Note: Descriptions are shown in the official language in which they were submitted.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
AMIDO MACROLIDES
BACKGROUND OF THE INVENTION
Erythromycins are macrolide antibiotics, glycosylated polyketides originally
discovered in 1952 in the metabolic products of a strain of Streptomyces
erythreus,
now classified as Sacclaaropolyspora erythraea. The antibiotic occurs in
various
forms, designated A, B, C, and D. Since their discovery, many have worked to
prepare derivatives of the molecule to improve or modify its properties. The
focus of
much of this work involved chemical modification of the naturally produced
erythromycin molecule. For example, clarithromycin is a semi-synthetic
antibiotic
that is made by chemically methylating the hydroxyl group at C-6.
X Y Z
e2 erythromycin A OH OH OMe
erythromycin B H OH OMe
erythromycin C OH OH OH
erythromycin D H H OH
clarithromycin OH OMe OMe
~OH
Azalides, such as azithromycin, are erythromycin derivatives where the C-9
ketone has been replaced with a N-CH2 unit through a Beckmann rearrangement.
See, for example, O'Connell et al., "Azalides and methods of making same,"
U.S.
Patent No. 6,270,768, incorporated herein by reference.
Ketolides are erythromycin derivatives where the C-3 cladinose sugar is
chemically removed and the resulting free hydroxyl group converted into a keto
group. For example, Phan et al., "2-Halo-6-O-substituted ketolide derivatives
," U.S.
Patent No. 6,124,269 describes ketolides with a cyclic carbamate group at C-11
and
C-12 and an O-alkylaryl group at C-6. Agouridas et al., "Erythromycin
compounds,"
U.S. Patent No. 5,635,485 also describes ketolides with a cyclic carbamate
group at
C-11 and C-12 but which have a -OMe group at C-6 and an alkylaryl group at the
carbamate nitrogen.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-2-
The complexity of the macrolide molecule has limited medicinal chemistry
efforts to produce derivatives of the naturally occurring erythromycins and
their
precursors. Recently, the discovery and isolation of modular polyketide
synthases
("PKS's") have expanded the scope of macrolide structures that may be made.
PKS's
are multifunctional enzymes that catalyze the formation of the polyketide
chains
through repeated reactions between acylthioesters.
The Sac. erytlzraea PKS, known as 6-deoxyerythronolide B synthase (DEBS),
is an assembly of three multifunctional proteins encoded by the eryAl, eryAll,
and
eryAlll genes and is described in Katz et al., "Recombinant DNA method for
producing erythromycin analogs," U.S. Patent No. 5,824,513; Katz et al.,
"Method of
directing biosynthesis of specific polyketides," U.S. Patent No. 6,004,787;
Katz et
al., "Polyketide derivatives and recombinant methods for making same," U.S.
Patent
Nos. 6,060,234, 6,063,561, and 6,200,813; each of which is incorporated herein
by
reference. DEBS produces the polyketide macrolactone 6-deoxyerythronolide B,
which is processed by additional tailoring enzymes present in Sac. erythraea
to make
erythromycins A-D. The collective assembly of the PKS gene and the genes for
the
tailoring enzymes are referred to as the biosynthetic gene cluster. The
organization of
the gene cluster is described in Summers et al., "Polyketide-associated sugar
biosynthesis genes," U.S. Patent No. 5,998,194, incorporated herein by
reference.
Recombinant methods using vectors encoding a variety of PKS's, including
the PKS from Sac. erythraea, to make novel polyketides are described in Khosla
et
al., "Recombinant production of novel polyketides," U.S. Patent Nos.
5,672,491,
5,830,750, 5,962,290, 6,022,731, and 6,077,696; Khosla et al., "Recombinant
combinatorial genetic library for the production of novel polyketides," U.S.
Patent
No. 5,712,146; Khosla et al., "Method to produce novel polyketides," U.S.
Patent
Nos. 6,066,721, 6,221,641, and 6,261,816; Barn et al., "Production of
polyketides in
bacteria and yeast," U.S. Patent Nos. 6,033,883 and 6,258,566 and PCT
publication
WO 98/27203; Khosla et al., "Biosynthesis of polyketide synthase substrates,"
PCT
publication WO 01/27305; and Santi et al., "Heterologous production of
polyketides,"
PCT publication WO 01/31035; each of which is incorporated herein by
reference.
Leadlay et al., "Erythromycins and process for their preparation, " U.S.
Patent No.
6,271,255, incorporated herein by reference, describes additional methods for
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-3-
modifying the loading domain and thus varying the nature of the starter units
that
initiate polyketide synthesis. Methods for making polyketides in a cell-free
system
are described, for example by Khosla et al., "Synthesis of polyketides from
diketides," U.S. Patent No. 6,080,555; Khosla et al., "Cell-free synthesis of
polyketides," U.S. Patent No. 6,274,560, and PCT Publication No. WO 97/02358,
each of which is incorporated herein by reference. Erythromycin analogues
where the
naturally occurring ethyl group at C-13 is replaced with other groups have
been
described, for example in Dirlam et al., "Novel macrolides," PCT publication
WO
99/35156; Jin, "Novel erythromycin derivatives," PCT publication WO 99/35157;
Ashley et al., "Synthesis of oligoketides," U.S. Patent 6,492,562; McMillen &
Kaneko, "Ketolide antibiotics," PCT publication WO 00/44761; Grant et al.,
"Ketolide antibacterials," PCT publication WO 00/62783; and Chu, "Anti-
infective
compounds," U.S. Patents 6,395,710 and 6,451,768 and PCT publication WO
01/49699; and Xue et al., "Mufti-plasmid approach to preparing large libraries
of
polyketides," PCT publication WO 00/63361; each of which is incorporated
herein by
reference.
Various macrolides are also disclosed in U.S. Patent No. 6,451,768 and U.S.
Patent No. 6,395,710 both of which are incorporated herein by reference in
their
entirety and for all purposes. United States Patent No. 6,492,562, also herein
incorporated by reference in its entirety for all purposes, describes methods
for
preparing various macrolides.
Due to the increase in the incidence of resistant strains to currently used
antibiotics, a need exists for novel compounds having antibiotic activity,
particularly
against resistant strains. The present invention fulfills this need by
providing novel
erythromycins, ketolides, and azalides.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-4-
SUMMARY OF THE INVENTION
The invention provides various compounds and pharmaceutically acceptable
salts thereof. Thus, in one embodiment, the invention provides compounds and
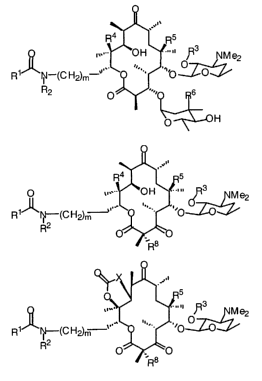
pharmaceutically acceptable salts thereof of the formula I
3 NMe2
."'O O O
R N-(CH~,n a
R2 .-, '~~..-. r,~
I
where:
Rl is C1-C8 substituted or unsubstituted alkyl, CZ-Cs substituted or
unsubstituted alkenyl, C2-Cs substituted or unsubstituted alkynyl, C4-C15
substituted
or unsubstituted aryl, CS-C2o substituted or unsubstituted arylalkyl, CS-C2o
substituted
or unsubstituted biarylalkyl, CS-C2o substituted or unsubstituted arylalkenyl,
or CS-CZo
substituted or unsubstituted arylalkynyl;
RZ is H, Cl-C4 substituted or unsubstituted alkyl, C2-C4 substituted or
unsubstituted alkenyl, or C2-C4 substituted or unsubstituted alkynyl;
. R3 is H, Cl-C4 alkanoyl, or benzoyl;
R4 is H or OH;
RS is H, OH, Cl-C4 alkoxy, C~-Cø alkenyloxy, or C2-C4 alkynyloxy;
R6 is OH or OMe; and
m = 0-2.
In some embodiments, the invention provides compounds of formula I where
R2 is H and m is 0. In other embodiments, the invention provides compounds of
formula I where R2 is H and m is 1. In yet other embodiments, the invention
provides
compounds of formula I where R2 is H and m is 2.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-5-
In still other embodiments, the invention provides compounds of formula I
which have any of the following structures:
v /
o
N~
H
~OH p~OH
O
O
H OMe
i I O "... ~OH . "HO NM~
N~''~ O ~~~ ~ 'O~
H
O ~''O OMe
~OH
O , ~ ~ O O
,N H~ N~H
or
In another aspect, the invention provides compounds of formula II and
pharmaceutically acceptable salts thereof
R4 ..' 5
OH ""' R3 M e2
Ng-(CH2)r~'~~.. ~'.. .,
O
O ~ $~O
R
II
where:
R3 is H, Cl-C4 alkanoyl, or benzoyl;
R4 is H or OH;
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-6-
Rs is H, C1-Cø alkoxy, C2-C4 alkenyloxy, or C2-C4 alkynyloxy;
Rs is H or F; and
m = 0-2.
In some embodiments, the invention provides compounds of formula II where
m is 0. In other embodiments, the invention provides compounds of formula II
where
m is 1. In yet other embodiments, the invention provides compounds of formula
II
where m is 2.
In a further embodiment, the invention provides compounds of formula II
which have any of the following structures:
Na. N n >
o~~
W2
N n Ns~
or
In yet another aspect, the invention provides compounds of formula III and
pharmaceutically acceptable salts thereof
C'I ~ 3 NMe2
R1~~~CH2)m
R2
III
where:
Rl is C1-C$ substituted or unsubstituted alkyl, C2-C8 substituted or
unsubstituted alkenyl, C2-C8 substituted or unsubstituted alkynyl, C4-Cls
substituted
or unsubstituted aryl, Cs-C2o substituted or unsubstituted arylalkyl, Cs-CZO
substituted
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
_7_
or unsubstituted biarylalkyl, CS-CZO substituted or unsubstituted arylalkenyl,
or CS-Coo
substituted or unsubstituted arylalkynyl;
R2 is H, Cl-C~. substituted or unsubstituted alkyl, C2-C4 substituted or
unsubstituted alkenyl, or C2-C4 substituted or unsubstituted alkynyl;
R3 is H, C1-C4 alkanoyl, or benzoyl;
R4isHorOH;
RS is H, C1-C4 alkoxy, C2-C4 alkenyloxy, or CZ-C4 alkynyloxy;
R8 is H or F; and
m = 0-2.
In some embodiments, the invention provides compounds of formula III in
which m is 0. In other embodiments, the invention provides compounds of
formula
III in which m is 1. In still other embodiments, the invention provides
compounds of
formula II in which m is 2. In some such embodiments, the invention provides
compounds of formula III where R2 is H and m is 0. In other such embodiments,
the
invention provides compounds of formula III where R2 is H and m is 1. In yet
other
such embodiments, the invention provides compounds of formula III where R2 is
H
and m is 2.
In yet other embodiments, the invention provides compounds of formula III in
which R1 is a C4-Cls substituted or unsubstituted aryl, CS-Czo substituted or
unsubstituted arylalkyl, CS-CZO substituted or unsubstituted biarylalkyl, CS-
C2o
substituted or unsubstituted arylalkenyl, or CS-C2o substituted or
unsubstituted
arylalkynyl; and RZ is H.
In still another aspect, the invention provides compounds of formula IV and
pharmaceutically acceptable salts thereof
3
OR NMe2
R1 N-(C i
R2
IV
where:
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
_g_
R1 is C1-C$ substituted or unsubstituted alkyl, C2-C8 substituted or
unsubstituted alkenyl, C2-C8 substituted or unsubstituted alkynyl, C4-Cls
substituted
or unsubstituted aryl, Cs-CZO substituted or unsubstituted arylalkyl, Cs-CZO
substituted
or unsubstituted biarylalkyl, Cs-CZO substituted or unsubstituted arylalkenyl,
or Cs-C2o
substituted or unsubstituted arylalkynyl;
R2 is H, Cl-C4 substituted or unsubstituted alkyl, C2-C4 substituted or
unsubstituted alkenyl, or CZ-C4 substituted or unsubstituted alkynyl;
R3 is H, C1-C4 alkanoyl, or benzoyl;
RS is H, C1-C4 alkoxy, C2-C4 alkenyloxy, or C2-C4 alkynyloxy;
R$isHorF;
X is O or NR7, wherein R7 is H, Cl-C4 alkyl, or CG-Coo arylalkyl; and
m = 0-2.
In some embodiments, the invention provides compounds of formula IV in
which R2 is H.
In still other embodiments, the invention provides compounds of formula IV
in which Rl is C6-Cls substituted or unsubstituted aryl, Cs-C2o substituted or
unsubstituted arylalkyl, Cs-CZO substituted or unsubstituted biarylalkyl, Cs-
CZo
substituted or unsubstituted arylalkenyl, or Cs-C2o substituted or
unsubstituted
arylalkynyl; and R2 is H.
In still other embodiments, the invention provides compounds of formula IV
in which Rl is C6-Cis substituted or unsubstituted aryl, Cs-C2o substituted or
unsubstituted arylalkyl, Cs-C2o substituted or unsubstituted biarylalkyl, Cs-
C2o
substituted or unsubstituted arylalkenyl, or Cs-C2o substituted or
unsubstituted
arylalkynyl; R2 is H; and X is NH.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-9-
In yet other embodiments, the invention provides compounds of formula IV
which have any of the following structures:
U,
~I o ~I o
N' ~ N' ~ O
H H
O
O
O~Ni
or
where R~ is H or F.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-10-
In yet other embodiments, the invention provides compounds of formulas I,
III, or IV in which R' is a group having any of the following formulas:
I\ ~ I/
I \ ~ cl I \ ~ I \
i / N / S Me0 / O
H
I\
\ ~ O~~ I w
O w ,N \ \ IN I \ i
I ~ O I ~ ~ CI /
/I
O w ~ _
o~ oI /I
I \ ~ s I \ H I \
I \ ~ i i i
/ O S~
s ~ I ~ ~ s
\ \1 / \ \I
s I ~N ~ I \ v
O°Nw \ ~ S i / S
I / I \ \
/ I I /N ,- S /
I
H I \ ~ O. \ I
N N~ I / I
\ I \ ~ / I o I \
/ I w ~ or
N
In one embodiment, the invention provides compounds of the formula V and
pharmaceutically acceptable salts thereof
R9
N
.OH
O ~,,. ~OH ...~~ NMe2
~ ,, HO
,.-
R1' _N-(C H2) = O -' O O
12
R ~~'O R6
~OH
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-11._
wherein
R1 is CI-C8 substituted or unsubstituted alkyl, C2-C$ substituted or
unsubstituted alkenyl, C2-C8 substituted or unsubstituted alkynyl, C4-C15
substituted
or unsubstituted aryl, CS-CZO substituted or unsubstituted arylalkyl, CS-C2o
substituted
or unsubstituted biarylalkyl, CS-C2o substituted or unsubstituted arylalkenyl,
or CS-C2o
substituted or unsubstituted arylalkynyl;
R2 is H, Cl-C4 substituted or unsubstituted alkyl, C2-C4 substituted or
unsubstituted alkenyl, or CZ-C4 substituted or unsubstituted alkynyl;
R4 is H or OH;
R~ is OH or OMe;
R9 is H or Cl-C4 alkyl; and
m=0-2.
In still other embodiments, the invention provides pharmaceutical
formulations and medicaments. Such pharmaceutical formulations and medicaments
include any of the compounds of the invention and a pharmaceutically
acceptable
carrier. In some embodiments, the pharmaceutical formulation or medicament
includes any compound of formulae I - V and a pharmaceutically acceptable
carrier.
In another embodiment, the invention provides a method of treating a bacterial
infection with compounds of the formulae I - V. A method of treating a
bacterial
infection includes administering a compound of formulae I-V to a patient in
need
thereof. In one embodiment, the bacterial infection results from Gram positive
bacteria, Gram negative bacteria or anaerobic bacteria. In another embodiment,
the
bacteria are Staphylococcus aureus, Streptococcus epiderrrzidis, Strep.
praeumo~2iae,
Strep. pyogenes, enterococci, Moraxella catarrhalis or Haemoplailus
influefazae. In
yet another embodiment, the bacterial infection is community-acquired
pneumonia,
acute exacerbated chronic bronchitis, acute sinusitis,
tonsillitis/pharyngitis, upper
respiratory tract infection, lower respiratory tract infection, skin
infection, soft tissue
infection, meningitis, hospital-acquired infection, bone infection or joint
infection.
In another embodiment, the invention provides a method of treating a gastric
motility disease. The method includes treating a patient with gastric motility
disease
with a compound of formula I. In one embodiment, the gastric motility disease
is
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-12-
gastro-esophageal reflux disease (GERD), post-operative ileus, diabetes, or
gastroparesis.
Other objects, features, and advantages of the present invention will become
apparent from the following detailed description. It should be understood,
however,
that the detailed description and the specific examples, while indicating some
preferred embodiments of the invention, are given by way of illustration only,
since
various changes and modifications whiting the spirit and scope of the
invention will
become apparent to those skilled in the art from this detailed description.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 shows a variety of 15-amidoerythromycins prepared according to the
methods of the invention.
Figure 2 shows some embodiments of additional 15-amidoerythromycins that
can be prepared according to the methods of the invention.
Figure 3 shows a synthetic procedure that may be used to prepare certain
macrolides according to the invention. Rl is as defined in the specification
and can
include any of the groups shown in Figures 1 and 2.
DETAILED DESCRIPTION OF THE INVENTION
The present invention relates to erythromycin derivatives, intermediates
thereto, and methods for their use in the treatment of disease. The inventive
compounds possess antibacterial activity against Gram positive, Gram negative,
and
anaerobic bacteria, and are useful as broad-spectrum antibacterial agents for
the
treatment of bacterial infections in humans and animals. These compounds are
effective against diverse strains including but not limited to Staphylococcus
aureus,
Streptococcus epidernzidis, Strep. pneuznoniae, Strep. pyogefzes, enterococci,
Moraxella catarrhalis and Haernoplzilus influefzzae. Exemplary infections that
may
be treated include community-acquired pneumonia, acute exacerbated chronic
bronchitis, acute sinusitis, tonsillitis/pharyngitis, upper and lower
respiratory tract
infections, skin and soft tissue infections, meningitis, hospital-acquired
infections, and
bone and joint infections. Certain of the inventive compounds also possess
prokinetic
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-13-
activity and are useful in the treatment of diseases of gastric motility,
including but
not limited to gastro-esophageal reflux disease (GERD), post-operative ileus,
diabetes, and gastroparesis.
Some of the crystalline forms for the compounds may exist as polymorphs and
as such are intended to be included in the present invention. In addition,
some of the
compounds may form solvates with water (i.e., hydrates) or common organic
solvents, and such solvates are also encompassed within the scope of this
invention.
For use in medicine, the salts of the compounds of this invention refer to non-
toxic "pharmaceutically acceptable salts." Other salts may, however, be useful
in the
preparation of compounds according to this invention or of their
pharmaceutically
acceptable salts. Suitable pharmaceutically acceptable salts of the compounds
include
acid addition salts which may, for example, be formed by mixing a solution of
the
compound with a solution of a pharmaceutically acceptable acid such as
hydrochloric
acid, sulfuric acid, fumaric acid, malefic acid, succinic acid, acetic acid,
benzoic acid,
citric acid, tartaric acid, carbonic acid, phosphoric acid, glucoheptonic
acid,
lactobionic acid, and dodecylsulfonic acid.
The present invention includes within its scope prodrugs of the compounds of
this invention. In general, such prodrugs will be functional derivatives of
the
compounds that are readily convertible in vivo into the required compound.
Examples
of prodrugs of the inventive compounds include but are not limited to 2'-O-
esters
such as acetates, propionates, hemisuccinates, stearates, and the like. Thus,
in the
methods of treatment of the present invention, the term "administering" shall
encompass the treatment of the various disorders described with the compound
specifically disclosed or with a compound which may not be specifically
disclosed,
but which converts to the specified compound i~a vivo after administration to
the
patient. Conventional procedures for the selection and preparation of suitable
prodrug
derivatives are described, for example, in "Design of Prodrugs", ed. H.
Bundgaard,
Elsevier, 195.
Listed below are definitions of various terms used to describe this invention.
These definitions apply to the terms as they are used throughout this
specification,
unless otherwise limited in specific instances, either individually or as part
of a larger
group.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-14-
When a particular group is "substituted," that group may have one or more
substituents, preferably from one to five substituents, more preferably from
one to
three substituents, most preferably from one to two substituents,
independently
selected from the list of substituents. It is understood that substituents and
substitution
patterns on the compounds of this invention can be selected by one of ordinary
skill in
the art to provide compounds that are chemically stable and that can be
readily
synthesized by techniques known in the art as well as those methods set forth
herein.
Examples of suitable substituents include alkyl, alkenyl, alkynyl, aryl, halo,
trifluoromethoxy, trifluoromethyl, hydroxy, alkoxy, cycloalkyloxy,
heterocyclooxy,
alkanoyl, alkanoyloxy, amino, alkylamino, aralkylamino, cycloalkylamino,
heterocycloamino, dialkylamino, alkanoylamino, thin, alkylthio,
cycloalkylthio,
heterocyclothio, ureido, nitro, cyano, carboxy, caroboxylalkyl, carbamyl,
alkoxycarbonyl, alkylthiono, arylthiono, alkylsulfonyl, sulfonamindo, aryloxy,
and
the like, in addition to those otherwise specified herein. The substituent may
be
further substituted, for example, by halo, hydroxy, alkyl, alkoxy~ aryl,
substituted aryl,
substituted alkyl, substituted aralkyl, and the like.
Under standard nomenclature used throughout this disclosure, the terminal
portion of the designated side chain is described first, followed by the
adjacent
functionality toward the point of attachment. Thus, for example, a
"arylalkylcarboxamido" substituent refers to a group of the formula
(aryl)- (alkyl
The term "therapeutically effective amount" as used herein, means that amount
of active compound or pharmaceutical agent that elicits the biological or
medicinal
response in a tissue system, animal or human that is being sought by a
researcher,
veterinarian, medical doctor or other clinician, which includes alleviation of
the
symptoms of the disease or disorder being treated.
The term "alkyl" refers to straight or branched chain hydrocarbons, optionally
substituted. "Alkenyl" refers to a straight or branched chain hydrocarbon with
at least
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-15-
one carbon-carbon double bond. "Alkynyl" refers to a straight or branched
chain
hydrocarbon with at least one carbon-carbon triple bound.
The terms "halogen," "halo," or "halide" refer to fluorine, chlorine, bromine
and iodine.
The term "cycloalkyl" refers to optionally substituted, saturated cyclic
hydrocarbon ring systems, preferably containing 1 to 3 rings and 3 to 7
carbons per
ring which may be further fused with an unsaturated C3-C7 carbocyclic ring.
Exemplary groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl,
cycloheptyl, cyclooctyl, cyclononyl, cyclodecyl, and adamantyl. Exemplary
substituents include one or more alkyl groups or one or more groups described
above
as alkyl substituents.
The term "aryl" refers to aromatic monocyclic, fused bicyclic, or fused
polycyclic hydrocarbon or heterocyclic groups having 1 to 20 carbon atoms in
the
ring portions, such as phenyl, naphthyl, pyrrolyl, indolyl, pyrazolyl,
pyrazolinyl,
imidazolyl, oxazolyl, isoxazolyl, thiazolyl, thiadazolyl, isothiazolyl, furyl,
thienyl,
oxadiazolyl, pyridinyl, N-oxo-pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl,
tetrazinyl,
triazinyl, triazolyl, benzothiazolyl, benzoxazolyl, benzothienyl, quinolinyl,
quinolinyl-
N-oxide, isoquinolinyl, benzimidazolyl, benzofuryl, chromonyl, coumarinyl,
cinnolinyl, quinoxalinyl, indazolyl, benzisothiazolyl, benzisoxazolyl,
benzodiazinyl,
benzofurazanyl, benzothiopyranyl, benzpyrazolyl, indolinyl, isochromanyl,
isoindolinyl, naphthyridinyl, phthalazinyl, purinyl, quinazolinyl, and the
like. Aryl
groups may be substituted.
The term "biaryl" refers to a combination of two bonded, nonfused aryl groups
as defined above. Exemplary biaryl groups include biphenyl, furylphenyl,
phenylfuryl, thienylphenyl, phenylthienyl, pyridylphenyl, phenylpyridyl,
furylpyridyl,
pyridylfuryl, pyrrolylpyridyl, pyridylthienyl, isoxazolylthienyl,
isoxazolylphenyl, and
the like.
The above defined groups may be substituted by one or more substituents.
Illustrative examples of substituents include but are not limited to alkyl,
alkenyl,
alkynyl, aryl, halo, haloalkyl, hydroxy, alkoxy, haloalkoxy, cycloalkoxy, oxo,
aryloxy, alkanoyl, alkanoyloxy, amino, alkylamino, dialkylamino, arylamino,
diarylamino, alkylarylamino, sulfonamido, carbonyl, carboxyl, alkoxycarbonyl,
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-16-
amidocarbonyl, oxoallcyl, cyano, nitro, carbamyl, guanidine, amidino,
sulfonyl, and
the like.
Compounds of the Invention and Their Preparation
In one aspect of the invention, compounds of formula (I) are provided
R4 R5
O ~"." OH .,~,~ Rs NMe2
R1"N-(CH~m ~,~_~ '~i0 O
R2 ..
wherein Rl is Cl-C8 substituted or unsubstituted alkyl, C2-C8 substituted or
unsubstituted alkenyl, C2-C8 substituted or unsubstituted alkynyl, C4-C15
substituted
or unsubstituted aryl, CS-CZO substituted or unsubstituted arylalkyl, CS-CZO
substituted
or unsubstituted biarylalkyl, CS-C2o substituted or unsubstituted arylalkenyl,
or CS-CZo
substituted or unsubstituted arylalkynyl; RZ is H, C1-C4 substituted or
unsubstituted
alkyl, C2-C4 substituted or unsubstituted alkenyl, or C2-C4 substituted or
unsubstituted
alkynyl; R3 is H, C1-C4 alkanoyl, or benzoyl; R4 is H or OH; RS is H, OH, Cl-
C4
alkoxy, C2-C4 alkenyloxy, or C2-C4 alkynyloxy; R6 is OH or OMe; and m is 0-2.
In one embodiment of the invention, compounds of formula (I) are provided
wherein Rl is C6-Cls substituted or unsubstituted arylalkyl, C6-Cls
substituted or
unsubstituted biarylalkyl, C6-Cls substituted or unsubstituted arylalkenyl, or
C6-Cls
substituted or unsubstituted arylalkynyl.
In one embodiment of the invention, compounds of formula (I) are provided
wherein R' = H; R3 = H; R4 = H or OH; R5 = OH or alkoxy; R6 = OMe; and m = 0.
In another embodiment of the invention, compounds of formula (I) are
provided wherein R2 = H; R3 = H; R4 = H or OH; RS = OH or alkoxy; R6 = OMe;
and
m = 1.
In another embodiment of the invention, compounds of formula (I) are
provided wherein RZ = H; R3 = H; R4 = H or OH; RS = OH or alkoxy; R~ = OMe;
and
m=2.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-17-
In another embodiment of the invention, compounds of formula (I) are
provided wherein Rl is C4-C15 substituted or unsubstituted aryl, CS-Coo
substituted or
unsubstituted arylalkyl, or CS-Caosubstituted or unsubstituted biarylalkyl;
and RZ is H.
In another embodiment of the invention, compounds of formula (I) having the
structures
o
lMez
O /
HO OMe
O ~M~ \ I O ,,. OH ~~HO NM~
..,,0__
and ~ 0~~~~~0 OMe
are provided.
In another aspect of the invention, methods for the preparation of compounds
of formula (I) are provided. In one embodiment of the invention, certain
embodiments of the compounds of formula (I) wherein R2 = Ii are prepared from
azidoerythromycins as illustrated in Scheme 1.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-18-
SCHEME 1
0
1. Me3P
R .,~ R R R 2. R1 COzH, HOBt
H "'' NMe O ,. 'OH .,.. ~ a NM~ EDCI
".. 2
N3-(CHg)m~"~ O '~. .,~ O O > Ng-(CHp)m~~... O '" 'v0 O >
O ~~'O R6 O R6
~OH ~OH
O O
~I MeOH ~
R1~ ~ e2 > R1"NH-(CH2)n,~~,.
The azidoerythromycin is treated with an acylating reagent, for example acetic
anhydride, propionic anhydride, benzoic anhydride, and the like, at ambient
temperature in an inert solvent such as ethyl acetate, dichloromethane, or
acetonitrile,
to produce the 2'-O-aryl azidoerythromycin. This is detailed below in Example
5.
The azide is then reduced using a phosphine such as trimethylphosphine or
triphenylphosphine in a solvent such as tetrahydrofuran (THF) or a mixture of
THF
and dichloromethane, and the intermediate phosphinimine is reacted with a
carboxylic
acid, a carbodiimide such as 1-[3-(dimethylamino)propyl]-2-ethylcarbodiimide
hydrochloride (EDCI) or dicyclohexyl-carbodiimide, and a coupling adjuvant
such as
1-hydroxybenzotriazole (HOBt), N-hydroxysuccinimide (HOSu), or 1-hydroxy-7-
azabenzotriazole (HABt), to produce the 2'-O-acyl-amidoerythromycin. A general
procedure for this conversion is given below in Example 6. The 2'-O-acyl group
is
cleaved by heating in methanol, optionally in the presence of added
triethylamine.
Examples 7 - 45 below provide details of the preparation of compounds of
formula (I)
according to this method.
In another embodiment of the invention, the azidoerythromycin is treated with
trimethylphosphine, and the resulting phosphinimine is reacted with a
carboxylic acid
and a coupling agent such as O-benzotriazol-1-yl-N,N,N',N'-tetramethyluronium
hexafluorophosphate (HBTU), O-(7-azabenzotriazol-1-yl)-N,N,N',N'-
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-19-
tetramethyluronium hexafluorophosphate (HATU), or a similar coupling agent
which
incorporates the coupling agent and adjuvant into one molecule.
The carboxylic acids used in the preparation of the inventive compounds
include alkanoic acids, alkenoic acids, alkynoic acids, N-protected amino
acids andlor
peptides such as N-BOC, N-Cbz, and N-FMOC amino acids andlor peptides, benzoic
acids, heterocyclic carboxylic acids, biaryl carboxylic acids, arylacetic
acids,
biarylacetic acids, and the like, each of which may be substituted by a
variety of
groups.
In one embodiment of the invention, the carboxylic acids used in the
preparation of the inventive compounds are biarylacetic acids. One means of
preparing these biarylacetic acids is via a Suzuki coupling as illustrated in
Scheme 2.
SCHEME 2
Pd catalyst / I ~ 1. NaOH
C02M a + ~ > I COzM a > O I ~ C02H
(HO)2B ~ 2. HCI
A haloarylacetic ester is coupled with an arylboronic acid in the presence of
a
palladium catalyst and base to provide the biarylacetic ester. The ester is
subsequently saponified to provide the biarylacetic acid. Example 47 below
provides
a detailed procedure for this method as applied to the synthesis of 3-(2-
furyl)phenylacetic acid. Suitable palladium catalysts include
tetrakis(triphenylphosphine)palladium, palladium on carbon with
triphenylphosphine,
and similar sources of palladium(0). An alternate means of preparing these
biarylacetic acids is through hydrolysis of the corresponding nitriles,
prepared via
Suzuki coupling of an arylboronic acid with a haloarylacetonitrile. The
nitrile is
hydrolyzed to the acid using a mixture of sodium hydroxide and hydrogen
peroxide.
In another embodiment of the invention, certain embodiments of the
compounds of formula (I) wherein R' = alkyl, alkenyl, or alkynyl are prepared
from
azidoerythromycins as illustrated in Scheme 3.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-20-
SCHEME 3
1. Me3P
2. R2X RiC02H
' 3. H O EDCI, HOBt
R2NH-(CHI >
N3-( CHp) m~" >
O
MeOH ~
> R1' 'N-(C
Rz
The 2'-O-acyl azidoerythromycin is reacted with trimethylphosphine to
generate the phosphinimine, which is then alkylated by reaction with an alkyl
halide
or alkyl sulfonate, R2X. Suitable alkylating agents include but are not
limited to
methyl iodide, methyl bromide, methyl triflate, ethyl tosylate, ethyl
triflate, ethyl
iodide, allyl bromide, and propargyl bromide. The resulting phosphonium salt
is
hydrolyzed, and the product amine is acylated with the carboxylic acid,
R1COOH, and
a coupling reagent such as EDCI and HOBt, HATU, or the like, to provide a
compound of formula (I) wherein R2 = alkyl, alkenyl, or alkynyl and R3 =
alkanoyl or
benzoyl. Removal of the 2'-O-acyl group by methanolysis provides the compound
of
formula (I) wherein R2 = alkyl, alkenyl, or alkynyl and R3 = H.
In one aspect of the invention, methods for preparing azidoerythromycins are
provided. In one embodiment of the invention, the method comprises the steps
of (1)
chemically synthesizing a racemic chlorinated diketide thioester; (2) growing
a
culture of a first organism in the presence of the racemic chlorinated
diketide thioester
so as to produce a chlorinated erythronolide; (3) partially purifying the
chlorinated
erythronolide; (4) chemically converting the chlorinated erythronolide into
the azido
erythronolide; (5) growing a culture of a second organism in the presence of
the azido
erythronolide, so as to produce an azidoerythromycin; and (6) purifying the
azidoerythromycin.
In one embodiment of the invention, a method for preparing 15-
azidoerythromycin is provided. This method comprises the steps of (1)
chemically
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-21-
synthesizing racemic (2S*,3R*)-5-chloro-3-hydroxy-2-methylpentanoyl N-
propionylcysteamine thioester; (2) growing a culture of a first organism in
the
presence of racemic (2S*,3R*)-5-chloro-3-hydroxy-2-methylpentanoyl N-
propionylcysteamine thioester so as to produce 15-chloro-6-deoxyerythronolide
B; (3)
partially purifying the 15-chloro-6-deoxyerythronolide B so produced; (4)
chemically
converting the 15-chloro-6-deoxyerythronolide B into 15-azido-6-
deoxyerythronolide
B; (5) growing a culture of a second organism in the presence of the 15-azido-
6-
deoxyerythronolide B, so as to produce 15-azidoerythromycin; and (6) purifying
the
15-azidoerythromycin.
In another embodiment of the invention, a method for preparing 14-azido-14-
desmethylerythromycin is provided. This method comprises the steps of (1)
chemically synthesizing racemic (2S*,3R*)-4-chloro-3-hydroxy-2-methylbutanoyl
N-
propionylcysteamine thioester; (2) growing a culture of a first organism in
the
presence of racemic (2S*,3R*)-4-chloro-3-hydroxy-2-methylbutanoyl N-
propionylcysteamine thioester so as to produce 15-chloro-6-deoxyerythronolide
B; (3)
partially purifying the 14-chloro-14-desmethyl-6-deoxyerythronolide B so
produced;
(4) chemically converting the 14-chloro-14-desmethyl-6-deoxyerythronolide B
into
14-azido-14-desmethyl-6-deoxyerythronolide B; (5) growing a culture of a
second
organism in the presence of the 14-azido-14-desmethyl-6-deoxyerythronolide B,
so as
to produce 14-azido-14-desmethylerythromycin A; and (6) purifying the 14-azido-
14-
desmethylerythromycin A.
In another embodiment of the invention, a method for preparing 15-
(azidomethyl)-erythromycin is provided. This method comprises the steps of (1)
chemically synthesizing racemic (2S*,3R*)-4-chloro-3-hydroxy-2-methylhexanoyl
N-
propionylcysteamine thioester; (2) growing a culture of a first organism in
the
presence of racemic (2S*,3R*)-4-chloro-3-hydroxy-2-methylhexanoyl N-
propionylcysteamine thioester so as to produce 15-(chloromethyl)-6-
deoxyerythronolide B; (3) partially purifying the 15-(chloromethyl)-6-
deoxyerythronolide B so produced; (4) chemically converting the 15-
(chloromethyl)-
6-deoxyerythronolide B into 15-(azidomethyl)-6-deoxyerythronolide B; (5)
growing a
culture of a second organism in the presence of the 15-(azidomethyl)-6-
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-22-
deoxyerythronolide B, so as to produce 15-(azidomethyl)-erythromycin ; and (6)
purifying the 15-(azidomethyl)-erythromycin .
The synthesis of racemic thioesters is described in Ashley et al., "Synthesis
of
oligoketides," U.S. Patent 6,492,562, incorporated herein by reference. In
brief, the
trichlorotitanium enolate of 3-propionyl-2-benzoxazolone is reacted with an
aldehyde
so as to produce the aldol adduct. The aldol adduct is converted into the
thioester by
reaction with the sodium salt of N-propionylcysteamine. This process is
exemplified
in detail in Example 1 below. When the aldehyde used is 3-chloropropanal, the
thioester so produced is (2S*,3R*)-5-chloro-3-hydroxy-2-methylpentanoate N-
propionylcysteamine thioester. When the aldehyde used is chloroacetaldehyde,
the
thioester so produced is (2S*,3R*)-4-chloro-3-hydroxy-2-methylbutanoate N-
propionylcysteamine thioester. When the aldehyde used is 4-
chlorobutyraldehyde, the
thioester so produced is (2S*,3R*)-6-chloro-3-hydroxy-2-methylhexanoate N-
propionylcysteamine thioester.
The preparation of 13-substituted 6-deoxyerythronolide B compounds is
described in Khosla et al., U.S. Patent Nos. 6,080,555, 6,274,560, 6,066,721,
and
6,261,816; and Ashley et al., U.S. Patent 6,492,562; each of which is
incorporated
herein by reference. In one embodiment, 15-chloro-6-deoxyerythronolide B is
prepared by a method in which racemic (2S*,3R*)-5-chloro-3-hydroxy-2-
methylpentanoyl N-propionylcysteamine thioester is provided to a 6-
deoxyerythronolide B synthase ("DEBS") that is unable to act on its natural
substrate,
propionyl CoA, due to a mutation in the ketosynthase domain of module 1 of
DEBS.
This recombinant DEBS can be expressed in the natural host that normally
produces
erythromycin, Saccharopolyspora erythraea, or the entire PKS gene cluster can
be
inserted by plasmid in a suitable host such as S. coelicolor (see e.g.,
Jacobsen et al,
Science 277: 367-369 (1997), incorporated herein by reference) or S. lividans
which
has been modified to delete its endogenous actinorhodin polyketide synthesis
mechanism. In a preferred embodiment, the host is S. coelicolor CH999/pJRJ2,
which expresses a mutant 6-DEB synthase having an inactivated module 1
ketosynthase.
A cell free system as described in Khosla et al., "Synthesis of polyketides
from diketides," U.S. Patent No. 6,080,555; Khosla et al., "Cell-free
synthesis of
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-23-
polyketides," U.S. Patent 6,274,560; and PCT Publication No. WO 97/02358, each
of
which is incorporated herein by reference, may also be employed by producing
the
relevant PKS proteins recombinantly and effecting their secretion or lysing
the cells
containing them. A typical cell-free system would include the appropriate PKS,
NADPH and an appropriate buffer and substrates required for the catalytic
synthesis
of polyketides.
In another embodiment of the invention, the appropriate thioester diketide
substrates are provided to PKS enzymes other than the 6-dEB synthase of
Saccharopolyspora erythraea. Other PKS enzymes include the 6-dEB synthase of
Micronaoraospora megalomicea and its KS 1 ° derivative described in
McDaniel &
Volchegursky, "Recombinant megalomicin biosynthetic genes," PCT publication WO
01/27284; the oleandolide PKS and its KS 1 ° derivative described in
Betlach et al.,
"Recombinant oleandolide polyketide synthase," U.S. Patent 6,251,636; and the
narbonolide PKS and its KS 1 ° derivative described in McDaniel et al.,
"Heterologous
expression of narbonolide PKS," U.S. Patent 6,503,741 and in Betlach et al.,
"Nucleic
acids encoding narbonolide polyketide synthase enzymes from streptomyces
narbonensis," U.S. Patent No. 6,303,767; each of which is incorporated herein
by
reference.
In another embodiment of the invention, the appropriate thioester diketide
substrates are provided to any of the above PKS enzymes in a host cell that is
capable
of performing one or more of the post-PKS hydroxylation andlor glycosylation
steps
leading to the erythromycins. Thus, a chlorinated erythronolide B is prepared
by
providing a chlorinated thioester diketide to a strain comprising both DEBS
and a 6-
hydroxylase, for example the eryF hydroxylase from Sac. erytlaraea. The
production
of 14-chloro-14-desmethylerythronolide B is described in Example 72 below.
The preparation of 15-chloro-6-deoxyerythronolide B is detailed in Example 2
below. Using the same methods but starting with the appropriate diketide
thioesters
described above, 14-chloro-14-desmethyl-6-deoxyerythronolide B and 15-
(chloromethyl)-6-deoxyerythronolide B are prepared as described below in
Examples
71 and 73.
In one embodiment of the invention, the chlorinated erythronolide so produced
is reacted with sodium azide and sodium iodide in DMSO so as to produce the
azido
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-24-
erythronolide by displacement of the chloride with azide. This is detailed in
the
Examples below. Thus, 15-azido-6-deoxyerythronolide B is prepared from 15-
chloro-6-deoxyerythronolide B (Example 3), 14-azido-14-desmethyl-6-
deoxyerythronolide B is prepared from 14-chloro-14-desmethyl-6-
deoxyerythronolide
B (Example 74), 15-(azidomethyl)- 6-deoxyerythronolide B is prepared from 15-
(chloromethyl)- 6-deoxyerythronolide B (Example 75), 15-azido-erythronolide B
is
prepared from 15-chloro-erythronolide B, 14-azido-14-desmethyl-erythronolide B
is
prepared from 14-chloro-14-desmethyl-erythronolide B, and 15-(azidomethyl)-
erythronolide B is prepared from 15-(chloromethyl)-erythronolide B.
In another embodiment of the invention, the azidoerythronolide prepared
according to one of the above methods is then added to a culture of a
bioconversion
organism capable of glycosylating at the C-3 and C-5 positions, and optionally
hydroxylating at C-6, hydroxylating at the C-12 position and/or methylating a
3-O-
mycarosyl group, depending on the strain and fermentation conditions employed.
In a
preferred embodiment, the bioconversion organism is Saccharopolyspora
erythraea
K39-14V, a strain in which the module 1 ketosynthase of the native DEBS genes
has
been inactivated as described in Santi et al., "Hosts for the biosynthesis of
polyketides," PCT publication WO 01/83803, incorporated herein by reference.
I~39-
14V contains the enzyme activities needed to convert an erythronolide into the
erythromycin A derivative, i.e., hydroxylation at C-6, addition of mycarose to
the 3-
OH, addition of desosamine to the 5-OH, hydroxylation at C-12, and methylation
of
the mycarose. In one embodiment of the invention, K39-14V is supplied with 15-
azido-6-deoxyerythronolide B under fermentation conditions wherein 15-
azidoerythromycin A is the predominant product. This is detailed below in
Example
4. In another embodiment of the invention, K39-14V is supplied with 15-azido-6-
deoxyerythronolide B under fermentation conditions wherein 15-
azidoerythromycin B
is the predominant product, for example under conditions of limited oxygen. In
another embodiment of the invention, K39-14V is supplied with 14-azido-14-
desmethyl-6-deoxyerythronolide B under fermentation conditions wherein 14-
azido-
14-desmethylerythromycin A is produced. In another embodiment of the
invention,
I~39-14V is supplied with 14-azido-14-desmethyl-6-deoxyerythronolide B under
fermentation conditions wherein 14-azido-14-desmethylerythromycin B is
produced
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-25-
(Example 76). In another embodiment of the invention, K39-14V is supplied with
15-
(azidomethyl)-6-deoxyerythronolide B under fermentation conditions wherein 15-
(azidomethyl)-erythromycin A is produced. In another embodiment of the
invention,
K39-14V is supplied with 15-(azidomethyl)-6-deoxyerythronolide B under
fermentation conditions wherein 15-(azidomethyl)-erythromycin B is produced
(Example 77). In another embodiment of the invention, a bioconversion strain
that
lacks an active 12-hydroxylase, encoded by the eryK gene or a homologue, is
used to
convert the azido-6-deoxyerythromycin into the azidoerythromycin B. In another
embodiment of the invention, the azidoerythromycin B compounds are produced
using a mutant of K39-14V wherein the eryK gene encoding the 12-hydroxylase
has
been inactivated or deleted.
The resulting azidoerythromycins are isolated as described for 15-
azidoerythromycin A in Example 4 below. The cells are removed from the
fermentation by centrifugation, and the broth is extracted to remove the
azidoerythromycins. In one embodiment of the invention, the extraction is
performed
using absorption onto a solid resin, such as XAD or HP20. The
azidoerythromycins
are subsequently eluted from the resin with an organic solvent such as
methanol,
ethanol, acetone, acetonitrile, or ethyl acetate. In another embodiment of the
invention, the extraction is performed by mixing the broth with an immiscible
organic
solvent in which the azidoerythromycin is soluble, such as ethyl acetate,
dichloromethane, or chloroform. After concentration of the extract, the
residue is
subjected to chromatography to provide purified azidoerythromycins.
In another embodiment of the invention, compounds of formula (I) wherein RS
= alkoxy are provided. In one embodiment, these compounds are prepared from
azidoerythromycins as illustrated in Scheme 4 and exemplified in Example 80.
The
azidoerythromycin is first treated with hydroxylamine, typically in the
presence of an
acid catalyst such as acetic acid and in a mixture of water and isopropanol as
solvent,
to produce the 9-oxime. This procedure is exemplified in detail with 15-
azidoerythromycin A in Example 48 below. The 9-oxime is protected, for example
using an acetal such as l,l-diisopropoxycyclohexane (I~IPCH) or 2,2-
dimethoxypropane in the presence of a mild acid catalyst such as pyridinium p-
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-2G-
toluenesulfonate (PPTS). This procedure is exemplified in detail with 15-
azidoerythromycin A 9-oxime in Example 49 below. Other protecting groups, such
as
trialkylsilyl ethers, can also be used to protect the 9-oxime. The 2'- and 4"-
OH
groups are next protected, for example as trimethylsilyl ethers by treatment
with a
mixture of trimethylsilylinudazole and chlorotrimethylsilane, as described
below in
Example 50.
SCHEME 4
n U~ 1. DIPOH,
NH20H " OH ~~~HO~ NM~e2 PPTS >
N3-(Oh'I?)m I > N3 (CH~m ~ O ~''O-'Oy 2. TMSCI,
AcOH, PrOH TMSIm
O ''O OMe
OH ~OH
Y
MeBr R4 Me AcOH
N3 (CFI~m ".1 fCO~BU > , OH .", TMS NM~ >
ACN
N3 (OH2)m O ." ..,,0 O
O '
O OTMS
:.YO'Uti ..,0~~ Na2S204 ,".:Y 'OH ."~y NM~
Ns (CHa~m ' > ., HO~
~, HC02H N3 (CH?~m O 'O~
O OMe
O ~~''O OMe
~OH
OH
In one embodiment of the invention, the 6-OH is methylated using methyl
bromide in the presence of potassium tert-butoxide or similar base to provide
embodiments of the compounds of formula (I) wherein RS = OMe. This process is
exemplified below in Example 51. In another embodiment of the invention, the 6-
OH
is allylated using allyl bromide in the presence of potassium tent-butoxide or
similar
base to provide embodiments of the compounds of formula (I) wherein RS = O-
allyl.
In another embodiment of the invention, the 6-OH is allylated using allyl tent-
butyl
carbonate in the presence of a palladium catalyst, for example palladium
acetate and
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
_27_
triphenylphosphine, to provide embodiments of the compounds of formula (I)
wherein
RS = O-allyl.
The 9-oxime and 2'- and 4"-OH groups are deprotected by treatment with
acetic acid in acetonitrile. The 9-oxime can be cleaved, for example using
sodium
hydrosulfite and formic acid, to provide embodiments of compounds of formula
(I)
wherein RS = alkoxy. These steps are exemplified below in Examples 52 and 53.
In another aspect of the invention, compounds of formula (II) are provided
~3
M e2
i
wherein R3, R4, and m are as defined above; RS is H, C1-Cø alkoxy, CZ-C4
alkenyloxy,
or C2-C4 alkynyloxy; and R8 is H or F.
In one embodiment of the invention, compounds of formula (II) wherein m = 0
are provided.
In another embodiment of the invention, compounds of formula (II) wherein m
= 1 are provided.
In another embodiment of the invention, compounds of formula (II) wherein m
= 2 are provided.
In one embodiment of the invention, the compounds of formula (II) wherein
R4 = OH, RS = Cl-C4 alkoxy, and m = 0 are provided.
In another embodiment of the invention, the compounds of formula (II)
wherein R4 = OH, RS = C1-C4 alkoxy, and m = 1 are provided.
In another embodiment of the invention, the compounds of formula (II)
wherein R4 = OH, RS = C1-C4 alkoxy, and m = 2 are provided.
In another embodiment of the invention, the compound of formula (II)
wherein R4 = H, RS = C1-C4 alkoxy, and m = 0 are provided.
In another embodiment of the invention, the compound of formula (II)
wherein R4 = H, RS = C1-C4 alkoxy, and m = 1 are provided.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
_~8_
In another embodiment of the invention, the compound of formula (II)
wherein R4 = H, RS = C1-C4 alkoxy, and m = 2 are provided.
In another embodiment of the invention, the compounds of formula (II)
wherein R3 = acetyl or benzoyl, R4 = OH, RS = C1-C4 alkoxy, and m = 0 are
provided.
In another embodiment of the invention, the compounds of formula (II)
wherein R3 = acetyl or benzoyl, R4 = OH, RS = C1-C4 alkoxy, and m = 1 are
provided.
In another embodiment of the invention, the compounds of formula (II)
wherein R3 = acetyl or benzoyl, R4 = OH, RS = C1-C4 alkoxy, and m = 2 are
provided.
In another embodiment of the invention, the compounds of formula (II)
wherein R3 = acetyl or benzoyl, R4 = H, RS = C1-C4 alkoxy, and m = 0 are
provided.
In another embodiment of the invention, the compounds of formula (II)
wherein R3 = acetyl or benzoyl, R4 = H, RS = C1-C4 alkoxy, and m = 1 are
provided.
In another embodiment of the invention, the compounds of formula (II)
wherein R3 = acetyl or benzoyl, R4 = H, RS = C1-C4 alkoxy, and m = 2 are
provided.
In another embodiment of the invention, the compounds of formula (II) having
the formulas
N~
3
O
H Me
N3\":, 'OH ;,IO O~~ N ~ Ns~
O O ~ and
~F
are provided.
In another aspect of the invention, methods for the preparation of compounds
of formula (II) are provided. In one embodiment of the invention, certain
embodiments of the compounds of formula (II) are prepared from compounds of
formula (I) wherein RS = alkoxy as illustrated in Scheme 5.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-29-
SCHEME 5
BZ2o
..w ~ PPTS
s .,.. >
N ~''~ H20, acetone
NCS, Me2S
> ,...
..., az
N ~,1, Et3N N ~,;
The cladinose is removed from the compound of formula (I) by treatment with
a mild acid in the presence of water, for example using PPTS in a mixture of
acetone
and water as exemplified below in Example 54, or using aqueous HCI. After
protection of the 2'-OH by treatment with benzoic anhydride in the absence of
additional base (Example 55), the 3-OH is oxidized to the ketone (Example 56).
Suitable oxidation methods include but are not limited to the Corey-Kim (N-
chlorosuccinimide, dimethylsulfide, triethylamine), Swern (oxalyl chloride,
DMSO,
triethylamine), Moffat (dicyclohexylcarbodiimide, DMSO), and modified Pfizer-
Moffat (1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide, pyridinium
trifluoroacetate, DMSO) conditions.
In another embodiment of the invention, compounds of formula (II) wherein
R$ = F are prepared from compounds of formula (II) wherein R$ = H as
illustrated in
Scheme 6. The compound of formula (II) wherein R8 = H is treated with a base,
for
example potassium tart-butoxide, and a fluorinating agent such as N-
fluorobenzenesulfonimide (NFBS) to yield the compound of formula (II) wherein
R8
= F. A representative method for fluorination in given in Example 60.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-30-
SCHEME 6
1. KOtBu
e2 > e2
N; 2. NFBS N
In another embodiment of the invention, the 2'-O-benzoyl protecting group is
removed from the compound of formula (II) wherein R3 = benzoyl by heating in
methanol containing triethylamine to provide the compound of formula (II)
wherein
R3 = H.
In another aspect of the invention, compounds of formula (III) are provided
2
Ri"N- CFi
i ( 2)r
R2
wherein R1, R2, R3, R4, R8, and m are as defined above and Rs is H, C1-C4
alkoxy, C2-
C4 alkenyloxy, or C2-C4 alkynyloxy.
In one embodiment of the invention, compounds of formula (III) are provided
wherein Rl is C~-CIS substituted or unsubstituted arylalkyl, C6-Cls
substituted or
unsubstituted biarylalkyl, C6-Cls substituted or unsubstituted arylalkenyl, or
C6-Cls
substituted or unsubstituted arylalkynyl.
In one embodiment of the invention, compounds of formula (III) wherein R2 is
H and Rs is alkoxy are provided.
In another embodiment of the invention, compounds of formula (III) are
provided wherein Rl is C4-Cls substituted or unsubstituted aryl, Cs-
CZOSUbstituted or
unsubstituted arylalkyl, or Cs-C2osubstituted or unsubstituted biarylalkyl; R~
is H; and
Rs is alkoxy.
In another embodiment of the invention, compounds of formula (III) are
provided wherein m = 0.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-31-
In another embodiment of the invention, compounds of formula (III) are
provided wherein m = 1.
In another embodiment of the invention, compounds of formula (III) are
provided wherein m = 2.
In another embodiment of the invention, compounds of formula (III) are
provided wherein R2 is H; R3 is acetyl or benzoyl; R4 is H or OH; RS is
alkoxy; and m
= 0.
In another embodiment of the invention, compounds of formula (III) are
provided wherein RZ is H; R3 is acetyl or benzoyl; R4 is H or OH; RS is
alkoxy; and m
= 1.
In another embodiment of the invention, compounds of formula (III) are
provided wherein R2 is H; R3 is acetyl or benzoyl; R4 is H or OH; RS is
alkoxy; and m
= 2.
In another embodiment of the invention, compounds of formula (III) are
provided wherein R2 is H; R3 is H; R4 is H or OH; R$ is alkoxy; and m = 0.
In another embodiment of the invention, compounds of formula (III) are
provided wherein RZ is H; 8315 H; R4 is H or OH; RS is alkoxy; and m = 1.
In another embodiment of the invention, compounds of formula (III) are
provided wherein R2 is H; R3 is H; R4 is H or OH; RS is alkoxy; and m = 2.
In another embodiment of the invention, compounds of formula (III) having
the structures
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-32-
0
o ~
HO Me / O
/ O ,. OH ..,~ NMe2
I . He
N~,.... O °. .O-
H . / O
O O
/ O NMe2
/I o ~ wI I
and ~ O
are provided.
In another aspect of the invention, methods for the preparation of compounds
of formula (III) are provided. In one embodiment of the invention, compounds
of
formula (III) are prepared from compounds of formula (II) wherein R3 = benzoyl
as
illustrated in Scheme 7.
S CHEME 7
1. MesP
2. R~ C02H, HOBt
EDCI O
N3- ~ R~~H-(CH2)m
The compound of formula (II) is treated with trimethylphosphine, and the
resulting phosphinimine is reacted with a carboxylic acid, a coupling agent
such as a
carbodiimide, and a coupling adjuvant such as HOBt, HABt, or HOSu as discussed
above and exemplified below in Example 65.
In another embodiment of the invention, the compound of formula (II) is
treated with trimethylphosphine, and the resulting phosphinimine is reacted
with a
carboxylic acid and a coupling agent such as O-benzotriazol-1-yl-N,N,N',N'-
tetramethyluronium hexafluorophosphate (HBTU), O-(7-azabenzotriazol-1-yl)-
N,N,N',N'-tetramethyluronium hexafluorophosphate (HATU), or a similar coupling
agent which incorporates the coupling agent and adjuvant into one molecule.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-33-
In another embodiment of the invention, the 2'-O-benzoyl protecting group is
removed from the compound of formula (III) wherein R3 = benzoyl by heating in
methanol optionally containing triethylamine to provide the compound of
formula
(III) wherein R3 = H. This is exemplified below in Example 66.
In another embodiment of the invention, certain embodiments of the
compounds of formula (III) wherein RZ = alkyl, alkenyl, or alkynyl are
prepared from
compounds of formula (II) as illustrated in Scheme 8. The compound of formula
(II)
wherein R3 = alkanoyl or benzoyl is reacted with trimethylphosphine to
generate the
phosphinimine, which is then alkylated by reaction with an alkyl halide or
alkyl
sulfonate, R2X. Suitable alkylating agents include but are not limited to
methyl iodide,
methyl bromide, methyl triflate, ethyl tosylate, ethyl triflate, ethyl iodide,
allyl
bromide, and propargyl bromide.
SCHEME 8
0
0
1. Me3P R4 . R5 t
R R 2. R2X , OH .,.. Bz NMep R COpH
H ~~ B'~ NMeg 3. Hp0 RzNH_(CH~m , .. , ~ G4.~/~, EDCI, HOBt
> W ' O ~" ' O~ >
Ng-(CH~m-~ O ~~~~ ~'O
O
O O ;Rg
~RB
O
Ra ' Rs
MeOH > O ,. OH .~" NM
~ J,~ HO~/~,
Rt"N-(CH~,n= Et3N Rt"N-(CH~rt,= O ~' ''O~
R2 R2
O
rR8
The resulting phosphonium salt is hydrolyzed, and the product amine is
acylated with the carboxylic acid, R1COOH, and a coupling reagent such as EDCI
and
HOBt, HATU, or the like, to provide a compound of formula (III) wherein R2 =
alkyl,
alkenyl, or alkynyl and R3 = alkanoyl or benzoyl. Removal of the 2'-O-acyl
group by
methanolysis provides the compound of formula (III) wherein RZ = alkyl,
alkenyl, or
alkynyl and R3 = H.
In another aspect of the invention, compounds of formula (IV) are provided
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-34-
_ ~s M e2
O
Ri N- C H
~ 2 C 2)m
R
wherein Rl, R2, R3, R8, and m are as defined above and RS is H, C1-C4 alkoxy,
C2-C4
alkenyloxy, or CZ-C4 alkynyloxy, and X is O or NR7, wherein R7 is H, C1-C4
alkyl, or
CG-C2o arylalkyl.
In one embodiment of the invention, compounds of formula (IV) are provided
wherein Rl is C6-Cls substituted or unsubstituted arylalkyl, C~-Cls
substituted or
unsubstituted biarylalkyl, C6-Cls substituted or unsubstituted arylalkenyl, or
C~-Cls
substituted or unsubstituted arylalkynyl.
In another embodiment of the invention, compounds of formula (IV) are
provided wherein m = 0.
In another embodiment of the invention, compounds of formula (IV) are
provided wherein m = 1.
In another embodiment of the invention, compounds of formula (IV) are
provided wherein m = 2.
In another embodiment of the invention, compounds of formula (IV) are
provided wherein R' = H.
In one embodiment of the invention, compounds of formula (IV) are provided
wherein R' = H; R3 = H; RS = OMe; X = O; and m = 0.
In another embodiment of the invention, compounds of formula (IV) are
provided wherein R2 = H; R3 = H; RS = OMe; X = O; and m = 1.
In another embodiment of the invention, compounds of formula (IV) are
provided wherein R2 = H; R3 = H; RS = OMe; X = O; and m = 2.
In another embodiment of the invention, compounds of formula (IV) are
provided wherein RZ = H; R3 = H; RS = OMe; X = NH; and m = 0.
In another embodiment of the invention, compounds of formula (IV) are
provided wherein R~ = H; R3 = H; RS = OMe; X = NH; and m = 1.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-35-
In another embodiment of the invention, compounds of formula (IV) are
provided wherein RZ = H; R3 = H; RS = OMe; X = NH; and m = 2.
In another embodiment of the invention, compounds of formula (IV) are
provided wherein Rl is C~-C15 substituted or unsubstituted aryl, CS-CZO
substituted or
unsubstituted arylalkyl, CS-CZO substituted or unsubstituted biarylalkyl, CS-
C2o
substituted or unsubstituted arylalkenyl, or CS-C2o substituted or
unsubstituted
arylalkynyl; and R2 is H.
In another embodiment of the invention, compounds of formula (IV) are
provided wherein Rl is C~-Cls substituted or unsubstituted aryl, CS-C2o
substituted or
unsubstituted arylalkyl, CS-CZO substituted or unsubstituted biarylalkyl, CS-
CZo
substituted or unsubstituted arylalkenyl, or CS-C2o substituted or
unsubstituted
arylalkynyl; RZ is H; and X is NH.
In another embodiment of the invention, compounds of formula (IV) having
the structures
O~ i
/ o ~ / I
\ I / N N~
N H
H
S
/ I O ~ / I O
\ N~ N~ ~ N~
H H
O
O
/N~IO e20'N/~O e2
S Hi, ~ S
and
are provided.
In another embodiment of the invention, compounds of formula (IV) having
the structures are provided:
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-36-
0
O / H O O / 0 TH
O ", OMe O OMe
O "~ .~1H0 NM~ W I O ~,:.. ..,HC~e2
N~~" ~ O ~~~ 'O~ H O
H
O O O O
.- I~~~F
9e
e2 IC~~e2
'N --
H
e2
O
and
In another embodiment of the invention, compounds of formula (IV) having
the structures are also provided:
0
o H ,o ~ O
~N OMe and ~ / ~N
O ~'~ ~ OMe
O NMe O
O~N N~~~' O ''' 'iOH~ 2 ~ O~~ O~~ NMez
H O N~NW ~' O ~'~ 'iOHO
o-~~o H .
O ~ O
Rs
In another aspect of the invention, methods for the preparation of compounds
of formula (IV) are provided. In one embodiment of the invention, compounds of
formula (II) wherein R4 = OH and R3 = acetyl are first treated with 1,1-
carbonyldiimidazole and 4-(dimethylamino)pyridine to provide the 11,12-cyclic
carbonate, as illustrated in Scheme 9.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-37-
SCHEME 9
0
H Me Im2C0 1. MesP
DMAP
OH ", ~~ NMez > >
O .,. ...,0 ~ 2. Rt C02H
N3-(CH2)m Ng-(CH2) EDCI, HOBt
O' ~ ~O
MeOH
OII A° NMe2
R1~NH-(CH2) R
The resulting carbonate is then treated with trimethylphosphine, a carboxylic
acid, a coupling agent, and a coupling adjuvant as described above to yield
the
compound of formula (IV) wherein R3 = acetyl and X = O. In another embodiment,
the compound of formula (IV) wherein R3 = H and X = O is prepared by treatment
of
the compound of formula (IV) wherein R3 = acetyl and X = O with methanol at 50
°C.
In another embodiment, compounds of formula (II) wherein R4 = OH and R3 =
benzoyl are first treated with methanesulfonic anhydride in pyridine to
produce the
11-O-mesylate as illustrated in Scheme 10. A representative procedure for
mesylation
is given in Example 57. The 11-O-mesylate is treated with 1,8-
diazabicyclo[5.4.0]undec-7-ene (DBU) to produce the 10,11-anhydrocompound, as
detailed in Example 58. Treatment with l,l-carbonyldiimidazole and a strong
base
such as sodium hydride produces the 12-O-imidazolide, which is reacted with an
amine, R7-NH2, to produce the 11-amino-11-deoxy-11,12-cyclic carbamate, a
compound of formula (IV) wherein R3 = benzoyl and X = NR7. A representative
procedure for carbamate formation when X = NH is given in Example 59. When R7
=
alkyl, it is usually necessary to perform the reaction at elevated
temperature, for
example 50 °C, in order to provide the (IOR) diastereomer.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
_38_
SCHEME 10
Ms20 DBU
e2 >
N3 (CH2)m pyridine N3 (CH~m
1. Im2C0, NaH 1. Me3P
> >
2. R~NH2 >B~e2 2. RtC02H
N3 (CH~m N3-(CHI O EDCI, HOBt
MeOH
O O
J.~ Et3N
Ri"NH-(CH~J Rt~Nf~-(CHI
In another embodiment of the invention, a compound of formula (IV) wherein
R3 = acetyl or benzoyl and R8 = F is prepared from a compound of formula (IV)
wherein R3 = acetyl or benzoyl and R8 = H by treatment with potassium tert-
butoxide
and NFBS. A representative procedure for fluorination is given in Example 60.
In another embodiment of the invention, a compound of formula (IV) wherein
X = NR7 is prepared according to the method illustrated in Figure 3. An
azidoerythromycin wherein R4 is OH and RS is alkoxy, alkyenyloxy, or
alkynyloxy,
prepared for example according to the method illustrated in Scheme 4 above, is
protected as the 2',4"-diacetate or 2',4"-dibenzoate. Treatment with 1,1-
carbonyldiimidazole and a strong base, for example sodium
bis(trimethylsilyl)amide
(NHS) in an inert solvent such as tetrahydrofuran at a temperature between -
10°C and 30 °C, followed by treatment with an amine, for example
ammonia, and
subsequently potassium tert-butoxide provides the 11,12-cyclic carbamate. The
cladinose is removed, for example by treatment with aqueous acid, and the
resulting
3-OH is subjected to hydrogenolysis to reduce the azide to the amine.
Hydrogenolysis is performed in a solvent such as methanol in the presence of a
metal
catalyst, such as palladium or palladium supported on an inert material such
as
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-39-
charcoal, and an acid, such as HCI, acetic acid, or similar, under a moderate
pressure
of hydrogen gas, such as that provided by a hydrogen balloon. The acid may be
generated i~z situ, for example by addition of chlorotrimethylsilane to
methanol. The
resulting ammonium salt is condensed with a carboxylic acid R1COOH as
described
above. Subsequent to amide formation, the 3-OH is oxidized to the 3-oxo group,
for
example using the Dess-Martin periodinane, Swern oxidation, Corey-Kim
oxidation,
or similar. Finally, the 2'-protecting group is removed by methanolysis. This
procedure is exemplified in detail in Example ~ 1 below.
In another embodiment of the invention, the compound of formula (IV)
wherein R3 = benzoyl is treated with methanol and triethylamine at 50
°C to provide
the compound of formula (IV) wherein R3 = H and X = NR7. A representative
procedure for debenzoylation is given in Example 62.
In another aspect of the invention, compounds of formula (V) are provided
p9
O lM e2
R1' -N-(CH~,~
R2
wherein R1, R2, R4, RG and m are as defined above; and R9 is H or C1-C4 alkyl.
In one embodiment of the invention, compounds of formula (V) are provided
wherein Rl is C6-Cls substituted or unsubstituted arylalkyl, CG-C15
substituted or
unsubstituted biarylalkyl, C6-C15 substituted or unsubstituted arylalkenyl, or
C6-Cls
substituted or unsubstituted arylalkynyl.
In another embodiment of the invention, compounds of formula (V) are
provided wherein m = 0.
In another embodiment of the invention, compounds of formula (V) are
provided wherein m = 1.
In another embodiment of the invention, compounds of formula (V) are
provided wherein m = 2.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-40-
In one embodiment of the invention, compounds of formula (V) are provided
wherein RZ = H; R4 = OH; R~ = OMe; R~ = Me; and m = 0.
In another embodiment of the invention, compounds of formula (V) are
provided wherein R2 = H; Rø = OH; R~ = OMe; R~ = Me; and m = 1.
In another embodiment of the invention, compounds of formula (V) are
provided wherein R2 = H; R4 = OH; R~ = OMe; R~ = Me; and m = 2.
In another embodiment of the invention, compounds of formula (V) are
provided wherein Rl is C4-C15 substituted or unsubstituted aryl, CS-CZO
substituted or
unsubstituted arylalkyl, or CS-CZO substituted or unsubstituted biarylalkyl.
In another aspect of the invention, methods for the preparation of compounds
of formula (V) are provided. In one embodiment of the invention, an
azidoerythromycin 9-oxime described above is subjected to a Beckmann
rearrangement as illustrated in Scheme 11.
SCHEME 11
N
Ra
TsCI . O ~ ~ NM~ NaBH4
H~ MeOH
N~ NaHC03 N ~'~~~ O ,~~ ~~~0 O
acetone/water O~''O R6
~~OH
HN
R4 OH
OH ~~'O~~ HCHO ~ e2
HC02H N
O~ ~''O R6
~~OH
Treatment of the azidoerythromycin 9-oxime with a sulfonyl chloride, for
example benzenesulfonyl chloride or p-toluenesulfonyl chloride (TsCl) in the
presence of sodium bicarbonate, yields the 6,9-cyclic iminoether as described
for the
preparation of azalides derived from erythromycin A in Kobrehel et al., "11-
Aza-10-
deoxo-10-dihydroerythromycin A and derivatives thereof as well as a process
for their
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-41-
production," U.S. Patent No. 4,328,334, incorporated herein by reference. The
oximinoether is then reduced, for example using sodium borohydride, under
conditions wherein the azide group remains intact. The resulting azalide is
then
alkylated, for example methylated using formaldehyde and formic acid as
described
for the production of azithromycin in Kobrehel & I~jokic, U.S. Patent No.
4,517,359,
incorporated herein by reference, to produce the azidoazalide.
In another embodiment of the invention, the azidoazalide is converted into an
embodiment of the compounds of formula (V) as illustrated in Scheme 12.
According
to this embodiment, the 2'-OH is protected, for example as the acetate or
benzoate,
and the azide is reduced by treatment with a phosphine, for example
trimethylphosphine or triphenylphosphine, and the amide is formed by
condensation
with carboxylic acid R1COOH as described above. Finally, the 2'-protecting
group is
removed by methanolysis.
S CHEME 12
Rs
".. ~ OH 1. Me3P
Ac2O OH ~~~ Ac
N ~,,. > " . ~ >
N ~"... O .. ,,O O 2. R1 C02H,
'''O R6 EDCI, HOBt
~OH
~OH
Rs Rs
'N
R4 " OH
O "... pH ... ~ c NM~ MeOH > O~ "..
R1~N~"~ O ... ..,,0 R1J''N~"
H H
~~''O R6
~OH ~OH
In another embodiment, the azidoazalide is converted into an embodiment of the
compounds of formula (V) by protecting the 2'-OH as the acetate or benzoate,
reducing the azide to the amine by hydrogenolysis and forming the amide by
condensation of the aminoazalide with R1COOH as described above.
Methods of Use
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-42-
This invention further provides a method of treating bacterial infections, or
enhancing the activity of other anti-bacterial agents, in warm-blooded
animals, which
comprises administering to the animals a compound of the invention alone or in
admixture with a diluent or in the form of a medicament according to the
invention.
When the compounds are employed for the above utility, they may be
combined with one or more pharmaceutically acceptable carriers, e.g.,
solvents,
diluents, and the like, and may be administered orally in such forms as
tablets,
capsules, dispersible powders, granules, or suspensions containing for
example, from
about 0.5% to 5% of suspending agent, syrups containing, for example, from
about
10% to 50% of sugar, and elixers containing, for example, from about 20% to
50%
ethanol, and the like, or parenterally in the form of sterile injectable
solutions or
suspensions containing from about 0.5% to 5% suspending agent in an isotonic
medium. These pharmaceutical preparations may contain, for example, from about
0.5% up to about 90% of the active ingredient in combination with the carrier,
more
usually between 5% and 60% by weight.
Compositions for topical application may take the form of liquids, creams or
gels, containing a therapeutically effective concentration of a compound of
the
invention admixed with a dermatologically acceptable carrier.
In preparing the compositions in oral dosage form, any of the usual
pharmaceutical media may be employed. Solid carriers include starch, lactose,
dicalcium phosphate, microcrystalline cellulose, sucrose, and kaolin, while
liquid
carriers include sterile water, polyethylene glycols, non-ionic surfactants
and edible
oils such as corn, peanut and sesame oils, as are appropriate to the nature of
the active
ingredient and the particular form of administration. Adjuvants customarily
employed in the preparation of pharmaceutical compositions may be
advantageously
included, such as flavoring agents, coloring agents, preserving agents, and
antioxidants, for example, vitamin E, ascorbic acid, BHT and BHA.
The active compounds may also be administered parenterally or
intraperitoneally. Solutions or suspensions of these active compounds as a
free base
or pharmacologically acceptable salt can be prepared in water suitably mixed
with' a
surfactant such as hydroxypropyl-cellulose. Dispersions can also be prepared
in
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-43-
glycerol, liquid polyethylene glycols and mixtures thereof in oils. Under
ordinary
conditions of storage and use, these preparations may contain a preservative
to
prevent the growth of microorganisms.
The pharmaceutical forms suitable for injectable use include sterile aqueous
solutions or dispersions and sterile powders for the extemporaneous
preparation of
sterile injectable solutions or dispersions. In all cases, the form must be
sterile and
must be fluid to the extent that easy syringability exists. It must be stable
under the
conditions of manufacture and storage and must be preserved against the
contaminating action of microorganisms such as bacteria and fungi. The carrier
can
be a solvent or dispersion medium containing, for example, water, ethanol,
polyol
(e.g., glycerol, propylene glycol, and liquid polyethylene glycol), suitable
mixtures
thereof, and vegetable oils.
The effective dosage of active ingredient employed may vary depending on
the particular compound employed, the mode of administration and the severity
of the
condition being treated. However, in general, satisfactory results are
obtained when
the compounds of the invention are administered at a daily dosage of from
about 0.1
mg/kg to about 400 mglkg of animal body weight, preferably given once a day,
or in
divided doses two to four times a day, or in sustained release form. For most
large
mammals, including humans, the total daily dosage is from about 0.07 g to 7.0
g,
preferably from about 100 mg to 1000 mg. Dosage forms suitable for internal
use
comprise from about 100 mg to 500 mg of the active compound in intimate
admixture
with a solid or liquid pharmaceutically acceptable carrier. This dosage
regiment may
be adjusted to provide the optimal therapeutic response. For example, several
divided
doses may be administered daily or the dose may be proportionally reduced as
indicated by the exigencies of the therapeutic situation.
The production of the above-mentioned pharmaceutical compositions and
medicaments is carried out by any method known in the art, for example, by
mixing
the active ingredients) with the diluent(s) to form a pharmaceutical
composition (e.g.,
a granulate) and then forming the composition into the medicament (e.g.,
tablets).
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-44-
A detailed description of the invention having been provided above, the
following examples are given for the purpose of illustrating the present
invention and
shall not be construed as being a limitation on the scope of the invention or
claims.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-45-
EXAMPLE 1
(~)-(2S 3R'~)-5-chloro-3-hydroxy-2-methylpentanoate N-propionylcysteamine
thioester
H
H
C SAN
O
3-Chloropropanal
Anhydrous hydrogen chloride was bubbled into a stirred solution of freshly
distilled acrolein (11.2 g) and dicinnamylacetone (5 mg) in 100 mL of CH2C12
at 0 °C
until a red color persisted. IH-NMR analysis of a 20 uL-aliquot diluted into
500 uL of
CDC13 showed 95% conversion to 3-chloropropanal, and integration relative to
the
CH2C12 peak showed the reaction solution to contain 1.8 M 3-chloropropanal.
(+_)-3-f(2S*,3R~)-5-chloro-3-h droxy-2-meth~pentanoyll-~-benzoxazolone
A vigorously stirred solution of 3-propionyl-2-benzoxazolone (19.1 g) in 200
mL of CH2C12 at 0 °C was treated with titanium tetrachloride (12.0 mL)
followed by
slow addition of triethylamine (16.7 mL). After stirring for 30 minutes, the
dark red
solution was treated with 60 mL of 1.8 M 3-chloropropanal in CHZCl2 for 30
minutes,
then quenched by addition of 200 mL of 2 N HCI. The phases were separated, and
the organic phase was filtered through a pad of silica gel. The aqueous phase
was
extracted with ether, and the extract was used to wash the pad of silica. The
combined silica eluate was concentrated under reduced pressure, resulting in
crystallization. The white crystals were collected by vacuum filtration and
dried to
provide 19.9 g of product; mp = 116 -117 °C.
(~)-(2S*.3R*)-5-chloro-3-hey-2-meth~pentanoate N-propion~ysteamine
thioester
A 4.37 M solution of sodium methoxide in methanol (66.54 mL) was added to
a stirred solution of N,S-dipropionylcysteamine (55.04 g) in 224.5 mL of
methanol
under inert atmosphere. After 25 minutes, glacial acetic acid (13.32 mL) was
added
followed by solid (~)-3-[(2S*,3R*)-5-chloro-3-hydroxy-2-methylpentanoyl]-2-
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-46-
benzoxazolone (75.0 g). After an additional 10 minutes, glacial acetic acid
(9.38 mL)
was added and the mixture was concentrated under reduced pressure. The residue
was diluted with 1,2-dichloroethane and reconcentrated under reduced pressure.
The
residue was dissolved in ethyl acetate and filtered through silica gel,
washing the
silica with additional ethyl acetate. The eluate was concentrated and the
residue was
chromatographed on 500 g of silica gel, eluting with a gradient from 20% ethyl
acetate in CH2Clz to 100% ethyl acetate. The product-containing fractions were
pooled and evaporated. The residue was triturated with hexanes, concentrated,
and
allowed to crystallize, yielding 53.0 g of pure product. 13C-NMR (CDCl3, 100
MHz):
8 203.4, 174.5, 69.0, 53.5, 41.8, 38.9, 36.9, 29.6, 28.8, 11.5, 9.8.
EXAMPLE 2
H
A seed culture of Streptonayces coelicolor CH999/pJRJ2 was made by
inoculating 1 mL of frozen mycelium into a 250 mL baffled flask containing 50
mL
of FKA medium (corn starch, 45 g/L; corn steep liquor, 10 g/L; dried,
inactivated
brewers yeast, 10 g/L; and CaC03, 1 g/L), 0.050 mL of 50 mg/mL thiostrepton in
DMSO (filter sterilized), and 0.500 mL of 50% Antifoam B. The flask was
incubated
at 30 °C with shaking at 175 rpm for 48 hours (Innova floor shaker).
The culture was
transferred into a 2.8-L baffled flask containing 500 mL of FKA medium, 0.500
mL
of 50 mg/mL thiostrepton in DMSO, and 5 mL of 50% Antifoam B, and the flask
was
incubated at 30 °C with shaking at 175 rpm for 48 hours.
A 10-L stirred tank bioreactor (B. Braun) was autoclaved, filled with 5 L of
sterile FKA medium and 5 mL of 50 mg/mL thiostrepton in DMSO, and then
inoculated with 500 mL (10%) of seed culture. The bioreactor was run for 24
hours at
15-chloro-6-dEB
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-47-
30 °C with stirring at 600 rpm, sparged with air at 1.33 LPM, and the
pH was
maintained at 6.50 by automated addition of 2.5 N NaOH and 2.5 N H2S04.
Three liters of the above culture was used to inoculate a 100-L bioreactor
containing 55 L of sterile FKA medium with 2 g/L Tastone 310 added prior to
sterilization. The fermentor agitation rate was set at a tip speed of 2.5 m/s,
the
temperature was maintained at 30°C, the pH was controlled at pH 6.5 by
automated
addition of 2.5 N NaOH and 2.5 N H2S04, and the airflow was set at 0.4 vvm.
Foaming was controlled by automated addition of 50% Antifoam B. During the
fermentation, the dissolved oxygen was maintained at >_ 50% air saturation by
cascade control using the agitation rate (tip speed of 2.5-3.0 m/s) and back
pressure
(0.1-0.4 bar) in that order. After 24 hours post-inoculation, a 400 g/L
solution of (~)-
(2S*,3R*)-5-chloro-3-hydroxy-2-methylpentanoate N-propionylcysteamine
thioester
in DMSO was added to a final concentration of 1 g/L. Titers of 15-chloro-6-
deoxyerythronolide B were monitored by HPLC, and the culture was harvested by
centrifugation when a maximum titer was reached.
The 15-chloro-6-deoxyerythronolide B was isolated by solid phase extraction.
The broth was clarified by centrifugation and loaded onto a column containing
HP-20
resin (Rohm and Haas) at a concentration of 1 L resin/20 g 15-chloro-6-
deoxyerythronolide B. The column was then equilibrated with 5 column volumes
of
water at a flow rate of 2-4 mL/cm2-min. The loaded resin was washed with 2
column
volumes of water followed by 2 column volumes of 30% methanol in water. The 15-
chloro-6-deoxyerythronolide B was eluted from the resin with methanol. The
fractions containing 15-chloro-6-deoxy-erythronolide B were identified by HPLC
with ELSD detection.
The methanol fractions containing 15-chloro-6-deoxyerythronolide B were
pooled, and the volatiles were removed under reduced pressure. The dried
solids
were extracted with 3-5 L of methanol and filtered to yield a solution
containing 6-10
mg/rnL 15-chloro-6-deoxyerythronolide B, which was diluted with an equal
volume
of water. This solution was loaded onto a column of HP20SS (1 L resin/20 g of
15-
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-48-
chloro-6-deoxyerythronolide B), which was then washed with 2 column volumes of
50% aqueous methanol. The 15-chloro-6-deoxyerythronolide B was then eluted
with
70% methanol in water, and the fractions were analyzed by HPLC. Product-
containing fractions were pooled and evaporated to dryness to yield 19.07 g of
15-
chloro-6-deoxyerythronolide B of 88% purity. 13C-NMR (CDCI~, 100 MHz): 8
213.7,
178.0, 79.3, 76.3, 71.6, 70.7, 43.8, 43.2, 41.0 (2C), 39.5, 37.6, 37.5, 35.4,
35.0, 16.6,
14.6, 13.3, 9.4, 6.9, 6.2.
EXAMPLE 3
15-azido-6-dEB
N ~ H
A mixture of 15-chloro-6-dEB (19.07 g, ca. 88% pure), sodium iodide (5.95
g), and sodium azide (10.31 g) in 60 mL of dimethylsulfoxide was heated at 50
°C for
3 days. The mixture was cooled to ambient temperature and diluted with ethyl
acetate. The solution was washed with water, and the aqueous phase was
extracted
with ethyl acetate. The combined organic phases were washed with brine, dried
over
MgS~4, filtered, and evaporated. The residue was purified by silica gel
chromatography using ethyl acetate/hexanes, yielding 13.96 g of 15-azido-6-
dEB.
isC-NMR (CDC13, 100 MHz): 8 213.6, 178.0, 79.3, 76.4, 71.8, 70.7, 48.5, 43.9,
43.2,
41.2, 39.6, 37.7, 37.5, 35.6, 31.7, 16.6, 14.7, 13.4, 9.3, 6.9, 6.3.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-49-
EXAMPLE 4
~M e2
A seed culture of Sacclzaropolyspora erytlzraea K39-14V was made by
inoculating a 1 mL aliquot of frozen mycelium into each of three 250 mL
baffled
flasks containing 50 mL of Vl medium (corn starch, 16 g/L; corn dextrin, 10
g/L;
soya meal flour, 15 g/L; corn steep liquor, 5 glL; soy bean oil, 6 g/L,;
sodium chloride,
2.5 g/L; ammonium sulfate, 1 g/L; and CaC03, 4 g/L) and 0.100 mL of Antifoam
B.
The flasks were incubated at 34 °C with shaking at 175 rpm for 48 hours
(Innova floor
shaker). Each culture was transferred into a 2.8-L baffled flask containing
500 mL of
Vl medium and 1 mL of Antifoam B, and the flasks were incubated at 34
°C with
shaking at 175 rpm for 48 hours.
Three 10-L stirred tank bioreactors (B. Braun) were autoclaved, and each
filled with 10 L of sterile 50% F1 medium (corn starch, 17.5 g/L; corn dextrin
(type
3), 16 g/L; soy meal flour, 16.5 g/L; calcium carbonate, 4 g/L, corn steep
liquor, 6
g/L; soy bean oil, 3 glL; sodium chloride 2.5 g/L; and ammonium sulfate, 1
g/L). The
fermentor agitation rate was set at a tip speed of 2-4 m/s, the pH was
controlled at pH
7.0 by automated addition of 2.5 N NaOH and 2.5 N H2S04, the temperature was
maintained at 34°C, and the airflow was set at 0.15 vvm. Foaming was
controlled by
automated addition of 50% Antifoam B. Each fermentor was inoculated with a 500-
mL seed culture prepared above. During the fermentation, the dissolved oxygen
was
maintained at >_ 80% air saturation by cascade control using agitation rate
(tip speed
of 2-4 m/s)airflow (0.15-0.5 vvm), and oxygen enrichment in that order. After
24
hours post-inoculation, a continuous 2 g/L/day dextrin feed (150 g/L dextrin
in
deionized water) was initiated, and a 4% w/v solution of 15-azido-6-
15-azidoerythromycin A
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-50-
deoxyerythronolide B was added to a final concentration of 250-300 mg/L of 15-
azido-6-deoxyerythronolide B. Samples were analyzed by HPLC every 12 hours.
After cessation of 15-azidoerythromycin production (ca. 60 hours), the culture
was
harvested by centrifugation.
The 15-azido-erythromycin A was isolated by solid phase extraction. The
broth was adjusted to pH 9 using 2.5 N NaOH, clarified by centrifugation, and
loaded
onto a column containing HP-20 resin (Rohm and Haas) at a concentration of 1 L
resin/20 g 15-azidoerythromycin A. The column was then equilibrated with 5
column
volumes of water at a flow rate of 2-4 mL/cm2-min. The loaded resin was washed
with 2 column volumes of water. The 15-azido-erythromycin A was eluted from
the
resin with 5 column volumes of methanol. The fractions containing 15-azido-
erythromycin A were identified by HPLC, pooled, and the volatiles were removed
under reduced pressure. The dried solids were mixed with 800 mL of acetone and
3.2-L of hexane for 20 minutes. The mixture was then filtered using a #4
Whatman
filter paper. The solids were extracted twice in this manner, and the
filtrates were
combined and evaporated.
The crude product was dissolved in methanol and diluted with an equal
volume of water. This solution was loaded onto a column of HP20SS (1 L
resin/20 g
of 15-azido-erythromycin A), which was then washed successively with 1 column
volume of 50% aqueous methanol, 3 column volumes of 3:2 methanol/water, 3
column volumes of 7:3 methanol/water, 10 column volumes of 4:1 methanol/water,
and finally 5 column volumes of 100% methanol. The fractions were analyzed by
HPLC. Product-containing fractions were pooled and evaporated to dryness to
yield
13 g of 15-azidoerythromycin A of 95% purity. 13C-NMR (CI~C13, 100 MHz): b
221.9, 175.4, 103.2, 96.3, 83.3, 79.8, 78.0, 75.0, 74.4, 73.0, 72.6, 70.9,
69.0, 68.6,
65.6, 65.6, 49.5, 49.1, 45.2, 44.7, 40.3 (2C), 39.6, 38.5, 37.7, 34.9, 28.6,
27.8, 27.0,
21.5, 21.4, 18.7, 18.2, 16.2, 15.6, 12.0, 9.1.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-51-
EXAMPLE 5
2'-O-acetyl-15-azidoerythromycin A
e2
A solution of 15-azidoerythromycin A in CH2C12 was treated with acetic
anhydride for 1 hour at ambient temperature, then the solution was
concentrated to
dryness. The residue was chromatographed on silica gel using acetone/hexanes +
1 %
Et3N. 13C-NMR (CDCl3, 100 MHz): ~ 222.0, 175.3, 170.0, 100.9, 96.1, 83.3,
79.6,
77.9, 75.0, 74.4, 73.1, 72.7, 71.7, 68.6, 68.4, 65.7, 63.5, 49.4, 49.1, 45.1,
44.6, 40.7
(2C), 39.4, 38.1, 37.7, 34.9, 30.3, 29.7, 27.8, 27.0, 21.5, 21.2, 18.6, 18.1,
16.3, 15.5,
12.0, 9Ø
EXAMPLE 6
General procedure for preparation of 15-amidoerythromycins
To a solution of 2'-O-acetyl-15-azidoerythromycin A (0.050 g, 0.062 mmol,
1.0 eq) in dichloromethane or tetrahydrofuran (1.0 ml) is added
trimethylphosphine
(0.31 ml of a 1M solution in THF, 0.306 mmol, 5.0 eq). The solution is stirred
at
room temperature for 45 minutes before transferring to a solution of the
carboxylic
' acid (0.092 mmol, 1.5 eq), 1-[(3-(dimethylamino)propyl]-1-ethylcarbodiimide
hydrochloride (0.019 g, 0.099 mmol, 1.6 eq) and 1-hydroxybenzotriazole (0.017
g,
0.124 mmol, 2.0 eq) in dichloromethane or tetrahydrofuran (1.0 ml) also
stirred at
room temperature for 45 minutes. The resulting solution is stirred at room
temperature for 3 to 14 hours before partitioning between ethyl acetate (10
ml) and
NaHC03 (10 ml). The aqueous phase is extracted with ethyl acetate (3 x 10 ml)
and
the combined organics further washed with brine (25 ml) before drying
(Na2SOq.),
filtering, and concentrating under reduced pressure. The residue is dissolved
in
methanol (2 ml) and stirred at 50 °C for 14 hours before concentrating
under reduced
pressure. Silica gel chromatography (0-X10 min 50% acetone-hexane, 1% Et3N;
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-52-
1020 min 60% acetone-hexane, 1% Et3N; 20-X30 min 70% acetone-hexane, 1~/0
Et3N; 30+ min 80% acetone-hexane, 1 % Et3N) yields the 15-amido compound as a
white solid.
EXAMPLE 7
15-(6-quinolinecarboxamido)erythromycin A
e2
'H
N
H
This compound was prepared according to the method of Example 6 using
quinoline-6-carboxylic acid. 13C-NMR (CDCl3, 100 MHz): ~ 222.2, 177.0, 166.7,
151.8, 149.3, 137.2, 132.6, 129.9, 127.7, 127.6, 127.3, 121.8, 103.2, 96.3,
83.1, 79.4,
78.0, 75.1, 74.4, 73.0, 72.6, 70.8, 69.0 (2C), 65.7 (2C), 49.5, 45.1, 44.8,
40.3 (2C),
40.2, 38.4, 37.9, 36.6, 34.9, 28.6, 28.4, 27.0, 21.5, 21.4, 18.7, 18.0, 16.3,
15.5, 11.9,
9Ø ES-MS: m/z 905 [M+H]+.
EXAMPLE 8
15-(4-quinolinecarboxamido)erythromycin A
I \ ~~.;,~OH ...,, Me2
~~ H(~~7~~
N / H~'' O
O ~~° Me
O~H
This compound was prepared according to the method of Example 6 using
quinoline-4-carboxylic acid. 13C-NMR (CDCl3, 100 MHz): S 221.8, 176.7, 167.4,
149.9, 148.5, 142.2, 129.9, 129.7, 127.6, 125.4, 124.6, 118.6, 103.2, 96.3,
83.2, 79.5,
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-53-
78.0, 74.9, 74.4, 72.9, 72.6, 70.8, 69.0, 68.9, 65.6, 49.5, 44.7, 40.3, 39.9,
38.4, 37.9,
36.6, 34.9, 29.2, 28.6, 26.8, 21.5, 18.7, 18.0, 16.3, 15.5, 11.9, 9Ø ES-MS:
fnlz 905
[M+H]+, 747, 453.
EXAMPLE 9
15-(3-indoleacetamido)erythromycin A
H
N
°2
/ ~ ~ N~
H
This compound was prepared according to the method of Example 6 using 3-
indoleacetic acid. 13C-1VMR (CDCl3, 100 MHz): 8 222.0, 176.1, 171.6, 136.5,
127.1,
124.1, 122.3, 119.8, 118.7, 111.4, 108.9, 103.2, 96.3, 83.3, 79.5, 76.7, 75.0,
74.3,
72.6, 72.5, 70.8, 69.0, 68.8, 65.7, 65.5, 49.5, 45.1, 44.7, 40.2, 39.9, 38.0,
36.0, 34.9,
33.4, 29.7, 28.5, 28.2, 26.9, 21.5, 21.4, 18.6, 18.1, 16.4, 15.4, 11.9, 9Ø
ES-MS: m/z
907 [M+H]+, 749, 454.
EXAMPLE 10
15-(phenylacetamido)erythromycin A
H OH
,,.. OH ,..,, Me2
,~, HO
N O--.~7 C)
H
Me
O~H
This compound was prepared according to the method of Example 6 using
phenylacetic acid. 13C-NMR (CDCl3, 100 MHz): ~ 176.1, 170.9, 135.0, 129.4,
128.9,
127.2, 103.2, 96.3, 83.2, 79.6, 78.0, 75.1, 74.3, 72.7, 72.6, 70.8, 69.0,
68.8, 65.6, 49.5,
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-54-
45.2, 44.7, 43.8, 40.3, 38.5, 37.9, 36.3, 34.9, 29.7, 28.7, 28.4, 26.9, 21.5,
21.4, 18.6,
18.2, 16.2, 15.5, 11.9, 9.1. ES-MS: m/z 868 [M+H]+.
EXAMPLE 11
15-(phenylpropionamido)erythromycin A
r OH ""~ Me2
H(
H
O ~' Me
O~H
This compound was prepared according to the method of Example 6 using
hydrocinnamic acid. 13C-NMR (CDC13, 100 MHz): S 222.0, 176.5, 172.0, 141.0,
128.5, 128.4, 126.1, 103.2, 96.3, 83.2, 79.5, 78.0, 75.1, 74.3, 72.8, 72.6,
70.8, 69.9,
68.9, 65.6, 49.5, 45.2, 44.7, 40.3, 40.0, 38.4, 37.9, 35.8, 34.9, 31.7, 28.6,
28.4, 26.9,
21.5, 21.4, 18.6, 18.1, 16.2, 15.5, 11.9, 9Ø ES-MS: m/z 882 [M+H]+.
EXAMPLE 12
15-(2-quinoxalinecarboxamido)erythromycin A
H
This compound was prepared according to the method of Example 6 using 2-
quinazolinecarboxylic acid. 13C-NMR (CDC13, 100 MHz): b 222.1, 175.9, 163.2,
143.9, 143.5, 140.3, 131.5, 130.7, 129.7, 129.5, 103.0, 96.4, 83.5, 79.7,
77.9, 75.0,
74.4, 73.1, 72.6, 70.8, 68.8, 65.7, 65.6, 60.4, 49.5, 45.1, 44.8, 40.3, 39.8,
38.5, 37.9,
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-55-
36.7, 34.9, 29.7, 29.2, 28.7, 26.9, 22.7, 21.5, 21.3, 18.6, 18.2, 16.3, 15.7,
11.9, 9.2.
ES-MS: m/z 906 [M+H]+.
EXAMPLE 13
15-(4-(4-chlorophenyl)benzamido)erythromycin A
"", NMe2
.,,.
' Me
~~H
This compound was prepared according to the method of Example 6 using 4-
(4-chlorophenyl)benzoic acid. 13C-NMR (CDCl3, 100 MHz): S 168.8, 142.7, 134.1,
133.6, 129.0, 128.4, 127.6, 127.0, 103.1, 96.3, 83.1, 79.4, 78.0, 75.2, 74.3,
72.9, 70.8,
69.1, 65.5, 60.4, 49.5, 45.1, 44.8, 40.2, 38.4, 37.9, 36.3, 34.9, 29.7, 28.6,
28.3, 27.0,
21.5, 21.4, 21.0, 18.6, 18.0, 16.3, 15.5, 11.9, 9Ø ES-MS: m/z 964 [M+H]+.
EXAMPLE 14
15-(5-phenylpentanamido)erythromycin A
JM e2
This compound was prepared according to the method of Example 6 using 5-
phenylpentanoic acid. 13C-NMR (CDC13, 100 MHz): 8 178.7, 172.8, 151.7, 142.3,
128.4, 128.3, 125.7, 102.9, 101.8, 94.5, 85.7, 79.8, 78.1, 75.9, 75.0, 74.8,
73.0, 70.7,
70.3, 68.9, 65.9, 49.6, 44.4, 43.6, 40.3, 36.7, 35.9, 34.6, 31.1, 30.5, 28.6,
28.2, 26.5,
25.4, 21.6, 21.4, 18.2, 16.4, 14.7, 13.0, 11.8. ES-MS: fyzlz 892 [M+H]+.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-56-
EXAMPLE 15
15-(4-(4-chlorophenyl)phenylacetamido)erythromycin A
C
NMe2
Ni\
H
H
This compound was prepared according to the method of Example 6 using 4-
(4-chlorophenyl)phenylacetic acid. 13C-NMR (CDC13, 100 MHz): b 222.8, 176.2,
170.8, 139.2, 138.8, 134.5, 133.4, 129.9, 128.9, 128.3, 127.4, 103.1, 96.3,
83.2, 79.5,
78.0, 75.0, 74.3, 72.6, 70.8, 69.0, 68.9, 65.6, 65.6, 49.5, 45.1, 44.7, 43.4,
40.2, 39.9,
38.4, 36.2, 34.8, 29.7, 28.3, 26.9, 21.5, 21.4, 18.6, 18.1, 16.2, 15.5, 11.9,
9Ø ES-MS:
mlz 978 [M+H]+.
EXAMPLE 16
15-((1,2-benzisoxazol-3-yl)acetamido)erythromycin A
O e2
N~
H
This compound was prepared according to the method of Example 6 using 2-
(1,2-benzisoxaol-3-yl)acetic acid. 13C-NMR (CDCl3, 100 MHz): 8 176.3,166.9,
163.2, 153.7, 130.1, 123.7, 122.1, 121.3, 109.8, 103.2, 96.3, 79.6, 78.0,
74.9, 74.4,
72.9, 72.6, 69.0, 68.8, 65.5, 49.5, 45.1, 44.7, 40.3, 39.9, 38.5, 38.0, 36.6,
34.9, 33.6,
29.7, 28.6, 28.4, 26.8, 21.5, 21.4, 18.6, 18.2, 16.2, 15.5, 11.9, 9.1. ES-MS:
m/z 909
[M+H]+.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-57-
EXAMPLE 17
15-(2-(2-furyl)phenylacetamido)erythromycin A
H H
O ~~~.. OOH ,..~" Me2
i,,~ ~
H
O ~h~ Me
OH
This compound was prepared according to the method of Example 6 using 2-
(2-furyl)phenylacetic acid. 13C-NMR (100 MHz, CDC13): 8 222.1, 175.8, 170.9,
152.9, 142.5, 131.9, 131.5, 130.8, 128.3, 128.3, 127.7, 111.5, 108.8, 103.1,
96.3, 83.2,
79.5, 77.9, 75.1, 74.2, 72.7, 72.6, 70.8, 69.0, 68.8, 65.7, 65.6, 49.5, 45.1,
44.6, 42.7,
40.2, 39.9, 38.5, 37.9, 36.3, 34.9, 29.7, 28.7, 28.4, 27.0, 21.5, 21.4, 18.6,
18.1, 16.1,
15.5, 11.8, 9.1. ES-MS: m/z 934 [M+H]+.
EXAMPLE 18
15-(3-(2-furyl)phenylacetamido)erythromycin A
H
This compound was prepared according to the method of Example 6 using 3-
(2-furyl)phenylacetic acid. 13C-NMR (CDCl3, 100 MHz): ~ 222.1, 176.1, 170.7,
153.7, 142.1, 134.0, 131.9, 131.1, 129.8, 124.3, 111.6, 105.0, 103.1, 96.3,
83.1, 79.4,
77.9, 77.2, 75.1, 74.2, 72.6, 70.8, 69.0, 68.9, 65.6, 49.5, 45.1, 44.7, 43.6,
43.4, 40.3,
38.4, 37.9, 36.2, 34.8, 30.4, 29.7, 28.6, 28.3, 27.0, 21.5, 21.4, 18.6, 18.1,
16.2, 15.4,
11.8, 9Ø ES-MS: rnlz 934 [M+H]+.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-5 ~-
EXAMPLE 19
15-(4-(2-furyl)phenylacetamido)erythromycin A
~)
OH
This compound was prepared according to the method of Example 6 using 3-
(2-furyl)phenylacetic acid.
EXAMPLE 20
15-(4-phenylbutyramido)erythromycin A
N
H
~~H
This compound was prepared according to the method of Example 6 using 4-
phenylbutyric acid.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
EXAMPLE 22
15-(3-(3-quinoline)propionamido)erythromycin A
-59-
EXAMPLE 21
15-(benzyloxyacetamido)erythromycin A
HO OH
/ ".... OOH ".,, NMe2
", HO
'O OMe
~OH
This compound was prepared according to the method of Example 6 using
benzyloxyacetic acid.
,,'
H OH
OH ..." M
.,
HO
~ N~...., ,, ..~0
H _
NJ O ~' Me
O H
e2
This compound was prepared according to the method of Example 6 using 3-
(3-quinolyl)propionic acid.
EXAMPLE 23
15-((3,4,5-trimethoxyphenyl)oxalamido)erythromycin A
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-60-
M
e2
M
H
This compound was prepared according to the method of Example 6 using
(3,4,5-trimethoxybenzoyl)formic acid.
EXAMPLE 24
15-((4-methyl-5-phenyloxazol-2-yl)acetamido)erythromycin A
~H
"'' NMe2
~Q O O
~'O OMe
OOH
This compound was prepared according to the method of Example 6 using (4-
methyl-5-phenyloxazol-2-yl)acetic acid.
EXAMPLE 25
15-(3-quinolinecarboxamido)erythromycin A
NMe2
N~
J H
N
H
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-61-
This compound was prepared according to the method of Example 6 using
quinoline-3-carboxylic acid.
EXAMPLE 26
15-(3-pyridinecarboxamido)erythromycin A
NM e2
H
This compound was prepared according to the method of Example 6 using
pyridine-3-carboxylic acid.
EXAMPLE 27
15-(5-indolecarboxamido)erythromycin A
e2
w _
N
H
This compound was prepared according to the method of Example 6 using
indole-5-carboxylic acid.
~~H
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-62-
EXAMPLE 28
15-(3-indolecarboxamido)erythromycin A
e2
HN
H
This compound was prepared according to the method of Example 6 using
indole-3-carboxylic acid.
EXAMPLE 29
This compound was prepared according to the method of Example 6 using 4-
(2-furyl)benzoic acid.
15-(4-(2-furyl)benzamido)erythromycin A
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-63-
EXAMPLE 30
15-(4-biphenylacetamido)erythromycin A
i
H OH
/ ~~".. OH ...,, Me2
.., HC~
N~''.,. O ,' .4~0-
Me
O~H
This compound was prepared according to the method of Example 6 using 4-
biphenylacetic acid.
EXAMPLE 31
15-(3-(3-furyl)phenylacetamido)erythromycin A
This compound was prepared according to the method of Example 6 using 3-
(3-furyl)phenylacetic acid.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-64-
EXAMPLE 32
15-(3-(3-thienyl)phenylacetamido)erythromycin A
'2
This compound was prepared according to the method of Example 6 using 3-
(3-thienyl)phenylacetic acid.
EXAMPLE 33
15-(3-(2-thienyl)phenylacetamido)erythromycin A
S /
JMe2
N~
H
This compound was prepared according to the method of Example 6 using 3-
(2-thienyl)phenylacetic acid.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-65-
EXAMPLE 34
15-(2-(2-thienyl)phenylacetamido)erythromycin A
HO OH
'~~~., ~H ",,, NMe2
O
v
H
O ~~'O OMe
O OH
This compound was prepared according to the method of Example 6 using 2-
(2-thienyl)phenylacetic acid.
EXAMPLE 35
15-(3-(2-pyrrolyl)phenylacetamido)erythromycin A
M e2
IC~
H
This compound was prepared according to the method of Example 6 using 3-
(2-pyrrolyl)phenylacetic acid.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-66-
EXAMPLE 36
15-(3-(4-pyridyl)phenylacetamido)erythromycin A
e2
This compound is prepared according to the method of Example 6 using 3-(4-
pyridyl)phenylacetic acid.
EXAMPLE 37
15-(3-(2-furyl)pyridyl-5-acetamido)erythromycin A
H OH
".~w ~OH
~,,, .,~0 O
N~~~~ ,_~~C~e2
N
H
O ~~" Me
O~H
This compound is prepared according to the method of Example 6 using 3-(2-
furyl)pyridyl-5-acetic acid.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-67-
EXAMPLE 38
15-(3-(2-thienyl)pyridyl-5-acetamido)erythromycin A
OH
~OH ...,~ NMe2
O
O OMe
~~OH
This compound is prepared according to the method of Example 6 using 3-(2-
thienyl)-pyridyl-5-acetic acid.
EXAMPLE 39
15-(3-(2-pyrrolyl)pyridyl-5-acetamido)erythromycin A
...,, Me2
' O OH~''O ~~~/~J~
Me
O~H
This compound is prepared according to the method of Example 6 using 3-(2-
pyrrolyl)-pyridyl-5-acetic acid.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-68-
EXAMPLE 40
e2
"~O H
This compound is prepared according to the method of Example 6 using 5-
phenylthienyl-2-acetic acid.
EXAMPLE 41
e2
O H
This compound is prepared according to the method of Example 6 using 5-(2-
pyridyl)-thienyl-2-acetic acid.
15-(5-phenylthienyl-2-acetamido)erythromycin A
15-(5-(2-pyridyl)thienyl-2-acetamido)erythromycin A
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-69-
EXAMPLE 42
15-(5-(3-isoxazolyl)thienyl-2-acetamido)erythromycin A
O.N ~ ~ O ,l/e2
S
H
This compound is prepared according to the method of Example 6 using 5-(3-
isoxazolyl)-thienyl-2-acetic acid.
EXAMPLE 43
15-(3-(5-phenylthien-2-yl)propionamido)erythromycin A
This compound was prepared according to the method of Example 6 using 3-
(5-phenylthien-2-yl)propionic acid.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-70-
EXAMPLE 44
15-(3-(5-(2-pyridyl)thien-2-yl)propionamido)erythromycin A
NMe2
H
This compound is prepared according to the method of Example 6 using 3-(5-
(2-pyridyl)thien-2-yl)propionic acid.
EXAMPLE 45
15-(3-(5-(3-isoxazolyl)thien-2-yl)propionamido)erythromycin A
O' \ \ I H~
This compound is prepared according to the method of Example 6 using 3-(5-
(3-isoxazolyl)-thien-2-yl)propionic acid.
EXAMPLE 46
General Preparation of Biaryl Carboxylic Acids
1. Biaryl Carboxylic Ester
To a solution of the haloaryl carboxylic ester (1.0 mmol) in tetrahydrofuran
(15 mL) is added the boronic acid (1.3 mmol) and 2 M aq. sodium carbonate (1
mL).
The solution is degassed and sonicated three times before adding
tetrakis(triphenylphosphine)-palladium (0.1 mmol). The mixture is degassed and
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-71-
sonicated a further three times before stirring at 50°C for 14 hours.
After cooling to
room temperature the solution is concentrated under reduced pressure. The
product
ester is purified by column chromatography.
2. Biaryl Carboxylic Acid
To a solution of the biaryl carboxylic ester (1.0 mmol) in acetone (15 mL) is
added 30°70 aq. potassium hydroxide (2.5 mL). The mixture is stirred at
room
temperature for 16 hours before diluting with 1 N NaOH (50 ml), and washing
with
CH2C12 (75 ml). The aqueous phase is acidified with 1 N HCl (75 ml) and the
organics extracted with CH~C12 (3 x 100 ml). The extract is dried (Na2SOq.),
filtered,
and concentrated under reduced pressure to yield the biaryl carboxylic acid.
EXAMPLE 47
3-(2-furyl)phenylacetic acid
COO H
1. Methyl 3-(2-fur~phenylacetate
To a solution of bromophenylacetic acid methyl ester (0.075 g, 0.328 mmol,
1.0 eq) in tetrahydrofuran (5.0 ml) was added 2-furanboronic acid (0.048 g,
0.426
mmol, 1.3 eq) and sodium carbonate (0.328 ml of a 2M soln in H20). The
solution
was degassed and sonicated three times before adding
tetrakis(triphenylphosphine)palladium (0.038 g, 0.033 mmol, 0.1 eq). The
mixture
was degassed and sonicated a further three times before stirring at
50°C for 14 hours.
After cooling to room temperature the solution was concentrated under reduced
pressure. Column chromatography (silica, 20% EtOAc/hexane) yielded 3-
furanylphenylacetic acid methyl ester (0.047 g, 67°70) as a white
solid; 1H-NMR
(CDC13, 400 MHz): b 7.60 (1H, d, J = 1.6 Hz), 7.47 (1H, t, J = 1.0 Hz), 7.34
(1H, t, J
= 7.5 Hz), 7.19 (2H, m), 6.65 (1H, d, J = 3.6 Hz), 6.48 (1H, dd, J = 3.6, 1.6
Hz), 3.71
(3H, s), 3.66 (2H, s).
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-72-
2. 3-(2-Fur~phen~acetic acid
To a solution of 3-(2-furyl)phenylacetic acid methyl ester (0.047 g, 0.217
mmol, 1.0 eq) in acetone (3.0 ml) was added potassium hydroxide (0.5 ml of a
30%
solution in H20). The mixture was stirred at room temperature for 16 hours
before
diluting with 1 N NaOH (10 ml), and washing with CH2C12 (15 ml). The aqueous
phase was acidified with 1 N HCl (15 ml) and the organics extracted with
CH2Cl2 (3
x 20 ml). The extract was dried (Na2SOq.) and concentrated under reduced
pressure to
yield 3-(2-furyl)phenylacetic acid (0.043 g, 100%) as a white solid; 1H-NMR
(CDC13,
400 MHz): 8 7.60 (1H, br s), 7.46 (1H, t, J = 1.0 Hz), 7.34 (1H, t, J = 7.5
Hz), 7.19
(2H, m), 6.65 (1H, d, J = 3.2 Hz), 6.46 (1H, m), 3.67 (2H, s).
EXAMPLE 48
15-azidoerythromycin A 9-oxime
H
A mixture of 15-azidoerythromycin A (4.0 g) and 50% aqueous
hydroxylamine (5.0 mL) in 10 mL of isopropanol was treated with 1.6 mL of
acetic
acid and stirred at 50 °C for 15 hours. The mixture was cooled to
ambient
temperature, treated with 20 mL of sat. aq. NaHC03, and concentrated under
reduced
pressure to an aqueous slurry, which was extracted with chloroform. The
extract was
washed with sat. aq. NaHCO3 and brine, then dried over Na2S04, filtered, and
evaporated to yield 4.2 g of product. ES-MS: f12~Z 791 [M+H]+.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-73-
EXAMPLE 49
15-azidoerythromycin A 9-oxime IPCH ketal
e2
A mixture of 15-azidoerythromycin A 9-oxime (4.2 g), 1,1-
diisopropoxycyclohexane (6.8 mL), and pyridinium p-toluenesulfonate (2.45 g)
in 14
mL of dichloromethane was stirred at ambient temperature for 16 hours. The
mixture
was diluted with dichloromethane and washed with sat. aq. NaHC03 and brine,
then
dried over Na2SOø, filtered, and evaporated . The product was purified by
silica gel
chromatography (70:30 acetone/hexane + 1% Et3N) to yield 4.0 g of product. 13C-
NMR (100 MHz, CDCl3): & 174.6, 104.2, 103.0, 96.3, 83.1, 78.0, 75.3, 74.0,
73.0,
72.7, 70.9, 70.2, 68.8, 65.5, 63.4, 61.9, 49.5, 49.2, 40.3, 36.0, 35.0, 27.0,
26.7, 25.7,
25.4, 24.7, 24.3, 23.4, 22.9, 21.5, 18.6, 18.5, 16.3, 15.8, 14.4, 9.2; ES-MS:
nz/z 931
[M+H]+, 791.
b~oH
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-74-
EXAMPLE 50
2',4"-bis-O-(trimethylsilyl)-15-azidoerythromycin A 9-oxime IPCH ketal
..
~H ...n ~ MS NMe2
.'
N~~°~'. '"~O O
Me
A solution of chlorotrimethylsilane (0.08 mL) and trimethylsilylimidazole
(1.57 mL) in 4 mL of dichloromethane was added to a solution of 15-
azidoerythromycin A 9-oxime IPCH ketal (4.0 g) in 12 mL of dichloromethane.
After
5 minutes, 100 mL of ethyl acetate was added and the solution was washed with
sat.
aq. NaHC03 and brine, then dried over Na2SO4, filtered, and evaporated to give
4.2 g
of product. 13C-NMR (100 MHz, CDC13): 8 175.4, 170.5, 135.2, 104.0, 102.6,
101.0,
96.6, 94.7, 80.8, 75.4, 73.9, 73.2, 73.1, 70.2, 67.6, 65.0, 64.9, 63.1, 61.7,
49.6, 49.1,
44.4, 40.8, 35.9, 26.9, 25.6, 24.6, 24.2, 23.3, 22.9, 21.9, 21.6, 19.2, 18.3,
16.2, 14.3,
9.6, 0.9, 0.8; ES-MS: m/z 1075 [M+H]+.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-75-
EXAMPLE 51
2',4"-bis-O-(trimethylsilyl)-6-O-methyl-15-azidoerythromycin A 9-oxime IPCH
ketal
e2
N-
A mixture of 2',4"-bis-O-(trimethylsilyl)-15-azidoerythromycin A 9-oxime
IPCH ketal (4.2 g), 2 M methyl bromide in ether (3.9 mL), 7 mL of THF, and 7
mL of
DMSO was cooled on ice. A mixture of 1.0 M potassium tart-butoxide in DMSO
(7.8
mL) and DMSO (7 mL) was added by syringe pump at a rate of 4 ml/hour, and the
reaction was monitored by thin-layer chromatography (15% acetone in hexane,
pretreating with ammonia vapor). The addition was continued for 3.5 hours
before
addition of sat. aq. NaHC03 (100 mL) and extracting the mixture with ethyl
acetate.
The extract was washed sequentially with NH4Cl/NaCl, water, and brine, then
dried
over Na2S04, filtered, and evaporated to yield 4.0 g of product. 13C-NMR (100
MHz,
CDCl3): & 175.7, 169.8, 103.6, 102.5, 101.2, 96.2, 80.8, 79.1, 77.9, 73.6,
73.3, 73.1,
72.9, 69.7, 67.2, 65.1, 62.8, 61.8, 51.1, 49.7, 49.1, 40.9, 36.0, 26.5, 25.7,
25.6, 24.7,
24.5, 24.4, 23.4, 22.9, 22.2, 22.0, 20.2, 19.5, 18.6, 16.2, 15.1, 9.6, 1.0,
0.9. ES-MS:
fnlz 1089 [M+H]+, 791.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-76-
EXAMPLE 52
6-O-methyl-15-azidoerythromycin A 9-oxime
"~OH
.
HO OMe
,,., 'OH ",,, NMe2
,,, HO~
_..''' n ~-. '"~n_ _O_ i
'O OMe
~OH
A solution of 2',4"-bis-O-(trimethylsilyl)-6-O-methyl-15-azidoerythromycin
A 9-oxime IPCH ketal (4.0 g) in 50 mL of acetonitrile was treated with water
(25 mL) ,
and acetic acid (30 mL), the mixture was stirred for 16 hours at ambient
temperature,
and then concentrated under reduced pressure. The residue was concentrated
from
isopropanol and toluene to remove volatiles, then chromatographed on silica
gel (1:1
acetonelhexanes + 1°Io Et3N) to yield 2.4 g of product. 13C-NMR (100
MHz, CDC13):
8 175.4, 171.2, 169.3, 102.7, 96.0, 80.3, 78.6, 78.2, 78.0, 77.5, 73.8, 72.8,
72.6, 71.3, ,
68.6, 65.5, 65.2, 60.4, 51.1, 49.4, 49.0, 45.7, 44.9, 40.3, 39.2, 25.3, 21.4,
21.4, 21.0,
20.0, 18.6, 18.6, 16.1, 15.7, 15.0, 9.1. ES-MS: m/z 805 [M+H]+.
EXAMPLE 53
6-O-methyl-15-azidoerythromycin A
H
Y, -OH ..~~~ Me2
/[\ HO
\~V ~ ~~
Me
H
A solution of 15-azidoerythromycin A 9-oxime (2.4 g) in 25 mL of ethanol
was treated with 25 mL of water, sodium hydrosulfite (4.8 g), and formic acid
(0.77
ml) and heated to 80 °C. After 4 hours, the mixture was cooled to
ambient
temperature, adjusted to pH 10 using 6 N NaOH, and extracted with ethyl
acetate.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
_77_
The extract was washed with brine, dried over NaaS04, filtered, and evaporated
to
yield 2.4 g of product. 13C-NMR (100 MHz, CDCl3): ~ 221.3, 175.6, 102.8, 96.1,
80.7, 78.4, 78.3, 78.0, 74.0, 72.7, 72.7, 70.9, 68.8, 65.8, 65.6, 50.6, 49.5,
49.1, 45.2,
45.0, 40.2, 39.4, 37.1, 34.9, 28.6, 27.7, 21.5, 19.7, 18.7, 17.9, 16.0, 15.6,
12.3, 9Ø
ES-MS: m/z 790 [M+H]+.
EXAMPLE 54
3-O-descladinosyl-6-O-methyl-15-azidoerythromycin A
e2
A mixture of 6-O-methyl-15-azidoerythromycin A (2.0 g) and pyridinium p-
toluenesulfonate (3.82 g) in 20 mL of acetone and 5 mL of water was stirred at
50 °C
for 44 hours, then cooled to ambient temperature and treated with sat. aq.
NaHC03
and extracted with ethyl acetate. The extract was washed with brine, dried
over
Na2S04, filtered, and evaporated. The residue was chromatographed on silica
gel to
yield product (0.5 g) and unreacted starting material, which was recycled
through the
procedure. 13C-NMR (100 MHz, CDCl3): 8 220.5, 174.7, 106.4, 88.1, 78.7, 78.0,
74.0, 72.7, 70.6, 70.0, 69.4, 65.6, 49.5, 49.0, 48.8, 45.4, 44.4, 40.4, 40.2,
38.7, 35.4,
29.2, 28.2, 28.0, 21.2, 21.0, 18.7, 18.3, 17.7, 16.2, 15.1, 15.0, 12.5, 8.2.
ES-MS: m/z
632 [M+H]+.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
_78_
EXAMPLE 55
2'-O-benzoyl-3-O-descladinosyl-6-O-methyl-15-azidoerythromycin A
a2
N~
To a solution of 3-O-descladinosyl-6-O-methyl-15-azidoerythromycin A (0.6
g, 0.951 mmol, 1.0 eq) in dichloromethane (10 ml) was added benzoic anhydride
(0.32 g, 1.426 mmol, 1.5 eq) and the mixture stirred at room temperature for
14 hours.
Sat. aq. NaHC03 (30 ml) was added and the organics extracted with CH2C12 (3 x
30
ml). The combined organics were dried (Na~S04), filtered, and concentrated
under
reduced pressure to yield a pale yellow oil, which was taken on without
further
purification; ES-MS: rnlz 736 [M+H]+.
EXAMPLE 56
2'-O-benzoyl-3-des(cladinosyloxy)-3-oxo-6-O-methyl-15-azidoerythromycin A
N
N-Chlorosuccinirnide (0.191 g, 1.427 mmol, 1.5 eq) was added to a solution of
dimethyl sulfide (0.085 ml, 1.712 mmol, 1.8 eq) and dichloromethane (5 ml) at -
15
°C. After stirring at -15 °C for 15 minutes, 2'-O-benzoyl-3-O-
descladinosyl-6-O-
methyl-15-azidoerythromycin A (0.951 mmol, 1.0 eq) in dichloromethane (5 ml)
was
added dropwise and the resulting solution was stirred at -15 °C for 30
minutes before
adding triethylamine (0.132 ml, 0.951 mmol, 1.0 eq). The mixture was warmed to
room temperature over 40 minutes before diluting with EtOAc (100 ml) and
washing
with sat. aq. NaHC03 (2 x 100 ml) and brine (100 ml). The organics were dried
(Na2SOq.), filtered, and concentrated under reduced pressure. The product was
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-79-
purified by silica gel chromatography (0~ 10 min 20% acetone-hexane, 1 % Et3N;
10-X20 min 30% acetone-hexane, 1% Et3N; 20+ min 40% acetone-hexane, 1%
Et3N) to provide the product (0.5 g, 72% over two steps) as a white solid; 13C-
NMR
(100 MHz, CDC13): b 220.8, 205.0, 169.0, 165.2, 132.8, 130.4, 129.7, 128.3,
101.3,
74.2, 73.6, 71.9, 69.1, 68.9, 63.6, 53.5, 50.7, 49.5, 49.1, 46.4, 44.8, 41.5,
40.7, 39.0,
37.2, 31.3, 28.2, 27.8, 21.0, 19.4, 17.5, 16.2, 14.3, 12Ø ES-MS: m/.z 734
[M+H]+.
EXAMPLE 57
2'-O-benzoyl-3-des(cladinosyloxy)-3-oxo-6-O-methyl-11-O-methanesulfonyl-15-
azidoerythromycin A
N
3
e2
To a solution of 2'-O-benzoyl-3-des(cladinosyloxy)-3-oxo-6-O-methyl-15-
azidoerythromycin A (0.5 g, 0.683 mmol, 1.0 eq) in pyridine (7.0 eq) was added
methanesufonyl chloride (0.26 ml, 3.415 mmol, 5.0 eq) dropwise. The resulting
solution was stirred at room temperature for 16 hours before diluting with
ethyl
acetate (100 ml) and washing with sat. aq. NaHC03 (2 x 70 ml) and brine (70
ml).
The organics were dried (Na2SO4), filtered, and concentrated under reduced
pressure
to yield product as a brown solid (0.5 g) which was taken on without further
purification.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-80-
EXAMPLE 58
2'-O-benzoyl-10,11-anhydro-3-des(cladinosyloxy)-3-oxo-6-O-methyl-15-
N
3
e2
To a solution of 2'-O-benzyl-3-des(cladinosyloxy)-3-oxo-6-O-methyl-11-O-
methanesulfonyl-15-azidoerythromycin A (0.683 mmol, 1.0 eq) in acetone (7.0
ml)
was added 1,8-diazabicyclo[5.4.0]undec-7-ene (0.510 ml, 3.415 mmol, 5.0 eq)
and the
solution stirred at room temperature for 8 hours before concentrating under
reduced
pressure. Purification by silica gel chromatography (0-X10 min 25%
acetone-hexane, 1 % Et3N; 10-X20 min 30% acetone-hexane, 1 % Et3N; 2030
min 35% acetone~exane, 1% Et3N; 30+ min 40% acetone-hexane, 1% Et3N)
yielded the product (0.25 g, 50% over two steps) as a white solid; 13C-NMR
(100
MHz, CDC13): ~ 206.8, 176.4, 169.4, 165.1, 141.5, 139.2, 132.7, 130.5, 129.7,
128.3,
102.1, 78.4, 72.7, 72.0, 69.2, 63.7, 51.0, 50.4, 49.0, 47.1, 41.5, 40.7, 40.1,
38.3, 31.6,
31.2, 29.0, 28.2, 22.6, 21.5, 21.0, 18.6, 14.5, 14.2, 13.6. ES-MS: frilz 716
[M+H]+.
EXAMPLE 59
2'-O-benzoyl-11-amino-11-deoxy-3-O-des(cladinosyloxy)-3-oxo-6-O-methyl-15-
azidoerythromycin A 11,12-cyclic carbamate
n
e2
To a solution of 2'-O-benzoyl-10,11-anhydro-3-O-des(cladinosyloxy)-3-oxo-
6-O-methyl-15-azidoerythromycin A (0.246 g, 0.344 mmol, 1.0 eq) in
tetrahydrofuran
azidoerythromycin A
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-81-
(3.0 ml) at -15 °C was added sodium hydride (0.028 g of a 60% soln. in
oil, 0.688
mmol, 2.0 eq). The solution was stirred at -15 °C for 15 minutes before
adding a
solution of 1,1-carbonyldiimidazole (0.167 g, 1.032 mmol, 3.0 eq) in
tetrahydrofuran
(3.0 ml) dropwise. The solution was stirred at -15 °C for 15 minutes
before warming
to room temperature over a period of 40 minutes. Sat. aq. NaHC03 (25 ml) was
added followed by ethyl acetate (50 ml), and the solution was partitioned. The
organics were further washed with sat. aq. NaHC03 (25 ml) and brine (25 ml)
before
drying (Na2SOq.), filtering, and concentrating under reduced pressure. The
residue
was dissolved in a mixture of acetonitrile and tetrahydrofuran (10:1, 4 ml)
and conc.
ammonium hydroxide (4 ml) was added. The mixture was stirred at room
temperature for 16 hours before diluting with ethyl acetate (50 ml) and
washing with
sat. aq. NaHC03 (2 x 25 ml) and brine (30 ml). The organics were dried
(Na~SOq.),
filtered, and concentrated under reduced pressure. Silica gel chromatography
(0-X10
min 25% acetone-hexane, 1% Et3N; 10-X20 min 30% acetone~exane, 1% Et3N;
2030 min 35% acetone-hexane, 1% Et3N; 30+ min 40% acetone~exane, 1°70
Et3N) yielded the product as a white solid; ES-MS: nz/z 759 [M+H]+.
EXAMPLE 60
2'-O-benzoyl-11-amino-11-deoxy-3-des(cladinosyloxy)-2-fluoro-3-oxo-6-O-
methyl-15-azidoerythromycin A 11,12-cyclic carbamate
~~.Z~e2
O
A solution of 2'-O-benzoyl-11-amino-11-deoxy-3-des(cladinosyloxy)-3-oxo-
6-O-methyl-15-azidoerythromycin A 11,12-cyclic carbamate (1.0 eq) in THF is
cooled on ice and treated with a 1 M solution of potassium tart-butoxide in
THF (1.0
eq.), followed by N-fluoro-benzenesulfonimide (1.2 eq). After 1 hour, the
mixture is
warmed to ambient temperature and poured into a mixture of ethyl acetate and
sat. aq.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
_82_
NH4.C1. The organic phase is washed with brine, dried over MgS04, filtered,
and
evaporated. The product is purified by silica gel chromatography
(acetone/hexanes +
1 % Et3N).
EXAMPLE 61
2'-O-benzoyl-11-amino-11-deoxy-3-des(cladinosyloxy)-3-oxo-6-O-methyl-15-(3
(2-furyl)phenylacetamido)erythromycin A 11,12-cyclic carbamate
- O H
O / ~ ,,,,
OMe
.,~~~ Bz NMe2
O N~~~,,. ~.. .
O
H
O'~
To a solution of 2'-O-benzoyl-1 f-amino-11-deoxy-3-des(cladinosyloxy)-3-
oxo-6-O-methyl-15-azidoerythromycin A 11,12-cyclic carbamate (0.050 g, 0.062
mmol, 1.0 eq) in dichloromethane (1.0 ml) was added trimethylphosphine (0.31
ml of
a 1M solution in THF, 0.306 mmol, 5.0 eq). The solution was stirred at room
temperature for 45 minutes before transferring to a solution of the carboxylic
acid
(0.092 mmol, 1.5 eq), 1-[(3-(dimethylamino)propyl]-1-ethylcarbodiimide
hydrochloride (0.019 g, 0.099 mmol, 1.6 eq) and 1-hydroxybenzotriazole (0.017
g,
0.124 mmol, 2.0 eq) in dichloromethane (1.0 ml) also stirred at room
temperature for
45 minutes. The resulting solution was stirred at room temperature for 14
hours
before partitioning between ethyl acetate (10 ml) and NaHC03 (10 ml). The
aqueous
phase was extracted with ethyl acetate (3 x 10 ml) and the combined organics
further
washed with brine (25 ml) before drying (Na2SOq.), filtering, and
concentrating under
reduced pressure. The product was purified by silica gel chromatography using
acetone/hexane + 1 % Et3N.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-83-
EXAMPLE 62
11-amino-11-deoxy-3-des (cladinosyloxy)-3-oxo-6-O-methyl-15-(3-(2-
furyl)phenylacetamido)erythromycin A 11,12-cyclic carbamate
Me
' NMe2
HC~
O
A mixture of 2'-O-benzoyl-11-amino-11-deoxy-3-des(cladinosyloxy)-3-oxo-
6-O-methyl-15-(3-(2-furyl)phenylacetamido)erythromycin A 11,12-cyclic
carbamate
and triethylamine in methanol was heated at 50 °C overnight, then
evaporated to
dryness. The product was purified by silica gel chromatography
(acetonelhexanes +
1 % Et3N).
EXAMPLE 63
2'-O-benzoyl-11-amino-11-deoxy-3-des(cladinosyloxy)-2-fluoro-3-oxo-6-O-
methyl-15-(3-(2-furyl)phenylacetamido)erythromycin A 11,12-cyclic carbamate
~s
O e2
N~
H
To a solution of 2'-O-benzoyl-11-amino-11-deoxy-3-des(cladinosyloxy)-2-
fluoro-3-oxo-6-O-methyl-15-azidoerythromycin A 11,12-cyclic carbamate (0.050
g,
0.062 mmol, 1.0 eq) in dichloromethane or tetrahydrofuran (1.0 ml) is added
trimethylphosphine (0.31 ml of a 1M solution in TIFF, 0.306 mmol, 5.0 eq). The
solution is stirred at room temperature for 45 minutes before transfernng to a
solution
of the carboxylic acid (0.092 mmol, 1.5 eq), 1-[(3-(dimethylamino)propyl]-1-
ethylcarbodiimide hydrochloride (0.019 g, 0.099 mmol, 1.6 eq) and 1-
hydroxybenzotriazole (0.017 g, 0.124 mmol, 2.0 eq) in dichloromethane or
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-84-
tetrahydrofuran (1.0 ml) also stirred at room temperature for 45 minutes. The
resulting solution is stirred at room temperature for 14 hours before
partitioning
between ethyl acetate (10 ml) and NaHC03 (10 ml). The aqueous phase is
extracted
with ethyl acetate (3 x 10 ml) and the combined organics further washed with
brine
(25 ml) before drying (Na2SOq.), filtering, and concentrating under reduced
pressure.
The product is purified by silica gel chromatography using acetone/hexane + 1
%
Et3N.
EXAMPLE 64
11-amino-11-deoxy-3-des(cladinosyloxy)-2-fluoro-3-oxo-6-O-methyl-15-(3-(2-
furyl)phenylacetamido)erythromycin A 11,12-cyclic carbamate
e2
A solution of 2'-O-benzoyl-11-amino-11-deoxy-3-des(cladinosyloxy)-3-oxo-
6-O-methyl-15-(3-(2-furyl)phenylacetamido)erythromycin A 11,12-cyclic
carbamate
and triethylamine in methanol is heated at 50 °C overnight, then
evaporated to
dryness. The product is purified by silica gel chromatography (acetonelhexanes
+ 1%
Et3N).
EXAMPLE 65
2'-O-benzoyl-3-des(cladinosyloxy)-3-oxo-6-O-methyl-15-
(3-(2-furyl)phenylacetamido)erythromycin A
O /
OMe
0O
H
O,
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-85-
To a solution of 2'-O-benzoyl-3-des(cladinosyloxy)-3-oxo-6-O-methyl-15-
azidoerythromycin A (0.045 g, 0.062 mrnol, 1.0 eq) in dichloromethane or
tetrahydrofuran (1.0 ml) is added trimethylphosphine (0.31 ml of a 1M solution
in
THF, 0.306 mmol, 5.0 eq). The solution is stirred at room temperature for 45
minutes
before transferring to a solution of the carboxylic acid (0.092 mmol, 1.5 eq),
1-[(3-
(dimethylamino)propyl]-1-ethylcarbodiimide hydrochloride (0.019 g, 0.099 mmol,
1.6 eq) and 1-hydroxybenzotriazole (0.017 g, 0.124 mmol, 2.0 eq) in
dichloromethane
or tetrahydrofuran (1.0 ml) also stirred at room temperature for 45 minutes.
The
resulting solution is stirred at room temperature for 14 hours before
partitioning
between ethyl acetate (10 ml) and NaHC03 (10 ml). The aqueous phase is
extracted
with ethyl acetate (3 x 10 ml) and the combined organics further washed with
brine
(25 ml) before drying (Na2SOq.), filtering, and concentrating under reduced
pressure.
The product is purified by silica gel chromatography using acetone/hexane + 1
%
Et3N.
EXAMPLE 66
3-des(cladinosyloxy)-3-oxo-6-O-methyl-15-
(3-(2-furyl)phenylacetamido)erythromycin A
Me
NMe2
HC~
O
A mixture of 2'-O-benzoyl-3-des(cladinosyloxy)-3-oxo-6-O-methyl-15-
(3-(2-furyl)phenylacetamido)erythromycin A and triethylamine in methanol is
heated
at 50 °C overnight, then evaporated to dryness. The product is purified
by silica gel
chromatography (acetone/hexanes + 1% Et3N).
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-86-
EXAMPLE 67
2'-O-acetyl-3-des(cladinosyloxy)-3-oxo-6-O-methyl-15-
(3-(2-furyl)phenylacetamido)erythromycin A 11,12-cyclic carbonate
O /
Me
O ' " ,4c NMe2
N
H
To a solution of 2'-O-acetyl-3-des(cladinosyloxy)-3-oxo-6-O-methyl-15-
azidoerythromycin A 11,12-cyclic carbonate (0.043 g, 0.062 mmol, 1.0 eq) in
dichloromethane or tetrahydrofuran (1.0 ml) is added trimethylphosphine (0.31
ml of
a 1M solution in THF, 0.306 mmol, 5.0 eq). The solution is stirred at room
temperature for 45 minutes before transferring to a solution of the carboxylic
acid
(0.092 mmol, 1.5 eq), 1-[(3-(dimethylamino)propyl]-1-ethylcarbodiimide
hydrochloride (0.019 g, 0.099 mmol, 1.6 eq) and 1-hydroxybenzotriazole (0.017
g,
0.124 mmol, 2.0 eq) in dichloromethane or tetrahydrofuran (1.0 ml) also
stirred at
room temperature for 45 minutes. The resulting solution is stirred at room
temperature for 14 hours before partitioning between ethyl acetate (10 ml) and
NaHC03 (10 ml). The aqueous phase is extracted with ethyl acetate (3 x 10 ml)
and
the combined organics further washed with brine (25 ml) before drying
(Na2SOq.),
filtering, and concentrating under reduced pressure. The product is purified
by silica
gel chromatography using acetone/hexane + 1 % Et3N.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
_g7_
EXAMPLE 68
3-des(cladinosyloxy)-3-oxo-6-O-methyl-15-
(3-(2-furyl)phenylacetamido)erythromycin A 11,12-cyclic carbonate
O /
O
e2
A solution of 2'-O-acetyl-3-des(cladinosyloxy)-3-oxo-6-O-methyl-15-
(3-(2-furyl)phenylacetamido)erythromycin A 11,12-cyclic carbonate in methanol
is
heated at 50 °C for 16 hours, then evaporated to dryness. The product
is purified by
silica gel chromatography (acetonelhexane + 1% Et3N).
EXAMPLE 69
(~)-(2S* 3R*)-4-chloro-3-hydroxy-2-methylbutanoate N-propionylcysteamine
thioester
OH O H
O
This is prepared according to the method of Example 1, substituting
chloroacetaldehyde in place of 3-chloropropanal.
EXAMPLE 70
(~)-(2S ;3Rx-)-4-chloro-3-hydroxy-2-methylbutanoate
N-propionylcysteamine thioester
OH O H
O
This is prepared according to the method of Example 1, substituting
chlorobutyraldehyde in place of 3-chloropropanal.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
_88_
EXAMPLE 71
14-chloro-14-desmethyl-6-deoxyerythronolide B
H
This is prepared according to the method of Example 2, substituting (~)-
(2S*,3RD')-4-chloro-3-hydroxy-2-methylbutanoate N-propionylcysteamine
thioester in
place of (~)-(2S*,3R*)-5-chloro-3-hydroxy-2-methylpentanoate N-
propionylcysteamine thioester.
EXAMPLE 72
14-chloro-14-desmethyl-erythronolide B
H
~O H
This is prepared according to the method of Example 2, substituting (~)-
(2S~,3RD)-4-chloro-3-hydroxy-2-methylbutanoate N-propionylcysteamine thioester
in
J
place of (~)-(2S*,3R*)-5-chloro-3-hydroxy-2-methylpentanoate N-
propionylcysteamine thioester, and substituting Streptomyces coelicolor
CH999/p23-
55/pJRJ2, a strain containing the eryF gene on the chromosome along with
plasmid
pJRJ2, in place of Str-eptomyces coelicolor CH999/pJRJ2. 13C-NMR
(CDCl3+CD30D, 100 MHz): 8 218.6, 176.2, 81.1, 79.2, 74.9, 73.0, 69.7, 44.7,
43.7,
43,7, 39.7, 38.6, 38.1, 36.0, 25.8, 17.4, 14.6, 9.2, 8.6, 6.9.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-~9-
EXAMPLE 73
15-(chloromethyl)-6-deoxyerythronolide B
)H
This is prepared according to the method of Example 2, substituting (~)-
(2S*,3R*)-6-chloro-3-hydroxy-2-methylhexanoate N-propionylcysteamine thioester
in place of (~)-(2S*,3RD)-5-chloro-3-hydroxy-2-methylpentanoate N-
propionylcysteamine thioester.
EXAMPLE 74
14-azido-14-desmethyl-6-deoxyerythronolide B
,,,'
r
~OH
~~'OH
O~ 'OOH
This is prepared according to the method of Example 3, substituting 14-
chloro-14-desmethyl-6-deoxyerythronolide B in place of 15-chloro-6-
deoxyerythronolide B.
EXAMPLE 75
15-(azidomethyl)-6-deoxyerythronolide B
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-90-
N3~ )H
This is prepared according to the method of Example 3, substituting 15-
(chloromethyl)-6-deoxyerythronolide B in place of 15-chloro-6-
deoxyerythronolide
B.
EXAMPLE 76
14-azido-14-desmethyl-erythromycin A
NMe2
J ~~a
N3~~,, '~
Me
O~H
This is prepared according to the method of Example 4, substituting 14-azido-
14-desmethyl-6-deoxyerythronolide B or 14-azido-14-desmethylerythronolide B in
place of 15-azido-6-deoxyerythronolide B.
EXAMPLE 77
e2
b ~~H
15-(azidomethyl)-erythromycin A
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-91-
This is prepared according to the method of Example 4, substituting 15-
(azidomethyl)-6-deoxyerythronolide B in place of 15-azido-6-deoxyerythronolide
B.
EXAMPLE 78
15-((6-methoxy-1-benzofuran-3-yl)acetamido)erythromycin A
~e2
H
This compound was prepared according to the method of Example 6 using 2-
(6-methoxy-1-benzofuran-3-yl)acetic acid. 13C-NMR (100 MHz, CDC13): & 222.1,
176.3, 169.8, 158.3, 156.5, 142.3, 120.8, 119.8, 113.8, 111.8, 103.1, 96.5,
83.1, 79.4,
78.0, 75.0, 74.2, 72.6, 70.8, 69.0, 68.8, [65.6, 65.6] (2C), 55.7, 49.5, 46.0,
45.1, 44.6,
40.3, 40.0, 38.4, 37.9, 36.2, 34.9, 31.8, 29.7, 28.6, 28.3, 26.9, 21.5, 21.4,
18.6, 18.1,
16.1, 15.3, 11.8, 9Ø
EXAMPLE 79
15-((5-chlorobenzo[b]thiophen-3-yl)acetamido)erythromycin A
I ,,,,
HO OH
O ~"... OOH ...n NMe2
S
HC~
i N~v.,. ..e. .n.0- O
H
O ~''O OMe
~OH
This compound was prepared according to the method of Example 6 using 2-
(5-chlorobenzo[b]thiophen-3-yl)acetic acid. 13C-NMR (100 MHz, CDC13): 8 222.0,
169.4, 139.8, 138.6, 130.8, 129.0, 126.9, 125.0, 123.8, 121.5, 103.1, 96.3,
83.1, 79.4,
77.3, 75.1, 74.2, 72.7, 72.6, 70.8, 69.0, 68.9, 49.5, 45.1, 44.6, 40.3, 44.6,
40.3, 40.0,
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-92-
38.4, 37.9, 36.5, 36.2, 34.9, 29.7, 29.3, 28.6, 28.4, 26.9, 21.5, 21.4, 18.6,
18.1, 16.1,
15.4, 11.9, 9Ø
EXAMPLE 80
6-O-methyl-15-azidoerythromycin A
(a) Formation of 15-azidoer~thromycin A 9-oxime. A suspension of 15-azido-
erythromycin A (33.0 g, 90% purity, 42.5 mmol) in 90 ml of 2-propanol was
treated
with 32.0 ml of 50% aqueous hydroxylamine and stirred while acetic acid (10.30
ml)
was added and the mixture was stirred for 15 hours at 50~C. The reaction can
be
followed by thin-layer chromatography (10:1:0.05/CHC13/MeOH/MHq.OH). Upon
cooling to ambient temperature, saturated NaHC03 was added and the mixture was
concentrated in vacuo to remove isopropanol. The resulting aqueous mixture was
extracted three times with 300-ml portions of CHC13. The organic extracts were
combined, washed with saturated NaHCO3, water, and brine, then dried over
MgSO4,
filtered, and concentrated to yield 34 g of crude product. Analysis by LC/MS
revealed no clean separation of E and Z oximes, [M+H]+ = 791. 13C NMR (CDC13,
100 MHz) S 175.1, 171.3, 103.1, 96.4, 83.4, 80.1, 78.1, 75.3, 74.2, 73.2,
72.6, 71.1,
70.6, 68.7, 65.5, 65.4, 49.5, 49.1, 44.6, 40.2, 39.0, 37.8, 35.1, 32.6, 29.7,
29.4, 29.2,
27.6, 27.0, 25.5, 22.6, 21.5, 21.3, 18.6, 16.4, 15.9, 14.3, 9.2.
(b) Oxime protection. The crude oxime from step (a) above (34.0 g, 42.5 mmol)
was suspended in 120 ml of CH2C12 and treated with 1,1-diisopropoxycyclohexane
(51.0 ml, 246.8 mmol, 6 eq.) and pyridinium p-toluenesulfonate (24.8 g, 98.8
mmol, 2
eq.) for 15 hours at ambient temperature. The mixture was diluted with 600 ml
of
CH2Cl2, and then washed sequentially with saturated NaHCO3, water, and brine.
The
organic phase was dried with MgS04, filtered, and evaporated to yield brown
syrup.
Chromatography on silica gel (gradient from 2:1 to 1:1 hexanes/acetone + 1%
Et3N)
yielded 32 g of product. 13C NMR (CDCl3, 100 MHz) 8 174.6, 170.2, 104.2,
103.0,
96.3, 83.1, 78.0, 77.4, 75.2, 74.0, 73.0, 72.6, 70.9, 70.2, 68.8, 65.5, 63.4,
61.9, 49.5,
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-93- ,
49.2, 44.5, 40.3, 39.2, 37.8, 36.0, 35.0, 33.4, 33.0, 28.6, 27.7, 27.0, 26.7,
25.7, 25.4,
24.7, 24.3, 23.4, 22.9, 21.5, 18.6, 18.5, 16.3, 15.8, 14.4, 9.2.
(c) Trimethylsil lad tion. A solution of 15-azidoerythromycin A 9-[O- (1-
isopropoxy-cyclohexyl)]oxime (32.00 g, 34.4 mmol) in 120 ml of CH2C12 was
cooled
on ice under inert atmosphere and treated with a solution of
chlorotrimethylsilane
(0.628 m1,4.95 mmol, 0.15 eq.)) and 1-trimethylsilylimidazole (12.1 ml, 82.6
mmol,
2.4 eq.) in 30 ml of CH2C12. After 5 minutes, the reaction was diluted with 1L
of
ethyl acetate and washed sequentially with saturated NaHC03, water, and brine.
The
organic phase was dried with MgS04, filtered, and evaporated. The crude
product
was purified by silica gel chromatography (gradient from hexanes to 10:1
hexanes/acetone + 1 % Et3N), yielding 29 g of product. 13C NMR (CDCI3, 100
MHz)
~ 175.5, 170.5, 104.1, 102.7, 96.8, 81.4, 80.9, 79.2, 75.6, 74.0, 73.3, 73.2,
70.3, 67.8,
65.2, 65.0, 63.2, 49.7, 49.2, 44.5, 41.0, 38.3, 35.8, 34.4, 33.6, 33.2, 31.6,
29.8, 28.1,
26.7, 25.4, 24.3, 22.9, 22.6, 22.1, 21.7, 19.3, 18.4, 16.3, 15.1, 14.4, 14.1,
9.7, 1.0, 0.9.
(d) Meth lay tion. To a solution of 29 g of fully protected oxime from step
(c)
above (27.0 mmol) in a mixture of 55 ml of freshly distilled THF and 55 ml of
dry
DMSO was added 27.05 ml of a 2N solution of methyl bromide in ether (54 mmol,
2
eq.). The resulting solution was cooled to 5~C and put under nitrogen before
addition
of 54 ml of tBuOK (54 mmol, 2 eq.) diluted with 55 ml of DMSO using a syringe
pump over a 3h period. When thin-layer chromatographic analysis (5:1 toluene
/acetone, NH40H) showed no starting material remaining, 200 ml of sat.aq.
NaHC03
was added, and the mixture was extracted with 1 L of ethyl acetate. The
organic
extract was washed sequentially with saturated NaHC03, water, and brine. The
organic phase was dried with MgSOø, filtered, and evaporated. The crude
product
was purified by silica gel chromatography (gradient from hexanes to 10:1
hexanes/acetone + 1% Et3N), yielding 30 g of product. 13C NMR (CDCl3, 100 MHz)
8 175.7, 169.8, 103.7, 102.6, 96.2, 80.8, 79.1, 78.6, 77.9, 73.7, 73.3, 73.1,
73.0, 69.7,
65.1, 65.1, 62.8, 51.1, 49.7, 49.1, 45.3, 41.0, 39.8, 37.7, 35.8, 34.6, 33.6,
33.1, 29.6,
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-94-
28.0, 26.5, 25.6, 24.5, 24.4, 22.9, 22.2, 22.0, 20.2, 19.5, 18.6, 16.2, 15.8,
15.1, 9.6,
1.0, 0.8.
(e) Removal of silyl and acetal protection. A solution of the above product 6-
O-
methyl-2', 4"-bis-O-trimethylsilyl-15-azidoerythromycin A 9-[O-(1-isopropoxy-
cyclohexyl)]-oxime in 150 ml of acetonitrile was treated with 75 ml of water
and 90
ml of acetic acid, and stirred for 18 hours at ambient temperature. The
mixture was
concentrated after addition of 2-propanol, then repeatedly after addition of
toluene.
Chromatography of the residue on silica gel (gradient from 2:1 to 1:1
hexanes/acetone
+ 1%o Et3N) gave 20 g of 6-O-methyl-15-azidoerythromycin A 9-oxime as white
solid.
13C NMR (CDC13, 100 MHz) b 175.4, 169.7, 102.8, 96.1, 80.4, 78.8, 78.4, 78.0,
73.8,
73.0, 72.7, 71.1, 70.0, 68.6, 65.6, 65.5, 51.1, 49.5, 49.1, 45.7, 45.0, 40.3,
39.3, 37.4,
34.9, 32.6, 29.0, 27.8, 25.3, 21.4, 20.0, 19.8, 18.6, 16.1, 15.7, 15.0, 10.6,
9.1.
(f) Deoximation. A solution of 6-O-methyl-15-azidoerythromycin A 9-oxime (20
g, 25.1 mmol) and sodium hydrosulfite (85°70, 52.0 g, 254 mmol, 10 eq.)
in 350 ml of
1:1 ethanol/water was placed under inert atmosphere. Formic acid (4.82 ml,
127.8
i
mmol, 5 eq.) was added dropwise, and the mixture was stirred at 80~C for 4.5
hours.
After cooling to ambient temperature, the reaction was adjusted to pH 10 with
6 N
NaOH and extracted three times with 250-ml portions of ethyl acetate. The
organic
extracts were combined and washed sequentially with saturated NaHCO3, water,
and
brine. The organic phase was dried with MgS04, filtered, and evaporated to
yield 16
g of 6-O-methyl-15-azidoerythromycin A. 13C NMR (CDC13, 100 MHz) b 221.0,
175.6, 102.8, 96.1, 80.6, 78.4, 78.3, 77.9, 74.0, 72.7, 72.6, 70.9, 68.7,
68.7, 65.8, 65.6,
50.6, 49.5, 49.0, 45.2, 44.9, 40.2, 39.4, 39.4, 37.1, 34.8, 28.7, 27.6, 25.3,
21.4, 19.7,
18.7, 17.9, 16.0, 15.6, 12.4, 9Ø
EXAMPLE 81
2'-O-benzoyl-6-O-methyl-3-descladinosyl-11-amino-11-deoxy-3-15-
azidoerythromycin A 11,12-cyclic carbamate
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-95-
(a) Benzoxlation of the 2' and 4" h d'r roxYl r~ oupS. To a solution of 14 g
of 6-O-
methyl-15-azidoerythromycin A (17.7 mmol, 1.0 eq.) in a mixture of 28 ml of
tetrahydrofuran and 112 ml of ethyl acetate was added 11.62 g of benzoic
anhydride
(51.4 mmol, 2.9 eq.), 2.17 g of 4-(dimethylamino)pyridine (17.7, leq.), and
7.28 ml of
triethylamine (52.3 mmol, 3 eq.) sequentially at ambient temperature. The
resulting
solution was stirred for 3 days. Analysis by thin-layer chromatography (5:1
toluene/acetone, NHøOH) showed less than 10% starting material remaining.
After
addition of 200 ml of sat. NaHC03 the resulting mixture was extracted with 2 X
600
ml of ethyl acetate. The organic extracts were combined and washed
sequentially with
saturated NaHC03, water, and brine. The organic phase was dried with MgS04,
filtered, and evaporated to give a slight yellow solid. Column purification on
silica gel
(8:1 hexane/acetone, +1% Et3N to 4:1 hexane/acetone, +1% Et3N) yielded 12 g of
the
2',4"-dibenzoate product. 13C NMR (CDC13, 100 MHz) 8 220.9, 175.2, 166.1,
165.3,
133.3, 132.6, 130.8, 129.8, 129.6, 129.6, 128.3, 128.2, 100.2, 95.7, 80.3,
78.8, 78.2,
77.6, 73.8, 72.9, 72.7, 72.4, 68.7, 67.5, 63.6, 63.5, 50.3, 49.6, 48.9, 46.2,
45.1, 44.6,
40.8, 38.8, 38.5, 37.1, 35.2, 31.6, 27.6, 21.2, 19.8, 18.4, 17.7, 16.0, 15.5,
12.2, 9.3.
(b) Carbamate formation. To a solution of 12 g (12.0 mmol) of the 2',4"-
dibenzoate from step (a) above in a mixture of 42 ml of freshly distilled THF
and 15
ml of dry DMF was added 8.8 g of solid 1,1-carbonyldiimidazole (54.3, 4.5
eq.). The
resulting mixture was stirred under nitrogen for 20 min. at ambient
temperature before
8 ml of a 2 N solution of sodium bis(trimethylsilyl)amide (NaI~V>DS) in THF
(16
mmol, 1.3 eq.) was added dropwise. The resulting mixture was stirred overnight
under
nitrogen. The temperature of the mixture was lowered to -15~C, and liquid NH3
was
added in by condensation of anhydrous ammonia gas using a dry ice condenser.
The
mixture was capped and stirred for 2h at -15~C. Liquid NH3 was dripped in
above
mixture again before the mixture was capped, and stirred for another 2h at
OeC. The
mixture was capped and stirred at O~C for another lh after liquid NH3 was
dripped in
the mixture for the third time. After the temperature of above mixture was
raised to
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-96-
room temperature, 15 ml of a 1 N solution of potassium tert-butoxide in THF
(15
mmol, 1.2 eq.) was added in dropwise, and the mixture was stirred overnight
under
nitrogen. Then the mixture was diluted with sat. NaHC03, and extracted with 2
X 600
ml of ethyl acetate. The organic extracts were combined and washed
sequentially
with saturated NaHC03, water, and brine. The organic phase was dried with
MgS04,
filtered, and evaporated to give a slight yellow solid, which was used for
next step
reaction without further purification.
(c) Cladinose removal. The crude product from step (b) was dissolved in 120 ml
of EtOH and 120 ml of 2N aqueous HCI, and the resulting solution was heated at
45~C for 4h. The solution was cooled to room temperature, and the pH was
adjusted
to 9 by adding 4N NaOH. The mixture was extracted with 2 X 500 ml of ethyl
acetate, the organic extracts were combined, and then washed sequentially with
saturated NaHC03, water, and brine. The organic phase was dried with MgS04,
filtered, and evaporated. The crude product was purified by silica gel
chromatography (gradient from 3:1 hexanes/acetone + 1 % Et3N, to 1:1
hexanes/acetone + 1°Io Et3N), yielding 5.7 g of product. 13C NMR
(CDCl3, 100 MHz)
~ 217.7, 174.8, 265.3, 158.0, 132.6, 130.6, 129.7, 128.2, 99.7, 83.4, 80.7,
77.9, 77.5,
72.1, 71.9, 68.9, 63.2, 57.9, 49.5, 48.4, 45.2, 43.9, 40.7, 38.7, 37.0, 35.8,
31.9, 28.5,
21.0, 19.2, 18.0, 14.8, 13.7, 13.3, 7.7.
EXAMPLE 82
2'-O-benzoyl-6-O-methyl-3-descladinosyl-11-amino-11-deoxy-3-15-
aminoerythromycin A 11,12-cyclic carbamate hydrochloride
400 mg of 10°70 activated palladium on carbon was weighed out into a
round
bottom flask under nitrogen, and 50 ml of methanol was added in slowly
followed by
sequential addition of 400 mg of 2'-O-benzoyl-6-O-methyl-3-descladinosyl-11-
amino-11-deoxy-3-15-azidoerythromycin A 11,12-cyclic carbamate (0.53 mmol)
(Example 81) and 0:13 mL (2 eq.) of trimethylsilyl chloride. The atmosphere in
the
flask was then purged using a hydrogen balloon three times before the flask
was
closed and stirred vigorously under a hydrogen atmosphere overnight. The
mixture
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-97-
was filtered, and the solid was washed with 3 X 50 ml of methanol. The
combined
methanol solution was evaporated under vacuum to give 380 mg of the amine
hydrochloride, which was used without purification for the next reaction.
EXAMPLE 83
General procedure for 15-amidoketolide formation from 2'-O-benzoyl-6-O-
methyl-3-descladinosyl-11-amino-11-deoxy-3-15-aminoerythromycin A 11,12-
cyclic carbamate hydrochloride
(a) To a solution of 26 mg of amine hydrochloride salt from Example 82, 1.3
eq.
of R1COOH, and 1.2 eq. of 1-hydroxybenzotriazole in 2 ml of dry
dimethylformamide was added 1.2 eq. Of 1-[3-(dimethylamino)propyl]-3-
ethylcarbodiimide hydrochloride, and 3 eq. of triethylamine at ambient
temperature.
The mixture was stirred overnight before it was diluted with 30 ml of sat
NaHC03,
and extracted with 50 ml of CH2Clz. The organic extract was washed
sequentially
with saturated NaHC03, water, and brine. The organic phase was dried with
MgSO~,
filtered, and evaporated to give a slight yellow solid, which was used for
next step.
reaction without further purification.
(b) To the solution of the 3-hydroxyl compound from Step (a) above in 2 ml of
CH2C12 was added an excess amount of NaHC03 and the Dess-Martin periodinane (4
eq.), and the resultant mixture was stirred at ambient temperature for 30 min.
The
mixture was treated with 15 mL of saturated aqueous sodium thiosulfate for 10
minutes, then diluted with sat. NaHC03, and extracted with CH2Ch, the organic
extract was washed sequentially with saturated NaHC03, water, and brine. The
organic phase was dried with MgS04, filtered, and evaporated to give a slight
yellow
solid, which was used for next step reaction without further purification.
(d) The compound from step (b) above was dissolved in 2 ml of methanol and
heated
at 70~C under stirring overnight. Evaporation of the methanol under vacuum
gave
a yellow solid, which was purified by flash column to provide final 15-
amidoketolide.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-98-
(e)
EXAMPLE 84
11-amino-11-deoxy-3-descladinosyloxy-3-oxo-15-(3-(2-
furyl)phenylacetamido)-erythromycin A 11,12-cyclic carbamate
This compound was prepared according to the methods of Example 83, using
3-(2-furyl)phenylacetic acid.
1H NMR (CDCl3, 400 MHz) 8 7.53 (m, 2H), 7.45 (d, J = l.BHz, 1H), 7.40 (t, J
= 7.6Hz, 1H), 7.12 (d, J = 7.6Hz , 1H), 6.65(d, J = 3.3Hz, 1H), 6.45 (d,d,~J =
l.BHz,
J = 3.3Hz ,1H), 5.92 (s, 1H), 5.64 (s, 1H), 4.89 (d, J = 5.6Hz, 1H), 4.25 (d,
J =
7.2Hz, 1H), 4.13 (d, J = 8.OHz, 1H), 3.75 (q, J = 6.4Hz, 1H), 3.63 (s, 1H),
3.53 (m,
5H), 3.13 (m, 1H), 2.96 (m, 1H), 2.83 (m, 1H), 2.71 to 2.30 (m, 6H), 2.22 (s,
6H),
2.01 to 1.57 (m, 5H), 1.40 (s, 3H), 1.34 (d, J = 6.8Hz, 3H), 1.33 (s, 3H),
1.24 (d, J =
7.6Hz, 3H), 1.21 (d, J = 6.OHz, 3H), 1.09 (d, J = 6.4Hz, 3H), 1.08 (d, J =
6.OHz, 3H).
13C NMR (CDC13, 100 MHz) ~ 217.8, 203.9, 170.7, 169.4, 157.4, 153.4,
142.1, 135.2, 131.4, 129.3, 128.2, 124.7, 122.6, 111.6, 105.4, 103.8, 82.9,
79.2, 77.9,
72.9, 70.2, 69.5, 65.8, 57.8, 51.0, 49.1, 48.0, 44.4, 43.6, 40.1, 39.7, 37.3,
36.6, 29.2,
28.1, 21.1, 19.3, 17.6, 16.5, 16.5, 14.2, 13.5, 13.2.
EXAMPLE 85
11-amino-11-deoxy-3-descladinosyloxy-3-oxo-15-(3-quinolylacetamido)-
erythromycin A 11,12-cyclic carbamate
This compound was prepared according to the methods of Example 83, using
3-quinolyl-acetic acid.
1H NMR (CDC13, 400 MHz) S 8.78 (s, 1H), 8.07 (s, 1H), 8.01 (d, J = 8.4Hz,
1H), 7.77 (d, J = 8.OHz, 1H), 7.62 (t, J = 8.OHz , 1H), 7.48(d, J = 7.6Hz,
1H), 6.54 (s,
1H), 5.74 (s, 1H), 4.94 (d, d, J = 2.4Hz, J = lOHz, 1H), 4.25 (d, J = 7.2Hz,
1H), 4.12
(d, J = 8.OHz, 1H), 3.75 (q, J = 6.8Hz, 1H), 3.70 to 3.42 (m, 6H), 3.16 (m,
1H), 2.95
(m, 1H), 2.80 (m, 2H), 2.60 to 2.50 (m, 2H), 2.45 (s, 3H), 2.28 (s, 6H), 1.97
(m, 1H),
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-99-
1.78 to 1.56 (m, 4H), 1.40 (s, 3H), 1.33 (d, J = 6.8Hz, 3H), 1.26 (s, 3H),
1.23 (d, J =
8.OHz, 3H), 1.20 (d, J = 6.4Hz, 3H), 1.08 (d, J = 6.8Hz, 3H), 1.06 (d, J =
6.4Hz,
3H).
i3C N~ (CDCl3, 100 MHz) 8 217.9, 203.9, 169.9, 169.6, 157.5, 151.6,
147.2, 136.0, 129.2, 129.0, 127.9, 127.8, 127.7, 126.7, 103.8, 83.0, 79.4,
77.9, 72.9,
70.2, 69.5, 65.7, 57.8, 51.0, 49.2, 48.2, 44.4, 40.6, 40.2, 39.8, 37.3, 35.7,
29.1, 28.0,
21.1,19.4,17.5,16.7,16.7,14.2,13.6,13.2.
EXAMPLE 86
11-amino-11-deoxy-3-descladinosyloxy-3-oxo-15-(2-([1,2,4]-triazol-1-yl)pyrid-
5-ylacetamido)erythromycin A 11,12-cyclic carbamate
This compound was prepared according to the methods of Example 83, using
2-([1,2,4]-tetrazol-1-yl)pyrid-5-ylacetic acid.
1H NMR (CDC13, 400 MHz) ~ 9.11 (s, 1H), 8.34 (s,lH), 8.11(d, 1H), 7.86 (m,
2H), 6.21 (s, 1H), 5.92 (s, 1H), 4.82 (d, d, J = 2.OHz, J = 10.4Hz, 1H), 4.25
(d, J =
7.6Hz, 1H), 4.10 (d, J = 8.8Hz, 1H), 3.75 (q, J = 6.4Hz, 1H), 3.71 to 3.46 (m,
6H),
3.16 (m, 1H), 2.96 (m, 1H), 2.82 (m, 1H), 2.71 to 2.40 (m, 3H), 2.49 (s, 3H),
2.32 (s,
6H), 2.02 to 1.96 (m, 1H), 1.82 to 1.55 (m, 4H), 1.43 (s, 3H), 1.37 (d, J =
7.OHz, 3H),
1.27 (s, 3H), 1.24 (d, J = 4.OHz, 3H), 1.23 (d, J = 6.4Hz, 3H), 1.08 (d, J =
6.8Hz,
3H), 1.10 (d, J = 6.8Hz, 6H).
isC NMR (CDC13, 100 MHz) ~ 217.7, 203.7, 169.9, 169.5, 157.5, 148.6,
148.4, 141.4, 140.0, 130.5, 129.6, 113.2, 82.9, 79.5, 78.0, 72.8, 70.2, 69.5,
65.8, 57.8,
51.0, 49.0, 48.3, 44.3, 40.1, 39.9, 39.7, 37.3, 35.4, 29.2, 28.9, 28.1, 21.1,
19.4, 17.5,
17.1, 14.2, 13.6, 13.2.
EXAMPLE 87
11-amino-11-deoxy-3-descladinosyloxy-3-oxo-15-(4-(3-pyridyl)imidazol-1-
ylacetamido)erythromycin A 11,12-cyclic carbamate
This compound was prepared according to the methods of Example 83, using
4-(3-pyridyl)imidazol-1-ylacetic acid.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-100-
EXAMPLE 88
Preparation of 3-(2-furanyl)phenylacetic acid
(a) Methyl 3-bromophenylacetate. To a solution of 0.710 g of 3-
bromophenylacetic acid (3.28 mmol) in 5 ml of methanol was added
trimethylsilyl-
diazomethane dropwise until the solution became yellow, then acetic acid was
added
dropwise until the solution was clear again. The solvent was then evaporated
under
vacuum to give the methyl ester of the bromophenylacetic acid, which was used
for
next step reaction without further purification.
(b) Methyl 3-(2-furan~phenylacetate. The above ester, 2-furanboronic acid
(1.44g, 12.78 mmol, 3.9 eq.), and tetrakis(triphenylphosphine)palladium (0.38
g, 0.33
mmol) were weighed out into a round bottom flask. The mixture was degassed for
30
min. before 50 ml of freshly distilled THF and 2 mL of degassed saturated
aqueous
sodium carbonate were added in. The resulted mixture was heated at 50~C
overnight.
After cooling to room temperature, the solution was concentrated under vacuum.
Column purification (gradient from 5:1 hexanes/EtOAc, to 4:1 hexanes/EtOAc)
yielded 0.40 g 3-furanylphenylacetic acid methyl ester.
(c) 3-(2-furanyl)~hen~lacetic acid. To a solution of the methyl 3-(2-
furanyl)phenylacetate from step (b) in 5 ml of methanol was added 10 ml of 1N
NaOH aqueous solution, the resulting solution was stirred overnight before
diluting
with 40 of ml 1N NaOH aqueous solution and washing with 2 X 30 ml CH2C12. The
aqueous solution was then acidified with 2N HCI, and extracted with 3 X 30 ml
of
CHZC12. The organic extracts were combined and were dried with MgS04,
filtered,
and evaporated to give 0.32 g of the product as a golden-colored solid.
EXAMPLE 89
General preparation of aryl- and heteroaryl-acetic acids
To a microwave reaction vial was added 30 mg of palladium(II) acetate (0.13
mmol, 13 mol°7o), 73 mg (0.18 mmol, 18%) of (2-dicyclohexanylphosphino)
-2'-
(N,N-dimethyl)biphenyl, and the vial was degassed under nitrogen for 15 min.
before
adding 2 ml of degassed dry toluene. The resulting solution was stirred for 10
min.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-101-
before adding 1.5 ml of a 1 N solution of potassium tart-butoxide in THF and
di-
tbutyl malonate (3 mmol, 3 eq.) under nitrogen. The resulting mixture was
stirred for
min. before the aryl bromide or heteroaryl bromide(1 mmol) was added in. The
mixture was heated in a microwave reactor at 180eC for 3 min. repeatedly until
no
5 starting material remained by thin-layer chromatographic analysis (5:1
toluene/acetone). Then methanol was added to the mixture before it was
concentrated
under vacuum. The mixture was then partially redissolved into CH2C12, and
filtered.
The filtrate was concentrated, and purified by silica gel column (from 15:1
hexanes/acetone to 5:1 hexanes/acetone) to yield around 200 mg of a yellow
solid. To
10 the solid in 10 ml of toluene was added 1 ml of trifluoroacetic acid, and
the resulting
solution was heated at 100~C for 3h. The solvent was evaporated under vacuum
to
yield around 80 mg of final product.
Compounds prepared according to this general procedure include:
(a) 3-quinolylacetic acid, using 3-bromoquinoline; and
(b) 2-([1,2,4]-triazol-1-yl)pyrid-5-yl-acetic acid, using 2-([1,2,4]-tetrazol-
1-
yl)- 5-bromo-pyridine.
EXAMPLE 90
Preparation of 4-(3-pyridyl)imidazol-1-ylacetic acid
To a solution of 55 mg of 4-(3-pyridyl)imidazole (0.38 mmol, 1.0 eq.) in 2 ml
of dry of dimethylformamide was added 15.5 mg of NaH (60%) (0.38 mmol, 1.0
eq.),
and the mixture was stirred vigorously for 10 min. before dropwise addition of
0.05
ml of tart-butyl bromoacetate (0.34 mmol, 0.9 eq.). The resulting solution was
heated
at 70~C for 30 min before it was diluted with 20 ml of water, and extracted
with 3 X
30 ml of ethyl acetate. The organic extracts were combined and washed
sequentially
with water, and brine. The organic phase was dried with MgS04, filtered, and
evaporated to give 60 mg of a slight yellow solid.
To a solution of above solid in 5 ml of CH2CI2 was added 5 ml of
trifluoroacetic acid, and the resulting solution was stirred at room
temperature for 3h.
The solvent was then evaporated under vacuum to give 35 mg of final product.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-102-
EXAMPLE 91
11-amino-11-deoxy-3-descladinosyloxy-2-fluoro-3-oxo-15-(3-(2-
furyl)phenylacetamido)-erythromycin A 11,12-cyclic carbamate
Me
" NMe2
O
To a solution of 11-amino-11-deoxy-3-descladinosyloxy-3-oxo-15-(3-(2-
furyl)phenylacetamido)-erythromycin A 11,12-cyclic carbamate in freshly
distilled
tetrahydrofuran (0.2 mmol/ml) was added 3.5 eq. of tBuOK (1N solution in THF)
dropwise at -78~C under N2. The temperature of the resulting solution was
allowed to
slowly rise to -40~C to -20~C, and the solution was stirred at this
temperature for 10
min before a THF solution of N-fluorobenzenesulfonimide (0.6 mmol/ml) was
added
dropwise. The temperature of the resulting solution was then allowed to rise
to O~C to
-10~C, and the mixture was stirred at this temperature for 4 h before it was
diluted
with 100 ml of ethyl acetate and 30 ml of sat. aq. NaHC03. The organic layer
was
separated, washed sequentially with water and brine, then dried with MgS04,
filtered,
and concentrated under vacuum. The crude product was purified by silica gel
chromatography (2:1 hexaneslacetone + 1% Et3N).
Subsequently debenzoylation by heating in methanol at 50 °C and
column
purification provided final product.
EXAMPLE 92
Microbiological Activity
Minimum inhibitory concentrations ("MICs") were determined by the NCCLS
broth microdilution procedure for susceptibility testing for bacteria that
grow
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-103-
aerobically (National Committee for Clinical Laboratory Standards, 1997.
Methods
for dilution antimicrobial susceptibility tests for bacteria that grow
aerobically, 4"' ed.
Approved standard. NCCLS Document M7-A4. National Committee for Clinical
Laboratory Standards, Villanova, PA.). Stock solutions were prepared on the
day of
the test and appropriate aliquots were added to cation adjusted Mueller-Hinton
broth
(CAMHB) or Haemophilus test media. Two-fold serial dilutions were prepared and
added to wells in microtiter plates. Final test concentrations ranged from 16
to 0.015
ug/ml. Broth cultures of bacteria inoculated from growth on overnight plates
for all
test bacteria except Streptococcus pneuzzzoniae and Haemophilus izzfluenzae
were
incubated at 35°C and then adjusted to the Kirby Bauer standard and
diluted in
CAMHB to achieve a final inoculum concentration of approximately 5x105 CFU/ml.
Inocula for S. pzzeufnozziae and H. influenzae were prepared by directly
suspending
colonies from an overnight plate, adjusting the turbidity and diluting as
above. S.
pneumoniae media was supplemented with 2.5°10 lysed horse blood. All
plates were
incubated in ambient air at 35°C for 20-24 h for S. pneumoniae and
Haenzoplzilus
znfluezzzae and 16-20 h for all other bacteria. The MIC endpoints were
determined by
reading the lowest concentration of test compound that completely inhibited
the
growth of the test bacteria. Results for compounds described in the above
Examples
are listed in Table 1.
CA 02471383 2004-06-21
WO 03/061671 PCT/US03/01398
-104-
TABLE 1
Example S. aureusS. pneumo S. pneumo S. pneumo H. influenzae
OC4172 OC9132 OC4438 ATCC6301 ATCC49766
Erythromycin0.5 0.06 8 0.06 2
A
7 8 2 >16 nd >16
8 16 1 >16 nd >16
g 4 0.25 >16 nd >16
1 0.25 >16 nd 8
11 2 nd 16 0.25 8
13 4 nd 8 0.5 >16
14 8 nd 8 0.25 >16
1 nd 8 0.12 4
17 1 nd 4 0.06 2
18 0.5 nd 4 0.06 2
19 1 nd , 4 0.12 2
4 0.5 16 nd 8
21 1 nd 16 0.12 4
23 8 1 >16 nd >16
8 2 >16 nd >16
26 16 2 >16 nd >16
27 16 2 >16 nd >16
29 8 nd >16 1 >16
1 0.06 8 nd 4
84 0.06 0.25 0.25 0.015 0.5
85 1 0.25 4 0.25 8
86 0.5 0.25 2 0.06 2
87 8 1 8 1 > 16
nd = not
determined.
5 The invention having now been described by way of written description, those
of skill in the art will recognize that the invention can be practiced in a
variety of
embodiments, and that the foregoing description and examples, while describing
the
best mode contemplated by the inventors, is for purposes of illustration and
not
limitation of the following claims. All references cited herein, including
patents,
10 patent applications, PCT publications, papers, text books, and the like,
and the
references cited therein, to the extent that they are not already, are hereby
incorporated herein by reference in their entirety.