Note: Descriptions are shown in the official language in which they were submitted.
CA 02473422 2006-07-18
-1-
PREPAR.ATION OF 5-CHLOROIMIDAZOLES
This application is a division of Canadian Patent
Application Serial No. 2,117,209. The claims of the present
application are directed to a process for the production of
5-chloroimidazole-4-carbaldehydes. However, for a ready
understanding of the overall invention, including all
features which are inextricably bound up in one and the same
inventive concept, the teachings of those features claimed in
Canadian Patent Application Serial No. 2,117,209 are all
retained herein.
Field of The Invention
The present invention relates to novel 5-
chloroimidazoles, a process of preparing the 5-
chloroimidazoles, and a process of converting some of the 5-
chloroimidazoles to the corresponding 5-chloroimidazole-4-
carbaldehydes.
Backaround of The Invention
Several methods for the production of 5-
chloroimidazole-4-carbaldehydes are known.
U.S. Patent No. 4,355,040 describes a process
according to which 2-amino-3,3-dichloroacrylonitrile is
reacted with an aldehyde to the corresponding azomethine
intermediate product and further with a hydrogen halide and
water to the corresponding 2-substituted-5-haloimidazole-4-
carbaldehyde. Experimental data is lacking in the patent. A
great drawback of the synthesis is that the starting
material, 2-amino-3,3-dichloroacrylonitrile, has to be
produced from dichloroacetonitrile by its reaction with
hydrogen cyanide/sodium cyanide. The extremely toxic
reactants and the safety measures associated therewith that
are required just for the preparation of the starting
CA 02473422 2006-07-18
-2-
material, make the entire process unsuitable for industrial-
scale production.
In another embodiment, U.S. Patent No. 4,355,040
discloses a 3-stage process wherein, an amidine hydrochloride
is cyclized under high NH3 pressure with dihydroxyacetone,
the imidazole alcohol is halogenated and finally oxidized to
aldehyde.
It has now been revealed that pressures of over
20 bars are necessary for the cyclization reaction.
The oxidation of the alcohol is achieved
according to U.S. Patent No. 4,355,040 in the presence of
chromium oxide. It will be appreciated by those skilled in
the art that oxidation with heavy metal oxides, that are not
generally recyclable, is no longer justifiable in view of
current ecological concerns and requirements.
Summary of The Invention
According to one aspect of the present invention,
which is claimed in Canadian Patent Application Serial No.
2,117,209, there is provided a 2-substituted-5-
chloroimidazole of the general formula (Ia):
~Cl
Ia
H
wherein R represents n-butyl, 2-butenyl or 3-butenyl.
According to another aspect of the present
invention, which is claimed in Canadian Patent Application
Serial No. 2,117,209, there is provided a process for the
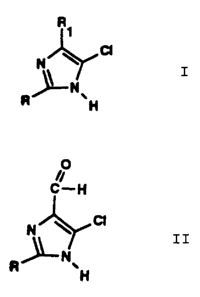
production of a 5-chloroimidazole of the general formula (I):
q I
;0-
N A '
N
M
CA 02473422 2004-08-04
-3-
wherein R represents hydrogen, a straight-chain or branched
Cl to C. alkyl group, a straight-chain or branched C2 to C6
alkenyl group, a cyclopropyl-, cyclobutyl-, cyclopentyl-, or
cyclohexyl- group, or a benzyl or phenyl group optionally
substituted with one or more of halogen atoms, straight-chain
or branched Clto C6 alkyl groups, nitro, or amino groups, and
R1 represents hydrogen, a straight-chain or branched Clto C6
alkyl group, a cyclopropyl-, cyclobutyl-, cyclopentyl-, or
cyclohexyl- group, or a benzyl or pheny:L group optionally
substituted with one or more of halogen atoms, straight-chain
or branched Clto C. alkyl groups, nitro, or amino groups, -
C02R3 or -(CHz) -CO2R3, wherein n is from 1 to 4 and R3
represents a straight-chain or branched Clto C. alkyl group,
comprising, in a first step, reacting a glycine ester
hydrohalide of the general formula (III):
R' NHa HX
C02R2 I I I
wherein R1 has the above-mentioned meaning, R2 represents a
straight-chain or branched Clto C6 alkyl group and X
represents a halogen atom, with an imidic acid ester of the
general formula (IV):
R OR
4
NH
IV
wherein R has the above-mentioned meaning and R4 represents a
straight-chain or branched Clto C6 alkyl group, in the
CA 02473422 2006-07-18
-4-
presence of a base, to give the corresponding 3,5-
dihydroimidazol-4-one of the general formula (V):
O
N V
R~SI-N
wherein R and R1 have the above-mentioned meanings, and, in a
second step, chlorinating the corresponding 3,5-
dihydroimidazol-4-one (V).
According to a further aspect of the present
invention, there is provided a process for the preparation of
a 5-chloroimidazole-4-carbaldehyde of the general formula
(II) :
0
C--H
II
N Ar p
A~N
H
wherein R represents hydrogen, a straight-chain or branched
Clto C6 alkyl group, a straight-chain or branched C2 to C6
alkenyl group, a cyclopropyl-, cyclobutyl-, cyclopentyl-, or
cyclohexyl- group, or a benzyl or phenyl group optionally
substituted with one or more of halogen atoms, straight-chain
or branched Clto C6 alkyl groups, nitro, or amino groups.
The process includes reacting a 5-chloroimidazole of formula
(Ia), wherein R is as defined immediately above, with
phosphorus oxychloride or phosgene in the presence of N,N-
dimethylformamide.
The 5-chloroimidazoles (I) and (Ia) and the 5-
chloroimidazole-4-carbaldehydes (II) are important starting
materials for the production of anti-hypertensive
pharmaceutical agents (U.S. Patent No. 4,355,040) and
herbicidal compounds (German OS 2804435).
CA 02473422 2006-07-18
-5-
Detailed Description of The Invention
For the preparation of a 5-chloroimidazole of the
general formula (I) according to the present invention, in a
first step, a glycine ester hydrohalide of the general
formula (III) :
R~ NH~ NX
COZ RZ I I I
wherein R1 has the above-mentioned meaning, R2 is an alkyl
group and X is a halogen atom, is reacted with an imidic acid
ester of the general formula (IV):
on
IV
NH
wherein R has the above-mentioned meaning and R4 is an alkyl
group, in the presence of a base, to the corresponding 3,5-
dihydroimidazol-4-one of the general formula (V):
R
O
N
~'\"_N%
R
wherein R and R1 have the above-mentioned meanings.
With reference to the substituents, namely R, R1,
R2, R3 and R4 it will be understood that the indicated groups
have the following meanings.
An alkyl group is a straight-chain or branched C1-
C6 alkyl group, such as, methyl, ethyl, n-propyl, isopropyl,
n-butyl, sec-butyl, tert-butyl, pentyl or hexyl groups. The
CA 02473422 2004-08-04
-6-
preferred alkyl group is one of the mentioned C1-C4-alkyl
groups. The n-butyl group is the preferred R alkyl group
substituent.
An alkenyl group is a straight-chain or branched
C1-C6-alkenyl group, such as, 1-propenyl, 2-propenyl, 1-
butenyl, 2-butenyl, 3-butenyl, pentenyl and its isomers, or
hexenyl and its isomers. The preferred R alkenyl group
substituents are 2-butenyl and 3-butenyl.
Suitable representatives of cycloalkyl groups are
cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl groups.
Both the benzyl group and the phenyl group can
contain substituents, such as, the above-mentioned alkyl
groups, halogen atoms, nitro groups or amino groups.
Suitable halogens are chlorine, bromine or
iodine. Preferably, the halogen is chlorine.
Suitably, the glycine ester hydrohalide (III) is
reacted in the presence of a base, suitably at a pH of from
about 7 to 12, preferably from about 9 to 11, with the imidic
acid ester (IV). The glycine ester hydrohalides (III) are
commercially available stable compounds. Suitable bases are
alkali hydroxides, such as, sodium hydroxide or potassium
hydroxide, or alkali alcoholates, such as, sodium or
potassium methylate, ethylate or tert-butylate.
Advantageously, the base is dissolved in a suitable solvent.
Especially suitable solvents are aliphatic alcohols, such as
methanol or ethanol. The imidic acid ester (IV) is suitably
added in the form of a solution in an inert solvent, such as,
aromatic solvents, including toluene and chlorobenzene, or
the above-mentioned aliphatic alcohols.
Advantageously, the reaction of the glycine ester
hydrohalide (III), imidic acid ester (IV) and base takes
place in the stoichiometric ratio of 1:1:1. A suitable
reaction temperature is in the range of from about -20 C to
50 C, preferably from about 0 C to 25 C.
CA 02473422 2006-07-18
-7-
After a reaction time of a few hours, the
corresponding 3,5-dihydroimidazol-4-one (V) can be isolated
by a method known to those skilled in the art, such as by
simple filtration, in yields greater than 95 percent.
Advantageously, the resulting reaction mixture is
prepared without isolation of the 3,5-dihydroimidazol-4-one
(V) for further processing to the corresponding 5-
chloroimidazole (I) (one-reactor process).
The first step of the process according to the
present invention represents a tremendous improvement over
the known process according to R. Jacquier et al Bull. Soc.
Chim. France, 1040; 1971, wherein the free glycine ester is
reacted with an imidic acid ethyl ester in the absence of a
solvent to the corresponding 3,5-dihydroimidazol-4-one. A
disadvantage of the known process is the fact that the free
glycine ester is very unstable and, therefore, must be newly
synthesized and isolated for every reaction. According to
the known process, after a reaction time of 24 hours and
more, yields of only 30 to 48% could be obtained.
In the second step, the 3,5-dihydroimidazol-4-one
(V) is chlorinated to the corresponding 5-chloroimidazole
(I). Suitably the chlorination takes place with thionyl
chloride or phosphorus oxychloride, advantageously with an
excess of the chlorinating agent of from about 10 to 300%, at
a reaction temperature in the range of from about 20 C to
110 C. In this step, the chlorinating agent can also serve
as the solvent so that, generally, an additional solvent is
not necessary. Preferably, phosphorus oxychloride is used as
the chlorinating agent. The resultant 5-chloroimidazole (I)
can be isolated with a high purity from the reaction mixture
in a manner known to those skilled in the art, preferably by
extraction.
Preferred 5-chloroimidazoles of the general
formula (I) are those wherein R represents n-butyl, 2-butenyl
or 3-butenyl.
CA 02473422 2006-07-18
-8-
The starting material for the further reaction
according to the present invention to a 5-chloroimidazole-4-
carbaldehyde (II) is a 5-chloroimidazole (I), wherein R1 is
hydrogen. The reaction to the desired 5-chloroimidazole-4-
carbaldehyde (II) takes place according to the present
invention with phosphorus oxychloride or phosgene in the
presence of N,N-dimethylformamide. Suitably the molar ratio
of the 5-chloroimidazole (I) to phosphorus oxychloride or
phosgene to N,N-dimethylformamide is in the range of from
about 1:1:1 to 1:5:5, preferably at about 1:3:3. The
reaction temperature is suitably in the range of from about
50 C to 130 C. Optionally, in the presence of an additional
inert solvent, it is possible in the one-reactor process to
conduct the reaction in the solvent of the first step.
The isolation of the resultant 5-chloroimidazole-
4-carbaldehyde (II) from the reaction mixture takes place
advantageously in a manner known to those skilled in the art
by extraction with a suitable solvent.
The following Examples illustrate the present
invention.
EXAMPLE 1
Production of 2-n-butyl-3,5-dihydroimidazol-4-one
31.71 g (0.25 mol) of glycine methyl ester
hydrochloride was added to a solution of 10.1 g (0.25 mol)
sodium hydroxide in methanol at 0 C. After 15 minutes, 126.5
g of a 22.8 % solution of pentanimidic acid methyl ester in
chlorobenzene was added over a period of 5 minutes, dropwise,
to the resulting white suspension. The light yellow
suspension was stirred for 4 hours at room temperature and
diluted with chlorobenzene (100 ml). The methanol was
distilled off at a temperature of 26 C and at a pressure of
from 30 to 50 mbar. The resulting orange suspension was
diluted with methylene chloride (100 ml) and then filtered.
After removal of the solvent from the filtrate, 34.08 g(97 a)
CA 02473422 2006-07-18
-9-
of 2-n-butyl-3,5-dihydroimidazol-4-one (content >95%,
according to GC and 1H-NMR) was obtained.
Production of 2-n-butyl-5-chloro-lH-imidazole
2-n-Butyl-3,5-dihydroimidazol-4-one (14.02 g, 0.1
mol) was added in portions over a period of 15 minutes to
POC13 (50 ml) at 95 C. The solution was heated for 2 hours
at 100 C, cooled and poured over 400 g of ice. The mixture
was adjusted to pH 7 with 255 ml of 30% sodium hydroxide
solution and extracted three times with 500 ml aliquots of
ethyl acetate. The combined organic phases were dried over
MgSO4, filtered and concentrated by evaporation with a rotary
evaporator. After purification of the residue by column
chromatography, 2-n-butyl-5-chloro-lH-imidazole (5.52 g,
34.7%) was obtained in a high yield (>98%, according to GC
and 1H-NMR). The product had a melting point of from about
85 to 87 C. Other data regarding the product were:
1H-NMR (CDC13) b 0.91 (3H, t, J = 7.5 Hz),
1.36 (2H, sextet, J = 7.5 Hz),
1.68 (2H, q, J = 7.5 Hz),
2.70 (2H, t, J = 7.5 Hz),
6.83 (1H, s),
10.65 (1H, br. s).
EXAMPLE 3
Production of 2-n-butyl-5-chloroimidazole-4-carbaldehyde from
2-n-butyl-5-chloro-lH-imidazole
N,N-dimethylformamide (1.46 g, 20 mmol) was added
to a solution of 2-n-butyl-5-chloro-lH-imidazole (1.60 g, 10
mmol) in POC13 (3.07 g, 20 mmol) and chlorobenzene (20 ml)
heated to 95 C. The mixture was stirred for 3.5 hours at
98 C. Further portions of POC13 (1.53 g, 10 mmol) and N,N-
dimethylformamide (0.73 g, 10 mmol) were added thereafter.
After another 2.5 hours at 98 C the mixture was cooled and
poured over ice (40 g). After 15 minutes, the mixture was
CA 02473422 2006-07-18
-10-
adjusted to pH 7 with 11 ml of 30% sodium hydroxide solution
and extracted three times with 100 ml aliquots of ethyl
acetate. The combined organic phases were dried over MgSO4,
filtered and concentrated by evaporation. 2-n-Butyl-5-
chloroimidazole-4-carbaldehyde was obtained in a yield of 1.3
g (70%).