Note: Descriptions are shown in the official language in which they were submitted.
CA 02457671 2009-10-27
23804-665
1
PROCESS FOR THE PREPARATION OF A
14-HYDROXYNORMORPHINONE DERIVATIVE
The invention relates to a process for the production of 14-
hydroxynormorphinone derivatives,
to a new synthetic route for producing noroxymorphone, as well as to new
intermediates in said
route.
Noroxymorphone is a key intermediate for the production of important medicinal
opioids, such
as naltrexone and naloxone. The common starting material for the production of
these opioids is
thebaine from which they are readily synthesized. However, thebaine has only a
low natural
abundance in poppy heads and opium. As the supply of thebaine is limited and
the demand is
increasing, many alternative approaches have been made for the preparation of
14-
hydroxymorphine derivatives. See for example EP 0,158,476, US 5,922,876, and
the references
cited therein.
Further, in an attempt to remove the requirement for (the preparation of)
thebaine, Coop et aL
(Tetrahedron 55 (1999), 11429-11436; WO 00/66588) recently described an
oxidative method
for the production of 14-hydroxycodeinone in a yield of 51% from codeinone,
using Co(OAc)3
as the metallic oxidant in acetic acid at room temperature. Other oxidative
conditions with
metallic oxidants, such as Co(OAc)3 under other conditions, FeC13, Co(OAc)2 in
combination
with several cooxidants, Ru04, Mn(OAc)3, Cu(OAc)2, and others, proved to be
not very useful
according to Coop.
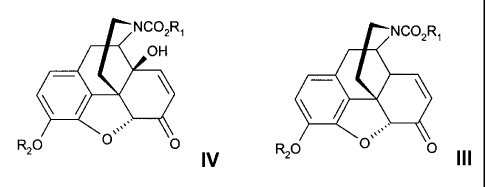
Surprisingly, and in spite of the findings of Coop, it has now been found that
in the production of
14-hydroxynormorphinone derivatives of formula IV from compounds of formula
III cobalt (Il)
salts can be used as efficient oxidants when the reaction is performed in the
presence of a mild
base and oxygen or air is used as cooxidant. Therefore, the invention relates
to a process for the
preparation of a 14-hydroxynormorphinone derivative of formula IV
ANC02R1
OH
R20 0`` O .
N
CA 02457671 2009-10-27
23804-665
2
comprising reacting the compound of formula III,
NC02'
R20 0 0 III
with a cobalt (II) oxidant in the presence of a mild base and air or oxygen as
the
cooxidant; wherein R, is (1-7C)alkyl optionally substituted with one or more
chlorines (such as 1,1,1-trichioroethyl), butenyl, vinyl, benzyl, phenyl or
naphthyl;
and R2 is benzyl or benzyl substituted with one or more (1-6C)alkoxy group or
benzyl substituted with one or more halogen.
According to one aspect of the present invention, there is provided a process
for
preparation of a 14-hydroxynormorphinone derivative of formula IV
NC02Ri
OH
R20 0 IV
comprising reacting a compound of formula III,
A ,
R20 0 0 III
with a cobalt (II) oxidant in the presence of a mild base and air or
oxygen as cooxidant;
CA 02457671 2009-10-27
23804-665
2a
wherein R, is (1 C-7C)alkyl optionally substituted with one or more
substituents wherein each substituent independently is chlorine, butenyl,
vinyl,
benzyl, phenyl or naphthyl;
and R2 is benzyl, benzyl independently substituted with one or more
(1 C-6C)alkoxy group or benzyl independently substituted with one or more
halogen.
According to another aspect of the present invention, there is provided a
morphinone derivative wherein the derivative is the compound of the formula
III as
described herein.
According to still another aspect of the present invention, there is provided
a
process for preparation of a compound of formula III, wherein the compound of
formula III is as described herein, the process comprising reactively
contacting a
morphine derivative of formula II
NCOZRi
R20 0 OH 11
wherein R, and R2 are as described herein, for the compound of
formula III, with an oxidizing agent effective for oxidizing allylic hydroxy
groups to
form keto groups.
According to yet another aspect of the present invention, there is provided a
process for production of noroxymorphone, comprising:
(a) a reaction step wherein a morphinone derivative of formula III as
described herein is oxidized into a 14-hydroxynormorphinone derivative of
formula IV as described herein,
(b) deprotecting the 3-position and reducing the double bond at the
7,8-position of the 14-hydroxynormorphinone derivative of formula IV to form a
3,14-hydroxynormorphinone derivative of formula V,
CA 02457671 2009-10-27
23804-665
2b
AOH ,
HO O O V
(c) wherein R, is as defined for the derivative of formula IV; and
hydrolyzing the 3,14-hydroxynormorphinone derivative of formula V into
noroxymorphone of formula VI,
AOH 5
HO o VI.
According to a further aspect of the present invention, there is provided a
process
for production of noroxymorphone, comprising:
(a) a reaction step comprising the oxidation of a compound of
formula II as described herein to form a morphinone derivative of formula III
as
described herein,
(b) a reaction step wherein a morphinone derivative of formula III as
described herein is oxidized into a 14-hydroxynormorphinone derivative of
formula IV as described herein,
(c) deprotecting the 3-position and reducing the double bond at the
7,8-position of the 14-hydroxynormorphinone derivative of formula IV to form a
3,14-hydroxynormorphinone derivative of formula V,
5'O V
CA 02457671 2009-10-27
23804-665
2c
(d) wherein R, is as defined for the derivative of formula IV; and
hydrolyzing the 3,14-hydroxynormorphinone derivative of formula V into
noroxymorphone of formula VI,
AOH H
O O O
VI.
According to yet a further aspect of the present invention, there is provided
a
process for production of noroxymorphone comprising
(a) converting morphine having the formula I
ANCH3
HO O OH I
by reaction with a haloformate ester of the formula X-C(=O)OR,,
wherein R, is as described herein and X is a halogen,
followed by a reaction with R2-X, wherein X is a halogen and R2 is as
described herein, to form a morphine derivative of formula II as described
herein;
(b) oxidizing the morphine of formula II to form a morphinone
derivative of formula III according to the process as described herein;
(c) oxidizing the morphinone derivative of formula III to form a 14-
hydroxynormorphinone derivative of formula IV according to the process as
described herein;
(d) deprotecting the 3-position and reducing the double bond at the
7,8-position of the 14-hydroxynormorphinone derivative of formula IV to form a
3,14-hydroxynormorphinone derivative of formula V,
CA 02457671 2009-10-27
23804-665
2d
AOH R~
HO 0 O V;
(e) and hydrolyzing the 3,14-hydroxynormorphinone derivative of
formula V into noroxymorphone of formula VI,
ACH H
O OO
VI.
The oxidation process of the present invention is an efficient process with
good
yields, which are significantly improved when compared to the process
described
by Coop et al.
The cobalt (II) oxidant according to the present invention may be selected
from a
range of cobalt (II) salts, such as CoF2, CoCl2, CoBr2, Co(II)sulfate,
Co(II)nitrate,
Co(II)acetate, Co(II)propionate, and the like, and mixtures thereof. The
preferred
oxidant in the process of this invention is Co(OAc)2 and the preferred
cooxidant is
air. The reaction mixture of this oxidation process is a heterogeneous system;
the
oxidant dissolves only in minor amounts in the organic solvent that is used.
The
amount of cobalt (II) salts used is not very critical, as long as the system
is
heterogeneous, and a skilled person will know to choose sufficient amounts
thereof. The cooxidant is introduced into the reaction mixture by bubbling it
through the solution, while stirring.
A person skilled in the art is aware what type of base are meant with the term
mild
bases, however preferred bases are sodium acetate, potassium acetate, sodium
phosphate and potassium phosphate. Most preferred is sodium acetate.
Preferably R1 is (1-7C)alkyl, and most preferred is ethyl. For R2 benzyl is
most
preferred. The oxidation process according to the present invention is
performed
CA 02457671 2009-10-27
23804-665
2e
in an organic solvent well-suited for dissolution of this type of compounds,
preferably (1-4C)alcohols or mixtures thereof. Preferred is ethanol.
CA 02457671 2004-02-11
WO 03/018588 PCT/EP02/09280
3
The reaction temperature is usually higher than room temperature, and may be
chosen dependent
on the boiling point of the solvent used. However, the temperature may not be
higher than about
100 C in order to keep the oxygen sufficiently in solution.
In the terms (1-7C)alkyl, (1-6C)alkoxy and (1-4C)alcohols the alkyl group is a
branched or
unbranched alkyl group having 1 to 7, 1 to 6 or 1 to 4 carbon atoms,
respectively, such as
methyl, ethyl, isopropyl, t-butyl, heptyl and the like.
The compound of formula III may suitably prepared by methods well known in the
art.
Preferably, the process for the preparation of a compound of formula III
comprises reactively
contacting a morphine derivative of formula II
NCOZRI
R20 C~ SOH II
with an oxidizing agent effective for oxidizing allylic hydroxy groups to form
keto groups, where
a morphinone compound of the formula III is prepared. Preferably, the
oxidizing agent is sodium
dichromate. Preferably Rl is ethyl. For R2 benzyl is most preferred.
The new process of this invention may conveniently be used in the production
of
noroxymorphone. Therefore, another aspect of this invention is a process for
the production of
noroxymorphone, comprising a reaction step wherein a morphinone compound of
formula III is
oxidized into the 14-hydroxynormorphinone derivative of formula IV. In
particular preferred is
the process further comprising the oxidation of a morphine derivative of
formula II into the
compound of formula III as described above.
Especially preferred is a process for the production of noroxymorphone
comprising the steps:
(a) converting morphine having the formula I
CA 02457671 2004-02-11
WO 03/018588 PCT/EP02/09280
4
NCH3
HO 0 "OH
by reaction with a haloformate ester of the formula X-C(=O)ORI, wherein RI is
as previously
defined and X is a halogen (F, Cl, Br or I, preferably Cl),
followed by a reaction with R2-X, wherein X (preferably Cl) and R2 are as
previously defined, to
form a morphine derivative of formula II;
(b) oxidizing the morphine of formula II to form a morphinone derivative of
formula III
according to the previously described process;
(c) oxidizing the morphinone derivative of formula III to form a 14-
hydroxynormorphinone
derivative of formula IV according to the previously described process;
(d) deprotecting the 3-position and (at the same time) reducing the double
bond at the 7,8-
position of the 14-hydroxynormorphinone derivative of formula IV to form a
3,14-hydroxynor-
morphinone derivative of formula V, using methods well known in the art for
such type of
reaction, e.g. using hydrogen and palladium-carbon as a catalyst,
cERI
H0 0 O V;
(e) and hydrolyzing the 3,14-hydroxynormorphinone derivative of formula V into
noroxymorphone of formula VI, using methods well known in the art for such
type of
hydrolysis, e.g. using sulfuric acid,
NH
OH
HO 0 VI.
In the process for the production of noroxymorphone, the novel intermediates
of formula II, III
and IV form each another aspect of the present invention. The intermediates of
formula II, III
CA 02457671 2004-02-11
WO 03/018588 PCT/EP02/09280
and IV are in particular preferred wherein R, is ethyl. Also preferred are
intermediates of
formula II, III and IV wherein R2 is benzyl. Most preferred are the
intermediates of formula II,
III and IV wherein R, is ethyl and R2 is benzyl.
5
The invention is further illustrated by the following example.
EXAMPLE 1
The underlined numbers refer to the numbers of the structures of Scheme I. (Bn
= benzyl).
(5a, 6a) (benzyloxy)-7 8-didehvdro-4 5-epox-6-_hydroxW orphinan-17-carboxylic
acid
ethylester (2)
Morphine (1, 8 g) was dissolved in 80 ml of toluene and the solution was dried
by azeotropic
distillation of water. Sodium carbonate (15 g) and sodium hydrogen carbonate
(6 g) were added
and the solution was again dried by azeotropic distillation. Ethyl
chloroformate (30 g) was
slowly and in portions added over a period of approximately 4 h at 78 C.
Completion of the
reaction was checked with TLC. The excess of reagent and the salts were
dissolved by addition
of water. The layers were separated and the toluene layer was washed with
water. The toluene
solution was evaporated to dryness and the residue was dissolved in 70 ml of
ethanol. The 3-
carboxylic acid ethyl ester group was saponified by 6 g potassium hydroxide
(dissolved in 18 ml
of ethanol), and 5 g potassium carbonate at 55 C. The pH was checked (in a 1:1
dilution in
water) and was >11. To this basic solution 5 g benzylchloride was added and
the reaction was
performed for 4 h at 75 C. The product was precipitated by the addition of
water (70 ml),
filtered, washed with water and dried. The yield of product (2) was 10 g. 1H
NMR (600 MHz,
CDC13) 5 1.29 (m, 3H), 1.92 (m, 2H), 2.52 (s, 1H), 2.72 (m, 2H), 2.85 (m, 1H),
3.01 (m, 1H),
4.01 (m, 1H), 4.17 (m, 3H), 4.87 (d, 111), 4.89 (d, 1H), 5.09 (d, 1H), 5.18
(d, 1H), 5.29 (t, 111),
5.72 (t, IH), 6.53 (d, 1H), 6.75 (d, 1H), 7.37 (m, 5H).
(5a)-3-(benzylox)-7 8-didehvdro-4 5-epoxy-6-oxomorphinan-17-carboxylic acid
ethylester (3)
A solution of Jones reagent was prepared by dissolving 7,5 g sodium
dichromate.2H20 in 22 ml
water and 6 ml sulfuric acid. Compound (2) (7,5 g) was dissolved in 60 ml
trichloro ethylene and
CA 02457671 2004-02-11
WO 03/018588 PCT/EP02/09280
6
28 ml water was added. The pH was adjusted to 5 with sulfuric acid. The
mixture was heated
under reflux and the Jones reagens was slowly added over a period of 1 h The
oxidation was
continued for another 1,5 h under reflux. The excess of oxidant was destroyed
with 6 ml 2-
propanol. The layers were separated and the organic layer was washed with 10%
sodium
hydrogen carbonate solution and water and dried with sodium sulfate. The
solution was
evaporated to dryness and the residue was dissolved in ethanol. Yield: - 9 g
product (3). 1H
NMR (200 MHz, CDC13) 6 1.28 (m, 3H), 1.92 (m, 2H), 2.8 (m, 2H), 2.9 (m, 111),
3.05 (m, 1H),
4.02 (m, 1H), 4.19 (m, 2H), 4.72 (s, 111), 5.03 (m, 1H), 5.18 (s, 2H), 6.12
(dd, 1H), 6.57 (d,
1H), 6.64 (m, 1H), 6.74 (d, 1H), 7.34 (m, 5H).
(5a)-3-(benzyloxy-7,8-didehydro-4,5-epoxy-14-hydroxy-6-oxomorphinan-17-
carboxylic acid
ethylester (4)
The solution of product (3) in ethanol (9 g in 135 ml) was heated to 60 C, 2,6
g cobalt (II)
acetate and 0,5 g sodium acetate were added and air was bubbled through the
solution under
vigorous stirring. The reaction was followed with TLC. After completion of the
reaction the
solution was treated with charcoal (0,3 g) and filtered. The solution was
distilled to volume and
this concentrated solution (6,3 g (4) in 53 ml of ethanol) was transferred to
the next step. 1H
NMR of 4 (360 MHz,'CH30H-d4) 6 1.28 (m, 311), 1.55 (m, 1H), 2.52 (m, 111),
2.74 (m, 111),
2.92 (m, 2H), 4.05 (m, 11-1), 4.15 (m, 2H), 4.64 (m, 111), 4.72 (s, 1H), 4.85
(m, 1H), 5.1 (s, 2H),
6.05 (d, 1H), 6.6 (d, 1H), 6.76 (d, 1H), 6.91 (m, 1H), 7.3 (m, 511).
(5a)-4,5-epoxy-3,14-dihydroxy-6-oxomorphinan-17-carboxylic acid ethylester (5)
To the solution of the previous step 6 ml of acetic acid was added. The
product (4) was reduced
with hydrogen and palladium carbon (5%) as a catalyst (0,9 g) at 20 C and
normal pressure.
After filtration and evaporation of ethanol 5,4 g of crude product (5) was
obtained. The product
was recrystallized from 2 parts (w/v) of ethyl acetate to obtain 4,7 g product
(5).
(5a -4,5-epoxy-3,14-dihydroxymorphinan-6-one (noroxymorphone) (6)
Product (5) (4,7 g) was dissolved in 28 ml of water and 5,6 ml of sulfuric
acid and refluxed for
approx. 24 h. The product was precipitated at pH = 9 by dilution with water
and 4,6 g of crude
product (6) was obtained after filtration and drying. The product was purified
by dissolution in
CA 02457671 2004-02-11
WO 03/018588 PCT/EP02/09280
7
ethanol, precipitation from this solvent at pH = 2, dissolution in water,
charcoal treatment and
precipitation at pH = 9. 1H NUR (400 MHz, DMSO-d6) 8 1.17 (m, 1H), 1.41 (m,
1H), 1.72 (m,
1H), 2.07 (m, 1H), 2.29 (m, 1H), 2.36 (m, 1H), 2.62 (m, 1H), 3.9 (m, 4H), 4.68
(s, 1H), 6.52
(d, 1H), 6.56 (d, 1H).
CA 02457671 2004-02-11
WO 03/018588 PCT/EP02/09280
8
SCHEME 1
NCH3 NCO2C2H5
HO 0 OH BnO 0 OH
1 2
NCOAH5 NCOAH5
OH
BnO OO BnO O``,` O
4 3
NCOAH5 NH
OH OH
HO OO HO OO
6