Note: Descriptions are shown in the official language in which they were submitted.
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
TRIOXANE DERIVATIVES
The present invention relates to certain compounds containing a trioxane
moiety
that have potent antimalarial activity and antiturnour activity.
Artemisinin (1), which is also known as qinghaosu, is a tetracyclic 1,2,4-
trioxane
occurring in Artemisia au.nua. Artemisinin and its derivatives
dihydroartemisinin (DHA)
(2), artemether (3) and sodium artesunate (4) are used routinely in the
treatment of
malaria and have been found to be particularly effective against cerebral
malaria.
4 5 H a H = H = H _
.,,~Oz - ,,,v0 ,,,v0 .,v0
O ~Oli, 8a 8 O Oim O ~ O O/~~ ~ i
O/~.
H =~2~2a ~.,H H __ '~iH H .,H H ~~H
O s O O~~ O _
to '
O OH OCH3 Na+-O II
O
1 2 3 4
Different mechanisms of action have been proposed to account fox the
antimalarial action of artemisinin and its derivatives (Posner et al., J. Am.
Chem. Soc.
1996, 118; 3537; Posner et al., J. Am. Chem. Soc. 1995, 117, 5885; Posner et
al., J. Med.
Chem. 1995, 38, 2273). Whilst the mechanism of action of artemisinin as an
antimalarial
has not been unequivocally established, it has been shown that the peroxide
linkage is
essential for antimalarial activity.
Certain artemisinin derivatives containing a peroxide moiety have also been
tested
for biological activity other than antimalarial activity. For example, the
cytoxicity to
Ehrlich ascites tumour cells of artemisinin, dihydroartemisinin, artemisitene,
arteether,
ethylperoxyartemisitene and an ether dimer of artemisinin has been
demonstrated
(Beekman et al., PlaytotlZer. Res., 1996, 10, 140; Woerdenberg et al., J. Nat.
Prod., 1993,
56, 849).
1
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
Selective cancer cell cytoxicity from exposure to dihydroartemisinin and
holotransferrin, a non heme iron transport protein saturated with iron, has
also been
disclosed (Lai et al., Ca~icer Lett., 1995, 91, 41 and US Patent No. 5578637),
with the
drug combination being approximately 100 times more effective on molt-4 cells
than
lymphocytes.
It is known that some biologically active molecules contain chemical groups
which enable them to bind to DNA. The method by which DNA binding occurs
depends
upon the overall structure of the molecule and the nature of the chemical
groups
contained within the molecule. For instance, the major and minor grooves of
the double
helical DNA are occupied by water under physiological conditions. However,
certain
oligopeptidic compounds such as netropscin and disamycin can displace water
molecules
and form strong hydrogen bonds with hydrophilic groups along the DNA strands.
Alternatively, some compounds contain groups which are capable of
intercalating
with DNA. Intercalators are compounds which insert between the bases of DNA.
Well
characterised examples of intercalators are provided by anthracyclines, such
as
adriamycin and daunomycin, which are used for the treatment of cancer, and
acridines,
such as amascrine, which is used for treating acute leukaemia and malignant
lymphomas.
The antiturnour activity is associated with the intercalating property of
these compounds.
Naturally occurring polyamines, such as the tetra-amine spermine (5) and the
triamine spermidine (6) occur in cells at micromolar concentrations, and may
even rise to
millimolar levels in certain cancer cells (Tabor & Tabor, Afzn. Rev. Biocheni,
1984, 53,
749). The biosynthetic building blocks for these and closely related
polyamines are the
alpha amino acids ornithine and lysine, affording the diamines putrescine (7)
(1,4-
diaminobutane) and cadaverine (8) (1,5-diamino pentane) respectively.
2
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
H2N~N~N~.NH2 H2N~H~NH2
H
6
H2N~NH2 H2N NH2
7 8
In recent years, it has been established that polyamines and polyamine amides
have potential as novel therapeutic lead compounds in the design of anti-
tumour agents.
Other workers, (Bergeron et al., Med. Chem., 1987, 31, 1183, Bergeron et al.,
CahceY
Res., 1989, 49, 2959) have addressed the usefulness of polyamines in cancer
chemotherapy. In more recent studies, polyarnines have been identified as
novel leads for
the design of antidiarrhoeal agents and antimalarials and as ion chelators.
There is ever increasing realisation of the biological effects of polyamines,
particularly in cellular pxocesses, including growth and replication (Heby &
Persson,
Tre~.ds Bio. Sci., 1990, 15, 153). Thus, it is not surprising that polyamine
conjugates
continue to be the focus of significant attention as potential anticancer
agents. It has been
shown that a polyamine transporter specifically mediates the uptake of
extracellular
polyamines into cells (Seiler & Dezeure, Int. J. Biochem., 1990, 22, 211), and
rapidly
dividing tumour cells require large quantities of polyamines. Consequently,
this
polyamine transporter is up-regulated in tumour cells more so than in normal
cells (Seiler
et al., CatzceY Res., 1990, 50, 5077). Polyamines bind to DNA via either the
major or the
minor groove (Rodger et al., Biopolymers, 1994, 34, 1583, Rogers et al., Bio,
org. Med.
Chem., 1995, 3, 861) and it is thought that endogenous polyamines also effect
chromatin
stability and structure (Basu et al., Bioclzem.J., 1992, 282, 723). Taking
these aspects into
account when designing polyamine based anticancer agents, there exists a
potential
uptake mechanism with selectivity for cancer cells (Cohen & Smith, Bioclzem.
Soc.
TYans., 1990, 18, 743) and two possible modes of cytotoxicity. This
cytotoxicity may be
mediated either by DNA binding and hence disruption of transcription
(Feuerstein et al.,
3
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
Nucleic Acids Res., 1990, 18, 1271), or by interference with polyamine
biosynthetic
pathways thereby modulating the cellular concentrations of endogenous
polyamines.
To date, some of the simplest and most effective synthetic polyamines to
display
anticancer activity have been developed by Porter, Bergeron and their co-
workers. They
initially found activity with spermidine and spermine analogues which are N-
alkylated
(Porter et al., Cancer Res., 1982, 42, 4072, Porter et al., Cancer Res., 1985,
45, 2050).
Further studies showed the best analogues to be tetra-amines which have been
bis-
ethylated on the terminal, primary amines (e.g. compounds 9, 10 and 11)
(Bergeron et al.,
J. Med. Chern., 1987, 31, 1183, Porter et al., Cancer Res., 1987, 47, 2821).
H
/~N~N~N~Nw/ /~H~H~N~H~
H H H
9 DENSPM 10 DENSPM
H
~N~N~N N~
H H H
11 DENSPM
These compounds are recognised and taken into cells by the polyamine
transporter. Once inside the cells, they deplete intracellular pools by down-
regulating the
enzyme ornithine decarboxylase (ODC), the first enzyme in the polyamine
synthesis
pathway, and up regulating the spermine-spermidine N1-acetyltransferase (SSAT)
enzyme which works in the back conversion pathway (Bergeron et al., Cancer
Res.,
1989, 49, 2959). The cytotoxic effects of the analogues DENSPM (9), DESPM (10)
and
DEHSPM (11) in the in vitro culture of L1210 cells, over 96 h, were l.3uM,
0.2uM and
0.06~.M respectively.
Many polyamine or polyamide moeities, for example desferrioxamine B (12), are
low molecular weight ion chelating compounds. They facilitate iron
solublization and
transport.
4
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
12
Another approach to the development of antitumour compounds is the covalent
linking of cytotoxic agents, whose activity is mediated through direct
interaction with
DNA, to a polyamine. The resulting conjugate will be transported into the cell
through
the polyamine transport mechanism (if recognised) and the polyamine should
further aid
DNA binding of the cytotoxic component as its DNA target site. In line with
this, Cullis
has demonstrated that polyamines conjugated to the nitrogen mustard
chlorambucil
increase the efficiency of DNA alkylation at the N7 of guanine by factors in
the range of
103 to 104 (Cullis et al., J. Am. Chem. Soc., 1995, 117, X033).
It has now been discovered that artemisinin and synthetic trioxane derivatives
can
be chemically modified by the attachment of a polyamine residue to form
analogues of
arternisinin and synthetic trioxane derivatives which exhibit antimalarial,
cytotoxic and
antitumour activity.
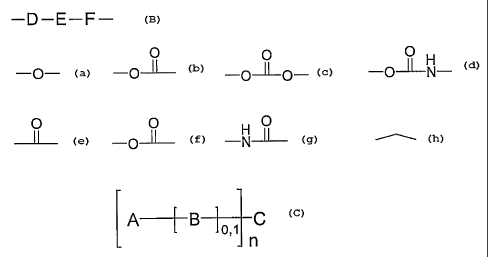
According to a first aspect of the present invention therefore there is
provided a
compound of general formula 13
A--f -B o,~ C
n
13
or a pharmaceutically acceptable salt thereof,
in which:
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
n = an integer of from 1 to 4;
A represents a trioxane-containing residue;
B represents a group having the general formula:
-D-E-F-
in which:
D is linked to A and represents an atom or group selected from the
following:
O O O H
-o- -oJ.l- -o~o- -p-L~-N- ,
E represents a bivalent, optionally substituted organic radical; and
F is linked to C and represents a group selected from the following:
O O O
-N-ll- ~ ; and
C represents a group containing at least two nitrogen atoms.
In a preferred embodiment of the present invention, A represents the following
trioxane-containing residue:
u....
H
With regard to the optionally substituted organic radical E, this preferably
comprises an organic radical of 2-50 carbon atoms, more preferably 1-20 carbon
atoms.
The optionally substituted organic radical E may comprise, for example, an
optionally
6
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
substituted alkyl, aryl, acyl, heteroalkyl or heteroacyl group. The organic
radical E may
optionally be substituted by groups including, but not limited to, primary,
secondary and
tertiary amines; halogen-containing groups, such as bromide, chloride and
fluoride;
alcohols and derivatives thereof, including ethers and esters; and carboxylic
acids and
derivatives thereof, including esters and amides. Examples of organic radicals
comprising Group E include -CH2-CHZ-, p-phenylene and pyridine.
With regard to group C, this may be, for example, a natural or synthetic
polyamine residue and is preferably of 2-50 carbon atoms. Preferably also,
group C
comprises. at least two amino groups, each of which is independently a primary
or
secondary group, after linking to group F through the same or different amino
groups of
the polyamine.
Those salts comprising pharmaceutically acceptable salts as referred to herein
will
be readily apparent to a skilled person. These salts include, but are not
limited to acetate,
adipate, besylate, bromide, camsylate, chloride, citrate, edisylate, estolate,
fumarate,
gluceptate, gluconate, glucuronate, hippurate, hyclate, hydrobromide,
hydrochloride,
iodide, isethionate, lactate, lactobionate, maleate, mesylate, methylbromide,
methylsulfate, napsylate, nitrate, oleate, pamoate, phosphate,
polygalacturonate, stearate,
succinate, sulfate, sulfosalicylate, tannate, tartrate, terephthalate,
tosylate, triethiodide,
benzathine, calcium, diolamine, meglumine, olamine, potassium, procaine,
sodium,
tromethamine, and zinc.
Apart from imparting selectivity against cancer cells, the incorporation of
amine
functionality into an endoperoxide was seen as a useful strategy for enhancing
antimalarial activity, since the amine peroxide should be concentrated in the
acidic
vacuole of the malaria parasite by ion-trapping (Vennerstrom et al., J. Med.
Chern., 1989,
32, 64, O' Neill et al., J. Med. Chem., 1996, 39, 4511). Intraparasitic
accumulation in the
haem "rich" food vacuole is considered to be key to the action of all basic
quinoline
antimalarial agents (O' Neill, Phartn. e~ Therapeutics, 1998, 77, 29). For
example,
chloroquine is a dibasic drug with pI~s of 8.1 (quinoline ring nitrogen) and
10.2
7
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
(diethylamino side chain) and accumulates in acidic vesicles to the square of
the
monobasic antimalarials such as mefloquine. Experiments on the pH gradient
between
the external medium and the parasite food vacuole have shown that the value is
around
2.2. On this basis, Ginsburg and co-workers suggested that chloroquine would
be
expected to accumulate 2.5 x 104 compared with 160 fold for the mono-basic
antimalarials such as mefloquine (Ginsburg et al., Biochem. Phar»zacol., 1989,
38, 2645).
It follows that the introduction of two basic amino groups into an artemisinin
derivative
would be expected to increase significantly the cellular accumulation of drug
in the
ferrous rich parasite food vacuole. This approach should provide analogues
with
increased antimalarial potency, since more drug will be available for
reductive
endoperoxide bioactivation to radical species.
One preferred embodiment of the present invention provides compounds having
the structure 13n.
13n, G = substituted aryl ring
Specific examples of such compounds are included in Chart 1.
s
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
CHART 1- C-10-Ether Linked Diamino Analogues
N
R1 / \ N I i
R2
13a-13d (R2= H, N02, F, CF3)
.""O
O O""..
R = / \ ~N~
N
~O~R_1 13e
R1= \ / ~~.R2
~N ~
13f-13j (R2=H,p-N02,p-Cl,rn-CFg,
p-F
This series of preferred compounds was prepared as shown in Scheme 1.
Dihydroartemisinin (2) was coupled with 1,4-benzenedimethanol to give the
corresponding alcohol (12a) in high yield with excellent diastereoselectivity
((3/oc 5:1).
The alcohol was then converted into the mesylate (12b) in high yield by
treatment with
mesyl chloride and triethylamine. The key mesylate was then allowed to react
with a
range of diamino nucleophiles to provide compounds (13a-13d). As shown in
Scheme 1,
DHA was also coupled with 1,3-benzenedimethanol to provide the alcohol which
could
then be transformed into target analogues (13f-13j) using the same chemistry
described
for the para substituted analogues.
9
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
Scheme 1
~N w
HN~ I ~
_ R2
1. BF3.Et20
2 ~ --
\ ene
HO OH
2. MsCl
12a (R3= H) 13a-13d (R2= H, N02, F, CF3)
12b (R3= Ms)
1. BF3. Et20 -
HO / \ benzene
OH
HN N I \ Ph
2. MsCl
Benzene 13e
_ ~N~ H
/ R2
.. O
O 0.,..
benzene
O~ / \ ~N
\~N~ I = R2
M ~s
13f 13j (R2= H, p-NO2, pCl, m-CF3, p-F)
Thus, and in accordance with a further aspect of the present invention there
is provided a
process for the production of a compound of general formula 13 as hereinbefore
defined,
said process comprising the steps of coupling dihydroartemisinin with
benzenedimethanol, converting the resultant alcohol into the corresponding
sulfonate by
treatment with a sulfonyl halide, and reacting said sulfonate with a diamino
nucleophile.
Most preferably, the resultant alcohol is converted into the corresponding
mesylate by treatment with mesyl chloride.
to
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
This process is particularly advantageous with regard to the production of
trioxane
derivatives of the type exemplified by compounds 13a-13j.
Further preferred compounds of the present invention were prepared according
to
Schemes 2 to 5 as shown below. Dihydroartemisinin (2) was coupled with a
variety of
alcohol methyl esters. These were then hydrolysed to give carboxylic acids,
which were
coupled with a number of polyamines and amines. Under standard conditions
compounds
with two artemisinin groups in the molecule were formed. If an excess of amine
was used
at 0°C, this gave the desired monomers.
.",o ..",o ..".o
0 Om" O AgC104 O On.,. 0 On".
0 HO \ / OMe DM~ O O
_ _
OH 0 O
\~ \
O OMe O OMe
O~~".
2.5% IfOH O iqR a
Met O
O
O OH
Scheme 2
.,~no 0
O On".
b p
O
O
OH
Scheme 3
18
11
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
.",~o
O Oi",.
EDCI
O H2N~NH2 HOBT, DCM'
O
O OH
Scheme 4
H
N ~~
,,,,
.""o
0 0.,.,.
~NHZ EDC~ O
O H2N HOBT, DCM ; 'O~ ~ \
O O O I H
O ~~N~/\/~NHZ
I'v~~~I I('O
/ I 21
OOH
Scheme 5
According to a further aspect of the present invention therefore there is
provided a
process for the production of a compound of general formula 13 as hereinbefore
described, said process comprising the steps of coupling dihydroartemisinin
with an
alcohol methyl ester, hydrolysing the resultant compound to produce the
corresponding
carboxylic acid, and coupling said carboxylic acid with a polyamine or amine.
12
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
Alternatively, a further aspect of the invention provides a process for the
production of a compound of general formula 13 as hereinbefore described, said
process
comprising the steps of coupling dihydroartemisinin with an anhydride, forming
a
carboxylic acid, and coupling said carboxylic acid with a polyamine or amine.
Preferably, coupling of the carboxylic acid and polyamine or amine is carried
out
at a temperature of between -20 and +40 ° C.
These processes are particularly advantageous with regard to the production of
trioxane derivatives of the type exemplified by Examples 20 to 35.
The antimalarial activity of the new compounds was assessed using two strains
of
P. falciparum from Thailand: (a) the uncloned I~1 strain which is known to be
CQ
resistant and (b) the HB3 strain which is sensitive to all antimalarials.
Parasites were
maintained in continuous culture using the method of Trager and Jenson (J.
Parasitol.,
1977, 63, 883-886). Cultures were grown in flasks containing human
erythrocytes (2-5%)
with parasitemia in the range of 1% to 10% suspended in RPMI 1640 medium
supplemented with 25 mM HEPES and 32 mM NaHC03, and 10% human serum
(complete medium). Cultures were gassed with a mixture of 3% 02, 4% C02 and
93% N2.
Antimalarial activity was assessed with an adaption of the 48-h sensitivity
assay of
Desjardins et al. (Aratimicrob. Agents. Chemother., 1979, 16, 710-718) using
[3H~-
hypoxanthine incorporation as an assessment of parasite growth. Stock drug
solutions
were prepared in 100% dimethylsulphoxide (DMSO) and diluted to the appropriate
concentration using complete medium. Assays were performed in sterile 96-well
microtitre-plates, each plate containing 200 ~,1 of parasite culture (2%
parasitemia, 0.5%
haematocrit) with or without 10 ~,l drug dilutions. Each drug was tested in
triplicate and
13
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
parasite growth was compared to control wells (which constituted 100 %
parasite growth).
After 24-h incubation at 37°C, 0.5 ~,Ci hypoxanthine was added to each
well. Cultures
were incubated for a further 24 h before they were harvested onto filter-mats,
dried for 1 h
at 55°C and counted using a Wallac 1450 Microbeta Trilux Liquid
scintillation and
luminescence counter. ICso values were calculated by interpolation of the
probit
transformation of the log dose - response curve.
The results of these experiments axe contained in Table 1, which shows the
IC50
of compounds of the present invention versus the K1 strain of P. falciparum
ifi vitro.
Table 1
Compound Number IC50 (nM) SD
(13a) 8.2 2.7
(13b) 138.1 11.1
(13c) 6.3 0.7
(13d) 12.5 1.6
(13e) 3.8 0.1
(13f) 6.6 0.7
(13g) 4.3 0.30
(13h) 13.3 0.6
(13i) 22.5 1.9
(13j) 12.6 17
Artemether 6.5 2
Chloroquine 255 10
14
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
Table 2 shows the IC50 of compounds of the invention versus the HB3 strain of
P. falciparu»z in vitro.
Table 2
Compound Number IC50 (nM) SD
(13c) 2.3 0.7
(13d) 8.1 1.2
(13e) 1.8 0.2
(13f) 3.4 0.7
(13g) 4.3 0.30
(13j) 12.6 17
Artemether 9.2 0.2
The anticancer activity of these compounds of the present invention was also
assessed, by NCI 3-cell line anticancer assay. In this protocol, each cell
line is inoculated
and preincubated on a microtiter plate. Test agents are then added at a single
concentration and the culture incubated for 48 hours. End-point determinations
are made
with sulforhodamine B, a protein-binding dye. Results for each test agent are
reported as
the percentage of growth of the treated cells when compared to the untreated
control
cells. Compounds which reduce the growth of any one of the cells lines to 32%
or less
(negative numbers indicate cell kill) are passed on for evaluation in the full
panel of 60
cell lines over a 5-log dose range.
The results of these assays are shown in Table 3 below.
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
Table 3
Compound Number ConcentrationNCI-H460 MCF7 SF-268
(Lung) (Breast)(CNS)
(13h) 1.OOE-04Molar22 11 31
(13i) 1.OOE-04Molar15 10 29
(13c) 1.OOE-04Molar12 13 27
According to a further aspect of the present invention there is provided a
compound of general formula 13 as hereinbefore defined, or a pharmaceutically
acceptable salt thereof, for use as a medicament.
Compounds of the present invention may be used particularly, but not
exclusively,
as medicaments for the treatment of malaria or cancer. Whilst the currently
preferred use
of peroxides is for treatment, it cannot be ruled out that these compounds
would have a
use in the prophylaxis of malaria.
A therapeutically effective non-toxic amount of a compound of general formula
13 as hereinbefore defined may be administered in any suitable manner,
including orally,
parenterally (including subcutaneously, intramuscularly and intravenously), or
topically.
The administration will generally be carried out repetitively at intervals,
for example once
or several times a day.
The amount of the compound of general formula 13 that is required in order to
be effective as an antimalarial or anticancer agent for treating human or
animal subjects
will of course vary and is ultimately at the discretion of the medical or
veterinary
practitioner treating the human or animal in each particular case. The factors
to be
considered by such a practitioner, e.g. a physician, include the route of
administration and
pharmaceutical formulation; the subject's body weight, surface area, age and
general
condition; and the chemical form of the compound to be administered.
16
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
In daily treatment, for example, the total daily dose may be given as a single
dose, multiple doses, e.g. two to six times per day, or by intravenous
infusion for any
selected duration.
The compound of general formula 13 may be presented, for example, in the
form of a tablet, capsule, liquid (e.g. syrup) or injection.
While it may be possible for the compounds of general formula 13 to be
administered alone as the active pharmaceutical ingredient, it is preferable
to present the
compounds in a pharmaceutical composition.
According to a further aspect of the present invention therefore there is
provided a
pharmaceutical composition containing a compound of general formula 13 as
hereinbefore defined, or a pharmaceutically acceptable salt thereof, as an
active
ingredient.
Such pharmaceutical compositions for medical use will be formulated in
accordance with any of the methods well known in the art of pharmacy for
administration
in any convenient manner. The compounds of the invention will usually be
admixed with
at least one other ingredient providing a compatible pharmaceutically
acceptable additive,
carrier, diluent or excipient, and may be presented in unit dosage form.
The carriers) must be pharmaceutically acceptable in the sense of being
compatible with the other ingredients of the formulation and not deleterious
to the
recipient thereof.
The possible formulations include those suitable for oral, rectal, topical and
parenteral (including subcutaneous, intramuscular and intravenous)
administration or for
administration to the lung or another absorptive site such as the nasal
passages.
All methods of formulation in making up such pharmaceutical compositions
will generally include the step of bringing the compound of general formula 13
into
association with a carrier which constitutes one or more accessory
ingredients. Usually,
the formulations are prepared by uniformly and intimately bringing the
compound of
17
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
general formula 13 into association with a liquid carrier or with a finely
divided solid
carrier or with both and then, if necessary, shaping the product into desired
formulations.
Formulations of the present invention suitable for oral administration may be
presented as discrete units such as capsules, cachets, tablets or lozenges,
each containing
a predetermined amount of the compound of general formula 13; as a powder or
granules;
or a suspension in an aqueous liquid or non-aqueous liquid such as a syrup, an
elixir, an
emulsion or a draught. The compound of general formula 13 may also be
presented as a
bolus, electuary or paste.
A tablet may be made by compression or moulding, optionally with one or more
accessory ingredients. Compressed tablets may be prepared by compressing, in a
suitable
machine, the compound of general formula 13 in a free-flowing form such as a
powder or
granules, optionally mixed with a binder, lubricant, inert diluent, surface
active or
dispersing agent. Moulded tablets may be made by moulding, in a suitable
machine, a
mixture of the powdered compound of general formula 13 with any suitable
carrier.
A syrup may be made by adding the compound of general formula 13 to a
concentrated, aqueous solution of a sugar, for example sucrose, to which may
be added
any desired accessory ingredient. Such accessory ingredients) may include
flavourings,
an agent to retard crystallisation of the sugar or an agent to increase the
solubility of any
other ingredient, such as a polyhydric alcohol, for example glycerol or
sorbitol.
Formulations for rectal administration may be presented as a suppository with
a
usual carrier such as cocoa butter.
Formulations suitable for parental administration conveniently comprise a
sterile aqueous preparation of the compound of general formula 13 which is
preferably
isotonic with the blood of the recipient.
In addition to the aforementioned ingredients, formulations of this invention,
for
example ointments, creams and the like, may include one or more accessory
ingredients,
for example a diluent, buffer, flavouring agent, binder, surface active agent,
thickener,
18
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
lubricant and/or a preservative (including an antioxidant) or other
pharmaceutically inert
excipient.
The compounds of this invention may also be made up for administration in
liposomal formulations which can be prepared by methods well-known in the art.
A further aspect of the present invention provides the use of a compound of
general formula 13 as hereinbefore defined, or a pharmaceutically acceptable
salt thereof,
in the manufacture of a medicament for the treatment or prophylaxis of
malaria.
A further aspect of the present invention provides the use of a compound of
general formula 13 as hereinbefore defined, or a pharmaceutically acceptable
salt thereof,
in the manufacture of a medicament for the treatment of cancer.
It has further surprisingly been found that exposure of tumour cells to iron
potentiates the anticancer effect of the compounds of the present invention.
This finding
is supported by the observation that cultured cancer cell lines were more
readily
destroyed by compounds of the present invention following exposure to culture
medium
having an elevated iron concentration.
According to a further aspect of the present invention therefore there is
provided a
product containing a first compound of general formula 13 as hereinbefore
defined, or a
pharmaceutically acceptable salt thereof, and a second, iron-containing,
compound as a
combined preparation for simultaneous, separate or sequential use in the
treatment of
cancers.
Preferably, the first and second compounds are used sequentially, the second,
iron-containing, compound being used first.
The first compound may be presented in any of the forms described above.
Administration of the first compound may be in any suitable manner, including
intravenously, intraarterially, intralesionally, topically, intracavitarily or
orally. Any
19
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
suitable dosage of the compound may be used. Preferably, a dosage within the
range of
0.1 to SOOmg/kg body weight is used, more preferably within the range of 0.5
to
300mg/kg body weight, such as 1 to 50 mg/kg body weight.
With regard to the iron-containing compound, this may take any suitable form.
Preferred agents for enhancing intracellular iron levels for use in the
present invention
include pharmaceutically acceptable iron salts and iron complexes. Iron salts
useful in
the present invention include ferrous fumarate, ferrous sulphate, ferrous
carbonate,
ferrous citrate, ferrous gluconate, ferrous lactate and ferrous maleate. Iron
complexes
useful in the present invention include ferrocholinate, ferroglycine sulphate,
dextran iron
complex, peptonized iron, iron sorbitex, saccharated iron, iron complexed with
iron
binding proteins and glycoproteins such as the holoferritins and
holotransferrins.
The iron-containing compound may be presented in any of the forms described
above in relation to the compound of general formula 13. Administration of the
iron-
containing compound may be achieved via any of the possible routes of
administration of
the first compound. The first and second compounds may be administered via the
same
or different routes.
The iron-containing compound may be used ay any appropriate dosage, but is
preferably used at a dosage within the range of 0.01 to 1000 mg iron/kg body
weight.
The product of the invention may further comprise one or more other agents
known to be useful in the treatment of tumours. Such agents may include, for
example,
androgen inhibitors, antiestrogens, antimetabolites and cytotoxic agents.
According to a further aspect of the present invention there is provided a
method
of treatment of malaria which comprises administering to an animal in need of
such
treatment a therapeutically effective amount of a compound of general formula
13 as
hereinbefore defined, or a pharmaceutically acceptable salt thereof.
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
In terms of suitable dosages for the treatment of malaria, the preferred
amount of
compounds of the present invention is between l0mg to 5g, preferably 50 to
1000mg,
administered over a period of 2-5 days, alone or in combination with other
antimalarial
drugs, such as, for example, the class II blood schizonticides or halofantrine
(Looaeesuwan, Am. J. Trop. Med., 1999, 60, 238).
According to a further aspect of the present invention there is provided a
method
of treatment of cancer which comprises administering to an animal in need of
such
treatment a therapeutically effective amount of a compound of general formula
13 as
hereinbefore defined, or a pharmaceutically acceptable salt thereof.
The method may further comprise the simultaneous, separate or sequential
administration to the said animal an effective amount of an iron-containing
compound as
hereinbefore described.
The invention will now be illustrated by the following non-limiting examples.
Examples
Experimental Section
Merck Kieselgel 60 F 254 pre-coated silica plates for TLC were obtained from
BDH,
Poole, Dorset, U.K. Column chromatography was carried out on Merck 9385 silica
gel.
Infra red (IR) spectra were recorded in the range 4000-600 cm 1 using a Perkin
Elmer 298
infrared spectrometer. Spectra of liquids were taken as films. Sodium chloride
plates
(nujol mull) , solution cells (dichloromethane) and KBr discs were used as
indicated.
1H NMR spectra were recorded using a Perkin Elmer R34 (220 MHz) and Bruker
(300 MHz and 200 MHz) spectrometers. Solvents are indicated in the text and
tetramethylsilane was used as the internal reference. Mass spectra were
recorded at 70eV
21
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
using a VG7070E mass spectrometer. The samples were introduced using a direct-
insertion probe. In the text the parent ion (M+) is given, followed by peaks
corresponding to major fragment losses with intensities in parentheses.
General Procedure 1. Synthesis of Ether Derivatives
Dihydroartemisinin (2.00 g, 7.04 mmol) was dissolved in anhydrous diethyl
ether (200
mL) under N2. BF3.Et20 (1.03 mL, 8.10 mmol) was added to the solution,
followed by
the appropriate (hydroxymethyl) benzyl alcohol (1.46 g, 10.56 mmol). The
mixture was
allowed to stir at room temperature for 20 h and then quenched with water. The
organic
phase was washed with Na2S04 solution (30 % w/v), dried over MgS04, filtered
and the
solvent was removed under reduced pressure to the give the crude product as an
oil.
Purification by silica gel chromatography using ethyl acetate/nhexane (40/60)
as the
eluent gave the corresponding ether products.
10(3-[[4-(Hydroxymethyl)benzyl]oxy]dihydroartemisinin (12a)
This compound was prepared using general procedure 1 to give the product as a
colourless syrup (78 % yield): 1H NMR (300 MHz, CDC13) 8 7.20-7.08 (4 H, m,
aromatic), 5.44 (1 H, s), 4.88 (1 H, d, J = 3.80 Hz), 4.85 (1 H, d, J = 12.19
Hz), 4.69 (2
H, s), 4.51 (1 H, d, J = 12.19 Hz), 2.67 (1 H, sex), 2.38 (1 H, dt, J = 13.46,
3.98 Hz),
2.07-1.20 (10 H, m), 1.46 (3 H, s) and 0.94 (6 H, d, J = 7.17 Hz); 13C NMR (75
MHz,
CDC13) 8 140.20, 137.88, 127.59, 127.05, 104.21, 101.45, 88.09, 81.18, 69.57,
65.11,
52.62, 44.45, 37.43, 36.46, 34.64, 30.94, 26.16, 24.69, 24.52, 20.32 and
13.06; IR (thin
film)/crri 1 3476, 2924, 1612, 1516, 1458, 1377, 1194, 1101, 1011, 876 (O-O)
and 826
(O-O); MS m/z (CI) [M + NH4]+ 422 (8), 359 (100), 284 (39), 221 (96) and 138
(33).
10(3-[[3-(Hydroxymethyl)benzyl]oxy]dihydroartemisinin
This compound was prepared using general procedure 1 to give the product as a
colourless solid (68 % yield): m.p. 118-120 °C; 1H NMR (300 MHz, CDC13)
8 7.34-7.26
22
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
(4 H, m, aromatic), 5.40 (1 H, s), 4.92 (1 H, d, J = 3.80 Hz), 4.88 (1 H, d, J
= 12.20 Hz),
4.70 (2 H, s), 4.55 (1 H, d, J = 12.20 Hz), 2.67 (1 H, sex), 2.38 (1 H, dt, J
= 13.50, 3.80
Hz), 2.07-1.20 (10 H, m), 1.45 (3 H, s), 0.96 (3 H, d, J = 6.00 Hz) and 0.88
(3 H, d, J =
7.60 Hz); 13C NMR (75 MHz, CDC13) 8 141.05, 138.93, 128.64, 126.80, 126.15,
104.21,
101.65, 88.06, 81.17, 69.90, 65.37, 52.62, 44.47, 37.43, 36.46, 34.65, 30.97,
26.16, 24.70,
24.51, 20.31 and 13.07; IR (Nujol)/crri 13507, 2924, 1611, 1462, 1378,
1227,1192, 1104,
1011, 874 (O-O) and 823 (O-O); Anal. C23Hsa06 requires C 68.29 %, H 7.97 %,
found C
68.01%,H8.14%.
General Procedure 2. Synthesis of C-10 Oxo Derivatives
To a solution of the appropriate (hydroxymethyl) benzyl alcohol (0.20 g, 0.50
mmol) in
anhydrous DCM (10 mL) under NZ was added triethylamine (0.08 mL, 0.55 mmol),
followed by mesyl chloride (0.06 mL, 0.74 mmol) at 0 °C. The mixture
was stirred at 0
°C for 2 h and then quenched with water (10 mL). The organic phase was
extracted with
DCM (3 x 10 mL) and then dried over MgS04, filtered and the solvent was
removed
under reduced pressure. The crude mesylate and the appropriate substituted
piperazine
derivative (4.11 mmol) were dissolved in anhydrous benzene (10 mL) under NZ
atmosphere. The mixture was heated at reflux for 5 h. After allowing to cool
to room
temperature, the mixture was quenched with saturated NaHC03 solution and the
organic
phase was extracted with diethyl ether (3 x 10 mL). The organic extracts were
washed
with brine, dried over MgS04, filtered and the solvent was removed under
reduced
pressure. Purification by silica gel chromatography using ethyl
acetate/nhexane (40/60) as
the eluent gave the corresponding piperazine products.
10(3-[[4-[Phenylpiperazyl)methyl]benzyl]oxy]dihydroartemisinin (13a)
This compound was prepared from 1-phenyl piperazine using general procedure 2
to give
the product as a yellow oil (84 % yield): 1H (300 MHz, CDCl3) b 7.30-7.28 (4
H, d, J =
4.5 Hz, aromatic), 7.00-6.90 (5 H, m, aromatic), 5.45 (1 H, s), 4.90 (1 H, d,
J = 3.80 Hz),
4.86 (1 H, d, J = 12.20 Hz), 4.50 (1 H, d, J =12.20 Hz), 3.53 (2 H, s, CH2),
2.69 (1 H, m,
23
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
CH), 2.51 (8 H, m, CH2), 2.38 (1 H, dt, J = 13.32, 4.12 Hz), 2.07-1.20 (10 H,
m), 1.45 (3
H, s, CH3), 0.95-0.93 (6 H, 2 x CH3); isC (75 MHz, CDC13) 8 129.34, 128.28,
127.19,
104.16, 101.51, 88.08, 81.18, 69.67, 62.98, 62.68, 52.92, 52.66, 44.49, 37.43,
36.49,
34.67, 30.97, 26.19, 24.71, 24.52, 20.31, 13.06; LC/MS (NH3); m/z 563[M + H+,
(100)],
279 (17).
10[3-[[4-[(4-Nitrophenylpiperazyl)methyl]benzyl]oxy]dihydroartemisinin (13b)
This compound was prepared from 1-(4-nitrophenyl)piperazine using general
procedure 2
to give the product as an orange solid (68 % yield): 1H (300 MHz, CDCl3) ~
8.12 (2 H, d,
J = 9.48 Hz, aromatic), 7.35-7.27 (4 H, m, aromatic), 6.82 (2 H, d, J = 9.48
Hz,
aromatic), 5.47 (1 H, s), 4.91 (1 H, d, J = 3.90 Hz), 4.90 (1 H, d, J = 12.50
Hz), 4.53 (1
H, d, J = 12.50 Hz), 3.58 (2 H, s), 3.47-3.42 (4 H, m), 2.68 (1 H, m), 2.64-
2.59 (4 H, m),
2.38 (1 H, dt, J = 13.32, 4.12 Hz), 2.07-1.20 (10 H, m), 1.46 (3 H, s) and
0.95 (6 H, d, J =
6.70 Hz); 13C (75 MHz, CDC13) ~ 154.96, 129.17, 127.34, 125.99, 112.70,
104.19,
101.43, 88.09, 81.16, 69.54, 62.61, 52.63, 52.48, 47.08, 44.46, 37.46, 36.47,
34.67, 30.94,
26.18, 24.71, 24.53, 20.31 and 13.06; IR (Nujol)/crri 1 2927, 1597, 1493,
1462, 1377,
1327, 1251, 1231, 1099, 1010, 876 (O-O) and 828 (O-O); MS m/z (EI) [M]+ 593
(1), 264
(98), 218 (34), 104 (100) and 56 (58).
10[3-[[4-[(4-Fluorophenylpiperazyl)methyl]benzyl]oxy]dihydroartemisinin (13c)
This compound was prepared from 1-(4-fluorophenyl)piperazine using general
procedure
2 to give the product as a yellow oil (64 % yield): 1H (300 MHz, CDCl3) 8 7.35-
7.28 (4
H, m, aromatic), 6.99-6.85 (4 H, m, aromatic), 5.46 (1 H, s), 4.91 (1 H, d, J
= 3.90 Hz),
4.90 (1 H, d, J = 12.45 Hz), 4.52 (1 H, d, J =12.45 Hz), 3.58 (2 H, s), 3.16-
3.11 (4 H, m),
2.68-2.63 (5 H, m), 2.38 (1 H, dt, J = 13.50, 3.90 Hz), 2.07-1.20 (10 H, m),
1.46 (3 H, s),
0.95 (3 H, d, J = 6.00 Hz) and 0.94 (3H, d, J = 6.60 Hz); 13C (75 MHz, CDC13)
b 141.05,
138.93, 128.64, 126.80, 126.15, 104.21, 101.65, 88.06, 81.17, 69.90, 65.37,
52.62, 44.47,
37.43, 36.46, 34.65, 30.97, 26.16, 24.70, 24.51, 20.10 and 13.07; IR (thin
film)/crri 1
24
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
2945, 1633, 1510, 1455, 1374, 1359, 1240, 1142, 1099, 1011, 876 (O-O) and 825
(O-O);
HRMS (EI) C33H43FN2O5 [M~+ requires 566.31561, found 566.31493.
10(i-[[4-[(4-
Trifluoromethylphenylpiperazyl)methyl]benzyl]oxy]dihydroartemisinin
(13d)
This compound was prepared from 1-(4-trifluoromethyl phenyl)piperazine using
general
procedure 2 to give the product as a yellow oil (64 % yield): 1H, (300 MHz,
CDCl3) 8
7.35-7.28 (5 H, m, aromatic), 7.10-7.03 (3 H, m, aromatic), 5.46 (1 H, s)~
4.92 (1 H, d, J
=12.30 Hz), 4.88 (1 H, d, J = 3.80 Hz), 4.52, (1 H, d, J = 12.30 Hz), 3.61 (2
H, m, CH2),
3.27 (4 H, m, CH2), 2.70-2.65 (5 H, m, CH2), 2.39 (1 H, m, CH2), 2.07-1.20 (10
H, m),
1.45 (3 H, s, CH3), 0.97-0.94 (6 H, d, 2 xCH3); 13C (75 MHz, CDCl3) 8 129.59,
129.32,
127.33, 118.78, 112.29, 104.19, 101.50, 88.09, 81.17, 69.61, 62.58, 52.73,
48.55, 44.48,
37.46, 36.48, 34.67, 30.96,26.18, 24.71, 24.54, 20.31, 13.06; IR (thin
film)/crri 1 (2925),
(1454), (1136), (1011). LC/MS (NH3); m/z 618 [M + H+, (100)], 603 (100), 333
(7).
10(3-[[4-[(Benzylpiperazyl)methyl]benzyl]oxy]dihydroartemisinin (13c)
This compound was prepared from 1-benzylpiperazine using general procedure 2
to give
the product as a yellow oil (72 % yield): 1H NMR (300 MHz, CDC13) 8 7.32-7.20
(9 H,
m, aromatic), 5.45 (1 H, s), 4.91 (1 H, d, J = 3.90 Hz), 4.90 (1 H, d, J =
12.50 Hz), 4.52
(1 H, d, J = 12.50 Hz), 3.54 (4 H, br s), 2.68 (1 H, m), 2.54-2.49 (8 H, m),
2.38 (1 H, dt, J
= 14.10, 3.90 Hz), 2.07-1.20 (10 H, m), 1.45 (3 H, s), 0.94 (3 H, d, J = 5.70
Hz) and 0.94
(3H, d, J = 7.2 Hz); 13C (75 MHz, CDCl3) 8 129.34, 128.28, 127.19, 104.16,
101.51,
88.08, 81.18, 69.67, 62.98, 62.68, 52.92, 52.66, 44.49, 37.43, 36.49, 34.67,
30.97, 26.19,
24.71, 24.52, 20.31 and 13.06; IR (thin film)/cni 1 2938, 1609, 1495, 1457,
1374, 1344,
1227, 1099, 1010, 876 (O-O) and 826 (O-O).
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
10/3-[[3-[(Phenylpiperazyl)methyl]benzyl]oxy]dihydroartemisinin (l3fj
This compound was prepared from 1-phenylpiperazine using general procedure 2
to give
the product as a brown foam (59 % yield): 1H NMR (300 MHz, CDC13) 8 7.30-7.22
(4 H,
m, aromatic), 6.94-6.85 (5 H, m, aromatic), 5.47 (1 H, s), 4.91 (1 H, d, J =
3.70 Hz), 4.90
(1 H, d, J = 12.20 Hz), 4.55 (1 H, d, J = 12.20 Hz), 3.59 (2 H, br s), 3.27-
3.21 (4 H, m),
2.69-2.61 (5 H, m), 2.38 (1 H, dt, J = 13.30, 3.80 Hz), 2.07-1.20 (10 H, m),
1.46 (3 H, s),
0.95 (3 H, d, J = 7.40 Hz) and 0.94 (3H, d, J = 6.00 Hz); 13C (75 MHz, CDC13)
~ 139.10,
129.17, 128.38, 116.16, 104.19, 101.40, 88.10, 81.18, 69.68, 52.65, 44.47,
37.45, 36.48,
34.69, 30.96, 26.19, 24.71, 24.53, 20.32 and 13.10; IR (Nujol)/cni 1 2925,
1601, 1504,
1455, 1375, 1228, 1101, 1013, 875 (O-O) and 825 (O-O); HRMS (EI) C33H44N2O5
[M]+
requires 548.32501, found 548.32604.
10(3-[[3-[(4-Nitrophenylpiperazyl)methyl]benzyl]oxy]dihydroartemisinin (13g)
This compound was prepared from 1-(4-nitrophenyl)piperazine using general
procedure 2
to give the product as an orange foam (85 % yield): 1H (300 MHz, CDC13) 8 8.13
(2 H, d,
J = 9.50 Hz, aromatic), 7.36-7.25 (4 H, m, aromatic), 6.82 (2 H, d, J = 9.50
Hz,
aromatic), 5.47 (1 H, s), 4.91 (1 H, d, J = 5.00 Hz), 4.90 (1 H, d, J = 12.30
Hz), 4.56 (1
H, d, J = 12.30 Hz), 3.61 (2 H, s), 3.49-3.43 (4 H, m), 2.71 (1 H, rn), 2.69-
2.63 (4 H, m),
2.39 (1 H, dt, J = 13.87, 3.98 Hz), 2.07-1.20 (10 H, m), 1.45 (3 H, s) and
0.94 (6H, d, J =
7.40 Hz); 13C (75 MHz, CDCl3) b 151.00, 128.50, 128.00, 125.99, 112.79,
104.00,
101.00, 88.09, 81.50, 69.56, 60.38, 52.61, 52.20, 46.96, 44.20, 37.47, 36.45,
34.50, 30.93,
26.00, 24.70, 24.55, 20.32 and 13.00; IR (Nujol)/crri 1 2923, 1598, 1506,
1456, 1378,
1328, 1248, 1099, 1010, 875 (O-O) and 826 (O-O); MS m/z (EI) [M~+ 593 (1), 264
(43),
219 (18), 105 (100) and 56 (30).
10(3-[[3-[(4-Chlorophenylpiperazyl)methyl]benzyl]oxy]dihydroartemisinin (13h)
This compound was prepared from 1-(4-chlorophenyl)piperazine using general
procedure
2 to give the product as a brown foam (64 % yield): 1H (300 MHz, CDCl3) b 7.32-
7.06 (6
26
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
H, m, aromatic), 6.83 (2 H, d, J = 9.06, aromatic), 5.47 (1 H, s), 4.92 (1 H,
d, J = 4.10
Hz), 4.90 (1 H, d, J = 12.01 Hz), 4.55 (1 H, d, J = 12.01 Hz), 3.61 (2 H, br
s), 3.22-3.17
(4 H, m), 2.69-2.61 (5 H, m), 2.39 (1 H, dt, J =13.50, 3.85 Hz), 2.07-1.20 (10
H, m), 1.46
(3 H, s) and 0.94 (3H, d, J = 6.60 Hz); 13C (75 MHz, CDC13) 8 138.50, 129.03,
128.44,
117.35, 104.19, 101.37, 88.09, 81.17, 69.64, 52.81, 49.03, 44.45, 37.45,
36.47, 34.68,
30.94, 26.19, 24.71, 24.53, 20.32 and 13.10; IR (Nujol)/crri 1 2927, 1616,
1496, 1459,
1378, 1225, 1102, 1032, 874 (O-O) and 815 (O-O); MS m/z (EI) [M]+ 422 (1), 300
(26),
193 (19), 131 (14) and 105 (100).
10(3-[[3-[(3-
Trifluoromethylphenylpiperazyl)methyl]benzyl]oxy]dihydroartemisinin
(13i)
This compound was prepared from 1-[(3-trifluoromethyl)phenyl]piperazine using
general
procedure 2 to give the product as a brown foam (66 % yield): 1H (300 MHz,
CDC13) ~
7.37-7.23 (4 H, rn, aromatic), 7.10-7.06 (4 H, m, aromatic), 5.47 (1 H, s),
4.92 (1 H, d, J
= 3.98 Hz), 4.90 (1 H, d, J = 12.16 Hz), 4.52 (1 H, d, J = 12.16 Hz), 3.58 (2
H, s), 3.16-
3.11 (4 H, m), 2.68-2.63 (5 H, m), 2.38 (1 H, dt, J =13.48, 4.02 Hz), 2.07-
1.20 (10 H, m),
1.45 (3 H, s), 0.95 (3 H, d, J = 7.20 Hz) and 0.94 (3H, d, J = 6.00 Hz); 13C
NMR (75
MHz, CDC13) 8 151.49, 138.65, 129.59, 128.40, 118.74, 115.81, 112.22, 104.19,
101.38,
88.09, 81.17, 69.68, 62.81, 52.80, 52.64, 48.64, 44.46, 37.46, 36.46, 34.69,
30.95, 26.19,
24.71, 24.53, 20.31 and 13.10; IR (Nujol)/crri 1 2921, 1612, 1496, 1454, 1350,
1228,
1120, 1011, 875 (O-O) and 826 (O-O); MS m/z (EI) [M]+ 616 (1), 334 (11), 227
(7), 105
(100) and 56 (10).
10(3-[[3-[(4-Fluorophenylpiperazyl)methyl]benzyl]oxy]dihydroartemisinin (13j)
This compound was prepared from 1-(4-fluorophenyl)piperazine using general
procedure
2 to give the product as an off-white foam (69 % yield): 1H (300 MHz, CDC13) 8
7.36-
7.27 (4 H, m, aromatic), 6.98-6.84 (4 H, m, aromatic), 5.47 (1 H, s), 4.91 (1
H, d, J = 3.43
Hz), 4.90 (1 H, d, J = 12.29 Hz), 4.54 (1 H, d, J =12.29 Hz), 3.58 (2 H, s),
3.12 (4 H, t, J
= 4.81 Hz), 2.63 (1 H, m), 2.62 (4 H, t, J = 13.80 Hz), 2.38 (1 H, dt, J =
13.40, 3.90 Hz),
27
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
2.07-1.20 (10 H, m), 1.45 (3 H, s), 0.94 (3 H, d, J = 6.00 Hz) and 0.93 (3 H,
d, J = 6.00
Hz); 13C (75 MHz, CDC13) 8 148.04, 138.62, 128.40, 126.34, 117.89, 115.70,
115.40,
104.19, 101.36, 88.09, 81.18, 69.65, 62.82, 52.99, 52.64, 50.07, 44.46, 37.45,
36.47,
34.69, 30.95, 26.19, 24.71, 24.53, 20.32 and 13.10; IR (Nujol)/crri 1 2924,
1510, 1460,
1377, 1229, 1160, 1102, 1012, 875 (O-O) and 826 (O-O); HRMS (EI) C33Hq.3FN2O5
[M~+
xequires 566.31561, found 566.31437; C33H4sFNzOs requires C 69.97 %, H 7.60 %,
N
4.97 %, found C 69.67 %, H 7.72 %, N 4.82 %.
Procedure 1. Synthesis of ~3 methyl-p-[(10-
dihydroartemisininoxy)methyl]benzoate
(14)
A solution of dihydroartemisinin (5 g, 17.6 mmol) in anhydrous dichloromethane
(40 ml)
was stirred at room temperature. Methyl 4-(hydroxymethyl)-benzoate (6.7 g, 40
mmol)
and silver perchlorate (0.73 g, 3.52 mrnol) were added. The reaction was
cooled to -78 °C
and stirred under N2. Trimethylsilyl triflate was added dropwise and the
reaction mixture
was stirred under N2 at -78 °C for 2 hours. The reaction was quenched
with triethylamine
(20 ml) and allowed to warm to room temperature. Any resulting precipitate was
removed by filtration and the reaction mixture was evaporated to dryness.
Purification of
the crude mixture by flash column chromatography, using 10% ethyl acetate/n-
hexane as
the eluent, gave the (3 (14) and a-isomers in 47% and 14% yield respectively.
Data for [3-isomer 14 1H NMR (250 MHz, CDCl3) 7.95 (2H, d, J = 8.3 Hz,
aromatic),
7.31 (2H, d, J = 8.3 Hz, aromatic), 5.38 (1H, s), 4.89 (1H, d, J = 13.2 Hz, AB
coupling),
4.85 (1H, d, J = 3.7 Hz), 4.50 (1H, d, J =13.2 Hz, AB coupling), 3.84 (3H, s,
OMe), 2.62
(1H, m, CH), 2.28 (1H, dt, J =13.4, 4.0 Hz), 2.00-1.16 (10H, m), 1.45 (3H, s,
CH3), 0.91-
0.86 (6H, 2 x CH3).
Procedure 2. Synthesis of (3-artelinic acid (15)
A suspension of 14[3 (3.65 g, 8,42 mmol) in aqueous potassium hydroxide and
methanol
(2.5 % KOH/MeOH, 1/1 mixture, 350 rnl) was stirred for 24 hours at room
temperature.
28
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
After this time the methanol was removed in vacuo and the aqueous mixture was
cooled
to 0 °C. The mixture was acidified to pH 2 by dropwise addition of
dilute hydrochloric
acid, then extracted with diethyl ether (3 x 200 ml). The organic extracts
were dried over
anhydrous MgS04 and the solution was evaporated to dryness. This gave pure
product 2
as a white foam in a 92% yield: 1H NMR (250 MHz, CDC13) 8.90 (2H, d, J = 8.3
Hz,
aromatic), 7.44 (2H, d, J = 8.3 Hz, aromatic), 5.47 (1H, s), 5.00 (1H, d, J =
13.3 Hz, AB
coupling), 4.90 (1H, d, J = 3.4 Hz), 4.62 (1H, d, J = 13.3 Hz, AB coupling),
2.72 (1H, m,
CH), 2.38 (1H, dt, J = 13.4, 4.0 Hz), 2.08-1.20 (11H, m), 1.47 (3H, s, CH3),
1.01-0.95
(6H, 2 x CH3).
Synthesis of Example 16
16
This compound was prepared from methyl 3-hydroxybenzoate using procedure 1 and
procedure 2 to give the product as a yellow foam (77 % yield): 1H NMR (250
MHz,
CDC13) 7.80 (1H, s, aromatic), 7.75 (1H, m, aromatic), 7.43-7.40 (2H, 2 x d, J
= 5.4 Hz,
aromatic), 5.59 (1H, d, J = 2.8 Hz), 5.51 (1H, s), 2.86 (1H, rn, CH), 2.40
(1H, dt, J =
13.2, 3.9 Hz), 2.08-1.20 (11H, m), 1.47 (3H, s, CH3), 1.07-0.98 (6H, 2 x CH3).
Synthesis of Example 17
29
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
This compound was prepared from methyl 4-hydroxybenzoate using procedure 1 and
procedure 2 to give the product as a yellow foam (45 % yield).
Hydrolysis of the ester was carried out by the same procedure as for the
synthesis of 15
but using a solvent mix of 2.5 % aqueous KOH, MeOH and THF in a 2/1/1 ratio:
1H
NMR (250 MHz, CDC13) 8.07 (2H, d, J = 8.7 Hz, aromatic), 7.18 (2H, d, J = 8.7
Hz,
aromatic), 5.62 (1H, d, J = 3.2 Hz), 5.46 (1H, s), 2.85 (1H, m, CH), 2.39 (1H,
dt, J =
14.2, 4.0 Hz), 2.13-1.20 (11H, m), 1.46 (3H, s, CH3), 1.05-0.95 (6H, 2 x CH3).
Procedure 3. Synthesis of 10 succinyl dihydroartemisinin (18)
A solution of dihydroartemisinin (1.5 g, 5.28 mmol) in anhydrous
dichloromethane (75
ml) was stirred at room temperature. Succinic anhydride (0.63 g, 6.33 mmol)
and triethyl
amine (3.7 ml, 26.4 mmol) were added and the solution was stirred at room
temperature
for 1 hour. The reaction mixture was washed with aqueous citric acid (pH 2, 2
x 20 ml),
dried over MgS04 and evaporated to dryness. Purification of the crude mixture
by flash
column chromatography, using 2-5% methanol/dichloromethane as the eluent, gave
18 in
a 65 % yield: 1H NMR (250 MHz, CDCl3) 5.73 (1H, d, J = 9.9 Hz), 5.36 (1H, s),
2.62
(2H, m), 2.64 (4H, dd, J = 4.4, 1.9 Hz), 2.30 (1H, dt, J = 13.3, 4.0 Hz), 1.99-
1.18 (10H,
m), 1.36 (3H, s, CH3), 0.89 (3H, d, J = 5.8 Hz, CH3), 0.78 (3H, d, J = 7.1 Hz,
CH3).
Synthesis of Example 19
.",.o
0 0.,,..
0
0 0
0
'OH
19
This compound was prepared from phthalic anhydride using procedure 3 to give
19 in a
70 % yield: 1H NMR (250 MHz, CDC13) 7.78 (2H, m, aromatic), 7.52 (2H, m,
aromatic),
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
5.91 (1H, d, J = 9.8), 5.56 (1H, s), 2.58 (1H, m, CH), 2.30 (1H, dt, J = 14.1,
3.9 Hz),
2.00-1.20 (11H, m), 1.36 (3H, s, CH3), 0.91-0.84 (6H, 2 x CH3).
Procedure 4. Synthesis of Example 20
A solution of 15 (100 mg, 0.24 mmol), 1-(3-dimethylaminopropyl)-3-
ethylcarbodiimide
hydrochloride (46 mg, 0.24 mmol) and 1-hydroxybenzotriazole (32 mg, 0.24 mmol)
in
dichloromethane (2 ml) was stirred at room temperature. 1,5-diaminopentane
(.14 ul, 0.12
mmol) was added and the reaction was stirred for 2 hours at room temperature.
After this
time the reaction was washed with water (3 x 20 ml),the organic extracts dried
over
MgS04 and evaporated to dryness. Purification of the crude mixture, using
preparative
HPLC (C18, Luna, 100 x 21.2 mm, 10 micron), gave 20 in a 14 % yield: 1H NMR
(250
MHz, CDC13) 7.68 (4H, d, J = 8.2 Hz, aromatic), 7.28 (4H, d, J = 8.2 Hz,
aromatic), 6.31
(2H, m, amide) 5.37 (2H, s), 4.89 (2H, d, J = 12.9 Hz, AB coupling), 4.83 (2H,
s), 4.49
(2H, d, J =12.9 Hz, AB coupling), 3.40 (4H, m) 2.61 (2H, m, CH), 2.26 (2H, dt,
J = 13.5,
4.0 Hz), 2.08-1.16 (26H, m), 1.38 (6H, s, CH3), 0.89-0.83 (12H, 2 x CH3).
Procedure 5. Synthesis of Example 21
A solution of 1,4-diaminobutane (0.17 g, 1.9 mmol) in dichloromethane (6 ml)
was
cooled to 0 °C. A pre-formed solution of 15 (200 mgs, 0.47 mmol), 1-(3-
dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (92 mg, 0.47 mmol) and
1-hydroxybenzotriazole (65 mg, 0.47 mmol) in dichloromethane (2 ml) was added
dropwise over 1 hour. The reaction mixture was stirred at 0°C for a
further hour, washed
with water (3 x 20 ml), the organic extracts dried over MgSOø and evaporated
to dryness.
Purification of the crude mixture by flash column chromatography, using 50%
methanol/dichloromethane as the eluent, gave product 21 in a 21% yield: 1H NMR
(250
MHz, CDC13) 7.70 (2H, d, J = 8.2 Hz, aromatic), 7.28 (2H, d, J = 8.2 Hz,
aromatic), 7.00
(1H, m, amide) 5.37 (1H, s), 4.85 (1H, d, J = 12.7 Hz, AB coupling), 4.8 (1H,
s), 4.48
(1H, d, J = 12.7 Hz, AB coupling), 3.38 (2H, m) 2.60 (2H, m, CH, NH), 2.26
(1H, dt, J =
14.3, 4.0 Hz), 2.01-1.15 (17H, m), 1.38 (3H, s, CH3), 0.89-0.83 (6H, 2 x CH3).
31
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
Synthesis of Example 22
This compound was prepared from compound 15 and 1,4-diaminobutane, using a
similar
procedure to 4. Purification of the crude material by preparative HPLC gave
the product
in a 31 % yield: 1H NMR (250 MHz, CDCl3) 7.80 (4H, d, J = 8.2 Hz, aromatic),
7.39
(4H, d, J = 8.2 Hz, aromatic), 6.61 (2H, m, amide) 5.46 (2H, s), 4.95 (2H, d,
J = 12.7 Hz,
AB coupling), 4.91 (2H, s), 4.57 (2H, d, J = 12.7 Hz, AB coupling), 3.52 (4H,
m) 2.69
(2H, m, CH), 2.34 (2H, dt, J =13.6, 3.9 Hz), 2.10-1.24 (24H, m), 1.46 (6H, s,
CH3), 0.98-
0.94 (12H, 2 x CH3).
Synthesis of Example 23
N~
N~ J
23
This compound was prepared from compound 15 and 1,4-bis(3-aminopropyl)-
piperazine,
using a similar procedure to 4 to give the product in a 21 % yield: 1H NMR
(250 MHz,
CDCl3) 8.12 (2H, m, amide), 7.80 (4H, d, J = 8.2 Hz, aromatic), 7.36 (4H, d, J
= 8.2 Hz,
aromatic), 5.45 (2H, s), 4.92 (2H, d, J = 12.8 Hz, AB coupling), 4.88 (2H, s),
4.57 (2H, d,
J =12.8 Hz, AB coupling), 3.57 (4H, m) 2.70-1.19 (40H, m) 1.45 (6H, s, CH3),
0.95-0.92
(12H, 2 x CH3).
32
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
Synthesis of Example 24
NON
24
This compound was prepared from compound 15 and 3,3'-diamino-N-
methyldipropylamine, using a similar procedure to 4 to give the product in a
27 % yield:
1H NMR (250 MHz, CDCl3) 7.56 (4H, d, J = 8.2 Hz, aromatic), 7.16 (4H, d, J =
8.2 Hz,
aromatic), 5.24 (2H, s), 4.72 (2H, d, J =13.3 Hz, AB coupling), 4.69 (2H, s),
4.36 (2H, d,
J = 13.3 Hz, AB coupling), 3.33 (4H, m) 2.47 (2H, m) 2.3 (4H, t, J = 6.3 Hz),
2.15 (2H,
dt, J = 14.2, 3.9 Hz), 2.07 (3H, s, NMe), 1.87-1.03 (26H, m) 1.25 (6H, s,
CH3), 0.76-0.70
(12H, 2 x CH3).
Synthesis of Example 25
,,,,
o,a
.0~0 o w
W
0
This compound was prepared from compound 15 and benzylamine, using a similar
procedure to 4 to give the product in a 20 % yield: 1H NMR (250 MHz, CDC13)
7.79 (2H,
d, J = 8.2 Hz, aromatic), 7.44-7.28 (7H, m, aromatic), 6.46 (1H, m, amide)
5.46 (1H, s),
4.95 (1H, d, J = 12.8 Hz, AB coupling), 4.90 (1H, s), 4.66 (2H. d, J = 5.63
Hz, benzylic
CH2), 4.57 (1H, d, J = 12.8 Hz, AB coupling), 2.68 (1H, m, CH), 2.38 (1H, dt,
J = 13.54,
3.93 Hz), 2.08-1.24 (10H, m), 1.46 (3H, s, CH3), 0.97-0.89 (6H, 2 x CH3).
33
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
Synthesis of Example 26
26
This compound was prepared from compound 15 and tris(2-aminoethyl)amine, using
a
similar procedure to 4 to give the product in a 13 % yield: 1H NMR (250 MHz,
CDC13)
7.69 (6H, d, J = 8.1 Hz, aromatic), 7.18 (6H, d, J = 8.1 Hz, aromatic), 5.43
(3H, s), 4.86
(3H, d, J =12.4 Hz, AB coupling), 4.86 (3H, s), 4.50 (3H, d, J = 12.4 Hz, AB
coupling),
3.56 (6H, m), 2.77 (6H, m), 2.67 (3H, m) 2.37 (3H, dt, J = 13.8, 3.8 Hz), 1.93-
1.21 (33H,
m) 1.46 (9H, s, CH3), 0.96-0.93 (18H, 2 x CH3).
Synthesis of Example 27
0 0 0 .
o ;
N I ,o,d
~N \
/ ~N
_O, ~ ~ I H 0
0 p 0
27
This compound was prepared from compound 17 and 1,4-diaminobutane, using a
similar
procedure to 4 to give the product in a 17 % yield: 1H NMR (250 MHz, CDC13)
7.79 (4H,
d, J = 8.6 Hz, aromatic), 7.14 (4H, d, J = 8.6 Hz, aromatic), 6.66 (2H, m,
amide), 5.57
34
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
(2H, d, J = 3.3 Hz), 5.46 (2H, s), 3.50 (4H, m), 2.83 (2H, m, CH), 2.39 (2H,
dt, J = 13.9,
3.8 Hz), 2.07-1.22 (24H, m), 1.44 (6H, s, CH3), 1.04-0.93 (12H, 2 x CH3).
Synthesis of Example 28
2s
This compound was prepared from compound 17 and 1,5-diaminopentane, using a
similar
procedure to 4 to give the product in a 25 % yield: 1H NMR (250 MHz, CDC13)
7.63 (4H,
d, J = 8.8 Hz, aromatic), 7.03 (4H, d, J = 8.8 Hz, aromatic), 6.14 (2H, m,
amide), 5.46
(2H, d, J = 3.2 Hz), 5.34 (2H, s), 3.35 (4H; m), 2.71 (2H, m, CH), 2.26 (2H,
dt, J = 13.6,
4.0 Hz), 1.95-1.11 (26H, m), 1.32 (6H, s, CH3), 0.93-0.84 (12H, 2 x CH3).
Synthesis of Example 29
''° o
.. ~ o 0 0
O,~~ ~ \ I Fib ~N~N I / ~O O
O~O~O U
O
29
This compound was prepared from compound 17 and 1,4-bis(3-aminopropyl)-
piperazine,
using a similar procedure to 4 to give the product in a 28 % yield: 1H NMR
(250 MHz,
CDC13) 7.97 (2H, m, amide), 7.79 (4H, d, J = 8.7 Hz, aromatic), 7.15 (4H, d, J
= 8.7 Hz,
aromatic), 5.56 (2H, d, J = 3.4 Hz), 5.47 (2H, s), 3.58 (4H, m), 2.85 (2H, m,
CH), 2.58
(10H, m), 2.40 (2H, dt, J = 14.0, 3.8 Hz), 2.08-1.15 (26H, m), 1.44 (6H, s,
CH3), 1.05-
0.97 (12H, 2 x CH3).
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
Synthesis of Example 30
0 0
O,Oo' \ I H~ I ~H ~ / O,O
O~O~O Ow, O O
This compound was prepared from compound 17 and 3,3'-diamino-N-
methyldipropylamine, using a similar procedure to 4 to give the product in a
17 % yield:
1H NMR (250 MHz, CDC13) 7.78 (4H, d, J = 8.7 Hz, aromatic), 7.33 (2H, m,
amide),
7.15 (4H, d, J = 8.7 Hz, aromatic), 5.56 (2H, d, J = 3.1 Hz), 5.46 (2H, s),
3.53 (4H, m),
2.83 (2H, m, CH), 2.51 (4H, m), 2.51-1.11 (33H, m), 1.44 (6H, s, CH3), 1.04-
0.96 (12H,
2 x CH3).
Synthesis of Example 31
,,,,, o
o_o,~
o w
31
This compound was prepared from compound 17 and benzylamine, using a similar
procedure to 4 to give the product in a 54 % yield: 1H NMR (250 MHz, CDC13)
7.76 (2H,
d, J = 8.8 Hz, aromatic), 7.38-7.28 (5H, m, aromatic), 7.16 (2H, d, J = 8.8
Hz, aromatic),
6.37 (1H, m, amide), 5.56 (1H, d, J = 3.3 Hz), 5.46 (1H, s), 4.65 (2H, d, J =
5.6 Hz,
benzylic), 2.84 (1H, m, CH), 2.39 (1H, dt, J = 13.3, 4.0 Hz), 2.08-1.22 (10H,
m), 1.45
(3H, s, CH3), 1.04-0.96 (6H, 2 x CH3).
36
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
Synthesis of Example 32
,,,,
0
O,o~ ~ I N ~ o o_ °
' O~O 0 \ ~H I ~0~~
O
32
This compound was prepared from compound 16 and 1,4-diaminobutane, using a
similar
procedure to 4 to give the product in a 23 % yield: 1H NMR (250 MHz, CDCl3)
7.57-7.21
(8H, m, aromatic), 6.92 (2H, m, amide), 5.60 (2H, d, J = 3.2 Hz), 5.48 (2H,
s), 3.46 (4H,
m), 2.80 (2H, m, CH), 2.37 (2H, dt, J = 13.9, 3.6 Hz), 2.05-1.22 (24H, m),
1.38 (6H, s,
CH3), 1.04-0.95 (12H, 2 x CH3).
Synthesis of Example 33
,,,,
,,,.
O,~~~ ~ / I N N I / O,O
O~O O \ ~ O~~'~, O O
O O
33
This compound was prepared from compound 16 and 1,5-diaminopentane, using a
similar
procedure to 4 to give the product in a 28 % yield: 1H NMR (250 MHz, CDC13)
7.35-6.98
(8H, m, aromatic), 6.5 (2H, m, amide), 5.39 (2H, d, J = 3.2 Hz), 5.27 (2H, s),
3.21 (4H,
m), 2.61 (2H, m, CH), 2.17 (2;H, dt, J = 13.9, 4.0 Hz), 1.85-1.02 (26H, m),
1.19 (6H, s,
CH3), 0.83-0.75 (12H, 2 x CH3).
Synthesis of Example 34
,,,, o
0 0 0 .
N~ ~N~H I
O.O~O~ H N
O
O ..","
34
37
CA 02469224 2004-06-03
WO 03/048167 PCT/GB02/05531
This compound was prepared from compound 16 and 1,4-bis(3-aminopropyl)-
piperazine,
using a similar procedure to 4 to give the product in a 42 % yield: 1H NMR
(250 MHz,
CDC13) 8:03 (2H, m, amide), 7.55-6.23 (8H, m, aromatic), 5.56 (2H, d, J = 3.1
Hz), 5.46
(2H, s), 3.53 (4H, m), 2.81 (2H, m, CH), 2.53-1.20 (42H, m), 1.41 (6H, s,
CH3), 1.03-
0.94 (12H, 2 x CH3).
Synthesis of Example 35
,,,,
O~O~~ ~O N~N~O O O
O O O I I H I I .O.O
O O
..."i
This compound was prepared from compound 18 and 1,4-diaminobutane, using a
similar
procedure to 4 to give the product in a 14 % yield: 1H NMR (250 MHz, CDC13)
6.20 (2H,
m, amide), 5.77 (2H, d, J = 9.9 Hz), 5.44 (2H, s), 3.25 (4H, m), 2.76 (4H, m,
CH), 2.57-
1.26 (32H, m), 1.43 (6H, s, CH3), 0.97 (6H, d, J = 5.7 Hz, CH3), 0.85 (6H, d,
J = 7.09).
It is to be understood that the present invention is not intended to be
restricted to
the details of the above examples and embodiments, which are described by way
of
example only.
38