Note: Descriptions are shown in the official language in which they were submitted.
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
MAGNETO-OPTICAL BIO-DISCS AND SYSTEMS
INCLUDING RELATED METHODS
CROSS-REFERENCE TO RELATED APPLICATIONS
This application is a continuation-in-part of U.S. Application Serial No.
10/099,266 filed March 14, 2002 which is a continuation-in-part of U.S.
Application
Serial No. 09/997,741 filed November 27, 2001 which claimed the benefit of
priority
from U.S. Provisional Application Serial No. 60/253,283 filed November 27,
2000; U.S.
Provisional Application Serial No. 60/253,958 filed November 28, 2000; and
U.S.
Provisional Application Serial No. 60/272,525 filed March 1, 2001.
This application also claims the benefit of priority from U.S. Provisional
Application Serial No. 60/355,644 filed February 5, 2002; U.S. Provisional
Application
Serial No. 60/356,982 filed February 13, 2002; U.S. Provisional Application
Serial No.
60/358,479 filed February 19, 2002; U.S. Provisional Application Serial No.
60/372007 filed April 11, 2002; U.S. Provisional Application Serial No.
60/388,132
filed June 12, 2002; and U.S. Provisional Application Serial No. 60/408,227
filed
September 4, 2002.
Each of the above utility and provisional applications is herein incorporated
by
reference in its entirety.
BACKGROUND OF THE INVENTION
1. Field of the Invention
This invention relates in general to molecular and cellular biomagnetic assays
~ and, in particular, to molecular and cellular biomagnetic assays conducted
on optical
bio-discs. The invention further relates to magneto-optical (MO) analysis
discs and
M~ drive systems, hereinafter referred to as magneto-optical bio-disc systems
(MOBDS). More specifically, but without restriction to the particular
embodiments
hereinafter described in accordance with the best mode of practice, this
invention
relates to biomagnetic methods, including immunomagnetic methods, for
detection
and selective manipulation of specific target cells in cell populations and
solutions of
cell populations, using magnetic particles or beads, and to magnetically
guided neurite
growth and nerve regeneration.
1
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
2. Discussion of the Related Art
There is a significant need to make diagnostic assays and forensic assays of
all
types faster and more local to the end-user. Ideally, clinicians, patients,
investigators,
the military, other health care personnel, and consumers should be able to
test
themselves for the presence of certain factors or indicators in their systems,
and for
the presence of certain biological material at a crime scene or on a
battlefield. At
present, there are a number of silicon-based chips with nucleic acids and/or
proteins
attached thereto, which are commercially available or under development. These
chips are not for use by the end-user, or for use by persons or entities
lacking very
specialized expertise and expensive equipment.
SUMMARY OF THE INVENTION
The present invention relates to performing biomagnetic assays and laboratory
analysis, and particularly to using magnetic, paramagnetic, or
superparamagnetics
particles, herein referred to as bio-magnetic or magnetic particles or beads,
on optical
bio-discs, including, but not limited to, CD, CD-R, DVD, DVD-R, and MO discs.
The
biomagnetic assays of the present invention may include, for example,
immunomagnetic assays, and molecumagnetic assays such as assays using DNA
and RNA, implemented on non-magnetic and magnetic platforms. The non-magnetic
platforms may include, for example, microtiterplates and non-magneto-optical
bio-
discs systems. The magnetic platform includes, for example, the magneto-
optical bio-
disc system (MOBDS). Assays conducted using the MOBDS are herein referred to
as
MO bio-magnetic assays (MOBMA) and assays using non-magneto-optical bio-discs
are herein referred to as optical disc bio-magnetic assays (ODBMA). The ODBMA
may be carried out, for example, using a modified optical disc drive having a
controllable electromagnet associated therewith. The invention includes
methods for
preparing assays, methods for performing assays, methods for performing
laboratory
or clinical analysis, discs for performing assays or analysis, and related
detection
systems.
The biological sample can include blood, serum, plasma, cerebrospinal fluid,
breast aspirate, synovial fluid, pleural fluid, perintoneal fluid, pericardial
fluid, urine,
saliva, amniotic fluid, semen, mucus, a hair, feces, a biological particulate
suspension,
a single-stranded or double-stranded nucleic acid molecule, a cell, an organ,
a tissue,
2
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
or a tissue extract, or any other sample that includes a target that may be
bound to a
magnetic particle through chemical or biological processes. Further details
relating to
other aspects associated with the selection and detection of various targets
is
disclosed in, for example, commonly assigned co-pending U.S. Provisional
Patent
Application Serial No. 60/278,697 entitled "Dual Bead Assays for Detecting
Medical
Targets" filed March 26, 2001, which is incorporated herein by reference in
its entirety.
The target of interest can include tumor cells, bacteria, virus, or a target
agent
molecule such as a nucleic acid characteristic of a disease, or a nucleotide
sequence
specific for a person, or a nucleotide sequence or an antigenic determinant
specific for
an organism or cell type, which may be a bacterium, a virus, a mycoplasm, a
fungus, a
plant, or an animal. The target agent can include a nucleic acid molecule or
antigenic
determinant associated with cancer. The target nucleic acid molecule can
include a
nucleic acid, which is at least a portion of a gene selected from the group
consisting of
HER2neu, p52, p53, p21, and bcl-2. The target agent can be an antibody that is
present only in a subject infected with HIV-1, a viral protein antigen, or a
protein
characteristic of a disease state in a subject. The methods and apparatus of
the
present invention can be used for determining whether a subject is infected by
a virus,
whether nucleic acid obtained from a subject exhibits a single nucleotide
mutation
(SNM) relative to corresponding wild-type nucleic acid sequence, or whether a
subject
expresses a protein of interest, such as a bacterial protein, a fungal
protein, a viral
protein, an HIV protein, a hepatitis C protein, a hepatitis B protein, or a
protein known
to be specifically associated with a disease.An example of a dual bead
experiment
detecting a nucleic acid target is presented below in Example 1.
According to another aspect of the invention, there is provided multiplexing
methods wherein more than one target agent (e.g., tens, hundreds, or even
thousands
of different target agents) can be identified on one optical analysis disc.
Multiple
capture agents can be provided in a single chamber together in capture fields,
or
separately in separate capture fields. Different reporter beads can be used to
be
distinguishable from each other, such as beads that fluoresce at different
wavelengths
or different size reporter beads. Experiments were performed to identify two
different
targets using the multiplexing technique. An example of one such assay is
discussed
below in~Example 2.
3
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
In accordance with yet another aspect, the invention includes an optical disc
with a substrate, a capture layer associated with the substrate, and a capture
agent
bound to the capture layer, such that the capture agent binds to a dual bead
complex.
Multiple different capture agents can be used for different types of dual bead
complexes. The disc can be designed to allow for some dual bead processing on
the
disc with appropriate chambers and fluidic structures, and can be pre-loaded
with
reporter and capture beads so that only a sample needs to be added to form the
dual
bead complex structures.
ODBMA Cell Analysis
According to one aspect of the current invention, there is provided a disc and
disc drive system for performing bead or bead-cell assays. The disc drive can
include
an electromagnet for performing the isolation process, and may include
appropriate
light source control and detection for the type of reporter beads used. The
disc drive
can be optical or magneto-optical.
For processing performed on the disc, the drive may advantageously include an
electromagnet, and the disc preferably has a mixing chamber, a waste chamber,
and
capture area. In this embodiment, the sample is mixed with beads in the mixing
chamber, a magnetic field is applied adjacent the mixing chamber, and the
sample not
held by the magnet is directed to the waste chamber so that all magnetic
beads,
whether bound into a dual bead complex or unbound, remain in the mixing
chamber.
The magnetic beads are then directed to the capture area. One of a number of
different valuing arrangements can be used to control the flow.
The Ma neq to-Optical Bio-Disc S stem MOBDS~
In another aspect of the present invention, a MOBMA is performed using MO
discs made for use with biological samples and used in conjunction with a disc
drive,
such as a magneto-optical (MO) disc drive, that can selectively form magnetic
domains or regions on a disc. In this MOBMA aspect of the present invention,
magnetic domains can be formed in the MO disc in a highly controllable and
precise
manner. These domains may be employed advantageously to magnetically bind bio-
magnetic particles like magnetic beads, including unbound magnetic capture
beads,
and magnetic complexes including dual bead complexes with magnetic capture
4
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
beads, magnetic bead-cell complexes, or any biological or chemical complex
having at
least one magnetic particle or a magnetic property associated therewith so
that these
complexes may be bound using a magnet or magnetic domains. The MO disc drive
can write to selected locations on the disc, and then use an optical reader to
detect
features located at those domains or regions. The domains can be selectively
erased,
thereby allowing individual beads and complexes to be selectively released.
Biomedical applications related to using the MOBDS for analysis is described
below.
In still another aspect of the invention, there is provided a method of using
a
bio-disc and drive including forming magnetic domains or regions on the
optical bio
disc or medical CD. This method includes providing magnetic beads to the discs
so
that the beads bind at the magnetic domains. The method preferably further
includes
detecting at the locations where the magnetic beads bind biological samples,
preferably using reporter beads that are detectable, such as by fluorescence
or optical
event detection. The method can be formed in multiple stages in terms of time
or in
terms of location through the use of multiple chambers. The domains are
selectively
written at pre-determined locations on the MO bio-disc and a sample is moved
over
the magnetic domains in order to capture magnetic beads or magnetic complexes.
The regions can then be selectively erased and the magnetic beads or magnetic
complexes released individually and relocated if desired. This method allows
many
different tests to be performed at one time, and can allow a level of
interactivity
between the user and the disc drives such that additional tests can be created
during
the testing process. Further details relating to magneto-optical recording,
precise
creation of magnetic regions on a magneto-optical disc, and magneto-optical
detection
methods are disclosed in, for example, U. S. Patent No. 6,212,136 to Maeda at
al., U.
S. Patent No. 5,329,503 to Ohmori et al., U. S. Patent No. 4,985,881 to Saito
et al., U.
S. Patent No. 4,843,604 to Fujiwara et al., and U. S. Patent No. 4,748,606 to
Naito et
al. All of which are hereby incorporated by reference in their entireties.
The MO bio-disc may also be optimized for different types of MOBMA
applications including, for example, optimization of the optical properties of
the
magneto-optic stack of the MO bio-disc so that an incident tracking and
detection
beam of electromagnetic radiation is allowed to partially pass through the
reflective
layer and a transmitted beam detected using a separate detector and components
of
the operative layer of the MO bio-disc may be modified such that the magnetic
field
5
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
and strength of the magnetic field generated on the MO bio-disc is large and
strong
enough to capture and distinguish between magnetic complexes having different
types
of magnetic beads attached thereto. Furthermore, different types of
electromagnetic
radiation sources may be used in conjunction with the MOBDS including, but not
limited to, infra-red, red, blue, and fluorescent type optical sources. The
magnetic
beads and magnetic complexes may be detected, for example, optically by
analysis of
the characteristics of the reflected and/or transmitted beam, as discussed
below, for
example, in conjunction with Figs. 28A, 28B, 29A and 29B, or by fluorescence
using a
fluorescent type optical source in conjunction with a fluorescent detector and
fluorescent labelled target molecules or cells.
The bio-magnetic bead assays according to the present invention may be
implemented, in a genetic assay, herein referred to as the molecumagnetic
assay,
with magnetic capture beads and fluorescent reporter beads. These bio-magnetic
beads or particles are coated with capture probes and reporter probes
respectively.
The capture probes and reporter probes are complementary to a target sequence
but
not to each other. The capture beads are mixed with varying quantities of
target DNA.
Unbound target is removed from the solution by magnetic concentration of the
magnetic beads. Fluorescent reporter beads are then allowed to bind to the
captured
target DNA. Unbound reporter beads are removed by magnetic concentration of
the
magnetic beads. Thus, only in the presence of the target sequence, the
magnetic
capture beads bind to fluorescent reporter beads, resulting in a dual bead
assay.
Capture Probe Binding
A number of different surface chemistries and different methods for binding
the
probes to the beads were investigated including covalently conjugating the
capture or
reporter probe onto carboxylated capture beads and reporter beads,
respectively, via
EDC conjugation. One observed result using the EDC conjugation method of
attaching the probes on the beads was non-covalent attachment of the probes.
This
limitation was overcome by the development of a method for attaching the
probes
using partially double stranded DNA probes and by selection of an appropriate
bead
type with high conjugation efficiency. The use of double stranded probes in
the
conjugation process reduces the non-covalent attachment of probes to beads
significantly. By using appropriate bead type and conjugation conditions, the
covalent
6
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
conjugation efficiency was as high as 95%. Details relating to DNA probe
conjugation
onto solid surfaces is disclosed in, for example, commonly assigned U.S.
Patent
Application No. 10/087,547, entitled "Methods for Decreasing Non-Specific
Binding of
Beads in Dual Bead Assays Including Related Optical Bio-discs and Disc Drive
Systems", filed February, 28, 2002, which is herein incorporated by reference
in its
entirety.
The use of magnetic beads in the capture of target DNA speeds up the washing
steps and facilitates the separation steps between bound and unbound
significantly.
Furthermore, when the target concentration is limiting, each target molecule
may
hybridize to one reporter bead. Due to its size, a single target molecule is
not
detectable by any existing technologies. However, a 1 p,m or larger reporter
bead can
be easily detected and quantified by various methods. Therefore, the bead
assay
increases the sensitivity of the target capture tremendously.
Bio-Disc Drive and Related Signal Processing Systems
In yet another principal aspect, the present invention also involves
implementing the methods recited above on an analysis disc, modified optical
disc,
MOBDS, or a bio-disc. A bio-disc drive assembly may be employed to rotate the
disc,
read and process any encoded information stored on the disc, and analyze the
test
samples in a flow channel of the bio-disc. The bio-disc drive is thus provided
with a
motor for rotating the bio-disc, a controller for controlling the rate of
rotation of the
disc, a processor for processing return and/or transmitted signals form the
disc, and
an analyzer for analyzing the processed signals. The rotation rate of the
motor is
controlled to achieve the desired rotation of the disc. The bio-disc drive
assembly may
also be utilized to write information to the bio-disc either before or after
the test
material in the flow channel and target zones is interrogated by a read beam
of the
drive and analyzed by an analyzer. The bio-disc may include encoded
information for
controlling the rotation rate of the disc, providing processing information
specific to the
type of test to be conducted, and for displaying the results on a monitor
associated
with the bio-drive.
The various embodiments of the apparatus and methods of the present
invention can be designed for use by an end-user, inexpensively, without
specialized
expertise and expensive equipment. The system can be made portable, and thus
7
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
usable in remote locations where traditional diagnostic equipment may not
generally
be available. Other related aspects applicable to components of this assay
system ,
and signal acquisition methods are disclosed in commonly assigned and co-
pending
U.S. Patent Application Serial No. 10/038,297 entitled "Dual Bead Assays
Including
Covalent Linkages For Improved Specificity And Related Optical Analysis Discs"
filed
January 4, 2002; U.S. Provisional Application Serial No. 60/272,525 entitled
"Biological Assays Using Dual Bead Multiplexing Including Optical Bio-Disc and
Related Methods" filed March 1, 2001; and U.S. Provisional Application Serial
Nos.
60/275,643, 60/314,906, and 60/352,270 each entitled "Surface Assembly for
Immobilizing Capture Agents and Dual Bead Assays Including Optical Bio-Disc
and
Methods Relating Thereto" respectively filed March 14, 2001, August 24, 2001,
and
January 30, 2002. All of these applications are herein incorporated by
reference in
their entirety.
Other features and advantages will become apparent from the following
detailed description, drawing figures, and technical examples.
BRIEF DESCRIPTION OF THE DRAWING FIGURES
Further objects of the present invention together with additional features
contributing thereto and advantages accruing therefrom will be apparent from
the
following description of preferred embodiments of the present invention which
are
shown in the accompanying drawing figures with like reference numerals
indicating
like components throughout, wherein:
Fig. 1 is a perspective view of an optical disc system according to the
present
invention;
Fig. 2 is a block and pictorial diagram of an optical reading system according
to
embodiments of the present invention;
Figs. 3A, 3B, and 3C are respectively exploded, top, and perspective views of
a
reflective disc according to embodiments of the present invention;
Figs. 4A, 4B, and 4C are respectively exploded, top, and perspective views of
a
transmissive disc according to embodiments of the present invention;
Fig. 5A is a partial longitudinal cross sectional view of the reflective
optical bio-
disc shown in Figs. 3A, 3B, and 3C illustrating a wobble groove formed
therein;
8
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
Fig. 5B is a partial longitudinal cross sectional view of the transmissive
optical
bio-disc illustrated in Figs. 4A, 4B, and 4C showing a wobble groove formed
therein
and a top detector;
Fig. 6A is a partial radial cross-sectional view of the disc illustrated in
Fig. 5A;
Fig. 6B is a partial radial cross-sectional view of the disc illustrated in
Fig. 5B;
Figs. 7A, 8A, 9A, and 10A are schematic representations of a capture bead, a
reporter bead, and a dual bead complex as utilized in conjunction with genetic
assays;
Figs. 7B, 8B, 9B, and 10B are schematic representations of a capture bead, a
reporter bead, and a dual bead complex as employed in conjunction with
immunochemical assays;
Fig. 11 A is a pictorial representation of one embodiment of a method for
producing genetic dual bead complex solutions;
Fig. 11 B is a pictorial representation of one embodiment of a method for
producing immunochemical dual bead complex solutions;
Fig. 12A is a pictorial representation of another embodiment of a method for
producing genetic dual bead complex solutions;
Fig. 12B is a pictorial representation of another embodiment of a method for
producing immunochemical dual bead complex solutions;
Fig. 13 is a longitudinal cross sectional view illustrating the disc layers in
combination with a mixing or loading chamber;
Fig. 14 is a view similar to Fig. 13 showing the mixing chamber loaded with
dual
bead complex solution;
Figs. 15A and 15B are radial cross sectional views of the disc and target zone
illustrating one embodiment for binding of reporter beads to capture agents in
a
genetic assay;
Figs. 16A and 16B are radial cross sectional views of the disc and target zone
showing another embodiment for binding of reporter beads to capture agents in
a
genetic assay;
Fig. 17 is radial cross sectional view of the disc and target zone
illustrating one
embodiment for binding of capture beads to capture agents in a genetic assay;
Fig. 18 is radial cross sectional view of the disc and target zone depicting
another embodiment for binding of capture beads to capture agents in a genetic
assay;
9
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
Figs. 19A, 19B, and 19C are partial cross sectional views illustrating one
embodiment of a method according to this invention for binding the reporter
bead of a
dual bead complex to a capture layer in a genetic assay;
Figs. 20A, 20B, and 20C are partial cross sectional views showing one
embodiment of a method according to the present invention for binding the
reporter
bead of a dual bead complex to a capture layer in a immunochemical assay;
Figs. 21 A, 21 B, and 21 C are partial cross sectional views illustrating
another
embodiment of a method according to this invention for binding the reporter
bead of a
dual bead complex to a capture layer in a genetic assay;
Figs. 22A, 22B, and 22C are partial cross sectional views presenting another
embodiment of a method according to the invention for binding the reporter
bead of a
dual bead complex to a capture layer in a immunochemical assay;
Figs. 23A and 23B are partial cross sectional views depicting one embodiment
of
a method according to the present invention for binding the capture bead of a
dual
bead complex to a capture layer in a genetic assay;
Figs. 24A and 24B are partial cross sectional views showing another
embodiment of a method according to this invention for binding the capture
bead of a
dual bead complex to a capture layer in a genetic assay;
Figs. 25A-25D illustrate a method according to the present invention for
detecting the presence of target DNA or RNA in a genetic sample utilizing an
optical
bio-disc;
Figs. 26A-26D illustrate another method according to this invention for
detecting
the presence of target DNA or RNA in a genetic sample utilizing an optical bio-
disc;
Figs. 27A-27D illustrate a method according to the present invention for
detecting the presence of a target antigen in a biological test sample
utilizing an
optical bio-disc;
Fig. 28A is a graphical representation of an individual 2.1 um reporter bead
and
a 3 um capture bead positioned relative to the tracks of an optical bio-disc
according
to the present invention;
Fig. 28B is a series of signature traces derived from the beads of Fig. 28A
utilizing a detected signal from the optical drive according to the present
invention;
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
Fig. 29A is a graphical representation of a 2.1 um reporter bead and a 3 um
capture bead linked together in a dual bead complex positioned relative to the
tracks
of an optical bio-disc according to the present invention;
Fig. 29B is a series of signature traces derived from the dual bead complex of
Fig. 29A utilizing a detected signal from the optical drive according to this
invention;
Fig. 30A is a bar graph showing results from a dual bead assay according to
the present invention;
Fig. 30B is a graph showing a standard curve demonstrating the detection limit
for fluorescent beads detected with a flourimeter;
Fig. 30C is a pictorial representation demonstrating the formation of the dual
bead complex;
Fig. 31 is a bar graph showing the sensitivity of disc drive detection using a
dual bead complex;
Fig. 32 is a schematic representation of combining beads for dual bead assay
multiplexing according to embodiments of the present invention;
Fig. 33A is a schematic representation of a fluidic circuit according to the
present invention utilized in conjunction with a magnetic field generator to
control
movement of magnetic beads;
Figs. 33B-33D are schematics of a first fluidic circuit that implements the
valuing structure of FIG. 33A according to one embodiment of fluid transport
aspects
of the present invention;
Figs. 34A-34C are schematics of a second fluidic circuit that implements the
valuing structure of FIG. 33A according to another embodiment of the fluid
transport
aspects of this invention;
~ Fig. 35 is a perspective view of the magnetic field generator and a disc
including one embodiment of a fluidic circuit employed in conjunction with
magnetic
beads according to this invention;
Figs. 36A, 36B, and 36C are plan views illustrating a method of separation and
detection for dual bead assays using the fluidic circuit shown in Fig. 35;
Fig. 37 is a perspective view of a magneto-optical bio-disc showing magnetic
domains or regions, magnetically bound capture beads, and the formation of
dual
bead complexes according to another aspect of the present invention;
11
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
Fig. 38 shows the use of ligation to form a covalent bond between the capture
and reporter probes;
Fig. 39 is a bar graph showing the results from a genetic test detected by an
enzyme assay in a ligation experiment;
Fig. 40 is a bar graph comparing the number of beads bound as a function of
target concentration using 2.1 ~m reporter beads with and without ligation;
Fig. 41 is a bar graph comparing the number of beads bound as a function of
target concentration using a 39mer bridge with and without ligation; ,
Fig. 42A is schematic representation of various probe structures including DNA
sequences for use in a dual bead complex employing cleavable or displaceable
spacers according to the present invention;
Fig. 42B is pictorial diagrammatic representation showing a cleavable spacer
connecting a dual bead complex prior to binding of a target;
Fig. 42C is a view similar or Fig. 42B illustrating the cleavable spacer
including
a Notl connecting the dual bead complex after target binding;
Fig. 42D is a view similar to Fig. 42C depicting the dual bead complex after
target binding and after cleavage by Notl;
Fig. 43A is pictorial diagrammatic representation showing a displaceable
spacer connecting a dual bead complex prior to binding of a target;
Fig. 43B is a view similar to Fig. 43A illustrating initial binding of a
displacement
probe to the displaceable spacer connecting the dual bead complex after target
binding;
Fig. 43C is a view similar to Fig. 43B depicting complete displacement of the
displacement probe connecting the dual bead complex in the presence of target
mediated binding;
Fig. 44 is a pictorial representation of cleavable spacers covalently attached
to
a capture bead according to the present invention;
Fig. 45 is a view similar to Fig. 44 showing thiol groups attached to the
cleavable spacers binding covalently to a metallic reporter bead;
Fig. 46A is a pictorial representation of a pair of dual bead complexes bound
together by a cleavable spacer before target binding;
Fig. 46B is a view similar to Fig. 46A showing the dual bead complexes bound
together by the cleavable spacer after target binding and without target
binding;
12
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
Fig. 46C is a view similar to Fig. 46B showing one of the dual bead complexes
dissociated after enzyme cleavage and the other held together by the presence
of the
target;
Fig. 47A is a pictorial presentation of a dual bead complex formed by a pair
of
cleavable spacers and use of a bridge bound to a target;
Fig. 47B is a view similar to Fig. 47A after target binding including the
bridge
resulting in a double helix containing two breaks;
Fig. 47C is a view similar to Fig. 47B after restriction digestion of the
cleavable
spacers and ligation of the breaks in the double helix;
Fig. 48A is a pictorial representation of two dual bead complexes each joined
together by a pair of cleavable spacers as implemented in an immunochemical
assay
prior to target antigen binding;
Fig. 48B is a view similar to Fig. 48A showing the dual bead complexes bound
together by the cleavable spacer with and without target binding;
Fig. 48C is a view similar to Fig. 48B illustrating one of the dual bead
complexes dissociated after enzyme digestion and the other held together by
the
presence of the target;
Fig. 49 is a schematic presenting a method for evaluating a solid phase for
covalent conjugation of a probe;
Fig. 50 is a schematic detailing various steps in the quantification of
covalently-
bound and non-covalently bound probes to a solid substrate;
Fig. 51 A is a graphic presentation of experimental results of various tests
of
magnetic bead carriers for covalent linkage of a probe;
Fig. 51 B is a graphic presentation of experimental results of various tests
of
fluorescent bead carriers for covalent linkage of a probe;
Fig. 52A is a pictorial representation illustrating the structural differences
between single-stranded and double-stranded DNA that are relevant to their use
as
probes;
Fig. 52B is a graphic presentation of results of an experiment designed to
evaluate the binding properties of single-stranded and double-stranded DNA to
a solid
phase;
Fig. 53A is graphic presentation of enzyme assay results of a screen of two
different capture beads for use in a dual bead assay, these results indicating
that both
13
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
of the tested beads bind a similar amount of target regardless of whether the
probe is
bound covalently or non-covalently;
Fig. 53B is a graphic presentation of results of a dual bead assay designed to
examine the number of reporter beads captured by two different capture beads,
these
results indicate that covalent bonding of the probe to the capture bead
greatly
improves assay sensitivity;
Fig. 54 is a graphic presentation demonstrating that the introduction of PEG
linkers into probes significantly improves target mediated binding;
Fig. 55 is a bar graph presentation illustrating probe density determination
employing 3~,m beads;
Fig. 56 is a bar graph presentation demonstrating the pretreatment of the
beads
with various detergents including salmon sperm DNA which reduced nonspecific
binding by over 10 fold;
Fig. 57 is a bar graph presentation showing the range of detection of the dual
bead assay;
Fig. 58 is a bar graph illustrating the use of NaCI in varying concentrations
and
the related non-specific binding;
Fig. 59 is a bar graph presentation showing increasing EDTA concentration and
the related non-specific binding;
Fig. 60 is a bar graph presentation depicting an increasing NaCI concen-
tration
and the related non-specific binding;
Figs. 61 A and 61 B are bar graph presentations illustrating an increasing
concentration of MgCl2 and related non-specific binding;
Fig. 62 is a pictorial schematic representation showing the use of probe
blocking agents to increase the sensitivity of the bead assay;
Fig. 63 is a bar graph presentation illustrating the effect of incubation time
during a hybridization reaction;
Fig. 64 is a bar graph presentation showing a mixing method directed to
increasing efficiency in dual bead binding;
Figs. 65A and 65B together comprise a pictorial representation of another
embodiment of a method for producing genetic dual bead complex solutions
related to
the method discussed in connection with Fig. 11 A;
14
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
Figs. 66A and 66B taken together form a pictorial representation of another
embodiment of a method for producing immunochemical dual bead complex
solutions
similar to that shown in Fig 11 B;
Figs. 67A and 67B together show a pictorial representation of yet another
embodiment of a method for producing genetic dual bead complex solutions, this
method being related to the method illustrated in Fig. 12A;
Figs. 68A and 68B taken together illustrate a pictorial representation of
still
another embodiment of a method for producing immunochemical dual bead complex
solutions which is similar to that shown in Fig. 12B;
Fig. 69 is a bar graph presentation demonstrating the effect of DNAseI
digestion in absence of reporter beads;
Fig. 70 is a bar graph presentation showing the efficiency of dual bead assay
by the effect of DNAseI enzymes digestion;
Fig. 71 is a schematic representation of separation of reporter beads from
capture beads by enzyme digestion and physical or chemical treatments;
Fig. 72 is a bar graph presentation showing dual bead complexes prior to and
after washing in a basic solution;
Fig. 73A is a bar graph presentation illustrating dual bead complexes prior to
and after washing in a 7M urea solution;
Fig. 73B is a bar graph presentation representing dual bead complexes prior to
and after washing in a 7M urea solution including the detection of dissociated
reporter
beads after the urea wash;
Fig. 74 is a bar graph presentation demonstrating the use of 1.5M guanidine
isothiocyanate as a denaturing agent during dual bead assay; and
Fig. 75 is a bar graph presentation showing the varying concentrations of
guanidine isothiocyanate employed as a denaturing agent during dual bead
assay;
Fig. 76 is a top plan view of a portion of a magneto-optical bio-disc having
fluidic circuits; and
Figs. 77A-77E are plan views illustrating a method of separating and testing
cells using the fluidic circuit shown in Fig. 76.
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
The following description of the present invention relates to optical analysis
discs, disc drive systems, optical disc biomagnetic assays, and assay
chemistries and
techniques. The invention further relates to alternate magneto-optical drive
systems,
MO bio-discs, MO bio-disc systems, MO biomagnetic assays, and related
processing
methods.
Disc Drive System and Related Optical Analysis Discs
With reference now to Fig. 1, there is shown a perspective view of an optical
analysis disc, optical bio-disc, or medical CD 110 for use in an optical disc
drive 112.
Drive 112, in conjunction with software in the drive or associated with a
separate
computer, can cause images, graphs, or output data to be displayed on display
monitor 114. As indicated below, there are different types of discs and drives
that can
be used including, but not limited to, magneto-optical discs and magneto-
optical disc
drives. The disc drive can be in a unit separate from a controlling computer,
or
provided in a bay within a computer. The device can be made as portable as a
laptop
computer, and thus usable with battery power and in remote locations not
generally
served by advanced diagnostic equipment. The drive is preferably a
conventional
drive with minimal or no hardware modification, but can be a dedicated bio-
disc or
medical CD drive. Further details regarding these types of drive systems and
related
signal processing methods are~disclosed in, for example, commonly assigned and
co-
pending U.S. Patent Application Serial No. 09/378,878 entitled "Methods and
Apparatus for Analyzing Operational and Non-operational Data Acquired from
Optical
Discs" filed August 23, 1999; U.S. Provisional Patent Application Serial No.
60/150,288 entitled "Methods and Apparatus for Optical Disc Data Acquisition
Using
Physical Synchronization Markers" filed August 23, 1999; U.S. Patent
Application
Serial No. 09/421,870 entitled "Trackable Optical Discs with Concurrently
Readable
Analyte Material" filed October 26, 1999; U.S. Patent Application Serial No.
09/643,106 entitled "Methods and Apparatus for Optical Disc Data Acquisition
Using
Physical Synchronization Markers" filed August 21, 2000; U.S; and U.S. Patent
Application Serial No. 10/043,688 entitled "Optical Disc Analysis System
Including
Related Methods For Biological and Medical Imaging" filed January 10, 2002.
These
applications are herein incorporated by reference in their entirety.
16
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
Optical bio-disc 110 for use with embodiments of the present invention may
have any suitable shape, diameter, or thickness, but preferably is implemented
on a
round disc with a diameter and a thickness similar to those of a compact disc
(CD), a
recordable CD (CD-R), CD-RW, a digital versatile disc (DVD), DVD-R, DVD-RW, a
magneto-optical disc, or other standard optical disc format. The disc may
include
encoded information, preferably in a known format, for performing,
controlling, and
post-processing a test or assay, such as information for controlling the .
rotation rate
and direction of the disc, timing for rotation, stopping and starting, delay
periods,
locations of samples, position of the light source, and power of the light
source. Such
encoded information is referred to generally here as operational information.
The disc may be a reflective disc, as shown in Figs. 3A-3C, a transmissive
disc,
Figs. 4A-4C, or some combination of reflective and transmissive. In a
reflective disc,
an incident light beam is focused onto the disc (typically at a reflective
surface where
information is encoded), reflected, and returned through optical elements to a
detector
on the same side of the disc as the light source. In a transmissive disc,
light passes
through the disc (or portions thereof) to a detector on the other side of the
disc from
the light source. In a transmissive portion of a disc, some light may also be
reflected
and detected as reflected light.
Fig. 2 shows an optical disc reader system 116. This system may be a
conventional reader for CD, CD-R, DVD, MO, or other known comparable format, a
modified version of such a drive, or a completely distinct dedicated device.
The basic
components are a motor for rotating the disc, a light system for providing
light, and a
detection system for detecting light.
With reference now generally to Figs. 2-4C, a light source 118 provides light
to
optical components 120 to produce an incident light beam 122. In the case of
reflective disc 144, Figs. 3A-3C, a return beam 124 is reflected from either
reflective
surface 156, 174, or 186, Figs. 3C and 4C. Return beam 124 is provided back to
optical components 120, and then to a bottom detector 126. In this type of
disc, the
return beam may carry operational information or other encoded data as well as
characteristic information about the investigational feature or test sample
under study.
For transmissive disc 180, Figs. 4A-4C, some of the energy from the incident
beam 122 will undergo a light/matter interaction with an investigational
feature or test
sample and then proceed through the disc as a transmitted beam 128 that is
detected
17
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
by a top detector 130. For a transmissive disc including a semi-reflective
layer 186
(Fig. 4C) as the operational layer, some of the energy from the incident beam
122 will
also reflect from the operational layer as return beam 124, which carries
operational
information or stored data. Optical components 120 can include a lens, a beam
splitter, and a quarter wave plate that changes the polarization of the light
beam so
that the beam splitter directs a reflected beam through the lens to focus the
reflected
beam onto the detector. An astigmatic element, such as a cylindrical lens, may
be
provided between the beam splitter and detector to introduce astigmatism in
the
reflected light beam. The light source can be controllable to provide variable
wavelengths and power levels over a desired range in response to data
introduced by
the user or read from the disc. This controllability is especially useful when
it is
desired to detect multiple different structures that fluoresce at different
wavelengths.
Now with continuing reference to Fig. 2, it is shown that data from detector
126
and/or detector 130 is provided to a computer 132 including a processor 134
and an
analyzer 136. An image or output results can then be provided to a monitor
114.
Computer 132 can represent a desktop computer, programmable logic, or some
other
processing device, and also can include a connection (such as over the
Internet) to
other processing and/or storage devices. A drive motor 140 and a controller
142 are
provided for controlling the rotation rate and direction or rotation of disc
the 144 or
180. Controller 142 and the computer 132 with processor 134 can be in remote
communication or implemented in the same computer. Methods and systems for
reading such a disc are also shown in Gordon, U.S. Patent No. 5,892,577, which
is
incorporated herein by reference.
The detector can be designed to detect all light that reaches the detector, or
light only at specific wavelengths, through its design or an external filter.
By making
the detector controllable in terms of the detectable wavelength, beads or
other
structures that fluoresce at different wavelengths can be separately detected.
A hardware trigger sensor 138 may be used with either a reflective disc 144 or
transmissive disc 180. Triggering sensor 138 provides a signal to computer 132
(or to
some other electronics) to allow for the collection of data by processor 134
only when
incident beam 122 is on a target zone or inspection area. Alternatively,
software read
from a disc can be used to control data collection by processor 134
independent of
any physical marks on the disc. Such software or logical triggering is
discussed in
18
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
further detail in commonly assigned and co-pending U.S. Provisional
Application
Serial No. 60/352,625 entitled "Logical Triggering Methods And Apparatus For
Use
With Optical Analysis Discs And Related Disc Drive Systems" filed January 28,
2002,
which is herein incorporated by reference in its entirety.
The substrate layer of the optical analysis disc may be impressed with a
spiral
track that starts at an innermost readable portion of the disc and then
spirals out to an
outermost readable portion of the disc. In a non-recordable CD, this track is
made up
of a series of embossed pits with varying length, each typically having a
depth of
approximately one-quarter the wavelength of the light that is used to read the
disc.
The varying lengths and spacing between the pits encode the operational data.
The
spiral groove of a recordable CD-like disc has a detectable dye rather than
pits.
Furthermore, in the MO disc, the data is stored in magnetic domains created on
the
MO disc. This is where the operation information, such as the rotation rate,
is
recorded. Depending on the test, assay, or investigational protocol, the
rotation rate
may be variable with intervening or consecutive periods of acceleration,
constant
speed, and deceleration. These periods may be closely controlled both as to
speed
and time of rotation to provide, for example, mixing, agitation, or separation
of fluids
and suspensions with agents, reagents, antibodies, or other materials.
Different
optical analysis disc, medical CD, and bio-disc designs that may be utilized
with the
present invention, or readily adapted thereto, are disclosed, for example, in
commonly
assigned, co-pending U.S. Patent Application Serial No. 09/999,274 entitled
"Optical
Bio-discs with Reflective Layers" filed on November 15, 2001; U.S. Patent
Application
Serial No. 10/005,313 entitled "Optical Discs for Measuring Analytes" filed
December
7, 2001; U.S. Patent Application Serial No. 10/006,371 entitled "Methods for
Detecting
Analytes Using Optical Discs and Optical Disc Readers" filed December 10,
2001;
U.S. Patent Application Serial No. 10/006,620 entitled "Multiple Data Layer
Opfical
Discs for Detecting Analytes" filed December 10, 2001; and U.S. Patent
Application
Serial No. 10/006,619 entitled "Optical Disc Assemblies for Performing Assays"
filed
December 10, 2001, which are all herein incorporated by reference in their
entirety.
Numerous designs and configurations of an optical pickup and associated
electronics may be used in the context of the embodiments of the present
invention.
Further details and alternative designs for compact discs and readers are
described in
Compact Disc Technology, by Nakajima and Ogawa, IOS Press, Inc. (1992); The
19
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
Compact Disc Handbook, Digital Audio and Compact Disc Technology, by Baert et
al.
(eds.), Books Britain (1995); and CD-Rom Professional's CD-Recordable
Handbook:
The Complete Guide to Practical Desktop CD, Starrett et al. (eds.),
ISBN:0910965188
(1996); all of which are incorporated herein in their entirety by reference.
The disc drive assembly is thus employed to rotate the disc, read and process
any encoded operational information stored on the disc, and analyze the
liquid,
chemical, biological, or biochemical investigational features in an assay
region of the
disc. The disc drive assembly may be further utilized to write information to
the disc
either before, during, or after the material in the assay zone is analyzed by
the read
beam of the drive. In alternate embodiments, the disc drive assembly is
implemented
to deliver assay information through various possible interfaces such as via
Ethernet
to a user, over the Internet, to remote databases, or anywhere such
information could
be advantageously utilized. Further details relating to this type of disc
drive interfacing
are disclosed in commonly assigned co-pending U.S. Patent Application Serial
No.
09/986,078 entitled "Interactive System For Analyzing Biological Samples And
Processing Related Information And The Use Thereof " filed November 7, 2001,
which
is incorporated herein by reference in its entirety.
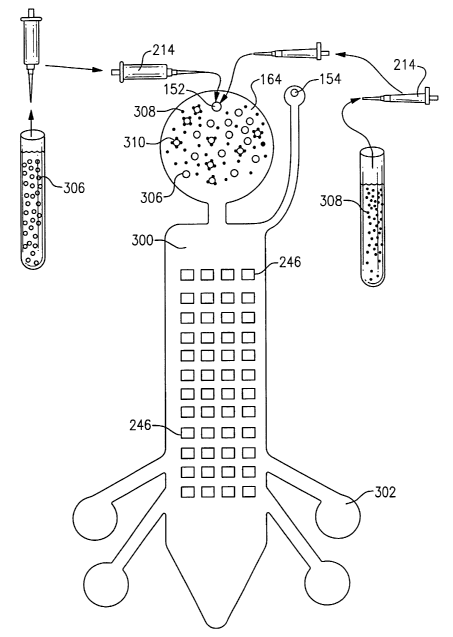
Referring now specifically to Figs. 3A, 3B, and 3C, the reflective disc 144 is
shown with a cap 146, a channel layer 148, and a substrate 150. The channel
layer
148 may be formed by a thin-film adhesive member. Cap 146 has inlet ports 152
for
receiving samples and vent ports 154. Cap 146 may be formed primarily from
polycarbonate, and may be coated with a cap reflective layer 156 on the bottom
thereof. Reflective layer 156 is preferably made from a metal such as aluminum
or
gold.
. Channel layer 148 defines fluidic circuits 158 by having desired shapes cut
out
from channel layer 148. Each fluidic circuit 158 preferably has a flow channel
160 and
a return channel 162, and some have a mixing chamber 164. A mixing chamber 166
can be symmetrically formed relative to the flow channel 160, while an off-set
mixing
chamber 168 is formed to one side of the flow channel 160. Fluidic circuits
158 are
rather simple in construction, but a fluidic circuit can include other
channels and
chambers, such as preparatory regions or a waste region, as shown, for
example, in
U.S. Patent No. 6,030,581 entitled "Laboratory in a Disc" which is
incorporated herein
by reference. These fluidic circuits can include valves and other fluid
control
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
structures such as those alternatively employed herein and discussed in
further detail
in connection with Figs. 33A-33D, 34A-34C, 35, and 36A-36C. Channel layer 148
can
include adhesives for bonding to the substrate and to the cap.
Substrate 150 has a plastic layer 172, and has target zones 170 formed as
openings in a substrate reflective layer 174 deposited on the top of layer
172. In this
embodiment, reflective layer 174, best illustrated in Fig. 3C, is used to
encode
operational information. The reflective layer 174 is not limited to a single
layer but
may also be several stacks of reflective layers on the substrate 150 such as
optical
stacks on an MO disc, for example. Plastic layer 172 is preferably formed from
polycarbonate. Target zones 170 may be formed by removing portions of the
substrate reflective layer 174 in any desired shape, or by masking target zone
areas
before applying substrate reflective layer 174. The substrate reflective layer
174 is
preferably formed from a metal, such as aluminum, gold, or magnetic alloys,
and can
be configured with the rest of the substrate to encode operational information
that is
read with incident light, such as through a wobble groove or through an
arrangement
of pits. Light incident from under substrate 150 thus is reflected by layer
174, except
at target zones 170, where it is reflected by layer 156. Target zones are
where
investigational features are detected. If the target zone is a location where
an
antibody, strand of DNA, or other material that can bind to a target is
located, the
target zone can be referred to as a capture zone.
With reference now particularly to Fig. 3C, optical disc 144 is cut away to
illustrate a partial cross-sectional perspective view. An active layer 176 is
formed over
substrate reflective layer 174. Active layer 176 may generally be formed from
nitrocellulose, polystyrene, polycarbonate, gold, activated glass, modified
glass, or a
modified polystyrene such as, for example, polystyrene-co-malefic anhydride.
In this
embodiment, channel layer 148 is situated over active layer 174.
In operation, samples can be introduced through inlet ports 152 of cap 146.
When rotated, the sample moves outwardly from inlet port 152 along active
layer 176.
Through one of a number of biological or chemical reactions or processes,
detectable
features, referred to as investigational features, may be present in the
target zones.
Examples of such processes are shown in the incorporated U.S. Patent No.
6,030,581
and in commonly assigned, co-pending U.S. Patent Application No. 09/988,728
entitled "Methods And Apparatus For Detecting And Quantifying Lymphocytes With
21
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
Optical Biodiscs" filed November 16, 2001; and U.S. Patent Application No.
10/035,836 entitled "Surface Assembly For Immobilizing DNA Capture Probes And
Bead-Based Assay Including Optical Bio-Discs And Methods Relating Thereto"
filed
December 21, 2001, both of which are herein incorporated by reference in their
entireties.
The investigational features captured within the target zones, by the capture
layer with a capture agent, may be designed to be located in the focal plane
coplanar
with reflective layer 174, where an incident beam is typically focused in
conventional
readers. Alternatively, the investigational features may be captured in a
plane spaced
away from the focal plane. The former configuration is referred to as a
"proximal" type
disc, and the latter a "distal" type disc.
Referring to Figs. 4A, 4B, and 4C, it is shown that one particular embodiment
of
the transmissive optical disc 180 includes a clear cap 182, a channel layer
148, and a
substrate 150. The clear cap 182 includes inlet ports 152 and vent ports 154
and is
preferably formed mainly from polycarbonate. Trigger marks 184 may be included
on
the cap 182. Channel layer 148 has fluidic circuits 158, which can have
structure and
use similar to those described in conjunction with Figs. 3A, 3B, and 3C.
Substrate 150
may include target zones 170, and preferably includes a polycarbonate layer
172.
Substrate 150 may, but need not, have a thin semi-reflective layer 186
deposited on
top of layer 172. Semi-reflective layer 186 is preferably significantly
thinner than
substrate reflective layer 174 on substrate 150 of reflective disc 144 (Figs.
3A-3C).
The semi-reflective layer 186 is not limited to a single layer but may also be
several
stacks of semi-reflective layers on the substrate 150 such as optical stacks
on an MO
disc, for example. Semi-reflective layer 186 is preferably formed from a
metal, such
as aluminum, gold, or magnetic alloys, but is sufficiently thin to allow a
portion of an
incident light beam to penetrate and pass through layer 186, while some of the
incident light is reflected back. A gold film layer, for example, is 95%
reflective at a
thickness greater than about 700 A, while the transmission of light through
the gold
film is about 50% transmissive at approximately 100 A.
Fig. 4C is a cut-away perspective view of transmissive disc 180. The semi-
reflective nature of layer 186 makes its entire surface potentially available
for target
zones, including virtual zones defined by trigger marks or encoded data
patterns on
the disc. Target zones 170 may also be formed by marking the designated area
in the
22
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
indicated shape or alternatively in any desired shape. Markings to indicate
target
zone 170 may be made on semi-reflective layer 186 or on a bottom portion of
substrate 150 (under the disc). Target zones 170 may be created by silk
screening
ink onto semi-reflective layer 186.
An active layer 176 is applied over semi-reflective layer 186. Active layer
176
may be formed from the same materials as described above in conjunction with
layer
176 (Fig. 3C) and serves substantially the same purpose when a sample is
provided
through an opening in disc 180 and the disc is rotated. In transmissive disc
180, there
is no reflective layer, on the clear cap 182, comparable to reflective layer
156 in
reflective disc 144 (Fig. 3C).
Referring now to Fig. 5A, there is shown a cross sectional view taken across
the tracks of the reflective disc embodiment 144 (Figs. 3A-3C) of the bio-disc
110 (Fig.
1 ) according to the present invention. As illustrated, this view is taken
longitudinally
along a radius and flow channel of the disc. Fig. 5A includes the substrate
150 that is
composed of a plastic layer 172 and a substrate reflective layer 174. In this
embodiment, the substrate 150 includes a series of grooves 188. The grooves
188
are in the form of a spiral extending from near the center of the disc toward
the outer
edge. The grooves 188 are implemented so that the interrogation or incident
beam
122 may track along the spiral grooves 188 on the disc. This type of groove
188 is
known as a "wobble groove". The groove 188 is formed by a bottom portion
having
undulating or wavy side walls. A raised or elevated portion separates adjacent
grooves 188 in the spiral. The reflective layer 174 applied over the grooves
188 in this
embodiment is, as illustrated, conformal in nature. Fig. 5A also shows the
active layer
176 applied over the reflective layer 174. As shown in Fig. 5A, the target
zone 170 is
formed by removing an area or portion of the reflective layer 174 at a desired
location
or, alternatively, by masking the desired area prior to applying the
reflective layer 174.
As further illustrated in Fig. 5A, the plastic adhesive member or channel
layer 148 is
applied over the active layer 176. Fig. 5A also shows the cap portion 146 and
the
reflective surface 156 associated therewith. Thus, when the cap portion 146 is
applied
to the plastic adhesive member 148 including the desired cut-out shapes, the
flow
channel 160 is thereby formed.
Fig. 5B is a cross sectional view, similar to that illustrated in Fig. 5A,
taken
across the tracks of the transmissive disc embodiment 180 (Figs. 4A-4C) of the
bio-
23
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
disc 110 (Fig. 1 ) according to the present invention. This view is taken
longitudinally
along a radius and flow channel of the disc. Fig. 5B illustrates the substrate
150 that
includes the thin semi-reflective layer 186. This thin semi-reflective layer
186 allows
the incident or interrogation beam 122, from the light source 118 (Fig. 2), to
penetrate
and pass through the disc to be detected by the top detector 130, while some
of the
light is reflected back in the form of the return beam 124. The thickness of
the thin
semi-reflective layer 186 is determined by the minimum amount of reflected
light
required by the disc reader to maintain its tracking ability. The substrate
150 in this
embodiment, like that discussed in Fig. 5A, includes the series of grooves
188. The
grooves 188 in this embodiment are also preferably in the form of a spiral
extending
from near the center of the disc toward the outer edge. The grooves 188 are
implemented so that the interrogation beam 122 may track along the spiral.
Fig. 5B
also shows the active layer 176 applied over the thin semi-reflective layer
186. As
further illustrated in Fig. 5B, the plastic adhesive member or channel layer
148 is
applied over the active layer 176. Fig. 5B also shows the clear cap 182. Thus,
when
the clear cap 182 is applied to the plastic adhesive member 148 including the
desired
cut-out shapes, the flow channel 160 is thereby formed and a part of the
incident
beam 122 is allowed to pass therethrough substantially unreflected. The amount
of
light that passes through can then be detected by the top detector 130.
Fig. 6A is a view similar to Fig. 5A but taken perpendicularly to a radius of
the
disc to illustrate the reflective disc and the initial refractive property
thereof when
observing the flow channel 160 from a radial perspective. In a parallel
comparison
manner, Fig. 6B is a similar view to Fig. 5B but taken perpendicularly to a
radius of the
disc to represent the transmissive disc and the initial refractive property
thereof when
observing the flow channel 160 from a radial perspective. Grooves 188 are not
seen
in Figs. 5A and 5B since the sections are cut along the grooves 188. Figs. 6A
and 6B
show the presence of the narrow flow channel 160 that is situated
perpendicular to the
grooves 188 in these embodiments. Figs. 5A, 5B, 6A, and 6B show the entire
thickness of the respective reflective and transmissive discs. In these views,
the
incident beam 122 is illustrated initially interacting with the substrate 150
which has
refractive properties that change the path of the incident beam as shown to
provide
focusing of the beam 122 on the reflective layer 174 or the thin semi-
reflective layer
186.
24
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
The fluidic circuits 158 may also be configured in the equi-radial or "e-rad"
format disclosed in commonly assigned and copending U.S. Provisional
Application
Serial No. 60/353,014 entitled "Optical Discs Including Equi-Radial and/or
Spiral
Analysis Zones and Related Disc Drive Systems and Methods" filed January 29,
2002,
which is hereby incorporated by reference in its entirety.
Assay Chemistries and Dual Bead Formation
Referring now to Figs. 7A-10A and 7B-10B, there is shown a capture bead 190,
a reporter bead 192, and the formation of a dual bead complex 194. Capture
bead
190 can be used in conjunction with a variety of different assays including
biological
assays such as immunoassays (Figs. 7B-10B), molecular assays, and more
specifically genetic assays (Figs. 7A-10A). In the case of immunoassays,
antibody
transport probes 196 are conjugated onto the beads. Antibody transport probes
196
include proteins, such as antigens or antibodies, implemented to capture
protein
targets. In the case of molecular assays, oligonucleotide transport probes 198
would
be conjugated onto the beads. Oligonucleotide transport probes 198 include
nucleic
acids such as DNA or RNA implemented to capture genetic targets.
As shown in Fig. 7A, a target agent such as target DNA or RNA 202, obtained
from a test sample, is added to a capture bead 190 coated with oligonucleotide
transport probes 198. In this implementation, transport probes 198 are formed
from
desired sequences of nucleic acids. Aspects relating to DNA probe conjugation
onto
solid phase of this system of assays are discussed in further detail in
commonly
assigned and co-pending U.S. Provisional Application Serial No. 60/278,685
entitled
"Use of Double Stranded DNA for Attachment to Solid Phase to Reduce Non-
Covalent
Binding" filed March 26, 2001. This application is herein incorporated by
reference in
its entirety.
As shown in Fig. 7B, a target agent such as target antigen 204 from a test
sample is added to a capture bead 190 coated with antibody transport probes
196. In
this alternate implementation, the transport probes 196 are formed from
proteins such
as antibodies.
Capture bead 190 has a characteristic that allows it to be isolated from a
material suspension or solution. The capture bead may be selected based upon a
desired size, and a preferred way to make it isolatable is for it to be
magnetic.
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
Fig. 8A illustrates the binding of target DNA or RNA 202 to complementary
transport probes 198 on capture bead 190 in the genetic assay implementation
of the
present invention. Fig. 8B shows an immunoassay version of Fig. 8A, transport
probes 196 can alternatively include antibodies or antigens for binding to a
target
protein 204.
Fig. 9A shows a reporter bead 192 coated with oligonucleotide signal probes
206 complementary to target agent 202 (see Fig. 8A). Reporter bead 192 is
selected
based upon a desired size and the material properties for detection and
reporting
purposes. In one specific embodiment a 2.1 um polystyrene bead is employed.
Signal probes 206 can be strands of DNA or RNA to capture target DNA or RNA.
Fig. 9B illustrates a reporter bead 192 coated with antibody signal probes 208
that bind to the target agent 204 as shown in Fig. 8B. Reporter bead 192 is
selected
based upon a desired size and the material properties for detection and
reporting
purposes. This may also preferably include a 2.1 um polystyrene bead. Signal
probes 208 can be antigens or antibodies implemented to capture protein or
glycoprotein targets.
Fig. 10A is a pictorial representation of a dual bead complex 194 that can be
formed from capture bead 190 with probe 198, target agent 202, and reporter
bead
192 with probe 206. Probes 198 and 206 conjugated on capture bead 190 and
reporter bead 192, respectively, have sequences complementary to the target
agent
202, but not to each other. Further details regarding target agent detection
and
methods of reducing non-specific binding of target agents to beads are
discussed in
commonly assigned and co-pending U.S. Provisional Application Serial No.
60/278,106 entitled "Dual Bead Assays Including Use of Restriction Enzymes to
Reduce Non-Specific Binding" filed March 23, 2001; and U.S. Provisional
Application
Serial No. 60/278,110 entitled "Dual Bead Assays Including Use of Chemical
Methods
to Reduce Non-Specific Binding" also filed March 23, 2001, which are both
incorporated herein by reference in their entirety.
Fig. 10B is a pictorial representation of the immunoassay version of a dual
bead complex 194 that can be formed from capture bead 190 with probe 196,
target
agent 204, and reporter bead 192 with probe 208. Probes 196 and 208 conjugated
on
capture bead 190 and reporter bead 192, respectively, only bind to the target
agent
202, and not to each other.
26
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
In an alternative embodiment of the current system of assays, the efficiency
and specificity of target agent binding may be enhanced by using a clcavable
spacer
or a displaceable spacer that temporarily links the reporter bead 192 and
capture bead
190. The dual bead complex formed by the clcavable spacer or displaceable
spacer
essentially places the transport probe and the signal probe in close proximity
to each
other thus allowing more efficient target binding to both probes. Once the
target agent
is bound to the probes, the spacer may then be cleaved permitting the bound
target
agent to retain the dual bead structure. The use of clcavable spacers in dual
bead
assay systems is~ disclosed in further detail in commonly assigned and co-
pending
U.S. Provisional Application Serial No. 60/278,688 entitled "Dual Bead Assays
Using
Clcavable Spacers to Improve Specificity and Sensitivity" filed March 26,
2001, which
is herein incorporated in its entirety by reference. The use of clcavable
spacers and
displaceable spacers are also described below in conjunction with Figs. 42A-
42D and
43A-43C.
With reference now to Fig. 11A, there is illustrated a method of preparing a
molecular assay using a "single-step hybridization" technique to create dual
bead
complex structures in a solution according to one aspect of the present
invention.
This method includes 5 principal steps identified consecutively as Steps I,
II, III, IV,
and V.
In Step I of this method, a number of capture beads 190 coated with
oligonucleotide transport probes 198 are deposited into a test tube 212
containing a
buffer solution 210. The number of capture beads 190 used in this method may
be,
for example, on the order of 10E+07 and each on the order of 1 um or greater
in
diameter. Capture beads 190 are suspended in hybridization solution and are
loaded
into the test tube 212 by injection with pipette 214. The preferred
hybridization
solution is composed of 0.2M NaCI, 1 OmM MgCl2, 1 mM EDTA, 50mM Tris-HCI, pH
7.5, and 5X Denhart's mix. A desirable hybridization temperature is 37 degrees
Celsius. In a preliminary step in this embodiment, transport probes 198 are
conjugated to 3 um magnetic capture beads 190 by EDC conjugation. Further
details
regarding conjugation methods are disclosed in commonly assigned U.S.
Provisional
Application Serial No. 60/271,922 entitled, "Methods for Attaching Capture DNA
and
Reporter DNA to Solid Phase Including Selection of Bead Types as Solid Phase"
filed
February 27, 2001; and U.S. Provisional Application Serial No. 60/277,854
entitled
27
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
"Methods of Conjugation for Attaching Capture DNA and Reporter DNA to Solid
Phase" filed March 22, 2001, both of which are herein incorporated by
reference in
their entirety.
As shown in Step II, target DNA or RNA 202 is added to the solution.
Oligonucleotide transport probes 198 are complementary to the DNA or RNA
target
agent 202. The target DNA or RNA 202 thus binds to the complementary sequences
of transport probe 198 attached to the capture bead 190 as shown in Fig. 8A.
With reference now to Step III, there is added to the solution 210 reporter
beads 192 coated with oligonucleotide signal probes 206. As also shown in
Figs. 9A
and 10A, signal probes 206 are complementary to the target DNA or RNA 202. In
one
embodiment, signal probes 206, which are complementary to a portion of the
target
DNA or RNA 202, are conjugated to 2.1 um fluorescent reporter beads 192.
Signal
probes 206 and transport probes 198 each have sequences that are complementary
to the target DNA 202, but not complementary to each other. After adding
reporter
beads 192, the dual bead complex 194 is formed such that the target DNA 202
links
capture bead 190 and reporter beads 192. With specific and thorough washing,
there
should be minimal non-specific binding between reporter bead 192 and capture
bead
190. The target agent 202 and signal probe 206 are preferably allowed to
hybridize
for three to four hours at 37 degrees Celsius.
In this embodiment and others, it was found that intermittent mixing (i.e.,
periodically mixing and then stopping) produced greater yield of dual bead
complex
than continuous mixing during hybridization. Thus when this step is performed
on-
disc, the disc drive motor 140 and controller 142, Fig. 2, may be
advantageously
employed to periodically rotate the disc to achieve the desired intermittent
mixing.
This may be implemented in mixing protocols encoded on the disc that rotate
the disc
in one direction, then stop the disc, and thereafter rotate the disc again in
the same
direction in a prescribed manner with a preferred duty cycle of rotation and
stop
sessions. Alternatively, the encoded mixing protocol may rotate the disc in a
first
direction, then stop the disc, and thereafter rotate the disc again in the
opposite
direction with a preferred duty cycle of rotation, stop, and reverse rotation
sessions.
These features of the present invention are discussed in further detail in
connection
with Figs. 33A and 35.
28
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
As next shown in Step IV of Fig. 11A, after hybridization, the dual bead
complex 194 is separated from unbound reporter beads in the solution. The
solution
can be exposed to a magnetic field to capture the dual bead complex structures
194
using the magnetic properties of capture bead 190. The magnetic field can be
encapsulated in a magnetic test tube rack 216 with a built-in magnet 218,
which can
be permanent or electromagnetic to draw out the magnetic beads and remove any
unbound reporter beads in the suspension. Note that capture beads not bound to
reporter beads will also be isolated. Alternatively, this magnetic removal
step may be
performed on-disc as shown in Figs. 33A, 35, and 36A-36C.
The purification process illustrated in Step IV includes the removal of
supernatant containing free-floating particles. Wash buffer is added into the
test tube
and the bead solution is mixed well. The preferred wash buffer for the one
step assay
consists of 145mM NaCI, 50mM Tris, pH 7.5, 0.1 % SDS, 0.05% Tween, 0.25%
NFDM, and lOmM EDTA. Most of the unbound reporter beads 182, free-floating
DNA, and non-specifically bound particles are agitated and removed from the
supernatant. The dual bead complex can form a matrix of capture beads, target
sequences, and reporter beads, wherein the wash process can further assist in
the
extraction of free floating particles trapped in the lattice structure of
overlapping dual
bead particles. Further details relating to other aspects associated with
methods of
decreasing non-specific binding of reporter beads to capture beads are
disclosed in,
for example, commonly assigned U.S. Provisional Application Serial No.
60/272,134
entitled "Reduction of Non-Specific Binding in Dual Bead Assays by Selection
of Bead
Type and Bead Treatment" filed February 28, 2001; and U.S. Provisional
Application
Serial No. 60/275,006 entitled "Reduction of Non-Specific Binding in Dual Bead
Assays by Selection of Buffer Conditions and Wash Conditions" filed March 12,
2001.
Both of these applications are herein incorporated by reference in their
entirety.
The last principal step shown in Fig. 11A is Step V. In this step, once the
dual
bead complex has been washed approximately 3-5 times with wash buffer
solution,
the assay mixture may be loaded into the disc and ready to be analyzed.
Fig. 11 B illustrates an immunoassay using a "single-step antigen binding"
method, similar to that in Fig. 11A, to create dual bead complex structures in
a
solution. This method similarly includes 5 principal steps. These steps are
respectively identified as Steps I, II, II I, IV, and V in Fig. 11 A.
29
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
As shown in Step I, capture beads 190, e.g., on the order of 10E+07 in number
and each on the order of 1 um or above in diameter, which are coated with
antibody
transport probes 196 are added to a buffer solution 210. This solution may be
that
same as that employed in the method shown in Fig. 11 A or alternatively may be
specifically prepared for use with immunochemical assays. The antibody
transport
probes 196 have a specific affinity for the target antigen 204. The transport
probes
196 bind specifically to epitopes within the target antigen 204 as also shown
in Fig.
8B. In one embodiment, antibody transport probes 196 that have an affinity for
a
portion of the target antigen may be conjugated to 3 um magnetic capture beads
190
via EDC conjugation. Alternatively, conjugation of the transport probes 196 to
the
capture bead 190 may be achieved by passive adsorption.
With reference now to Step II shown in Fig. 11 B, the target antigen 204 is
added to the solution. The target antigen 204 binds to the antibody transport
probe
196 attached to the capture bead 190 as also shown in Fig. 8B.
As illustrated in Step III, reporter beads 192 coated with antibody signal
probes
208 are added to the solution. Antibody signal probes 208 specifically binds
to the
epitopes on target antigen 204 as also represented in Figs. 9B and 10B. In one
embodiment, signal probes 208 are conjugated to 2.1 um fluorescent reporter
beads
192. Signal probes 208 and transport probes 196 each bind to specific epitopes
on
the target antigen, but not to each other. After adding reporter beads 192,
the dual
bead complex 194 is formed such that the target antigen 204 links capture bead
190
and reporter bead 192. With specific and thorough washing, there should be
minimal
non-specific binding between reporter bead 192 and capture bead 190.
In Step IV, after the binding in Step III, the dual bead complex 194 is
separated
from unbound reporter beads in the solution. The solution can be exposed to a
magnetic field to capture the dual bead complex structures 194 using the
magnetic
properties of capture bead 190. The magnetic field can be encapsulated in a
magnetic test tube rack 216 with a built-in magnet 218, which can be permanent
or
electromagnetic to draw out the magnetic beads and remove any unbound reporter
beads in the suspension. Note that capture beads not bound to reporter beads
will
also be isolated. Alternatively, as indicated above, this magnetic removal
step may
also be performed on-disc as shown in Figs. 33A, 35, and 36A-36C.
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
The purification process of Step IV includes the removal of supernatant
containing free-floating particles. Wash buffer is added into the test tube
and the bead
solution is mixed well. Most of the unbound reporter beads 182, free-floating
protein
samples, and non-specifically bound particles are agitated and removed from
the
supernatant. The dual bead complex can form a matrix of capture beads, target
antigen, and reporter beads, wherein the wash process can further assist in
the
extraction of free floating particles trapped in the lattice structure of
overlapping dual
bead particles.
The last principal step in Fig. 11 B is Step V. In this step, once the dual
bead
. complex has been washed approximately 3-5 times with wash buffer solution,
the
assay mixture is loaded into the disc and is thereby in condition to be
analyzed.
Fig. 12A shows an alternative genetic assay method referred to here as a "two-
step hybridization" to create the dual bead complex which has 6 principal
steps.
Generally, capture beads are coated with oligonucleotide transport probes 198
complementary to DNA or RNA target agent and placed into a buffer solution. In
this
embodiment, transport probes that are complementary to a portion of target
agent are
conjugated to 3 um magnetic capture beads via EDC conjugation. Other types of
conjugation of the oligonucleotide transport probes to a solid phase may be
utilized.
These include, for example, passive adsorption or use of streptavidin-biotin
interactions. These 6 main steps according to this method of the present
invention are
consecutively identified as Steps I, II, III, IV, V, and VI in Fig. 12A.
More specifically now with reference to Step I shown in Fig. 12A, capture
beads
190, suspended in hybridization solution, are loaded from the pipette 214 into
the test
tube 212. The preferred hybridization solution is composed of 0.2M NaCI, lOmM
MgCl2, 1 mM EDTA, 50mM Tris-HCI, pH 7.5, and 5X Denhart's mix. A desirable
hybridization temperature is 37 degrees Celsius.
In Step II, target DNA or RNA 202 is added to the solution and binds to the
complementary sequences of transport probe 198 attached to capture bead 190.
In
one specific embodiment of this method, target agent 202 and the transport
probe 198
are allowed to hybridize for 2 to 3 hours at 37 degrees Celsius. Sufficient
hybridization, however, may be achieved within 30 minutes at room temperature.
At
higher temperatures, hybridization may be achieved substantially
instantaneously.
31
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
As next shown in Step III, target agents 202 bound to the capture beads are
separated from unbound species in solution by exposing the solution to a
magnetic
field to isolate bound target sequences by using the magnetic properties of
the capture
bead 190. The magnetic field can be enclosed in a magnetic test tube rack 216
with a
built-in magnet permanent 218 or electromagnet to draw out the magnetic beads
and
remove any unbound target DNA 202 free-floating in the suspension via pipette
extraction of the solution. As with the above methods, in the on-disc
counterpart
hereto, this magnetic removal step may be performed as shown in Figs. 33A, 35,
and
36A-36C. A wash buffer is added and the separation process can be repeated.
The
preferred wash buffer after the transport probes 198 and target DNA 202
hybridize,
consists of 145mM NaCI, 50mM Tris, pH 7.5, and 0.05% Tween. Hybridization
methods and techniques for decreasing non-specific binding of target agents to
beads
are further disclosed in commonly assigned and co-pending U.S. Provisional
Application Serial No. 60/278,691 entitled "Reduction of Non-Specific Binding
of Dual
Bead Assays by Use of Blocking Agents" filed March 26, 2001. This application
is
herein incorporated by reference in its entirety.
Referring now to Step IV illustrated in Fig. 12A, reporter beads 192 are added
to the solution as discussed in conjunction with the method shown in Fig. 11
A.
Reporter beads 192 are coated with signal probes 206 that are complementary to
target agent 202. In one particular embodiment of this method, signal probes
206,
which are complementary to a portion of target agent 202, are conjugated to
2.1 um
fluorescent reporter beads 192. Signal probes 206 and transport probes 198
each
have sequences that are complementary to target agent 202, but not
complementary
to each other. After the addition of reporter beads 192, the dual bead complex
structures 190 are formed. As would be readily apparent to one of skill in the
art, the
dual bead complex structures are formed only if the target agent of interest
is present.
In this formation, target agent 202 links magnetic capture bead 190 and
reporter bead
192. Using the preferred buffer solution, with specific and thorough washing,
there is
minimal non-specific binding between the reporter beads and the capture beads.
Target agent 202 and signal probe 206 are preferably allowed to hybridize for
2-3
hours at 37 degrees Celsius. As with Step II discussed above, sufficient
hybridization
may be achieved within 30 minutes at room temperature. At higher temperatures,
the
hybridization in this step may also be achieved substantially instantaneously.
32
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
With reference now to Step V shown in Fig. 12A, after the hybridization in
Step
IV, the dual bead complex 194 is separated from unbound species in solution.
The
solution is again exposed to a magnetic field to isolate the dual bead complex
194
using the magnetic properties of the capture bead 190. Note again that the
isolate will
include capture beads not bound to reporter beads. As with Step III above in
the on-
disc counterpart hereto, this magnetic separation step may be performed as
shown in
Figs. 33A, 35, and 36A-36C.
A purification process to remove supernatant containing free-floating
particles
includes adding wash buffer into the test tube and mixirig the bead solution
well. The
preferred wash buffer for the two-step assay consists of 145mM NaCI, 50mM
Tris, pH
7.5, 0.1 % SDS, 0.05% Tween, 0.25% NFDM, and 1 OmM EDTA. Most unbound
reporter beads, free-floating DNA, and non-specifically bound particles are
agitated
and removed from the supernatant. The dual bead complex can form a matrix of
capture beads, target agents, and reporter beads, wherein the wash process can
further assist in the extraction of free floating particles trapped in the
lattice structure of
overlapping dual bead particles. Other related aspects directed to reduction
of non-
specific binding between reporter bead, target agent, and capture bead are
disclosed
in, for example, commonly assigned U.S. Provisional Application Serial No.
60/272,243 entitled "Mixing Methods to Reduce Non-Specific Binding in Dual
Bead
Assays" filed February 28, 2001; and U.S. Provisional Application Serial No.
60!272,485 entitled "Dual Bead Assays Including Linkers to Reduce Non-Specific
Binding" filed March 1, 2001, which are incorporated herein in their entirety.
The final principal step shown in Fig. 12A is Step VI. In this step, once the
dual
bead complex 194 has been washed approximately 3-5 times with wash buffer
solution, the assay mixture is loaded into the disc and analyzed.
Alternatively, during
this step, the oligonucleotide signal and transport probes may be ligated to
prevent
breakdown of the dual bead complex during the disc analysis and signal
detection
processes. Further details regarding probe ligation methods are disclosed in
commonly assigned and co-pending U.S. Provisional Application Serial No.
60/278,694 entitled "Improved Dual Bead Assays Using Ligation" filed March 26,
2001, which is herein incorporated in its entirety by reference.
In accordance with another aspect of this invention, Fig. 12B shows an
immunoassay method, similar to those discussed in connection with the
immunoassay
33
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
method of Fig. 11 B and following the steps of the genetic assay of Fig. 12A.
This
method is also referred to here as a "two-step binding" to create the dual
bead
complex in an immunochemical assay. As with the method shown in Fig. 12A, this
method includes 6 main steps. In general, capture beads coated with antibody
transport probes that specifically bind to epitopes on target antigens are
placed into a
buffer solution. In one specific embodiment, antibody transport probes are
conjugated
to 3 um magnetic capture beads. Different sized magnetic capture beads may be
employed depending on the type of disc drive and disc assembly utilized to
perform
the assay. These 6 main steps according to this alternative method of the
invention
are respectively identified as Steps I, II, III, IV, V and VI in Fig. 12B.
With specific reference now to Step I shown in Fig. 12B, capture beads 190,
suspended in buffer solution 210, are loaded into a test tube 212 via
injection from
pipette 214.
In Step II, target antigen 204 is added to the solution and binds to the
antibody
transport probe 196 attached to capture bead 190. Target antigen 204 and the
transport probe 196 are preferably allowed to bind for 2 to 3 hours at 37
degrees
Celsius. Shorter binding times are also possible.
As shown in Step III, target antigen 204 bound to the capture beads 190 is
separated from unbound species in solution by exposing the solution to a
magnetic
field to isolate bound target proteins or glycoproteins by using the magnetic
properties
of capture bead 190. The magnetic field can be enclosed in a magnetic test
tube rack
216 with a built-in magnet permanent 218 or electromagnet to draw out the
magnetic
beads and remove any unbound target antigen 204 free-floating in the
suspension via
pipette extraction of the solution. A wash buffer is added and the separation
process
can be repeated.
As next illustrated in Step IV, reporter beads 192 are added to the solution
as
discussed in conjunction with the method shown in Fig. 11 B. Reporter beads
192 are
coated with signal probes 208 that have an affinity for the target antigen
204. In one
particular embodiment of this two-step immunochemical assay, signal probes
208,
which bind specifically to a portion of target agent 204, are conjugated to
2.1 um
fluorescent reporter beads 192. Signal probes 208 and transport probes 196
each
bind to specific epitopes on the target agent 204, but do not bind to each
other. After
the addition of reporter beads 192, the dual bead complex structures 190 are
formed.
34
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
As would be readily apparent to those skilled in the art, these dual bead
complex
structures are formed only if the target antigen of interest is present. In
this formation,
target antigen 204 links magnetic capture bead 190 and reporter bead 192.
Using the
preferred buffer solution, with specific and thorough washing, there is
minimal non-
specific binding between the reporter beads and the capture beads. Target
antigen
204 and signal probe 208 are allowed to hybridize for 2-3 hours at 37 degrees
Celsius.
As with Step II discussed above, sufficient binding may be achieved within 30
minutes
at room temperature. In the case of immunoassays temperatures higher than 37
degrees Celsius are not preferred because the proteins will denature.
Turning next to Step V as illustrated in Fig. 12B, after the binding shown in
Step
IV, the dual bead complex 194 is separated from unbound species in solution.
This is
achieved by exposing the solution to a magnetic field to isolate the dual bead
complex
194 using the magnetic properties of the capture bead 190 as shown. Note again
that
the isolate will include capture beads not bound to reporter beads.
A purification process to remove supernatant containing free-floating
particles
includes adding wash buffer into the test tube and mixing the bead solution
well. Most
unbound reporter beads, free-floating proteins, and non-specifically bound
particles
are agitated and removed from the supernatant. The dual bead complex can form
a
matrix of capture beads, target agents, and reporter beads, wherein the wash
process
can further assist in the extraction of free floating particles trapped in the
lattice
structure of overlapping dual bead particles.
The final main step shown in Fig. 12B is Step VI. In this step, once the dual
bead complex 194 has been washed approximately 3-5 times with wash buffer
solution, the assay mixture is loaded into the disc and analyzed.
As with any of the other methods discussed above, the magnetic removal or
separation steps in the method shown in Fig. 12B may be alternatively
performed on-
disc using the disc, fluidic circuits, and apparatus illustrated in Figs. 33A-
33D, 34A-
34C, 35, and 36A-36C.
With reference now to Fig. 13, there is shown a cross sectional view
illustrating
the disc layers (similar to Fig. 6) of the mixing or loading chamber 164.
Access to the
loading chamber 164 is achieved by an inlet port 152 where the dual bead assay
preparation is loaded into the disc system.
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
Fig. 14 is a view similar to Fig. 13 showing the mixing or loading chamber 164
with the pipette 214 injection of the dual bead complex 194 onto the disc. In
this
example, the complex includes reporters 192 and capture bead 190 linked
together by
the target DNA or RNA 202. The signal DNA 206 is illustrated as single
stranded DNA
complementary to the capture agent. The discs illustrated in Figs. 13 and 14
may be
readily adapted to other assays including the immunoassays and general
molecular
assays discussed above which employ, alternatively, proteins such as antigens
or
antibodies implemented as the transport probes, target agents, and signal
probes
accordingly.
Fig. 15A shows the flow channel 160 and the target or capture zone 170 after
anchoring of dual bead complex 194 to a capture agent 220. The capture agent
220
in this embodiment is attached to the active layer 176 by applying a small
volume of
capture agent solution to the active layer 176 to form clusters of capture
agents within
the area of the target zone 170. In this embodiment, the capture agent
includes biotin
or BSA-biotin. Fig. 15A also shows reporters 192 and capture beads 190 as
components of a dual bead complex 194 as employed in the present invention. In
this
embodiment, anchor agents 222 are attached to the reporter beads 192. The
anchor
agent 222, in this embodiment, may include streptavidin or Neutravidin. So
when the
reporter beads 192 come in close proximity to the capture agents 220, binding
occurs
between the anchor probe 222/206 and the capture agent 220, via biotin-
streptavidin
interactions, thereby retaining the dual bead complex 194 within the target
zone 170.
At this point, an interrogation beam 224 directed to the target zone 170 can
be used to
detect the dual bead complex 194 within the target zone 170.
The embodiment of the present invention illustrated in Figs. 15A and 15B, may
alternatively be implemented on the transmissive disc shown in Figs. 4A-4C,
5B, and
6B.
Fig. 15B is a cross sectional view similar to Fig. 15A illustrating the
entrapment
of the reporter bead 192 within the target zone 170 after a subsequent change
in disc
rotational speed. The change in rotational speed removes the capture beads 190
from
the dual bead complex 194, ultimately isolating the reporter bead 192 in the
target
zone 170 to be detected by the interrogation or read beam 224.
Fig. 16A is a cross sectional view, similar to Fig. 15A, that illustrates an
alternative embodiment to Fig. 15A wherein the signal probes 206 or anchor
agents
36
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
222, on the reporter beads 192, directly hybridize to the capture agent 220.
Fig. 16A
shows the flow channel 160 and the target or capture zone 170 after anchoring
of dual
bead complex 194 with the capture agent 220. The capture agent 220 in this
embodiment is attached to the active layer 176 by applying a small volume of
capture
agent solution to the active layer 176 to form clusters of capture agents
within the area
of the target zone 170. Alternatively, the capture agent 220 may be attached
to the
active layer using an amino group that covalently binds to the active layer
176. In this
embodiment, the capture agent includes DNA. Fig. 16A also shows reporters 192
and
capture beads 190 as components of a dual bead complex 194 as employed in the
present invention. In this embodiment, anchor agents 222 are attached to the
reporter
beads 192. The anchor agent 222 in this embodiment may be a specific sequence
of
nucleic acids that are complimentary to the capture agent 220 or the
oligonucleotide
signal probe 206 itself. So when the reporter beads 192 come in close
proximity to
the capture agents 220, hybridization occurs between the anchor agent 222 and
the
capture agent 220 thereby retaining the dual bead complex 194 within the
target zone
170. In an alternate embodiment, the signal probe 206 serves the function of
anchor
agent 222. At this point, an interrogation beam 224 directed to the target
zone 170
may be used to detect the dual bead complex 194 within the target zone 170.
Fig. 16B illustrates the embodiment in Fig. 16A after a subsequent change in
disc rotational speed. The change in rational speed removes the capture bead
190
from the dual bead complex 194, ultimately isolating the reporter bead 192 and
the
target DNA sequence 202 in the target zone 170 to be detected by an
interrogation
beam 224.
The embodiment of the present invention depicted in Figs. 16A and 16B, may
alternatively be implemented on the transmissive disc illustrated in Figs. 4A-
4C, 5B,
and 6B.
Referring now to Fig. 17, there is shown an alternative to the embodiment
illustrated in Fig. 15A. In this embodiment, anchor agents 222 are attached to
the
capture beads 190 instead of the reporter beads. The anchor agent 222 in this
embodiment may include streptavidin or Neutravidin. As in Fig. 15A, the target
zone
170 is coated with a capture agent 220. The capture agent may include biotin
or BSA-
biotin. Fig. 17 also shows reporters 192 and capture beads 190 as components
of a
dual bead complex 194 as employed in the present invention. When the capture
37
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
beads 190 come in close proximity to the capture agents 220, binding occurs
between
the anchor probe 222 and the capture agent 220, via biotin-streptavidin
interactions,
thereby retaining the dual bead complex 194 within the target zone 170. At
this point,
an interrogation beam 224 directed to the target zone 170 can be used to
detect the
dual bead complex 194 within the target zone 170. The embodiment of the
present
invention shown in Fig. 17, may alternatively be implemented on the
transmissive disc
illustrated in Figs. 4A-4C, 5B, and 6B.
Fig. 18 is an alternative to the embodiment illustrated in Fig. 16A. In this
embodiment, anchor agents 222 are attached to the capture beads 190 instead of
the
reporter beads. In this embodiment the transport probes 198, or an anchor
agent 222
on the capture bead 190, directly hybridizes to the capture agent 220. In this
embodiment, the capture agent 220 includes specific sequences of nucleic acid.
The
anchor agent 222 in this embodiment may be a specific sequence of nucleic
acids that
are complimentary to the capture agent 220 or the oligonucleotide signal
transport
probe 198 itself. So when the capture beads 190 come in close proximity to the
capture agents 220, hybridization occurs between the anchor agent 222 and the
capture agent 220 thereby retaining the dual bead complex 194 within the
target zone
170. At this point, an interrogation beam 224 directed to the target zone 170
can be
used to detect the dual bead complex 194 within the target zone 170. The
embodiment of the present invention illustrated in Fig. 18, may alternatively
be
implemented on the transmissive disc shown in Figs. 4A-4C, 5B, and 6B.
Figs. 19A-19C are detailed partial cross sectional views showing the active
layer 176 and the substrate 174 of the present bio-disc 110 as implemented in
conjunction with the genetic assays discussed herein. Figs. 19A-19C
illustrates the
capture agent 220 attached to the active layer 176 by applying a small volume
of
capture agent solution to the active layer 176 to form clusters of capture
agents within
the area of the target zone. The bond between capture agent 220 and the active
layer
176 is sufficient so that the capture agent 220 remains attached to the active
layer 176
within the target zone when the disc is rotated. Figs. 19A and 19B also depict
the
capture bead 190 from the dual bead complex 194 binding to the capture agent
220 in
the capture zone. These dual bead complexes are prepared according to the
methods
such as those discussed in Figs. 11A and 12A. The capture agent 220 includes
biotin
and BSA-biotin. In this embodiment, the reporter bead 192 anchors the dual
bead
38
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
complex 194 in the target zone via biotin/streptavidin interactions.
Alternatively, the
target zone may be coated with streptavidin and may bind biotinylated reporter
beads.
Fig. 19C illustrates an alternative embodiment which includes an additional
step to
those discussed in connection with Figs. 19A and 19B. In this preferred
embodiment,
a variance in the disc rotations per minute may create a centrifugal force
great enough
to break the capture beads 190 away from the dual bead complex 194 based on
the
differential size and/or mass of the bead. ~ Although there is a shift in the
rotation
speed of the disc, the reporter bead 192 remains anchored to the target zone.
Thus,
the reporter beads 192 are maintained within the target zone and detected
using an
optical bio-disc or medical CD reader.
The embodiment of the present invention discussed in connection Figs. 19A-
19C, may be implemented on the reflective disc illustrated in Figs. 3A-3C, 5A,
and 6A
or on the transmissive disc shown in Figs. 4A-4C, 5B, and 6B.
Figs. 20A, 20B, and 20C illustrate an alternative embodiment to the
embodiment discussed in Figs. 19A-19C. Figs. 20A-20C show detailed partial
cross
sectional views of a target zone implemented in conjunction with
immunochemical
assays. Figs. 20A and 20B also depict the capture bead 190 from the dual bead
complex 194 binding to the capture agent 220 in the capture zone. The capture
agent
220 includes biotin and BSA-biotin. These dual bead complexes may be prepared
according to methods such as those discussed in Figs. 11 B and 12B. In this
embodiment, the reporter bead 192 anchors the dual bead complex 194 in the
target
zone via biotin/streptavidin interactions. The embodiment of the present
invention
discussed with reference to Figs. 20A-20C, may be implemented on the
reflective disc
depicted in Figs. 3A-3C, 5A, and 6A or on the transmissive disc shown in Figs.
4A-4C,
5B, and 6B.
Referring now to Figs. 21 A, 21 B, and 21 C, there is shown detailed partial
cross
sectional views of a target zone including the active layer 176 and the
substrate 174 of
the present bio-disc 110 as implemented in conjunction with the genetic assays
discussed herein. Figs. 21A-21C illustrate the capture agent 220 attached to
the
active layer 176 by use of an amino group 226 that is an integral part of the
capture
agent 220. As indicated, the capture agent 220 is situated within the target
zone. The
bond between the amino group 226 and the capture agent 220, and the amino
group
226 and the active layer 176 is sufficient so that the capture agent 220
remains
39
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
attached to the active layer 176 within the target zone when the disc is
rotated. The
preferred amino group 226 is NH2. A thiol group may alternatively be employed
in
place of the amino group 226. In this embodiment of the present invention, the
capture agent 220 includes the specific sequences of amino acids that are
complimentary to anchor agent 222 or oligonucleotide signal probe 206 which
are
attached to the reporter bead 192.
Fig. 21 B depicts the reporter bead 192 of the dual bead complex 194, prepared
according to methods such as those discussed in Figs. 11 A and 12A, binding to
the
capture agent 220 in the target zone. As the dual bead complex 194 flows
towards
the capture agent 220 and is in sufficient proximity thereto, hybridization
occurs
between the anchor agent 222, or oligonucleotide signal probe 206, and the
capture
agent 220. Thus, the reporter bead 192 anchors the dual bead complex 192
within
the target zone.
Fig. 21 C illustrates an alternative embodiment that includes an additional
step
to those discussed in connection with Figs. 21 A-21 B. In this preferred
embodiment, a
variance in the disc rotations per minute may create enough centrifugal force
to break
the capture beads 190 away from the dual bead complex 194 based on the
differential
size and/or mass of the bead. Although there is a shift in the rotation speed
of the
disc, the reporter bead 192 with the target DNA sequence 202 remains anchored
to
the target zone. In either case, the reporter beads 192 are maintained within
the
target zone as desired.
The embodiment of the present invention discussed with reference to Figs.
21 A-21 C, may be implemented on the reflective disc shown in Figs. 3A-3C, 5A,
and
6A or on the transmissive disc illustrated in Figs. 4A-4C, 5B, and 6B.
Figs. 22A, 22B, and 22C illustrate an alternative embodiment to the
embodiment discussed in Figs. 21 A-21 C. Figs. 22A-22C show detailed partial
cross
sectional views of a target zone implemented in conjunction with
immunochemical
assays. Figs. 22A and 22B also depict the reporter bead 192 from the dual bead
complex 194, prepared according to methods such as those discussed in Figs. 11
B
and 12B, binding to the capture agent 220 in the capture zone. In this
embodiment,
the capture agent 220 includes antibodies bound to the target zone by use of
an
amino group 226 that is made an integral part of the capture agent 220.
Alternatively,
the capture agents 220 may be bound to the active layer 176 by passive
absorption,
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
and hydrophobic or ionic interactions. In this embodiment, the reporter bead
192
anchors the dual bead complex 194 in the target zone via specific antibody
binding.
As with the embodiment illustrated in Fig. 21 C, Fig. 22C shows an alternative
embodiment that includes an additional step to those discussed in connection
with
Figs. 22A-22B. In this alternative embodiment, a variance in the disc
rotations per
minute may create enough centrifugal force to break the capture beads 190 away
from
the dual bead complex 194 based on the differential size and/or mass of the
bead.
Although there is a shift in the rotation speed of the disc, the reporter bead
192 with
the target antigen 204 remains anchored to the target zone. In either case,
the
reporter beads 192 are maintained within the target zone as desired. The
embodiment of the present invention described in conjunction with Figs. 22A-
22C,
may be implemented on the reflective disc illustrated in Figs. 3A-3C, 5A, and
6A or on
the transmissive disc shown in Figs. 4A-4C, 5B, and 6B.
Figs. 23A and 23B are detailed partial cross sectional views showing the
active
layer 176 and the substrate 174 of the present bio-disc 110 as implemented in
conjunction with the genetic assays. Figs. 23A and 23B illustrate an
alternative
embodiment to that discussed in Figs. 19A and 19B above. In contrast to the
embodiment in Figs. 19A and 19B, in the present embodiment, the anchor agent
222
is attached to the capture bead 190 instead of the reporter bead 192. Fig. 23B
illustrates the capture bead 190, from the dual bead complex 194, binding to
the
capture agent 220 in the capture zone. The capture agent 220 includes biotin
and
BSA-biotin. In this embodiment, the capture bead 190 anchors the dual bead
complex
194 in the target zone via biotin/streptavidin interactions.
The embodiment of the present invention discussed with reference to Figs. 23A
and 23B, may be implemented on the reflective disc illustrated in Figs. 3A-3C,
5A, and
6A or on the transmissive disc shown in Figs. 4A-4C, 5B, and 6B.
With reference now to Figs. 24A and 24B, there is presented detailed partial
cross sectional views showing the active layer 176 and the substrate 174 of
the
present bio-disc 110 as implemented in conjunction with the genetic assays.
Figs.
23A and 23B illustrate an alternative embodiment to that discussed in Figs. 21
A and
21 B above. In contrast to the embodiment in Figs. 21A and 21 B, in the
present
embodiment, the anchor agent 222 is attached to the capture bead 190 instead
of the
reporter bead 192. Fig. 23B illustrates the capture bead 190, from the dual
bead
41
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
complex 194, binding to the capture agent 220 in the capture zone. The capture
agent
220 is attached to the active layer 176 by use of an amino group 226 that is
made an
integral part of the capture agent 220. As indicated, the capture agent 220 is
situated
within the target zone. The bond between the amino group 226 and the capture
agent
220, and the amino group 226 and the active layer 176 is sufficient so that
the capture
agent 220 remains attached to the active layer 176 within the target zone when
the
disc is rotated. In this embodiment of the present invention, the capture
agent 220
includes the specific sequences of amino acids that are complimentary to the
anchor
agent 222 or oligonucleotide transport probe 198 which are attached to the
capture
bead 190. In this embodiment, the capture bead 190 anchors the dual bead
complex
194 in the target zone via hybridization between the capture agent 220 and the
anchor
agent or the transport probe 198.
The embodiment of the present invention shown in Figs. 24A and 24B, may be
implemented on the reflective disc illustrated in Figs. 3A-3C, 5A, and 6A or
on the
transmissive disc depicted in Figs. 4A-4C, 5B, and 6B.
Disc Processing Methods
Turning now to Figs. 25A-25D, there is shown the target zones 170 set out in
Figs. 21 A-21 C and Figs. 24A-24B in the context of a disc, using as an input
the
solution created according to methods such as those shown in Figs. 11A and
12A.
Fig. 25A shows a mixing/loading chamber 164, accessible through an inlet port
152, and leading to a flow channel 160. Flow channel 160 is pre-loaded with
capture
agents 220 situated in clusters. Each of the clusters of capture agents 220 is
situated .
within a respective target zone 170. Each target zone 170 can have one type of
capture agent or multiple types of capture agents, and separate target zones
can have
one and the same type of capture agent or multiple different capture agents in
multiple
capture fields. In the present embodiment, the capture agent can include
specific
sequences of nucleic acids that are complimentary to anchor agents 222 on
either the
reporter 192 or capture bead 190.
In Fig. 25B, a pipette 214 is loaded with a test sample of DNA or RNA that has
been sequestered in the dual bead complex 194. The dual bead complex is
injected
into the flow channel 160 through inlet port 152. As flow channel 160 is
further filled
with the dual bead complex from pipette 214, the dual bead complex 194 begins
to
42
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
move down flow channel 160 as the disc is rotated. The loading chamber 164 can
include a break-away retaining wall 228 so that complex 194 moves down the
flow
channel at one time.
In this embodiment, anchor agents 222, attached to reporter beads 192, bind to
the capture agents 220 by hybridization, as illustrated in Fig. 25C. In this
manner,
reporter beads 192 are retained within target zone 170. Binding can be further
facilitated by rotating the disc so that the dual bead complex 194 can slowly
move or
tumble down the flow channel. Slow movement allows ample time for additional
hybridization. After hybridization, the disc can be rotated further at the
same speed or
faster to clear target zone 170 of any unattached dual bead complex 194, as
illustrated in Fig. 25D.
An interrogation beam 224 can then be directed through target zones 170 to
determine the presence of reporters, capture beads, and dual bead complex, as
illustrated in Fig. 25D. In the event no target DNA or RNA is present in the
test
sample, there will be no dual bead complex structures, reporters, or capture
beads
bound to the target zones 170, but a small amount of background signal may be
detected in the target zones from non-specific binding. In this case, when the
interrogation beam 224 is directed into the target zone 170, a zero or low
reading
results, thereby indicating that no target DNA or RNA was present in the
sample.
The speed, direction, and stages of rotation, such as one speed for one period
followed by another speed for another period, can all be encoded in the
operational
information on the disc. The method discussed in connection with Figs. 25A-25D
may
also be performed on the transmissive disc illustrated in Figs. 4A-4C, 5B, and
6B
using a system with the top detector 130.
Figs. 26A-26D show the target zones 170 including the capture chemistries
discussed in Figs. 19A-19C and Figs. 23A-23B. This method uses as an input the
solution created according to methods shown in Figs. 11 A and 12A. Figs. 26A-
26D
illustrate an alternative embodiment to that discussed in Figs. 25A-25D
showing a
different bead capture method described in further detail below.
Fig. 26A shows a mixing/loading chamber 164, accessible through an inlet port
152, and leading to a flow channel 160. Flow channel 160 is pre-loaded with
capture
agents 220 situated in clusters. Each of the clusters of capture agents 220 is
situated
within a respective target zone 170. Each target zone 170 can have one type of
43
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
capture agent or multiple types of capture agents, and separate target zones
can have
one and the same type of capture agent or multiple different capture agents in
multiple
capture fields. In the present embodiment, the capture agent can include
specific
biotin and BSA-biotin that has affinity to the anchor agents 222 on either the
reporter
192 or capture bead 190. The anchor agents may include streptavidin and
Neutravidin.
In Fig. 26B, a pipette 214 is loaded with a test sample of DNA or RNA that has
been sequestered in the dual bead complex 194. The dual bead complex is
injected
into the flow channel 160 through inlet port 152. As flow channel 160 is
further filled
with the dual bead complex from pipette 214, the dual bead complex 194 begins
to
move down flow channel 160 as the disc is rotated. The loading chamber 164 can
include a break-away retaining wall 228 so that complex 194 moves down the
flow
channel at one time.
In this embodiment, anchor agents 222, attached to reporter beads 192, bind to
the capture agents 220 by biotin-streptavidin interactions, as illustrated in
Fig. 26C. In
this manner, reporter beads 192 are retained within target zone 170. Binding
can be
further facilitated by rotating the disc so that the dual bead complex 194 can
slowly
move or tumble down the flow channel. Slow movement allows ample time for
additional binding between the capture agent 220 and the anchor agent 222.
After
binding, the disc can be rotated further at the same speed or faster to clear
target
zone 170 of any unattached dual bead complex 194, as illustrated in Fig. 26D.
An interrogation beam 224 can then be directed through target zones 170 to
determine the presence of reporters, capture beads, and dual bead complex, as
illustrated in Fig. 26D. In the event no target DNA is present in the test
sample, there
will be no dual bead complex structures beads bound to the target zones 170. A
small
amount of background signal may be detected in the target zones from non-
specific
binding. In this case, when the interrogation beam 224 is directed into the
target zone
170, a zero or low reading results, thereby indicating that no target DNA or
RNA was
present in the sample.
The speed, direction, and stages of rotation, such as one speed for one period
followed by another speed for another period, can all be encoded in the
operational
information on the disc.
44
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
The method discussed in conjunction with Figs. 26A-26D was illustrated on a
reflective disc such as the disc shown in Figs. 3A-3C, 5A, and 6A. This method
may
also be performed on the transmissive disc shown in Figs. 4A-4C, 5B, and 6B
using a
system with the top detector 130.
Referring next to Figs. 27A-27D there is shown a series of cross sectional
side
views illustrating the steps of yet another alternative method according to
the present
invention. Figs. 27A-27D show the target zones 170 including the capture
mechanisms discussed in connection with Figs. 22A-22C. This method uses an
input
the solution created according to the preparation methods shown in Figs. 11 B
and
12B. Figs. 27A-27D illustrate an immunochemical assay and an alternative bead
capture method.
Fig. 27A shows a mixing/loading chamber 164, accessible through an inlet port
152, and leading to a flow channel 160. Flow channel 160 is pre-loaded with
capture
agents 220 situated in clusters. Each of the clusters of capture agents 220 is
situated
within a respective target zone 170. Each target zone 170 can have one type of
capture agent or multiple types of capture agents, and separate target zones
can have
one and the same type of capture agent or multiple different capture agents in
multiple
capture fields. In the present embodiment, the capture agent can include
antibodies
that specifically bind to epitopes on the anchor agents 222 on either the
reporter 192
or capture bead 190. Alternatively, the capture agent can directly bind to
epitopes on
the target antigen 204 within the dual bead complex 194. The anchor agents 222
can
include the target antigen, antibody transport probe 196, the antibody signal
probe
208, or any antigen, bound to either the reporter bead 192 or the capture bead
190,
that has epitopes than can specifically bind to the capture agent 220.
In Fig. 27B, a pipette 214 is loaded with a test sample of target antigen that
has
been sequestered in the dual bead complex 194. The dual bead complex is
injected
into the flow channel 160 through inlet port 152. As flow channel 160 is
further filled
with the dual bead complex from pipette 214, the dual bead complex 194 begins
to
move down flow channel 160 as the disc is rotated. The loading chamber 164 may
include a break-away retaining wall 228 so that complex 194 moves down the
flow
channel at one time.
In this embodiment, anchor agents 222, attached to reporter beads 192, bind to
the capture agents 220 by antibody-antigen interactions, as illustrated in
Fig. 27C. In
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
this manner, reporter beads 192 are retained within target zone 170. Binding
can be
further facilitated by rotating the disc so that the dual bead complex 194 can
slowly
move or tumble down the flow channel. Slow movement allows ample time for
additional binding between the capture agents 220 and the anchor agent 222.
After
binding, the disc can be rotated further at the same speed or faster to clear
target
zone 170 of any unattached dual bead complex 194, as illustrated in Fig. 27D.
An interrogation beam 224 can then be directed through target zones 170 to
determine the presence of reporters, capture beads, and dual bead complex, as
illustrated in Fig. 27D. In the event no target antigen is present in the test
sample,
there will be no dual bead complex structures, reporters, or capture beads
bound to
the target zones 170, but a small amount of background signal may be detected
in the
target zones from non-specific binding. In this case, when the interrogation
beam 224
is directed into the target zone 170, a zero or low reading results, thereby
indicating
that no target was present in the sample.
The speed, direction, and stages of rotation, such as one speed for one period
followed by another speed for another period, can all be encoded in the
operational
information on the disc.
The methods described in Figs. 25A-25D, 26A-26D, and 27A-27D are
implemented using the reflective disc system 144. As indicated above, it
should be
understood that these methods and any other bead or sphere detection may also
be
carried out using the transmissive disc embodiment 180, as described in Figs.
4A-4G,
5B, and 6B. It should also be understood that the methods described in Figs.
11 A-
11 B, 12A-12B, 25A-25D, 26A-26D, and 27A-27D are not limited to creating the
dual
bead complexes outside of the optical bio-discs but may include embodiments
that
use "in-disc" or "on-disc" formation of the dual bead complexes. In these on-
disc
implementations the dual bead complex is formed within the fluidic circuits of
the
optical bio-disc 110. For example, the dual bead formation may be carried out
in the
loading or mixing chamber 164. In one embodiment, the beads and sample are
added
to the disc at the same time, or nearly the same time. Alternatively, the
beads with the
probes can be pre-loaded on the disc for future use with a sample so that only
a
sample needs to be added.
The beads would typically have a long shelf life, with less shelf life for the
probes. The probes can be dried or lyophilized (freeze dried) to extend the
period
46
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
during which the probes can remain in the disc. With the probes dried, the
sample
essentially reconstitutes the probes and then mixes with the beads to produce
dual
bead complex structures can be performed.
In either case, the basic process for on-disc processing includes: (1)
inserting
the sample into a disc with beads with probes; (2) causing the sample and the
beads
to mix on the disc; (3) isolating, such as by applying a magnetic field, to
hold the dual
bead complex and move the non-held beads away, such as to a region referred to
here as a waste chamber; and (4) directing the dual bead complexes (and any
other
material not moved to the waste chamber) to the capture fields. The detection
process can be the same as one of those described above, such as by event
detection or fluorimetry.
In addition to the above, it would be apparent to those of skill in the art
that the
disc surface capturing techniques and the linking techniques for forming the
dual bead
complexes illustrated in Figs. 25A-25D, 26A-26D, and 27A-27D may be
interchanged
to create alternate variations thereof. For example, the inventors have
contemplated
that the capture agents 220 as implemented to include specific sequences of
nucleic
acids may be used to capture dual bead complexes formed by either DNA
hybridization as illustrated in Fig. 10A or the antibody-antigen interactions
shown in
Fig. 10B. Similarly, capture agents 220 as implemented to include antibodies
may be
employed to capture dual bead complexes formed by either the DNA hybridization
method shown in Fig. 10A or the antibody-antigen interactions illustrated in
Fig. 10.B.
And also, capture agents 220 as implemented to include biotin or BSA-biotin
may be
similarly utilized to capture dual bead complexes formed by either the DNA
hybridization techniques illustrated in Fig. 10A or the antibody-antigen
interactions
depicted in Fig. 10B. Other combinations including different anchor agents to
perform
the binding function with the capture agent, are readily apparent from the
present
disclosure and are thus specifically provided for herein.
Detection and Related Signal Processing Methods and Apparatus
The number of reporter beads, target cells, or particles bound in the capture
field or zone can be detected in a qualitative manner, and may also be
quantified by
the optical disc reader.
47
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
The test results of any of the test methods described above can be readily
displayed on monitor 114 (Fig. 1 ). The disc according to the present
invention
preferably includes encoded software that is read to control the controller,
the
processor, and the analyzer as shown in Fig. 2. This interactive software is
implemented to facilitate the methods described herein and the display of
results.
Fig. 28A is a graphical representation of an individual 2.1 um reporter bead
192
and a 3 um capture bead 190 positioned relative to tracks A, B, C, D, and E of
an
optical bio-disc or medical CD according to the present invention.
Fig. 28B is a series of signature traces, from tracks A, B, C, D, and E,
derived
from the beads of Fig. 28A utilizing a detected signal from the optical drive
according
to the present invention. These graphs represent the detected return beam 124
of the
reflective disc illustrated in Figs. 5A and 6A for example, or the transmitted
beam 128
of the transmissive disc illustrated in Figs, 5B and 6B. As shown, the
signatures for a
2.1 um reporter bead 190 are sufficiently different from those for a 3 um
capture bead
192 such that the two different types of beads can be detected and
discriminated.
This detection method is not limited to beads or bead complexes but may also
be
used to detect other objects such as cells or bead-cell complexes. As would be
apparent to one of skill in the art, signature traces from a 1 um magnetic
bead and an
8 um cell in an MO bio-disc, for example, would be sufficiently different so
as to
distinguish these particles from one another. A sufficient deflection of the
trace signal
from the detected return beam as it passes through a bead is referred to as an
event.
Fig. 29A is a graphical representation of a 2.1 um reporter bead and a 3 um
capture bead linked together in a dual bead complex positioned relative to the
tracks
A, B, C, D, and E of an optical bio-disc or medical CD according to the
present
invention.
Fig. 29B is a series of signature traces, from tracks A, B, C, D, and E,
derived
from the beads of Fig. 29A utilizing a detected signal from the optical drive
according
to the present invention. These graphs represent the detected return beam 124
of a
reflective disc 144 or transmitted beam 128 of a transmissive disc 180. As
shown, the
signatures for a 2.1 um reporter bead 190 are sufficiently different from
those for a 3
um capture bead 192 such that the two different types of beads can be detected
and
discriminated. A sufficient deflection of the trace signal from the detected
return beam
or transmitted as it passes through a bead is referred to as an event. The
relative
48
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
proximity of the events from the reporter and capture bead indicates the
presence or
absence the dual bead complex. As shown, the traces for the reporter and the
capture bead are right next to each other indicating the beads are joined in a
dual
bead complex.
Alternatively, other detection methods can be used. For example, reporter
beads can be fluorescent or phosphorescent. Detection of these reporters can
be
carried out in fluorescent or phosphorescent type optical disc readers. Other
signal
detection methods are described, for example, in commonly assigned co-pending
U.S.
Patent Application Serial No. 10/008,156 entitled "Disc Drive System and
Methods for
Use with Bio-Discs" filed November 9, 2001, which is expressly incorporated by
reference; U.S. Provisional Application Serial Nos. 60/270,095 filed February
20, 2001
and 60/292,108, filed May 18, 2001; and the above referenced U.S. Patent
Application
Serial No. 10/043,688 entitled "Optical Disc Analysis System Including Related
Methods For Biological and Medical Imaging" filed January 10, 2002.
Fig. 30A is a bar graph of data generated using a fluorimeter showing
concentration-dependent target detection using fluorescent reporter beads.
This
graph shows the molar concentration of target DNA versus the number of
detected
beads. The dynamic range of target detection shown in the graph is 10E-16 to
10E-
10 Molar (moles/liter). While the particular graph shown was generated using
data
from a fluorimeter, the results may also be generated using a fluorescent type
optical
disc drive.
Fig. 30B presents a standard curve demonstrating that the sensitivity of a
fluorimeter is approximately 1000 beads in a fluorescent dual bead assay. The
sensitivity of any assay depends on the assay itself and on the sensitivity of
the
detection system. Referring to Figs. 30A-30C, various studies were done to
examine
the sensitivity of the dual bead assay using different detection methods,
e.g., a
fluorimeter, and bio-disc or medical CD detection according to the present
invention.
As stated above and shown in Fig. 30B, the sensitivity of a fluorimeter is
approximately 1000 beads in a fluorescent dual bead assay. In contrast, Fig.
30A
shows that even at 10E-16 Molar (moles/liter), a sufficient number of beads
over zero
concentration can be detected to sense the presence of the target. With a
sensitivity
of 10E-16 Molar, a dual bead assay represents a very sensitive detection
method for
49
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
DNA that does not require DNA amplification (such as through PCR) and can be
used
to detect even a single bead.
In contrast to conventional detection methods, the use of a medical CD or bio
disc coupled with a CD-reader or optical bio-disc drive (Fig. 1 ) improves the
sensitivity
of detection. For example, while detection with a fluorimeter is limited to
approximately 1000 beads (Fig. 30B), use of a bio-disc coupled with CD-reader
may
enable the user to detect a single bead with the interrogation beam as
illustrated in
Figs. 29A, 29B, and 30C. Thus, the bioassay system provided herein improves
the
sensitivity of dual bead assays significantly.
The detection of single beads using an optical bio-disc or medical CD is
discussed in detail in conjunction with Figs. 28A and 28B. Fig. 28B shows the
signal
traces of each bead as detected by the medical CD or bio-disc reader. Dual
bead
complexes may also be identified by the bio-disc reader using the unique
signature
traces collected from the detection of a dual bead complex as shown in Figs.
29A and
29B. Different optical bio-disc platforms, including but not limited to the
reflective and
the transmissive disc formats illustrated respectively in Figs. 3C and 4C, may
be used
in conjunction with the reader device for detection of beads.
Fig. 30C is a pictorial representation demonstrating the formation of the dual
bead complex linked together by the presence of the target in a genetic assay.
Sensitivity to within one reporter molecule is possible with the present dual
bead
assay quantified with a bio-CD reader shown in Figs. 1 and 2 above. Similarly,
the
dual bead complex formation may also be implemented in an immunochemical assay
format as illustrated above in Figs. 7B, 8B, 9B, 1 OB, 11 B, and 12B.
Fig. 31 shows data generated using a fluorimeter illustrating the
concentration
dependent detection of two different targets. Target detection was carried out
using
two different methods (the single and the duplex assays). In the single assay,
the
capture bead contains a transport probe specific to a single target and a
reporter
probe coated with a signal probe specific to the same target is mixed in a
solution
together with the target. In the duplex assay, the capture bead contains two
different
transport probes specific to two different targets. Experimental details
regarding the
use of the duplex target detection method are discussed in further detail in
Example 2.
Mixing different reporter beads (red and green fluorescent or silica and
polystyrene
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
beads, for example) containing signal probes specific to one of the two
targets, allows
the detection of two different targets simultaneously.
Detection of the dual bead duplex assay may be carried out using a magneto-
optical disc system described below. Figs. 32 and 37 illustrate the formation
and
binding of various dual bead complexes onto an optical disc which may be
detected by
an optical bio-disc drive (Fig. 2), a magneto-optical disc system, a
fluorescent disc
system, or any similar device. Unique signature traces of a dual bead complex
collected from an optical disc reader are shown in Fig. 29B above. The traces
from
Fig. 29B further illustrate that different bead types can be detected by an
optical disc
reader since different beads show different signature profiles.
Multiplexing, Magneto-Optical and Magnetic Discs Systems
The use of a dual bead assay in the capture of targets allows for the use in
multiplexing assays. This type of multiplexing is achieved by combining
different sizes
of magnetic beads with different types and sizes of reporter beads. Thus,
different
target agents can be detected simultaneously. As indicated in Fig. 32, four
sizes of
magnetic capture beads, and four sizes of three types of reporter beads
produce up to
48 different types of dual bead complex. In a multiplexing assay, probes
specific to
different targets are thus conjugated to capture beads. Reporter beads having
different physical and/or optical properties, such as fluorescence at
different
wavelengths, allow for simultaneous detection of different target agents from
the same
biological sample. As indicated in Figs. 28A, 28B, 29A, and 29B, small
differences in
size can be detected by detecting reflected or transmitted light.
Multiple dual bead complex structures for capturing different target agents
can
be carried out on or off the disc. The dual bead suspension is loaded into a
port on
the disc. The port is sealed and the disc is rotated in the disc reader.
During
spinning, free (unbound) reporter beads are spun off to a periphery of the
disc while
the magnetic capture beads and magnetic bead complexes or dual beads are
captured in a magnetic field, in magnetic domains on the MO disc, for example.
The
reporter beads detecting various target agents are thus localized in capture
regions.
In this manner, the presence of a specific target agent can be detected, and
the
amount of a specific target agent can be quantified by the disc reader.
51
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
Fig. 33A is a general representation of an optical disc according to another
aspect of the present invention. The disc 110 illustrated in Fig. 33A may be
employed
to practice the methods corresponding generally to the single-step methods of
Figs.
11A and 11B illustrated above or the related single step methods described
below in
connection with Figs. 65A-65B and 66A-66B. The sample and beads can be added
at
one time or successively but closely in time. Alternatively, the beads can be
pre-
loaded into a portion of the disc. These materials can be provided to a mixing
chamber 164 that can have a breakaway wall 228 (see Fig. 25A), which holds in
the
solution within the mixing chamber 164. Mixing the sample and beads on the
disc
would be accomplished through rotation at a rate insufficient to cause the
wall to break
or the capillary forces to be overcome.
The disc can be rotated in one direction, or it can be rotated alternately in
opposite directions to agitate the material in a mixing chamber. The mixing
chamber
is preferably sufficiently large so that circulation and mixing is possible.
The mixing
can be continuous or intermittent.
Fig. 33B shows one embodiment of a rotationally-directionally-dependent valve
arrangement that uses a movable component for a valve. The mixing chamber
leads
to an intermediate chamber 244 that has a movable component, such as a ball
246.
In the non-rotated state, the ball 246 may be kept in a slight recessed
portion, or
chamber 244 may have a gradual V-shaped tapering in the circumferential
direction to
keep the ball centered when there is no rotation.
Referring to Figs. 33C and 33D in addition to Figs. 33A and 33B, when the disc
is rotated clockwise (Fig. 33C), ball 246 moves to a first valve seat 248 to
block
passage to detection chamber 234 and to allow flow to waste chamber 232, shown
in
Fig. 33A. When the disc is rotated counter-clockwise (Fig. 33D), ball 246
moves to a
second valve seat 250 to block a passage to waste chamber 232 and to allow
flow to
detection chamber 234.
Figs. 34A-34C show a variation of the prior embodiment in which the ball is
replaced by a wedge 252 that moves one way or the other in response to
acceleration
of the disc. The wedge 252 can have a circular outer shape that conforms to
the
shape of an intermediate chamber 244. The wedge is preferably made of a heavy
dense material relative to chamber 244 to avoid sticking. A coating can be
used to
promote sliding of the wedge relative to the chamber.
52
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
When the disc is initially rotated clockwise as shown in Fig. 34B, the angular
acceleration causes wedge 252 to move to block a passage to detection chamber
234
and to allow flow to waste chamber 232. When the disc is initially rotated
counter-
clockwise, Fig. 34C, the angular acceleration causes wedge 252 to block
passage to
waste chamber 232 and allows flow to detection chamber 234. During constant
rotation after the acceleration, wedge 252 remains in place blocking the
appropriate
passage.
In another embodiment of the present invention where the capture beads are
magnetic, a magnetic field from a magnetic field generator or field coil 230
can be
applied over the mixing chamber 164 to hold the dual bead complexes and
unbound
magnetic beads in place while material without magnetic beads are allowed to
flow
away to a waste chamber 232. This technique may also be employed to aid in
mixing
of the assay solution within the fluidic circuits or channels before any
unwanted
material is washed away. At this stage, only magnetic capture beads, unbound
or as
part of a dual bead complex, remain. The magnetic field is released, and the
dual
bead complex with the magnetic beads is directed to a capture and detection
chamber
234.
The process of directing non-magnetic beads to waste chamber 232 and then
magnetic beads to capture chamber 234 can be accomplished through the
microfluidic
construction and/or fluidic components. A flow control valve 236 or some other
directing arrangement can be used to direct the sample and non-magnetic beads
to
waste chamber 232 and then to capture chamber 234. A number of embodiments for
rotationally dependent flow can be used. Further details relating to the use
of flow
control mechanisms are disclosed in commonly assigned co-pending U.S. Patent
Application Serial No. 09/997,741 entitled "Dual Bead Assays Including Optical
Biodiscs and Methods Relating Thereto" filed November 27, 2001, which is
herein
incorporated by reference in its entirety.
Fig. 35 is a perspective view of a disc including one embodiment of a fluidic
circuit employed in conjunction with magnetic beads and the magnetic field
generator
230 according to the present invention. Fig. 35 also shows the mixing chamber
164,
the waste chamber 232, and the capture chamber 234. The magnetic field
generator
230 is positioned over disc 110 and has a radius such that as disc 110
rotates,
magnetic field generator 230 remains over mixing chamber 164, and is radially
spaced
53
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
from chambers 232 and 234. As with the prior embodiment discussed above, a
magnetic field from the magnetic field generator 230 can be applied over the
mixing
chamber 164 to hold the dual bead complexes and/or unbound magnetic beads in
place while additional material is allowed to enter the mixing chamber 164.
The
method of rotating the disc while holding magnetic beads in place with the
magnetic
field generator 230 may also be employed to aid in mixing of the assay
solution within
the mixing chamber 164 before the solution contained therein is directed
elsewhere.
Figs. 36A-36C are plan views illustrating a method of separation and detection
for dual bead assays using the fluidic circuit shown in Fig. 35. Fig. 36A
shows an
unrotated optical disc with a mixing chamber 164 shaped as an annular sector
holding
a sample with dual bead complexes 194 and various unbound reporter beads 192.
The electromagnet is activated and the disc is rotated counter-clockwise (Fig.
36B), or
it can be agitated at a lower rpm, such as 1 X or 3X. Dual bead complexes 194,
with
magnetic capture beads, remain in mixing chamber 164 while the liquid sample
and
the unbound reporter beads 192 move in response to angular acceleration to a
rotationally trailing end of mixing chamber 164. The disc is rotated in the
counter-
clockwise direction illustrated in Fig. 36B with sufficient speed to overcome
capillary
forces to allow the unbound reporter beads in the sample to move through a
waste
fluidic circuit 238 to waste chamber 232. At this stage in the process, the
liquid will not
move down the capture fluidic circuit 240 because of the physical
configuration of the
fluidic circuit as illustrated.
As illustrated next in Fig. 36C, the magnet is deactivated and the disc is
rotated
clockwise. Dual bead complexes 194 move to the opposite trailing end of the
mixing
chamber 164 in response to angular acceleration and then through a capture
fluidic
circuit 240 to the capture chamber 234. At this later stage in the process,
the dual
bead solution will not move down the waste fluidic circuit 238 due to the
physical
layout of the fluidic circuit, as shown. The embodiment shown in Figs. 36A-36C
thus
illustrates directionally-dependent flow as well as rotational speed dependent
flow.
In this embodiment and others in which a fluidic circuit is formed in a region
of
the disc, a plurality of regions can be formed and distributed about the disc,
for
example, in a regular manner to promote balance. Furthermore, as discussed
above,
instructions for controlling the rotation can be provided on the disc.
Accordingly, by
reading the disc, the disc drive can have instructions to rotate for a
particular period of
54
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
time at a particular speed, stop for some period of time, and rotate in the
opposite
direction for another period of time. In addition, the encoded information can
include
control instructions such as those relating to, for example, the power and
wavelength
of the light source. Controlling such system parameters is particularly
relevant when
fluorescence is used as a detection method.
In yet another embodiment, a passage can have a material or configuration that
can seat or dissolve either under influence from a laser in the disc drive, or
with a
catalyst pre-loaded in the disc, or such a catalyst provided in the test
sample. For
example, a gel may solidify in the presence of a material over time, in which
case the
time to close can be set sufficiently long to allow the unbound capture beads
to flow to
a waste chamber before the passage to the waste chamber closes. Alternatively,
the
passage to the waste chamber can be open while the passage to the detection
chamber is closed. After the unbound beads are directed to the waste chamber,
the
passage to the direction chamber is opened by energy introduced from the laser
to
allow flow to the detection chamber.
With reference now generally to Fig. 37, it is understood that magneto-optic
recording is an optical storage technique in which magnetic domains or areas
are
written into a thin film by heating it with a focused laser in the presence of
an external
magnetic field. The presence of these domains is then detected by the same
laser
from differences in the polarization of the reflected light between the
different magnetic
domains in the layer (Kerr rotation). By switching either the magnetic field
for constant
high laser power, or modulating the laser power with a constant magnetic
field, a data
pattern can be written into the layer. Many magneto-optic storage systems have
been
introduced into the market, including both computer data storage systems and
audio
systems (most notably MiniDisc). Descriptions of the current status of this
field can be
found in "The Principles of Optical Disc Systems", Bouwhuis et. al. 1985 (ISBN
0-
85274-785-3); "Optical Recording, A Technical Overview" A.B. Marchant 1990
(ISBN
0-201-76247-1 ); and "The Physical Principles of Magneto-Optical Recording",
M.
Mansuripur 1995 (ISBN 0521461243). All of these documents are herein
incorporated
by reference in their respective entireties.
Moving now specifically to Fig. 37, there is illustrated yet another
embodiment
of the optical disc 110 for use with a multiplexing dual bead assay. In this
case, a
disc, such as one used with a magneto-optical drive, has magnetic domains or
regions
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
242 that can be selectively written and erased with a magnetic head. Hereafter
this
type of disc will generally be referred to as a "magneto-optical bio-disc" or
an "MO bio-
disc". A magneto-optical disc drive, for example, can create magnetic domains
242 as
small as 1 um by 1 um spuare. The close-up section of the magnetic domain 242
shows the direction of the magnetic field with respect to adjacent regions.
The ability to selectively write to small areas in a highly controllable
manner to
make them magnetic allows capture areas to be created in desired locations.
These
magnetic capture areas or domains can be formed in any desired configuration
or
location in one fluidic chamber or in multiple fluidic chambers. These areas
capture
and hold magnetic beads when applied over the disc. The domains can be
selectively
erased if desired, thereby allowing them to be made non-magnetic and allowing
the
beads to be released.
In one configuration of a magnetic bead array according to this aspect of the
present invention, a set of three radially oriented magnetic capture regions
243 are
shown, by way of example, with no beads attached to the magnetic capture
regions in
the columns illustrated therein. With continuing reference to Fig. 37, there
is shown a .
set of four columns in Section A with individual magnetic beads magnetically
attached.
to the magnetic areas in a magnetic capture region. Another set of four
columns
arrayed in Section B is shown after binding of reporter beads to form dual
bead
complexes attached to specific magnetic domains or areas, with different
columns
having different types of reporter beads. As illustrated in Section B, some of
the
reporter beads utilized vary in size to thereby achieve the multiplexing
aspects of the
present invention as implemented on a magneto-optical bio-disc or MO medical
disc.
In Section C, a single column of various dual bead complexes is shown as
another
example of multiplexing assays employing various bead sizes individually
attached at
separate magnetic areas.
In a method of using such a magneto-optical bio-disc, the write head in an MO
drive is employed to create magnetic domains, and then a sample can be
directed
over that domain to capture magnetic beads provided in the sample. After
introduction
of the first sample set, other magnetic domains may also be created and
another
sample set can be provided to the newly created magnetic capture region for
detection. Thus detection of multiple sample sets may be performed on a single
disc
at different time periods. The magneto-optical drive also allows the
demagnetization
56
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
of the magnetic domains or capture regions to thereby release and isolate the
magnetic beads if desired. Thus this system provides for the controllable
capture,
detection, isolation, and release of one or more specific target molecules
from a
variety of different biochemical, chemical, or biological samples.
As described above, a sample can be provided to a fluidic chamber on a disc.
Alternatively, a sample could be provided to multiple chambers that have sets
of
different beads. In addition, a series of chambers can be created such that a
sample
can be moved by rotational motion from one chamber to the next, and separate
tests
can then be performed in each chamber.
With such an MO bio-disc, a large number of tests can be performed at one
time and can be performed interactively. In this manner, when a test is
performed and
a result is obtained, the system can be instructed to create a new set of
magnetic
regions for capturing the dual bead complex. Regions can be created one at a
time or
in large groups, and can be performed in successive chambers that have
different pre-
loaded beads. Other processing advantages can be obtained with an MO bio-disc
that has writeable magnetic regions. For example, the "capture agent" is
essentially
the magnetic field created by the magnetic region on the disc and therefore
there is no
need to add an additional biological or chemical capture agent.
Instructions for controlling the locations for magnetic regions written or
erased
on the MO bio-disc, and other information such as rotational speeds, stages of
rotation, waiting periods, wavelength of the light source, and other
parameters can be
encoded on and then read from the disc itself. As would be readily apparent to
one of
ordinary skill in the art given the disclosure provided herein, the MO bio-
disc illustrated
in Fig. 37 may include any of the fluidic circuits, mixing chambers, flow
channels,
detection chambers, inlet ports, or vent ports employed in the reflective and
transmissive discs discussed above. Illustrative examples of the use of the MO
bio-
disc according to this aspect of the present are provided below in Examples 5
and 6.
Thus in summary, the following embodiments of the magneto-optical aspects of
the present invention have been contemplated by the inventors and are herein
described in detail. Firstly, there is provided a method of performing a
genetic dual
bead assay in association with a magneto-optical bio-disc. This method
includes the
steps providing a plurality of magnetic capture beads having covalently
attached
transport probes, providing a plurality of reporter beads having covalently
attached
57
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
specific sequences of DNA, preparing a sample containing target DNA molecules
to
be tested for DNA sequences complementary to the specific DNA sequences, and
loading the capture beads into a magneto-optical bio-disc via an inlet port
provided
therein. The magneto-optical bio-disc has a magnetic capture layer. This
method
further includes loading the sample and the plurality of reporter beads into
the bio-
disc, rotating the bio-disc to facilitate hybridization of any target DNA
present in the
sample to the specific sequences of DNA on the reporter beads and to the
transport
probes to form dual bead complexes, interrogating a number of the magnetic
capture
beads with an incident beam of radiant energy to determine whether each of the
number of magnetic capture beads has formed a dual bead complex, magnetizing
specific regions of the magnetic capture layer to bind thereto a plurality of
the dual
bead complexes, and quantitating the plurality of the dual bead complexes.
The method may include the further steps of rotating the disc to direct any
unbound beads into a waste chamber and then de-magnetizing the specific
regions of
the magnetic capture layer to thereby release a number of the plurality of the
dual
bead complexes. Thereafter the disc may be rotated to direct the released
number of
dual bead complexes to an analysis area for further processing so that the
released
number of dual bead complexes are sequestered in the analysis area. The
analysis
area may be an analysis chamber having agents that react with the sequestered
dual
bead complexes.
According to a second embodiment of the magneto-optical aspects of the
present invention there is provided another method of performing a dual bead
assay in
association with a magneto-optical bio-disc. This other method includes the
steps of
providing a plurality of magnetic capture beads having attached transport
probes,
providing a plurality of reporter beads having attached signal probes, and
loading the
capture beads into a magneto-optical bio-disc via an inlet port provided
therein. The
magneto-optical bio-disc has a magnetic capture layer. This second method
further
includes loading a sample containing a target and the plurality of reporter
beads into
the bio-disc, rotating the bio-disc to facilitate binding of the target and
the reporter
beads to the magnetic capture beads to form dual bead complexes, interrogating
a
number of the magnetic capture beads with an incident beam of radiant energy
to
determine whether each of the number of magnetic capture beads has formed a
dual
bead complex, magnetizing specific regions of the magnetic capture layer to
bind
58
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
thereto a plurality of the dual bead complexes, and quantitating the plurality
of the dual
bead complexes.
This method may similarly include the further step of rotating the disc to
direct
any unbound beads into a waste chamber and then de-magnetizing the specific
regions of the magnetic capture layer to thereby release a number of the
plurality of
the dual bead complexes. It is also an aspect of this method to then rotate
the disc to
direct the released number of dual bead complexes to an analysis area for
further
processing so that the released number of dual bead complexes are sequestered
in
the analysis area. The analysis area may include a reaction chamber havina
aaents
that react with the sequestered dual bead complexes.
In accordance with a third embodiment of the magneto-optical aspects of the
present invention there is provided a method of performing a multiplexed dual
bead
assay in association with a magneto-optical bio-disc. This multiplexing method
includes the steps of (1 ) providing at least two groups of differently sized
magnetic
capture beads, each group having magnetic capture beads of the same size and
having a different specific type of transport probe associated with each
group; (2)
providing a plurality of reporter beads having attached at least two different
types of
signal probes; and (3) loading the capture beads into a magneto-optical bio-
disc via an
inlet port provided therein. As in the above MO bio-disc methods, this magneto-
optical
bio-disc has a magnetic capture layer. The method also includes (4) loading a
sample
containing at least one target and the plurality of reporter beads into the
bio-disc; (5)
rotating the bio-disc to facilitate binding of the target and the reporter
beads to the
magnetic capture beads to form dual bead complexes; (6) interrogating a number
of
the magnetic capture beads with an incident beam of radiant energy to
determine
whether each of the number of magnetic capture beads has formed a dual bead
complex; and (7) determining the size of the magnetic bead in the dual bead
complex.
This particular method concludes with the steps of (8) magnetizing specific
regions of
the magnetic capture layer to bind thereto a plurality of the dual bead
complexes; and
(9) quantitating the plurality of the dual bead complexes.
According to one aspect of this specific method, the step of quantitating may
advantageously include quantitating the plurality of the dual bead complexes
according to the size of the magnetic capture bead. The method may include the
further step of rotating the disc to direct any unbound beads into a waste
chamber and
59
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
then de-magnetizing the specific regions of the magnetic capture layer to
thereby
release a number of the plurality of the dual bead complexes containing same-
sized
magnetic capture beads. The method may further include rotating the disc to
direct
the released number of same-sized dual bead complexes to an analysis area for
further processing so that the released number of same-sized dual bead
complexes
are sequestered in the analysis area. The analysis area may include a reaction
chamber having agents that react with the sequestered same-sized dual bead
complexes. In one particular embodiment hereof, the signal probe is a specific
sequence of DNA.
According to yet a fourth embodiment of the magneto-optical aspects of the
present invention there is provided another principal method of performing a
multiplexed dual bead assay in association with a magneto-optical bio-disc.
This
additional dual bead multiplexing method includes the steps of (1 ) providing
at least
two groups of different types of reporter beads, each group having reporter
beads of
the same type and having a different specific type of signal probe associated
with
each group; (2) providing a plurality of magnetic capture beads having
different types
of transport probes attached thereto; and (3) loading the capture beads into a
magneto-optical bio-disc via an inlet port provided therein. As in the above
MO bio-
diac methods, this particular magneto-optical bio-disc has a magnetic capture
layer.
The method continues with the additional steps of (4) loading a sample to be
tested for
at least one target and the plurality of reporter beads into the bio-disc; (5)
rotating the
bio-disc to facilitate binding of any target present in the sample to the
reporter beads
and to the magnetic capture beads to form dual bead complexes; and (6)
interrogating
a number of the reporter beads with an incident beam of radiant energy to
determine
whether each of the number of reporter beads has formed a dual bead complex.
This
particular embodiment of the present method then concludes with (7)
determining the
type of the reporter bead in the dual bead complex; (8) magnetizing specific
regions of
the magnetic capture layer to bind thereto a plurality of the dual bead
complexes; and
(9) quantitating the plurality of the dual bead complexes.
In one specific embodiment hereof, the step of quantitating includes
quantitating the plurality of the dual bead complexes according to the type of
reporter
bead. The method may further include the further step of rotating the disc to
direct
any unbound beads into a waste chamber and then, if desired, de-magnetizing
the
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
specific regions of the magnetic capture layer to thereby release a number of
the
plurality of the dual bead complexes containing same-type reporter beads. The
further
step of rotating the disc to direct the released number of same-type dual bead
complexes to an analysis area for further processing so that the released
number of
same-type dual bead complexes are sequestered in the analysis area may also be
performed.
As with the above methods, the analysis area may include a reaction chamber
having agents that react with the sequestered same-type dual bead complexes.
The
present invention further contemplates an optical bio-disc used to perform any
of the
above methods, and an optical bio-disc used to analyze any the dual bead
complexes
prepared according to the methods discussed above in connection with Figs. 11
A,
11 B, 12A, 12B or those methods discussed in detail below in conjunction with
Figs.
65A and 65B, 66A and 66B, 67A and 67B as well as 68A and 68B. Furthermore, the
MO bio-disc may be implemented in other biomagnetic assays including
immunomagnetic and molecumagnetic assays such as cellular capture and analysis
methods using the MO biodisc as discussed below.
Genetic Assays Usinq Ligation to Increase Assay Sensitivity
Referring to Fig. 38, there is shown the dual bead complex 194 held together
by the target DNA 202 through the covalently bound transport probes 198 and
signal
probes 206 on the capture bead 190 and the reporter bead 192, respectively. As
depicted in this figure, the 5' end of the signal probe 206 is held right next
to the 3' end
of the transport probe 198. This configuration allows the ligation of the 3'
and 5' ends
of the probes upon addition of ligase. Ligation of both probes only occurs in
the
presence of the target and it enhances the sensitivity of the assay by
increasing the
bond strength between the reporter and capture beads preventing the
dissociation of
the dual bead complex.
Referring now to Fig. 39, there is a bar graph illustrating the results from a
genetic test detected by an enzyme assay. A 3~,m capture bead bound to
transport
probes was used to capture the target in this test. Once the target was
captured, a
biotinylated reporter probe was introduced and allowed to bind to the target.
The
capture beads were then washed to remove unbound reporter probes. Ligase is
then
added to the solution to ligate the ends of the reporter and transport probes,
as shown
61
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
in Fig. 38. After a series of wash steps, streptavidinated-alkaline
phosphatase is
added to the bead solution and allowed to bind with the biotin on the reporter
probe.
The beads are again washed and a chromagen alkaline phosphatase substrate is
added to the bead solution. The intensity of the color formed by the alkaline
phosphatase and substrate reaction is then quantified using a
spectrophotometer.
The results from this quantification are shown in Fig. 39. The data presented
in this
figure indicates that there is approximately a 50% increase in signal when the
probes
are ligated. Thus the assay sensitivity is significantly increased by the
ligation step in
this experiment. Examples 3 and 4 discuss in detail the procedures followed in
carrying out a similar experiment.
Fig. 40 shows a bar graph from a genetic test using a ligation step
implemented
in a dual bead assay instead of an enzyme assay. The enzyme assay as discussed
in
Fig. 39, is used to verify the activity of ligase in a non-dual bead format,
which serves
as a control in the dual bead experiment. As with the enzyme assay, the same
Sum
capture beads bound to transport probes were used in the dual bead assay. The
reporter beads used in the dual bead assay were 2.1 um fluorescent beads. The
dual
beads were formed as discussed in either Fig. 11 A or 12A. The ligation step
is
implemented in Step V in Fig. 11A or Step VI in Fig. 12A where ligase is added
to the
dual bead complex solution and allowed to ligate the transport probes to the
signal
probes. The data shown in Fig. 40 indicates that ligation significantly
increases the
signal and sensitivity of the assay relative to the non-ligated control
treatment in Set 1
but not in Set 2.
Similarly, Fig. 41 is a bar graph showing the number of reporter beads bound
in
a dual bead complex using a 39mer bridge employing the same ligation step as
discussed in Fig. 40. As in Fig. 40, the data in Fig. 41 indicates that
ligation
significantly increases the sensitivity of the dual bead assay in both Sets 1
and 2. This
data demonstrates that the use of a 39mer bridge aids in the ligation process
thus
enhancing the signal from both Sets as implemented in the dual bead assay.
Dual Bead Assavs Using Cleavable Spacer or Displacement Probes
The use of cleavable spacers in dual bead assay increases the specificity of
the
assay. Indeed, in addition to complementary sequences to the target DNA, the
capture
probes and reporter probes contain sequences that are complementary to each
other.
62
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
This additional requirement enhances specificity to target capture.
Furthermore,
additional bonding between the capture bead and reporter beads via the
hydrogen
bonds between capture and reporter probes strengthen the interactions between
the
dual beads.
In this embodiment of the present invention, in the absence of a target, the
capture probe hybridizes to the reporter probes, resulting in the formation of
the dual
bead complexes as shown in 42B and 43A. As illustrated in Figs. 42B and 42C,
the
dual bead complexes are subjected to selective restriction enzyme digestion
after
target capture. The sequence specific digestion will selectively cleave the
hydrogen
bonds between the capture probes and reporter probes as depicted in Fig. 42D.
In the
absence of target, with the severance of the hydrogen bonds holding the
capture and
reporter probes, the dual beads dissociate from each other. In the presence of
target,
the capture and reporter beads remain bound via the target-mediated hydrogen
bonds
(Fig. 42D). The amount of target captured therefore is correlated with the
number of
dual beads remaining after enzyme digestion.
Alternatively, instead of restriction enzyme digestion, the bond holding the
capture probes and reporter probes can be unraveled by the use of a
displaceable
linker. The linker is detached using a displacement probe. In this case, the
reporter
probe contains a sequence that is partially complementary to the capture probe
resulting in a mismatched overhang as depicted in Fig. 43A. To dissociate the
capture
and reporter probes from each other, the complex is subjected to heat
treatment that
will initiate the melting of the reporter probe from the capture probe,
followed by
addition of a large excess of displacement probe. The higher concentration of
displacement probe and the tighter interactions between the displacement probe
and
the mismatched overhang which will result in the unraveling of the reporter
probe from
the capture probe as illustrated in Figs 43B and 43C. This will result in the
dissociation
of reporter beads from capture beads in the absence of target DNA.
More specifically, the dual bead assay according to the present invention may
be implemented using 3p,m magnetic capture beads and 2.1 lum fluorescent
reporter
beads. These beads are coated with transport probes and signal probes
respectively.
The transport probes and signal probes, in addition to being complementary to
a
target sequence, pUCl9 for example, contain sequences that are complementary
to
each other, as illustrated in Figs. 42A, 42B, 42C, and 43D. The sequences that
bind
63
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
the transport probe and the signal probes together are designed such that they
are
susceptible to the cleavage of very rare restriction enzymes including Not 1.
The use
or rare restriction enzymes and restriction sites prevents the accidental
cleavage of
the target DNA. The capture beads and reporter beads are mixed with varying
quantities of target DNA. After target capture, the DNA complex is subjected
to
restriction digestion by a rare restriction enzyme including Not 1. The
restriction
digestion by this enzyme will cleave the DNA sequence connecting the reporter
beads
to the capture beads. In the absence of target DNA, the reporter beads will be
dissociated from the capture beads and removed by magnetic concentration of
the
magnetic beads. Thus, only in the presence of the target sequence, the
magnetic
capture beads bind to fluorescent reporter beads, resulting in a dual bead
assay. The
introduction of clcavable spacers into the capture and reporter probes
improves the
specificity and the sensitivity of the dual bead significantly.
In an alternative embodiment of the present invention, a shorter overlap and a
mismatched overhang between the complementary sequences of probes on the
reporter bead and the capture bead (probe 1 and probe 2B), resulting in the
formation
of a displaceable linker, is used in conjunction with a displacement probe as
illustrated
in Figs. 43A and 438. The mismatched overhang on probe 2B is the site for
initial
binding of the displacement probe as shown in Fig. 42B. Once the displacement
probe binds to the overhang, the displacement probe proceeds to displace the
overlaying sequences between probe 1 and probe 2B which is depicted in Fig.
43C.
In the absence of target DNA, the reporter beads will be dissociated from the
capture
beads by the actions of the displacement probe and consequently removed by
magnetic concentration of the magnetic beads. Thus, only in the presence of
the
target sequence, the magnetic capture beads bind to fluorescent reporter
beads,
resulting in the non-dissociation of the dual bead complex.
The general operation of the clcavable spacer according to the present
invention can be understood more particularly by reference to Figs. 44, 45,
46A-46C,
47, 48A, 48B, and 49A-49C, which schematize two embodiments of the present
invention. With reference to Fig. 44, a capture bead is provided with a
derivatized
surface to which is attached a plurality of clcavable spacer molecules 256.
Each
spacer 256 including a cleavage site 258, a signal probe 206, and a transport
probe
198. As shown in Fig. 44, the transport probes include a thiol group which
reacts to
64
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
form a covalent bond with metallic elements as discussed in conjunction with
Fig. 45.
The capture bead, which may be porous or solid, can be selected from a variety
of
materials such as plastics, glass, mica, silicon, and the like.
The surface of the capture bead 190 or reporter bead 192 can be conveniently
derivatized to provide covalent bonding to each of the probes including the
cleavable
spacer molecule 256. Referring now to Fig. 45, there is shown metallic
reporter beads
that provide a convenient reflective signal-generating means for detecting the
presence of a target. Typical materials used in creating metallic beads are
gold,
silver, nickel, chromium, platinum, copper, and the like, with gold being
presently
preferred for its ability readily and tightly to bind e.g. via dative binding
to a free SH
group at the signal responsive end of the cleavable spacer. The metal beads
may be
solid metal or may be formed of plastic, or glass beads or the like, on which
a coating
of metal has been deposited. Also, other reflective materials can be used
instead of
metal. The presently preferred gold spheres bind directly to the thio group of
the signal
probe 206.
As depicted in Figs. 44 and 45, the transport probe 198 is attached covalently
at the amino end via an amide linkage. The cleavable spacer molecule includes
the
cleavage site 258 that is susceptible to cleavage during the assay procedure,
by
chemical or enzymatic means, heat, light or the like, depending on the nature
of the
cleavage site. Chemical means are presently preferred with a siloxane cleavage
group, and a solution of sodium fluoride or ammonium fluoride, exemplary,
respectively, of a chemical cleavage site and chemical cleaving agent. Other
groups
susceptible to cleaving, such as ester groups or dithio groups, can also be
used.
Dithio groups are especially advantageous if gold spheres are added after
cleaving
the spacer. Alternatively, the cleavage site may be a restriction site for
cleavage using
restriction enzymes. Restriction cleavage is the preferred method when
performing
genetic or immunochemical assays. Spacers may contain two or more cleavage
sites
to optimize the complete cleavage of all spacers.
Nucleic Acid Assays Using Cleavable S~~acers
In one aspect of the invention, the transport and signal probes are adapted to
bind complementary strands of nucleic acids that may be present in a test
sample.
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
The complementary oligonucleotides comprise members of a specific binding
pair, i.e.,
one oligonucleotide will bind to a second complementary oligonucleotide.
As is shown more particularly in Figs. 46A through 46C, schematizing one
embodiment of the invention, cleavable spacer molecules 256 including the
transport
probes 198 and signal probes 206 located at different sites on the surface of
the
capture bead 190 and reporter bead 192. As illustrated in Fig. 46A,
oligonucleotide
target agents 202 are located in close proximity to the transport probes 198
and signal
probes 206. In the event these target agents are complimentary to both probes,
hybridization occurs between the target agent 202, transport probe 198, and
the
reporter probes 206 to form a double helix as is shown in Fig. 46B. If there
is no
complementarity between the target agent 202 and the probes, there is no
binding
between those groups as is further illustrated in Fig. 46B where no double
helix if
formed.
When the cleavage site 258 is cleaved, but for the binding by the double helix-
coupled oligonucleotides, the reporter beads 192 will be free of the capture
bead 190
and dissociated therefrom. This is illustrated more fully in Fig. 46C. The
presence or
absence of dual bead complexes 194 may then be detected by an incident light,
particularly an incident laser light.
Nucleic Acid Assays Using Cleavable Spacers and Ligation
With reference now to Fig. 47A, there is illustrated a schematic
representation
of an alternative embodiment employing a bridging agent 260. The bridging
agent 260
may include a relatively short oligonucleotide sequence for binding to a
portion of a
target such that when the target binds to the transport 198 and signal probes
206, the
bridging agent 260 acts as a bridge between the ends on the transport probe
198 and
the signal probe 206. This results in the formation of a double helix with two
breaks as
depicted in Fig. 47B.
Continuing on to the next step shown in Fig. 47C, there is shown a schematic
representation of the use of DNA ligase in conjunction with the cleavable
spacer in a
further embodiment of the nucleic acid detection embodiment of the present
invention.
The ligation procedure links the breaks in the double helix covalently. This
covalent
linkage increases the strength with which analyte-specific binding adheres the
dual
66
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
bead complex thus permitting, in this embodiment, increased stringency of wash
affording increased specificity of the assay.
It will be appreciated by those skilled in nucleic acid detection that the
cleavable
reflective signal elements of the present invention are particularly well
suited for
detecting amplified nucleic acids of defined size, particularly nucleic acids
amplified
using the various forms of polymerase chain reaction (PCR), ligase chain
reaction
(LCR), amplification schemes using T7 and SP6 RNA polymerase, and the like.
Immunoassays Using Cleavable Spacers
In a further embodiment of the invention shown in Figs. 48A through 48C, the
cleavable spacer 258 includes modified antibodies to permit an immunoassay.
The
modified antibodies may be attached non-covalently to the cleavable spacer 258
mediated by oligonucleotides that are covalently attached to the antibodies.
Use of
complementary nucleic acid molecules to effectuate non-covalent, combinatorial
assembly of supramolecular structures is described in further detail in co-
owned and
co-pending U.S. Patent Applications Serial No. 08/332,514, filed October 31,
1994;
08/424,874, filed April 19, 1995; and 08/627,695, filed March 29, 1996,
incorporated
herein by reference. In another embodiment, antibodies can be attached
covalently to
the cleavable spacer using conventional cross-linking agents, either directly
or through
linkers.
The antibody probes include an antibody transport probe 196 bound to the
capture bead 190 and an antibody signal probe 208 bound to the reporter bead
192.
Both beads and probes are held together by the cleavable spacer 258. The
antibody
transport probe 196 and the antibody signal probe 208 have affinity to
different
epitopic sites of an antigen of interest.
With further reference to the immunoassay schematized in Figs. 48A-48C,
upon application of a test solution containing target antigen 204 or a non-
specific
target agent 200 to the collection of dual bead complexes 194 as illustrated
in Fig.
48A, target antigen 204 binds to the antibody transport probe 196 and the
antibody
signal probe 208 as shown in Fig. 48B. This binding prevents decoupling of the
dual
bead complex 194 when the cleavage site 258 is cleaved, such as, for example,
by
contact with a chemical cleaving agent. In contrast, the second cleavable
signal
element, which was not bound by the non-specific target agent 200 because the
lack
67
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
of binding affinity of the antibodies to the target agent 200, allow the dual
bead
complexes to dissociate as illustrated in Fig. 48C.
Presence and absence of the dual bead complex 194 may then be detected as
reflectance or absence of reflectance of incident light, particularly incident
laser light.
As should be apparent, coupling of antibodies as depicted permits the
adaptation of standard immunoassay chemistries and immunoassay geometries for
use with the cleavable spacers in the dual bead assay of the present
invention. Some
of these classical immunoassay geometries are further described in U.S. Pat.
No.
5,168,057, issued Dec. 1, 1992, incorporated herein by reference. Other
immunoassay geometries and techniques that may usefully be adapted to the
present
invention are disclosed in Diamandis et al. (eds.), Immunoassay, AACC Press
(July
1997); Gosling et al. (eds.), Immunoassay: Laboratory Analysis and Clinical
Applications, Butterworth-Heinemann (June 1994); and Law (ed.), Immunoassay: A
Practical Guide, Taylor & Francis (October 1996), the disclosures of which are
incorporated herein by reference. Thus, it should be apparent that the direct
detection
of analytes schematized in Figs. 48A-48C is but one of the immunoassay
geometries
adaptable to the cleavable spacer type dual bead assay and assay devices of
the
present invention.
The present invention will prove particularly valuable in immunoassays
screening for human immunodeficiency viruses, hepatitis a virus, hepatitis B
virus,
hepatitis C virus, and human herpes viruses.
It will further be appreciated that antibodies are exemplary of the broader
concept of specific binding pairs, wherein the antibody may be considered the
first
member of the specific binding pair, and the antigen to which it binds the
second
member of the specific binding pair. In general, a specific binding pair may
be defined
as two molecules, the mutual affinity of which is of sufficient avidity and
specificity to
permit the practice of the present invention. Thus, the cleavable spacer of
the present
invention may include other specific binding pair members as side members. In
such
embodiments, the first side member of the cleavable signal element includes a
first
member of a first specific binding pair, the second side member of the
cleavable
spacer includes a first member of a second specific binding pair, wherein said
second
member of said first specific binding pair and said second member of said
second
specific binding pair are connectably attached to one another, permitting the
formation
68
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
of a tethering loop of the general formula: first member of first specific
binding pair-
second member of first specific binding pair-second member of second specific
binding pair-first member of second specific binding pair.
Among the specific binding pairs well known in the art are biologic receptors
and their natural agonist and antagonist ligands, proteins and cofactors,
biotin and
either avidin or streptavidin, alpha spectrin and beta spectrin monomers, and
antibody
Fc portions and Fc receptors.
Methods for DNA Conjugation onto Solid Phase
Successful conjugation of probes to a solid phase such as a bead or a bio-
disc,
is an important step for the dual bead assays of the invention. In certain
embodiments
of the invention, probes are attached covalently to the beads. Efficiency of
the
covalent conjugation depends on the type of bead utilized and the specific
conjugation
method employed.
As illustrated in Fig. 49, a systematic method to evaluate the use of a solid
phase for probe conjugation is presented. The methodology identifies covalent
linkages that improve specificity of a dual bead assay. This approach can be
used to
evaluate treatment of solid phase (i.e., coating of a solid surface such as
the surface
of a bead or a surface on a biodisc) to see whether the treatment improves the
solid
phase conjugation efficiency. As a first step, probes are tagged with an
appropriate
molecule for detection and measurement of the amount of probe bound at a later
time.
By way of non-limiting example, a biotin moiety (B) can be attached at the 3'
end of a
DNA probe. Next, the probe is conjugated in the presence or absence of a cross-
linking agent, e.g., EDC (1-Ethyl 3-3 dimethylaminopropyl carbodiimide-HCI).
In the
presence of a cross-linking agent, a probe will be conjugated both covalently
and non-
covalently. Alternatively, in the absence of the cross-linking agent, a probe
will only
be absorbed to the bead non-covalently. After the appropriate washing steps
are
performed, a detection agent is added that binds specifically to the biotin
molecule
previously tagged to the probe. For example, streptavidin-alkaline phosphatase
(S-
AP) is added to the probe-bound beads, and the S-AP binds specifically to the
biotinylated probes. Next, alkaline phosphatase substrate is added to the
sample.
This substrate develops color upon loss of a phosphate group, and the
intensity of the
color correlates with the amount of probes bound to the beads. After an
appropriate
69
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
incubation period, the solution is isolated and the optical density of the
solution at an
appropriate wavelength is determined with a spectrophotometer or microtiter
plate
reader.
Referring to Fig. 50, there is illustrated conjugation of an oligonucleotide
probe
onto a carboxylated bead. Conjugation of probes may be carried out covalently
or
non-covalently. In a dual bead assay, covalent probe conjugation is preferred
over
non-covalent conjugation as discussed in further detail in connection with
Figs. 51 A,
51 B, 53A, and 53B. This conjugation process is performed prior to Step I of
the dual
bead assay as presented in Figs. 11 A, 11 B, 12A, and 12B. The amount of probe
covalently bound to the solid surface may be evaluated by determining the
amount of
probe that binds to the solid phase covalently and non-covalently, i.e., non-
specifically,
in the presence and absence of a crosslinking agent (e.g., EDC). The
percentage of
non-covalently bound probe can be determined according to the formula 100% *
N/T,
and the percentage of covalently bound probe can be determined by the formula
100% * (T-N)/T, wherein "T" represents the total amount of signal obtained in
the
presence of a cross-linking agent (i.e., the total amount of covalently and
noncovalently bound probe) and "N" represents the total amount of signal
obtained
when no crosslinking agent is used. Alternatively, the amount of probes
conjugated
covalently can be obtained directly if all non-covalently bound probes are
removed
prior to the addition of the S-AP. This can be conveniently achieved by
heating the
beads to 70°C prior to the step of adding the S-AP. If the percentage
of non-
covalently bound probe is less than 20%, the beads being tested can be used as
solid
phase for covalent conjugation. Results of an application of this methodology
are
presented in Figs. 51 A, 51 B, and 55 (see Example 7 for details).
As depicted in Figs. 51 A and 51 B, the 1.8 ~,m, 2.1 Vim, and 3 p,m beads
provide
suitable solid phase for covalent probe conjugation with at least 75%
conjugation
efficiency. The 1-2 ~,m beads, however, may not be suitable for covalent
conjugation
of probes due to their low covalent conjugation efficiency of less than 21 %.
Various embodiments of the invention utilize nucleic acid molecules as probes.
Fig. 52A shows the structural differences between single stranded and double
stranded DNA in order to illustrate how the single stranded DNA can more
readily bind
non-covalently to a solid phase. Single-stranded DNA has hydrophobic base side
chains that can readily absorb to a solid phase non-covalently. In contrast,
with
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
double-stranded DNA hydrophobic base interaction with a solid phase does not
generally occur and non-covalent or non-specific binding is limited in
comparison to a
single-stranded DNA molecule. Thus, in various embodiments of the invention,
double stranded DNA can be utilized in place of single-stranded DNA, thereby
enhancing DNA binding to a solid phase by covalent linkage (Fig. 52B). After
covalent
binding of one of the strands of the double-stranded DNA probe to the solid
phase, the
non-covalently bound strand may be removed by heating the sample to
70°C in the
appropriate buffer. Under these conditions, the double stranded DNA are
separated,
and only single strand DNA probe that is covalently attached to the bead
ramain and
is used to capture the target. Experimental details regarding the use of
double
stranded DNA for covalent probe conjugation is described in further detail
below in
Example 8.
In various embodiments of the invention, heat treatment can be used to
selectively remove non-covalently bound probes) from a solid phase. This
method is
useful when, for example, despite all optimizations with respect to the type
of the solid
phase, treatment of the solid phase, and the use of double stranded DNA, non-
covalent binding to the solid phase is still problematic. The conditions for
the heat
treatment have been optimized; the optimal buffer consists of: 2%BSA, 50 mM
Tris-
HCI, 145 mM NaCI, 1 mM MgCl2, 0.1 mM ZnCl2. The treatment is done at a
temperature less than or equal to approximately 70°C, since at higher
temperatures,
the magnetic beads can lose their magnetic properties.
In other embodiments of the invention, the methodology presented herein to
determine optimal conditions to obtain covalent linkages that improve
specificity of a
dual bead assay can be applied to a disc surface that is used as a solid
phase.
Similarly, the invention provides in other embodiments analogous to those
described
herein above to evaluate solid surfaces for protein binding. For example, such
an
application would be useful where the probe utilized is an antigen or
antibody.
Referring now to Fig. 53A, there is shown a bar graph of results collected
from
an enzyme assay detecting targets bound to probes on two different capture
beads for
use in a dual bead assay. As illustrated above in Fig 51A, the 1-2 ~,m beads
have a
covalent binding efficiency up to 20% and the rest of the probes bind non-
covalently
and the covalent binding efficiency of the 3 ~,m beads is between 75-85%. The
data
shown in Fig. 53A indicates that both of the tested beads bind a similar
amount of
71
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
target regardless of whether the probe is bound covalently or non-covalently.
This
suggests that covalent binding is not necessary in an enzyme assay format.
In contrast to Fig. 53A, Fig. 53B represents results of a dual bead assay
designed to examine the number of reporter beads captured by the same capture
beads used in Fig. 53A. The results shown in Fig. 53B indicate that covalent
binding
of the probe to the capture bead is necessary to enhance the sensitivity of
the assay.
In this particular embodiment of the present invention, the 3~,m capture bead
contains
more covalently bound probes than the 1-2~,m beads, as mentioned above. This
allows the retention of the reporter bead in the dual bead complex since
covalently
tethered probes on the capture bead have higher bond strength than non-
covalently
bound probes.
As mentioned in the summary of the invention above, the surface of the beads
or solid phase may be uneven which limits the probe accessibility to the
target in
solution. Probe linkers may be used to extend the length of the probes to
increase
probe target accessibility as discussed with reference to Fig. 52A.
With reference now to Fig. 54, there is presented data collected from a dual
bead assay showing enhanced target binding using PEG as a linker. Linkers may
increase the assay sensitivity by approximately 50% or more. The use of
linkers also
decreases non-specific reporter bead binding to the capture beads. In this
embodiment of the present invention, probes are attached to a solid phase by
way of a
linker molecule. The use of a linker molecule makes the probe longer and more
rigid.
These two properties increase the accessibility of the probe(s), and,
therefore,
maximize the efficiency of target capture and the sensitivity of the dual bead
assay.
As known to those skilled in the art, various linker molecules can be used
that satisfy
the criteria described herein. By way of non-limiting example, bovine serum
albumin
(BSA) or polyethylene glycol (PEG) can be used as linker molecules. In certain
embodiments of the invention, the linker can be a series of 3 to 10 PEG
molecules
that are attached covalently to the 5' end of a DNA probe. Details relating to
the use
of PEG as a linker molecule are described below in Example 9.
With reference now to Fig. 55, there is shown a bar graph demonstrating
determination of percent covalent probe density on 3~,m Spherotech beads.
These
graphs represent signals generated from an enzyme assay using biotinylated
probes
and streptavidin-linked alkaline phosphatase enzyme reactions. As discussed
with
72
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
reference to Fig. 50, the covalent conjugation efficiency can be calculated by
determining the total amount of probes bound to non heat-treated beads. A
separate
aliquot of the beads is then heated to remove the non-covalently bound probes
and
the amount of covalent probes is then determined using the enzyme assay as
described in Example 7 below. With these data, the percentage of covalent
probe
binding can then be determined using the following formula: H/T*100 where H
represents signal from heat treated beads and T is the total signal from the
non-heat
treated beads.
Fig. 56 is a bar graph presentation demonstrating the pretreatment of the
beads
with various blocking agents including detergents. Decreasing non-specific
bead
binding is critical in the dual bead assay since the assay sensitivity is
inversely related
to the baseline signal which is the non-specific binding of the reporter beads
to the
capture beads. Thus the lower the baseline, the more sensitive the assay
becomes.
As illustrated, the use of salmon sperm DNA worked best in reducing the
nonspecific
binding relative to the other blocking agents tested in this experiment.
Salmon sperm
' DNA blocking reduced non-specific binding by approximately 10 fold. Salmon
sperm
DNA is, therefore, a preferred method for blocking non-specific bead binding
in one '
aspect of the present invention. Other blocking agents may also be used
including
BSA, Denhardt's solution, and sucrose. Preferably, beads should be blocked by
an
appropriate blocking agent after conjugation and heat treatment as shown in
Fig. 50 or
prior to Step I in Figs. 11 A, 11 B, 12A, and 12B above to increase the dual
bead assay
sensitivity.
Methods for Decreasing Non-Specific Bead Binding
As discussed above, Fig. 56 is a bar graph presentation demonstrating the
pretreatment of the beads with various blocking agents including detergents to
decrease non-specific binding of the beads. Decreasing non-specific bead
binding is
critical in the dual bead assay since the assay sensitivity is inversely
related to the
baseline signal which is the non-specific binding of the reporter beads to the
capture
beads. Thus the lower the baseline the more sensitive the assay becomes. As
illustrated, the use of salmon sperm DNA worked best in reducing the
nonspecific
binding relative to the other blocking agents tested in this experiment.
Salmon sperm
DNA blocking reduced non-specific binding by approximately 10 fold. Salmon
sperm
73
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
DNA is, therefore, a preferred method for blocking non-specific bead binding
in one
aspect of the present invention. Other blocking agents may also be used
including
BSA, Denhardt's solution, and sucrose. Preferably, beads should be blocked by
an
appropriate blocking agent prior to Step I in Figs. 11 A, 11 B, 12A, and 12B,
above, to
increase the dual bead assay sensitivity.
Fig. 57 is a bar graph of data generated using a fluorimeter showing a
concentration dependent target detection using fluorescent reporter beads in a
dual
bead assay. This graph shows the picomolar concentration of target DNA versus
number of beads bound in a dual bead complex. The dynamic range of target
detection shown in this graph is 0.25 pM to 2500 pM (picomoles/liter). While
this
particular graph was generated using data from a fluorimeter, the results may
also be
generated .using a fluorescent type optical disc drive. Experimental details
from an
experiment related to detection of a range of target concentrations is
discussed in
detail in Example 10.
Referring now to Figs. 58, 59, 60, 61 A, and 61 B, here are shown data from
experiments performed to determine the optimal concentration or ionic strength
of
various salts in the hybridization buffer or assay buffer. The salt
concentration in the
assay buffer. needs to be optimized in order to increase hybridization
efficiency or
binding efficiency and decrease non-specific bead binding between capture and
reporter beads resulting in lower signal to noise ratio which increases the
sensitivity of
the assay. In general, the data presented in these figures show that 40mM
EDTA,
300mM NaCI, 30mM MgCl2 are the optimal salt concentrations for use in one
embodiment of the dual bead assay.
With specific reference to Fig. 58, there is a bar graph presentation showing
data collected from an experiment using various concentrations of NaCI in the
bead
buffer and the related non-specific binding as a result of changes in the
ionic strength
of the buffer. Based on the results presented in Fig. 58, the optimal bead
buffer
concentration of sodium chloride for use in the dual bead assay is 0.2M since
non
specific bead binding is minimal at this NaCI concentration.
Now referring to Fig. 59, there is shown a bar graph illustrating the effect
of
increasing EDTA concentration on the dual bead assay sensitivity using
different
target concentrations. Fig. 59 also shows the related non-specific binding as
affected
by the concentration of EDTA in the assay buffer. The optimal EDTA buffer
74
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
concentration, based on the data presented, for use in the hybridization
buffer is
40mM since signal generated from the dual bead assay was highest at this
concentration.
Similarly Fig. 60 presents a bar graph presentation showing the effect of
increasing NaCI concentration on the dual bead assay sensitivity using
different target
concentrations. The non-specific bead binding data related to optimization of
the
buffer concentration of NaCI is represented in Fig. 58.. As shown in Fig. 60,
the
optimal NaCI concentration for hybridization as implemented in a dual bead
assay is
0.3M NaCI. A detailed description of the experimental procedure used to
generate this
data is discussed below in Example 11.
Turning now to Figs. 61 A and 61 B, here are bar graph presentations showing
the effect of increasing the concentration of MgCl2 in the assay buffer on the
dual bead
assay sensitivity and an enzyme assay sensitivity, respectively. Data from
these
figures indicate that a concentration of 30mM MgCl2 in the hybridization
buffer is
~ optimal for increasing the signal generated and the assay sensitivity.
According to the
data shown in Figs. 61 A and 61 B, the enzyme assay appears to be more
sensitive
than the dual bead assay in the 30mM MgCl2 treatment. This conclusion is based
on
the difference in signal within the treatment group from the various target
concentrations. Thus as illustrated, the slope of the concentration curve in
the 30mM
MgCl2 group of the enzyme assay of Fig. 61 B is steeper than the corresponding
curve
in Fig. 61A. Example 12 describes in detail the procedure for carrying out an
experiment relating to Fig. 61 A.
Referring next to Fig. 62, there is shown a pictorial representation of the
use of
probe blocking agents to increase the sensitivity of the bead assay. The probe
blocking agent used in this particular example is a biotinylated DNA that is
complimentary to the probe on the bead. The amount of probe blocking agent
used to
block excess probes on the bead is such that a pre-determined fraction of
probes are
blocked by the blocking agent. The use of the probe blocking agent in dual
bead
assay increases the sensitivity of the assay in that it enhances the
probability of target
binding to a single capture and reporter bead in a dual bead assay. This may
increase the sensitivity of the dual bead assay up to one target per dual bead
complex. The use of the biotinylated probe blocking agent allows for the
quantitation
of the blocking efficiency of the probe blocking agent for optimization of the
assay.
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
The amount of biotinylated probes bound to the beads may be quantitated by an
enzyme assay using streptavidinated or neutravidinated enzymes including
streptavidin-alkaline phosphatase (S-AP) and their appropriate substrates. The
choice
of enzyme and substrate for use in this test is dictated by the type of
detection
desired. In general, a colorimetric test is performed wherein the enzyme-
substrate
reaction produces color that is quantified by a spectrophotometer.
Alternatively,
streptavidinated or neutravidinated fluorescent tags may also be used which
may be
quantified using a fluorimeter or a Fluorimager. Both the colorimetric and
fluorescent
quantitation may also be carried out using the appropriate optical disc reader
as
shown in Figs. 1 and 2.
Fig. 63 shows a bar graph presentation of data illustrating the effect
incubation
time on the signal generated and the assay sensitivity using different target
concentrations during a hybridization reaction in a dual bead assay. The data
shows
that 2 hours is the minimum incubation time required to generate the maximum
signal
and sensitivity for the dual bead assay and that a 4 hour or overnight
hybridization is
not necessary.
Similarly, Fig. 64 shows a bar graph of data collected illustrating the effect
of
incubation time and mixing on the hybridization. efficiency and the assay
sensitivity
using different target concentrations in a hybridization reaction as
implemented on a
dual bead assay. As in Fig. 63, Fig. 64 also shows that 2 hours is an optimal
time for
hybridization and extending the hybridization time does not increase the
signal
generated. In addition, mixing significantly increased the hybridization
efficiency after
2 hours of hybridization relative to control. Example 14, presented below,
explains the
details regarding the experiment performed to generate the data shown in Figs.
63
and 64.
Dual Bead Preparation Methods Including
Removal of Non-Specifically Bound Complexes
The following preparation methods are more particular alternative embodiments
of the corresponding methods described above in connection with Figs. 11 A, 11
B,
12A, and 12B.
With reference now to Figs. 65A and 65B, there is illustrated a method of
preparing a molecular assay using a "single-step hybridization" technique to
create
dual bead complex structures in a solution according to one aspect of the
present
76
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
invention. This method is similar to the method discussed above in connection
with
Fig. 11A. The present method includes eight principal steps identified
consecutively
as Steps I, II, III, IV, V, VI, VII, and VIII.
In Step I of this method, a number of capture beads 190 coated with
oligonucleotide transport probes 198 are deposited into a test tube 212
containing a
buffer solution 210. The number of capture beads 190 used in this method may
be,
for example, on the order of 10E+07 and each on the order of 1 um or greater
in
diameter. Capture beads 190 are suspended in hybridization solution and are
loaded
into the test tube 212 by injection with pipette 214. The preferred
hybridization
solution is composed of 0.2M NaCI, lOmM MgCl2, 1 mM EDTA, 50mM Tris-HCI, pH
7.5, and 5X Denhart's mix. A desirable hybridization temperature is 37 degrees
Celsius. In a preliminary step in this embodiment, transport probes 198 are
conjugated to 3 um magnetic capture beads 190 by EDC conjugation. Further
details
regarding conjugation methods are disclosed in commonly assigned U.S.
Provisional
Application Serial No. 60/271,922 entitled, "Methods for Attaching Capture DNA
and
Reporter DNA to Solid Phase Including Selection of Bead Types as Solid Phase"
filed
February 27, 2001; and U.S. Provisional Application Serial No. 60/277,854
entitled
"Methods of Conjugation for Attaching Capture DNA and Reporter DNA to Solid
Phase" filed March 22, 2001, both of which are herein incorporated by
reference in
their entirety.
As shown in Step II, target DNA or RNA 202 is added to the solution.
Oligonucleotide transport probes 198 are complementary to the,DNA or RNA
target
agent 202. The target DNA or RNA 202 thus binds to the complementary sequences
of transport probe 198 attached to the capture bead 190 as shown in Fig. 8A.
With reference now to Step III, there is added to the solution 210 reporter
beads 192 coated with oligonucleotide signal probes 206. As also shown in
Figs. 9A
and 10A, signal probes 206 are complementary to the target DNA or RNA 202. In
one
embodiment, signal probes 206, which are complementary to a portion of the
target
DNA or RNA 202, are conjugated to 2.1 um fluorescent reporter beads 192.
Signal
probes 206 and transport probes 198 each have sequences that are complementary
to the target DNA 202, but not complementary to each other. After adding
reporter
beads 192, the dual bead complex 194 is formed such that the target DNA 202
links
capture bead 190 and reporter beads 192. With specific and thorough washing,
there
77
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
should be minimal non-specific binding between reporter bead 192 and capture
bead
190. The target agent 202 and signal probe 206 are preferably allowed to
hybridize
for three to four hours at 37 degrees Celsius.
In this embodiment and others, it was found that intermittent mixing (i.e.,
periodically mixing and then stopping) produced greater yield of dual bead
complex
than continuous mixing during hybridization. Thus when this step is performed
on-
disc, the disc drive motor 140 and controller 142, Fig. 2, may be
advantageously
employed to periodically rotate the disc to achieve the desired intermittent
mixing.
This may be implemented in mixing protocols encoded on the disc that rotate
the disc
in one direction, then stop the disc, and thereafter rotate the disc again in
the same
direction in a prescribed manner with a preferred duty cycle of rotation and
stop
sessions. Alternatively, the encoded mixing protocol may rotate the disc in a
first
direction, then stop the disc, and thereafter rotate the disc again in the
opposite
direction with a preferred duty cycle of rotation, stop, and reverse rotation
sessions.
These features of the present invention are discussed in further detail in
connection
with Figs. 33A and 35.
As next shown in Step IV of Fig. 65A, after hybridization, the dual bead
complex 194 is separated from unbound reporter beads in the solution. The
solution
can be exposed to a magnetic field to capture the dual bead complex structures
194
using the magnetic properties of capture bead 190. The magnetic field can be
encapsulated in a magnetic test tube rack 216 with a built-in magnet 218,
which can
be permanent or electromagnetic to draw out the magnetic beads and remove any
unbound reporter beads in the suspension. Note that capture beads not bound to
reporter beads will also be isolated. Alternatively, this magnetic removal
step may be
performed on-disc as shown in Figs. 33A, 35, and 36A-36C.
The purification process illustrated in Step IV includes the removal of
supernatant containing free-floating particles. Wash buffer is added into the
test tube
and the bead solution is mixed well. The preferred wash buffer for the one
step assay
consists of 145mM NaCI, 50mM Tris, pH 7.5, 0.1 % SDS, 0.05% Tween, 0.25%
NFDM, and lOmM EDTA. Most of the unbound reporter beads 182, free-floating
DNA, and non-specifically bound particles are agitated and removed from the
supernatant. The dual bead complex can form a matrix of capture beads, target
sequences, and reporter beads, wherein the wash process can further assist in
the
78
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
extraction of free floating particles trapped in the lattice structure of
overlapping dual
bead particles. Further details relating to other aspects associated with
methods of
decreasing non-specific binding of reporter beads to capture beads are
disclosed in,
for example, commonly assigned U.S. Provisional Application Serial No.
60/272,134
entitled "Reduction of Non-Specific Binding in Dual Bead Assays by Selection
of Bead
Type and Bead Treatment" filed February 28, 2001; and U.S. Provisional
Application
Serial No. 60/275,006 entitled "Reduction of Non-Specific Binding in Dual Bead
Assays by Selection of Buffer Conditions and Wash Conditions" filed March 12,
2001.
Both of these applications are herein incorporated by reference in their
entirety.
The next step in Fig. 65A is Step V. In this step, once the dual bead complex
has been washed approximately 3-5 times with wash buffer solution, restriction
enzymes, urea, acids (preferably strong acids), or bases (preferably strong
bases)
may be added to the dual bead solution, as illustrated. The dual bead
complexes are
thus dissociated by the actions of these dissociation agents thereby releasing
the
reporter beads 192 from the capture beads 190 as shown in Fig. 65B, Step VI.
After the dissociation of the dual bead structure, the capture beads 190 are
separated from the, now unbound, reporter beads 192 in the solution, as shown
in
Step VII. The solution can be exposed to a magnetic field to capture the
magnetic
capture beads 190. The magnetic field can be encapsulated in a magnetic test
tube
rack 216 with a built-in magnet 218, which can be permanent or electromagnetic
to
draw out the magnetic beads and remove any unbound reporter beads in the
suspension. Note that non-dissociated dual bead complexes not separated during
Step VI will also be removed from the solution. During step VII, the
supernatant
containing the released reporter beads are collected using a pipette 214. The
assay
mixture may then be loaded into the disc 144 or 180 and analyzed using an
optical
bio-disc or medical CD reader, as illustrated in Step VIII. Either a
transmissive bio-
disc 180 or a reflective bio-disc 144 may be used to analyze the reporter
beads.
Details relating to the reflective and transmissive optical bio-discs are
discussed
above in conjunction with Figs. 3A-3C and 4A-4C, respectively. The optical bio-
disc
reader and other alternative disc readers that may be used to analyze optical
bio-discs
are described in detail above in connection with Figs. 1 and 2. If the
reporter beads
are fluorescent, the reporter beads isolated in Step VII may also be
quantified using a
79
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
fluorimeter or any similar fluorescent type analyzer including fluorescent
optical disc
readers.
Figs. 66A and 66B taken together illustrate an immunoassay using a "single-
step antigen binding" method, similar to that in Figs. 65A and 65B, to create
dual bead
complex structures in a solution. This method similarly includes eight
principal steps
and is related to the method discussed above in connection with Fig. 11 B. The
eight
steps of,this method are respectively identified as Steps I, II, III, IV, V,
VI, VII, and VIII
in Figs. 66A and 66B.
As shown in Step I, Fig. 66A, capture beads 190, e.g., on the order of 1 OE+07
in number and each on the order of 1 um or above iri diameter, which are
coated with
antibody transport probes 196 are added to a buffer solution 210. This
solution may
be the same as that employed in the method shown in Figs. 65A and 65B or
alternatively may be specifically prepared for use with immunochemical assays.
The
antibody transport probes 196 have a specific affinity for the target antigen
204. The
transport probes 196 bind specifically to epitopes within the target antigen
204 as also
shown in Fig. 8B. In one embodiment, antibody transport probes 196 which have
an
affinity for a portion of the target antigen may be conjugated to 3 um
magnetic capture
beads 190 via EDC conjugation. Alternatively, conjugation of the transport
probes 196
to the capture bead 190 may be achieved by passive adsorption.
With reference now to Step II shown in Fig. 66A, the target antigen 204 is
added to the solution. The target antigen 204 binds to the antibody transport
probe
196 attached to the capture bead 190 as also shown in Fig. 8B.
As illustrated in Step III, reporter beads 192 coated with antibody signal
probes
208 are added to the solution. Antibody signal probes 208 specifically binds
to the
epitopes on target antigen 204 as also represented in Figs. 9B and 10B. In one
embodiment, signal probes 208 are conjugated to 2.1 um fluorescent reporter
beads
192. Signal probes 208 and transport probes 196 each bind to specific epitopes
on
the target antigen, but not to each other. After adding reporter beads 192,
the dual
bead complex 194 is formed such that the target antigen 204 links capture bead
190
and reporter bead 192. With specific and thorough washing, there should be
minimal
non-specific binding between reporter bead 192 and capture bead 190.
In Step IV, after the binding in Step III, the dual bead complex 194 is
separated
from unbound reporter beads in the solution. The solution can be exposed to a
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
magnetic field to capture the dual bead complex structures 194 using the
magnetic
properties of capture bead 190. The magnetic field can be encapsulated in a
magnetic test tube rack 216 with a built-in magnet 218, which can be permanent
or
electromagnetic to draw out the magnetic beads and remove any unbound reporter
beads in the suspension. Note that capture beads not bound to reporter beads
will
also be isolated. Alternatively, as indicated above, this magnetic removal
step may
also be performed on-disc as shown in Figs. 33A, 35, and 36A-36C.
The purification process of Step IV includes the removal of supernatant
containing free-floating particles. Wash buffer is added into the test tube
and the bead
solution is mixed well. Most of the unbound reporter beads 182, free-floating
protein
samples, and non-specifically bound particles are agitated and removed from
the
supernatant. The dual bead complex can form a matrix of capture beads, target
antigen,. and reporter beads, wherein the wash process can further assist in
the
extraction of free floating particles trapped in the lattice structure of
overlapping dual
bead particles.
The next step in Fig. 66A is Step V. In this step, once the dual bead complex
has been washed approximately 3-5 times with wash buffer solution. Urea,
acids, or
bases may be added to the dual bead solution as dissociation agents, using a
pipette
214 as illustrated. The acids or bases employed herein are preferably strong
acids or
bases, respectively. The dual bead complexes are thus dissociated by the
actions of
these dissociation agents thereby releasing the reporter beads 192 from the
capture
beads 190 as shown in Fig. 66B, Step VI.
After the dissociation of the dual bead structure, the capture beads 190 are
separated from the, now unbound, reporter beads 192 in the solution, as shown
in
Step VII. The solution can be exposed to a magnetic field, either on-disc or
off-disc, to
capture the magnetic capture beads 190. In the preparatory off-disc method
shown
here, the magnetic field can be encapsulated in a magnetic test tube rack 216
with a
built-in magnet 218, which can be permanent or electromagnetic to draw out the
magnetic beads and remove any unbound reporter beads in the suspension. Note
that non-dissociated dual bead complexes not separated during Step VI will
also be
removed from the solution. In Step VII, the supernatant containing the
released
reporter beads are collected using a pipette 214. The assay mixture may then
be
loaded directly into the disc and analyzed using an optical bio-disc reader,
as
81
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
illustrated in Step VIII. Either a transmissive bio-disc 180 or a reflective
bio-disc 144
may be used to analyze the reporter beads. Details relating to the reflective
and
transmissive optical bio-discs are discussed in detail in connection with
Figs. 3A-3C
and 4A-4C, respectively. The optical bio-disc reader and other alternative
disc
readers that may be used to analyze optical bio-discs are described in detail
above
with reference to Figs. 1 and 2. If the reporter beads are fluorescent, the
reporter
beads isolated in Step VII may also be quantified using a fluorimeter or any
similar
fluorescent type analyzer including fluorescent optical disc readers.
Figs. 67A and 67B taken together show an alternative genetic assay method
referred to here as a "two-step hybridization". This method is a modified
embodiment
of the method discussed above in connection with Fig. 12A. The present method
has
nine principal steps directed to creating the dual bead complex. Generally,
capture
beads are coated with oligonucleotide transport probes 198 complementary to
DNA or
RNA target agent and placed into a buffer solution. In this embodiment,
transport
probes which are complementary to a portion of target agent are conjugated to
3 um
magnetic capture beads via EDC conjugation. Other type of conjugation of the
oligonucleotide transport probes to a solid phase may be utilized. These
include, for
example, passive adsorption or use of streptavidin-biotin interactions. The
nine main
steps according to this method of the present invention are consecutively
identified as
Steps I, II, III, IV, V, and VI in Fig. 67A, and Steps VII, VIII, and IX in
Fig. 67B.
More specifically now with reference to Step I shown in Fig. 67A, capture
beads
190, suspended in hybridization solution, are loaded from the pipette 214 into
the test
tube 212. The preferred hybridization solution is composed of 0.2M NaCI, lOmM
MgCl2, 1 mM EDTA, 50mM Tris-HCI, pH 7.5, and 5X Denhart's mix. A desirable
hybridization temperature is 37 degrees Celsius.
In Step II, target DNA or RNA 202 is added to the solution and binds to the
complementary sequences of transport probe 198 attached to capture bead 190.
In
one specific embodiment of this method, target agent 202 and the transport
probe 198
are allowed to hybridize for 2 to 3 hours at 37 degrees Celsius. Sufficient
hybridization, however, may be achieved within 30 minutes at room temperature.
At
higher temperatures, hybridization may be achieved substantially
instantaneously.
As next shown in Step III, target agents 202 bound to the capture beads are
separated from unbound species in solution by exposing the solution to a
magnetic
82
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
field to isolate bound target sequences by using the magnetic properties of
the capture
bead 190. The magnetic field can be enclosed in a magnetic test tube rack 216
with a
built-in magnet permanent 218 or electromagnet to draw out the magnetic beads
and
remove any unbound target DNA 202 free-floating in the suspension via pipette
extraction of the solution. As with the above methods, in the on-disc
counterpart
hereto, this magnetic removal step may be performed as shown in Figs. 33A, 35,
and
36A-36C. A wash buffer is added and the separation process can be repeated.
The
preferred wash buffer after the transport probes 198 and target DNA 202
hybridize,
consists of 145mM NaCI, 50mM Tris, pH 7.5, and 0.05% Tween. Hybridization
methods and techniques for decreasing non-specific binding of target agents to
beads
are further disclosed in commonly assigned and co-pending U.S. Provisional
Application Serial No. 60/278,691 entitled "Reduction of Non-Specific Binding
of Dual
Bead Assays by Use of Blocking Agents" filed March 26, 2001. This application
is
herein incorporated by reference in its entirety.
Referring now to Step IV illustrated in Fig. 67A, reporter beads 192 are added
to the solution as discussed in conjunction with the method shown in Fig. 65A.
Reporter beads 192 are coated with signal probes 206 that are complementary to
target agent 202. In one particular embodiment of this method, signal probes
206,
which are complementary to a portion of target agent 202, are conjugated to
2.1 um
fluorescent reporter beads 192. Signal probes 206 and transport probes 198
each
have sequences that are complementary to target agent 202, but not
complementary
to each other. After the addition of reporter beads 192, the dual bead complex
structures 190 are formed. As would be readily apparent to one of skill in the
art, the
dual bead complex structures are formed only if the target agent of interest
is present.
In this formation, target agent 202 links magnetic capture bead 190 and
reporter bead
192. Using the preferred buffer solution, with specific and thorough washing,
there is
minimal non-specific binding between the reporter beads and the capture beads.
Target agent 202 and signal probe 206 are preferably allowed to hybridize for
2-3
hours at 37 degrees Celsius. As with Step II discussed above, sufficient
hybridization
may be achieved within 30 minutes at room temperature. At higher temperatures,
the
hybridization taking place in this step may also be achieved substantially
instantaneously.
83
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
With reference now to Step V shown in Fig. 67A, after the hybridization in
Step
IV, the dual bead complex 194 is separated from unbound species in solution.
The
solution is again exposed to a magnetic field to isolate the dual bead complex
194
using the magnetic properties of the capture bead 190. Note again that the
isolate will
include capture beads not bound to reporter beads. As with Step III above in
the on-
disc counterpart hereto, this magnetic separation step may be performed as
shown in
Figs. 33A, 35, and 36A-36C.
A purification process to remove supernatant containing free-floating
particles
includes adding wash buffer into the test tube and mixing the bead solution
well. The
preferred wash buffer for the two-step assay consists of 145mM NaCI, 50mM
Tris, pH
7.5, 0.1 % SDS, 0.05% Tween, 0.25% NFDM, and 1 OmM EDTA. Most unbound
reporter beads, free-floating DNA, and non-specifically bound particles are
agitated
and removed from the supernatant. The dual bead complex can form a matrix of
capture beads, target agents, and reporter beads, wherein the wash process can
further assist in the extraction of free floating particles trapped in the
lattice structure of
overlapping dual bead particles. Other related aspects directed to reduction
of non-
specific binding between reporter bead, target agent, and capture bead are
disclosed
in, for example, commonly assigned U.S. Provisional Application Serial No:
601272,243 entitled "Mixing Methods to Reduce Non-Specific Binding in Dual
Bead
Assays" filed February 28, 2001; and U.S. Provisional Application Serial No.
60/272,485 entitled "Dual Bead Assays Including Linkers to Reduce Non-Specific
Binding" filed March 1, 2001, which are incorporated herein in their entirety.
The next step shown in Fig. 67A is Step VI. In this step, once the dual bead
complex 194 has been washed approximately 3-5 times with wash buffer solution,
the
assay mixture is loaded into the disc and analyzed. Alternatively, during this
step, the
oligonucleotide signal and transport probes may be ligated to prevent
breakdown of
the dual bead complex during the disc analysis and signal detection processes.
Further details regarding probe ligation methods are disclosed in commonly
assigned
and co-pending U.S. Provisional Application Serial No. 601278,694 entitled
"Improved
Dual Bead Assays Using Ligation" filed March 26, 2001, which is herein
incorporated
in its entirety by reference. In yet another embodiment of the present
invention,
restriction enzymes, urea, acids, or bases may be added using a pipette 214 to
the
dual bead solution, as illustrated in Step VI of Fig. 67A. The dual bead
complexes are
84
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
dissociated by the actions of these dissociation agents thus releasing the
reporter
beads 192 from the capture beads 190 as shown in Fig. 67B, Step VII. The acids
or
bases utilized herein are preferably strong acids or bases, respectively.
After the dissociation of the dual bead structure, the capture beads 190 are
separated from the, now unbound, reporter beads 192 in the solution, as shown
in
Step VIII. The solution can be exposed to a magnetic field to capture the
magnetic
capture beads 190. The magnetic field can be encapsulated in a magnetic test
tube
rack 216 with a built-in magnet 218, which can be permanent or electromagnetic
to
draw out the magnetic beads and remove any unbound reporter beads in the
suspension. Note that non-dissociated dual bead complexes not separated during
Step VIII will also be removed from the solution. During Step VIII, the
supernatant
containing the released reporter beads are collected using a pipette 214. The
assay
mixture may then be loaded into the disc and analyzed using an optical bio-
disc
reader, as illustrated in Step IX. Either a transmissive bio-disc 180 or a
reflective bio-
disc 144 may be used to analyze the reporter beads. Details relating to the
reflective
and transmissive optical bio-discs are discussed in detail in conjunction with
Figs. 3A-
3C and 4A-4C, respectively. The optical bio-disc reader and other alternative
disc
readers that may be used to analyze optical bio-discs are described in detail
above
with reference to Figs. 1 and 2. If the reporter beads are fluorescent, the
reporter
. beads isolated in Step VIII may also be quantified using a fluorimeter or
any similar
fluorescent type analyzer including fluorescent optical disc readers.
Experiments
performed using dissociation agents to release the reporter beads from the
dual bead
complex are described in detail below in Examples 15 and 16.
In accordance with another aspect of this invention, Figs. 68A and 68B taken
together show an immunoassay method, similar to those discussed in connection
with
Figs. 66A and 66B, and following the techniques of the genetic assay in Figs.
67A and
67B. This method is also referred to here as a "two-step binding" to create
the dual
bead complex in an immunochemical assay. This method is a related and more
specific embodiment of the method illustrated above in Fig. 12B. As with the
method
shown in Figs. 67A and 67B, this method includes nine main steps. In general,
capture beads coated with antibody transport probes which specifically binds
to
epitopes on target antigen are placed into a buffer solution. In one specific
embodiment, antibody transport probes are conjugated to 3 um magnetic capture
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
beads. Different sized magnetic capture beads may be employed depending on the
type of disc drive and disc assembly utilized to perform the assay. The nine
main
steps according to this alternative method of the invention are respectively
identified
as Steps I, II, III, IV, V and VI in Fig. 68A and Steps VII, VIII, and IX in
Fig. 68B.
With specific reference now to Step I shown in Fig. 68A, capture beads 190,
suspended in buffer 210 solution, are loaded into a test tube 212 via
injection from
pipette 214.
In Step II, target antigen 204 is added to the solution and binds to the
antibody
transport probe 196 attached to capture bead 190. Target antigen 204 and the
transport probe 196 are preferably allowed to bind for 2 to 3 hours at 37
degrees
Celsius. Shorter binding times are also possible.
As shown in Step III, target antigen 204 bound to the capture beads 190 are
separated from unbound species in solution by exposing the solution to a
magnetic
field to isolate bound target proteins or glycoproteins by using the magnetic
properties
of the capture bead 190. The magnetic field can be enclosed in a magnetic test
tube
rack 216 with a built-in magnet permanent 218 or electromagnet to draw out the
magnetic beads and remove any unbound target antigen 204 free-floating in the
suspension via pipette extraction of the solution. A wash buffer is added and
the
separation process can be repeated.
As next illustrated in Step IV, reporter beads 192 are added to the solution
as
discussed in conjunction with the method shown in Fig. 66A. Reporter beads 192
are
coated with signal probes 208 that have an affinity for the target antigen
204. In one
particular embodiment of this two-step immunochemical assay, signal probes
208,
which bind specifically to a portion of target agent 204, are conjugated to
2.1 um
fluorescent reporter beads 192. Signal probes 208 and transport probes 196
each
bind to specific epitopes on the target agent 204, but do not bind to each
other. After
the addition of reporter beads 192, the dual bead complex structures 190 are
formed.
As would be readily apparent to those skilled in the art, these dual bead
complex
structures are formed only if the target antigen of interest is present. In
this formation,
target antigen 204 links magnetic capture bead 190 and reporter bead 192.
Using the
preferred buffer solution, with specific and thorough washing, there is
minimal non-
specific binding between the reporter beads and the capture beads. Target
antigen
204 and signal probe 208 are allowed to hybridize for 2-3 hours at 37 degrees
Celsius.
86
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
As with Step II discussed above, sufficient binding may be achieved within 30
minutes
at room temperature. In the case of immunoassays temperatures higher than 37
degrees Celsius are not preferred because the proteins will denature.
Turning next to Step V as illustrated in Fig. 68A, after the binding shown in
Step
IV, the dual bead complex 194 is separated from unbound species in solution.
This is
achieved by exposing the solution to a magnetic field to isolate the dual bead
complex
194 using the magnetic properties of the capture bead 190 as shown. Note again
that
the isolate will include capture beads not bound to reporter beads.
A purification process to remove supernatant containing free-floating
particles
includes adding wash buffer into the test tube and mixing the bead solution
well. Most
unbound reporter beads, free-floating proteins, and non-specifically bound
particles
are agitated and removed from the supernatant. The dual bead complex can form
a
matrix of capture beads, target agents, and reporter beads, wherein the wash
process
can further assist in the extraction of free floating particles trapped in the
lattice
structure of overlapping dual bead particles.
The last step shown in Fig. 68A is Step VI. In this step, once the dual bead
complex 194 has been washed approximately 3-5 times with wash buffer solution,
urea, acids (preferably strong acids), or bases (preferably strong bases) may
be
added to the dual bead solution using a pipette 214, as illustrated in Step VI
of Fig.
68A. The dual bead complexes are dissociated by the actions of these
dissociation
agents thus releasing the reporter beads 192 from the capture beads 190 as
shown
next in Step VII of Fig. 68B.
After the dissociation of the dual bead structure, the capture beads 190 are
separated from the, now unbound, reporter beads 192 in the solution, as shown
in
Step VIII of Fig. 68B. The solution can be exposed to a magnetic field to
capture the
magnetic capture beads 190. The magnetic field can be encapsulated in a
magnetic
test tube rack 216 with a built-in magnet 218, which can be permanent or
electromagnetic to draw out the magnetic beads and remove any unbound reporter
beads in the suspension. Note that non-dissociated dual bead complexes not
separated during Step VII will also be removed from the solution. During Step
VIII in
this method, the supernatant containing the released reporter beads 192 are
collected
using a pipette 214. The assay mixture may then be loaded into the disc and
analyzed using an optical bio-disc reader, as illustrated in Step IX. Either a
87
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
transmissive bio-disc 180 or a reflective bio-disc 144 may be used to analyze
the
reporter beads. Details relating to the reflective and transmissive optical
bio-discs are
discussed in detail with reference to Figs. 3A-3C and 4A-4C, respectively. The
optical
bio-disc reader and other alternative disc readers that may be used to analyze
optical
bio-discs or medical CDs are described in detail above in conjunction with
Figs. 1 and
2. The reporter beads isolated in Step VIII may also be quantified using a
fluorimeter
or any similar fluorescent type analyzer including fluorescent optical disc
readers.
As with any of the other methods discussed above, the magnetic removal or
separation steps in the method shown in Figs. 68A and 68B may be alternatively
performed on-disc using the disc, fluidic circuits, and apparatus illustrated
in Figs.
33A-33D, 34A-34C, 35, 36A-36C, and 37.
Use of Dissociation Agents to Increase Assay
Sensitivity and Decrease Non-specific Bead Binding
Moving along now to Fig. 69, there is shown a bar graph presentation
demonstrating the DNAseI digestion efficiency in the absence of reporter
beads. In
this experiment, biotinylated target DNA 202 was hybridized to transport
probes 198
on the magnetic capture bead 190 as illustrated above in Fig. 8A. After
hybridization,
streptavidinated alkaline phosphatase (S-AP) was added to the assay mix and
allowed
to bind with the biotin on the target DNA. Following a series of wash steps,
an S-AP
chromagen substrate was added to an aliquot of the assay solution and the
amount of
bound target was quantified colorimetrically using a spectrophotometer. At the
same
time, an aliquot of equal volume to that taken above was incubated in buffer .
containing DNAseI. After incubation, the assay mix was washed and S-AP was
added
to the solution and allowed to bind to residual targets that were not digested
by
DNAseI. The observations showed a high DNAse digestion activity as manifested
in
the difference in signal between the control and the DNAseI digestion
treatments.
Details regarding an experiment similar to the one discussed here is described
in
detail below in Example 15.
Referring now to Fig. 70, there is shown a bar graph illustration of data
collected from an experiment similar to that discussed in Fig. 69. In this
experiment,
dual bead complexes were used instead of the magnetic capture bead alone as
described in Fig. 69. In this case, the target 202 is situated between the
capture bead
190 and reporter bead 192 as shown above in Fig. 10A. After formation of the
dual
88
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
bead complex, as described in Fig. 65A, Steps I-V, the amount of reporter
beads in
the supernatant collected in Step IV (Fig. 65A) and the reporter beads bound
to the
capture beads in the assay mix shown in Step V (Fig. 65A) were quantitated
using a
fluorimeter. DNAseI was then added to the assay mix and allowed to cleave the
target bound to the probes in the dual bead complex, as represented in Step VI
in Fig.
65B, thereby releasing the reporter bead from the magnetic capture beads. The
next
step involved the isolation of the capture beads and collection of the
supernatant
containing newly released fluorescent reporter beads as shown in Step VII in
Fig. 65B.
The signal from the reporter beads in the supernatant was quantified using a
fluorimeter. The undissociated reporter beads were also quantified by a
fluorimeter.
Data collected from this experiment shows the DNAseI enzyme digestion was not
as
efficient in a dual bead complex relative to that in a single bead set-up as
described in
Fig. 69. The decrease in DNAse digestion activity may be due to steric
hindrance
from the beads in the dual bead complex blocking DNAse access to the target.
Experimental details regarding the use of restriction enzyme digestion as
implemented
in a dual bead assay is discussed in further detail below in Example 15. The
fluorescent reporter beads collected in this assay may also be quantified
using an
optical bio-disc or medical CD with a fluorescent type optical disc reader or
any similar
device as discussed above.
The sensitivity of any assay depends on the ratio of signal over noise. The
sensitivity of the dual bead assay relies on the minimization of non-specific
binding
between capture beads and reporter beads. The non-specific interactions
between
the dual beads in the absence of targets are so stable that stringent washing
cannot
eliminate them. The contribution of the non-specific dual beads, however, can
be
negated by the exclusive detection and quantification of target-mediated dual
beads.
As shown in Fig. 71, the dual bead complexes can be separated by enzyme
digestion
(DNAse, restriction enzymes) or by chemical and physical treatments (heat,
urea,
base, or acid treatment). Furthermore, the quantification of reporter beads in
the
presence of a large excess of unbound capture beads may reduce the sensitivity
of
the detection assay. Therefore, the separation of reporter beads from capture
beads
will facilitate the quantification 'of reporter beads by the bio-disc reader
and thus
increase the sensitivity of the assay.
89
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
With more particular reference now to Fig. 71, there is a schematic
representation of separation of reporter beads from capture beads in a dual
bead
complex by enzyme digestion and physical or chemical treatments. Fig. 71 shows
a
summary of the dual bead formation and dissociation using various dissociation
agents as discussed in conjunction with Figs. 65A, 65B, 67A, and 67B. After
the
released reporter beads are collected, the beads are quantified by any one of
several
methods including a fluorimeter, a fluorimager, a fluorescent type optical
disc reader
system, a CD-R type optical disc system or any device capable of detecting
micro-
spheres or fluorescence. The current preferred method of detection is the use
of an
optical bio-disc or medical CD system as discussed in detail in connection
with Figs. .1,
2, 3A, 3B, 3C, 4A, 4B, and 4C.
With reference next to Fig. 72, there is a bar graph presentation showing
separation of reporter beads from capture beads using high pH washes at
various
target concentrations similar to that described in Fig. 70. The dual bead
complex is
formed as described in Steps I-VI in Fig. 67A. Once the dual beads are formed
and
isolated, a strong base is added to the dual bead solution as shown in Step VI
(Fig.
67A). After a brief incubation, the dual beads are dissociated by the actions
of the
base that disrupts the hydrogen bonding between the target and the probes
thereby
releasing the fluorescent reporter beads and the magnetic capture beads from
the
dual bead complex as shown in Step VII (Fig. 67B). The next step is to isolate
the
capture beads as described in Step VIII of Fig. 67B. The isolated capture
beads will
also contain non-dissociated dual bead complexes. This isolate was quantified
by a
fluorimeter in this particular experiment. The data shown in Fig. 72
illustrate 100%
dissociation of dual bead complexes at target concentrations from 1 x 1 O-'6 M
to 1 x
10-'4 M. Details of an experiment performed using base as a dissociation agent
is
described in detail below in Example 16. The reporter beads may also be
quantified
by any one of several methods including a fluorimager, a fluorescent type
optical disc
reader system, a CD-R type optical disc system or any device capable ~ of
detecting
microspheres or fluorescence. The preferred method of detection is the use of
an
optical bio-disc or medical CD system as discussed in detail in conjunction
with Figs.
1, 2, 3A, 3B, 3C, 4A, 4B, and 4C.
Referring now to Fig. 73A, there is a bar graph illustration of data collected
from
an experiment using urea as a denaturing or dissociation agent. Details of
this
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
experiment are similar to those discussed in conjunction with Fig. 69. As in
Fig. 69,
biotinylated target DNA 202 was hybridized to transport probes 198 on the
magnetic
capture bead 190 as illustrated above in Fig. 8A. After hybridization,
streptavidinated
alkaline phosphatase (S-AP) was added to the assay mix and allowed to bind
with the
biotin on the target DNA 202. Following a series of wash steps, an S-AP
chromagen
substrate was added to an aliquot of the assay solution and the amount of
bound
target was quantified colorimetrically using a spectrophotometer. At the same
time, an
aliquot of equal volume to that taken above was incubated in buffer containing
7M
urea. After incubation, the assay mix was washed and S-AP was added to the
solution and allowed to bind to residual targets, still bound to transport
probes on the
capture bead, that were not denatured by the 7M urea. The results thereof
showed a
relatively efficient denaturation activity as revealed by the difference in
signal between
the control and the 7M urea treatments. Details regarding an experiment
similar to the
one discussed here is described in detail below in Example 16.
Referring next to Fig. 73B, there is a bar graph illustrating data collected
from
an experiment using urea as a dissociation agent in a dual bead assay. Details
of this
experiment are similar to that discussed in conjunction with Fig. 70. As in
Fig. 70, dual
bead complexes were used instead of the magnetic capture bead alone as
described
in Fig. 73A. In this case, the target is situated between the capture and
reporter
beads as shown above in Fig. 10A. After formation of the dual bead complex, as
described in Fig. 67A, Steps I-VI, the amount of reporter beads 192 in the
supernatant
collected in Step V (Fig. 67A) and the reporter beads 192 bound to the capture
beads
190 in the assay mix shown in Step VI (Fig. 67A) were quantitated using a
fluorimeter.
A predetermined amount of urea was then added to make the final urea
concentration
in the assay solution 7M. This resulted in denaturation of the target bound to
the
probes in the dual bead complex, as represented in Step VII in Fig. 67B, thus
releasing the reporter bead from the magnetic capture beads. The next step
involved
the isolation of the capture beads 190 and collection of the supernatant
containing
newly released fluorescent reporter beads 192 as shown in Step VIII in Fig.
67B. The
signal from the reporter beads in the supernatant was quantified using a
fluorimeter.
The undissociated reporter beads were also quantified by a fluorimeter. Data
collected from this experiment shows the 7M urea denaturation was more
efficient in a
dual bead complex assay relative to using DNAseI as a dissociation agent as
91
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
described in Fig. 70. The increase in dissocaition may be due to the lack of
steric
hindrance from the beads in the dual bead complex since urea is a
significantly
smaller molecule than DNAse. Experimental details regarding the use of 7M urea
denaturation as implemented in a dual bead assay is discussed in further
detail in
Example 16. The fluorescent reporter beads collected in this assay may also be
quantified using an optical bio-disc or medical CD with a fluorescent type
optical disc
reader or any similar device as discussed above.
Use of DNA Denaturing Agents to Improve DNA Target Detection
It is a principal aspect of the invention to further modify the dual bead
assays to
detect medical targets. In real samples, the DNA targets are double-stranded
and very
long. The ability of the dual bead assay, as well as for any other DNA
diagnostic
assays, to detect sequences of clinical interest within the whole genome
relies first on
the specificity of the probes for the sequence of interest and second on the
use of very
strong detergent to keep the DNA target in the denatured, single-stranded,
form for
captu re.
The success of the dual bead assays in detecting sequences of clinical
interest
relies primarily on the design of the probes. Given the complexity and
degeneracy of
the human genome, the probes designed to detect sequences of clinical interest
have
to be unique to the diagnostic sequence and yet common enough to recognize
mutants of the sequences. The design of the probes using computer software
allows
comparison of sequences to existing sequences in the data bank such as Blast
search. Once probes specific for the sequence of interest have been designed,
the
major modification introduced to the dual bead assays includes the use of a
denaturing agent in the hybridization buffer to prevent re-annealing of
complementary
sequences of the target DNA. This allows hybridization between the target and
probes.
The present invention is also addressed at implementing the methods recited
above on to an analysis disc, modified optical disc, medical CD, or a bio-
disc. A bio
disc or medical CD drive assembly, such as those discussed above with
reference to
Figs, 1 and 2, may be employed to rotate the disc, read and process any
encoded
information stored on the disc, and analyze the DNA samples in the flow
channel of
the bio-disc or medical CD. The bio-disc drive is thus provided with a motor
for
92
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
rotating the bio-disc, a controller for controlling the rate of rotation of
the disc, a
processor for processing return signals form the disc, and an analyzer for
analyzing
the processed signals. The rotation rate of the motor is controlled to achieve
the
desired rotation of the disc. The bio-disc drive assembly may also be utilized
to write
information to the bio-disc either before, during, or after the medical test
material in the
flow channel and target zones is interrogated by the read beam of the drive
and
medically diagnosed by the analyzer. In this embodiment of the present
invention, the
analyzer may advantageously include specialized diagnostic software to thereby
provide a medical expert system. The bio-disc may similarly include
corresponding
medical expert system software and encoded information for controlling the
rotation
rate of the disc, providing processing information specific to the type of DNA
test to be
conducted, and for displaying the results on a monitor associated with the bio-
drive.
In a preferred embodiment of this invention, guanidine isothiocyanate is used
as a typical denaturing agent. Data collected from an experiment using 1.5M ,
guanidine isothiocyanate as denaturing agent is illustrated in Fig. 74. The
experimental procedure followed for this experiment is described in detail
below in
Example 17. Fig. 74 further illustrates that the assay sensitivity is
significantly
increased if an appropriate amount of denaturing agent is used in a
hybridization
experiment. In this particular assay, biotinylated target DNA 202 was
hybridized to
transport probes 198, in the presence of 1.5M guanidine isothiocyanate, on the
magrietic capture bead 190 as illustrated above in Fig. 8A. After
hybridization,
streptavidinated alkaline phosphatase (S-AP) was added to the assay mix and
allowed
to bind with the biotin on the target DNA 202. Following a series of wash
steps, an S-
AP chromagen substrate was added to the assay solution, ample time was
allotted for
color development, and the amount of bound target was quantified
colorimetrically
using a spectrophotometer.
An appropriate amount of guanidine isothiocyanate is necessary to prevent re-
annealing of the complementary sequences of the target DNA while allowing
hybridization between the target and the probes. At high concentrations,
however,
guanidine isothiocyanate prevents any hybridization. To determine the
appropriate
buffer concentration of guanidine isothiocyanate for use in a dual bead assay,
a
titration of guanidine isothiocyanate was performed. The data from this
titration
experiment is shown in Fig. 75. As illustrated in Fig. 75, the optimal
hybridization
93
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
buffer concentration of guanidine isothiocyanate is 1.5M since the addition of
1.5M
guanidine isothiocyanate showed the highest difference in signal between the
O.OM
and 1.0x10-1°M target concentration.
MO Bio-Magnetic Assays (MOBMA) for Selectings Detecting, and
Manipulating Specific Cell Populations using the MO Bio-Disc System (MOBDS)
A further aspect of the present invention relates to a magnetic method for
detection of specific target cells in cell populations and solutions of cell
populations,
using magnetic particles or beads as described above and referred to as the
MOBMA.
In one embodiment of the MOBMA aspect of the current invention, the magnetic
beads are coated with one or more binding agents or capture probes to thereby
form
bio-magnetic particles or beads. Further details related to attaching binding
agents
onto a solid support including magnetic particles is disclosed in commonly
assigned
and co-pending U.S. Patent Application Serial No. 101194,396 entitled "Multi-
Purpose
Optical Analysis Disc For Conducting Assays and Various Reporting Agents for
use
Therewith" filed July 12, 2002, which is incorporated by reference in its
entirety herein.
The binding agents may include, for example, antibodies recognizing Fc
portions of
target cell associating antibodies directed to specific antigen determinants
on cell
membranes. Capture, detection, manipulation, and quantitation of target cells
are
carried out using the MOBDS of the present invention. The captured cells may
also
be magnetically manipulated or moved from one analysis, separation, or testing
chamber to another, on the MO bio-disc, to facilitate cellular testing. For
example,
captured target cancer cells may ebe magnetically aliquoted into equal cell
numbers to
different analysis chambers containing cancer therapeutic agents to determine
the
effect of these agents on a cellular level. Specific examples of applications
of this
method are described below.
IncubAation of the cell suspension with a mild detergent andlor second set of
antibodies or antibody fragments, pre-labeled or not with fluorescent agents,
metallocolloids, bio-magnetic particles, radioisotopes, biotin complexes or
certain
enzymes allowing visualization, may dramatically increase the specificity and
sensitivity of said method. The method can further be used to dynamically
isolate,
purify, manipulate, and quantify target cells magnetically captured on
magnetic
capture zones on the MO bio-disc. Further details relating to a method for
detection of
94
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
specific target cells in specialized or mixed cell population and solutions
containing
mixed cell populations is disclosed in, for example, U.S. Patent Serial No.
6,184,043
to Fodstad and Kvalheim, which is herein incorporated by reference in its
entirety.
The binding agents may bind one or more chemical/biochemical entities
together by affinity. In affinity binding, a pair of binding partners, which
for example
are attached to the substances to be linked, bind to each other_ when brought
in
contact. A molecule on a cell surface may represent one of the binding
partners, in
such a linkage. Several such binding partner systems are known, such as, for
example, antigen-antibody, enzyme-receptor, ligand-receptor interactions on
cells,
biotin-avidin binding, hapten/anti-hapten binding pairs, and oligonucleotide
complementary sequence binding, of which antigen-antibody binding is most
frequently used.
Methods are known in which one of the binding partners is attached to an
insoluble support, such as magnetic particles or beads, and by which isolation
of
target cells in a mixed cell population is performed as negative isolation or
positive
isolation. In a negative isolation procedure the unwanted cells can be removed
from
the cell preparation by incubating the cells with antibody-coated magnetic
beads (bio-
magnetic beads or particles) specific for the unwanted cells. Following the
incubation
the resulting cell-bead complex can be removed or isolated magnetically,
leaving the
wanted target cells behind. In a positive isolation procedure, on the other
hand, the
wanted target cells are removed from the mixed cell population using magnetic
beads
coated with antibodies (bio-magnetic beads) directed to the cells of interest.
An object of the MOBMA aspect of the present invention is to detect and study
specific target cells for diagnostic purposes. The test samples may include
blood,
bone marrow, ascites fluid, and cells from various tissues or tumors. The
present
invention represents a sensitive detection system and method for detecting a
variety
of cell types, such that a high number of cells can be readily screened using
the
MOBDS and the procedure is rapid and simple. Furthermore, the present MOBDS
may be used for isolation of cells for biochemical, biological and
immunological
examination, and for studying of specific genes at the nucleotide or protein
level. In
addition, the isolated or captured cell-bead complexes may be released by
disrupting
or removing the magnetic field within the magnetic capture zone, on the MO bio-
disc,
where cells of interest are located, using the associated MO drive and the
cells
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
cultured or used for in vitro cytotoxicity studies without the need for
cleaving the cell-
bead complex.
Another embodiment of the MOBMA aspect of the present invention includes
immunomagnetic positive isolation of target cells, normal or pathogenic, in a
mixed
cell population or physiological solution. In this embodiment of the present
invention,
a linkage is created between a specific target cell and an insoluble support
such as a
magnetic bead or particle. The particle is either coated with an anti-cell
antibody
directed to specific antigen determinants on the membranes of the target
cells, or the
particles are coated with a polyclonal anti-mouse or anti-human antibody
capable of
binding to the Fc-portions of a specific anti-cell antibody (primary antibody)
directed to
the antigen determinants on the membrane of the target cell. Instead of using
the
polyclonal anti-mouse/anti-human antibody for coating the particles, a
monoclonal rat
anti-mouse/anti-human antibody may be used. The use of a monoclonal antibody
may reduce the risk for possible cross-reactions with non-target cells in
solutions.
Furthermore, incubation of the cell suspension with a mild detergent and/or
second set
of antibodies or antibody fragments that bind to antigenic determinants on one
or more
cell types among the captured cells, in a mixed cell capture, pre-labeled with
fluorescent agents, magnetic particles, metallocolloids, bio-magnetic
particles,
radioisotopes, biotin-complexes or certain enzymes allowing visualization,
will
dramatically increase the specificity of the method of this embodiment.
A detailed application of the immunomagnetic positive isolation embodiment
described above is presented below using cancer cells as the target cells for
detection
and isolation. This embodiment is, however, not limited to cancer cells and
the
disclosure shall not liri~it the method to this particular field of use since
the method is
suitable for use with a range of cytological applications including assays
directed to,
for example, cells of the immune system such as natural killer cells,
monocytes, T-
cells, B-cells, and subtypes thereof including CD4+ and CD8+ cells; bacterial
cells
(Chandler, et al., Int. J. of Food Micr., 70: 143-154, 2001, and Yu, J. of
Immuno.
Mthds., 218: 1-8, 1998); and peroxisomes (Luers, et al., Electrophoresis, 19:
1205-
1210, 1998).
In the management of cancer patients, the staging of the disease with regards
to whether it is localized or if metastatic spread has occurred to other
tissues is of
utmost importance for the choice of therapeutic regimens for the individual
patient.
96
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
Malignant cells spread by direct invasion into the surrounding tissue, through
the
lymphatics, or by the distribution of tumor cells in the blood to distant
organs, including
the bone marrow the central nervous system, and the cerebrospinal fluid.
Detection of
metastatic tumor cells has, until recently, relied on morphological methods
using light
and electron microscopy on biopsied tumor specimens, on smears of bone marrow
and peripheral blood, and on slides prepared after cytocentrifugation of
various body
fluids. Since the advent of monoclonal antibodies recognizing antigens
predominantly
expressed on the surface of different types of malignant cells, the
identification of
metastatic cells has, to an increasing extent, also involved
immunocytochemistry and
immunofluorescence. Thus, slides prepared from biopsied tumors or
cytocentrifugates are treated with monoclonal antibodies, and the binding of
these to
the tumor cells is visualized colorimetrically or by fluorescence. The latter
method
requires the use of a fluorescence microscope, alternatively preparing a cell
suspension and use of a flow cytometer.
The previous methods suffer from limited sensitivity andlor specificity, and
is
usually laborious and time consuming, also requiring a high degree of
expertise.
Flowcytometric examinations also involve expensive equipment.
The morphological methods for the detectiori of tumor cells in blood and bone
marrow are much less sensitive than methods involving immunocytochemistry and
immunofluorescence (Beiske, et al., Am. J. Pathology 141 (3), September 1992).
Also
the latter methods are, however, inadequate in cases where the tumor cells
represent
less than 1 % of the total number of cells in a test sample. Flow cytometry
may
provide better sensitivity than the methods involving the use of a microscope,
but
requires the availability of a high number of cells, and also involves several
technical
difficulties. Furthermore, aggregation of cells may cause problems with flow
cytometry, and this method does not provide possibilities to distinguish
between
labeled tumor cells and non-specifically fluorescing normal cells.
The present invention allows for a very sensitive detection of, for example,
metastatic tumor cells, since a high number of cells can readily be screened
and the
cell-bead complexes are easily detected and quantitated by the MOBDS. The
monoclonal antibodies used for the detection of target cells in the present
invention
are selected such that these antibodies bind with sufficient specificity to,
for example,
tumor cells and not to non-target cells present in mixed cell suspensions or
test
97
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
sample. The test sample may include, for example blood, bone marrow, and other
solutions containing tumor manifestations, such that all cells with attached
beads
represent the target cells. The target cells are then captured on specific
magnetized
areas or magnetic domains in a magneto-optical (MO) bio-disc and quantitated
using
the associated magneto-optical drive and software. In addition, the procedure
is rapid
and simple, and can be performed by any investigator without the need for
expensive
and sophisticated equipment such as a flow cytometer. Further details relating
to the
MOBDS are disclosed in, for example, commonly assigned co-pending U.S. Patent
Application Serial No. 10/099,256 entitled "Dual Bead Assays Using Cleavable
Spacers and/or Ligation to Improve Specificity and Sensitivity Including
Related
Methods and Apparatus" filed March 14, 2002; and the above referenced U.S.
Patent
Application Serial No. 10/099,266 entitled "Use ,of Restriction Enzymes and
Other
Chemical Methods to Decrease Non-Specific Binding in Dual - Bead Assays and
Related Bio-Discs, Methods, and System Apparatus for Detecting Medical
Targets"
filed March 14, 2002. Details relating to magneto-optical recording, precise
creation of
magnetic regions on a magneto-optical disc, and magneto-optical detection
methods
are described above in conjunction with Fig. 37 and in, for example,
Tsunashima,.
Magneto Optical Recording, J. Phys. D: Appl. Phys. 34, R87-8102, 2001, and in
Coombs, Differential Phase Contrast and Magneto-optic Edge Detection, Applied
Optics, 34(29), 6723-6729, 1995, which are both incorporated by reference in
their
entireties as if fully repeated herein.
As mentioned above, the immunomagnetic embodiment of the MOBMA aspect
of the present invention involves the binding of capture antibodies including
monoclonal antibodies, e.g. of murine or human origin, that specifically
recognize
antigens present on tumor cells, but not antigens on normal cells, or for
other
purposes to specified sub-populations of normal cells, to magnetic particles
or beads
to thereby form bio-magnetic particles. The capture antibodies may bind
directly to
antigens on the target cell or the Fc-portion of primary antibodies or cell
binding
antibodies that bind to the tumor cells. The cell binding antibodies may be of
the IgG
or IgM type or a fragment of IgG or IgM antibodies. The capture or primary
antibodies
may be antibodies directed against groups of antigen determinants including,
for
example, tumor associated glycoprotein 72 (TAG 72) antigen (CC49 antibody)
(Barjer
et al., Gynec. Onco. 82: 57-63, 2001 ), CD56/NCAM antigen (MOC-1
antibody)(Speirs
98
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
et al., J. Histochem. Cytochem., 41 (9):1303-10, 1993), epithelial cell
surface antigen
(BER-EP4 antibody) (Borgen et al., J. Hematother., 6(2):103-114, 1997, and
Ziggeuner et al. J. Urol., 164: 1834-1837, 2000), Cluster 2 epithelial antigen
(MOC-31
antibody) (Rye et al., Am J. Patho., 150(1 ): 99-106, 1997, and Ree et al.,
Int. J.
Cancer, 97: 28-33, 2002), Cluster 2 (MW 40 kD) antigen (NrLulO antibody)
(Myklebust
et al., Br. J. Cancer Suppl. 14: 49-53, 1991), HMW-melanoma-associated antigen
(225.28S antibody) (Dell'Erba et al., Anticancer Res., 21 (2A):925-930, 2001
), 80 kD,
Sarcoma-associated antigen (TP-1 & TP-3 antibodies) (Bruland, et al., Cancer
Res.,
48: 5302-5309, 1988), cytokeratin antigens (pan-anti-CK antibody) (Bilkenroth
et al.,
Int. J. Cancer, 92: 577-582, 2001 ), mucin antigens TAG 12 (2E11 ) (Diel et
al., J. Natl.
Cancer Inst., 88(22):1652-8, 1996), and EGF-receptor antigen (425.3 antibody)
(Merck). The 425.3 antibody is directed towards antigens in both normal and
malignant cells. The capture antibodies can furthermore be directed against
growth
factor receptors, for example EGF-receptor, PDGF (A and B) receptor, insuline
receptor, insuline-like receptor, transferrin receptor, NGF and FGF receptors,
group of
integrins, other adhesion membrane molecules and MDR proteins in both normal
cells
and abnormal cells, and antigens present on subpopulations of normal cells, in
addition to oncogenic products, expressed on the membranes of normal and
malignant cells and on malignant cells alone, for example Neu/erb B2/HER2. As
for
the malignant cells, these may be breast, ovarian and lung carcinoma cells,
melanoma, sarcoma, glioblastoma, cancer cells of the gastrointestinal tract
and the
reticuloendothelial system, or the target cells may be associated with non-
neoplastic
diseases, such as cardiovascular, neurological, pulmonary, autoimmune,
gastrointestial, genitourinary, reticuloendothelial and other disorders.
Furthermore,
the malignant cell population may be located in bone marrow, peripheral blood,
come
. from pleural and peritoneal effusions and other body fluid compartments,
such as
urine, cerebrospinal fluid, semen, lymph or from solid tumors in normal
tissues and
organs, for example liver, lymph nodes, spleen, lung, pancreas, bone tissue,
the
central nervous system, prostatic gland, skin and mucous membranes. A partial
list of
the antigen determinants and the corresponding antibodies or antibody
fragments that
may be used in conjunction with the present improved invention is presented
below in
Table 1.
99
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
In cases where the density of the target cells is low, for example, malignant
cells or the target cells representing a very low fraction of the total number
of cells
(__<1 %), the target cells can be positively separated from non-target cells
and pre-
concentrated using a magnet prior to analysis in the MOBDS. The isolated
target
cells, can then be enumerated using the MOBDS and the fraction of target cells
relative to the total number of cells in the initial cell suspension can be
calculated.
Drug Sensitivity Assays on the MOBDS
As mentioned above, the MOBDS may be used to isolate, manipulate, and
study target cells in vitro. One assay that can be performed on target cells
is a
cytotoxicity, chemosensitivity, drug sensitivity, or drug resistance assay to
measure
drug-induced cell death following drug exposure. In this method, the target
cells may
be, for example, collected from a malignant tumor biopsy, bacteria, plant,
virus, and
unicellular organisms. The target cells are immunomagnetically isolated in one
or
more chambers in the MO bio-disc, as described above, to thereby constitute an
isolate. Once the target cells are isolated, a pre-determined number of cells,
from the
isolate, are magnetically moved or manipulated to various chambers within the
MO
bio-disc including test chambers. These chambers may contain drugs including,
but
not limited to, chemotherapeutic agents, antibiotics, or antiviral,
individually or in
combination, at various pre-determined dosages. The chemotherapeutic agents
may
include, for example, breast cancer, lung cancer, brain cancer, liver cancer,
and
ovarian cancer drugs such as Cisplatin, Topotecan, Taxol, Gemcitabine (1),
Mitomycin-C, Navelbine, Nitrogen Mustard, 5-Fluorouracil, Doxorubicin,
Etoposide,
Trimetrexate, and various combinations thereof including, but not limited to,,
Cisplatin
and Topotecan, Cisplatin and Taxol, Cisplatin and Gemcitabine (1 ), Cisplatin
and
Nitrogen Mustard, and Cisplatin and 5-Fluorouracil. The isolated cells are
then
incubated at pre-determined conditions in the various concentrations and
combinations of drugs. After incubation, the number of live cells are counted,
using
the MOBDS, and the IC50 (drug concentration in which 50% of the cells die as a
result
~30 of exposure a drug or a combination of drugs) from all the drugs are
calculated to
determine the best therapy regimen for the patient. This test is very
important since
drugs found active in vitro are approximately 10 times more likely to be
clinically
effective relative to drugs found inactive in vitro. In vivo drug sensitivity
assays are
100
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
well know in the art (Bosanquet et al., Leukemia 16(6):1035-44, 2002;
Nagourney et
al., J. Clin. Onco. 18:11, 2245-49, 2000; Bosanquet et al., Br. J. Haem.,
106:71-77,
1999; and Cortazar et al., J. Clin. Onco., 17:5, 1625-31, 1999). The assays of
the
prior art are mostly performed using a microtiter plate and the cell counting
and
analysis done using a microscope or colorimetrically using a microtiter plate
reader by
preferential staining of the live or dead cells, to determine the number of
live cells.
The present invention automates the process in that the cell selection,
manipulation,
incubation, analysis, and IC50 determination is performed using the MOBDS
without
the need for a separate analysis device such as a microscope or a microtiter
plate
reader. The automated process increases the efficiency and the accuracy of
these
drug sensitivity assays.
Sample Preparation
Steps for performing the above described immunomagnetic embodiment of the
present invention may differ depending on type of tissues to be examined
including,
for example:
a) cells from solid tissue or needle tumor biopsies are separated mechanically
or with mild enzymatic treatment to generate a suspension of single cells, to
which the primary, specific antibodies or antibody fragments are added
directly or after washing the cell suspension in phosphate buffered saline or
culture medium with or without serum, such as fetal calf serum, bovine,
horse, pig, goat, or human serum;
b) cells in pleural or ascitic effusion, cerebrospinal fluid, urine, lymph or
body
fluids such as effusions in the joints of patients with various forms of
arthritis, the specific capture antibodies or antibody fragments are either
added to the samples directly, or after centrifugation with or without
washings before or after the cells in the samples are spun down and
brought back into suspension; and
c) mononuclear cells from blood or bone marrow aspirate are isolated by
gradient centrifugation and the capture antibodies are added to the isolated
mononuclear cells on or before washing and resuspension.
101
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
TABLE 1
LIST OF RELEVANT ANTIGENS AND
EXAMPLES OF ASSOCIATED ANTIGEN-BINDING ANTIBODIES
ANTIGENS MONOCLONAL ANTIBODIES
Adhesion molecules
Fibronectin receptor (a5~1 integrin)Pierce 36114, BTC 21/22
Calbiochem 341649
Integrin a3~ 1 M-Kiol 2
Vitronectin receptor (ay(33 integrin)TP36.1, BTC 41/42
Integrin a2 Calbiochem 407277
Integrin a3 Calbiochem 407278
Integrin a4 Calbiochem 407279
Integrin a5 Calbiochem 407280
Integrin aV Calbiochem 407281
Integrin ~2 Calbiochem 407283
Integrin (34 Calbiochem 407284
Gpll(3111a 8221
ICAM-I (CD54) C57-60, CL203.4, RR 1/1
VCAM-1 Genzyme 2137-01
ELAM-1 Genzyme 2138-01
E-selectin BBA 8
P-selectin/GMP-140 BTC 71/72
LFA-3 (CD58) TS 2/9
CD44 BM 1441 272, 25.32
CD44-variants 11.24, 11.31, 11.10
N-CAM(CD56) MOC-1
H-CAM BCA9
L-CAM BM 1441 892
N-CAM TURA-27
MACAM-1 NRI-M9
E-cadherin BTC 111, HECD-1, 6F9
P-cadherin NCC-CAD-299
Tenascin BM 1452 193, Calbiochem 580664
Thrombospondin receptor (CD36) BM 1441 264
VLA-2 A1.43
102
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
TABLE 1-continued
LIST OF RELEVANT ANTIGENS AND
EXAMPLES OF ASSOCIATED ANTIGEN-BINDING ANTIBODIES
ANTIGENS MONOCLONAL ANTIBODIES
Laminin receptor
HNK-1 epitope HNK-1
Carbohydrate antigens
T-antigen HHB, HT-8
Tn-antigen TKH6, BaGs2
Sialyl Tn TKH-2
Gastrointestinal cancer associated antigenCA 19-9
(MW 200 kD)
Carcinoma associated antigen C-50
Ley MLuC1, BR96, BR64 ,
di-Lea, tri-Le' B3
Dimeric Le' epitope NCC-ST-421
H-type 2 g1
CA15-3 epitope CA15-3 =
CEA I-9, I-14, I-27, II-10, I-46,
Calbiochem 250729
Galb1-4GIcNac (nL4,6,8) 1 B2
H-II BE2
A type 3 HH8
Lacto-N-fucopentanose III (CD15) PM-81
Glycolipids
GD3 ME 36.1, R24
GD2 ME36.1, 3F8, 14.18
Gb3 38-13
GM3 M2590
GM2 MKI-8, MKI-16,
FucGM~ 1 D7, F12
Growth factor receptors
EGF receptor 425.3, 2.E9, 225
c-erbB-2 (HER2) BM 1378 988, 800 E6
PDGFa receptor Genzyme 1264-00
PDGF(i receptor Sigma P 7679
Transferrin receptor OKT 9, D65.30
103
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
TABLE 1-continued
LIST OF RELEVANT ANTIGENS AND
EXAMPLES OF ASSOCIATED ANTIGEN-BINDING ANTIBODIES
ANTIGENS MONOCLONAL ANTIBODIES
NGF receptor BM 1198 637
IL-2 receptor (CD25) BM 1295 802, BM 1361 937
c-kit BM 428 616, 14 A3, ID9.3D6
TNF-receptor GEnzyme 1995-01, PAL-M1
NGF receptor
Melanoma anti--pens
High molecular weight antigen (HMW 9.2.27, NrMLS, 225.28,
250.000)
763.74, TP41.2, IND1
MW105 melanoma-associated glycoproteinME20
100 kDa antigen (melanoma/carcinoma) 376.96
gp113 MUC18,
p95-100 PAL-M2
Sp75 15.75
gr 100-107 NKI-bereb
MAA K9.2
MW 125 kD (gp125) Mab 436
Sarcoma antigens
TP-1 and TP-3 epitope TP-1, TP-3
MW 200 kD 29-13, 29.2
MW 160 kD 35-16, 30-40
Carcinoma markers
MOC-31 epitope (cluster 2 epithelial MOC-31, NrLulO
antigen)
MUC-1 antigens (such as DF3-epitope MUC-1, DF3, BCP-7 to -10
(gp290 kD))
MUC-2 and MUC-3 PMH1
LUBCRU-G7 epitope (gp 230 kD) LUBCRU-G7
Prostate specific antigen BM 1276 972
Prostate cancer antigen E4-SF
Protate high molecular antigen MW > PD41
400 kD
Polymorphic epithelial mucins BM-2, BM-7, 12-H-12
Prostate specific membran antigen (Cyt-356)7E11-C5
Human milk fat globulin Immunotech HMFG-1, 27.1
42 kD breast carcinoma epitope B/9189
104
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
TABLE 1-continued
LIST OF RELEVANT ANTIGENS AND
EXAMPLES OF ASSOCIATED ANTIGEN-BINDING ANTIBODIES
ANTIGENS MONOCLONAL ANTIBODIES
MW > 106 mucin TAG-72, CC-49, CC-83
Ovarian carcinoma OC125 epitope (MW OC125
750 kD)
Pancreatic HMW glycoprotein DU-PAN-2
Colon antigen Co17-1 A (MW 37000) 17-1 A
G9-epitope (colon carcinoma) G9
Human colonic sulfomucin ~ 91.9H
MW 300 kD pancreas antigen MUSE11
GA 733.2 GA733, KS1.4
TAG 72 B72.3, CC49, CC83
Undefined Oatl, SM1
Pancreatic cancer-associated MUSE 11
Pancarcinoma CC49
Prostate adenocarcinoma-antigen PD 41
MW 150-130 kD adenocarcinoma of the AF-10
lung
gp160 lung cancer antigen (Cancer Res.anti gp160
48, 2768, 1988)
MW 92 kD bladder carcinoma antigen 3G2-C6
MW 600 kD bladder carcinoma antigen C3
Bladder carcinoma antigen (Cancer Res.AN43, BB369
49, 6720, 1989)
CAR-3 epitop MW > 400 kD AR-3
MAM-6 epitope (C15.3) 115D8
High molecular ovarian cancer antigen OVX1, OVX2
Mucin epitope la3 la3
Hepatocellular carcinoma antigen MW KM-2
900 kD
Hepernal epitope (gp43) HepatocellularHepema-1
carc. ag
O-linked mucin containing N-glycolylneuraminic3E1.2
acid
MW 48 kD colorectal carcinoma antigen D612
MW 71 kD breast carcinoma antigen BCA 227
16.88 epitope (colorectal carcinoma 16.88
antigen)
CAK1 (ovarian cancers) K1
Colon specific antigen p Mu-1, Mu-2
Lung carcinoma antigen MW 350-420 kD DF-L1, DF-L2
gp54 bladder carcinoma antigen T16
105
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
TABLE 1-continued
LIST OF RELEVANT ANTIGENS AND
EXAMPLES OF ASSOCIATED ANTIGEN-BINDING ANTIBODIES
ANTIGENS MONOCLONAL ANTIBODIES
gp85 bladder carcinoma antigen T43
gp25 bladder carcinoma antigen T138
Neuroblastoma antigens
Neuroblastoma-associated, such as UJ13AUJ13A
epitope
Glioma antigens
Mel-14 epitope Mel-14
Head and neck cancer antigens
MW 18-22 kD antigen E48
HLA-antigens
HLA Class 1 TP25.99
HLA-A VF19LL67
HLA-B H2-149.1
HLA-A2 KS1
H LA-ABC W 6.32
HLA-DR, D0, DP Q 5/13, B 8.11.2
a2 -microglobulin NAMB-1
Apoptosis receptor
Apo-1 epitope Apo 1
Various
125 Plasminogen activator antigens & receptorsRabbit polyclonal
p-glycoprotein C219, MRK16, JSB-1,
265/F4
cathepsin D CIS-Diagnostici, Italy
biliary epithelial antigen HEA 125
neuroglandular antigen (CD63) ME491, NKI-C3, LS62
CD9 TAPA-1, R2, SM23
pan-human cell antigen pan-H
106
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
In another embodiment of the MOBDS aspect of the present invention, target
cells may be separated by a two dimensional or dual parameter immunomagnetic
cell
separation method such as that described in, for example, Partington, et al.,
A Novel
Method of Cell Separation Based on Dual Parameter Immunomagnetic Cell
Selection,
J. of Immuno. Mthds., 223: 195-205, 1999, which is incorporated by reference
in its
entirety herein. In this embodiment, the tagging and separation of target
cells with bio
magnetic particles may be performed within the fluidic circuits in the MO bio-
disc. This
approach takes advantage of size differences between commercially available
immunomagnetic beads and/or particles and their differing properties in terms
of
attraction to magnetic fields of various strengths. For example, the first
step of
separation may be the positive selection of cells utilising 50nm beads. Cells
isolated
in the first step--still rosetted with the 50nm beads--can then be subjected
to further
positive or negative selection using larger beads, for example, M280 or M450
Dynabeads, without the need for prior bead removal. The strength of the
magnetic
field during the second separation step is modulated such that the magnetic
force
generated on the MO disc magnetic domain is sufficient to attract only the
larger
Dynabeads but not the 50nm beads. This dual parameter embodiment thus provides
a better method for isolating and purifying cells of interest.
In yet another embodiment of the MOBDS aspect of the present invention,
target cells may be labeled or tagged with magnetic particles by ballistic
transfer. The
ballistic transfer technology employs a cold gas shock wave to accelerate
microprojectiles that carry matter into cells by mechanical force. This
embodiment of
the present invention relates to a method that enables separation of target
cells
rendered magnetically susceptible and can thus be separated by retaining them
in a
strong magnetic field. The tagged cells may then be captured, analyzed,
manipulated,
and quantitated using the MOBDS. Further details relating to ballistic tagging
of cells
with magnetic particles is disclosed in, for example, U.S. Patent Serial No.
6,348,338
to Wittig, which is herein incorporated in its entirety by refererice.
A further embodiment of the current invention is using the MOBDS to perform
cellular analysis, manipulation, and quantitation by immunomagnetic cell
selection, as
described above, and aligning the target cells in various pre-determined
configurations
on the MO bio-disc such as alignment of cells on the tracks of the MO bio-
disc. In this '
embodiment, cells are magnetically sorted and aligned on the MO bio-disc, and
107
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
analyzed using the MO disc drive and associated software. Related methods and
systems for immunomagnetic cellular analysis are described in, for example,
Tibbe et
al., Cytometry, 43:31-37, 2001, and Tibbe et al., Cytometry, 47:163-172, 2002,
both of
which are herein incorporated by reference in their entireties. These methods
of the
prior art require specialized devices and systems including fluorescence
detection.
The current invention circumvents the need for these specialized devices in
that a
standard MO disc drive may be used to perform the immunomagnetic cell
selection,
manipulation, detection, quantitation, and analyses described above.
Molecular Applications of the MOBDS Biomagnetic Assay
The isolated target cells may be characterized for the presence of specific
biochemical and biological features. Of particular importance will be the use
of such
cells for studies in molecular biology. In contrast to the above-cited methods
of the
prior art, the present method allows studies and growth of the target cells
without
performing a cleavage of the magnetic particle-target cell linkage. For
several
purposes it may be of interest to examine specific genes in a pure population
of target
cells at the DNA, mRNA, and protein level, both in tumor biopsies as well as
in tumor
cells present in blood, bone marrow and other body fluids, for example urine,
cerebrospinal fluid, semen, lymph, or from otherwise normal tissues and
organs, for
example liver, lymph nodes, spleen, lung, pancreas, bone tissues, central
nervous
system, prostatic gland, skin and mucous membranes, and in other areas of
cytological assays.
With the methods of prior art, signals obtained on Southern, Northern and
Western blots represent the normal cells as well as the tumor cells in the
biopsy. If a
single cell suspension is first prepared from the tumor material, and the
tumor cells are
then positively immunomagnetically detected and separated using the MOBDS of
the
present invention, any gene studies performed on this material would represent
the
target cells only. This also relates to, for example, malignant cells present
in
mammalian tissues including bone marrow, peripheral blood, pleural and
peritoneal
effusions, and other body fluids, for example urine, cerebrospinal fluid,
semen and
lymph. Studies involving polymerase chain reaction (PCR) and microarray
methodologies will also gain increased specificity and reliability when
performed on
108
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
pure target cell populations obtained using the methods and apparatus of the
present
invention.
Neurobiological Assays on the MO Bio-Disc System (MOBDS)
In still another embodiment of the MOBDS aspect of the present invention,
neurons may be isolated and stimulated to grow and regenerate in biological
matrix or
solution in a MO bio-disc. The biological matrix may be formed from a gel or
any
biologically compatible material that provides a solid support for the cells
while
allowing passage of essential nutrients and gases to enable survival of the
cells. The
biological matrix also allows growth and movement of the cells and its
components
such as axon and dendrite movement within the matrix. The direction and rate
of
growth of the axons, dendrites, and cells may be controlled by a magnetic
field on the
MO bio-disc, generated by the MO drive. In this embodiment, neurites, for
example,
may be provided with magnetic nanoparticles or beads that are absorbed or
actively
incorporated into the neurons and their axons. Arravs of these neurons are
then
aligned in predetermined locations within the biological matrix on the MO bio-
disc. ,
The aligned neurons or neurites are then exposed to a magnetic field that is
displaced
to thereby physically move the magnetic particle-loaded neurons and their
axons
along a desired axis for bridging the gap or gaps between the neurons in the
array.
Since magnetic regions generated on the MO disc are relatively small and can
be
precisely controlled, use of the magnetically directed neurite growth
embodiment of
the present invention allows a more precise control of neurite growth relative
to
methods of the prior art. Magnetically directed neurite growth in vitro and
nerve
regeneration in vivo are well known in the art (Moorman et al., Brain Res.
Bulletin,
35(5-6):419-422, 1994, Dubey et al., Exp. Neuro:, 158:338-350, 1999, Macias et
al.,
Bioelectromagnetics, 21:272-286, 2000, Shah et al., Bioelectromagnetics,
22:267-271,
2001, and U.S. Patent No. 6,132,360 to Halpern, all of which are herein
incorporated
by reference in their entireties). This embodiment of the present invention is
directed
at using the MOBDS to study precise magnetic control of neurite growth in
vitro and at
creating neural networks in vitro.
Referring now to Fig. 76, there is illustrated a top plan view of a portion of
a
magneto-optical bio-disc having fluidic circuits including an inlet port 152,
a mixing
chamber 164, a separation or analysis chamber 300, and testing chambers 302.
The
109
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
magneto-optical bio-disc may include one or more components of the bio-discs
described above in conjunction with Figs. 3A to 3C, 4A to 4C, 33A to 33C, 35,
36A-
36C, and 37.
With reference next to Figs. 77A-77E, there are shown plan views illustrating
a
method of separation and testing of cells in the fluidic circuit shown in Fig.
76. In this
particular method of the present invention, test cells 306 are loaded into the
mixing
chamber 164 through the inlet port 152 using a pipette 214 (Fig. 77A). Bio-
magnetic
particles 308 are then loaded into the mixing chamber 164 using a pipette 214.
The
bio-magnetic particles are coated with binding agents specific for surface
markers on
the cells of interest in the sample. The binding agents may include, for
example,
antibodies outlined above in Table 1. The cells 306 and the bio-magnetic
particles are
then incubated for a sufficient time to allow binding of the bio-magnetic
particles 308 to
the cells of interest through binding of the binding agents and the cell
surface markers.
After incubation, cells of interest or target cells are thus labeled with bio-
magnetic
particles resulting in a labeled cell 310. This renders the labeled cells 310
susceptible
to magnetic manipulation. Magnetic domains or regions 246 may also be formed
in
the separation chamber 300 before of after the incubation using a magneto-
optical
drive. Details relating to the formation of magnetic domains 246 on magneto-
optical
bio-discs are discussed above in conjunction with Fig. 37. Once the incubation
is
complete, the disc is rotated at a pre-determined speed and duration, using
the
magneto-optical drive, to move the suspension containing unlabeled cells 306,
bio-
magnetic particles 308, and labeled cells 310 into the separation chamber 300
(Fig.
77B). As the suspension moves through the separation chamber, the labeled
cells
310 and free or unattached bio-magnetic particles 308 magnetically bind to the
magnetic domains 243 within separation chamber 300. The disc is then rotated
at
another pre-determined speed and time to move unbound and unlabeled cells 306
in
the suspension to the bottom of the separation chamber 300 (Fig. 77C). The
magnetic domains 246 are then sequentially erased and formed to selectively
release
the magnetically bound labeled cells 310 (Fig. 77D). The labeled cells 310 are
then
guided magnetically to one or more testing chambers 302 by sequentially
erasing and
forming the magnetic domains 246 (Fig. 77E). The testing chamber 302 may be
pre-
loaded with a test solution having a test agent including, but not limited to,
chemotherapeutic agents, antibiotics, and anti-viral medications. The labeled
cells
110
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
310 may then be incubated with the test agent. A beam of electromagnetic
radiation
may then be scanned through the testing chamber 302 to quantify the live and
apototic cells and thereby determine the sensitivity of the cells when exposed
to the
test agent. Apototic cells are characterized by membrane blebbing, cell
shrinkage,
protein fragmentation, chromatin condensation and DNA degradation, all of
which alter
the optical properties of the cell rendering it distinguishable from non-
apototic cells
using an optical disc reader.
Other Implementations of the Current invention
This invention or different aspects thereof may be readily implemented in or
adapted to many of the discs, assays, and systems disclosed in the following
commonly assigned and co-pending patent applications: U.S. Patent Application
Serial No. 09/378,878 entitled "Methods and Apparatus for Analyzing
Operational and
Non-operational Data Acquired from Optical Discs" filed August 23, 1999; U.S.
Provisional Patent Application Serial No. 60/150,288 entitled "Methods and
Apparatus
for Optical Disc Data Acquisition Using Physical Synchronization Markers"
filed August
23, 1999; U.S. Patent Application Serial No. 09/421,870 entitled "Trackable
Optical
Discs with Concurrently Readable Analyte Material" filed October 26, 1999;
U.S.
Patent Application Serial No. 09/643,106 entitled "Methods and Apparatus for
Optical
Disc Data Acquisition Using Physical Synchronization Markers" filed August 21,
2000;
U.S. Patent Application Serial No. 09/999,274 entitled "Optical Bio-discs with
Reflective Layers" filed on November 15, 2001; U.S. Patent Application Serial
No.
09/988,728 entitled "Methods And Apparatus For Detecting And Quantifying
Lymphocytes With Optical Biodiscs" filed on November 20, 2001; U.S. Patent
Application Serial No. 09/988,850 entitled "Methods and Apparatus for Blood
Typing
with Optical Bio-discs" filed on November 19, 2001; U.S. Patent Application
Serial No.
09/989,684 entitled "Apparatus and Methods for Separating Agglutinants and
Disperse Particles" filed November 20, 2001; U.S. Patent Application Serial
No.
09/997,741 entitled "Dual Bead Assays Including Optical Biodiscs and Methods
Relating Thereto" filed November 27, 2001; U.S. Patent Application Serial No.
09/997,895 entitled "Apparatus and Methods for Separating Components of
Particulate Suspension" filed November 30, 2001; U.S. Patent Application
Serial No.
10/005,313 entitled "Optical Discs for Measuring Analytes" filed December 7,
2001;
111
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
U.S. Patent Application Serial No. 10/006,371 entitled "Methods for Detecting
Analytes
Using Optical Discs and Optical Disc Readers" filed December 10, 2001; U.S.
Patent
Application Serial No. 10/006,620 entitled "Multiple Data Layer Optical Discs
for
Detecting Analytes" filed December 10, 2001; U.S. Patent Application Serial
No.
10/006,619 entitled "Optical Disc Assemblies for Performing Assays" filed
December
10, 2001; U.S. Patent Application Serial No. 10/020,140 entitled "Detection
System
For Disc-Based Laboratory And Improved Optical Bio-Disc Including Same" filed
December 14, 2001; U.S. Patent Application Serial No. 10/035,836 entitled
"Surface
Assembly For Immobilizing DNA Capture Probes And Bead-Based Assay Including
Optical Bio-Discs And Methods Relating Thereto" filed Dec. 21, 2001; U.S.
Patent
Application Serial No. 10/038,297 entitled "Dual Bead Assays Including
Covalent
Linkages For Improved Specificity And Related Optical Analysis Discs" filed
January
4, 2002; U.S. Patent Application Serial No. 10/043,688 entitled "Optical Disc
Analysis
System Including Related Methods For Biological and Medical Imaging" filed
January
10, 2002; U,.S. Provisional Application Serial No. 60/348,767 entitled
"Optical Disc
Analysis System Including Related Signal Processing Methods and Software"
filed
January 14, 2002; U.S. Patent Application Serial No. 10/086,941 entitled "
Methods
For DNA Conjugation Onto Solid Phase Including Related Optical Biodiscs and
Disc
Drive Systems" filed February 26, 2002; U.S. Provisional Application Serial
No.
60/363,949 entitled " Methods for Differential Cell Counts Including
Leukocytes and
Use of Optical Bio-Disc for Performing Same" filed March 12, 2002; U.S.
Provisional
Application Serial No. 60/382,944 entitled "Methods and Apparatus for Use in
Detection and Quantitation of Cell Populations and Use of Optical Bio-Disc for
Performing Same" filed May 24, 2002; and U.S. Provisional Application Serial
No.
60/384,205 entitled "Optical Disc Systems For Determining The Concentration Of
Cells Or Particles In A Sample And Methods Relating Thereto" filed May 30,
2002. All
of these applications are herein incorporated by reference in their
entireties. They
thus provide background~and related disclosure as support hereof as if fully
repeated
herein.
112
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
Experimental Details
While this invention has been described in detail with reference to the
drawing
figures, certain examples and further illustrations of the invention are
presented below.
EXAMPLE 1
The two-step hybridization method demonstrated in Fig. 12A was used in
performing the dual bead assay of this example.
A. Dual Bead Assay
In this example, the dual assay in carried out to detect the gene sequence DYS
that is present in male but not in female. The assay is comprised of 3p
magnetic and
capture beads coated with covalently attached capture probe; 2.1 p fluorescent
reporter beads coated with a covalently attached sequence specific for the DYS
gene,
and target DNA molecule containing DYS sequences. The target DNA is a
synthetic
80 oligonucleotide sequence. The capture probe and reporter probes are 40
nucleotides in length and are complementary to DYS sequence but not to each
other.
The specific methodology employed to prepare the assay involved treating
1x10' capture beads and 2x10'reporter beads in 100 microgram per milliliter
Salmon
Sperm DNA for 1 hr. at room temperature. This pretreatment will reduce non-
covalent
binding between the capture and reporter beads in the absence of target DNA as
shown in Fig. 38. The capture beads were concentrated magnetically with the
supernatant being removed. A 100 microliter volume of the hybridization buffer
(0.2 M
NaCI, 1 mM EDTA, 10 mM MgCl2, 50 mM Tris HCI, pH 7.5, and 5X Denhart's
mixture,
10 microgram per milliliter denatured salmon sperm DNA) were added to the
capture
beads and the beads were re-suspended. Various concentrations of target DNA
ranging from 1, 10, 100, 1000 femtomoles were added while mixing at
37°C for 2
hours. The beads were magnetically concentrated and the supernatant containing
target DNA was removed. A 100 microliter volume of wash buffer (145mM NaCI, 50
mM Tris pH 7.5, 0.1 % SDS, 0.05 % Tween, 0.25 % NFDM, 10 mM EDTA) was added
and the beads were re-suspended. The beads were magnetically concentrated and
the supernatant was again removed. The wash procedure was repeated twice.
113
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
A 2x10' amount of reporter beads in 100 microliter hybridization buffer ( 0.2
M
NaCI, 1 mM EDTA, 10 mM MgCl2, 50 mM Tris HCI, pH 7.5, and 5X Denhart's
mixture,
microgram per milliliter denatured salmon sperm DNA) were then added to washed
capture beads. The beads were re-suspended and incubated while mixing at
37°C for
5 an additional 2 hours. After incubation the capture beads were concentrated
magnetically, and the supernatant containing unbound reporter beads were
removed.
A 100 microliter volume of wash buffer (145mM NaCI, 50 mM Tris pH 7.5, 0.1 %
SDS,
0.05 % Tween, 0.25 % NFDM, 10 mM EDTA) was added and the beads were re-
suspended. The beads were magnetically concentrated and the supernatant was
10 again removed. The wash procedure was repeated twice.
After the final wash, the beads were re-suspended in 20 microliters of binding
buffer (50 mM Tris, 200mM NaCI, 10 mM MgCl2, 0.05% Tween 20, 1 % BSA). A 10
microliter volume was loaded on to the disc that was prepared as described
below in
Part B of this example.
B. Preparation of the Disc
A gold disc was coated with malefic anhydride polystyrene. An amine DNA
sequence complementary to the reporter probes (or capture agent) was
immobilized
on to the discrete reaction zones on the disc. Prior to sample injection, the
channels
were blocked with a blocking buffer (50mM Tris, 200mM NaCI, 10 mM MgCl2, 0.05%
Tween 20, 1 % BSA, 1 % sucrose) to prevent non-covalent binding of the dual
bead
complex to the disc surface. A perspective view of the disc assembly showing
capture
agents 220, the capture zones 170, and fluidic circuits as employed in the
present
invention is illustrated in detail in Figs. 25A-25D. Alternatively, if the
reporter beads
are coated with Streptavidin, a capture zone could be created with the capture
agent
such as BSA Biotin which could be immobilized on to the disc (pretreated with
Polystyrene) by passive absorption. A perspective view of the disc assembly
showing
the use of biotin capture agents is presented in Figs. 26A-26D. Various
methods for
use in this type of anchoring of beads onto the disc are also shown in Figs.
15A-15B,
17, 19A-19C, and 23A-23B.
114
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
C. Capture of Dual Bead Complex Structure on the Disc
A 10 microliter volume of the dual bead mixture prepared as described in Part
A
above was loaded in to the disc chamber and the injection ports were sealed.
To
facilitate hybridization between the reporter probes on the reporter beads and
the
capture agents, the disc was centrifuged at low speed (less than 800 rpm) upto
,15
minutes. The disc was read in the CD reader at the speed 4X (approx. 1600 rpm)
for
5 minutes. Under these conditions, the unbound magnetic capture beads were
centrifuged away from the capture zone. The magnetic capture beads that were
in the
dual bead complex remained bound to the reporter beads in the capture zone.
The
steps involved in using the disc to capture and analyze dual bead complexes
are
presented in detail in Figs. 25A-25D, 26A-26D, and 27A-27D.
D. Quantification of Dual Bead Complex Structures
The amount of target DNA captured could be enumerated by quantifying the
number of capture magnetic beads and the number of reporter beads since each
type
of bead has a distinct signature.
EXAMPLE 2
The following example is directed to dual bead multiplexing and related assays
as discussed above with reference to, for example, Fig. 32.
A. Dual Bead Assay Multiplexing
In this example, the dual bead assay is carried out to detect two DNA targets
simultaneously. The assay is comprised of 3pm magnetic capture bead. One
population of the magnetic capture bead is coated with capture probes 1 which
are
complementary to the DNA target 1, another population of magnetic capture
beads is
coated with capture probes 2 which are complementary to the DNA target 2.
Alternatively two different types of magnetic capture beads may be used. There
are
two distinct types of reporter beads in the assay. The two types may differ by
chemical composition (for example Silica and Polystyrene) andlor by size.
Various
combinations of beads that may be used in a multiplex dual bead assay format
are
depicted in Fig. 32. One type of reporter bead is coated with reporter probes
1, which
are complementary to the DNA target 1. The other reporter beads are coated
with
115
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
reporter probes 2, which are complementary to the DNA target 2. Again the
capture
probes and the reporter probes are complementary to the respective targets but
not to
each other.
The specific methodology employed to prepare the dual bead assay
multiplexing involved treating 1 x10' capture beads and 2x10 reporter beads in
100
pg/ml salmon sperm DNA for 1 hour at room temperature. This pretreatment will
reduce non-covalent binding between the capture and reporter beads in the
absence
of target DNA. The capture beads were concentrated magnetically with the
supernatant being removed. A 100 microliter volume of the hybridization buffer
(0.2 M
NaCI, 1 mM EDTA, 10 mM MgCl2, 50 mM Tris HCI, pH 7.5, and 5X Denhart's
mixture,
10 microgram per milliliter denatured salmon sperm DNA) were added and the
beads
were re-suspended. Various concentrations of target DNA ranging from 1, 10,
100,
1000 femto moles were added to the capture beads suspension. The suspension
was
incubated while mixing at 37°C for 2 hours. The beads were magnetically
concentrated and the supernatant containing target DNA was removed. A 100
microliter volume of wash buffer (145mM NaCI, 50 mM Tris pH 7.5, 0.1 % SDS,
0.05
Tween, 0.25 % NFDM, 10 mM EDTA) was added and the beads were re-suspended.
The beads were magnetically concentrated and the supernatant was again
removed.
The wash procedure was repeated twice.
A 2x10' amount of reporter beads in 100 microliter hybridization buffer (0.2 M
NaCI, 1 mM EDTA, 10 mM MgCl2, 50 mM Tris HCI, pH 7.5, and 5X Denhart's
mixture,
10 microgram per milliliter denatured salmon sperm DNA) were then added to
washed
capture beads. The beads were re-suspended and incubated while mixing at
37°C for
an additional 2 hours. After incubation the capture beads were concentrated
magnetically, and the supernatant containing unbound reporter beads were
removed.
A 100 microliter volume of wash buffer (145mM NaCI, 50 mM Tris pH 7.5, 0.1 %
SDS,
0.05 % Tween, 0.25 % NFDM, 10 mM EDTA) was added and the beads were re-
suspended. The beads were magnetically concentrated and the supernatant was
again removed. The wash procedure was repeated twice.
After the final wash, the beads were re-suspended in 20 microliters of binding
buffer (50 mM Tris, 200mM NaCI, 10 mM MgCl2, 0.05% Tween 20, 1 % BSA). A 10
microliter volume of this solution was loaded on to the disc that was prepared
as
described in below in Part B of this example.
116
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
B. Disc Preparation
A gold disc was coated with malefic anhydride.polystrene as described.
Distinct
reaction zones were created for two types of reporter beads. Each reaction
zone
consisted of amine DNA sequences complementary to the respective reporter
probes
(or capture agents). Prior to sample injection, the channel were blocked with
a
blocking buffer (50mM Tris, 200mM NaCI, 10 mM MgCl2, 0.05% Tween 20, 1 % BSA,
1 % sucrose) to prevent non-covalent binding of the dual bead complex to the
disc
surface. Alternatively, magnetic beads employed in a multiplexing dual bead
assay
format may be detected using a magneto-optical disc and drive. The chemical
reaction zones, in the magnetic disc format, are replaced by distinctly spaced
magnetic capture zones as discussed in conjunction with Fig. 37, see below
Examples
5 and 6.
C. Capture of Dual Bead Complex Structures on the Disc
A 10 microlitre volume of the dual bead mixture prepared as described above in
Part A of this example, was loaded in to the disc chamber and the injection
ports were
sealed. To facilitate hybridization between the reporter probes on the
reporter beads
and the capture agents, the disc was centrifuged at low speed (less than 800
rpm) for
up to 15 minutes. The disc was read in the CD reader at the speed 4X (approx.
1600
rpm) for 5 minutes. Under these conditions, the unbound magnetic capture beads
were centrifuged to the bottom of the channels. The reporter beads bound to
the
capture zone via hybridization between the reporter probes and their
complementary
agent.
D. Quantification of Dual Bead Complex Structures
The amount of target DNA 1 and 2 captured could be enumerated by
quantifying the number of the respective reporter beads in the respective
reaction
zones.
117
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
EXAMPLE 3
The sensitivity of the dual bead assay depends on the strength of the target
mediated-bonds holding the dual beads together. The dual beads are held
together
by hydrogen bonds. The strength of the bond would increase significantly if
the bond
holding the dual beads is covalent. For this purpose, after target capture, a
ligation
reaction is carried out to create a covalent bond between the capture and
reporter
probes as illustrated above in Fig. 38. The 5' end of the reporter probe
carries a
phosphate group which is required in the ligation reaction.
Ligation Experiment: The assay is comprised of 3pm magnetic capture beads
(Spherotech, Libertyville, IL) coated with covalently attached capture probes;
2.1 pm
fluorescent reporter beads (Molecular Probes, Eugene, OR) coated with a
covalently
attached sequence specific for the DYS gene, and target DNA molecules
containing
DYS sequences. The target DNA is a synthetic 80 oligonucleotide long. The
capture
probes and reporter probes are 40 nucleotides in length and are complementary
to the
DYS sequence but not to each other.
The specific methodology employed to prepare the assay involved treating
1 x10' capture beads and 2x10' reporter beads in 1 OONg/ml salmon sperm DNA
for 1
hour at room temperature. This pre-treatment will reduce the non-specific
binding
between the capture and reporter beads in the absence of target DNA. The
capture
beads were concentrated magnetically with the supernatant being removed. Then
100p1 of the hybridization buffer (0.2M NaCI, 1 mM EDTA, lOmM MgCl2, 50mM Tris-
HCI, pH 7.5 and 5X Denhart's mix, l0~glml denatured salmon sperm DNA) was
added and the beads were resuspended. Various concentration of target DNA
ranging from 1, 10, 100, and 1000 femtomoles were added to the capture bead
suspensions. The beads suspension was incubated while mixing at 37 degrees
Centigrade for 2 hours. The beads were magnetically concentrated and the
supernatant containing unbound target DNA was removed. One hundred microliters
of
wash buffer (145mM NaCI, 50mM Tris, pH 7.5, 0.1 % SDS, 0.05% Tviieen, 0.25%
NFDM (Non Fat Dried Milk), lOmM EDTA) was added and the beads were
resuspended. The beads were magnetically concentrated and the supernatant was
again removed. The wash procedure was repeated twice.
A 2x10' amount of reporter beads in 100N1 hybridization buffer (0.2M NaCI,
1 mM EDTA, 1 OmM MgCl2, 50mM Tris-HCI, pH 7.5 and 5X Denhart's mix, 1 ONg/ml
118
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
denatured salmon sperm DNA) was then added to washed capture beads. The beads
were resuspended and incubated while mixing at 37 degrees Centigrade for an
additional 2 hours. After incubation, the capture beads were concentrated
magnetically, and the supernatant containing unbound reporter beads were
removed.
One hundred microliters of wash buffer (145mM NaCI, 50mM Tris, pH 7.5, 0.1 %
SDS,
0.05% Tween, 0.25% NFDM (Non Fat Dried Milk), IOmM EDTA) was added and the
beads were resuspended. The beads were magnetically concentrated and the
supernatant was again removed. The wash procedure was repeated twice.
After the final wash, the beads were resuspended in 20N1 of binding buffer
(50mM Tris, 200mM NaCI, lOmM MgCl2, 0.05% T-20, 1 % BSA). Then l0pl was
loading onto the bio-disc which was prepared as described above in Example 2,
Part
B.
A. Preparation of Capture Beads
The specific methodology employed to prepare the above assay involved
treating 1 x10' capture beads and 2x10' reporter beads in 1 OOpg/ml salmon.
sperm
DNA for 1 hour at room temperature. This pre-treatment will reduce the non-
specific
binding between the capture and reporter beads in the absence of target DNA.
The
capture beads were concentrated magnetically with the supernatant being
removed.
Then 1 OONI of the hybridization buffer (0.2M NaCI, 1 mM EDTA, 1 OmM MgCl2,
50mM
Tris-HCI, pH 7.5 and 5X Denhart's mix, l0pg/ml denatured salmon sperm DNA) was
added and the beads were resuspended. Various concentrations of target DNA
ranging from 1, 10, 100, and 1000 femtomoles were added to the capture bead
suspensions. The beads suspension was incubated while mixing at 37 degrees
Centigrade for 2 hours. The beads were magnetically concentrated and the
supernatant containing unbound target DNA was removed. One hundred microliters
of wash buffer (145mM NaCI, 50mM Tris, pH 7.5, 0.1 % SDS, 0.05% Tween, 0.25%
NFDM (Non Fat Dried Milk), lOmM EDTA) was added and the beads were
resuspended. The beads were magnetically concentrated and the supernatant was
again removed. The wash procedure was repeated twice.
119
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
8. Hybridization to Target DNA or Bridging Sequence
Various concentration of target DNA at concentrations Omole, 1 E-14, 1 E-13,
1 E-12, and 1 E-11 moles were added to the capture bead suspensions. The beads
suspension was incubated while mixing at 37 degrees Centigrade for 2 hours.
The
beads were magnetically concentrated and the supernatant containing unbound
target
DNA was removed. One hundred microliters of wash buffer (145mM NaCI, 50mM
Tris, pH 7.5, 0.1 % SDS, 0.05% Tween, 0.25% NFDM (Non Fat Dried Milk), 1 OmM
EDTA) was added and the beads were resuspended. The beads were magnetically
concentrated and the supernatant was again removed. The wash procedure was
repeated twice. The capture beads were re-suspended in 50~,L of 40mM NaCI
solution.
C. Hybridization to Reporter Probes or Reporter Beads
A 2x10' amount of reporter beads or 100 pmoles of reporter probes in 100p1
hybridization buffer (0.2M NaCI, 1 mM EDTA, 1 OmM MgCl2, 50mM Tris-HCI, pH 7.5
and 5X Denhart's mix, l0pg/ml denatured salmon sperm DNA ) was then added to
washed capture beads. The beads were resuspended and incubated while mixing at
37 degrees Centigrade for an additional 2 hours. After incubation, the capture
beads
were concentrated magnetically, and the supernatant containing unbound
reporter
beads or unbound reporter probes were removed. One hundred microliters of wash
buffer (145mM NaCI, 50mM Tris, pH 7.5, 0.1 % SDS, 0.05% Tween, 0.25% NFDM
(Non Fat Dried Milk), lOmM EDTA) was added and the beads were resuspended.
The beads were magnetically concentrated and the supernatant was again
removed.
The wash procedure was repeated twice.
D. Ligation Reactions
A 10 ~,L volume of the 10X ligation buffer (final concentration 66mM Tris, pH
7.6, 6.6mM MgCl2, 100mM DTT, 66~,M ATP) and 4 units ligase (concentrations 10
units per ~,L) was added to the bead suspensions. The ligation reaction was
carried
out for 2 hours at room temperature. The bead suspensions were washed 3 times
with wash buffer (145mM NaCI, 50mM Tris, pH 7.5, 0.2 %SDS, 0.05 % Tween 20,
0.25 % NFDM). In the control tube, no ligase was added.
120
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
E. Enzyme Assays ,
The amount of reporter probe was directly correlated with the amount of target
DNA captured. Therefore, one way to quantify the target captured was to
quantify the
amount of reporter probe. The rationale for this assay is that the reporter
probe was
biotinylated. The concentrations of the reporter probe therefore could be
determined
by an enzyme assay wherein the enzyme Streptavidin-Alkaline phosphatase binds
to
the biotin moiety. A chromogenic substrate for Alkaline phosphatase, p-
nitrophenyl
phosphate, was used as reporter. This colorless substrate is hydrolyzed by
alkaline
phosphatase to a yellow product which has an absorbance at 405nm. The beads
were washed with 100p1 of CDB (2% BSA, 50mM Tris-HCI, pH 7.5, 145mM NaCI, 1.0
mM MgCl2, 0,1 mM ZnCl2, 0.05% NaN3) and incubated with 100N1 of 250ng/ml
Streptavidin-Phosphatase for 1 hour at 37°C. The beads were washed 3
times with
wash buffer (145mM NaCI, 50mM Tris, pH 7.5, 0.05% Tween) to get rid of unbound
S-
AP. The beads were incubated with 100N1 of the S-AP substrate p-nitrophosphate
at
3.7mg/ml in 0.1 M Tris, pH 10, 2mM MgCl2 for 5-15 minutes at room temperature.
The
color development of the supernatant was monitored at 405nm. The intensity of
the
color is directly correlated with the amount of the biotinylated reporter
probe and thus
the amount of target captured.
F. Dual Bead Assays
The amount of reporter beads was directly correlated with the amount of target
captured. Therefore, one way to quantify the target captured was to quantify
the
amount of reporter beads. After hybridization and ligation, the beads were re-
suspended in 200pL PBS and the amount of reporter beads was quantified by the
fluorimeter Fluoromax-2 at Ex = 500nm, Slit = 2.0; Em = 530nm, Slit = 2Ø
Alternatively, the number of fluorescent reporter beads can be quantified by
the bio-
CD reader as described above.
121
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
EXAMPLE 4
The use of cleavable spacers in dual bead assay increases the specificity of
the
assay. The following example is directed to a dual bead assay using cleavable
spacers.
A. Design of Capture and Reporter Probes
The design of capture probes and reporter probes is critical in the success of
the dual bead assay using cleavable spacers. The capture probes and reporter
probes contain 3 branches as illustrated above in Fig. One branch of the
reporter or
capture probes participates in the target capture. Several linkers (PEG
groups) are
introduced into the capture or reporter probes to minimize coiling of the
probe and to
increase target capture efficiency. The second branch of the capture or
reporter
probes contains 3 linkers followed by a biotin at the end. Other functional
groups such
as carboxyl or amine could also be used. The biotin participates in the
conjugation of
the capture or reporter probes onto the solid phase. The third branch of the
capture
probe hybridizes to the reporter probe.
When restriction enzyme digestion is the method of choice for cleaving the
capture and reporter probes, a restriction site is introduced into the
sequences of the
probes. The choice of restriction site is important in that it has to be
unique (not
common) so that only the sequence holding the capture and reporter probes (and
not
the target DNA) is cleaved. The formation of the capture and reporter probes
in the
presence of the target is shown above in Fig. 42C.
When displacement of the reporter probe is the method of choice for cleaving
the capture and reporter probes, the sequence on the reporter probes that
participates
in the hybridization with the capture probe is relatively short (about 10
nucleotides).
The remaining sequence is not complementary to the capture probe and therefore
will
be available for the displacement probe to hybridize. This is generally
illustrated
above in Figs. 43A and 43B to show hybridization of capture probe (Probe 1 )
to
reporter probe (Probe 2B). In this example, the probes used were synthesized
by
Biosource of Camarillo, CA.
122
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
B. Immobilization of Capture Probe onto Streptavidin Beads
1. Preparation of capture beads: The first step in the assay is the
conjugation of
the capture probe onto a solid phase. In this example, 2.8pm magnetic beads
coated
with streptavidin from Dynal were used as the solid phase. Typically, 6.7x10'
Dynal
beads were used per conjugation. The beads were resuspended in 200N1 of
binding
and washing buffer (lOmM Tris-HCI, pH 7.5, 1 mM EDTA, 2M NaCI). The beads were
magnetically concentrated and the supernatant was removed. The wash procedure
was repeated twice.
2. Conjugation of capture probes onto capture beads: The magnetic beads
were resuspended in 400p1 binding and washing buffer (lOmM Tris-HCI, pH 7.5, 1
mM
EDTA, 2M NaCI) to a final concentration of 5pg of beads/pl. Then 600 picomoles
of
capture probes in water was added to the bead suspension. The final salt
concentration in the mixture is 1 M NaCI. It should be noted that high salt is
required
for efficient conjugation. The mixture was incubated at 37 degrees Centigrade
for 2 to
4 hours with occasional mixing. The beads were then magnetically concentrated
and
the supernatant was removed. The beads were washed 3 times with binding and
washing buffer (1 OmM Tris-HCI, pH 7.5, 1 mM EDTA, 2M NaCI).
3. Determination of conjugation efficiency; The optical density of the
supernatant before and after conjugation was measured at 260nm to quantify the
amount of capture probes conjugated. Typically, over 50% of the capture probes
were
conjugated onto the streptaividin beads. The density of probes was from 0.5
x106 to
1 x106 probes/bead. Table 2 below presents a listing of an example for the
determination of conjugation efficiency of biotinylated probe onto
Streptavidin coated
magnetic beads.
4. Blocking of remaining streptavidin sites on the bead: The beads were
incubated in 400p1 of PBS containing 2mg/ml biotin for 1 hour on a rotating
mixer to
block all remaining streptavidin sites on the Dynal magnetic beads. The
magnetic
beads were washed 3 times with binding and washing buffer (lOmM Tris-HCI, pH
7.5,
1 mM EDTA, 2M NaCI) and resuspended in 1000p1 hybridization buffer (0.2M NaCI,
1 OmM MgCl2, 1 mM EDTA, 50mM Tris, pH 7.5).
123
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
TABLE 2
Conjugation of Biotinylated Capture Probe onto
Streptavidin Coated Magnetic Beads
1. Number of beads used: 1.2 0 beads-
2. Number of streptavidin molecules per bead: 7 x 10 molecules/bead
3. Amount of biotinylated capture probe 1 bound to 1 mg
of bead: 127
pmoles or 8 x 1 O13 molecule
4. Number of biotin probes/bead: 8 x 106 molecules/bead
5. All free streptavidin binding sites were saturated with
biotin
C. Hybridization of Capture Probe to Reporter Probes
1. Hybridization: Out of the 1000p1 bead suspension, 400p1 was mixed with
400p1 TE buffer containing 1 nanomole of reporter probe 2A, 400N1 was mixed
with
400p1 TE buffer containing 1 nanomole of reporter probe 2B, 200N1 was mixed
with
200p1 TE (Tris-EDTA) as a negative control. The hybridization was carried out
at 37°C
for 2 hours.
2. Washing: Following hybridization, the magnetic beads were washed 3X with
wash buffer (145mM NaCI, 50mM Tris, pH 7.5, 0.05% Tween).
3. Determination of hybridization efficiency' Here 50p1 out of 800N1 was
assayed for the hybridization efficiency. The rationale for this assay is that
the
reporter probes 2A and 2B were biotinylated. The concentrations of these
probes
therefore could be determined by an enzyme assay wherein the enzyme
Streptavidin
Alkaline phosphatase binds to the biotin moiety. A chromogenic substrate for
Alkaline
phosphatase, p-nitrophenyl phosphate, was used as reporter. This colorless
substrate
is hydrolyzed by alkaline phosphatase to a yellow product which has an
absorbance at
405nm. The beads were washed with 100p1 of CDB (2% BSA, 50mM Tris-HCI, pH
7.5, 145mM NaCI, 1.OmM MgCl2, 0,1 mM ZnCl2, 0.05% NaN3) and incubated with
100N1 of 250ng/ml Streptavidin-Phosphatase for 1 hour at 37°C. The
beads were
washed 3 times with wash buffer (145mM NaCI, 50mM Tris, pH 7.5, 0.05% Tween)
to
get rid of unbound S-AP. The beads were incubated with 100N1 of the S-AP
substrate
p-nitrophosphate at 3.7mg/ml in 0.1 M Tris, pH 10, 2mM MgCl2 for 5-15 minutes
at
room temperature. The color development of the supernatant was monitored at
124
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
405nm. The intensity of the color is directly correlated with the amount of
the
biotinylated reporter probe 2A or 2B hybridized.
At this point, the reporter probes could be attached to another solid phase
via
their biotin moiety. For this alternate dual bead assay, a different type of
streptavidin
coated beads, i.e. polystyrene or fluorescent, is added to the bead
suspension,
resulting in the formation of the dual bead complexes. If the solid phase is
the surface
of the bio-disc, then the mixture of capture and reporter probes is incubated
on a
streptavidin coated disc surface.
D. Hybridization of Probes to Target DNA
1. Hybridization: In this example, the target DNA was a single stranded 80mer
oligonucleotide. Various concentrations of target DNA ranging from 0, 1, and
1000
picomoles were added to the bead suspensions. The beads suspensions were
incubated while mixing at 37 degrees Centigrade for 2 hours.
2. Washing: The beads were magnetically concentrated and the supernatant
containing unbound target DNA was removed. One hundred microliters of wash
buffer
(145mM NaCI, 50mM Tris, pH 7.5, 0.1 % SDS, 0.05% Tween, 0.25% NFDM (Non Fat
Dried Milk), lOmM EDTA) was added and the beads were resuspended. The beads
were magnetically concentrated and the supernatant was again removed. The wash
procedure was repeated twice.
E. Distinction of Target-Mediated Capture by
Restriction Enzyme Digestion or by Probe Displacement
1. Restriction enzyme di es~ The restriction enzyme site that was
introduced in the capture and reporter probes was NOT1. This restriction
enzyme site
is rare and in this model system is not found in any other sites. The beads
were
resuspended in 400p1 CDB (2% BSA, 50mM Tris-HCI, pH 7.5, 145mM NaCI, 1.0 mM
MgCl2, 0,1 mM ZnCl2, 0.05% NaN3). The bead suspension was aliquoted into seven
tubes, one control and 6 digestion tubes. The enzyme NOT1 was prepared
according
to the manufacturer's specifications. Then 5 units of enzyme were added to the
each
digestion tubes in a total volume of 100p1. Water was added to the control
tube. The
digestion was carried out for 3-4 hours at 37°C.
2. Displacement of the reporter probe by the displacement ~arobe: The beads
were resuspended in 400N1 CDB (2% BSA, 50mM Tris-HCI, pH 7.5, 145mM NaCI, 1.0
125
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
mM MgCl2, 0,lmM ZnCl2, 0.05% NaN3). The bead suspension was aliquoted into two
tubes, one control and one displacement tube. The beads were heated for 5
minutes
at 55°C in 200N1 of 6xSSC, 1 mM EDTA. The heat treatment was used to
induce the
melting of the reporter probe 2B from the capture probe. At this point, a 10
fold
excess of displacement probe was added to the bead suspension and the mixture
was
incubated at 37°C for several hours Water was added to the control
tube.
F. Quantification of Target Captured by Enzyme Assay
The amount of reporter probe remaining after the restriction enzyme digestion
or probe displacement was directly correlated with the amount of target DNA
captured.
Therefore, one way to quantify the target captured was to quantify the amount
of
remaining reporter probe. The rationale for this assay is that the reporter
probes 2A
and 2B were biotinylated. The concentrations of these probes therefore could
be
determined by an enzyme assay wherein the enzyme Streptavidin-Alkaline
phosphatase binds to the biotin moiety.
A chromogenic substrate for Alkaline phosphatase, p-nitrophenyl phosphate,
was used as reporter. This colorless substrate is hydrolyzed by alkaline
phosphatase
to a yellow product which has an absorbance at 405nm. The beads were washed
with
100N1 of CDB (2% BSA, 50mM Tris-HCI, pH 7.5, 145mM NaCI, 1.0 mM MgCl2, 0,1 mM
ZnCl2, 0.05% NaN3) and incubated with 100N1 of 250ng/ml Streptavidin-
Phosphatase
for 1 hour at 37°C. The beads were washed 3 times with wash buffer
(145mM NaCI,
50mM Tris, pH 7.5, 0.05% Tween) to get rid of unbound S-AP. The beads were
incubated with 100N1 of the S-AP substrate p-nitrophosphate at 3.7mg/ml in 0.1
M Tris,
pH 10, 2mM MgCl2 for 5-15 minutes at room temperature. The color development
of
the supernatant was monitored at 405nm. The intensity of the color is directly
correlated with the amount of the biotinylated reporter probe 2A or 2B
hybridized.
G. Quantification of Target Captured by Dual Bead Assay
In the case when the reporter probes are immobilized on another solid phase
such as fluorescent or polystyrene streptavidin coated beads, the amount of
target
captured could be quantified by dual bead assay. The number of reporter beads
remaining following restriction enzyme digestion or probe displacement could
be
126
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
enumerated by the fluorimeter (for fluorescent beads) or by the bio-CD reader
since
each type of bead has a distinct signal signature.
EXAMPLE 5
The following example illustrates a dual bead assay carried out on a
magnetically writable and erasable analysis disc such as the magneto-optical
bio-disc
110 discussed in conjunction with Fig. 37.
In this example, the dual bead assay is carried out to detect the gene
sequence
DYS which is present in male but not female. The assay is comprised of 3pm
magnetic capture beads (Spherotech, Libertyville, IL) coated with covalently
attached
transport probes; 2.1 pm fluorescent reporter beads (Molecular Probes, Eugene,
OR)
coated with a covalently attached sequence specific for the DYS gene, and
target
DNA molecules containing DYS sequences. The target DNA is a synthetic 80
oligonucleotides long. The transport probes and reporter probes are 40
nucleotides in
length and are complementary to the DYS sequence but not to each other.
The specific methodology employed to prepare the assay involved treating
1 x10' capture beads and 2x10' reporter beads in 1 OOpg/ml salmon sperm DNA
for 1
hour at room temperature. This pre-treatment will reduce the non-specific
binding
between the capture and reporter beads in the absence of target DNA.
After pretreatment with salmon sperm DNA, the capture beads are loaded
inside the MO bio-disc via the injection port. The MO bio-disc contains
magnetic
regions created by the magneto optical drive. The capture beads thus are held
within
specific magnetic regions on the MO bio-disc.
The sample containing target DNA and reporter beads in 200N1 hybridization
buffer (0.2M NaCI, 1 mM EDTA, 1 OmM MgCl2, 50mM Tris-HCI, pH 7.5 and 5X
Denhart's mix, lONg/ml denatured salmon sperm DNA) is then added to the MO bio-
disc via the injection port. The injection port is then sealed. The magnetic
field is
released. The disc is rotated at very low speed (less than 800rpm) in the
drive to
facilitate hybridization of target DNA and reporter beads to the capture
beads. The
temperature of the drive is kept constant at 33 degrees Centigrade. After 2
hours of
hybridization, the magnetic field is created by the magneto optical drive. At
this stage,
only magnetic capture beads, unbound or as part of a dual bead complex, remain
on
the MO bio-disc. Unbound target and reporter beads are directed to a waste
chamber
127
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
by any of the mechanisms described above. Two hundred microliters of wash
buffer
(145mM NaCI, 50mM Tris, pH 7.5, 0.1 % SDS, 0.05% Tween, 0.25% NFDM (Non Fat
Dried Milk), lOmM EDTA) is then added. The magnetic field is released and the
disc
is rotated at low speed (less than 800rpm) for 5 minutes to remove any non-
specific
binding between the capture beads and reporter beads. The magnetic field is
then
reapplied. The wash buffer is directed to the waste chamber by any of the
mechanisms described above. The wash procedure is repeated twice.
At this stage, only magnetic capture beads, unbound or as part of a dual bead
complex, remain. The magnetic field is released and the dual bead complexes
are
directed to a detection chamber. The amount of target DNA captured is then
enumerated by quantifying the number of capture magnetic beads and the number
of
reporter beads since each type of bead has a distinct signature as illustrated
above in
Figs. 28A, 28B, 29A, and 29B.
EXAMPLE 6
In this example, a dual bead assay using the multiplexing techniques described
above in connection with Figs. 32 and 37 is carried out on a magnetically
writable and
erasable analysis disc such as the MO bio-disc 110 discussed with reference to
Fig.
37.
The dual bead assay is carried out to detect 2 or more DNA targets
simultaneously. The assay is comprised of 3pm magnetic capture beads
(Spherotech,
Libertyville, IL). One population of the magnetic capture beads is coated with
transport probes 1 which are complementary to the DNA target 1. Another
population
of the magnetic capture beads is coated with transport probes 2 which are
complementary to the DNA target 2. Alternatively, 2 or more different types of
magnetic capture beads may be used. There are two or more distinct types of
reporter beads in the assay. The reporter beads may differ by chemical
composition
(for example silica and polystyrene) and/or by size. One type of reporter
beads is
coated with reporter probes 1, which are complementary to the DNA target 1.
The
other reporter beads are coated with reporter probes 2, which are
complementary to
the DNA target 2. Again, the transport probes and reporter probes are
complementary
to the respective targets but not to each other.
128
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
The specific methodology employed to prepare the dual bead assay
multiplexing involved treating 1x10' capture beads and 2x10' reporter beads in
100pg/ml salmon sperm DNA for 1 hour at room temperature. This pre-treatment
will
reduce the non-specific binding between the capture and reporter beads in the
absence of target DNA.
After pretreatment with salmon sperm DNA, the capture beads are loaded in
the MO bio-disc. The magnetic field is applied to create distinct magnetic
zones for
specific capture beads. The capture beads can be held on the MO bio-disc at a
density of 1 capture bead per 10Nm2. The surface area usable for bead
deposition on
the MO bio-disc is approximately 3 x 109Nm2. The capacity of the MO bio-disc
for 3Nm
beads at the given density is about 3 x 10$ beads.
The sample containing the targets DNA of interest is mixed with different
types
of reporter beads in 200N1 hybridization buffer (0.2M NaCI, 1 mM EDTA, 1 OmM
MgCl2,
50mM Tris-HCI, pH 7.5 and 5X Denhart's mix, lONg/ml denatured salmon sperm
DNA)
and added to the MO bio-disc via the injection port. The injection port is
then sealed.
The magnetic field is released. The disc is rotated at very low speed (less
than
800rpm) in the drive to facilitate hybridization of targets DNA and reporter
beads to the
different types of capture beads. The temperature of the drive is kept
constant at 33
degrees Centigrade. After 2 to 3 hours of hybridization, the magnetic field is
regenerated by the magneto optical drive. At this stage, only magnetic capture
beads,
unbound or as part of dual bead complexes, remain on the MO bio-disc. Unbound
targets and reporter beads are directed to a waste chamber by any of the
mechanisms
described above. Two hundred microliters of wash buffer (145mM NaCI, 50mM
Tris,
pH 7.5, 0.1% SDS, 0.05% Tween, 0.25% NFDM (Non Fat Dried Milk), lOmM EDTA) is
then added. The magnetic field is released and the disc is rotated at low
speed (less
than 800rpm) for 5 minutes to remove any non-specific binding between the
capture
beads and reporter beads. The magnetic field is then reapplied. The wash
buffer is
directed to the waste chamber by any of the mechanisms described above. The
wash
procedure is repeated twice.
At this stage, the magnetic field is released and the dual bead complexes are
directed to a detection chamber. The amount of different types of target DNA
can be
enumerated by quantifying the number of corresponding capture magnetic beads
and
129
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
reporter beads since each type of bead has a distinct signature as shown above
in
Figs. 28A, 28B, 29A, and 29B.
EXAMPLE 7
This experiment was performed to determine the amount of covalently
conjugated probe on different beads to determine which bead type is best for
covalent
probe linking.
A. Conjugation
Magnetic beads (1-2 pm) from Polysciences, magnetic beads (3 pm) from
Spherotech, fluorescent beads (1.8 pm) from Polysciences and fluorescent beads
(2.1
pm) from Molecular Probes were evaluated in this example. Approximately 5x10$
beads were used per conjugation reaction. The beads were washed and
resuspended in 0.05 M MES buffer (2-N-morpholeno-ethanesulphonic acid), pH 6.0
and activated for 15 minutes by the addition of 0.1 M EDC (1- ethyl 3-3
dimethylaminopropyl carbodimide-HCI). After activation, the pH of the bead
solution
was adjusted to ~7.5 with NaOH. Then 0.5 nanomoles of biotinylated probes were
added to the solution. The probes were allowed to conjugate for 2-3 hours at
room
temperature on a rotating mixer. The beads were then magnetically concentrated
and
the supernatant was collected. To estimate the amount of biotinylated probes
bound
to the beads, the optical density (at 260nm) of the supernatant was measured
before
and after the conjugation.
B. Determination of Covalent Conjugation Efficiency
Typically 1 to 5x10' beads, conjugated with biotinylated probes as discussed
above, were used in the determination of covalent conjugation efficiency of
the
probes. These beads were washed three times in wash buffer and were
resuspended
in 200p1 CDB (145mM NaCI, 50mM Tris HCI, 2% BSA, 1 mg/ml MgCl2, 0.1 mM ZnCl2,
0.05% NaN2). The beads were then magnetically concentrated, and the
supernatant
was removed. The beads were resuspended in 100p1 CDB containing 550ng/ml
streptavidin-alkaline phosphatase (S-AP) and incubated for 1 hour at
37° C to allow
sufficient time for the streptavidin to bind to the biotin on the probe.
Following
incubation with S-AP, the beads were magnetically concentrated, and the
supernatant
130
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
containing unbound S-AP was removed. The beads were washed three times in wash
buffer. Next 100p1 of p-nitrophenylphosphate (pNPP), a substrate for alkaline
phosphatase at a concentration of 3.7 mg/ml in 0.1 M Tris- HCI, pH 10 was
added to
the beads at fixed time intervals to minimize the variation due to difference
in
incubation time. The incubation time with the substrate was varied (from 2 min
upto
30 min) as needed to obtain reliable OD at 405 nm since time for color
development
varies depending upon the concentration of probe. The optical density obtained
from
a spectrophotometer at 405nm wavelength was proportional to the amount of
probes
bound to the beads.
The results of the experiment are presented in Figs. 51 A and 51 B. As
indicated, 87% of the probes that bound to the 1-2 pm magnetic beads from
Polysciences were non covalently bound, as compared to 15% of non-covalently
bound probes on the 3 pm Spherotech beads.
Referring to Figs. 53A and 53B, data showing a correlation between the
covalent conjugation efficiency and the sensitivity of the dual bead assay is
presented.
These results indicate that with higher covalent conjugation efficiency, the
more
sensitive and specific the dual bead assay is. The amount of covalenty bound
probes
may be calculated by repeating steps in this Part B after performing the steps
in Part
C below. The calculation of the amount of covalent binding is presented in
Fig. 55.
C. Heat Treatment in Removal of Non-Covalently Pound Probes
After determining which bead type has the desired covalent conjugation
efficiency, the steps in Parts A and B above may be repeated using non-
biotinylated
probes and the appropriate bead type for use in a dual bead assay.
Following conjugation, the non-covalently bound probes could be selectively
removed by heat treatment of the beads. For this purpose, up to 3x10' beads
were
resuspended in 100p1 of CDB solution heated at 70°C for 10 minutes. The
beads
were then immediately magnetically concentrated and the supernatant was
removed.
The beads were washed twice in wash buffer and once in CDB and re-suspended in
1 OOpI CDB. At this point the beads are now ready for use in a dual beads
test.
131
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
EXAMPLE 8
Experiments were also done to evaluate the use of double stranded DNA
during probe conjugation to increase the covalent conjugation efficiency .of
the DNA
probe on the solid phase.
A. Formation of Double Stranded DNA
The capture probe utilized was 40 nucleotides in length and contained an
aminogroup (NH2) at the 5' end and several chains of PEG (polyethylene glycol)
linker.
The strand complimentary to the aminated probe used in this experiment was 40
nucleotides in length and contained a biotin group at the 5' end. A
hybridization
reaction was carried out with an excess of complementary probes under
stringent
conditions at 37°C.
B. Conjugation of Double-Stranded DNA Probe onto Beads
Magnetic beads (1-2pm) from Polysciences, magnetic beads (3pm) from
Spherotech, fluorescent beads (l.8pm) from Polysciences and fluorescent beads
(2.1 pm) from Molecular Probes were evaluated in this example. Approximately
5x10$
were used per conjugation reaction. The beads were washed and resuspended in 1
ml of 0.05 M MES buffer (2-N-morpholeno-ethanesulphonic acid), pH 6.0 and
activated for 15 minutes by the addition of 0.1M EDC (1- ethyl 3-3
dimethylaminopropyl carbodimide -HCI). After activation, the pH was adjusted
to ~7.5
with NaOH. A volume of 0.5 nanomoles of probes were then added to the
solution.
The probe conjugation was carried out for 2-3 hours at room temperature on a
rotating
mixer. The beads were then magnetically concentrated and the supernatant was
removed. To estimate the amount of probes bound to the beads, the optical
density at
260nm of the supernatant was measured before and after the conjugation.
After the conjugation, all unreacted carboxyl groups on the beads were blocked
with 1 ml 0.1 M Tris- HCI pH 7.5 for 1 hour at room temperature on a mixer.
The
beads were then blocked for 30 minutes in 1 ml of l0mglml BSA in PBS at room
temperature on the mixer to block any unspecific protein binding sites. After
blocking,
the beads were washed three times with PBS and resuspended in storage buffer
(PBS
with 10 mglml BSA, 5% glycerol, 0.1 % sodium azide).
132
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
C. Determination of Covalent Conjugation Efficiency
An aliquot of 2x10 $ magnetic beads was taken out from the above conjugated
beads and pre-treated with 0.1 mg/ml salmon sperm DNA for 1 hour at room
temperature. The beads were then washed 3 times in wash buffer and resuspended
in 200,1 CDB. Then 200 picomoles of blocking probes and 100p,1 of
hybridization
buffer were added to the bead solution. The blocking probes were allowed to
hybridize for two hours at 37°C. After hybridization, the beads were
magnetically
concentrated and the supernatant was removed. The beads were then washed three
times in wash buffer using by magnetic concentration. The beads were
resuspended
with 100p,1 of buffer containing 550ng/ml streptavidin-alkaline phosphatase (S-
AP) and
incubated for 1 hour at 37° C. Following incubation with S-AP, the
beads were
magnetically concentrated, and the supernatant containing unbound S-AP was
removed. The beads were washed three times in wash buffer. Next 1001 of p-
nitrophenylphosphate (pNPP), a substrate for alkaline phosphatase at a
concentration
of 3.7 mg/ml, was added to the beads at fixed time intervals to minimize the
variation
due to difference in incubation time. The time for color development varies
depending
upon the concentration of probe. The incubation time with the substrate was
varied
from 2 min up to 30 min as needed to obtain reliable OD at 405 nm. The optical
density at 405nm was proportional to the amount of probes bound to the beads.
The
results from one of these double stranded conjugation experiments are
presented in
Figs. 52A and 52B above.
D. Use of Heat Treatment to Separate Complimentary Strands from Capture Probes
An aliquot of 100p1 of beads were heated for 10 min. at 70°C.
Magnetically
concentrate the beads and take out the supernatant promptly. Wash once in hot
wash
buffer and once in CDB. Then resuspend in CDB.
EXAMPLE 9
Experiments were also conducted to test the use of linkers of longer spacers
to
increase the efficiency of conjugation and the accessibility and rigidity of
the probes
attached to a solid phase. In these experiments, the capture and reporter
probes
were 40 nucleotides in length. These synthetic nucleotide sequences were
specific to
the analyte of interest. In this example, the 5' end of the capture probe and
3' end of
133
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
the reporter probe contained conjugated 3 polyethylene glycol moieties. These
covalently bound linkers were introduced to the probes during probe synthesis.
Data
collected from one of these experiments are depicted in Fig. 54 above. As
shown in
Fig. 54, the use of linkers significantly increases the sensitivity of the
dual bead assay.
The beads used in this particular assay were 3p,m magnetic beads from
Spherotech and 2.1 ~,m reporter beads from Molecular Probes. The probes were
covalently conjugated to the beads as described above. An aliquot of 2x10' of
probe
conjugated capture beads and 6x10' of reporter beads per assay were washed
three
times with PBS. After washing, the beads were pretreated with 100 p,g/ml of
salmon
sperm DNA in water for one hour at room temperature. The beads were washed
three
times in wash buffer (0.145M NaCI, 50 mM Tris-HCI pH 7.5, 0.5% Tween-20), once
with hybridization buffer ( 50mM Tris-HCI pH 7.5, 0.1 M NaCI, 1 OmM MgCI, 1 mM
EDTA pH 7.5) and re-suspended in hybridization buffer containing 1 OO~g/ml
DNA, and
5X Denhart's mixture.
The two-step hybridization method, as presented in Fig. 12A, was employed in
performing the dual bead assay of this example. Different concentrations of a
single
target were used including Control (0 femtomole), 10 femtomole, 1 femtomole,
0.1
femtomole, 0.01 femtomole, 0.001 femtomole, 0.0001 femtomole diluted in
hybridization buffer containing 100p,g/ml of salmon sperm DNA and 5X Denhart's
solution. The various target solutions were then mixed with the capture beads
and
incubated at 37° C for 2 hours to allow ample time for target
hybridization to the
capture probe on the beads. After hybridization the hybridized capture beads
were
washed three times with wash buffer, once with hybridization buffer, and re-
suspended in 1001 hybridization buffer including 100~,g/ml DNA, and 5X
Denhart's
mixture. The capture bead solution, containing hybridized target, was then
mixed with
100p,1 of reporter beads and incubated at 37°C for 2 hours while
continuously mixing.
Then washed 6 times with new wash buffer (145mM NaCI, 50mM Tris-HCI pH 7.5, 05
Tween 20, 0.1 % SDS, 0.25% NFDM) and once with PBS. The washed solution
containing the dual bead complexes was then re-suspended with 250,1 PBS. The
fluorescent signal from the reporter beads were then quantified using a
fluorimeter.
Results showed that when 3 PEG linkers were introduced into the capture
probe, it lowered the background in dual bead assays and improved the assay
sensitivity significantly as compared to probes without linkers.
134
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
EXAMPLE 10
This study was performed to elucidate the assay sensitivity and range of
detection of a genetic dual bead assay; the results are shown in Fig. 57.
A. Preparation of Capfure and Reporter Beads
The beads used in this experiment were magnetic capture beads (3um
Spherotech) and yellow-fluorescent reporter beads (1 um Polysciences) each
covalently conjugated with DNA transport probes and DNA signal probes,
respectively. Approximately 1x10' capture beads and 2x10' reporter beads were
used for this experiment. These beads were washed 3X with PBS and resuspended
in 1 ml water containing 100 ~,g/ml digested salmon sperm DNA. The bead
solutions in
the salmon sperm DNA mixture were then incubated for 1 hour at room
temperature.
After incubation the beads were washed 3x with wash buffer (145mM NaCI, 50mM
Tris, pH 7.5, 0.05% Tween) and 1 X with hybridization buffer (0.1 M NaCI, 1
OmM
MgCl2, 1 mM EDTA, 50mM Tris, pH 7.5). The beads were then resuspended in
hybridization buffer (containing 100p,g/ml of DNA).
B. Dual Bead Assay
A serial dilution of DNA target agents were prepared containing: 100
femtomole, 10 femtomole, 1 femtomole, 0.1 femtomole, 0.01 femtomole, and 0
femtomoles (negative control). Equal amounts of capture beads were then mixed
with the various solutions of target and incubated at 37°C for 2 hours
to let target
hybridized to the 5' capture probe on the beads. After incubation, the bead
solutions
were washed 3X with the wash buffer (145mM NaCI, 50mM Tris, pH 7.5, 0.05%
Tween) and 1 X with hybridization buffer (0.1 M NaCI, 1 OmM MgCl2, 1 mM EDTA,
50mM
Tris, pH 7.5), then resuspended in hybridization buffer (containing 100 ~,g/ml
salmon
sperm DNA).
Reporter beads were mixed with the capture bead solution and incubated at
37°C for 2 hours in a rotating mixer. After incubation the bead
solutions were washed
6X with 0.5 ml of wash buffer (145mM NaCI, 50mM Tris, pH 7.5, 0.05% Tween, 0.1
SDS, 0.25% NFDM) and resuspended in 250.1 water. The number of reporter beads
135
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
bound were then quantified by a fluorimeter at Excitation = 500nm, Emission =
530nm,
Slit = Ex-2, Em-2, and integration time of 0.1 second.
EXAMPLE 11
This study was performed to determine the optimal salt concentration in the
hybridization buffer for use in a genetic dual bead type assay.
A. Preparation of Capture and Reporter Beads
The beads used in this experiment were magnetic capture beads (3um
Spherotech) and yellow-fluorescent reporter beads beads (2.1 um from Molecular
Probes) each covalently conjugated with DNA transport probes and DNA signal
probes, respectively. The beads were washed 1 X with hybridization buffer (0.1
M
NaCI, 10 mM MgCl2, 1 mM EDTA, 50mM tris, pH 7.5). The beads were pretreated
with
0.1 % CHAPS and salmon sperm DNA for 1 hour at room temp. The beads were then
washed 3X with wash buffer (145mM NaCI, 50 mM Tris, pH 7.5, 0.1 % SDS, 0.05%
Tween, 0.25% NFDM), 1X with respective hybridization buffer (145mM NaCI, lOmM
MgCl2, 1 mMEDTA, 1 OOp,g/ml salmon sperm DNA, 50 mM Tris, pH 7.5). After
washing, the beads were resuspended in hybridization buffer.
8. Dual Bead Assay
v~ The magnetic capture beads, prepared in section A, were divided into 24
l0ul
aliquots. Six sets of aliquots were diluted in hybridization solution
containing various
concentrations of NaCI (OmM, 145mM, 300mM, and 400mM). The target DNA mixture
was made in the hybridization buffer, of varying salt concentrations, in the
following
concentrations: 1 OOfmole, 1 Ofmole, 1 fmole, 0.1 fmole, 0.01 fmole, 0
femtomole
(negative control). The various target solutions were then mixed with their
respective
bead solutions according to the salt concentration of the hybridization buffer
and
incubated at 37°C for 2 hours to let target to hybridize to the 5'
transport probe on the
capture beads. After hybridization, the assay solutions were washed 3X with
wash
buffer containing the appropriate amount of NaCI required for each treatment
group
(OmM, 0.145mM, 0.3mM, 0.4mM NaCI), 1 X with the appropriate hybridization
buffer.
The beads were then resuspended in their respective hybridization buffers
containing
the appropriate amount of NaCI.
136
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
A 100,1 volume of reporter beads in hybridization buffer containing various
NaCI concentrations (OmM, 0.145mM, 0.3mM, 0.4mM NaCI), were added to the
appropriate assay solution, so that the same concentration of NaCI was
maintained
within the different treatment groups. These assay solutions were then
incubated at
37°C for 2 hours in a rotating mixer. After incubation, the various
solutions were then
washed 6X with 0.5 ml of wash buffer containing the appropriate amount of NaCI
and
once with water also containing ~ NaCI at concentrations equal to that in each
respective hybridization buffer (OmM, 0.145mM, 0.3mM, 0.4mM NaCI). The various
dual bead solutions were then resuspended in 250,1 water. The number of
reporter
beads bound were then quantified using a fluorimeter at Excitation = 500nm,
Emission
= 530nm, Slit = Ex-2, Em-2 and an integration time of 0.1 second. The results
from
this assay are shown in above Fig. 60. Detection of the dual bead complex may
be
carried out using the optical disc system described as described in
conjunction with
Fig. 2., a magneto-optical disc system, a fluorescent disc system, or any
similar
device. Unique signature traces of a dual bead complex collected from an
optical disc
reader are shown above in Fig. 29B.
EXAMPLE 12
In this case, magnetic capture beads (3~,m) from Spherotech and yellow-
fluorescent (2.1 ~,m) reporter beads from Molecular Probes were evaluated.
This study
was performed to determine the optimal MgCl2 concentration in the
hybridization
buffer for use in a genetic dual bead type assay.
A. Preparation of Capture and Reporter Beads
The magnetic capture beads and yellow-fluorescent reporter beads were each
covalently conjugated with DNA transport probes and DNA signal probes,
respectively. After conjugation, the beads were washed 1 X with hybridization
buffer
(0.1 M NaCI, 10 mM MgCl2, 1 mM EDTA, 50mM Tris, pH 7.5). The beads were
pretreated with 100~,g/ml salmon sperm DNA for 1 hour at room temp. The beads
were then washed 3X with wash buffer (145mM NaCI, 50 mM Tris, pH 7.5, 0.1 %
SDS,
0.05% Tween, 0.25% NFDM), 1X with hybridization buffer (145mM NaCI, lOmM
MgCl2, 1 mM EDTA, 1 OO~g/ml salmon sperm DNA, 50 mM Tris, pH 7.5). After
washing, the beads were resuspended in hybridization buffer.
137
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
B. Dual Bead Assay
The magnetic capture beads, as prepared in Part A, were divided into 24 l0ul
aliquots. Six sets of aliquots were diluted in hybridization solution
containing various
concentrations of MgCl2 (OmM, lOmM, 20mM, and 30mM). The target DNA mixture
was made in hybridization buffer containing varying amounts MgCl2, in the
following
concentrations: 1 OOfmole, 1 Ofmole, 1 fmole, 0.1 fmole, 0.01 fmole, Ofmole
(negative
control). The various target solutions were then mixed with their respective
bead
solutions according to the salt concentration of the hybridization buffer and
incubated
at 37°C for 2 hours to let target to hybridize to the 5' transport
probe on the capture
beads. After hybridization, the assay solutions were washed 3X with wash
buffer
containing the appropriate amount of MgCl2 required for each treatment group
(OmM,
1 OmM, 20mM, 30mM MgCl2), 1 X with the appropriate hybridization buffer. The
beads
were then resuspended in their respective hybridization buffers containing the
appropriate amount of MgCl2.
A 100p,1 volume of reporter beads in hybridization buffer containing various
MgCl2 concentrations (OmM, lOmM, 20mM, 30mM MgCl2), were added to the
appropriate assay solution, so that the same concentration of MgCl2 was
maintained
within the different treatment groups. These assay solutions were then
incubated at
37°C for 2 hours in rotating mixer. After incubation, the various
solutions were then
washed 6X with 0.5 ml of wash buffer containing the appropriate amount of NaCI
and
once with water also containing MgCl2 at concentrations equal to that in each
respective hybridization. The various dual bead solutions were then
resuspended in
250,1 water. The number of reporter beads bound were then quantified by the
fluorimeter at Excitation = 500nm, Emission = 530nm, Slit = Ex-2, Em-2, and an
integration of 0.1 second. The results from this assay are shown in above Fig.
61A.
The dual bead complex may also be quantifed using using an optical disc reader
as
shown in Figs. 1 and 2.
138
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
EXAMPLE 13
The following experiment was performed to determine the effect of using a
probe blocking agent to reduce the density of probes on beads on the
sensitivity of the
dual bead assay.
A. Preparation of Capture and Reporter Beads
The magnetic capture beads (3um Spherotech) and yellow-fluorescent reporter
beads (2.1 um Polysciences) were each covalently conjugated with DNA transport
probes and DNA signal probes, respectively. After conjugation, the beads were
washed 1X with hybridization buffer (0.1 M NaCI, 10 mM MgCl2, 1mM EDTA, 50mM
tris, pH 7.5). The beads were pretreated with 100~,g/ml salmon sperm DNA for 1
hour
at room temp. The beads were then washed 3X with wash buffer (145mM NaCI, 50
mM Tris, pH 7.5, 0.1 % SDS, 0.05% Tween, 0.25% NFDM), 1 X with hybridization
buffer (1 OOmM NaCI, 1 OmM MgCl2, 1 mMEDTA, 1 OO~,g/ml salmon sperm DNA, 50 mM
Tris, pH 7.5). After washing, the beads were resuspended in hybridization
buffer.
B. Probe Blocking
A biotinylated transport blocking probe was diluted to the following final
concentrations: 500pmole, 50pmole, 35pmole, 30pmoles. A 13,1 (2x10') volume of
magnetic beads were used for each tube (5 tubes total). A 5~,1 amount of
blocking
probe, as prepared above, and 32 ~,I hybridization buffer was added to each
tube. The
blocking probes and the transport probes were then hybridized for 2 hours at
37°C.
After hybridization, the beads were washed 3X with wash buffer (145nM NaCI,
50nM
Tris, pH 7.5, 0.05% Tween) and resuspended in 100,1 CDB (2% BSA, 50mM Tris-
HCI,
pH7.5, 145mM NaCI, 1.OmM MgCl2, 0.1 mM ZnCl2, 0.05% NaN3). The reporter beads
were prepared in a similar fashion using a biotinylated reporter blocking
probe.
C. Probe Density Determination on Beads after Blocking Agent Treatment
An aliquot of each set of bead solution prepared in Part B was incubated with
100,1 of S-AP (1420ng/ml) for one hour at 37°C then washed 3X with wash
buffer.
After washing, a 100p.1 volume of S-AP substrate, p-nitrophenyl phosphate, at
a
concentration of 3.7mg/ml in 0.1 M Tris, 2mM MgCl2, pH 10 was added to the
bead
solution. After allowing sufficient time for color development, the solution
was
139
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
analyzed using a spectrophotometer (OD @ 405 nm). The amount of blocking
probes
on the beads was calculated from the absorbance at 405nm.
D. Dual Bead Assay
The beads, as prepared in Part B, were washed and resuspended in
hybridization buffer containing 100~g/ml salmon sperm DNA and 5X Denhart's
solution. A solution target DNA mixture was prepared in the hybridization
buffer with
the following concentrations: Ofmole-control, 1 Ofmole, 1 fmole, 0.1 fmole,
0.01 fmole,
0.001fmole, 0.0001fmole. The target solutions were then mixed with equal
amounts of
capture and reporter beads and incubated at 37°C for 2 hours. The
capture and
reporter beads having been blocked with the same amount of probe blocking
agent for
each set of assay mixture, i.e., add l0ul l0fmole target to 100u1 reporter and
capture
bead solution each having been blocked with 50pmole blocking probe. After
hybridization, the assay solution was washed 3X with wash buffer and 1 X with
hybridization buffer and resuspended in hybridization buffer (containing 100
~,g/ml
salmon sperm DNA and 5X Denhart's Mix). The assay solution was concentrated
and
resuspended in 250.1 water. The number of reporter beads bound were then
quantified by the fluorimeter at Excitation = 500nm, Emission = 530nm, Slit =
Ex-2,
Em-2, and an integration time of 0.1 seconds.
EXAMPLE 14
The following experiment was performed to determine the optimal hybridization
incubation time of a genetic dual bead assay. The results from this experiment
are
shown above in Figs. 63 and 64.
A. Preparation of Capture and Reporter Beads
The beads used in this experiment were 25,1 of capture beads (3~,m
carboxylated magnetic particles at a concentration of 1.5x10' beads/p,l) with
5'
transport probes attached by covalent conjugation and 4001 of reporter beads
(2~,m
YF beads at a concentration of 6.6x106 beads/~,I). These beads were washed 3X
with
PBS and pretreated with 1 OO~g/ml salmon sperm DNA and 0.1 % CHAPS for 1 hour
at
room temperature. The beads were then washed 3X with wash buffer (145mM NaCI,
50mM Tris, pH 7.5, 0.05% Tween) and 1 X with hybridization buffer (50mM Tris-
HCI
140
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
pH 7.5, 1 OmM MgCl2, 0.1 M NaCI, 1 mM EDTA). The capture beads were then
resuspended in 250p.1 hybridization buffer, and the reporter beads in 400p.1
hybridization buffer.
B. Dual Bead Assay
A set of target DNA solutions are made in hybridization solution with the
following concentrations: 0 picomole, 1 picomole, 10 picomole, 100 picomole
target.
A test sample was prepared containing l0p,l capture beads, l5p,l of reporter
beads,
1 p,l salmon sperm DNA and 74 p,l target solution all in hybridization buffer.
Aliquots of
this test sample were analyzed at various incubation times 30min, 1 hr, 2hr,
3hr, 4hr,
and overnight. One set was incubated at 37° C without mixing and the
other set was
mixed on a rotating mixer.
The sample aliquots were washed 6X with 0.5m1 wash buffer (145mM NaCI,
50mM Tris, pH 7.5, 0.05% Tween, 0.1 % SDS, 0.25% NFDM) and then resuspended
in 202 ~,I PBS. The number of reporter beads bound were then quantified by the
fluorimeter at Excitation = 450nm, Emission = 480nm, Slit = Ex-1.365, Em-1.05,
integration time = 0.1 second.
EXAMPLE 15
After formation of the dual bead complexes, as discussed in connection with
Fig. 67A the reporter beads can be separated from the capture beads in a DNA
dependent procedure. The dual bead complexes are subjected to DNAases
(enzymes that specifically cut DNA). This treatment separates the reporter
beads
from the capture beads by cutting the DNA that holds them together. Thus, the
non-
target mediated dual beads will not be affected. The reporter beads that are
released
after the DNAse treatment are indicative of the amount of target DNA present
in the
sample. In this experiment, the DNAseI effect in a dual bead assay was
evaluated.
The results from this experiment is shown in Figs. 69 and 70.
A. Dual Bead Assay
The dual bead assay was carried out as described previously in Example 1,
Part A. Briefly, the assay is comprised of 3pm magnetic capture beads
(Spherotech,
Libertyville, IL) coated with covalently attached transport probes; 2.1 Nm
fluorescent
141
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
reporter beads (Molecular Probes, Eugene, OR) coated with a covalently
attached
reporter probes, and target DNA molecules of interest. In this example, the
target
DNA is a synthetic 80 oligonucleotides long. The transport probes and reporter
probes are 40 nucleotides in length and are complementary to the target DNA
but not
to each other.
The specific methodology employed to prepare the assay involved treating
1 x10' capture beads and 2x10' reporter beads in 100pg/ml salmon sperm DNA for
1
hour at room temperature. This pre-treatment will reduce the non-specific
binding
between the capture and reporter beads in the absence of target DNA. The
capture
beads were concentrated magnetically with the supernatant being removed. A
100N1
volume of the hybridization buffer (0.2M NaCI, 1 mM EDTA, 1 OmM MgCl2, 50mM
Tris-
HCI, pH 7.5 and 5X Denhart's mix, lONg/ml denatured salmon sperm DNA) were
added and the beads were resuspended. Various 'concentration of target DNA
ranging from 1, 10, 100 and 1000 femtomoles were added to the capture bead
suspensions. The beads suspension was incubated while mixing at 37 degrees
Centigrade for 2 hours. The beads were magnetically concentrated and the
supernatant containing unbound target DNA was removed. One hundred microliters
of wash buffer (145mM NaCI, 50mM Tris, pH 7.5, 0.1 % SDS, 0.05% Tween, 0.25%
NFDM (Non Fat Dried Milk), lOmM EDTA) was added and the beads were
resuspended. The beads were magnetically concentrated and the supernatant was
again removed. The wash procedure was repeated twice.
A 2x10' amount of reporter beads in 100p1 hybridization buffer (0.2M NaCI,
1 mM EDTA, 1 OmM MgCl2, 50mM Tris-HCI, pH 7.5 and 5X Denhart's mix, 1 Og/ml
denatured salmon sperm DNA) were then added to washed capture beads. The
beads were resuspended and incubated while mixing at 37 degrees Centigrade for
an
additional 2 hours. After incubation, the capture beads were concentrated
magnetically, and the supernatant containing unbound reporter beads were
removed.
One hundred microliters of wash buffer (145mM NaCI, 50mM Tris, pH 7.5, 0.1 %
SDS,
0.05% Tween, 0.25% NFDM (Non Fat Dried Milk), lOmM EDTA) was added and the
beads were resuspended. The beads were magnetically concentrated and the
supernatant was again removed. The wash procedure was repeated twice.
142
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
B. DNAseI Assays
DNAseI was selected for this purpose because it is not sequence specific.
Following washing, the dual bead complexes were resuspended in 87.5pL water.
Ten
(10) units of DNAseI (2.5pL) and 10pL of DNAseI reaction buffer (40mM Tris-
HCI,
1 OmM MgS04, 1 mM CaCl2) were added to the re-suspended beads. The digestion
reaction was carried out for 1 hour at 37°C. After digestion, the
capture beads were
concentrated magnetically and the supernatant containing reporter beads was
removed. The magnetic capture beads were washed 2 times with 100N1 water. The
washed water was combined with the supernatant. The number of reporter beads
was quantified by the fluorimeter Fluoromax-2 at excitation 500nm, emission
530nm
and slit sizes 2Ø See Figs. 69 and 70 for results from this experiment.
Alternatively,
the number of fluorescent reporter beads can be quantified by the bio-disc
reader as
described above.
EXAMPLE 16
In this example, the dual bead complexes are separated by physical or
chemical treatments. The dual bead assay was carried out as described above in
Example 15. Following washing, the bead products were washed 5 times with the
wash buffer (145mM NaCI, 50mM Tris, pH 7.5, 0.1 % SDS, 0.05% Tween, 0.25%
NFDM (Non Fat Dried Milk), lOmM EDTA) and divided into four sets.
1. Control: No treatment, the beads were washed twice with 200pL wash buffer.
2. Acid Wash: The beads were washed twice with 200pL wash buffer
containing 0.1 M acetic acid (pH 4).
3. Basic Wash: The beads were washed twice with 200pL wash buffer
containing 0.1 M sodium bicarbonate (pH 9).
4. Urea: The beads were washed twice with 200pL wash buffer containing 7M
urea.
After the physical or chemical treatment, the capture beads were concentrated
magnetically, and the supernatant containing released reporter beads was
saved.
The beads were washed 3 times with wash buffer. The wash was also saved. The
magnetic capture beads were re-suspended in 400pL wash buffer. The amount of
reporter beads in the supernatants and in the solution of capture beads were
quantified by the fluorimeter Fluoromax-2 at Ex = 500nm, Slit = 2.0; Em =
530nm, Slit
143
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
= 2Ø Alternatively, the number of fluorescent reporter beads can be
quantified by the
bio-disc reader as described above.
As evident by this example, high pH washes can dissociate the reporter beads
from the capture beads at low target concentrations. As shown by the
experimental
results in Fig. 72, the basic wash completely dissociated the reporter beads
from
capture beads at low target concentrations.
The results of this experiment also established that a 7M urea treatment
efficiently dissociates reporter beads from capture beads without
significantly
compromising the sensitivity. As illustrated by the experimental results
present in the
bar graphs of Figs. 73A and 73B, urea treatment efficiently dissociates
reporter beads
from capture beads.
EXAMPLE 17
In Examples 15 and 16 discussed above, the target DNA is single stranded.
When clinical samples are used, the DNA is double stranded and therefore the
hybridization buffer requires a denaturing reagent such as guanidine
isothiocyanate.
The concentration of the denaturing reagent used in the assay is a major
contributor in
the specificity and sensitivity of the dual bead assay. In this example, the
dual bead
assay to detect HSV was carried out in the presence of 1.5M guanidine
isothiocyanate.
A. Preparation of Capture Beads
The dual bead assay is comprised of 3pm magnetic capture beads
(Spherotech, Libertyville, IL) coated with covalently attached 5' HSV
transport probes
and 2.1 ~,m fluorescent reporter beads from Molecular Probes (Eugene, OR)
conjugated to the 3' HSV reporter probes and target DNA molecules of interest.
In
this example, the target was a double-strand PCR product containing the HSV
gene
sequence, amplified for 30 cycles and Qiagen column purified. The transport
probes
and reporter probes are 40 nucleotides in length and are complementary to the
target
DNA but not to each other.
The specific methodology employed to prepare the assay involved treating
1 x10' capture beads and 2x10' reporter beads in 1 OOpg/ml salmon sperm DNA
for 1
hour at room temperature. This pre-treatment will reduce the non-specific
binding
144
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
between the capture and reporter beads in the absence of target DNA. The
capture
beads were concentrated magnetically with the supernatant being removed. The
capture beads were resuspended in 600~,L of hybridization buffer (1.5 GuSCN,
8mM
EDTA, 40mM Tris, pH7.5) containing 5X Denhart's mix, and 10~g/ml salmon sperm
DNA.
B. Preparation ~f Target DNA
The target was a double-strand PCR product, amplified for 30 cycles and
Qiagen column purified. The target was diluted to appropriate concentrations,
and
heated at 95°C for 5 minutes to denature the double strand then quickly
chilled on ice.
C. Hybridization with the Target DNA
A total of 12.5~,L of the chilled target was added to the 100~,L of the
pretreated
capture beads. Various concentrations of target DNA ranging from 0, 1 O-16, 10-
'5, 10-
'4, 1O-'3, and 10-'2 moles were added to the capture bead suspensions. The
beads
suspension was incubated while mixing at 37 degrees Centigrade for 2 hours.
The
beads were magnetically concentrated and the supernatant containing unbound
target
DNA was removed. One hundred microliters of wash buffer (145mM NaCI, 50mM
Tris, pH 7.5, 0.1 % SDS, 0.05% Tween, 0.25% NFDM (Non Fat Dried Milk), 1 OmM
EDTA) was added and the beads were resuspended. The beads were magnetically
concentrated and the supernatant was again removed. The wash procedure was
repeated twice.
D. Dual Bead Assay
A 2x107 amount of reporter beads in 100p1 hybridization buffer (1.5 GuSCN,
8mM EDTA, 40mM Tris, pH7.4) containing 5X Denhart's mix and lONg/ml denatured
salmon sperm DNA ) were then added to washed capture beads. The beads were
resuspended and incubated while mixing at 37 degrees Centigrade for an
additional 3
hours. After incubation, the capture beads were concentrated magnetically, and
the
supernatant containing unbound reporter beads were removed. One hundred
microliters of wash buffer (145mM NaCI, 50mM Tris, pH 7.5, 0.1 % SDS, 0.05%
Tween, 0.25% NFDM (Non Fat Dried Milk), lOmM EDTA) was added and the beads
145
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
were resuspended. The beads were magnetically concentrated and the supernatant
was again removed. The wash procedure was repeated twice.
E. Quantification of Target DNA
The dual bead complexes were resuspended in 250N1 PBS and the amount of
target was quantified by fluorescence measurement of the reporter beads using
the
fluorimeter Fluoromax-2 at Ex = 500nm, Slit = 2.0; Em = 530nm, Slit = 2Ø
Alternatively, the number of fluorescent reporter beads can be quantified by
use of the
optical bio-disc reader as described above.
EXAMPLE 1 ~
The following example illustrates a dual bead assay carried out on a
magnetically writable and erasable analysis disc such as the magneto-optical
bio-disc
110 discussed in conjunction with Fig. 37.
In this example, the dual bead assay is carried out to detect the gene
sequence
DYS which is present in male but not female. The assay is comprised of 3pm
magnetic capture beads (Spherotech, Libertyville, IL) coated with covalently
attached
transport probes; 2.1 Nm fluorescent reporter beads (Molecular Probes, Eugene,
OR)
coated with a covalently attached sequence specific for the DYS gene, and
target
DNA molecules containing DYS sequences. The target DNA is a synthetic 80
oligonucleotides long. The transport probes and reporter probes are 40
nucleotides in
length and are complementary to the DYS sequence but not to each other.
The specific methodology employed to prepare the assay involved treating
1 x10' capture beads and 2x10' reporter beads in 1 OOpg/ml salmon sperm DNA
for 1
hour at room temperature. This pre-treatment will reduce the non-specific
binding
between the capture and reporter beads in the absence of target DNA.
After pretreatment with salmon sperm DNA, the capture beads are loaded
inside the MO bio-disc via the injection port. The MO bio-disc contains
magnetic
regions created by the magneto optical drive. The capture beads thus are held
within
specific magnetic regions on the MO bio-disc.
The sample containing target DNA and reporter beads in 200p1 hybridization
buffer (0.2M NaCI, 1 mM EDTA, 1 OmM MgCl2, 50mM Tris-HCI, pH 7.5 and 5X
Denhart's mix, l0pg/ml denatured salmon sperm DNA) is then added to the MO bio-
146
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
disc via the injection port. The injection port is then sealed. The magnetic
field is
released. The disc is rotated at very low speed (less than 800rpm) in the
drive to
facilitate hybridization of target DNA and reporter beads to the capture
beads. The
temperature of the drive is kept constant at 33 degrees Centigrade. After 2
hours of
hybridization, the magnetic field is created by the magneto optical drive. At
this stage,
only magnetic capture beads, unbound or as part of a dual bead complex, remain
on
the MO bio-disc. Unbound target and reporter beads are directed to a waste
chamber
by any of the mechanisms described above. Two hundred microliters of wash
buffer
(145mM NaCI, 50mM Tris, pH 7.5, 0.1 % SDS, 0.05% Tween, 0.25% NFDM (Non Fat
Dried Milk), lOmM EDTA) is then added. The magnetic field is released and the
disc
is rotated at low speed (less than 800rpm) for 5 minutes to remove any non-
specific
binding between the capture beads and reporter beads. The magnetic field is
then
reapplied. The wash buffer is directed to the waste chamber by any of the
mechanisms described above. The wash procedure is repeated twice.
At this stage, only magnetic capture beads, unbound or as part of a dual bead
complex, remain. The magnetic field is released and the dual bead complexes
are
directed to a detection chamber. The amount of target DNA captured is then
enumerated by quantifying the number of capture magnetic beads and the number
of
reporter beads since each type of bead has a distinct signature as illustrated
above in
Figs. 28A, 28B, 29A, and 29B.
EXAMPLE 19
In this example, a dual bead assay using the multiplexing techniques described
above in connection with Figs. 32 and 37 is carried out on a magnetically
writable and
erasable analysis disc such as the MO bio-disc 110 discussed with reference to
Fig.
37.
The dual bead assay is carried out to detect 2 or more DNA targets
simultaneously. The assay is comprised~of 3pm magnetic capture beads
(Spherotech,
Libertyville, IL). One population of the magnetic capture beads is coated with
transport probes 1 which are complementary to the DNA target 1. Another
population
of the magnetic capture beads is coated with transport probes 2 which are
complementary to the DNA target 2. Alternatively, 2 or more different types of
magnetic capture beads may be used. There are two or more distinct types of
147
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
reporter beads in the assay. The reporter beads may differ by chemical
composition
(for example silica and polystyrene) and/or by size. One type of reporter
beads is
coated with reporter probes 1, which are complementary to the DNA target 1.
The
other reporter beads are coated with reporter probes 2, which are
complementary to
the DNA target 2. Again, the transport probes and reporter probes are
complementary
to the respective targets but not to each other.
The specific methodology employed to prepare the dual bead assay
multiplexing involved treating 1 x10' capture beads and 2x10' reporter beads
in
100Ng/ml salmon sperm DNA for 1 hour at room temperature. This pre-treatment
will
reduce the non-specific binding between the capture and reporter beads in the
absence of target DNA.
After pretreatment with salmon sperm DNA, the capture beads are loaded in
the MO bio-disc. The magnetic field is applied to create distinct magnetic
zones for
specific capture beads. The capture beads can be held on the MO bio-disc at a
density of 1 capture bead per 10Nm2. The surface area usable for bead
deposition on
the MO bio-disc is approximately 3 x 109Nm2. The capacity of the MO bio-disc
for 3Nm
beads at the given density is about 3 x 10$ beads.
The sample containing the targets DNA of interest is mixed with different
types
of reporter beads in 200p1 hybridization buffer (0.2M NaCI, 1 mM EDTA, 1 OmM
MgCl2,
50mM Tris-HCI, pH 7.5 and 5X Denhart's mix, lONg/ml denatured salmon sperm
DNA)
and added to the MO bio-disc via the injection port. The injection port is
then sealed.
The magnetic field is released. The disc is rotated at very low speed (less
than
800rpm) in the drive to facilitate hybridization of targets DNA and reporter
beads to the
different types of capture beads. The temperature of the drive is kept
constant at 33
degrees Centigrade. After 2 to 3 hours of hybridization, the magnetic field is
regenerated by the magneto optical drive. At this stage, only magnetic capture
beads,
unbound or as part of dual bead complexes, remain on the MO bio-disc. Unbound
targets and reporter beads are directed to a waste chamber by any of the
mechanisms
described above. Two hundred microliters of wash buffer (145mM NaCI, 50mM
Tris,
pH 7.5, 0.1 % SDS, 0.05% Tween, 0.25% NFDM (Non Fat Dried Milk), lOmM EDTA) is
then added. The magnetic field is released and the disc is rotated at low
speed (less
than 800rpm) for 5 minutes to remove any non-specific binding between the
capture
beads and reporter beads. The magnetic field is then reapplied. The wash
buffer is
148
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
directed to the waste chamber by any of the mechanisms described above. The
wash
procedure is repeated twice.
At this stage, the magnetic field is released and the dual bead complexes are
directed to a detection chamber. The amount of different types of target DNA
can be
enumerated by quantifying the number of corresponding capture magnetic beads
and
reporter beads since each type of bead has a distinct signature as shown above
in
Figs. 28A, 28B, 29A, and 29B.
EXAMPLE 20
A. Separation of T Helper Cells in AIDS Patients using Monoclonal CD4
Antibodies
Attached to Paramagnetic Beads
A sample (whole blood or mononuclear cells) is injected into the
mixing/loading
chamber where it is mixed with anti-CD4 coated paramagnetic beads (bio-
magnetic
particles). After a 15-minute incubation, to allow binding of the paramagnetic
beads to
CD4+ cells, magnetic domains are created within the flow channel or analysis
chamber using the laser in the MO drive. The tagged CD4+ cells will then bind
to
these magnetic domains such that when the disc is rotated at a pre-determined
speed
and time untagged cells and cellular components will move down the flow
channel
toward a waste chamber. The number of CD4+ cells bound to the magnetic domains
is then quantified by the MO reader. The number of CD4+ cells thus determined
will
be indicative of the heath status of the patient.
B. Manipulation of the Seleeted T helper Cells
After the CD4+ cells have been separated from other cellular components, the
magnetic domains are then erased. The disc is then rotated at a pre-determined
speed and direction to cause the newly released CD4+ cells to move to a flow
channel
into different testing chambers where the cells could be exposed to different
drug
treatments that could reduce their susceptibility to HIV destruction. The
design of the
fluidic circuits and the speed and direction of rotation of the disc can be
determined by
one of skill in the art by no more than routine experimentation.
149
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
EXAMPLE 21
Detection of Cancer Cells using Carcinoma Marker Antibodies such as MOC-31 or
NrLu10 Attached to Paramagnetic Beads
A sample (single cell suspension prepared for example from the biopsied
tissue) is loaded into the mixing chamber of the MO bio-disc or MO analysis
disc.
Paramagnetic beads are then coated with MOC-31 capture antibodies to thereby
form
bio-magnetic beads. These bio-magnetic beads are then loaded in to the mixing
chamber containing the sample. After a 15-minute incubation, magnetic domains
are
created within the mixing chamber in the MO disc to bind magnetically tagged
or
labeled cells. The disc is then rotated at a pre-determined speed and duration
to
remove unlabelled cells. The labeled or tagged carcinoma cells, which are
bound to
the paramagnetic fields, will be immobilized in the magnetic field whereas
other cells
will move down the flow channel toward a waste chamber. The number of cancer
cells can be quantified by the reader. Further details related to
quantification of
particles and cells within fluidic circuits are disclosed in, for example,
commonly
assigned and co-pending U.S. Patent Application Serial No. 10/241,512 entitled
"Methods for Differential Cell Counts Including Related Apparatus and Software
Performing Same" filed September 11, 2002; and U.S. Patent Application Serial
No.
10/279,677 entitled "Segmented Area Detector for Biodrive and Methods Relating
Thereto" filed October 24, 2002. Both of which are herein incorporated by
reference in
their entireties.
The magnetic field is turned off and the rotation of the disc will cause the
identified cancels cells to move to various testing chambers on the MO disc
where the
cancer cells will be exposed to different anticancer drugs. The efficacy of
the drug can
be determined by quantifying the number of live cells or dead cells since
apoptotic
cells have a different signature trace than live cells.
150
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
EXAMPLE 22
Detection of Tuberculosis (TB) using Magnetic Beads Coated with Specific TB
Probes
A sample containing DNA fragments is injected into the mixing/loading chamber
where it is mixed with paramagnetic beads or bio-magnetic particles coated
with DNA
probes for one or more species of TB. The sample and the probes are then
allo~ived to
hybridize for about 1 hour in the disc with intermittent mixing by rotating
the disc
clockwise then counter clockwise in the reader. Magnetic domains are then
created in
the mixing chamber where the magnetic beads are captured. The mixing chamber
is
then washed to remove unbound DNA fragments. The magnetic domains are then
erased and the disc is rotated to cause the magnetic particles to be released
and
move to an analysis chamber where the captured specific TB DNA sequence could
be
amplified for downstream applications.
EXAMPLE 23
The interactions of different neurons in the brain to generate specific action
potentials are still unclear. Interactions of specific neurons in the brain
can be studied
on the MO disc. In various incubation chambers, neurons carrying different
cellular
markers can be isolated using paramagnetic beads coated with antibodies
directed
against specific cellular surface markers.
The neurons isolated in one chamber can then be manipulated, moved and
mixed with the neurons isolated in a different chamber using magnetic field on
the
disc. To determine whether there is "communication" between the 2 types of
neurons,
the production of an action potential (K+ or Ca 2+) could be monitored.
Concluding Summary
All patents and other publications mentioned in this specification are
incorporated herein in their entireties by reference.
While this invention has been described in detail with reference to certain
preferred embodiments and technical examples, it should be appreciated that
the
present invention is not limited to those precise embodiments or examples.
Rather, in
view of the present disclosure, which describes the current best mode for
practicing
151
CA 02468245 2004-05-25
WO 03/046511 PCT/US02/38021
the invention, many modifications and variations would present themselves to
those of
skill in the art without departing from the scope and spirit of this
invention.
For example, any of the off-disc preparation procedures may be readily
performed on-disc by use of suitable fluidic circuits employing the methods
described
herein. Also, any of the fluidic circuits discussed in connection with the
reflective and
transmissive discs may be readily adapted to the MO bio-disc. In addition, the
scope
of the present invention is not solely limited to the formation of only dual
bead
complexes. The methods and apparatus hereof may be readily applied to the
creation
of multi-bead assays. For example, a single capture bead may bind multiple
reporter
beads. Similarly, a single reporter bead may bind multiple capture beads.
Furthermore, linked chains of multi-bead or dual bead complexes may be formed
by
target mediated binding between capture and reporter beads. The linked chains
may
further agglutinate to thereby increase detectability of a target agent of
interest.
The scope of the invention is, therefore, indicated by the following claims
rather than by the foregoing description. All changes, modifications, and
variations
coming within the meaning and range of equivalency of the claims are to be
considered within their scope.
152