, acetic anhydride/
lutidine/ THF, 2 mL, Cap B, N-methylimidazole/ THF, Perspective Biosystems
Inc.) and allowed to shake on a shaker for 2 h. Filtered and washed with CH3CN
and Et20. Dried in vacuo and loading capacity was determined by standard
procedure. Final loading 52.62 ~,mol/g.
EXAMPLE 4
5'-O-DMT-L-N4-benzoyl-2'-deoxyadenosine-3'-O-[(2-cyanoethyl)-N,N
diisopropylphosphoramidite] (5).
1-Chloro-5,3-bis(tolyl)-2-deoxy L-ribose is prepared as described in [Jung,
M.E. et. al. Tetrahedron Lett. 1990, 31, 6983-6986; Gosselin, G. et. al.
Tety~ahed~on Lett. 1997, 3~, 4199-4202, Nucleosides & Nucleotides 1998,17,
1731-1738]. This is then coupled with N4-benzoyl adeiune under Vorbruggen
condition to give the N4-benzoyl-5',3'-tolyl-1-adenosine. Deprotection of the
tolyl
group with methylamine gives L-adenosine. It is then converted into N4-benzoyl
L-adenosine under transient protection conditions in the presence of benzoyl
chloride, TMSCl, pyridine and aqueous ammonia. 5'-Tritylation in presence of
DMTCI, in pyridine and phosphitylation at the 3'-position gives compound 5.
EXAMPLE 5
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-62-
5'-O-DMT-L lV4-benzoyl-5-methyl-2'-deoxycytidine-3'-O-[(2-cyanoethyl)-
N,N diisopropylphosphoramidite] (6)
Compound 2 is converted into 5'-O-DMT-L-5-methylcytidine according to
literature procedure [Divakar K. J. et. al. J. Chem. Soc. Perk.
Ts°ayas. 1 1982, 1171-
1176]. It is then converted into 1V4-benzoyl derivative according to
literature
procedure [Bhat, V. et. al. Nucleosides Nucleotides 1989, 8, 179-183]. This is
then phosphitylated at the 3'- position to give compound 6.
EXAMPLE 6
5'-O-DMT-L-N2-isobutyryl-2'-deoxyguanosine-3'-O-[(2-cyanoethyl)-N,N
diisopropylphosphoramidite] (7)
1-Chloro-5,3-bis(tolyl)-2-deoxy L-ribose is prepared as described in Jung,
M. E. et. al Tet~ahed~oya Lett. 1997, 38, 4199-4202 and Gosselin, G. et. al.
Nucleosides & Nucleotides 1998,17, 1731-1738. This is then coupled with 4-
chloro-2-aminopyrrolo [2,3-d]pyrimidine [prepared according to the procedure
described by Davoll, J. et. al. J. Chem. Soc. 1960, 131-138] under NaH and
acetonitrile [Ramasamy, K. et. al. J. Hetr-ocyclic Claem., 1988, 25, 1893-
1897].
This is then treated with aqueous ammonia at 80 °C to give L-2'-
deoxyguanosine.
This is converted into L-NZ-isobutyryl-2'-deoxyguanosine under transient
protection conditions in presence of isobutyryl chloride, pyridine and TMSCI
[Ti,
G. S. et. al. J. Am. Chefra. Soc.,1982,104, 1316-1319]. This is then converted
into 5'-O-DMT-L-N'-isobutyryl-2'-deoxyguanosine in the presence of DMTCI,
DMAP and pyridine followed by phosphitylation at 3'-position to give compound
7.
EXAMPLE 7
5'-O-DMT-L-5-(1-propynyl)uridine-3'-O-[(2-cyanoethyl) N,N
diisopropylphosphamidite] (8)
1-Chloro-5,3-bis(tolyl)-2-deoxy L-ribose is prepared as described in Jung,
M. E. et. al Tety~alaedroh Lett. 1997, 38, 4199-4202 and Gosselin, G. et, al.
Nucleosides & Nucleotides 1998,17, 1731-1738. This is then coupled with 5-
iodouracil under Vorbruggen condition to give the L-5-(iodo)-5',3'-tolyl-1-
uridine.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-63-
This is then coupled with propyne [as described in Switzer C. et. al., Bioorg.
Med.
Claena. Lett.1996, 6, 815-818] to give L-5-(propynyl)-5',3'-tolyl uridine.
Deprotection of protecting groups at 5' and 3' position gives L-5-
(propynyl)uridine. This compound is converted into the 5'-O-DMT compound
with DMTCl, DMAP and pyridine followed by phosphitylation to give the title
compound 8.
EXAMPLE 8
5'-O-DMT-L-5-(1-propynyl)cytidine-3'-O-[(2-cyanoethyl)-N,N
diisopropylphosphoramidite] (9)
5'-O-DMT-L-5-(1-propynyl)uridine (prepared following the procedure
described for compound 7) is converted into 5'-O-DMT-L-5-(1-propynyl)cytidine
according to literature procedure [Divakar K. J, et. al. J. Cheyrr. Soc. Perk.
Ti-aas.
1 1982, 1171-1176]. This is phosphitylated at the 3'-position to give compound
9.
EXAMPLE 9
5'-O-DMT-L-3(2-deoxy-(3-D-erythro-pentofuranosyl)(9I)-1H-pyrimido[5,4-
b]benzoxazin-2(3H)-one-3'-O-[(2-cyanoethyl)-N,N
diisopropylphosphoramidite] (10)
L-5-Bromouridine is obtained from 5-bromo uridine and 1-Chloro-5,3-
bis(tolyl)-2-deoxy L-ribose under Vorbruggen conditions. This is converted
into
5,3-bis (tolyl)-L-3-(2-deoxy-(3-D-erythro-pentofuranosyl)(9I)-1H-pyrimido[5,4-
b]benzoxazin-2(3H)-one according to literature procedure [Lin, K-Y. et. al J.
Arra.
Clze~a. Soc. 1995,117, 3873-3874, Matteucci, M. D. et. al. 94-US10536] . This
is
then deprotected with methyl amine, tritylated at 5' position and
phosphitylated at
3' position to give compound 10.
EXAMPLE 10
5'-O-DMT-L-1V4-benzoyl-2'-deoxyadenosine-3'-O-succinyl CPG (11).
5'-O-DMT-L-IV4-benzoyl-2'-deoxyadenosine (prepared as described in the
synthesis of compound 5) is converted into 3'-O-succinyl derivative in the
presence of succinic anhydride and DMAP in dichloroethane at 60 °C. The
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-64-
succinyl derivative is coupled to amino alkyl CPG in presence of 2-(1H-
benzotriazole)-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate and N
methylmorpholine in DMF to give compound 11.
EXAMPLE 11
5'-O-DMT-L 1V4-benzoyl-5-methyl-2'-deoxycytidine-3'-O-succinyl CPG (12).
5'-O-DMT-L-N4-benzoyl-5-methyl-2'-deoxycytidine (prepared as
described in the synthesis of compound 6) is converted into 3'-O-succinyl
derivative in the presence of succinic anhydride and DMAP in dichloroethane at
60 °C. The succinyl derivative is coupled to amino alkyl CPG in
presence of 2-'
(1H-benzotriazole)-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate and N
methylmorpholine in DMF to yield the compound 12.
EXAMPLE 12
5'-O-DMT-L-N2-isobutyryl-2'-deoxyguanosine-3'-O-succinyl CPG (13).
5'-O-DMT-L-N2-isobutyryl-2'-deoxyguanosine (prepared as described in
the synthesis of compound 7) is converted into 3'-O-succinyl derivative in the
presence of succinic anhydride and DMAP in dichloroethane at 60 °C. The
succinyl derivative is coupled to amino alkyl CPG in presence of 2-(1H-
benzotriazole)-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate and N
methylmorpholine in DMF to yield the compound 13.
EXAMPLE 13
5'-O-DMT-L-5-(1-propynyl)uridine-3'-O-succinyl CPG (14).
5'-O-DMT-L-5-(1-propynyl)uridine (prepared as described in the synthesis
of compound 8) is converted into 3'-O-succinyl derivative in the presence of
succinic anhydride and DMAP in dichloroethane at 60 °C. The succinyl
derivative is coupled to amino alkyl CPG in presence of 2-(1H-benzotriazole)-1-
yl)-1,1,3,3-tetramethyluronium tetrafluoroborate and N methylmorpholine in
DMF to yield the compound 14
EXAMPLE 14
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-65-
5'-O-DMT-L-5-(1-propynyl)cytidine-3'-O-succinyl CPG (15).
5'-O-DMT-L-5-(1-propynyl)cytidine (prepared as described in the
synthesis of compound 8) is converted into 3'-O-succinyl derivative in the
presence of succinic anhydride and DMAP in dichloroethane at 60 °C. The
succinyl derivative is coupled to amino alkyl CPG in the presence of 2-(1H-
benzotriazole)-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate and N
methylmorpholine in DMF to yield the compound 15.
EXAMPLE 15
5'-O-DMT-L-3(2-deoxy-[3-D-erythro-pentofuranosyl)(9I)-1H-pyrimido[5,4-
b]benzoxazin-2(3H)-one-3'-O-succinyl CPG (16).
5'-O-DMT-L-3(2-deoxy-(3-D-erythro-pentofuranosyl)(9I)-1H-
pyrimido[5,4-b]benzoxazin-2(3H)-one (prepared as described in the synthesis of
compound 10) is converted into 3'-O-succinyl derivative in the presence of
succinic anhydride and DMAP in dichloroethane at 60 °C. The succinyl
derivative is coupled to amino alkyl CPG in presence of 2-(1H-benzotriazole)-1-
yl)-1,1,3,3-tetramethyluronium tetrafluoroborate and N methylmorpholine in
DMF to yield the compound 16.
EXAMPLE 16
Synthesis of oligonucleotides containing L-thymidine modification
The amidite 3 was dissolved in anhydrous acetonitrile to give a 0.1 M
solution and loaded on to a Expedite Nucleic Acid Synthesis system (Millipore
8909) to synthesize the oligonucleotides. The coupling efficiencies were more
than 98%. For the coupling of the modified amidite (3) coupling time was
extended to 10 min and this step was carried out twice. All other steps in the
protocol supplied by Millipore were used as such. After completion of the
synthesis the CPG was suspended in aqueous ammonia (30 wt %) and at room
temperature for 2 h to deprotect oligonucleotides form the CPG. Filtered the
CPG
and heated the filtrate at 55 °C for 6 h to complete the deprotection
of all
protecting groups. Ammonia was removed on a speed vac concentrator and then
the product was purified by High Performance Liquid Chromatography (HPLC,
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-66-
Waters, C-4, 7.8 X 300 mm, A = 50 mM triethylammonium acetate, pH = 7, B =
acetonitrile, 5 to 60 % B in 55 Min, Flow 2.5 mL/min., ~, = 260 nm).
Detritylation
with aqueous 80% acetic acid and evaporation followed by desalting by HPLC on
Waters C-4 column gave 2'-modified oligonucleotides (Table I).
Oligonucleotides were analyzed by HPLC, CGE and mass spectrometry.
EXAMPLE 17
Table I Oligonucleotides containing L-thymidines
ISIS Sequence Mass Mass HPLC
Calcd
No. ObservedRetention
Time
mm.
1207455' T*GC ATC CCC CAG GCC ACC 6591.06 6591.2923.40
AT*3'
(SEQ ID NO:1)
1217855' T*CCCGCTGTGATGCATT* 3' 6673.02 6673.8528.74
(SEQ ID N0:2)
1245855' T*CCGTCATCGCTCCTCAGGT* 7061.48 7061.6033.46
3'
(SEQ ID N0:3)
T* = L-Thymidine, All P = S, C° = 2'-O-MOE SMeC, A° = 2'-O-
MOE A, T° _
2'-O-MOE SMeLT, G° = 2'-O- MOE G, aWaters C-4, 3.9x300 mm, solvent A=50
mm
TEAAc, pH 7; Solvent B = CH3CN; gradient 5-60% B in 55 min; flow rate 1.5
mL/min, 7~ = 260 nm.
EXAMPLE 18
Table II. Tm values of L-thymidine modified oligonucleotides against RNA
ISIS Sequence Target ~Tm
# RNA
C C
8651 TGC ATC CCC CAG GCC ACC 68.7
AT
(SEQ ID N0:4)
1207455' T*GC ATC CCC CAG GCC 66.94 -1.76
ACC AT*
(SEQ ID N0:5)
5132 5' TCCCGCTGTGATGCATT 3' 60.6
(SEQ ID N0:6)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-67-
1217855' T*CCCGCTGTGATGCATT* 63.3 2.7
3'
(SEQ ID N0:7)
T* = L-Thyrnidine, All P = S, C° = 2'-O-MOE '1"1GC, A" = 2'-
O-MOE A, T° = 2'-O-MOE SMeLT, G° = 2'-O- MOE G.
In order to overcome the binding affinity loss due to the L-isomer
placement we also incorporated 2'-O-MOE (2'-O-(2-methoxyethyl) modification
in the L/D-chimera and evaluated the binding affinity of the resultant
chimeric
compound to RNA target. The Tm analysis indicated that incorporation of 2'-O-
MOE modification along with L-thymidine in the chimera compensates the
affinity loss due to L-thymidine towards RNA binding. Thus the designer
oligonucleotide construct consisting of combined L-thymidine caps, 2'-O-MOE
and 2'-deoxyphophorothioates provide favorable properties for superior
antisense
oligonucleotide drugs.
EXAMPLE 19
Table III L-D Chimeric oligonucleotide Gapmers, hemimers and Inverted
Gapmers
Entry Sequence Target Class
17 5 C*TAGATTCCACACTCTCGTC 3' Mur. MDM2Gapmer
(SEQ ID N0:8)
18 5 CTAGATTCCACACTCTCGTC' 3' Mur. MDM2Gapmer
(SEQ ID N0:9)
19 5' CTTAGATTCCACACTCTCGTC~3 Mur. MDM2Gapmer
(SEQ ID NO:10)
5' CCGGTACCCCAGGTTCTTCA* 3' Mur. A-raf3'-hemimer
(SEQ ID NO:11)
21, 5' C*CGGTACCCCAGGTTCTTCA* Mur. A-raf3'-hemimer
22 3'
(SEQ ID N0:12)
23 5' CTAGATTCCACACTCTCGTC -~ Mur. MDM2Inverted
3' gapmer
(SEQ ID N0:13)
24 5' C*TAGATTCCACACTCTCGTC 3' Mur. MDM2Inverted
gapmer
(SEQ ID N0:14)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-68-
25 5'C*TAGATTCCACACTCTCGTC* 3' Mur. MDM2Inverted
gapmer
(SEQ ID NO:15)
C* = L-Cytidine, A* = L-Adenosine, All P = S, C° = 2'-O-MOE SMeC,
A°
= 2'-O-MOE A, T° = 2'-D-MOE SMeU, Go = 2'-O- MOE G.
EXAMPLE 20
L - Nucleosides with Novel Nucleobases and Oligonucleotides derived
therefrom
DMTO g DMTO
_'~IJO
O O O
P.
NCO' ~
O
H
CPG
3. B=T 4. B=T
5. B = N4-benzoyl A 11. B = N4-benzoyl A
6. B = N4-benzoyl-5-methyl C 12. B = N4-benzoyl-5-methyl C
7. B = N2-isobutyryl G 13. B = N2-isobutyryl G
8. B = 5-(1-propynyl) U 14. B = 5-(1-propynyl) U
9. B = 5-(1-propynyl) C 15. B = 5-(1-propynyl) C
10. B = phenoxazine 16. B = phenoxazine
EXAMPLE 21
5'-O-DMT-2',3'-dideoxy-1V4-[4-(CPG-succinyl)methylester]benzoylcytidine
(29).
2',3'-dideoxycytidine 26 [Prepared according to the literature procedure
Horwitz, J. P. et. al. J. Org. Chena. 1967, 32, 817-818) is converted into 5'-
O-silyl
derivative in presence of TBDMSCl and pyridine. This is then treated with 4-
(hydroxymethyl)benzoylchloride in pyridine to give compound 27 (Scheme 3).
Compound 27 is treated with succinic anhydride and DMAP in 1,2-dichloroethane
to give the succinyl derivative. The succinyl derivative is coupled with
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-69-
axninoalkyl CPG in presence of TBTU and 4-methylmorpholine in DMF to give
28. Compound 28 is desilylated with triethylamine trihydrofluoride and
triethylamine in THF. It is then tritylated with DMTCI in pyridine and DMAP to
give compound 29.
Scheme 3 O ~ ~ OH
f
NH2 HN
~N ~N
N_ 'O I N- 'O
HO O 1. TBDMSCI, py TBDMSO-,~
26 2~ O ~ ~ OH 27
Ch
Py
1. Sccinic anhydride, DMAP,
1,2-dichloroethane
2. CPG, TBTU,
4-methylmorpholine
O
HN I ~ O
O
~ N / H CPG
~ O
N- 'O
TBDMSO O
v
28
1. TEA.3HF, TEA, THF
2. DMTCI, Py. DMAP
O
HN I ~ O
O
~ N / H CPG
N~O O
DMTO O
.,=
29
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-70-
EXAMPLE 22
5'-O-DMT-2',3'-dideoxy-1V4-[4-(CPG-succinyl)methylester]benzoyladenosine
(33).
2',3'-Dideoxyadenosine 30 [Prepared according to the literature procedure
Horwitz, J. P. et. al. J. O~g. Clzem. 1967, 32, 817-818] is converted into 5'-
O-silyl
derivative in presence of TBDMSCI and pyridine. This is then treated with 4-
(hydroxymethyl)benzoylchloride in pyridine to give compound 31 (Scheme 4).
Compound 31 is treated with succinic anhydride, DMAP in 1,2-dichloroethane to
give the succinyl derivative. The succinyl derivative is coupled with
aminoalkyl
CPG in presence of TBTU and 4-methylinorpholine in DMF to give 32.
Compound 32 is desilylated with triethylamine trihydrofluoride and
triethylamine
in THF. It is then tritylated with DMT chloride in pyridine and DMAP to give
compound 33. '
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-71-
Sc
N HZ
//N ~ ~ N
\N NJ 1. TBDMSCI, py
HO~
30 a, O ~ ~ OH 31
CI
Py
1. Sccinic anhydride, DMAP,
1,2-dichloroethane
2. CPG, TBTU,
4-methylmorpholine
O
HN ~ O
O
~N ~ ~ N \~H CPG
\N ~ O
TBDMSO~ N
32
1. TEA.3HF, TEA, THF
2. DMTCI, Py. DMAP
O
HN I ~ O
/N ~ \ N / O
CPG
N~J o
DMTO~ N
33
EXAMPLE 23
Synthesis of 2'-3'-dideoxy oligonucleotides
Oligonucleotides 34 (SEQ 1D N0:16) and 35 (SEQ ID N0:17) are
prepared accoding to the procedure used for the synthesis of componds 17-25
(SEQ )D NOs:B-15) using solid support 29 and 33 respectively.
EXAMPLE 24
Table IV 2; 3'-Dideoxy containing oligonucleotide Gapmers, hemimers
entry Sequence Target Class
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-72-
34 5 CTAGATTCCACACTCTCGTC'~ Mur. MDM2Gapmer
3'
(SEQ ID N0:16)
35 5' CCGGTACCCCAGGTTCTTCA* Mur. A-raf3'-hemimer
3'
(SEQ ID N0:17)
C* = 2,-3'-Dideoxycytidine, A* = 2,-3'-Dideoxyadenosine All P = S,
C° _
2'-O-MOE SMeC, Ao = 2'_O-MOE A, T° = 2'-O-MOE SM''IJ, G° = 2'-O-
MOE G.
EXAMPLE 25
5'-O-DMT-2',3'-dideoxy-2',3'-didehydro-1V4-[4-(CPG-succinyl)methylester]-
benzoylcytidine (39).
2',3'-Dideox-2',3'-didehydroycytidine 36 [prepared according to the
reported procedure, Chu, C. K. et. al. J. O~g. Claem. 1989, 54, 217-225] is
converted into 5'-O-silyl derivative in presence of TBDMSCI in pyridine. This
is
then treated with 4-(hydroxymethyl)benzoylchloride in pyridine to give
compound
37 (Scheme 5). Compound 37 is treated with succinic anhydride, DMAP in 1,2-
dichloroethane to give the succinyl derivative. The succinyl derivative is
coupled
with aminoalkyl CPG in presence of TBTU and 4-methylmorpholine in DMF to
give 38. Compound 38 is desilylated with triethylamine trihydrofluoride and
triethylamine in THF. It is then tritylated with DMT chloride in pyridine and
DMAP to give compound 39.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-73-
Scheme 5 O ~ ~ OH
NHZ HN
~N ~N
N~o I N~o
HO-~~ 1. TBDMSCI, py TBDMSO~
36 2. O ~ ~ OH 37
Ch
Py
1. Sccinic anhydride, DMAP,
1,2-dichloroethane
2. CPG, TBTU,
O 4-methylmorpholine
HN I ~ O
O
~ N / \~~~ H CPG
I ~ O
N_ 'O
TBDMSO~
38
1. TEA.3HF, TEA, THF
2. DMTCI, Py. DMAP
0
HN I ~ O
O
~ N / \\~~ H CPG
I O
N~O
DMTO-,~
39
EXAMPLE 26
5'-O-DMT-2',3'-didehydro-2',3'-dideoxy-1Vø-[4-(CPG-succinyl)methylester]-
benzoyladenosine (43).
2',3'-Dideoxy-2'-3'-didehydroadenosine 40 [prepared according to the
reported procedure, Chu, C. K. et. al. J. Org. Cherra. 1989, 54, 217-225] is
converted into 5'-O-silyl derivative in presence of TBDMSCI and pyridine. This
is then treated with 4-(hydroxyrnethyl)benzoylchloride in pyridine to give
compound 41 (Scheme 6). Compound 41 is treated with succinic anhydride,
DMAP in 1,2-dichloroethane to give the succinyl derivative. The succinyl
derivative is coupled with aminoalkyl CPG in the presence of TBTU and 4-
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
_74_
methylmorpholine in DMF to give 42. Compound 42 is desilylated with
triethylamine trihydrofluoride and triethylamine in THF. It is then tritylated
with
DMTCI in pyridine and DMAP to give compound 43.
Scheme 6
NHZ
~N I ~ N
\N J
HO-~ N 1. TBDMSCI, py TBDMSO
40 2, O / ~ OH 41
CI~
PY
1. Succinic anhydride,
DMAP,
1,2-dichioroethane
2. CPG, TBTU,
4-methylmorpholine
O
HN I ~ O
N I ~N / O
CPG
s O
~J
N N
TBDMSO~
42
1. TEA.3HF, TEA, THF
2. DMTCI, Py. DMAP
O
H N I ~. O
N I ~N / O
CPG
i O
~J
N N
DMTO-~~
43
EXAMPLE 27
Table V. 2,3'-Didehydro-2',3'-dideoxy modified nucleoside containing
Chimeric oligonucleotide Gapmers, hemimers
EntrySequence Target Class
44 5 CTAGATTCCACACTCTCGTC'' Mur. MDM2 Gapmer
3'
(SEQ m N0:18)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-75-
45 5' CCGGTACCCCAGGTTCTTCA* Mur. A-f~af3'-hemimer
3'
(SEQ ID N0:19)
C* = 2,3'-Didehydro-2',3'-dideoxycytidine, A* = 2,3'-Didehydro-2',3'-
dideoxyadenosine, All P = S, C° = 2'-O-MOE SMeC, A° = 2'_O-MOE
A, T° = 2'-
O-MOE SMeU, G° = 2'-O- MOE G.
EXAMPLE 28
5'-O-DMT-2',3'-dideoxy-2'-fluoro-1V4-[4-(CPG-succinyl)methylester]benzoyl-
cytidine (50).
2',3'-Dideoxy-2'-fluro uridine 46 [prepared as reported, Martin J. A. et. al.
J. Med. Chem. 1990, 33, 2137-2145] is converted into 2',3'-dideoxy-2'-
flurocytidine 47 (Scheme 7) according to the reported procedure [Reference :-
Divalcar, K. J. et. al. .I. Clzem. Soc. Pef-k. Ty-ans. 1 1982, 1171-1176].
Compound
47 is converted into 5'-O-silyl derivative in presence of TBDMSCl and
pyridine.
This is then treated with 4-(hydroxymethyl)benzoylchloride in pyridine to give
compound 48. Compound.48 is treated with succinic anhydride, DMAP in 1,2-
dichloroethane to give the succinyl derivative. The succinyl derivative is
coupled
with aminoalkyl CPG in presence of TBTU and 4-methylmorpholine in DMF to
give 49. Compound 49 is desilylated with triethylamine trihydrofluoride and
triethylamine in THF. It is then tritylated with DMTCl in pyridine and DMAP to
give compound 50.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-76-
0
Scheme 7
I NH
N- "-O
HO--~~
46 F O ~ ~ OH
NHZ HN
~N ~N
N~o I N~o
HO-~~ 1. TBDMSCI, py TBDMSO~
47 F 2. O ~ ~ OH 4g F
CI~
Py
1. Succinic anhydride, DMAP,
1,2-dichloroethane
2. CPG, TBTU,
4-methylmorpholine
O
H N ~. O
O
I ~ N H CPG
O
N~O
TBDMSO--~~
F
49
1. TEA.3HF, TEA, THF
2. DMTCI, Py. DMAP
O
HN ' ~ ' O
O
I ~ N ~ H CPG
~ O
N- 'O
DMTO-,~~
50 F
EXAMPLE 29
Table VI. 3'-Deoxy-2'-fluorocytidine Chimeric oligonucleotide Gapmers,
hemimers and Inverted Gapmers
EntrySequence Target Class
51 5 CTAGATTCCACACTCTCGTC~~t Mur. MDM2Gapmer
3'
(SEQ ID N0:20)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
- 77 _
C* = 2',3'-Dideoxy-2'-fluorocytidine, All P = S, C° = 2'-O-MOE
SMeC, A°
= 2'-O-MOE A, T° = 2'-O-MOE SMeU, G° = 2'-O- MOE G.
EXAMPLE 30
5'-O-DMT-2',3'-deoXy-3'-fluro lV4-[4-(CPG-succinyl)methylester]benzoyl-
cytidine (56).
2',3'-Dideoxy-3'-fluro uridine 52 [prepared according to thereported
procedure Zaitseva, G. V. et. al. Bioorg. K7Zim. 1988, 14, 1275-1281] is
converted
into 2',3'-dideoxy-3'-fluorocytidine 53 (Scheme 8) according to the reported
procedure [Reference:-. Divakar, K. J. et. al. J. Chem. Soc. Perk. Traps. 1
1982,
1171-1176. Compound 53 is converted into 5'-O-silyl derivative in presence of
TBDMSCI and pyridine. This is then treated with 4-(hydroxymethyl)benzoyl
chloride in pyridine to give compound 54. Compound 54 is treated with succinic
anhydride, DMAP in 1,2-dichloroethane to give the succinyl derivative. The
succinyl derivative is coupled with aminoalkyl CPG in presence of TBTU and 4-
methylmorpholine in DMF to give 55. Compound 55 is desilylated with
triethylamine trihydrofluoride and triethylamine in THF. It is then tritylated
with
DMT chloride in pyridine and DMAP to give compound 56.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-78-
1. TEA.3HF, TEA, THF
2. DMTCI, Py. DMAP
O
O
Scheme 8
NH
N ~O
HO-,~~
O ~ ~ OH
52
NHZ HN
~N ~N
N' \_O I N ~O
HO-,~~ 1. TBDMSCI, py TBDMSO~
2. O OH -
53 ~ ~ F 54
CI~
Py
1. Succinic anhydride, DMAP,
1,2-dichloroethane
2. CPG, TBTU,
4-methylmorpholine
TB
O
HN I ~ 0
O
~ N / \1~ H CPG
~ O
N~O
DMSO-,~ s
D
HN I ~ O
O
~N s \1~N CPG
~ O H
N_ 'O
MTO-
56
EXAMPLE 31
5 Table VII. 2',3'-Dideoxy-3'-fluorocytidine Chimeric oligonucleotide Gapmers
EntrySequence Target Class
57 5 CTAGATTCCACACTCTCGTC~4 Mur. MDM2Gapmer
3'
(SEQ ID N0:20)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-79-
C* = 2',3'-Dideoxy-3'-fluorocytidine, All P = S, C° = 2'-O-MOE
SMeC, A°
= 2'-O-MOE A, T° = 2'-O-MOE SMe~, Go = 2'_O_ MOE G.
EXAMPLE 32
5'-D-DMT-3'-deoxy-2'-O-[2-(methoxy)ethyl] 1V4-[4-(CPG-succinyl)methyl-
ester]benzoylcytidine (62)
5'-O-TBDMS-N4-benzoyl-5-methylcytidine 58 is synthesized according to
the literature procedure [Reese, C. B. et. al. Tetrahed~oh Lett. 1999, 55,
5635-
5640]. The compound 58 is then converted into 59 according to reported
procedure [Danel, K. et. al. J. Med. Claena. 1996, 39, 2427-2431]. Compound 59
is converted into 5'-O-silyl derivative in presence of TBDMSCl and pyridine.
This is then treated with 4-(hydroxymethyl)benzoylchloride in pyridine to give
compound 60. Compound 60 is treated with succinic anhydride, DMAP in 1,2-
dichloroethane to give the succinyl derivative. The succinyl derivative is
coupled
with aminoalkyl CPG in presence of TBTU and 4-methylmorpholine in DMF to
give 61. Compound 61 is desilylated with triethylamine trihydrofluoride and
triethylamine in THF. It is then tritylated with DMTCl in pyridine and DMAP to
give compound 62.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-80-
NHBz Scheme 9
~N
O N~O 1. Dehydroxylation TBDMS
TBDMSO
2. Methanolic ammonia
OH O~O~CH3 CH3
58 n ,-, nu 59
H
~N
~N~O 1. Succinic anhydride, DMAP,
1. TBDMSCI, py TgDMSO O 1,2-dichloroethane '
2. CPG, TBTU,
O OH 4-methyimorpholine
2. ~ ~ 60 O~O~CH3
CI~
Py
O
HN ~ O
O
~ N H CPG
~ O
N"O
TBDMSO--~~
p~O~CH3
61
1. TEA.3HF, TEA, THF
2. DMTCI, Py. DMAP
O
HN I ~ O
O
~ N ~ . H CPG
N- 'O O
DMTO--~~
O~.O~CH3
62
E~~AMPLE 33
Table VIII. 3'-Deoxy-2'-O-[2-(methoxy)ethyl]-5-methylcytidine Chimeric
oligonucleotide Gapmers
EntrySequence Target Class
63 5 CTAGATTCCACACTCTCGTC"~ Mur. MDM2Gapmer
3'
(SEQ ID N0:21)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-81-
C* = 3'-Deoxy-2'-O-[2-(methoxy)ethyl]-5-methylcytidine, All P = S,
C° _
2'-O-MOE SMeC, Ao = 2'_O-MOE A, T° = 2'-O-MOE SMeU, G° = 2'-O-
MOE G.
EXAMPLE 34
N trifluroacetyl-pyrrolidine-2-(DMT)methanol-3-O-[(2-cyanoethyl)-N,N
diisopropylphosphoramidite] (67).
Compound 64 is synthesized according to the literature procedure [Huwe,
C. M. et. al. SyfZthesis,1997, l, 61-67]. It is then converted into
trifluromethyl
derivative 65 in presence of ethyl trifluroacetate in ethanol. Compound 65 is
tritylated to give compound 66. Compound 66 is phosphitylated to give the
compound 67.
EXAMPLE 35
3-O-(CPG-succinyl)-N trifluoroacetyl-pyrrolidine-2-(DMT)methanol (68).
Compound 66 is treated with succinic anhydride, DMAP in 1,2-
dichloroethane to give the succinyl derivative. The succinyl derivative is
coupled
with aminoalkyl CPG in presence of TBTU and 4-methylmorpholine in DMF to
give 68.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-82-
Scheme 10
CF3 CF3
H H H ~ DMT
N CF3COOEt N DMtCI, Py N
OH E H OH D P OrH
64 65 66
1. Succinic anhydride,
D2 dchloroethane
Q,~o 2. CPG, TBTU,
4-methylmorpholine
F3
N
67
68
EXAMPLE 36
Table IX. 3-hydroxy-2-pyrrolidinemethanol Chimeric oligonucleotide
Gapmers, hemimers
EntrySequence Target Class
69 5 CTAGATTCCACACTCTCGTB ~ 3' Mur. MDM2Gapmer
(SEQ ID N0:22)
70 5 B*CTAGATTCCACACTCTCGTB" 3' Mur. MDM2Gapmer
(SEQ ID N0:23)
71 5' CCGGTACCCCAGGTTCTTCAB * 3' Mur. A-raf3'-hemimer
(SEQ ID N0:24)
72 5' B*CCGGTACCCCAGGTTCTTCAB* Mur. A-raf3'-hemimer
3'
(SEQ ID N0:25)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-83-
B* = 3-hydroxy-2-pyrrolidinemethanol, All P = S, C° = 2'-O-MOE
SMeC,
A° = 2'-O-MOE A, T° = 2'-O-MOE SMeU, G° = 2'-O- MOE
G.
EXAMPLE 37
1-[2-(O-succinylCPG)-1-[2-hydroxy-1-(O-DMT-methyl)ethoxy] ethyl] cytosine
(76)
Compound 73 is prepared according to the reported procedure (Scheme
11) [Reference:- Bessodes, M. et. al. Tetrahedron Lett. 1985, 26(10), 1305-
1306].
This is converted into silylated compound in presence of 1,3-dichloro-1,1,3,3-
tetraisopropyldisiloxane in pyridine followed by benzoylation of exocyclic
amino
group with benzoic anhydride in DMF give compound 74. Compound 74 is
succinylated to give sucinyl derivative. The succinyl derivative is coupled
with
aminoalkyl CPG in presence of TBTU and 4-methylmorpholine in DMF to give
75. This is desilylated and tritylated to give compound 76.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-84-
Scheme 11
NHZ NHBz
HO O N O ~ N O
_ Si O
S~-O OH
73 ~ ~ 74
1. Succinic anhydride, DMAP,
1,2-dichloroethane
2. CPG, TBTU,
4-methylmorpholine
NHBz
1. TEA.3HF, TEA, THF
2. DMTCI, Py. DMAP
NHBz
C 'N
N- 'O
DMTO~p~
\J O1I
HO O
CPG
O
76
EXAMPLE 38
5 1-[2-O-(acetyl)-1-[2-[(Z-cyanoetliyl)-N,N diisopropylphosphoramidite]-1-(O-
DMT-methyl)ethoxy]ethyl]cytosine (79)
Compound 73 is silylated with 1,3-dichloro-1,1,3,3-
tetraisopropyldisiloxane in pyridine to give compound 77. This is then
acetylated
with acetyl chloride in pyridine to give compound 78. Compound 78 is
10 desilylated with TEA.3HF and TEA in THF. This is tritylated with DMTCl,
DMAP and pyridine followed by phosphitylation give compound 79.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-85-
Scheme 12
NH2 NHZ
wN wN
N~O ~ N' '-O
HO~O~ ~ ~ ~ _O~O
H'O~// '~'OH O~g;-O OH
73 ~ ~ 77
N HAc
~N
N"O
O~g;-O O\ /
~f ~(O
78
1. TEA.3HF, TEA, THF
2. DMTCI, Py. DMAP
3. Phosphitylation
NHAc
~N
N ~O
DMTO~O~
O O\ /
P ~I'(/
NC N(iPr)2 O
79
EXAMPLE 39
Table X.1-[2-hydroxy-1-[2-hydroxy-1-(hydroxymethyl)ethoxy]ethylcytosine
Chimeric oligonucleotide Gapmers
EntrySequence Target Class
80 5 CTAGATTCCACACTCTCGTC"~ Mur. MDM2 Gapmer
3'
(SEQ ID N0:26)
81 5 C~TAGATTCCACACTCTCGTC~ Mur. MDM2 Gapmer
3'
(SEQ ID N0:27)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-86-
C* = 1-[2-hydroxy-1-[2-hydroxy-1-(hydroxymethyl)ethoxy]ethylcytosine,
All P = S, C° = 2'-O-MOE SMeC, Ao = 2'-O-MOE A, T° = 2'-O-
MOE SMeLJ, G° _
2'-O- MOE G.
EXAMPLE 40
1-[2-O-(acetyl)-1-[2-[(2-cyanoethyl)-N,N diisopropylphosphoramidite]-1-(O-
DMT-methyl)thioethyl] ethyl] cytosine (86)
Compound 82 is synthesized according to literature procedure [Nake, T.
et. al. J. Am. Chem. Soc. 2000, 122, 7233-7243]. This is converted into 83 by
following a reported procedure for cleavage of vicinlal diols and subsequent
reduction of aldehyde thus obtained [Bessodes, M. et. al. TetraheclYOfx Lett.
1985,
26(10), 1305-1306]. Compound 83 is silylated with 1,3-dichloro-1,1,3,3-
tetraisopropyldisiloxane in pyridine to give compound 84. This is then
acetylated
with acetyl chloride in pyridien to give compound 85. Compound 85 is
1 S desilylated with TEA.3HF and TEA in THF. This is tritylated with DMTCl,
DMAP and pyridine followed by phosphitylation give compound 86.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
_ 87 _
Scheme 13
NH2 NH2
~N ~N
N~O I N' 'O
HO S HO S
OH OH HO OH
82 83
O
HN~Ph NH2
I I
N O ~ N O
~,O S ~ ~-O S
O~Si-O O ph O~Si-O OH
O 84
1. TEA.3HF, TEA, THF
2. DMTCI, Py. DMAP
3. Phosphitylation
NHBz
~N
I ~
N ~O
DMTO g
O O\ 'Ph
O-p ~II(~
NCB ~ p
N(iPr)2
86
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-88-
EXAMPLE 41
1-[2-(O-succinyl-CPG)-1-[2-hydroxy-1-(O-DMT-
methyl)thioethyl]ethyl]cytosine (89)
Compound 83 is converted into silylated compound in presence of 1,3-
dichloro-1,1,3,3-tetraisopropyldisiloxane in pyridine followed by benzoylation
of
exocyclic amino group with benzoic anhydride in DMF give compound 87.
Compound 87 is succinylated to give sucinyl derivative. The succinyl
derivative
is coupled with aminoalkyl CPG in presence of TBTU and 4-methylmorpholine in
DMF to give 88. This is desilylated followed by tritylation give compound 89
Scheme 14
NHZ NHBz
~N
N_ 'O
HO~g~ _
H'O/ \JOH
83 of
1. Suocinic anhydride, DMAP,
1,2-dichloroethane
2. CPG, TBTU,
4-methylmorpholine
NHBz
O
CPG
88
1. TEA.3HF, TEA, THF
2. DMTCI, Py. DMAP
NHBz
~N
N- 'O
DMTO~g~
\J O
HO
CPG
O
89
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-89-
EXAMPLE 42.
Table XI. 1-[2-hydroxy-1-[2-hydroxy-1-
(hydroxymethyl)thioethyl]ethylcytosine Chimeric oligonucleotide Gapmers
EntrySequence Target Class
90 5 CTAGATTCCACACTCTCGTC'~ Mur. MDM2 Gapmer
3'
(SEQ ID N0:28)
91 5 C'TAGATTCCACACTCTCGTCT Mur. MDM2 Gapmer
3'
(SEQ ID N0:29)
C* = 1-[2-hydroxy-1-[2-hydroxy-1-
(hydroxymethyl)thioethyl]ethylcytosine, All P = S, C° = 2'-O-MOE SMeC,
A° _
2'-O-MOE A, T° = 2'-O-MOE SMe-U, G° = 2'-O-MOE G.
EXAMPLE 43
5'-O-DMT-2',3'-dideoxy-3'-(N acetyl)amino lV4-[4-(CPG-
succinyl)methylester]benzoylcytidine (95).
Compound 92 is prepared according to the procedure reported in the
literature (Reference:-I~renitsky, T. A. et. al. J. Med. Chem. 1983, 26(6),
891-
895). This is then selectively tritylated with DMTCl and pyridine to give the
5'-
O-DMT derivative which is acetylated to give acetylated product. Selective
removal of the acetyl group at 1V4-position with aqueous ammonia at room
temperature gives compound 93. This is then treated with 4-
(hydroxymethyl)benzoyl chloride in pyridine to give compound 94. Compound
94 is treated with succinic anhydride, DMAP in 1,2-dichloroethane to give the
succinyl derivative. The succinyl derivative is coupled with aminoalkyl CPG in
presence of TBTU and 4-methylmorpholine in DMF to give 95.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-90-
Scheme 15
NH2 NH2
HO O N O N O
DMTO O
NH2 HN~O
92 93
O
~ i OH
O
HN
OH
~N
DMTO O N~O
HN~O
94
O
O
HN I / O
N H CPG
O
DMTO O N~O
HN\ 'O
EXAMPLE 44
5 Table XII. 2',3'-dideoxy-3'-(amino)cytidine Chimeric oligonucleotide
Gapmers
EntrySequence Target Class
96 5 CTAGATTCCACACTCTCGTC'~ Mur. MDM2 Gapmer
3'
(SEA ID N0:30)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-91-
C~k = 2',3'-dideoxy-3'-(amino)cytidine, All P = S, C° = 2'-O-MOE
$MeC,
A° = 2'-O-MOE A, T° = 2'-O-MOE SMeU, Go = 2'-O- 2'-O- MOE
G.
EXAMPLE 45
5'-O-DMT-2'-deoxy-3'-S-phenyl-3'-thio 1V4-[4-(CPG-succinyl)methylester]-
benzoylcytidine (101).
2'-Deoxy-3'-S-phenyl-3'-thiouridine 97 [prepared as reported in
Kawakami, H. et. al. Hete~ocycles, 1991, 32(12), 2451-2470] is converted into
2'-
deoxy-3-S-phenyl-3-thiocytidine 98 (Scheme 7) according to the reported
procedure [Divakar, K. J. et. al. J. Chem. Soc. Peek. Ti~aizs. 1 1982, 1171-
1176].
Compound 98 is converted into 5'-O-silyl derivative in presence of TBDMSCl
and pyridine. This is then treated with 4-(hydroxymethyl)benzoylchloride in
pyridine to give compound 99. Compound 99 is treated with succinic anhydride,
DMAP in 1,2-dichloroethane to give the succinyl derivative. The succinyl
derivative is coupled with aminoalkyl CPG in presence of TBTU and 4-
methylmorpholine in DMF to give 100. Compound 100 is desilylated with
triethylamine trihydrofluoride and triethylamine in THF. It is then tritylated
with
DMTCl in pyridine and DMAP to give compound 101.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-92-
0
Scheme 16
NH
N- '_O
HO p
y i
PhS~ 97
N H2
~N
N "-O
HO O 1.l
PhS~, 2,
98
Py
1. Succinic anhydride,
D MAP,
1,2-dichloroethane
2. CPG, TBTU,
O 4-methylmorpholine
HN ~ O
O
~ N H CPG
~ O
N ~O
TBDMSO-~~~
PhS~'
100
1. TEA.3HF, TEA, THF
2. DMTCI, Py. DMAP
O
HN I ~ O
O
~ N ~ H CPG
N~O O
DMTO-~~
PhS~ 101
EXAMPLE 46
Table XiII. 2'-deoxy-3'-S-phenyl-3'-thiocytidine Chimeric oligonucleotide
Gapmers
Entry Sequence Target Class
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-93-
102 ~ 5
C°T°A°G°A°TTCCACACTCT°C°G.degr
ee.T°CT 3 ~ Mur. MDM2 ~ Gapmer
(SEQ ID N0:31)
C* = 2'-deoxy-3'-S-phenyl-3'-thiocytidine, All P = S, C° = 2'-O-
MOE
SMeC' Ao = 2'-O-MOE A, T° = 2'-O-MOE SM''~(J, G° = 2'-O- 2'-
O- MOE G.
EXAMPLE 47
5'-O-DMT-3'-deoxy-2'-S-phenyl-2'-thio 1V4-[4-(CPG-succinyl)methylester]-
benzoylcytidine (107).
3'-Deoxy-2'-S-phenyl-2'-thiouridine 103 [prepared as reported ,
I~awakami, H. et. al. Heterocycles, 1991, 32(12), 2451-2470] is converted into
2',3'-dideoxy-2'-flurocytidine 104 (Scheme 17) according to the reported
procedure [Divakar, K. J. et. al. J. Chen2. Soc. Perk. Traps. 1 1982, 1171-
1176].
Compound 104 is converted into 5'-O-silyl derivative in presence of TBDMSCI
and pyridine. This is then treated with 4-(hydroxyrnethyl)benzoylchloride in
pyridine to give compound 105. Compound 105 is treated with succinic
anhydride, DMAP in 1,2-dichloroethane to give the succinyl derivative. The
succinyl derivative is coupled with aminoalkyl CPG in presence of TBTU and 4-
methylinorpholine in DMF to give 106. Compound 106 is desilylated with
triethylamine trihydrofluoride and triethylamine in THF. It is then tritylated
with
DMTCI in pyridine and DMAP to give compound 107.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-94-
0
Scheme 17
~NH
N- 'O
HO-,~
103 ~SPh O ~ ~ OH
NHZ HN~
~N I ~N
N- 'O N- 'O
HO~ 1. TBDMSCI, py TBDMSO~
OH ~S'Ph
~SPh O
104 2' C'~~ 105
Py
1. Succinic anhydride,
DMAP,
1,2-dichloroethane
2. CPG, TBTU,
O 4-methylmorpholine
HN I ~ O
O
I ~ N / \~~ H CPG
O
N ~O
TBDMSO-~~
~SPh
106
1. TEA.3HF, TEA, THF
2. DMTCI, Py. DMAP
O
HN I ~ 0
O ~ ~
I ~ N ~ ~~~H CPG
I IO
N~O
DMTO
'~~SPh
107
EXAMPLE 48
Table XIV. 3'-deoxy-2'-S-phenyl-2'-thiocytidine Chimeric oligonucleotide
Gapmers
EntrySequence Target Class
108 5 CTAGATTCCACACTCTCGTC"~ Mur. MDM2 Gapmer
3'
(SEQ ID N0:32)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-95-
C* = 3'-deoxy-2'-S-phenyl-2'-thiocytidine, All P = S, C° = 2'-O-
MOE
SMeC' Ao = 2~_O-MOE A, T° = 2'-O-MOE SMeU, Go = 2'_O- 2'-O- MOE G.
EXAMPLE 49
5'-O-DMT-1 [2,3-deoxy-2-N-morpholino-(3-D-glycero-pent-2-enofuranosyl]-
cytosine lV4-[4-(CPG-succinyl)methylester]benzoyl (113).
1 [2,3-Deoxy-2-N-morpholino-(3-D-glycero-pent-2-enofuranosyl]uracil 109
[prepared as reported in Kandasamy, S. et. al. Tetrahedroh,1996, 52(13), 4877-
4882] is converted into 2',3'-dideoxy-2'-flurocytidine 110 (Scheme 18)
according
to the reported procedure [Divakar, I~. J. et. al. J. Chem. Soc. Perk. Traps.
1
1982,1171-1176]. Compound 110 is converted into 5'-O-silyl derivative in
presence of TBDMSCI and pyridine. This is then treated with 4-(hydroxymethyl)-
benzoylchloride in pyridine to give compound 111. Compound 111 is treated
with succinic anhydride, DMAP in 1,2-dichloroethane to give the succinyl
derivative. The succinyl derivative is coupled with aminoalkyl CPG in presence
of TBTU and 4-methylmorpholine in DMF to give 112. Compound 112 is
desilylated with triethylamine trihydrofluoride and triethylamine in THF. It
is
then tritylated with DMT chloride in pyridine and DMAP to give compound 113.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-96-
0
Scheme 18
NH
N- 'O
HO-~~
i~
109 NV OH
NHZ
~N
N~O
HO-~ 1. TBDMSCI, py
110 /NCO 2. O
CI
Py
1. Succinic anhydride,
DMAP,
1,2-dichloroethane
2. CPG, TBTU,
O 4-methyimorpholine
HN I ~ O
0
~ N ~ ~~ H CPG
O
N~O
TBDMSO-~~
N~
~O
112
1. TEA.3HF, TEA, THF
2. DMTCI, Py. DMAP
O
HN I ~ O
O ~ ~
~ N ~ ~~'~H CPG
~ IIO
N- 'O
DMTO-,~
I~N~
113
EXAMPLE 50
Table XV.1 [2,3-deoxy-2-N-morpholino-(3-D-glycero-pent-2-
enofuranosyl]cytosine Chimeric oligonucleotide Gapmers
Entry Sequence Target Class
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-97-
114 5 CTAGATTCCACACTCTCGTC"~ Mur. MDM2 Gapmer
3
(SEQ ID N0:33)
C* = 1 [2,3-deoxy-2-N-morpholino-(3-D-glycero-pent-2-
enofuranosyl]cytosine, All P = S, C° = 2'-O-MOE SMeC, A° = 2'-O-
MOE A, T° _
2'-O-MOE SM°CT, G° = 2'-O- 2'-O- MOE G.
EXAMPLE 51
Preparation of CPG Resin Substituted With 9-(Aminoethoxy)phenoxazine
Nucleoside (G-Clamp), G-Clamp Succinate 154
After drying at 50 °C in vacuo overnight, the G-clamp 2'-
deoxynucleoside
(152, 0.51 g, 0684 mmol) was dissolved in anhydrous DCM/Pyr (5:1) and 0.103 g
(1.03 mmol) succinic anhydride were added to the solution. Subsequently 41.5
mg (0.34 mmol) DMAP in 1 mL DMF were added and the mixture was stirred
overnight. After completion of the reaction (TLC) the solvent was evaporated
in
vacuo and the remaining yellow oil was dissolved in DCM, washed twice with
10% aq. NAHCO3, 10% aq. citrate and brine. After drying over NaZS04 the
organic phase was evaporated in vacuo to yield a yellow solid (0.45 g, 75%).
MS
(HR-FAB) m/z 897.256 (M + Na)+.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-98-
T
D
i
R ii
NH
DMTO N O
O
O
O
II N
O H
CPG
151,153,155: R = H;
152,154,156: R = OCH~CH2NHCOCF3
EXAMPLE 52
G-Clamp-Succinyl-LCAA-CPG 156
131 mg (0.15 mmol) G-clamp succinate were dissolved in DMF and 68 ~,L
(0.4 mmol) DIEA were added. Subsequently a solution of 57 mg (0.15 mmol)
HATU in DMF was added to the mixture under stirring. Stirring was continued
for about 1 min in order to allow pre-activation before the mixture was added
to 1
g of LCAA-CPG (initial loading: 115 ,umol/g) and the suspension was shaken
overnight. Subsequently the resin was washed 3 times each with DMF, DCM and
CH3CN and the unreacted amino groups of the resin were capped by shaking the
resin with 0.24 mL (2 mmol) ethyl trifluoroacetate and 0.28 ml (2 mmol) TEA in
5 ml MeOH. Finally the resin was washed with MeOH, CH3CN and DCM and
dried in vacuo. The loading with G-clamp succinate was determined by DMT
assay (final loading: 65 ~mol/g).
R
/
O
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-99-
EXAMPLE 53
2'-deoxy phenoxazine CPG
2'-deoxy phenoxazine CPG was synthesized following the procedures
illustrated in example 52 above.
EXAMPLE 54
Oligonucleotide Synthesis
Solid phase syntheses of oligonucleotides containing G-clamp and
phenoxazine units were carried out using standard phosphoramidite chemistry
and
an Applied Biosystems (Perkin Elmer Corp.) DNA/RNA synthesizer 380B.
Cleavage and deprotection of the oligonucleotides was performed using a
solution
of 40% aq. MeNHz and 28-30% aq. NH3 (1:l) at r.t. for 4 h. The
oligonucleotides
were purified by reversed phase HPLC using a 306 Piston Pump System, a 811 C
Dynamic Mixer, a 170 Diode Array Detector and a 215 Liquid Handler together
with the Unipoint Software from Gilson (Middleton, Wi). The HPLC conditions
were as follows: Column: Waters Deltapak C1$ reversed phase (300x3.9 mrn, 15
,u, 300 ~); Solvent A: 0.1 M NH40Ac in H20; solvent B: 0.1 M NH40Ac in
CH3CN/H20 (80:20); Gradient: 0-40 min 0-50% B. After chromatographic
purification the oligonucleotides were desalted by RP-HPLC, lyophilized, and
stored at -20°C.
EXAMPLE 55
Guanidinylation on Solid Support
As outlined in Scheme 21, we have used two different strategies to
introduce the guanidinuim moiety. One strategy is the selective deprotection
of
the primary amino group followed by guanidinylation on the solid support (A).
In
the case of the 2'-O-(aminohexyl) function the allyloxycarbonyl (Alloc)
protecting group was selectively removed by treating the support-bound
oligonucleotides with 1.0 mL of 10 mg Pda[(Ph-CH=CH)2C0]3 and 26 mg P(Ph)3
in a solution of 1.2 M nBuNH2/ HCOOH in THF at 50°C for 1.5 h. After
the
removal of Alloc, the support-bound oligonucleotides were washed with DCM,
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-100 -
acetone, sodium N,N-diethyldithiocarbamate (ddtc Nab), HaO, acetone, DCM,
diethyl ether and dried in vacuo. Prior to guanidinylation, the resin was
suspended in a solution of 10% DIEA in DMF, shaken for 5 min, and washed with
DMF followed by DCM. Subsequently, a 1.0 M solution of 1H-pyrazole-1-
carboxamidine hydrochloride and DIEA in DMF was added to the support-bound
oligonucleotides and the suspension was shaken at r.t. for 5 h. For final
deprotection and cleavage of the oligonucleotides, the resin was treated with
conc.
aqueous ammonia at 55°C for 1 h. After separation from the CPG support
and
evaporation of ammonia, the aqueous solution was filtered through a 0.45 ~.m
Nylon-66 filter and stored frozen at -20°C for further analysis.
Scheme 20
g ~ z CI-
O
~ NH NH2
O , I / NH
NH O w
N~o ~. I ~
O O O N O
O
O NH NH2
~+
O~ I O NHS CI- , P-O-
O OiI
O.~
Scheme 20.
Modified nucleotides 2'-O-(guanidinylhexyl)-5-methyluridine (A); 9-
guanidinylethoxy phenoxazine nucleotide (B)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-101-
A
0
NH
H
N O
i, ii O~ iii
HR O O~~NH~ Ha
R = ~O~ CI_
O
NI'HZ CI'
NHR I W O~NHZ ~NHa
~NH ,
H ~ O ~ y
i O O N O ii
~'OH O~'OH
v
R=
CF3
Scheme 21.
(A) Reaction conditions: (i) 1.0 mL of 10 mg Pdz[(Ph-CH=CH)ZCO]3, 26
mg P(Ph)3 in 1.2 M nBuNH2/HCOOH in THF, 50°C, 1.5 h; (ii) washing with
DCM, acetone, sodium N,N-diethyldithiocarbamate (ddtc Na+), H20, acetone,
DCM, diethyl ether; (iii) 1.0 M of 1H-pyrazole-1-carboxamidine hydrochloride
and DIEA in DMF, r.t., 5 h. (B) (i) 40% aq. CH3-NHZ/conc. aq. NH3 (1:1),
55°C,
1h; (ii) 1.0 M 1H-pyrazole-1-carboxamidine hydrochloride in 1.0 M aq. Na2C03,
r.t., 3 h for ON-3, ON-4 and 55°C, 12 h for ON-5, ON-6, respectively.
EXAMPLE 56
Guanidinylation of Completely Deprotected Oligonucleotide in Solution
The base-labile trifluoroacetyl group (Tfa), which is compatible with the
conditions of oligonucleotide synthesis and deprotection, was chosen for
protection of the primary amino group of G-clamp. The oligonucleotides were
deprotected and cleaved from the solid support prior to guanidinylation by
using a
1:1 mixture of 40% aqueous CH3-NHa and conc. aqueous ammonia (AMA),
which prevents the formation of acyl- or acrylonitrile adducts with the highly
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-102-
nucleophilic primary amino group. To avoid transamination at cytosine during
the
deprotection step, N-acetyl- instead of N-benzoyl-protected C was used for
oligonucleotide synthesis. After the oligomers were purified by RP-HPLC and
analyzed by ES-MS, the primary amino group of G-clamp was guanidinylated by
treating the oligonucleotides with 1-2 ~mol of 2 mmol (297 mg) of 1H-pyrazole-
1-carboxamidine hydrochloride in 2 mL of a 1.0 M aqueous NaZC03 solution at
r.t. for 3 h. Subsequently, the oligonucleotides were purified by gel
chromatography (Sephadex G25) followed by RP-HPLC and analyzed by
capillary gel electrophoresis (CGE) and electrospray mass spectrometry (ES-
MS).
The guanidynyl-modified oligonucleotides synthesized during this study are
summarized in Table XVI.
Interestingly, in the case of self complementary sequences, such as ON-5
(SEQ ID N0:38) or ON-6 (SEQ ID N0:39), the conditions described above
yielded only a small fraction of guanidinyl G-clamp oligomer. Apparently, the
double-stranded structure of these palindromic oligonucleotides with the
primary
amino group being involved in base pairing interaction with complementary
guanine prohibited guanidinylation. W order to disrupt hydrogen bond
interaction
and to prevent duplex formation, the reaction was carried out at elevated
temperature of 55°C and extended reaction time of about 12 h. Using
these
conditions, complete guanidinylation of the amino groups of ON-5 (SEQ ID
NO:38) and ON-6 (SEQ ID N0:39) was achieved without causing any detectable
side reactions.
Guanidinylation of the primary amino groups slightly increased the
hydrophobicity of the corresponding oligomers, which could be detected by RP-
HPLC analysis as a minor change in the retention time. The T", data of ON-3 in
comparison to the unmodified G-clamp ON-2 (SEQ ID N0:35) show a decrease
in hybridization affinity towards complementary RNA and DNA of 5.9 and
5.7°K,
respectively (Table XVII). These findings, which seem to be contradictory to
the
formation of the additional hydrogen bonding between guanidinyl G-clamp and a
complementary guanine, could be explained by another structural detail
observed
by crystallographic X-ray analysis of the duplex of self complementary ON-5
(SEQ ID N0:38) [Wilds, C. J.; Maier, M. A.; Tereshko, V.; Manoharan, M.; Egli,
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-103-
M. ira pf~eparatiora]. The modified base pairs C* and G showed some buckling
relative to the other base pairs in the duplex, which might be a consequence
of
altered steric requirements for accommodating the guanidinium-ethoxy moiety
within the geometric boundaries of both the Watson-Crick and Hoogsteen-type
hydrogen bonds. It can be assumed that the out-of plane distortion is
responsible
for the loss of affinity observed for the guanidinyl-modified ON-3 (SEQ ID
N0:36) compared to the parent G-clamp containing ON-2 (SEQ ID N0:35).
In summary, two methods for postsynthetic modification of
oligonucleotides have been developed, which involve the conversion of primary
amino functions into guanidinium groups by using 1H-pyrazole-1-carboxamidine
hydrochloride. For reaction on the solid support, the amino groups were
protected
by Alloc, which can be selectively removed without cleaving the
oligonucleotide
from the support, and the guanidinylation was carried out in 10% DIEA in DMF.
On the other hand, primary amino groups were protected with Tfa, which can be
readily removed under the conditions of oligonucleotide deprotection and
cleavage, for postsynthetic guanidinylation in aqueous solution. Using these
methods several modified oligonucleotides bearing guanidinium moieties, facing
either the minor or major groove, have been prepared and analyzed.
EXAMPLE 57
Table XVl. Oligonucleotide Sequence and Guanidinyl Modification.
OLIGO Sequence 5' -~ ModiPcation MW~~,~ MWfound
3'
ON-1 TTT TU*T TTT all PO; U*: 2'-O- 3281.6 3281.7
T
(SEQ ID NO:34) liexylguanidinyl-USme
ON-2 TCT CC*C TCT all PO; C* = 2.'-deoxy-3039.1 3039.4
C
(SEQ ID N0:35) G-clamp
ON-3 TCT CC*C TCT all PO; C* = 2'-deoxy-3081.1 3080.8
C
(SEQ ID N0:36) guanidinyl G-clamp
ON-4 CTC GTA CCC* all PO; C* = 2'-deoxy-5553.7 5552.1
TCC
CGG TCC (SEQ guanidinyl G-clamp
ID
N0:37)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-104-
ON-5 GC*G TAUM ACGC all PO; UM = 2'-MOE-U""';3293.3 3292.8
(SEQ ID N0:38) C* = 2'-deoxy-
guanidino G-clamp
ON-6 GCG TAUM AC*GC all PO; UM = 2'-MOE-U'";3293.3 3293.0
(SEQ ID N0:39) C* = 2'-deoxy-
guanidino G-clamp
EXAMPLE 58
Table XVII. T"t Data of ON-3 (SEQ )D N0:36) in comparison to the parent G-
clamp-modified ON-2 (SEQ IDN0:35).
ON Modification Target Strand"Tm ~T",/modb
ON-2 G-Clamp RNA 70.8 18.4
ON-3 Guanidinyl RNA 64.9 , 12.5
G-clamp
ON-2 G-Clamp DNA 59.2 22.1
ON-3 Guanidinyl DNA 53.5 16.4
G-clamp
" Sequence: 5'-AAAAA GAG AGG GAG A (SEQ ID NO:40); " vs. parent DNA.
E~~AMPLE 59
Guanidinyl G-clamp modification
The guanidinyl G-clamp modification was designed to allow for additional
hydrogen bonds to the 06 and N7 Hoogsteen binding sites of guanosine (Figure
1B). Binding studies of DNA oligomers containing a single unit to a RNA target
revealed an increase in the melting temperature of 16°C relative to the
wildtype
DNA, slightly lower than the ~T", observed for the original G-clamp
modification.
To investigate the structural properties of this modification we determined
the X-
ray crystal structure of a modified decamer duplex with the sequence
GC*GTATMOEACGC (SEQ ID NO:41), where C* is the guanidino G-clamp and a
2'-O-methoxyethyl thymine is TMOE (Figure 1C). Altmann, K.-H.; Dean, N. M.;
Fabbro, D.; Freier, S. M.; Geiger, T.; Haner, R.; Husken, D.; Martin, P.;
Monia, B.
P.; Miiller, M.; Natt, F.; Nicklin, P.; Phillips, J.; Pieles, U.; Sasmor, H.;
Moser H.
E. Chinaia 1996, 50, 16~-176; Teplova, M.; Minasov, G.; Tereshko, V.; Inamati,
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-105-
G. B.; Coolc, P. D.; Manoharan, M.; Egli. M. Nature Struct. Biol. 1999, 6, 535-
539. The synthesis and purification of the oligonucleotides was carried out
according to standard procedures. Crystals of this decamer duplex were grown
by
the hanging drop vapor diffusion method using commercially available screens
(Hampton Research, Laguna Niguel, CA) [Hanging drop vapor diffusion: a 2 ~,L
droplet (1.2 mM DNA, 5 % MPD, 20 mM Na cacodylate pH 6.0, 6 mM spermine
~ 4 HCI, 40 mM NaCI, 6 mM KCI, 10 mM MgCl2 was equilibrated against a
reservoir of 1 mL 35% v/v MPD. Space group P212121; cell dimensions a = 24.52
1~, b = 43.02 ~, c =46.68 ~]. Data collection was performed synchrotron source
[A crystal (0.7 x 0.2 x 0.2 mm) was picked up .from a droplet with a nylon
loop
and transferred into a cold NZ stream (120 K). High- and low- resolution data
sets
were collected on the 5-ID beam line (~= 0.97810 of the DND-CAT at the
Advanced Photon Source, Argonne, IL, using a MARCCD detector. Data were
integrated and merged with DENZO/SCALEPACKI°. The overall Rmerge for
all
reflections between 20 and 1 ~ was 4.7 % (Otwinowski, Z.; Minor, W. Methods
Efazymol. 1997, 276, 307-326) and data collection and refinement statistics
are
listed in Table XVIII. The structure was solved by molecular replacement using
the DNA decamer as the initial model and refined with the programs CNS1~' and
SHELX-9713. After monitoring the Rfree using 10% of the reflections and
° 20 reaching 22 %, all reflections were included in the final rounds
of isotropic
refinement; Brunger, A. T. Crystallography & NMR System (CNS), Version 0.9,
Yale University, New Haven, CT, 1998 [Sheldrick, G. M; Schneider, T. R.
Methods Enzynaol. 1997, 277, 319-343; Egli, M.; Tereshko, V.; Teplova, M.;
Minasov, G.; Joachimiak, A.; Sanishvili, R.; Weeks, C. M.; Miller, R.; Maier,
M.
A.; An, H. Y.; Cook, P. D.; Manoharan, M. Biopolyme~s: Nucleic Acids Sciences
1998, 48, 234-252; Clarke, N. D.; Beasner, L. J.; Goldberg, H. R.; Berkower,
C.;
Pabo, C. O. Science 1991, 254, 267-270; Rich, A. In The Chemical Bond:
Sty°uctu~e afzd Dyfaatraics; Zewail, A. Ed.; Academic Press, New York,
1992; pp
31-86; Pabo, C. O.; Sauer, R. T. Anrau. Rev. Biochem. 1992, 61, 1053-1059;
Lin,
K.-Y.; Jones, R. J.; Matteucci, M. D. J. Am. ClZena. Soc. 1995,117, 3873-
3874].
The overall structure of this duplex is A-form as a result of 2'-O-
methoxyethyl thymine units at positions 6 and 16 in the duplex. An A-form
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-106 -
environment is desirable to study the structure of nucleic acid modifications
for
antisense purposes. As illustrated in the case of base pair C12*-G9 (Figure
2),
electron density around the heterocycles clearly shows the two Hoogsteen-type
hydrogen bonds formed between the amino and imino nitrogens of the tethered
guanidinium and 06 and N7 of guanosine, respectively. The hydrogen bond
lengths are 2.88 ~ and 2.861 and the lengths of the corresponding hydrogen
bonds in base pair C2*-G19 are 2.92 ~ and 2.87 ~, respectively. The quality of
the electron density around individual atoms of the phenoxazine ring and
tethered
group demonstrate that this modification is well ordered and does not assume
random conformations. There is some buckling of modified base pairs relative
to
the other base pairs in the duplex. This out-of plane distortion of the base
pair
between the G-clamp and G may be a consequence of the requirement to optimize
the geometry of both the Watson-Crick and Hoogsteen-type hydrogen bonds
within the geometric boundaries provided by a guanidinium-ethoxy moiety. In
addition, the observed arrangements help avoid a steric contact between 06 of
G
and the ethoxy-linker oxygen of the G-clamp (Figures 1 and 2).
Presence of the G-clamp results in a considerable improvement of intra-
strand stacking at the GpC* step compared with stacking between cytosine and
the
5'-adjacent base (G1 and Gl l, respectively). The overlap between Gl and C2*
is
depicted in Figure 3. While the "cytosine core" displays relatively little
stacking
to the guanosine base, the remainder of the phenoxazine ring system virtually
covers the entire guanosine base. However, while stacking between G-clamp and
the base to the 5'-side is improved, stacking to the 3'-adjacent base is not
affected
by incorporation of the modified base.
Placement of the positively charged guanidinium moiety in the center of
the major groove, a site of strong negative potential, likely results in a
siguficant
electrostatic contribution to stability. Moreover, the guanidinium group and
phosphates from opposite strands are relatively closely spaced. The average
distance between the imino nitrogens of C~= and 02P oxygens of phosphates is
5.8
1~. Although too long for direct salt bridges, water molecules link
guanidinium
and phosphate groups. In the case of C12*, single water molecules mediate
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-107 -
contacts between a water bound between guanidinium imino nitrogens and 02P
oxygens of residues C8 and G9.
Interactions between positively charged amines and the Hoogsteen binding
site of guanosine are well k~iown. For example, X-ray crystallographic studies
of
the 7~ repressor bound to duplex DNA revealed specific contacts between a
lysine
and the 06 position of G [Clarke, N. D.; Beamer, L. J.; Goldberg, H. R.;
Berkower, C.; Pabo, C. O. Science 1991, 254, 267-270; Rich, A. In The Chemical
Bond: Structure afad Dynamics; Zewail, A. Ed.; Academic Press, New York,
1992, 31-86; Pabo, C. O.; Sauer, R. T. Annu. Rev. Biochem.1992, 61, 1053-
1059].
The present structure of the guanidyl G-clamp is similar to the bidentate
hydrogen
bonding of the arginine fork with the N7 and 06 positions of guanine in
protein-
nucleic acids interactions [Clarke, N. D.; Beamer, L. J.; Goldberg, H. R.;
Berkower, C.; Pabo, C. O. Science 1991, 254, 267-270; Rich, A. In The Chemical
Bohd: St~uctuYe and Dynamics; Zewail, A. Ed.; Academic Press, New York,
1992, 31-86; Pabo, C. O.; Sauer, R. T. Aranu. Rev. Biochem. 1992, 61, 1053-
1059].
The observed structure reveals some buckling of the C*-G base pair, presumably
due to sterics as a consequence of the extended guanidinylethoxy spacer arm. A
comparison of the T", data of the G-clamp and guanidino G-clamp revealed that
guanidinylation appears to have only a slight effect on overall stability.
Two crucial stabilizing factors of this modification are an increase in the
number of hydrogen bonds and improved stacking interactions. Additional
contributions to stability are favorable electrostatic interactions and well-
ordered
water networks. It is difficult to discern if one of these contributions plays
a more
important role than the others. Binding studies of oligomers with the
phenoxazine
moiety alone showed moderate increases in T"t of 2-7°C [Lin, K.-Y.;
Jones, R. J.;
Matteucci, M. D. J. Am. Chem. Soc. 1995,117, 3873-3874]. Stability was
increased most when several phenoxazine groups were clustered together on the
same strand, allowing for tricyclic-tricyclic stacking interactions. In the
case of an
acyclic G-clamp modification, no enhancement in binding was observed. Only
when both the phenoxazine and tethered amino group were present was a drastic
improvement in binding observed [Lin, K.-Y.; Matteucci, M. D. J. Arn. Chem.
Soc. 1998, 120, 8531-8532; Flanagan, W. M.; Wolf, J. J.; Olson, P.; Grant, D.;
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-108-
Lin, K.; Wagner, R. W.; Matteucci, M. D. Pr°oc. Natl. Acad. Sci. USA
1999, 96,
3513-3518]. Clearly, hydrogen bonds from the guanidinium group maintain the
guanidine G-clamp modification in a position that allows stacking interactions
and
formation of stable water networks. This is the first report of a single base
pair
within a nucleic acid duplex combining Watson-Crick and Hoogsteen binding to a
total number of five hydrogen bonds
EXAMPLE 60
Reflection Data and Refinement Statistics.
Table XVIII
ResolutionN (unique)Mean % complete R-factors
[1l ~(~]
10.00-3.001073 26.90 98.8 0.175
3.00-2.50 768 31.51 99.9 0.182
2.50-2.00 1722 34.38 100.0 0.180
2.00-1.80 1288 36.70 100.0 0.154
1.80-1.60 2005 30.33 99.9 0.153
1.60-1.40 3314 27.90 100.0 0.166
1.40-1.20 5804 24.68 100.0 0.179
1.20-1.10 4680 20.08 100.0 0.187
1.10-1.00 6666 14.35 99.6 0.200
All data 27320 23.63 99.6 0.175
'~R-factor = Ejtxt ~ F'(lz.kl)o F(hkl)~ ~ l E~tkr F(hkl)~; no Q cutoff was
used.
EXAMPLE 61
Synthesis of G-clamp Modified Oligonucleotides targeting c-raf'lVIessage
Sequence (5'-3') Backbone Modification
ATG-CAT-TCT-GCC-CCC-AAG-GA P=S (SEQ m N0:42)
ATG-C*AT-TCT-GCC-CCC-AAG-GA P=S (SEQ ID N0:43)
ATG-CAT-TC*T-GCC-CCC-AAG-GA P=S (SEQ ID N0:44)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-109-
ATG-CAT-TCT-GC~C-CCC-AAG-GA P= S
(SEQ ID N0:45)
ATG-CAT-TCT-GCC*-CCC-AAG-GA P= S (SEQ m N0:46)
ATG-CAT-TCT-GCC-C*CC-AAG-GA P= S (SEQ m N0:47)
ATG-CAT-TCT-GCC-CC*C-AAG-GA P= S
(SEQ m N0:48)
ATG-CAT-TCT-GCC-CCC*-AAG-GA P= S (SEQ m N0:49)
C* = G-clamp modification.
EXAMPLE 62
Ih vivo Stability of Modified MDM-2 Oligonucleotides
Table XIX 2'-deoxy Oligonucleotides for in vivo Stability Evaluation
Sequence (5'-3') Target Backbone
CTA GAT TCC ACA CTC TCG TC MDM-2 P=S
(SEQ m NO:50)
C*TA GAT TCC ACA CTC TCG TC MDM-2 P=S
(SEQ 117 NO:51)
CTA GAT TCC ACA CTC TCG TC* MDM-2 P=S
(SEQ m N0:52)
C*TA GAT TCC ACA CTC TCG TC* MDM-2 P=S
(SEQ m N0:53)
C* = G-clamp modification.
The in. vivo stability of selected modified oligonucleotides synthesized
is determined in BALB/c mice. Following a single i.v. administration of 5
mg/kg
of oligonucleotide, blood samples are drawn at various time intervals and
analyzed by CGE.
Fox each oligonucleotide tested, 9 male BALB/c mice (Charles River,
Wilmington, MA) weighing about 25 g are used. Following a one week
acclimatization the mice received a single tail-vein injection of
oligonucleotide (5
mg/kg) administered in phosphate buffered saline (PBS), pH 7Ø One retro-
orbital bleed (either at 0.25, 0.5, 2 or 4 h post-dose) and a terminal bleed
(either
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-110 -
1, 3, 8, or 24 h post-dose) are collected from each group. The terminal bleed
(approximately 0.6-0.8 mL) is collected by cardiac puncture following
lcetamine/xylazine anasthesia. The blood is transferred to an EDTA-coated
collection tube and centrifuged to obtain plasma. At termination, the liver
and
kidneys are collected from each mouse. Plasma and tissue homogenates are used
for analysis to determine intact oligonucleotide content by CGE. All samples
are
immediately frozen on dry ice after collection and stored at -80°C
until analysis.
The CGE analysis indicated the relative nuclease resistance of G-
clasnp modification containing oligomers compared to the parent MDM-2
(uniformly 2'-deoxy-phosphorothioate oligonucleotide targeted to mouse MDM-
2). Because of the nuclease resistance of the G-clamp modification, the
modified
oligonucleotides are found to be more stable in plasma, while ISIS 11061 (SEQ
ID N0:42) was not. Similar observations are noted in kidney and liver tissue.
This implies that G-clamp modifications offer excellent nuclease resistance in
plasma, kidney and liver against exonucleases and endonucleases. Thus
oligonucleotides with longer durations of action can be designed by
incorporating
both the G-clamp modification and other analogous motifs into their structure.
A
plot of the percentage of full length oligonucleotide remaining intact in
plasma
one hour following administration of an i.v. bolus of 5 mg/kg oligonucleotide
is
determined to evaluate the stability in plasma.
A plot of the percentage of full length oligonucleotide remaining
intact in tissue 24 hours following achninistration of an i.v. bolus of 5
mg/kg
oligonucleotide is determined. CGE traces of test oligonucleotides and a
standard phosphorothioate oligonucleotide in both mouse liver samples and
mouse kidney samples after 24 hours axe evaluated. There is a greater amount
of
intact oliogonucleotide for the oligonucleotides of the invention as compared
to
the standard of the parent unmodified. The maximum stability is seen when both
5' and 3' ends are capped with C*.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-111 -
EXAMPLE 63
Control of c-raf Message an bEND Cells using G-clamp Modified
Oligonucleotides
ISIS # Sequence (5'-3') Backbone Sequence ID
NO:
11061 ATG-CAT-TCT-GCC-CCC-AAG-GA P= S (SEQ )D NO:42)
----- ATG-C*AT-TCT-GCC-CCC-AAG-GA P= S (SEQ D7 N0:43)
----- ATG-CAT-TC*T-GCC-CCC-AAG-GA P= S (SEQ ~ N0:44)
----- ATG-CAT-TCT-GC*C-CCC-AAG-GA P= S (SEQ lD N0:45)
----- ATG-CAT-TCT-GCC*-CCC-AAG-GA P= S (SEQ ID N0:46)
----- ATG-CAT-TCT-GCC-C*CC-AAG-GA P= S (SEQ ID N0:47)
----- ATG-CAT-TCT-GCC-CC*C-A.AG-GA P= S (SEQ ID N0:4~)
----- ATG-CAT-TCT-GCC-CCC*-AAG-GA P= S (SEQ ID NO:49)
C* = G-clamp modification
Tn order to assess the activity of some of the oligonucleotides, an in
vitro cell culture assay is used that measures the cellular levels of c-raf
expression
in bEND cells.
Cells and Reageizts
The bEnd.3 cell line, a brain endothelioma, is obtained from Dr.
Werner Risau (Max-Planck Institute). Opti-MEM, trypsin-EDTA and DMEM
with high glucose are purchased from Gibco-BRL (Grand Island, NY).
Dulbecco=s PBS is purchased from Irvine Scientific (Irvine, CA). Sterile, 12
well tissue culture plates and Facsflow solution are purchased from Becton
Dickinson (Mansfield, MA). Ultrapure formaldehyde is purchased from
Polysciences (Warrington, PA). NAP-5 columns are purchased from Pharmacia
(Uppsala, Sweden).
Oligofzucleotide Treatr~aefat
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-112 -
Cells are grown to approximately 75 % confluency in 12 well plates
with DMEM containing 4.Sg/L glucose and 10 % FBS. Cells are washed 3 times
with Opti-MEM pre-warmed to 37°C. Oligonucleotide is premixed with a
cationic lipid (Lipofectin reagent, (GIBCOBRL) and, serially diluted to
desired
concentrations and transferred on to washed cells for a 4 hour incubation at
37°C.
Media is then removed and replaced with normal growth media for 24 hours for
northern blot analysis of mRNA.
NoYthern Blot Analysis
For determination of mRNA levels by Northern blot analysis, total
RNA is prepared from cells by the guanidinium isothiocyanate procedure [Monia
et al., PYOC. Natd. Acad. Sci. USA, 1996, 93, 15481-15484] 24 h after
initiation of
oligonucleotide treatment. Total RNA is isolated by centrifugation of the cell
lysates over a CsCl cushion. Northern blot analysis, RNA quantitation and
normalization to G3PDH mRNA levels are done according to the reported
procedure [Dean and McKay, Pr-oc. Natl. Acad. Sci. USA,1994, 91, 11762-
11766].
In bEND cells the G-clamp oligonucleotides showed reduction of c-
oaf message activity as a function of concentration. The fact that these
modified
oligonucleotides retained activity promises reduced frequency of dosing with
these oligonucleotides which also show increased in vivo nuclease resistance.
All
G-clamp modified oligonucleotides retained the activity of the parent 11061
oligonucleotide (SEQ ID N0:42) and improved the activity even further.
EXAMPLE 64
Compound 201 (R' = CN, n =1, Scheme 22a, Table XX).
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-113 -
R"CH2(CHp)~CH20H NHS ! MeOH
PhaP, DEAD / MeCN RT, 72 h
RT, 24 h
246 247 201 R"= CN
R = H, OAc or OCHZCHZOCH3 R = H, OAc or OCHZCHZOCH3 248 R" = NHCbz
R" = NHCbz or CN; n = 0 - 4 R = H, OH or OCHZCH20CH3.
n=0, 1,2,3or4
Scheme 22a
Table XX
General structureEntryX
201 O~CN (Where,
n = 1, 2,
3 or 4)
202 NHCOCF3 4Where,
n=0, 1, 2,
3 or4; m=0,1,2,3
or
0 "n C
OCF3
X ~~ ~
203 ~NHCOCF3 (Where,
n =0, 1, 2,
3 or 4; m
O~~
~C
HN ~ OC
OC
3 3 =0,1,2,3or4;1=0,1,2,3or4)
N ~ O 204 0~0~ NHR' (Where,
n = 0, 1,
2, 3 or 4and
R' = H, Me,
El or any
R or s
n
a-amino acid
~ or a peptide
derived either
from R or
s or
from both R
and s a-amino
acids)
N
DMTO O
205 O~SR' (Where,
n = 0, 1,
2, 3 or 4and
R' =Acetyl,
benzyl, Me,
Et, H)
OH R
R = H or OH OII
or OCH2CHZOCH3
or2'-modified 206 o~N~NHR'
n H
S (where, n = 0, 1,
2, 3 or 4
~ R'=H, Me, Et, iPr,
benzyl or
207 0~ H- ' NHR' CH2(CHz)mNHCOCF3 and
m =1, 2, 3, 4 or
5)
NHz
208 ~NHR'
O~
H
O
NH2
209 0~ (Where n
=1, 2, 3 or
4
n NHz
EXAMPLE 65
Compound 248 (R' = NHCbz, n = 0, Scheme 22).
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-114 -
Compound 248 is prepared from compound 246 (1 mmol) and benzyl N
(2-hydroxyethyl)carbamate (1 mmol) according to the literature procedure [Lin
and Matteucci, J. Am. Cher~2. Soc., 1998,120, 8531- 8532].
EXAMPLE 66
Compound 249 (n = 0, Scheme 22b).
Compound 248 (1 mmol) upon treatment with DMT-Cl (1 molar eq.) in
pyridine yields the corresponding 5'-O-DMT derivative. The DMT derivative is
stirred with ethyl trifluoroacetate in presence of TEA to obtain N
trifluoroacetyl-
5'-O-DMT derivative of compound 248. Free 3'-hydroxy functional group of the
product obtained is reacted with acetic anhydride in anhydrous pyridine to
obtain
the completely protected nucleoside 249.
1. DMT-CI / Py HCOONHyJ Pd-C (10 %)
_ >
2. CFaCOOEt , DIEA 1 DCM 10 min
3. Ar201 Py
~Ac
248 (n = 0, 1, 2, 3 or 4) 249 (n = 0, 1, 2, 3 or 4) 250 (n = 0, 1, 2, 3 or 4)
Scheme 226
EXAMPLE 67
Compound 250 (n = 0, Scheme 22b).
A suspension of compound 249 (1 mmol) and ammonium formate (5
mmol) in ethyl acetate is deoxygenated under argon and 10 % palladium on
charcoal (10 mol %) is added into the suspension under argon. The reaction
mixture is stirred for 10 min at ambient temperature to obtain compound 250.
EXAMPLE 68
Compound 206 (n = 0, R = Me, Scheme 22c, Table XX).
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-115 -
Compound 250 (1 mmol) in anhydrous THF is stirred with 1,1'-carbonyl-
diimidazole (CDI, 1 mmol) under argon at ambient temperature for 2 h. After 2
h,
the reaction mixture is cooled on an ice bath and anhydrous methylamine gas is
bubbled through the reaction mixture for 10 min. The resulting mixture is
stirred
for 30 min to obtain compound 206.
RF-
CDI, RNHz / THF Thio-CDI, RNHz / THF
E
206 (See Table 1 for 250 (n = 0, 1, 2, 3 or 4j 207 (See Table 1 for
definition of R' and n) definition of R' and n)
1. CbzHN-CHZCHz-OSOz-CH3/ DIEA
Phosphitylation 2. HCOONH41 Pd-C (10 % ) Phosphitylation
3. CF3COOEt l DIEA
RH RI
202 (n = o, 1,2, 3 or 4
206a and m=0, 1, 2, 3 or4) 207a
Phosphitylation
Scheme 22c
IO
202a (n = o, 1,2, 3 or 4
and m = 0, 1, 2, 3 or 4)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-116 -
EXAMPLE 69
Compound 206a (n = 0, R = Me, Scheme 22c).
Phosphitylation of the 3'-hydroxy group of compound 206 as described in
Example 2 for the synthesis of compound 3 yields compound 206a.
EXAMPLE 70
Compound 207 (n = 0, R = Me, Scheme 22c, Table XX).
Compound 207 is obtained from compound 250, 1,1'-
thiocarbonyldiimidazole and methylamine under similar reaction conditions as
described for the synthesis of compound 206 in Example 68.
EXAMPLE 71
Compound 207a (n = 0, R = Me, Scheme 22c).
Phosphitylation of 3'-hydroxy group of compound 207 as described in
Example 2 for the synthesis of compound 3 yields compound 207x.
EXAMPLE 72
Compound 202 (n = 0, m = 0, Scheme 22c, Table XX).
Compound 250 (1 mmol) is stirred with N benzyloxycarbonyl-2,-amino-
ethanol-O-methane sulfonate (1 mmol) in presence DIEA in anhydrous DCM
overnight. The secondary amine thus obtained is subj ected to transfer
hydrogenation as described in Example 59 to remove the benzyloxycarbonyl
protection. The unprotected amine is then stirred with ethyl trifluoroacetate
in
presence of DIEA in DCM to obtain the desired compound 202.
EXAMPLE 73
Compound 202a (n = 0, m = 0, Scheme 22c).
Phosphitylation of compound 202 as described in Example 2 for the
synthesis of compound 3 yields compound 202a.
EXAMPLE 74
Compound 208a (n = 0, Scheme 22d, Table XX).
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-117 -
Compound 250 (1 mmol) and TEA (1 mmol) are added into a solution of
compound A (1 mmol, Scheme 1d) and the resulting mixture is stirred at ambient
temperature to obtain compound 208x.
II SMeO
NC~o~N~H~CN
A
TEA / DMF, RT, 4 h
250 (n = 0, 1, 2, 3 or 4) 208a (n = 0, 1, 2, 3 or 4)
Phosphitylalion
Scheme 22d 208b (n = 0, 1, 2, 3 or 4)
EXAMPLE 75
Compound 208b (n = 0, Scheme 22d).
Phosphitylation of compound 208a as described in Example 2 yields
compound 208b.
EXAMPLE 76
Compound 201a (n =1, Scheme 22e).
Reaction of compound 201 with DMTCI in pyridine yields compound
201 a.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-118 -
NCO
n HN ~ I
N~ O
~ DMT-CI / Py NHa~ NN4Cl
HO
OH
201 (n=1,2,3or4) 201a(n=1,2,3or4) 209 (n=1,2,3or4)
OI O1'
NC~O~O-N
OO
Scheme 22e
EXAMPLE 77
Phosphitylalion
E
209b (n = 1, 2, 3 or 4) 209a (n = 1, 2, 3 or 4)
Compound 209 (n =1, Scheme 22e, Table XX).
Compound 201 a is treated with ammonia and ammonium chloride in THF
at elevated temperature under pressure to obtain compound 209 [Granik, Russ.
Chem. Rev.,1983, 52, 377-393].
EXAMPLE 78
Compound 209a (n =1, Scheme 22e).
2-Cyanoethoxycarbonyloxysuccinimide (2 mmol) and DIEA are added
into a solution of compound 209 (1 mmol) in DCM and the resulting mixture is
stirred at ambient temperature to obtain compound 209a.
EXAMPLE 79
Compound 209b (n = 0, Scheme 22e).
Phosphitylation of compound 209a as described in Example 2 for the
synthesis of compound 3 yields compound 209b.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
- 119 -
EXAMPLE 80
Compound 252 (Scheme 23a).
Phenoxazine nucleoside 252 with desired tether X is synthesized in five
steps from 5-bromo-3'-O-TBDMS-5'-O-DMT-dU (251) according to the
literature procedure by [Lin and Matteucci J. Anz. Claem. Soc., 1998, 120,
8531-
8532].
EXAMPLE 81
Compound 253 (Scheme 23a).
OII X X
~~Br H I N
Ac0-i Q ,N ~ ~~O ~~O
EtOH/PhsP/OEPD/OIEA
DMTO~N M.cru,Rr DMrO E-, to _N
OTBDM \~~-'-' ~~J~ ~YS
OTBOMS OTBDMS
251 252 253
(SeeTabIeXVIfordefntionofX) (SeeTabIeXVIfordefintionotX)
Hy,SiTMG/Fy~
RT, 72 h
NCO X
G
HN HN
O O
DMrO ~~ E r.TeAF DtvnO
2.Phosphilylellon
0 oTBDMs
NC~O.P~N~pr)z
254
21Ba (n = 1, 2, 3, or 4) (See Table XVI for defintion of X)
Scheme 23a
Reaction of compound 252 (1 mmol) with ethanol (1 mmol) under
Mitsunobu alkylation condition (Ph3P and DEAD 1 mmol each) in presence of
DIEA in acetonitrile yields compound 253.
EXAMPLE 82
Compound 254 (Scheme 23a).
Compound 253 (1 mmol) after thorough drying over Pa05 under vacuum is
taken in a reaction vessel under argon. TMG (10 mmol) in anhydrous pyridine,
placed on a freezing bath, is saturated with anhydrous HAS for 45 min. After
45
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-120-
min, the resulting solution is transferred into the precooled vessel
containing
compound 253 under argon and is sealed. The sealed vessel is then brought to
ambient temperature and is stored at ambient temperature for 3 days. Bubbles
off
the H2S into a chlorox bath and removes pyridine from the reaction mixture
under
vacuum. The residue after standard work up and purification yields compound
254.
EXAMPLE 83
Compound 210a (n =1, Scheme 23a, Table XXI).
Compound 254 (X = O-(CHa)3-CN) is treated with TBAF in THF to
remove the 3'- O - TBDMS group. The resulting 3'-OH group is subjected to
phosphitylation under the conditions described in Example 2 to obtain compound
210a.
Table XXI
General structureEntryX
210 O~R (Where,R=NHz(NHP),CNorOH(OP),n=1,2,3or4
and P is protecting
group)
211 O~N~NHCOCF3
(Where,n=0,1,2,3or4;m=0,1,2,3
or
n
m
COC 3
4)
X /~~
212 O~N~N~NHCOCF3
(Where n=0,1,2,3or4;m
n COC
m COCF
t
3
HN 3
= 0,1,2,3 or
4; I = 0,1,2,3
or 4)
N 213 O~O'NHR' (Where,n=0,1,2,3or4andR'=H,Me,EtoranyROrs-
~ I O
I n a-amino acid
~ or a peptide
delved either
from tt or
s or
N from both R
DMTO S and s-amino
acids)
214 O~SR' (Where,
n = 0, 1,
2, 3 or 4and
R' = Acetyl,
benzyi, Me,
Et, H)
OH R
R = H or OH 0
or OCH2CH20CH3 ~
or 2' - modified ~
215 O~H
NHR'
~,, (where, n = 0 1, 2,
3 or 4
~ R = H, Me, Et, ~Pr,
~ benzy! or
216 O~H CHz(CHz)mNHCOCF3 and
NHR' m =1, 2, 3, 4 or
O 5)
~z
217 O~
NHR'
H
O+
Hz
218 O~ ~ ~ (Wheren=1,2,3or4
~
NHz
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-121 -
EXAMPLE 84
Compound 210b (n = 0, Scheme 23b, Table XXI).
Compound 254 (1 mmol, n = 0, Scheme 23b) is stirred with TBAF in THF
to remove the 3'-O-protection. The resulting product is subjected to transfer
hydrogenation using ammonium formate and Pd-C (10 %) in ethyl acetate (See
Example 67 for details) to remove the benzyloxycarbonyl protection from the
side
chain moiety. The free amine thus formed and the ring nitrogen are then
protected
as trifluoroacetamide by stirnng the compound (1 mmol) with ethyl
trifluoroacetate (10 mmol) in pyridine at ambient temperature. Finally the
trifluoroacetamide derivative obtained is phosphitylated as described in
Example
2 for the synthesis of compound 3 to obtain the desired phosphoramidite 210b.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-122-
1.TBAF1THF
2. HCOONHd / Pd-C (10 % )
3. CF~COOEt / Py
4. Phosphitylatioin
254 (n = 0, 1, 2, 3 or 4) 210b (n = 0, 1, 2, 3 or 4)
1. CF~COOEt / Py
2. HCOONHy/ Pd-C (10~)
RI-
1. CDI, RNHz 1. CDI, RNHz
2.TBAF/THF 2.TBAF/THF
E
3. CF3COOEt , DIEA / DCM 3. CF~COOEt , DIEA / DCM
4. Phosphitylatfoin 4. Phosphitylatloin
255 (n = 0, 1, 2, 3 or 4)
RI
216a (n = 0, 1, 2, 3 or 4) 215a (n = 0, 1, 2, 3 or 4)
1. CbzHN-CHZCHz-OSOZCH3
2. HCOONHq / Pd-C (10%)
3. CF3COOEt / Py
F3COCHN~
1.TBAF/THF
2. Phosphitylatioin
256 (n = 0, 1, 2, 3 or 4 and
m=0,1,2,3or4)
Scheme 23b
211a (n = 0, 1, 2, 3 or 4 and
m=0,1,2,3or4)
1
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-123-
EXAMPLE 85
Compound 255 (n = 0, Scheme 23b).
Compound 254 (n = 0, 1 mmol) is stirred with ethyl trifluoroacetate (5
mmol) in pyridine at ambient temperature. The trifluoroacetamide formed after
purification is stirred with ammonium formate (10 mmol) in the presence of Pd-
C
(10 %) in ethyl acetate as described in Example 67 to obtain compound 255.
EXAMPLE 86
Compound 215a (n = 0, R = Me, Scheme 23b, Table XXI).
Compound 255 (1 mmol) is reacted with CDI and methylamine as
described in Example 68. The urea derivative thus obtained is stirred with
TBAF
in THF to,remove 3'=O-protection. After deprotection of 3'-O- TBDMS, the
resulting product is trifluoroacetylated at the ring nitrogen by stirring it
with
excess ethyl trifluoroacetate in anhydrous pyridine. Phosphitylation of the
trifluoroacetamide derivative under the conditions described in Example 2 for
the
synthesis of compound 3 yields compound 215x.
EXAMPLE 87
Compound 216a (n = 0, R = Me, Scheme 23b, Table XXI).
Compound 216a is synthesized from compound 255, l,l'-thiocarbonyl-
diimidazole and methylamine as described in Example 86 for the synthesis of
compound 215x.
EXAMPLE 88
Compound 256 (m = 0, n = 0, Scheme 23b).
Compound 256 is prepared from compound 255 (1 mmol) and N benzyl-
oxycarbonyl-2-aminoethanol-O-methane sulfonate (1 mmol) as described in
Example 72.
EXAMPLE 89
Compound 211a (m = 0, n = 0, Scheme 23b, Table XXI).
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-124-
Compound 256 is stirred with TBAF in THF to remove the TBDMS
protection on the 3'-OH group. After deprotection, the 3'-OH group is
phosphitylated as described in Example 2 for the synthesis of compound 3 to
obtain compound 211 a.
EXAMPLE 90
Compound 257 (n = 0, Scheme 23c).
Compound 257 is obtained from compound 255 under the conditions
described in Example 74.
EXAMPLE 91
Compound 217a (n = 0, Scheme 23c, Table XXI).
Compound 217a is prepared from compound 257 as described in Example
89 for the preparation of compound 2.11 a.
HzN~O
(CEOC)HN N~O
F~COCN~ O SMeO NCEOC HN
O NCB
N O N~H~CN N~ I D
DMTO ~N
DMTO ~N
TEAI ~MF, RT, 4 h
OTBDMS ~
OTBDMS
255 (n = 0, 1, 2, 3 or 4) 257 (n = 0, 1, 2, 3 or 4)
1.TBAF/THF
2. PhosphlhAallon
Scheme 23c 217a (n = 0, 1, 2, 3 or 4)
EXAMPLE 92
Compound 258 (n =1, Scheme 23d).
Compound 258 is synthesized from compound 252 as described in
Examples 77 and 78.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-125-
1. NHa. NHQCI
2, _
OH, PhaP, DEAC
~~0~0-N~
O
252 (n=1,2,3or4) 258 (n=1,2,3or4) 259 (n=1,2,3or4)
HzS-TMGIPy
i.TBAF1THF
E
2. Phosphitylation
260 (n = 1, 2, 3 or 4)
218a(n=1,2,3or4)
Scheme 23d
EXAMPLE 93
Compound 218a (n =1, Scheme 23d, Table XXT).
The phosphoramidite 218a is synthesized from compound 258 under
identical conditions described in Examples 81 and 83 for the preparation of
compound 2IOa from compound 253.
EXAMPLE 94
Compound 262 (Scheme 24).
2-Amino-3-mehtoxy-benzenethiol [moue et. al., Chem. Pha~m. Bull.,
1997, 45, 1008-1028] is reacted with BoczO in presence of NaHC03 and
subsequently with AcZO in pyridine to obtain compound 262.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-126-
SH SCOCHs SCOCHs SCOCH~
NHa 1, ecc,o, N_eHCO, ~ NHBocR.oH,~h,P
~ NHBoc TM~'~ - I 'EAD ~ NHBoc
I ~
I
/ 2.ACa0/PY / / ,R
/.
OCHs OCH, OH O
261 262 263 264
SCOCH3
NHa.HX
I /
.R
O
265
Scheme24
EXAMPLE 95
Compound 263 (Scheme 24).
After thorough drying over PZOS under vacuum, compound 262 in
anhydrous dichloromethane is treated with TMS-I for 5 min to obtain compound
263.
EXAMPLE 96
Compound 264 (Scheme 24).
Tether of choice is attached to the hydroxyl function of compound 263 in
presence of Ph3P and DEAD (Mitsunobu alkylation) to obtain compound 264.
EXAMPLE 97
Compound 265 (Scheme 24).
Compound 264 is stirred with TFA in DCM for 30 min to obtain
compound 265.
EXAMPLE 98
Compound 267 (Scheme 25a).
Compound 267 is synthesized from compound 266 and compound 265
according to reported procedures [Lin et. al., J. Am. Chem. Soc., 1995,117,
3873-
3874].
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-127-
0
Br
~~ 1. POCI3 Triazole, TEA NH3
/ MeOH, RT 72 H
O N 2. 265, D
ACO-I " 2. DMT-CI / Py
, SCOCH3
NHZ.HX
OAc R I /
X
265
266 267 268
(See Table XVIt for definition of X)
Scheme 25a
EXAMPLE 99
Compound 268 (Scheme 25b, Table XXII). Tricyclic nucleoside 268 is prepared
from compound 267 according to the reported procedure [Lin and Matteucci, J.
Am. Chem. Soc.,1998,120, 8531- 8532].
zsa
219 - 227 (See Table
XVII for details)
Scheme 25b
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-128-
Table XXII
General structureEntry X
219 O~R (Where,R=CN,NNz(NHP)orOH(OP),
n=1,2,3or4
and P is
protecting
group )
220 ~NHCOCF3
(Where,n=0,1,2,3or4;m=0,1,2,3
or
O~C
O
a 4)
221 0 1-In N
m N~NHCOCF3
(Where,n=0,1,2,3or4;m
COC~COCF
3 =0,1,2,3ar4;1=0,1,2,3or4)
HN
~~ NHR' (Where,
i 22 n = 0, 1,
S 2, 3 or
4and R'
= H, Me
Et or any
a or s -
0~0~
N a-amino acid
I or a peptide
~ derived
either from
R or s or
from both
R and s
a-amino
acids)
DMTO ~
N
223 O~SR' (Where,
n =0, 1,
2, 3 or4and
R'=Acetyl,
benzyl,
Me, Et,
H)
O~ R
N~O~P'N(~Pr)z 0II
~
R= H or OH or 224 o~H
OCHZCH~OCH3 NHR'
or 2' - modified
S (where, n = 0, 1,
2, 3 or 4
I' R' = H, Me, Et, iPr,
~ benzyl or
225 O~H CHz(CHz)mNHCOCF3 and
NHR' m =1, 2, 3, 4 or
O 5)
NHz
226 ~NHR'
O~
H
NHz
227 0~ (Wheren=1,2,3or4
n NHz
1~
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-129-
Table XXIII
General structureEntryX
228 O~R (Where,
R=CN, NHZ(NHP)
or OH (OP),
n = 1, 2,
3 ar4
and P is protecting
group )
229 ~NHCOCF3 Where,
n =0, 1, 2,
3 or4; m=0,1,2,3
or
O~~
OCF
X
230 O~N~N~NHCOCF3
(Where n =0,
1, 2, 3 or4;
m
HN ~ COCF3 COCF3
=0,1,2,3or4;1=0,1,2,3or4)
N/ 231 p~O~NHR'(Where,n=0,1,2,3or4andR'=H,Me,EtoranyROrS-
S
I a-amino acid
or a peptide
derived either
from R or
s or
DMTO ~N from both R
and s a-amino
acids)
232 O~SR' (Where,
n = 0, 1,
2, 3 or 4and
R' = Acetyl,
benzyl, Me,
Et, H)
O~ R
NC~O~P~Nb Pr)2
0
R = H or OH
or OCH2CH~OCH3 33 NHR'
O~
or2'-modified H
S (where, n = 0, 1,
2, 3 or 4
II R' = H, Me, Et, iPr,
~y benzyl ar
~
234 O~H CHz(CHz)mNHCOCFa and
NHR' m =1, 2, 3, 4 or
5)
NHz
235 O~
~NHR'
H
O+
NHZ
236 O~ (Where n
= 0, 1, 2,
3 or 4
n NHZ
EXAMPLE 100
Compound 219 (X = O[CHZ]3CN, Scheme 25b, Table XXII).
Compound 268 (X = O[CHZ]3CN, Scheme 25a) is phosphitylated under
the conditions described in Example 2 to obtain compound 219.
EXAMPLE 101
Compound 220 [X = O(CH2)ZN(COCF3)[CH2]N(H)COCF3], Scheme 25b,
Table XXII).
Compound 220 (as specified) is prepared from compound 268 (X =
O[CHZ]2NHCbz) and N benzyloxycarbonyl aminoethanol-O-methylsulfonate as
described in Examples 67, 72 and 73.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
- 130 -
EXAMPLE 102
Compound 224 (X = O[CHZ]2NHCONHCH3, Scheme 25b, Table XXII).
Compound 224 (as specified) is synthesized from compound 268 (X =
O[CHZ]2NHCbz), CDI and methylamine as described in Examples 67, 68 and 69.
EXAMPLE 103
Compound 225 (X = O[CH2]2NHCSNHCH3, Scheme 25b, Table XXII).
Compound 224 (as specified) is synthesized from compound 268 (X =
O[CHZ]ZNHCbz), 1,1'-thiocarbonyldiimidazole and methylamine as described in
Examples 67, 70 and 71.
EXAMPLE 104
Compound 226 (X = O[CHZ]2NHC[NH]NH3, Scheme 25b, Table XXII).
Compound 226 (as specified) is synthesized from compound 268 (X =
O[CHZ]2NHCbz) and compound A (See Scheme 22d) as described in Examples
67, 74 and 75
EXAMPLE 105
Compound 227 (X = O[CHZ]3CH2C[NH]NH3, Scheme 25b, Table XXII).
Compound 227 (as specified) is synthesized from compound 268 (X =
O[CHZ]3CN) as described in Examples 77, 78 and 79.
EXAMPLE 106
Compound 270 (Scheme 26).
Alkylation of hydroxyl function of compound 269 [Bigge, C. F. et. al.,
PCT Int. Appl. (1997), 280pp CODEN PIXXD2 WO 9723216 A1 19970703]
using tether of choice (as defined in Table XXIV) in presence of Ph3P and DEAD
yields compound 270.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-131 -
EXAMPLE 107
Compound 271 (Scheme 26).
Compound 271 (1 mmol) is dissolved in ethyl acetate containing 10
acetic acid, the resulting solution after deoxygenation is mixed with 10 mol
OBn pgn OH
N~2 R~OH, PH3P, DEAD ~ ~ N~2 Hz/ Pd-C, HOAC ~ ~ NH2.HX
OH OR OR
269 270 271
Scheme 26
percentage of Pd-C (10 %) subjects to catalytic hydrogenation under pressure
to
obtain compound 271.
EXAMPLE 108
Compound 273 (Scheme 27a).
Compound 272 is obtained from compound 266 and compound 271 as
described in Examples 88 and 98.
0
Br
~~ 1, POCK Triazole, TEA NH3 / MeOH, RT 72
H
O N 2 271, DIES
ACO OH 2, DMTCI l Py
~ NH2.HX
OAc R I /
271
X
266 272 273
(See Table XIX for definition of X)
Scheme 27a
EXAMPLE 109
Compound 237 (X = O[CH2]2N(Phthaloyl), Scheme 27b, Table XXI~.
Phosphitylation of compound 273 (X = O[CHZ]ZN(Phthaloyl), Scheme
27a) under identical conditions described in Example 2 yields compound 237.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-132-
Table XXIV
General structureEntryX
237 O~R (Where,R=CN,NHz(NHP)orOH(OP),
n=1,2,3or4
and P is
protecting
group )
238 ~NHCOCF3
(Where.n=0,1,2,3or4;m=0,1,2,3
or
O~~
X O
s 4)
/ I ~~
W 239 O~COC~COC~NHCOCF3
(Where n=0,1,2,3or4;m
H = 0,1,2,3
or4;l =
0,1,2,3
or4)
N~ Z
240 O~O~NHR'(Where,n=0,1,2,3or4andR'=H,Me,EtoranyROrs-
a-amino acid
or a peptide
derived
either from
R or s or
DMTO ~ N from both
R and s
a-amino
acids)
241 O~SR' (Where,
n = 0,1,
2, 3 or
4and R'
= Acetyl,
benryl,
Me, El,
H)
O~ R
NCO. P, N(iPr)z
O
R = H or OH II
or OCHZCHZOCH3 ~
or2'-modified 242 NHR'
o~H
Y,Z=O; or Y,Z=S,orY=O
andZ=Sort=SandZ= SI' (where,n=0,1,2,3or4
O
R' = H, Me, Et, iPr,
~ benzyl or
243 NHR' CHz(CHz)mNHCOCFa
0~ H and m = 1, 2, 3,
O 4 or 5)
NIIHz
244 ~NHR'
O~
H
O
~Hz
245 p~ (Wheren=0,1,2,3or4
~~ ln~ ~NHz
EXAMPLE 110
Compound 238 (X = O[CHZ]2N f COCF3} [CHZ]2NH f COCF3}, Scheme 27b,
Table XXI~.
Compound 273 (X = O[CHZ]2N f Phthaloyl~, Scheme 27a) is treated with
hydrazine to remove the phthaloyl protection from the side chain. The
corresponding free amine thus formed is reacted with N benzyloxycarbonyl
aminoethanol-O-methane sulfonate in presence of base as described in Example
64, followed by phosphitylation (Example 2) yields compound 238.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-133-
~~i~~"z
273
237 - 245 (See Table
XIX for details)
Scheme27b
EXAMPLE 111
Compound 242 (X = O[CHZ]2NHCONHCH3, Scheme 27b, Table XXII).
Compound 273 (X = O[CHZ]2N{Phthaloyl}, Scheme 27a) is treated with
hydrazine to remove the phthaloyl protection from the side chain. The desired
compound 242 is obtained by reacting the free amino group formed with CDI and
methylamine as described in Examples 68 and 69.
EXAMPLE 112
Compound 243 (X= O[CHZ]2NHCSNHCH3, Scheme 27b, Table XXII).
Compound 273 (X = O[CH2]2N{Phthaloyl}, Scheme 27a) is treated with
hydrazine to remove the phthaloyl protection from the side chain. The desired
compound 243 is obtained by reacting the free amino group formed with 1,1'-
thiocarbonyldiimidazole and methylamine as described in Examples 68 and 69.
EXAMPLE 113
Compound 244 (X = O[CHZ]aNHC~NH}NH3, Scheme 27b, Table XXII).
Compound 273 (X = O[CH2]zN f Phthaloyl}, Scheme 27a) is treated with
hydrazine to remove the phthaloyl protection from the side chain. The desired
compound 243 is prepared from the amino compound and compound A (See
Scheme 22d) as described in Examples 67, 74 and 75.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-134-
EXAMPLE 114
Compound 245 (X = O[CHZ]3CH2C[NH]NH3, Scheme 27b, Table XXII).
Compound 227 (as specified) is synthesized from compound 273 (X =
O[CH2]3CN) as described in Examples 77, 7~ and 79.
EXAMPLE 115
Compound 284 (Scheme 28).
Compound 283 prepared according to the literature procedure [Pal, B. C.
et. al., Nucleosides & Nucleotides, 1988, 7, 1-21] is stirred with BocaO in
presence of Na.HC03 in aqueous methanol to protect the ring nitrogen as Boc.
The Boc proteted nucleoside is then acetylated in anhydrous pyridine to obtain
compound 284.
COON COON
HN~ BocN
S S
I ~I
1. BoclO, aq. NaHCO~
HON ~ AcO~N RNH2, HATU, DIEA
2. AczO/ Py
I[[ I~~~O\/77I
OH OAc
283 284 285
1. TFA
2. LiOH
Phosphitylation 7. DMT~CI / Py
2. CF3COOEt, DIE4
mPrm 287 286
274-282
(See Table XX for details)
Scheme 28
EXAMPLE 116
Compound 285 (R = [Phthaloyl]N[CH2]3-, Scheme 28).
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-135-
N (phthaloyl)ethylenediamine is coupled to the carboxyl group of
compound 284 in the presence of HATLT and HOAT under peptide coupling
conditions to obtain compound 285.
EXAMPLE 117
Compound 286 (R = [Phthaloyl]N[CHZ]3-, Scheme 28).
Compound 285 is subjected to TFA treatment in dichloromethane for 30
min to remove the Boc protection. After deblocking the ring nitrogen, the
resulting compound is stirred in aqueous THF containing 0.1 M LiOH at O
°C to
obtain compound 286 (as specified).
EXAMPLE 118
Compound 287 (R = [Phthaloyl]N[CH2]3-, Scheme 28).
Compound 287 (1 mmol) in anhydrous pyridine is treated with DMT-Cl (1
mmol) in presence of DMAP (10 mol %) to obtain the corresponding 5'-O-DMT
derivative. After dimethoxytritylation, the resulting product is stirred with
excess
of ethyl trifluoroacetate in presence of DIEA in anhydrous dichloromethane to
obtain compound 287.
EXAMPLE 119
Compound 274 (R = [Phthaloyl]N[CHI]3-, Scheme 28, Table XX'~.
Phosphitylation of compound 287 under the conditions described in
Example 2 for the synthesis of compound 3 yields compound 274.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-136-
Table
XXV
General structureEntryX
274 HN~R
(Where,R=CN,NHz(NHP)orOH(OP),
n=1,2,3or4
and
P is
protecting
group
)
275 ~ NHCOCF3
4Where,
n =
0,
1,
2,
3 or
4;
m =0,1,2,3
or
HN~~
OCF
)
COX
276 HN~N
N~NHCOCF3
(Where,
n =D,1,
2,
3 or4;
m
n COC~COCF
i
HN 3
=0,1,2,3or4;1=0,1,2,3or4)
S
N~ 277 HN~O~NHR'
(Where,n=0,1,2,3or4andR'=H,Me,EtoranyROrs-
a-amino
DMTO acid
N or
a peptide
derived
either
from
rt
or
s or
from
both
R and
s a-amino
acids)
~ 278 HN~SR'
(Where,
n =
0,
1,
2,
3 or
4and
R'
=Acetyl,
benzyl,
Me,
Et,
H)
O~ R
NC~O~ P~ N(iPr)z
O
R = H or OH ~IyI
or OCHaCH20CH3 ~
or2'-modified 279 HN~H
NHR'
Y,Z=O; or Y,Z=S,orY=O
andZ=Sort=SandZ= (where,n=0,1 2,3or4
O
R' = H, Me, Et, iPr,
benzyl or
280 HN~H CHz(CHz)mNHCOCF3
NHR' and m =1, 2, 3,
O 4 or 5)
NHz
281 HN~
~NHR'
H
O
NHz
282 HN~ (Wheren=0,1,2,3or4
n NHz
EXAMPLE 120
Nuclease resistance of oligonucleotides with selected modifications
Phenoxazine 151 and G-clamp 152 nucleosides were prepared by
modifying previously published procedures [Lin, K.-Y.; Jones, R. J.;
Matteucci,
M. J. Am. Chern. Soc. 1995,117, 3873-3874; Lin, K.-Y.; Matteucci, M. J. Am.
Chena. Soc. 1998,120, 8531-8532]. The succinates 153 and 154 and the
corresponding substituted solid supports 155 and 156 were prepared as outlined
in
Scheme 19. Using the CPG supports, the two cytidine analogs 151 and 152 were
incorporated at the 3'-terminus of two model oligonucleotides 157 and 158,
respectively, with the sequence TlBdC* (dC* = phenoxazine (SEQ m N0:62) or
G-clamp deoxyribonucleoside (SEQ ID N0:63)). Solid phase oligonucleotide
syntheses was earned out using standard phosphoramidite chemistry.
Deprotection of G-clamp containing oligonucleotide 158 was performed with a
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-137-
1:1 solution of MeNH2 (40%, aq.) and NH3 (28-30%, aq.) at r.t. for 4 h. The
oligonucleotides were purif ed and desalted by reversed phase HPLC.
Snake venom phosphodiesterase (SVPD) and bovine intestinal mucosal
phosphodiesterase (BIPD), were utilized as the hydrolytic enzymes for iya
vitro
nuclease resistance studies. Both enzymes predominantly exhibit 3 '
exonuclease
activity. An unmodied l9mer oligothymidylate (oligonucleotide 159) (SEQ ID
N0:64) was used as a control. Oligonucleotide samples were incubated with
SVPD (2.5 units/~.mol substrate) or BIPD (0.55 units/~mol substrate) in 50 mM
Tris-HCl, 8 mM MgCl2 buffer, pH 7.5 at 37°C. At certain time points
aliquots of
10 ~.l were removed and heated in boiling water for 2 min to inactivate the
enzyme. Subsequently, the samples were desalted by membrane dialysis against
Nanopure deionized water using Millipore 0.025 ~m VS membranes and stored
frozen until they were analysed. The progress of enzymatic degradation was
monitored by capillary gel electrophoresis (CGE).
The results of the nuclease resistance study with SVPD as the hydrolytic
enzyme are shown in Figure 4. As expected, the unmodified control
oligonucleotide 159 (insert) was degraded rapidly by sequential removal of the
terminal nucleotides. Under the applied conditions the tli2 for this
oligonucleotide
was reached at about 3 min. After 20 min of incubation the full length
oligomer
was almost completely degraded to a series of shorter fragments. In contrast,
the
modified oligonucleotides 157 and 158 bearing the heterocyclic modifications
at
their 3' end were not significantly degraded even after an incubation time of
8 h.
According to the degradation rates and the CGE profiles, there is no
significant
difference in the 3' exonuclease resistance of these two oligomers. Very
similar
results for the nuclease resistance against BIPD as the hydrolytic enzyme were
obtained fox both modified oligonucleotides 157 and 158.
In a second set of experiments, the inhibitory effects of phenoxazine and
G-clamp oligonucleotides on the nuclease activity was investigated. Unmodified
oligonucleotide 159 was incubated with BIPD and the degradation of a l9mer
oligothymidylate with 5' labeled with fluorescein was followed under the
presence of various amounts of oligonucleotides 157 and 158, respectively.
Oligonucleotide samples were incubated with BIPD (0.55 units/~.mol substrate)
in
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-138-
50 mM Tris-HCI, 8 mM MgCl2, pH 7.5 at 37°C. At certain time points
aliquots of
~1 were withdrawn and diluted directly into 200 ~L dH20 before CGE
analysis. The influence of the modified oligonucleotides on the nucleolytic
activity was determined~by looking at the overall velocity of the enzymatic
5 reaction. Therefore, all products of degradation were quantified at each
time
point, weighted considering their stage of degradation (n-x) and summarized to
obtain the number of hydrolyzed linkages. The velocity of the enzymatic
reaction
was determined graphically from the number of hydrolyzed phosphodiester
linkages as a function of the incubation time.
10 This second part of our study was driven by the question why
oligonucleotides bearing these tricyclic base modifications at their 3'
terminus
exhibit such extraordinary nuclease resistance. Therefore it was intended to
determine whether or not they are recognized as a substrate, i.e. whether or
not
they are bound to the active site of the enzyme and are capable to affect the
degradation of a natural DNA fragment. In Figure 5, the velocity of the
enzymatic
degradation of unmodified oligonucleotide 159 is depicted as a function of the
concentration of oligonucleotide 157 and 158. From the diagram it is obvious
that
the presence of the modified oligonucleotides has a distinct inhibitory effect
on
the enzymatic reaction. Again, no significant difference is detectable between
the
two derivatives phenoxazine and G-clamp. Both are capable to slow down the
degradation process of oligonucleotide 159 at concentrations above 0.2 ~,M.
The
IC50 values are reached at about 0.5 pM and at concentrations of 5 ~M and
higher
the enzymatic reaction is almost completely prohibited.
EXAMPLE 121
Nuclease resistance of oligonucleotides with selected modifications
Phenoxazine 151 and G-clamp 152 nucleosides were prepared by
modifying previously published procedures [Lin, K.-Y.; Jones, R. J.;
Matteucci,
M. J. Afn. Chem. Soc. 1995,117, 3873-3874; Lin, K.-Y.; Matteucci, M..l. Am.
Clae~ri. Soc. 1998,120, 8531-8532]. The succinates 153 and 154 and the
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-139-
corresponding substituted solid supports 155 and 156 were prepared as outlined
in
Scheme 19. Using the CPG supports, the two cytidine analogs 151 and 152 were
incorporated at the 3' terminus of two model oligonucleotides 157 and 158,
respectively, with the sequence TlBdC* (dC* = phenoxazine (SEQ ID N0:62) or
G-clamp deoxyribonucleoside (SEQ ID N0:63)). Solid phase oligonucleotide
syntheses was carried out using standard phosphoramidite chemistry.
Deprotection of G-clamp containing oligonucleotide 158 was performed with a
1:1 solution of MeNH2 (40%, aq.) and NH3 (28-30%, aq.) at r.t. for 4 h. The
oligonucleotides were purified and desalted by reversed phase HPLC.
Snake venom phosphodiesterase (SVPD) and bovine intestinal mucosal
phosphodiesterase (BIPD), were utilized as the hydrolytic enzymes for ih vitro
nuclease resistance studies. Both enzymes predominantly exhibit 3 '
exonuclease
activity. An unmodied ~ 9mer oligothymidylate (oligonucleotide 159) (SEQ ID
N0:64) was used as a control. Oligonucleotide samples were incubated with
SVPD (2.5 units/~mol substrate) or BIPD (0.55 units/qmol substrate) in 50 mM
Tris-HCI, 8 mM MgCl2 buffer, pH 7.5 at 37°C. At certain time points
aliquots of
10 ~l were removed and heated in boiling water for 2 min to inactivate the
enzyme. Subsequently, the samples were desalted by membrane dialysis against
Nanopure deionized water using Millipore 0.025 ~.m VS membranes and stored
frozen until they were analysed. The progress of enzymatic degradation was
monitored by capillary gel electrophoresis (CGE).
The results of the nuclease resistance study with SVPD as the hydrolytic
enzyme are shown in Figure 4. As expected, the unmodified control
oligonucleotide 159 (insert) was degraded rapidly by sequential removal of the
terminal nucleotides. Under the applied conditions the tli2 for this
oligonucleotide
was reached at about 3 min. After 20 min of incubation the full length
oligomer
was almost completely degraded to a series of shorter fragments. In contrast,
the
modified oligonucleotides 157 and 158 bearing the heterocyclic modifications
at
their 3' end were not significantly degraded even after an incubation time of
8 h.
According to the degradation rates and the CGE profiles, there is no
significant
difference in the 3' exonuclease resistance of these two oligomers. Very
similar
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-140-
results for the nuclease resistance against BIPD as the hydrolytic enzyme were
obtained for both modified oligonucleotides 157 and 158.
In a second set of experiments, the inhibitory effects of phenoxazine and
G-clamp oligonucleotides on the nuclease activity was investigated. Umnodified
oligonucleotide 159 was incubated with BIPD and the degradation of a l9mer
oligothymidylate with 5' labeled with fluorescein was followed under the
presence of various amounts of oligonucleotides 157 and 158, respectively.
Oligonucleotide samples were incubated with BIPD (0.55 units/qmol substrate)
in
50 mM Tris-HCI, 8 mM MgCl2, pH 7.5 at 37°C. At certain time points
aliquots of
10 ~,1 were withdrawn and diluted directly into 200 ~.L dH20 before CGE
analysis. The influence of the modified oligonucleotides on the nucleolytic
activity was determined by looking at the overall velocity of the enzymatic
reaction. Therefore, all products of degradation were quantified at each time
point, weighted considering their stage of degradation (n-x) and summarized to
obtain the number of hydrolyzed linkages. The velocity of the enzymatic
reaction
was determined graphically from the number of hydrolyzed phosphodiester
linkages as a function of the incubation time.
This second part of our study was driven by the question why
oligonucleotides bearing these tricyclic base modifications at their 3'
terminus
exhibit such extraordinary nuclease resistance. Therefore it was intended to
determine whether or not they are recognized as a substrate, i.e. whether or
not
they are bound to the active site of the enzyme and are capable to affect the
degradation of a natural DNA fragment. In Figure 5, the velocity of the
enzymatic
degradation of umnodified oligonucleotide 159 is depicted as a function of the
concentration of oligonucleotide 157 and 158. From the diagram it is obvious
that
the presence of the modified oligonucleotides has a distinct inhibitory effect
on
the enzymatic reaction. Again, no significant difference is detectable between
the
two derivatives phenoxazine and G-clamp. . Both are capable to slow down the
degradation process of oligonucleotide 159 at concentrations above 0.2 ~M. The
IC50 values are reached at about 0.5 ~M and at concentrations of 5 ~.M and
higher
the enzymatic reaction is almost completely prohibited.
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-141 -
The nuclease resistance data demonstrate that, despite their natural
phosphodiester backbones, both heterocyclic modifications provide an almost
complete protection against 3' exonuclease attack. Obviously the enzyme is not
capable to digest oligonucleotides, which contain the modified nucleobases
phenoxazine or G-clamp at their 3' terminus. The observed high nuclease
stability could principally have various reasons. Either the bulky heterocycle
moieties simply prevent the enzyme from binding to the 3'-terminus by steric
hindrance, meaning that the oligonucleotides are not recognized as a
substrate, or
they bind to the active site of the enzyme without being hydrolyzed, which
would
directly affect the enzyme's activity. The observed decrease in the velocity
of the.
enzymatic degradation of a natural DNA fragment indicates that
oligonucleotides
containing phenoxazine and G-clamp residues are able to bind to the enzyme's
active site. Hydrolysis of the 3' terminal nucleotide phosphodiester linkage,
however, is prevented due to the presence of the unnatural tricyclic base
moieties.
The dose-dependence of the inhibitory effects with IC50 values of about 0.5
~.MoI
suggests that the binding of the modified oligonucleotides is competitive and
reversible.
There is no detectable difference between the nuclease resistance of
oligonucleotides 157 and 158 indicating that the observed stabilizing effect
is
mainly due to presence of the bulky heterocycles. With the present data,
however,
it remains unclear to what extent the positively charged amino tether of the G-
clamp moiety contributes to the nuclease resistance of oligonucleotide 158. In
previous studies it has been shown that cationc modifications of the sugar
moieties, such as 2'-O-aminoalkyl, can efficiently protect phosphodiester
oligonucleotides from enzymatic degradation [Manoharan, M.; Tivel, K. L.;
Anrade, L. I~., Cook, P. D. Tetralaedron Lett. 1995, 36, 3647-3650; Teplova,
M.;
Wallace, S. C.; Tereshko, V.; Minasov, G.; Syrnons, A. M.; Cook, P. D.;
Manoharan, M.; Egli, M. PNAS 1999, 96, 14240-14245]. Crystal structure studies
of a complex formed between a 2'-aminopropyl modified oligonucleotide and an
exonuclease (DNA polymerise I Klenow fragment) demonstrate that the
aminopropyl residue prevents binding of a metal ion, which is needed to
catalyze
hydrolysis of the 3 ' phosphodiester linkage. The amino tether of a G-clamp
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-142-
residue, however, protrudes into the major groove, while the 2' modification
points into the shallow groove of a duplex. Whether or not the positive charge
of
the latter can interfere with the metal binding of an exonuclease remains to
be
investigated.
EXAMPLE 122
Degradation by SYPD
Oligonucleotides, at a final concentration of 2 ~,M, were incubated with
snake venom phosphodiesterase (.005 U/ml) in 50 mM Tris-HCl, pH 7.5, 8 mM
MgCl2 at 37°C. The total reaction volume was 100 qL. At each time point
10 ~,L
aliquots of each reaction mixture were placed in a 500 ~,L microfuge tube and
put
in a boiling water bath for two minutes. The sample was then cooled on ice,
quick
spun to bring the entire volume to the bottom of the tube, and desalted on a
Millipore .025 micron filter disk (Bedford, MA) that was floating in water in
a 60
mm petrie dish. After 30-60 minutes on the membrane the sample was diluted
with 200 ~L distilled H2O and analyzed by gel-filled capillary
electrophoresis.
The oligonucleotide and metabolites were separated and analyzed using the
Beckman PACE MDQ capillary electrophoresis instrument using a 100 ~m m 30
cm coated capillary (Beckman No. 477477) with eCAP ssDNA 100-R gel
(Beckman No. 477621) and Tris-Borate Urea buffer (Beckman No. 338481). The
samples were injected electrokinetically using a field strength of between 5-
10 kV
for a duration of between 5 and 10 seconds. Separation wash achieved at
40°C
with an applied voltage of lSkV. The percentage of full length oligonucleotide
was calculated by integration using Caesar v. 6 software (Senetec Software,
New
Jersey) followed by correction for differences in extinction coefficient for
oligonucleotides of different length.
EXAMPLE 123
In T~ivo nuclease stability and binding affinity properties of
L/D-oligonucleotide chimera
Naturally occurring D-Oligonucleotides are degraded by nucleases very
rapidly whereas enatiomeric L-DNA oligomers have enhanced resistance to the
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
-143-
action of nucleasesl. However L-DNA have been found to hybridize either weakly
or not at all with natural RNA and DNA. Damha and Capobianco [Damha, M. J.;
Giannaris, P. A., Marfey, P. Biochemistry, 1994, 33, 7877-7885; Capobinaco, m.
L.; Garbesi, A.; Arcamone, F.; Maschera, B.; Palu, G. Nucleic Acids Syrnp.
Series
1991, 24, 274] independently have shown that chimeric L/D -oligomers with
terminal L-units provided adequate duplex forming capability and excellent
enzymatic stability in human serum [Damha, M. J.; Giannaris, P. A., Marfey, P.
Biochemistry, 1994, 33, 7877-7885; Capobinaco, m. L.; Garbesi, A.; Arcamone,
F.; Maschera, B.; Palu, G. Nucleic Acids Symp. Series 1991, 24, 274].
Here we report the in vivo nuclease stability of L/D-oligonucleotide
chimera in mouse. We synthesized the phosphoramidite and CPG derived from
L-thymidine, wluch was synthesized from a novel route [Jung, E. M.; Xu, Y.
Tetralaedf-on Lett. 1997, 24, 4199-4202]. A 20 mer phosphorothioate
oligonucleotide ISIS-120745 (antisense to mouse ICAM-1) was capped with L-2'-
deoxy thymidine at 3' and 5'-positions. The oligonucleotide was then
administered IV bolus into BalbC mouse. After 24 h. mouse was sacrificed and
the oligonucleotide was isolated from different organs. Percentage of full-
length
oligonucleotide present in different organs were analyzed by CGE. From all the
major organs >90 % of the intact L-thymidine capped oligonucleotide was
isolated where as the parent oligonucleotide was degraded completely (Figures
6
and 7).
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
SEQUENCE LISTTNG
<110> Isis Pharmaceuticals, Inc.
<120> Nuclease Resistant Chimeric Oligonucleotides
<130> ISIS5075
<150> 60/302,682
<151> 2001-07-03
<150> 09/996,292
<151> 2001-11-28
<160> 55
<170> PatentIn version 3.1
<210> 1
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (20)
<223> All P=S
<220>
<221> misc_feature
<222> (1). (1)
<223> N= L-Thymidine
<220>
<221> misc_feature
<222> (20) .(20)
<223> N= L-Thymidine
<400> 1
ngcatccccc aggccaccan 20
<210> 2
<211> 17
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely 'synthetic sequence
<220>
<221> misc_feature
<222> (1) . (17)
<223> All P=S
<220>
<221> misc_feature
<222> (1). (1)
<223> N= L-Thymidine
<220>
<221> misc_feature
<222> (2). (3)
Page 1
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<223> N= 2'-0-MOE SmeC
<220>
<221> misc_feature
<222> (15) . (15)
<223> N= 2'-0-MOE A
<220>
<221> misc_feature
<222> (16) . (16)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (17) . (17)
<223> N= L-Thymidine
<400> 2
nnncgctgtg atgcnnn 17
<210> 3
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (20)
<223> All P=S
<220>
<221> misc feature
<222> (1)..(1)
<223> N= L-Thymidine
<220>
<221> misc_feature
<222> (2). (3)
<223> N= 2'-O-MOE 5meC
<220>
<221> misc_feature
<222> (13) . (24)
<223> N= 2'-O-MOE ~meC
<220>
<221> misc_feature
<222> (15) . (15)
<223> N= 2'-0-MOE 5meU
<220>
<221> misc_feature
<222> (16) . (16)
<223> N= 2'-O-MOE 5meC
<220>
<221> misc feature
Page 2
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<222> (17)..(17)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (18) .(19)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (20) .(20)
<223> N= L-Thymidine
<400> 3
nnngtcatcg ctnnnnnnnn 20
<210> 4
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc feature
<222> (1).-(20)
<223> All P=S
<400> 4
tgcatccccc aggccaccat 20
<210> 5
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1) . (20)
<223> All P=S
<220>
<221> misc_feature
<222> (1). (1) '
<223> N= L-Thymidine
<220>
<221> misc_feature
<222> (20) .(20)
<223> N= L-Thymidine
<400> 5
ngcatccccc aggccaccan 20
<210> 6
<211> 17
<212> DNA
Page 3
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (17)
<223> All P=S
<400> 6
tcccgctgtg atgcatt 17
<210> 7
<211> 17
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (17)
<223> All P=S
<220>
<221> misc_feature
<222> (1) . (1)
<223> N= L-Thymidine
<220>
<221> misc_feature
<222> (2). (3)
<223> N= 2'-0-MOE SmeC
<220>
<221> misc_feature
<222> (15) .(15)
<223> N= 2'-O-MOE A
<220>
<221> misc feature
<222> (16) .~. (16)
<223> N= 2'-O-MOE Smell
<220>
<221> misc_feature
<222> (17) .(17)
<223> N= L-Thymidine
<400> 7
nnncgctgtg atgcnnn 17
<210> 8
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
Page 4
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<220>
<221> misc feature
<222> (1) .~. (20)
<223> All P=S
<220>
<221> misc_feature
<222> (1). (1)
<223> N= L-Cytidine
<220>
<221> misc_feature
<222> (2). (2)
<223> N= 2'-O-MOE Smell
<220>
<221> misc_feature
<222> (3). (3)
<223> N= 2'-O-MOE A
<220>
<221> misc feature
<222> (4) .~, (4)
<223> N= 2'-O-MOE G
<220>
<221> misc feature
<222> (5) .-. (5)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (16) . (16)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (17) . (17)
<223> N= 2'-O-MOE SmeC
<220>
<221> misc_feature
<222> (18) . (18)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (19) . (19)
<223> N= 2'-O-MOE Smell
<220>
<221> misc feature
<222> (20)x.(20)
<223> N= 2'-O-MOE 5meC
<400> 8
nnnnnttcca cactcnnnnn 20
<210> 9
Page 5
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (20)
<223> All P=S
<220>
<221> misc_feature
<222> (1). (1)
<223> N= 2'-O-MOE SmeC
<220>
<221> misc feature
<222> (2)..(2)
<223> N= 2'-O-MOE Smell
<220>
<221> misc_feature
<222> (3). (3)
<223> N= 2'-0-MOE A
<220>
<221> misc feature
<222> (4) .~. (4)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (5). (5)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (16) .(16)
<223> N= 2'-O-MOE Smell
<220>
<221> misc_feature
<222> (17) .(17)
<223> N= 2'-0-MOE SmeC
<220>
<221> misc_feature
<222> (18) .(18)
<223> N= 2'-0-MOE G
<220>
<221> misc_feature
<222> (19) .(19)
<223> N= 2'-0-MOE 5meU
<220>
<221> misc_feature
<222> (20) .(20)
<223> N= L-Cytidine
Page 6
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<400> 9
nnnnnttcca cactcnnnnn
<210> 10
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (20)
<223> All P=S
<220>
<221> misc_feature
<222> (1). (1)
<223> N= L-Cytidine
<220>
<22I> misc_feature
<222> (2). (2)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (3). (3)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (4). (4)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (5). (5)
<223> N= 2'-0-MOE A
<220>
<221> misc_feature
<222> (16) .(16)
<223> N= 2'-O-MOE Smell
<220>
<221> misc_feature
<222> (17) . (17)
<223> N= 2'-O-MOE SmeC
<220>
<221> misc_feature
<222> (18) .(18)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (19) .(19)
Page 7
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<223> N= 2'-0-M0E Smell
<220>
<221> misc_feature
<222> (20) .(20)
<223> N= L-Cytidine
<400> 10
nnnnnttcca cactcnnnnn
<210> 11
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (201
<223> All P=S
<220>
<221> misc_feature
<222> (10) .(10)
<223> N= 2'-0-MOE 5meC
<220>
<221> misc feature
<222> (11)-.(11)
<223> N= 2'-0-MOE A
<220>
<221> misc_feature
<222> (12)..(13)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (14) . (14)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (16) . (16)
<223> N= 2'-O-MOE SmeC
<220>
<221> misc_feature
<222> (17) .(18)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (19) .(19)
<223> N= 2'-O-MOE SmeC
<220>
<221> misc feature
Page 8
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<222> (20)..(20)
<223> N= L-Adenosine
<400> 11
ccggtacccn nnnntnnnnn
<210> 12
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (20)
<223> All P=S
<220>
<221> misc_feature
<222> (1). (1)
<223> N= L-Cytidine
<220>
<221> misc_feature
<222> (10) .(10)
<223> N= 2'-0-MOE SmeC
<220>
<221> misc_feature
<222> (11) .(11)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (12) .(13)
<223> N= 2'-0-M0E G
<220>
<221> misc_feature
<222> (14) .(14)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (16) .(16) '
<223> N= 2'-0-MOE SmeC
<220>
<221> misc_feature
<222> (17) .(18)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (19) .(19)
<223> N= 2'-0-MOE 5meC
<220>
Page 9
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<221> misc_feature
<222> (20) . (20)
<223> N= L-Adenosine
<400> 12
ncggtacccn nnnntnnnnn
<210> 13
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1)..(20)
<223> All P=S
<220>
<221> misc_feature
<222> (9). (9)
<223> N= 2'-O-MOE SmeC
<220>
<221> misc_feature
<222> (10) .(10)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (11) . (11)
<223> N= 2'-O-MOE 5meC
<220>
<221> misc_feature
<222> (12) . (12)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (20) .(20)
<223> N= L-Cytidine
<400> 13
ctagattcnn nnctctcgtn
<210> 14
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (20)
<223> All P=S
Page 10
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<220>
<221> misc_feature
<222> (1). (1)
<223> N= L-Cytidine
<220>
<221> misc_feature
<222> (9). (9)
<223> N= 2'-O-MOE 5meC
<220>
<221> misc_feature
<222> (10) .(10)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (11) .(11)
<223> N= 2'-O-MOE SmeC
<220>
<221> misc_feature
<222> (12) .(12)
<223> N= 2'-O-MOE A
<400> 14
ntagattcnn nnctctcgtc
<210> 15
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<22l> misc_feature
<222> (1). (20)
<223> All P=S
<220>
<221> misc_feature
<222> (1). (1)
<223> N= L-Cytidine
<220>
<221> misc_feature
<222> (9). (9)
<223> N= 2'-O-MOE 5meC
<220>
<221> misc_feature
<222> (10) .(10)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (11) .(11)
<223> N= 2'-O-MOE 5meC
Page 11
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<220>
<221> misc_feature
<222> (12) .(12)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (20) . (20)
<223> N= L-Cytidine
<400> 15
ntagattcnn nnctctcgtn
<210> 16
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1) . (20)
<223> All P=S
<220>
<221> misc_feature
<222> (1). (1)
<223> N= 2,-3'-Dideoxycytidine
<220>
<221> misc_feature
<222> (2). (2)
<223> N= 2'-0-MOE 5meU
<220>
<221> misc_feature
<222> (3). (3)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (4). (4)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (5). (5)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (16) .(16)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (17) . (17)
<223> N= 2'-O-MOE SmeC
Page 12
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<220>
<221> misc_feature
<222> (18) .(18)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (19) .(19)
<223> N= 2'-0-MOE 5meU
<220>
<221> misc_feature
<222> (20) .(20)
<223> N= 2, 3'-Dideoxycytidine
<400> 16
nnnnnttcca cactcnnnnn
<210> 17
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (20)
<223> All P=S
<220>
<221> misc_feature
<222> (10) .(10)
<223> N= 2'-0-MOE 5meC
<220>
<221> misc_feature
<222> (11) .(11)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (12) .(13)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (14) .(14)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (16) .(16)
<223> N= 2'-O-MOE SmeC
<220>
<221> misc feature
<222> (17)-.. (18)
Page 13
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<223> N= 2'-0-MOE Smell
<220>
<221> misc_feature
<222> (19) .(19)
<223> N= 2'-O-MOE 5meC
1<220>
<221> misc_feature
<222> (20) . (20)
<223> N= 2',-3'-Dideoxyadenosine
<400> 17
ccggtacccn nnnntnnnnn
<210> 18
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (20)
<223> All P=S
<220>
<221> misc_feature
<222> (1) . (1)
<223> N= 2'-O-MOE 5meC
<220>
<221> misc_feature
<222> (2). (2)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (3). (3)
<223> N= 2'-0-MOE A
<220>
<221> misc_feature
<222> (4). (4)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (5). (5)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (16) . (16)
<223> N= 2'-O-MOE 5meU ,
<220>
<221> misc feature
Page 14
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<222> (17)..(17)
<223> N= 2'-O-MOE 5meC
<220>
<221> misc_feature
<222> (18) .(18)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (19) .(19)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (20) .(20)
<223> N= 2'-3'-Didehydro-2', 3'-dideoyxcytidine
<400> 18
nnnnnttcca cactcnnnnn
<210> 19
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (20)
<223> All P=S
<220>
<221> misc_feature
<222> (I0) .(10)
<223> N= 2'-0-MOE 5meC
<220>
<221> misc_feature
<222> (11) .(11)
<223> N= 2'-O-MOE A
<220>
<221> mist feature
<222> (12) .~. (13)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (14) .(15)
<223> N= 2'-O-MOE 5meU
<220>
<221> mist feature
<222> (16)-.. (16)
<223> N= 2'-0-MOE SmeC
<220>
Page 15
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<221> misc_feature
<222> (17) .(18)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (19) .(19)
<223> N= 2'-O-M0E 5meC
<220>
<221> mist feature
<222> (20) .~. (20)
<223> N= 2',-3'-Didehydro-2',3'-dideoxyadenosine
<400> 19
ccggtacccn nnnnnnnnnn 20
<210> 20
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (20)
<223> All P=S
<220>
<221> mist feature
<222> (1) .~. (1)
<223> N= 2'-O-MOE SmeC
<220>
<221> misc_feature
<222> (2) . (2)
<223> N= 2'-O-MOE Smell
<220>
<221> misc_feature
<222> (3). (3)
<223> N= 2'-O-MOE A
<220>
<221> misc_featute
<222> (4) . (4)
<223> N= 2'-O-MOE G
<220>
<221> mist feature
<222> (5) .~. (5)
<223> N= 2'-0-MOE A
<220>
<221> misc_feature
<222> (16) .(16)
<223> N= 2'-O-MOE 5meU
Page 16
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<220>
<221> misc_feature
<222> (17) . (17)
<223> N= 2'-O-MOE 5meC
<220>
<221> misc_feature
<222> (18) . (18)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (19) .(19)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc feature
<222> (20)..(20)
<223> N= 2'-3'-Dideoxy-3'-fluorocytidine
<400> 20
nnnnnttcca cactcnnnnn 20
<210> 21
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (20)
<223> All P=S
<220>
<221> misc_feature
<222> (1). (1)
<223> N= 2'-O-MOE 5meC
<220>
<221> misc_feature
<222> (2). (2)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (3). (3)
<223> N= 2'-0-MOE A
<220>
<221> misc_feature
<222> (4). (4)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (5). (5)
<223> N= 2'-O-MOE A
Page 17
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<220>
<221> misc_feature
<222> (16) .(16)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (17) .(17)
<223> N= 2'-O-MOE 5meC
<220>
<221> misc_feature
<222> (18) .(18)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (19) .(19)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (20) .(20)
<223> N= 3'-Deoxy-2'-O-[2-(methoxy)ethyl]-5-methylcytidine
<400> 21
nnnnnttcca cactcnnnnn 20
<210> 22
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (20)
<223> All P=5
<220>
<221> misc_feature
<222> (1). (1)
<223> N= 2'-O-MOE 5meC
<220>
<221> misc_feature
<222> (2). (2)
<223> N= 2'-O-MOE Smell
<220>
<221> misc_feature
<222> (3) . (3)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (4). (4)
<223> N= 2'-O-MOE G
Page 18
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<220>
<221> misc_feature
<222> (5) . (5)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (16) .(16)
<223> N= 2'-0-MOE 5meU
<220>
<221> misc_feature
<222> (17) .(17)
<223> N= 2'-0-MOE SmeC
<220>
<221> misc_feature
<222> (18) .(18)
<223> N= 2'-0-MOE G
<220>
<221> misc_feature
<222> (19) .(19)
<223> N= 2'-0-MOE Smell
<220>
<221> misc_feature
<222> (20) .(20)
<223> N= 3-hydroxy-2-pyrrolidinemethanol
<400> 22
nnnnnttcca cactcnnnnn 20
<210> 23
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1) . (21)
<223> All P=S
<220>
<221> misc_feature
<222> (1). (1)
<223> N= 3-hydroxy-2-pyrrolidinemethanol
<220>
<221> misc_feature
<222> (2). (2)
<223> N= 2'-0-MOE 5meC
<220>
<221> misc_feature
<222> (3) . (3)
Page 19
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<223> N= 2'-0-MOE 5meU
<220>
<221> misc_feature
<222> (4). (4)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (5). (5)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (6). (6)
<223> N= 2'-0-MOE A
<220>
<221> misc_feature
<222> (17) .(17)
<223> N= 2'-0-MOE Smell
<220>
<221> misc_feature
<222> (18) .(18)
<223> N= 2'-0-MOE SmeC
<220>
<221> misc_feature
<222> (19) .(19)
<223> N= 2'-0-MOE G
<220>
<221> misc_feature
<222> (20) .(20)
<223> N= 2'-0-MOE 5meU
<220>
<221> misc_feature
<222> (21) .(21)
<223> N= 3-hydroxy-2-pyrrolidinemethanol
<400> 23
nnnnnnttcc acactcnnnn n 21
<210> 24
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (21)
<223> All P=S
<220>
<221> misc feature
Page 20
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<222> (10)..(10)
<223> N= 2'-0-MOE SmeC
<220>
<221> misc_feature
<222> (11) .(11)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (12) .(13)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (14) .(15)
<223> N= 2'-O-MOE Smell
<220>
<221> misc_feature
<222> (16) . (16)
<223> N= 2'-0-MOE 5meC
<220>
<221> misc_feature
<222> (17) .(18)
<223> N= 2'-0-MOE Smell
<220>
<221> misc_feature
<222> (19) .(19)
<223> N= 2'-0-MOE SmeC
<220>
<221> misc_feature
<222> (21) .(21)
<223> N= 3-hydroxy-2-pyrrolidinemethanol
<400> 24
ccggtacccn nnnnnnnnna n 21
<210> 25
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (22)
<223> All P=S
<220>
<221> misc_feature
<222> (1). (1)
<223> N= 3-hydroxy-2-pyrrolidinemethanol
<220>
Page 21
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<221> misc_feature
<222> (11) .(11)
<223> N= 2'-O-MOE SmeC
<220>
<221> misc_feature
<222> (12) .(12)
<223> N= 2'-0-MOE A
<220>
<221> misc_feature
<222> (13) .(14)
<223> N= 2'-0-MOE G
<220>
<221> misc_feature
<222> (15) .(16)
<223> N= 2'-0-MOE 5 meU
<220>
<221> misc_feature
<222> (17) .(17)
<223> N= 2'-O-MOE 5 meC
<220>
<221> misc_feature
<222> (18) .(19)
<223> N= 2'-O-MOE 5 meU
<220>
<221> misc_feature
<222> (20) .(20)
<223> N= 2'-0-MOE 5 meC
<220>
<221> misc_feature
<222> (22) .(22)
<223> N= 3-hydroxy-2-pyrrolidinemethanol
<400> 25
nccggtaccc nnnnnnnnnn an 22
<210> 26
<211> 20
<212> DNA
<213> Artificial'Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (20)
<223'> All P=S
<220>
<221> misc_feature
<222> (1) . (1)
<223> N= 2'-O-MOE SmeC
Page 22
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<220>
<221> misc feature
<222> (2)..(2)
<223> N= 2'-0-MOE Smell
<220>
<221> misc_feature
<222> (3). (3)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (4). (4)
<223> N= 2'-0-MOE G
<220>
<221> misc_feature
<222> (5) . (5)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (16) . (16)
<223> N= 2'-0-MOE Smell
<220>
<221> misc_feature
<222> (17) .(17)
<223> N= 2'-O-MOE 5meC
<220>
<221> misc_feature
<222> (18) .(18)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (19) . (19)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (20) .(20)
<223> N= 1-[2-hydroxy-1-[2-hydroxy-1-
(hydroxymethyl)ethoxy]ethylcytosine
<400> 26
nnnnnttcca cactcnnnnn 20
<210> 27
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1) . (20)
<223> All P=S
Page 23
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<220>
<221> misc_feature
<222> (1). (1)
<223> N= 1-[2-hydroxy-1-[2-hydroxy-1-
(hydroxymethyl)ethoxy]ethylcytosine
<220>
<221> misc_feature
<222> (2). (2)
<223> N= 2'-O-MOE Smell
<220>
<221> misc_feature
<222> (3). (3)
<223> N= 2'-0-MOE A
<220>
<221> misc_feature
<222> (4). (4)
<223> N= 2'-0-MOE G
<220>
<221> misc_feature
<222> (5). (5)
<223> N= 2'-0-MOE A
<220>
<221> misc_feature
<222> (16) .(16)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (17) .(17)
<223> N= 2'-O-MOE SmeC
<220>
<221> misc_feature
<222> (18) . (18)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (19) . (19)
<223> N= 2'-O-MOESmeU
<220>
<221> misc_feature
<222> (20) . (20)
<223> N= 1-[2-hydroxy-1-[2-hydroxy-1-
(hydroxymethyl)ethoxy]ethylcytosine
<400> 27
nnnnrittcca cactcnnnnn 20
<210> 28
<211> 20
<212> DNA
Page 24
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (20)
<223> All P=S
<220>
<221> misc_feature
<222> (1). (1)
<223> N= 2'-O-MOE SmeC
<220>
<221> misc_feature
<222> (2). (2)
<223> N= 2'-O-MOE Smell
<220>
<221> misc_feature
<222> (3) . (3)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (4). (4)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (5). (5)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (16) . (16)
<223> N= 2'-O-MOE Smell
<220>
<221> misc_feature
<222> (17) .(17)
<223> N= 2'-O-MOE SmeC
<220>
<221> misc_feature
<222> (18) .(18)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (19) .(19)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (20) .(20)
<223> N= 1-[2-hydroxy-1-[2-hydroxy-1-
(hydroxymethyl)ethoxy]ethylcytosine
Page 25
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<400> 28
nnnnnttcca cactcnnnnn 20
<210> 29
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence .
<220>
<221> misc_feature
<222> (1). (20)
<223> All P=S
<220>
<221> misc_feature
<222> (1). (1)
<223> N= 1-[2-hydroxy-1-[2-hydroxy-1-
(hydroxymethyl)ethoxy]ethylcytosine
<220>
<221> misc_feature
<222> (2). (2)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (3). (3)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (4) . (4)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (5). (5)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (16) . (16)
<223> N= 2'-O-MOE Smell
<220>
<221> misc_feature
<222> (17) .(17)
<223> N= 2'-O-MOE 5meC
<220>
<221> misc_feature
<222> (18) .(18)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (19) .(19)
Page 26
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<223> N= 2'-O-MOE Smell
<220>
<221> misc_feature
<222> (20) .(20)
<223> N= 1-[2-hydroxy-1-[2-hydroxy-1-
(hydroxymethyl)ethoxy]ethylcytosine
<400> 29
nnnnnttcca cactcnnnnn 20
<210> 30
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (20)
<223> All P=S
<220>
<221> misc_feature
<222> (l) . (1)
<223> N= 2'-O-MOE 5meC
<220>
<221> misc_feature
<222> (2)..(2)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (3) . (3)
<223> N= 2'-0-M0E A
<220>
<221> misc_feature
<222> (4) . (4) °
<223> N= 2'-0-MOE G
<220>
<221> misc_feature
<222> (5) . (5) '
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (16) .(16)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (17) .(17)
<223> N= 2'-0-MOE 5meC
<220>
Page 27
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<221> misc_feature
<222> (18) .(18)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (19) .(19)
<223> N= 2'-0-MOE 5meU
<220>
<221> misc_feature
<222> (20) .(20)
<223> N= 2',3'-dideoxy-3'-(amino)cytidine
<400> 30
nnnnnttcca cactcnnnnn 20
<210> 31
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (20)
<223> All P=S
<220>
<221> misc_feature
<222> (1). (1)
<223> N= 2'-O-MOE 5meC
<220>
<221> misc_feature
<222> (2). (2)
<223> N= 2'-0-MOE 5meU
<220>
<221> misc_feature
<222> (3). (3)
<223> N= 2'-O-MOE A
<220>
<221> misc_featu~e
<222> (4). (4)
<223> N= 2'-O-M0E G
<220>
<221> misc_feature
<222> (5). (5)
<223> N= 2'-0-MOE A
<22d>
<221> misc_feature
<222> (16) . (16)
<223> N= 2'-O-MOE Smell
Page 28
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<220>
<221> misc_feature
<222> (17) .(17)
<223> N= 2'-O-MOE SmeC
<220>
<221> misc_feature
<222> (18) .(18)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (19) .(19)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (20) .(20)
<223> N= 2'-deoxy-3'-S-phenyl-3'-thiocytidine
<400> 31
nnnnnttcca cactcnnnnn 20
<210> 32
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (20)
<223> All P=S
<220>
<221> misc_feature
<222> (1). (1)
<223> N= 2'-O-MOE SmeC
<220>
<221> misc_feature
<222> (2). (2)
<223> N= 2'-O-MOE Smell
<220>
<221> misc_feature
<222> (3). (3)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (4). (4)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (5). (5)
<223> N= 2'-O-MOE A
Page 29
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<220>
<221> misc_feature
<222> (16) . (16)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (17) . (17)
<223> N= 2'-O-MOE 5meC
<220>
<221> misc_feature
<222> (18) . (18)
<223> N= 2'-O-MOE G
<220>
<221> mist feature
<222> (19)..(19)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (20) .. (20)
<223> N= 3'-deoxy-2'-S-phenyl-2'-thiocytidine
<400> 32
nnnnnttcca cactcnnnnn 20
<210> 33
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (20)
<223> All P=S
<220>
<221> misc_feature
<222> (1). (1)
<223> N= 2'-O-MOE 5meC
<220>
<221> misc_feature
<222> (2). (2)
<223> N= 2'-O-MOE Smell
<220>
<221> misc_feature
<222> (3). (3)
<223> N= 2'-O-MOE A
<220>
<221> misc_~feature
<222> (4). (4)
<223> N= 2'-O-MOE G
Page 30
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<220>
<221> misc_feature
<222> (5). (5)
<223> N= 2'-O-MOE A
<220>
<221> misc_feature
<222> (16) .(16)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (17) .(17)
<223> N= 2'-O-MOE SmeC
<220>
<221> misc_feature
<222> (18) .(18)
<223> N= 2'-O-MOE G
<220>
<221> misc_feature
<222> (19) .(19)
<223> N= 2'-O-MOE 5meU
<220>
<221> misc_feature
<222> (20) . (20)
<223> N= 1[2,3-deoxy-2-N-morpholino
-B-D-gylcero-pent-2-enofuranosyl]
cytosine
<400> 33
nnnnnttcca cactcnnnnn 20
<210> 34
<211> 10
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc feature
<222> (5)..(5) '
<223> 2'-O-hexylguanidinyl-U 5me
<400> 34
ttttnttttt 10
<210> 35
<211> 10
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
Page 31
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<221> misc_feature
<222> (5). (5)
<223> N= 2'-deoxy-G-clamp
<400> 35
tctcnctctc 10
<210> 36
<211> 10
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (5) . (5)
<223> N= 2'-deoxy-guanidinyl G-clamp
<400> 36
tctcnctctc 10
<210> 37
<211> 18
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (9) . (9)
<223> N= 2'-deoxy-guanidinyl G- clamp
<400> 37
ctcgtaccnt cccggtcc 18
<210> 38
<211> 10
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature'
<222> (2). (2)
<223> N= 2'-deoxy-guanidino G-clamp
<220>
<221> misc_feature
<222> (6). (6)
<223> N= 2'-MOE-U 5me
<400> 38
gngtanacgc 10
<210> 39
<211> 10
Page 32
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (6) . (6)
<223> N= 2'-MOE- U 5me
<220>
<221> misc_feature
<222> (8). (8)
<223> N= 2'-deoxy-guanidine G-clamp
<400> 39
gcgtanangc 10
<210> 40
<211> 15
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<400> 40
aaaaagagag ggaga 15
<210> 41
<211> 10
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (2) . (2)
<223> N= guanidine G-clamp
<220>
<221> misc_feature
<222> (6). (6)
<223> N= 2'-0-methoxyethyl thymine
<400> 41
gngtanacgc 10
<210> 42
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1) . (20)
<223> All P=S
Page 33
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<400> 42
atgcattctg cccccaagga 20
<210> 43
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (20)
<223> All P=S
<220>
<221> misc_feature
<222> (4) . (4)
<223> N= G-clamp modification
<400> 43
atgnattctg cccccaagga 20
<210> 44
<2l1> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1) . (20)
<223> All P=S
<220>
<221> misc_feature
<222> (8) . (8)
<223> N= G-clamp modification
<400> 44
atgcattntg cccccaagga 20
<210> 45
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (20)
<223> All P=S
<220>
<221> misc_feature
<222> (11) .(11)
<223> N= G-clamp modification
Page 34
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<400> 45
atgcattctg nccccaagga 20
<210> 46
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1) . (20)
<223> All P=S
<220>
<221> misc_feature
<222> (12) .(12)
<223> N= G-clamp modification
<400> 46
atgcattctg cncccaagga 20
<210> 47
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc feature
<222> (1).-(20)
<223> All P=S
<220>
<221> misc_feature
<222> (13) .(13)
<223> N= G-clamp modification
<400> 47
atgcattctg ccnccaagga 20
<210> 48
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1) . (20)
<223> All P=S
<220>
<221> misc_feature
<222> (14) .(14)
Page 35
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<223> N= G-clamp modification
<400> 48
atgcattctg cccncaagga 20
<210> 49
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (20)
<223> All P=S
<220>
<221> misc_feature
<222> (15) .(15)
<223> N= G-clamp modification
<400> 49
atgcattctg ccccnaagga 20
<210> 50
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<400> 50
ctagattcca cactctctcg tc 22
<210> 51
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (1)
<223> N= G-clamp'modification
<400> 51
ntagattcca cactctcgtc 20
<210> 52
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc feature
Page 36
SUBSTITUTE SHEET (RULE 26)
CA 02452458 2003-12-30
WO 03/004602 PCT/US02/20934
<222> (20)..(20)
<223> N= G-clamp modification
<400> 52
ctagattcca cactctcgtn 20
<210> 53
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (1). (1)
<223> N= G-clamp modification
<220>
<221> misc_feature
<222> (20) . (20)
<223> N= G-clamp modification
<400> 53
ntagattcca cactctcgtn 20
<210> 54
<211> 19
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (19) . (19)
<223> N= phenoxazine
<400> 54
tttttttttt ttttttttn 19
<210> 55
<211> 19
<212> DNA
<213> Artificial Sequence
<220>
<223> Completely synthetic sequence
<220>
<221> misc_feature
<222> (19) . (19)
<223> N= G-clamp modification
<400> 55
tttttttttt ttttttttn 19
Page 37
SUBSTITUTE SHEET (RULE 26)