Note: Descriptions are shown in the official language in which they were submitted.
CA 02469953 2007-09-04
Imidazo[2,1-b]-1,3,4-thiadiazole Sulfonamides
FIELD OF THE INVENTION
This invention relates to sulfonamide compounds useful in the prevention of
neuronal cell
loss or in the treatment of nerve cell or axonal degradation.
BACKGROUND OF THE INVENTION
Various neurotrophins characterized by Neuronal Growth Factor (NGF), brain
derived
growth factor (BDNF), neurotrophin-3 (NT-3), and others (NT-4, CNTF, GDNF, IGF-
1),
have been identified as key survival factors for neurons. NGF plays a critical
role in the
development and maintenance of cholinergic forebrain neurons of the CNS and
neurons of
the peripheral nervous system (PNS); neurons of the PNS are characterized as
small fiber
sensory neurons associated with pain and temperature sensation, in addition to
neurons of
the sympathetic ganglia and dorsal root ganglia (SCGs and DRGs, respectively).
BDNF
plays a role in motor neuron survival. Both BDNF and NT-3 are expressed in the
CNS
and serve similar purposes in multiple subsets of cortical and hyppocampal
neurons;
neurons of the CNS are characterized by those found in the brain, spinal
chord, and eye.
The removal of these, and related trophic factors from in vitro cellular media
results in the
degradation of the axonal processes, leading to apoptosis of cultured neurons.
Localized tissue loss of NGF, or reduced axonal retrograde transport of NGF to
the cell
body, have been causally implicated in the development of peripheral
neuropathies and
neuropathic pain regularly observed in diabetes and HIV patients. Several
double blind
Phase II clinical trials have found that the systemic administration of
recombinant human
NGF (rhNGF) (US 5,604,202) displayed beneficial effects on neuropathic pain,
physiology, and cognition related to these diseases (Apfel, S. C. et. aL JAMA,
248(17),
2215-2221; Apfel, S. C. Neurology 51, 695-702, 1998; McAurthur, J. C. etal.
Neurology
54, 1080-1088, 2000). Side effects related to rhNGF treatment included
injection site
pain, hyperalgesia, and other pain related symptoms. Despite these symptoms, a
large
number of patients continued rhNGF treatment after unblinding.
CA 02469953 2007-09-04
Various chemotherapeutic drugs such as TaxolTm, cisplatin, vinblastine, and
vincristine,
cause dose dependent peripheral neuropathies, characterized by peripheral pain
and loss of
function. In many cases these neuropathies effectively limit the amount, and
duration, of
chemotherapy given to patients. For example, upwards of 50% of patients
receiving
TaxolTm chemotherapy experience severe, and cumulative, peripheral
neuropathies. The
progression of the neuropathy necessitates the use of a dosing regime which is
characterized by three cycles of fourteen days of TaxolTm treatment, followed
by 14 days
of recovery. Regression of the neuropathy is often observed between treatment
cycles and
following the final treatment. The degree and duration of recovery varies
largely between
patients. In addition to peripheral neuropathies, cisplatin treatment
invariably results in
some form of auditory loss, especially in children, due to neuronal damage in
the inner
ear, with minimal recovery of the neurons after completion of treatment.
SUMMARY OF THE INVENTION
The invention relates to imidazo[2,1-b]thiadiazole sulfonamides, which are
useful in the
treatment of neurodegenerative diseases of the CNS and/or PNS, for the
inhibition of
various serine-threonine protein kinases, phosphatases, CA, for inhibiting the
degradation,
dysfunction, or loss of neurons of the CNS and/or PNS, or enhancing the
phenotype of
neuronal cell types and preserving the axonal function of neuronal and
synaptic processes
of the CNS and/or of the PNS or altering signal transduction.
Also included are selected methods for the preparation of these compounds.
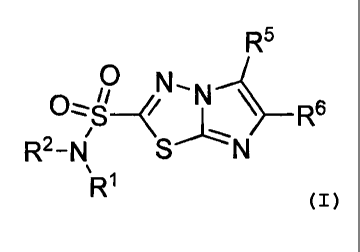
The imidazo[2,1-b]-1,3,4-thiadiazole sulfonamide derivatives and precursors of
the
present invention include compounds of the Formula 1:
R5
0 N ki
I r
R2-N
R1 (I)
2
CA 02469953 2007-09-04
or pharmaceutically acceptable salts thereof wherein:
Wand R2 are individually selected from the group consisting of H, lower alkyl,
substituted
lower alkyl, and fluoroalkyl;
R5 is selected from the group consisting of II, halogen, cyano, azide,
thiocyanate, formyl,
lower alkyl, substituted lower alkyl, fluoroalkyl, aralkyl, substituted
aralkyl, aryl,
substituted aryl, heteroaryl, and substituted heteroaryl;
R6 is selected from the group consisting of H, lower alkyl, substituted lower
alkyl,
fluoroalkyl, substituted fluoroalkyl, aryl, substituted aryl, heteroaryl,
substituted
heteroaryl, adamantly, coumarinyl, and substituted coumarinyl; or R6 is
represented by W:
R7 R8
X7--X8
W = X9-R9 ) n
x11.y.1
R11 R1
wherein:
n represents 0 or 1;
the ring system containing X7-X11 represents a 5 or 6 membered aromatic or
heteroaromatic ring system, in which X7-X11 are independently selected from
the group
consisting of C, N, S, and 0;
when any one of X7-X11 independently represents C, a respective R7-R11 is
independently
selected from the group consisting of:
a) H, halogen, nitro, cyano, lower alkyl, substituted lower alkyl,
fluoroalkyl,
aralkyl, substituted aralkyl, aryl, substituted aryl, heteroaryl, substituted
heteroaryl, acyl, lower alkylcarbonyl, substituted lower alkylcarbonyl,
arylcarbonyl, substituted arylcarbonyl, heteroarylcarbonyl, or substituted
heteroarylcarbonyl;
3
CA 02469953 2011-10-28
b) SO2NR16R17 wherein R16 and R17 are independently selected from the group
consisting of lower alkyl, substituted lower alkyl, aralkyl, substituted
aralkyl,
heteroaralkyl, substituted heteroaralkyl aryl, substituted aryl, heteroaryl,
and
substituted heteroaryl, or wherein R16 and R17 are joined to form an alkyl,
substituted alkyl, heteroalkyl, or substituted heteroalkyl ring system;
SOJR18 wherein m=0, 1 or 2, and wherein R18 is selected from the group
consisting of lower alkyl, substituted lower alkyl, aralkyl, substituted
aralkyl,
= heteroaralkyl, substituted heteroaralkyl, aryl, substituted aryl,
heteroaryl, and
substituted heteroaryl;
d) )CR19 wherein X is defined as S or 0, and R19 is defined as alkyl,
substituted
alkyl, fluoroalkyl, aryl, substituted aryl, heteroaryl, substituted
heteroaryl, acyl,
lower alkylcarbonyl, substituted lower alkylcarbonyl, arylcarbonyl,
substituted
arylcarbonyl, heteroarylcarbonyl, substituted heteroarylcarbonyl, lower
alkylaminocarbonyl, arylaminocarbonyl, or substituted arylaminocarbonyl;
NR14--tt 15
wherein R14 and R15 are defined as lower alkyl joined to form an
alkyl, substituted alkyl, heteroalkyl, or substituted heteroalkyl ring system;
and
CO2R2 wherein R2 is defined as H, lower alkyl, substituted lower alkyl,
aralkyl, substituted aralkyl, aryl, substituted aryl, heteroaryl, substituted
heteroaryl, or NR217'K.22,
wherein R21 and R22 are independently selected from
the group consisting of lower alkyl, aralkyl, aryl;
wherein when any one of X7-X11 represents N, that nitrogen is attached to the
adjacent
atoms by either one single and one double bond (as in pyridinyl systems), or
by two single
bonds (as in indolyl or imidazolyl systems);
wherein when any one of X7-X" represents N, and that nitrogen is attached to
the adjacent
atoms by one single and one double bond, the respective R7-R11 represents a
lone pair;
when any one of X7-X" represents N, and that nitrogen is attached to the
adjacent atoms
by two single bonds (as in indolyl or imidazolyl systems), the respective R7-
R" is selected
from the group consisting of H, lower alkyl, substituted lower alkyl, aralkyl,
substituted
4
CA 02469953 2007-09-04
aralkyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, SO2R18,
wherein R18 is
defined as in c), CORI 8, wherein le8 is defined as in c);
when n=0, R7 and R8, or R8 and R9 are combined to form a fused 5,6, or 7
membered
alkyl, substituted alkyl, heteroalky, substituted heteroalkyl, heteroaralkyl,
substituted
heteroaralkyl, aryl, substituted aryl, heteroaryl, or heteroaryl ring system;
when n=1 and X9 represents C, R7 and R8, or R8 and R1 are combined to form a
fused 5,
6, or 7 membered alkyl, substituted alkyl, heteroalky, substituted
heteroalkyl, aryl,
substituted aryl, or heteroaryl ring system; and
any one of R7-R11 represents a lone pair when the respective X7-X" represents
S or 0;
with the proviso that compounds 1, 4, 10, 14, 20, 60, 72, 105, 109, 111, 114,
124, 126,
127, 133, and 153 are excluded.
The invention relates to sulfonamide compounds of Formula I and the use of
compounds
of Formula I (including those noted within the proviso excluding the actual
compounds
themselves) for the prevention of neuronal cell loss or the treatment of nerve
cell or axonal
degradation, in either the central or peripheral nervous systems (CNS and PNS,
respectively). The invention is useful in prevention or treatment of
conditions leading to
or resulting from such diseases as Alzheimer's, Huntington's, Parkinson's,
muscular
dystrophy, diabetes, HIV, from ischemic insults such as stroke in the brain
(CNS), retinal
ganglion loss following acute ocular stroke or hypertension as in glaucoma,
and from
infection by viruses such as Hepatitis C and Herpes Simplex. Further, the
invention
provides compounds for use in treatment of neuropathies resulting from chemo-
therapeutic agents used in the treatment of HIV and proliferative disease such
as cancer,
for the treatment of inflammatory diseases.
In order to identify compounds which mimic the positive effects of NGF on
peripheral
neurons, but which lack the inherent difficulties associated with the use of
recombinant
5
CA 02469953 2007-09-04
human proteins and the rhNGF related hyperalgesia, we have developed several
in vitro
screens using a variety of neurotoxic insults. PNS neurons such as the
superior cervical
ganglion (SCG) and dorsal root ganglion (DRG) undergo apoptosis when subjected
to
NGF withdrawal. Treatment with chemotherapeutic agents such as TaxolTm,
cisplatin,
vinblastine, vincristine, and anti-viral agents such as D4T, also induce
neuronal apoptosis.
Similarly, neurons of the CNS, such as cortical neurons, are sensitive to
various
neurotoxic agents such as fl-amyloid, NMDA, osmotic shock, TaxolTm and
cisplatin.
Additionally, retinal ganglion (RG) neurons subjected to hypoxia undergo
apoptosis.
Compounds which protect neurons from neurotoxic insults such as those
mentioned above
will be useful in the treatment of the peripheral neuropathies observed in
diseases such as
diabetes and HIV. Compounds which protect neurons from chemotherapeutic
toxicity, if
given prior to concurrently with, or following, chemotherapeutic treatment
will allow for
the use of increasing concentrations of chemotherapeutics and/or extend the
duration of
chemotherapy treatments. Alternatively, enhanced recovery will be observed if
such
compounds are given during the recovery stages, and post treatment. These
compounds
will also be useful in the treatment of neurodegenerative diseases of the CNS,
such as AD,
PD, HD, stroke, MS, macular degeneration, glaucoma, optical stroke and retinal
degeneration, and the like.
We have shown that compounds of Formula I protect SCG neurons from several
neurotoxic insults, including NGF withdrawal and treatment with
chemotherapeutics such
as TaxolTm, cisplatin, and vincristine. Compounds of Formula I also protect
cortical motor
neurons from malonate induced death.
R5 R7 R8
0 N._ k7-X8
/LK N
s \ R6 w = )--( ( ?(9-R9 )
R2 ¨N S N txi
R1 R11 `R10
6
CA 02469953 2007-09-04
When such agents are administered to mice treated with TaxolTm, either before
during or
after a two week dosing period, marked improvements are observed in the
animal's
general health, weight gain, and gait, as compared to animals treated with
TaxolTm alone.
Additionally, compounds of Formula I aid in the regeneration of neurons
damaged as a
result of sciatic nerve crush.
Selected examples from Formula I have been previously described. Their uses
include
anti-bacterial agents (Gadad, A. K. EurJ. Med. Chem., 35(9), 853-857, 2000),
anti-
proliferative agents (Gadad, A. K. India. Arzneim.-Forsch., 49(10), 858-863,
1999), and as
carbonic anhydrase (CA) inhibitors (Barnish, I. T., et. al. J Med. Chem.,
23(2), 117-121,
1980; Barnish, I. T. et. al GB 1464259, abandoned; Supuran, C, T. Met.-Based
Drugs
2(6), 331-336, 1995 ¨ Co(II), Cu (II), Zn(II) complexes of compound 1).
Barnish et al.
demonstrated that certain compounds reduced the number and intensity of
electroshock
induced seizures in rats. This anti-seizure activity was linked to increased
cerebral blood
flow, attributed to the ability of these compounds to inhibit CA. No direct
evidence of
neuronal protection as a result of these compounds has been previously
demonstrated in
vitro or in vivo (ie. histology, neuronal cell count, etc.).
We have found that various aryl sulfonamide CA inhibitors do not protect SCG
neurons
from apoptosis. These finding indicate that the neuroprotection mediated by
compounds
represented by Formula I is independent of their CA activity. Additionally, we
have
prepared several synthetic derivatives of represented by Formula I which
display reduced
CA inhibition inhibit CA, while retaining their neuroprotective capabilities.
The invention relates to synthetic routes for preparation of compounds
represented by
Formula I, and methods for the functionalization of compounds represented by
Formula I.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 illustrates the protection of SCG neurons from TaxolTm induced
killing provided
by Compound 1 (AEG 3482).
7
CA 02469953 2007-09-04
Figure 2 illustrates the protection of cortical motor neurons from malonate
killing in the
presence of compound 91. 350 uM slices of P1 rat motor cortex were treated
with
malonate and incubated in media for 14 days, before malonate and drug were
added. Part
(a) shows control motor neurons, and illustrates large diamond-shaped neurons;
part (b)
shows malonate treatment alone, which results in killing with a complete loss
of neurons;
and part (c) shows 90% rescue of cortical motor neurons in the presence of
compound 91
(1 uM) and malonate.
Figure 3 illustrates the co-treatment of H460 and 0V2008 cancer cell line with
Taxollm
and Compound 1. H460 and 0V2 008 cells were treated with TaxolTm and/or
TaxolTm +
compound 1.
Figure 4 illustrates weight loss induced by TaxolTm in Spraugue Dawley rats
treated with
50% HPDC vehicle (veh/veh), compound 1 dissolved in 50% HPDC at 1, 5, or 10
mg/kg
(veh/1 , veh/5, veh/10, respectively), or TaxolTm (9 mg/kg) + compound I
dissolved in
50% HPDC at 1, 5, and 10 mg/kg (Tax/1, Tax/5, Tax/10) according to the dosing
regime
described in Example 160.
Figure 5 illustrates that gait disturbance in rats induced by TaxolTm was
reduced with
compound 1.
Figure 6 illustrates that compound 1 caused a reversal in HIM wave disturbance
induced
by TaxolTm, as indicated by H-reflex amplitude.
Figure 7 illustrates sciatic nerve recovery after crush injury, as measured by
inner toe
spread in male Spraugue Dawley rats treated with either vehicle control,
compound 1 or
compound 76.
8
CA 02469953 2007-09-04
Figure 8 illustrates the effect of intravitreal compound 1, followed by
subsequent daily
injections on protection of RGs after ocular stroke. Compound 1, given post
stroke,
protects the RG population allowing for normal conductance.
Figure 9 illustrate the neuroprotection of cortical neurons provided by
Compound 76 from
amyloid beta 25-35 toxicity. Top (a) shows control untreated cultures display
low level
annexin V staining; middle (b) shows 48 hour treatment with amyloid beta
peptide results
in the appearance of apoptotic cells; and bottom (c) illustrates co-treatment
with 10 uM
Compound 76 prevents the occurrence of annex in V stained cells.
DETAILED DESCRIPTION
The compounds represented by Formula (1) may be referred to herein
interchangeably as
as Compound (I). Compounds referred to herein by number (such as compound 1 or
compound 76) refer to the compounds outlined as Examples 1 to 153.
In the definitions of the groups of Formula I, lower alkyl means a straight-
chain or
branched alkyl group having 1 to 8 carbon atoms, such as methyl, ethyl,
propyl, iso-
propyl, n-butyl, iso-butyl, sec-butyl, tert-butyl, pentyl, iso-amyl,
neopentyl, 1-ethylpropyl,
hexyl, and octyl. The lower alkyl moiety of lower alkoxy, lower alkylsulfonyl,
lower
alkoxylcarbonyl, lower alkylaminocarbonyl has the same meaning as lower alkyl
defined
above. The acyl moiety of the acyl and the acyloxy group means a straight-
chain or
branched alkanoyl group having 1 to 6 carbon atoms, such as formyl, acetyl,
propanoyl,
butyryl, valeryl, pivaloyl and hexanoyl, and arylcarbonyl group described
below, or a
heteroarylcarbonyl group described below. The aryl moiety of the aryl, the
arylcarbonyl
and arylaminocarbonyl groups means a group having 6 to 16 carbon atoms such as
phenyl,
biphenyl, naphthyl, or pyrenyl. The heteroaryl moiety of the heteroaryl and
the
heteroarylcarbonyl groups contain at least one hetero atom from 0, N, and S,
such as
pyridyl, pyrimidyl, pyrroleyl, furyl, benzofuryl, thienyl, benzothienyl,
triazolyl, quinolyl, iso-quinolyl, benzoimidazolyl, thiazolyl, benzothiazolyl,
oxazolyl, and
indolyl. The aralkyl moiety of the aralkyl and the aralkyloxy groups having 7
to 15 carbon
9
CA 02469953 2007-09-04
atoms, such as benzyl, phenethyl, benzhydryl, and naphthylmethyl. The
heteroaralkyl
moiety of the heteroaralkyl and the heteroaralkyloxy groups having 7 to 15
carbon such as
pyridylmethyl, quinolinylmethyl, and iso-quinolinylmethyl. The substituted
lower alkyl
group has 1 to 3 independently-substitutuents, such as hydroxyl, lower
alkyloxy, carboxyl,
lower alkylcarbonyl, nitro, amino, mono- or di-lower alkylamino, dioxolane,
dioxane,
dithiolane, and dithione. The lower alkyl moiety of the substituted lower
alkyl, and the
lower alkyl moeity of the lower alkoxy, the lower alkoxycarbonyl, and the mono-
and di-
lower alkylamino in the substituents of the substituted lower alkyl group have
the same
meaning as lower alkyl defined above. The substituted aryl, the substituted
heteroaryl ,
the substituted aralkyl, and the substituted heteroaralkyl groups each has 1
to 5
independently-selected substituents, such as lower alkyl, hydroxy, lower
alkoxy, carboxy,
lower alkoxycarbonyl, nitro, amino, mono or di-lower alkylamino, azido, and
halogen.
The lower alkyl moiety of the lower alkyl, the lower alkoxy, the lower
alkylamino, and the
mono- and di-lower alkylamino groups amoung the susbtituents has the same
meaning as
lower alkyl defined above. The heterocyclic group formed with a nitrogen atom
includes
rings such as pyrrolyl, piperidinyl, piperidino, morpholinyl, morpholino,
thiomorpholino,
N-methylpiperazinyl, indolyl, and isoindolyl. The cycloalkyl moeity means a
cycloalkyl
group of the indicated number of carbon atoms, containing one or more rings
anywhere in
the structure, such as cycloalkyl groups include cyclopropyl,
cyclopropylmethyl,
cyclobutyl, cyclopentyl, cyclohexyl, 2-norbornyl, 1-adamantyl and the like.
The
fluoroalkyl moiety means a lower fluoroalkyl group in which one or more
hydrogens of
the corresponding lower alkyl group, as defined above, is replaced by a
fluorine atom,
such as CH2F, CHF2, CF3, CH2CF3, and CH2CH2CF3.
Some of the compounds described herein contain one or more chiral centres and
may thus
give rise to diastereomers and optical isomers. The present invention is meant
to
comprehend such possible diastereomers as well as their racemic, resolved and
enantiomerically pure forms, and pharmaceutically acceptable salts thereof.
The term "subject" or "patient" as used herein may refer to mammals including
humans,
primates, horses, cows, pigs, sheep, goats, dogs, cats and rodents.
CA 02469953 2007-09-04
The pharmaceutical compositions of the invention are administered to subjects
in effective
amounts. An effective amount means that amount necessary to delay the onset
of, inhibit
the progression of, halt altogether the onset or progression of or diagnose
the particular
condition or symptoms of the particular condition being treated. In general,
an effective
amount for treating a neurological disorder is that amount necessary to affect
any
symptom or indicator of the condition In general, an effective amount for
treating
neuropathies and neuropathic pain will be that amount necessary to favorably
affect
mammalian cancer cell proliferation in situ. When administered to a subject,
effective
amounts will depend, of course, on the particular condition being treated; the
severity of
the condition; individual patient parameters including age, physical
condition, size and
weight; concurrent treatment; frequency of treatment; and the mode of
administration.
These factors are well known to those of ordinary skill in the art and can be
addressed with
no more than routine experimentation. It is preferred generally that a maximum
dose be
used, that is, the highest safe dose according to sound medical judgment.
A variety of administration routes are available. The particular mode selected
will depend,
of course, upon the particular condition being treated, the particular drug
selected, the
severity of the condition being treated and the dosage required for
therapeutic efficacy.
The methods of this invention, generally speaking, may be practiced using any
mode of
administration that is medically acceptable, meaning any mode that produces
effective
levels of the active compounds without causing clinically unacceptable adverse
effects.
Such modes of administration include oral, rectal, sublingual, topical, nasal,
transdermal,
intradermal or parenteral routes. The term "parenteral" includes subcutaneous,
intravenous, intramuscular, or infusion. Oral routes are preferred.
Dosage may be adjusted appropriately to achieve desired drug levels, locally
or
systemically. Generally, daily oral doses of active compounds will be from
about 0.01
mg/kg per day to 1000 mg/kg per day. It is expected that IV doses in the range
of about 1
to 1000 mg/m2 per day will be effective. In the event that the response in a
subject is
11
CA 02469953 2007-09-04
insufficient at such doses, even higher doses (or effective higher doses by a
different, more
localized delivery route) may be employed to the extent that patient tolerance
permits.
The compositions may conveniently be presented in unit dosage form and may be
prepared by any of the methods well known in the art of pharmacy. All methods
include
the step of bringing the conjugates of the invention into association with a
carrier that
constitutes one or more accessory ingredients. In general, the compositions
are prepared
by uniformly and intimately bringing the compounds into association with a
liquid carrier,
a finely divided solid carrier, or both, and then, if necessary, shaping the
product.
Compositions suitable for oral administration may be presented as discrete
units such as
capsules, cachets, tablets, or lozenges, each containing a predetermined
amount of the
active compound. Other compositions include suspensions in aqueous liquors or
non-
aqueous liquids such as a syrup, an elixir, or an emulsion.
Other delivery systems can include time-release, delayed release or sustained
release
delivery systems. Such systems can avoid repeated administrations of the
active
compounds of the invention, increasing convenience to the subject and the
physician.
Many types of release delivery systems are available and known to those of
ordinary skill
in the art. They include polymer based systems such as polylactic and
polyglycolic acid,
.polyanhydrides and polycaprolactone; nonpolymer systems that are lipids
including sterols
such as cholesterol, cholesterol esters and fatty acids or neutral fats such
as mono-, di and
triglycerides; hydrogel release systems; silastic systems; peptide based
systems; wax
coatings, compressed tablets using conventional binders and excipients,
partially fused
implants and the like. In addition, a pump-based hardware delivery system can
be used,
some of which are adapted for implantation.
A long-term sustained release implant also may be used. "Long-term" release,
as used
herein, means that the implant is constructed and arranged to deliver
therapeutic levels of
the active ingredient for at least 30 days, and preferably 60 days. Long-term
sustained
release implants are well known to those of ordinary skill in the art and
include some of
12
CA 02469953 2007-09-04
the release systems described above. Such implants can be particularly useful
in treating
solid tumors by placing the implant near or directly within the tumor, thereby
affecting
localized, high-doses of the compounds of the invention.
When administered, the Formulations of the invention are applied in
pharmaceutically
acceptable compositions. Such preparations may routinely contain salts,
buffering agents,
preservatives, compatible carriers, and optionally other therapeutic
ingredients. When used
in medicine the salts should be pharmaceutically acceptable, but non-
pharmaceutically
acceptable salts may conveniently be used to prepare pharmaceutically
acceptable salts
thereof and are not excluded from the scope of the invention. Such salts
include, but are
not limited to, those prepared from the following acids: hydrochloric,
hydrobromic,
sulphuric, nitric, phosphoric, maleic, acetic, salicylic, p-toluenesulfonic,
tartaric, citric,
methane sulfonic, formic, malonic, succinic, naphthalene-2-sulfonic, benzene
sulfonic,
and the like. Also, pharmaceutically acceptable salts can be prepared as
alkaline metal or
alkaline earth salts, such as sodium, potassium or calcium salts.
Suitable buffering agents include: phosphate buffers, acetic acid and a salt
(1-2% WN);
citric acid and a salt (1-3% WN); and phosphoric acid and a salt (0.8-2% WN),
as well as
others known in the art.
Suitable preservatives include benzalkonium chloride (0.003-0.03% WN);
chlorobutanol
(0.3-0.9% WN); parabens (0.01-0.25% WN) and thimerosal (0.004-0.02% WN), as
well
as others known in the art.
Suitable carriers are pharmaceutically acceptable carriers. The term
pharmaceutically
acceptable carrier means one or more compatible solid or liquid filler,
dilutants or
encapsulating substances that are suitable for administration to a human or
other animal.
The term "carrier" denotes an organic or inorganic ingredient, natural or
synthetic, with
which the active ingredient is combined to facilitate the application. The
components of
the pharmaceutical compositions are capable of being commingled with the
molecules of
the present invention, and with each other, in a manner such that there is no
interaction
13
CA 02469953 2007-09-04
which would substantially impair the desired pharmaceutical efficacy. Carrier
Formulations suitable for oral, subcutaneous, intravenous, and intramuscular
administration etc., are those which are known in the art.
The compounds of the invention may be delivered with other therapeutic agents.
The
invention additionally includes co-administration of compound I of the
invention with
other compounds known to be useful in treating neurodegenerative diseases,
typified by
but not limited to, acetylcholinesterase inhibitors for treating AD, such as
tacrine,
doneprizil, and rivastigmin, and L-dopa for treating PD, and ACE inhibitors
and insulin
for the treatment of diabetes.
In the case of peripheral neuropathy induced by a toxic agent, compound I
would be
delivered separately before, simultaneously with (ie. in the form of anti-
cancer coctails,
see below), or after exposure to the toxic agent. Preferably, compound I and
the
chemotherapeutic agent are each administered at effective time intervals,
during an
overlapping period of treatment in order to prevent or restore at least a
portion of the
neurological function destroyed by the neurotoxic or chemotherapeutic agent.
The
chemotherapeutic can be any chemotherapeutic agent that causes neurotoxicity,
such as
dideoxyinosine, deoxy cytizine, D4T, cisplatin, etoposide, vincristine,
epithilone or its
derivatives, or TaxolTm/TaxoterTm and derivatives thereof, which are
representative of the
classes of agents induce neuropathies.
By "toxic agent" or "neurotixic agent" is meant a substance that through its
chemical
action injures, impairs, or inhibits the activity of a component of the
nervous system. The
list of neurotoxic agents that cause neuropathies is lengthy (see a list of
candidate agents
provided in Table 1). Such neurotoxic agents include, but are not limited to,
neoplastic
agents such as vincristine, vinblastine, cisplatin, TaxolTm, or dideoxy-
compounds, eg.,
dideoxyinosine; alcohol; metals; industrial toxins involved in occupational or
environmental exposure; contaminants in food or medicinals; or over-doses of
vitamines
or therapeutic drugs, eg. Antibiotics such as penicillin or chloramphenicol,
or mega-doses
of vitamins A, D, or B6.
14
CA 02469953 2011-10-28
Table 1
Neurotoxic Agents
AGENT ACTIVITY AGENT ACTIVITY
acetazolimi de diuretic imipramine antidepressant
_
acrylamide flocculant, grouting agent indomethacin anti-
inflammatory
adriamycin antineoplastic inorganic lead toxic metal in paint,
etc.
alcohol (ie. ethanol) solvent, recreational drug iso-niazid
antituberculousis
almitine respiratory stimulant lithium
antidepressant
amiodarone antiarrthymic methylmercury industrial waste
_
amphotericin antimicrobial metformin antidiabetic
_
arsenic herbicide, insecticide methylhydrazine
synthetic intermediate
aurothioglucose antirheumatic metronidazole _
antiprotozoal
barbiturates anticonvulsive, sedative misonidazole _ .
radtosensitizer
buckthorn toxic berry nitrofurantoin urinary antiseptic
carbamates insecticide nitrogen mustard antineoplastic, nerve
gas
carbon disulfide industrial applications nitous oxide
anesthetic
_
chloramphenicol antibacterial organophosphates insecticides
chloroquine antimalarial ospolot anticonvulsant
chlorestyramine antihyperlipoproteinemic penicillin
antibacterial
cisplatin antineoplastic perhexiline antiarrhythmic
clioquinol amebicide, antibacterial perhexiline maleate
antiarrythmic
colestipol anti hyperlipoproteinemic phenytoin
anticonvulsant
colchicine gout suppressant platinum drug component
colistin antimicrobial primidone anticonvulsant -
cycloserine antibacterial procarbazine antineoplastic
cytarabine antineoplastic pyridoxine vitamin B6
dapsone dermatological including sodium cyanate
antisickling
leprosy
dideoxycytidine anatineoplastic streptomycin antimicrobial
dideoxyinosine antineoplastic sulphonamides antimicrobial
dideoxythymidine antiviral suramin anteneoplastic
disulfiram antialcohol tamoxifen antineoplastic
doxorubicin antineoplastic TaxolTm antineoplastic
CA 02469953 2007-09-04
ethambutol antibacterial thalidomide antileprous
ethionamide antibacterial thallium rat poison
glutethimide sedative, hypnotic triamterene diuretic
gold antirheumatic trimethyltin toxic metal
hexacarbons solvents L-trypophan health food
additive
hormonal contraceptives vincristine antineoplastic
hexatriethylolmelamine fireprooing, crease vinblastine
antineoplastic
proofing
hydralazine antihypertensive vindesine antineoplastic
hydroxychloroquine antirheumatic vitamine A or D mega doses
Several neurotoxic agents and protocols may be used to induce apoptosis in SCG
neurons.
Several of these insults include the withdrawal of trophic support (for
example NGF),
treatment with neurotoxic chemotherapeutics such as TaxolTm, cisplatin,
vincristine, or
vinblastine, and treatment with neurotoxic anti-virals such as D4T. Selected
compounds
represented by Formula I have been found to inhibit apoptosis induced by the
above
insults.
Neurotrophins are critical to the growth, development, and survival of small
fiber neurons
of the PNS. SCG neurons are neurons of the PNS that undergo apoptosis upon NGF
withdrawal. In a typical experiment SCG neurons are cultured in the presence
of NGF,
which induces survival and neurite out-growth. After 5 days the NGF is removed
by
either the addition of anti-NGF polyclonal antibody (Sigma) or by repeated
washings (4
times) with NGF free media, resulting in the apoptosis of up to 90% of the
neurons after
48 hours, as measured by MTS staining. The addition of selected compounds of
Formula I
to the final cellular media provides upwards of 100% protection, at drug
concentrations
ranging from 3 to 50 ptM (see Example 154).
TaxolTm is regularly used in breast cancer chemotherapy. In cancer cells
TaxolTm binds to
the cyto-skeletal protein tubulin, thereby inhibiting normal microtubular
assembly and
inducing cellular apoptosis. Despite its potency as an anti-tumour agent,
TaxolTm is also
16
CA 02469953 2007-09-04
toxic to neurons, inducing dose limiting peripheral neuropathies. The addition
of TaxolTm
(100 ng/mL) to cultured SCG neurons induces the degradation or loss of upwards
of 80 %
of the neurons. The addition of selected compounds of Formula Ito the cellular
media,
concurrently with TaxolTm, protects upwards of 100% of the neurons, at drug
concentrations ranging from 3 to 50 1\4 (see Example 155 and Figure 1).
The mechanism of Cisplatin's anti-cancer action is not fully understood, but
is believed to
involve DNA binding and cleavage. Cisplatin is highly toxic to neurons. The
addition of
cisplatin (3 g/mL) to cultured SCG neurons induces apoptosis of upwards of
80% of the
neurons. The addition of selected compounds of Formula Ito the cellular media,
concurrently with cisplatin, protects upwards of 100% of the neurons, at drug
concentrations ranging from 1 to 50 M (see Example 156).
Similarly, vincristine and vinblastine are commonly used anti-tumour agents
whose mode
of action involve tubulin binding. As above, the addition of vincristine (100
ng/mL) to
cultured SCG neurons induces apoptosis of upwards of 80% of the neurons. The
addition
of selected compounds of Formula Ito the cellular media, concurrently with
vincristine,
protects upwards of 100% of the neurons, at drug concentrations ranging from 1
to 50 M
(see Example 157).
Various neurodegenerative diseases are related to the cellular or functional
loss of motor
neurons of the CNS and PNS. ALS is a characterized by motor neuron loss as a
result of
mitochondrial dysfunction, which can be mimicked in culture by the addition of
malonate
to organotypic brain slices. P1 rat motor cortex brain slices were cultured
for 2 weeks
prior to drug and malonate addition. After an additional two weeks the slices
were fixed
and stained with SMI-32 antibody which selectively stains motor neurons found
in layer V
of the cortex. Compound 91 protected upwards of 80 % of these labeled motor
neurons at
a drug concentration of 1 M (Example 158).
Taken together, compound of Formula I display remarkable neuroprotective
capabilities,
against a wide range of insults in both the CNS and the PNS. One of the
intended uses of
17
CA 02469953 2007-09-04
these agent is in the conjugation with chemotherapeutic agents. If compounds
represented
by Formula I were to protect cancer cells from the same chemotherapeutic
agents, it would
have limited value. Two pieces of evidence suggest these compounds do not
protect
cancer cells from chemotherapeutics. Selected compounds represented by Formula
I have
previously been shown to be anit-proliferative (Gadad, A. K. India. Arzneim.-
Forsch.,
49(10), 858-863, 1999), suggesting these compounds will be beneficial when
used in
conjunction with other chemotherapeutic agents. Additionally, we have shown
that
compound 1 displays no protection when human ovarian carcinoma cells (0V2008)
and
human lung carcinoma cells (1-1460) were treated with TaxolTm and/or cisplatin
(see
Example 159 and Figure 3).
Compound I and several of its derivatives have been reported to be potent
inhibitors of
carbonic anhydrase (CA) (Barnish, I. T., et. al. J. Med. Chem., 23(2), 117-
121, 1980). CA
plays an important role in maintaining both intra- and extra-cellular pH
levels. In an effort
to determine whether the neuroprotective profile of compound 1 was due to CA
inhibition,
a number of well-known, cell permeable, aryl sulfonamide CA inhibitors were
evaluated
against the TaxolTm killing of SCGs. Dorzolamide, (Ponticello, G. S., et. al
J. Med.
Chem., 1987, 30, 591) aminobenzolamide N-acetylaminobenzolamide,
acetazolamide, and
methazolamide (see Marten, T. H..1 Glaucoma, 1995, 4, 49) all failed to
significantly
inhibit TaxolTm induced killing of SCGs at concentrations as high as 50 M.
Additionally, the ability of compounds represented by Formula Ito inhibit CAII
varied
greatly depending upon the substitution patterns found on the sulfonamide or
the C6
position (see Example 163). For example, compound 137 is the N-methyl
derivative of
compound 1, compound 137, displays a 100 fold decrease in CAII activity
(CAII(50) 2.06
!AM and 250 nM, respectively) while retaining a similar IC(50) against Taxol
(7 M each).
Similarly, compound 77 is a poor CAII inhibitor (IC(50) 6.3 12M), but displays
a more
potent against Taxol killing of SCGs (IC (50) 2 M). Based on these results it
is clear that
although compounds of Formula I are known CA inhibitors, the primary mechanism
by
which it is protecting neurons appears to be independent of CA inhibition.
18
CA 02469953 2007-09-04
Adenovirus overexpression of Erkl and Erk2, two members of the MAP kinase
family of
signaling proteins, have been shown to stimulate neuronal out-growth and the
formation of
new synaptic connections in primary neurons of the PNS and CNS. Additionally,
the Erks
protect cultured neurons from a number of insults including neurotrophin
withdrawal
(Bonni, A., etal., Science, 1999, 286, 1358-1362). A dramatic increase in Erk
activity
was observed in both PC12 cells and in primary cultures of sympathetic neurons
when
treated with compound I. The activity of Akt, however, remained unchanged when
both
PC12 cells and SCGs were treated with compound 1. Akt is activated by NGF and
has
been demonstrated to be neuroprotective in both PNS and CNS neurons. Compound
1,
therefore, protects neurons by activating a subset of NGF-stimulated signaling
pathways.
TaxolTm commonly causes dose dependent peripheral neuropathies during cancer
treatment. When treated with TaxolTm (9 mg/kg in Cremophor EL and ethanol)
twice
weekly for 3 weeks, Sprague Dawley rats displayed acute symptoms of
chemotoxicity,
characterized by reduced appetite, weight loss, gait disturbance (a general
marker of
TaxolTm induced peripheral neuropathy), and general poor health (see Example
160). For
example, over a thirteen day period control animals gained an average of 50 g,
whereas the
TaxolTm treated animals displayed no weight gain (see Figure 4). All of the
TaxolTm
treated animals developed peripheral neuropathies, characterized by 'tip toe
walking'. The
extent of this neuropathy was analyzed by quantifying the refracted light
captured by a
video camera as the animals walked over a glass plate. This data was analyzed
by
Northern Eclipse software. The TaxolTm treated animals displayed a 46 %
reduction in
foot-pad contact with the glass plate, as compared to control animals (see
Example 160).
When compound 1 (5 mg/kg) was given with TaxolTm (9 mg/kg) on a bi-weekly
schedule,
the animals displayed greatly improved health. This was characterized by
normal weight
gain, as compared to control (Figure 4), and a reduction in the severity of
the peripheral
neuropathies; a 23 % loss in foot pad contact was observed, as compared to a
46 % loss in
the animals treated with TaxolTm alone (see Example 160 and Figures 5 and 6).
No acute
signs of toxicity were observed in animals in acute toxicity studies with
compound 1 alone
(1, 5, and 10 mg/kg for 3 weeks).
19
CA 02469953 2007-09-04
The sciatic nerve crush model is a representative model of axonal repair and
regeneration.
The sciatic nerve is physically crushed with forceps at the mid-thigh; only
the right leg is
injured, the left leg serving as a control. The axons die from the crush point
to their point
of innervation. Functional loss of the axons is rapidly observed as the
animals drag their
right leg and the toes of the right leg no longer spread. Recovery is observed
in
approximately 28 days as the animals regain use of their right leg. More
quantitative
measurements of recovery include toe spread measurements between the digits 1
and 5
and digits 2 and 4, gait analysis and electrical conductivity from the toes to
the injury site
(see Example 161).
Rats were subjected to the crush injury and treated with either vehicle
control or
compounds 1, 76 or 111 (1 and 10 mg/kg). Functional recovery was measured as
above
and improved recovery was observed when the animals were treated with
compound. For
example, increase toe spread was observed for those animal treated with
compound (see
Figure 7).
Various diseases which result in loss of vision are related to increased inter-
ocular
pressure and ocular stroke or ischemia. Loss of the retinal ganglion (RG)
occur during
ischemic insult and in diseases such as diabetes and glaucoma. A model of
inter-ocular
ischemia involves an invasive increase in ocular pressure which results in the
collapse of
the central retinal artery. Retinal ischemia is confirmed by whitening of the
iris and loss
of red reflex. The inter-ocular pressure is normalized after 30 minutes. This
procedure is
performed on the right eye and the left eye serves as a control. Compound was
given
either by intra-vitrial injection or via SC injections at 10 mg/kg (see
Example 162).
The health of the RG neurons was assessed by means of histological staining of
retinal
slices and electro-retinogram (ERG) recordings. Histology of the control
animals showed
almost complete loss of the RG layer, where as animals treated with compound 1
showed
healthy RG layers. Similarly, significant improvements were observed in the
ERG for
those animals treated with compound verses vehicle control animals (see Figure
8). This
CA 02469953 2007-09-04
protection was observed for both the animals which received intra-vitrial
injections and
those that were treated systemically (SC).
Alzheimer's disease is one of the biggest unmet medical needs in neurology.
One of the
main areas of AD research has been deposition and neurotoxicity of amyloid
beta peptide
fragments. Amyloid peptides are potently toxic to cortical neurons and
protection of the
cortical neurons would be a very desirable therapeutic target. We establish
mixed
neuronal/glial cortical cultures from postnatal rat pups. Amyloid beta
peptides are
potently toxic to neurons in these cultures. Exposure to 10 uM 25-35 amyloid
beta
increased the number of apoptotic cells compared to control. Compound 76
prevented the
appearance of annexinV positive cells indicating that it protected in vitro
against the
amyloid beta peptide.
For any of the compounds having the structure of Formula I which bear
similarity to those
known in the art, the use of these compounds for treatment and/or prevention
of
neurological disorders, cancer, inflammation, or symptoms related thereto are
encompassed by the invention.
21
CA 02469953 2007-09-04
Examples of Formula I are provided below in Table 2. These compounds are
referred to
throughout the disclosure as their corresponding example number.
Table 2: Compounds
Example STRUCTURE
1 o rN X 4110
o¨\s\s
---* )=-N
-- \
NH2
0 N.......
H2N¨g4 1 \ 111
2 II
0 S N
F
O NNH2N--g-- \
3 .
II
O S------N
F
O N¨N N F
S---
IV-----
H2N
F
0 ,
0-g_N r? \ F
H2NI
S-----N
F
F
6N-N N * F
(?\s---0=N F F
NI-12
22
CA 02469953 2007-09-04
F (CH3
F
7 N¨N N . S
0
F
--S F
NH2
F *
F
S
8 N¨N" .
0 \
\s_.--B 2=N
F F
0-- \ \ S
NH2
F (--0
F 0 Nõ)
9 N¨N, ' F
I-12N, õ.4 )--------N F
CA S
IiCI
N¨N N
0 ii
S
,
H2,N, \\ S
0
CI
NH2 N.,
11 0=14 1 \ II CI
II
0 S N
40 a
12 0 iNi¨N N
\\ ,--- )=N a
--S a
0-- \ s
NH2
13 0\\ ii¨N\ *
Br
,--N
--S¨ N
0-- \ S
NH2
23
CA 02469953 2007-09-04
0 N-N N = Br
14 oH \_N
H2N
N -N
15 H2N S N
Me0
0 N
=/
16 H2N
H3C/
o C:1CH,
17 N-N
o44 \_N
H2N
0¨CH,
0
rj Ipt
18 /
H2N
H,C-0
BrH
H3C-0 CH3
19 0
0 N-N
)--=N
S
NH2
NH2
20 0=1--CN 0
s
0
0"-N
NH2
21 0=14 N
0
0 SN
24
CA 02469953 2007-09-04
OH
0 ,
22 0 NINil \ = OH
H2N S¨"--N
0.--c)
23 . o
0 N¨N N
--/4
,., ,µ,,,\s\ 0)=-N
. 121 \ \ \ V
0
N-N N *
24 0
\\ )¨
NH2 F--NF
F
F¨.../.....
F
25 * 0
0----S\---S-2¨
NH2
OH
26
1H2 N...... /
/
0=1-- 1 \ 'I,0
II ---
0 S N
0
0).LCH,
27 ti1H2 ri N-__N
0=*LS \ = 0
8 s .rsJ
N,
NH2 N......
28 0=14 1 \ 40 0 /-1
0 S N
0 N__
II ri
29 H2N_-1
1 ....._ F
0\f___
0 S----N\ = F
F
CA 02469953 2007-09-04
/CH3
0-r
30 NH2
o=¨ L\
0 S N
=
31 0
N142
0=14 \ 0
o S N
32 NH2
0=1-- 4.
0Il S N
(N)
33 - .111
N"\_NN
CH,SO,H
S
0 CH,SO,H
CH3
0 OH
NH2 N.....
34 0=14 II * 07-1
0
CH,
0
NH2 N
\ = 0
g S
,CH3
0
NH2 N..õ2_1
0=14 1 \ 0
0 S N
36
O N
e N
H2N
37 I
NO2
26
CA 02469953 2007-09-04
.1CI
N¨N N
38
--S
S 0
NH2
II
N,N 0
39 H N¨S
2 I I
0 OH
N =
oµS
NH,
/-
41
0 x
--S
S
NH
NH2
42 0=14 ¨N
11
0
= \s\
N-N N CH3
43
O\ s
NH2
II
44
0 N_NN
0
0-- S
NH2
CH3
450 N-N N= \-CH2
0-- S
NH2
27
CA 02469953 2007-09-04
N¨N CH3
46 o
\\sX )=N
0¨ S
NH,
yH2
47 0=S¨ 11 CH3
I I
CH3
N¨N
48 0\\
0=S S N CH3
H2N
CH3
49
N¨" CFFI3
0 n
)=N
--S
S
NH,
1-1.3C CH3
CH
50=N¨N CH3
)=N =CH3
S
NH,
Ad
51
N¨N N*
C\kss )=N
S H3C
NI-I2
F F
52 0 N¨N * F
)=¨N
¨5
0¨ S
NH,
28
CA 02469953 2007-09-04
CI
53 0 N-N
S
NH2
o N-N
54 =
--S
0-- S
NH2
H3C CH3
CH,
NH2
55 0=1--CNO OH
OH S
CH,
H3C CH3
CH3
NH2
I N,
56
0 S.-"N
CH3CH3
CH,
H3C 4110 c
H3
57
N-N QH3C
0
\
- s )=N
H3C
0-- S
NH2
N N ¨z,0 H3
/ p
58 H2N H
\N-4,
CH3
59
0
II
// N"µ
H2N¨S ( F
\ /
0
29
CA 02469953 2007-09-04
N¨N4g
60 o
--S
S
NH,
61 14111
N¨N
S
NH,
O N¨N
s041Ik
62 0.,11
I -
H2N
0
63
H2N'
Br 0¨CH,
NH2
I
64 s--L'N' 1110
CH,
65 0 N¨N
¶s)=--N
H2N
=
66 NHk N-
04
011 S
CA 02469953 2007-09-04
0 N__
044 211,_\ = NO
67 H2 N S N
0 N.....
A4 Nti \
0 = NI-0
68
H2NI S2N
z-z-
N 0
69 N¨N 0
c\'\
.-- S s
0' \
NH2
0 N
0:.-14 :II .__.\ 4. NH2
70 i
H2N S N
IIa11
N_N .N 0 o01-4 \
--S
0--- \ S
NH, I
Bµ011
0 N-N N .
H,NI V--- =
F
WI F
F
0----µR\SCI-S)---N =F 414 F
NH2
31
CA 02469953 2007-09-04
= OH
74 N-N N- *
0
\\
-s j4 ),N
NH2
*
75 N-N N *
0
\\ ..__ )_,N o\CH3
--S
0--- \ S
NH2
=
N
76 0 N- N, liti 0
/
\\ --II, )=N H3C
--S
0-- \ S
NH2
0µ
CH,
77 N-N N . *
0
NH,
78 N-N N = 0
F---N F
H2N--- \\ S
0
0 N___
II b 1( \
79 II , # # \
0 - N
0-j
F F
F
80 0 N
II //
H2N-S- --- III \ = =
II
0 S-N
32
CA 02469953 2007-09-04
81
0 N-N
)--=N
\ S
NH2
F F
= F
82 0 N-N N
)=-N
\ S
NH2
83
N-N =
)=N F F
sN
H2N S
0
S
84 0 N-N
H2N---\\ S
0
0 N
H N¨g4
85C
2 II ,N
0 6
NH2
\
11
0 S N
86
=
H,C,0
87 C\ 0
N¨N N =
k
NH2
33
CA 02469953 2007-09-04
II //
H N¨S-- ...õ..L \ = 0
2 II
0 S N
88
=
F
0
N-N N . . F
89 0 ,
(311,k
S
/ S F
H2N
0 NI,
H2N14 1 \ . 0
II
90 0 S N
6
-N
0 N
Fl2N¨I4 1 \ = 0
oil S N
91
CI
N--N \
H2N¨S4 .. j... \ . 0
II
92 0 S N
= CI
CI
? N.--,N \
H2N¨S I \ = 0 Br
II
93 0 S'N
=
0 N,_
=
, ¨ 0
94 8 S N
. Br
34
CA 02469953 2007-09-04
Br
95 .
0
i 1
0 S N \ C
H4¨
H2N¨S¨ ),... \ * 0
11
O S N
96
0
1-130¨N\
CH3
O N,...
H3N¨ IA-- 7 \ * 0
II
O S'----N
97
=
CH3
H3C
li N-...N µ
* 0
11 s---IN
0
98
0
0
/
H3C
* 0
N¨N i
N--
0
0----S\ S
NH2 Oil
100 0
N¨N N . 0, F
i
0 1 i
S----4=s)--=N F
I
H2N
¨0 CH,
O N,...
.(
101 H2N-1-- 1 \ = 0/ \ o
8 S N
O N,...
0,ThrOH
H2 N¨g--</ I \
=
102 II
O S N 0
CA 02469953 2007-09-04
CH3
0 N
= 0
103 0
H2N S N
0
H3C
CI
CH3 * cH3
104 N-N
Cl\\ A )=N
õ-s s
o
NH2
105 H2 N-
0
CIH
106 0 N¨N X \ I
)=N
S
NH2
HI-BrH
,N
107
¨S
S
NH2
N.%)
N¨N
108 ()\\ )-"="N
--S s
\
NH2
109 N¨N 0
)=N
--S s 0
NH2
36
CA 02469953 2007-09-04
\=
NC1
110 N-,.m . 1
0õ A ------N 0
--S S
0-- \
NH2
,.__-,4
1\11H2 *
111 0 )____
II s N 0
0
0 N S
112 H2N¨S .1.z:,,.... %... j
I I
0 S N
S.
113 \ I
N-N '..`- \
0 \\S)LS*-.-.LN
H2N
BrH
N¨N/) n
1 0
.¨\\s---
14 )---=N S----N N+
NH2 0
Fisc =
115 N-N N
, /
0 11 \ S
\\ N
0-"SrNSr-
NH2
116 NH2 N___ =
I
0=S 1 \
II \ S
0 S N
37
CA 02469953 2007-09-04
117 =
I
11
0SN
0-N
=
118
0 N-N
S
NH,
0-N CH
\ 3
N-N
0
119 0
--S-
S
NH2
0=
NH2 _1\13 c1\11
120 H
o S N N
H3C
S-\\
121 o
--
0--SS
NH2
fl,CõtN
122 0 N-N Ns3--CH3
--S
0-- S
NH,
CI
NH
123 0=1
ii
38
CA 02469953 2007-09-04
BH
Br =124 0 N¨N
\\s,
0-- \ S
NH,
Br
NH m
2 (1.=_-)/
125 0=1-4
0
Br
126 0 N¨N
)¨N
0-- S
NH,
Br
CI
N¨N N
127 0
\\
,s-
0-- s
NH2
Br
0
128 0_11 N N
-Sy Br
H2N
Br
0
N
129 /
H,N SN
Br H3C/0
Br
130 04 \ *kik
H2N S N
39
CA 02469953 2007-09-04
131 0 N-N aN =
\
oss\ s)=N
NH2
BrH
132 N-N
Br
= *
CL A=N
--S
0-= S
NH2
Br
NN"0
133 0 S
0
)=N
H2 N S
0
Br
134 H2N¨S4 1 \ F
0
BrH
135
9
11.
H
2 s-
136
S.
H2N S N
0 N¨N N =
137
H3C¨N
CA 02469953 2007-09-04
F
F
N-M" 1. F
o\\ ___,. "..\____N F
138
F
NI
i
HC
0 N¨N 'N, =
139
S---,,
CH,
F
F
140 F
0 n \
\\ ---4c 7---N F
--s F
0-- \ S
1-1,?L'0H,
I N
O õ
zz._4 7 \ *
'-i---Fi cS N
H,/il3)
H,C/O
Ili11
III0 N-
4 s---1-'-----N
H,C1
0
H,C
1-13C
0 N,
044 NI \ .
H30---N\I Szls-N
CH3 Br
Br
0-41-141 \ =
Hp-NI Br
\s--.1:---N
\CH,
41
CA 02469953 2007-09-04
O N
n 114 N \ =
.-...--:z j______
145 /
s
LI r..-- N S N
e 13%.= \
CH3 OH
0 N
0_-__sii_\/, y \ .
H30¨N\ c ,,
0
/
146 ,, -
,L.,CH3 0 $
O rµL
\ .
¨S-
147 / S===.N
it
H3C¨N
\cH3
Na 0 N,
148 \N¨ I4 1 \ =
ii
0 S N
Na 0 N--
II ____
\N¨S¨ \ . =
149 II c
O - N
0¨CH3
Na On N¨N
\,g4 1 \ = *
II
150 0
F
F F
F F
0 N,
151 H2N--1¨ NI1 \ = N=NEN
11 ---1.----:
0 S N
F F
F
F
0 1µ11µi
152 0 p¨N, N
F
--S F
N
/
H,C CH3
42
CA 02469953 2007-09-04
SYNTHETIC PROCEDURES
5-Amino-1,3,4-thiadiazole-2-sulfonamide, intermediate El, was prepared by the
acid
hydrolysis of acetazolamide (Aldrich). Selected 2-bromoethanones were
purchased from
either Aldrich Chemical Co. or from Maybridge Inc.
Various acetophenones were readily prepared by the following protocols. A
selection of
4-phenoxyacetophenones were prepared under standard Ullmann condensation
conditions
by heating 4-fluoroacetophenone with the appropriate phenol and K2CO3 in DMF
or
DMAc.
0 OH 0
+ R K2CO3, solvent
heat
0 B(OH)2
[10 8R Pd Ln, base
solvent, heat
Selected 4'-arylacetophenones were prepared by Suzuki coupling of either 3'-
or 4'-
haloacetophenone with an arylboronic acid, or 4-acetylbenzeneboronic acid with
an
arylbromide, using an appropriate palladium catalyst, base, and solvent
system. These
products may be obtained using alternative coupling partners; ie. Suzuki
coupling between
aryl bromides and aetophenone boronic acids, the use of flouroboranate salts,
the use of
Stille couplings between aryl bromides and arylstannanes, etc..
Various acetophenones were a-brominated using bromine or pyridinium tribromide
in an
appropriate solvent system.
43
CA 02469953 2007-09-04
The imidazo[2,1-b]-1,3,4-thiadiazole sulfonamides were prepared according to
literature
procedures. For example, compound 1 was prepared in good yield by refluxing
intermediate Al, with an 2-bromoacetophenone, intermediate BI, in either
alcohol or 1,4-
dioxane, for 48 hours.
N¨N
H2N, j/ + 101 Br solvent
Ns ¨NH2
0/ µ0 reflux H2N S"-L-N
Al B1
Compound I was either mono- or dialkylated by the treatment of compound 1 with
the
appropriate alcohol (1 or 2 equiv), triphenyphosphine, and DIAD or polymer
supported
DIAD to yield compounds such as 137 and 139. Alternatively, N-alkylation may
be
accomplished using Mel and NaF/alumina () as base, for the conversion of Ito
137.
01.
0
_<
H2N S N ROH,
DIAD, Ph3P
1 THF
ROC!, DIPEA, THF
or
Mel, NaF/alumina
CH2CN
0 N.õ. 1. Me0H 0
R,
11 1110
DIAD, Ph3P N..õ \
N v
RiR2N
0 2. PrNH2
R=4-NO2Ph or R=CH3 137; R1=H,
R=CH3
139; R1=R2=cH3
Selective mono alklylation may be accomplished by alkylation of the N-acyl
derivatives of
1, followed by lkylation using Mitsunobu conditions, as above, followed by de-
acylation
with PrNH2, to provide the mono N-methyl derivative 137. This last series of
reactions
also works with solid supported chemistry.
Compound 1 was readily functionalized at the imidazole methine position by
treatment
with Na0C1 or Br2, to provide compounds 123 and 124, respectively.
44
CA 02469953 2007-09-04
X
0
0 NI\ 441 _ Na0C1 or Br2 es 9 N .. = I
*... .1
____________________________________________ , k.) 7 \ .
H2N S N solvent H2N
123, X =CI
1 124, X=Br
Demethylation of intermediate D with BBr3 provides the phenolic compound 145.
Acylation of compound 145 with benzoyl chloride provides compound 146.
o_ 0 N__ BBr3 0 N
l_ y \ 411 ol_ --rsii \ 11
-S.-
OH
D 145 1
PhOCI, or (R0)20, Et3N, THF
0 N
0 1 \ lit
\ o-
146 Ph
In several cases the requisite 2-bromoacetophenones were commercially
available. In
other cases they were prepared by the treatment of an appropriately
substituted
acetophenone with bromine, in an appropriate solvent, as exemplified below.
Acylation of
4-aminoacetophenone was followed by bromination in Me0H to provide
intermediate
A69. Condensation of intermediate A69 with intermediate El, yielded the
desired
compound 69.
45
CA 02469953 2007-09-04
N¨N
0 0 j/ El
1) Ac20 B H2NS02
NH2 ¨
1
2) Br2, Me0H r NHBn
.1 Me0H, reflux
A69
0 N., HCI n N-N
41,
NHBn =
NH2
H2N H20 H2N S N
69 70
Treatment of compound 69 with methanolic HC1 provided compound 70.
Several a-bromoketones were prepared by bromination of the appropriate enol
silyl ether.
Therefore, deprotonation of either 4'-piperidenylaceophenone (A67) or 4'-
morpholinoacetophenone (A68) with LiHMDS, silation with TMSC1, and quenching
with
N-bromosuccinamide, yielding the desired a-bromoketone intermediates B67 and
B68,
respectively, as shown below.
0 0
1) LIHMDS, THF
Br
101 R 23 TNMBsSChleat
A67 and A88 B67 or B68
El 0 N
\ =
solvent, reflux H2N S N
67, R= 68, R= Lo
Condensation of B67 and B68 with El, provided compounds 67 and 68,
respectively.
Treatment of selected aryl ketones with bromine or pyridinium perbromide also
provided
the desired 2-bromoacetophenones, which were again condensed with 2-amino-
1,3,4-
46
CA 02469953 2007-09-04
thiadiazole-5-sulfonamide to provide the desired 6-aryl-imidazo[2,1-b]-1,3,4-
thiadiazole-
2-sulfonamides, as shown below for compound 58.
0
Pyr Br3
Br
NHAc solvent NHAc
A58 A58
N-N El
H2NS02¨ j/ Ns -NH2 0 N__
solvent, reflux H21 S N NHAc
58
Compound 151 was prepared using the following strategy. 2',3',4',5', 6'-
Pentafluoroacetophenone, A6, was treated with sodium azide, followed by
bromide, to
provide 2-bromo-4'-azido-2',3',5',6'-tetrafluoroacetophenone, A151 (Keana, J.
F. W.;
Cal, S. X. J. Org. Chem., 1990, 55, 3640).
0 F 0 F
=
F 1) NaN3 Br
A151
A6 2) Br2
N3
N-N
j/ F F
H2NS02-Ths'NE12 El N I
\
N3
solvent, reflux H2N S"-A-N
F F
151
Condensation of A151 with El to provided compound 151.
47
CA 02469953 2007-09-04
SELECTED COMPOUND SYNTHESIS
GENERAL PREPARATIVE METHODS
Commercially available acetophenones, 2-haloacetophenones (Intermediates A and
B,
respectively), phenols (Intermediate C), and benzeneboronic acids
(Intermediate D) were
purchased from either Aldrich Chemical Company, Lancaster, Maybridge Inc, or
FisherScientific. The remainder of starting materials were obtained from
Aldrich
Chemical Company. 5-Amino-1,3,4-thiadiazole-2-sulfonamide, intermediate El,
was
prepared by the acid hydrolysis of acetazolamide (Aldrich).
Method A: Bromination of acetophenones with bromine
The appropriate acetophenone (Intermediate A) was dissolved in diethyl ether,
methylene
chloride, or chloroform, and cooled to 0 'C. Bromine (1.1 equiv) was dissolved
in either
methylenechloride or diethyl ether and added to the solution of acetophenone
via a
dropping funnel. After the addition of bromine was complete 2 drops of acetic
acid were
added and the solution was warmed to room temperature. Solvent was removed
under
reduced pressure to provide crude 2-bromoacetophenone (Intermediate B) which
was
generally used without further purification.
Method B: Bromination of acetophenones with pyridinium tribromide
The appropriate acetophenone (Intermediate A) was dissolved in acetic acid and
treated
with pyridinium tribromide (1.1 equiv). The solution was stirred until all
solid had
reacted, the solvent was removed under reduced pressure and the residue was
extracted
with an appropriate solvent, washing with water. The organic layer was dried
over
anhydrous magnesium sulphate, filtered, and the solvent removed under reduced
pressure
to provide the title compounds, which was generally used without further
purification.
Method C: Condensation of 2-bromoacetophenone with 2-amino-1,3,4-thiadiazole
-5-sulfonamide
48
CA 02469953 2007-09-04
The appropriate 2-nomoacetophenone and 2-amino-1,3,4-thiadiazole-5-sulfonamide
(1.0
equiv) were refluxed in 1,4-dioxane or an appropriate alcohol for 12-60 hrs.
The resulting
solution was cooled on ice and the resulting precipitate was collected by
filtration to
provide the title compound as crystalline solid. If no solid was observed the
solvent was
removed under reduced pressure and the title compounds were purified by silica
gel
chromatography, trituration, or recrystallization from an appropriate solvent.
Example 1: 6-Phenylimidazo[2,1-b]-1,3,4-thiadiazole-2-sulfonamide
2-Bromoacetophenone (4.00 g, 20.0 mmol) and 2-amino-1,3,4-thiadiazole-5-
sulfonamide
(3.60 g, 20.0 mmol) were refluxed in ethanol (150 mL) for 60 hrs. The
resulting solution
was cooled on ice and the resulting precipitate was collected by filtration to
provide
compound 1 as a white crystalline solid (2.50 g, 44 %). I H NMR (200MHz, DMSO-
d6)
8.89 (s, 1H), 8.72 (br s, 2H), 7.90 (d, 2H), 7.43 (t, 2H), 7.32 (t, 1H).
Example 2: 6-(2-Fluorophenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
Compound 2 was prepared by the bromination of 2'-fluoroacetophenone with
bromine,
according to Method A, followed by condensation with 2-amino-1,3,4-thiadiazole-
5-
sulfonamide hydrochloride, according to Method C, to provide a white solid. 'H
NMR
(200 MHz, DMSO-d6) 8 8.75 (br s, 2H), 8.6 (d, 1 H, j=3.6 Hz), 8.1 (m, 1H), 7.
3 (m, 3H).
Example 3: 6-(3-Fluorophenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-sulfonamide
Compound 3 was prepared by the bromination of 3'-fluoroacetophenone with
bromine,
according to Method A, followed by condensation with 2-amino-1,3,4-thiadiazole-
5-
sulfonamide hydrochloride, according to Method C, to provide a white solid
NMR (200 MHz, DMSO-d6) 5 8.95 (s, 1H), 8.74 (s, I H), 7.73 (m, 2H), 7. 5 (m,
1H),
7.1 (m, 1H).
Example 4: 6-(4-Fluorophenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
2-Bromo-4'-fluoroacetophenone (1.08 g, 5.0 mmol) 2-amino-1,3,4-thiadiazole-5-
sulfonamide hydrochloride (900 mg, 5.0 mmol) were refluxed in ethanol (25 mL)
for 48
hrs. The resulting solution was cooled on ice and the resulting precipitate
was collected
49
CA 02469953 2007-09-04
by filtration to provide compound 4 as a white crystalline solid (17 mg). 1H
NMR
(200MHz, DMSO-d6) 8 8.87 (s, 1H), 8.74 (br s, 2H), 7.40 (m, 2H), 7.28 (m, 2H).
Example 5: 6-(3,4-Difluorophenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
2-Chloro-3',4'-difluoroacetophenone (190 mg, 1.0 mmol) and 2-amino-1,3,4-
thiadiazole-
5-sulfonamide (150 mg, 1.0 mmol), and CETAB (437 mg, 1.20 mmol) were refluxed
in
dioxane (5 mL) for 48 hrs. The solvent was removed under reduced pressure and
the
resulting solid was purified by silica gel chromatography, eluting with 1:1
hexane/ethyl
acetate, to provide compound 5 (173 mg, 57%) as a white crystalline solid. 11-
1NMR
(200MHz, acetone-d6) 6 8.41 (d, 1H), 8.26 (m, 1H), 7.92 (br s, 2H), 7.24-7.08
(m, 2H).
Example 6: 6-(2,3,4,5,6-Pentafluorophenyl)imidazo[2,1-b]-1,3,4-
thiadiazole-2-
sulfonamide
2-Bromo-1-(pentafluorophenypethan-1-one (2.89 g, 10.0 mmol) and 2-amino-1,3,4-
thiadiazole-5-sulfonamide (1.80 g, 10.0 mmol) were refluxed in ethanol (20 mL)
for 60
hrs. Solvent was evaporated and the crude solid was purified by flash
chromatography
using 20 : 80 : 0.1 ethyl acetate : hexanes : acetic acid to provide compound
6 as white
needles (125 mg, 3.4 %). 1H NMR (200MHz, DMSO-d6) 8 8.82 (s, 1H), 8.78 (s,
2H).
Example 7: 6-(4-Ethylthio-2,3,5,6-tetrafluorophenypimidazo[2,1-1,]-1,3,4-
thiadiazole-2-sulfonamide
2',3',4',5',6'-Pentafluoroacetophenone (2.5 mmol) was heated with ethanethiol
(2.5
mmol) in THF (5 mL). Solvent was removed underreduced pressure to provide the
desired compounds as a white solid. Pyridinium tribromide (920 mg, 2.5 mmol))
was
added and the mixture stirred for 16 hours. Solvent was removed under reduced
pressure
and 2-amino-1,3,4-thiadiazole-5-sulfonamide (450 mg, 2.5 mmol) was added and
solution
was refluxed for 48 hours. The solution was cooled to room temperature and
filtered to
provide compound 7 as a white solid (173 mg, 17%). IFINMR (200 MHz, DMSO-d6) 6
8.82 (s, 1H), 8.79 (br s, 21-1), 3.00 (quart, .1=8.21-1z, 2H), 1.98 (t,
J=8.2Hz, 31-1).
50
CA 02469953 2007-09-04
Example 8: 6-(4-Benzylthio-2,3,5,6-tetrafluorophenyl)imidazo[2,1-b]-
1,3,4-
thiadiazole-2-sulfonamide
Compound 8 was prepared according to the procedure described for compound 7,
using
benzylmercaptan in the place of ethanethiol, to provide compound 8 as a white
solid (204
mg, 16%). IFINMR (200 MHz, DMSO-d6) 8 8.83 (s, 1H), 8.79 (br s, 2H), 7.23 (s,
5H),
4.21 (s, 2H).
Example 9: 6-(4-Morpholino-2,3,5,6-tetrafluorophenypimidazo[2,1-b]-
1,3,4-
thiadiazole-2-sulfonamide
Compound 6 (100 mg) was dissolved in 1 ml of DMSO and lml of morpholine was
added,
the solution was heated to 90 C for 2 hrs. The solution was allowed to cool
down to room
temperature and ethyl acetate was added. The solution was washed twice with
water and
once with brine. The organic layer was separated dried over magnesium sulfate
and
evaporated under reduced pressure. The residue was purified by silica gel
chromatography using a 20% to 50% ethyl acetate in hexanes gradient to give a
white
solid (30 mg). 11-1 NMR (200 MHz, CDC13) 8 8.05 (s, IH), 3.76 (m, 4 H), 3.27
(m 4 H).
Example 10: 6-(4-Chlorophenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
Compound 10 was obtained from Talon.
Example 11: 6-(3,4-Dichlorophenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
2-Bromo-3',4'-dichloroacetophenone (267 mg, 1.0 mmol) and 2-amino-1,3,4-
thiadiazole-
5-sulfonamide hydrochloride (180 mg, 1.00 mmol) were refluxed in ethanol (20
mL) for
48 hrs. The resulting solution was cooled on ice and the resulting precipitate
was
collected by filtration to provide compound 11 as a white crystalline solid
(91 mg, 26 %).
'H NMR (200MHz, DMSO-d6) 6 9.01 (s, 1H), 8.74 (s, 2H), 8.13 (d, 1H), 7.89 (dd,
1H),
7.70 (d, 11-1).
Example 12: 6-(2,3,4-trichlorophenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
Compound 12 was prepared by bromination of 2',3',4'-trichloroacetophenone with
bromine according to Method A, and condensation of the corresponding 2-
51
CA 02469953 2007-09-04
bromoacetophenone with 2-amino-1,3,4-thiadiazole-5-sulfonamide, according to
Method
C, to yield compound 12 as a white solid (22% yield). 114 NMR (200MHz, DMSO-
d6) 8
9.00 (s, 1H), 8.74 (br s, 21-1), 8.07 (d, J=8.5Hz, 2H), 7.74 (d, J=8.5Hz, 2H).
Example 13: 6-(3-bromophenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-sulfonamide
Compound 13 was prepared by bromination of 3'-bromoacetophenone with bromine
according to Method A, and condensation of the corresponding 2-
bromoacetophenone
with 2-amino-1,3,4-thiadiazole-5-sulfonamide, according to Method C, to yield
compound
13 as a white solid. 1H NMR (200MHz, DMSO-d6) 8 8.94 (d, J=1.3Hz, 1H), 8.74
(br s,
2H), 8.08 (d, J=1.1Hz, 1H), 7.90 (dd, J=1.4, 7.7Hz, 1H), 7.48 (d, J=7.7.Hz,
111), 7.38 (t,
J=8.6Hz, 1H).
Example 14: 6-(4-Bromopheny1)-imidazo [2,1-b]-1,3,4-thiadiazole-2-
sulfonam ide
2-Bromo-4'-bromoacetophenone (2.78 g, 10.0 mmol) and 2-amino-1,3,4-thiadiazole-
5-
sulfonamide (1.80 g, 12.0 mmol) were refluxed in 1,4-dioxane (25 mL) for 16
hrs. The
resulting solution was cooled on ice and the resulting precipitate was
collected by filtration
to provide compound 14 as a white crystalline solid (3.60 g). H NMR (200MHz,
DMSO-
d6) 8 8.92 (s, 1H), 8.75 (br s, 214), 7.85 (d, 2H), 7.62 (d, 2H).
Example 15: 6-(2-Methoxyphenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
2-Bromo-2'-methoxyacetophenone (916 mg, 4.0 mmol) and 2-amino-1,3,4-
thiadiazole-5-
sulfonamide (720 mg, 4.0 mmol) were refluxed in ethanol (20 mL) for 48 hrs.
The
resulting solution was cooled on ice and the resulting precipitate was
collected by filtration
to provide compound 15 as a white crystalline solid. 1H NMR (200MHz, DMSO-d6)
8
9.68 (br s, 1H), 8.78 (br s, 2H), 8.12 (d, 1H), 7.34 (t, 1H), 7.11 (d, 1H),
7.05 (t, 11-1), 3.96
(s, 3H).
Example 16: 6-(3-Methoxyphenyl)imidazo[2,1-1]-1,3,4-thiadiazole-2-
sulfonamide
2-Bromo-3'-methoxyacetophenone (1.00 g, 4.37 mmol) and 2-amino-1,3,4-
thiadiazole-5-
sulfonamide (786 mg, 4.37 mmol) were refluxed in 1,4-dioxane (25 mL) for 48
hrs. The
52
CA 02469953 2007-09-04
resulting solution was cooled on ice and the resulting precipitate was
collected by filtration
to provide compound 16 as a white crystalline solid (375 mg, 28 %).
11-1NMR (200MHz, DMSO-d6) 6 8.89 (s, 111), 8.73 (br s, 2H), 7.46 (s, 211),
7.33 (t,
J=8.1Hz, 1H), 6.88 (d, J=7.3Hz, 1H), 3.80 (s, 3H). 13C NMR (50MHz, DMSO-d6) 6
164.3,
159.8, 146.7, 145.2, 134.8, 130.0, 117.4, 113.7, 111.4,110.3, 55.1.
Example 17: 6-(4-Methoxyphenyl)imidazo[2,1-17]-1,3,4-thiadiazole-2-
sulfonamide
2-Bromo-4'-methoxyacetophenone (2.29 g, 10.0 mmol) and 2-amino-1,3,4-
thiadiazole-5-
sulfonamide (1.80 g, 12.0 mmol) were refluxed in 1,4-dioxane (25 mL) for 24
hrs. The
resulting solution was cooled on ice and the resulting precipitate was
collected by filtration
to provide compound 17 as a white crystalline solid (2.65 g, 86 %). 1H NMR
(200MHz,
DMSO-d6) 6 8.83 (s, 1H), 8.00 (d, 2H), 7.13 (d, 2H), 3.88 (s, 31-1).
Example 18: 6-(2,5-Dimethoxyphenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
2-Bromo-2',5'-dimethoxyacetophenone (261 mg, 1.00 mmol) and 2-amino-1,3,4-
thiadiazole-5-sulfonamide (180 mg, 1.20 mmol) were refluxed in 1,4-dioxane (7
mL) for
48 hrs. The resulting solution was cooled on ice and the resulting precipitate
was
collected by filtration to provide compound 18 as a white crystalline solid
(15.5 mg, 5 %).
Ill NMR (200M1-Iz, DMSO-d6) 68.72 (br s, 2H), 8.60 (s, 1H), 7.70 (d, 1H), 7.04
(d, 111),
6.87 (dd, 1H), 3.89 (s, 3H), 3.74 (s, 3H).
Example 19: 6-(2,4-Dimethoxyphenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
2-Bromo-2',4'-dimethoxyacetophenone (259 mg, 1 mmol) and 2-amino-1,3,4-
thiadiazole-
2-sulfonamide (180 mg, 1 mmol) were refluxed in ethanol for 5 days. After
cooling the
resulting precipitate was filtered and washed with methanol, providing 19 (56
mg) as a
beige powder. 1H NMR (200MHz, DMSO-d6) 6 8.69 (br s, 2H), 8.47 (s, 1H), 8.06
(d,
J=8.8 Hz, 1H), 6.66 (s, 111), 6.62 (d, J=2.4 Hz, 1H), 3.93 (s, 3H), 3.80 (s,
3H).
Example 20: 6-(1,3-Benzodioxo1-5-yl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
53
CA 02469953 2007-09-04
1-(1,3-benzodioxo1-5-y1)-2-bromoethan-1-one (100 mg, 0.41 mmol) and 2-amino-
1,3,4-
thiadiazole-5-sulfonamide (74 mg, 0.41 mmol) were refluxed in ethanol (5 mL)
for 30 hrs.
The resulting solution was cooled on ice and the resulting precipitate was
collected by
filtration to provide compound 20 as a pale yellow powder (40 mg, 44 %). H NMR
(200MHz, DMSO-d6) 6 8.75 (s, 1H), 8.69 (s, 2H), 7.43 (m, 2H), 6.97 (d, J = 8.6
Hz, 1H),
6.04 (s, 2H).
Example 21: 6-(3,4-dihydro-2H-1,5-benzodioxepin-7-yl)imidazo[2,1-b]-
1,3,4-
thiadiazole-2-sulfonamide
2-Bromo-1-(3,4-dihydro-2H-1,5-benzodioxepin-7-yl)ethan-1-one (542 mg, 2 mmol)
and
2-amino-1,3,4-thiadiazole-5-sulfonamide (360 mg, 2 mmol) were refluxed in
ethanol
(10m1) for 60 hours. The resulting mixture was cooled on ice and the resulting
precipitate
collected by suction filtration, giving 21 (310 mg) as a yellow powder. IH NMR
(200MHz, DMSO-d6) 6 8.79 (s, 11-1), 8.71 (br s, 2H), 7.49 (s, 1H), 7.45 (d,
J=7.9Hz, 114),
7.00 (d, J=7.9Hz, 1H), 4.2-4.0 (m, 4H), 2.10 (t, J=4.9Hz, 2H). I3C NMR (50MHz,
DMSO-
d6) 6 163.9, 151.4, 151.0, 146.2, 145.1, 128.9, 122.0, 120.1, 118.2, 110.7,
70.6, 31.5.
Example 22: 6-(3,4-Dihydroxyphenyl-imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
2-Chloro-3',4'-dihydroxyacetophenone (186 mg, 1.0 mmol) and 2-amino-1,3,4-
thiadiazole-5-sulfonamide (150 mg, 1.0 mmol), and CETAB (10 mg) were refluxed
in
dioxane (5 mL) for 48 hrs. The solvent was removed under reduced pressure and
the
resulting solid was purified by silica gel chromatography, eluting with 1:1
hexane/ethyl
acetate, to provide compound 22 (11 mg, 4%) as a white crystalline solid. 'H
NMR
(200MHz, acetone-d6) 6 8.38 (s, 1H), 8.10 (br s, 211), 7.84 (br s, 2H), 7.44
(d, 1H), 7.39
(dd, 1H), 6.88 (d, 1H).
Example 23: 6-(2-spiro(cyclohexyl)benzo-1,3-dioxo1-5-yl)imidazo[2,1,-11]-1,3,4-
thiadiazole-2-sulfonamide.
Compound 23 was prepared by the bromination of 6-(2-spiro(cyclohexyl)benzo-1,3-
dioxo1-5-yl)ethanone with bromine, according to Method A, followed by
condensation
54
CA 02469953 2007-09-04
with 2-amino-1,3,4-thiadiazole-5-sulfonamide hydrochloride, according to
Method C, to
provide a white solid.
Example 24: 6-(3-trifloromethoxyphenyl)imidazo[2,1-b]-1,3,4-thiadiazole-
2-
sulfonamide
Compound 24 was prepared by bromination of 3'-trifluoromethoxyacetophenone
with
bromine according to Method A, and condensation of the corresponding 2-
bromoacetophenone with 2-amino-1,3,4-thiadiazole-5-sulfonamide, according to
Method
C, to yield compound 24 as a white solid (22% yield). 1H NMR (200MHz, DMSO-d6)
6
9.01 (s, 1H), 8.74 (br s, 2H), 7.92 (d, J=7.6Hz, 1H), 7.91 (s, 1H), 7.57 (t,
J=7.6Hz, 1H),
7.30 (d, J=7.6Hz, 1H).
Example 25: 6-(4-trifloromethoxyphenyl)imidazo[2,1-1)]-1,3,4-
thiadiazole-2-
sulfonamide
Compound 25 was prepared by bromination of 4'-trifluoromethoxyacetophenone
with
bromine according to Method A, and condensation of the corresponding 2-
bromoacetophenone with 2-amino-1,3,4-thiadiazole-5-sulfonamide, according to
Method
C, to yield compound 25 as a white solid (22% yield). 1H NMR (200MHz, DMSO-d6)
6
8.93 (s, 1H), 8.73 (s, 2H), 8.00 (d, J=8.8Hz, 2H), 7.43 (d, J=8.4Hz, 2H).
Example 26
Compound 26 was prepared in a manner similar to compound 31.
NMR (200 MHz, DMSO-d6) 6 = 3.71 (t, 2H, J = 4.6 Hz), 4.00 (t, 2H, J = 4.9 Hz),
6.99
(d, 2H, J = 8.3 Hz), 7.81 (d, 2H, J = 8.2 Hz), 8.67 (s, 2H), 8.73 (s, 1H)
Example 27
Step 1: Compound 26 (472 mg, 1.39 mmol), di-tert-butyldicarbonate (383 jtL,
1.67
mmol), triethylamine (194 pit, 1.39 mmol), DMAP (20 mg, 0.16 mmol) were added
to
DMF (5 mL) and stirred at RT under N2 for 45 min. The volatiles were removed
under
reduced pressure and the contents washed with H20 and Et0Ac. The organic layer
was
collected, dried over MgSO4. 11-1 NMR (200MHz, CDC13) 6 1.23 (s ,9H), 3.73 (t,
2H, J =
CA 02469953 2007-09-04
5.4 Hz), 4.00 (t, 2H, J = 5.6 Hz), 7.02 (d, 2H, J = 8.8 Hz), 7.82 (d, 2H, J =
8.4 Hz), 8.69 (s,
1H).
Step 2 : The material from step 1 (627 mg, 1.39 mmol), acetic anhydride (158
pt, 1.67
mmol), triethlyamine (233 L, 1.67 mmol), DMAP (21 mg, 0.17 mmol) were added
to
DMF (5 mL) and stirred under N2 for 3 h. The volatiles were removed under
reduced
pressure and the contents washed with H20 and Et0Ac. The organic layer was
collected,
dried over MgSO4. 1H NMR (200MHz, CDC13) 6 1.20 (s, 9H), 2.08 (s, 3H), 4.09
(b, 2H),
4.40 (b, 2H), 6.90 (d, 2H, J = 8.8 Hz), 7.62 (d, 2H, J = 8.4 Hz).
Step 3 : The material from Step 3 was dissolved in 10 mL TFAJCH2C12 (1:1) and
stirred
for 30 min at RT. The volatiles were removed under reduced pressure and the
contents
washed with NaHCO3 (aq) and Et0Ac. The organic layer was collected, dried over
MgSO4. The product was recrytallized from ethanol. 111 NMR (200MHz, DMSO-d6) 6
2.03 (s, 3H), 4.20 (b, 2H), 4.31 (b, 2H), 7.02 (d, 2H, J = 8.8 Hz), 7.82 (d,
2H, J = 8.4 Hz),
8.69 (s, 2H), 8.76 (s, 1H).
Example 28 :
Step 1 : 2-Bromoethanol, K2CO3 and 4-hydroxy acetophenone were refluxed
together in
Me0H. The solvent was removed under reduced pressure and the residue treated
to
standard ethyl acetate/water work-up to provide a white semi-sold.. 1H NMR
(200 MHz,
CDC13) 6 2.55 (s, 3H), 3.65 (t, 2H, J = 6.1 Hz), 4.35, t, 2H, J = 6.4 Hz),
6.94 (d, 2H, J =
9.5 Hz), 7.93 (d, 2H, J = 8.8 Hz).
Step 2 : The material from Step 1 (50 mg, 0.216 mmol), NaN3 (20 mg, 0.307
mmol) were
dissolved in acetone (5 mL) and H20 (0.5 mL) and heated to reflux with
stirring for 16 h.
Solvent was removed and the desired compounds was obtained in quantitative
yield. 1H
NMR (200 MHz, CDCI3) 6 2.55 (s, 3H), 3.63 (t, 2H, J = 4.6 Hz), 4.21 (t, 2H, J
= 5.19
Hz), 6.95 (d, 2H, J = 8.85 Hz), 7.94 (d, 211, J = 8.9 Hz).
Step 3 :The material from Step 2 was brominated according to Method A and
purified on
silica gel (4:1, CH2C12:Hexanes) to give 1-(4-(2-azidoethoxy)pheny1)-2-bromo
ethanone.
1H NMR (200 MHz, CDC13) 6 3.63 (t, 2H, J = 4.9 Hz), 4.21 (t, 2H, J = 5.2 Hz),
4.40 (s,
2H), 6.95 (d, 2H, J = 9.2 Hz), 7.94 (d, 2H, J = 8.6 Hz).
56
CA 02469953 2007-09-04
Step 4 : 1-(4-(2-azidoethoxy)pheny1)-2-bromo ethanone was condensed with 1,3,4-
thiadiazole-2-sulfonamide according to Method C yielding an off yellow solid.
1H NMR
(200 MHz, DM50-d6) 6 3.65 (t, 2H, J = 4.0 Hz), 4.20 (t, 2H, J = 4.3 Hz), 7.02
(d, 2H, J
8.2 Hz), 7.83 (d, 21-1, J = 8.2 Hz), 8.71 (s, 2H), 8.76 (s, 1H).
Example 29:
Step 1 : 3-Bromo-1,1,1-trifluoropropane, K2CO3 and 4'-hyrdoxyacetophenone were
refluxed together in Me0H for 16 hours. Volatiles were removed under reduced
pressure
and the residue subjected to standard ethyl acetate/water work-up. 1H NMR (200
MHz,
CDCI3) 6 2.48-2.70 (m, 5H), 4.19 (t, 2H, J = 6.4 Hz), 6.87 (d, 2H, J = 8.8
Hz), 7.87 (d, 2H,
9.2 Hz).
Step 2: 4'-(3,3,3-trifluoropropoxy)acetophenone was brominated
according to
Method A. 1H NMR (200 MHz, CDC13) 6 2.48-2.70 (m, 211), 4.27 (t, 211, J = 6.4
Hz), 4.40
(2, 2H) 6.96 (d, 2H, J = 8.8 Hz), 7.98 (d, 2H, 9.2 Hz).
Step 3: 2-Bromo-4'-(3,3,3-trifluoropropoxy)acetophenone was condensed with
5-
Amino-1,3,4-thiadiazole-2-sulfonamide according to Method C, yielding an off
yellow
solid. 1H NMR (200 MHz, DMSO-d6) 6 2.65-2.85 (m, 211), 4.23 (t, 2H, J = 6.2
Hz), 7.02
(d, 2H, J = 8.6 Hz), 7.83 (d, 2H, J = 8.6 Hz), 8.71 (s, 211), 8.76 (s, 1H).
Example 30:
Step 1 : 1-Bromo-2-(2-methoxyethoxy)ethane, K2CO3 and 4'-hyrdoxyacetophenone
were
refluxed together in Me0H for 16 hours. Volatiles were removed under reduced
pressure
and the residue subjected to standard ethyl acetate/water work-up. 1H NMR (200
MHz,
CDC13) 6 2.55(s, 3H) 3.39 (s, 3H), 3.56 (t, 2H, J = 4.0 Hz), 3.71 (t, 211, J =
4.6 Hz), 3.88
(t, 2H, 4.3 Hz), 4.21 (t, 2H, J = 4.9 Hz), 6.94 (d, 2H, J = 8.8. Hz), 7.92 (d,
2H, J = 8.2 Hz).
Step 2 : The material from Step 1 was brominated according to Method A. 11-1
NMR (200
MHz, CDC13) 6 3.40 (s, 3H), 3.56 (t, 2H, J = 4.0 Hz), 3.71 (t, 211, J = 4.6
Hz), 3.88 (t, 2H,
4.3 Hz), 4.21 (t, 2H, J = 4.9 Hz), 4.40 (s, 214), 6.94 (d, 2H, J = 8.8. Hz),
7.92 (d, 211, J =
8.2 Hz).
57
CA 02469953 2007-09-04
Step 3: The Material from Step 2 was condensed with 2-amino-1,3,4-thiadiazole-
2-
sulfonamide according to Method C to provide a yellow solid. 11-1 NMR (200
MHz,
DMSO-d6) 6 3.23 (s, 31-1), 3.47 (b, 211), 3.57 (b, 2H), 3.73 (b, 2H), 4.11 (b,
2H), 7.00 (d,
214, J = 8.2 Hz), 7.81 (d, 2H, J = 8.6 Hz), 8.70 (s, 2H), 8.74 (s, 1H).
Example 31:
Step 1: 4-Hydroxyacetophenone (500 mg, 3.67 mmol), K2CO3 (510 mg,
3.69
mmol) and benzy1-2-bromoethyl ether (580 uL, 3.67 mmol) were suspended in
ethanol (25
mL). The mixture was heated to reflux with stirring for 21 h. The volatiles
were removed
under reduced pressure and the contents washed with H20 and Et0Ac. The organic
layer
was collected, dried over MgSO4 and purified on silica gel (1:3 Et0Ac/Hexanes)
yielding
4'-(2-Benzyloxyethoxy)acetophenone as a white crystalline solid (600 mg, 61%).
11-1
NMR (200 MHz, CDC13) 6 2.55 (s, 3H), 3.85 (t, 2H, J = 4.9 Hz), 4.21 (t, 211, J
= 4.9 Hz),
4.64 (s, 2H), 6.95 (d, 2H, J = 8.9 Hz), 7.35 (b, 511), 7.93 (d, 2H, J = 8.8
Hz).
Step 2: 4-(2-Benzyloxyethoxy)acetophenone (447 mg, 1.65 mmol) was brominated
using
Method A to yield a yellow oil (51% conversion). 11-1 NMR (200 MHz, CDC13) 8
3.85 (t,
2H, J = 4.9 Hz), 4.21 (t, 2H, J = 4.9 Hz), 4.64 (s, 2H), 4.80 (s, 211), 6.95
(d, 2H, J = 8.9
Hz), 7.35 (b, 5H), 7.93 (d, 2H, J = 8.8 Hz).
Step 3: 6-(4'-(2-Benzyloxyethoxy)pheny1)-imidazo[2,1-b]-1,3,4-thidiazole-2
sulfonamide
The crude material from Step 2 (299 mg, 0.86 mmol) was condensed with 2-amino-
1,3,4-
thiadiazole-2-sulfonamide using Method C in 2-propanol, yielding a yellow
solid (140 mg,
38%). 1H NMR (200 MHz, DMSO-d6) 6 3.77 (b, 211), 4.18 (b, 2H), 4.55 (s, 2H),
7.01 (d,
2H, J = 8.5 Hz), 7.33 (b, 5H) 7.81 (d, 211, J = 8.5 Hz), 8.69 (s, 2H), 8.74
(s, 1H).
Example 32:
Step 1: 4-Hydroxyacetophenone (500 mg, 3.67 mmol), K2CO3 (510 mg, 3.69 mmol)
and
benzy1-3-bromopropyl ether (547 AL, 3.67 mmol) were suspended in ethanol (25
mL).
The mixture was heated to reflux with stirring for 21 h. The volatiles were
removed under
reduced pressure and the contents washed with H20 and Et0Ac. The organic layer
was
collected, dried over MgSO4 and purified on silica gel (1:3 Et0Ac/Hex)
yielding 4'-(2-
Benzyloxyethoxy)acetophenone as a white crystalline solid (737 mg, 71%). 'H
NMR (200
58
CA 02469953 2007-09-04
MHz, CDC13) 6 2.08-2.14 (m, 2H), 2.56 (s, 3H), 3.67 (t, 211, J = 6.1 Hz), 4.16
(t, 2H, J =
6.1 Hz), 4.53 (s, 2H), 6.92 (d, 2H, J = 8.5 Hz), 7.31 (s, 5H), 7.93 (d, 2H, J
= 8.9 Hz).
Step 2: 4'-(3-Benzyloxy)propoxyacetophenone (447 mg, 1.65 mmol) was brominated
using Method A to provide a yellow oil (82 % conversion). 1H NMR (200 MHz,
CDC13) 6
2.08-2.14 (m, 2H), 3.67 (t, 2H, J = 6.1 Hz), 4.16 (t, 2H, J = 6.1 Hz), 4.40
(s, 2H), 4.53 (s,
211), 6.92 (d, 2H, J = 8.5 Hz), 7.93 (d, 2H, J = 8.9 Hz)
Step 3: The crude material from step 3 (671 mg, 1.85 mmol) was condensed with
2-
amino-1,3,4-thiadiazole-2-sulfonamide using Method C (2-propanol) provided a
yellow
solid (85 mg, 10 %). 1H NMR (200 MHz, DMSO-d6) 6 1.96-2.06 (m, 2H), 3.58 (t,
2H, J =
6.4 Hz), 4.08 (t, 2H, J = 5.8 Hz), 4.47 (s, 2H), 6.98 (d, 2H, J = 8.5 Hz),
7.30 (s, 2H), 7.80
(d, 2H, J = 8.5 Hz), 8.69 (s, 211), 8.73 (s, 1H).
Example 33: 6-(4-(2-Morpholinoethoxy)phenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide bis(methanesulfonic acid)
6-(4-(2-Morpholinoethoxy)phenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-sulfonam
ide was
prepared according to Method C.114 NMR (200 MHz, DMSO-d6) 6 8.70 (s, 1H), 7.82
(8.2Hz, 2H), 7.05 (d, J=8.2Hz, 2H), 4.34 (m, 4H), 3.81 (m, 4H), 3.48 (m, 2HH),
3.26 (m,
2H).
6-(4-(2-Morpholinoethoxy)phenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-sulfonamide
(100
mg) was suspended in Me0H (2 mL) and treated with methanesulfonic acid (100
uL).
Diethyl ether (10 mL) was added and the resulting solid was filtered and
washed with
diethyl ether to provide compound 33. 1H NMR (200 MHz, D20) 8 8.09, 7.53 (d,
J=6.7Hz,
211), 6.97 (d, J=6.7Hz, 211), 4.43 (s, 2H), 4.20 (m, 214), 3.96 (br t, 2H),
3.70 (m, 4H), 3.35
(m, 2H), 2.80 (br s, 4H).
Example 34:
Compound 34 was prepared in a manner similar to that described for compound
32.
'H NMR (200 MHz, DMSO-d6) 6 3.71 (t, 2H, J = 4.9 Hz), 3.82 (s, 2H), 3.98 (t,
2H, J =
4.6 Hz), 7.01 (d, 1H, J = 8.2 Hz), 7.40-7.48 (m, 3H), 8.71 (s, 2H), 8.79 (s,
1H).
Example 35:
59
CA 02469953 2007-09-04
Compound 35 was prepared in a manner similar to compound 32.
114 NMR (200 MHz, DMSO-d6) 6 8.78 (s, 111), 8.67 (s, 2H), 7.48 (s, 1H), 7.40
(d,
J=8.6Hz, 1H), 7.34 (s, 51-1), 7.02 (d, J=8.6Hz, 1H), 4.56 (s, 2H), 4.14 (br s,
2H), 3.83 (s,
3H), 3.77 (br s, 2H).
Example 36:
Compound 36 was prepared in a manner similar to compound 32.
'H NMR (200 MHz, DMSO-d6) 6 8.78 (s, 1H), 8.70 (s, 2H), 7.47 (s, 1H), 7.42 (d,
J=8.2Hz, 1H), 7.30 (s, 5H), 7.01 (d, J=8.2Hz, 1H), 4.47 (s, 2H), 4.06 (t,
J=6.1Hz, 2H),
3.80 (s, 3H), 3.59 (t, J=6.3Hz, 2H), 1.99 (t, J=6.I Hz, 2H).
Example 37: 6-(3-Nitrophenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
2-Bromo-3'-nitroacetophenone (224 mg, 1.0 mmol) and 2-amino-1,3,4-thiadiazole-
5-
sulfonamide (180 mg, 1.20 mmol) were refluxed in 1,4-dioxane (7 mL) for 48
hrs. The
resulting solution was cooled on ice and the resulting precipitate was
collected by filtration
to provide compound 37 as a yellow crystalline solid (54 mg, 15 %). 1H NMR
(200MHz,
DMSO-d6) 8 9.12 (s, 1H), 8.75 (br s, 2H), 8.70 (t, 1H), 8.31 (d, 1H), 8.14 (d,
1H), 7.72 (t,
1H).
Example 38: 6-(3-nitro-4-chlorophenyl)imidazo[2,1 -b]-1,3,4-thiadiazole-2-
sulfonamide
Compound 38 was prepared by bromination of 3-nitro-4-chloroacetophenone with
bromine according to Method A, and condensation of the corresponding 2-
bromoacetophenone with 2-amino-1,3,4-thiadiazole-5-sulfonamide, according to
Method
C, to yield compound 38 as a white solid (22% yield). 1H NMR (200MHz, DMSO-d6)
6
9.09 (s, 1H), 8.77 (s, 2H), 8.53 (s, 1H), 8.18 (d, J=6.9Hz, 2H), 7.86 (d,
J=8.5Hz, 2H).
Example 39:
Step 1: 4-Acetylbenzoic acid (1.00 g, 6.09 mmol) was suspended in
methanol
(10mL). Hydrochloric acid (500 L) was added. The reaction mixture was
refluxed
overnight. The resulting suspension was cooled to ¨10 C, filtered and the
solid washed
CA 02469953 2007-09-04
with cold methanol (3 x 2 mL) to provide methyl 4-acetylbenzoate as a white
solid (799
mg, 74%). 1H NMR (200MHz, CDC13) 68.12 (d, J = 8.9Hz, 2H), 8.01 (d, J = 8.9Hz,
2H),
3.95 (s, 3H), 2.65 (s, 3H).
Step2
Methyl 4-acetylbenzoate (200 mg, 1.12 mmol) was suspended in chloroform
(5mL) and treated with pyridinium tribromide (359 mg, 1.12mmol). The reaction
mixture
was stirred overnight. One half equivalent of pyridinium tribromide (179 mg,
0.56 mmol)
was added to the reaction mixture and stirred for two days. The solvent was
removed
under reduced pressure. Standard aqueous/ethyl acetate workup provided a brown
solid,
which was identified as a 8:12:3 mixture of starting material, methyl 4-(2-
bromoacetyl)benzoate and methyl 4-(2.2-dibromoacetyl)benzoate compound. 1H NMR
(200MHz, DMSO-d6) 6 8.08 (d, J = 6.7Hz, 4H), 4.98 (s, 211), 3.87 (s, 311).
Step 3
Methyl 4-(2-bromoacetyl)benzoate (100 mg, 0.39 mmol) and 5-amino-1,3,4-
thiadiazole-2-sulfonamide (70mg, 0.39 mmol) were refluxed together in methanol
(10 mL)
for 48 hours. The resulting suspension was cooled to ¨10 C, filtered and the
solid washed
with cold methanol (3 x 2 mL) to provide compound 39 as a white solid (12.9
mg, 9.35%).
11-1NMR (200MHz, DMSO-d6) 6 9.03 (s, 1H), 8.70 (br s, 2H), 8.03 (s, 4H), 3.85
(s, 3H).
Example 40: 6-(4-carboxyphenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
Step 1: 4-
Acetylbenzoic acid (186 mg, 1.14 mmol) was dissolved in warm acetic
acid (5 mL) and treated with bromine (58 mL, 1.14 mmol). The solution was
stirred
overnight before being cooled on ice. The resulting solid was filtered, washed
with 1:1
methanol/water (3 x 10 mL) and dried in vacuo to provide 4-(2-
bromoacetyl)benzoic acid
as a white solid (102 mg). 111 NMR (200MHz, DMSO-d6) 8 8.07 (s, 4H), 4.98 (s,
211).
Step 2: 4-(2-
bromoacetyl)benzoic acid (102 mg) and 5-amino-1,3,4-thiadiazole-2-
sulfonamide (75 mg, 0.42 mmol) were refluxed together in methanol (20 mL) for
48
hours. The resulting suspension was cooled to ¨10 C, filtered and the solid
washed with
cold methanol (3 x 5 mL) to provide compound 40 as a white crystalline solid
(16 mg). 114
NMR (200MHz, DMSO-d6) 6 9.02 (s, 11-1), 8.00 (s, 4H).
Example 41: 6-(3-
cyanophenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-sulfonamide
61
CA 02469953 2007-09-04
3-(2-bromoacetyl)benzonitrile (100 mg, 0.45 mmol) and 2-amino-1,3,4-
thiadiazole-5-
sulfonamide (80 mg, 0.45 mmol) were refluxed in ethanol (10 mL) for 60 hrs.
The
resulting solution was cooled on ice and the resulting precipitate was
collected by filtration
to provide compound 41 as a white crystalline solid (78 mg, 57 %). 11-1NMR
(200M1-Iz,
DMS0-(16) 8 9.03 (s, 1H), 8.76 (s, 2H), 8.32 (s, 1H), 8.23 (d, J = 7.6 Hz,
1H), 7.71 (m,
2H); '3C NMR (50 MHz, DMSO) 8 164.9, 145.8, 144.6, 134.6, 131.3, 130.2, 129.4,
128.3,
118.7, 112.4, 112.1.
Example 42: 6-(4-cyanophenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
4-(2-bromoacetyl)benzonitrile (448 mg, 2 mmol) and 2-amino-1,3,4-thiadiazole-5-
sulfonamide (360 mg, 2 mmol) were refluxed in ethanol for 60 hours. The
resulting
mixture was cooled on ice and the precipitate collected by suction filtration
to provide 42
(300 mg) as a white powder. 111 NMR (200M1-Iz, DMSO-d6) 8 9.07 (s, 1H), 8.77
(br s,
2H), 8.09 (d, 2H), 7.90 (d, 2H).
Example 43: 6-(4-(methylsulfonyl)phenyl)imidazo[2,1
sulfonamide
2-Bromo-144-(methylsulfonyl)phenyllethan-1-one (100 mg, 0.36 mmol) and 2-amino-
1,3,4-thiadiazole-5-sulfonamide (65 mg, 0.36 mmol) were refluxed in ethanol (5
mL) for
60 hrs. The resulting solution was cooled on ice and the resulting precipitate
was
collected by filtration to provide compound 43 as a white powder (55 mg, 43
%). 1H
NMR (200MHz, DMSO-d6) 8 9.08 (s, 1H), 8.76 (s, 2H), 8.15 (d, J = 8.1 Hz, 2H),
7.96 (d,
J = 8.1 Hz, 2H), 3.23 (s, 311).
Example 44: 6-(4-(phenylmethylsulfonyl)phenyl)imidazo[2,1-b]-1,3,4-
thiadiazole-2-
sulfonamide
Compound 44 was prepared by bromination of 4'-
(phenylmethylsulfonyl)acetophenone
with bromine according to Method A, and condensation of the corresponding 2-
bromoacetophenone with 2-amino-1,3,4-thiadiazole-5-sulfonamide, according to
Method
C, to yield compound 44 as a white solid. 114 NMR (200MHz, DMSO-d6) 8 9.06 (s,
1H),
8.74 (s, 2H), 8.02 (m ,6H), 7.65 (m, 3H).
62
CA 02469953 2007-09-04
Example 45:
Compound 44 was prepared by according to Methods A and C, to yield compound 45
as a
white solid. 1H NMR (200MHz, DMSO-d6) 6 9.08 (s, 114), 8.65 (s, 2H), 8.08 (d,
J=8.0 Hz,
2H), 7.85 (d, J=8.0Hz, 2H), 3.18 (quart, J=7.6Hz, 2H), 1.05 (t, J=7.6Hz, 3H).
Example 46: 6-(4-Pentylphenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
2-Bromo-1-(4-pentylphenyl)ethan-1-one (269 mg, 1.0 mmol) and 2-amino-1,3,4-
thiadiazole-5-sulfonamide (180 mg, 1.0 mmol) were refluxed in ethanol (10 mL)
for 48
hrs. The resulting solution was cooled on ice and the resulting precipitate
was collected
by filtration to provide compound 46 as a white powder (180 mg, 51 %). 'H NMR
(200MHz, DMSO-d6) 6 8.81 (s, 114), 8.71 (s, 2H), 8.79 (d, J = 8.2 Hz, 2H),
7.24 (d, J = 8.2
Hz, 2H), 2.54 (t, J = 7.0 Hz, 2H), 1.57 (quintet, J = 7.6 Hz, 2H), 1.27 (m,
4H), 0.85 (t, J =
6.7 Hz, 3H); '3C NMR (50 MHz, DMSO) : 6 164.0, 147.1, 145.3, 142.5, 130.9,
128.9,
125.2, 110.7, 34.9, 30.9, 30.6, 22.0, 14Ø
Example 47: 6-(4-Methylphenyl)imidazo[2,1-1]-1,3,4-thiadiazole-2-
sulfonamide
2-Bromo-1-(4-methylphenyl)ethan-lone (213 mg, I mmol) and 2-amino-1,3,4-
thiadiazole-
5-sulfonamide (148 mg, 1 mmol) were refluxed in ethanol (10 mL) for 60 hours.
Solvent
was removed under reduced pressure. The suspension was cooled to ¨4 C,
filtered and
washed with cold methanol (3 x 5 mL), to provide compound 47 (118 mg, 42 %) as
a
white powder. 1H NMR (200MHz, DMSO-d6) 6 8.80 (s, 1H), 8.71 (s, 2H), 7.78 (d,
214),
7.23 (d, 2H), 2.31 (s, 3H).
Example 48: 6-(2,4-dimethylphenyl)imidazo[2,1 -b]-1,3,4-thiadiazole-2-
sulfonamide
2-Bromo-1-(2,4-dimethylphenypethan-1-one (227 mg, 1 mmol) and 2-amino-1,3,4-
thiadiazole-2-sulfonamide (180 mg, 1 mmol) were refluxed in ethanol for 5
days. The
volatiles were removed in vacuo. The residue was purified by column
chromatography on
silica using 30% ethyl acetate/1% acetic acid in hexane as eluant.
Recrystallization from
dichloromethane gave 48 (30 mg) as a white powder. 'H NMR (200MHz, DMSO-d6)
63
CA 02469953 2007-09-04
6 8.71 (br s, 2H), 8.52 (s, 1H), 7.70 (d, J=7.6 Hz, 1H), 7.10 (s, 1H), 7.06
(s, 1H), 2.46(s,
3H), 2.29 (s, 3H).
Example 49: 6-(4-(phenylmethylsulfonyl)phenyl)imidazo[2,1-b]-1,3,4-
thiadiazole-2-
sulfonamide
Compound 49 was prepared by bromination of 4'-tert-butylacetophenone with
bromine
according to Method A, and condensation of the corresponding 2-
bromoacetophenone
with 2-amino-1,3,4-thiadiazole-5-sulfonamide, according to Method C, to yield
compound
49 as a white solid. 1H NMR (200MHz, DMSO-d6) 6 8.81 (s, 1H), 8.70 (s, 2H),
7.81 (d,
J=7.9Hz, 2H), 7.44 (d, J=8.2Hz, 2H), 1.29 (s, 9H).
Example 50
Compound 50 was prepared according to Method A and Method C, to yield compound
50
as a white solid. 1H NMR (200MHz, DMSO-d6) 6 8.72 (br s, 2H), 8.54 (s, 1H),
7.80 (s,
1H), 7.18 (s, 1H), 3.05 (t, 2H), 1.95 (t, 211), 1.15 (s, 9H).
Example 51:
MS (m/z) M =249.10
Example 52: 6-(4-(Trifluoromethyl)phenyl)imidazo[2,1 -b]-1,3,4-thiadiazole-
2-
sulfonamide
2-Bromo-1-[4-(trifluoromethyl)phenyliethan-1-one (534 mg, 2.0 mmol) and 2-
amino-
1,3,4-thiadiazole-5-sulfonamide (360 mg, 2.0 mmol) were refluxed in ethanol
(10 mL) for
60 hrs. The resulting solution was cooled on ice and the resulting precipitate
was
collected by filtration to provide compound 52 as a white powder (270 mg, 39
%). 1H
NMR (200MHz, DMSO-d6) 6 9.05 (s, 1H), 8.74 (s, 2H), 8.10 (d, J = 8.0 Hz, 2H),
7.80 (d,
J = 8.0 Hz, 2H).
Example 53: 6-(5-chloro-2-trifluoromethylphenyl)imidazo[2,1 -b]-1,3,4-
thiadiazole-2-
sulfonamide
64
CA 02469953 2007-09-04
Compound 51 was prepared by bromination of 5'-chloro-4'-
trifluoromethylacetophenone
with bromine according to Method A, and condensation of the corresponding 2-
bromoacetophenone with 2-amino-1,3,4-thiadiazole-5-sulfonamide, according to
Method
C, to yield compound 53 as a white solid. IHNMR (200MHz, DMSO-d6) 8 9.04 (s,
1H),
8.78 (s, 2H), 8.47 (s, 1H), 7.82 (d, J=8.6Hz, 1H), 7.72 (d, J=8.6Hz, 1H).
Example 54: 6-(3,5-di(trifluoromethyl)phenyl)im idazo[2,1-b]-1,3,4-
thiadiazole-2-
sulfonamide
2-Bromo-143,5-di(trifluoromethyl)phenyljethan-1 -one (670 mg, 2.0 mmol) and 2-
amino-
1,3,4-thiadiazole-5-sulfonamide (360 mg, 2.0 mmol) were refluxed in ethanol
(10 mL) for
60 hrs. The resulting solution was cooled on ice and the resulting precipitate
was
collected by filtration to provide compound 54 as a white powder (292 mg, 70
%). IH
NMR (200MHz, DMSO-d6) 6 9.26 (s, 1H), 8.77 (s, 211), 8.54 (s, 2H), 8.04 (s,
1H).
Example 55: 6-(3,4-Di-tert-buty1-4-hydroxyphenyl)imidazo[2,1-b]-1,3,4-
thiadiazole-2-sulfonamide
2-Bromo-1-(3,4-di-tert-buty1-4-hydroxyphenypethan-1 -one (327 mg, 1 mmol) and
2-
amino-1,3,4-thiadiazole-5-sulfonamide (148 mg, 1 mmol) were refluxed in
ethanol (10
mL) for 60 hours. Solvent was removed under reduced pressure. The resulting
solid was
suspended in methanol (5 mL) and stirred for 30 minutes prior to suction
filtration,
washing twice for cold methanol (2 mL), to provide compound 55 (93 mg, 24 %)
as a
white powder. I H NMR (200MHz, DMSO-d6) 8 8.72 (s, 1H), 8.68 (s, 2H), 7.63 (s,
2H),
1.41 (s, 9H).
Example 56: 6-(5,5,8,8-Tetramethy1-5,6,7,8-tetrahydronaphthalen-2-
yl)imidazo[2,1-b]-1,3,4-thiadiazole-2-sulfonamide
2-Bromo-1-(5,5,8,8-tetramethy1-5,6,7,8-tetrahydronaphthalen-2-yl)ethan-1-one
(250 mg,
0.81 mmol) and 2-amino-1,3,4-thiadiazole-5-sulfonamide (146 mg, 0.81 mmol)
were
refluxed in ethanol (10 mL) for 60 hours. Solvent was evaporated under reduced
pressure
and the resulting solid suspended in ethanol (3 m1). The precipitate was
collected by
suction filtration and washed with ethanol to provide compound 56 (45 mg) as
an off
CA 02469953 2007-09-04
white solid. 'H NMR (200MHz, DMSO-d6) 8 8.8 (s, 1H), 8.6 (br s, 2H), 7.8 (br
s, 1H), 7.6
(dd, 1H), 7.3 (d, 1H), 1.6 (s, 4H), 1.3 (s, 61-1), 1.2 (s, 6H).
Example 57:
11-1NMR (200MHz, DMSO-d6) 8 8.73 (s, 2H), 8.48 (s, 1H), 7.75 (s, 1H), 7.18 (s,
1H), 5.42
(br s, 4H), 2.21 (s, 3H), 1.63 (s, 3H), 1.23 (s, 91-1).
Example 58: 6-(4-(S-1-acetam idoethyl)phenyl)imidazo [2,1-b]-1,3,4-
thiadiazole-
2-sulfonamide
2-Bromo-4'-(S-1-acetamidoethyl)acetophenone (426 mg, 1.5 mmol) and 2-amino-
1,3,4-
thiadiazole-5-sulfonamide (222 mg, 1.5 mmol) were refluxed in ethanol (10 mL)
for 60
hours. The resulting solution was cooled to ¨4 C for 2 hours and the
resulting solid was
filtered, washing twice for cold methanol (2 mL), to provide compound 58 (172
mg, 34 %)
as white crystals. 1H NMR (200MHz, DMSO-d6) 8 8.82 (s, 1H), 8.70 (s, 2H), 8.28
(d,
1H), 7.83 (d, 2H), 7.34 (d, 2H), 4.91 (dt, 11-1), 1.83 (s, 3H), 1.33 (d, 31-
1).
Example 59: 6-(4-(Trifluoromethyl imidazo[2,1-b]-1,3,4-thiadiazole-
2-
sulfonamide
3-bromo-1,1,1-trifluoropropan-2-one (534 mg, 2.0 mmol) and 2-amino-1,3,4-
thiadiazole-
5-sulfonamide (360 mg, 2.0 mmol) were refluxed in ethanol (10 mL) for 60 hrs.
The
resulting solution was cooled on ice and the resulting precipitate was
collected by filtration
to provide compound 59 as a white powder (270 mg, 39 %). 11-1 NMR (200MHz,
DMSO-
d6) 8 9.05 (s, 1H), 8.74 (s, 2H).
Example 60: 6-(1-adamantyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-sulfonamide
1-(1-Adamanty1)-2-bromoethan-1-one (514 mg, 2.0 mmol) and 2-amino-1,3,4-
thiadiazole-
5-sulfonamide (360 mg, 2.0 mmol) were refluxed in ethanol (10 mL) for 60 hrs.
The
resulting solution was cooled on ice and the resulting precipitate was
collected by filtration
to provide compound 60 as a white powder (120 mg, 18 %). 1H NMR (200MHz, DMS0-
d6) 68.64 (s, 2H), 8.03 (s, 1H), 2.03 (m, 3H), 1.90 (m, 6H), 1.72 (m, 6H).
66
CA 02469953 2007-09-04
Example 61: 6-(1-Naphthyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
Compound 61 was prepared by bromination of 1-acetylnapthylene with bromine
according
to Method A, followed by condensation of the corresponding bromide with 2-
amino-1,3,4-
thiadiazole-5-sulfonamide, according to Metod C, to yield an off white solid.
1H NMR
(200MHz, DMSO-d6) 5 9.25 (br s, 2H), 8.79 (s, 1H), 8.77 (s, 114), 8.63 (dd,
1H), 7.97 (m,
2H), 7.78 (dd, J=1.2, 7.0Hz, 1H), 7.61-7.49 (m, 2H).
Example 62: 6-(2-Naphthyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
2-Bromonaphthone (2.50 g, 10.0 mmol) and 2-amino-1,3,4-thiadiazole-5-
sulfonamide
(1.80 g, 1.2 mmol) were refluxed in1,4-dioxane (20 mL) for 96 hrs. The
resulting solution
was cooled on ice and the resulting precipitate was collected by filtration to
provide
compound 62 as a tan crystalline solid (2.36 g, 38 %) in two crops. 1H NMR
(200MHz,
DMSO-d6) 8 8.99 (s, 1H), 8.74 (br s, 2H), 8.42 (s, 1H), 8.05 (d, 1H), 7.96-
7.89 (m, 2H),
7.51 (m, 2H).
Example 63: 6-(8-Bromo-7-methoxylnaphth-2-yl)imidazo[2,1-b]-1,3,4-
thiadiazole-2-
sulfonamide
Step 1: 2-Acetyl-6-methoxyacetophenone (1.00 g, 5.0 mmol) was
dissolved in
methanol (5 mL) and was treated with bromine (500 JAL, 10.0 mmol). The
reaction was
stirred at room temperature for 2 hours before the volatiles were removed in
vacuo to
provide a 95:5 mixture of 2,7'-dibromo-6'-methoxylnaphone and 2-acety1-6-
methoxyacetophenone. This crude mixture was advanced to the next step without
further
purification. 'H NMR (200MHz, DMSO-d6) 8 8.75 (s, 1H), 8.20 (d, 1H), 8.10 (d,
1H),
8.07 (dd, 1H), 7.64 (d, 1H), 5.01 (s, 2H), 4.03 (s, 3H).
Step 2: To the crude mixture obtained above was added 5-amino-1,3,4-
thiadiazole-
2-sulfonamide (740 mg, 5.0 mmol) and methanol (20 mL). The resulting
suspension was
refluxed for 48 hours, cooled on ice and the solid filtered off, to provide
compound 63 as a
white solid (230 mg). 'H NMR (200MHz, DMSO-d6) 8 8.95 (s, 1H), 8.76 (d, 11-1),
8.44 (s,
1H), 8.11 (s, 2H), 8.02 (d, 1H), 7.52 (d, 1H), 3.99 (s, 3H).
Example 64: 6-pyrenylimidazo[2,1-b]-1,3,4-thiadiazole-2-sulfonamide
67
CA 02469953 2007-09-04
1-(Bromoacety1)-pyrene (646mg, 2 mmol) and 2-amino-1,3,4-thiadiazole-5-
sulfonamide
(360mg, 2 mmol) were refluxed in ethanol (20 ml) for 60 hrs. Solvent was
removed under
reduced pressure. The resulting solid was purified by silica gel
chromatography, eluting
with solvent gradient of 30-100% ethyl acetate/hexane, to afford compound 64
as a
brownish orange solid (4.5 mg). 'H NMR (200MHz, DMSO-d6) 6 9.0 (s, 1H), 8.8
(s, 2H),
8.50-8.00 (m, 9H).
Example 65: 5-Methy1-6-phenyl-imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
2-Bromopropiophenone (1.07 mg, 5.00 mmol) and 2-amino-1,3,4-thiadiazole-5-
sulfonamide hydrochloride (900 mg, 5.0 mmol) were refluxed in ethanol (25 mL)
for 48
hrs. The resulting solution was cooled on ice and the resulting precipitate
was collected
by filtration to provide compound 65 as a white crystalline solid (100 mg). 1H
NMR
(200MHz, DMSO-d6) 6 8.75 (br s, 3H), 7.75 (d, 2H), 7.45 (t, 2H), 7.30 (t, 1H),
2.65 (s,
3H).
Example 66: 5,6-Diphenyl-imidazo[2,1-b]-1,3,4-thiadiazole-2-sulfonamide
2-Desyl bromide (550 mg, 2 mmol) and 2-amino-1,3,4-thiadiazole-5-sulfonamide
(360
mg, 2 mmol) were refluxed in ethanol (20m1) for 60 hrs. Solvent was removed
under
reduced pressure. Purification by silica gel chromatography, eluting with
30:0.1:70 ethyl
acetate/ acetic acid/ hexane, and recrystallization from dichloromethane gave
compound
66 as a white crystalline solid (175 mg). 1H NMR (200MHz, DMSO-d6) 6 8.8-8.6
(s, 2H),
7.7-7.4 (m, 7H), 7.4-7.2 (m, 3H).
Example 67: 6-(4-
Piperidinophenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-sulfonamide
Stepl: 4'-
Piperidinoacetophenone (203 mg, 1.00 mmol) was dissolved in THF (5
mL) and treated with lithium bis(trimethylsilyl)amide (1.10 mL, 1.0M in THF,
1.10
mmol). The solution was stirred for 30 minutes prior to the addition of
chlorotrimethylsilane (140 1AL, 1.10 mmol). After stirring for an additional
30 minutes N-
bromosuccinamide (300 mg, 1.73 mmol) was added and the mixture was refluxed
from 4
hours. Standard aqueous/ethyl acetate workup provided a yellow solid which was
further
purified by silica gel chromatography, eluting with 3:1 hexane/ethyl acetate,
to provide 2-
68
CA 02469953 2007-09-04
bromo-4'-piperidinoacetophenone as an off white solid (209 mg, 74 %). 1H NMR
(200MHz, CDCI3) 8 8.75 (d, 2H), 6.84 (d, 2H), 4.61 (s, 2H), 3.40 (m, 4H), 1.68
(m, 6H).
Step 2: 2-Bromo-4'-piperidinoacetophenone (209 mg, 0.74 mol) and 5-
amino-
1,3,4-thiadiazole-2-sulfonamide (220 mg, 1.48 mmol) were suspended in 1,4-
dioxane (10
mL) and refluxed for 48 hours. The solvent was removed under reduced pressure
and the
residue was purified by silica gel chromatography, eluting with 1:1
hexane/ethyl acetate,
to provide compound 67 as a yellow solid (8.0 mg, 2.7 %). 1H NMR (200MHz, DMSO-
d6) 8 8.68 (s, 3H), 7.75 (d, 2H), 6.98 (d, 211), 3.23 (m , 4H), 1.57 (m, 61-
1).
Example 68: 6-(4-Morpholinophenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
Step 1: 4'-Morpholinoacetophenone (218 mg, 1.0 mmol) was dissolved
in THF (5
mL) and treated with lithium bis(trimethylsilyl)amide (1.1 mL, 1.0M in THF,
1.1 mmol).
The solution was stirred for 30 minutes prior to the addition of
chlorotrimethylsilane (140
pit, 1.1 mmol). After stirring for an additional 30 minutes N-bromosuccinamide
(300 mg,
1.73 mmol) was added and the mixture was refluxed from 4 hours. Standard
aqueous/ethyl acetate workup provided a yellow solid, which was identified as
a 3:1
mixture of 2-bromo-4'-morpholinoacetophenone and starting material. 1H NMR
(200MHz, CDC13) 8 7.86 (d, 2H), 6.84 (d, 2H), 4.62 (s, 2H), 3.84 (t, 4H), 3.32
(t, 4H).
Step 2: The crude 2-Bromo-4'-morpholinoacetophenone from above and 5-
amino-
1,3,4-thiadiazole-2-sulfonamide (100 mg, 0.66 mmol) were suspended in 1,4-
dioxane (10
mL) and refluxed for 48 hours. The solvent was removed under reduced pressure
and the
residue was purified by silica gel chromatography, eluting with 1:1
hexane/ethyl acetate,
to provide compound 68 as a yellow solid (23 mg). 1H NMR (200MHz, DMSO-d6) 6
8.67
(s, 3H), 7.74 (d, 2H), 6.99 (d, 211), 3.75 (t, 4H), 3.13 (t, 4H).
Example 69: 6-(4-Benzoylamidophenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
Step 1: 4-Aminoacetophenone (1.35 g, 10.0 mmol) was dissolved in
dichloromethane (10 mL) and treated with benzoyl chloride (1.74 mL, 15.0
mmol). The
mixture was stirred for 16 hours at which time a white precipitate had formed.
The solid
was removed by filtration, washing with dichloromethane (3 x 20 mL) to provide
4-
69
CA 02469953 2007-09-04
acetamidoacetophenone as a white sold (2.74 g). 1H NMR (200MHz, DMSO-d6) 6
10.53
(s, 1H), 7.92 (s, 7H), 7.54 (m, 3H), 2.50 (s, 3H).
Step 2: 4-Benzoylamidoacetophenone (2.55 g) was dissolved in acetic
acid (25
mL) and was treated with pyridinium tribromine (3.00 g, 8.0 mmol). The
reaction was
stirred at room temperature for 24 hours before the volatiles were removed in
vacuo to
provide 2-bromo-4'-benzoylamidoacetophenone. This crude mixture was advanced
to the
next step without further purification. 1H NMR (200MHz, DMSO-d6) 6 10.61 (s,
1H),
8.96 (d, 2H), 8.61 (t, 1H), 8.15-8.00 (m, 7H), 4.86 (s, 2H).
Step 3: To the crude mixture obtained above was added 5-amino-1,3,4-
thiadiazole-
2-sulfonamide (1.50 mg, 10.0 mmol) and methanol (20 mL). The resulting
suspension
was refluxed for 48 hours. The solvent was removed under reduced pressure and
the
residue was triturated from acetone to provide compound 69 as a yellow solid
(120 mg).
11-1NMR (200M1-Iz, DMSO-d6) 6 10.35 (s, 1H), 8.82 (s, 1H), 8.70 (br s, 2H),
7.92 (d, 2H),
7.82 (s, 5H), 7.54 (d, 2H).
Example 70: 6-(4-Aminophenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
Compound 66 (60 mg) was suspended in methanol (10 mL) and treated with 6N HC1
(1
mL). The suspension was refluxed for 16 hours until all solids had dissolved.
The solvent
was removed under reduced pressure and the residue was suspended in water (10
mL),
neutralized with saturated aqueous NaHCO3. The resulting precipitate was
extracted with
ethyl acetate to provide compound 70 as a yellow solid (15 mg). 'NMR (200W-1z,
DMSO-d6) 6 8.65 (br s, 2H), 8.53 (s, 1H), 7.58 (d, 21-1), 6.58 (d, 2H), 5.32
(br s, 2H).
Example 71:
Compound 77 was prepared by the bromination of 4-acetylphenylboronic acid
according
to Method A to yield the desired a-bromoacetophenone, which was condensed with
2-
amino-1,3,4-thiadiazole-5-sulfonamide according to Method C, to yield compound
71 as a
white solid. 1H NMR (200MHz, DMSO-d6) 6 8.89 (s, 1H), 8.73 (s, 2H), 8.05 (br
s, 2H),
7.86 (s, 4H).
Example 72: 6-(Biphenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-sulfonamide
CA 02469953 2007-09-04
2-Bromo-4'-phenylacetophenone (1.38 g, 5.0 mmol) and 2-amino-1,3,4-thiadiazole-
5-
sulfonamide hydrochloride (0.90 g, 5.0 mmol) were refluxed in ethanol (20 mL)
for 60
hrs. The resulting solution was cooled on ice and the resulting precipitate
was collected
by filtration to provide compound 72 as a white solid (0.75 g, 45 %). 1H NMR
(200 MHz,
DMSO-d6) 5 8.95 (s, 114), 8.74 (s, 2H), 8.00 (d, J = 8.6 Hz, 2H), 7.74 (m,
4H), 7.44 (m,
3H).
Example 73: 6-(4-(2,3,4,5,6-tetrafluorophenyl)phenypimidazo[2,1-b]-1,3,4-
thiadiazole-
2-sulfonamide
Step 1: 4'-Bromoacetophenone and 2,3,4,5,6-tetrafluorobenzene boronic (5.0
mmol) acid, K2CO3 (10 mmol), and PdC12(PPh)2 (0.1 equiv) were refluxed in
toluene for
16 hours. The solvent was removed under reduced pressure and the residue was
purified
by silica gel chromatography, eluting with 4:1 hexane/ethyl acetate, to
provide 4%
(2,3,4,5,6-tetrafluorophenylacetophenone as a white solid.
Step 2: 4'-(2,3,4,5,6-Tetrafluorophenylacetophenone was dissolved in
diethyl ether
and treated with bromine (5 mmol). Solvent was removed under reduced pressure
to
provide 2-bromo-4'-(2,3,4,5,6-tetrafluorophenylacetophenone, which was used
without
further purification.
Step 3: 2-bromo-4'-(2,3,4,5,6-tetrafluorophenylacetophenone and 2-
amino-1,3,4-
thiadiazole-5-sulfonamide hydrochloride (0.90 g, 5.0 mmol) were refluxed in
ethanol (20
mL) for 60 hrs. The resulting solution was cooled on ice and the resulting
precipitate was
collected by filtration to provide compound 73 as a white solid (0.75 g, 45
%). 1H NMR
(200 MHz, DMSO-d6) 6 8.99 (s, 1H), 8.74 (s, 2H), 8.05 (d, J=8.6Hz, 2H), 7.57
(d,
J=8.6Hz, 2H).
Example 74: 6-(4-(hydroxymethylphenyl)phenyl)imidazo[2,1-14-1,3,4-thiadiazole-
2-sulfonamide
Compound 74 was prepared in a manner similar to compound 73.
1H NMR (200 MHz, DMSO-d6) 6 8.93 (s, 1H), 8.73 (s, 2H), 7.98 (d, J=8.21-1z,
2H), 7.73
(d, J=8.2Hz, 2H), 7.67 (d, J=8.2Hz, 2H), 7.39 (d, J=8.2Hz, 2H), 4.53 (s, 2H).
71
CA 02469953 2007-09-04
Example 75: 6-(4-(2-methoxyphenyl)phenyl)imidazo[2,1 -b]-1,3,4-thiadiazole-
2-sulfonamide
Compound 75 was prepared in a manner similar to compound 73.
IHNMR (200MHz, DMSO-d6) 5 8.89 (s, 1H), 8.73 (s, 2H), 7.92 (d, J=8.3Hz, 2H),
7.53
(d, J=8.31-1z, 21-1), 7.31 (m, 2H), 7.10 (d, J=8.5Hz, 1H), 7.02 (t, J=7.3Hz,
1H), 3.76 (s, 3H).
Example 76: 6-(4-(3-methoxyphenyl)phenyl)imidazo[2,1-b]-1,3,4-thiadiazole-
2-sulfonamide
Compound 76 was prepared in a manner similar to compound 73.
IHNMR (200MHz, DMSO-d6) 5 8.94 (s, 1H), 8.73 (s, 2H), 7.97 (d, J=8.2Hz, 2H),
7.74
(d, J=8.3Hz, 2H), 7.34 (t, J=7.3Hz, 1H), 7.26 (d, J=7.3Hz, 1H), 7.23 (s, 1H),
6.92 (d,
J=7.3Hz, 1H), 3.81 (s, 3H).
Example 77: 6-(4-(4-trifluoromethylphenyl)phenyl)imidazo[2,1 -b]-1,3,4-
thiadiazole-
2-sulfonamide
Compound 77 was prepared in a manner similar to compound 73.
IHNMR (200MHz, DMSO-d6) 5 8.91 (s, 1H), 8.73 (s, 2H), 7.96 (d, J=8.6Hz, 2H),
7.70
(d, J=8.8Hz, 2H), 7.66 (d, J=8.6Hz, 2H), 7.02 (d, J=8.8HZ, 211), 3.80 (s, 3H).
Example 78: 6-(4-(3-trifluoromethoxyphenyl)phenyl)imidazo[2,1 -b]-1,3,4-
thiadiazole-
2-sulfonamide
Compound 78 was prepared in a manner similar to compound 73.
II-1 NMR (200MHz, DMSO-d6) 6 9.00 (s, 11-1), 8.75 (s, 2H), 8.01 (d, J=8.0Hz,
2H), 7.79
(m, 31-1), 7.69 (s, 1H), 7.59 (t, J=8.1Hz, 1H), 7.35 (d, J=7.9Hz, 1H).
Example 79: 6-(4-(hydroxymethylphenyl)phenyl)imidazo[2,1
2-sulfonamide
Compound 79 was prepared in a manner similar to compound 73. MS (m/z) M+1 =
401
Example 80: 6-(4-(2-trifluoromethylphenyl)phenyDimidazo[2,1-b]-1,3,4-
thiadiazole-
2-sulfonamide
72
CA 02469953 2007-09-04
Compound 80 was prepared in a manner similar to compound 73.
1H NMR (200MHz, DMSO-d6) 5 8.93 (s, 1H), 8.71 (s, 2H), 7.95 (d, 211), 7.77 (d,
2H),
7.66 (t, 1H), 7.60 (t, 1H), 7.35 (m, 2H).
Example 81: 6-(4-(3-trifluoromethylphenyl)phenypimidazo[2,1-14-1,3,4-
thiadiazole-
2-sulfonamide
Compound 81 was prepared in a manner similar to compound 73.
1H NMR (200MHz, DMSO-d6) 5 8.98 (s, 1H), 8.74 (s, 2H), 8.02 (m, 4H), 7.84 (d,
J=8.211z, 2H), 7.70 (m, 2H).
Example 82: 6-(4-(4-trifluoromethylphenyl)phenypimidazo[2,1 -b]-1,3,4-
thiadiazole-
2-sulfonamide
Compound 82 was prepared in a manner similar to compound 73.
1H NMR (200MHz, DMSO-d6) 5 8.97 (s, 111), 8.73 (s, 2H), 8.05-7.80 (m, 8H).
Example 83: 6-(4-(3,5-ditrifluoromethylphenyl)phenyl)imidazo[2,1 -b]-1,3,4-
thiadiazole-
2-sulfonamide
Compound 83 was prepared in a manner similar to compound 73.
1H NMR (200M1-1z, DMSO-d6) 5 9.02 (s, 1H), 8.75 (s, 2H), 8.38 (s, 2H), 8.01
(m, 5H).
Example 84: 6-(thiophen-2-yl-phenyl)im idazo[2,1-b]-1,3,4-thiadiazole-
2-sulfonamide
Compound 84 was prepared in a manner similar to compound 73.
1H NMR (200M1-Iz, DMSO-d6) 5 8.92 (s, 1H), 8.70 (s, 2H), 7.94 (d, J-8.2Hz,
2H), 7.73
(d, J=8.2Hz, 211), 7.56 (m, 211), 7.15 (m, 1H).
Example 85: 6-(thiophen-3-yl-phenyl)imidazo[2,1 -b]-1,3,4-thiadiazole-
2-sulfonamide
Compound 85 was prepared in a manner similar to compound 73.
73
CA 02469953 2007-09-04
11-1NMR (200M1-Iz, DMSO-d6) 8 8.79 (s, 1H), 8.72 (s, 2H), 7.88 (d, J=8.211z,
2H), 7.16 (t,
J=8.2Hz, 111), 7.03 (d, J=8.2Hz, 211), 6.49 (d, J=8.211z, 1H), 6.38 (s, 1H),
6.27 (d,
J=7.3Hz, 11-1), 2.86 (s, 6H).
Example 86: 6-(4'-methoxybiphen-3-yl)imidazo[2,1-4.1,3,4-thiadiazole-
2-sulfonamide
Compound 86 was prepared in a manner similar to compound 73.
1H NMR (200MHz, DMSO-d6) 8 8.98 (s, 1H), 8.72 (s, 2H), 8.14 (s, 111), 7.84 (d,
J=7.3Hz,
1H), 7.67 (d, .1-8.511Z, 2H), 7.54 (m, 2H), 7.05 (d, J=8.9Hz, 2H), 2.80 (s,
3H).
Example 87: 6-(4-phenoxyphenyl)imidazo[2,1 -b]-1,3,4-thiadiazole-
2-sulfonamide
Step 1: 4-Phenoxyacetopheneone was prepared by refluxing 4-
fluoroaceotone and
phenol in DMAc for 16 hours. The solvent was removed under reduced pressure
and the
reside subjected to standard ethyl acetate/water work-up and the resulting
material purified
by silica gel chromatography.
Step 2: 4-Phenoxyacetophenone was brominated with bromine according
to
Method A to provide the desired a-bromoacetophenone, which was condensed with
2-
amino-13,4,-thiadiazole-5-sulfonamide according to method C, to provide
compound 87
as an off white solid.. 111 NMR (200MHz, DMSO-d6) 8 8.82 (s, 1H), 8.71 (s,
211), 7.90 (d,
J=7.9Hz, 211), 7.40 (t, J=7.3Hz, 2H), 7.07 (t, J=7.3Hz, 111), 7.08-7.03 (m,
611).
Example 88: 6-(4-chlorophenoxyphenyl)imidazo[2,1-b]-1,3,4-thiadiazole-
2-sulfonamide
Compound 88 was prepared in a manner similar to compound 87.
1H NMR (200MHz, DMSO-d6) 6 8.81 (s, 11-1), 8.72 (s, 2H), 7.89 (d, J=8.5Hz,
2H), 7.23
(m, 211), 7.11-7.01 (m, 4H).
Example 89: 6-(4-(3,4-difluorophenoxy)phenyl)imidazo[2,1-b]-1,3,4-thiadiazole-
2-sulfonamide
74
CA 02469953 2007-09-04
Compound 89 was prepared in a manner similar to that described for compound
87. MS
(m/z) M+1 = 409.
Example 90: 6-(4-(4-azaphenoxy)phenyl)imidazo[2,1-17]-1,3,4-thiadiazole-
2-sulfonamide
Compound 90 was prepared in a manner similar to that described for compound
87. 1H
NMR (200MHz, DMSO-d6) 6 9.02, 8.76 (s, 2H), 8.41 (d, J=8.6Hz, 2H), 8.10 (d,
J=8.6Hz,
2H), 7.71 (d, J=8.6Hz, 211), 7.01 (d, J=8.6Hz, 2H).
Example 91: 6-(4-chlorophenoxyphenyl)imidazo[2,1-b]-1,3,4-thiadiazole-
2-sulfonamide
Compound 91 was prepared in a manner similar to compound 87.
1H NMR (200MHz, DMSO-d6) 6 8.84 (s, 1H), 8.72 (s, 2H), 7.92 (d, J=8.5Hz, 2H),
7.44
(d, J=8.5H2, 21-1), 7.08 (m, 4H).
Example 92: 6-(3,4-dichlorophenoxyphenyl)imidazo[2,1-b]-1,3,4-thiadiazole-
2-sulfonamide
Compound 92 was prepared in a manner similar to compound 87.
1H NMR (200MHz, DMSO-d6) 8 8.86 (s, 1H), 8.72 (s, 2H), 7.94 (d, J=8.5Hz, 2H),
7.63
(d, J=8.9Hz, 1H), 7.34 (d, J=2.7Hz, 1H), 7.16 (d, J=8.9Hz, 2H), 7.04 (dd,
1J=8.9Hz,
2J=2.8Hz, 1H).
Example 93: 6-(2-bromophenoxyphenyl)imidazo[2,1-b]-1,3,4-thiadiazole-
2-sulfonamide
Compound 93 was prepared in a manner similar to compound 87.
'1-I NMR (200MHz, DMSO-d6) 6 8.82 (s, 1H), 8.72 (s, 2H), 7.90 (d, J=8.2Hz,
2H), 7.74
(d, J=7.911z, 1H), 7.37 (t, J=7.5Hz, 1H), 7.15 (t, J=8.4Hz, 2H), 7.00 (d,
J=8.5Hz, 2H).
Example 94: 6-(3-bromophenoxyphenyl)imidazo[2,1-b]-1,3,4-thiadiazole-
2-sulfonamide
CA 02469953 2007-09-04
Compound 94 was prepared in a manner similar to compound 87. MS (m/z) M+1 =
452,
M+2 = 454.
Example 95: 6-(4-bromophenoxyphenypimidazo[2,1-b]-1,3,4-thiadiazole-
2-sulfonamide
Compound 95 was prepared in a manner similar to compound 87. 1H NMR (200 MHz,
DMSO-d6) 8 6.98-7.19 (m, 4H), 7.55 (d, 2H, J = 8.5 Hz), 7.92 (d, 2H, J = 8.5
Hz), 8.71 (s,
2H), 8.84 (s, 1H).
Example 96: 6-(3-dimethylaminophenoxyphenyl)imidazo[2,1-b]-1,3,4-thiadiazole-
2-sulfonamide
Compound 96 was prepared in a manner similar to compound 87.
1H NMR (200MHz, DMSO-d6) 6 8.79 (s, 1H), 8.72 (s, 2H), 7.88 (d, J=8.2Hz, 2H),
7.16 (t,
J=8.2Hz, 1H), 7.03 (d, J=8.2Hz, 2H), 6.49 (d, J=8.2Hz, 1H), 6.38 (s, 1H), 6.27
(d,
J=7.3Hz, 111), 2.86 (s, 6H).
Example 97: 6-(4-(4-iso-propylphenoxy)phenyl)imidazo[2,1-b]-1,3,4-thiadiazole-
2-sulfonamide
Compound 97 was prepared in a manner similar to compound 87.
1H NMR (200MHz, DMSO-d6) 5 8.81 (s, 1H), 8.72 (s, 2H), 7.89 (d, J=8.9Hz, 2H),
7.26
(d, J=8.51-1z, 2H), 7.03 (d, J=8.9Hz, 2H), 6.97 (d, J=8.5Hz, 2H), 2.88
(septet, J=6.9Hz,
11-1), 1.19 (d, J=6.7Hz, 6H).
Example 98: 6-(4-(4-methoxyphenoxy)phenyl)imidazo[2,1 -b]-1,3,4-thiadiazole-
2-sulfonamide
Compound 98 was prepared in a manner similar to that described for compound
87.1H
NMR (200MHz, DMSO-d6) 5 8.79 (s, 11-1), 8.71 (s, 2H), 7.84 (d, J=8.811z, 2H),
7.06-6.94
(m, 4H), 3.74 (s, 3H).
Example 99: 6-(4-(4-nitrophenoxy)phenyl)imidazo[2,1-b]-1,3,4-thiadiazole-
2-sulfonamide
76
CA 02469953 2007-09-04
Compound 99 was prepared in a manner similar to compound 87.
1H NMR (200MHz, DMSO-d6) 8 8.91 (s, 1H), 8.73 (s, 2H), 8.25 (d, J=8.9Hz, 2H),
8.00
(d, J=8.2Hz, 2H), 7.27 (d, J=8.51-lz, 2H), 7.18 (d, J=8.91-1z, 2H).
Example 100: 6-(4-(3,4-difluorophenoxy)phenyl)imidazo[2,1-b]-1,3,4-thiadiazole-
2-sulfonamide
Compound 100 was prepared in a manner similar to compound 87. MS (m/z) M+1 =
409.
Example 101:
Step 1: 4-Hydroxyacetophenone (5.00 g, 36.7 mmol) was dissolved in THF (100
mL) and cooled on ice. Sodium bis(trimethylsilyl)amide (40.5 mL, 1.0M in THF,
40.5
mmol) was added and the solution was warmed to room temperature. After
stirring for 2
hours the solution was cooled on ice and ethyl bromoacetate (6.14 mL, 55.1
mmol) was
added. The solution was stirred over night. Standard aqueous workup provided
the
desired ester as a clear oil, which was dissolved in 3:2:1 THF/methanol/LOM
NaOH (36
mL). After stirring over night the solution was diluted with diethyl ether and
water. The
aqueous layer was separated, washed with diethyl ether, and acidified. The
resulting solid
was extracted with ethyl acetate to provide 2-(4-acetylphenoxy)acetic acid as
a white solid
(2.91 g).
Step 2: 2-(4-acetylphenoxy)acetic acid (2.81 g, 14.5 mmol) was dissolved in
acetic
acid (100 mL) and treated with bromine (740 1.1,L, 14.5 mmol) and stirred 48
hours.
Bromine (370 mL, 7.25 mmol) was added and solution was stirred for an
additional 16
hours. Volatiles were removed under reduced pressure to provide a brown solid
which
was titurated with diethyl ether to provide 2-(4-(2-bromoacetyl)phenoxy)acetic
acid (1.82
g) as a light brown solid.
Step 3: 2-(4-(2-Bromoacetyl)phenoxy)acetic acid (1.00 g, 3.66 mmol)
and 5-
amino-1,3,4-thiadiazole-2-sulfonamide (659 mg, 3.66 mmol) were refluxed
together in
methanol (20 mL) for 48 hours. The resulting suspension was cooled to -10 C,
filtered
and the solid washed with cold methanol (3 x 5 mL) to provide compound 101 as
a white
crystalline solid (578 mg, 44 %). 114 NMR (200MHz, DMSO-d6) 8 8.69 (s, 1H),
7.65 (d,
2H), 7.02 (d, 2H), 4.83 (s, 2H), 3.70 (s, 311), 2.64 (s, 3H).
77
CA 02469953 2007-09-04
Example 102:
Compound 101 (50 mg, 0.14 mmol) was dissolved in 3:2:1 THF/methanol/1M NaOH (9
mL) and stirred over night. The resulting solution was diluted with ethyl
acetate and
water. The aqueous layer was washed with ethyl acetate and acidified to yield
a white
suspension. The suspension was extracted with ethyl acetate, the organic layer
was dried
over anhydrous MgSO4, filtered, and the solvent removed under reduced pressure
to
provide compound 102 as a white solid (10.2 mg). Ili NMR (200MHz, DMSO-d6) 6
8.74
(s, 111), 7.80 (d, J=8.8Hz, 2H), 6.96 (d, J=8.8Hz, 211), 4.66 (s, 2H).
Example 103:
4-(2-Bromopropionyl)phenylacetic acid (256 mg, 1.0 mmol) and 5-amino-1,3,4-
thiadiazole-2-sulfonamide (185 mg, 1.22 mmol) were refluxed together in
methanol (20
mL) for 48 hours. The resulting suspension was cooled to ¨10 C, filtered and
the solid
washed with cold methanol (3 x 5 mL) to provide compound 103 as a white
crystalline
solid (22 mg, 7 %). 111NMR (200MHz, DMSO-d6) 5 8.69 (s, 1H), 7.65 (d, 2H),
7.02 (d,
2H), 4.83 (s, 2H), 3.70 (s, 3H), 2.64 (s, 3H).
Example 104: 6-(3-chloro-4-methylpheny1)-5-methylimidazo[2,1-b]-
1,3,4-
thiadiazole-2-sulfonamide
2-Bromo-1-(3-chloro-4-methylphenyppropan-1 -one (262 mg, 1.0 mmol) and 2-amino-
1,3,4-thiadiazole-5-sulfonamide (180 mg, 1.0 mmol) were refluxed in ethanol
(10 mL) for
5 days. The resulting solution was concentrated and the crude material was
purified by
column chromatography on silica gel, eluting with 25:75 ethyl acetate/hexanes
to provide
compound 104 as a yellow powder (38 mg, 11 %). IHNMR (200MHz, DMSO-d6) 6 8.72
(s, 2H), 7.74 (m, 1H), 7.59 (m, 1H), 7.48 (m, 1H), 2.68 (s, 3H), 2.39 (s, 3H).
Example 105: 6-(2-Pyridyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
2-(2-bromoacetyl)pyridine (2.5 g, 12.4 mmol) and 2-amino-1,3,4-thiadiazole-5-
sulfonamide (2.2 g, 12.4 mmol) were refluxed in methanol (75 mL) for 48 hrs.
After
evaporation of methanol IM sodium hydroxide (25 mL) was added and the
resulting
78
CA 02469953 2007-09-04
solution was washed with ether (3 x 20 mL). The aqueous layer was acidified to
a pH of 7
with 1M hydrochloric acid and extracted with ethyl acetate (3 x 25 mL). The
solid
obtained from the organic layers was recrystallized in acetone to provide
compound 105 as
a light brown powder (84 mg, 2.4 %). 1H NMR (200MHz, DMSO-d6) 6 8.82 (s, 1H),
8.74
(br s, 2H), 8.58 (d, J = 5.5 Hz, 1H), 7.92 (m, 2H), 7.33 (m, 1H).
Example 106: 6-(2-Pyridypimidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide HC1
Compound 105 (25 mg) was dissolved in methanol and HC1 gas was bubbled through
for
30 seconds. Volatiles were removed under reduced pressure to provide a white
solid
(99%). 1H NMR (200MHz, D20-d6) 6 8.86 (s, 1H), 8.57 (d, J=5.8Hz, 1H), 8.50 (t,
J=7.5Hz, 1H), 8.27 (d, J=8.2Hz, 11-1), 7.80 (t, J=6.7Hz, 1H).
Example 107: 6-(4-Pyridyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
2-Bromo-1-(4-pyridiny1)-1-ethanone hydrobromide (100 mg, 0.356 mmol) and 2-
amino-
1,3,4-thiadiazole-5-sulfonamide (64 mg, 0.356 mmol) were refluxed in 1,4-
dioxane (5
mL) for 48 hours. The resulting solid was isolated by filtration and
recrystallized from
methanol to provide compounds 107 as a brown solid (129 mg, 42 % yield). 1H
NMR
(200MHz, DMSO-d6) 6 9.52 (s, 1H), 8.90 (d, 2H), 8.84 (s, 2H), 8.39 (d, 2H).
Example 108: 6-(2-Pyrimidenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
Stepl: Acetylpyrazine (244 mg, 2.0 mmol) was suspended in glacial
acetic acid (10
mL) and treated with pyridinium tribromide (640 mg, 2.0 mmol). The reaction
mixture
was stirred overnight. The solvent was removed under reduced pressure and the
residue
was purified by silica gel chromatography, eluting with 1:1 hexane/ethyl
acetate, to
provide 2-(2-bromoacetyl)pyrazine as a brown solid (154 mg, 38%). 11-INMR
(200MHz,
DMSO-d6) 69.16 (d, J = 1.5Hz, 1H), 8.94 (d, J = 2.4Hz, 1H), 8.82 (dd, J = 1.5,
2.4Hz,
1H), 4.99 (s, 2H).
Step 2 2-(2-Bromoacetyl)pyrazine (154 mg, 0.764 mmol) and 5-amino-
1,3,4-
thiadiazole-2-sulfonamide (138 mg, 0.764 mmol) were refluxed together in
methanol (10
mL) for 48 hours. The resulting suspension was cooled to ¨10 C, filtered and
the solid
washed with cold methanol (3 x 2 mL) to provide compound 108 as a brown solid
(6.6
79
CA 02469953 2007-09-04
mg, 3.1%). 111 NMR (200MHz, DMSO-d6) 8 9.19 (s, 1H), 8.98 (s,1H), 8.78 (s, 21-
1), 8.65
(d, J = 1.51-1z, 1H), 8.59 (d, J = 2.7Hz, IH).
Example 109: 6-(coumaran-2-yl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
2-(2-Bromoacetyl)coumaran (mg, 1.0 mmol) and 5-amino-1,3,4-thiadiazole-2-
sulfonamide
(180 mg, 1.0 mmol) were refluxed together in methanol (10 mL) for 72 hours.
The
resulting suspension was cooled to ¨10 C, filtered and the solid washed with
cold
methanol (3 x 2 mL) to provide compound 109 as a white solid (15.6 mg, 5.4%).
'H NMR
(200MHz, DMSO-d6) 8 8.79 (s, 1H), 8.65 (br s, 1H), 8.61 (s, 1H), 7.81 (d,
J=7.6Hz, 1H),
7.58 (m, 1H), 7.38 (m, 2H). 13C NMR (50MHz, DMSO-d6) 8 164.7, 158.5, 152.5,
139.9,
137.5, 131.9, 128.9, 124.8, 119.8, 119.1, 116.0, 114.6, 96.7.
Example 110:
6-(ChloroacetyI)-2-H-1,4-benzoxazin-3(4H)-one (248 mg, 1.1 mmol), n-Bu4NI (405
mg,
1.1 mmol) and 5-amino-1,3,4-thiadiazole-2-sulfonamide (148 mg, 1.0 mmol) were
refluxed together in methanol (12 mL) for 4 days. The resulting precipitate
was isolated
by filtration, washing with cold methanol, to provide compounds 110 as a white
solid (58
mg). 'H NMR (200MHz, DMSO-d6) 8 10.84 (s, 1H), 8.79 (s, 3H), 7.48 (s, 1H),
7.42 (d,
2H), 7.00 (d, 2H), 4.58 (s, 2H).
Example 111: 6-(Benzo[b]furan-2-ypimidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
1-(1-benzofuran-2-y1)-2-bromoethan-1-one (100 mg, 0.41 mmol) and 2-amino-1,3,4-
thiadiazole-5-sulfonamide (74 mg, 0.41 mmol) were refluxed in ethanol (5 mL)
for 30 hrs.
The resulting solution was cooled on ice and the resulting precipitate was
collected by
filtration to provide compound 111 as an off white powder (47 mg, 33 %).
NMR
(200MHz, DMSO-d6) 8 8.86 (s, 11-1), 8.77 (br s, 21-1), 7.65 (m, 2H), 7.29 (m,
3H).
Example 112: 6-(2-Thiopheny1)-imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
2-Acetylthiophene (252 mg, 2.0 mmol) was dissolved in acetic acid (5 mL) and
treated
with bromine (100 jiL, 2.0 mmol). The solution was stirred overnight before
the volatiles
CA 02469953 2007-09-04
were removed under reduced pressure to provide a white solid, which contained
a 3:1
mixture of (2-bromoacetyl)thiophene and starting material. This crude mixture
was
refluxed in methanol (10 mL) with 5-amino-1,3,4-thiadiazole-2-sulfonamide (300
mg, 2.0
mmol) for 5 days. The resulting solid was filtered, washing with methanol (3 x
5 mL) to
provide compounds 112 as a light pink solid (60.5 mg). 1H NMR (200MHz, DMSO-
d6) 6
8.70 (s, 1H), 7.48 (m, 2H), 7.11 (t, 1H).
Example 113: 6-(5-Phenylthiophen-2-y1)-imidazo[2,1 -M-1,3,4-thiadiazole-2-
sulfonamide
2-Bromo-1-(5-pheny1-2-thieny1)-1-ethanone (100 mg, 0.36mmol) and 2-amino-1,3,4-
thiadiazole-2-sulfonamide were refluxed in ethanol for 120 hours. The
volatiles were
removed in vacuo. The residue was purified by column chromatography on silica
gel
using 20% ethyl acetate/I% acetic acid in hexane followed by 30% ethyl
acetate/1% acetic
acid in hexane as eluant. Triturating with diethyl ether provided compound 113
(7 mg) as
an orange solid. 'FT NMR (200MHz, DMSO-d6) 8 8.82 (s, 114), 8.70 (br s, 2H),
7.70-7.20
(m, 7H).
Example 114: 6-(5-Nitro-2-thiopheny1)-imidazo[2,1-b]-1,3,4-
thiadiazole-2-
sulfonamide hydrobromide
Example 115: 6-(3-Methylbenzo[b]thiophen-2-yDimidazo[2,1-11-1,3,4-
thiadiazole-2-sulfonamide
2-Bromo-1-(3-methylbenzo[b]thiophen-2-ypethan-1 -one (125 mg, 0.5 mmol) and 2-
amino-1,3,4-thiadiazole-5-sulfonamide (74 mg, 0.5 mmol) were refluxed in
ethanol (10
mL) for 72 hrs. The resulting solution was cooled on ice and the resulting
precipitate was
collected by filtration to provide compound 115 as a white crystalline solid
(63 mg, 39 %).
1H NMR (200MHz, DMSO-d6) 6 8.80 (s, 1H), 8.78 (s, 2H), 8.94 (dd, 1H), 8.83
(dd, 1H),
7.42 (dt, 1H), 7.37 (dt, 1H), 2.58 (s, 311).
Example 116: 6-(Benzo[b]thiophen-3-yDimidazo[2,1-b]-1,3,4-
thiadiazole-2-
sulfonamide
81
CA 02469953 2007-09-04
1-Benzo[b]thiophen-3-y1-2-bromoethan-l-one (125 mg, 0.5 mmol) and 2-amino-
1,3,4-
thiadiazole-5-sulfonamide (74 mg, 0.5 mmol) were refluxed in ethanol (10 mL)
for 72 hrs.
The resulting solution was cooled on ice and the resulting precipitate was
collected by
filtration to provide compound 116 as a white crystalline solid (63 mg, 39 %).
114 NMR
(200MHz, DMSO-d6) 6 8.91 (s, 1H), 8.74 (br s, 211), 8.52 (d, 1H), 8.13 (s,
1H), 8.05 (d,
1H), 7.50-7.42 (m, 2H).
Example 117: 6-(3-Phenylisoxazol-5-y0imidazo[2,1-b]-1,3,4-
thiadiazole-2-
sulfonamide
Prepared according to Method C. 'H NMR (200MHz, DMSO-d6) 6 9.03 (s, 1H), 8.79
(s,
2H), 7.95 (m, 3H), 7.53 (m, 2H), 7.36 (s, 3H).
Example 118: 6-(5-Methy1-3-phenylisoxazol-4-yl)imidazo[2,1-b]-1,3,4-
thiadiazole-2-sulfonamide
2-Bromo-1-(5-methy1-3-phenylisoxazol-4-yl)ethan-1-one (100 mg, 0.36 mmol) and
2-
amino-1,3,4-thiadiazole-5-sulfonamide (65 mg, 0.36 mmol) were refluxed in
ethanol (5
mL) for 60 hrs. Solvent was evaporated and the crude solid was purified by
flash
chromatography using 35 : 65 ethyl acetate : hexanes to provide compound 118
as a
yellowish powder (64 mg, 50 %). 'H NMR (200MHz, DMSO-d6) 6 8.73 (br s, 2H),
8.25
(s, 111), 7.58 (m, 2H), 7.45 (m, 311), 2.56 (s, 3H).
Example 119: 6-(Ethyl isoxazol-5-y1-3-carboxylate)imidazo[2,1-b]-
1,3,4-
thiadiazole-2-sulfonamide
Ethyl 5-(2-bromoacetyl)isoxazole-3-carboxylate (100 mg, 0.38 mmol) and 2-amino-
1,3,4-
thiadiazole-5-sulfonamide (70 mg, 0.38 mmol) were refluxed in ethanol (5 mL)
for 60 hrs.
The resulting solution was cooled on ice and the resulting precipitate was
collected by
filtration to provide compound 119 as an orange powder (21 mg, 16 %). 1H NMR
(200MHz, DMSO-d6) 6 9.14 (s, 1H), 8.82 (s, 211), 7.16 (s, 1H), 4.39 (q, J =
7.0 Hz, 211),
1.34 (t, J = 7.1 Hz, 3H).
82
CA 02469953 2007-09-04
Example 120: 6-(5-
methyl-l-pheny1-1H-pyrazol-4-yl)imidazo[2,1-b]-1,3,4-
thiadiazole-2-sulfonamide
2-Bromo-1-(5-methyl-l-pheny1-1H-pyrazol-4-yl)ethan-1-one (100 mg, 0.36 mmol)
and 2-
amino-1,3,4-thiadiazole-5-sulfonamide (65 mg, 0.36 mmol) were refluxed in
ethanol (5
mL) for 45 hrs. Solvent was evaporated and the solid was recrystallized from
ethanol to
provide compound 120 as a beige powder (30 mg, 28 %). 1H NMR (200MHz, DMSO-d6)
6 8.71 (s, 2H), 8.55 (s, 1H), 7.98 (s, 1H), 7.52 (m, 5H), 2.56 (s, 3H).
Example 121: 6-(Thiaxo1-2-yl)imidazo[2,1-b]-1,3,4-thiadiazole-2-sulfonamide
Stepl 2-Acetylthiazole (400 4, 3.86 mmol) was suspended in
chloroform (10 mL)
and treated with pyridinium tribromide (1.23 g, 3.86 mmol). The reaction
mixture was
stirred for two days. The solvent was removed under reduced pressure. Standard
aqueous/ethyl acetate workup provided a dark orange solid, which was
identified as a 8:1
mixture of 2-(2-bromoacetyl)thiazole and starting material. 1H NMR (200MHz,
DMSO-
d6) 6 8.30 (d, J = 2.1Hz, I H), 8.18 (d, J = 2.1Hz, 1H), 4.93 (s, 2H).
Step 2 2-(2-Bromoacetyl)thiazole (206 mg, 1.0 mmol) and 5-amino-1,3,4-
thiadiazole-2-sulfonamide (180mg, 1.0 mmol) were refluxed together in methanol
(10 mL)
for 72 hours. The resulting suspension was cooled to ¨10 C, filtered and the
solid washed
with cold methanol (3 x 2 mL) to provide compound 121 as a white solid (15.6
mg, 5.4%).
1H NMR (200MHz, DMSO-d6) 6 8.90 (s, 114), 8.74 (br s, 2H), 7.90 (d, J = 3.1Hz,
1H),
7.75 (d, J = 3.3Hz, 1H).
Example 122: 6-(2,4-Thiaxo1-5-yl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
Compoudn 122 was prepared according to the method described for compound 103
to
provide compound 122 as an off white solid. 1H NMR (200MHz, DMSO-d6) 6 8.74
(s,
2H), 8.65 (s, 1H), 2.63 (s, 3H), 2.47 (s, 3H).
Example 123: 5-Chloro-
6-phenyl-imidazo[2,1-b]-1,3,4-thiadiazole-sulfonamide
Compound 1 (250 mg, 0.891 mmol) was dissolved in 10:1 THF/water (22 mL) and
treated
with 40 % sodium hypochlorite (1 mL). The solution was stirred for 3 hours
before the
83
CA 02469953 2007-09-04
volatiles were removed under reduced pressure to provide a light yellow solid
(310 mg,
98%). 11-1 NMR (200MHz, DMSO-d6) 6 7.96 (m, 2H), 7.43 (t, 2H), 7.39 (t, IH).
Example 124: 5-Bromo-6-phenyl-imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
hydrobromide
Compound 1(5.00 g, 17.8 mmol) in AcOH (200 mL) was treated with bromine (0.96
mL,
18.7 mmol). The solution was stirred overnight and the formation of a white
precipitate
was observed. The solvent was evaporated and the solid was suspended in Me0H
(50
mL). That suspension was put in the fridge for one hour and filtered to
provide compound
124 as a white powder (4.8 g, 76 % after 3rd crop). '1-I NMR (200 MHz, DMSO) :
5 8.82
(s, 2H), 7.98 (d, J = 8.2 Hz, 2H), 7.46 (m, 3H).
Example 125: 5-Bromo-6-(2-pyridyl)imidazo[2,1-1]-1,3,4-thiadiazole-2-
sulfonamide hydrobromide
Compound 105(50 mg, 0.18 mmol) was suspended in acetic acid (5 mL) and treated
with
bromine (10 4, 0.20 mmol). After stirring overnight the volatiles were removed
under
reduced pressure and the solid was dried under vacuum to provide compound 125
as a
yellow solid (54 mg, 84 %). 1H NMR (200MHz, DMSO-d6) 6 8.84 (s, 2H), 8.67 (d,
J = 4.6
Hz, 1H), 8.02 (m, 21-1), 7.43 (m, 1H); 13C NMR (50 MHz, DMSO) 5 166.9, 147.1,
146.0,
142.6, 137.9, 124.8, 122.4, 98.7.
Example 126: 5-Bromo-6-(4-nitrophenyl)imidazo[2,1-1]-1,3,4-
thiadiazole-2-
sulfonamide hydrobromide
Example 127: 5-Bromo-6-(4-chlorophenyl)imidazo[2,14+1,3,4-thiadiazole-2-
sulfonamide hydrobromide
Example 128: 5-Bromo-6-(4-bromophenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide hydrobromide
84
CA 02469953 2007-09-04
Compound 14 (100 mg, 0.278 mmol) was suspended in acetic acid (5 mL) and
treated
with neat bromine (16 AL, 0.306 mmol). The reaction mixture was stirred
overnight and
volatiles were removed under reduced pressure to provide compounds 128 as a
light
orange solid (143 mg, 99%). 1H NMR (200MHz, DMSO-d6) 8 8.82 (s, 2H), 7.92 (d,
2H),
7.68 (d, 2H).
Example 129: 5-Bromo-6-(2-bromo3-methoxyphenypimidazo[2,1-1+1,3,4-
thiadiazole-
2-sulfonamide hydrobromide
Prepared according to the method described for compound 128.
Example 130: 5-Bromo-6-(2-naphthypimidazo[2,1-1+1,3,4-thiadiazole-2-
sulfonamide
hydrobromide
Compound 62 (192 mg, 0.582 mmol) was suspended in acetic acid (10 mL) and
treated
with neat bromine (31 L, 0.612 mmol). After stirring overnight the volatiles
were
removed under reduced pressure, to provide compound 130 as a tan solid (279
mg, 93 %).
1H NMR (200MHz, DMSO-d6) 8 8.83 (br s, 21-1), 8.42 (s, 1H), 8.04 (d, 1H), 7.98-
7.80 (m,
3H), 7.48 (m, 2H).
Example 131: 5-Chloro-6-(biphenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
hydrobromide
Compound 131 was prepared according to the method described for compound 123
to
provide a white solid.
Example 132: 5-Bromo-6-(biphenyl)im idazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
hydrobromide
Compound 72 (1.00 g, 2.80 mmol) was suspended in AcOH (20 mL) and treated with
bromine (172 pt, 3.4 mmol). The solution was stirred overnight and the
formation of a
white precipitate was observed. The solvent was evaporated and the solid was
dissolved in
10 mL Me0H. That suspension was put in the fridge for one hour to increase
precipitation.
Successive precipitations and filtrations provided compound 132 as a yellow
powder (1.10
CA 02469953 2007-09-04
g, 79 %). 1H NMR (200 MHz, DMSO) 6 8.82 (s, 1H), 8.11 (d, J = 8.5 Hz, 211),
7.82 (d, J
= 8.6 Hz, 2H), 7.74 (d, J = 8.5 Hz, 2H), 7.45 (m, 31-1).
Example 133: 5-Bromo-6-(5-nitrothiophen-2-yDimidazo[2,1-b]-1,3,4-
thiadiazole-2-
sulfonamide hydrobromide
Example 134: 5-Bromo-6-trifluoromethylimidazo[2,1 -b]-1,3,4-
thiadiazole-2-
sulfonamide
Compound 59 (430 mg, 1.59 mmol) was suspended in acetic acid (10 mL) and
treated
with bromine (244 L, 4.76 mmol). After stirring stirring overnight the
volatiles were
removed under reduced pressure to provide a 1:1 mixture of compounds 59 and
134. The
above process was repeated to provide compound 134 as a white solid (477 mg).
'H NMR
(200MHz, DMSO-d6) ö 8.89 (s, 2H).
Example 135: 5-Thiopheny1-6-(2-naphthypimidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
Compound 130 (100 mg, 0.204 mmol) and mercaptobenzene (28 uL, 0.269 mmol) were
combined in THF (10 mL) and refluxed for 16 hours, followed by 72 hours at
room
temperature. Volatiles were removed under reduced pressure and the resulting
residue
was triturated with diethyl ether to provide compound 135 as a yellow solid
(62 mg, 69 %
yield). 1H NMR (200MHz, DMSO-d6) 8 8.82 (br s, 2H), 8.58 (s, 1H), 8.19 (d,
1H), 7.92
(d, 111), 7.83 (m, 2H), 7.53 (m, 211), 7.38-7.12 (m, 511).
Example 136: 5-(S-(2-Thio-5-amino-1,3,4-thiadiazoly1)-6-(2-
naphthypimidazo[2,1-M-
1,3,4-thiadiazole-2-sulfonamide
Compound 130 (102 mg, 0.208 mmol) and 5-amino-1,3,4-thiadiazole-2-thiol (37
mg,
0.275 mmol) were combined in a 2:1 mixture of ethyl acetate/methanol (15 mL)
and
refluxed for 16 hours, followed by 72 hours at room temperature. Volatiles
were removed
under reduced pressure and the resulting residue was recrystallized from
methanol to
provide compound 136 as a yellow solid (32 mg, 34 % yield). 1H NMR (200MHz,
86
CA 02469953 2007-09-04
DMSO-d) 5 8.86 (br s, 2H), 8.66 (s, 1H), 8.28 (d, 1H), 8.02 (d, 1H), 7.98 (m,
214), 7.57
(m, 2H), 7.36 (br s, 2H).
Example 137: 6-Phenyl-imidazo[2,1-14-1,3,4-thiadiazole-2-N-methylsulfonamide
Compound 1 (110 mg, 0.50 mmol), methanol (30 mg, 0.5 mmol), and
triphenylphosphine
(130 mg, 0.5 mmol) were combined in THF (5 mL). This solution was added to a
reaction
vessel containing polymer supported DIAD (500 mg, 0.50 mmol). After being
shaken
overnight, the solid resin was removed by filtration and the filtrate was
concentrated under
reduced pressure. The resulting semi-solid was purified by silica gel
chromatography,
eluting with 10% ethyl acetate/hexane, to provide compound 137 as a white
solid. 1H
NMR (200M1-Iz, acetone-d6) 8 8.58 (s, 1H), 7.98 (d, 21-1), 7.42 (t, 2H), 7.31
(t, 1H), 2.93
(s, 3H).
Example 138: 6-(2,3,4,5,6-pentafluorophenyl)imidazo[2,1 -b]-1,3,4-
thiadiazole-2-N-
methylsulfonamide
MS (m/z) M+1 = 385.
Example 139: 6-Phenyl-imidazo[2,1-b]-1,3,4-thiadiazole-2-N,N-
dimethylsulfonamide
Compound 1 (140 mg, 0.5 mmol), methanol (64 mg, 2.0 mmol), and
triphenylphosphine
(525 mg, 2.0 mmol) were combined in THF (2 mL) and treated with DIAD (200 4,
2.0
mmol). The resulting solution was stirred over night. The resulting solid was
filtered and
washed with THF (2 x 3 mL) to provide compound 139 as a white crystalline
solid. 1H
NMR (200M1-Iz, DMSO-d6) 5 8.92 (s, 1H), 8.91 (d, 2H), 7.44 (t, 2H), 7.34 (t,
1H), 2.93 (s,
6H).
Example 140: 6-(2,3,4,5,6-pentafluorophenyl)imidazo[2,1-b]-1,3,4-
thiadiazole-2-N,N-
dimethylsulfonamide
Compound 140 was prepared in a manner similar to compound 137. iH NMR (200MHz,
DMSO-d6) 5 8.86 (t, 1H), 2.95 (s, 6H).
Example 141: 6-(3-Methoxyphenyl-imidazo[2,1-1+1,3,4-thiadiazole-2-N,N-
diethylsulfonamide
87
CA 02469953 2007-09-04
Compound 16 (50 mg, 0.162 mmol), ethanol (28 L, 0.486 mmol), and
triphenylphosphine (127 mg, 0.86 mmol) were combined in THF (10 mL) and
treated with
DIAD (96 L, 0.468 mmol). The resulting solution was stirred over night.
Solvent was
removed under reduced pressure and the resulting semi-solid was triturated
with diethyl
ether to provide compound 141 as a white crystalline solid (19.6 mg, 33 %). 1H
NMR
(200MHz, CDCI3) 6 8.06 (s, 1H). 7.38 (d, 2H), 7.35 (t, 1H), 6.88 (d, 1H), 3.88
(s, 3H),
3.45 (q, 411), 1.35 (t, 6H).
Example 142: 6-(3-Methoxyphenyl)imidazo[2,1-1]-1,3,4-thiadiazole-2- N,N-
dibutylsulfonamide
Compound 16 (50 mg, 0.162 mmol), butanol (44 p.L, 0.486 mmol), and
triphenylphosphine (127 mg, 0.486 mmol) were combined in THF (10 mL) and
treated
with DIAD (96 4, 0.486 mmol). The resulting solution was stirred over night.
The
solvent was removed under reduced pressure and the resulting semi-solid was
purified by
silica gel chromatography, eluting with 10 % ethyl acetate/hexane, to provide
compound
142 as a light yellow solid (49 mg, 72 %). 111 NMR (200MHz, CDC13) 6 8.08 (s,
111),
7.38 (d, 2H), 7.35 (t, I H), 3.89 (s, 3H), 3.34 (t, 4H), 1.67 (m, 4H), 1.36
(m, 3H), 0.93 (t,
6H).
Example 143: 6-(4-Bromophenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-N,N-
dimethylsulfonamide
Compound 143 was prepared from compound 14 according to the method described
for
compound 142.
Example 144: 6-(4-Bromophenyl)imidazo[2, I -b]-1,3,4-thiadiazole-2-/V,N-
dimethylsulfonamide hydrobromide
6-(4-Bromophenypimidazo[2,1-14-1,3,4-thiadiazole-2-N,N-dimethylsulfonamide
(100 mg,
0.258 mmol) was suspended in acetic acid (5 mL) and treated with neat bromine
(18 L,
0.315 mmol). The reaction mixture was stirred overnight and volatiles were
removed
88
CA 02469953 2007-09-04
under reduced pressure to provide compounds 144 as a light orange solid (140
mg, 99%).
1H NMR (200MHz, DMSO-d6) 8 7.84 (d, 211), 7.63 (d, 211), 2.93 (s, 6H).
Example 145: 6-(3-Hydroxyphenyl-imidazo[2,1-1]-1,3,4-th iadiazole-2-N,N-
dimethylsulfonamide
Step 1: Compound 16 (212 mg, 0.851 mmol), methanol (103 L. 2.55
mmol), and
triphenylphosphine (669 mg, 2.55 mmol) were combined in THF (10 mL) and
treated with
DIAD (502 uL, 2.55 mmol). The resulting solution was stirred over night.
Solvent was
removed under reduced pressure and the resulting semi-solid was triturated
with methanol
to provide white crystalline solid (244 mg, 85 %). 'H NMR (200MHz, DMSO-d6) 8
8.12
(s, 1H), 7.40 (dd, 1H), 7.38 (d, 1H), 7.33 (t, 1H), 6.88 (ddd, 1H), 3.87 (s,
3H), 3.02 (s, 6H).
Step 2: The above compound (420 mg, 1.24 mmol) was suspended in
methylenechloride (10 mL) and treated with a BBr3 (6.20 mL, 1.0M in CH2C12,
6.20
mmol). The reaction mixture was stirred overnight before being quenched with
water (1
mL), followed by saturated NaHCO3 (10 mL). The resulting mixture was diluted
with
ethyl acetate (20 mL) and subjected to standard workup. The organic layer
provided a off
yellow solid which was further purified by recrystallization from methanol to
provide
compounds 145 as a gray solid (14 mg, 32 %). 11-INMR (200MHz, DMSO-d6) 8 9.50
(br
s, 1H), 8.83 (s, IH), 7.30 (m, 2H), 7.25 (t, 1H), 6.71 (dd, 1H), 2.93 (s,
614).
Example 146: 6-(3-Benzoyloxyphenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-N,N-
dimethylsulfonamide
Compound 145 (20 mg, 0.062 mmol) was dissolved in TI-IF (2 mL) and treated
with
triethylamine (10 mL, 0.068 mmol) followed by benzoyl chloride (9 mL, 0.068
mmol).
The reaction mixture was stirred for 4 hours before a second equiv of
triethylamine and
benzoyl chloride were added. Standard aqueous workup and purification by
silica gel
chromatography, eluting with 30% ethyl acetate/hexane, provided compound 146
as a
white solid (16 mg, 62 %). '14 NMR (200MHz, CDCI3) 8 8.22 (m, 21-1), 8.12 (s,
1H),
7.77-7.60 (m, 3H), 7.58-7.44 (m, 3H), 7.22 (m, 1H).
89
CA 02469953 2007-09-04
Example 147: 6-(2-Naphthyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-/V,N-
dimethylsulfonamide
Compound 62 (330 mg, 1.0 mmol), methanol (180 ilL, 4.40 mmol), and
triphenylphosphine (1.15 g, 4.40 mmol) were combined in THF (10 mL) and
treated with
DIAD (1.90 mL, 4.40 mmol). The resulting solution was stirred over night. The
solvent
was removed under reduced pressure and the resulting semi-solid was triturated
with
diethyl ether, to provide compound 147 as a white crystalline solid (268 mg,
75 %). IFI
NMR (200MHz, DMSO-d6) 5 9.02 (s, 1H), 8.45 (s, 1H), 8.07-7.83 (m, 31-1), 7.56
(m, 2H),
2.91 (s, 6H).
Example 148: 6-Phenyl-imidazo[2,1-b]-1,3,4-thiadiazole-2-sulfonamide
sodium salt
Compound 1 (200 mg, 0.71 mmol) was added to a solution of sodium hydroxide (28
mg,
0.71 mmol) in 4:1 Me0H/H20 (5 mL). The solution was stirred overnight at room
temperature before the solvent was removed under reduced pressure to provide
compound
148 as a white solid (235 mg, 99%). 'H NMR (200 MHz, DMSO-d6) 5 8.59 (s, 1H),
7.85
(d, J = 8.2 Hz, 2H), 7.32 (m, 3H).
Example 149: 6-3'methoxybiphen-4-yl-imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
sodium salt
Compound 149 was prepared from compound 76 according to the method described
for
compound 148; white solid (96%). IFI NMR (200 MHz, DMSO-d6) 5 8.65 (s, 1H),
7.93
(d, J=8.8Hz, 2H), 7.70 (d, J=8.8Hz, 2H), 7.36 (m, 2H), 7.24 (s, 1H), 6.90 (d,
J=6.4Hz,
1H), 3.81 (s, 3H).
Example 150: 6-3'trifluoromethylbiphen-4-yl-imidazo[2,1-b]-1,3,4-
thiadiazole-2-
sulfonamide sodium salt
Compound 150 was prepared from compound 81 according to the method described
for
compound 148; white solid (96%). 1H NMR (200 MHz, DMSO-d6) 5 8.71 (s, 1H),
7.98
(m, 4H), 7.79 (d, J=8.2Hz, 2H), 7.69 (m, 2H).
90
CA 02469953 2007-09-04
Example 151: 6-(4-Azido-2,3,5,6-tetraflourophenyl)imidazo[2,1-b]-
1,3,4-
thiadiazole-2-sulfonamide
2-Bromo-4'-azido-2',3',5',6'-tetrafluoroacetophenone (Keana, J. F. W.; Cal, S.
X. J. Org.
Chem., 1990, 55, 3640) (353 mg, 1 mmol) and 2-amino-1,3,4-thiadiazole-5-
sulfonamide
(148 mg, 1 mmol) were refluxed in ethanol (10 mL) for 60 hours. The resulting
solid was
filtered, washing twice for cold methanol (2 mL), to provide compound 151(102
mg, 25
%) as a white powder. 1H NMR (200MHz, DMSO-d6) 8 8.81 (t, J=2.0Hz, 1H), 8.79
(br s,
2H).
Example 152: 6-(4-azido-2,3,5,6-pentafluorophenyl)imidazo[2,1-b]-1,3,4-
thiadiazole-
2-N,N-dimethylsulfonamide
Compound 152 was prepred in a manner similar to compound 137. 1H NMR (200MHz,
DMSO-d6) 6 8.22 (t, 1H), 3.07 (s, 6H).
Example 153: 6-(4-Nitrophenyl)imidazo[2,1-b]-1,3,4-thiadiazole-2-
sulfonamide
2-Bromo-4'-nitroacetophenone (2.44 g, 10.0 mmol) and 2-amino-1,3,4-thiadiazole-
5-
sulfonamide (1.50 g, 10.0 mmol) were refluxed in 1,4-dioxane (20 mL) for 48
hrs. The
resulting solution was cooled on ice and the resulting precipitate was
collected by filtration
to provide compound 153 as a white crystalline solid (2.40 g, 67 %). 1H NMR
(200MHz,
DMSO-d6) 6 9.18 (s, 11-1), 8.69 (br s, 1H), 8.35 (d, 2H), 8.16 (d, 2H).
Example 154: Protection of SCG neurons from anti-NGF killing
SCG neurons were isolated from day 1 neonatal Sprague Dawley rats, plated at a
cell
density of 5,000 cells/well, and incubated in Biowhittaker Utraculture
containing 1%
Penstrep, 1% L-glutamine, 0.7% ARAC, 3% rat serum, and NGF (50 ng/mL, Calomone
Labs), at 37 C, under a 5% CO2 atmosphere. After 4 days the cells were
treated with anti-
NGF antibody (Sigma). At this time compound was added and the cells were
maintained
serum and NGF free for 48 hours, at which time viability was assessed using
Alamar Blue
(Medicorp) staining.
Table 3a summarizes selected 1C(50) values from compounds tested using this
protocol.
91
CA 02469953 2007-09-04
Table 3a
Rescue from anti-NGF killing of SCG neurons
Compoun IC(50) Compound IC(50) Compound IC(50)
d (uM)* (uM)* (uM)*
1 22 81 5 98 17
2 20 82 25 99 7
3 10 84 >30 106 7
9 8 85 >30 111 7
21 22 86 17 148 22
23 25 87 10 149 7
26 23 88 10 150 7
34 >30 89 10
35 >30 91 7
36 17 92 7
72 17 93 7
74 20 94 10
75 10 95 7
76 5 96 20
79 7 97 7
* +/- 1 uM
Example 155: In Vitro Protection of SCG neurons from Taxol killing
SCG neurons were isolated from day 1 neonatal Sprague Dawley rats, plated at a
cell
density of 10,000 cells/well, and incubated in Biowhittaker Utraculture
containing 1%
Penstrep, 1% L-glutamine, 0.7% ARAC, 3% rat serum, and NGF (50 ng/mL, Calomone
Labs) at 37 C, under a 5% CO2 atmosphere. After 5 days the cells were treated
with
compound and TaxolTm (50 ng/mL). Viability was assessed 48 hours later using
MIS
(Promega) staining.
Table 3b summarizes selected IC(50) values from compounds tested using this
protocol.
Table 3b
Rescue from anti-NGF killing of SCG neurons
Compounds IC(50) (uM)* Compound IC(50) (uM)* Compound IC(50) (uM)*
92
CA 02469953 2007-09-04
1 7 47 6 81 3
4 5 48 5 82 2
5 49 5 87 5
6 3 50 7 95 3
5 7 4 51 5 99 2
8 5 52 3 101 >30
11 3 53 5 102 >30
12 5 54 10 103 >30
13 7 55 15 104 7
14 5 56 3 105 7
10 57 6 107 20
16 5 58 30 108 5
17 3 59 10 109 7
18 20 60 25 110 >30
15 19 15 61 10 111 2
15 62 15 112 5
21 7 63 7 113 7
22 25 64 10 114 7
24 7 65 15 115 7
20 25 5 66 20 116 7
26 7 67 10 117 3
10 68 7 118 17
31 10 70 22 120 7
32 10 71 >30 121 2
25 37 10 72 10 122 8
38 3 73 2 123 >30
23 74 7 124 3
41 3 75 5 125 3
42 3 76 2 128 2
30 46 7 77 2 129 7
93
CA 02469953 2007-09-04
130 2 141 >30 153 5
131 3 142 >30
132 1 143 10
134 1 144 15
135 7 145 20
136 5 146 20
137 10 147 17
138 2 148 7
139 10 151 2
140 5 152 2
Figure 1 illustrates the protection provided by compound 1 (referred to here
as AEG3482)
against TaxolTm induced killing. P1 Sprague Dawley rat SCG neurons were
cultured and
incubated with NGF (50 ng/mL) for 5 days. Addition of TaxolTm (50 ng/mL)
resulted in a
72% loss in viability as measured by MTS staining. Co-treatment with compound
1
resulted in 100% protection at 10 uM, with an 1050 of 7 uM.
Example 156: In Vitro Protection of SCG neurons from cisplatin
killing
SCG neurons were isolated from day 1 neonatal Sprague Dawley rats, plated at a
cell
density of 10,000 cells/well, and incubated in Biowhittaker Utraculture
containing 1%
Penstrep, 1% L-glutamine, 0.7% ARAC, 3% rat serum, and NGF (50 ng/mL, Calomone
Labs) at 37 C, under a 5% CO2 atmosphere. After 5 days the cells were treated
with
compound and cisplatin (3 g/mL). Viability was assessed 48 hours later using
MTS
(Promega) staining.
Table 4: Protection of SCG neurons against cisplatin killing
Entry Compound icso ( 1 04)
1 1 5
Example 157: In Vitro Protection of SCG neurons from Vincristine killing
94
CA 02469953 2007-09-04
SCG neurons were isolated from day 1 neonatal Sprague Dawley rats, plated at a
cell
density of 10,000 cells/well, and incubated in Biowhittaker Utraculture
containing 1%
Penstrep, 1% L-glutamine, 0.7% ARAC, 3% rat serum, and NGF (50 ng/mL, Calomone
Labs) at 37 C, under a 5% CO2 atmosphere. After 5 days the cells were treated
with
compound and vincristine (50 ng/mL). Viability was assessed 48 hours later
using MTS
(Promega) staining.
Table 5: Protection of SCG neurons against Vincristine killing
Entry Compound IC50 ( 1 P.M)
1 1 10
Example 158: Protection of motor neurons in layer V of the motor
cortex
350 uM slices of P1 rat motor cortex were obtained using a McIlwain tissue
chopper
(Mickle Laboratory Engineering Co., England). Slices were cultured in 50%
Neurobasal,
25% HBSS, 25% Horse serum, 1% penicillin/streptomycin, 2mM glutamine, 6.4mg/mL
glucose for 2 weeks. Neuronal death was initated by addition of 5mM malonate.
Test
compounds were added coincident with malonate and slices were cultured for an
additional two weeks. Slices were fixed in 4% paraformaldehyde and stained
with SMI-
32 antibody (Sternberger monoclonals, Maryland). Large SMI positive cells with
apical
dendrites residing in layer V of the the cortex were identified as motor
neurons and
counted. Malonate treatment greatly reduced the SMI-positive motor neuron
count.
Figure 2 illustrates the protection of cortical motor neurons from malonate
killing. Slices
of P1 rat motor cortex (350 uM) were treated with malonate and incubated in
media for
14 days, before malonate and drug were added. Part (a) shows control motor
neuons.
Large sized diamond-shaped neurons are visible; part (b) shows malonate
treatment alone,
which results in killing with a complete loss of neurons; and part (c) shows
90% rescue of
cortical motor neurons in the presence of compound 91(1 uM) and malonate. In
Part C,
large diamond-shaped neurons are again visible.
CA 02469953 2007-09-04
Example 159: Co-treatment of 1-1460 and 0V2008 cell line with
TaxolTm and
Compound 1
H460 and 0V2008 cells were plated and incubated for 48 hours. Compound 1
and/or
compound 1 and TaxolTm were added. Viability was determined after 24 hours,
staining
with MTT (Promega).
Figure 3 illustrates the co-treatment of H460 and OV 2008 cell lines with
TaxolTm and
compound 1. 1-1460 lung carcinoma and 0V2008 ovarian carcinoma cells were
treated
with TaxolTm (50 nM) and/or TaxolTm (50 nM) + compound 1 (noted as AEG 03482)
at
levels of 5, 10, and 20 uM. Compound 1 did not protect H460 or 0V2008 cells
from
TaxolTm induced apoptosis.
Example 160: Protection of Sprague Dawley rats from TaxolTm induced
neuropathies
Adult Sprague Dawley rats were treated with TaxolTm (IP, 9 mg/kg in Cremophor
EL and
ethanol) twice weekly for 3 weeks (J. Neuro-Oncology (1999) 41: 107-116).
Compound
was administered 1 hour prior to TaxolTm treatment (IP, 1, 5 and 10 mg/kg in
hydroxypropyl-p-cyclodextrin). TaxolTm treated control animals were treated
with saline
solution at the same time of Compound treated animals. Non-treated control
animals were
treated with saline solution as above. Weight gain was measured every second
day,
starting at Day 1. Gait analysis was measured by quantifying the refracted
light captured
by a video camera as the animals walked over a glass plate, 2 days after the
final TaxolTm
treatment (Physiology and Behavior (1994), 55(4): 723-726; Med. Sci. Res.
(1988) 16:
901-902). This data was analyzed by Northern Eclipse software. HIM wave
recovery was
analyzed using standard procedures 2 days after the final TaxolTm treatment
(Muscle
Nerve (1998) 21: 1405-1413; Annals of Neurology (1998) 43 (1): 46-55).
Figure 4 shows weight loss induced by TaxolTm. Male Spraugue Dawley rats were
treated
with 50% HPDC vehicle (veh/veh), compound 1 dissolved in 50% HPDC at 1, 5, or
10
mg/kg (veh/1 , veh/5, veh/10, respectively), or TaxolTm (9 mg/kg) + compound 1
dissolved
96
CA 02469953 2007-09-04
in 50% HPDC at 1, 5, and 10 mg/kg (Tax/1, Tax/5, Tax/10) according to the
dosing
regime described in Example 160. Weight measurements were made ever other day.
Figure 5 shows gait disturbance induced by TaxolTm with compound 1. Two days
after
the completion of drug treatments animal walking gait was analyzed according
to a) total
imprint area, and b) total number of contact points. Compound 1 prevented
TaxolTm
induced gait disturbance.
Figure 6 illustrates the effect of Compound 1 on H-reflex amplitude, a
measurement of
HIM wave disturbance induced by TaxolTm. Two days after the completion of drug
treatments, the dorsal root ganlia and attached nerves were dissected
bilaterally from L4
and LS and their H/M wave conductance measured. Compound 1 caused a reversal
in
HIM wave disturbance induced by TaxolTm
Example 161: Sciatic Nerve Crush Injury Model
Male Sprague Dawley rats were anaesthetized (halothane and buprenorphine) and
the right
hind leg was blunt dissected to expose the sciatic nerve at mid-thigh. The
nerve was
crushed twice for a total of 30 seconds using No.7 Dumont jeweller's forceps.
The
incision is sutured and the animals are allowed to recover for 28 days.
Functional
recovery was measured by gait, nerve conductance and toe spread
measurementsbetween
digits land 5 and digits 2 and 4.
Figure 7 illustrates sciatic nerve recovery after crush injury, as indicated
by inner toe
spread. Male Spraugue Dawley rats were subjected to sciatic nerve crush and
treated with
either vehicle control, compound 1 or compound 76 (noted as AEG 33764).
Compounds
1 and 76 induced increased recovery in toe spread area.
Example 162: Optical Stroke Model
The right eye of each rat was dilated fully using 1% tropacamide and 2.%
pheylephrine
hydrochloride (Alcon Canada). A single drop of 0.5% proparacaine (Alcon) was
used as a
topical anesthetic. The anterior chamber of the right eye was cannulated with
a 30-gauge
97
CA 02469953 2007-09-04
needle connected to a saline reservoir and a manometer to monitor intraocular
pressure.
Intraocular pressure was raised to 110 mm Hg by raising the saline reservoir
for 60
minutes. This increase in pressure collapses the central retinal artery.
Retinal ischemia
was confirmed by whitening of the iris and loss of red reflex. After 60
minutes of
ischemia , the intraocular pressure was normalized and the needle withdrawn. A
33- blunt
needle (Hamilton) was inserted through the corneal puncture, maneuvered around
the lens
displacing it medially, and advanced into the intravitrial space. A 2 1.1I,
volume of drug or
vehicle (50% HPCD) was injected into the vitreous of the eye. The needle was
withdrawn
and maxitrol (Alcon) was applied to the cornea to prevent infection.
Alternatively, drug
was given subcutaneously before or after the insult, for a period of up to 14
days.
Optical function after 24 hrs, 28 hrs and 7 days was assessed using ERG
measurements
and histological staining of the RG layer.
Figure 8 shows protection of RGs by compound 1 after ocular stroke. Ocular
stroke was
induced in the right eye of rats resulting in almost complete loss of the RG
population, as
seen here by a loss in reactivity of the optic fiber to stimulation. Compound
1 was
delivered intravitreally followed by subsequent daily injections for 1 week
post-ischemia
(post stroke). Compound 1, given post stroke, protects the RG population
allowing for
normal conductance.
Example 163: CA II inhibition
CA II inhabition was measured using the protocol described by Pocker, Y.'
Stone,
J.Biochemishy 1967, 6, 668. The IC (50)s if selected compound represented by
Formula I
are listed in Table 6.
Table 6 : CA II inhibition by compounds represented by formula I
Compounds IC(50) (uM) Compound IC(50) (uM) Compound IC(50) (uM)
1 0.250 21 5.32 49 0.466
98
CA 02469953 2007-09-04
4 0.217 22 0.153 50 0.938
0.192 24 0.613 72 1.61
6 0.164 25 0.302 72 2.19
7 0.581 37 0.199 74 1.68
5 8 1.47 38 0.577 75 0.441
11 1.80 39 0.154 76 0.526
12 1.59 40 1.52 81 1.58
13 4.05 41 0.346 82 5.43
14 0.198 42 0.272 87 0.128
15 0.152 43 0.886 99 0.914
16 0.150 44 0.619 105 0.150
17 0.179 45 0.166 111 53.2
18 0.337 46 0.601 137 2.06
19 0.373 47 0.361 139 11.3
20 0.404 48 0.288 142 60.3
Example 164: Neuroprotection of Cortical Neurons in the Presence of beta-
Amyloid
Primary neuronal/glial cortical cultures were established from postnatal day 1
Sprague
Dawley rats. Cerbral cortices were isolated and dissociated with 0.25% trypsin
for 20
minutes at 37 degrees. The tissue was then triturated in PBS containing 0.1%
bovine
serum albumin and 0.2mg/m1DNAse. Cells were plated in poly D-lysine coated 96
well
plates at a density of 1e6 cells per mL. Cultures were maintained at 37
degrees in 5%
CO2/95% air for 2 weeks in Neurobasal (Gibco) supplemented with B27,
glutamine, and
penicillin/streptomycin. 5ng/mL AraC was added after 48 hours. After 2 weeks
cells
were exposed to 10 uM 25-35 amyloid beta peptide with and without 10 uM
compound
76. After 2 days of treatment apoptotic cells were detected with Cy3-
conjugated annexin
V (Sigma).
Figure 9 shows the protection of provided by Compound 76 from amyloid beta 25-
35
toxicity. Mixed neuronal/cortical cultures were obtained from P1 rat cortex.
After 2
99
CA 02469953 2007-09-04
weeks in vitro cells were exposed to 10 uM 25-35 amyloid beta peptide. Top (a)
shows
control untreated cultures display low level annexin V staining. Middle (b)
shows 48 hour
treatment with amyloid beta peptide results in the appearance of apoptotic
cells which
stain with annexin V on the cell periphery. Bottom (c) illustrates co-
treatment with 10 uM
Compound 76 prevents the occurrence of annex in V stained cells.
100