Note: Descriptions are shown in the official language in which they were submitted.
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
CYSTEINE MUTANTS AND METHODS FOR DETECTING LIGAND BINDING TO
BIOLOGICAL MOLECULES
BACKGROUND
The drug discovery process usually beings with massive functional screening of
compound libraries
to identify modest affinity leads (Kd ~ 1 to 10 pM) for subsequent medicinal
chemistry
optimization. However, not all targets of interest are amenable to such
screening. In some cases,
an assay that is amenable to high throughput screening is not available. In
other cases, the target
can have multiple binding modes such that the results of such screens are
ambiguous and difficult to
interpret. Still in other cases, the assay conditions for high throughput
screening are such that they
are prone to artifacts. As a result, alternative methods for ligand discovery
are needed that to not
necessarily rely on functional assays. The present invention provides such
methods.
SUMMARY
The present invention relates generally to variants of target biological
molecules ("TBMs") and to
methods of making and using the same to identify ligands of TBMs. More
specifically, the
invention relates to individual variant TBMs and sets of variant TBMs, each of
which represents a
modified version of a protein of interest where a thiol has been introduced at
or near a site of
interest. Ligands of TBMs are identified in part through the formation of a
covalent bond between
a potential ligand and a reactive thiol on the TBM.
DESCRIPTION OF THE FIGURES
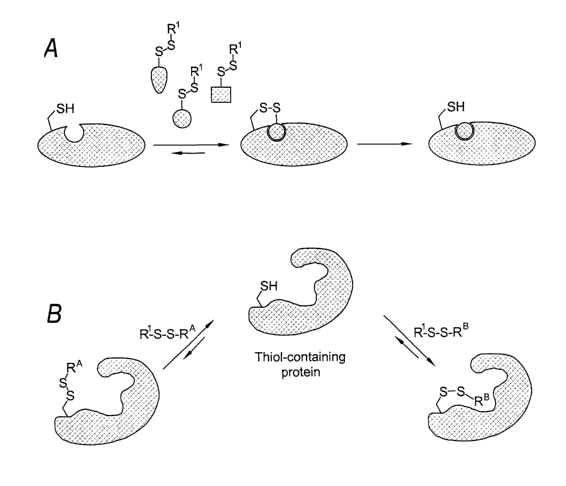
Figure 1 schematically illustrates one embodiment of the tethering method
wherein the target is a
protein and the covalent bond is a disulfide. A thiol-containing protein is
reacted with a plurality of
ligand candidates. A ligand candidate that possesses an inherent binding
affinity for the target is
identified and a ligand is made comprising the identified binding determinant
(represented by the
circle).
Figure 2 is a representative example of a tethering experiment. Figure ZA is
the deconvoluted mass
spectrum of the reaction of thymidylate synthase ("TS") with a pool of 10
different ligand
candidates with little or no binding affinity for TS. Figure 2B is the
deconvoluted mass spectrum of
the reaction of TS with a pool of 10 different ligand candidates where one of
the ligand candidates
possesses an inherent binding affinity to the enzyme.
Figure 3 shows three illustrative examples of the distribution pattern of the
residues that are each
mutated to a cysteine. Figure 3A is an example where the residues are
distributed about a single
site of interest. The structure is of the core domain of HIV integrase with
the portion comprising
the site of interest shaded in dark gray. Figure 3B is an example where the
residues are distributed
about two sites of interest. The structure is of the human interleukin-1
receptor with the portions
1
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
comprising the two sites of interested shaded in dark gray. Figure 3C is an
example where the
residues are distributed throughout the surface of a protein. The structure is
the trimeric structure of
human TNF-a.
Figure 4 shows the side chain rotamers of cysteines in A) (3-sheets and B) a-
helices.
DESCRIPTION OF THE PREFERRED EMBODIMENTS
The present invention relates generally to variants of target biological
molecules ("TBMs") and to
methods of making and using the same to identify ligands of TBMs.
Unless defined otherwise, technical and scientific terms used herein have the
same meaning as
commonly understood by one of ordinary skill in the art to which this
invention belongs.
References, such as Singleton et al., Dictionary of Microbiology and Molecular
Biology 2nd ed., J.
Wiley & Sons (New York, NY 1994), and March, Advanced Organic Chemistry
Reactions,
Mechanisms and Structure 4th ed., John Wiley & Sons (New York, NY 1992),
provide one skilled
in the art with a general guide to many of the terms used in the present
application.
Definitions
The definition of terms used herein include:
The term "aliphatic" or "urisubstituted aliphatic" refers to a straight,
branched, cyclic, or polyeyclic
hydrocarbon and includes alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl,
and cycloalkynyl
moieties.
The term "alkyl" or "unsubstituted alkyl" refers to a saturated hydrocarbon.
The term "alkenyl" or "unsubstituted alkenyl" refers to a hydrocarbon with at
least one carbon-
carbon double bond.
The term "alkynyl" or "unsubstituted alkynyl" refers to a hydrocarbon with at
least one carbon-
carbon triple bond.
The term "aryl" or "unsubstituted aryl" refers to mono or polycyclic
unsaturated moieties having at
least one aromatic ring. The term includes heteroaryls that include one or
more heteroatoms within
the at least one aromatic ring. Illustrative examples of aryl include: phenyl,
naphthyl,
tetrahydronaphthyl, indanyl, indenyl, pyridyl, pyrazinyl, pyrimidinyl,
pyrrolyl, pyrazolyl,
imidazolyl, thiazolyl, oxazolyl, isooxazoly, thiadiazolyl, oxadiazolyl,
thiophenyl, furanyl,
quinolinyl, isoquinolinyl, and the like.
2
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
The term "substituted" when used to modify a moiety refers to a substituted
version of the moiety
where at least one hydrogen atom is substituted with another group including
but not limited to:
aliphatic; aryl, alkylaryl, F, Cl, I, Br, -OH; -NO2; -CN; -CF3; -CHZCF3; -
CHZCI; -CHZOH;
-CHZCHZOH; -CHZNH2; -CHZSOZCH3; -ORX; -C(O)R'; -COOR'; -C(O)N(R")2; -OC(O)R";
-OCOOR'; -OC(O)N(R")z; -N(R")i; -S(O)ZR'; and -NR"C(O)Rx where each occurrence
of R" is
independently hydrogen, substituted aliphatic, unsubstituted aliphatic,
substituted aryl, or
unsubstituted aryl. Additionally, substitutions at adjacent groups on a moiety
can together form a
cyclic group.
I0 The term "antagonist" is used in the broadest sense and includes any ligand
that partially or fully
blocks, inhibits or neutralizes a biological activity exhibited by a target,
such as a TBM. In a
similar manner, the term "agonist" is used in the broadest sense and includes
any ligand that mimics
a biological activity exhibited by a target, such as a TBM, for example, by
specifically changing the
function or expression of such TBM, or the efficiency of signaling through
such TBM, thereby
altering (increasing or inhibiting) an already existing biological activity or
triggering a new
biological activity.
The term "ligand" refers to an entity that possesses a measurable binding
affinity fox the target. In
general, a ligand is said to have a measurable affinity if it binds to the
target with a Ka or a K; of
less than about 100 mM, preferably less than about 10 mM, and more preferably
less than about 1
mM. In preferred embodiments, the Iigand is not a peptide and is a small
molecule. A Iigand is a
small molecule if it is less than about 2000 daltons in size, usually less
than about 1500 daltons in
size. In more preferred embodiments, the small molecule ligand is less than
about 1000 daltons in
size, usually less than about 750 daltons in size, and more usually less than
about 500 daltons in
2.5 size.
The term "ligand candidate" refers to a compound that possesses or has been
modified to possess a
reactive group that is capable of forming a covalent bond with a complimentary
or compatible
reactive group on a target. The reactive group on either the ligand candidate
or the target can be
masked with, for example, a protecting group.
The term "polynucleotide", when used in singular or plural, generally refers
to any
polyribonucleotide or polydeoxribonucleotide, which may be unmodified RNA or
DNA or
modified RNA or DNA. Thus, for instance, polynucleotides as defined herein
include, without
limitation, single- and double-stranded DNA, DNA including single- and double-
stranded regions,
single- and double-stranded RNA, and RNA including single- and double-stranded
regions, hybrid
molecules comprising DNA and RNA that may be single-stranded or, more
typically, double-
3
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
stranded or include single- and double-stranded regions. In addition, the term
"polynucleotide" as
used herein refers to triple-stranded regions comprising RNA or DNA or both
RNA and DNA. The
strands in such regions may be from the same molecule or from different
molecules. The regions
may include all of one or more of the molecules, but more typically involve
only a region of some
of the molecules. One of the molecules of a triple-helical region often is an
oligonucleotide. The
term "polynucleotide" specifically includes DNAs and RNAs that contain one or
more modified
bases. Thus, DNAs or RNAs with backbones modified for stability or for other
reasons are
"polynucleotides" as that term is intended herein. Moreover, DNAs or RNAs
comprising unusual
bases, such as inosine, or modified bases, such as tritylated bases, are
included within the term
"polynucleotides" as defined herein. In general, the term "polynucleotide"
embraces all chemically,
enzymatically and/or metabolically modified forms of unmodified
polynucleotides, as well as the
chemical forms of DNA and RNA characteristic of viruses and cells, including
simple and complex
cells.
The phrase "protected thiol" as used herein refers to a thiol that has been
reacted with a group or
molecule to form a covalent bond that renders it less reactive and which may
be deprotected to
regenerate a free thiol.
The phrase "reversible covalent bond" as used herein refers to a covalent bond
that can be broken,
preferably under conditions that do not denature the target. Examples include,
without limitation,
disulfides, Schiff bases, thioesters, coordination complexes, boronate esters,
and the like.
The phrase "reactive group" is a chemical group or moiety providing a site at
which a covalent
bond can be made when presented with a compatible or complementary reactive
group. Illustrative
examples are -SH that can react with another -SH or -SS- to form a disulfide;
an -NHZ that can react
with an activated -COOH to form an amide; an -NHZ that can react with an
aldehyde or lcetone to
form a Schiff base and the like.
The phrase "reactive nucleophile" as used herein refers to a nucleophile that
is capable of forming a
covalent. bond with a compatible functional group on another molecule under
conditions that do not
denature or damage the target. The most relevant nucleophiles are thiols,
alcohols, and amines.
Similarly, the phrase "reactive electrophile" as used herein refers to an
electrophile that is capable
of forming a covalent bond with a compatible functional group on another
molecule, preferably
under conditions that do not denature or otherwise damage the target. The most
relevant
electrophiles are imines, carbonyls, epoxides, aziridies, sulfonates,
disulfides, activated esters;
activated carbonyls, and hemiacetals.
4
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
The phrase "site of interest" refers to any site on a target on which a ligand
can bind. For example,
when the target is an enzyme, the site of interest can include amino acids
that make contact with, or
lie within about 10 Angstroms (more preferably within about 5 Angstroms) of a
bound substrate,
inhibitor, activator, cofactor, or allosteric modulator of the enzyme. When
the enzyme is a
protease, the site of interest includes the substrate binding channel from S6
to S6', residues involved
in catalytic function (e.g. the catalytic triad and oxy anion hole), and any
cofactor (e.g. metal such
as Zn) binding site. When the enzyme is a protein kinase, the site of interest
includes the substrate-
binding channel in addition to the ATP binding site. When the enzyme is a
dehydrogenease, the
site of interest includes the substrate binding region as well as the site
occupied by NAD/NADH.
When the enzyme is a hydralase such as PDE4, the site of interest includes the
residues in contact
with CAMP as well as the residues involved in the binding of the catalytic
divalent cations.
The terms "target," "Target Molecule," and "TM" are used interchangeably and
in the broadest
sense, and refer to a chemical or biological entity for which the binding of a
ligand has an effect on
the function of the target. The target can be a molecule, a portion of a
molecule, or an aggregate of
molecules. The binding of a ligand may be reversible or irreversible. Specific
examples of target
molecules include polypeptides or proteins such as enzymes and receptors,
transcription factors,
ligands for receptors such growth factors and cytokines, immunoglobulins,
nuclear proteins, signal
transduction components (e.g., kinases, phosphatases), polynucleotides,
carbohydrates,
glycoproteins, glycolipids, and other macromolecules, such as nucleic acid-
protein complexes,
chromatin or ribosomes, lipid bilayer-containing structures, such as
membranes, or structures
derived from membranes, such as vesicles. The definition specifically includes
Target Biological
Molecules ("TBMs") as defined below.
A "Target Biological Molecule" or "TBM" as used herein refers to a single
biological molecule or a
plurality of biological molecules capable of forming a biologically relevant
complex with one
another for which a small molecule agonist or antagonist has an effect on the
function of the TBM.
In a preferred embodiment, the TBM is a protein or a portion thereof or that
comprises two or more
amino acids, and which possesses or is capable of being modified to possess a
reactive group that is
capable of forming a covalent bond with a compound having a complementary
reactive group.
Preferred TBMs include: cell surface and soluble receptors and their ligands;
steroid receptors;
hormones; immunoglobulins; clotting factors; nuclear proteins; transcription
factors; signal
transduction molecules; cellular adhesion molecules, co-stimulatory molecules,
chemokines,
molecules involved in mediating apoptosis, enzymes, and proteins associated
with DNA and/or
RNA synthesis or degradation.
5
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
Many TBMs are those participate in a receptor.-ligand binding interaction and
can be either member
of a receptor-ligand pair. Illustrative examples of growth factors and their
respective receptors
include those for: erythropoietin (EPO), thrombopoietin (TPO), angiopoietin
(ANG), granulocyte
colony stimulating factor (G-CSF), granulocyte macrophage colony stimulating
factor (GM-CSF),
epidermal growth factor (EGF), heregulin-a and heregulin-j3, vascular
endothelial growth factor
(VEGF), placental growth factor (PLGF), transforming growth factors (TGF-c~
and TGF-~), nerve
growth factor (NGF), neurotrophins, fibroblast growth factor (FGF), platelet-
derived growth factor
(PDGF), bone morphogenetic protein (BMP), connective tissue growth factor
(CTGF), hepatocyte
growth factor (HGF), and insulin-like growth factor 1 (IGF-1). Illustrative
examples of hormones
and their respective receptors include those for: growth hormone, prolactin,
placental lactogen
(LPL), insulin, follicle stimulating hormone (FSH), luteinizing hormone (LH),
and neurokinin-1.
Illustrative examples of cytokines and their respective receptors include
those for: ciliary
neurotrophic factor (CNTF), oncostatin M (OSM), TNF-a; CD40L, stem cell factor
(SCF);
interleukin-1, interleukin-2, interleukin-4, interleulein-5, interleulcin-6,
interleukin-8, interleulcin-9,
interleukin-13, and interleukin-18.
Other TBMs include: cellular adhesion molecules such as CD2, CDlla, LFA-l, LFA-
3, ICAM-5,
VCAM-1, VCAM-5, and VLA-4; costimulatory molecules such as CD28, CTLA-4, B7-l;
B7-2,
ICOS, and B7RP-1; chemokines such as RANTES and MIPlb; apoptosis factors such
as APAF-1,
p53, bax, bak, bad, bid, and c-abl; anti-apoptosis factors such as bcl2, bcl-
x(L), and mdm2;
transcription modulators such as AP-1 and AP-2; signaling proteins such as
TRAF-1, TRAF-2,
TRAF-3, TRAF-4, TRAF-5, and TRAF-6; and adaptor proteins such as grb2, cbl,
shc, nek, and crk
Enzymes are another class of preferred TBMs and can be categorized in numerous
ways including
as: allosteric enzymes; bacterial enzymes (isoleucyl tRNA synthase, peptide
deformylase, DNA
gyrase, and the like); fungal enzymes (thymidylate synthase and the like);
viral enzymes (HIV
integrase, HSV protease, Hepatitis C helicase, Hepatitis C protease,
rhinovirus protease and the
like); lcinases (serinelthreonine, tyrosine, and dual specificity);
phosphatases (serine/threonine,
tyrosine, and dual specificity); and proteases (aspartyl, cysteine, metallo,
and serine proteases).
Notable subclasses of enzymes include: kinases such as Lck, Syk, Zap-70, JAK,
FAK, ITK, BTK,
MEK, MEKK, GSK-3, Raf, tgf (3-activated kinase-1 (TAK-1), PAK-l, cdlc4, Akt,
PKC A, IKK j3,
IKK-2, PDK, ask, nik, MAPKAPK, p90rsk, p70s6k, and PI3-K (p85 and p110
subunits);
phosphatases such as CD45, LAR, RPTP-a, RPTP-~, Cdc25A, kinase-associated
phosphatase, map
kinase phosphatase-1, PTP-1B, TC-PTP, PTP-PEST, SHP-1 and SHP-2; caspases such
as caspases-
3S 1, -3, -7, -8, -9, and -11; and cathespins such as cathepsins B, F, K, L,
S, and V. Other enzymatic
targets include: BACE, TALE, cytosolic phospholipase A2 (cPLA2), PARP, PDE I-
VII, Rac-2,
CD26, inosine monophosphate dehydrogenase, 15-lipoxygenase, acetyl CoA
carboxylase,
6
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
adenosylmethionine decarboxylase, dihydroorotate dehydrogenase, leukotriene A4
hydrolase, and
nitric oxide synthase.
Variants of TBMs
The present invention relates generally to variants of target biological
molecules ("TBMs") and to
methods of making and using the same to identify ligands of the TBMs. In
preferred embodiments,
the TBMs are proteins and the variants are cysteine mutants thereof wherein a
naturally occurring
non-cysteine residue of a TBM is mutated into a cysteine residue. The non-
native cysteine provides
a reactive group on the TBM for use in tethering.
Tethering is a method of ligand identification that relies upon the formation
of a covalent bond
between a reactive group on a target and a complimentary reactive group on a
potential ligand, and
is described in U.S. Patent No. 6,335, 155, PCT Publication Nos. WO 00!00823
and WO 02/42773,
Erlanson et al., Proc. Nat. Acad. Sci. LISA 97: 9367-9372 (2000), and U.S.
Serial No. 10/121,216
entitled METHODS FOR LIGAND DISCOVERY by inventors Daniel Erlanson, Andrew
Braisted,
and James Wells (corresponding PCT Application No. US02/13061). The resulting
covalent
complex is termed a target-ligand conjugate. Because the covalent bond is
formed at a pre-
determined site on the target (e.g., a native or non-native cysteine), the
stoichiometry and binding
location are known fox ligands that are identified by this method.
Once formed, the ligand portion of the target-ligand conjugate can be
identified using a number of
methods. In preferred embodiments, mass spectroscopy is used. The target-
ligand can be detected
directly in the mass spectrometer or fragmented prior to detection.
Alternatively, the ligand can be
liberated from the target-ligand conjugate within the mass spectrophotometer
and subsequently
identified. In other embodiments, alternate detection methods are used
including to but not limited
to: chromatography, labeled probes (fluorescent, radioactive, etc.), nuclear
magnetic resonance
("NMR"), .surface plasmon resonance (e.g., BIACORE), capillary
electrophoresis, X-ray
crystallography and the like. In still other embodiments, functional assays
can also be used when
the binding occurs in an area essential for what the assay measures.
A schematic representation of one embodiment of the tethering method where the
target is a protein
and the covalent bond is a disulfide is shown in Figure 1. A thiol containing
protein is reacted with
a plurality of ligand candidates. In this embodiment, the ligand candidates
possess a masked thiol
in the fornl of a disulfide of the formula -SSR' where R' is unsubstituted C,-
C,o alkyl, substituted
C~-Clo alkyl, unsubstituted aryl or substituted aryl. In certain embodiments,
R' is selected to
enhance the solubility of the potential ligand candidates. As shown, a ligand
candidate that
possesses an inherent binding affinity for the target is identified and a
corresponding ligand that
7
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
does not include the disulfide moiety is made comprising the identified
binding determinant
(represented by the circle).
Figure 2 illustrates two representative tethering experiments where a target
enzyme, E. coli
thymidylate synthase, is contacted with ligand candidates of the formula
O
Rc~~'yH2
H
wherein R° is the variable moiety among this pool of library members
and is unsubstituted aliphatic,
substituted aliphatic, unsubstituted aryl, or substituted aryl. Like all TS
enzymes, E, coli TS has an
active site cysteine (Cys146) that can be used for tethering. Although the E.
coli TS also includes
four other cysteines, these cysteines are buried and were found not to be
reactive in tethering
experiments. For example, in an initial experiment, wild type E. coli TS and
the C146S mutant
(wherein the cysteine at position 146 has been mutated to serine) were
contacted with cystamine,
HZNCH2CHZSSCHZCHZNH2. The wild type TS enzyme reacted cleanly with one
equivalent of
cystamine while the mutant TS did not react indicating that the cystamine was
reacting with and
was selective for Cys 146.
Figure 2A is the deconvoluted mass spectrum of the reaction of TS with a pool
of 10 different
ligand candidates with little or no binding affinity for TS. In the absence of
any binding
interactions, the equilibrium in the disulfide exchange reaction between TS
and an individual ligand
candidate is to the unmodified enzyme. This is schematically illustrated by
the following equation.
TS-Cysl.~s-SH + R~~N~~~H2 ~ TS-Cys~ns-SS~/N~Rc -~- TS-Cyslas-SS~H2
H ~ ~O
As expected, the peak that corresponds to the unmodified enzyme is one of two
most prominent
peaks in the spectrum. The other prominent peak is TS where the thiol of Cys
146 has been
modified with cysteamine. Although this species is not formed to a significant
extent for any
individual library member, the peak is due to the cumulative effect of the
equilibrium reactions for
each member of the library pool. When the reaction is run in the presence of a
thiol-containing
reducing agent such as 2-mercaptoethanol, the active site cysteine can also be
modified with the
reducing agent. Because cysteamine and 2-mercaptoethanol have similar
molecular weights, their
respective disulfide bonded TS enzymes are not distinguishable under the
conditions used in this
experiment. The small peaks on the right correspond to discreet library
members. Notably, none of
these peaks are very prominent. Figure 2A is characteristic of a spectrum
where none of the ligand
candidates possesses an inherent binding affinity for the target.
8
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
Figure 2B is the deconvoluted mass spectrum of the reaction of TS with a pool
of 10 different
ligand candidates where one of the ligand candidates possesses an inherent
binding affinity to the
enzyme. As can be seen, the most prominent peak is the one that corresponds to
TS where the thiol
of Cys146 has been modified with the N tosyl-D-proline compound. This peak
dwarfs all others
including those corresponding to the unmodified enzyme and TS where the thiol
of Cys146 has
been modified with cysteamine. Figure 2B is an example of a mass spectrum
where tethering has
captured a moiety that possesses a strong inherent binding affinity for the
desired site.
The representative tethering experiments of Figure 2 were performed on a TBM
that already
possessed a naturally occurring cysteine at a site of interest (Cys146 located
in the active site of the
E. coli TS enzyme). However, because TBMs do not always possess a naturally
occurring cysteine
at or near a site of interest, the present invention provides cysteine mutant
variants of TBMs as well
as methods for making the same.
Thus, in one aspect of the present invention, a set comprising at least one
cysteine mutant of a
protein TBM is provided wherein a naturally occurring non-cysteine residue at
or near a site of
interest is mutated to a cysteine residue. In one embodiment, the set
comprises a plurality of
cysteine mutants of a protein TBM wherein each mutant has a different
naturally occurring non-
cysteine residue that is mutated to a cysteine residue. In another embodiment,
the set comprises at
least three cysteine mutants of a protein TBM wherein each mutant has a
different naturally
occurring non-cysteine residue that is mutated to a cysteine residue. In yet
another embodiment,
the set comprises at least five cysteine mutants of a protein TBM wherein each
mutant has a
different naturally occurring non-cysteine residue that is mutated to a
cysteine residue. In still yet
another embodiment, the set comprises at least ten cysteine mutants of a
protein TBM wherein each
mutant has a different naturally occurnng non-cysteine residue that is mutated
to a cysteine residue.
In another aspect of the present invention, methods are provided for
identifying residues that are
suitable for mutating into cysteines. In preferred embodiments, a model or an
experimentally
derived three-dimensional structure (e.g., X-ray or 3D NMR) of a TBM is used
to help identify
residues that are suitable for mutating into cysteines. If a structure of the
TBM of interest in
unavailable, then a three-dimensional structure of a related or homologous TBM
can be used as a
stand-in. Once suitable residues are identified using the stand-in structure,
then methods lrnown in
the art, such as sequence alignment, are used to identify the corresponding
residues in the TBM of
interest. In general, the methods described below for identifying suitable
residues for mutating into
cysteines can be used alone or in any combination with each other.
9
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
In one method, the local backbone conformation of a candidate residue is
determined and a
database of experimentally solved structures is searched for examples of a
disulfide-bonded
cysteine having the same or similar local backbone conformation as the
candidate residue. Any
combination of a residue's backbone atoms (N, Ca, C and O) can be used to
determine the local
conformation. The likelihood that the TBM accepts the cysteine mutation
improves as more
examples are found in a database of known disulfide-bonded cysteines in the
same or similar local
backbone conformation. Experimentally solved structures are available from
many sources
including the Protein Databank ("PDB") which can be found on the Internet at
http://www.rcsb.or
and the Protein Structure Database which can be found on the Internet at
http://www.pcs.com. Lists
of unique, high-resolution protein chains (grouped by structures having a
certain resolution and R-
factor) that can be used to compile a database of experimentally solved
structures are found on the
Internet at htt~//www fccc edu/research/labs/dunbrack/culledpdb.htinl. In
general, the local
environment of a candidate residue includes the candidate residue itself and
at least one residue
preceding or following the candidate residue in sequence. A conformation is
considered the same
or similar if the root mean square deviation ("RMSD") of the atoms being
compared is less than or
equal to aboutl.0 Angstrom2, more preferably, less than or equal to about 0.75
Angstromz, and even
more preferably, less than or equal to about 0.5 Angstrom2.
In one embodiment, the method comprises:
a) obtaining a set of coordinates of a three dimensional structure of a
protein TBM
having n number of residues;
b) selecting a candidate residue i on the three dimensional structure of the
TBM
wherein the candidate residue i is the ith residue where i is a number between
1 and h and residue i
is not a cysteine;
c) selecting a residue j where residue j is adjacent to residue i in sequence;
d) determining a candidate reference value wherein the candidate reference
value is a
spatial relationship between residue i and residue j;
e) obtaining a database comprising sets of coordinates of disulfide-containing
protein
fragments wherein each fragment comprises at least a disulfide-bonded cysteine
and a first adjacent
residue where the disulfide-bonded cysteine and the first adjacent residue
share the same sequential
relationship as residue i and residue j;
f) determining a comparative reference value for each fragment wherein the
comparative reference value is the corresponding spatial relationship between
the disulfide-bonded
cysteine and the first adjacent residue as the candidate reference value is
between residue i and j;
and,
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
g) determining a score wherein the score is a measure of the number of
fragments in
the database that possess a comparative reference value that is the same or
similar to the candidate
reference value.
In another embodiment, the method further comprises
selecting a residue k where residue Ic is adjacent to residue i in sequence
and 7t is not j; and
wherein
the candidate reference value is a spatial relationship between residue i,
residue j,
and xesidue k;
each fragment comprises at least a disulfide-bonded cysteine, a first adjacent
residue, and a second adjacent residue where the disulfide-bonded cysteine and
the first and second
adjacent residues share the same sequential relationship as residue i, residue
j, and residue lc; and
the comparative reference value is the corresponding spatial relationship
between
the disulfide bonded cysteine, the first adjacent residue, and the second
adjacent residue as the
candidate reference value is between residue i, residue j, and residue k.
In another embodiment, the method comprises:
a) obtaining a set of coordinates of a three dimensional structure of a
protein TBM
having ra number of residues;
b) selecting a candidate residue i on the three dimensional structure of the
TBM
wherein the candidate residue i is the ith residue where i is a number between
1 and iz and residue i
is not a cysteine;
c) selecting residue j and residue Ic wherein residue j and residue Jz are
both adjacent in
sequence to residue i;
2,5 d) determining a candidate reference value wherein the candidate reference
value is a
spatial xelationship of at least one backbone atom from each of residue i,
residue j, and residue k;
e) obtaining a database comprising sets of coordinates of disulfide-containing
protein
fragments wherein each fragment comprises at least a disulfide-bonded
cysteine, a first adjacent
xesidue, and a second adjacent residue where the disulfide-bonded cysteine,
the first adjacent
residue, and the second adjacent residue share the same sequential
relationship as residue i, residue
j, and residue k;
f) determining a comparative reference value for each fragment wherein the
comparative reference value is the corresponding spatial relationship between
the disulfide-bonded
cysteine, the first adjacent residue, and the second adjacent residue as the
candidate reference value
is between residue i, residue j, and residue k; and,
11
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
g) determining a score wherein the score is a measure of the number of
fragments in
the database that possess a comparative reference value that is the same or
similar to the candidate
reference value.
In another embodiment the spatial relationship comprises a dihedral angle. In
yet another
embodiment, the spatial relationship comprises a pair of phi psi angles. In
another embodiment, the
spatial relationship comprises a distance between atoms of two residues. An
illustrative example of
a computer algorithm for identifying disulfide bonded pairs in a database such
as the PDB and
matching them with a residue that is a candidate far cysteine mutation is
described in Example 1.
In another method, a site of interest is defined on a TBM and suitable
residues for cysteine mutation
are identified based on the location of the residue from the site of interest.
In one embodiment, a
suitable residue is a non-cysteine residue that is located within the site of
interest. In another
embodiment, a suitable residue is a non-cysteine residue that is located
within about 5 t~ from the
site of interest. In yet another embodiment, a suitable residue is a non-
cysteine residue that is
located within about 10 ~ from the site of interest. For the purposes of these
measurements, any
non-cysteine residue having at least one atom falling within about 5 ~ or
about 10 1~ respectively
from any atom of an amino acid within the site of interest is a suitable
residue for mutating into a
cysteine. A TBM can have one or multiple sites of interests. In some cases, a
TBM has one site of
interest and the set of residues that are each being mutated to a cysteine is
clustered around this site
of interest. In other cases, a TBM has at least two different sites of
interest and the set of residues
that are each being mutated to a cysteine is clustered around the at least two
different sites of
interest. Still in other cases, a TBM either does not possess a distinct site
of interest or possesses
multiple sites of interests such that the set of residues that are being
mutated to a cysteine is
dispersed throughout the protein surface. Figure 3 shows three illustrative
examples of the
distribution pattern of the residues that are each mutated to a cysteine
Tn another method, solvent accessibility is calculated for each non-cysteine
residue of a TBM and
used to identify suitable residues for cysteine mutation. Solvent
accessibility can be calculated
using any number of known methods including using standard numeric methods
(Lee, B. &
Richards, F. M. J. Mol. Biol 55:379-400 (1971); Shrake, A. & Rupley, J. A. J.
Mol. Biol. 79:351-
371 (1973)) and analytical methods (Connolly, M. L. Science 221:709-713
(1983); Richmond, T. J.
J. Mol. Biol. 178:63-89 (1984)). In one embodiment, suitable residues for
mutation include
residues where the combined surface area of the residue's atoms is equaled to
or greater than about
3 S 20 h2. In another embodiment, suitable residues for mutation include
residues where the combined
surface area of the residue's atoms is equaled to or greater than about 30
t~z. In yet another
12
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
embodiment, suitable residues for mutation include residues where the combined
surface area of the
residue's atoms is equaled to or greater than about 40 A2.
In another method, suitable residues for cysteine mutation are identified by
hydrogen bond analysis.
Tn one embodiment, a suitable residue is a non-cysteine residue that does not
participate in any
hydrogen bond interaction. In another embodiment, a suitable residue is a non-
cysteine residue
whose side chain does not participate in any hydrogen bond interaction. Tn yet
another
embodiment, a suitable residue is a non-cysteine residue whose side chain does
not participate in a
hydrogen bond interaction with a backbone atom.
In another method, suitable residues for cysteine mutation are identified by
rotamer analysis. In
one embodiment, the method comprises:
a) obtaining a three dimensional structure of a TBM having n number of
residues and
a site of interest;
b) selecting a candidate residue i that is at or near the site of interest
wherein the
candidate residue i is the ith residue where i is a number between 1 and n and
residue i is not a
cysteine;
c) generating a set of mutated TBM structures wherein each mutated TBM
structure
possesses a cysteine residue instead of residue i and wherein the cysteine
residue is placed in a
standard rotamer conformation; and,
d) evaluating the set of mutated TBM structures.
In another embodiment, a standard rotamer conformation for cysteine comprises
the set of cysteine
rotamers enumerated by Ponders and Richards as described by Ponder, J. W. and
Richards, F. M. J.
Mol. Biol. 193: 775-791 (1987).
In another embodiment, a standard rotamer conformation for cysteine comprises
a chil angle
selected from the group consisting of about 60°, about 180°, and
about 300° and a chit angle
selected from the group consisting of about 60°, about 120°,
about 180°, about 270°, and about
300°.
In another embodiment, the method further comprises determining whether
residue i is part of an a-
helix or a (3-sheet and then selecting a standard rotamer conformation based
on the assigned
secondary structure. As shown in Figure 4, a different set of rotamers is
preferred depending on the
secondary structure that is assigned to the cysteine. Residue i is considered
to be part of an a-helix
if the phi psi angles of residues i-1, i, and i-~-1 are about 30030°
and 31530° respectively, and is
considered to be part of a (3-sheet if the phi psi angles of residues i-1, i,
and i+1 are about 24030°
13
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
and 12030°. If residue i is part of an a-helix, then a standard rotamer
conformation for cysteine
comprises a chil chit pair selected from the group consisting of about
180° and about 60°; about
180° and about 270°; and about 300° and about
300°. If residue i is part of an (3-helix, there a
standard rotamer conformation for cysteine comprises a chil chit pair selected
from the group
consisting of about 180° and about 60°; about 180° and
about 180°; about 180° and about 270°; and
about 300° and about 300°.
In another embodiment, the set of mutated TBM structures are evaluated based
upon whether an
unfavorable steric contact is made. A residue is considered to be a suitable
candidate for cysteine
mutation if it can be substituted with at least one cysteine rotamer for which
no unfavorable steric
contact is made. An unfavorable steric contact is defined as interatomic
distances that are less than
about 80% of the sum of the van der Waals radii of the participating atoms. In
one variation, only
the backbone atoms of the TBM are considered for the purposes of determining
whether the
rotamers malce an unfavorable contact with the TBM. In another variation, the
backbone atoms and
C~ of the TBM are considered for the purposes of determining whether the
rotamers make an
unfavorable contact with the TBM.
In another embodiment, the set of mutated TBM structures are evaluated based
on a force field
calculation. Illustrative force field methods are described by, for example,
Weiner, S. J. et al. .J.
Comput. Chem. 7: 230-252 (1986); Nemethy, G. et al. J. Phys. Chem. 96: 6472-
6484 (1992); and
Brooks, B.R, et al. J. Conaput. Cherra. 4: 187-217 (1983). All minimized
conformations within
about 10 kcal/mol or more preferably within about 5 kcal/mol, of the lowest-
energy conformation
are considered accessible.
In another embodiment, each mutated TBM structure possesses a cysteine that is
capped with a S-
methyl group (side chain is -CHzSSCH3) instead of residue i and wherein the
capped cysteine
residue is placed in a standard rotamer conformation for cysteine. A residue
is considered to be a
suitable candidate for cysteine mutation if it can be substituted with at
least one rotamer that places
the methyl carbon of the S-methyl group closer to the site of interest than
the C~,
In addition to adding one or more cysteines to a site of interest, it may be
desirable to delete one or
more naturally occurring cysteines (and replacing them with alanines for
example) that are located
outside of the site of interest. These mutants wherein one or more naturally
occurring cysteines are
deleted or "scrubbed" comprise another aspect of the present invention.
Various recombinant,
chemical, synthesis and/or other techniques can be employed to modify a target
such that it
possesses a desired number of free thiol groups that are available for
tethering. Such techniques
include, for example, site-directed mutagenesis of the nucleic acid sequence
encoding the target
14
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
polypeptide such that it encodes a polypeptide with a different number of
cysteine residues.
Particularly preferred is site-directed mutagenesis using polymerase chain
reaction (PCR)
amplification (see, for example, U.S. Pat. No. 4,683,195 issued 28 July 1987;
and Current Protocols
In Molecular Biology, Chapter 15 (Ausubel et al., ed., 1991). Other site-
directed mutagenesis
techniques are also well known in the art and are described, for example, in
the following
publications: Ausubel et al., supra, Chapter 8; Molecular Cloning: A
Laboratory Manual., 2nd
edition (Sambrook et al., 1989); Zoller et al., Methods Enzymol. 100:468-500
(1983); Zoller &
Smith, DNA 3:479-488 (1984); Zoller et al., Nucl. Acids Res., 10:6487 (1987);
Brake et al., Proc.
Natl. Acad. Sci. USA 81:4642-4646 (1984); Botstein et al., Science 229:1193
(1985); Kunkel et al.,
Methods Enzymol. 154:367-82 (1987), Adelman et al., DNA 2:183 (1983); and
Carter et al., Nucl.
Acids Res., 13:4331 (1986). Cassette mutagenesis (Wells et al., Gene, 34:315
[1985]), and
restriction selection mutagenesis (Wells et al., Philos. Trans. R. Soc. London
SerA, 317:41S [1986])
may also be used.
Amino acid sequence variants with more than one amino acid substitution may be
generated in one
of several ways. If the amino acids are located close together in the
polypeptide chain, they may be
mutated simultaneously, using one oligonucleotide that codes for all of the
desired amino acid
substitutions. If, however, the amino acids are located some distance from one
another (e.g.
separated by more than ten amino acids), it is more difficult to generate a
single oligonucleotide
that encodes all of the desired changes. Instead, one of two alternative
methods may be employed.
In the first method, a separate oligonucleotide is generated for each amino
acid to be substituted.
The oligonucleotides are than annealed to the single-stranded template DNA
simultaneously, and
the second strand of DNA that is synthesized from the template will encode all
of the desired amino
acid substitutions. The alternative method involves two or more rounds of
mutagenesis to produce
the desired mutant.
The invention is further illustrated by the following, non-limiting examples.
Unless otherwise
noted, all the standard molecular biology procedures are performed according
to protocols
described in (Molecular Cloning: A Laboratory Manual, vols. 1-3, edited by
Sambrook, J., Fritsch,
E.F., and Maniatis, T., Cold Spring Harbor Laboratory Press, 1989; Current
Protocols in Molecular
Biology, vols. 1-2, edited by Ausbubel, F., Brent, R., Kingston, R., Moore,
D., Seidman, J.G.,
Smith, J., and Struhl, K., Wiley Interscience, 1987).
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
EXAMPLE 1
This example provides an illustrative computer algorithm written in FORTRAN
for identifying
disulfide pairs from the PDB that align with potential tethering mutants. A
stepwise description of
the program and the source code are described below.
First, a user supplies the name of the PDB file for the template protein, the
residues of the fragment
to match, and the relative position of the cysteine within that fragment.
Preferred values are 1-2
residues N- and C-terminal to a potential mutant site. For example, if residue
Glu 200 of PTP1B is
a candidate residue, then the user would specify the fragment from residues
198 to 202 with the
cysteine at relative position 3.
Second, the program reads the template file, extracts the coordinates of the
N,Ca,C,O atoms for the
template residues, and determines the values of ~ (C'-N-Ca C torsion) and ~ (N-
Ca C-N') for each
of the template residues
Third, the program generates a "residue filter" based on the template ~/~
values. This filter is used
to identify contiguous segments of a test protein that have ~/~r values
matching those of the
template residues to within a coarse (~60°) tolerance. The filter also
requires that the fragment must
contain a cysteine at the appropriate position. In the PTP1B example above,
the filter would
identify 5-residue fragments of a test protein that roughly matched the
backbone conformations of
residues 198-202 of PTP1B and contained a cysteine in position 3.
Fourth, the rest of the program operates iteratively on a user-supplied list
of test proteins provided
in a simple text file. In one embodiment, this file contains approx. 2500
culled PDB chains. For
each test structure:
a) The program reads the coordinates, determines the sequence and ~/1~ values
for
each residue, and identifies any contiguous chains that match the residue
filter specified in step (3).
b) The program checks to see that the cysteine residue in this fragment is
participating
in a disulfide bond. This is done by simple distance-and angle-based searching
from the Sr atom.
Fragments containing unpaired cysteines are rejected.
c) For each fragment, the N,Ca,C,O atoms of the backbone are overlaid onto the
corresponding atoms from the template molecule (e.g. 198-202 of PTP1B). If the
backbone fits
with an RMSD within a user-specified tolerance (typically 0.5-0.75 A), the
overlaid coordinates of
this fragment along with its disulfide-bound partner are written to a file in
PDB format. A log file is
maintained of each "hit", along with its RMSD value. The hits are viewed with
a graphic program
like Insight II or PyMOL.
16
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
Source Code
P
c
parameter(MAX HITS = 10000)
S C
$INCLUDE tk.inc
$INCLUDE tk functions.inc
$INCLUDE rsm.inc
$INCLUDE rsm functions.inc
c
Record /hndlrec/ data handle, fragment handle, template handle
Record /atom rec/ AtomRec
Record /res rec/ ResRec
Record /res filter/ FragmentFilter(MAX_RMS ATOMS),
1S TemplateFilter(MAX_RMS_ATOMS)
Record /vec/ TemplateVecArray, FragmentVecArray, T1, T2
Dimension TemplateVecArray(MAX_RMS ATOMS),
FragmentVeCArray(MAX_RMS ATOMS)
c
-Integer*4 numTemplateRes, TemplateResList(MAX HITS), numHitRes,
HitResList(MAX_HITS), numTemplateVec,
CysIndex, FrameIndex, numSS, SS-1(MAX_RES), SS-2(MAX-RES),
min element, max_element, num_res,
icnt, jcnt, numFragAtom, FragAtomList(MAX RES),
2.S FragAtomIndex(MAX_RES),ires, fires, icys, cys_idx, jcys,
iatom, jatom, LISTin, PDBout, LOGout, len_name, len_root
Real*8 temp min, temp max, R2(3, 3), RMS cutoff, RMSwalue,
RMS_WT(MAX_RES), angle_tol
Character listfile*80, full name*80, file-path*80, file name*80,
file_root*80, file_ext*80,
structure_name*15, full_structure name*23, first resnumber*7,
charl*1, char3*1, dine*80,
token*80
C
3S LISTin = 9
PDBout = 10
LOGout = 11
FrameIndex = 1
RMS_cutoff = 0.5
angle tol = 60.
do fires = 1, MAX_RES
RMS_WT(ires) - 1.0
end do
c
4S c...Get template information.
c
write (6,'(/, " Enter template PDB filename : " ,$)')
read (5,'(a)') tline
if (.not.readPDBFile(tline, template_handle)) then
S0 write (6,'(" ERROR: Unable to read template PDB file ***** " )')
return
end if
if (get num total residues(template handle, num_res)) continue
c...get template residue numbers and convert to residue indeces
S5 10 write (6,'(5x, " Enter beginning, ending template residues . " ,$)')
read (5,'(a)') tline
if (.not.get token(tline, token)) goto 10
do icnt = 1, num_res
if (getResData(template_handle, FrameIndex, icnt, ResRec))
60 continue
1'7
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
if (ljust(ResRec.residue number)) continue
if (compstr(ResRec.residue-number, token)) then
fires = icnt
goto 20
$ end if
end do
write (6, ' ( "ERROR: Unable to find residue ", a50) ' ) token
goto 10
20 if (.not.get token(tline, token)) goto 10
do icnt = l,~num_res
if (getResData(template handle, FrameIndex, icnt, ResRec))
continue
if (ljust(ResRec.residue number)) continue
if (compstr(ResRec.residue number, token)) then
1$ fires = icnt
goto 30
end if
end do
write (6,'(" ERROR: Unable to find residue " ,a50)') token
goto to
continue
c
numTemplateRes = fires - fires + l
do icnt = Z, numTemplateRes
2$ TemplateResList(icnt) = fires + icnt-1
end do
if (numTemplateRes .eq. 1) then
cys-idx = 1
else
30 write (6,'(5x, " Enter relative position of cysteine : " $)')
read(5,*) cys idx
end if
write (6, ' (5x, ' ' Enter the RMS cutoff : ' ' , $) ' )
read (5,*) RMS cutoff
3$ c -
c...Collect template residue atoms for fitting (N/CA/C/O).
c
numTemplateVec = 0
do icnt = 1, numTemplateRes
fires = TemplateResList(icnt)
if (.not.getAtomOfRes(template handle, FrameIndex, fires, 'N',
AtomRec)) then
write (6,'(" ERROR: Unable to get N of template residue
",i4)') fires
4$ call exit
else
numTemplateVec = numTemplateVec + 1
TemplateVecArray(numTemplateVec) = AtomRec.vector
end if
if (. not.getAtomOfRes(template handle, Framelndex, fires, 'CA',
AtomRec)) then
write (6,'(" ERROR: Unable to get CA of template residue
" ,i4)') fires
call exit
$$ else
numTemplateVec = numTemplateVec + 1
TemplateVecArray(numTemplateVec) = AtomRec.vector
end i f
if (.not.getAtomOfRes(template handle, FrameIndex, fires, 'C',
AtomRec)) then
18
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
write (6,'(" ERROR: Unable to get C of template residue
" , i4 ) ' ) fires
call exit
else
$ numTemplateVec = numTemplateVec + 1
TemplateVecArray(numTemplateVec) = AtomRec.vector
end if
if (.not.getAtomOfRes(template_handle, FrameIndex, fires, 'O',
AtomRec)) then
write (6,'(" ERROR: Unable to get 0 of template residue
" , i4 ) ' ) fires
call exit
else
numTemplateVec = numTemplateVec + 1
1$ TemplateVecArray(numTemplateVec) = AtomRec.vector
end if
end do
C
c...Construct residue filter based on internal angles from the template.
c
if (.not. initializeResFilter(FragmentFilter, MAX_RMS_ATOMS)) then
write(6, '(2X, " ERROR: Unable to make residue-filter
record" ) r )
call exit
end if
FragmentFilter(1).seq-len = numTemplateRes
FragmentFilter(1).start_residue = 2
do icnt = l, numTemplateRes
fires = TemplateResList(icnt)
if (.not.GetR.esData(template'handle, FrameTndex, fires, ResRec))
then
fires
write (6,'(" ERROR: Unable to get record for residue " ,i4)')
call exit
3$ end if
FragmentFilter(icnt).phi_val = ResRec.phi_val
FragmentFilter(icnt).phi_tol = angle tol
FragmentFilter(icnt).psi_val = ResRec.psi_val
FragmentFilter(icnt).psi_tol = angle_tol
end do
FragmentFilter(cys_idx).residue name = 'CYS'
if (returnTrajectory(template_handle)) continue
C
call getenv ('RSM PDB_LISTFILE', listfile)
4$ if (listfile.eq.'~') then
write (6,'(/, " Enter structure listfile : " ,$)')
read (5,'(a)') listfile
end if
open (file=listfile, unit=LISTin, status="old")
SO C
write (6,'(/, " Enter output logfile : " ,$)~)
read (5,'(a)') tline
open (file=tline, unit=LOGout, status="unknown")
write (6,'(" Enter output PDBfile : ",$)')
$$ read (5,'(a)') tline
open (file=tline, unit=PDBout, status="unknown')
C
c...Main loop
c
60 50 read (LISTin,'(a)',end=999) full_name
if (full name(1:l).eq.'#') goto 50
19
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
if (parse filename(full name, file_path, file name, file root,
file_ext)) continue
len name = index(file root,' ') - 1
c
if (.not. readPDBFile(full_name, data handle)) then
write(6, '(2X, " **Unable to read -PDB file ")')
go to 100
end if
C
c...Select only fragments containing cysteines.
c
if (selectResByFilter(data handle, FrameIndex, FragmentFilter,
numHitRes, HitResList)) continue
if (numHitRes .eq. 0) goto 100
c
c...Get list of cysteines participating in disulfide bonds.
C
call find_disulfide_pairs(data handle, FrameIndex, MAX_RES, numSS,
SS_1, SS_2)
if (numSS .eq. 0) goto l00
c
c...Loop through fragments. Test whether: (a) cys idx'th residue is
participating in a disulfide and
c (b) whether the fragment has an acceptable RMS overlap with the
template coordinates.
C
do 90, icnt = 1, numHitRes
icys = HitResList(icnt) + cys_idx - 1
jcys = 0
do jcnt = 1, numSS
if (SS_l(jcnt).eq.icys) then
jcys = SS_2 (jcnt)
else if (SS 2(jcnt) .eq.icys) then
joys = SS-1(jcnt)
end if
end do
if (joys .eq. 0) goto 90
c
c...Extract coordinates for RMS test
c
numFragAtom = 0
do jcnt = 1, numTemplateRes
fires = HitResList(icnt) + jcnt - 1
if (.not.getAtomOfRes(data handle, FrameIndex, fires, 'N',
4$ AtomRec)) then
write (6,'(" ERROR: Unable to get N of fragment residue
",i4)') fires
goto 90
else
numFragAtom = numFragAtom + 1
FragAtomList(numFragAtom) = AtomRec.index
end if
if (.not.getAtomOfRes(data handle, FrameIndex, fires, 'CA',
AtomRec)) then
write (6,'(" ERROR: Unable to get CA of fragment residue
",i4)') fires
goto 90
else
numFragAtom = numFragAtom + 1
FragAtomList(numFragAtom) = AtomRec.index
end if
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
if (. not.getAtomOfRes(data handle, FrameIndex, fires, 'C',
AtomRec)) then
write (6,'(" ERROR: Unable to get C of fragment residue
",i4)') fires
goto 90
else
numFragAtom = numFragAtom + 1
FragAtomList(numFragAtom) = AtomRec.index
end if
if (. not.getAtomOfRes(data handle, FrameIndex, fires, '0',
AtomRec)) then
",i4)') fires
write (6,'(" ERROR: Unable to get O of fragment residue
goto 90
else
numFragAtom = numFragAtom + 1
FragAtomList(numFragAtom) = AtomRec.index
end if
do iatom = Z, numFragAtom
jatom = FragAtomList(iatom)
if (. not.getAtomData(data-handle, FrameIndex, jatom,
AtomRec)) then
write (6,'(" ERROR: Unable to get record for fragment
atom ",i6)') jatom
goto 90
else
FragmentVecArray(iatom) = AtomRec.vector
end if
end do
end do
C
c...RMS Fit to template.
C
call RMS_FIT(numTemplateVec, TemplateVecArray, FragmentVecArray,
RMS WT, RMS_VALUE, t1, t2, r2)
t2.x = -1.0 * t2.x
t2.y = -1.0 * t2.y
t2.z = -1.0 * t2.z
if (RMS VALUE ,gt. RMS cutoff) goto 90
c
c...Extract remaining atoms for fragment.
C
if (. not.getAtomOfRes(data'handle, FrameIndex, icys,'CB',
AtomRec)) then
write (6,'(" ERROR: Unable to get CB of fragment residue
" ,i4)') icys
goto 90
else
numFragAtom = numFragAtom + 1
$0 FragAtomList(numFragAtom) = AtomRec.index
end if
if (. not.getAtomOfRes(data'handle, FrameIndex, icys,'SG',
AtomRec)) then
write (6,'(" ERROR: Unable to get CB of fragment residue
",i4)') icys
goto 90
else
numFragAtom = numFragAtom + 2
FragAtomList(numFragAtom) = AtomRec.index
end if
21
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
if (. not.getAtomOfRes(data_handle, FrameIndex, jcys, 'CA',
AtomRec)) then
write (6,'(" ERROR: Unable to get CA of fragment residue
",i4)') joys
S goto 90
else
numFragAtom = numFragAtom + 1
FragAtomList(numFragAtom) = AtomRec.index
end if
if (. not.getAtomOfRes(data_handle, FrameIndex, jcys, 'CB',
AtomRec)) then
write (6,'(" ERROR: Unable to get CB of fragment residue
",i4)') jcys
goto 90
1S else
numFragAtom = numFragAtom + 1
FragAtomList(numFragAtom) = AtomRec.index
end if
if (. not.getAtomOfRes(data_handle, FrameIndex, jcys, 'SG',
2,0 AtomRec) ) then
write (6,'(" ERROR: Unable to get CB of fragment residue
",i4)') joys
goto 90
else
ZS numFragAtom = numFragAtom + 1
FragAtomList(numFragAtom) = AtomRec.index
end if
call index_int_array(numFragAtom, FragAtomList, FragAtomIndex)
call reorder int array(numFragAtom, FragAtomList, FragAtomIndex)
30 c
c...Construct fragment object and apply transformations.
C
if (getResData(data handle, 1, icys, ResRec)) continue
if (ResRec.ChainID.ne.' ') then
35 first_resnumber = ResRec.ChainID //
ResRec.residue_number(1:6)
else
first_resnumber = ResRec.residue_number(1:6)
end if
40 full_structure_name =
file root(l:len name)//'-'//first resnumber
c
if (make trj_from-atom_list(data handle, INT ONE, INT ONE,
numFragAtom, FragAtomList,
45 . fragment handle)) continue
call rsm_translate_frame(fragment_handle, INTONE, t2)
call rsm_rotate_frame(fragment handle, INT ONE, r2)
call rsm_translate_frame(fragment_handle, INTONE, t1)
call append_fragment(fragment handle, full_structure name,
SO PDBout, .FALSE.)
write (LOGout,'(a22,1x,f5.2)') full_structure name, RMS_value
if (returnTrajectory(fragment handle)) continue
c
90 end do
SS 100 if (returnTrajectory(data~handle)) continue
goto 50
999 close(LISTin)
close(PDBout)
close(LOGout)
60 call exit
end
22
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
EXAMPLE 2
CLONING AND MUTAGENESIS OF HUMAN IL-2
Interleukin-2 (IL-2) (accession number SWS P01585) is a cytolcine with a
predominant role in the
proliferation of activated T helper lymphocytes. Mitogenic stimuli or
interaction of the T cell
receptor complex with antigen/MHC complexes on antigen presenting cells causes
synthesis and
secretion of IL-2 by the activated T cell, followed by clonal expansion of the
antigen-specific cells.
These effects are known as autocrine effects. In addition, IL-2 can have
paracrine effects on the
growth and activity of B cells and natural killer (NK) cells. These outcomes
are initiated by
interaction of IL-2 with its receptor on the T cell surface. Disruption of the
IL-2/IL-2R interaction
can suppress immune function, which has a number of clinical indications,
including graft vs. host
disease (GVHD), transplant rejection, and autoimmune disorders such as
psoriasis, uveitis,
rheumatoid arthritis, and multiple sclerosis. There is structural information
available of the C12SA
mutant [3INK, Mc Kay, D. B. & Brandhuber, B. J., Science 257: 412 (1992)].
Cloning of Human IL-2
Numbering of the wild type and mutant IL-2 residues follows the convention of
the first amino acid
residue (A) of the mature protein being residue number 1 independent of any
presequence e.g. met
for the E. coli produced protein [see Taniguchi, T., et al., Nature 302: 305-
310 (1983) and Devos,
R., et al., Nucleic Acids Res. 11: 4307-4323 (1983)].
The DNA sequence encoding human Interleukin-2 (IL-2) was isolated from plasmid
pTCGF-11
(ATCC). PCR primers were designed to contain resfiriction endonuclease sites
NdeI and XhoI for
subcloning into a pRSET expression vector (Invitrogen).
IL2 GGAATTCCATATGGCACCTACTTCAAGTTCTACAAAGAAAACA SEQID NO:1
Forward
IL2 CCGCTCGAGTCAAGTTAGTGTTGAGATGATGCTTTGACA SEQ ID NO:2
Reverse
Double-stranded IL-2/pRSET was prepared by the following procedure. The PCR
product
containing the IL-2 sequence and pRSET were both cut with restriction
endonucleases (1 ~l PCR
product, 1 p1 each endonuclease, 2 pM appropriate lOx buffer, 15 ~,l water;
incubated at 37 C for 2
hours). The products of nuclease cleavage were isolated from an agarose gel
(1% agarose, TAE
buffer) and ligated together using T4 DNA Iigase (80 ng IL-2 sequence, 160 ng
pRSET vector, 4 ItI
SX ligase buffer [300 mM Tris pH 7.5, 50 mM MgCl2, 20% PEG 8000, 5 mM ATP, S
mM DTT], 1
p,1 ligase; incubated at 15 C for 1 hour). 10 ~,l of the ligase reaction
mixture was transformed into
23
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
XL1 blue cells (Stratagene) (10 ~,l reaction mixture, 10 ~l SX KCM [0.S M KCI,
0.15 M CaCl2,
0.25 M MgCl2], 30 ~l water, SO p1 PEG-DMSO competent cells; incubated at 4 C
for 20 minutes,
25 C for 10 minutes), and plated onto LB/agar plates containing 100 p,g/ml
ampicillin. After
incubation at 37 C overnight, single colonies were grown in 5 ml 2YT media for
18 hours. Cells
were then isolated and double-stranded DNA extracted from the cells using a
Qiagen DNA
miniprep kit.
Generation of IL-2 Cys Mutations
Site-directed mutants of IL-2 were prepared by the single-stranded DNA method
(modification of
Kunkel, T. A., Proc. Natl. Acad. Sci. U. S. A. 83: 488-492 (1985).
Oligonucleotides were designed
to contain the desired mutations and 1 S-20 bases of flanking sequence.
The single-stranded form of the IL-2/pRSET plasmid was prepared by
transformation of double-
stranded plasmid into the CJ236 cell line (1 p1 IL-2/pRSET double-stranded
DNA, 2 ~,1 2x KCM
salts, 7 ~1 water, 10 p1 PEG-DMSO competent CJ236 cells; incubated at 4 C for
20 minutes and 2S
IS C for 10 minutes; plated on LB/agar with 100 pg/ml ampicillin and incubated
at 37 C overnight).
Single colonies of CJ236 ceps were then grown in SO ml 2YT media to midlog
phase; S p,1 VCS
helper phage (Stratagene) were then added and the mixture incubated at 37 C
overnight. Single-
stranded DNA was isolated from the supernatant by precipitation of phage (1/5
volume 20% PEG
8000/2.5 M NaCI; centrifuge at 12K for 1 S minutes.). Single-stranded DNA was
then isolated from
phage using Qiagen single-stranded DNA kit. Sequencing identified a leucine-2S
to serine
mutation, which was corrected by mutagenesis using the "SZSL" oligonucleotide.
525L TAATTCCATTCAAAATCATCTGTA SEQ ID NO:3
Mutagenic Oligonucleotides
N30C GGTGAGTTTGGGATTCTTGTAACAATTAATTCCATTCAAAATCATCTG SEQID NO:4
Y31C GGTGAGTTTGGGATTCTTACAATTATTAATTCCATTC SEQID NO:S
K32C GGTGAGTTTGGGATTACAGTAATTATTAATTCC SEQ ID NO:6
N33C CCTGGTGAGTTTGGGACACTTGTAATTATTAATTCC SEQID NO:7
K3SC GCATCCTGGTGAGACAGGGATTCTTGTAATTATTAATTCC SEQID NO:8
R38C CTTAAATGTGAGCATACAGGTGAGTTTGGGATTC SEQ ID NO:9
F42C GGGCATGTAAAACTTACATGTGAGCATCCTGG SEQ ID NO:10
24
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
K43C CTTGGGCATGTAAAAACAAAATGTGAGCATCC SEQ ID NO:11
Y4SC GGCCTTCTTGGGCATACAAAACTTAAATGTGAGC SEQ ID NO:12
E68C CTCAAACCTCTGGAGTGTGTGCTAAATTTAGC SEQ ID NO:13
L72C GTTTTTGCTTTGAGCACAA.TTTAGCACTTCCTCC SEQ ID NO:14
N77C CCTGGGTCTTAAGTGAAAACATTTGCTTTGAGCTAAATTTAGC SEQ ID NO:1S
Y31C K43C
GGGCATGTAAAAACAAAATGTGAGCATCCTGGTGAGTTTGGGATTCTTACAATTATTAATTCC
SEQ ID N0:16
S There was an additional double mutant made, L72C K43C, using the
oligonucleotides
corresponding to K43C and L72C single mutants (SEQ ID NO:11 and SEQ ID N0:14
respectively).
Site-directed mutagenesis was accomplished as follows: Mutagenesis
oligonucleotides were
dissolved to a concentration of 10 OD and phosphorylated on the S' end (2 ~1
oligonucleotide, 2 ~l
10 mM ATP, 2 p.1 lOX Tris-magnesium chloride buffer, 1 ~l 100 mM DTT, 10 p.1
water, 1 ~1 T4
PNK; incubate at 37 C for 4S minutes.). Phosphorylated oligonucleotides were
then annealed to
single-stranded DNA template (2 ~l single-stranded plasmid, 1 ~1
oligonucleotide, 1 ~.1 lOx TM
buffer, 6 ~.1 water; heat at 94 C for 2 minutes, SO C for S minutes, cool to
room temperature).
1 S Double-stranded DNA was then prepared from the annealed
oligonucleotide/template (add 2 ~1 10X
TM buffer, 2 ~12.S mM dNTPs, 1 ~l 100 rnM DTT, 1.S ~1 10 mM ATP, 4 ~l water,
0.4 ~l T7 DNA
polymerase, 0.6 ~.1 T4 DNA ligase; incubate at room temperature for 2 hours).
E. eoli (XLl blue,
Stratagene) was then transformed with the double-stranded DNA (1 ~,1 double-
stranded DNA, 10 ~1
Sx KCM, 40 ~tl water, SO ~l DMSO competent cells; incubate 20 minutes at 4 C,
10 minutes at
room temperature), plated onto LB/agar containing 100 ~,g/ml ampicillin, and
incubated at 37 C
overnight. Approximately four colonies from each plate were used to inoculate
S ml 2YT
containing 100 pg/ml ampicillin; these cultures were grown at 37 C for 18-24
hours. Plasmids
were then isolated from the cultures using Qiagen miniprep kit. These plasmids
were sequenced to
determine which IL-2/pRSET clones contained the desired mutation.
2S
Sequencing primers
Forward primer, "T7" AATACGACTCACTATAG SEQ ID N0:17
Reverse primer, "RSET TAGTTATTGCTCAGCGGTGG SEQ ID N0:18
REV"
2S
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
Expression of IL-2 Mutants
Mutant proteins were expressed as follows: IL-2/pRSET clones containing the
mutation were
transformed into BL21 DE3 pLysS cells (Invitrogen) (1 p1 double-stranded DNA,
2 ~,l Sx KCM, 7
Etl water, 10 p.1 DMSO competent cells; incubate 20 minutes at 4 C, 10 minutes
at room
temperature), plated onto LB/agar containing 100 ~g/ml ampicillin, and
incubated at 37 C
overnight. 10 ml cultures in 10 ml 2YT with 100 ~g/ml ampicillin were grown
overnight from
single colonies. 100 ml 2YT/ampicillin (100 ~g/ml) was inoculated with these
overnight cultures
and incubated at 37 C for 3 hours. This culture was then added to 1.5 L
2YT/ampicillin (100
pg/ml) and incubated until late-log phase (absorbance at 600 nm ~0.8), at
which time IPTG was
added to a anal concentration of 1 mM. Cultures were incubated at 37 G for
another 3 hours and
then cells were pelleted (10 Krpm, 10 minutes) and frozen at -20 C overnight.
IL-2 mutants were then purified from the frozen cell pellets. First, cells
were lysed in a
microfluidizer (100 ml Tris EDTA buffer, 3 passes through a Microfluidizer
[Microfluidics 110S])
and inclusion bodies were isolated by precipitation (10 Krpm, 10 minutes).
Following cell lysis, 50
E~1 of cell material was saved for analysis by SDS-PAGE. All mutants expressed
as determined by
gel but several (e.g. E68C) precipitated on refolding. Inclusion bodies were
then resuspended in 45
ml guanidine HCl and spun at 10 Krpm for 10 minutes. The supernatant was added
to refolding
buffer (45 ml guanidine HCI, 36 ml Tris pH 8, 231 mg cysteamine, 46 mg
cystamine, 234 ml water)
and incubated at room temperature for 3-5 hours. The mixture was then spun at
10 Krpm for 20
minutes. and the supernatant dialyzed 4-5 times in 5 volumes of buffer (10 mM
ammonium acetate
pH 6, 25 mM NaCI). The protein solution was then filtered through cellulose
and injected onto an
S Sepharose fast flow column (2.5 cm diameter x 14 cm long) at 5 ml/min. The
protein was then
eluted using a gradient of 0 - 75% Buffer B over 60 minutes (Buffer A: 25 mM
NH40Ac, pH 6, 25
mM NaCI; Buffer B: 25 mM NH4OAc, pH 6, 1 M NaCI). Purified protein was then
exchanged
into the appropriate buffer for the TETHER assay (typically 100 mM Hepes, pH
7.4). Average
yields were 0.5 to 4 mg/L culture.
EXAMPLE 3
CLONING AND MUTAGENESIS OF HUMAN IL-4
IL-4 (accession number SWS P05112) is a cytokine that is critical for early
immune response and
allergic response; its interaction with the IL-4R is involved in the
generation of Th2 cells. IL-4
recruits and activates B-cells that produce IgE (immunoglobulin E),
eosinophils, and mast cells.
These cells in turn tag and attack parasites in skin and in mucosal tissues
and eject them from these
tissues. The role of the IL-4/IL4R interaction in immune and allergic
responses suggests that
disruption of this interaction may alleviate such conditions as asthma,
dermatitis, conjunctivitis, and
26
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
rhinitis. 'There are crystal structures of IL-4 in isolation and in co-complex
with a receptor
molecule [1HIK, Muller, T. & Buehner, M., JMoI Biol 247: 360-372 (1995); with
receptor alpha,
IIAR, Hage, T., et aL, Cell 97: 271-281 (1999)].
Cloning of human IL-4
Numbering of the wild type and mutant IL-4 residues follows the convention of
the first amino acid
residue (H) of the mature protein being residue number 1 independent of any
presequence e.g. met
for the E. coli produced protein [Yokota, T., et al., Proc. Natl. Acad. Sci.
U. S. A. 83: 5894-5898
(1986)]. IL-4 lacking the secretion signal and containing an additional N-
terminal methionine was
expressed intracellularly in E. coli from the Sunesis RSET.IL4 plasmid.
The DNA sequence encoding human interleukin-4 (IL4) was isolated by PCR from
the plasmid
pcD-hIL-4 (ATCC Accession No. 57592) using PCR primers:
IL4 ForRse 5' GGGTTTCATATGCACAAGTGCGATATCACCTT SEQ ID N0:19
IL4 RevRse 5' CCGCTCGAGTCAGCTCGAACACTTTGAATA SEQ ID N0:20
These primers correspond to extracellular domain of the protein and which were
designed to
contain restriction endonuclease sites Nde I and XhoI for subcloning into a
pRSET vector
(Invitrogen). The PCR reaction was purified on a Qiaquick PCR purification
column (Qiagen).
The PCR product containing the IL4 sequence was out with restriction
endonucleases (41 p1 PCR
product, 2 p.1 each endonuclease, 5 p,1 appropriate lOx buffer; incubated at
37 C for 90 minutes).
The pRSET vector was cut with restriction endonucleases (6 ~g DNA, 4 p1 each
endonuclease, 10
p1 appropriate lOx buffer, water to 100 p1; incubated at 37 C for 2 hours; add
2 p,1 CIP and
incubated at 37 C for 45 minutes). The products of nuclease cleavage were
isolated from an
agarose gel (1% agarose, TBE buffer) and ligated together using T4 DNA ligase
(200 ng pRSET
vector, 150 ng IL4 PCR product, 4 p.l Sx ligase buffer [300 mM Tris pH 7.5, 50
mM MgCl2, 20%
PEG 8000, 5 mM ATP, 5 mM DTT], 1 ~1 Iigase; incubated at IS C for 1 hour). 101
of the ligation
reaction was transformed into XL1 blue cells (Stratagene) (10 p1 reaction
mixture, 10 p,1 Sx KCM
[0.5 M KCI, 0.15 M CaCh, 0.25 M MgCl2], 30 p1 water, 50 p1 PEG-DMSO competent
cells;
incubated at 4 C for 20 minutes, 25 C for IO minutes), and plated onto LB/agar
plates containing
100 pg/ml ampicillin. After incubation at 37 C overnight, single colonies were
grown in 3 ml 2YT
media for 18 hours. Cells were then isolated and double-stranded DNA extracted
from the cells
using a Qiagen DNA miniprep kit.
27
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
Generation of IL-4 Cysteine Mutations
Mutations were generated using as previously described [Kunlcel, T. A., et
al., Metlzods Ehzymol.
154:367-82 (1987)]. DNA oligonucleotides used are shown below and were
designed to hybridize
with sense strand DNA from plasmid. Sequences were verified using primers with
SEQ ID N0:17
and SEQ ID N0:18.
Mutagenic Oligonucleotides
QgC TTGATGATCTCACATAAGGTGA SEQ ID NO:21
E9C AGTTTTGATGATACACTGTAAGGTGAT SEQID NO:22
K12C GCTGTTCAAAGTGCAGATGATCTCCTG SEQID NO:23
S16C CTGCTCTGTGAGGCAGTTCAAAGT SEQID NO:24
K37C CAGTTGTGTTACAGGAGGCAGCAAAG SEQID NO:2S
N38C CCTTCTCAGTTGTGCACTTGGAGGC SEQID NO:26
K42C GCAGAAGGTTTCACACTCAGTTGTG SEQID NO:27
QS4C GGCTGTAGAAACACCGGAGCACAGTCG SEQID NO:28
Q78C GAATCGGATCAGACACTTGTGCCTGTG SEQID NO:29
R81C GCCGTTTCAGGAAGCAGATCAGCTGC SEQID NO:3O
R8SC CCTGTCGAGACATTTCAGGAATCG SEQID NO:31
R88C CCCAGAGGTTGCAGTCGAGCCG SEQID NO:32
N89C CCCAGAGGCACCTGTCGAGCCG SEQ ID NO:33
N97C CACAGGACAGGAACACAAGCCCGCC SEQID NO:34
K1O2C CTGGTTGGCTTCACACACAGGACAGG SEQID NO:3S
K117C CTCTCATGATCGTGCATAGCCTTTCC SEQID NO:36
R121C GAATATTTCTCACACATGATCGTC SEQID NO:37
Expression of IL-4 Mutants
BL21 DE3 cells (Stratagene) were transformed with RSET.IL4 plasmids containing
the described
cysteine mutations and plated onto LB agar containing 100 ~.g/ml ampicillin.
After overnight
28
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
growth fresh individual colonies were used to inoculate a 37 C overnight shake
flask culture with
30 ml 2YT (with 50 p,g/ml ampicillin) media. In the morning this overnight
culture was used to
inoculate 1.5 L of 2YT/ampicillin (50 ~.g/ml), which was further cultured at
37 C and 200 rpm in a
4.0 L dented bottom shake flask. When the optical density of the culture at ~,
= 600 reached 0.8 it
was induced to produce IL-4 protein by the addition of 1 mM IPTG. After 4 more
hours of
incubation the cultures were harvested, the cells pelleted by centrifugation
at 7K rpm for 10 minutes
(K-9 Komposite Rotor), and frozen at-20 C.
The cell pellet was then thawed and resuspended in I00 ml of 10 mM Tris pH 8,
50 mM NaCI and
1 mM EDTA. This solution was kept chilled and run through a microfluidizer
twice (model 1105
Microfluidics Corp, Newton Massachusetts), and centrifuged at 7K rpm for 15
minutes). The pellet
containing the IL-4 inclusion bodies was then resuspended in a 50 ml solution
of 5 M guanidine
HCI, 50 mM Tris pH 8, 50 mM NaCI, 2.5 mM reduced glutathione, and 0.25 mM
oxidized
glutathione, and incubated for one hour at room temperature with gentle
mixing. The solubilized
protein solution was then centrifuged at 7.5K rpm for 15 minutes and the
supernatant 0.45 ~m
filtered to remove insoluble debris.
The IL-4 was refolded by slowly adding the filtered solution to 9 volumes (450
ml) of 50 mM Tris
pH 8, 50 mM NaCI, 2.5 mM reduced glutathione and 0.25 mM oxidized glutathione
over a 30
minute period. The resulting solution was further incubated with slow stirring
for 3 hours at room
temperature, then placed in a 3000 mwco dialysis bag and exchanged 3 times
against 20 L of 0.5x
PBS (phosphate-buffered saline).
The refolded mutant proteins were then purified using a Hi-S Column Cartridge
(Bio-Rad). After
clarifying the protein solution by centrifugation and filtration it was loaded
onto the column at a 5
ml/min flow rate. The column was next washed with buffer A (0.5x PBS) for 15-
20 minutes, and
1.5 minute 7.5 ml fractions were collected over a 0-100% gradient between
Buffer A and Buffer B
(PBS, 1M NaCI). The fractions that contained the IL-4 protein as determined by
SDS-PAGE and
optical density as 280 nm were pooled, concentrated with a 5K mwco filter, and
their buffer
exchanged to PBS. This solution was then 0.2 pm filtered, frozen in ethanol
dry ice bath, and
stored at
-80 C.
EXAMPLE 4
CLONING AND MUTAGENESIS OF HUMAN TUMOR NECRO IS FACTOR-ALPHA fTNF
..
29
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
Tumor necrosis factor-a (TNF-a) (accession number SWS P01375) is a cytokine
produced mainly
by activated macrophages, and it plays a critical role in immune responses
including septic shock,
inflammation, and cachexia. This protein can interact with two receptors, TNF
Rl and TNF R2.
These two receptors share no similarity in their intracellular domains, which
suggests that they are
involved in different signal transduction pathways. A structure of TNF-a is
available [1TNF, Eck,
M. J., et al., JBiol ClZem 264: 17595-17605(1989)]; TNF-a is an elongated beta
sheet, and it forms
a trimer. Mutation of some of the intersubunit residues of the trimer
indicates that they form part of
the binding site to the receptor. However, there is no structure of TNF bound
to a receptor to date.
Cloning of human TNF-a
The DNA sequence encoding human Tumor Necrosis Factor (TNF) was isolated by
PCR from the
plasmid pUC-RI-4large (ATCC #65947) using PCR primers listed below
corresponding to
extracellular domain of the protein and which were designed to contain
restriction endonuclease
sites Nde I and XhoI for subcloning into a pRSET vector (Invitrogen).
TNF RSET For GGGTTTCATATGGTCCGTTCATCTTCTCGAAC SEQ ID N0:38
5'
TNF RSET Rev CCGCTCGAGTCACAGGGCAATGATCCCAA SEQ ID N0:39
5'
The PCR reaction was purified on a Qiaquick PCR purification column (Qiagen).
The PCR product
containing the TNF sequence was cut with restriction endonucleases (41 p1 PCR
product, 2 ~l each
endonuclease, 5 ~1 appropriate lOx buffer; incubated at 37 C for 90 minutes).
The pRSET vector
was cut with restriction endonucleases (6 p.g DNA, 4 p.1 each endonuclease, 10
p.1 appropriate lOx
buffer, water to 100 p.1; incubated at 37 C for 2 hours; added 2 p1 CIP and
incubated at 37 C for 45
minutes). The products of nuclease cleavage were isolated from an agarose gel
(1% agarose, TBE
buffer) and ligated together using T4 DNA ligase (200 ng pRSET vector, 150 ng
TNF PCR product,
4 p1 5x ligase buffer [300 mM Tris pH 7.5, 50 mM MgCl2, 20% PEG 8000, 5 mM
ATP, 5 mM
DTT], 1 ~1 ligase; incubated at 15 C for 1 hour). 10 p1 of the ligation
reaction was transformed
into XL1 blue cells (Stratagene) (10 ~,1 reaction mixture, 10 p1 5x KCM [0.5 M
KCI, 0.15 M CaClz,
0.25 M MgCla], 30 ~1 water, 50 ~tl PEG-DMSO competent cells; incubated at 4 C
for 20 minutes,
25 C for 10 minutes), and plated onto LB/agar plates containing 100 p.g/ml
ampicillin. After
incubation at 37 C overnight, single colonies were grown in 3 ml 2YT media for
18 hours. Cells
were then isolated and double-stranded DNA extracted from the cells using a
Qiagen DNA
miniprep kit. Sequencing of TNF genes was accomplished using primers having
SEQ ID N0:17
and SEQ ID NO:18.
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
Generation of TNF-a Cysteine Mutations
Mutations were generated using as previously described [Kunlcel, T, A., et
al., Methods Entzymol.
154: 367-82 (1987)]. DNA oligonucleotides used are shown below and were
designed to hybridize
with sense strand DNA from plasmid. Sequences of the mutants were verified
using primers with
S SEQ ID NO:17 and SEQ ID N0:18.
Mutagenic Oligonucleotides
R32C GAGGGCATTGGCGCAGCGGTTCAGCCAC SEQ ID
NO:40
A33C CAGGAGGGCATTGCACCGGCGGTTCAG SEQID NO:41
N34C GGCCAGGAGGGCACAGGCCCGGCGGTTC SEQID NO:42
R44C CAGCTGGTTATCACACAGCTCCACGCC SEQID NO:43
Q47C TGGCACCACCAGGCAGTTATCTCTCAG SEQID NO:44
T72C GAGGAGCACATGGCAGGAGGGGCAGCC SEQID NO:4S
H73C GGTGAGGAGCACACAGGTGGAGGGGCAG SEQID NO:46
L7SC GGTGTGGGTGAGGCACACATGGGTGGAG SEQID NO:47
T77C GCTGATGGTGTGGCAGAGGAGCACATG SEQID NO:48
V91C CAGAGAGGAGGTTGCACTTGGTCTGGTAG SEQID NO:49
N92C GGCAGAGAGGAGGCAGACCTTGGTCTG SEQID NO:SO
S95C GCTCTTGATGGCACAGAGGAGGTTGAC SEQID NO:Sl
E104C CCTCAGCCCCCTCTGGGGTGCACCTCTGGCAGGGG SEQID NO:S2
T1OSC CCTCAGCCCCCTCTGGGCACTCCCTCTGGCAGGGG SEQID NO:S3
ElO7C GGCCTCAGCCCCGCATGGGGTCTCCCTCTGGC SEQID NO:S4
EIIOC CCAGGGCTTGGCGCAAGCCCCCTCTGGGG SEQID NO:SS
AlllC ATACCAGGGCTTGCACTCAGCCCCCTC SEQID NO:S6
K112C GGGTAGTTTCTGGCAAAATATGGCTTG SEQ ID NO:S7
QI25C CACCCTTCTCCAGGCAGAAGACCCCTCC SEQ ID NO:S$
31
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
R138C GCTGAGATCAATTGTCCCGACTATCTC SEQ ID NO:59
E146C GACCTGCCCAGAGCAGGCAAAGTCGAG SEQ ID N0:60
S147C GTAGACCTGCCCACACTCGGCAAAGTC SEQ ID NO:61
Expression of TNF-a Mutant Proteins
BL21 DE3 cells (Stratagene) were transformed with RSET TNF-a plasmids
containing the
described cysteine mutations and plated onto LB agar containing 100 ~g/ml
ampicillin. After
overnight growth fresh individual colonies were used to inoculate a 37 C
overnight shalce flask
culture with 30 ml 2YT (with 50 ~g/ml ampicillin) media. In the morning this
overnight culture
was used to inoculate 1.5 L of 2YT/ampicillin (50 ~,g/ml), which was further
cultured at 37 C and
200 rpm in a 4.0 L dented bottom shake flask. When the optical density of the
culture at ~. = 550
reached 0.8 it was induced to produce TNF-a protein by the addition of 1 mM
IPTG. After 4 more
hours of incubation the cultures were harvested, the cells pelleted by
centrifugation at 7K rpm for
10 minutes (K-9 Komposite Rotor), and frozen at -20 C.
The cell pellet was then thawed and resuspended in 100 ml of 25 mM ammonium
acetate pH 6, 1
mM DTT and 1 mM EDTA. This solution was kept chilled and run through a
microfluidizer twice
(model 1105 Microfluidics Corp, Newton Massachusetts), centrifuged at 9K rpm
for 15 minutes to
remove insoluble material and further clarified by 0.45 ~m filtration. This
solution was then loaded
onto an S-Sepharose ff Column (Bio-Rad) column at a 5 ml/min flow rate. The
flow rate was then
increased to 7.5 mL/min for the following steps. The column was next washed
with Buffer A (0.2
M ammonium acetate pH 6, 1 mM DTT) until the ODZSO approached zero (15-20
minutes), and
fractions were collected over a 0-100% gradient in 60 minutes between Buffer A
and Buffer B (1 M
ammonium acetate pH 6, 1 mM DTT). The fractions that contained the TNF-a
protein as
determined by SDS-PAGE and optical density at 280 nm were pooled and placed in
a 3000 mwco
dialysis bag and dialyzed overnight at 4 C against 4 L of 10 mM Tris pH 7.5,
10 mM NaCI, and 1
mM DTT. The dialyzed protein solution was then clarified by centrifuging at
13.5K rpm for 10
minutes filtering through a 0.2 ~m filter.
The mutant proteins were then loaded onto a Q-Sepharose Column (Bio-Rad) at a
5 ml/min flow
rate. The flow rate was increased to 7.5 mLlmin for the following steps. The
column was next
washed with Buffer A (10 mM Tris pH 7.5, 10 mM NaCI, 1 mM DTT) until the ODZBO
approached
zero (15-20 minutes), and fractions were collected over a 0-100% gradient in
40 minutes between
Buffer A and Buffer B (10 mM Tris pH 7.5, 0.5 M NaCI, 1 mM DTT). The fractions
that contained
the TNF-a protein as determined by SDS-PAGE and optical density at 280 nm were
pooled and
32
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
concentrated with a SK mwco filter, and their buffer exchanged to PBS. This
solution was then 0.2
pm filtered, frozen in ethanol dry ice bath, and stored at -80 C.
EXAMPLE 5
CLONING AND MUTAGENESIS OF HUMAN INTERLEUKIN-1 RECEPTOR TYPE I fIL 1RI)
Binding of the IL-1 receptor (accession number SWS P14778) to IL-lalpha or IL-
lbeta is another
important mediator of immune and inflammatory responses. This interaction is
controlled by at
least three mechanisms. Firstly, the protein IL-R2 binds to IL-lalpha and IL-
lbeta but does not
signal. Secondly, proteolytically processed IL-1R1 and IL-1R2 are soluble and
bind to IL-1 in
circulation. Finally there exists a natural IL-1R antagonist called IL-lra,
that functions by binding
IL-1R1 and thereby blocking IL-1Rl binding of IL-lalpha and IL-lbeta.
Inhibition of these
interactions with an orally available small molecule would be desirable in
treatment of diseases
such as rheumatoid arthritis, autoimmune disorders, and ischemia. Two
structures of IL-1R have
been solved [with a antagonist peptide, 1GOY, Vigers, G. P. A., et aL, J.
Biol. Cl2em. 275:36927-
36933 (2000); with receptor antagonist, LIRA, Schreuder, H., et al., Nature
386: 194-200 (1997)].
Cloning of human TL-1 receptor type I
The IL-1 receptor has three regions: an N-terminal extracellular region, a
transmembrane region,
and a C-terminal cytoplasmic region. The extracellular region itself contains
three irnmunoglobin
Like C2-type domains. The constructs used here contain the two N-terminal
domains of the
extracellular region. Numbering of the wild type and mutant IL1R residues
follows the convention
of the first amino acid residue (L) of the mature protein being residue number
1 after processing of
the signal sequence [Sims, J. E., et al., Pi~oc. Natl. Acad. Sci. U. S. A. 86:
8946-8950 (1989)]. The
sequence of the 2 domain protein is shown below as SEQ ID N0:62.
1 LEADKCKERE EKIILVSSAN EIDVRPCPLN PNEHKGTITW YKDDSKTPVS TEQASRIHQH
61 KEKLWFVPAK VEDSGHYYCV VRNSSYCLRT KISAKFVENE PNLCYNAQAI FKQKLPVAGD
121 GGLVCPYMEF FKNENNELPK LQWYKDCKPL LLDNIHFSGV KDRLIVMNVA EKHRGNYTCH
181 ASYTYLGKQY PTTRVIEFTT LEENK
In brief, cysteine mutants were made in the context of a 2 domain receptor and
a 2 domain receptor
with a his tag. In addition, the constructs possessed a mutation at a
glycosylation site, and one
construct possessed a mutation at a glycosylation site in addition to a
deletion at the C-terminal
residue of the 2 domain region. The assembly of these constructs is described
below.
The DNA sequence encoding human Interleukin-1 receptor (IL1R) was isolated by
PCR from a
HepG2 cDNA library (ATCC) using PCR primers (ILlRsigintFor 5'; ILlRintRev 5')
corresponding to the signal sequence and the end of the extracellular domain
of the protein.
33
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
ILlRsigintFor TTACTCAGACTTATTTGTTTCATAGCTCTA SEQ ID N0:63
ILlRiritReV GAAATTAGTGACTGGATATATTAACTGGAT SEQ ID NO:C)4
The appropriate sized band was isolated from an agarose gel and used as the
template for a second
round of PCR using oligos (ILlRsigFor; IL1R319Rev), which were designed to
contain restriction
endonuclease sites EcoRI and XhoI for subcloning into a pFBHT vector.
ILlRsig For CCGGAATTCATGAAAGTGTTACTCAGACTTATTTGTTTC SEQ ID
NO:65
IL1R319 Rev CCGCTCGAGTCACTTCTGGAAATTAGTGACTGGATATATTAA SEQ ID
NO:G6
The pFBHT vector is modified from the original pFastBacl(Gibco/BRL) by cloning
the sequence
for TEV protease followed by (His) tag and a stop signal into the XhoI and
HinDIII sites. The
PCR product containing the IL1R sequence was cut with restriction
endonucleases (41 ~tl PCR
product, 2 ~1 each endonuclease, 5 ~tl appropriate lOx buffer; incubated at 37
C for 90 minutes).
The pFBHT vector was cut with restriction endonucleases (6 ~,g DNA, 4 ~,1 each
endonuclease, 10
~l appropriate lOx buffer, water to 100 q1; incubated at 37 C for 2 hours; add
2 ~.l CIP and
incubated at 37 C for 45 minutes). The products of nuclease cleavage were
isolated from an
a.garose gel (1% agarose, TBE buffer) and ligated together using T4 DNA ligase
(200 ng pFBHT
vector, 150 ng IL1R PCR product, 4 ~1 5x ligase buffer [300 mM Tris pH 7.5, 50
mM MgCl2, 20%
PEG 8000, 5 mM ATP, 5 mM DTT], 1 ~1 ligase; incubated at 15 C for 1 hour). 10
~.1 of the
ligation reaction was transformed into XL1 blue cells (Stratagene) (10 ~l
reaction mixture, 10 ~1 Sx
I~CM [0.5 M ICI, 0.15 M CaClz, 0.25 M MgCl2], 30 ~.1 water, 50 ~l PEG-DMSO
competent cells;
incubated at 4 C for 20 minutes, 25 C for 10 minutes), and plated onto LB/agar
plates containing
100 ~g/ml ampicillin. After incubation at 37 C overnight, single colonies were
grown in 3 ml 2YT
media for 18 hours. Cells were then isolated and double-stranded DNA extracted
from the cells
using a Qiagen DNA miniprep kit.
A 2-domain version of IL1R was created by PCR using the 3-domain IL1R-FBHT
clone as a
template. PCR was performed using the primers ILlRsigFor (SEQ ID N0:65)
corresponding to the
signal sequence, in addition to one of the following two reverse primers. The
reverse primers are
ILIR2Drevstop-Xho, which corresponds to the end of the second extracellular
domain of the
protein with a stop signal, and ILIR2Drev-Xho, which corresponds to the end of
the second
34
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
extracellular domain of the protein without a stop signal to create a fusion
with the TEV protease
site and the His tag.
ILIR2Drevstop-Xho CCGCTCGAGTCATCATTTGTTTTCCTCTAGAGTAATAAA SEQID
N0:67
ILIR2Drev-Xho CCGCTCGAGTCATTTGTTTTCCTCTAGAGTAATAAA SEQ ID
N0:68
The PCR primers contain restrictions sites (EcoRI at the 5'end and XhoI at the
3' end), which were
used to ligate the 2-domain version into the pFBHT vector. The PCR product
containing the
IL1R2D sequence was cut with restriction endonucleases (41 p.1 PCR product, 2
~l each
endonuclease, 5 p.1 appropriate lOx buffer; incubated at 37 C for 90 minutes).
The products of
nuclease cleavage were isolated from an agarose gel (1% agarose, TBE buffer)
and ligated together
using T4 DNA ligase (200 ng pFBHT vector, 150 ng IL1R2D PCR product, 4 ~.I 5x
ligase buffer
[300 mM Tris pH 7.5, 50 mM MgCl2, 20% PEG 8000, 5 mM ATP, 5 mM DTT], 1 ~tl
ligase;
incubated at 15 C for 1 hour). 10 ~cl of the ligation reaction was transformed
into XLl blue cells
(Stratagene) (10 ~1 reaction mixture, 10 ~1 Sx KCM [0.5 M KCI, 0.15 M CaCIz,
0.25 M MgCIz], 30
p1 water, 50 p.1 PEG-DMSO competent cells; incubated at 4 C for 20 minutes, 25
C for 10
minutes), and plated onto LB/agar plates containing 100 ~cg/ml ampicillin.
After incubation at 37
C overnight, single colonies were grown in 3 ml 2YT media for 18 hours. Cells
were then isolated
and double-stranded DNA extracted from the cells using a Qiagen DNA miniprep
leit.
Additionally, the two glycosylation sites within IL1R2D, N83 and N176, were
each individually
mutated to a histidine, in order to make a more homogeneous protein. Each of
these single mutants
were made in the context of the 2-domain protein without a his tag (sILlRd2-
FB) and the 2-domain
protein with a his tag (sILlRd2-FBHT). Mutation was accomplished by PCR using
two sets of
primers to make two fragments, followed by stitching together of the fragments
using the outside
primers ILlRsigFor (SEQ ID N0:65) and either ILIR2Drevstop-Xho (SEQ ID N0:67)
or
ILlR2Drev-Xho (SEQ ID N0:68) as described below. Brief descriptions of the 2-
domain
glycosylation mutants and their construction follow.
The construct fox the N83H mutant without a his tag is referred to as sILIR2D-
N83H-FB, and it
was created using ILlRsigFor (SEQ ID N0:65) and N83HR (SEQ ID N0:69) along
with N83HF
(SEQ ID N0:70), and ILIR2Drevstop-Xho (SEQ ID N0:67)
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
N83HR GAGGCAGTAAGATGAATGTCTTACC SEQ ID N0:69
N83HF CTATTGCGTGGTAAGACATTCATCTT SEQ ID NO:70
The construct for the N83H mutant with a his tag is referred to as sILIR2D-
N83H-FBHT and was
created using ILlRsigFor (SEQ ID N0:65), and N83HR (SEQ ID N0:69) along with
N83HF (SEQ
ID N0:70) and ILIR2Drev-Xho (SEQ ID N0:68).
The construct for the N176H mutant without a his tag is referred to as sILIR2D-
N176H-FB and it
was created using ILlRsigFor (SEQ ID N0:65), N176HR (SEQ ID N0:71), N176HF
(SEQ ID
N0:72), and ILIR2Drevstop-Xho (SEQ ID N0:67).
N176HR ATGACAAGTATAGTGCCCTCTATGCTTTTCACG SEQID NO:71
N176HF GCTGAAAAGCATAGAGGGCACTATACTTGTCAT SEQID N0:72
The construct for the N176H mutant with a his tag is referred to as sILIR2D-
N176H-FBHT.and it
was created using ILlRsigFor (SEQ ID N0:65), and N176HR (SEQ ID N0:71), along
with
N176HF (SEQ ID N0:72), and ILIR2Drev-Xho (SEQ ID N0:68).
The PCR products were isolated from and agarose gel and PCR was used to sew
the two fragments
together using the ILlRsigFor (SEQ ID N0:65) and ILIR2Drevstop-Xho (SEQ ID
N0:67) or
ILIR2Drev-Xho primers (SEQ ID N0:68). The PCR products containing the IL1R2D
sequences
mutated at the glycosylation site were cut with restriction endonucleases (41
p1 PCR product, 2 ~1
each endonuclease, 5 p.1 appropriate lOx buffer; incubated at 37 C for 90
minutes). The products
of nuclease cleavage were isolated from an agarose gel (1% agarose, TBE
buffer) and ligated
together using T4 DNA ligase (200 ng pFBHT vector, 150 ng IL1RZD PCR product,
4 p1 Sx ligase
buffer [300 mM Tris pH 7.5, 50 mM MgCl2, 20% PEG 8000, 5 mM ATP, 5 mM DTT], 1
~l ligase;
incubated at 15 C for 1 hour). 10 p1 of the ligation reaction was transformed
into XLl blue cells
(Stratagene) (10 p.1 reaction mixture, 10 ~1 Sx KCM [0.5 M KCI, 0.15 M CaCl2,
0.25 M MgCl2], 30
2.5 p,1 water, 50 ~1 PEG-DMSO competent cells; incubated at 4 C for 20
minutes, 25 C for 10
minutes), and plated onto LB/agar plates containing 100 pg/ml ampicillin.
After incubation at 37
C overnight, single colonies were grown in 3 ml 2YT media for 18 hours. Cells
were then isolated
and double-stranded DNA extracted from the cells using a Qiagen DNA miniprep
kit. The
subsequent plasmids are referred to as sILIR2D-N83H-FB or sILIR2D-N83H-FBHT
and as
sILIR2D-N176H-FB or as sILIR2D-N176H-FBHT.
36
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
Finally, an additional construct was made using the sILIR2D-N83H-FB construct.
The additional
construct contains the 2-domain IL1R receptor without a his tag and with two
mutations: a N83H
glycosylation mutation and a deletion of the C-terminal residue (K205). This
construct is named
sIL1R2D2M-FB, and was made using the K205de1 oligonucleotide.
K205de1 CTCGAGTCATCAGTTTTCCTCTAG SEQ ID NO:73
Generation of IL-1RI Cysteine Mutations
Site-directed mutants of IL1R2D were prepared by the single-stranded DNA
method [modification
of Kunkel, T. A., Proc. Natl. Acid. Sci. U. S. A. 82: 488-492 (1985)].
Oligonucleotides were
designed to contain the desired mutations and 15-20 bases of flanking
sequence.
The single-stranded form of the IL1R2D (sILIR2D-FBHT, sILIR2D-N176H-FB/FBHT,
sILIR2D-
N83H-FB/FBHT, sIL1R2D2M-FB) plasmid was prepared by transformation of double-
stranded
plasmid into the CJ236 cell line (1 ~l IL1R-FB double-stranded DNA, 2 p.1 2x
KCM salts, 7 ~tI
water, 10 p1 PEG-DMSO competent CJ236 cells; incubated at 4 C for 20 minutes
and 25 C for 10
minutes; plated on LB/agar with 100 pg/m1 arnpicillin and incubated at 37 C
overnight). Single
colonies of CJ236 cells were then grown in 50 rnl 2YT media to midlog phase;
10 p1 VCS helper
phage (Stratagene) were then added and the mixture incubated at 37 C
overnight. Single-stranded
DNA was isolated from the supernatant by precipitation of phage (1/5 volume
20% PEG 8000/2.5
M NaCI; centrifuge at 12K for 15 minutes.). Single-stranded DNA was then
isolated from phage
using Qiagen single-stranded DNA kit.
Site-directed mutagenesis was accomplished as follows. Oligonucleotides were
dissolved to a
concentration of 10 OD and phosphorylated on the 5' end (2 p.1
oligonucleotide, 2 ~1 10 mM ATP,
2 p.1 lOx Tris-magnesium chloride buffer, 1 p.1 100 mM DTT, IO ~l water, I p1
T4 PNK; incubate at
37 C fox 45 minutes). Phosphorylated oligonucleotides were then annealed to
single-stranded
DNA template (2 p.1 single-stranded plasmid, I p.1 oligonucleotide, 1 ~.I lOx
TM buffer, 6 p.1 water;
heat at 94 C for 2 minutes, 50 C for 5 minutes, cool to room temperature).
Double-stranded DNA
was then prepared from the annealed oligonucleotide/template (add 2 ~.1 lOx TM
buffer, 2 ~,1 2.5
mM dNTPs, 1 ~cl 100 mM DTT, 1.5 w1 10 mM ATP, 4 p.1 water, 0.4 p.1 T7 DNA
polymerise, 0.6 q1
T4 DNA ligase; incubate at room temperature for two hours). E. coli (XL1 blue,
Stratagene) was
then transformed with the double-stranded DNA (1 p.1 double-stranded DNA, 10
p.1 Sx KCM, 40 p,1
water, 50 u1 DMSO competent cells; incubate 20 minutes at 4 C, 10 minutes at
room temperature),
plated onto LBlagar containing 100 p,g/ml ampicillin, and incubated at 37 C
overnight.
Approximately four colonies from each plate were used to inoculate 5 ml 2YT
containing 100
37
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
~,glml ampicillin; these cultures were grown at 37 C for 18-24 hours. Plasmids
were then isolated
from the cultures using Qiagen miniprep kit. These plasmids were sequenced to
determine which
ILIR2D-FB clones contained the desired mutation.
Sequencing of IL1R2D genes was accomplished as follows. The concentration of
plasmid DNA
was quantitated by absorbance at 280 nm. 800 ng of plasmid was mixed with
sequencing reagents
(8 p,1 DNA, 3 p.1 water, 1 ~.1 sequencing primer, 8 ~.1 sequencing mixture
with Big Dye [Applied
Biosystems]). The sequencing primers used were FB Forward and FB Reverse,
shown below.
FB Forward TATTCCGGATTATTCATACC SEQ ID N0:74
FB Reverse CCTCTACAAATGTGGTATGGC SEQ ID N0:75
The mixture was then run through a PCR cycle (96 C, 10 s; 50 C, S s; 60 C 4
minutes; 25 cycles)
and the DNA reaction products were precipitated (20 ~,I mixture, 80 p,1 75%
isopropanol; incubated
minutes at room temperature, pelleted at 14 K rpm for 20 minutes; wash with
250 ~.l 70%
ethanol; heat 1 minute at 94 C). The precipitated products were then suspended
in Template
15 Suppression Buffer (TSB, Applied Biosystems) and the sequence read and
analyzed by an Applied
Biosystems 310 capillary gel sequencer. In general, 3 out of 4 of the plasmids
contained the desired
mutation. A listing of the constructs and their mutants) is given below,
although any cysteine
mutants can be made in any of the given contexts.
Construct Mutant s
sILIR2D-N83H-FB El IC, II3C, VI6C, Q108C, I110C, K112C, K114C, VI17C,
VI24C, YI27C, E129C
sILIR2D-N83H-FBHT ElIC, I13C, V16C, Q108C, I110C, K112C, Q1I3C, K114C,
VI17C, V124C, Y127C, E129C
sIL 1 R2D-N 176H-FB E 11 C
sILIR2D-N176H-FBHT E11C, VI6C, V124C, E129C
sIL1R2D2M-FB E11C, KI2C, I13C, A107C, K112C, V124C, Y127.
Mutagenic Oligonucleotides
EIIC T~TTATTTTACATTCACGTTCC SEQID NO:76
38
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
K12C CACTAAAATTATACATTCTTCACGTTC SEQID NO:77
I13C TGACACTAAAATACATTTTTCTTCACG SEQID NO:78
VICC ATTTGCAGATGAACATAAAATTATTT SEQID NO:79
A107C ~TATGGCTTGGCAATTATAACATAAG SEQID NO:80
Q108C CTTAAATATGGCGCATGCATTATAACA SEQID NO:81
I110C GTTTCTGCTTAAAGCAGGCTTGTGCATT SEQ ID NO:82
K112C GGGTAGTTTCTGACAAAATATGGC SEQID NO:83
Q113C AACGGGTAGTTTACACTTAAATATGGC SEQID NO:84
K114C CTGCAACGGGTAGGCACTGCTTAAATATG SEQID NO:85
V117C CTCCGTCTCCTGCACAGGGTAGTTTCTG SEQ ID NO:BC)
V124C CATATAAGGGCAACAAAGTCCTCC SEQ ID NO:87
Y127C CTCCATACAAGGGCACACAAG SEQ ID NO:$8
E129C TTTAAAAAAACACATATAAGGGCA SEQ ID NO:89
Expression of IL-1 R mutant proteins
All IL1R-FB/FBHT plasmids were site-specifically transposed into the
baculovirus shuttle vector
(bacmid) by transforming the plasmids into DHlObac (GibcoBRL) competent cells
as follows: 1
~l DNA at 5 ng/~1, 10 ~.l 5x KCM [0.5 M KCl, 0.15 M CaClz, 0.25 M MgCl2], 30
~,1 water was
mixed with 50 p1 PEG-DMSO competent cells, incubated at 4 C for 20 minutes, 25
C for 10
minutes, add 900 p,1 SOC and incubate at 37 C with shaking for 4 hours, then
plated onto LB/agar
plates containing 50 pg/ml kanamycin, 7 ~g/ml gentamycin, 10 pg/ml
tetracycline, 100 ~g/ml
Bluo-gal, 10 pg/ml IPTG. After incubation at 37 C for 24 hours, large white
colonies were picked
and grown in 3 ml 2YT media overnight. Cells were then isolated and double-
stranded DNA was
extracted from the cells as follows: pellet was resuspended in 250 ~.l of
Solution 1 [15 mM Tris-
HCl (pH 8.0), 10 mM EDTA, 100 ~g/ml RNase A]. 250 ~l of Solution 2 [0.2 N
NaOH, 1% SDS]
was added, mixed gently and incubated at room temperature for 5 minutes. 250
~.1 3 M potassium
acetate was added and mixed, and the tube placed on ice for 10 minutes. The
mixture was
centrifuged 10 minutes at 14,OOOx g and the supernatant transferred to a tube
containing 0.8 ml
isopropanol. The contents of the tube were mixed and placed on ice for 10
minutes; centrifuged 15
39
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
minutes at 14,000x g. The pellet was washed with 70% ethanol and air-dried and
the DNA
resuspended in 40 p1 TE.
The bacmid DNA was used to transfect Sf9 cells. Sf7 cells were seeded at 9 x
105 cells per 35 mm
well in 2 ml of Sf 900 II SFM medium containing O.Sx concentration of
antibiotic-antimycotic and
allowed to attach at 27 C for 1 hour. During this time, 5 ~1 of bacmid DNA was
diluted into 100
~1 of medium without antibiotics, 6 ~1 of CeIIFECTIN reagent was diluted into
100 ~1 of medium
without antibiotics and then the 2 solutions were mixed gently and allowed to
incubate for 30
minutes at xoorn temperature. The cells were washed once with medium without
antibiotics, the
medium was aspirated and then 0.8 ml of medium was added to the lipid-DNA
complex and
overlaid onto the cells. The cells were incubated for 5 hours at 27 C, the
transfection medium was
removed and 2 ml of medium with antibiotics was added. The cells were
incubated for 72 hours at
27 C and the virus was harvested from the cell culture medium.
The virus was amplified by adding 0.5 ml of virus to a 50 ml culture of Sf~
cells at 2 x lOG cells/ml
and incubating at 27 C for 72 hours. The virus was harvested from the cell
culture medium and
this stock was used to express the various IL1R constructs in High-Five cells.
A 1 L culture of
High-Five cells at 1 x 10~ cells/ml was infected with virus at an approximate
MOI of 2 and
incubated for 72 hours. Cells were pelleted by centrifugation and the
supernatant was loaded onto
an IL1R antagonist column at 1 ml/min, washed with PBS followed by a wash with
Buffer A (0.2
M NaOAc pH 5.0, 0.2 M NaCI). The protein was eluted from the column by running
a gradient
from 0-100% of Buffer B (0.2 M NaOAc pH 2.5, 0.2 M NaCI) in 10 minutes
followed by 15
minutes of 100% Buffer B at 1 ml/min collecting 2 ml fractions in tubes
containing 300 ~.l of
unbuffered Tris. The appropriate fractions were pooled, concentrated and
dialyzed against 5 L of
50 mM Tris pH 8.0, 100 mM NaCl at 4 C and filtered through a 0.2 ~m filter.
EXAMPLE 6
CLONING AND MUTAGENESIS OF HUMAN CASPASE-3 fCASP-3)
Caspase-3 (accession number SWS P42574) is one of a series of caspases
involved in the apoptosis
3U of cells. It exists as the inactive proform, and can be processed by
caspases 8, 9, or 10 to form a
small subunit and a large subunit, which heterodimerize to constitute the
active form. Caspases that
are substrates for caspase-3 in the cascade are caspase-6, caspase-7 and
caspase-9. Caspase-3 has
been shown to be the important for the cleavage of amyloid-beta precursor
protein 4A. This
cleavage has been linked to the deposition of Abeta peptide deposition and
death of neurons in
Alzheimers disease and hippocampal neurons following ischemic and exitoxic
brain injury. There
is a crystal structure available for caspase-3 [1CP3, Mittl, P. R., et al.,
JBiol CIZerya 272:6539-6547
(1997)].
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
Cloning of Human Caspase-3
The human version of caspase-3 (also known as Yama, CPP32 beta) was cloned
directly from
Jurkat cells (Clone E6-1; ATCC). Briefly, total RNA was purified from Jurleat
cells growing at 37
C/5% COz using Tri-Reagent (Sigma). Oligonucleotide primers were designed to
allow DNA
encoding the large and small subunits of Caspase-3/Yama/CPP32 to be amplified
by polymerase
chain reaction (PCR). Briefly, DNA encoding amino acids 28-175 (encompassing
most of the large
subunit) was directly amplified from 1 ~.g total RNA using Ready-To-Go-PCR
Beads
(Amersham/Pharmacia) and the following oligonucleotides:
casp-3 large for TTCCATATGTCTGGAATATCCCTGGACAACAGTTA SEQ ID NO:90
carp-3 large rev AAGGAATTCTTAGTCTGTCTCAATGCCACAGTCCAG SEQ ID N0:91
DNA 'encoding amino acids 176-277 (encompassing most of the small subunit) was
directly
amplified from 1 p,g total RNA using Ready-To-Go-PCR Beads
(Amersham/Pharmacia) and the
following oligonucleotides:
carp-3 small for TTCCATATGAGTGGTGTTGATGATGACATGGCG SEQ ID N0:92
casp-3 small rev AAGGAATTCTTAGTGATAAAAATAGAGTTCTTTTGTGAG SEQ ID N0:93
Amplified DNA corresponding to either the large subunit or the small subunit
of caspase-3 was
then cleaved with the restriction enzymes EcoRI and NdeI and directly cloned
using standard
molecular biology techniques into pRSET-b (Invitrogen) digested with EcoRI and
NdeI. [See e.g.,
Tewari. M., et al., Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-
inhabitable
protease that cleaves the death substrate poly (ADP-ribose) polymerase, Cell
81: 801-809 (1995)x.
Generation of Casp-3 Cys Mutations
Plasmids containing DNA encoding either the large or small subunits of Caspase-
3 were separately
transformed into E. cola K12 CJ236 cells (New England BioLabs) and cells
containing each
2S construct were selected by their ability to grow on ampicillin containing
agar plates. Overnight
cultures of the large and small subunits were individually grown in 2YT
(containing 100 p.g/mL of
ampicillin) at 37 C. Each culture was diluted 1:100 and grown to A~oo = 0.3-
0.6. A 1.5 mL
sample of each culture was removed and infected with 10 ~.L of phage VCS-M13
(Stratagene),
shaken at 37 C for 60 minutes, and an overnight culture of each was prepared
with 1 mL of the
infected culture diluted 1:100 in 2YT with 100 ~g/mL of ampicillin and 20
~g/ml of
41
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
chloramphenicol and grown at 37 C. Cells were centrifuged at 3000 rcf for 10
minutes and 1/5
volume of 20%PEG/2.5 M NaCI was added to the supernatant. Samples were
incubated at room
temperature for 10 minutes and then centrifuged at 4000 rcf for 15 minutes.
The phage pellet was
resuspended in PBS and spun at 15 I~ rpm for 10 minutes to remove remaining
particulate matter.
Supernatant was retained, and single~stranded DNA was purified from the
supernatant following
procedures for the QIA prep spin M13 kit (Qiagen).
Mutagenic Oligonucleotides
Cysteine mutations in the small subunit were made with the corresponding
primers:
Y204C TCGCCAAGAACAATAACCAGG ~ SEQ ID N0:94
S2O9C GCCATCCTTACAATTTCGCCA SEQ ID NO:95
W214C CTGGATGAAACAGGAGCCATC SEQ ID NO:96
S251C AGCGTCAAAGCAAAAGGACTC SEQ ID NO:97
F256C CTTTGCATGACAAGTAGCGTC SEQ ID NO:98
Cysteine mutations in the large subunit were made with the corresponding
primers:
M61C CCGAGATGTACATCCAGTGCT SEQ ID NO:99
T62C AGACCGAGAACACATTCCAGT SEQ ID
N0:100
S65C ATCTGTACCACACCGAGATGT SEQ ID
NO:101
H121C TTCTTCACCACAGCTCAGAAG SEQ ID
N0:102
L168C GCCACAGTCACATTCTGTACC SEQ ID
N0:103
Approximately 100 pmol of each primer was phosphorylated by incubating at 37 C
for 60 minutes
in buffer containing 1X TM Buffer (0.5 M Tris pH 7.5, 0.1 M MgClz ), 1 mM ATP,
5 mM DTT,
and SLT T4 I~inase (NEB). I~inased primers were annealed to the template DNA
in a 20 ~,L reaction
volume (~50 ng kinased primer, 1X TM Buffer, and 10-50 ng single-stranded DNA)
by incubation
at 85 C for 2 minutes, 50 C for 5 minutes, and then at 4 C for 30-60 minutes.
An extension
cocktail (2 mM ATP, 5 mM dNTPs, 30 mM DTT, T4 DNA ligase (NEB), and T7
polymerase
(NEB)) was added to each annealing reaction and incubated at room temperature
for 3 hours.
42
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
Mutagenized DNA was transformed into E. coli XL1-Blue cells, and colonies
containing plasmid
DNA selected were for by growth on LB agar plates containing 100 ~.g/ml
ampicillin. DNA
sequencing was used to identify plasmids containing the appropriate mutation.
Expression of Casp-3 Mutant Proteins
Plasmid DNA encoding cysteine mutations in the large subunit were transformed
into Codon Plus
BL21 Cells and plasmid DNA encoding cysteine mutations in the small subunit
were transformed
into BL21 (DE3) pLysS Cells. Codon Plus BL21 Cells containing plasmids
encoding wild-type
and cysteine mutated versions of the large subunit were grown in 2YT
containing 150 ~,g/mL of
ampicillin overnight at 37 C and immediately harvested. BL21 pLysS cells
containing plasmids
encoding wild-type and cysteine mutated versions of the small subunit were
grown in 2YT at 37 C
with 150 ~,g/mL of ampicillin until A~oo = 0.6. Cultures were subsequently
induced with 1mM
IPTG and grown for an additional 3-4 hours at 37 C. After harvesting cells by
centrifuging at 4K
rpm for 10 minutes, the cell pellet was resuspended in Tris-HCl (pH 8.0)/5 mM
EDTA and micro
fluidized twice. Inclusion bodies were isolated by centrifugation at 9K rpm
for 10 minutes and then
resuspended in 6 M guanidine hydrochloride. Denatured subunits were rapidly
and evenly diluted
to 100 ~g/mL in renaturation buffer (100 mM Tris/KOH (pH 8.0), 10% sucrose,
0.1% CHAPS,
0.15 M NaCI, and 10 mM DTT) and allowed to renature by incubation at room
temperature for 60
minutes with slow stirring.
Renatured proteins were dialyzed overnight in buffer containing 10 mM Tris (pH
8.5), 10 mM
DTT, and 0.1 mM EDTA. Precipitate was removed by centrifuging at 9K rpm for 15
minutes and
filtering the supernatant through a 0.22 pm cellulose nitrate filter. The
supernatant was then loaded
onto an anion-exchange column (UnoS Q-Column (BioRad)), and correctly folded
caspase-3
protein was eluted with a 0-0.25 M NaCI gradient at 3 mL/min. Aliquots of each
fraction were
electrophoresed on a denaturing polyacrylamide gel and fractions containing
Caspase-3 protein
were pooled.
EXAMPLE 7
CLONING AND MUTAGENESIS OF HUMAN PROTEIN TYROSINE PHOSPHATASE-1B
PTP-1B
PTP-1B (accession number SWS P18031) is a tyrosine phosphatase that has a C-
terminal domain
that is associated to the endoplasmic reticulum (ER) and a phosphatase domain
that faces the
cytoplasm. The proteins that it dephosphorylates are transported to this
location by vesicles. The
activity of PTP-1B is regulated by phosphorylation on serine and protein
degradation. PTP-lBis a
negative regulator of insulin signaling, and plays a role in the cellular
response to interferon
stimulation. This phosphatase may play a role in obesity by decreasing the
sensitivity of organisms
to leptin, thereby increasing appetite. Additionally, PTP-1B plays a role in
the control of cell
43
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
growth. A crystal structure has been solved for PTP-1B [1PTY, Puius, Y. A., et
al., Proc Natl Acad
Sci USA 94: 13420-13425 (1997)].
Cloning of human PTP-1B
Full length human PTP-1B is 435 amino acids in length; the protease domain
comprises the first
288 amino acids. Because truncated portions of PTP-1B comprising the protease
domain is fully
active, various truncated versions of PTP-1B are often used. A cDNA encoding
the first 321 amino
acids of human PTP-1B was isolated from human fetal heart total RNA
(Clontech).
Oligonucleotide primers corresponding to nucleotides 91 to 114 (For) and
complementary to
nucleotides 1030 to 1053 (Rev) of the PTP-1B cDNA [Genbank M31724.1, Chernoff,
J., et al.,
Proc. Natl. Acid. Sci. U. S. A. 87: 2735-2739 (1990)] were synthesized and
used to generate a DNA
using the polymerise chain reaction.
Forward GCCCATATGGAGATGGAAAAGGAGTTCGAG SEQ ID
N0:104
Rev GCGACGCGAATTCTTAATTGTGTGGCTCCAGGATTCGTTT SEQ ID
NO:105
The primer Forward incorporates an NdeI restriction site at the first ATG
codon and the primer Rev
inserts a UAA stop codon followed by an EcoRI restriction site after
nucleotide 1053. cDNAs were
digested with restriction nucleases NdeI and EcoRI and cloned into pRSETc
(Invitrogen) using
standard molecular biology techniques. The identity of the isolated cDNA Was
verified by DNA
sequence analysis (methodology is outlined in a later paragraph).
A shorter cDNA, PTP-1B 298, encoding amino acid residues 1-298 was generated
using
oligonuclotide primers Forward and Rev2 and the clone described above as a
template in a
polymerise chain reaction.
Rev2 TGCCGGAATTCCTTAGTCCTCGTGGGAAAGCTCC SEQ ID
N0:106
Generation of PTP-1B Cysteine Mutants
Site-directed mutants of PTP-1B (amino acids 1-321), PTP-1B 298 (amino acids 1-
298) and PTP-
1B 298-2M (with Cys32 and Cys92 changed to Ser and Val, respectively) were
prepared by the
single-stranded DNA method (modification of Kunkel, 1985). 298-2M was made
with the
following oligonucleotides.
44
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
C32S CTTGGCCACTCTAGATGGGAAGTCACT SEQID
N0:107
C92V CCAAAAGTGACCGACTGTGTTAGGCAA SEQID
N0:108
Oligonucleotides were designed to contain the desired mutations and 12 bases
of flanleing sequence
on each side of the mutation. The single-stranded form of the PTP-1B/pRSET,
PTP-1B 298/pRSET
and PTP-1B 298-2M/pRSET plasmid was prepared by transformation of double-
stranded plasmid
into the CJ236 cell line (1 ~,1 double-stranded plasmid DNA, 2 ~.1 Sx KCM
salts, 7 ~tl water, 10 p1
PEG-DMSO competent CJ236 cells; incubated on ice for 20 minutes followed by 25
C for 10
minutes; plated on LB/agar with 1.00 pg/ml ampicillin and incubated at 37 C
overnight). Single
colonies of CJ236 cells were then grown in 100 ml 2YT media to midlog phase; 5
~,l VCS helper
phage (Stratagene) were then added and the mixture incubated at 37 C
overnight. Single-stranded
DNA was isolated from the supernatant by precipitation of phage (1/5 volume
20% PEG
8000/2.5M NaCI; centrifuge at 12K for 15 minutes). Single-stranded DNA was
then isolated from
phage using Qiagen single-stranded DNA lcit.
Site-directed mutagenesis was accomplished as follows. Oligonucleotides were
dissolved in TE (10
mM Tris pH 8.0, 1mM EDTA) to a concentration of 10 OD and phosphorylated on
the 5' end (2 p1
oligonucleotide, 2 p.1 10 mM ATP, 2 ~,1 lOx Tris-magnesium chloride buffer, 1
p1 100 mM DTT,
12.5 p.1 water, 0.5 0,1 T4 PNK; incubate at 37 C for 30 minutes).
Phosphorylated oligonucleotides
were then annealed to single-stranded DNA template (2 ~1 single-stranded
plasmid, 0.6 ~.l
oligonucleotide, 6.4 p.1 water; heat at 94 C for 2 minutes, slow cool to room
temperature). Double-
stranded DNA was then prepared from the annealed oligonucleotide/template (add
2 p.1 lOx TM
buffer, 2 ~.1 2.5 mM dNTPs, 1 ~,1 100 mM DTT, 0.5 ~1 10 mM ATP, 4.6 ~tl water,
0.4 p1 T7 DNA
polymerase, 0.2 ~,1 T4 DNA ligase; incubate at room temperature for two
hours). E. coli (XI,1 blue,
Stratagene) were then transformed with the double-stranded DNA (5 p1 double-
stranded DNA, 5 p1
Sx KCM, 15 p.1 water, 25 p1 PEG-DMSO competent cells; incubate 20 minutes on
ice, 10 min. at
room temperature), plated onto LB/agar containing 100 pg/ml ampicillin, and
incubated at 37 C
overnight. Approximately four colonies from each plate were used to inoculate
5 ml 2YT
containing 100 pg/ml ampicillin; these cultures were grown at 37 C for 18-24
hours. Plasmids
were then isolated from the cultures using Qiagen miniprep lcit. These
plasmids were sequenced to
determine which clones contained the desired mutation.
A listing of the constructs and the single mutations to cysteine made in each
context is given below.
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
PTP-1B 321 H25C, D29C, R47C, D48C, SSOC, K120C, M258C
PTP-1B 298 H25G, D29C, D48C, SSOC, K120C, M258C, F280C
PTP-1B 298-2M E4C, E8C, H25C, A27C, D29C, K36C, Y46C, R47C, D48C,
V49C, SSOC, FSZC, K120C, S151C, Y152C, T178C, D181C,
F182C, E186C, S187C, A189C, K197C, E200C, L272C, E276C,
I218C, M258C, Q262C, V287C
However, it should be understood that any of the site-directed mutants may be
made in any
construct of PTP-1B. For example, another construct is another truncated
version of PTP-1B
having residues 1-382, shown as SEQ ID N0:109 below.
1 MEMEKEFEQI DKSGSWAAIYQDTRHEASDFPCRVAKLPKNKNRNRYRDVSPFDHSRIKLH
61 QEDNDYINAS LIKMEEAQRSYILTQGPLPNTCGHFWEMVWEQKSRGVVMLNRVMEKGSLK
121 CAQYWPQKEE KEMIFEDTNLKLTLISEDIKSYYTVRQLELENLTTQETREILHFHYTTWP
l81 DFGVPESPAS FLNFLFKVRESGSLSPEHGPVVVHCSAGIGRSGTFCLADTCLLLMDKRKD
1O 241 PSSVDTKKVL LEMRKFRMGLIQTADQLRFSYLAVIEGAKFIMGDSSVQDQWKELSHEDLE
301 PPPEHIPPPP RPPKRILEPHNGKCREFFPNHQWVKEETQEDKDCPTKEEKGSPLNAAPYG
361 IESMSQDTEV RSRWGGSLRGA
Mutagenic Oligonucleotides
E4C CTCGAACTCCTTGCACATCTCCATATG SEQID
NO:110
E8C CTTGTCGATCTGGCAGAACTCCTTTTC SEQID
NO:111
H25C GTCACTGGCTTCACATCGGATATCCTG SEQID
N0:112
A27C TGGGAAGTCACTGCATTCATGTCGGAT SEQID
N0:113
D29C TCTACATGGGAAGCAACTGGCTTCATG SEQID
NO: l I4
K36C GTTCTTAGGAAGACAGGCCACTCTACA SEQID
NO:115
Y46C ACTGACGTCTCTGCACCTATTTCGGTT SEQ ID
N0:116
R47C GGGACTGACGTCACAGTACCTATTTCG SEQID
N0:117
46
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
D48C AAAGGGACTGACGCATCTGTACCTATT SEQID
N0:118
V49C GTCAAAGGGACTGCAGTCTCTGTACCT SEQID
N0:119
SSOC CTATGGTCAAAGGGACAGACGTCTCTGTACC SEQID
N0:120
F52C CCGACTATGGTCACAGGGACTGACGTC SEQID
N0:121
K12OC GTATTGTGCGCAACATAACGAACCTTT SEQID
N0:122
S151C CACTGTATAATAGCACTTGATATCTTC SEQID
N0:123
Y152C GTCGCACTGTATAACATGACTTGATATC SEQID
N0:124
T178C CAAAGTCAGGCCAGCAGGTATAGTGGAA SEQID
N0:125
D181C AGGGACTCCAAAGCAAGGCCATGTGGT SEQID
N0:126
E186C GAATGAGGCTGGTGAGCAAGGGACTCCAAAG SEQID
N0:127
S187C GAATGAGGCTGGGCATTCAGGGACTCC SEQID
N0:128
A189C GTTCAAGAATGAGCATGGTGATTCAGG SEQID
N0:129
K197C CTGACTCTCGGACGCAGAAAAGAAAGTTC SEQID
N0:130
E200C GAGTGACCCTGAGCATCGGACTTTGAAAAG SEQID
N0:131
M258C CTGGATCAGCCCACACCGAAACTTCCT SEQID
N0:132
Q262C CTGGTCGGCTGTACAGATCAGCCCCAT SEQID
N0:133
L272C CTTCGATCACAGCGCAGTAGGAGAAGCG SEQID
N0:134
E276C GAATTTGGCACCGCAGATCACAGCCAG SEQID
N0:135
47
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
I281C AGAGTCCCCCATGCAGAATTTGGCACC SEQ ID
NO;136
V287C CCACTGATCCTGGCAGGAAGAGTCCCC SEQID
N0:137
Besides mutations to cysteines, mutations removing naturally occurring
cysteines can also be made.
For example, two different "scrubs" of Cys 215 were made in the PTP-1B 298-2M
context using
the following oligonueleotides:
C215A GATGCCTGCACTGGCGTGCACCACAAC SEQID
N0;138
C215S GATGCCTGCACTGGAGTGCACCACAAC SEQID
N0:139
In the PTP-1B 298 context, two quadruple mutants were made using the C92A
oligonucleotide
shown below. They are C32S, C92A, V287C, C215A, which used SEQ ID N0:107 SEQ
ID
N0:140 SEQ ID N0:137 and SEQ ID N0:138 and C32S, C92A, E276C, C215A, which
used SEQ
ID N0:107, SEQ ID N0:140 SEQ ID N0:135 and SEQ ID N0:138.
C92A CCAAAAGTGACCGGCTGTGTTAGGCAA SEQID
N0:140
Sequencing of PTP-IB clones was accomplished as follows. The concentration of
plasmid DNA
was quantitated by absorbance at 280 nm. 1000 ng of plasmid was mixed with
sequencing reagents
(1 p,g DNA, 6 p1 water, 1 p1 sequencing primer at 3.2 pm/p.l, 8 ~l sequencing
mixture with Big Dye
[Applied Biosystems]). The sequencing primers are SEQ ID N0:17 and SEQ ID
N0:18. The
mixture was then run through a PCR cycle (96 C,10 s; 50 C, 5 s; 60 C 4
minutes; 25 cycles) and
the DNA reaction products were precipitated (20 p1 mixture, 80 p1 75%
isopropanol; incubated 20
minutes at room temperature then pelleted at 14 K rpm for 20 minutes; wash
with 250 p1 75%
isopropanol; heat 1 minute at 94 C). The precipitated products were then
resuspended in 20 ~1
TSB (Applied Biosystems) and the sequence read and analyzed by an Applied
Biosystems 310
capillary gel sequences. In general, 1/4 of the plasrnids contained the
desired mutation.
Expression of Cysteine Mutants of PTP-1B
Mutant proteins were expressed as follows. PTP-1B clones were transformed into
BL21 codon plus
cells (Stratagene) (1 p,1 double-stranded DNA, 2 ~,l 5x KCM, 7 ~1 water, 10
~,1 DMSO competent
cells; incubate 20 minutes at 4 C, 10 minutes at room temperature), plated
onto LB/agar containing
100 ~.g/ml ampicillin, and incubated at 37 C overnight. 2 single colonies were
picked off the
48
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
plates or from frozen glycerol stocks of these mutants and inoculated in 100
ml 2YT with 50 p,g/ml
carbenicillin and grown overnight at 37 C. 50 ml from the overnight cultures
were added to 1.5 L
of 2YT/carbenicillin (50 ~.g/ml) and incubated at 37 C for 3-4 hours until
late-log phase
(absorbance at 600 nm ~0.8-0.9). At this point, protein expression was induced
with the addition of
IPTG to a final concentration of 1 mM. Cultures were incubated at 37 C for
another 4 hours and
then cells were harvested by centrifugation (7K rpm, 7 minutes) and frozen at-
20 C.
PTP-1B proteins were purified from the frozen cell pellets as described in the
following. First, cells
were lysed in a microfluidizer in 100 ml of buffer containing 20 mM MES pH
6.5, 1 mM EDTA, 1
mM DTT, and 10% glycerol buffer (with 3 passes through a Microfluidizer
[Microfluidics 110S])
and inclusion bodies were removed by centrifugation (10K rpm, 10 minutes).
Purification of all
PTP-1B mutants was performed at 4 C. The supernatants from the centrifugation
were filtered
through 0.45 ~.m cellulose acetate (5 ~1 of this material was analyzed by SDS-
PAGE) and loaded
onto an SP Sepharose fast flow column (2.5 cm diameter x 14 cm long)
equilibrated in Buffer A (20
mM MES pH 6.5, 1 mM EDTA, 1 mM DTT, 1% glycerol) at 4 ml/min.
The protein was then eluted using a gradient of 0 - 50% Buffer B over 60
minutes (Buffer B: 20
mM MES pH 6.5, 1 mM EDTA, 1 mM DTT, 1 % glycerol, 1 M NaCI). Yield and purity
was
examined by SDS-PAGE and, if necessary, PTP-1B was further purified by
hydrophobic interaction
chromatography (HIC). Protein was supplemented with ammonium sulfate until a
final
concentration of 1.4 M was reached. The protein solution was filtered and
loaded onto an HIC
column at 4 ml/min in Buffer A2: 25 mM Tris pH 7.5, 1 mM EDTA, 1.4 M
(NHø)~SO4, 1 mM
DTT. Protein was eluted with a gradient of 0 - 100% Buffer B over 30 minutes
(Buffer B2: 25 mM
Tris pH 7.5, 1 mM EDTA, 1 mM DTT, 1% glycerol). Finally, the purified protein
was dialyzed at
4 C into the appropriate assay buffer (25 mM Tris pH 8, 100 mM NaCI, 5 mM
EDTA, 1 mM DTT,
1% glycerol). Yields varied from mutant to mutant but typically were within
the range of 3-20
mg/L culture.
EXAMPLE 8
CLONING AND MUTAGENESIS -OF HUMAN IMMUNODEFICIENCY VIRUS 1NTEGRASE
HIV IN
HIV IN is one of three key enzyme targets of the human immunodeficiency virus;
it removes two
nucleotides from each 3' end of the originally blunt viral DNA, and inserts
the viral DNA into the
host DNA by strand transfer. The integration process is completed by host DNA
repair enzymes.
HIV IN has three distinct domains: the N-terminal domain, the catalytic core
domain, and the C-
terminal domain. Although the X-ray crystal structures of each of these
isolated domains have been
solved, it is not yet clear how they interact with each other. Integration is
absolutely essential for
49
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
the replication of the virus and progression of disease, and thus integrase
inhibitors can be used in
the treatment of HIV/AIDS. Structures of core domain of integrase are
available [lEXQ, Chen, J.
C. -H., et al., Proc. Natl. Acad. Sci. U. S. A. 97: 8233-8238 (2000); 1BL3,
Maignan, S., et al., JMoI
Biol 282:359-368 (1998); in complex with tetraphenyl arsonium, 1HYZ and 1HYV,
Molteni, V., et
al., Acta Crystallogr D Bio Crystallog., 57:536-544 (2001)].
Cloning of HIV IN
Numbering of the wild type and mutant HIV-1 integrase residues follows the
convention of the first
amino acid residue of the mature protein being residue number 1, and the HIV-1
integrase catalytic
core domain being comprised of residues S2-210 [Leavitt, A. D., et al., J Biol
Che~i 268: 2113-
2119 (1993)].
A plasmid construct, pT7-7 HT-INtetta, encoding the HIV integrase core domain
(residues SO-212),
having an N-terminal 6x histidine tag and thrombin cleavable linker, and C56S,
W131D, F139D,
and F18SK mutations in the pT7-7 (Novagen) vector background [Chen, J. C. -H.,
et al., Proc. Natl.
Acad. Sci. U. S. A. 97: 8233-8238 (2000)] was obtained from Dr. Andy Leavitt
at UCSF. Upon
comparison of the crystal structure of this core domain variant [Chen, J. C. -
H., et al., Proc. Natl.
Acad. Sci. U. S. A. 97: 8233-8238 (2000)] to other integrase core structures,
it was noted that the
F139D mutation, designed to increase solubility of the protein, caused a
rotation of the side chain
that transmitted a distortion to the catalytically important Asp116. The
mutation was therefore
reverted to the wild-type phenylalanine residue by Quiclechange mutagenesis
(Stratagene),
following manufacturer's instructions and using SEQ ID N0:141 and SEQ ID
N0:142.
D139F1-int GTATCAAACAGGAATTCGGTATCCCGTACAAC SEQ ID N0:141
D139F2-int GTTGTACGGGATACCGAATTCCTGTTTGATACC SEQ ID N0:142
This generated pT7-7 HT-INm, encoding the triple mutant (C56S, W131D, F185K)
of the integrase
core, SEQ ID NO: I43.
52 GQVDSSPGIW QLDCTHLEGK VILVAVHVAS GYIEAEVIPA ETGQETAYFL LKLAGRWPVK
112 TIHTDNGSNF TGATVRAACD WAGTKQEFGI PYNPQSQGVV ESMNKELKKI IGQVRDQAEH
3O 172 LKTAVQMAVF IHNKKRKGGI GGYSAGERIV DIIATDIQT
In preparation for making cysteine mutations at tethering sites, the two wild-
type cysteines, (C130
and C65) were replaced by alanine residues and the DNA encoding the His-tagged
INm core
domain transferred into the pRSET A vector, containing an F1 origin of
replication that allows
preparation of single-stranded plasmid DNA, and thus mutagenesis by the
Kunlcel method [Kunkel,
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
T. A., et al., Methods Enzymol. 204: 125-139 (1991)]. Replacement of 0130 by
alanine was
accomplished by cassette mutagenesis, using the double stranded cassette
composed of SEQ ID
N0:144 and SEQ ID N0:145. The cassette, containing the appropriate overhangs
at each end, was
ligated into pT7-7 HT-INS; digested with BsiWI and EcoRI.
C130A cassette 1 GTACGTGCTGCAGCCGACTGGGCTGGTATCAAACAGG SEQ ID N0:144
C130A cassette 2 GAATTCCTGTTTGATACCAGCCCAGTCGGCTGCAGCAC SEQ ID N0:145
The C65A mutation was carried out independently by Quickchange mutagenesis on
pT7-7 HT-INM
using SEQ ID N0:146 and SEQ ID N0:147.
C65A1-irit ATCTGGCAACTGGACGCGACTCACCTCGAGGGT SEQ ID NO:146
C65A2-int ACCCTCGAGGTGAGTCGCGTCCAGTTGCCAGAT SEQ ID N0:147
The DNA encoding HT-C130A integrase core domain was subcloned into the pRSET A
vector by
PCR cloning. SEQ ID N0:14~ and SEQ ID N0:149 were used as PCR primers, and the
resulting
amplified product was digested with NdeI and Hind III, and ligated into pRSET
A that had been
digested with the same enzymes, to generate pRSET-HT-C130A-INS;.
C130 rsetF GGAGATATACATATGCACCACCATCACC SEQ ID N0:148
C130 rsetR ATCATCGATGATAAGCTTCCTAGGTCTGG SEQ ID N0:149
A BamHI fragment of pT7-7 HT-C65A-INm containing the C65A mutation was ligated
into
pRSET-HT-C130A-INtp, to generate pRSET-HT-IN~e",p,ate. This plasmid served as
a template for
further Kunlcel mutagenesis to introduce cysteine substitutions at positions
chosen for tethering.
SEQ ID N0:17 was used for sequencing.
Mutagenic Oligonucleotides
Q62C GTGAGTCGCGTCCAGGCACCAGATACCCGG SEQ ID
N0:150
D64C CTCGAGGTGAGTCGCGCACAGTTGCCAGATAC SEQ ID
NO:151
T66C CTTTACCCTCGAGGTGACACGCGTCCAGTTGCC SEQ ID
N0:152
51
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
H67C GGATAACTTTACCCTCGAGGCAAGTCGCGTCCAGTTG SEQ ID
N0:153
L68C AACTTTACCCTCGCAGTGAGTCGCGTCCA SEQ ID
N0:154
K71C GCAACCAGGATAACGCAACCCTCGAGGTG SEQID
N0:155
E92C CAGTTTCCTGACCAGTGCAGGCCGGGATAACTTC SEQ ID
N0:156
H114C GGATCCGTTGTCAGTGCAGATGGTTTTAACCGGC SEQ ID
N0:157
D116C GTTGGATCCGTTGCAAGTGTGGATGGTTTTAACCG SEQ ID
N0:158
N120C CGGTAGCACCAGTGAAGCAGGATCCGTTGTCAGTG SEQ ID
N0:159
N144C CACCCTGAGACTGCGGGCAGTACGGGATACCGA SEQID
N0:160
Q148C CATAGATTCAACAACACCGCAAGACTGCGGGTTGT SEQ ID
N0:161
I151C GCTCTTTGTTCATAGATTCGCAAACACCCTGAGA SEQ ID
N0:162
E152C GCTCTTTGTTCATAGAGCAAACAACACCCTGAGA SEQ ID
N0:163
N155C CCGATGATTTTTTTGAGCTCTTTGCACATAGATTCAACAACSEQ ID
N0:164
K156C CCGATGATTTTTTTGAGCTCGCAGTTCATAGATTC SEQ ID
N0:165
K159C CCTGACCGATGATTTTGCAGAGCTCTTTGTTCAT SEQ ID
N0:166
G163C CCTGATCACGAACCTGGCAGATGATTTTTTTG SEQ ID
N0:167
Q168C GGTTTTCAGGTGTTCAGCGCAATCACGAACCTGA SEQ ID
N0:168
T174C GCCATCTGAACCGCGCATTTCAGGTGTTCAGCC SEQ ID
N0:169
Expression of IN Cysteine Mutants
pT7-7 and pRSET integrase core domain expression plasmids were transformed
into BL2lstar E.
coli (Invitrogen) by standard methods, and a single colony from the resulting
plate was used to
inoculate 250 mL of 2x YT broth containing 100 ~.g/mL ampicillin. Following
overnight growth at
37 C, the cells were harvested by centrifugation at 4K rpm and resuspended in
100 mL 2YT/amp.
40 mL of the washed cells was used to inoculate 1.5 L of the same media, and
after growth at 37 C
to an OD at 600 nm of between 0.5 and 0.8, the culture was moved to 22 C and
allowed to cool.
IPTG was added to a final concentration of 0.1 mM and expression continued 17-
19 h at 22 C.
Cells were harvested by centrifugation at 4K rpm. Cell pellets were
resuspended in 100 mL Wash 5
buffer (Wash 5: 20 mM Tris-HCI, 1 M MgClz, 5 mM imidazole, 5 mM (3-
mercaptoethanol, pH 7.4)
52
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
and lysis was accomplished by sonication for 1 minute, repeated a total of 3
times with 2 minutes
rest between. Cell debris was removed by centrifugation at 14K rpm followed by
filtration.
Integrase core domain was purified by affinity chromatography on Ni-NTA
superflow resin
(Qiagen) at 4 C. After loading the cell lysate, the column was washed with
Wash 40 buffer (Wash
40: 20 mM Tris-HCl, 0.5 M NaCI, 40 mM imidazole, 5 mM (3-mercaptoethanol, pH
7.4) and His-
tagged IN core domain eluted with E400 buffer (E400: 20 mM Tris-HCI, 0.5 M
NaCI. 400 mM
imidazole, 5 mM (3-mercaptoethanol). The purified enzyme was dialyzed versus
20 mM Tris, 0.5
M NaCI, 2.5 mM CaClz, 5 mM (3-mercaptoethanol, pH 7.4 at 4 C , and aliquoted
into 1,5 mL tubes.
Biotinylated thrombin (Novagen) (2U thrombin/mg of protein) was added and the
tubes rotated
overnight at 4 C, followed by thrombin removal using streptavidin-agarose
resin (Novagen) and
separation of His-tagged protein and peptides from the cleaved material by
passage through a
second column of Ni-NTA sepharose fast-flow. Purified, cleaved integrase core
domain was
dialyzed against 20 mM Tris-HCI, 0.5 M NaCI, 3 mM DTT, and 5% glycerol, pH
7.4, and stored at
-20 C. Protein concentrations were determined by absorbance at 280 nm after
desalting on NAP-5
columns (Pharmacia), using E28o'~° _ (1.174), and molecular weights
confirmed by ESI mass
spectrometry (Finnigan).
EXAMPLE 9
HUMAN BETA-SITE AMYLOID PRECURSOR PROTEINCLEAV1NG ENZYMEl fBACEl)
BACE1 (accession number SWS 56817) is a typel integral glycoprotein that is an
aspartic protease.
Found mostly in the Golgi, BACEl cleaves the amyloid precursor protein to form
the Abeta
peptide. A strong association has been shown between deposition of this
peptide on the cerebrum
and Alzheimer's disease; therefore BACEl is one of the primary targets for
this disease. A crystal
structure of BACE1 has been solved [1FKN, Hong, L. et al., Science 290:150-153
(2000)].
Cloning of Human BACE1
The proprotease domain gene sequence (bases 64-1362, amino acid residues 22-
454) was subcloned
from pFBHT into the E. coli expression vector pRSETC by PCR, to create pB22,
which served as a
template for mutagenesis to incorporate cysteine tethering sites. For a
description of pFBHT, a
modified pFastBac plasmid, see example 4 above. The subcloning was
accomplished as follows.
The cDNA encoding full-length human BACE1, bases 1-1551, starting from the
initiator Met codon
and including an extra 48 bases of mRNA transcript following the stop codon
[Vassar, R., et al.,
Science 286: 735-741 (1999)] was obtained by a combination of PCR cloning of
the 3' 1425 bases
from human cDNA libraries, and synthesis of the remaining 5' 126 bases by
serial overlapping
PCR. All PCR reactions were performed using Advantage2 polymerase (Clontech)
according to
manufacturers instructions. A fragment spanning bases 126-374 was obtained by
PCR from a
53
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
human cerebral cortex library and SEQ ID N0:170 and SEQ ID N0:171; a fragment
spanning
bases 339-770 was obtained by PCR from a Stratagene Unizap XR human brain cDNA
library, and
SEQ ID N0:172 and SEQ ID N0:173; and the 3' end fragment, spanning bases 735-
1551, was
obtained by PCR from a human brain library, using SEQ ID NO:174 and SEQ ID
N0:175. The
three fragments, having 35 by of overlap at the junctions, were gel purified
and combined in one
PCR reaction, using primers to the ends (SEQ ID N0:170 and SEQ ID NO:176) to
amplify the 126-
1551 product.
FOr2 GCTGCCCCGGGAGACCGACGAAGA SEQ ID NO:17O
midRev2 CGGAGGTCCCGGTATGTGCTGGAC SEQ ID N0:171
midFor CCAGAGGCAGCTGTCCAGCACATA SEQ ID N0:172
midRevl TCCCGCCGGATGGGTGTATACCAG SEQID N0:173
BACE14 GTACACAGGCAGTCTCTGGTATACACC SEQ ID NO:174
BACE11 GTGTGGTCCAGGGGAATCTCTATCTTCTG SEQ ID NO:17S
RACES GTCATCGTCTCGAGTCACTTCAGCAGGGAGATGTCATCAG SEQ ID NO:176
The 126-1551 piece, and the subsequent elongated products, were used as a
templates for serial
overlapping PCR reactions, to add the remaining 5' -126 bases using SEQ ID
N0:177, SEQ ID
NO:178 and SEQ ID N0:179 as forward primers, with SEQ ID N0:176 always at the
reverse
primer.
BACE fi112
CGGCTGCCCCTGCGCAGCGGCCTGGGGGGCGCCCCCCTGGGGCTGCGGCTGCCCCGGGAG
SEQ ID N0:177
BACE filll
ATGGGCGCGGGAGTGCTGCCTGCCCACGGCACCCAGCACGGCATCCGGCTGCCCCTGCGC
2p SEQ ID N0:178
BACE for-EcoRI
CCGGAATTCATGGCCCAAGCCCTGCCCTGGCTCCTGCTGTGGATGGGCGCGGGAGTG
SEQ ID N0:179
SEQ ID N0:179 and SEQ ID N0:176 contained EcoRI and XhoI restriction sites,
respectively, and
digestion of the PCR product, along with the Baculovirus expression vector,
pFBHT, with the same
54
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
enzymes was followed by gel purification and ligation of the resulting DNA
fragments, yielding the
construct, pFBHT-BACE. This construct was used as a template for PCR
amplification of bases 1-
1362, corresponding to the preproBACE soluble protease domain, using SEQ ID
N0:180 and SEQ
ID N0:181.
S
proFor-Nde CGCCATATGGCGGGAGTGCTGCCTGCCCACGGC SEQ ID N0:180
BACErev-RI CCGGAATTCTCAGGTTGACTCATCTGTCTGTGGAAT SEQ ID N0:181
SEQ ID N0:180 and SEQ ID NO:181 contained NdeI and EcoRI restriction sites,
respectively, and
digestion of the PCR product, along with the E. coli expression vector,
pRSETC, with the same
enzymes was followed by gel purification and ligation of the resulting DNA
fragments led to the
construct pB 1. Vector pB 1 was then used as a template fox Kunlcel
mutagenesis (Kunkel, T. A., et
al., Metlae~ds Enzymol. 154:367-382 [1987]) to delete the BACE presequence
(bases 1-63),
producing the construct pB22. pB22 served as a template for mutagenesis to
incorporate cysteine
tethering sites, using either the Kunkel method or a Quiclcchange mutagenesis
kit (Stratagene).
Mutagenenic Oligonucleotides
L91C GCCTGTATCCACGCAGATGTTGAGCGT SEQID
N0:182
T133C CTTGCCCTGGCAGTAGGGCACATACCA SEQ ID
N0:183
Q134C TTCCCACTTGCCGCAGGTGTAGGGCAC SEQID
NO:184
F169C CGTTGATGAAGCACTTGTCTGATTCGC SEQID
NO:185
I171C GTTGGAGCCGTTGCAGAAGAACTTGTC SEQ ID
NO:186
R189C GGAGTCGTCAGGACAGGCAATCTCAGC SEQ ID
N0:187
Y2S9C GATGACCTCATAACACCACTCCCGCCG SEQID
N0:188
N294C GGGCAAACGAAGGCAGGTGGTGCCACT SEQ ID
N0:189
R296C TTTCTTGGGCAAACAAAGGTTGGTGGT SEQ ID
N0:190
T390C CATAACAGTGCCGCAGGATGACTGTGA SEQ ID
N0:191
V393C AACAGCTCCCATACAAGTGCCCGTGGA SEQID
N0:192
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
Expression of Human BACEI Mutants
pB22 was transformed into BL2lstar E. coli (Invitrogen) by standard methods,
and a single colony
from the resulting plate was used to inoculate 50 mL of 2xYT broth containing
100 ~.g/mL
ampicillin. Following overnight growth at 37 C, 40 mL of the culture was used
to inoculate 1.5 L
of the same media, and after growth at 37 C to an OD at 600 nm of between 0.5
and 0.8, IPTG was
added to a final concentration of 1.0 mM and expression continued 3 h at 37 C.
Cells were
harvested by centrifugation at 4K rpm. Cell pellets were resuspended in 100 mL
buffer TE (10 mM
Tris-HCl, 1 mM EDTA, pH 8.0) and lysis was accomplished using a French Press
microfluidizer
(two passages). The crude extract, containing BACE1 as insoluble inclusion
bodies, was
centrifuged at 14K rpm for 15 minutes, and the resulting pellet washed by
resuspension in PBS (10
mM sodium phosphate, 150 mM NaCI, pH 7.4) followed by centrifugation at 14K
rpm for 20
minutes. Washed inclusion body pellets were solubilized in 50 mM CAPS, 8 M
urea, 1 mM
EDTA, and 100 mM (3-mercaptoethanol, pH 10, and remaining insoluble debris
removed by
centrifugation at 20K rpm for 30 minutes. BACEl was refolded by slow injection
of the urea-
solubilized protein to between 50 and 100 volumes of rapidly stirred water, or
10 mM NazC03, pH
10, followed by incubation at room temperature for 3-7 days. When BACE1
enzymatic activity no
longer increased over time, the pH of the refolding solution was adjusted to
8.0 by addition of 5
mM (final concentration) Tris-HCI, and loaded onto a Q-Sepharose column.
Protein was eluted
using a linear gradient of 0 to 500 mM NaCI in 10 mM Tris-HCI, pH 8Ø BACEl
was further
purified by S-Sepharose chromatography at pH 4.5. Purified enzyme was dialyzed
versus 20 mM
Tris, 0.125 M NaCI, pH 7.2 at 4 C, and stored at 4 C. Protein concentrations
were determined by
absorbance at 280nm, using s28o'°~° _ (0.74).
EXAMPLE 10
CLONING AND MUTAGENESIS OF MITO EN-ACTIVATED PROTEIN
KINASEIEXTRACELLULAR SIGNAL-REGULATED K1NASE K1NASE fMEK)
Mek-1 (accession number SWS Q02750) is a dual specificity lcinase that plays a
key role in cellular
proliferation and survival in response to mitogenic stimuli. Melc-1 is the
central component of a
three-kinase cascade commonly called a MAP kinase cascade. This Raf Melc-Erk
Icinase cascade
transmits information from cell surface receptors (e.g. EGFR, HER2, PDGFR,
FGFR, IGF, etc.) to
the nucleus. This pathway is upregulated in approximately 30% of all tumor
types, either through
overexpression of specific cell surface receptors (e.g. HER2 in breast
cancers) or through activating
mutations in Ras, a key upstream component of this pathway. Disruption of Mek-
1 function has
dramatic anti-tumor effects, both in cell culture and in animals. Mele-2
(accession number SWS
P3G507) is a dual specificity kinase that is both highly homologous (79%
identity) to Mek-1 and
coordinately expressed with Mek-1. Thus, Mek-1 and Mek-2 represent attractive
targets for the
56
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
development of novel anti-cancer therapeutics. There are no crystal structures
to date for Melc-1 or
Melc-2.
Cloning of human Melc-1 and Mek-2
Numbering of the wild type and mutant Mek-1 and Mek-2 residues begins at their
respective amino
termini, with residue number 1 being the initiation methionine, according to
the NCBI reported
sequences (NCBI accession number L05624 for Melc-1 and NCBI accession number
HUMMEK2F
for Mek-2). All standard cloning and mutagenesis steps were carried out
according to the
recommendations of the enzyme manufacturer.
The DNA encoding human Mek-1 was isolated from plasmid pUSE MEK1 (Upstate
Biotechnology) and inserted into plasmid pGEX-4T-1 (Amersham) in frame with
GST as follows.
First, pUSE MEKl was digested with NotI (New England Biolabs), the 3' overhang
filled in with
the Klenow fragment of DNA polymerase (New England Biolabs), and the 1193 by
product
encoding MEK1 was isolated from an agarose gel. pGEX-4T-1 was linearized by
digestion with
EcoRI (New England Biolabs) and the 3' overhang similarly filled in with the
Klenow fragment of
DNA polymerase (New England Biolabs). The MEK1 and pGEX-4T-1 DNA fragments
were then
ligated with T4 Iigase and amplified in E. coli strain Top 10F' (Invitrogen)
to generate plasmid
pGEX-MEKl .
The DNA encoding human Mek-2 was isolated from plasmid pUSE MEK2 (Upstate
Biotechnology) and inserted into plasmid pGEX-4T-1 (Amersham) in frame with
GST as follows.
First, pUSE MEK2 was digested with NotI (New England Biolabs), the 3' overhang
filled in with
the Klenow fragment of DNA polymerase (New England Biolabs), and the 1213 by
product
encoding MEK2 was isolated from an agarose gel. pGEX-4T-1 was linearized by
digestion with
EcoRI (New England Biolabs) and the 3' overhang similarly filled in with the
Klenow fragment of
DNA polymerase (New England Biolabs). The MEK2 and pGEX-4T-1 DNA fragments
were then
ligated with T4 ligase and amplified in E. coli strain ToplOF' (Invitrogen) to
generate plasmid
pGEX-MEK2.
Generation of Mek-1 and Mek-2 Cysteine Mutants
All mutagenesis steps were performed using long range PCR. Reactions contained
the parent
plasmid (2 ng/~1), sense strand mutant primer (0.5 ~,M), and antisense strand
mutant primer (0.5
~.M) that are unique to each reaction. In addition, all reactions contained
dNTPs (25 uM) and Pfu
polymerase (0.05 Units/ql; Stratagene). Reactions were incubated for one
minute at 95 C followed
by 16 cycles of (0.5 minutes at 95 C, 1 minute at 55 C, and 2 minutes at 68 C
) and a final 10
minutes at 6S C. Parent plasmid DNA was then digested with DpnI (New England
Biolabs) and
57
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
the remaining linear PCR product was transformed into E. coli strain TopIOF'
(Invitrogen).
Mutagenized plasmid DNA, the result of in vivo recombination and subsequent
amplification, was
purified using QIAquick (Qiagen) columns and verified by sequencing.
First, a 6xHIS epitope tag was introduced into pGEX-MEK1, at the carboxy
terminus of MEKl, to
generate pGEX-MEK1-HIS using the sense and antisense oligonucleotides MEK1-
6HIS-s and
MEK1-6HIS-as, resepectively. Similarly, a 6xHIS epitope tag was introduced
into pGEX- MEK2,
at the carboxy terminus of MEK2, to generate pGEX-MEK2-HIS using the sense and
antisense
oligonucleotides, IVIEK2-6HIS-s and MEK2-6HIS-as, resepectively.
MEK1-6HIS-s
CACGCTGCCAGCATCGGCGTCGACCCAACCCTGGTT
CCGCGTGGATCCCATCACCATCACCATCACTGAGCG
GCCAATTCCCGG
SEQ ID N0:193
MEK1-6HIS-as
CCGGGAATTGGCCGCTCAGTGATGGTGATGGTGATG
GGATCCACGCGGAACCAGGGTTGGGTCGACGCCGAT
GCTGGCAGCGTG
SEQ ID N0:194
MEK2-6HIS-s
ACGCGTACTGCAGTGGGCGTCGACCCAACCCTGGTT
~S CCGCGTGGATCCCATCACCATCACCATCACTGAGCG
GCCAATTCCCGG
SEQ ID N0:195
MEK2-6HIS-as
CCGGGAATTGGCCGCTCAGTGATGGTGATGGTGATG
30 GGATCCACGCGGAACCAGGGTTGGGTCGACGCCCAC
TGCAGTACGCGT
SEQ ID N0:196
Subsequently, 16 individual mutations were introduced into pGEX-MEK1-HIS.
Similarly, the
35 analogous 16 individual mutations were introduced into pGEX-MEK2-HIS. Each
of these
mutations introduces a cysteine into the MEK1 or MEK2 protein, and each is
named according to
the resultant amino acid substitution. For example, primer pair MEK1-N78C-
sense and MEK1-
N78C-antisense were used to introduce a cysteine in place of N78 of MEK1,
generating pGEX-
MEK1/N78C-HIS.
58
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
Mutagenic Oligonucleotides
MEKI-N78C-S GAGCTGGGGGCTGGCTGCGGCGGTGTGGTGTTCSEQID
N0:197
MEK1-N78C-as G~CACCACACCGCCGCAGCCAGCCCCCAGCTCSEQ ID .
N0:198
MEK1-G79C-S CTGGGGGCTGGCAATTGCGGTGTGGTGTTCAAGSEQID
N0:199
MEK1-G79C-as CTTGAACACCACACCGCAATTGCCAGCCCCCAGSEQ ID
N0:200
MEK1-I107C-S GAGATCAAACCCGCATGCCGGAACCAGATCATASEQ ID
N0:201
MEK1-I107C-asTATGATCTGGTTCCGGCATGCGGGTTTGATCTCSEQ ID
N0:202
MEK1-R108C-S ATCAAACCCGCAATCTGCAACCAGATCATAAGGSEQID
N0:203
MEK1-R1O8C-aSCCTTATGATCTGGTTGCAGATTGCGGGTTTGATSEQ ID
N0:204
MEK1-I111C-S GCAATCCGGAACCAGTGCATAAGGGAGCTGCAGSEQ ID
N0:205
MEK1-II l CTGCAGCTCCCTTATGCACTGGTTCCGGATTGCSEQ ID
IC-as
N0:206
MEKl-EI 14C-S~CCAGATCATAAGGTGCCTGCAGGTTCTGCATSEQ ID
N0:207
MEK1-E114C-asATGCAGAACCTGCAGGCACCTTATGATCTGGTTSEQ ID
N0:208
MEK1-L118C-S AGGGAGCTGCAGGTTTGCCATGAGTGCAACTCTSEQ ID
N0:209
MEKl-LI 18C-aSAGAGTTGCACTCATGGCAAACCTGCAGCTCCCTSEQ ID
N0:2I0
MEK1-V127C-S ~CTCTCCGTACATCTGCGGCTTCTATGGTGCGSEQ ID
N0:211
MEK1-V127C-asCGCACCATAGAAGCCGCAGATGTACGGAGAGTTSEQ ID
N0:212
MEK1-M143C-s GAGATCAGTATCTGCTGCGAGCACATGGATGGASEQ ID
N0:213
MEKI-M143C- TCCATCCATGTGCTCGCAGCAGATACTGATCTCSEQ ID
as N0:214
MEK1-S15OC-S CACATGGATGGAGGTTGCCTGGATCAAGTCCTGSEQ ID
N0:215
MEK1-S150C-asCAGGACTTGATCCAGGCAACCTCCATCCATGTGSEQ ID
N0:2I6
MEK1-L180C-s ~GGCCTGACATATTGCAGGGAGAAGCACAAG SEQ ID
N0:217
MEK1-L180C-asCTTGTGCTTCTCCCTGCAATATGTCAGGCCTTTSEQ ID
N0:218
MEK1-I186C-5 AGGGAGAAGCACAAGTGCATGCACAGAGATGTCSEQ ID
N0:219
59
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
MEKl-I186C-asGACATCTCTGTGCATGCACTTGTGCTTCTCCCTSEQ ID
N0:220
MEKl-K192C-S ATGCACAGAGATGTCTGCCCCTCCAACATCCTASEQ ID
N0:221
MEK1-K192C-asTAGGATGTTGGAGGGGCAGACATCTCTGTGCATSEQ ID
N0:222
MEK1-S194C-S AGAGATGTCAAGCCCTGCAACATCCTAGTCAACSEQ ID
N0:223
MEKl-S194C-asGTTGACTAGGATGTTGCAGGGCTTGACATCTCTSEQ ID
N0:224
MEKI-LI97C-s AAGCCCTCCAACATCTGCGTCAACTCCCGTGGGSEQ ID
NO:225
MEK1-L197C-asCCCACGGGAGTTGACGCAGATGTTGGAGGGCTTSEQ ID
N0:226
MEK1-V211C-S CTCTGTGACTTTGGGTGCAGCGGGCAGCTCATCSEQID
N0:227
MEKl-V211C-asGATGAGCTGCCCGCTGCACCCAAAGTCACAGAGSEQ ID
N0:228
MEK2-N82C-SGAGCTGGGCGCGGGCTGCGGCGGGGTGGTCACCSEQID
N0:229
MEK2-N82C-asGGTGACCACCCCGCCGCAGCCCGCGCCCAGCTCSEQ ID
N0:230
MEK2-G83C-SCTGGGCGCGGGCAACTGCGGGGTGGTCACCAAASEQ ID
N0:23
I
MEK2-G83C-asTTTGGTGACCACCCCGCAGTTGCCCGCGCCCAGSEQ ID
NO:232
MEK2-I111C-sGAGATCAAGCCGGCCTGCCGGAACCAGATCATCSEQ ID
N0:233
MEK2-I111C-asGATGATCTGGTTCCGGCAGGCCGGCTTGATCTCSEQ ID
N0:234
MEK2-RI12C-SATCAAGCCGGCCATCTGCAACCAGATCATCCGCSEQ ID
N0:235
MEK2-R112C-asGCGGATGATCTGGTTGCAGATGGCCGGCTTGATSEQ ID
N0:236
MEK2-II GCCATCCGGAACCAGTGCATCCGCGAGCTGCAGSEQ ID
15C-s
N0:237
MEK2-II CTGCAGCTCGCGGATGCACTGGTTCCGGATGGCSEQ ID
15C-as
N0:23
8
MEK2-E118C-SAACCAGATCATCCGCTGCCTGCAGGTCCTGCACSEQ ID
N0:23
9
MEK2-E118C-asGTGCAGGACCTGCAGGCAGCGGATGATCTGGTTSEQ ID
N0:240
MEK2-LI22C-sCGCGAGCTGCAGGTCTGCCACGAATGCAACTCGSEQ ID
NO:241
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
MEK2-L122C-asCGAGTTGCATTCGTGGCAGACCTGCAGCTCGCGSEQ ID
N0:242
MEK2-V131C-SAACTCGCCGTACATCTGCGGCTTCTACGGGGCCSEQ ID
N0:243
MEK2-V131C-asGGCCCCGTAGAAGCCGCAGATGTACGGCGAGTTSEQ ID
N0:244
MEK2-M147C-SGAGATCAGCATTTGCTGCGAACACATGGACGGCSEQ ID
N0:245
MEK2-MI47C- GCCGTCCATGTGTTCGCAGCAAATGCTGATCTCSEQID
as N0:246
MEK2-SI54C-SCACATGGACGGCGGCTGCCTGGACCAGGTGCTGSEQ ID
N0:247
MEK2-S154C-asCAGCACCTGGTCCAGGCAGCCGCCGTCCATGTGSEQ ID
N0:248
MEK2-L184C-SCGGGGCTTGGCGTACTGCCGAGAGAAGCACCAGSEQ ID
N0:249
MEK2-L184C-aSCTGGTGCTTCTCTCGGCAGTACGCCAAGCCCCGSEQ ID
N0;250
MEK2-I190C-SCGAGAGAAGCACCAGTGCATGCACCGAGATGTGSEQ ID
N0:251
MEK2-I190C-asCACATCTCGGTGCATGCACTGGTGCTTCTCTCGSEQ ID
N0:252
MEK2-KI96C-SATGCACCGAGATGTGTGCCCCTCCAACATCCTCSEQ ID
N0:253
MEK2-K196C-asGAGGATGTTGGAGGGGCACACATCTCGGTGCATSEQ ID
N0:254
MEK2-S198C-SCGAGATGTGAAGCCCTGCAACATCCTCGTGAACSEQ ID
N0:255
MEK2-S198C-asGTTCACGAGGATGTTGCAGGGCTTCACATCTCGSEQ ID
N0:256
MEK2-L201C-5AAGCCCTCCAACATCTGCGTGAACTCTAGAGGGSEQ ID
N0:257
MEK2-L201C-asCCCTCTAGAGTTCACGCAGATGTTGGAGGGCTTSEQ ID
N0:25
8
MEK2-V215C-SCTGTGTGACTTCGGGTGCAGCGGCCAGCTCATASEQID
N0:259
MEK2-V215C-aSTATGAGCTGGCCGCTGCACCCGAAGTCACACAGSEQ ID
N0:260
Sequencing primers
pGEX forward GGGCTGGCAAGCCACGTTTGGTG SEQ ID
N0:261
pGEX reverse CCGGGAGCTGCATGTGTCAGAGG SEQ ID
N0:262
61
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
Expression of Mek-1 and Mek-2 mutants
Mutant alleles of Mek-1 and Mek-2 were expressed in E. coli and purified
essentially as described
for Mek-1 [by McDonald, O. B., et al., Analytical Biochern. 268: 3I8-329
(I999)]. Plasmids
S containing the mutant Mek-1 and Mek-2 alleles were transformed into BL21 DE3
pLysS cells
(Invitrogen) according to manufacturer's suggestions. Cultures were grown
overnight at 37 C
from single colonies in 100 ml 2YT medium supplemented with 100 ~glml
ampicillin and 100
~,g/ml chloramphenicol. This culture was then added to 1.5 L 2YT supplemented
with 100 ~,g/ml
ampicillin to achieve an ODGO° of approximately 0.05 and then grown to
an OD~oo of approximately
0.7 at 30 C. Expression was induced with the addition of IPTG to a final
concentration of 1 mM
and the culture was incubated for four hours at 2S C. Cells were pelleted in a
Sorfall GSA rotor at
6I~ rpm for 15 minutes and stored at -80 C.
Mek-1 and Mek-2 mutants were purified from cells by first resuspending cell
pellets in ice cold
1S PBS containing 0.5% Triton X-100 and incubating on ice for 4S minutes,
followed by extensive
sonication. Lysates were clarified by centrifugation in a Sorvall GSA rotor at
12K rpm for one
hour. Fusion proteins were first purified on Ni-NTA resin (Qiagen) according
to manufacturer's
suggestions, followed by further purification on glutathione agarose as
described [by McDonald, O.
B., et al., Analytical Biocherra. 268: 318-329 (1999)]. Epitope tags were
removed with thrombin
cleavage and aliquots of purified protein were stored at -80 C in TBS
containing 10% glycerol.
EXAMPLE 11
CLONING AND MUTAGENESIS OF HUMAN CATHEPSIN S ICATS~
Cathepsin S (accession number SWS P25774) is a thiol protease located
primarily in the lysosome.
2S This enzyme plays roles in antigen presentation by processing of the MHC-II
antigen receptor; thus
inhibitors to the enzyme could be used for diseases such as inflammation and
autoimmunity such as
rheumatoid arthritis, multiple sclerosis, asthma and organ rejection. It has
also been reported that
cats is present in increased levels in the Alzheimer's disease and Down
Syndrome brain compared
with normal brain. A structural model of cathepsin S [1BXF, Fengler, A. &
Brandt W., Protein
Eng 11:1007-1013(1998)] and a crystal structure of the C25S mutant
[Turkenburg, J. P. et al. Acta
Crystallogr D Biol Crystallog 58: 4S 1-4SS (2002)] are available.
Cloning of human cats
The DNA sequence encoding human cathepsin S (cats) was isolated by PCR from
the plasmid
pDualGC (Stratagene #EOI089) using PCR primers listed below corresponding to
the protein N-
and C-termini. These primers were designed to contain restriction endonuclease
sites EcoRI and
XhoI, for subcloning into a modified pFastBac vector, pFBHT (c.~ example 4
above). SEQ ID
G2
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
NO: 263 was used with SEQ ID NO: 264 and SEQ ID NO 265 to make cats with and
without a
6xhis tag, respectively.
5' Cats EcoRI CCGGAATTCATGAAACGGCTGGTTTGTGTGCT SEQ ID N0:263
3' Cats XhoI CCCCGCTCGAGGATTTCTGGGTAAGAGGGAAAG SEQ ID N0:264
3'CatS XhoI stop CCCCGCTCGAGCTAGATTTCTGGGTAAGAGGGAAA SEQ ID N0:265
S The PCR reaction was purified on a Qiaquick PCR purification column
(Qiagen). The PCR product
containing the cats sequence was cut with restriction endonucleases (42 ~l PCR
product, 1 ~1 each
endonuclease, 5 ~tl appropriate lOx buffer; incubated at 37 C for 3 hours).
The pFBHT vector was
cut with restriction endonucleases (5 pg DNA, 1 ~1 each endonuclease, 3 ~l
appropriate lOx buffer,
water to 30 ~1; incubated at 37 C for 3 hours; added 1 ~1 CIP and incubated at
37 C for 6Q
minutes). The products of nuclease cleavage were isolated from an agarose gel
(1% agarose, TBE
buffer) and ligated together using T4 DNA ligase (50 ng pFBHT vector and 50 ng
catS PCR
product in 10 ~,1, 10 ~.l 2x ligase buffer (Roche), 1 ~.l ligase, incubated at
25 C for 15 minutes). 1 ~l
of the ligation reaction was transformed into Library Efficiency Chemically
Competent DHSa cells
(Invitrogen) (1 ~l ligation reaction, 100 p1 competent cells; incubated at 4 C
for 30 minutes, 42 C
for 45 seconds, 4 C for 2 minutes, then 900 ~1 SOC media was added and
incubated for 1 hour
with shaking at 225 rpm at 37 C), and plated onto LB/agar plates containing
100 ~.g/ml ampicillin.
After incubation at 37 C overnight, single colonies were grown in 3 ml LB
media containing 100
ug/ml ampicillin for 8 hours. Cells were then isolated and double-stranded DNA
extracted from the
cells using a Qiagen DNA miniprep lcit. Sequencing of cats gene was
accomplished using
M13/pUC Forward and Reverse Amplification Primers (Invitrogen # 18430-017).
Generation of Cats Cysteine Mutations
Mutations were generated using as previously described [Kunlcel T. A., et al.,
Methoels_Erazynaol.
154: 367-382 (1987)]. DNA oligonucleotides used are shown below and were
designed to
hybridize with sense strand DNA from plasmid. Sequences were verified using
primers with SEQ
ID NO:74 and SEQ ID N0:75.
Mutagenic Oligonucleotides
Y1 SC CACAAGAACCTTGACATTTCACTTCAGT SEQ ID NO:266
K64C CACCATTGCAGCCACAGTTTCCATATTT SEQ ID NO:267
63
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
N67C CATGAAGCCACCACAGCAGCCTTTGTT SEQ ID NO:268
T72C CTGGAAAGCCGTGCACATGAAGCCACC SEQ ID N0:269
E11SC GCCATAAGGAAGGCAAGTGTACTTTGA SEQ ID NO:Z7O
R141C GAAAGAAGGATGACACGCATCTACACC SEQ ID NO:271
F146C ACTTCTGTAGAGGCAGAAAGAAGGATG SEQ ID NO:272
F211C TGGGTAAGAGGGACAGCTAGCAATCCC SEQID NO:273
Scrub mutations of the cysteines were also made using the following
oligonucleotides.
C12A CACTTCAGTAACAGCCCCTTTCTCTCTC SEQ ID NO:274
C12Y CACTTCAGTAACATACCCTTTCTCTCTC SEQ ID NO:275
C25S CACTGAAAGCCCAGGAAGCACCACAAGA SEQ ID N0:27G
C110A CAGTGTACTTTGAAGCTGTGGCAGCACG SEQ ID NO:277
Expression of Cats Mutant Proteins
All Cats-FBHT plasmids were site-specifically transposed into the baculovirus
shuttle vector
(bacmid) by transforming the plasmids into DHlObac (Gibco/BRL) competent cells
as follows: I
~,1 DNA at 5 ng/pl, lOpl Sx KCM [0.5 M KCI, 0.15 M CaClz, 0.25 M MgCl2], 30 p1
water was
mixed with 50 p,1 PEG-DMSO competent cells, incubated at 4 C for 20 minutes,
25 C for 10
minutes, added 900 p.1 SOC and incubated at 37 C with shaking for 4 hours,
then plated onto
LB/agar plates containing 50 ~.glml kanamycin, 7 ~.glml gentamycin, 10 pg/ml
tetracycline, 100
~,g/rnl Bluo-gal, 10 ~g/ml IPTG. After incubation at 37 C for 24 hours, large
white colonies were
picked and grown in 3 ml 2YT media overnight. Cells were then isolated and
double-stranded
DNA was extracted from the cells as follows: pellet was resuspended in 250 ~l
of Solution 1 [15
mM Tris-HCl (pH 8.0), 10 mM EDTA, 100 ~,g/ml RNase A]. Added 250 ~,l of
Solution 2 [0.2 N
NaOH, 1% SDS] mixed gently and incubated at room temperature for 5 minutes.
Added 250 p,1 3
M potassium acetate, mixed and placed on ice for 10 minutes. Centrifuged 10
minutes at 14,OOOx g
and transferred supernatant to a tube containing 0.8 ml isopropanol. Mix and
place on ice for 10
minutes. Centrifuge 15 minutes at 14,OOOx g, wash with 70% ethanol, air dry
pellet and
resuspended DNA in 40 ~l TE.
64
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
The bacmid DNA was used to transfect Sf9 cells. Sf9 cells were seeded at 9 x
lOs cells per 3S mrn
well in 2 ml of Sf 900 II SFM medium containing O.Sx concentration of
antibiotic-antimycotic and
allowed to attach at 27 C for 1 hour. During this time, 5 ~,l of bacmid DNA
was diluted into 100
~1 of medium without antibiotics, 6 ~l of CeIIFECTIN reagent was diluted into
100 ~l of medium
without antibiotics and then the 2 solutions were mixed gently and allowed to
incubate for 30
minutes at room temperature. The cells were washed once with medium without
antibiotics, the
medium was aspirated and then 0.8 ml of medium was added to the lipid-DNA
complex and
overlaid onto the cells. The cells were incubated for 5 hours at 27 C, the
transfection medium was
removed and 2 ml of medium with antibiotics was added. The cells were
incubated for 72 hours at
27 C and the virus was harvested from the cell culture medium.
The virus was amplified by adding 1.0 ml of virus to a 50 ml culture of Sf~
cells at 2 x 10~ cells/ml
and incubating at 27 C for 72 hours. The virus was harvested from the cell
culture medium and
this stock was used to express the various cats constructs in High-Five cells.
A 1 L culture of
High-Five cells at 2 x 10~ cells/ml was infected with virus at an approximate
MOI of 2 and
incubated for 72 hours. Cells were pelleted by centrifugation and the
supernatant was dialyzed
against 20 L Load buffer (50 mM NaHZP04, pH 8.0, 300 mM NaCI, 10 mM
imidazole), filtered and
loaded onto a Ni-NTA (Superflow Ni-NTA, Qiagen) column at 1 ml/min, washed
with Load buffer
at 2 ml/min and eluted with 50 mM NaH2P04, pH 8.0, 300 mM NaCI, 250 mM
imidazole.
EXAMPLE 12
CASPASE-1
Caspase-1 (accession number SWS P25774), like other caspases exists as an
inactive proform, and
is proteolytically processed into a large subunit and a small subunit, which
then combine to form
the active enzyme. An important substrate of caspase-1 is the proform of
interleukin-1 (beta).
Caspase-1 produces the active form of this cytokine, which plays a role in
processes such as
inflammation, septic shock and wound healing. Additionally, active capase-1
induces apoptosis,
and plays a role in the progression Huntington's disease. The structure of
caspase-1 has been
solved [1BMQ, Okamoto, Y., et al., Chern Phar~rn Bull (Tolcyo), 47:11-21
(1999)].
IL1313
IL-13 (accession number SWS P35225), which is produced mainly by activated Th2
cells, shows
structural and functional similarities to IL-4. Like IL-4, it increases the
secretion of
immunoglobulin E by B cells and is involved in the expulsion of parasites. In
addition, IL-13
downregulates the production of cytokines including IL-lb, IL-6, TNF-alpha and
IL-8 by
stimulated monocytes. IL-13 also prolongs monocyte survival, increases the
expression of MHC
class II and CD23 on the surface of monocytes, and increases expression of
CD23 on B cells.
6S
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
Furthermore, IL-2 and IL-13 synergize in the regulation interferon-gamma
synthesis. Due to these
effects, IL-13 plays a role in conditions such as allergy and asthma. In
particular, a polymorphism
at position 130 (Q) increases the risk of asthma development. The structure of
IL-13 has been
solved by nuclear magnetic resonance (NMR) [1GA3, Eissenmesser, E. Z. et al.,
J. Mol. Biol. 310:
231-241 (2001)].
CD40L
CD40L (accession number SWS P29965) is a protein that is found in two forms, a
transmembrane
form and also an active, proteolytically processed, extracellular soluble
form. The transmembrane
form is expressed on the surface of CD4+ T lymphocytes. Like other members of
the TNF family,
it is forms a homotrimer. CD40L mediates the proliferation of B cells,
epithelial cells, fibroblasts,
and smooth muscle cells. Binding of CD40L to the CD40 receptor on T cells
provides a critical
signal for isotype class switching and production of immunoglobulin
antibodies. Defects in CD40L
lead to an elevation in IgM levels, and an deficiency in all other
immunoglobulin subtypes.
Inhibitors to CD40L would find use in the treatment of autoimmune disease and
graft rejection. In
addition, reduced interaction between CD40L and its receptor reduces the
degree of tau
hyperphosphorylation in a mouse model of Alzheimer's disease. The crystal
structure of CD40L
has been solved [lALY, Karpusas, M., et al., Structure 3:1031-1039(1995),
erratum in Str~ueture
3:1046 (1995)].
HUMAN B-CELL ACTIVATING FACTOR (BAFFI
A member of the TNF superfamily, BAFF (accession number SWS Q9Y275) is a
homotrimer and
found in both transmembrane and soluble forms. The transmembrane form is
processed by the
furin family of proprotein convertases. BAFF is upregulated by interferon-
gamma and
downregulated by PMA/ionomycin treatment. BAFF binds to three different
receptors. When it
binds to the B-cell specific receptor (BAFFR), it promotes survival of B-cells
and the B-cell
response. Furthermore, both BAFF and a proliferation-inducing ligand (APRIL)
bind to the
receptors transmembranc activator and CAML interactor (TACT) and B cell
maturation antigen
(BCMA), forming a 2 ligands-2 receptors pathway that is responsible for
stimulation of T-cell and
B-cell function and humoral immunity. Inhibitors of BAFF would serve as
therapeutics for
autoimmune diseases characterized by abnormal B-cell activity, such as
systemic lupus
erythematosis (SLE) and rheumatoid arthritis (RA). A structure of the soluble
protein is available
[1JH5, Liu, Y., et al., Cell, 108: 383-394 (2002)].
TUMOR SUPPRESSOR P53
P53 (accession number SWS P04637), a transcription factor that can suppress
tumor growth, binds
DNA as a homotetramer and is activated by phosphorylation of a serine residue.
There are two
66
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
mechanisms of tumor suppression, depending upon the cell type: induction of
growth arrest and
activation of apoptosis. PS3 controls cell growth by regulating expression of
a set of genes; for
example, it increases the transcription of an inhibitor of cyclin-dependent
kinases. Apoptosis
results from the p53-mediated stimulation of Bax or Fas expression, or the
decrease in Bcl2
expression. P53 is mutated or inactivated in about 60% of known cancers, and
is also often
overexpressed in a variety of tumor tissues. Reversible inhibitors of pS3
could be used as an
adjunct to conventional radio- and chemotherapy to prevent damage to nornlal
tissues during
treatment and its severe side effects. Such an inhibitor was shown to protect
mice from lethal doses
of radiation without the promotion of tumor formation. There is a crystal
structure of human p53
bound to Xerzopus laevis mdm2 protein [lYCQ, Kussie, P. H., et al., Scie~zce
274: 948-9S3 (1996)].
P53-BINDING PROTEIN MDM2
In response to DNA damage, p53 increases the transcription of the protein mdm2
(accession
number SWS Q00987). In a form of negative feedback, mdm2 inhibits pS3-induced
cell cycle
1 S arrest and apoptosis by two means. Firstly, mdm2 binds the transcriptional
activation domain of
pS3, reducing its transcriptional activation activity. Secondly, in the
presence of ubiquitin E1 and
E2, mdm2 serves as an ubiquitin protein ligase E3 for both itself and pS3. The
ubiquitination of
pS3 allows its export from the nucleus to the proteasome, where it is
destroyed. There are eight
isoforms of mdm2 that are produced by alternative splicing. They are mdm2,
mdm2-A, mdm2-Al,
mdm2-B, mdm2-C, mdm2-D, mdm2-E, and mdm2-alpha. Of these, mdm2-A, mdm2-B, mdm2-
C,
mdm2-D, and mdm2-E are observed in human cancers but not in normal tissues.
Mdm2
amplification has also been observed in certain tumor types, including soft
tissue sarcoma,
osteosarcoma, and glioblastoma. These tumors often contain wild type pS3.
Small molecule
inhibitors of mdm2 could promote the proapoptotic activity of the wild type
pS3 and find use in
cancer therapy. The structure of ~e~zopus laevis mdm2 in complex with human
pS3 has been
solved [IYCR, Kussie, P. H. et al., Science 274: 948-9S3 (1996)].
BB CL-XX
BcI-x (accession number SWS Q07817) is a member of the Bcl2 family of proteins
and has two
major isoforms produced by alternative splicing, bcl-x(L), bcl-x(S). The Iong
isoform, bcl-x(L) is
found in long-lived postmitotic cells and inhibits apoptosis, whereas the
short isoform, bcl-x(S), is
found in cells with a high turnover rate and promotes apoptosis. The long
isoform inhibits
apoptosis by binding to voltage-dependent anion channel (VDAC) and preventing
the release of
apopotosis activator cytochrome c from fihe mitochondrial membrane. This
antiapoptotic activity is
3S dependent upon the BH4 (bcI-2 homology) domain of Bcl-x(L); binding of this
protein to other
Bcl2 family members is dependent upon the BH1 and BH2 domains. Expression of
Bcl-x(L) has
been observed to be expressed primarily by the neoplastic cells in a majority
of lymphoma cases.
67
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
Inhibition of bcl-x(L) expression in several cell lines resulted in apoptosis.
Thus, due to its
antiapoptotic effects, bcl-x(L) is a target for cancer therapeutics.
Interestingly, binding of Bcl-x(L)
to another Bcl2 family member, the proapoptotic protein Bax, results in an
increase in apoptosis
(see below). A crystal structure of Bcl-x(L) has been solved [1MAZ, Muchmore,
S. W., et al.
Nature 381: 335=341 (1996)].
BAX
Bax [accession number SWS Q07812 (BAX alpha); SWS Q07814 (BAX beta); SWS
Q07815
(BAX gamma); SWS P55269 (BAX delta)] promotes apoptosis by binding to the
antiapoptotic
protein bcl-x(L), inducing the release of cytochrome c, and activating caspase-
3. Bax has several
isoforms produced by alternative splicing; some are membrane bound and others
axe cytoplasmic.
The BH3 domain of Bax is necessary for its binding to members of the anti-
apoptotic Bcl2 family.
Defects in Bax are observed in some cell lineages from hematopoietic cancers.
Bax agonists could
be used in cancer therapies, while Bax inhibitors could be used to counteract
neuronal cell death
resulting from ischemia, spinal cord injury, Parkinson's disease and
Alzheimer's disease. An NMR
structure of BAX has been solved (1F16, Suzuki, M., et al., Cell 103:645-654
(2000)].
CDC25A
CDC25A (accession number SWS P30304) is a dual-specificity phosphatase also
lrnown as M-
phase inducer phosphatase 1 (MPIl). Induced by cyclin B, CDC25A is required
for progression of
the cell cycle, and induces mitosis in a dosage-dependent manner. CDC25
directly
dephosphorylates CDC2, thereby decreasing its activity. It has also been
demonstrated in vitro that
CDC25 dephosphorylates CDK2 in complex with cyclinE. Elevated levels of CDC25
can trigger
uncontrolled cell growth and are linked with increased mortality in breast
cancer patients.
Activated CDC25A is also observed in degenerating neurons of the Alzheimer's
diseased brain. A
structure of the catalytic core has been solved [1C25, Fauman, F. B., et al.,
Cell 93: 617-625
(1998)].
CD28
CD28 (accession number SWS P10747) is a disulfide-linked homodimeric
transmembrane protein
expressed on activated B-cells and a subset of T-cells. This protein can bind
three others: B7-1,
B7-2, and CTLA-4. The interaction of CD28 with B7-1 and B7-2 present on the
surface of antigen
presenting cells (APCs) results in a co-stimulation of naive T-cell
activation, whereas subsequent
interaction of the same B7-1 and B7-2 molecules with CTLA-4 leads to an
attenuation of the T-cell
stimulation. CD28-associated signaling pathways are important therapeutic
targets for autoimmune
disease, graft vs. host disease (GVHD), graft rejection, and promotion of
immunity against tumors.
The structure of CD28 has not been solved to date.
68
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
B7
There are 2 B7 proteins: B7-1 (accession number SWS P33681), also known as
CD80, and B7-2
(accession number SWS P42081), also known as CD86. Both are highly
glycosylated
transmembrane proteins expressed on activated B-cells. ~ Early events in
immune response are
controlled by the interactions of these molecules with CD28 and CTLA-4 (see
above). Thus B7-1
and B7-2 make significant targets for therapeutics treating autoimmune
disease. A structure of the
soluble form of B7-1 has been solved [1DR9, Ikemizu, S., et al., Immunity 12:
51-60 (2000)] in
addition to a structure of B7-1 in complex with CTLA-4 [1I8L, Stamper, C. C.,
et al., Nature 410:
608-611 (2001)]. In addition, a structure of B7-2 in complex with CTLA-4 has
been solved [1I85,
Schwartz, J.-C. D., et al., Nature 410: 604-608 (2001)].
The immune system comprises in part the complement cascade, which is a set of
more than 20
proteins. CSa is one of these complement proteins; it is a cytolcine-like
activation product of C5.
CSa effects inflammation, and specifically has a role in the recruitment of
neutrophils in response to
bacterial infection. In sepsis, the life threatening spread of bacterial
toxins through the blood, the
effects of CSa are exhausted, due to an overexposure of the neutrophils to
excessive amounts of this
complement protein. Furthermore, expression levels of CSa receptor (accession
number SWS
P21730) are increased in certain vital organs during sepsis. Thus inhibitors
of CSa or the CSa
receptor could help in treating sepsis. Inhibitors of CSa could also be used
in the treatment of
bullous pemphigoid, the most common autoimmune blistering disease. Another
effect of CSa is its
synergy with the Abeta peptide to promote secretion of IL-1 and IL-6 in human
macrophage-like
THP-1 cells; CSa may therefore be involved in the pathogenesis of Alzheimer's
disease. Although
the structure of CSa has been solved by NMR [1KJS, Zhang, X, et al., Proteins
28: 261-267
(1997)], there is no structure of the CSa receptor to date.
AKT
Akt is an important component of the signaling pathway of growth factor
receptors. There are three
highly related Akt genes, Akt 1-3 (accession numbers SWS P31749, Aktl; SWS
P31751, Akt2;
SWS Q9Y243), which show compensatory effects for one another. However, they
have different
expression patterns, suggesting that each may have unique functions as well.
Each Alct is activated
by phosphorylation of multiple residues and is activated by the kinase ILK.
Binding of activated
Akt to PI3K (phosphatidyl inositol 3-Icinase) causes the translocation of the
active Akt to the
plasma membrane. Akt has pleiotropic effects leading to cell survival.
Additionally, Akt
amplification and elevated levels of Akt have been found in some types of
cancers. A crystal
structure of the kinase domain of Akt2, also known as PKB-(3, has recently
been obtained [Yang, J.,
et al., Molecular Fell 9: 1227-1240 (2002)]."
69
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
CD45
CD45 (accession number SWS P08575) is a receptor protein tyrosine phosphatase
that is primarily
Located in the plasma membrane of leukocytes; it has several isoforms
differing in the extracellular
domain, the significance of which is presently unknown. Substrates for CD45
include the kinases
lyc, fyn, and other src kinases. Additionally, CD45 engages in noncovalent
interactions with the
lymphocyte phosphatase associated protein (LPAP). CD45 is critical for
activation through the
antigen receptor on T cells and B cells, and may also be important for the
antigen-mediated
activation in other leukocytes. Dimerization of CD45 disables its function.
Inhibitors of CD45
could be used to prevent allograft rejection. There is no structure of CD45 to
date.
TYROSINE KINASE-TYPE CELL SURFACE RECEPTOR HER2
HER-2 (accession number SWS P04626), otherwise lrnown as ErbB2 is a receptor
tyrosine kinase
that is related to EGFR (ErbB 1). Although there are no known ligands for HER-
2 in isolation,
when HER-2 dimerizes with other members of the ErbB family, i.e., ErbBl, ErbB3
and ErbB4, the
dimeric complex can bind to a number of ligands. These Iigands include
heregulins, EGF,
betacellulin, and NRG, although binding depends upon which ErbB proteins are
in the heterodimer.
Ligand binding increases the phosphorylation of HER-2, and effects subsequent
intracellular
signaling steps. HER-2 is frequently overexpressed in breast cancer cells, and
this overexpression
may mediate their proliferation. Breast cancer cells overexpressing HER-2 are
also more
responsive to HER-2 inhibitors. HER-2 is also implicated in a number of other
cancers, such as
ovarian, prostate, lung, fallopian tube, osteosarcoma, and childhood
medulloblastoma. The
structure of this receptor has not yet been solved.
HUMAN GLYCOGEN SYNTHASE K1NASE-3 fGSK-3~
GSK-3 (accession numbers SWS P49840, GSK-3a; SWS P49841, GSK-3~3) is involved
in the
hormonal control of Myb, glycogen synthase, and c jun. The phosphorylation of
c jun by GSK-3
decreases the affinity of c-jun for DNA. . Additionally, GSK-3 is
phosphorylated by ILK-1 and
Akt-1. Phosphorylation by Aktl causes the inhibition of catalytic activity of
GSK-3, which
normally phosphorylates cyclin D, thereby targeting cyclin D for destruction.
The net effect of this
phosphorylation of GSK-3 is the promotion of cell survival. Increased GSK-3
activity has been
found in tissue from diabetic patients, consistent with its role in the
development of insulin
resistance. Furthermore, GSK-3~ is overexpressed in the Alzheimer's disease
brain, and this
overexpression is associated with tau protein hyperphosphorylation, a hallmark
of the disease.
Finally, the effects of some mood-stabilizing drugs such as lithium appear to
be mediated by
inhibition of GSK. Therefore it is possible that GSK-3 inhibitors would
increase the effectiveness
of some psychoactive drugs. There is a structure available for GSK-3(3 [IH$F,
Dajani,. R., et al.,
Cell 105: 721-732 (2001)].
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
ALPHA-E/BETA-7
The protein complex alpha-E/beta-7 is a transrnembrane integrin that plays a
role in lymphocyte
migration and homing. Specifically, the complex serves as a receptor for E-
cadherin. Alpha-E
(accession number SWS P38570) is made up of two subunits, a and a, the oe-
subunit itself is
composed of a light chain and a heavy chain linked by a disulfide bond.
Likewise, beta-7
(accession number SWS P26010) is also composed of a- and (I- subunits. The
alpha-E/beta-7
complex normally mediates the adhesion of infra-epithelial T lymphocytes to
mucosal epithelial cell
layers; it also plays a role in the dissemination of non-Hodgkin's lymphoma.
Furthermore, a
possible mechanism of inflammation involves migration of lymphocytes from the
gut epithelium to
other parts of the body. Changes in alpha-E/beta-7 levels have been observed
in a variety of
diseases. Elevated levels of this integrin have been observed in patients with
Systemic Lupus
Erythematosus (SLE), in the lung epithelium of patients with interstitial lung
disease, and in the
sinovial fluid of patients with rheumatoid arthritis. Altered patterns of
alpha-E/beta-7 expression
have been observed in patients with Crohn's disease, and antibodies to this
complex were shown to
prevent immunization-induced colitis in a mouse model. Hence, inhibitors to
this complex would
be valuable in the treatment of inflammation, especially mucosal inflammation.
'There are no
structures available for alpha-E or beta-7.
TISSUE FACTOR
Human tissue factor (accession number SWS P13726), also lrnown as
thromboplastin, is an integral
transmembrane protein that is normally located at the extravascular cell
surface. Upon injury to the
skin, tissue factor is exposed to blood and complexes with the active form of
coagulation enzyme
Factor VII, known as Factor VIIA (see below). Tissue factor can bind both the
inactive and active
forms of coagulation Factor VII, and is an obligate cofactor for Factor VIIA
in triggering the
coagulation cascade. Furthermore, since Tissue Factor plays a major role in
thrombosis, inhibition
of this factor would be expected to decrease the risk for clinical outcomes of
tlu-ombosis such as
atherosclerosis, arterial occlusion, stroke, and myocardial infarction. A
structure of the
extracellular domain of tissue factor has been solved [2HFT, Muller, Y. A., et
al., J Mol Biol
256:144-159 (1996)].
FACTOR VII
Factor VII (accession number SWS P08709) is the zymogen (inactive precursor)
form of the serine
protease coagulation Factor VIIa. More than 99% of this protease circulates in
the inactive single-
chain form; upon cleavage of an Arg-Ile peptide bond by one of several
factors, the active two-
chain form is produced. This two-chain form comprises a heavy chain and a
light chain, linked by a
disulfide bond. Enzymatic carboxylation of Glu residues in Factor VII, which
is dependent upon
vitamin K, allows the protein to bind calcium. In the presence of calcium and
the cofactor human
71
CA 02454246 2004-O1-14
WO 03/014308 PCT/US02/24921
tissue factor (see above), Factor VIIa cleaves Factor X and Factor IX to
produce their respective
active forms, which propagate the coagulation cascade. Defects in Factor VII
can lead to bleeding
disorders, where recombinant Factor VIIa finds use as a treatment. Conversely,
some
polymorphisms of the Factor VII gene have been associated with an increased
risk for myocardial
infarction, which is often caused by blood clots. Factor VII inhibitors are
expected to find use in
preventing heart disease. A structure of the zymogen form of factor VII in
complex with an
inhibitory peptide has been solved [1JBU, Eigenbrot, C., et al., Structure
9:627-636 (2001)].
72