Note: Descriptions are shown in the official language in which they were submitted.
CA 02474645 2011-01-13
-1-
COMPOSITIONS AND METHODS FOR
SYSTEMIC INHIBITION OF CARTILAGE DEGRADATION
Field of the Invention
The present invention relates to therapeutic compositions and methods for the
protection of articular cartilage.
Background of the Invention
Diseases and conditions that cause the destruction of cartilage within the
joints poses a significant public health concern, particularly in view of the
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-2-
demographics of an aging population. The articular cartilage of the joint
represents a
complex system of many different molecules. Multiple mechanisms are involved
in
the degradation of articular cartilage in arthritides such as rheumatoid
arthritis (RA)
and osteoarthritis (OA). OA, a non-inflammatory arthritis, is the most common
form
of joint disease, and is second only to cardiovascular disease as a cause of
early
retirement and disability. Some individuals exhibit OA in a single or limited
number
of joints, such as may result from traumatic injury due to accident or
surgery. Many
other individuals suffer from OA in multiple joints due to wear and tear
associated
with aging or with athletic or occupational activity over an extended period
of time.
RA is the most common form of inflammatory arthritis, affecting 3% of women
and
1% of men. The majority of RA patients have symptoms in multiple joints,
especially the small joints of the hand, the elbows, the wrists and. the
shoulders.
The destruction of hyaline articular cartilage is the hallmark of OA and
disabling RA. Although various therapeutic approaches may provide relief of
symptoms, no therapeutic regimen has been proven to retard progression of
articular
cartilage degradation. The progressive deterioration and loss of articular
cartilage
leads to an irreversible impairment of joint motion. These changes in
cartilage are
the final pathogenic events that are common to osteoarthritis (OA) and
rheumatoid
arthritis (RA).
Cartilage destructive processes may also be associated with or initiated by
surgical procedures of the joint. Arthroscopy is a surgical procedure in which
a
camera, attached to a remote light source and video monitor, is inserted into
an
anatomic joint (e.g., 'knee, shoulder, etc.) through a small portal incision
in the
overlying skin and joint capsule. Through similar portal incisions, surgical
instruments may be placed in the joint, their use guided by arthroscopic
visualization.
As arthroscopists' skills have ' improved, an increasing number of operative
procedures, once performed by "open" surgical technique, now can be
accomplished
arthroscopically. Such procedures include, for example, partial meniscectomies
and
ligament reconstructions in the knee, shoulder acromioplasties and rotator
cuff
debridements and elbow synovectomies. As a result of widening surgical
indications
and the development of small diameter arthroscopes, wrist and ankle
arthroscopies
also have become routine.
Throughout each arthroscopy, physiologic irrigation fluid (e.g., normal saline
or lactated Ringer's) is flushed continuously through the joint, distending
the joint
capsule and removing operative debris, thereby providing clearer intra-
articular
visualization. -U.S. Patent 4,504,493 to Marshall discloses an isomolar
solution of
CA 02474645 2005-04-29
-3-
glycerol in water for a non-conductive and optically clear irrigation solution
for
arthroscopy. Conventional physiologic irrigation fluids do not provide
analgesic,
anti -inflammatory or anti-cartilage degradation effects.
Summary of the Invention
An object of the present invention is to provide compositions and methods
for systemic inhibition of cartilage degradation. In accordance with an aspect
of
the present invention, there is provided a targeted drug delivery system for
the
protection of cartilage, comprising a plurality of chondroprotective agents
contained within a delivery vehicle, the delivery vehicle being coupled to an
antibody or antibody fragment that is specific to an antigenic determinant
localized within the joint, the plurality of chondroprotective agents
including at
least one anabolic chondroprotective agent and at least one inhibitor of
cartilage
catabolism, each being included in therapeutically effective amounts such that
the
plurality of chondroprotective agents both inhibit cartilage catabolism and
promote cartilage anabolism.
In accordance with another aspect of the present invention, there is
provided a targeted drug delivery system for the protection of cartilage,
comprising a plurality of chondroprotective agents, wherein at least one of
the
chondroprotective agents is contained within a delivery vehicle that is
targeted to
the joint, the plurality of chondroprotective agents including at least one
anabolic
chondroprotective agent and at least one inhibitor of cartilage catabolism,
the
plurality of chondroprotective agents each being included in therapeutically
effective amounts such that the plurality of chondroprotective agents both
inhibit
cartilage catabolism and promote cartilage anabolism.
In accordance with another aspect of the present invention, there is
provided a targeted drug delivery system for the protection of cartilage,
comprising a plurality of chondroprotective agents, wherein at least one of
the
CA 02474645 2005-04-29
-3a-
chondroprotective agents is contained within a delivery vehicle that is
targeted to
a molecule, cell or structure of hyaline cartilage, the plurality of
chondroprotective agents including at least one anabolic chondroprotective
agent
and at least one inhibitor of cartilage catabolism, the plurality of
chondroprotective agents each being included in therapeutically effective
amounts
such that the plurality of chondroprotective agents both inhibit cartilage
catabolism and promote cartilage anabolism.
In accordance with another aspect of the present invention, there is
provided a targeted drug delivery system for the protection of cartilage,
comprising a therapeutically effective amount of at least one
chondroprotective
agent, which is an anabolic chondroprotective agent or an inhibitor of
cartilage
catabolism, contained within targeted immunoparticles that are coupled to
antibodies or antibody fragments that are specific to an antigenic determinant
localized within the joint.
In accordance with another aspect of the present invention, there is
provided a targeted drug delivery system for the protection of cartilage,
comprising a therapeutically effective amount of anabolic chondroprotective
agent contained within a delivery vehicle that is targeted to the joint.
In accordance with another aspect of the present invention, there is
provided a targeted drug delivery system for the protection of cartilage,
comprising a therapeutically effective amount of anabolic chondroprotective
agent contained within a delivery vehicle that is targeted to a molecule, cell
or
structure of hyaline cartilage.
In accordance with another aspect of the present invention, there is
provided a method of protecting cartilage in a patient, comprising: delivering
to
the patient in need thereof a targeted drug delivery system comprising a
plurality
of chondroprotective agents contained within a delivery vehicle, the delivery
vehicle being coupled to an antibody or antibody fragment that is specific to
an
antigenic determinant localized within the joint, the plurality of
chondroprotective
CA 02474645 2005-04-29
-3b-
agents including at least one anabolic chondroprotective agent and
at least one inhibitor of cartilage catabolism, each being included in
therapeutically effective amounts such that the plurality of chondroprotective
agents both inhibit cartilage catabolism and promote cartilage anabolism.
In accordance with another aspect of the present invention, there is
provided a method of protecting cartilage in a patient, comprising:
concurrently
administrating to the patient in need thereof a plurality of chondroprotective
agents, wherein at least one of the chondroprotective agents is contained
within a
delivery vehicle that is targeted to the joint, the plurality of
chondroprotective
agents including at least one anabolic chondroprotective agent and at least
one
inhibitor of cartilage catabolism, the plurality of chondroprotective agents
each
being included in therapeutically effective amounts such that the plurality of
chondroprotective agents both inhibit cartilage catabolism and promote
cartilage
anabolism.
In accordance with another aspect of the present invention, there is
provided a method of protecting cartilage in a patient, comprising:
concurrently
administrating to the patient in need thereof a plurality of chondroprotective
agents, wherein at least one of the chondroprotective agents is contained
within a
delivery vehicle that is targeted to a neoepitope associated with the
degeneration
of articular cartilage, the plurality of chondroprotective agents including at
least
one anabolic chondroprotective agent and at least one inhibitor of cartilage
catabolism, the plurality of chondroprotective agents each being included in
therapeutically effective amounts such that the plurality of chondroprotective
agents both inhibit cartilage catabolism and promote cartilage anabolism.
In accordance with another aspect of the present invention, there is
provided a method of protecting cartilage in a patient, comprising:
concurrently
administrating to the patient in need thereof a plurality of chondroprotective
agents, wherein at least one of the chondroprotective agents is contained
within a
delivery vehicle that is targeted to molecules, cells or structures of hyaline
CA 02474645 2005-04-29
-3c-
cartilage, the plurality of chondroprotective agents including at least one
anabolic
chondroprotective agent and at least one inhibitor of cartilage catabolism,
the
plurality of chondroprotective agents each being included in therapeutically
effective amounts such that the plurality of chondroprotective agents both
inhibit
cartilage catabolism and promote cartilage anabolism.
The present invention provides methods and compositions for reducing or
preventing destruction of articular cartilage iii a joint, by administering a
combination of two or more metabolically active chondroprotective agents.
Metabolically active agents include, ' but are not limited to, compounds that
act
directly or indirectly to modulate or alter the biological, biochemical or
biophysical
state of a cell, including agents that alter the electrical potential of the
plasma
membrane, the ligand binding or enzymatic activity of cellular receptors,
intracellular
or extracellularly located enzymes, protein protein interactions, RNA-protein
interactions, or DNA protein interactions. In* one aspect of the present
invention
pharmaceutical compositions of metabolically active chondroprotective agents
are
provided that are based upon a combination of at least two agents that act
simultaneously on distinct molecular targets-
Representative chondroprotective - agents include, for example:
(1) antagonists of receptors for the interleukin-l family of proteins,
including, for
example, IL-lP, IL-17 and IL-18; (2) antagonists of the tumor necrosis factor
(TNF)
receptor family, including, for example, TNF-Rl; (3).agonists for interleukin
4, 10
and 13 receptors; (4) agonists for the. TGF-(3 receptor superfamily,
including, for
example, BMP-2, BMP-4 and BMP-7;. (5) inhibitors of COX-2; (6) inhibitors of
the
MAP Rinse family, including, for example, p38 MAP kinase; (7) inhibitors of
the
matrix metalloproteinases (MM!!) family of proteins, including, for example,
MMP-3
and MMP-9; (8) inhibitors of. the NF-xB family of proteins, including, for
example,
the p50/p65 dieter complex with IxB; (9) inhibitors of the nitric oxide
synthase
(NOS) family, including, for example, iNOS;. (10) agonists and antagonists of
integrin receptors, including, for example, agonists of avj33 integrin; (11)
inhibitors
of the protein kinase C (PKC) family; (12) inhibitors of the protein tyrosine
ldnase
family, including, for example, the src subfamily, (13) modulators of protein
tyrosine,
phosphatases; and (14) inhibitors : of protein =src - homology 2 (SH2)
domains.
Additional chondroprotective agents include other growth factors, ,such as by
way of
example insulin-like growth factors (e.g., IGF-1) and fibroblast growth
factors (e-g.,
bFGF).-
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-4-
In a preferred embodiment, at least one agent is a cytokine or growth factor
receptor agonist that directly provides anti-inflammatory activity and/or
promotes
cartilage anabolic processes, also referenced herein as an "anabolic agent,"
and at
least a second agent is a receptor antagonist or enzyme inhibitor that acts to
inhibit
cartilage catabolic processes and that may also inhibit pro-inflammatory
processes,
also referenced herein as an "inhibitor of cartilage catabolism" or "catabolic
inhibitory agent". As used herein, the term "chondroprotective agents" is
intended to
include both anabolic agents and inhibitors of cartilage catabolism.
In this embodiment of the invention, at least a first chondroprotective agent
is
an anti-inflammatory/anabolic cytokine, which act functionally to suppress the
role
of pro-inflammatory cytokines in the joint, promote cartilage matrix synthesis
and
inhibit matrix degradation. These receptor agonists include, for example,
specific
anti-inflammatory and anabolic cytokines, such as the interleukin (IL)
agonists (e.g.,
IL-4, IL-10 and IL-13) and specific members of the transforming growth factor-
a
superfamily (e.g., TGF(3 and BMP-7), insulin-like growth factors (e.g., IGF-1)
and
fibroblast growth factors (e.g., bFGF). At least a second chondroprotective
agent is
drawn from a class of cartilage catabolic inhibitors that include receptor
antagonists
or enzyme inhibitors that acts to inhibit and reduce the activity or the
expression of a
pro-inflammatory molecular target (e.g., the IL-1 receptor antagonists, TNF-a
receptor antagonists, cyclooxygenase-2 inhibitors, MAP kinase inhibitors,
nitric
oxide synthase (NOS) inhibitors, and nuclear factor kappaB (NFKB) inhibitors).
The
second chondroprotective agent may also be selected from inhibitors of matrix
metalloproteinases that inhibit cartilage catabolism, cell adhesion molecules,
including integrin agonists and integrin antagonists, that inhibit cartilage
catabolism,
intracellular signaling inhibitors, including protein kinase C inhibitors and
protein
tyrosine kinase inhibitors, that inhibit cartilage catabolism, and inhibitors
of SH2
domains that inhibit cartilage catabolism.
Articular cartilage is a specialized extracellular matrix that is produced and
maintained by metabolically active articular chondrocytes. The maintenance of
a
normal, healthy extracellular matrix reflects a dynamic balance between the
rate of
biosynthesis and incorporation of matrix components, and the rate of their
degradation and subsequent loss from the cartilage into the synovial fluid.
While the
regulatory mechanisms that underlie the matrix homeostasis are not well
understood,
they are clearly altered in inflammatory joint diseases and in response to
joint trauma
such that the rate of matrix breakdown exceeds the rate of new synthesis of
matrix
components. Matrix homeostasis is generally regarded to represent a dynamic
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-5-
balance between the effects of catabolic cytokines and anabolic cytokines
(including
growth factors). The optimal combination of therapeutic agents useful for
cartilage
protection shifts the dynamic matrix equilibrium through accelerating the
synthetic
rate and simultaneously inhibiting the rate of breakdown, thus maximizing
anabolic
processes and promoting repair.
Catabolic cytokines, such as IL-1(3 and TNF-a act at specific receptors on
chondrocytes to induce production of MMPs that induce matrix degradation while
the degradation is inhibited by anabolic cytokines such as TGF-(3, BMP-2 and
IGF-l.
Hence, a therapeutic approach that is based only upon inhibiting,catabolic
processes
(such as a combination of an MMP inhibitor and an IL-1 antagonist) is not
optimal
for cartilage repair since anabolic agents are needed to induce or accelerate
biosynthesis and assembly of components for matrix production. Secondly, the
multiplicity of catabolic cytokines (IL-1, TNF, IL-17, IL-18, LIF) that
contribute to
cartilage matrix destruction indicate it will not be practical to block all
the catabolic
cytokine activity. Conversely, an approach that relies only upon use of
anabolic
agents, such as IGF-l, BMP-2 or BMP-7, is not optimal since it does not
address the
counter-regulatory role of the catabolic cytokines. TGF-(3, BMP-2 and IGF-1
also
act at specific receptors to induce chondrocytes to produce matrix components,
which is inhibited by IL-10, TNF-a, IL-17 and LIF. Therefore, the optimal
therapeutic combination for chondroprotection is believed to be composed of at
least
one anabolic agent and one -inhibitor of cartilage catabolism.
In one aspect of the present invention, a plurality of chondroprotective
agents
are administered via a systemic route to a patient at risk of articular
cartilage
degradation. The plurality of agents that are administered systemically
include at
least one agent that promotes cartilage anabolic activity and at least one
agent that
inhibits cartilage catabolism. Each . agent is included in a sufficient amount
to
provide a combination that is therapeutically effective when the solution is
delivered
to the joint of a patient to both inhibit cartilage catabolic processes and to
promote
cartilage anabolic processes. Additionally, one or more agents that act to
inhibit pain
and/or inflammation may be administered with the chondroprotective agents.
Systemic administration of the plurality of chondroprotective agents may be
preferred when a patient is at risk of cartilage degradation, or suffers from
degenerative disease, at multiple joints simultaneously.
In order to minimize adverse or unwanted systemic effects, in one aspect of a
systemically delivered embodiment of the invention, a therapeutic strategy is
to
deliver the combination of agents in a carrier or delivery vehicle that is
targeted to
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-6-
the joint. In a preferred targeted embodiment, the at least one anabolic
chondroprotective agent and/or the at least one catabolic inhibitory
chondroprotective
agent, and preferably both the anabolic and catabolic inhibitory
chondroprotective
agents, may be encapsulated within a delivery vehicle, such as a nanosphere. A
targeting antibody or antibody fragment is coupled to the nanosphere. The
antibody
or antibody fragment is specific to a targeted antigenic determinant that is
localized
within the joint. A therapeutic method of the present invention includes
systemically
administering this targeted, encapsulated composition of one or more
chondroprotective agents to a patient at risk of cartilage degradation,
preferably by
intravascular, intramuscular, subcutaneous or inhalational administration.
In a further aspect of the present invention, compositions for systemic
administration are provided that include a plurality of chondroprotective
agents,
including at least one agent that promotes cartilage anabolic activity and At
least one
agent that inhibits cartilage catabolism. Additionally, one or more agents
that act to
inhibit pain and/or inflammation may be included in the compositions. All
agents are
included at a dosage sufficient to provide a cartilage protective therapeutic
effect at a
joint or joints when administered systemically. Methods of manufacturing a
medicament including such a composition for use in treating a patient at risk
of
cartilage degradation are also provided.
In order to target such systemically administered compositions to the joint,
the at least one anabolic chondroprotective agent and/or the at least one
catabolic
inhibitory chondroprotective agent, and preferably both the anabolic and
catabolic
'inhibitory chondroprotective agents, are encapsulated within a delivery
vehicle, such
as a nanosphere, to which is coupled an antibody or antibody fragment that is
specific
to an antigenic determinant that is localized within the joint. A method is
also
provided for manufacturing such a medicament including an encapsulated
chondroprotective agent(s) coupled to an antibody or antibody fragment, such
antibody or antibody fragment being targeted to an antigenic determinant that
is
localized within the joint, for use in treating a patient at risk of cartilage
degradation.
In a different aspect of the invention, a composition which includes one or
preferably multiple metabolically active chondroprotective agents together
with one
or more agents for the inhibition of pain, inflammation, or the like, or more
preferably a multiple agent combination of anabolic agents and inhibitors of
catabolism, in a pharmaceutically effective carrier may be prepared for intra-
articular
delivery directly to the joint of a patient. While systemic delivery of the
chondroprotective compositions of the present invention may be preferred for
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-7-
diseases or conditions affecting multiple joints, local delivery of the
compositions of
the present invention may be preferred in other instances. Such instances may
include the treatment of patients with a cartilage degenerative condition or
diseases
affecting only a single or limited number of joints, periprocedural
administration
associated with an operative or interventional procedure at a joint, or in
instances
where undesirable side effects may be associated with systemic administration.
In
this local delivery aspect of the invention, such compositions are delivered
locally by
infra-articular injection (including for the treatment of cartilage
degenerative diseases
such as osteoarthritis or rheumatoid arthritis) or via infusion, including
administration periprocedurally (i.e., preoperatively and/or intraoperatively
and/or
postoperatively) during surgical arthroscopic procedures.
This local delivery aspect of the present invention provides a solution
constituting a mixture of multiple agents in low concentrations directed at
inhibiting
locally the mediators of pain, inflammation, and cartilage degradation in a
physiologic electrolyte carrier fluid. The invention also provides a method
for
perioperative delivery of the irrigation solution containing these agents
directly to a
surgical site, where it works locally at the receptor and enzyme levels to
preemptively limit pain, inflammation, and cartilage degradation at the site.
Due to
the local perioperative delivery method of the present invention, a desired
therapeutic
effect can be achieved with lower doses of agents than are necessary when
employing other methods of delivery (i.e., intravenous, intramuscular,
subcutaneous
and oral).
The anti-pain and/or anti-inflammation agents and/or anti-cartilage
degradation agents in the solution include agents selected from multiple
classes of
receptor antagonists and agonists and enzyme activators and inhibitors, each
class
acting through a differing molecular mechanism of action for pain and/or
inflammation inhibition and/or cartilage degradation.
In addition to the anti-cartilage degradation agent(s), the compositions of
the
inventions may include anti-pain and/or anti-inflammation agents.
Representative
agents for the inhibition of pain and/or inflammation include, for example:
(1) serotonin receptor antagonists; (2) serotonin receptor agonists; (3)
histamine
receptor antagonists; (4) bradykinin receptor antagonists; (5) kallikrein
inhibitors;
(6) tachykinin receptor antagonists, including neurokininl and neurokininz
receptor
subtype antagonists; (7) calcitonin gene-related peptide (CGRP) receptor
antagonists;
(8) interleukin receptor antagonists; (9) inhibitors of enzymes active in the
synthetic
pathway for arachidonic acid metabolites, including (a) phospholipase
inhibitors,
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-8-
including PLAZ isoform inhibitors and PLC isoform inhibitors, (b)
cyclooxygenase
inhibitors, and (c) lipooxygenase inhibitors; (10) prostanoid receptor
antagonists
including eicosanoid EP-1 and EP-4 receptor subtype antagonists and
thromboxane
receptor subtype antagonists; (11) leukotriene receptor antagonists including
leukotriene B4 receptor subtype antagonists and leukotriene D4 receptor
subtype
antagonists; (12) opioid receptor agonists, including -opioid, 5-opioid, and
x-opioid
receptor subtype agonists; (13) purinoceptor antagonists including P2X
receptor
antagonists and P2y receptor antagonists; and (14) calcium channel
antagonists.
Each of the above agents functions either as an anti-inflammatory agent and/or
as an
anti-nociceptive (i.e., anti-pain or analgesic) agent. The selection of agents
from
these classes of compounds is tailored for the particular application.
The present invention also provides a method for manufacturing a
medicament compounded in one aspect of the invention as a dilute irrigation
solution
for use in continuously irrigating an operative site, typically at the site of
a joint of a
patient, during an arthroscopic operative procedure. In this local delivery
embodiment of the invention, the method entails dissolving in a physiologic
electrolyte carrier fluid at least one anti-cartilage degradation agent and
preferably
one or more anti-pain/anti-inflammatory agents, and for some applications anti-
cartilage degradation agents, each agent included at a concentration of
preferably no
more than about 100,000 nanomolar,'more preferably no more than about 25,000
nanomolar, and most preferably no more than about 10,000 nanomolar.
A method of the local delivery aspect of the present invention provides for
the
delivery of a dilute combination of multiple receptor antagonists and agonists
and
enzyme inhibitors and activators directly to a wound or operative site, during
therapeutic or diagnostic procedures for the inhibition of cartilage
degradation, pain,
and/or inflammation. Since the active ingredients in the solution are being
locally.
applied directly to the operative tissues in a continuous fashion, the drugs
may be
used efficaciously at extremely low doses relative to those doses required for
therapeutic effect when the same drugs are delivered orally, intramuscularly,
subcutaneously or intravenously. As used herein, the term "local" encompasses
application of a drug in and around a wound or other operative site, and
excludes
oral, subcutaneous, intravenous and intramuscular administration. The term
"continuous" as used herein encompasses uninterrupted application, repeated
application at frequent intervals, and applications which are uninterrupted
except for
brief cessations such as to permit the introduction of other drugs or agents
or
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541 PCT
-9-
procedural equipment, such that a substantially constant predetermined
concentration
is maintained locally at the wound or operative site.
The advantages of low dose applications of agents in accordance with this
aspect of the invention are three-fold. The most important is the absence of
systemic
side effects that often limit the usefulness of these agents. Additionally,
the agents
selected for particular applications in the solutions of the present invention
are highly
specific with regard to the mediators and mediation targets on which they
work. This
specificity is maintained by the low dosages utilized. Finally, the cost of
these active
agents per procedure is low.
The advantages of local administration of the agents via irrigation or other
fluid application in accordance with this aspect of the invention are the
following:
(1) local administration guarantees a known concentration at the target site,
regardless of interpatient variability in metabolism, blood flow, etc.; (2)
because of
the direct mode of delivery, a therapeutic concentration is obtained
instantaneously
and, thus, improved dosage control is provided; and (3) local administration
of the
active agents directly to a wound or operative site also substantially reduces
degradation of the agents through systemic processes (e.g., first- and second-
pass
metabolism) that would otherwise occur if the agents were given orally,
intravenously, subcutaneously or intramuscularly. This is particularly true
for those
active agents that are proteins and peptides, which are metabolized rapidly.
Thus,
local administration permits the use of compounds or agents which otherwise
could
not be employed therapeutically. For example, some agents in the following
classes
are peptidic: bradykinin receptor antagonists; tachykinin receptor
antagonists; opioid
receptor agonists; CGRP receptor antagonists; and interleukin receptor
antagonists,
TNF-receptor antagonists; TGF-(3 receptor agonists; BMP-2 and BMP-7 receptor
agonists; IL4, IL10 and IL-13 receptor agonists; and integrin receptor
agonists and
antagonists. Local, continuous delivery to the wound or operative site
minimizes
drug degradation or metabolism while also providing for the continuous
replacement
of that portion of the agent that may be degraded, to ensure that a local
therapeutic
concentration, sufficient to maintain receptor occupancy or enzymatic
saturation, is
maintained throughout the duration of the operative procedure.
Local administration of the solution perioperatively throughout a surgical
procedure in accordance with this aspect of the present invention produces a
preemptive analgesic, anti-inflammatory and cartilage protective effect. As
used
herein, the term "perioperative" encompasses application intraprocedurally,
pre- and
intraprocedurally, intra- and postprocedurally, and pre-, intra- and
postprocedurally.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
1U541YCT
-10-
To maximize the preemptive anti-inflammatory, analgesic (for certain
applications)
and cartilage protective (for certain applications) effects, the solutions of
the present
invention are most preferably applied pre-, intra- and postoperatively. By
occupying
the target receptors or inactivating or activating targeted enzymes prior to
the
initiation of significant operative trauma locally, the agents of the present
solution
modulate specific pathways to preemptively inhibit the targeted pathologic
process.
If inflammatory mediators and processes are preemptively inhibited in
accordance
with the present invention before they can exert tissue damage, the benefit is
more
substantial than if given after the damage has been initiated.
Inhibiting more than one pain, inflammatory or cartilage degradation
mediator by application of the multiple agent solutions of the present
invention has
been shown to dramatically reduce the degree of inflammation and pain, and
theoretically should provide a cartilage protective effect. The irrigation
solutions of
the present invention include combinations of drugs, each solution acting on
multiple
receptors or enzymes. The drug agents are thus simultaneously effective
against a
combination of pathologic processes, including pain and inflammation, and loss
of
cartilage homeostasis. The action of these agents is considered to be
synergistic, in
that the multiple receptor antagonists and inhibitory agonists'of the present
invention
provide a disproportionately increased efficacy in combination relative to the
efficacy
of the individual agents. The synergistic action of several of the agents of
the present
invention are discussed below, by way of example.
Used perioperatively, the solution should result in a clinically significant
decrease in operative site pain and inflammation, and of cartilage
degradation,
relative to currently-used irrigation fluids, thereby decreasing the patient's
postoperative analgesic (i.e., opiate) requirement and, where appropriate,
allowing
earlier patient mobilization of the operative site. No extra effort on the
part of the
surgeon and operating room personnel is required to use the present solution
relative
to conventional irrigation fluids. For optimum chondroprotection in accordance
with
this aspect of the invention, the solutions of the invention are administered
directly to
a joint prior to, during and/or after a surgical procedure.
In a further aspect of the invention, compositions for the protection of
cartilage including anabolic-promoting agents and catabolic inhibitory agents
are
provided. Such combinations such result in a state that is characterized by:
cartilage
anabolic activity equaling or exceeding cartilage catabolic activity; the
maintenance
of cartilage tissue so as to either maintain existing, or to increase,
cartilage volume;
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-11-
or an increase in the synthesis of cartilage matrix by articular chondrocytes
and in the
concomitant reduction in degradation of the cartilage matrix.
Brief Description of the Drawings
The present invention will now be described in greater detail, by way of
example, with reference to the accompanying drawings in which:
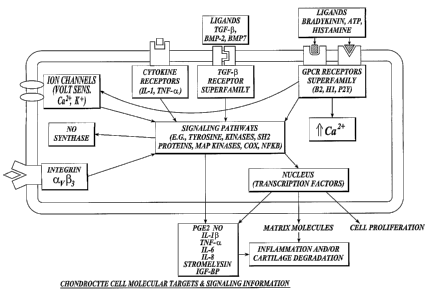
FIGURE 1 is a schematic overview of a chondrocyte cell showing molecular
targets and flow of signaling information leading to the production of
mediators of
inflammation and shifts in cartilage metabolism. The integration of extrinsic
signals
through several families of cell surface receptors, including cytokine
receptor such as
the interleukin-1 (IL-1) receptor family and the tumor necrosis factor (TNF)
receptor
family, the TGF- (3 receptor superfamily and integrins is shown to converge on
common intracellular signaling pathways that include major groups of protein
molecules that are therapeutic targets of drugs included in the solutions of
the present
invention (MAP kinases, PKC, tyrosine kinases, SH2 proteins, COX, PLA2 and NF-
KB. Activation of these signaling pathways controls chondrocyte expression of
a
number of inducible gene products, including IL-1, TNF- a, IL-6, IL-8 and
Stromelysin (MMP 3), and other mediators (nitric oxide (NO) and PGE2) which
may
lead to inflammation and/or cartilage degradation, or synthesis of matrix
molecules
and chondrocyte proliferation;
FIGURE 2 provides a schematic overview of a synoviocyte cell showing
molecular targets and flow of signaling information leading to the production
of
mediators of inflammation and shifts in cartilage metabolism. The integration
of
extrinsic signals through several families of cell surface receptors,
including cytokine
receptors which include the interleukin-1 (IL-1) receptor family and the tumor
necrosis factor (TNF) receptor family, the G-protein coupled receptors which
include
bradykinin, histamine and serotonin subtypes, and integrins is shown to
converge on
common intracellular signaling pathways that include major groups of protein
molecules that are therapeutic targets of drugs included in the solutions of
the present
invention (MAP kinases, PKC, tyrosine kinases, SH2 proteins, COX, PLA2 and NF-
KB-). Activation of these signaling pathways controls synoviocyte expression
of a
number of inducible gene products, including IL-1, TNF-a, IL-6, IL-8 and
Stromelysin (MMP-3), which may lead to inflammation and/or cartilage
degradation;
FIGURE 3 is a diagram of common signaling pathways in both chondrocyte
and synoviocyte cells, including key signaling proteins responsible for
"crosstalk"
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-12-
between GPCR activated receptor pathways and pro-inflammatory cytokine
pathways that lead to inflammation and. or cartilage degradation;
FIGURE 4 is a diagram of common signaling pathways in both chondrocyte
and synoviocyte cells, including key signaling proteins responsible for
"crosstalk"
between GPCR activated receptor pathways and pro-inflammatory cytokine
pathways. Specific molecular sites of action for some drugs in a preferred
chondroprotective solution of the present invention are identified;
FIGURE 5 is a diagram of molecular targets present on either chondrocytes
,or synoviocytes that promote an anabolic response of cartilage. Specific
sites of
action of some drugs in the preferred chondroprotective solution of the
present are
identified;
FIGURE 6 is a diagram ,of molecular targets present on either chondrocytes
or synoviocytes that promote a catabolic response in cartilage. Specific sites
of
action of some drugs in the preferred chondroprotective solution of the
present
invention are identified;
FIGURE 7 is a graphical representation of the production of prostaglandin E2
in synovial cultures by G-protein regulatory agonists following overnight
priming
with interleukin-1 (IL-l, IOU/ml). The cultures were stimulated for the
indicated
times with histamine (100 M, open bars), or bradykinin (1 M, closed bars),
and the
prostaglandin E2 released to the culture supernatant was determined as
described in
Study 1 herein below. The values shown are the mean the standard deviation
from
a representative experiment, and are corrected for basal prostaglandin E2
production
by unstimulated cultures;
FIGURE 8 is a graphical representation of the inhibition of prostaglandin E2
production in synovial cultures by ketoprofen. The cultures were primed
overnight
with IL-1 (lOU/ml) in the presence (shown as "^") or absence (shown as "Li" or
"011)
of the indicated concentrations of ketoprofen. After one day, prostaglandin E2
was
measured in the supernatants of cultures treated overnight with ketoprofen,
and the
remaining cultures were washed, incubated for 10 minutes with the indicated
concentrations of ketoprofen, and then prostaglandin E2 production was
measured in
response to a subsequent 3 minute challenge with histamine (100 M, 0) or
bradykinin (1 M, 0) in the continuing presence of the indicated amounts of
ketoprofen. The data shown are normalized to the maximum response obtained for
each agonist, respectively, and represent the mean the standard deviation
from
three experiments performed on different cell lines; and
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-13-
FIGURE 9 is a graphical representation of the effect of ketoprofen on IL-6
production by synovial cultures at 16 hours in the presence of the indicated
concentrations of IL-1 plus the added G-protein coupled receptor ligands.
Cultures
were incubated for 16 hours with IL-1 at the indicated concentration (o.3, 1.0
and 3.0
pg/ml) in the absence and presence of 0.75 M ketoprofen in experimental
growth
medium with one of the following additional receptor ligands: 1) isoproterenol
(ISO)
at 1.0 M to activate the camp pathway, or 2) histamine (HIS) at 100 M to
activate
the IP3/calcium pathway. Culture supernatants were collected and replaced with
fresh media aliquots containing the same agonist additions at 8 hour
intervals.
Following treatment, the supernatant medium corresponding to the treatment
interval
from 8 to 16 hours was collected and analyzed for IL-6.
Detailed Description of the Preferred Embodiment
The present invention provides methods and compositions for the protection
of cartilage. In a first embodiment, a method is provided for locally
administering to
a joint a composition including at least a first agent that acts to promote
cartilage
anabolic activity and at least a second agent that acts to inhibit cartilage
catabolism.
In a first aspect of this embodiment of the invention, such compositions are
locally
delivered by injection of the composition, which may include a sustained
release
delivery vehicle, into a joint. In a second aspect of this embodiment of the
invention,
the composition includes a liquid irrigation carrier and is locally and
perioperatively
delivered to the joint during an operative or interventional procedure.
In a second embodiment of the invention, a method is provided for
systemically administering to a patient a composition including at least a
first agent
that acts to promote cartilage anabolic activity and at least a second agent
that acts to
inhibit cartilage catabolism.
In a third embodiment of the invention, a method is provided for systemically
administering to a patient a composition including at least a first agent that
acts to
promote cartilage anabolic activity and/or at least a second agent that acts
to inhibit
cartilage catabolism, in which at least one of the agents is targeted to the
joint.
Before each of these embodiments is described in greater detail, and without
wishing
to be limited by theory, a rationale for chondroprotection in accordance with
the
present invention is set forth.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-14-
I. CHONDROPROTECTION RATIONALE
Recent advances in the understanding of the biochemistry and molecular
biology of inflammation and cartilage destruction have implicated a role for
numerous endogenous cytokines. Multiple pro-inflammatory mediators that have
been implicated in producing loss of cartilage in the. inflamed joint are the
cytokines,
TNF-a, IL-1, IL-6 and IL-8. Elevated levels of a number of these pro-
inflammatory
cytokines appear rapidly in the synovial fluid of acutely injured knee joints
and
remain elevated in patients for at least 4 weeks (Cameron, M.L. et al.,
"Synovial fluid
cytokine concentrations as possible prognostic indicators in the ACL-deficient
knee,"
Knee Surg. Sports Traumatol. Arthroscopy 2:38-44 (1994)). These cytokines are
produced locally in the joint from several activated cell types, including
synovial
fibroblasts, synovial macrophages, and chondrocytes. The locally produced
cytokines mediate pathophysiological events in acute and chronic inflammatory
conditions and are important autocrine and paracrine mediators of cartilage
catabolism. The actions of these cytokines are characterized by their ability
to cause
multiple effects on distinct cellular targets and by their ability to interact
in either a
positive or negative synergistic manner with other cytokines. IL-1 and TNF-a
are
particularly important because they also initiate chondrodestructive effects
by
disrupting the balance between the normal turnover and destruction of
cartilage
matrix components by modulating the activity of endogenous proteins (e.g.,
matrix
metalloproteinases (MMPs)) and tissue inhibitor of metalloproteinase (TIMP).
Cytokine control of cartilage homeostasis represents a highly regulated
balance
between active mediators acting on chondrocytes, which determines whether
matrix
degradation or repair occurs.
Injury to the joint frequently produces an inflammatory response within the
joint space that involves the synovial tissue and may lead to degradation of
articular
cartilage. Dramatic shifts in synovial and cartilage metabolism of the human
knee
have been described following joint injury and arthroscopic surgery (Cameron,
M.L.
et al., supra (1994) Cameron, M.L. et al., "The natural history of the
anterior cruciate
ligament-deficient knee: Changes . in synovial fluid cytokine and keratan
sulfate
concentrations," Am. J. Sports Med. 25:751-754 (1997)). Specific pro-
inflammatory
cytokine levels increase dramatically (up to 2-4 orders of magnitude) in knee
joint
synovial fluids during the acute inflammatory phase seen after anterior
cruciate
ligament (ACL) rupture. Significant changes also occur in concentrations of
cartilage matrix molecules due to overproduction of matrix metalloproteinases
(MMPs), such as collagenase and stromelysin-1, which are elevated in the
synovial
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-15-
fluid of patients after acute trauma (Lohmander, L.S. et al., "Temporal
patterns of
stromelysin-1 tissue inhibitor, and proteoglycan fragments in human knee joint
fluid
after injury to the cruciate ligament or meniscus," J. Orthopaedic Res. 12:21-
28
(1994)). Temporally, the changes in cytokines and cartilage matrix markers
(e.g.,
proteoglycans) in synovial fluid, which are correlated with cartilage
degeneration, are
maximal in the acute injury period but persist for extended periods (3 months
to one
year), declining slowly and remaining greater than pre-injury baseline levels.
Trauma due to arthroscopic surgery itself causes significant post-surgical
inflammation that reflects additional inflammatory activation of cells in the
joint,
including upregulation of cyclooxgenase-2 and other pro-inflammatory
cytokines. A
significant proportion (60-90%) of patients with rupture of the ACL show
radiographic changes of the knee indicative of osteoarthritis (OA) 10-15 years
after
injury (Cameron, M.L. et al., supra (1994)). Thus, the combined effects of
initial
joint injury and surgical trauma may induce a sustained inflammatory state and
associated changes in cartilage matrix metabolism which appear to be causative
factors resulting in the subsequent development of degenerative changes in
articular
cartilage and early development of osteoarthritis. The magnitude of this
health
problem is substantial since the total estimated number of arthroscopic
procedures
performed in the United States alone in 1996 was 1.8 million with an estimated
growth rate of approximately 10% per annum. Thus, it is desirable to provide a
pharmaceutical method to prevent degradation of articular cartilage within the
joint.
While post-surgical pain and inflammation are recognized as significant
clinical problems, current pharmacological treatment regimens for arthroscopic
surgery are only directed at acute postoperative analgesia. Existing surgical
treatment modalities do not address the chronic ' inflammatory state that is
induced
postoperatively and the need to inhibit cartilage destruction of the
operatively treated
joint. There is a clear need, therefore, to develop an effective, integrated
drug
therapy that will address both the acute and chronic aspects ~ of pain and
inflammation, as well as pathological changes in cartilage metabolism in the
injured
and operatively treated joint.
According to a first embodiment of this aspect of the invention, a method is
provided for reducing or preventing destruction of articular cartilage in a
joint, by
administering directly to the joint of a patient a composition which includes
one. or
preferably multiple metabolically active chondroprotective agents together
with one
or more agents for the inhibition of pain and/or inflammation, as previously
described, or preferably a combination of two or more metabolically active
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-16-
chondroprotective agents, at least one of which promotes cartilage anabolic
processes
and at least one of which is. an inhibitor of cartilage catabolic processes,
in a
pharmaceutically effective carrier for intra-articular delivery. Metabolically
active
agents include, but are not limited to, all compounds that act directly or
indirectly to
modulate or alter the biological, biochemical or biophysical state of a cell,
including
agents that alter the electrical potential of the plasma membrane, the ligand
binding
or enzymatic activity of cellular receptors, intracellular or extracellularly
located
enzymes, protein-protein interactions, RNA-proteins interactions, or DNA-
protein
interactions. For example, such agents may include receptor agonists that
initiate
signal transduction cascades, antagonists of receptors that inhibit signaling
pathways,
activators and inhibitors of intracellular or extracellular enzymes and agents
that
modulate the binding of transcription factors to DNA.
Suitable chondroprotective agents include, for example, (1) antagonists of
receptors for the interleukin-1 family of proteins, including, for example, IL-
1(3,
IL- 17 and IL- 18; (2) antagonists of the tumor necrosis factor (TNF) receptor
family,
including, for example, TNF-R1; (3) agonists for interleukin 4, 10 and 13
receptors;
(4) agonists for the TGF-(3 receptor superfamily, including, for example, BMP-
2,
BMP-4 and BMP-7; (5) inhibitors of COX-2; (6) inhibitors of the MAP kinase
family, including, for example, p38 MAP kinase; (7) inhibitors of the matrix
metalloproteinases (MMP) family of proteins, including, for example, MMP-3 and
MMP-9; (8) inhibitors of the NF-KB family of proteins, including, for example,
the
p50/p65 dimer complex with IKB; (9) inhibitors of the nitric oxide synthase
(NOS)
family, including, for example, iNOS; (10) agonists and antagonists of
integrin
receptors, including, for example, agonists of aVR3 integrin; (11) inhibitors
of the
protein kinase C (PKC) family; (12) inhibitors of the protein tyrosine kinase
family,
including, for example, the src subfamily; (13) modulators of protein tyrosine
phosphatases; and (14) inhibitors of protein src homology 2 (SH2) domains.
Other
suitable chondroprotective agents for use in the invention include other
growth
factors, such as by way of example insulin-like growth factors (e.g., IGF-1)
and
fibroblast growth factors (e.g., bFGF).
A first embodiment of the present invention provides a pharmacological
method of treating the injured or operatively treated joint using a
combination of
cartilage protective agents delivered locally to achieve maximal therapeutic
benefit.
A second embodiment of the present invention provides a pharmacological method
of providing therapeutic treatment by systemically administering a combination
of
cartilage protective agents. The use of a combination of chondroprotective
agents
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
2U541PCT
-17-
overcomes the limitations of existing therapeutic approaches that rely upon on
the
use of a single agent to block a multifactorial cartilage destructive process
in which a
shift between synthesis and degradation, in favor of catabolic processes has
occurred.
This aspect of the invention uniquely utilizes the approach of combining of
agents
that act simultaneously on distinct molecular targets to promote cartilage
anabolism
and inhibit unregulated or excess cartilage catabolic processes to achieve
maximum
inhibition of inflammatory processes and maintain cartilage homeostasis,
thereby
achieving a chondroprotective effect within the joint. Inhibition of a single
molecular target or biochemical mechanism known to induce cartilage
destruction
(catabolism), such as inhibiting interleukin-1 (IL-1) binding to the IL-1
receptor, will
likely not be optimal, since, for example, the actions of TNF-a mediated
through its
unique receptor shares many overlapping pro-inflammatory and cartilage
catabolic
functions with IL-1 and is also recognized as a major mediator of cartilage
destruction in the joint. Similarly, utilizing pharmaceutical agents that only
enhance
cartilage anabolic processes in the absence of inhibiting catabolic processes
will not
optimally counteract catabolic factors present within the injured joint.
Specifically, one aspect of the present invention provides pharmaceutical
compositions of metabolically active chondroprotective agents that are based
upon a
combination of at least two agents that act simultaneously on distinct
molecular
targets. In a representative embodiment, at least one agent is a cytokine or
growth
factor receptor agonist which directly provides anti-inflammatory activity
and/or
promotes cartilage anabolic processes and at least a second agent is a
receptor
antagonist or enzyme inhibitor that acts to inhibit pro-inflammatory and/or
cartilage
catabolic processes. A representative drug combination includes at least one
agent
drawn from a class of anti-inflammatory/anabolic cytokines that act
functionally to
suppress the role of pro-inflammatory cytokines in the joint, promote
cartilage matrix
synthesis and inhibit matrix degradation. These receptor agonists include, but
are not
limited to, specific anti-inflammatory and anabolic cytokines, such as the
interleukin
(IL) agonists (e.g., IL-4, IL-10 and IL-13) and specific members of the
transforming
growth factor-(3 superfamily (e.g., TGF[3 and BMP-7), insulin-like growth
factors
(e.g., IGF-1) and fibroblast growth factors (e.g., bFGF). At least a second
agent is
drawn from a class of receptor antagonists or enzyme inhibitors that acts to
inhibit
and reduce the activity or the expression of a pro-inflammatory molecular
target
(e.g., the IL-1 receptor antagonists, TNF-a receptor antagonists,
cyclooxygenase-2
inhibitors, MAP kinase inhibitors, nitric oxide synthase (NOS) inhibitors, and
nuclear
factor kappaB (NFKB) inhibitors). The metabolically active agents include both
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-18-
functional agonists and antagonists of receptors located on the surfaces of
cells, as
well as inhibitors of membrane bound or extracellularly secreted enzymes
(e.g.,
stromelysin and collagenase). In addition, many of the agents are directed at
novel
targets that are the intracellularly localized enzymes and transcription
factors that
transduce and integrate the signals of the surface receptors, including
inhibitors of the
enzymes NOS, COX-2, and mitogen-activated protein kinases (MAPK) and
inhibitors of protein-DNA interactions such as the transcription factor NFKB.
This
method allows the integrity of cartilage to be maintained by simultaneously
promoting cytokine-driven anabolic processes and inhibiting catabolic
processes.
The compositions of preferred embodiments of the present invention
constitute a novel therapeutic approach by combining multiple pharmacologic
agents
acting at distinct receptor and/or enzyme molecular targets. To date,
pharmacologic
strategies have focused on the development of highly specific drugs that are
selective
for individual receptor subtypes and enzyme isoforms that mediate responses to
individual signaling neurotransmitters and hormones. Furthermore, despite
inactivation of a single receptor subtype or enzyme, activation of other
receptor
subtypes or enzymes and the resultant signal transduction often can trigger a
cascade
effect. This explains the significant difficulty in employing a single
receptor-specific
drug to block a pathophysiologic process in which multiple signaling mediators
(e.g.,
cytokines, growth factors or eicosonoids) play a role. Therefore, targeting
only a
specific individual receptor subtype or isotype is likely to be ineffective.
In contrast to the standard approach to pharmacologic therapy, the therapeutic
approach of the present compositions is based on the rationale that a
combination of
drugs acting simultaneously on distinct molecular targets is highly effective
in the
inhibition of the full spectrum of events that underlie the development of a
pathophysiologic state. Furthermore, instead of targeting a specific receptor
subtype
alone, the compositions include drugs that target common molecular mechanisms
operating in different cellular physiologic processes involved in the
development of
pain, inflammation, and cartilage degradation (see FIGURE 1). In this way, -
the
cascading of additional receptors and enzymes in the nociceptive,
inflammatory, and
cartilage degradation pathways is minimized. In these pathophysiologic
pathways,
the compositions inhibit the cascade effect both "upstream" and "downstream".
An example of "upstream" inhibition is the cyclooxygenase antagonists in the
setting of pain and inflammation. The cyclooxygenase enzymes (COX, and COX2)
catalyze the conversion of arachidonic acid to prostaglandin H that is an
intermediate
in the biosynthesis of inflammatory and nociceptive mediators including
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-19-
prostaglandins, leukotrienes, and thromboxanes. The cyclooxygenase inhibitors
block "upstream" the formation of these inflammatory and nociceptive'
mediators.
This strategy precludes the need to block-the interactions of the seven
described
subtypes of prostanoid receptors with prostanoid products of the COX
biochemical
pathway. A similar "upstream" inhibitor. is aprotinin, a kallikrein inhibitor.
The
enzyme kallikrein, a serine protease, cleaves the high molecular weight
kininogens in
plasma to produce bradykinins, important mediators of pain and inflammation.
By
inhibition of kallikrein, aprotinin effectively inhibits the synthesis of
bradykinins,
thereby providing an effective "upstream" inhibition of these inflammatory
mediators.
The compositions of the invention may also make use of "downstream"
inhibitors to control the pathophysiologic pathways. In synoviocyte and
chondrocyte
preparations that have been treated with a variety of inflammatory cytokines
(e.g.,
IL-1(3 and TNFa) implicated in progressive articular cartilage degeneration,
MAP
kinase inhibitors produce a cartilage protective effect. The p38 MAP kinase is
a
point of conveyance in signaling pathways for multiple catabolic cytokines,
and its
inhibition prevents upregulation of multiple cellular products mediating
cartilage
degradation. The MAP kinase inhibitors, therefore, provide a significant
advantage
in the setting of joint inflammation by providing "downstream" cartilage
protective
effects that are independent of the physiologic combination of cytokine
receptor
agonists initiating the shift cartilage homeostasis.
II. LOCAL DELIVERY OF CHONDROPROTECTIVE COMPOSITIONS
Specific preferred embodiments of the solution of the present invention for
use in chondroprotection and surgical procedures preferably include a
combination of
agents that act simultaneously on distinct molecular targets to promote
cartilage
anabolism and inhibit unregulated or excess cartilage catabolic processes to
achieve
maximum inhibition of inflammatory processes and maintain cartilage
homeostasis,
thereby achieving a chondroprotective effect within the joint.
The irrigation and injectable solutions of one embodiment of the present
invention are dilute solutions of one or preferably more chondroprotective
agents
and, optionally, one or more pain and/or inflammation inhibitory agents in a
physiologic carrier. The carrier is a liquid solution, which as used herein is
intended
to encompass biocompatible solvents, suspensions, polymerizable and
non-polymerizable gels, pastes and salves, as well as sustained release
delivery
vehicles, such as microparticles, microspheres or nanoparticles composed of
proteins,
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-20-
liposomes, carbohydrates, synthetic organic compounds, or inorganic compounds.
Preferably the carrier is an aqueous solution that may include physiologic
electrolytes, such as normal saline or lactated Ringer's solution.
In each of the surgical solutions of a locally delivered embodiment of the
present invention, the agents are included in low concentrations in a liquid
or fluid
solution and are delivered locally in low doses relative to concentrations and
doses
required with systemic methods of drug administration to achieve the desired
therapeutic effect. As used herein, "liquid" or "fluid" is intended to
encompass
pharmaceutically acceptable, biocompatible solvents, suspensions,
polymerizable and
non-polymerizable gels, pastes and salves. Preferably the carrier is an
aqueous
solution that may include physiologic electrolytes, such as normal saline or
lactated
Ringer's solution. It is impossible or not practical to obtain an equivalent
therapeutic
effect by delivering similarly dosed agents via other (i.e., intravenous,
subcutaneous,
intramuscular or oral) routes of drug administration since drugs given
systemically
are subject to first- and second-pass metabolism. The concentration of each
agent is
determined in part based on its receptor dissociation constant, Kd or enzyme
inhibition constant, Ki. As used herein, the term dissociation constant is
intended to
encompass both the equilibrium dissociation constant for its respective
agonist-
receptor or antagonist-receptor interaction and the equilibrium inhibitory
constant for
its respective activator-enzyme or inhibitor-enzyme interaction. Each agent is
preferably included at a low concentration of 0.1 to 10,000 times Kd or Ki.
Preferably, each agent is included at a concentration of 1.0 to 1,000 times Kd
or Ki
and most preferably at approximately 100 times Kd or Ki. These concentrations
are
adjusted as needed to account for dilution in the absence of metabolic
transformation
at the local delivery site. The exact agents selected for use in the solution,
and the
concentration of the agents, varies in accordance with the particular
application, as
described below.
A solution in accordance with an aspect of the present invention can include
just a single or multiple chondroprotective agent(s), preferably multiple
chondroprotective agents at least one of which is an anabolic
chondroprotective agent
and at least one of which is an inhibitor of cartilage catabolism, or a
combination of
both chondroprotective agent(s) and pain and/or inflammation inhibitory
agents, at
low concentration. However, due to the aforementioned synergistic effect of
multiple agents, and the desire to broadly inhibit cartilage destruction, and
optionally
to block pain and inflammation, and it is preferred that multiple agents be
utilized.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-21-
The multiple drug combination can be delivered locally by intra-articular
injection or via infusion, including administration periprocedurally (i.e.,
preoperatively and/or intraoperatively and/or postoperatively) during surgical
arthroscopic procedures, alone or coupled with postoperative sustained
delivery, such
as by a regulated pump or use of a sustained release delivery vehicle.
Sustained
release delivery vehicles may include, but are not limited to, microparticles,
microspheres or nanoparticles composed of proteins, liposomes, carbohydrates,
synthetic organic compounds, or inorganic compounds. Thus, in some
embodiments,
the invention provides for a combination of agents to be delivered via
injection or
infusion, alone or together with analgesic and/or anti-inflammatory agents.
The rapid
onset of action achieved by direct, local delivery of the chondroprotective
agents at
or closely following the time of injury (e.g., perioperatively) has the
potential to
inhibit initial processes before they can trigger subsequent responses and
thereby
preemptively limit local tissue damage and the subsequent loss of cartilage.
Advantages of the present invention include: 1) a combination drag therapy
directed to the multifactorial causes of cartilage destruction during acute
and/or
chronic conditions; 2) the combination of chondroprotective agents may be
combined
with anti-inflammatory and analgesic agents; 3) local delivery of the drag
combination (where applicable) achieves an instantaneous therapeutic
concentration
of chondroprotective agents within the joint; 4) using an irrigation solution
periprocedurally (where applicable) provides continuous maintenance of drug
levels
within the joint in a therapeutically desirable range during an arthroscopic
surgical
procedure; 5) local delivery (for this embodiment of the invention) permits a
reduction in total drug dose and dosing frequency compared to systemic
delivery; 6)
local site-directed delivery to the joint (for this embodiment of the
invention) avoids
systemic toxicity and reduces adverse effects; and 7) direct, local delivery
to the joint
(for this embodiment of the invention) enables use of novel, pharmaceutically
active
peptides and proteins, including cytokines and growth factors, which may not
be
therapeutically useful if limited to systemic routes of administration.
From the molecular and cellular mechanisms of action defined for these
chondroprotective agents, these compounds are expected to exhibit
chondroprotective action when applied perioperatively in an'irrigation
solution (in
combination with other chondroprotective agents or in combination with other
anti-
pain and anti-inflammation agents described herein) or otherwise administered
directly to the joint via infusion or injection. In particular, these agents
are expected
to be effective drugs when delivered by an irrigation solution during an
arthroscopic
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-22-
surgical procedure. Each metabolically active chondroprotective agent may
preferably be delivered in combination with one or more other
chondroprotective
agents, including small molecule "drugs, peptides, proteins, recombinant
chimeric
proteins, antibodies, oligonucleotides or gene therapy vectors (viral and
nonviral), to
the spaces of the joint. For example, a drug such as a MAPK inhibitor can
exert its
actions on any cells associated with the fluid spaces of the joint and
structures
comprising the joint that are involved in the normal function of the joint or
are
present due to a pathological condition. These cells and structures include,
but are
not limited to: synovial cells, including both Type A fibroblast and Type B
macrophage cells; the cartilaginous components of the joint such as
chondroblasts
and chondrocytes; cells associated with bone, including periosteal cells,
osteocytes,
osteoblasts, osteoclasts; inflammatory cells including lymphocytes,
macrophages,
mast cells, monocytes, eosinophils; and other cells including endothelial
cells,
smooth muscle cells, fibroblasts and neural cells; and combinations of the
above.
This aspect of the present invention also provides for formulations of the
active therapeutic agent(s) that may be delivered in a formulation useful for
introduction and administration of the drug into the joint that would enhance
the
delivery, uptake, stability or. pharmacokinetics of the chondroprotective
agent(s).
Such a formulation may include, but is not limited to, microparticles,
microspheres,
nanospheres or nanoparticles composed of proteins, liposomes, carbohydrates,
synthetic organic compounds, or inorganic compounds. The present invention
provides for the delivery of a combination of chondroprotective agents, or one
or
preferably multiple chondroprotective agents with one or more anti-pain and/or
anti-inflammation agents present either as multiple pharmaceutically active
substances within a homogeneous vehicle (e.g., a single encapsulated
microsphere)
or as a discrete mixture of individual delivery vehicles (e.g., a group of
microspheres
encapsulating one or more agents). Examples of formulation molecules include,
but
are not limited to, hydrophilic polymers, polycations (e.g. protamine,
spermidine,
polylysine), peptide or synthetic ligands and antibodies capable of targeting
materials
to specific cell types, gels, slow release matrices (i.e., sustained delivery
vehicles,
soluble and insoluble particles) as well as formulation elements not listed.
In one aspect, the present invention provides for the local delivery of a
combination of two or more chondroprotective agents, or one or preferably
multiple
chondroprotective agents in combination with one or more anti-pain and/or
anti-inflammation agents, alone or in combination with one or more anti-pain
and/or
anti-inflammatory agents, via an irrigation solution, an infusion containing
the drugs
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-23-
which, are present at therapeutically effective low concentrations and which
enables
the drugs to be delivered directly to the affected tissue or joint. The drug-
containing
infusion or irrigation solution may be employed preoperatively and/or
intraoperatively and/or postoperatively in connection with a surgical
procedure or
may be administered at other times not related to surgical procedures.
Systemic
methods of drug delivery (e.g., intramuscular, intravenous, subcutaneous) have
required higher concentrations of drugs (and higher total dose) to be
administered to
the patient in order to achieve significant therapeutic concentrations in the
targeted
joint. Systemic administration may also result in high concentrations in
tissues other
than the targeted joint, which is undesirable and, depending on the dose, may
result
in adverse side effects. These systemic methods subject the drug to second-
pass
metabolism and rapid degradation, thereby limiting the duration of the
effective
therapeutic concentration. Because the combination of chondroprotective agents
(with or without one or more anti-pain and/or anti-inflammatory agents) are
administered directly to the joint by infusion or by irrigation, vascular
perfusion is
not required to carry the drug to the targeted tissue. This significant
advantage
allows for the local delivery of a lower therapeutically effective total dose
for a
variety of chondroprotective drugs.
A. Local Delivery Methods
The solutions of the present invention have application for a variety of
operative/interventional procedures, including surgical, diagnostic and
therapeutic
techniques. The combination of chondroprotective agents of the invention may
be
administered by injection or by irrigation. For solutions for injection, the
amount of
active ingredient that may be combined with the carrier materials to produce a
single
dosage form will vary depending upon the patient to be treated, the nature of
the
active agents in the solution and the particular mode of administration. It
will be
understood, however, that the specific dose level for any particular patient
will
depend upon a variety of factors including the activity of the specific
compound
employed, the age, body weight, general health, sex and diet of the patient,
time of
administration, route of administration, rate of excretion of the drug
combination,
and the severity of the particular disease undergoing therapy.
Injectable preparations, for example, sterile injectable aqueous or oleagenous
suspensions may be formulated according to the known art using suitable
dispersing
or wetting agents and suspending agents. The sterile injectable preparation
may also
be a sterile injectable solution or suspension in a nontoxic parenterally
acceptable
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-24-
diluent or solvent, for example, as a solution in propanediol. Among the
acceptable
vehicles and solvents that may be employed are water, Ringer's solution, and
isotonic
sodium chloride solution. In addition, sterile, fixed oils are conventionally
employed
as a solvent or suspending medium. For this purpose any biocompatible oil may
be
employed including synthetic mono- or diglycerides. In addition, fatty acids
such as
oleic acid find use in the preparation of injectables.
The solutions for injection of the invention may be administered in
connection with an arthroscopic surgical procedure or at any time determined
to be
desirable by a physician directing patient care.
The irrigation solutions of the invention may be perioperatively applied
during arthroscopic surgery of anatomic joints. As used herein, the term
"perioperative" encompasses application intraprocedurally, pre- and
intraprocedurally, intra- and postprocedurally, and pre-, intra- and
postprocedurally.
Preferably the solution is applied preprocedurally and/or postprocedurally as
well as
intraprocedurally. Such procedures conventionally utilize physiologic
irrigation
fluids, such as normal saline or lactated Ringer's, applied to the surgical
site by
techniques well known to those of ordinary skill in the art. The method of the
present invention involves substituting the anti-pain/anti-
inflammatory/chondroprotective irrigation solutions of the present invention
for
conventionally applied irrigation. fluids. The irrigation solution is applied
to the
wound or surgical site prior to the initiation. of the procedure, preferably
before tissue
trauma, and continuously throughout the. duration of the procedure, to
preemptively
block pain and inflammation, and cartilage degradation. As used herein
throughout,
the term "irrigation" is intended to mean the flushing of a wound or anatomic
structure with a stream of liquid. The term "application" is intended to
encompass
irrigation and other methods of locally introducing the solution of the
present
invention, such as introducing a gellable version of the solution to the
operative site,
with the gelled solution then remaining at the site throughout the procedure.
As used
herein throughout, the term "continuously" is intended to also include
situations in
which there is repeated and frequent irrigation -of wounds at a frequency
sufficient to
maintain a predetermined therapeutic local concentration of the applied
agents, and
applications in which there may be intermittent cessation of irrigation fluid
flow
necessitated by operating technique.
The concentrations listed above for each of the agents within the solutions of
the present invention are the concentrations of the agents delivered locally,
in the
absence ' of metabolic transformation, to the operative site in order to
achieve a
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-25-
predetermined level of effect at the operative site. It is understood that the
drug
concentrations in a given solution may need to be adjusted to account for
local
dilution upon delivery. Solution concentrations in the above embodiments are
not
adjusted to account for metabolic transformations or dilution by total body
distribution because these circumstances are avoided by local delivery, as
opposed to
oral, intravenous, subcutaneous or intramuscular application.
Arthroscopic techniques for which the present solution may be employed
include, by way of non-limiting example, partial meniscectomies and ligament
reconstructions in the knee, shoulder acromioplasties, rotator cuff
debridements,
elbow synovectomies, and wrist and ankle arthroscopies. The irrigation
solution is
continuously supplied intraoperatively to the joint at a flow rate sufficient
to distend
the joint capsule, to remove operative debris, and to enable unobstructed
intra-
articular visualization.
Suitable arthroscopic irrigation solutions for inhibition of cartilage
degradation and control of pain and inflammation during such arthroscopic
techniques are provided in Examples 1-3 herein below.
In each of the solutions of the present invention intended for local delivery,
the agents are included in low concentrations and are delivered locally in low
doses
relative to concentrations and doses required with systemic methods of drug
administration to achieve the desired therapeutic effect. It is impossible to
obtain an
equivalent therapeutic effect by delivering similarly dosed agents via other
(i.e.,
intravenous, subcutaneous, intramuscular or oral) routes of drug
administration since
drugs given systemically are subject to first- and second-pass metabolism and
are
often rapidly cleared from the system circulation.
Practice of the present invention should be distinguished from conventional
intra-articular injections of opiates and/or local anesthetics at the
completion of
arthroscopic or "open" joint (e.g., knee, shoulder, etc.) procedures. The
solutions of
this aspect of the present invention are used for continuous infusion
throughout the
surgical procedure to provide preemptive inhibition of pain and inflammation.
In
contrast, the high concentrations necessary to achieve therapeutic efficacy
with a
constant infusion of currently used local anesthetics can result in profound
systemic
toxicity.
Upon completion of the procedure of the present invention, it may be
desirable to inject or otherwise apply a higher concentration of the same
chondroprotective agent(s) and/or pain and/or inflammation inhibitors as used
in the
irrigation solution at the operative site, as an alternative or supplement to
opiates. It
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-26-
may also be desirable to deliver a sufficient amount of the solution to the
joint after
the surgical procedure so that a bolus of the solution remains in the synovial
capsule
of the patient following the surgical procedure.
As noted previously, the compositions of the present- invention including
multiple chondroprotective agents, including preferably at least one catabolic
inhibitory agent and at least one anabolic-promoting agent, may also be
adapted for
direct injection into an atomic joint. Preferably the agents are selected and
each
agent is included in a sufficient amount to provide a combination that is
therapeutically effective when the solution is delivered locally to the joint
of a patient
to both inhibit cartilage catabolic processes and to promote cartilage
anabolic
processes. Such compositions may be locally injected to provide a
chondroprotective
effect to a patient suffering from a chronic condition, such as osteoarthritis
or
rheumatoid arthritis, or an acute condition such as trauma from surgery or
accidental
injury. One suitable composition for local injection is provided in Example 4
below.
III. Systemic Administration of Chondroprotective Compositions
Embodiments of the present invention have thus far been described in terms
of local delivery of chondroprotective compositions, such as by intra-
articular
injection. Local administration has been described as having several
advantages over
systemic administration, including the avoidance of systemic side effects.
While
local delivery of the compositions of the present invention is preferred in
many
instances, it may not be practical for some cartilage degenerative conditions.
This is
particularly the case for patients suffering from chronic cartilage
degenerative
diseases in which multiple sites simultaneously are at risk of cartilage
degradation,
such as rheumatoid arthritis, polyarticular osteoarthritis and other
polyarthropathies.
For such patients, injection of chondroprotective compositions. into each or a
majority of their diseased sites (e.g., joints) may be painful, impractical,
costly or
otherwise dissuasive of treatment. The chondroprotective compositions
described
above for local delivery may, in accordance with another aspect of the present
invention, be adapted for administration via systemic routes. Systemic
delivery of
the compositions of the present invention is suitable for, but not limited to,
treatment
of patients with multiple sites at risk of cartilage degeneration. . In
addition to
polyarticular osteoarthritis and rheumatoid arthritis, discussed further
below, this
aspect of the present invention may be useful for treating other
noninflammatory and
inflammatory arthrititides including, but not limited to, neuropathic
arthropathy,
- acute rheumatic fever, ochronosis, systemic lupus erythematosus, juvenile
rheumatic
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
1U541YCT
-27-
arthritis, psoriatic arthritis, ankylosing spondylitis, and other
spondyloarthropathies
and crystalline arthropathies.
A. ARTHRITIS MECHANISMS
This aspect of the invention may be better appreciated with an understanding
of the mechanisms involved in the degradation of articular cartilage in
rheumatologic
arthropathies (e.g., rheumatoid arthritis (RA) and osteoarthritis (OA)). RA is
the
most common form of inflammatory arthritis, affecting approximately 3% of
women
and 1% of men. The majority of patients have multiple, and usually
symmetrical,
joint involvement, especially the small joints of the hands, the elbows, the
wrists and
the shoulders. OA is the most common form of joint disease and is second only
to
cardiovascular disease as a cause of early retirement and disability. OA
commonly is
polyarticular. The destruction of hyaline articular cartilage is the hallmark
of OA and
disabling RA.
Although various therapeutic approaches may provide relief of symptoms, no
treatment has been proven to retard progression of articular cartilage
degradation. In
OA, there may be either a suppression of normal chondrocyte functions or the
constitutive inability of these cells to match the rate of repair with the
increased rate
of degradation of the matrix. Various cytokines and inflammatory mediators
have
been shown to either create an imbalance in the synthetic functions of the
chondrocytes or, alternatively, to increase cartilage matrix catabolism by
upregulating various matrix-degrading enzymes, including the matrix
metalloproteinases (including collagenases).
To optimally treat diseases that involve cartilage degeneration, it is
expected
that a treatment regimen that stimulates anabolic processes and simultaneously
inhibits cartilage catabolism will be required. Thus, the therapeutic approach
described previously for the inhibition of cartilage destruction in joint
disease, based
upon a combination of a cartilage anabolic agent and an inhibitor of cartilage
catabolism, is expected to have utility in the treatment of arthritic
conditions such as
OA and RA. As noted above, practical considerations dictate that such diseases
that
simultaneously affect multiple sites in the body are best treated by systemic
administration of these therapeutic agents.
B. COMBINATIONS OF AGENTS
This aspect of the present invention thus provides compositions including
combinations of chondroprotective agents, and methods of systemic
administration
of such compositions. Agents that target differing receptors or molecular
targets are
utilized for a multifactorial approach, as described previously. Preferably
the
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-28-
therapeutic compositions of the present invention include at least one
chondroprotective agent that promotes cartilage anabolic activity, and at
least one
agent that inhibits cartilage catabolism. This combination is expected to
optimize
conditions for homeostasis, and to be preferable over conventional therapies
that
address only cartilage breakdown or more recent research to develop drugs that
address only cartilage synthesis. Suitable anabolic-promoting agents and
catabolic
inhibitory agents 'have been described above for local administration, and are
also
expected to be useful for the present systemic compositions. Aspects and
advantages
of the compositions of the present invention described above with respect to
local
delivery are to be understood to also apply, to the extent applicable, to the
systemic
embodiments of the invention.
Thus chondroprotective compositions of the present invention may suitably
include one or more of the following anabolic-promoting agents, by way of non-
limiting example: interleukin (IL) agonists (e.g., IL-4, IL-10, IL-13
agonists),
members of the transforming growth factor-a superfamily, including TGF-(3
agonists
(e.g., TGF(31, TGFJ32, TGF(33)' and bone morphogenetic protein agonists (e.g.,
BMP-2, BMP-4, BMP-6, BMP-7), insulin-like growth factors (e.g., IGF-1) and
fibroblast growth factors (e.g., bFGF), and fragments, deletions, additions,
amino,
acid substitutions, mutations and modifications that retain the biological
characteristics of these naturally occurring agents.
Chondroprotective compositions of the present invention may suitably
include one or more of the following inhibitors of cartilage catabolism, by
way of
nonlimiting example: IL-1 receptor antagonists, TNF-a receptor antagonists,
cyclooxygenase-2 specific inhibitors, MAP kinase inhibitors, nitric oxide
synthase
inhibitors, nuclear factor kB inhibitors, inhibitors of matrix
metalloproteinases, cell
adhesion molecules (including integrin agonists and integrin antagonists) that
inhibit
cartilage catabolism, intracellular signaling inhibitors (including protein
kinase C
inhibitors and protein tyrosine kinase inhibitors) that inhibit cartilage
catabolism, and
inhibitors of SH2 domains that inhibit cartilage catabolism.
As described with respect to previous embodiments, the at least one inhibitor
of cartilage catabolism in the systemic anabolic agent/catabolic inhibitory
agent'
combination may be a soluble receptor that inhibits cartilage catabolism, such
as a
soluble IL-1 receptor or a soluble tumor necrosis factor receptor. Specific
examples
include recombinant soluble human IL-1 receptors, soluble tumor necrosis
factor
receptors and chimeric rhTNFR:Fc. Examples of soluble tumor necrosis factors
useful for incorporation in the present invention include the functional TNF-a
CA 02474645 2011-01-13
-29-
antagonists disclosed in U.S. Patent No. 5,605,690 issued to Jacobs et al.,
while
examples of soluble human IL-1 receptors useful in the present invention
include
those disclosed in U.S. Patent No. 6,159,460 to Thompson et al.
Particularly promising
catabolic inhibitors useful for combination with anabolic agents for systemic
delivery
in accordance with this aspect of the present invention include IL-lra, TNFRI-
IgG1
fusion protein and inhibitors of matrix metalloproteinases.
As further described below, the chondroprotective compositions may also
include one or.more inhibitors of pain and/or inflammation or other
therapeutic
agents. Examples of chondroprotective compositions suitable for systemic
delivery
are provided in Examples 5 through 20 below.
C. SYSTEMIC DELIVERY
The present aspect. of the invention entails systemic delivery of
chondroprotective agents, and potentially anti-inflammatory and/or analgesic
agents
or other therapeutic agents, so as to provide therapeutic effect at multiple
articular
sites. As used herein, the terms "systemic delivery" and "systemic
administration"
are intended to include but are not limited to oral, intramuscular,
subcutaneous,
intravenous, inhalational, sublingual, buccal, topical, transdermal, nasal,
and other
routes of administration that effectively result in dispersement of the
delivered agent
to a single or multiple sites of intended therapeutic action. Preferred routes
of
systemic delivery for the present compositions are intravenous, intramuscular,
subcutaneous and inhalational. It will be appreciated that. the exact systemic
administration route for selected agents utilized in particular compositions
of the
present invention will be determined in part to account for the agent's
susceptibility
to metabolic transformation pathways associated with a given route of
administration. For example, peptidergic agents may be most suitably
administered
by routes other than oral.
The compositions of the present invention may be systemically administered
on a periodic basis at intervals determined to maintain a desired level of
therapeutic
effect. For example, compositions may be administered, such as by subcutaneous
injection, every two to four weeks. The dosage regimen will be determined by
the
physician considering various factors that may influence the action of the
combination of agents. These factors will include the cartilage site intended
for
treatment, the size of the joint (if appropriate), the amount of cartilage
tissue to be
treated, the site of cartilage damage, the condition of the damaged cartilage
at the
time of treatment, the patient's age, sex and weight, and other clinical
factors. The
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-30-
dosage for each individual agent will vary as a function of the other anabolic
and
catabolic inhibitory agents that are included in the composition, as well as
the
presence and nature of any drug delivery vehicle (e.g., a sustained release
delivery
vehicle). In addition, the dosage quantity may be adjusted to account for
variation in
the frequency of administration and the pharmacokinetic behavior of the
delivered
agent(s). Progress in the treatment of an individual can be monitored through
a
variety of methods known to those in the *art, including clinical assessment,
radiographic and magnetic resonance imaging, computed tomography, biochemical
markers and arthroscopic evaluation.
10- D. DELIVERY VEHICLES AND TARGETING
Methods of compounding compositions for various routes of systemic
administration are known and may be adapted for use with the present
compositions.
The chondroprotective agents and, if included, inflammation and pain
inhibitory
agents or other therapeutic agents are suitably compounded in a physiologic
carrier
or delivery vehicle, such as those described previously, as appropriate for a
given
route of systemic administration. In one aspect of the present invention,
systemic
delivery of these combinations of agents, or any component or components
thereof,
may be incorporated in or combined with a drug delivery vehicle such as a
sustained
release delivery vehicle and/or a depot.
As used herein, the term "delivery vehicle" is intended to include all
structures that contain, couple to or carry a therapeutic agent, such as
nanospheres
and other nanoparticles, microspheres and other microparticles, micelles and
liposomes, including such vehicles formed of proteins, lipids, carbohydrates,
synthetic organic compounds or inorganic compounds. Preferred delivery
vehicles
for the targeted systemic compositions of the present invention, decribed
further
below, are "particles," which is intended to include nanospheres and other
nanoparticles, microspheres and other microparticles, micelles, and other
delivery
vehicles, but excluding liposomes which are less preferred as also described
below.
The term "delivery system" is intended to refer to a delivery vehicle and one
or more
contained or coupled therapeutic agents.
The term "sustained release system" is intended to mean a delivery system
that provides for extended, enhanced or regulated delivery, duration or
availability of
any or all of the incorporated agents. Examples of sustained release systems
include
but are not limited to drug-containing microparticles, microspheres,
nanoparticles,
proteins, liposomes, carbohydrates, synthetic organic compounds, inorganic
compounds and injectable hydrogels such as that disclosed by U.S. Patent
CA 02474645 2011-01-13
-31-
Application Serial Number 09/861,182 to Jun Li et al.
Suitable sustained release systems are known for other pharmaceuticals
and may be adapted in accordance with the - present invention to deliver
chondroprotective agents at a relatively consistent therapeutic level, thereby
reducing
side effects and providing a longer duration of action when- compared to a
bolus
systemic delivery of agents.
The term "depot" as used herein is intended to mean a drug delivery system,
delivered at the site of intended action or remote from the site of intended
action,
which provides a reservoir of therapeutic agents for sustained release.
The individual agents may be compounded in a homogeneous mixture, may
be a mixture or administration of separate microparticles, microspheres, etc.,
or may
be concurrently and separately administered.
In order to minimize adverse or unwanted systemic effects, in the one aspect
of the invention, a therapeutic strategy is to deliver the combination of
agents in a
delivery vehicle that is preferentially targeted to a site or sites in the
body that
contain cartilage, in particular joints. As used herein, a "targeted delivery
vehicle" is
a delivery vehicle that can be used for the systemic delivery of a drug and
that is
adapted such that a greater quantity of the drug reaches the joint or desired
local site
of action than would otherwise reach the joint or desired local site of action
using a
non-adapted delivery vehicle or in the absence of a delivery vehicle (i.e.,
the drug is
preferentially localized to the joint because the targeted delivery vehicle
preferentially associates with molecules, cells or anatomic structures of the
joint).
Likewise, a "targeted delivery system" is such a targeted delivery vehicle
containing
one or more therapeutic drugs. A "targeted drug" is intended to refer to a
therapeutic
agent that is directly linked or coupled to a targeting structure. As used
herein, a.
systemically administered drug that has a "preferential effect" at a joint or
desired
local site of action exhibits greater pharmacological activity at the joint or
desired
local site of action than at the majority of other sites in the body.
1. TARGETING RATIONALE
In OA, there may be either a suppression of normal chondrocyte function or
the constitutive inability of these cells to match the rate of repair with the
increased
rate of degradation of the matrix. Various cytokines and inflammatory
mediators
have been shown to either create an imbalance in the synthetic function of the
chondrocytes or, alternatively, to increase cartilage matrix catabolism by
upregulating various matrix-degrading enzymes, including the matrix
metalloproteinases (including collagenases). Thus, the loss of integrity of
the
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-32-
cartilage extracellular matrix (CEM) is the result of a dynamic imbalance
between
the synthetic activities of the cartilage anabolic processes and catabolic
activities that
lead to degradation.
Systemic administration of cytokines, growth factors and other bioactive
molecules is often associated with serious side effects. For example,
pathological
effects have been correlated with the administration of systemic TGF-(31 and
other
factors. In addition, systemic delivery of an unprotected (naked) polypeptide,
often
preferable for poly joint arthropathies as previously noted, frequently is
limited by
problems with stability of protein agents due to rapid degradation and
inactivation of
the therapeutic protein in the circulation.
The breakdown of articular cartilage in OA and RA is likely to be related to
the synthesis and release of catabolic factors in the microenvironment of the
joint.
Prior studies have demonstrated that pro-inflammatory cytokines (e.g., IL-1)
and
inducible genes (e.g., NO synthase, COX-2, MMPs) are highly expressed in
synovial
membranes from patients with inflammatory arthritides. Similarly, these
mediators
and genes are frequently expressed in OA-affected chondrocytes. In both cases,
it is
a specialized micro environment of the joint that defines a pathophysiological
milieu
that critically affects the state of articular cartilage. For this reason, it
is desirable to
target therapeutic molecules to preferentially localize and act on their
intended
targets within the joint space. This aspect of the invention provides a
mechanism for
targeting a systemically administered anabolic chondroprotective agent and/or
a
systemically administered catabolic inhibitory chondroprotective agent, and
preferably both, to the joint for cartilage protection of the joint.
A preferred route of delivery is thus to target these protein factors to the
site
of action in the joint. To achieve clinical use, a safe method is needed for
the
delivery of these agents within the joints of patients in a sustained and
localized
manner. A biodegradable drug delivery vehicle that both protects and
stabilizes
systemically administered anabolic factors and/or catabolic inhibiting factors
while
outside the joint, and that simultaneously provides a unique method to target
the drug
delivery vehicle to the joint, is also desired.
Cartilage anabolic growth factors are potent mediators that are secreted by
the
body locally within the joint in minute quantities to elicit local biological
responses
in articular cartilage. Under normal physiologic conditions, appropriate
anabolic
growth factors are produced by chondrocytes within cartilage and synoviocytes
within other joint structures in sufficient concentrations to serve as the
necessary
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-33--
signal to maintain the cartilage in a healthy, stable state by influencing
cartilage
matrix metabolism.
2. ANTIBODY TARGETED DELIVERY VEHICLES
A preferred targeted chondroprotective composition of the present invention
includes at least one anabolic-promoting chondroprotective agent and/or at
least one
catabolic inhibitory chondroprotective agent, and preferably both an anabolic-
promoting chondroprotective agent and a catabolic inhibitory chondroprotective
agent, contained within a targeted delivery vehicle. The targeted delivery
vehicle
preferably comprises particles, and most preferably nanoparticles, that
encapsulate at
least one, and preferably all, of the chondroprotective agents. The particles
are
targeted to the joint by a targeting antibody or antibody fragment that is
coupled to
the particle, which antibody or fragment is specific for an antigenic
determinant that
is localized within the joint (i.e., preferentially expressed within the joint
relative to
most other locations within the body, preferably highly expressed within the
joint,
and more preferably that *is restricted in expression to the joint). The
antibody-targeted particles, also referred to herein as "targeted
immunoparticles," and
the chondroprotective agent(s) encapsulated therein are systemically
administered. A
portion of the targeted composition is taken up by the joint. The remainder of
the
composition is excreted and/or metabolized. Within the joint, the targeting
antibodies or fragments bind to the targeted antigen. Over time, the particles
degrade
within the joint, delivering a therapeutic concentration of the
chondroprotective
agent(s) locally within the joint in a sustained release manner over a
predetermined
period of time to act locally on the cells to be modulated (e.g., the primary
cells of
the joint) including the synoviocytes and the chondrocytes. The therapeutic
agents
may diffuse or be released into the synovial fluid to subsequently bind to the
surfaces
of the cells of the joint structures, undergo uptake or entry into cells of
the joint
structures, or directly act on cytokines and/or proteases that may be present
within
the synovial fluid.
The targeted compositions of this aspect of the present invention can be
targeted to the joints of a patient in accordance with the present invention,
without
knowledge of the specific molecular pathology that'underlies the joint
disease. The
targeting antibody ensures that the encapsulated agents are preferentially
localized
within the joint, and more preferably are localized in close proximity to or
are bound
to a constituent of the articular cartilage.
This aspect of the present invention thus provides a method to treat patients
suffering from inflammatory, non-inflammatory or other joint diseases
involving our
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-34-
or multiple joints by administering a pharmaceutical preparation including a
targeted
drug delivery vehicle, preferably containing both a cartilage anabolic agent
and an
anti-catabolic agent. Such.patients may suffer from OA, RA or other diseases
of the
joint such as noninflammatory and inflammatory. arthrititides including, but
not
limited to, neuropathic arthropathy, acute rheumatic fever, ochronosis,
systemic
lupus erythematosus, juvenile rheumatic arthritis, psoriatic arthritis,
ankylosing
spondylitis, and other spondyloarthropathies and crystalline arthropathies.
The
targeted treatment methods and compositions of. this aspect of the invention
are
particularly well suited for patients suffering from osteoarthritis. The
systemic
delivery of the combination of agents in a carrier that is targeted to the
joint enables
treatment of such conditions while minimizing adverse or unwanted systemic
effects.
A. CHARACTERISTICS AND IDENTIFICATION OF
TARGETS WITHIN THE JOINT
The present invention provides methods and compositions for targeting drugs
to the joint, and specifies preferred targets within the joint including
antigenic
determinants associated with molecules, cells and tissues of articular
cartilage and
other joint structures. Examples of such targets are selected from: collagens,
including collagen Type II and the minor collagens Type V, VI, IX, X and XI;
proteoglycans including large aggregating proteoglycans, aggrecan, decorin,
biglycan, fibromodulin and lumican; cartilage oligomeric matrix protein,
glycoprotein-39; proteoglycan chondroitin-sulfate and glycosaminoglycans;
macrophage synoviocytes and fibroblast synoviocytes; and chondrocytes.
In a preferred embodiment, the targeted immunoparticles react irreversibly,
bind in a reversible manner, or associate with specific components of the
articular
cartilage (also known as hyaline cartilage) within the joint. Other molecular
targets
within the joint may include components of the articular cartilage
extracellular
matrix, such as cartilage specific collagens, including collagen Type II, V,
VI, IX, X,
and XI, aggrecan and other small leucine-rich proteoglycans including decorin,
biglycan, fibromodulin and lumican. The proteoglycans are high molecular
weight
complexes of protein and polysaccharide and are found throughout the
structural
tissues of vertebrates, such as cartilage, but also are present on cell
surfaces.
Glycosaminoglycans (GAGs), the polysaccharide units in proteoglycans, are
polymers of acidic disaccharides containing derivatives of the amino sugars
glucosamine or galactosamine, and are useful targets. Cartilage oligomeric
matrix
protein (COMP) and glycoprotein-39 (HC-gp39), also termed YKL-40, similarly
are
useful targets.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
=35-
Articular cartilage contains several genetically distinct types of collagen
that
are useful in the present invention as molecular targets to which
immunoparticles,
including corresponding antibodies,, can, attach, thus permitting delivery of
the
encapsulated therapeutic agents to the site of antibody binding. Type II
collagen, the
primary collagen of articular cartilage, accounts for 90% to 95% of the total
collagen
content of articular or hyaline cartilage and forms the cross-banded fibrillar
structure
noted by electron microscopy. Type II collagen is also a unique and specific
marker
of hyaline cartilage. Hollander, et al., J. Clin. Invest. 93:1722 (1994);
Freed, L et al.,
Exp. Cell Res. 240:58 (1998). A major extracellular modification of the
collagen
molecules, which occurs after fibril formation, is the development of covalent
interfibrillar cross-links. Antibodies that bind to epitopes specific to Type
II collagen
have been described. Kafienah, W. et al., Tissue Engineering 8:817-826 (2002);
Kolettas, E, et al., Rheumatology 40:1146-1156 (2001). Type II collagen and
its
associated epitopes in articular cartilage represent preferred targets for the
present
invention.
For example, a monoclonal antibody to Type II collagen, isotype IgG1,
designated clone 6B3 (Linsenmayer, T.F. et al., Biochem. Biophys. Res. Commun.
92(2):440-6 (1980)) recognizes both al(II) and a3(XI) chains that have
identical
primary structures. In Western blotting, this mAb reacts with the TCA fragment
of
lathyritic Type II collagen after digestion with mammalian collagenase. It
also reacts
with pepsin-digested Type II collagen. Its epitope is localized in the triple
helix of
Type II collagen and it shows no cross-reaction with Type I or Type III
collagen.
Immunoblotting of cyanogen bromide (CNBr) peptides of collagen II shows that
this
mAb reacts with the CB 11 fragment, which is the site of immunogenic epitopes
25, along the intact Type II molecule.
In yet another example, a monoclonal antibody to Type II collagen (isotype
IgG1) (Miller, E.J., Biochemistiy 11:4903-4909 (1972); Glant, T.T., et al.,
Histochemistry 82:149-158 (1985a); British Journal of Haematology 90:757-766
(1995)) was developed using human cartilage specific CNBr-cleaved collagen II
as
the immunogen. This mAb, available commercially through Chemicon International
(Temecula, CA), reacts with both pepsin-solubilized and CNBr-cleaved human and
bovine. Type II collagen. No cross-reactivity is observed with collagen Types
I, III,
V and IX. . In a preferred embodiment of the present invention, the antibody
to
Type II collagen binds to the target epitope with a dissociation constant in
the range
of 0.1-10 nM.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-36-
The quantitatively minor collagens of articular cartilage also contribute to
the
structure of the matrix and serve as useful targets for the present invention.
For
example, Type IX collagen, a short nonfibrillar collagen (which contains a
glycosaminoglycan chain and is therefore also considered a proteoglycan) binds
covalently to Type II collagen fibrils and may help link fibrils together or
bind fibrils
to other matrix molecules. Type XI collagen, a -minor fibrillar collagen, may
be
involved in controlling the diameter of the Type II fibrils. Other collagens,
including
Type V and Type VI, may also form part of the matrix. These minor collagens of
articular cartilage may also function as targets for antibody-directed
immunoparticle
binding with appropriate antibodies.
In another embodiment of the invention, distinct types of proteoglycans
contained in articular cartilage are useful in the present invention as
molecular targets
for the binding of immunoparticles, thus permitting delivery of the
therapeutic agents
to the site of antibody binding. In articular cartilage, proteoglycans
constitute the
second largest portion of the solid phase, accounting for 5% to 10% of the wet
weight. The proteoglycans of the cartilage matrix consist primarily of large
aggregating (50% to 85%) and large nonaggregating (10% to 40%) proteoglycans.
Distinct small proteoglycans are also present.
The proteoglycans of cartilage that contribute most significantly to the
material properties of the tissue are the large, high-molecular weight
monomers
(molecular weight, 1-4 X 106). Structurally, the large proteoglycans consist
of an
extended protein core with several distinct regions: an N-terminal region with
two
globular domains (G1 and G2), a domain rich in keratan sulfate; a longer
domain rich
in chondroitin sulfate that may also contain some interspersed keratan sulfate
and
neutral oligosaccharide chains; and 'a C-terminal globular domain, G3.
Aggregates
are formed by many proteoglycan monomers binding to a chain of hyaluronate at
the
G1 globular domain. Each proteoglycan-hyaluronate bond is stabilized by a
separate
globular link protein (molecular weight, 41,000 to 48,000). The large size of
the
chondroitin sulfate-rich region (200-400 rim) and abundance of chains of the
"chondroitin-sulfate proteoglycan aggregate make this a preferred target for
targeted
immunoparticles of this aspect of the invention.
An additional target for antibodies or fragments thereof that are coupled to
immunoparticles of the present invention is provided by HC gp-39. Within the
joint,
fragments of HC gp-39 that have appropriate antigenic properties also are
sufficient
for targeting the drug delivery vehicle.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-37-
Immunoparticles may be targeted with antibodies or fragments thereof to
react irreversibly, bind in a reversible manner (most commonly), or associate
with
specific structures of the synovial membrane of the joint. The specialized
cells of the
joint that are preferred targets include the two principal cell types of the
synovium,
macrophage synoviocytes (Type A) and fibroblast synovioctyes (Type B).
Additional targets for antibodies or fragments thereof that are coupled to
immunoparticles of the present invention are chondrocytes. These cells are
known to
express a variety of proteins which are present on their surfaces and which
can serve
as epitopes for cellular targeting.
In another embodiment of the invention, the chondroitin-sulfate proteoglycan
associated with articular cartilage represents a preferred target for the
targeted drug
delivery system. Monoclonal antibodies useful for the present invention that
bind to
epitopes specific to chondroitin-sulfate proteoglycan have been described.
Morgan
Jr., A. et al., Hybridoma 1:27-36 (1981); Schrappe, M. et al., Cancer Res.
52:3838-
3844 (1992); Schrappe, M. et al., Cancer Res. 51:4986-93 (1991). One such
example is a mouse-anti-human chondroitin-sulfate proteoglycan monoclonal
antibody, designated clone 9.2.27 (IgG2a isotype). The 9.2.27 antibody
recognizes
the mature chondroitin sulfate proteoglycan core glycoprotein of 250 kDa as
well as
precursor polypeptides of 210, 220 and 240 kDa. A mouse anti-human aggregan
monoclonal antibody, clone 2A2.1, is also suitable for the present invention
and is
commercially available from United States Biological (Swampscott, MA). This
antibody does not react with chondroitin sulfate linkage regions. Transmission
electron microscopy indicates that it binds within the N-terminal portion of
the
chondroitin sulfate-attachment region.
B. TARGETING OF NEOEPITOPES ASSOCIATED
WITH CARTILAGE DEGENERATION
Biomolecular constituents of cartilage that may be either absent from normal
adult cartilage or present at very low levels, but that are elevated or more
highly
expressed in certain stages of either RA or OA, also may serve as -targets for
the
targeted drug delivery systems of the present invention. Preferred targets
associated
with cartilage degenerative conditions are neoepitopes that appear on
articular
cartilage of patients diagnosed with OA, RA or other degenerative joint
diseases,
such as neoepitopes of aggrecan or other cartilage proteoglycans, and
specifically
neoepitopes that are immunolocalized in the superficial layer of articular
cartilage of
such patients.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-38-
In one aspect of the invention, the targeting antibodies or antibody fragments
specifically bind to neoepitopes or cleavage sites of Type II collagen or Type
II
collagen fragments, particularly such neoepitopes or -cleavage sites generated
by the
individual or combined action of matrix metalloproteinase (MMP)-1, 3, 8 or 13,
or
other members of the MMP protein family, or a member of the A Disintegrin And
Metalloproteinase with Thrombospondin motifs' (ADMATS) protein family.
ADMATS are further described in Patent Applications WO 00/04917, EP 0 823 478
and U.S. 5,811,535 and in Tang, B. et al., FEBSLett. 445 (2-3):223-225 (1999).
Antibodies directed at specific epitopes that are defined by specified regions
of the Type II collagen structure are useful for targeting compositions of
this
invention. These structural regions are, in part, important in cartilage
affected by
degradation of Type II collagen that occurs in OA and RA. Degradation of Type
II
collagen results in fragments of the collagen protein that are released. from
the
cartilage matrix and appear in the synovial fluid, move into the circulation,
and are
eliminated through the urine. To have maximal utility, compositions of the
present
invention target epitopes that remain physically associated with the cartilage
matrix
rather than on released fragments.
Each of the collagenases, MMP-1 (EC 3.4.24.7), MMP-8 (EC 3.4.24.34), and
MMP-13 (EC 3.4.24.-) has the capacity to cleave triple-helical fibril-forming
Type II
collagen, giving rise to a large (3/4-length) amino-terminal fragment and a -
smaller
(1/4-length) carboxy-terminal fragment. Kafienah, W. et al., Biochem. J.
331:727-
732 (1998). They all initially cleave at a specific Gly-Leu/Ile bond to
generate the
characteristic % and % fragments. Specific collagenase isotypes have been
implicated in the pathologic loss of cartilage. Billinghurst, R. et al., J.
Clin. Invest.
99:1534-1545 (1997). For example, in an animal model of OA, focal areas of
collagenase 1 and collagenase 3 proteins have been localized to the
extracellular
matrix of OA lesion sites in the knee joint, coincident with 1/ collagen
cleavage.
Collagenase 3 protein was also abundant throughout the medial tibial cartilage
in the
diseased joints. Huebner, J. et al., Arthritis & Rheumatism 41:877-890 (1998).
Collagenase 1 (MMP-1) has been detected in synovium, synovial fluid, and
cartilage samples from humans with RA and with OA. Collagenase 3 (MMP-13)
cleaves collagen Type II at a rate that is 5-10 times faster than collagenase
1.
Significantly, MMP-13 has been identified in the syonvium of humans with RA
and
OA, as well as in human OA cartilage.
Fibrillar collagen can be damaged by helical cleavage, resulting in
denaturation, or by telopeptide cleavage, leading to removal of cross-links.
Two
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-39-
studies have demonstrated. the presence of active 'collagenases in cartilage
with
polyclonal antisera specific for cryptic epitopes within the helical region of
Type II
collagen, which are exposed upon collagen unwinding due to collagenase
cleavage,
and for a collagen neoepitope generated by collagenase. These findings
indicate that
collagenase 1, collagenase 3, or both = are involved in the cartilage
degradation
associated with various arthritides. Thus, the specific degradation of Type II
collagen from collagenase cleavage creates neoepitopes for antibody targeting
that
are useful for specific targeting aspects of this invention. These neoepitopes
will be
located within the N-terminal 3/4 fragment sequence of the alphal (II) chain,
which is
known to contain epitopes to AH12L3 and CB11B (recognized by antibody COL2-
3/4m).
In arthritides, the increased catabolism of the cartilage proteoglycan,
aggrecan, is one of the principal pathological processes that leads to the
degeneration
of articular cartilage. The consequent loss of sulphated glycosaminoglycans,
which
are intrinsic components of the aggrecan molecule, compromises both the
functional
and structural integrity of the cartilage matrix. Over a period of time, this
process
leads to cartilage degradation. In situ degradation of aggrecan is a
proteolytic
process involving cleavage at specific peptide bonds located within the core
protein.
The most well characterised enzymatic activities contributing to this process
are a
result of the specific action of metalloproteinases. In vitro aggrecanolysis
by matrix
metalloproteinases (MMPs) has been widely studied. However, it is now well
recognised that the principal proteinases responsible for aggrecan degradation
in situ
in articular cartilage are the aggrecanases, two recently identified isoforms
of which
are members of the A Disintegrin And Metalloproteinase with Thrombospondin
motifs' (ADAMTS) gene family. Monoclonal antibody technologies exist to
identify
novel' neoepitopes - on aggrecan or aggrecan degradation products.
Furthermore,
another aspect of this invention is the use of such monoclonal antibodies or
fragments thereof that bind to neoepitopes on aggrecan or aggrecan fragments
to
target nanoparticles containing a combination of an anabolic promoting agent
and a
catabolic inhibitor.
Temporal studies have established that aggrecanases are primarily responsible
for the catabolism and loss of aggrecan from articular cartilage in the
earlier stages of
arthritic joint diseases. Although a continuum, this process appears largely
to
precede collagen catabolism. At a later stage in the disease process, when
collagen
catabolism is occurring, there is evidence for MMP-mediated degradation of the
small proportion of aggrecan remaining in the cartilage.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
-40-
It has been possible to develop and use monoclonal antibody technologies to
identify catabolic neoepitopes on proteolytic degradation products to identify
specific
cleavage sites that are unique and characteristic for different families of
matrix
degrading enzymes. In these studies, several monoclonal antibodies were
characterized that specifically identified catabolic neoepitopes (new epitopes
created
as specific N- or C-terminal amino acid sequences of a proteolytic cleavage
product)
that were generated by the action aggrecanases (or MMPs) on the interglobular
(IGD)
domain of aggrecan. These antibodies have been used to monitor the proteolysis
of
aggrecan and link proteins. Furthermore, those skilled in the art will
recognize that
the use of antibodies that recognize neoepitopes on degradation products of
matrix
proteins generated during cartilage catabolism can be used to target
immunoparticles
to the joint, without departing from the scope of this invention.
In another aspect of the invention, the targeting antibodies or antibody
fragments specifically bind to neoepitopes or cleavage sites of aggrecan or
aggrecan
fragments within cartilage, particularly such neoepitopes or cleavage sites
generated
by the individual or combined action of MMP-1, 3, 8 or 13, or other members of
the
MMP protein family or a member of the ADMATS protein family. Examples of
monoclonal antibodies that recognize different structural epitopes or
neoepitopes
within aggrecan or aggrecan fragments are 8-A-4 or BC-3. MAb 2-B-6 has been
used to detect the large number of aggrecan degradation products that result
from
either aggrecanase, MMP or other proteolytic activities at many sites along
the core
protein of aggrecan. MAb 2-B-6 recognizes 4-sulphated unsaturated
dissaccharides
of chondroitin sulphate that are attached to these core protein fragments. A
related
antibody, MAb 3-8-3, has also been used to identify different deglycosylated
aggrecan metabolites containing 6-sulphated chondroiten sulfate
oligosaccharides.
MAb BC-3 recognizes the N-terminal neoepitope sequence defined by the amino
acid sequence, alanine-arginine, glycine (ARGxx...) generated after
aggrecanase
catabolism within the 1 GD domain of aggrecan.
The ADAMTS-4 gene (Genbank NM-005099) and ADAMTS-5 gene
(Genbank 007038) encode a disintegrin and a metalloproteinase with
thrombospondin motifs-4 and 5, which are members of the ADAMTS protein family.
Members of the family share several distinct protein modules, including a
propeptide
region, a metalloproteinase domain, a disintegrin-like domain, and a
thrombospondin
Type 1 (TS) motif. Individual members of this family differ in the number of C-
terminal TS motifs, and some have unique C-terminal domains. The enzyme
encoded by the ADAMTS-4 gene lacks a C-terminal TS motif. The enzyme encoded
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-41-
by the ADAMTS-5 gene contains 2 C-terminal TS motifs and functions as
aggrecanase to cleave aggrecan, a major proteoglycan of cartilage. Thus, both
of
these enzymes are responsible for the degradation of aggrecan and the
generation of
neoepitopes on aggrecan and aggrecan fragments (Tortorella, M., et al., J.
Biol.
Chem. 275(33):25791-25797 (2000); Tortorella, M. et al., J. Biol. Clzem.
275(24):18566-18573 (2000); Abbaszade, I. et al., J. Biol. Chem. 274(33):23443-
23450 (1999)).
In another embodiment of this invention for treatment of human cartilage in
the setting of OA, an antibody is used that targets the early biochemical
neoepitope
marker of OA, termed 3-B-3(-). The 3-B-3(-) epitope is an OA-related
phenotypic
change in the termini of the chondroitin sulfate (glycosaminoglycan) chains
of.
aggrecan.
C. TARGETING ANTIBODY CHARACTERISTICS
As used herein, the term "antibody" is intended to include immunoglobulin
molecules and immunologically active portions of immunoglobulin molecules
(i.e.,
molecules that contain an antigen binding site that specifically binds or
immunoreacts with an antigen). The term describes an immunoglobulin, whether
it
is naturally or synthetically produced. The proteins comprising the antibody
can be
derived from natural sources, or synthetically produced either in part or in
whole.
Examples of antibodies include all immunoglobulin subtypes and Fab and
F(ab')2,
scFv, Fv, dAb, Fd fragments, as well as fragments disclosed in U.S. Patent
5,534,254. ScFV refers to single-chain. minimum binding domains of an
immunoglobulin molecule. The term "antibody" is also intended to refer to an
antibody that functions in the extracellular space, within the plasma membrane
of a
cell, or in an intracellular region of a cell (e.g., the cytoplasm or nucleus)
to modulate
the expression or activity of one or more genes that regulate cartilage
metabolism.
Preferred antibodies for use in the present invention include humanized,
chimeric and
human monoclonal antibodies.
In the context of an antibody, the term "fragment" refers to any- sequence of
amino acids that is part of any targeted polypeptide defined above, having
common
relevant elements of origin, structure and mechanism and functional
equivalence to
the whole antigen for purposes of targeting within the present invention. The
calling
out of an antibody in the compositions and methods described herein is also
intended
to include the use of fragments of such antibodies.
A preferred embodiment of the present invention employs humanized or
human antibodies or fragments thereof that are covalently attached to the
surface of
CA 02474645 2011-01-13
-42-
the nanospheres or other particles in which the therapeutic chondroprotective
agents
of the present invention are encapsulated. Antibodies or fragments thereof are
preferred as the targeting molecules on the surface of particles in which
therapeutic
chondroprotective agents are encapsulated, because they must be sufficiently
stable
in vivo and exhibit a minimum potential of being removed from the surface of
the
particle by serum containing extracellular fluid proteins. It is envisioned
that fully
human monoclonal antibodies or humanized murine antibodies, which bind to any
molecules of the cartilage extracellular matrix or to cells of the joint, will
be most
useful as the type of joint-targeting antibodies that direct the delivery of
the
therapeutic agent(s) to be administered to human patients because they will
not
generate an immune response upon administration.
For example, a murine monoclonal antibody may be chimerized by
genetically recombining the nucleotide sequence encoding the murine Fv region
(i.e.,
containing the antigen binding sites) with the nucleotide sequence encoding a
human
constant domain region and an Fc region. Some murine residues may also be
retained within the human variable-region framework domains to ensure proper
target-site binding characteristics. Humanized antibodies for use in targeting
will be
recognized to have the advantage of decreasing the immunoreactivity of the
antibody
or polypeptide in the host recipient, and may be useful for increasing the in
vivo half-
life and reducing the possibility of adverse immune reactions to the
conjugated
antibody on the surface of the nanoparticle or other encapsulating particle.
It is highly advantageous to employ antibodies or their analogs with fully
human characteristics for treatment of human patients. Methods may be employed-
which are similar to those disclosed in U.S. Patents 6,075,181, 6,235,883 and
6,492,160 and Patent Application EP 1 167 537 Al.
Such methods previously have been
used to generate a variety of fully human antibodies against human IL-8 and
epidermal growth factor receptor. In the most preferred embodiments of the
present
invention, the antibody or antibody fragment binds to a target epitope with
dissociation constant in the range of 0.1 to10 nanomolar:
While fully human or humanized antibodies are preferred for use in the
present invention to treat human patients, it should also be understood that
the
methods and compositions of the present inventions are also useful in
veterinary
applications to treat other mammals susceptible to joint degeneration, (e.g.,
horses
and dogs).
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-43-
D. CHONDROPROTECTIVE AGENTS FOR
TARGETED DELIVERY
A preferred aspect of the targeted embodiment of the present invention will
include at least one chondroprotective agent that is encapsulated or contained
within
a nanoparticle or other delivery vehicle to which a targeting antibody,
antibody
fragment or other targeting structure is attached. The encapsulated or
contained
agent may be any one of anabolic chondroprotective agents or catabolic
inhibitory
agents disclosed herein having chemical or structural characteristics
rendering it
amenable to encapsulation or containment in a selected nanoparticle or other
delivery
vehicle. Additionally, agents that would otherwise be highly susceptible to
metabolic
degradation from systemic administration (e.g., proteins and peptides) or that
cause
harmful side effects if administered systemically without targeting are
preferable for
targeting. For example, each of the classes of anabolic chondroprotective
agents
disclosed herein below (including members of the transforming growth factor
(TGF)-
R superfamily, including TGF-(3 agonists and bone morphogenetic protein
agonists,
insulin-like growth factors and fibroblast growth factors) and certain of the
classes of
catabolic inhibitory agents disclosed herein below (Interleukin-1 receptor
antagonists
and TNF-a receptor antagonists) are proteins, and as such are well suited for
delivery
in targeted encapsulated form.
A most preferred targeted encapsulated composition of the present invention
will include both an anabolic chondroprotective agent and a catabolic
inhibitory
chondroprotective agent encapsulated in the same targeted immunoparticles (or
other
targeted particles), preferably both agents being proteins. Alternately, each
agent
may be separately encapsulated, and an admixture of the two (or more than two)
types of targeted particles may be administered, or less preferably the two or
more
targeted agents may be delivered separately, either concurrently or
sequentially, to
result in coincident presence of the agents within the joint.
In some instances either only the anabolic chondroprotective agent or the
catabolic inhibitory agent may be amenable to encapsulation, or one agent may
not
be associated with undesirable systemic side effects. In such instances, one
agent
may be delivered in encapsulated targeted form, while the other agent is
delivered in
non-targeted, non-encapsulated form, either together in the same dosage form
as an
admixture, or separately.
While not as preferred as administering both an anabolic agent and a catabolic
inhibitory agent, given all of the advantages provided by this combination
described
above, the delivery of a single chondroprotective agent (either anabolic-
promoting or
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-44-
catabolic inhibitory) encapsulated, within an immunoparticle that targets an
antigen
localized within the joint is also possible.
E. ENCAPSULATION OF AGENTS
The size of a substance is a major factor determining whether it can permeate
the wall of synovial capillaries and move from the systemic circulation into
the joint.
The maximum diameter of particles that can move across the synovial capillary
wall
is . generally regarded to be 50 nanometers. However, some studies of the
permeability of the synovial capillaries of the rat using lecitihin-coated
polystyrene
particles up to 240 nanometers in diameter can be transported across synovial
capillary walls via transcytosis. The present invention overcomes the
limitation
imposed by the synovial permeability barrier by preferably employing a class
of
encapsulation particles (e.g., nanoparticles (preferably nanospheres)), which
are
constrained in size distribution.
Sustained release dosage forms of the invention may comprise microparticles
and/or nanoparticles having therapeutic agents dispersed therein, or may
comprise
the therapeutic agent in pure, preferably crystalline, solid form: The
therapeutic
dosage forms of this aspect of the present invention may be of any
configuration
suitable for sustained release. Preferred sustained release therapeutic dosage
forms
of the present invention will have the following size, biodegradation and
biocompatibility characteristics.
The targeted delivery system of the present invention preferably utilizes
nanoparticles that are limited in size from about 5 nanometers to about 750
nanometers in diameter, with about 10 to about 500 nanometers being more
preferred, most preferably from about 20 to about 200 nanometers. Alternately
useful but less preferred, if demonstrated to result in sufficient
permeability in the
diseased state, are microparticles that range in size from about 1 micrometer
to about
100 micrometers in diameter, with about 1 to about 25 micrometers being more
preferred; more preferably from about 1 to about 10 micrometers.
Preferred particles are biodegradable structures that biodegrade and release
loaded drug at therapeutic levels over a period of time preferably between
from about
1 to about 150 days, preferably from about 7 to about 60 days, with from about
14 to
about 30 days being more preferred. It is understood by those in the art that
drug
release from nanoparticles and microparticles may occur by a combination of
physical processes, which include, but are not limited to, diffusion and
degradation
and may be described by complex kinetic processes that are unique to each
carrier
formulation and combination of anabolic and anti-catabolic therapeutic agents.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-45-
Preferred particles are biocompatible with targeted tissues of the joint and
the
local physiological environment into which the dosage form is administered,
including yielding biocompatible biodegradation products. Suitable
compositions
include biodegradable particles formulated from natural polymers, including
hyaluronan, chitosan, collagen, gelatin and alginate. These natural polymers
may be
combined with other polymers to produce copolymer particles composed of, for
example, chitosan and gelatin. Synthetic biodegradable poly(alpha-hydroxy
esters)
such as polylactic acid (PLLA), polyglycolic acid (PGA) and the copolymer PLGA
have been used successfully for the production of microparticles that
incorporate
protein therapeutics, such as human growth hormone. Another example of a
biodegradable polymer that may be suitable for use in preparing targeted
particles of
of the present invention are amphiphilic ABA triblock copolymers such as
poly(ethylene oxide):poly(3-hydroxybutyrate):poly(ethylene oxide).
In selecting a polymer nanoparticle system for use with selected
chondroprotective agents. of the present invention, it is also important to
ensure
adequate bioactivity of the encapsulated drug.
The use of targeted, biodegradable polymeric nanbspheres has the advantage
of providing selected differential release profiles for the encapsulated
therapeutic
agents. For some drug combinations, the optimal release kinetics may consist
of a
dual-release process, wherein each active agent demonstrates a different
sustained
release kinetic profile to provide the most optimal drug pharmacokinetics
within the
joint. Those skilled in the art will recognize, based on the disclosure
herein, that the
optimal release kinetics from the nanoparticles or microparticles will vary
for each
individual drug, and will also be a function of the amount of drug loaded into
particles during formulation, the size of the particles, and other
physiochemical
properties that are determined by the composition of the particles.
Quantitative
release rates for each drug from the encapsulating particles resident in the
joint will
be adjusted to obtain the optimal therapeutic concentration in the synovial
fluid and
intra-articular space to achieve the desired therapeutic concentrations. In
vitro
studies can be conducted to characterize the dual-release kinetics for the
sustained
release formulation in which each component (e.g., the anabolic drug and the
catabolic inhibitor) demonstrate sustained release over a period of 7-30 days,
by way
of example. Methods to quantitate the amount of each drug released into the
synovial fluid are "well known in the art, and may include measurements of
radioactively labeled drug. Alternatively, it is possible to covalently attach
fluorescent or other optical reporter molecules to prepare labeled drugs.
Those
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-46-
skilled in the art will recognize that many indirect methods for quantitation
exist,
such as ELISAs or mass-spectrometry, which are specific to each agent.
For practical reasons, it is difficult to achieve similar release kinetics for
two
drugs that vary substantially in size, as is the case for many of the anabolic
and
catabolic combinations. For example, in one preferred embodiment of this
invention,
it is desirable to deliver a catabolic inhibitor such as a p38 MAP kinase
inhibitor that
may be characterized by a molecular weight of 200-500 (for example, SB203580,
MW = 377) with an anabolic agent, such as human IL-10, which is 160 amino
acids
in length and has a MW of approximately 18 Kda. Independent control of the
sustained release rates for each agent can be achieved by varying the
structural
composition of the particles and/or by creating an admixture of two or more
immunoparticles. In the admixture, one set of particles is homogeneous with
respect
to the encapsulated anabolic agent, while a distinct set of particles is
homogeneous
with respect to the encapsulated catabolic inhibitor. The two sets of admixed
particles may vary in their respective sizes and polymeric composition, but
will be
characterized, if appropriate, by similar release rates for their active
agents, or by
release rates that are consistent, optimizing the local therapeutic effects of
each of the
encapsulated agents, respectively.
Liposomes are not preferred delivery vehicles for the targeted systemic
delivery of chondroprotective agents in accordance with the present invention.
Relative to nanospheres and other sustained release particle delivery systems,
liposomes have a short half-life within the circulatory system. Liposome drug
conjugates may be trapped in the liver and spleen, resulting in liposomal
breakdown
and release of the active agents. The agents are thus distributed systemically
in the
active state rather than being protected until localized within the joint. The
release of
agents from targeted liposomal delivery systems is not highly sustained and is
much
less localized than for targeted immunoparticle delivery systems. For these
reasons,
the use of particles (e.g., nanospheres) is highly preferred relative to the
use of
liposomes. None the less, for systemically delivered anabolic
chondroprotective
agents, or combinations of chondroprotective agents including an anabolic
agent, for
which targeting is highly desired, targeted liposomes may prove to be suitable
and
offer advantages relative to naked drug delivery.
F. COUPLING OF ANTIBODIES TO ENCAPSULATED
AGENTS
Representative "coupling" methods for linking the targeting antibody to the
sustained release nanoparticle through covalent or non-covalent bonds include
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-47-
chemical cross-linkers and heterobifunctional cross-linking compounds (i.e.,
"linkers") that react to form a bond between reactive groups (such as
hydroxyl,
amino, amido, or sulfhydryl groups) of the targeting antibody and other
reactive
groups (of a similar chemical nature) that are present on the surface of the
nanoparticle or other targeting vehicle. This bond formed between the
targeting
antibody and the particle or other delivery vehicle may include but is not
limited to
the following: a peptide bond, a disulfide bond, a thioester bond, an amide
bond and
a thioether bond
Direct conjugation of sustained release dosage forms to the targeting protein
(antibody) may disrupt recognition of the targeted molecule or cell by the
modified
targeting antibody. Ligand sandwich attachment techniques are useful
alternatives to
achieve attachment of the sustained release dosage form to the targeting
binding
proteins (antibodies). These techniques may involve the formation of a primary
peptide or protein shell using a protein that does not bind to the target cell
population. Binding protein is then bound to the primary peptide or protein
shell to
provide the resultant particle with functional binding protein/peptide. An
exemplary
ligand sandwich approach involves covalent attachment of avidin or
streptavidin to
the particles through functional groups as described above with respect to the
"direct"
binding approach. The binding protein is derivatized, preferably minimally,
via
functionalized biotin (e.g., through active ester, hydrazide, iodoacetal,
maleimidyl or
like functional groups). Ligand (i.e., binding peptide or
protein/functionalized
biotin) attachment to the available biotin binding sites of the
avidin/streptavidin
primary protein shell occurs through the use of a saturating amount of
biotinylated
protein/peptide.
3. ALTERNATIVE TARGETING AND PREFERENTIAL
EFFECT DELIVERY METHODS
Other methods of targeting the chondroprotective agents and combinations
thereof in the present invention, or in achieving preferential effectiveness
of such
agents or combinations at the joint relative to other sites of the body, are
also within
the scope of the present invention. For example, the two principle cell types
of the
synovium, macrophage synoviocytes (Type A) and fibroblast synoviocytes (Type
B),
and chondrocytes are known to express a variety of unique proteins that are
present
on the surfaces of these cells, and that could serve as epitopes for specific
cellular
targeting. Selecting agents, and preferably a combination of an anabolic-
promoting
agent and a catabolic inhibitory agent, that are directed to proteins that are
preferentially expressed in the joint, or that are preferentially expressed in
an
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-48-
inflamed or diseased joint, may preferentially increase local effect while
minimizing
unwanted systemic effects.
As discussed above, molecular targets within the joint may include
components of the cartilage extracellular matrix, such as cartilage specific
collagens,
including collagen Type II, IX, X, XI, aggrecan and other small leucine-rich
proteoglycans (e.g., decorin, biglycan, fibromodulin and lumican). Also
included as
targets are cartilage oligomeric matrix protein (COMP) and glycoprotein-39 (HC-
gp39), also termed YKL-40. A desirable target for use in this aspect of the
invention
would be a biochemical cartilage marker that may be either absent from normal
adult
cartilage, or present at very low levels, but that is present in certain
stages of either
RA or OA. Likewise, selection of chondroprotective agents, and preferably a
combination of an anabolic-promoting agent and a catabolic inhibitory agent
that are
specific to receptors that are upregulated in the diseased or inflamed state,
may also
preferentially increase local effect relative to other areas of the body.
As noted above, an encapsulated agent or agents may be targeted to one or
more structures within the joint by coupling the encapsulated agent to a
corresponding targeting antibody (or antibody fragment). Potentially such
antibodies
may also be coupled to the naked drugs themselves, using a linkage that is
cleaved
within the local environment of the joint to achieve targeting and delivery of
the
systemically administered drugs to the joint. Similarly, the chondroprotective
agents
that are included in an anabolic-promoting and catabolic inhibitory
composition of
the present invention may be selected such that they preferentially act on
sites within
the joint, relative to the rest of the body, thereby preferentially exerting
their effects
on synoviocytes and/or chondrocytes and/or components of the extracellular
matrix.
E. METHODS FOR SELECTION OF DELIVERY
VEHICLES AND DOSAGE
The specific nanosphere system and targeting antibody or fragment to be used
in accordance with the present invention to deliver selected agents, and the
precise
loading or dosage of therapeutic agents to include in the targeted
compositions, may
be determined analytically in accordance with the invention. The analytical
method
includes administering an antibody-labeled nanosphere (or other targeted
particle)
containing encapsulated therapeutic agents to a patient in need of such a
diagnostic
test, and subjecting the patient to imaging analysis to determine the location
of the
drug-containing nanospheres. The extent of deposition in the joint of a
patient may
then be determined by using imaging analysis. Imaging analysis is well known
in the
medical art, and includes, without limitation, x-ray analysis, magnetic
resonance
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-49-
imaging (MRI) or computed tomography (CT). In a preferred embodiment, the
joint-
targeting antibodies may be labeled with a detectable agent that can be imaged
in a
patient. For example, the targeting antibody may be labeled with a contrast
agent,
such as barium, which can be used for x-ray analysis, or a magnetic contrast
agent,
such as a gadolinium chelate, which can be detected using MRI or CT. Other
labeling agents include, without limitation, radioisotopes, such as 99Tc. In
additiona
to imaging analysis, a biopsy can be obtained from the patient to determine
the
presence and concentration of the particles in the joint (e.g., synovial
tissues).
The local delivery embodiment of the present invention was described in
terms of suitable concentrations for the therapeutic agents when delivered
locally,
sufficient to provide a predetermined level of inhibitory or therapeutic
effect at the
local delivery site (e.g., the joint). When delivered systemically, a greater
concentration or dosage of the agents will need to be administered. This
systemic
dosage and/or concentration is that which is required to result, after any
metabolic
transformation processes, in the supply of sufficient active agent(s) at the
desired
site(s) of potential cartilage degradation necessary to achieve a desired
level of local
therapeutic -effect. In particular, suitable therapeutic and preferred levels
for systemic
dosages and/or concentrations are those which result in the supply of active
agents at
the local site (e.g., the joint) at a concentration level that is within the
local delivery
therapeutic and preferred concentration ranges, respectively, previously
described.
For targeted sustained release delivery systems, a sufficient dosage or load
of
the agent is included in the composition to result in a local concentration at
the joint
or site of action over a predetermined sustained release period that achieves
a desired
level of therapeutic effect over the substantial duration of the desired
sustained
release period. Thus a sufficient dosage or load of the agent is included to
result in a
predetermined amount of the encapsulated agent that is taken up by the joint,
accounting for any metabolic transformation of the agent that occurs before
reaching
the joint or in the local environment of the joint. This predetermined amount
of
encapsulated agent reaching the joint will be determined in accordance with
the
disclosure contained herein such that as the nanosphere or other encapsulating
delivery system degrades, agent 'is released at the local site of action to
provide a
local concentration that is within the therapeutic concentration range for
that agent
during a desired period of sustained release (e.g., over a period of 1 day to
4 weeks,
more preferably between 1 day and 2 weeks).
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-50-
IV. Agents For The Inhibition Of Cartilage Degradation
The following is a description of exemplary classes of chondroprotective
agents, and exemplary drugs within each class, that are suitable for use in
the
compositions of the present invention. While not wishing to be limited by
theory, the
justification behind the selection of the various classes of agents that is
believed to
render the agents operative is also set forth.
1. INTERLEUKIN- 1 (IL-1) RECEPTOR ANTAGONISTS
The interleukin IL-1 exists in two forms, IL-la and IL-10, which are
polypeptides derived from separate gene products that share a similar spectrum
of
immunoregulatory and pro-inflammatory functions. IL-1 is a 17 kD polypeptide
that
can both act upon and be produced by a number of cell types in the joint,
including
synovial fibroblasts and macrophages, chondrocytes, endothelial. cells and
monocytes
and macrophages. There is substantial evidence that IL-1 plays a pivotal role
in joint
inflammation and in the pathophysiological loss of articular cartilage that
occurs in
the injured joint.
The actions of both forms of this cartilage destructive cytokine are mediated
by one of two IL-1 receptors (IL-1R), Type I IL-1 or Type II IL-1 receptors.
IL-1
receptors are structurally distinct and belong to a separate superfamily
characterized
by the presence of immunoglobulin binding domains. These receptors bear close
amino acid homology with other receptors containing immunoglobulin domains.
Expression of the larger Type I IL-1 receptor is present on T cells and
fibroblasts
while the smaller Type II IL-1 receptor is present on B cells, monocytes,
neutrophils,
-and bone marrow cells.
Type II IL-1 receptors bind IL-1 (3 with high affinity, but IL-1 (3 binding
does
not initiate intracellular signal transduction as it does upon binding to the
Type I IL-1
receptor. In contrast, the Type II -receptor serves as a precursor for a
soluble IL-1
binding factor that has been shown to be shed from cells and this soluble
receptor
acts as a physiological IL-10 antagonist. A naturally occurring IL-1 binding
protein
has been described which corresponds to the soluble external portion of the
Type II
receptor.
A naturally occurring secreted, soluble ligand that binds to IL-1 receptors,
alternatively referred to as the IL-1 receptor antagonist (sIL-1RA, IL-1Ra, IL-
lra),
has been cloned, sequenced and found to encode a 22 kD protein. IL-lRa
competitively inhibits the binding of IL-la and IL-1(3 to both Type I and II
IL-1
receptors. IL-iRa is a pure receptor antagonist since its binding to the
receptor does
not activate the cellular signal transduction machinery of membrane associated
IL-1
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-51-
receptors. Despite high affinity binding of this protein to the IL-1Rs, a 10-
100 fold
molar excess is required to inhibit IL-1 biological responses of cells that
express the
Type I IL-1R. Cells known to produce IL-1Ra include monocytes, neutrophils,
macrophages, synoviocytes and chondrocytes. IL-lRa has been shown to inhibit
PGE2 synthesis, induction of pro-inflammatory cytokines and MMPs, and nitric
oxide production. Secreted IL-lRa is released in vivo during experimentally
induced
inflammation. Importantly, IL-1Ra is expressed in synovial tissue and is
present in
normal human synovial fluid. In patients with knee injuries, levels of IL-lRa
in the
synovial fluid dramatically increase in the acute phase after injury, and
subsequently
decrease to below normal levels in sub-acute and chronic states. Thus, the IL-
lRa
has been shown to play a physiological role in responses of the joint to
injury.
IL-1 is considered the dominant cartilage destructive cytokine that plays a
pivotal role in joint destruction due to its ability to stimulate the
production of
degradative enzymes and pro-inflammatory cytokines by both chondrocytes and
synoviocytes. Moreover, IL-lfi is a potent inhibitor of proteoglycan and
collagen
synthesis by chondrocytes. At the cellular level, IL-in-induced responses of
synovial fibroblasts include increased production of PGE2, collagenase and
other
neutral proteases and the upregulation of pro-inflammatory cytokines, IL-6 and
IL-8.
IL-1, which is present in the joint fluid of patients with arthritic diseases,
stimulates chondrocytes to: 1) synthesize elevated amounts of enzymes such as
stromelysin, fibroblast and neutrophil collagenase and plasminogen activator,
and 2)
inhibit synthesis of plasminogen activator inhibitor-1 and TIMP. In addition,
IL-lP
is a potent inhibitor of the synthesis of matrix constituents such as Type II
collagen,
the predominant form of collagen in articular cartilage, and proteoglycans.
The
imbalance between the levels of inhibitors and proteases leads to an increase
in the
amount of active proteases. This increase, combined with a suppression of
matrix
biosynthesis, results in degradation of cartilage. In-animal studies,
injection of IL-1
into rabbit knee joints causes depletion of proteoglycan from the articular
cartilage.
Since IL-1 is, one of the key cytokines involved in the pathogenesis of
chronic
synovitis and cartilage degradation, reducing its production or blocking its
action
represents an appropriate strategy for new treatments to reduce synovial
inflammation and to provide a chondroprotective effect. A variety of
therapeutic
approaches for antagonizing the interaction of the agonist, IL-1, with its
natural
membrane bound receptor can be utilized which include: 1) naturally occurring
specific inhibitors of IL-1 activity that have been characterized to date,
including IL-
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-52-
1Ra and soluble IL-1 receptors; 2) anti-IL-1 Abs; and 3) small molecule
antagonists
which may be either peptidic or nonpetidic.
The ability to block the actions of this key cytokine will have effects on
many
cell types in the joint (e.g., synovial fibroblasts and chondrocytes), thus
inhibiting
subsequent pathological effects such as infiltration of inflammatory cells
into the
joint, synovial hyperplasia, synovial cell activation, as well as cartilage
breakdown
and inhibition of cartilage matrix synthesis. An IL-1 receptor antagonist
should
block the propagation of the inflammatory response by IL-1 and thereby
interrupt the
disease process. The therapeutic potential of a number of IL-1 receptor
antagonists
have been established in animal models of inflammation and arthritis (RA and
OA).
Patients suffering from RA have improved clinically following a subcutaneous
injection of IL-lRa or an intra-articular injection of soluble Type I IL-1R.
The effects of IL-1(3 and IL-lRa depend on their respective local
concentrations. In the supernatants of RA synovium pieces, IL-1(3 levels were
threefold higher than those of IL-1Ra. Thus, the spontaneous local production
of IL-
1Ra is not sufficient to inhibit IL-1J3 effects because a larger (10 to 100-
fold) molar
excess of IL-lRa is required to inhibit IL-1-induced biological responses in
cells that
express Type I IL-1R. Thus, high doses of IL-lRa have been used in vivo to
block
IL-1 in human volunteers in patients with RA. IL-lRa present locally in the
synovium provides a negative signal, down-regulating at least part of the IL-1-
mediated processes in synovitis, such as leukocyte accumulation in the
inflamed
tissue, PGE2 production and collagenase production by synovial cells. A
chondroprotective effect of IL-lRa has been demonstrated using direct
injection of
IL-lRa into the joint in a canine ACL model and by employing a gene therapy
approach based upon transfection of the IL-lRa gene into human synovial
fibroblasts.
The present invention discloses local and systemic delivery of an IL-1 soluble
receptor protein, which is comprised of an extracellular domain of a IL-1R,
and
which is capable of binding an IL-1 cytokine molecule in solution. In
particular, and
by. way of example, a soluble human IL-1 receptor (shulL-1R) polypeptide
comprising essentially the amino acid sequence 1-312, as disclosed within U.S.
Patent No. 5,319,071 and U.S. Patent No. 5,726,148, is disclosed herein for
use in
combination with one or more drugs chosen from either an anti-inflammatory
class,
anti-pain class, or chondroprotective class. Alternatively, the local or
systemic
delivery of a fusion protein consisting of the sIL-1R binding domain
polypeptide is
proposed for use to promote chondroprotection, as disclosed in U.S. Patent
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-53-
5,319,071. In addition, the local or systemic delivery of an IL-1 receptor
antagonist
as disclosed within U.S. Patent 5,817,306 is disclosed for use in the present
invention. The shulL-1R soluble receptor has been shown to bind IL-1 with
nanomolar affinity. Local delivery of a therapeutically effective'
concentration of an
IL-1R soluble receptor, such as shulL-1R, may occur by direct injection of the
joint
or in an irrigation solution (e.g., during an arthroscopic surgical procedure)
in
combination with one or more chondroprotective drugs, anti-inflammatory drugs,
or
anti-pain drugs and is disclosed herein as a cartilage protective agent when
applied
locally to tissues of the joint in a variety of inflammatory or
pathophysiological
,10 conditions. Alternately such agents may be delivered systemically, such as
in a
targeted systemic delivery system. Such treatment will preemptively inhibit IL-
1
stimulation of production of collagenase-1 and stromelysin-1. Employing a
wholly
different method based on gene delivery for local production of Type 1 soluble
receptors for IL-1 and/or TNF-a, it has been found that the presence of
soluble
15- receptors for these cytokines are able to confer protection to the rabbit
knee joint
during the acute inflammatory phase of antigen induced-arthritis.
IL-1 receptor antagonist peptides (11-15 amino acids) that bind specifically
with high affinity to the human Type I IL-1 receptor are suitable for use in
the
present invention as chondroprotective ~ agents. These small peptides provide
a
20 number of advantages over larger recombinant IL-1 soluble receptors or
recombinant
IL-Ira, including ease and cost of synthesis and the ability to penetrate
biological
barriers. Two of the most potent peptides, based on in vitro efficacy are: Ac-
FEWTPGWYQJYALPL-NH2 (AF12198, IC50=0.5-2nM) and Ac-FEWTPGWYQJY-
NH2 (AF 11567). AF 11567 is a truncated version of AF 12198, lacking the 4 C-
25 terminal residues and exhibiting slightly lower affinity for the Type I IL-
1 receptor
but possessing a similar plasma half-life of 2.3-2.6 hrs. Poor solubility and
rapid
metabolism appeared to limit the in vivo efficacy of AF12198 when administered
systemically via intravenous infusion. These limitations are in part overcome
through direct, local delivery methods such as injection into the intra-
articular joint
30 space or by inclusion in the surgical irrigation fluid or other infusion,
or through
systemic delivery using targeted delivery vehicles, as described above.
Examples of
IL-1 receptor antagonist agents suitable for the present invention are listed
below.
For all modes of local delivery (i.e., injection, infusion and irrigation) and
systemic
delivery (including though use of a targeted delivery system) the optimal dose
and/or
35 concentration of each suitable agent is that which is therapeutically
effective. As an
example, for each of the listed agents, the preferred and most preferred
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-54-
concentrations of an irrigation solution containing the listed agent are
provided.
Such concentrations are expected to be therapeutically effective. Similarly,
systemic
compositions in accordance with the present invention will suitably include a
dosage
or load of the agent sufficient to result in a local concentration at the
joint or site of
action within the listed therapeutic range. For targeted sustained release
delivery
systems, a sufficient dosage or load of the agent is included in the
composition to
result in a local concentration at the joint or site of action within the
listed therapeutic
range over a predetermined sustained release period.
TABLE 1
Therapeutic and Preferred Concentrations of Interleukin-1 Receptor Antagonists
Most Preferred Local
Local Delivery Delivery
Compound Therapeutic Concentrations
Concentrations (nM) )
rshulL-1R 0.2-2000 200
rhiL-Ira 0.2-2000 200
anti-ILI-antibody 0.2-2000 200
AF 12198 0.2-2000 200
AF 11567 0.2-2000 200
2. TUMOR NECROSIS FACTOR (TNF) RECEPTOR
ANTAGONISTS
TNF-a, a cytokine mainly produced by activated macrophages, has many
biological actions including transcriptional regulation of several genes that
are
mediated by specific TNF receptors, as well as immunoregulatory activities.
Originally, two different receptors termed TNF-R1 and TNF-R2 were cloned and
.characterized and also found to be produced as soluble receptors.
Receptors in this family are single transmembrane proteins with considerable
homology in their extracellular domains whereas their relatively short
intracellular
domains bear very little sequence homology. The actions of TNF are the result
of the
factor binding to cell surface receptors that are present on virtually all
cell types that
have been studied. Two receptors have been identified and cloned. One receptor
Type, termed TNFR-II (or Type A or 75kDa) encodes a. transmembrane protein of
439 amino acids and has an apparent molecular weight of 75kDa. The second
receptor type, termed TNFR-I (or Type B or 55 kDa) shows an apparent molecular
weight of 55 kDa and encodes a transmembrane protein of 426 amino acids. TNFR1
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-55-
contains an intracellular domain that can initiate signaling through the NF--
KB
pathway.
Both of the TNF receptors exhibit high affinity for binding TNFa. Soluble
TNF receptors (sTNFR) have been isolated and proved to arise as a result of
shedding of the extracellular domains of the membrane bound receptors. Two
types
of sTNFR have been identified and designated as sTNFR1 (TNF BPI) and sTNFRII
(TNF BPII). Both of these soluble receptor forms have been shown to represent
the
truncated forms of the two types of TNFR described above.
TNF-a plays a central role in the sequence of cellular and molecular events
underlying the inflammatory response and cartilage destruction. Many of the
effects
of TNF-a overlap with the pro-inflammatory effects of IL-1. Among the pro-
inflammatory actions of TNF-a is its stimulation of the release of other pro-
inflammatory cytokines including IL-1, IL-6 and IL-8. TNF-a also induces the
release of matrix metalloproteinases from neutrophils, fibroblasts and
chondrocytes
that degrade cartilage, in part through the stimulation of collagenase.
Furthermore,
TNF-a upregulates COX-2 in normal human articular chondrocytes and synovial
fibroblasts, resulting in increased PGE2 production.
This cytokine, along with IL-l, is considered to initiate and produce
pathological effects on cartilage in the joint, including leukocyte
infiltration, synovial
hyperplasia, synovial cell activation, cartilage breakdown and inhibition of
cartilage
matrix synthesis. In particular, during synovial inflammation, increased
levels of
TNF-a are found in synovial fluid of joints and increased production of TNF-(x
by
synovial cells occurs. Thus, systemic delivery, including in a targeted
delivery
system, or local delivery of a soluble TNF-a receptor in an irrigation
solution,
infusion, or injection will bind free TNF-a and function as an antagonist of
TNF
receptors in the surrounding tissue, thus. providing a cartilage protective
effect.
The present invention describes the use of functional antagonists of TNF-a
that act extracellularly to block interaction of the ligand with their cognate
membrane
receptors either by scavenging of available free ligand or by direct
competitive
interaction with the receptor itself, alone or in combination with other
agents to
provide a chondroprotective effect.. A variety of therapeutic approaches for
antagonizing the interaction of the agonist, TNF-a, with its natural membrane
bound
receptor can be utilized which include: 1) the use of naturally occurring
specific
inhibitors of TNF-a activity that have been characterized to date, including
soluble
TNF-a receptors; 2) the use of anti- TNF-a antibodies and 3) the use of small
molecule antagonists which may be either peptidic or nonpeptidic.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-56-
The present invention discloses the use of a chimeric soluble receptor (CSR)
protein, in which the extracellular domain of a TNF receptor, which possesses
binding activity for a TNF molecule, is covalently linked to a domain of an
IgG
molecule. In particular, and by way of first example, a chimeric polypeptide
(recombinant chimera) comprising the extracellular domain of the TNF receptor
extracellular polypeptide coupled to the CH2 and CH3 regions of a mouse IgGl
heavy chain polypeptide could be used, as disclosed in U.S. Patent No.
5,447,851.
The chimeric TNF soluble receptor (also termed the "chimeric TNF inhibitor" in
U.S.
Patent No. 5,447,851) has been shown to bind TNF-a with high affinity and has
been
demonstrated to be highly active as an inhibitor of TNF-a biological activity.
In
addition, a second example is a chimeric fusion construct comprised of the
ligand
binding domain of the TNF receptor with portions of the Fc antibody (termed Fc
fusion soluble receptors) that have been created for TNF-a receptors. The
present
invention also discloses the use of a soluble TNF receptor: Fc fusion protein,
or any
modified forms, as disclosed in U.S. Patent No. 5,605,690. The molecular form
of
the active soluble receptor fusion protein can be either monomeric or dimeric.
Existing studies establish that such a soluble TNF receptor:Fc fusion protein
retains
high binding affinity for TNF-a.
Within the context of defining soluble receptors as pharmacological
antagonists, the term soluble receptors includes, 'but is not limited to: (1)
soluble
receptors which correspond to naturally (endogenous) produced amino acid
sequences or. soluble fragments thereof consisting of an extracellular domain
of full-
length membrane receptor, (2) recombinant soluble receptors which are
truncated or
partial sequences of the full length, naturally occurring receptor amino acid
sequences which retain the ability to bind cognate ligand and retain
biological
activity and analogs thereof, and (3) chimeric soluble receptors which are
recombinant soluble receptors comprised of truncated or partial sequences
corresponding to a portion of the extracellular binding domain of the full
length
receptor amino acid sequences attached through oligomers (e.g., amino acids)
to a
sequence corresponding to a portion of an IgG polypeptide (e.g., IgG hinge and
Fc
domain) which retain biological activity and the ability to bind cognate
ligand.
Soluble, extracellular ligand-binding domains of cytokine receptors occur
naturally in body fluids and are thought to be involved in the regulation of
the
biologic activities of cytokines. The naturally occurring existence of
soluble,
truncated forms of a number of hematopoietic cytokine receptors has been
reported
(IL-1R, IL-4R, IL-6R, TNFR). For example, soluble TNFR is found at
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
ZU541 PCT
-57-
concentrations of about 1-2 ng/ml in the serum and urine of healthy subjects.
Lacking
signal transduction functions, these cytokine binding proteins arise as a
result of
alternative splicing of the mRNA for the complete receptor sequence (membrane-
bound form) or as a result of proteolytic cleavage and release of the membrane-
bound form of the receptor. Although the in vivo functions of these soluble
truncated
receptors are not fully established, they appear to act as physiological
antagonists of
their complementary endogenous cytokines. This antagonism occurs because (1)
scavenging of the free ligand through binding to its cognate soluble receptor
reduces
the effective free concentration available to the membrane-bound receptors and
(2)
actions of the cytokines are only produced subsequent to binding to cell
surface
'receptors.
The TNF-a soluble receptor will function as a natural antagonist of the TNF-
Rl and TNF-R2 by competing with these cell surface receptors for common pool
of
free ligand. Pharmacologically, the TNF soluble receptor will function as an
antagonist through its ability to decrease free ligand bioavailability rather
than by a
mechanism of competitive inhibition (i.e., competing with an endogenous ligand
for
a common binding site on a membrane receptor). Addition of a therapeutically
effective amount of the TNF soluble receptor to the joint should effectively
neutralize the biological activity of the ligand. Experiments in which
recombinant
soluble receptors have been administered in vivo have demonstrated the
capacity to
inhibit inflammatory responses and act as antagonists.
In this invention, agents suitable as chondroprotective agents for use in
combination with other chondroprotective, anti-pain and/or anti-inflammatory
agents
to inhibit cartilage destruction include soluble TNFR, the human chimeric
polypeptide (recombinant chimera) comprising the extracellular domain of the
TNF-
a receptor (p80) linked to the Fc portion of human IgGl, and the anti-TNF-a
antibody. For all modes of local delivery (i.e., injection, infusion and
irrigation) the
optimal dose and/or concentration of each suitable agent is that which is
therapeutically effective. ' As an example, for each of the listed agents, the
preferred
and most preferred concentrations of an irrigation solution containing the
listed agent
are provided, such concentrations expected to be therapeutically effective.
Similarly,
systemic compositions in accordance with the present invention will suitably
include
a dosage or load of the agent sufficient to result in a local concentration at
the joint or
site of action within the listed therapeutic range. For targeted sustained
release
delivery systems, a sufficient dosage or load of the agent is included in the
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-58-
composition to result in a local concentration at the joint or site of action
within the
listed therapeutic range over a predetermined sustained release period.
TABLE 2
Therapeutic and Preferred Concentrations TNF-Receptor Antagonists
Most Preferred
Local Delivery Local Delivery
Compound Therapeutic Concentrations
Concentrations (nM)
STNFR 0.1-2000 200
chimericrhTNFR:Fc 0.1-2000 200
anti-TNF-a antibody 0.2-2000 200
53. INTERLEUKIN RECEPTOR AGONISTS
Some cytokines are signaling glycoproteins that are important mediators of
synovial inflammation and cartilage destruction. Recent analysis of the
mechanism
of cartilage destruction suggests that not only is the absolute level of pro-
inflammatory master cytokine, IL-l, important in determining loss of
cartilage, but
that cytokine control of cartilage homeostasis is governed by the balance of
catabolic
and anabolic regulatory cytokines, and anabolic growth factors. If the balance
between IL-1(3 and IL-1 Ra production is altered in the inflammatory state in
favor of
IL-1(3, then it will contribute to the pathogenesis of chronic inflammatory
conditions
and cartilage destruction, such as is known to- occur after knee joint
surgery.
Potential therapeutic agents that would inhibit production of the pro-
inflammatory
cytokines at the sites of inflammation within the joint include the anti-
inflammatory
cytokines, IL-4, IL-10, and IL-13. These cytokines have been observed to
greatly
reduce articular cartilage destruction in vitro and in vivo via their effect
on a range of
pathways that reduce the impact of IL-1. Thus, anti-inflammatory cytokines
such as
IL-4, IL- 10, and IL- 13, may be useful in reducing inflammation by: 1)
reducing the
production of pro-inflammatory cytokines, and 2) inducing the production of
natural
anti-inflammatory-cytokines such as IL-iRa, as recently demonstrated in vivo
for IL-
4.
IL-4 appears to attenuate the inflammatory process in the synovium of
rheumatoid arthritis (RA) patients. In rheumatoid synovium, IL-4 has been
shown to
inhibit the production of pro-inflammatory cytokines by pieces of synovium, to
inhibit proliferation of synoviocytes and decrease bone resorption. IL-4 may
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
ZUS41YCT
-59-
promote a direct chondroprotective effect through suppression of matrix
metalloproteinase-3 (MMP-3) synthesis in human articular chondrocytes. A cell
culture system employing human articular chondrocytes was used to evaluate the
effect of IL-4 on IL-1-induced production of MMP-3 and tissue inhibitor of
metalloproteinase-1 (TIMP-1). It was found that IL-4 suppressed IL-1-
stimulated
MMP-3 protein and enzyme activity. In addition, IL-4 suppressed IL-1-induced
MMP-3 mRNA. Induction of iNOS can be inhibited by IL-4, IL-10 and IL-13.
Thus, IL-4 may be characterized as a protective mediator of joint destruction
seen in
inflammatory joint diseases.
Furthermore, the effects of IL-4 on the balance of IL-1 regulatory cytokine
levels have also been found to support a cartilage protective role. IL-4 and
IL-10
were found to suppress the production of inflammatory cytokines by freshly
prepared
rheumatoid synovial cells. While each interleukin was effective alone, the
combination of IL-4 and IL-10 synergistically inhibited the IL-1 and TNF-a
stimulated production of IL-6 and IL-8, without effects on cell viability. The
addition
of IL-4 to RA synovium cultures increased the production of IL-iRa and
decreased
that of IL-1(3. In=vivo treatment with IL-4 has recently been reported to
promote a
reduction in rat experimental arthritis by acting differentially on the IL-
1(3/IL-1 Ra
balance. IL-13, another cytokine that shares many properties with IL-4, also
induced
IL-iRa in RA synovium. Therefore, the systemic or local delivery of an IL-4
and IL-
13 combination may provide a synergistic therapeutic value.
IL-10 has a number of properties that indicate that it is a good candidate to
inhibit cartilage destruction. It inhibits both IL-1 and TNF-a release and
stimulates
TIMP-1 production while inhibiting MMP-2. The production of IL-10 inside the
RA
synovium has recently been reported and anti-inflammatory effects of IL-10
have
been characterized. IL-10 suppressed IL-1(3 production in an ex vivo RA model
using pieces of synovium, but to a lesser extent than IL-4.
A protective effect of IL-4 and IL-10 treatment on cartilage destruction has
been found in animal models of arthritis employing non-local methods of
delivery for
the cytokines. In a murine collagen-induced arthritis model, combination
treatment
of IL-4 and IL-10 produced substantial improvement. In addition to suppression
of
macroscopic signs of inflammation, combined treatment with IL-4 and IL- 10
also
reduced cellular infiltrates in the synovial tissue and caused pronounced
protection
against cartilage destruction. Moreover, levels of mRNA for TNF-a and IL-1
were
highly suppressed both in the synovial tissue and in the articular cartilage.
In
contrast, levels of IL-1 receptor antagonist (IL-iRa) mRNA remained elevated,
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
2U541YCT
-60-
which suggests that the mechanism of protection may be related to suppressed
production of TNF-a and IL-1, with concomitant up-regulation of the IL-1Ra/IL-
1
balance. These data are consistent with a dominant role of IL-10 in the
endogenous
suppression of the inflammatory response and destruction- of articular
cartilage, and a
combined treatment with IL-4 and IL-10 appears of potential therapeutic value.
The role of endogenous IL-4 and IL-10 and the therapeutic effect of addition
of these cytokines on joint inflammation and cartilage destruction in the
early stages
of the macrophage dependent murine streptococcal cell wall (SCW) arthritis
model
have also been investigated. It was demonstrated that endogenous IL- 10 plays
a role
in the regulation of SCW arthritis. Addition of exogenous IL-10 further
enlarged the
suppressive effect of endogenous IL-10. An even more pronounced effect was
found
with the combination of IL-4 and IL-10. The combination resulted in a reduced
swelling and an increase in chondrocyte proteoglycan synthesis. Treatment with
the
combination of IL-4 and IL-10 substantially diminished levels- of TNF-a, as
occurs
for IL-10 treatment alone, but also resulted in strongly reduced IL-1(3 levels
in the
synovium, an added effect of potential clinical benefit. Overall, the data is
consistent
with a role for both IL-4 and IL-10 as chondroprotective agents delivered
systemically or locally to joints to prevent cartilage destruction, and
indicates a
combination containing IL-4 and IL-10 may provide a greater potential
therapeutic
value than either agent alone.
Severe combined immunodeficient (SCID) mice were used as a model to
assess the effect of IL-4 or IL-10 injection on cartilage degradation and
mononuclear
cell (MNC) recruitment to human rheumatoid synovium in vivo. Human rheumatoid
synovium and cartilage from five rheumatoid arthritis patients were injected
with
recombinant human IL-4 (rhIL-4, 100 ng; rhlL-10, 100 ng), a combination of IL-
4
and IL-10, or TNF-alpha (1000 U), or phosphate-buffered saline twice a week
for 4
weeks. It was found that a combination of human IL-4 and IL-10 inhibited
cartilage
degradation and invasion by human synovial tissue, establishing the
chondroprotective properties of these interleukin agonists.
Human IL-13 has been cloned and sequenced and has been found to share
many of the properties of IL-4. IL-13 is about 25% homologous to IL-4. Like IL-
4,
IL-13 decreases the production of pro-inflammatory cytokines, including IL-1
and
TNF-a, by synovial fluid mononuclear cells.' IL-13 exhibits anti-inflammatory
effects in vivo and thus has therapeutic ' potential in the treatment of
cartilage
destruction in the joint.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-61-
Compounds useful as IL-4, IL-10 and IL-13 agonists include naturally
occurring human IL-4, IL-10 and IL-13, human recombinant IL-4 (r11IL-4), rhIL-
10,
and rhiL-13 as well as.partial sequences thereof, or peptide sequences which
have
been constructed using recombinant DNA techniques to recognize the IL-4, IL-10
and IL-13 receptors and are capable of activating these receptors on a cell
surface.
This specifically includes multispecific molecules comprised of an anti-Fc
receptor
portion and an anti-IL-4, anti-IL-10, and anti-IL-13 receptor portion, wherein
at least
one portion is constructed using recombinant DNA techniques. Within the
context of
defining interleukin agonists as pharmacological agonists, the term
interleukin
agonist includes, but is not limited to: (1) peptide sequences which
correspond to
naturally (endogenous) produced amino acid sequences or fragments thereof, (2)
recombinant interleukins which are truncated or partial sequences of the full
length
naturally occurring interleukin amino acid sequences which retain the ability
to bind
cognate receptor and retain biological activity and analogs thereof, and (3)
chimeric
interleukins which are recombinant polypeptides comprised of truncated or
partial.
sequences corresponding to a portion of the of the full length amino acid
sequences
attached through oligomers (e.g., amino acids) to a sequence corresponding to
a
portion of an IgG polypeptide (e.g., IgG hinge and Fc domain) which retain the
ability to bind the cognate receptor and retain biological activity.
Examples of interleukin agonists suitable for the present invention are listed
below. For all modes of local delivery (i.e., injection, infusion and
irrigation) the
optimal dose and/or concentration of each suitable agent is that which is
therapeutically effective. As an example, for each of the listed agents, the
preferred
and most preferred concentrations of an irrigation solution containing the
listed agent
are provided, such concentrations expected to be therapeutically effective.
Similarly,
systemic compositions in accordance with the present invention will suitably
include
a dosage or load of the agent sufficient to result in.a local concentration at
the joint or
site of action within the listed therapeutic range. For targeted sustained
release
delivery systems, a sufficient dosage or load of the agent is included in the
composition to result in a local concentration at the joint or site of action
within the
listed therapeutic range over a predetermined sustained release period.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-62-
TABLE 3
Therapeutic and Preferred Concentrations Interleukin A og nists
Local Delivery Preferred
Therapeutic Local Delivery
Compounds Concentrations Concentrations
(nanomolarl (nanomolar)
rhuman IL-4 0.5-5,000 5-500
rhuman IL-10 0.5-5,000 5-500
rhuman IL-13 0.5-5,000 5-500
4. TRANSFORMING GROWTH FACTOR-0.
SUPERFAMILY AGONISTS
Transforming growth factor-a (TGF-R) subfamily members are 25 kD
pleiotropic, multifunctional proteins capable of influencing a variety of
cellular
functions and are known to be involved in,tissue repair and remodeling. In
many
cases, it enhances the cell interaction with the extracellular matrix (ECM)
and
increases accumulation of ECM by stimulating production and secretion of ECM
proteins and protease inhibitors. TGF-(3 also has been shown to have
synergistic
interactions with other cytokines, generally showing anti-inflammatory
activities.
Multiple isoforms of TGF-(3 have been identified which share close amino acid
sequence homologies. TGF-(31, TGF-(32, and TGF- (33 have been found in human
tissue and are active in mammalian cells, although differing in binding
affinity.
Members of the TGF- (3 subfamily are potent modulators of chondrocyte
proliferation, differentiation and extracellular matrix accumulation. In
cartilage
organ cultures, TGF-01 regulates metabolism of proteoglycans and stimulates
collagen and glycosaminoglycan synthesis by rabbit articular chondrocytes.. In
addition, TGF-(31 increases TIMP expression in human articular chondrocytes
and
down-regulates expression of IL-1 receptors in articular cartilage.
Bone morphogenetic proteins (BMPs) are multifunctional regulators of cell
growth, differentiation and apoptosis that belong to the transforming growth
factor
(TGF)-(3 superfamily. More than a dozen members of the BMP protein family have
been identified in mammals, which can be subclassified into several groups
depending on their structures. BMP-2 and BMP-4 are highly similar to each
other.
BMP-5, BMP-6, osteogenic protein (OP)-1 (also called BMP-7), and OP-2/BMP-8
are structurally similar to each other. Growth-differentiation factor (GDF)-5
(also
termed cartilage-derived morphogenetic protein-1), GDF-6 (also cartilage-
derived
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-63-
morphogenetic protein-2), and GDF-7 form another related group. In contrast to
BMP-2, BMP-4, BMP-6, and OP-l/BMP-7, which induce bone and cartilage
formation in vivo, GDF-5, GDF-6, and GDF-7 more efficiently induce cartilage
and
tendon-like structures in vivo (Wolfinan et al., 1997).
Members of the TGF-(3 superfamily exert their effects via binding to two
types of serine/threonine kinase receptors, both of which are essential for
signal
transduction (Massague, 1998). The Type II receptors are constitutively active
kinases, which transphosphorylate Type I receptors upon ligand binding. The
Type I
receptors activate intracellular substrates such as Smad proteins and it is
through this
mechanism that specificity of intracellular signal transduction occurs. Seven
different Type I receptors have been isolated in mammals, which were
originally
termed activin receptor-like kinase (ALK)-1-ALK7. BMP Type IA receptor (BMPR-
IA or ALK-3) and BMP Type IB receptor (BMPR-IB or ALK-6) are structurally
similar to each other and specifically bind BMPs together with Type II
receptors.
ALK-2 has been shown to bind activin, but recent data revealed that it is a
Type I
receptor for certain BMPs (e.g., OP-1/BMP-7) (Macias-Silva et al., 1998). ALK-
1 is
structurally highly similar to ALK-2, but its physiological ligand is still
unknown.
ALK-5 and ALK-4 are Type I receptors for TGF-(3 (T(3R-I) and activin (ActR-
IB),
respectively. ALK-7 is structurally similar to ALK-4 and ALK-5, but its ligand
has
not been determined yet.
Naturally occurring TGF-(3 and BMP agonists as well as synthetic or human
recombinant (rh) agonists suitable for use in the cartilage-protective
solution of the
present invention may interact with any of the BMP receptors described above.
As
used herein, the term "TGF-(3 and BMP agonists" includes fragments, deletions,
additions, amino acid substitutions, mutations, and modifications thereof that
retain
the biological characteristics of the naturally occurring human TGF-(3 and BMP
agonist ligands. The TGF-f3 or BMP agonists may be used alone or in
synergistic
combination with other members of the TGF-(3 superfamily as anabolic cartilage
agents (chondrogenic or promoting cartilage matrix repair) or in combination
with
inhibitory agents that block cartilage catabolism.
Type I receptors function as downstream components of Type II receptors.
The specificity of the intracellular signals by Type I receptors is determined
by a
specific region in the serine/threonine kinase domain, termed the L45 loop.
Thus, the
structures of the L45 loop of BMPR-IA/ALK-3 and BMPR-IB/ALK-6 (BMPR-I
group) are, identical to each other, and they may transduce similar signals in
cells.
Similarly, the L45 loops of T(3R-I/ALK-5, ActR-IB/ALK-4, and ALK-7 (T(3R-I
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-64-
groups) are identical to each other, and they activate similar substrates
(Chen et al.,
1998). The L45 loops of ALK-1 and ALK-2 (ALK-1 group) are most divergent from
the other Type I receptors, but they activate substrates similar to that of
the Type I
receptors of the BMPR-I group (Armes et al., 1999).
Various proteins may transduce signals from the TGF-(3 and BMP
serine/threonine kinase receptors. Among them, the best-studied molecules are
proteins of the Smad family. Eight different Smad proteins have been
identified in
mammals, and these proteins are classified into three subgroups, (i.e.,
receptor-
regulated Smads (R-Smads), common partner Smads (Co-Smads), and inhibitory
.10 Smads). R-Smads are directly activated by Type I receptors, from complexes
with
Co-Smads, and translocate into the nucleus. The Smad heteromers bind to DNA
directly and indirectly via other DNA-binding proteins and thus regulate the
transcription of target genes. Smadl, Smad5, and Smad8 are activated by BMPs,
whereas Smad2 and Smad3 are activated by TGF-(3. For example, Smad2, in
combination with Smad4 that functions as a Co-Smad, is translocated to the
nucleus
where it activates the transcription of genes that mediate the biological
effects of
TGF. Smad6 and Smad7 are structurally distantly related to the other Smads and
act
as inhibitory Smads. It has been shown that BMPs induce new cartilage and bone
formation in vitro and in vivo and regulate chondrocyte growth and
differentiation.
Furthermore, these proteins are also implicated in the cartilage repair
process.
Various studies have shown that BMPs also promote and maintain the
chondrogenic
phenotype, which is indicated by their ability to stimulate proteoglycan
synthesis in
chick limb bud cells culture and fetal rat chondroblasts, as well as in rabbit
and
bovine articular chondrocytes. The importance of BMPs for cartilage and bone
formation has been proven by transgenic .approach in which specific BMP gene
knockouts were studied.
One member of the BMP family, osteogenic' protein (OP-1 or BMP-7),
appears particularly important for cartilage homeostasis under normal and
pathological conditions, such as during repair of cartilage. OP-1 appears to
be the
only member of the BMP family, along with cartilage-derived morphogenetic
proteins, which is expressed by adult articular chondrocytes (Chubinskaya, S.,
J. Histochemistiy and Cytochemistry 48:239-50 (2000)). OP-1 was originally
purified from bone matrix and shown to induce cartilage and bone formation.
The
human OP-1 gene has been cloned and biologically active recombinant OP-1
homodimers have been produced. Human recombinant OP-1 can stimulate synthesis
of aggrecan and collagen Type II by human articular chondrocytes in vitro. It
can
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-65-
also counteract the deleterious effects of IL-1 on the metabolism of these
chondrocytes and block bovine cartilage damage mediated by fibronectin
fragments.
This effect was demonstrated by studying the effects of recombinant human OP-1
on
the production of proteoglycan, prostaglandin E2, and IL-1 receptor antagonist
by
5. human articular chondrocytes cultured in the presence of interleukin- I
beta.
Treatment of human articular chondrocytes with OP-1 was effective in
overcoming
the down-regulation of proteoglycan synthesis induced by low doses of IL-1(3.
Furthermore, a study found that OP-1 stimulates the synthesis of hyaluronan
and
CD44, other molecules required for matrix assembly by human chondrocytes.
These
studies of the expression and regulation to OP-1 in human adult cartilage
suggest a
role for OP-1 in cartilage protection and repair and indicate that OP-1 can be
used as
a therapeutic agent that promotes cartilage anabolism and repair of human
articular
cartilage.
OP-1 (BMP-7) induces cartilage and bone formation when implanted at intra-
and extraskeletal sites in vivo. The influence of OP-1 on healing of full-
thickness
articular cartilage defects was investigated by drilling two adjacent holes
through
articular cartilage of rabbit knee joint. OP-1' induced articular cartilage
healing and
regeneration of the joint surface that contained cells resembling mature joint
chondrocytes.
These data suggest that one preferred embodiment of the solution useful for
the practice of the present invention for the prevention of cartilage
degradation and
maintaining biological homeostasis of articular. cartilage in humans after
surgical
trauma could include systemic or local application of a member of the TGF-(3
superfamily, preferably either TGF(32, BMP-7 (OP-1) or BMP-2, or an equivalent
agonist which acts through the. same receptors employed by these ligands. The
systemic or local delivery may occur in combination with a drug or drugs that
are
inhibitors of cartilage catabolic processes (e.g. such as MAP kinase
inhibitors, MMP
inhibitors or nitric oxide synthase inhibitors) and/or other agents for 'the
inhibition of
pain and inflammation.
Within the context of defining TGF-(3 and BMP agonists as pharmacological
agonists, the term TGF-(3 and BMP agonist includes, but is not limited to: (1)
peptide
sequences which correspond to naturally (endogenous) produced amino acid
sequences or fragments thereof, (2) recombinant TGF-(3s and BMPs which are
truncated or partial sequences of the full length naturally occurring TGF-(3
and BMP
amino acid sequences which retain the ability to bind cognate their respective
receptor and retain biological activity and analogs thereof, and (3) chimeric
TGF-(3s
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-66-
and BMPs which are recombinant polypeptides comprised of truncated or partial
sequences corresponding to a portion of the of the full length amino acid
sequences
attached through oligomers (e.g., amino acids), to a sequence corresponding to
a
portion of an IgG polypeptide (e.g., IgG hinge and Fc domain) which retain the
ability to bind the cognate receptor and retain biological activity.
Examples of TGF-(3 and BMP agonists suitable for the present invention are
listed below. For' all modes of local delivery (i.e., injection, infusion and
irrigation)
the optimal dose and/or concentration of each suitable agent is that which is
therapeutically effective. Asan example, for each of the listed agents, the
preferred
and most preferred concentrations of an irrigation solution containing the
listed agent
are provided, such concentrations expected to be therapeutically effective.
Similarly,
systemic compositions in accordance with the present invention will suitably
include
a dosage or load of the agent sufficient to result in a local concentration at
the joint or
site of action within the listed therapeutic range. For targeted sustained
release
delivery systems, a sufficient dosage or load of the agent is included in the
composition to result in a local concentration at the joint or site of action
within the
listed therapeutic range over a predetermined sustained release period.
A range of therapeutic concentrations for local delivery or local action in
the
surgical solution to the joint may be estimated from values of the
dissociation
constants (Kd) of each ligand for its cognate receptor. While these values
will vary
for particular cell types and tissues, the. following example is given for BMP-
4.
Binding experiments with 1251-BMP-4, revealed the presence of specific, high-
affinity binding sites with an apparent dissociation constant of 110 pM and
about
6000 receptors per cell. Therefore, at 11 nM BMP-4, binding of the ligand will
be
maximal and the available receptors will be fully occupied (saturated). The
presence
of functional receptors for BMP-4 on primary articular chondrocytes has been
demonstrated.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-67-
TABLE 4
Therapeutic and Preferred Concentrations TGF-(3 and BMP-Receptor A og nists
Local Delivery Most Preferred
Therapeutic Local Delivery
Compound Concentrations Concentrations
(nanomolar) (nanomolar
TGF-(31 0.05-500 0.5-100
TGF-02 0.05-500 0.5-100
BMP-2 0:1-2000 1-200
BMP-4 0.1-2000 1-200
BMP-7 (OP-1) 0.1-2000 1-200
5. CYCLOOXYGENASE-2 (COX-2) IN 03ITORS
Nonsteroidal anti-inflammatory drugs (NSAIDs) are widely used as
anti-inflammatory agents, but have not been specifically developed or
therapeutically
employed as chondroprotective agents. The direct molecular target for an NSAID
drug is the first enzyme in the prostaglandin synthetic pathway, referred to
either as
prostaglandin endoperoxide synthase or fatty acid cyclooxygenase. Two related
forms of cyclooxygenase, termed cyclooxygenase-1 or Type 1 (COX-1) and
cyclooxygenase-2 (COX-2) have been characterized. These isozymes are also
known as Prostaglandin G/ H Synthase (PGHS)-1 and PGHS-2. Both enzymes
catalyze the rate-limiting step in the formation of prostanoids that is the
conversion
of arachidonic acid to prostaglandin H2. COX-1 is present in platelets and
endothelial cells and exhibits constitutive activity. In contrast, COX-2 has
been
identified in endothelial cells, macrophages, fibroblasts and other cells in
the joint
and its expression is induced by pro-inflammatory cytokines, such as IL-1 and
TNF-
a.
Within the inflamed joint, COX-2 expression is upregulated and it has been
shown that large increases in activity of COX-2 occur concomitant with its
upregulation, leading to increased synthesis of prostaglandins which are
present in
the synovial fluid of patients suffering from inflammatory arthropathies.
Cellular
sources of prostaglandins (PGs) in the joint include activated chondrocytes,
Type A
and B synoviocytes and infiltrating macrophages. Cellular functions important
in
cartilage metabolism modulated by PGs include gene expression, extracellular
matrix
synthesis and proliferation. Because COX-2 is expressed in inflamed joint
tissue or
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-68-
after exposure to mediators. of inflammation (e.g., as a result of injury or
surgical
trauma), the use of a COX-2 inhibitor is expected to provide both anti-
inflammatory
and cartilage protective activity.
Cartilage destruction in inflammatory arthropathies can be triggered as a
consequence of joint injury and as a result of arthroscopic surgical
procedures.
Chondrocytes are the only cell type in articular cartilage and are known to
participate
in the breakdown of their own matrix through release of endogenous
inflammatory
mediators, including PGs. Studies have shown that COX-2 gene expression,
protein
synthesis, and PG release in normal human articular chondrocytes is rapidly
induced
by cytokines, including IL-1, TNF-a and IL-6. Levels of mRNA are detected as
early as 2 hours after cytokine induction, reach high levels at 6 hours and
show a
remarkably long duration of expression for at least 72 hours. Similarly, cell
culture
studies of IL-la and TNF-a activation of human synoviocytes have shown large
increases in expression of COX-2 and production of prostaglandin E2 (PGE2).
Treatment with a variety of NSAIDS, such as ketoprofen, abolishes the induced
PGE2 response. In a chondrocyte cell culture system, the specific COX-
2.inhibitor
compound NS-398 prevented the increase in PGE2 production induced by the
cytokines while COX-1 levels remained stable (Morisset, S., 1998, J.
Rheumatol.
25:1146-53). Thus, it can be deduced that blocking PG production by activated
chondrocytes, which is associated with expression of COX-2, can provide a
chondroprotective effect.
NSAIDs are commonly used in the treatment of patients with osteoarthritis or
rheumatoid arthritis, but their effects on articular cartilage metabolism in
the context
of these arthritic diseases remains a subject of debate. For instance, the
clinical
treatment of osteoarthritis and rheumatoid arthritis with NSAIDs is successful
in
reducing inflammation. However, it is thought that some NSAIDs which are not
selective for COX-2, primarily salicylates and indomethacin, accelerate
osteoarthritic
cartilage destruction by impairing proteoglycan synthesis by chondrocytes,
whereas
other NSAIDS. are thought to have a somewhat chondroprotective effect by
stimulating cartilage repair. Most studies have shown that NSAIDs have little
or no
effect on cartilage. Due to the current lack of use of this class of drugs in
the
.treatment of synovitis and cartilage destruction following traumatic joint
injury and
surgical trauma, the unique properties of each NSAID on the pathophysiological
mechanisms that contribute to cartilage destruction will need to be
established.
Since the two COX isozyrnes are pharmacologically distinct, isozyme-
specific (selective) cyclooxygenase inhibitors that are useful for anti-
inflammatory
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-69-
therapy have been developed and some of these same COX-2 inhibitors have been
tested in models of joint inflammation. However, the effects in vitro of the
COX-2
inhibitors on the synthesis and degradation of cartilage proteoglycans, as
well as
synovial production of IL-1, IL- 6, IL-8, and prostanoids, indicate that
certain
NSAIDs may vary considerably in their effects in vivo on cartilage and
synovial
production of interleukins and eicosonoids, such that the .integrated effects
of these
parameters may influence the outcome of COX-2 inhibitors on cartilage
integrity.
For example, some NSAIDS can accelerate joint damage in osteoarthritis by
enhancing the production of pro-inflammatory cytokines or inhibiting cartilage
proteoglycan synthesis. However, despite the possible variance in clinical
effect
among COX- 2 specific inhibitors, inhibition of COX-2 typically results in a
reduction of synovitis and an expected decrease in the risk of cartilage
damage.
A variety of biochemical and cellular and animal assays have been developed
to assess the relative selectivity of inhibitors for the COX-1 and COX-2
isoforms. In
general, a criteria for defining selectivity is the ratio of the. COX-1/COX-2
inhibitory
constants (or COX-2/COX-1) obtained for a given biochemical or cellular assay
system. The selectivity ratio accounts for different absolute IC50 values for
inhibition
of enzymatic activity that are obtained between microsomal and cellular assay
systems (e.g., platelet and macrophage., cell lines stably expressing
recombinant
human COX isozymes). Furthermore, inhibition of COX-2 mimics the inhibitory
effects triggered by chondroprotective (inhibitory) cytokines, such as IL-4,
which
down-regulate intracellular COX-2 synthesis. Comparison of the selectivity of
more
than 45 NSAIDs and selective COX-2 inhibitors (Can. J. Physiol. Pharma col.
75:1088-95 (1997)) showed the following rank-ordered relative selectivity for
COX-2 vs. COX-l: DuP 697 > SC-58451= celecoxib > nimesulide = meloxicam =
piroxicam = NS-398= RS-57067 >SC-57666 > SC-58125 > flosulide > etodolac >
L-745,337 > DFU-T-614, with IC50 values ranging from 7 nM to 17 M.
From the molecular and cellular mechanism of action defined for selective
COX-2 inhibitors, such as celecoxib and rotecoxib, as well as from animal
studies,
these compounds are expected to exhibit chondroprotective action when applied
perioperatively in an irrigation solution or in an injection directly to a
joint. In
particular, COX-2 inhibitors are expected to be effective drugs delivered in
an
irrigation solution during an arthroscopic surgical procedure or by direct.
injection
into a joint prior to, during or after a surgical procedure or other injury to
the joint.
Examples of COX-2 inhibitors suitable for the present invention are listed
below. For all modes of local delivery (i.e., injection, infusion and
irrigation) the
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-70-
optimal dose and/or concentration of each suitable agent is that which is
therapeutically effective. As an example, for each of the listed agents, the
preferred
and most preferred concentrations of an irrigation solution containing the
listed agent
are provided, such concentrations expected to be therapeutically effective.
Similarly,
systemic compositions in accordance with the present invention will suitably
include
a dosage or load of the agent sufficient to result in a local concentration
at"the joint or
site of action within the listed therapeutic range. For targeted sustained
release
delivery systems, a sufficient dosage or load of the agent is included in the
composition to result in a local concentration at the joint or site of action
within the
listed therapeutic range over a predetermined sustained release period.
TABLE 5
Therapeutic and Preferred Concentrations of C c~ygenase-2 Inhibitors
Local Delivery Most Preferred
Compounds Therapeutic Preferred Local Delivery
Concentrations (nM) Concentrations (nM)
rofecoxib (MK 966) 0.3-30,000 30-3,000
SC-58451 0.3-30,000 30-3,000
celecoxib (SC-58125) 0.3-30,000 30-3,000
meloxicam 0.5-50,000 50-5,000
nimesulide 0.5-50,000 50-5,000
diclofenac 0.3-30,000 30-3,000
NS-398 0.3-30,000 30-3,000
L-745,337 0.2-100,000 20-10,000
RS57067 0.2-100,000 20-10,000
SC-57666 0.2-100,000 -20-10,000
flosulide 0.2-100,000 20-10,000
6. MAP KINASE INHIBITORS
The mitogen-activated protein (MAP) kinases are a group of protein
serine/threonine kinases that are activated in response to a variety' of
extracellular
stimuli and function in transducing signals from the cell surface to the
nucleus. The
MAP kinase cascade is one of the major intracellular signaling pathways that
transmit signals from growth factors,, hormones and inflammatory cytokines to
intermediate early genes. In combination with other signaling pathways, these
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
1U 41YC'1'
-71-
activated mitogen-activated protein-kinases (MAPKs) differentially alter the
phosphorylation state and activity of transcription factors, and ultimately
regulate cell
proliferation, differentiation and cellular response to environmental stress.
For
example, a member of the MAPK family -(p38) mediates the major biochemical'
signal transduction pathways from the potent pro-inflammatory cytokines, IL-1
and
TNF-a, leading to induction of cyclooxygenase-2 (COX-2) in stimulated
macrophages, through cis-acting factors involved in the transcriptional
regulation of
the COX-2 gene.
The members of the MAP kinase class of agents are composed of at least
three families that are known to differ in sequence, size of the activation
loop,
activation by extracellular stimuli and participation in distinct signal
transduction
pathways. Prominent members among this family of MAP kinases include the
extracellular signal-regulated kinases (ERKs), ERK1 and ERK2 (p44MAPK and
p42MAPK, respectively); stress-activated protein kinase 1 (SAPK1) family which
is
also referred to as the JNK or jun N-terminal kinase family; and the p38 MAP
kinase
family which is also known as stress-activated kinase 2/3 (SAPK-2/3). The p38
kinases are activated by stresses, most notably pro-inflammatory cytokines.
Within
the p38 family, there are at least four distinct homologs (isotypes or
isoenzymes)
which standard nomenclature 'refers to either as SAPK2a, SAPK2b, SAPK2d,.
SAPK3, or p38 a, 13, S (SAPK4) and y, respectively. The inhibitors of MAP
kinases
useful for the practice of this invention may interact with any one or
combination of
the above MAP kinases. For specific MAP kinase inhibitors, the inhibitory
constants
characterized through assays of purified in vitro enzymes and in cellular
assays may
vary over a wide range of concentrations- and demonstrate utility in this
application.
Activation of p38 MAP kinase is mediated by dual phosphorylation of threonine
and
tyrosine residues. Both TNF-a and IL-1 treatment of cells has been shown to
rapidly
(within 5 min) increase phosphorylation and activate p38 MAP kinase.
Previous work has shown that small-molecule inhibitors can specifically
inhibit p38 MAP kinase (Lee, J. et al., Nature 372:739-746 (1994)) and produce
anti-
inflammatory effects at the biochemical level and in various animal models.
Cuenda
and coworkers (Cuenda, A. et al., FEBS Lett. 364:229 (1995)) showed that the
compound, SB203580 [4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-
pyridyl)imidazole] inhibited p38 in vitro (IC50 = 0.6 M), suppressed the
activation
of MAPK activating protein kinase-2 and prevented the phosphorylation of heat
shock protein (hsp) 27 in response to IL-1 and cellular stresses in vivo. The
kinase
selectivity of SB203580 inhibitory action for p38 was demonstrated by its
failure or
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-72-
at best weak inhibition of at least 15 other protein kinases in vitro,
including
members of the PKC, PKA, src and receptor tyrosine kinase families (Lee, J.,
Pharmacol. Ther. 82:389-397 (1999)). In cellular studies, pre-incubation with
SB
203580 has been shown to block the IL-1 and TNF-a induced phosphorylation of
the .
enyzme and subsequent IL-8 production. This supports the preemptive effect of
delivering the inhibitors during the surgical procedure.
The role of p38 mitogen-activated protein kinase (MAPK) in biochemical
inflammatory responses resulting in destruction of cartilage has been studied
using
SB203580, which specifically inhibits the enzyme. Actions of IL-1 that are
selectively controlled by p38 MAPK are the regulation of prostaglandin H
synthase-2
(COX-2), metalloproteinases, and IL-6 (Ridley, S. et al., J. bnmunol. 158:3165-
73
(1997)). In human fibroblasts and vascular endothelial cells, SB203580
inhibited
(IC50= 0.5 M) IL-1-induced phosphorylation of hsp 27 (an indicator of p38
MAPK
activity) in fibroblasts without affecting the other known IL-1-activated
protein
kinase pathways (p42/p44 MAPK, p54 MAPK/c-Jun N-terminal kinase). In addition,
SB203580 significantly inhibited IL-1-stimulated IL-6 (30 to 50% at 1 M) but
not.
IL-8 production from human fibroblasts and endothelial cells.
Importantly, SB203580 strongly inhibited IL-1-stimulated prostaglandin
production by fibroblasts and human endothelial cells. This was associated
with the
inhibition of the induction of COX-2 protein and mRNA. PGE2 contributes to
increased expression of matrix metalloproteinases that are important mediators
of
cartilage degradation. Both synovial fibroblasts and chondrocytes express the
COX-2
gene at high levels upon activation by cytokines and extracelluar stimuli. The
MAPK inhibitor provides chondroprotective activity due to its inhibitory
activity on
MAP kinases expressed in these and other cell types.
MAPK inhibitors are expected to be effective as cartilage protective agents
when applied systemically or locally to tissues of the joint in = a variety of
inflammatory or pathophysiological conditions. SB 203580 has been
characterized
in several pharmacological models in vivo and demonstrated to have activity
under
long term, oral dosing. SB203580 was found to inhibit the stimulation of
collagenase-l and stromelysin-1 production by IL-1 without affecting synthesis
of
TIMP-1. Furthermore, SB203580 prevented an. increase in IL-1-stimulated
collagenase-1 and stromelysin-1 mRNA. In a model of cartilage breakdown,
short-term IL-1-stimulated proteoglycan resorption and inhibition of
proteoglycan
synthesis were unaffected by SB 203580, while longer term collagen breakdown
was
prevented. In addition, SB203580 was found to be effective in inhibiting IL-
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
. 20541PCT
-73-
1-induced, nitric oxide production in bovine articular cartilage explants and
chondrocytes (Badger, 1998). These in vitro observations provide a basis for
cartilage protective activity of the MAP kinase inhibitor administered
systemically or
directly and locally to these tissues in the joint.
p38 MAP kinase is involved in TNF-induced cytokine expression, and drugs
which function as inhibitors of p38 MAP kinase activity block the production
of pro-
inflammatory cytokines (Beyaert, R. et al., EMBO J. 15:1914-23 (1996)). TNF-a
treatment of cells activated the p38 MAPK pathway as shown by increased
phosphorylation of p38 MAPK itself and activation of its substrate proteins.
Pretreatment of cells with SB203580 completely blocked TNF-a induced
activation
of MAPK activating protein kinase-2 and hsp27 phosphorylation. Under the same
conditions, SB203580 also completely inhibited TNF-a induced synthesis of IL-6
and expression of a reporter gene that was driven by a minimal promoter
containing
two NF-KB elements. Thus, these studies and related studies on other p38
inhibitors
show that the action of inhibitors, such as SB203580 and FR133605, on p38 MAPK
interfere selectively with TNF-a- and IL-1-induced activation of various
proteins
linked to the cartilage degradation. Thus, the selective inhibition of the MAP
kinase
signaling pathways of these key pro-inflammatory cytokines by inhibition of a
kinase
downstream of the receptor indicate that MAP kinase inhibitors may provide a
chondroprotective effect.
SB 203520 has been evaluated in several animal models of cytokine
inhibition and inflammatory disease. It was demonstrated to be a potent
inhibitor of
inflammatory cytokine production in vivo in both mice and rats with IC50
values of
15 to .25 mg/kg. SB 203580 possessed therapeutic activity in collagen-induced
'arthritis in DBA/LACJ mice with a dose of 50 mg/kg resulting in significant
inhibition of paw inflammation. Antiarthritic activity was also observed in
adjuvant-
induced arthritis in the Lewis-rat when SB203580 was administered p.o. at 30
and 60
mg/kg/day. Additional evidence was obtained for beneficial effects on bone
resorption with an IC50 of 0.6 M.
In summary, a,variety of biochemical, cellular and animal studies show that
p38 MAPK plays an important role in the regulation of responses to IL-1 and
TNF-a
and that it is involved in the regulation of mRNA levels of some
inflammatory-responsive genes, such as COX-2. Inhibitors of p38 block the
production of pro-inflammatory cytokines and inhibit the production of MMPs,
and
have been demonstrated to inhibit collagen breakdown in cartilage explants.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-74-
The use of MAPK inhibitor to block the actions of key pro-inflammatory
cytokines, such as IL-1 and TNF-a, will have beneficial effects on many cell
types in
the joint, including synovial fibroblasts, macrophages and chondrocytes, thus
inhibiting subsequent pathological effects such as infiltration of
inflammatory cells
into the joint, synovial hyperplasia, synovial cell activation, and cartilage
breakdown.
Thus, a MAPK inhibitor should block the propagation of the inflammatory
response
by the aforementioned cytokines', and thereby interrupt the disease process.
Examples of MAPK inhibitors suitable for the present invention are listed
below. For all modes of local delivery (i.e., injection, infusion and
irrigation) the
optimal dose and/or concentration of each suitable agent is that which is
therapeutically effective. As an example, for each of the listed agents, the
preferred
and most preferred concentrations of an irrigation solution containing the
listed agent
are provided, such concentrations expected to be therapeutically effective for
local
delivery. Similarly, systemic compositions in accordance with the present
invention
will suitably include a dosage or load of the agent sufficient to result in a
local
concentration at the joint or site of action within the listed therapeutic
range. For
targeted sustained release delivery systems, a sufficient dosage or load of
the agent is
included in the composition to result in a local concentration at the joint or
site of
action within the listed therapeutic range over a predetermined sustained
release
period.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-75-
TABLE 6
Therapeutic and Preferred Concentrations of MAP Kinase Inhibitors
Local Delivery Local Delivery
Therapeutic Preferred
Compounds Concentrations Concentrations
(nanomolar) (nanomolar)
SB 203580 0.5-50,000 50-5,000
SR 203580 iodo 0.5-50,000 50-5,000
SB 202190 0.2-20,000 20-2,000
SB 242235 0.2-10,000 20-1,000
SB 220025 0.2-10,000 20-1,000
RWJ 67657 0.3 30,000 30-3,000
RWJ 68354 0.9-90,000 90-9,000
FR133605 1-100,000 10-10,000
L-167307 0.5-50,000 50-5,000
PD 98059 0.1-10,000 10-1000
PD 169316 1-100,000 10-10,000
7. INHIBITORS OF MATRIX METALLOPROTEINASES
Destruction of articular cartilage is a common feature in joint diseases such
as
osteoarthritis and rheumatoid arthritis, but also occurs following injury to
the joint.
Pathophysiologically, a structural breakdown of proteoglycans and collagen is
observed, which impairs the biomechanical properties of cartilage. The
maintenance
of a normal, healthy extracellular matrix reflects a balance between the rate
of
biosynthesis and incorporation of matrix components, and the rate of their
degradation and subsequent loss from the cartilage into the synovial fluid. A
variety
of proteases have the potential to cleave cartilage and are involved in the
degradation
process, most notably the matrix metalloproteinases.
Matrix metalloproteinases (MMPs), or matrixins, are a family of at least 15
zinc endopeptidases that function extracellularly and play a key role in
pathological
degradation of tissue. Current nomenclature and alternative names for members
of
the MMP are provided in Table 7. Most MMPs are highly regulated and most are
not
constitutively expressed in normal tissues. However, pro-inflammatory
cytokines,
such as IL-1 and TNF-a, initiate transcription and expression. An imbalance
created
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-76-
by upregulation and activation of tissue-degrading MMPs is a primary causative
factor in the cartilage breakdown process during chronic inflammatory diseases
and
sustained synovial inflammatory responses subsequent to joint injury.
Cartilage
matrix metabolism has been studied in patients with either a meniscal injury
or
anterior cruciate ligament rupture in the knee. It was shown that
concentrations of
stromelysiri-1 (MW-3), collagenase, tissue inhibitor of metalloproteinases
(TIMP- '
1), and proteoglycan fragments increased in human knee synovial fluid after
traumatic knee injury. Temporally, these values increased immediately over
reference levels and remained significantly elevated (10-fold increase) over a
period
of one year. These changes likely drive the increase in the rn concentration
of
proteoglycan fragments that are observed in synovial fluid after knee ligament
injury.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-77-
TABLE 7
Matrix Metalloproteinases
MMP Alternative Names EC Number Substrates
MMP-1 Collagenase EC3.4.24.7 Collagens (I, II, II, VII, and
Fibroblast X);Gelatin; aggrecan;
Collagenase hyaluronidase-treated versican;
Interstitial proteoglycan link protein; large
Collagenase tenascin-C; a1- antitrypsin/a1 -
proteinase inhibitor (a1- AT); a1
antichymotrypsin ((x1- ACHYM);
a2 M; rat a1 M; pregnancy zone
protein; rat a1I3 (a1- inhibitor 3);
ovostatin; entactin; MBP; GST-
TNF/TNF peptide; L-selection; IL-
1(3; serum amyloid A; IGF-BP5;
IGF-BP3; MMP-2; MMP-13
MMP-2 72-kDa Gelatinase EC3.4.24.24 Collagens (I, IV, V, VI, X, XI, and
Gelatinase A XIV); Gelatin; elastin; fibronectin;
Type IV Collagenase laminin-1, laminin-5; gelactin-3;
Neutrophil aggrecan; decorin; hyaluronidase-
Gelatinase treated versican; proteoglycan link
protein; osteonectin; MBP; GST-
TNF/TNF peptide; IL-1(3; A(3i-ao ;
A(31o-2o ; APP695; a1- AT; prolysyl
oxidase fusion protein; IGF-BP5;
IGF-BP3; FGF Rl; MMP-1; MMP-
9; MMP-13
MMP-3 Stromelysin-1 EC3.4.24.17 Collagens (III, IV, V, IX); Gelatin;
Transin aggrecan; versican and
hyaluronidase-treated veriscan;
perlecan; decorin; proteoglycan link
protein; large tenascin-C;
fibronectin; laminin; entactin;
osteonection; elastin; casein; a1-
ACHYM; antithrombin-III; a2 M;
ovostain; Substance P; MBP; GST-
TNF/TNF peptide; IL-1J3; serum
amyloid A; IGF-BP3; fibrinogen
and cross-linked fibrin;
plasminogen; MMP-
1 "superactivation", MMP-2/TIMP-2
complex; MMP-7; MMP-8; MMP-
9; MMP-13
MMP-7 Matrilysin EC3.4.24.23 Collagen IV and X; Gelatin;
PUMP aggrecan; decorin; proteoglycan
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-78-
link protein; fribronectin and
laminin; insoluble fibronectin
fibrils; enactin; large and small
tenascin-C; osteonectin; (34 integrin;
elastin; casein; transferrin; MBP; a1
-AT; GST-TNF/TNF peptide;
plasminogen; MMP-1; MMP-2;
MMP-9; MMP-9/TIMP-1
MMP-8 Neutrophil EC3.4.24.34 Collagens (I, II, III, V, VII and X);
Collagenase Gelatin; aggrecan; a1 -AT; a1-
Collagenase I ACHYM; a2-antiplasmin;
fibronectin
MMP-9 92 kDa Gelatinase EC3.4.24.35 Collagens (IV, V, VII, X and XIV);
Gelatinase B Gelatin; elastin; galectin-3;
aggrecan; hyalurondise-treated
versican; proteoglycan link protein;
fibronectin; entactin; osteonectin;
a1-AT; MBP; GST-TNF/TNF
peptide; IL-1(3; AP 1-4o; plasminogen
MMP-10 Stromelysin-2 EC3.4.24.22 Collagens (III, IV and V); Gelatin;
casein; aggrecan; elastin;
proteoglycan link protein; MMP-1;
MMP-8
MMP-1 1 Stromelysin-3 EC3.4.24 Human enzyme: a1 -AT; a2 M;
casein, laminin, fibronectin, gelatin,
collagen IV and carboxymethylated
transferrin
MMP-12 Macrophage EC3.4.24 Collagen IV; Gelatin; elastin and x -
Metalloelastase elastin; casein; a1-AT; fibronectin;
vitronectin; laminin; enactin;
proteoglycan monomer; GST-TNF;
MBP; fibrinogen; fibrin;
plasminogen
MMP-13 Collagenase-3 EC3.4.24 Collagens (I, II and III, IV, IX, X
and XIV); Gelatin, al-ACHYM
and plasminogen activator inhibitor
2; aggrecan; perlecan; large
tenascin-C, fribronectin;
osteonectin;MMP-9
MMP-14 MT-MMP-1 EC3.4.24 Collagen (I, II and III); Gelatin,
casean, x-elastin, fribronectin,
laminin, vitronectin and
proteoglycans; large tenascin-C,
enactin; a1 AT, a2 M; GST-TNF;
MMP-2; MMP-13
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-79-
MMP-15 MT-MMP-2 Fibronectin, large tenascin-C,
entactin, laminin, aggrecan,
perlecan; GST-TNF; MMP-2
The MMP family of enzymes has been shown to be secreted from human
chondrocytes . and by cells of the synovium, such as synovial fibroblasts.
Furthermore, using in situ hybridization, it was shown that human synovium
synthesizes both stromelysin-1 and collagenase. Stromelysin-1 (MMP-3) is
capable
of degrading all of the components of the cartilage matrix. There is evidence
that
chondrocytes contribute to cartilage degradation by the release of the matrix-
degrading enzyme, collagenase-3. Upon activation by pro-inflammatory
cytokines,
MMPs are secreted from cells in a latent form, are activated extracellularly,
and are
inhibited by tissue inhibitors of metalloproteinases (TIMPs). The balance
between
the activities of MMPs and TIMPs is thought to be important for the
maintenance of
an intact cartilage matrix. Under pathological conditions such as
osteoarthritis and
rheumatoid arthritis, several studies have shown elevated amounts of MMPs,
resulting in an imbalance between MMPs and TIMPs that is considered to account
for the observed cartilage destruction.
The MMPs are regulated by cytokines, such as interleukin-1 (IL-1), and
growth factors that act on chondrocytes and synoviocytes to enhance their
protease
production. Other pro-inflammatory cytokines, such as IL-6, IL-8 and TNF-a,
also
upregulate the production of matrix-degrading enzymes. This leads to cartilage
destruction, which is usually assessed as the loss of sulfated
glycosaminoglycans
(GAGs) and the cleavage of collagen. IL-1, which is present in the joint fluid
of
patients with arthritic diseases, stimulates chondrocytes to synthesize
elevated
amounts of enzymes such as stromelysin, fibroblast and neutrophil collagenase,
and
plasminogen activator. In addition, IL-1 inhibits synthesis of plasminogen
activator
inhibitor-1 and TIMP, and also inhibits synthesis of matrix constituents such
as
collagen. The imbalance between the levels of inhibitors and enzymes leads to
an
increase in the amount of active proteases and, combined with a suppression of
matrix biosynthesis, results in cartilage degradation.
Using cartilage slices as an in vitro model, it has been shown that
collagenase
inhibitors can inhibit either the IL-1 or IL-8 stimulated invasion of
articular cartilage
by rheumatoid synovial fibroblasts (RSF). The collagenase inhibitors, 1, 1 0-o-
phenanthroline and phosphoramidon, substantially inhibited the
concentration-dependent penetration of cartilage by RSF cells at
concentrations of
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-80-
10-150 M. The selective effect of cytokines on the secretion of proteinases
demonstrates that synovial fibroblast-like cell-mediated articular degradation
is a
highly regulated process. Thus, the ability to inhibit protease activity and
associated
matrix degradation locally within the joint is expected to inhibit the
cartilage
. destruction process. The action of the inhibitors in the limited in vitro
system
suggests that therapeutic intervention using systemic or local delivery of
synthetic
MMP inhibitors' with appropriate pharmokinetics will be effective as
chondroprotective agents.
Examples of MMP inhibitors suitable for the present invention include U-
24522 ((R,S)-N-[2-[2-(hydoxylamino)-2-oxoethyl]-4-methyl-l-oxopentyl]-L-leucyl-
L-phenylalaniamide); BB2516; (N2-[35[Hydroxy-4-(N-hydroxyamino)-2R-isobutyl]-
L-leucine-Nl-methylamide, also -known as marimastat) peptides such as MMP
Inhibitor I and MMP-3 Inhibitor, and larger proteins such as TIMP-1 or
fragments
thereof, and are listed in the Table below: For all modes of local delivery
(i.e.,
injection, infusion and irrigation) the optimal dose and/or concentration of
each
suitable agent is that which is therapeutically effective. As an example, for
each of .
the listed agents, the preferred and most preferred concentrations of an
irrigation
solution containing the listed agent are provided, such concentrations
expected to be
therapeutically effective when delivered locally. Similarly, systemic
compositions in
accordance with the present invention will suitably include a dosage or load
of the
agent sufficient to result in a local concentration at the joint or site of
action within
the listed therapeutic range. For targeted sustained release delivery systems,
a
sufficient dosage or load of the agent is included in the composition to
result in a
local concentration at the joint or site of action within the listed
therapeutic range
over a predetermined sustained release period.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-81-
TABLE 8
Therapeutic and Preferred Concentrations of Matrix Metalloproteinases (MMPs)
Inhibitors
Local Delivery Most Preferred
Compounds Therapeutic Local Delivery
Concentrations Concentrations
(nanomolar) (nanomolar)
1. BB2516 0.2-2000 2-200
2. GM1489 0.2-400 2-100
3. GM6001 0.4-800 2-200
4. U-24522 0.2-2,000 20-200
minocycline 30-500,000 300-3,000
MMP Inhibitor I 0.3-3,000 3-600
4-Abz-Gly-Pro-D-Leu-D-Ala-NHOH
MMP-3 Inhibitor 0.5-5,000 5-500
Ac-Arg-Cys-Gly-Val-Pro-Asp-NH2
rhuman TIMP 1 0.5-5,000 5-500
rhuman TIMP2 0.3-3,000 3-600
8. INHIBITORS OF NUCLEAR FACTOR KAPPA B
NFxBB
Pro-inflammatory and cartilage-destructive cellular pathways are regulated by
extracellular and intracellular signaling mechanisms that are targets for
novel
therapeutic local and systemic drug delivery. The complete . molecular
signaling
mechanisms utilized by the pro-inflammatory cytokine interleukin-1 (IL-1) to
activate the transcription factor, nuclear factor kappaB (NFiB), are poorly
defined.
Nevertheless, a key molecule that is involved in intracellular signaling at
the level of
gene transcription is the pro-inflammatory transcription factor, (NFiB). NFiB
activity is mediated by a family of transcription factor subunits that bind to
DNA
either in the form of homodimers or heterodimers. These subunits are typically
present within the cytoplasm of cells in an inactive form due to the binding
of the
inhibitory subunit called IiB. Activation of IL-1 receptors, and other
extracellular
signals, induce degradation of IiB and concomitant dissociation of NFKB from
the
inhibitors, followed by translocation to the nucleus. NFiB, was found to be
involved
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-82-
in IL-1 induced expression and was capable of increasing pro-inflammatory COX-
2
protein expression in RA synovial fibroblasts.
The identification of NFxB as a key molecular target is based upon its role as
a common downstream signaling element regulating gene expression of several
critical inflammatory mediators linked to joint inflammation and cartilage-
destructive
pathways. The response of many genes (COX-2, collagenase, IL-6, IL-8) are
governed by promoters which contain both NFxB promoter elements. Activation of
NFKB mediates the induction of many, proteins central to the inflammatory
process,
such as cytokines, cell-adhesion molecules, metalloproteinases and other
proteins
that participate in the production of prostaglandins and leukotrienes (COX-2)
in
synoviocytes. Thus, this transcription factor represents a physiologically
significant
target in therapies directed to the injury responses of human synovial
fibroblasts,
human articular chondrocytes, as well as other cells in the joint.
Specifically, it has been shown that exposure of human rheumatoid synovial
fibroblasts (RSF) to interleukin lbeta (IL-lbeta) results in the coordinate up-
regulation of 85-kD phospholipase A2 (PLA2) and inducible cyclooxygenase (COX-
2). Together, these two enzymes promote. the subsequent biosynthesis of PGE2,
a
primary inflammatory mediator in the joint. Oligonucleotide decoys and
antisense
were used to demonstrate the participation of the (NFxB), in the regulation of
the
prostanoid-metabolizing enzymes. Antagonizing NFxB mRNA using anti-sense
oligonucleotide resulted in decreased binding to the COX gene promoter.
Hymenialdisine, a marine natural product, has recently been characterized as
an inhibitor of NFKB activation and exposure of IL-1-stimulated RSF-inhibited
PGE2 production in a concentration-dependent manner (IC50= 1 M). The
specificity
of the molecular target was shown through use of an analog, aldisine, and the
protein
kinase C inhibitor, RO 32-0432, which were inactive. Direct action of
hymenialdisine on IL-1-induced NFxB activation was demonstrated by a
significant
reduction (approximately 80%) in NFKB binding to the classical xB consensus
motif
and inhibition of stimulated p65 migration from the cytosol of treated cells.
As
expected for an inhibitor of NFxB transcriptional regulation, hymenialdisine-
treated
RSF did not transcribe the mRNAs for either COX-2 or PLA2 in response to IL-1.
Consequently, reduced protein levels for these enzymes and reductions in the
ability
to produce PGE2 were observed. Furthermore, IL-1-stimulated interleukin-8 (IL-
8)
production, which is known to be an NF-KB-regulated event, was also inhibited
by
hymenialdisine, whereas IL-1-induced production of vascular endothelial growth
factor, a non-NFiB-regulated gene, was not affected by exposure to
hymenialdisine.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-83-
Thus, hymenialdisine inhibits IL-1-stimulated synovial fibroblast formation of
PGE2
through its inhibitory effect on NF-KB activation. This provides a basis to
define its
use as a novel inhibitor to block the role of NF-KB in joint inflammation and
cartilage
destruction.
Examples of NF-KB inhibitors suitable for the present invention are listed
below. For all modes of local delivery (i.e., injection, infusion and
irrigation), the
optimal dose and/or concentration of each suitable agent is that which its
therapeutically effective. As an example, for each of the listed agents, the
preferred
and most preferred concentrations of an irrigation solution containing the
listed agent
are provided, such concentrations expected to be therapeutically effective
when
delivered locally. Similarly, systemic compositions in accordance with the
present
invention will suitably include a dosage or load of the agent sufficient to
result in a
local concentration at the joint or site of action within the listed
therapeutic range.
For targeted sustained release delivery systems, a sufficient dosage or load
of the
agent is included in the composition to result in a local concentration at the
joint or
site of action within the listed therapeutic range over a predetermined
sustained
release period.
TABLE 9
Therapeutic and Preferred Concentrations of Inhibitors of NFkB
Local Delivery Most Preferred
Therapeutic Local Delivery
Compounds Concentrations Concentrations
(nanomolar) (nanomolar)
Caffeic acid phenylethyl 1-100,000 50-20,000
ester (CAPE)
DM-CAPE 0.5-50,000 50-5,000
SN-50 peptide 0.1-100,000 100-20,000
hymenialdisine 1-100,000 100-10,000
pyrolidone dithiocarbamate 1-50,000 50-10,000
9. NITRIC OXIDE SYNTHASE INHIBITORS
Nitric oxide (NO) is a widespread intracellular and intercellular mediator
involved in the pathophysiological mechanisms of some connective tissue
diseases.
NO is formed from L-arginine by a family of enzymes, the NO synthases, which
are
localized intracellularly. Three isoforms of NO synthase have been cloned and
sequenced. Endothelial cell NO synthase (ecNOS) and brain NO synthase (bNOS)
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-84-
are constitutively active. A distinct isoform of NO synthase, inducible NOS
(iNOS-),
is found in many cell types, including chondrocytes. It is absent under basal
conditions, but is upregulated in response to pro-inflammatory mediators such
as IL-
1(3 and TNF-a. Recent findings show that IL-1 is a very potent stimulator of
chondrocyte NO synthesis and that IL-1 acts through its ability to upregulate
the
level of the iNOS. Within the joint, chondrocytes are the major source of NO
and
chondrocytic iNOS induced by pro-inflammatory cytokines is considered to
mediate
many'effects of IL-1 in, inflammatory arthropathies.
Drugs that specifically inhibit chondrocyte inducible NO synthase (iNOS)
may have a therapeutic role in the prevention of chondrodestruction that
occurs due
to joint injury (e.g., surgical procedures involving the joint). Evidence
supporting
such a beneficial therapeutic effect is based upon a substantial number of
studies
which have evaluated a variety of iNOS inhibitors for their ability to inhibit
inducible
NO synthase activity in cultured chondrocytes and explants of cartilage from
patients
with osteoarthritis. A class of compounds, termed S-substituted isothioureas,
have
been characterized as -potent inhibitors of NO biosynthesis in cartilage. S-
methyl
isothiourea and S-(aminoethyl) isothiourea were 2-4 times more potent than NG-
monomethyl-L-arginine, 5-10 times more potent than aminoguanidine and over 300
times more potent than N '-nitro-L-arginine and NW-nitro-L-arginine methyl
ester.
These isothiourea compounds provide a potent and relatively specific class of
inhibitors of iNOS in cartilage and thus are suitable for systemic or local
delivery in
accordance with aspects of the invention (Jang, D., Eur. J. Pharmacol. 312:341-
347(1996)).
The cartilage protective therapeutic potential of NO synthase inhibitors has
also been assessed using in vitro systems such as isolated chondrocytes to
define
effects on the cartilage matrix. Inhibition of endogenous NO production by NG -
monomethyl-L-arginine (L-NMMA), an established NO synthase inhibitor, leads to
the suppression of gelatinase, collagenase, and stromelysin production by IL-
1 j3-stimulated chondrocytes. Inhibition of NO production also partially
reduces the
increase in the lactate production that occurs from the exposure of
chondrocytes to
IL-1P3. Treatment of cartilage fragments with L-NMMA partially reverses the IL-
1(3
inhibitory effect of glycosaminoglycan synthesis, inhibits IL-1(3-stimulated
MMP
activities, and increases IL-1 receptor antagonist (IL-lra) production. NO can
also
modulate proteoglycan synthesis indirectly by decreasing the production of TGF-
(31
by chondrocytes exposed to IL-1p. It prevents autocrine-stimulated increases
in
TGF-(31, thus diminishing the anabolic effects of this cytokine in
chondrocytes.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-85-
A study has compared the potency of new aminoguanidine, S-
methylisothiourea (SMT), S-aminoethylisothiourea (AETU), L-NMMA and N-nitro-
L-arginine methyl ester (L-NAME) NOS inhibitors on the inhibitory effect of
recombinant human IL-1 responses on proteoglycan synthesis and NO production.
Different culture systems have been shown to respond in a concentration
dependent
manner to IL-1 [3 challenge with a large increase in NO production and a
marked
suppression of proteoglycan synthesis. The above NOS inhibitors (at 1 to 1000
M)
inhibited NO production by cartilage cells treated with IL-1(3 and had marked
effects
on restoring proteoglycan synthesis in chondrocytes. Therefore, if NO
production
can be blocked using a therapeutically effective concentration and dose, then
IL-1 (3
inhibition of proteoglycan synthesis will be prevented.
NO synthase is expressed in cartilage obtained from the joint of patients with
arthritic disease. In patients presenting either rheumatoid arthritis or
osteoarthritis,
increased levels of nitrite have been observed in the synovial fluid and it
has been
shown that a significant source of NO production in these patients is derived
from
articular cartilage. Furthermore, it has been found that sustained systemic
delivery of
L-NIL, a potent inhibitor of iNOS, reduces the progression of experimental OA
in
dogs (induced by sectioning of the ACL) and causes a substantial decrease in
IL-1(3,
PGE2, NO -and MMP production. These findings suggest that NO is a potent
regulator of the effects of IL-1(3 and contributes to the pathophysiology of
joint
diseases.
Thus, these in vitro and in vivo results indicate that specific inhibitors of
NO
syntheses are potential novel drugs for the clinical treatment of synovial
inflammation and can provide chondroprotective effects when delivered
systemically
or locally in combination with one or more drugs chosen from the anti-
inflammatory,
cartilage-protective, and anti-pain classes to treat a surgically treated
joint or other
injured joint.
Examples of NO synthase inhibitors suitable for the present invention are
listed below. For all modes of local delivery (i.e., injection, infusion and
irrigation),
30' the optimal dose and/or concentration of each suitable agent is that which
is
therapeutically effective. As an example, for each of the listed agents, the
preferred
and most preferred concentrations of an irrigation solution containing the
listed agent
are provided, such concentrations expected to be therapeutically effective
when
delivered locally. Similarly, systemic compositions in accordance with the
present
invention will suitably include a dosage or load of the agent sufficient to
result in a
local concentration at the joint or site of action within the listed
therapeutic range.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-86-
For targeted sustained release delivery systems, a sufficient dosage or load
of the
agent is included in the composition to result in a local concentration at the
joint or
site of action within the listed therapeutic range over a predetermined
sustained
release period. In one embodiment, the preferred NO synthase inhibitors for
inclusion in the solutions of the invention is 1400 W ((N-3-
(aminomethyl)benzyl)acetamidine), a selective, slow, tight binding inhibitor
of
iNOS, diphenyleneiodinium and 1,3-PBIT.
TABLE 10
Therapeutic and Preferred Concentrations of Nitric Oxide Synthase Inhibitors
Local Delivery Most Preferred
Therapeutic Local Delivery
Compounds Concentrations Concentrations
fAM-4)
N G -monomethyl-L-arginine 50-50,000 3,000
1400 W 0.1-1,000 1-20
diphenyleneiodium 0.1-1,000 1-100
S-methyl isothiourea 1-1,000 10-100
S-(aminoethyl)'isothiourea 1-1,000 10-100
L-N6-(1-iminoethyl)lysine 1-1,000 10-100
1,3-PBITU 0.5-500 5-100
2-ethyl-2-thiopseudourea 2-20,000 20-2,000
10. Cell Adhesion Molecules
1 Oa. Integrin Agonists and Antagonists
Integrins are heterodimer receptors located on the plasma membrane that
contain a and B subunits that bind ligands which are extracellular matrix
(ECM)
components or may be other large proteins, such as collagen, laminin,
vitronectin,
osteopontin (OPN) and fibronectin (FN). Degradation of the cartilage matrix is
regulated by chondrocytes through mechanisms that depend upon the interaction
of
these cells with the ECM. Chondrocyte gene expression is, in part, controlled
through cellular contacts involving the interaction of integrins with
components of
ECM in the environment surrounding the chondrocyte. Hence, integrins on
chondrocytes are involved in control of cartilage homeostasis, and this family
of
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-87-
receptors represents a class of therapeutic targets for preventing cartilage
degradation.
Human chondrocytes express an array of integrin receptors composed of
distinct a and B subunits, including a1J31, a5131, aV133 and lesser quantities
of others.
Of particular importance is the aV133 integrin, which is known to bind OPN.
The
aVB3 complex-specific function blocking monoclonal antibody (mAb) LM609 acts
as an agonist in a manner that is similar to the ligand, OPN: It attenuates
the
production of a number of proinflammatory and cartilage destructive mediators,
such
as IL-1, NO and PGE2. Thus, the agonistic mAb LM609 is thought to be suitable
for
use in the present invention.
In addition, two peptidomimetics, MK-383 (Merck) and RO 4483
(Hoffinann-LaRoche), have been studied in Phase II clinicals. Since these are
both
small molecules, they have a short half-life and high potency. However, these
seem
to also have less specificity, interacting with other closely related
integrins. These
peptidomimetics are also be suitable for use in the present invention.
TABLE 11
Therapeutic and Preferred Concentrations of Integrins
Local Delivery Therapeutic Local Delivery
Concentrations ([!g-/ml) Preferred
Class of Agent Concentrations (gg/ml
Integrins: S
aV(33 mAb LM 609 0.05-5,000 5-500
echistatin 0.1-10,000 100-1,000
11. Anti-chemotactic agents
Anti-chemotactic agents prevent the chemotaxis of inflammatory cells.
Representative examples of anti-chemotactic targets at which these agents
would act
include, but are not limited to, F-Met-Leu-Phe receptors, IL-8 receptors, MCP-
1
receptors, and MIP-1-I/RANTES receptors. Drugs within this class of agents are
early in the development stage, but it is theorized that they may be suitable
for use in
the present invention.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541 PCT
-88-
12. Intracellular Signaling Inhibitors
12a. Protein Kinase Inhibitors
i. Protein Kinase C (PKC) Inhibitors
Protein kinase C (PKC) plays a crucial role in cell-surface signal
transduction
for a number of physiological processes. PKC isozymes can be activated as
downstream targets resulting from initial activation of either G-protein
coupled
receptors (e.g., serotonin, bradykinin, etc.) or pro-inflammatory cytokine
receptors.
Both of these receptor classes play important roles in mediating cartilage
destruction.
Molecular cloning analysis has revealed that PKC exists as a large family
consisting of at least 8 subspecies (isozymes). These isozymes differ
substantially in
structure and mechanism for linking receptor activation to changes in the
proliferative response of specific cells. Expression of specific isozymes is
found in a
wide variety of cell types, including: synoviocytes, chondrocytes,
neutrophils,
myeloid cells, and smooth muscle cells. Inhibitors of PKC are therefore likely
to
effect signaling pathways in several cell types unless the inhibitor shows
isozyme
specificity. Thus, inhibitors of PKC can be predicted to be effective in
blocking the
synoviocyte and chondrocyte activation and may also have an anti-inflammatory
effect in blocking neutrophil activation and subsequent attachment. Several
inhibitors have been described and initial reports indicate an IC50 of 50 M
for
calphostin C inhibitory activity. G-6203 (also known as Go 6976) is a new,
potent
PKC inhibitor with high selectivity for certain PKC isotypes, with IC50 values
in the
2-10 M range. Concentrations of these and another drug, GF 109203X, also
known
as Go 6850 or bisindoylmaleimide I (available from Warner-Lambert), that are
believed to be suitable for local delivery use in the present invention are
set forth
below. Similarly, systemic compositions in accordance with the present
invention
will suitably include a dosage or load of the agent sufficient to result in a
local
concentration at the joint or site of action within the listed therapeutic
range. For
targeted sustained release delivery systems, a sufficient dosage or load of
the agent is
included in the composition to result in a local concentration at the joint or
site of
action within the listed therapeutic range over a predetermined sustained
release
period.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-89-
TABLE 12
Therapeutic and Preferred Concentrations of
Cartilage Destruction Inhibitory Agents
Local Delivery Therapeutic Local Delivery
Concentrations Preferred
Class of Agent (Nanomolar) Concentrations
(Nanomolar)
Protein Kinase C Inhibitors:
calphostin C 0.5-50,000 100-5,000
GF 109203X 0.1-10,000 1-1,000
G-6203 (Go 6976) 0.1-10,000 1-1,000
ii. Protein Tyrosine Kinase Inhibitors
Although there is a tremendous diversity among the numerous members of
the receptors tyrosine-kinase (RTK) family, the signaling mechanisms used by
these
receptors share many common features. Biochemical and molecular genetic
studies
have shown that binding of the ligand to the extracellular domain of the RTK
rapidly
activates the intrinsic tyrosine kinase catalytic activity of the
intracellular domain
(see FIGURE 5). The increased activity results in tyrosine-specific
phosphorylation
of a number of intracellular substrates that contain a common sequence motif.
Consequently, this causes activation of numerous "downstream" signaling
molecules
and a cascade of intracellular pathways that regulate phospholipid metabolism,
arachidonate metabolism, protein phosphorylation (involving mechanisms other
'than
protein kinases), calcium mobilization and transcriptional activation (see
FIGURE 2).
Growth-factor-dependent tyrosine kinase activity of the RTK cytoplasmic domain
is
the primary mechanism for generation of intracellular signals that lead to
cellular
proliferation. Thus, inhibitors have the potential to block this signaling and
thereby
prevent synoviocyte and chondrocyte activation.
Any of several related tyrphostin compounds have potential as specific
inhibitors of tyrosine kinase activity (IC50s in vitro in the 0.5-1.0 M
range), since
they have little effect on other protein kinases and other signal transduction
systems.
To date, only a few of the many tyrphostin compounds are commercially
available,
and suitable concentrations for these agents as used in the present invention
are set
forth below. In addition, staurosporine has been reported to demonstrate
potent
inhibitory effects against several protein tyrosine kinases of the src
subfamily and a
suitable concentration for this agent as used in the present invention for
local delivery
also is set forth below. Similarly, systemic compositions in accordance with
the
present invention will suitably include a dosage or load of the agent
sufficient to
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-90-
result in a local concentration at the joint or site of action within the
listed therapeutic
range. For targeted sustained release delivery systems, a sufficient dosage or
load of
the agent is included in the composition to result in a local concentration at
the joint
or site, of action within the listed therapeutic range over a predetermined
sustained
release period.
TABLE 13
Therapeutic and Preferred Concentrations of
Inhibitory Agents
Local Delivery Local Delivery
Therapeutic Preferred
Class of Agent Concentrations Concentrations
(Nanomolar) (Nanomolar)
Protein Kinase Inhibitors
lavendustin A 10-100,000 100-10,000
tyrphostin 0 10-100,000 100-10,000
AG1296
tyrphostin 10-100,000 100-10,000
AG1295
staurosporine 1-100,000 10-1,000
PD 158780 0.1-10,000 10-500
PD 174265 0.1-10,000 10-500
12b. Modulators of Intracellular Protein Tyrosine Phosphatases
Non-transmembrane. protein tyrosine phosphatases (PTPases) containing
src-homology2 SH2 domains are known and nomenclature refers to them as
SH-PTP1 and SH-PTP2. In addition, SH-PTP1 is also known as PTP 1 C, HCP or
SHP. SH-PTP2 is also known as PTP 1 D or PTP2C. Similarly, SH-PTP 1 is
expressed at high levels in hematopoietic cells of all lineages and all stages
of
differentiation, and the SH-PTP 1 gene has been identified as responsible for
the
motheaten (me) mouse phenotype and this provides a basis for predicting the
effects
of inhibitors that would block its interaction with its cellular substrates.
Stimulation
of neutrophils with chemotactic peptides is known to result in the activation
of
tyrosine kinases that mediate neutrophil responses (Cui, et al., J. Immunol.
(1994))
and PTPase activity modulates agonist induced activity by reversing the
effects of
tyrosine kinases activated in the initial phases of cell stimulation. Agents
that could
stimulate PTPase activity could have potential therapeutic applications as
anti-
inflammatory mediators.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-91-
These same PTPases have- also been shown to modulate the activity of certain
RTKs. They appear to counter-balance the effect of activated receptor kinases
and
thus may represent important drug targets. In vitro experiments show that
injection
of PTPase blocks insulin stimulated phosphorylation of tyrosyl residues on
endogenous proteins. Thus, activators of PTPase activity could serve to
reverse
activation of RTK-receptor action in restenosis, and are believed" to be
useful in the
solutions of the present invention. In addition, receptor-linked PTPases also
function
as extracellular ligands, similar to those of cell adhesion molecules. The
functional
consequences of the binding of a ligand to the extracellular domain have not
yet been
defined but it is reasonable to assume that binding would serve to modulate
phosphatase activity within cells (Fashena et al., Current Biology, 5;1367-
1369
(1995)) . Such actions could block adhesion mediated by other cell surface
adhesion
molecules (NCAM) and provide an anti-inflammatory effect. No drugs have been
developed yet for these applications.
12c. Inhibitors of SH2 Domains (src Homology2 Domains)
SH2 domains, originally identified in the src subfamily of protein tyrosine
kinases (PTKs), are noncatalytic protein sequences and consist of about 100
amino
acids conserved among a variety of signal transducing proteins (Cohen, et al.,
1995).
SH2 domains function as phosphotyrosine-binding modules and thereby mediate
critical protein-protein associations in signal transduction pathways within
cells
(Pawson, Nature, 573-580, 1995). In particular, the role of SH2 domains has
been
clearly defined as critical for receptor tyrosine kinase (RTK) mediated
signaling such
as in the case of the platelet-derived growth factor (PDGF) receptor.
Phosphotyrosine-containing sites on autophosphorylated RTKs serve as binding
sites
for SH2-proteins and thereby mediate the activation of biochemical signaling
pathways (see FIGURE 2) (Carpenter, G., FASEB J 6:3283-3289 (1992); Sierke,
S.et al., J. Biochein. 32:10102-10108 (1993)). The SH2 domains are responsible
for
coupling the activated growth-factor receptors to cellular responses that
include
alterations in gene expression, and ultimately cellular proliferation. Thus,
inhibitors
that will selectively block the effects of activation of specific RTKs
(excluding IGFR
and FGFR) expressed on the surface of synoviocytes are predicted to be
effective in
blocking cartilage degradation after arthroscopy procedures.
At least 20 cytosolic proteins have been identified that contain SH2 domains
and function in intracellular signaling. The distribution of SH2 domains is
not
restricted to a particular protein family, but found in several classes of
proteins,
protein kinases, lipid kinases, protein phosphatases, phospholipases, Ras-
controlling
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-92-
proteins and some transcription factors. Many of the SH2-containing proteins
have
known enzymatic activities while others (Grb2 and Crk) function as "linkers"
and
"adapters" between cell surface receptors and "downstream" effector molecules
(Marengere, L., et al., Nature 369:502-505 (1994). Examples of proteins
containing
SH2 domains with enzymatic activities that are activated in signal
transduction
include, but are not limited to, the src subfamily of protein tyrosine kinases
(src
(pp60`sr0), abl, lck, fyn, fgr and others), phospholipaseCy (PLCy),
phosphatidylinositol 3-kinase (PI-3-kinase), p21-ras GTPase activating protein
(GAP) and SH2 containing protein tyrosine phosphatases (SH-PTPases) (Songyang,
et al., Cell 72: 767-778 (1993). Due to the central role these various SH2-
proteins
occupy in transmitting signals from activated cell surface receptors into a
cascade of
additional molecular interactions that ultimately define cellular responses,
inhibitors
which block specific SH2 protein binding (e.g., c-src) are desirable as agents
with
potential therapeutic applications in cartilage protection.
In addition, the regulation, of many immune/inflammatory responses is
mediated through receptors that transmit signals through non-receptor tyrosine
kinases containing SH2 domains. T-cell activation via the antigen specific T-
cell
receptor. (TCR) initiates a signal transduction cascade leading to lymphokine
secretion and T-cell proliferation. One of the earliest biochemical responses
following TCR activation is an increase in tyrosine kinase activity. In
particular,
neutrophil activation is in part controlled through responses of the cell
surface
immunoglobulin G receptors. Activation of these receptors mediates activation
of
unidentified tyrosine kinases that are known to possess SH2 domains.
Additional
evidence indicates that several src-family kinases (lck, blk, fyn) participate
in signal
transduction pathways leading from cytokine and integrin receptors and hence
may
serve to integrate stimuli received from several independent receptor
structures.
Thus, inhibitors of specific SH2 domains have the potential to block many
neutrophil
functions and serve as anti-inflammatory mediators.
Efforts to develop drugs targeted to SH2 domains currently are being
conducted at the biochemical in vitro and cellular level. Should such efforts
be
successful, it is theorized that the resulting drugs would be useful in the
practice of
the present invention.
V. ADDITIONAL AGENTS
In addition to the chondroprotective agent(s) described above, locally and
systemically delivered compositions of the present invention may also include
other
therapeutic agents. For example, one or more anti-inflammatory or analgesic
agents
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
ZU541PCT
-93-
(also referred to herein as anti-pain agents) may be included. Suitable
examples of
anti-inflammatory and/or analgesic agents are further described in detail
below. As a
further example, the compositions of the present invention may include one or
more
disease modifying anti-rheumatic drugs (DMARDs), such as methotrexate,
sulfasalazine, gold compounds such as oral gold, gold sodium thiornalate and
aurothioglucose, azathioprine, cyclosporine, antimalarials, steroids,
colchicines,
cyclophosphanmide, hydroxychloroquine sulfate, leflunomide, minocycline and
penicillamine. The anti-inflammation, analgesic and/or DMARD agents may be
included in the compositions of the present invention, or may be administered
separately, either concurrently or sequentially.
Many of the aspects of the present invention regarding the previously
addressed benefits of local administration, targeted systemic administration,
and the
use of multiple agents in connection with chondroprotective agents also
applies to the
administration of other agents. Alleviating pain and suffering in
postoperative
patients is an area of special focus in clinical medicine, especially with the
growing
number of outpatient operations performed each year. The most widely used
systemic agents, cyclooxygenase inhibitors (e.g., ibuprofen) and opioids
(e.g.,
morphine, fentanyl), have significant side effects including gastrointestinal
irritation/bleeding and respiratory depression. The high incidence of nausea
and
vomiting related to opioids is especially problematic in the postoperative
period.
Therapeutic agents aimed at treating postoperative pain while avoiding
detrimental
side effects are not easily developed because the molecular targets for these
agents
are distributed widely throughout the body and mediate diverse physiological
actions.
Despite the significant clinical need to inhibit pain and inflammation, as
well as
cartilage degradation, methods for the delivery of inhibitors of pain,
inflammation,
and cartilage degradation at effective dosages while minimizing adverse
systemic
side effects have not been developed. As an example, systemic (i.e.,
intravenous,
oral, subcutaneous or intramuscular) methods of administration of opiates in
therapeutic doses frequently is associated with significant adverse side
effects,
including severe respiratory depression, changes in mood, mental clouding,
profound,
nausea and vomiting.
Prior studies have demonstrated the ability of endogenous agents, such as
serotonin (5-hydroxytryptamine, sometimes referred to herein as "5-HT"),
bradykinin
and histamine, to produce pain and inflammation. Sicuteri, F., et al.,
Serotonin-
Bradykinin Potentiation in the Pain Receptors in Man, Life Sci. 4: 309-316
(1965);
Rosenthal, S.R., Histamine as the Chemical Mediator for Cutaneous Pain, J.
Invest.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
LU5411'CT
-94-
Dermat. : 98-105 (1977); Richardson, B.P., et al., Identification of Serotonin
M-Receptor Subtypes and their Specific Blockade by a New Class of Drugs,-
Nature
316:, 126-131 (1985); Whalley, E.T., et al., The Effect of Kinin Agonists and
Antagonists, Naunyn-Schmiedeb Arch. Pharmacol. 36: 652-57 (1987); Lang, E., et
al., "Chemo-Sensitivity of Fine Afferents from Rat Skin In Vitro" J.
Neurophysiol.
887-901 (1990).
For example, 5-HT applied to a human blister base (denuded skin) has been
demonstrated to cause pain that can be inhibited by 5-HT3 receptor
antagonists.
Richardson et al., (1985). Similarly, peripherally-applied bradykinin produces
pain
that can be blocked by bradykinin receptor antagonists. Sicuteri et al., 1965;
Whalley et al., 1987; Dray, A., et al., "Bradykinin and Inflammatory Pain",
Trends
Neurosci. 16: 99-104 (1993). Peripherally-applied histamine produces
vasodilation,
itching and pain that can be inhibited by histamine receptor antagonists.
Rosenthal,
1977; Douglas, W.W., "Histamine and 5-Hydroxytryptamine (Serotonin) and their
Antagonists", in Goodman, L.S.,` et al., ed., The Pharmacological Basis of
Therapeutics, MacMillan Publishing Company, New York, pp. 605-638 (1985);
Rumore, M.M., et al., Analgesic Effects of Antihistaminics, Life Sci 36, pp.
403-416
(1985). Combinations of these three agonists (5-HT, bradykinin and histamine)
applied together have been demonstrated to display a synergistic pain-causing
effect,
producing a long-lasting and intense pain signal. Sicuteri et al., 1965;
Richardson et
al., 1985; Kessler, W., et al., "Excitation of Cutaneous Afferent Nerve
Endings In
Vitro by a Combination of Inflammatory Mediators and Conditioning Effect of
'Substance P," Exp. Brain Res. 91:467-476 (1992).
In the body, 5-HT is located in platelets and in central neurons, histamine is
found in mast cells, and bradykinin is produced from a larger precursor
molecule
during tissue trauma, pH changes and temperature changes. Because 5-HT can be
released in large amounts from platelets at sites. of tissue injury, producing
plasma
levels 20-fold greater than resting levels (Ashton, J.H., et al., "Serotonin
as a
Mediator of Cyclic Flow Variations in Stenosed Canine Coronary Arteries,"
Circulation 73:572-578 (1986)), it is possible that endogenous 5-HT plays a
role in
producing postoperative pain, hyperalgesia and inflammation. In fact,
activation of
platelets has been shown to result in excitation of peripheral nociceptors in
'vitro.
Ringkamp, M., et al., "Activated Human Platelets in Plasma Excite Nociceptors
in
Rat Skin, In Vitro," Neurosci. Lett. 170:103-106 (1994). Similarly, histamine
and
bradykinin also are released into tissues during trauma. Kimura, E., et al.,
"Changes
in Bradykinin Level in Coronary Sinus Blood After the Experimental Occlusion
of a
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
LuD41rui
-95-
Coronary Artery," Am Heart J. 85:635-647 (1973); Douglas, 1985; Dray of al.
(1993).
In addition, prostaglandins also are known to cause pain and inflammation.
Cyclooxygenase inhibitors (e.g., ibuprofen) are commonly used in non-surgical
and
postoperative settings toblock the production of prostaglandins, thereby
reducing
prostaglandin-mediated pain and inflammation. Flower, R.J., et al., Analgesic-
Antipyretics and Anti-Inflammatory Agents; Drugs Employed in the Treatment of
Gout, in Goodman, L.S., et al., ed., The Pharmacological Basis of
Therapeutics,
MacMillan Publishing Company, New York, pp. 674-715 (1985). Cyclooxygenase
inhibitors are associated with some adverse systemic side effects when applied
systemically. For example, indomethacin or ketorolac have well recognized
gastrointestinal and renal adverse side effects.
As discussed, 5-HT, histamine, bradykinin and prostaglandins cause pain and
inflammation. The various receptors through which these agents mediate their
effects on peripheral tissues have been known and/or debated for the past two
decades. Most studies have been performed in rats or other animal models.
However, there are differences in pharmacology and receptor sequences between
human and animal species.
Furthermore, antagonists of these mediators currently are not used for
postoperative pain treatment. A class of drugs, termed 5-HT and norepinephrine
uptake antagonists, which includes amitriptyline, has been used orally with
moderate
success for chronic pain conditions. However, the mechanisms of chronic versus
acute pain states are thought to be considerably different. In fact, two
studies in the
acute pain setting using amitriptyline perioperatively have shown no pain-
relieving
effect of amitriptyline. Levine, J.D. et al., "Desipramine Enhances Opiate
.Postoperative Analgesia, Pain 27:45-49 (1986); Derrick, J.M. et al., "Low-
Dose
Amitriptyline as an Adjunct to Opioids for Postoperative Orthopedic Pain: a
Placebo-Controlled Trial Period," Pain 52:325-30 (1993). In both studies the
drug
was given orally. The second study noted that oral amitriptyline actually
produced a
lower overall sense of well being in postoperative patients, which may be due
to the
drug's affinity for multiple amine receptors in the brain.
Amitriptyline, in addition to blocking the uptake of 5-HT and norepinephrine,
is a potent 5-HT receptor antagonist. Therefore, the lack of efficacy in
reducing
postoperative pain in the previously mentioned studies would appear to
conflict with
the proposal of a role for endogenous 5-HT in acute pain. There are a number
of
reasons for the lack of acute pain relief found with amitriptyline in these
two studies.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-96-
(1) The first study (Levine et al., 1986) used amitriptyline preoperatively
for one
week up until the night prior to surgery whereas the second study (Derrick et
al.,
1993) only used amitriptyline postoperatively. Therefore, the level of
amitriptyline
that was present in the operative site tissues during the actual tissue injury
phase, and
the time at which 5-HT is purported to be released, is unknown. (2)
Amitriptyline is
known to be extensively metabolized by the liver. With oral administration,
the
concentration of amitriptyline in the operative site tissues may not have been
sufficiently high for a long enough time period to inhibit the activity of
postoperatively released 5-HT in the second study. (3) Since multiple
inflammatory
mediators exist, and studies have demonstrated synergism between the
inflammatory
mediators, blocking only one agent (5-HT) may not sufficiently inhibit the
inflammatory response to tissue injury.
There have been a few studies demonstrating the ability of extremely high.
concentrations (1% - 3% solutions -- i.e., 10 - 30 mg per milliliter) of
histamine,
(H1) receptor antagonists to act as local anesthetics for surgical procedures.
This
anesthetic effect is not believed to be mediated via H1 receptors but, rather,
due to a
non-specific interaction with neuronal membrane sodium channels (similar to
the
action of lidocaine). Given the side effects (e.g., sedation) associated with
these high
"anesthetic" concentrations of histamine receptor antagonists, local
administration of
histamine receptor antagonists currently is not used in the perioperative
setting.
A. SYNERGISTIC INTERACTIONS DERIVED FROM
THERAPEUTIC COMBINATIONS OF ANTI-PAIN
AND/OR ANTI-INFLAMMATION AGENTS AND
CHONDROPROTECTIVE AGENTS
Given the complexity of the disease process associated with inflammation
and loss of cartilage homeostasis after arthroscopic therapeutic procedures
and the
multiplicity of molecular targets involved, blockade or inhibition of a single
molecular target is unlikely to provide adequate efficacy in preventing
cartilage
degradation and the development of osteoarthritis. Indeed, a number of animal
studies targeting different individual molecular receptors and or enzymes have
not
proven effective in animal models or have not yielded efficacy in clinical
trials to
date. Therefore, a therapeutic combination of drugs acting on distinct
molecular
targets and delivered locally or systemically appears desirable for clinical
effectiveness in the therapeutic approach to cartilage protection. As
described below,
the rationale for this synergistic molecular targeted therapy is derived from
recent
advances in understanding fundamental biochemical mechanisms by which
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-97-
synoviocyte and chondrocyte cells in the synovium and cartilage transmit and
integrate stimuli to which they are exposed during arthroscopic procedures.
The molecular switches responsible for cell signaling have been traditionally
divided into major discrete signaling pathways, each comprising a distinct set
of
protein families that act as transducers for a particular set of extracellular
stimuli and
mediating distinct cell responses. One such pathway transduces signals _ from
neurotransmitters and hormones through G-protein coupled receptors (GPCRs) to
produce contractile responses that include the production of inflammatory
mediators,
such as PGE2. The GPCRs couple to intracellular targets through activation of
trimeric G proteins (see FIGURE 2), Examples of signaling molecules involved
in
activation of synoviocytes and chondrocytes through the GPCR pathway are
histamine, bradykinin, serotonin and ATP. A second major pathway transduces
signals from pro-inflammatory cytokines, such as IL-1, through a kinase
cascade and
NF-KB protein into regulation of gene expression and the production of
catabolic
cytokines and other catabolic factors, including NO.
Signals transmitted from neurotransmitters and hormones stimulate either of
two classes of receptors: GPCRs, composed of seven-helix transmembrane
regions,
or ligand-gated ion channels. "Downstream" signals from both kinds of
receptors
converge on controlling the concentration of cytoplasmic Ca2+ (see FIGURE 3).
Each GPCR transmembrane receptor activates a specific class of trimeric G
proteins,
including Gq, Gi or many others. Gq subunits activate phospholipase Cy,
resulting in
activation of protein kinase C (PKC) and an increase in the levels of
cytoplasmic
calcium (FIGURE 3). In turn, elevated intracellular calcium leads to the
activation of
cPLA2 and the production of arachidonic acid (AA). The AA serves as a
substrate
for COX in both 'synoviocytes and chondrocytes, leading to the production of
PGE2.
PKC activation also results in activation of MAP kinase leading to activation
of NF-
B and, in cells and tissues which have been primed by exposure to pro-
inflammatory
cytokines, modulates increased gene expression of proteins involved in
cartilage
catabolism.
Pro-inflammatory cytokine signaling, such as mediated by both IL-1 and
TNF-a through their distinct cognate receptors, also converges on regulation
of cell
gene expression. - The signal transduction pathways utilized by these distinct
receptors, employ distinct kinases that are proximal to the receptors but the
signaling
pathways subsequently converge at the .level of MAP kinases (FIGURE 3 and 4).
Signal transduction depends upon phosphorylation of residues in a cascade of
kinases, including "downstream" enzymes such as p38 MAP kinase. Activation of
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-98-
the IL-1-receptor and TNF-receptor also leads to stimulation of MAP kinase,
common steps shared by the, Gq coupled GPCRs (see FIGURE 3). It is now
recognized that ligand-independent "crosstalk" can transactivate kinase
pathways in
response to costimulation of specific GPCRs and=cytokines such as IL-1,
leading to
synergistic cellular responses (see FIGURE 3). Thus, a combination of
selective
inhibitors which blocks transactivation of a common signaling pathway (as
shown in
FIGURES 1 and 2) leading to increased gene expression of pro-inflammatory
cytokines, iNOS, COX-2, and MMPs will act synergistically to prevent
inflammation
and cartilage degradation after arthroscopic surgical procedures.
B. ANTI-INFLAMMATION AND ANTI-PAIN AGENTS
Suitable classes of anti-inflammation and/or anti-pain agents for use in the
compositions and methods of the present invention include: (1) serotonin
receptor
antagonists; (2) serotonin receptor agonists; (3) histamine receptor
antagonists;
(4) bradykinin receptor antagonists; (5) kallikrein inhibitors; (6) tachykinin
receptor
antagonists, including neurokininl and neurokinin2 receptor subtype
antagonists; (7)
calcitonin gene-related peptide (CGRP) receptor antagonists; (8) interleukin
receptor
antagonists; (9) inhibitors of enzymes active in the synthetic pathway for
arachidonic
acid metabolites, including (a) phospholipase inhibitors, including PLAZ
isoform
inhibitors and PLC isoform inhibitors (b) cyclooxygenase inhibitors, and (c)
lipooxygenase inhibitors; (10) prostanoid receptor antagonists including
eicosanoid
EP-1 and EP-4 receptor subtype antagonists and thromboxane receptor subtype
antagonists; (11) leukotriene receptor antagonists including leukotriene B4
receptor
subtype antagonists and leukotriene D4 receptor subtype antagonists; (12)
opioid
receptor agonists, including -opioid, S-opioid, and K-opioid receptor subtype
agonists; (13) purinoceptor agonists and antagonists including P2X receptor
antagonists and P2y receptor antagonists; and (14) calcium channel
antagonists.
The following is a description of suitable drugs falling in each of the
aforementioned classes of anti-inflammation/anti-pain agents, as well as
suitable
concentrations for use in solutions, of the present invention intended to be
delivered
locally. Similarly, systemic compositions in accordance with the present
invention
will suitably include a dosage or load of the agent sufficient to result in a
local
concentration at the'joint or site of action within the listed therapeutic
range. For
targeted sustained release delivery systems, a sufficient dosage or load of
the agent is
included in the composition to result in a local concentration at the joint or
site of
action within the listed therapeutic range over a predetermined sustained
release
period. While not wishing to be limited by theory, the justification behind
the
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-99-
selection of the various classes of agents which is believed to render the
agents
operative is also set forth.
Each agent is preferably included at a low concentration of 0.1 to 10,000
times Kd or Ki, except for cyclooxygenase inhibitors, which may be required at
larger
concentrations depending on the particular inhibitor selected. Preferably,
each agent
is included at a concentration of 1.0 to 1,000 times Kd or Ki and most
preferably at
approximately 100 times Kd or Ki. These concentrations are adjusted as needed
to
account for dilution in the absence of metabolic transformation at the local
delivery
site. The exact agents selected for use in the solution, and the concentration
of the
agents, varies in accordance with the particular application, as described
below.
1. Serotonin Receptor Antagonists
Serotonin (5-HT) is thought to produce pain by stimulating serotonin2
(5-HT2) and/or serotonin3 (5-HT3) receptors on nociceptive neurons in the
periphery.
Most researchers agree that 5-HT3 receptors on peripheral nociceptors mediate
the
immediate pain sensation produced by 5-HT (Richardson et al., 1985). In
addition to
inhibiting 5-HT-induced pain, 5-HT3 receptor antagonists, by inhibiting
nociceptor
activation, also may inhibit neurogenic inflammation. Barnes P.J., et al.,
"Modulation of Neurogenic Inflammation: Novel Approaches to Inflammatory
Disease", Trends in Pharmacological Sciences 11: 185-189 (1990). A study in
rat
ankle joints, however, claims the 5-HT2 receptor is responsible for nociceptor
activation by 5-HT. Grubb, B.D., et al., "A Study of 5-HT-Receptors Associated
with Afferent Nerves Located in Normal and Inflamed Rat Ankle Joints", Agents
Actions 25: 216-18 (1988). Therefore, activation of 5-HT2 receptors also may
play a
role in peripheral pain and neurogenic inflammation.
One goal of the solution of the present invention is to block pain and a
multitude of inflammatory processes. Thus, 5-HT2 and 5-HT3 receptor
antagonists
are both suitably used, either individually or together, in the solution of
the present
invention, as shall be described subsequently. Amitriptyline (ElavilTM) is a
suitable
5-HT2 receptor antagonist for use in the present invention. Amitriptyline has
been
used clinically, for numerous years as an anti-depressant, and is found to
have
beneficial effects in certain chronic pain patients. Metoclopramide (ReglanTM)
is
used clinically as an anti-emetic drug, but displays moderate affinity for the
5-HT3
receptor and can inhibit the actions of 5=HT at this receptor, possibly
inhibiting the
pain due to 5-HT release from platelets. Thus, it also is suitable for use in
the present
invention.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
zUD41FU I A
-100-
Other suitable 5-HT2 receptor antagonists include imipramine, trazodone,
desipramine and ketanserin. = Ketanserin has been used clinically for its anti-
hypertensive effects. Hedner, T., et al., "Effects of a New Serotonin
Antagonist,
Ketanserin, in Experimental and Clinical Hypertension", Am JofH}pertension
317s-
23s (Jul. 1988). Other suitable 5-HT3 receptor antagonists include cisapride
and
ondansetron. Suitable serotoniniB receptor antagonists include yohimbine,
N-[-methoxy-3-(4-methyl-l-piperanzinyl)phenyl]-2'-methyl-4'-(5-methyl-1, 2, 4-
oxadiazol-3-yl)[1, 1-biphenyl]-4-carboxamide ("GR127935") and methiothepin.
Therapeutic and preferred concentrations for local delivery use of these drugs
in the
solution of an aspect the present invention are set forth in Table 44.
Similarly,
systemic compositions in accordance with the present invention will suitably
include
a dosage or load of the agent sufficient to result in a local concentration at
the joint or
site of action within the listed therapeutic range. For targeted sustained
release
delivery systems, a sufficient dosage or load of the agent is included in the
composition to result in a local concentration at the joint or site of action
within the
listed therapeutic range over a predetermined sustained release period.
TABLE 14
Therapeutic and Preferred Concentrations of Pain and/or Inflammation
Inhibitory
Agents
Local Delivery Local Delivery
Therapeutic Preferred
Class of Agent Concentrations Concentrations
(Nanomolar) (Nanomolar)
Serotonin2 Receptor Anta og nists:
Amitriptyline 0.1-1,000 50 - 500
MDL-11,939 0.1-1,000 50 - 500
AMI-193 0.1 - 2;000 50 - 500
Desipramine 0.1-15000 50 - 500
Ketanserin 0.1-1,000 50 - 500
Serotonin3 Receptor Antagonists:
Tropisetron 0.01 - 100 0.05 - 50
Metoclopramide 10 - 10,000 200 - 2,000
Cisapride 0.1-1,000 20 - 200
Ondansetron 0.1-1,000 20 - 200
SerotoninmB (Human 1Dfi) Antagonists:
Isamoltare 0.1-1,000 50 - 500
GR127935 0.1-1,000 10 - 500
Methiothepin 0.1 - 500 1 - 100
SB216641 0.2-2,000 2 - 200
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-101-
2. Serotonin Receptor A og nists
5-HT1A, 5-HT1B and 5-HT1D receptors are known to inhibit adenylate
cyclase activity. Thus including a low dose of these serotoninlA, serotoninlB
and
serotoninlD receptor agonists in the solution should inhibit neurons mediating
pain
and inflammation. The same action is expected from serotoninlE and serotonin1F
receptor agonists because these receptors also inhibit adenylate cyclase.
Buspirone is a suitable 1A receptor agonist for use in the present invention.
Sumatriptan is a suitable 1 A, 1 B, 1 D and 1 Preceptor agonist. A suitable 1
B and 1 D
receptor agonist is dihydroergotamine. A suitable 1E receptor agonist is
ergonovine.
Therapeutic and preferred concentrations for these receptor agonists when
delivered
locally are provided in Table 15. Similarly, systemic compositions in
accordance
with the present invention will suitably include a dosage or load of the agent
sufficient to result in a local concentration at the joint or site of action
within the
listed therapeutic range. For targeted sustained release delivery systems, a
sufficient
dosage or load of the agent is included in the composition to result in a
local
concentration at the joint or site of action within the listed therapeutic
range over a
predetermined sustained release period.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-102-
TABLE 15
Therapeutic and Preferred Concentrations of Pain and/or inflammation
Inhibitory
Agents
Local Delivery Local Delivery
Therapeutic Preferred
Class of Agent Concentrations Concentrations
(Nanomolar) (Nanomolar)
SerotoninlA A og nists:
Buspirone 1 - 1,000 10 - 200
Sumatriptan 1 - 1,000 10 - 200
Serotonin1B A og nists:
Dihydroergotamine 0.1-1,000 10 - 100
Sumatriptan 1 - 1,000 10 - 200
Naratriptan 1 - 1,000 10 - 200
Rizatriptan 1 - 1,000 10 - 200
Zolmitriptan 1 - 1,000 10 - 200
L-694,247 1 - 1,000 10 - 200
Serotonin,D A og nists:
Dihydroergotamine 0.1-1,000- 10 - 100
Sumatriptan 1 - 1,000 10 - 200
Naratriptan 1 - 1,000 10 - 200
Rizatriptan 1 - 1,000 10 - 200
Zolmitriptan 1 - 1,000 10 - 200
L-694,247 1 - 1,000 10 - 200
Serotonin,F A og nists:
Ergonovine 10 - 2,000 100 -1,000
Serotonin,F A og nists:
Sumatriptan '1 - 1,000 10 - 200
3. Histamine Receptor Antagonists
Histamine' receptors generally are divided into histamine, (H1) and
histamine2 (H2) subtypes. The" classic inflammatory response to the peripheral
administration of histamine is mediated via the. H, receptor. Douglas, 1985.
Therefore, the solution of the present invention preferably includes a
histamine H1
receptor antagonist. Promethazine (PhenerganTM) is a commonly used anti-emetic
drug that potently blocks H1 receptors, and .is suitable for use in the
present
invention. Interestingly, this drug also has been shown to possess local
anesthetic
effects but the concentrations necessary for this effect are several orders
higher than
that necessary to block H1 receptors, thus, the effects are believed to occur
by
different mechanisms. The histamine receptor antagonist concentration in the
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-103-
solution is sufficient to inhibit H1 receptors involved in nociceptor
activation, but not
to achieve a "local anesthetic" effect, thereby eliminating the concern
regarding
systemic side effects.
Other suitable H1 receptor antagonists include terfenadine, diphenhydramine,
amitriptyline, mepyramine and tripolidine. Because amitriptyline is also
effective as
a 'serotonin2 receptor antagonist, it has a dual function as used in the
present
invention. Suitable therapeutic and preferred concentrations for each of these
H1
receptor antagonists for local delivery are set forth in Table 16. Similarly,
systemic
compositions in accordance with the present invention will suitably include a
dosage
or load of the agent sufficient to result in a local concentration at the
joint or site of
action within the listed therapeutic range. For targeted sustained release
delivery
systems, a sufficient dosage or load of the agent is included in the
composition to
result in a local concentration at the joint or site of action within the
listed therapeutic
range over a predetermined sustained release period.
TABLE 16
Therapeutic and Preferred Concentrations of Pain and/or inflammation
Inhibitory
Agents
Local Delivery Local Delivery
Therapeutic Preferred
Class of Agent Concentrations Concentrations
(Nanomolar) (Nanomolar)
Histamine, Receptor Antagonists:
Promethazine 0.1-1,000 50 - 200
Diphenhydramine 0.1-1,000 50 - 200
Amitriptyline 0.1-1,000 50 - 500
Terfenadine 0.1-1,000 50 - 500
mepyramine (pyrilamine) 0.1-1,000 5 - 200
Tripolidine 0.01 - 100 5-20
4. Bradykinin Receptor Antagonists
Bradykinin receptors generally are divided into bradykinin1 (B1) and
bradykinin2 (B2) subtypes. Studies have shown that acute peripheral pain and
inflammation produced by bradykinin are mediated. by the B2 subtype whereas
bradykinin-induced pain in the setting of chronic inflammation is mediated via
the B1
subtype. Perkins, M.N., et al., "Antinociceptive Activity of the Bradykinin BI
and
B2 Receptor Antagonists, des-Arg9, [Leu8]-BK and HOE 140, in Two Models of
Persistent Hyperalgesia in the Rat", Pain 53: 191-97 (1993); Dray, A., et al.,
CA 02474645 2011-01-13
-104-
"Bradykinin and Inflammatory Pain", Trends Neurosci. 16: 99-104 (1993).
At present, bradykinin receptor antagonists are not used clinically. Some of
these drugs are peptides, and thus they cannot be taken orally, because they
would be
digested. Antagonists to B2 receptors block bradykinin-induced acute pain and
inflammation. Dray et al., 1993. B1 receptor antagonists inhibit pain in
chronic
inflammatory conditions. Perkins et al., 1993; Dray et al., 1993. Therefore,
depending on the application, the solution of the present invention preferably
includes either or both bradykinin B1 and B2 receptor antagonists. For
example,
arthroscopy is performed for both acute and chronic conditions, and thus an
irrigation
solution for arthroscopy could include both B 1 and B2 receptor antagonists.
Suitable bradykinin receptor antagonists for use in the present invention
include the following bradykininl receptor antagonists: the [des-Argl0]
derivative of
D-Arg-(Hyp3-Thi5-D-Tic7-Oic8)-BK ("the [des-ArglO] derivative of HOE 140",
available from " Hoechst Pharmaceuticals); and [Leu8] des-Arg9-BK. Suitable
bradykinin2 receptor antagonists include: [D-Phe7]-BK;
D-Arg-(Hyp3-Tbi5,8-D-Phe7)-BK ("NPC 349"); D-Arg-(Hyp3--D-Phe7)-BK ("NPC
567"); and D-Arg-(Hyp3-Thi5-D-Tic7-Oic8)-BK ("HOE 140"). These compounds
are more fully described in Perkins et al. 1993 and Dray
et al. 1993. Suitable therapeutic and preferred concentrations for local
delivery are provided in Table 17. Similarly, systemic compositions in
accordance
with the present invention will suitably include a dosage or load of the agent
sufficient to result in a local concentration at the joint or site of action
within the
listed therapeutic range. For targeted sustained release delivery systems, a
sufficient
dosage or load of the agent is included in the composition to result in a
local
concentration at the joint or site of action within the listed therapeutic
range over a
predetermined sustained release period.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-105-
TABLE 17
Therapeutic and Preferred Concentrations of Pain and/or inflammation
Inhibitory
Agents
Local Delivery Local Delivery
Therapeutic Preferred
Class of Agent Concentrations Concentrations
(Nanomolar) (Nanoniolarj,
Bradykinin1 Receptor Antagonists:
[Leu8] des-Arg9-BK 1 - 1,000 50 - 500
[des-Argl0] derivative of HOE 140 1 - 1,000 50 - 500
[leu9] [des-Arg10] kalliden 0.1 - 500 10 - 200
Bradykinin2 Receptor Antagonists:
[D-Phe7]-BK 100 - 10,000 200 - 5,000
NPC 349 1 - 1,000 50 - 500
NPC 567 1 - 1,000 50 - 500
HOE 140 1 - 1,000 50 - 500
5. Kallikrein Inhibitors
The peptide bradykinin is an important mediator of pain and inflammation, as
noted previously. Bradykinin is produced as a cleavage product by the action
of
kallikrein on high molecular weight kininogens in plasma. Therefore kallikrein
inhibitors are believed to be therapeutic in inhibiting bradykinin production
and
resultant pain and inflammation. A suitable kallikrein inhibitor for use in
the present
invention is aprotinin. Suitable concentrations for use in the solutions of
the present
invention when delivered locally are set forth below in Table 18. Similarly,
systemic
compositions in accordance with the present invention will suitably include a
dosage
or load of the agent sufficient to result in a -local concentration at the
joint or site of
action within the listed therapeutic range. For targeted sustained release
delivery
systems, a sufficient dosage or load of the agent is included in the
composition to
result in a local concentration at the joint or site of action within the
listed therapeutic
range over a predetermined sustained release period.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-106-
TABLE 18
Therapeutic and Preferred Concentrations of Pain and/or inflammation
Inhibitory
Agents
Local Delivery Local Delivery
Therapeutic Preferred
Class of Agent Concentrations Concentrations
(Nanomolar) (Nanomolar)
Kallikrein Inhibitor:
Aprotinin 0.1-1,000 .50 - 500
6. Tachykinin Receptor Antagonists
Tachykinins (TKs) are a family of structurally related peptides that include
substance P, neurokinin A (NKA) and neurokinin B (NKB). Neurons are the major
source of TKs in the periphery. An important general effect of TKs is neuronal
stimulation, but other effects include endothelium-dependent vasodilation,
plasma
protein extravasation, mast cell recruitment and degranulation and stimulation
of
inflammatory cells. Maggi, C.A., Gen. Pharmacol., 22: 1-24 (1991). Due to the
above combination of physiological actions mediated by activation of TK
receptors,
targeting of TK receptors is a reasonable approach for the promotion of
analgesia and
the treatment of neurogenic inflammation.
6a. Neurokinin1 Receptor Subtype Antagonists
Substance P activates the neurokinin receptor subtype referred to as NK1.
Substance P is an undecapeptide that is present in sensory nerve terminals.
Substance P is known to have multiple actions that produce inflammation and
pain in
the periphery after C-fiber activation, including vasodilation, plasma
extravasation
and degranulation of mast cells. Levine, J.D., et al., "Peptides and the
Primary
Afferent Nociceptor", J. Neurosci. 13: 2273 (1993). A suitable Substance P
antagonist is ([D-Pro9[spiro-gamma-lactam]Leu10,Trp11]physalaemin-(1-11)) ("GR
82334"). Other suitable antagonists for use in the present invention which act
on the
NK1 receptor are: 1-imino-2-(2-methoxy-phenyl)-ethyl)-7,7-diphenyl-4-
perhydroisoindolone(3aR,7aR) ("RP 67580"and 2S,3S-cis-3-(2-
methoxybenzylamino)-2-benzhydrylquinuclidine ("CP 96,345"). Suitable
concentrations for these agents when delivered locally are set forth in Table
19.
Similarly, systemic compositions in accordance with the present invention will
suitably include a dosage or load of the agent sufficient to result in a local
concentration at the joint or site of action within the listed therapeutic
range. For
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-107-
targeted sustained release delivery systems, a sufficient dosage or load of
the agent is
included in the composition to result in a local concentration at the joint or
site of
action within the listed therapeutic range over a predetermined sustained
release
period.
TABLE 19
Therapeutic and Preferred Concentrations of Pain and/or inflammation
Inhibitory
Agents
Local Delivery Local Delivery
Therapeutic Preferred
Class of Agent Concentrations. Concentrations
(Nanomolar) (Nanomolar)
Neurokininl Receptor Subtype Antagonists
GR 82334 1 - 1,000 10 - 500
CP 96,345 1-10,000 - 100-1,000
RP 67580 0.1-1,000 100-1,000
6b. Neurokinin2 Receptor Subtype Antagonists
Neurokinin A is a peptide which is colocalized in sensory neurons with
substance P and which also promotes inflammation and pain. Neurokinin A
activates
the specific neurokinin receptor referred to as NK2. Edmonds-Alt, S., et al.,
"A
Potent and Selective Non-Peptide Antagonist of the Neurokinin A (NK2)
Receptor",.
Life Sci. 50:PL101 (1992). Examples of suitable NK2 antagonists include: ((S)-
N-
methyl-N-[4-(4-acetylamino-4-phenylpiperidino)-2-(3,4-dichlorophenyl)butyl]-
benzamide ("( )-SR .48968"); Met-Asp-Trp-Phe-Dap-Leu ("MEN 10,627"); and
cyc(Gln-Trp-Phe-Gly-Leu-Met) ("L 659,877"). Suitable concentrations of these
agents for local delivery are provided in Table 20. Similarly, systemic
compositions
in accordance with the present invention will suitably include a dosage or
load of the
agent sufficient to result in a local concentration at the joint or site of
action within
the listed therapeutic range. For targeted sustained release delivery systems,
a
sufficient dosage or load of the agent is included in the composition to
result in a
local concentration at the joint or site of action within the listed
therapeutic range
over a predetermined sustained release period.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-108-
TABLE 20
Therapeutic and Preferred Concentrations of Pain and/or inflammation
Inhibitory
Agents
Local Delivery Local Delivery
Therapeutic Preferred
Class of Age Concentrations Concentrations
(Nanomolar) (Nanomolar)
Neurokinin2 Receptor Subtype
Antagonists:
MEN 10,627 1-1,000 10-1,000
L 659,877 10-10,000 100-10,000
( )-SR 48968 10-10,000 100-10,000
7. CGRP Receptor Antagonists
Calcitonin gene-related peptide (CGRP) is a peptide which is also colocalized
in sensory neurons with substance P, and which acts as a vasodilator and
potentiates
the actions of substance P. Brain, S., et al., Br. J Pharmacol. 99: 202
(1985). An
example of a suitable CGRP receptor antagonist is I-CGRP-(8-37), a truncated
version of CGRP. This polypeptide inhibits the activation of CGRP receptors.
Suitable concentrations for this agent when delivered locally are provided in
Table
21. Similarly, systemic compositions in accordance with the present invention
will
suitably include a dosage or load of the agent sufficient to result in a local
concentration at the joint or site of action within the listed therapeutic
range. For
targeted sustained release delivery systems, a sufficient dosage or load of
the agent is
included in the composition to result in a local concentration at the joint or
site of
action within 'the listed therapeutic range over a predetermined sustained
release
period.
TABLE 21
20' Therapeutic and Preferred Concentrations of Pain and/or inflammation
Inhibitory
Agents
Local Delivery Local Delivery
Therapeutic Preferred
Class of Agent Concentrations Concentrations
(Nanomolar) (Nanomolar)
CGRP Receptor Antagonist:
I-CGRP-(8-37) 1-1,000 10-500
tJ
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT '
-109-
8. Interleukin Receptor Antagonist
Interleukins are a family of peptides, classified as cytokines, produced by
leukocytes and other cells in response to inflammatory mediators. Interleukins
(IL)
may be potent hyperalgesic agents peripherally. Ferriera, S.H., et al., Nature
334:
698 (1988). An example of a suitable IL-1(3 receptor antagonist is Lys-D-Pro-
Thr,
which is a truncated version of IL-1(3. This tripeptide inhibits the
activation of IL-1(3
receptors. Suitable concentrations for this agent for local delivery are
provided in
Table 22. Similarly, systemic compositions in accordance with the present
invention
will suitably include a dosage or load of the agent sufficient to result in a
local
concentration at the joint or site of action within the listed therapeutic
range. For
targeted sustained release delivery systems, a sufficient dosage or load of
the agent is
included in the composition to result in a local concentration at the joint or
site of
action within the listed therapeutic range over a predetermined sustained
release
period.
TABLE 22
Therapeutic and Preferred Concentrations of Pain and/or Inflammation
Inhibitory Agents
Local Delivery Local Delivery
Therapeutic Preferred
Class of Agent Concentrations Concentrations
(Nanomolar) (Nanomolar)
Interleukin Receptor Antagonist:
Lys-D-Pro-Thr 1-1,000 10-500
9. Inhibitors of Enzymes Active in the Synthetic Pathway for Arachidonic Acid
Metabolites
9a. Phospholipase Inhibitors
The production of arachidonic acid by phospholipase A2 (PLA2) enzymes
(cPLA2, iPLA2, sPLA2) and phospholipase C (PLC) results in a cascade of
reactions
that produces numerous mediators of inflammation, know as eicosanoids. There
are
a number of stages throughout this pathway that can be inhibited, thereby
decreasing
the production of these inflammatory mediators. Examples of inhibition at
these
various stages are given below.
Inhibition of the enzyme PLA2 isoform inhibits the release of arachidonic acid
from cell membranes, and therefore inhibits the production of prostaglandins
and
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-110-
leukotrienes resulting in decreased inflammation and pain. Glaser, K.B.,
"Regulation
of Phospholipase A2 Enzymes: Selective Inhibitors and Their Pharmacological
Potential", Adv. Pharinacol. 32:31 (1995). An example of a suitable PLA2
isoform
inhibitor is manoalide. Inhibition of the phospholipase C, (PLC) isoform also
will
result in decreased production of prostanoids and leukotrienes, and,
therefore, will
result in decreased pain and inflammation. An example of a PLC, isoform
inhibitor
is 1-[6-((17(3-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl]-1H-pyrrole-
2,5-
dione. Suitable concentrations for this agent when delivered locally are
included in
Table 23. Similarly, systemic compositions in accordance with the present
invention
will suitably include a dosage or load of the agent sufficient to result in a
local
concentration at the joint or site of action within the listed therapeutic
range. For
targeted sustained release delivery systems, a sufficient dosage or load of
the agent is
included in the composition to result in a local concentration at the joint or
site of
action within the listed therapeutic range over a predetermined sustained
release
period.
TABLE 23
Therapeutic and Preferred Concentrations of Pain and/or inflammation
Inhibitory
Agents
Local Delivery Local Delivery
Therapeutic Preferred
Class of Agent Concentrations Concentrations
(Nanomolar) (Nanomolar)
Phospholipase Inhibitor:
Manoalide 100-100,000 500-10,000
aristolochic acid 40-400,000 400-40,000
ACA 10-100,000 100-10,000
HELSS 6-6,000 60-6,000
9b. Cyclooxygenase Inhibitors
Nonsteroidal anti-inflammatory drugs (NSAIDs) are widely used as
anti-inflammatory, anti-pyretic, anti-thrombotic and, analgesic agents. Lewis,
R.A.,
Prostaglandins and Leukotrienes, In: Textbook of Rheumatology, 3d ed. (Kelley
W.N., et al., eds.), p. 258 (1989). The molecular targets for these drugs are
Type I
and Type II cyclooxygenases (COX-1 and COX-2). These enzymes are also known
as Prostaglandin H Synthase (PGHS)-1 (constitutive) and -2 (inducible), and
catalyze
the conversion of arachidonic acid to Prostaglandin H that is an intermediate
in the
biosynthesis of prostaglandins and thromboxanes. The COX-2 enzyme has been
identified in endothelial cells, macrophages, and fibroblasts. This enzyme is
induced
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-111-
by IL-1 and TNF-a, and its expression is upregulated at sites of inflammation.
Constitutive activity of COX-1 and induced activity of COX-2 both lead to
synthesis
of prostaglandins that contribute to pain and inflammation.
Many NSAIDs currently on the market (diclofenac,"naproxen, indomethacin,
ibuprofen, etc.) are generally nonselective inhibitors of both isoforms of
COX, but
may show greater selectively for COX-1 over COX-2, although this ratio varies
for
the different compounds. Use of COX-1 and 2 inhibitors to block formation of
prostaglandins represents a better therapeutic strategy than attempting to
block
interactions of the natural ligands with the seven described subtypes of
prostanoid
receptors. Reported antagonists of the eicosanoid receptors (EP-1, EP-2, EP-3)
are
quite rare and only specific, high affinity antagonists of the thromboxane A2
receptor
have been reported. Wallace, J. et al. Trends in Pharin. Sci., 15:405-406
(1994).
Representative therapeutic and preferred concentrations of cyclooxygenase
inhibitors for local delivery use in the solution are provided in Table 24.
Similarly,
systemic compositions in accordance with the present invention will suitably
include
a dosage or load of the agent sufficient to result in a local concentration at
the joint or
site of action within the listed therapeutic range. For targeted sustained
release
delivery systems, a sufficient dosage or load of the agent is included in the
composition to result in a local concentration at the joint or site of action
within the
listed therapeutic range over a predetermined sustained release period.
TABLE 24
Therapeutic and Preferred Concentrations of Pain and/or inflammation
Inhibitory
Agents
Local Delivery Local Delivery
Therapeutic Preferred
Class of Agent Concentrations Concentrations
(Nanomolar) (Nanomolar)
Cyclooxygenase Inhibitors:
ketorolac 100 - 10,000 500 - 5,000
indomethacin 1,000 - 500,000 10,000 - 200,000
9c. Lipooxygenase Inhibitors
Inhibition of the enzyme lipooxygenase inhibits the production of
leukotrienes, such as leukotriene B4, which is known to be an important
mediator of
inflammation and pain. Lewis, R.A., Prostaglandins and Leukotrienes, In:
Textbook
of Rheumatology, 3d ed. (Kelley W.N., et al., eds.), p. 258 (1989). An example
of a
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-112-
5-lipooxygenase antagonist is 2,3,5-trimethyl-6-(12-hydroxy-5,10-dodecadiynyl)-
1,4-benzoquinone ("AA 861 "), suitable local delivery concentrations for which
are
listed in Table 25. Similarly, systemic compositions in accordance with the
present
invention will suitably include a dosage or load of the agent sufficient to
result in a
local concentration at the joint or site of action within the listed
therapeutic range.
For targeted sustained release delivery systems, a sufficient dosage or load
of the
agent is included in the composition to result in a local concentration at the
joint or
site of action within the listed therapeutic range over a predetermined
sustained
release period.
TABLE 25
Therapeutic and Preferred Concentrations of Pain and/or inflammation
Inhibitory
Agents
Local Delivery Local Delivery
Therapeutic Preferred
Class of Agent Concentrations Concentrations
(Nanomolar) (Nanomolar)
Lipooxygenase Inhibitor:
AA 861 100-10,000 500-5,000
Caffeic acid 500-50,000 2,000-20,000
10. Prostanoid Receptor Antagonists
Specific prostanoids produced as metabolites of arachidonic acid mediate
their inflammatory effects through activation of prostanoid receptors.
Examples of
classes of specific prostanoid antagonists are the eicosanoid EP-1 and EP-4
receptor
subtype antagonists and the thromboxane receptor subtype antagonists. A
suitable
prostaglandin E2 receptor antagonist is 8-chlorodibenz[b,f][1,4]oxazepine-
10(11H)-
carboxylic acid, 2-acetylhydrazide ("SC 19220"). A suitable thromboxane
receptor
subtype antagonist is [15-[la, 2(3(5Z), 3(3, 4a]-7-[3-[2-(phenylamino)-
carbonyl]
hydrazino] methyl]-7-oxobicyclo-[2,2,1]-hept-2-yl]-5-heptanoic acid ("SQ
29548").
Suitable concentrations for these agents when delivered locally are set forth
in
Table 26. Similarly, systemic compositions in accordance with the present
invention
will suitably include a dosage or load of the agent sufficient to result in a
local
concentration at the joint or site of action within the listed therapeutic
range. For
targeted sustained release delivery systems, a sufficient dosage or load of
the agent is
included in the composition to result in a local concentration at the joint or
site of
action within the listed therapeutic range over a predetermined sustained
release
period.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-113-
TABLE 26
Therapeutic and Preferred Concentrations of Pain and/or inflammation
Inhibitory
Agents
Local Delivery Local Delivery
Therapeutic Preferred
Class of Agent Concentrations Concentrations
(Nanomolar) (Nanomolar)
Eicosanoid EP-1 Antagonist:
SC 19220 100-10,000 500-5,000
11. Leukotriene Receptor Antagonists
The leukotrienes (LTB4, LTC4, and LTD4) are products of the
5-lipooxygenase pathway of arachidonic acid metabolism that are generated
enzymatically and have important biological properties. Leukotrienes are
implicated
in a number of pathological conditions including inflammation. Specific
antagonists
are currently being sought by many pharmaceutical companies for potential
therapeutic intervention in these pathologies. Halushka, P.V., et al., Annu.
Rev.
Phaf macol. Toxicol. 29:213-239 (1989); Ford-Hutchinson, A., Crit. Rev.
Immunol.
10:1-12 (1990). The LTB4 receptor is found in certain immune cells including
eosinophils and neutrophils. LTB4 binding to these receptors results in
chemotaxis
and lysosomal enzyme release thereby contributing to the process of
inflammation.
The signal transduction process associated with activation of the LTB4
receptor
involves G-protein-mediated stimulation of phosphotidylinositol (PI)
metabolism and
elevation of intracellular calcium (see FIGURE 2).
An example of a suitable leukotriene B4 receptor antagonist is SC (+)-(S)-7-
(3-(2-(cyclopropylmethyl)-3-methoxy-4-[(methylamino)-
carbonyl]phenoxy(propoxy)-3,4-dihydro- 8-propyl-2H-1-b enzopyran-2-propanoic
acid ("SC 53228"). Other suitable leukotriene B4 receptor antagonists include
[3-[-
2(7-chloro-2-quinolinyl)ethenyl]phenyl] [[3-(dimethylamino-3-oxopropyl)thio]
methyl]thiopropanoic acid ("MK 0571") and the drugs LY 66,071 and ICI 20,3219.
MK 0571 also acts as a LTD4 receptor subtype antagonist. Concentrations for
this
agent that are suitable for the practice of local delivery methods of the
present
invention are provided in Table 27. Similarly, systemic compositions in
accordance
with the present invention will suitably include a dosage or load of the agent
sufficient to result in a local concentration at the joint or site of action
within the
listed therapeutic range. For targeted sustained release delivery systems, a
sufficient
dosage or load of the agent is included in the composition to result in a
local
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-114-
concentration at the joint or site of action within the listed therapeutic
range over a
predetermined sustained release period.
TABLE 27
Therapeutic and Preferred Concentrations of Pain and/or inflammation
Inhibitory
Agents
Local Delivery Local Delivery
Therapeutic Preferred
Class of Agent Concentrations Concentrations
(Nanomolar) (Nanomolar)
Leukotriene B4 Antagonist:
SC 53228 100-10,000 500-5,000
12. Opioid Receptor A og nists
Activation of opioid receptors results in anti-nociceptive effects and,
therefore, agonists to these receptors are desirable. Opioid receptors include
the -,
8- and x-opioid receptor subtypes. The -receptors are located on sensory
neuron
terminals in the periphery and activation of these receptors inhibits sensory
neuron
activity. Basbaum, A.I., et al., "Opiate analgesia: How Central is a
Peripheral
Target?", N. Engl. J. Med. 325:1168 (1991). 8- and x-receptors are located on
sympathetic efferent terminals and inhibit the release of prostaglandins,
thereby
inhibiting pain and inflammation. Taiwo, Y.O., et al., "Kappa- and Delta-
Opioids
Block Sympathetically Dependent Hyperalgesia", J. Neurosei. 11:928 (1991).
The,
opioid receptor subtypes are members of the G-protein coupled receptor
superfamily.
Therefore, all opioid receptor agonists interact and initiate signaling
through their
cognate G-protein coupled receptor. Examples of suitable g-opioid receptor
agonists
are fentanyl and Try-D-Ala-Gly-[N-MePhe]-NH(CH2)-OH ("DAMGO"). An
example of a suitable S-opioid receptor agonist is [D-Pen2,D-Pen5]enkephalin
("DPDPE"). An example of a suitable r,-opioid receptor agonist is (trans)-3,4-
dichloro-N-methyl-N-[2-(1-pyrrolidnyl)cyclohexyl]-benzene acetamide
("U50,488").
Suitable concentrations for the local delivery of each of these agents are set
forth in
Table 28. Similarly, systemic compositions in accordance with the present
invention
will suitably include a dosage or load of the agent sufficient to result in a
local
concentration at the joint or site of action within the listed therapeutic
range. For
targeted sustained release delivery systems, a sufficient dosage or load of
the agent is
included in the composition to result in a local concentration at the joint or
site of
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-115-
action within the listed therapeutic range over a predetermined sustained
release
period.
TABLE 28
Therapeutic and Preferred Concentrations of Pain and/or inflammation
Inhibitory
Agents
Local Delivery Local Delivery
Therapeutic Preferred
Class of Agent Concentrations Concentrations
(Nanomolar) (Nanomolar)
-Opioid Agonist:
DAMGO 0.1-100 0.5-20
sufentanyl Ø01-50 1-20
fentanyl 0.1-500 10-200
PL 017 0.05-50 0.25-10
S-Opioid Agonist:
DPDPE 0.1-500 1.0-100
x-Opioid Agonist:
U50,488 0.1-500 1.0-100
13. Purinoceptor Antagonists
Extracellular ATP acts as a signaling molecule through interactions with P2
purinoceptors. One major class of purinoceptors are the P2x purinoceptors
which are
ligand-gated ion channels possessing intrinsic ion channels permeable to Nat,
K+,
and Ca2+. P2x receptors described in sensory neurons are important for primary
afferent neurotransmission and nociception. ATP is known to depolarize sensory
neurons and plays a role in nociceptor activation since ATP released from
damaged
cells stimulates P2X receptors leading to depolarization of nociceptive nerve-
fiber
terminals. The P2X3 receptor has a highly restricted distribution (Chen, C.C.,
et al.,
Nature, Vol. 377, pp. 428-431 (1995)) since it is selectively expressed in
sensory C-
fiber nerves that run into the spinal cord and many of these C-fibers are
known to
carry the receptors for painful stimuli. Thus, the highly restricted
localization of
expression for the P2X3 receptor subunits make these subtypes excellent
targets for
analgesic action (see FIGURES 3 and 7).
Calcium-mobilizing purine receptors, which belong to the G-protein receptor
superfamily, have been described on the surface of mammalian articular
chondrocytes. ATP was found to stimulate a dose-dependent, transient rise in
the
concentration of calcium ions in differentiated, primary chondrocytes.
Heterologous
desensitization experiments demonstrated that chondrocytes showed no
subsequent
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-116-
response to UTP after initial stimulation- with ATP. These results are
consistent with
the presence of a P2Y receptors of the cell surface of chondrocytes. Purine-
induced
calcium mobilization in passaged chondrocytes showed the same pharmacological
profile with respect to agonist sensitivity. ATP and UTP did not alter
cartilage
matrix synthesis as measured by rate of incorporation of [35S]sulfate into
glycosaminoglycan by cartilage explants or primary chondrocytes. Matrix
degradation, measured by release of glycosaminoglycan from cartilage explants,
was
also unaltered by either agonist. The presence of a functional P2Y purine
receptor on
the surface of primary articular chondrocytes enable concentrations of
extracellular
purines, such as ATP, to activate chondrocyte metabolism.
Other studies have defined the expression of both P1 and P2 purine receptor
genes by human articular chondrocytes and profiled ligand-mediated
prostaglandin
E2 release. The P2Y2 receptor agonists ATP and UTP stimulated a small release
of
PGE2 that was synergistically enhanced after pretreatment with human IL-1a.
PGE2
release in response to coaddition of ATP and UTP after IL-1 pretreatment was
mimicked by phorbol myristate acetate. The function of the P2Y2 receptor is to
increase IL-1-mediated PGE2 release, thereby promoting pain and inflammation
within the joint. Thus, the use of P2Y antagonists in the present invention
should
prevent activation of inflammatory mediator production by both synoviocytes
and
chondrocytes.
Suitable antagonists of P2x/ATP purinoceptors for use in the present
invention include, by way of example, suramin and pyridoxylphosphate-6-
azophenyl-2,4-disulfonic acid ("PPADS"). Suitable concentrations for the local
delivery of these agents are provided in Table 29. Similarly, systemic
compositions
in accordance with the present invention will suitably include a dosage or
load of the
agent sufficient to result in a local concentration at the joint or site of
action within
the listed therapeutic range. For targeted sustained release delivery systems,
a
sufficient dosage or load of the agent is included in the composition to
result in a
local concentration at the joint or site of action within the listed
therapeutic range
over a predetermined sustained release period.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-117-
TABLE 29
Therapeutic and Preferred Concentrations of Pain and/or inflammation
Inhibitory
A ents
Local Delivery Local Delivery
Therapeutic Preferred
Class of Agent Concentrations Concentrations
(Nanomolar)Nanomolar)
P2X and/or P2Y
Antagonists:
Suramin 100-100,000 1.0,000-100,000
PPADS 100-100,000 10,000-100,000
14. Ca2+ Channel Antagonists
Calcium channel antagonists are a distinct group of drugs that interfere with
the transmembrane flux of calcium ions required for activation of cellular
responses
mediating neuroinflammation. Calcium entry into synoviocytes and chondrocytes
is
a key event mediating activation of responses in these cells. Furthermore, the
role of
bradykinin, histamine, serotonin (SHT2) and neurokinin receptors (NK1 and NK2)
in
mediating the neuroinflammation signal transduction pathway includes increases
in
intracellular calcium, thus leading to activation of calcium channels on the
plasma
membrane. In many tissues, calcium channel antagonists, such as nifedipine,
can
reduce the release of arachidonic acid, prostaglandins, and leukotrienes that
are
evoked by various stimuli. Moncada, S. et al., Goodman's and Gilman's
Pharmacological Basis of Therapeutics, (7th ed.), MacMillan Publ. Inc., 660-5
(1995).
Finally, calcium channel antagonists and either tachykinin, histamine or
bradykinin antagonists exhibit synergistic effects in inhibiting
neuroinflammation.
The role of neurokinin receptors in mediating neuroinflammation has been
established. The neurokinin, (NK1) and neurokinin2 (NK2) receptor (members of
the
G-protein coupled superfamily) signal tran sduction pathway includes increases
in
intracellular calcium, thus leading to activation of calcium channels on the
plasma
membrane. Similarly, activation of bradykinin2 (BK2) receptors is coupled to
increases in intracellular calcium in synoviocytes and chondrocytes. Thus,
calcium
channel antagonists interfere with a common mechanism involving elevation of
intracellular calcium, part of which enters through L-type channels. This is
the basis
for synergistic interaction between calcium channel antagonists and
antagonists to
neurokinin, histamine, P2Y and bradykinin2 receptors.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-118-
Suitable calcium channel antagonists for the practice of the present invention
include nisoldipine, nifedipine, nimodipine, lacidipine, isradipine and
amlodipine.
Suitable concentrations for the local delivery of these agents are set forth
in Table 30.
Similarly, systemic compositions in accordance with the present invention will
suitably include a dosage or load of the agent sufficient to result in a local
concentration at the joint or site of action within the listed therapeutic
range. For
targeted sustained release delivery systems, a sufficient dosage or load of
the agent is
included in the composition to result in a local concentration at the joint or
site of
action within the listed therapeutic range over a predetermined sustained
release
period.
TABLE 30
Therapeutic and Preferred Concentrations of Spasm Inhibitory Agents
Local Delivery Local Delivery
Therapeutic Preferred
Class of Agent Concentrations Concentrations
(Nanomolar) (Nanomolar)
Calcium Channel Antagonists:
Nisoldipine 1-10,000 100-1,000
Nifedipine 1-10,000 100-5,000
Nimodipine 1-10,000 100-5,000
Lacidipine 1-10,000 100-5,000
Isradipine 1-10,000 100-5,000
Amlodipine 1-10,000 100-5,000
VI. EXAMPLES
The following are several formulations in accordance with the aspects of the
present invention suitable for irrigation in certain operative procedures
(Examples
1-3), and for systemic delivery, such as by intramuscular or subcutaneous
injection
(Examples 4-11) and are followed by a summary of three clinical studies
utilizing the
agents of the present invention.
EXAMPLE 1
Irrigation Solution for Arthroscopy
The following composition is suitable .for use in anatomic joint irrigation
during arthroscopic procedures. Each drug is solubilized in a carrier, fluid
containing
physiologic electrolytes, such as normal saline or lactated Ringer's solution,
as are
the remaining solutions described in subsequent examples.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-119-
Concentration
Class, of Agent Drug (Nanomolar)
MAP Kinase Inhibitor SB203580 200
Matrix Metalloproteinase U-24522 200
Inhibitor
TGF-(3 Agonist TGF-(32 200
EXAMPLE 2
Alternative Irrigation Solution for Arthroscopy
The following composition is an alternate formulation suitable for use in
anatomic joint irrigation during arthroscopic procedures.
Drug Concentration
Class of Agent (Nanomolar)
MAP Kinase Inhibitor SB203580 200
Nitric Oxide Synthase
Inhibitor L-NIL 1,000
Interleukin Receptor
Agonist IL-10 100
EXAMPLE 3
Alternate Irrigation Solution
The following drugs and concentration ranges in solution in a physiologic
carrier fluid are suitable for use in the present invention.
Concentration
Class of Agent Drug (Nanomolar)
MAP Kinase Inhibitor SB242235 200
Nitric Oxide Synthase L-NIL 10,000
Inhibitor
TGF-(3 Agonist TGF-(32 100
EXAMPLE 4
Chondroprotective Solution for Infection
The following composition is suitable for injection into an anatomic joint.
Each drug is solubilized in a carrier fluid containing physiologic
electrolytes, such as
normal saline or lactated Ringer's solution. A dosage of 20 ml of the solution
is
suitable for administration to a patient.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-120-
Concentration
Class of Agent Drug
BMP Receptor Agonist BMP-7 100 ng/ml
Nitric Oxide Synthase 1,3 PBIT 4.4 g/ml
Inhibitor
NFiB Agonist pyrrolidine- 16.4 g/ml
dithiocarbamate
EXAMPLE 5
Chondroprotective Composition for Systemic delivery
The following chondroprotective composition is suitable for systemic
delivery, such as by intramuscular or subcutaneous administration. Each drug
is
included in the composition at a concentration that will result in the
following
concentration at the site of intended action, and is solubilized in a
physiologic carrier
fluid or delivery system.
Concentration
Class of Agent Drug at Site of Action
(Nanomolar)
MAP Kinase Inhibitor SB203580 200
Matrix Metalloproteiinase U-24522 200
Inhibitor
TGF-(3 Agonist TGF-(32 200
EXAMPLE 6
Alternate Chondroprotective Composition for Systemic Delivery
The following chondroprotective composition is suitable for systemic
delivery, such as by intramuscular or subcutaneous administration. Each drug
is
included in the composition at a concentration sufficient to result in the
following
concentration at the site of intended action, and is solubilized in a
physiologic carrier
fluid or delivery system.
Concentration
Class of Agent Drug at Site of
Action
(Nanomolar)
MAP Kinase Inhibitor. SB203580 200
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-121-
Nitric Oxide Synthase
Inhibitor L-NIL 1,000
Interleukin Receptor
Agonist IL-10 100
EXAMPLE 7
Alternate Chondroprotective Composition for Systemic Delivery
The following chondroprotective composition is suitable for systemic
delivery, such as by intramuscular or subcutaneous administration. Each drug
is
included in the composition at a concentration sufficient to result in the
following
concentration at the site of intended action, and is solubilized in 'a
physiologic carrier
fluid or delivery system.
Concentration
Class of Agent Drug at Site of
Action
(Nanomolar)
MAP Kinase Inhibitor SB242235 200
Nitric Oxide Synthase L-NIL 10,000
Inhibitor
TGF-(3 Agonist TGF-(32 100
EXAMPLE 8
Alternate Chondroprotective Composition for Systemic Delivery
The following chondroprotective composition is suitable for systemic
delivery, such as by intramuscular or subcutaneous administration. Each drug
is
included in the composition at a concentration sufficient to result in the
following
concentration at the site of intended action, and is solubilized in a
physiologic carrier
fluid or delivery system.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-122-
Concentration
Class of Agent Drug at Site of
Action.
(Nanomolar)
Soluble TNF-a Receptor Etanerocept 250
(sTNFRII:Fc) (EnbrelTM,
Immunex)
MAP Kinase Inhibitor SB203580 500
TGF-(3 Agonist TGF-(32 200
EXAMPLE 9
Alternate Chondroprotective Composition for Systemic Delivery
The following chondroprotective composition is suitable for systemic
delivery, such as by intramuscular or subcutaneous administration. Each drug
is
included in the composition at a concentration sufficient to result in the
following
concentration at the site of intended action, and is solubilized in a
physiologic carrier
fluid or delivery system.
Concentration
Class of Agent Drug at Site of
Action
(Nanomolar)
IL-1 Receptor Anakinra 250
Antagonist (KineretTM,
(IL-1 Ra) Amgen)
MAP Kinase Inhibitor SB203580 500
TGF-(3 Agonist TGF-(32 200
EXAMPLE 10
Alternate Chondroprotective Composition for Systemic Delivery
The following chondroprotective composition is suitable for systemic
delivery, such as by intramuscular or subcutaneous administration. Each drug
is
included in the composition at a concentration sufficient to result in the
following
concentration at the site of intended action, and is solubilized in a
physiologic carrier
fluid or delivery system.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-123-
Concentration
Class of Agent Drug at Site of
Action
(Nanomolar)
MAP Kinase Inhibitor SB203580 500
IGF-1 IGF-1 250
BMP Agonist BMP-2 200
EXAMPLE 11
Alternate Chondroprotective Composition for Systemic Delivery
The following chondroprotective composition is suitable for systemic
delivery, such as by intramuscular or subcutaneous administration. Each drug
is
administered at the noted dosage, and is solubilized in a physiologic carrier
fluid or
delivery system.
Dosage
Class of Agent Drug (mg/kg/day)
Soluble TNF-a Receptor Etanerocept 0.5-1.0
(sTNFRII:Fc) (EnbrelTM,
Immunex)
MAP Kinase Inhibitor SB203580 30-60
TGF-(3 Agonist TGF-02 0.1-10
EXAMPLE 12
Alternate Chondroprotective Composition for Systemic Delivery
The following chondroprotective composition is suitable for systemic
delivery, such as by intramuscular or subcutaneous administration. Each drug
is
administered at the noted dosage, and is solubilized in .a physiologic carrier
fluid or
delivery system.
Dosage
Class of Agent Drug (mg/k /g dav)
IL-1 Receptor Anakinra 2.0
Antagonist (KineretTM,
(IL-1Ra) Amgen)
MAP Kinase Inhibitor SB203580 30-60
TGF-(3 Agonist TGF-(32 0.1-10
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-124-
EXAMPLE 13
Targeted Chondroprotective Drug Delivery System
The following chondroprotective composition is suitable for systemic
delivery, such as by intravenous, intramuscular, subcutaneous or inhalation
administration. The drugs are encapsulated within a DL-lactide/glycolide
copolymer
(PLGA) nanosphere, to which is coupled an anti-human Type II collagen
monoclonal
antibody. This antibody targets epitopes on human Type II collagen in
articular
cartilage. Each drug is included in the composition at a concentration
sufficient to
result in the following average concentration at the site of intended action
upon
degradation of the nanosphere and release of the agents over a period of
sustained
release.
Class of Agent Concentration
at Site of
Action
(Nanomolar)
Growth Factor IGF-1 250nM
MAP Kinase inhibitor SB220025 1000 nM
MMP inhibitor BB2516 .200 nM
(marimastat)
EXAMPLE 14
Targeted Chondroprotective Drug Delivery System
The following chondroprotective composition is suitable for systemic
delivery, such as by intravenous, intramuscular, subcutaneous or inhalation
administration. The drugs are encapsulated within a biodegradable PLA/PLGA co-
polymeric nanosphere, to which is coupled an anti-human aggrecan monoclonal
antibody. This antibody targets neoeptitopes on human aggrecan in articular
cartilage. Each' drug is included in the composition at a concentration
sufficient to
result in the following concentration at the site of intended action upon
degradation
of the nanosphere and release of the agents over a desired period of sustained
release.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-125-
Class of Agent Concentration
at Site of
Action
(Nanomolar)
Growth Factor IGF-1 250nM
MAP Kinase inhibitor SB220025 1000 nM
MMP inhibitor BB2516 200 nM
(marimastat)
EXAMPLE 15
Targeted Chondroprotective Drug Delivery System
The following chondroprotective composition is suitable for systemic
delivery, such as by intravenous, intramuscular, subcutaneous or inhalation
administration. The drugs are encapsulated within a chitosan/gelatin
nanosphere, to
which is coupled an anti-human Type II collagen monoclonal antibody. This
antibody targets neoepitopes on human Type II collagen. Each drug is included
in
the composition at a concentration sufficient to result in the following
concentration
at the site of intended action upon degradation of the nanosphere and release
of the
agents over a desired period of sustained release.
Class of Agent Concentration
at Site of
Action
(Nanomolar)
BMP Receptor agonist BMP-7 200
MAP Kinase Inhibitor S13203580. 500
EXAMPLE 16
Targeted Chondroprotective Drug Delivery S sti tem
The following chondroprotective composition is suitable for systemic
delivery, such as by intravenous, intramuscular, subcutaneous or inhalation
administration. The drugs are encapsulated within an albumin nanosphere, to
which
is coupled an anti-human Type II collagen monoclonal antibody. This antibody
targets neoeptitopes on human Type II collagen. Each drug is included in the
composition at a concentration sufficient to result in the following
concentration at
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-126-
the site of intended action upon degradation of the nanosphere and release of
the
agents over a desired period of sustained release.
Class of Agent Concentration
at Site of
Action
(Nanomolar)
BMP Receptor agonist BMP-7 200
MMP inhibitor BB2516 200
EXAMPLE 17
Targeted Chondroprotective Drug Delivery System
The following chondroprotective composition is suitable for systemic
delivery, such as by intravenous, intramuscular, subcutaneous or inhalation
administration. The drugs are encapsulated within a poly(lactide-co-
glycolide)/poly(ethyleneglycol) copolymer nanosphere, to which is coupled an
anti-
human Type II collagen monoclonal antibody.' This antibody targets
neoeptitopes on
human Type II collagen in articular cartilage. Each drug is included in the
composition at a concentration sufficient to result in the following
concentration at
the site of intended action upon degradation of the nanosphere and release of
the
agents over a desired period of sustained release.
Class of Agent Concentration
at Site of
Action
(Nanomolar).
BMP Receptor agonist BMP-7 200
MAP Kinase Inhibitor SB203580 500
EXAMPLE 18
Targeted Chondroprotective Drug Delivery System
The following chondroprotective composition is suitable for systemic
delivery, such as by intravenous, intramuscular, subcutaneous or inhalation
administration. The BMP receptor agonist, BMP-7 is encapsulated within a
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-127-
poly(lactide-co-glycolide) (PLGA) nanosphere to which is coupled an anti-human
Type II collagen monoclonal antibody. The IGF Receptor agonist, IGF-1 is
separately encapsulated within a chondroiten-6-sulfate/gelatin nanosphere to
which is
also coupled an anti-human aggrecan monoclonal antibody. The antibody used for
targeting each type of nanosphere binds to neoeptitopes on human Type II
collagen
and neoepitopes on aggrecan in articular cartilage. Each drug is included in
the
composition at a concentration sufficient to result in. the following average
concentration at the site of intended action upon degradation of the
nanosphere and
release of the agents over a desired period of sustained release.
Class of Agent Concentration
at Site of
Action
(Nanomolar)
BMP Receptor agonist BMP-7 200
IGF-Receptor agonist IGF-1 500
EXAMPLE 19
Targeted Chondroprotective Drug Delivery System
The following chondroprotective composition is suitable for systemic
delivery, such as by intravenous, intramuscular, subcutaneous or inhalation
administration. The drugs are encapsulated within an albumin nanosphere, to
which
is coupled an anti-human F(ab')2 fragment that binds to Type II collagen
monoclonal
antibody. This F(ab')2 antibody targets neoeptitopes on human Type II
collagen.
Each drug is included in the composition at a concentration sufficient to
result in the
following concentration at the site of intended action upon degradation of the
nanosphere and release of the agents over a desired period of sustained
release.
Class of Agent Concentration
at Site of
Action
(Nanomolar)
BMP Receptor agonist BMP-7 200
MMP inhibitor BB2516 200
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-128-
EXAMPLE 20
Targeted Chondroprotective Drug DeliverSystem
The following chondroprotective composition is suitable for systemic
delivery, such as by intravenous, intramuscular, subcutaneous or inhalation
administration. The drugs are encapsulated within an albumin nanosphere, to
which
is coupled an anti-human single-chain minimum binding domain of an
immunoglobulin molecule (scFV) that binds to Type II collagen monoclonal
antibody. This scFV antibody targets neoeptitopes on human Type II collagen.
Each
drug is included in the composition at a concentration sufficient to result in
the
following concentration at the site of intended action upon degradation of the
nanosphere and release of the agents over a desired period of sustained
release.
Class of Agent Concentration
at Site of
Action
(Nanomolar)
BMP Receptor agonist BMP-7 200
MMP inhibitor BB2516 200
STUDY 1
Synergistic stimulation of a rapid PGE2 burst upon exposure to IL-1 and GPCR
agonists.
Fibroblast-like synoviocytes exhibit characteristics of inflammatory cells and
seem to be crucial regulators of joint inflammation and cartilage degradation.
A
synoviocyte cell culture model system was used to characterize the synergistic
interactions between IL-1 and non-cytokine inflammatory mediators which are
important in modulating the destruction of joint tissue, including damage that
occurs
as a consequence of tissue injury during arthroscopic surgery. Experiments
were
conducted to investigate G-protein coupled receptor (GPCR) agonists
(histamine,
bradykinin and isoproteronol) on the regulation - of cytokine and prostanoid
production in cultured human synovial fibroblasts and to characterize the
activities of
ketoprofen in this system. The kinetics of induction of prostaglandin E2
(PGE2),
interleukin-6 (IL-6) and interleukin-8 (IL-8) in response to stimulation with
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-129-
interleukin-1 (IL-1) are described. The ability of GPCR ligands to potentiate
cytokine production following IL-1 priming was investigated.
In Studies 1-3, the following experimental methods and materials were
employed unless otherwise indicated.
1. Cell Culture; Synovial tissue was obtained from osteoarthritis patients
undergoing joint replacement surgery through the Clinical Research Center,
MacNeal
Hospital, and transported to the laboratory in Dulbecco's Modified Eagle's
Medium
(DMEM) containing penicillin (100 units/ml), streptomycin (100 g/ml), and
fungizone (0.25 gg/ml). The synovium was dissected and minced with scissors,
and
plated as explants in culture medium composed of DMEM containing L-glutamine
(2 mM), heat inactivated fetal bovine serum (10% v/v), plus antibiotics.
Cultures
were housed at 37 C in a humidified atmosphere of 5% CO2. Adherent synovial
cells grew out of the explants within 2-3 weeks, and were passaged by
trypsinization.
Seed cultures were fed twice weekly and were passaged' at confluency.
Experiments
were performed on cells from passages 3-8. Experimental cultures were plated
into
35 mm dishes at a density of 7.5 X 103 cells/cm2 in 2 ml culture medium.
Cultures
were grown to near confluency for experiments, and contained 2.3 + 0.3 X 105
cells
(mean + S.E.M.,n=3), and 104 + 13 g protein (n=10). The growth medium was
replaced twice weekly.
2. Experimental Treatments. One day prior to initiation of experimental
treatments, medium was changed to experimental growth medium composed of
DMEM containing 2% heat-inactivated fetal bovine serum, plus L-glutamine and
antibiotics as above, to render the cells quiescent. The next day,. cultures
were
primed by addition of specified concentrations of IL-.1 or additional ligands
to the
conditioned growth medium for 12-24 hr intervals, as indicated. In some
experiments, conditioned growth medium was collected for analysis following
priming with IL-1. ' Acute experimental treatments were performed after this
priming
interval, as follows. Cultures were removed from the incubator, washed three
times
with 2 ml aliquots of Locke's physiological buffer (LB composition in mM:
NaCl,
154; KC1, 2.6; KH2PO4, 2.15; K2HPO4, 0.85; MgCl2, 5; CaCl2, 2; D-glucose, 10;
HEPES, 10; pH 7.4, BSA, 0.1% w/v), and then equilibrated with an additional
aliquot of LB containing specified ligands for 10 min on a 37 bath. This
solution
was removed by aspiration and replaced with a fresh buffer aliquot containing
indicated ligands for specified time intervals at 37 . Pharmacological
inhibitors
typically were added during the 10 min preincubation interval, and agonists
plus the
specified inhibitors were present during the 3 min challenge interval.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-130-
3. Measurement of Prostaglandin E2. Following indicated treatment
protocols, aliquots of culture supernatant (1 ml) were collected and rapidly
frozen in
liquid nitrogen. Samples were stored at -80 until processing. Aliquots of
culture
supernatant were analyzed by competitive binding radioimmunoassay as specified
by
the manufacturer (Sigma Chemical Co.), using an antibody with equivalent
reactivity
toward prostaglandin E2 and El. For quantitation, a standard curve was
prepared
with each assay using fixed concentrations of [3H]prostaglandin E2, and
increasing
concentrations of authentic competing prostaglandin E2.
4. Measurement of IL-6. Production of the cytokine, IL-6, was also
measured in aliquots of supernatant culture media which had been stored frozen
at -
80 C. IL-6 was measured by sandwich ELISA with alkaline phosphatase
detection
as described by the manufacturer (Pharmingen) and quantitated using standard
curves
prepared with the respective pure recombinant human cytokines. Experimental
determinations were performed on duplicate cultures.
STUDY 2
Assays for F3Hlthymidine Incorporation and MTT
Synoviocyte cell lines were routinely evaluated for competence to proliferate
in response to IL-1, measured as incorporation of [3H]thymidine (Kimball &
Fisher,
1988). In this preparation, maximally effective concentrations of IL-1
stimulate
[3H]thymidine incorporation by 10-20 fold compared to quiescent cultures
maintained in 2% serum (data not shown).
1. Data analysis. Immunoassays were routinely performed in duplicate
aliquots from each culture. Experimental determinations were performed on
duplicate or triplicate cultures. Each experiment was repeated in at least two
cell
lines. Nonlinear regression curve fitting and statistical analyses were
performed
using Graph-PAD Prism software (San Diego, CA).
2. Materials. Cell culture: Cell culture media were obtained from Sigma or
Gibco/BRL. Fetal bovine serum was from Atlanta Biologicals Inc. (Norcross,
GA).
Drugs: Recombinant human interleukin-1 was obtained from Genzyme (Cambridge,
MA). Ketoprofen was provided by Omeros Medical Systems, Inc. (Seattle, WA).
Amitriptyline, forskolin, 5-hydroxytryptamine, isoproterenol, Bradykinin,
histamine,
and prostaglandin E2 were from Sigma. Radiochemicals: [3H]Prostaglandin E2,
was
obtained from American Radiolabeled Chemicals, Inc. (St. Louis, MO). All other
reagents were obtained in the highest purity available from standard
commercial
suppliers.
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-131-
The effect of GPCR agonists, histamine and bradykinin, on PGE2 production
in human synovial cells was measured with and without prior stimulation to IL-
1 to
assess the functional interactions between agonists mediating a common
pharmacological effect through these different classes of receptors. Overnight
exposure of cultured human synovial fibroblasts to IL-1 (10 U/ml) results in a
delayed (4 hrs) and sustained large enhancement of PGE2 production, which can
be
measured by radioimmunoassay as increased PGE2 in the culture supernatant. The
progressive increase in PGE2 production during prolonged IL-1 treatment (16-24
hr)
has been shown to arise from the coordinated upregulated expression of cPLA2
and
10. the COX-2 (Crofford, 1984, Hulkower'et al., 1984). Cultures that have been
primed
by overnight exposure to IL-1 respond to subsequent challenges with maximally
effective concentrations of histamine (100 M) or bradykinin (1 M) with
additional
rapid (minutes) and robust production of PGE2. Representative data for the
time
course of PGE2 production in response to histamine or bradykinin stimulation
are
shown in FIGURE 7. Under these conditions, histamine elicits a 5-10 fold
increase
in PGE2 production compared to IL-1 primed cells receiving no GPCR agonist
addition. Bradykinin elicits a 10-15 fold increase. The absolute quantity of
PGE2
produced during the brief 2 min agonist challenge approaches or exceed
quantities
that are cumulatively produced during the entire 18 hr IL-1 priming interval.
This is
remarkable insofar as FIGURE 7 shows that the vast majority of the histamine-
elicited burst in PGE2 production occurs within the initial 2 min period since
minimal additional accumulation is observed over the subsequent 60 min period.
The bradykinin-stimulated PGE2 response continues to increase (2-fold) over
the
same time period. In the absence of IL-1 priming, naive synoviocytes show no
detectable PGE2 production in response to stimulation with either GPCR agonist
alone. Under conditions of IL-1 priming, histamine and bradykinin both
synergistically potentiated PGE2 release.
Using cultured synovial fibroblasts from osteoarthritis patients, we found
time-dependent synergistic interactions between the pro-inflammatory cytokine,
IL-
1, and physiologically relevant G-protein coupled receptors on PGE2
production, and
evaluated the actions of target therapeutic agents. GPCR agonists acting
through
endogenous synoviocyte receptors that are coupled to increases in
intracellular
calcium, inositol phosphates and PKC signaling pathways rapidly and
dramatically
amplify PGE2 production in cells previously primed by IL-1.. COX inhibitors
effectively attenuated both the agonist-elicited rapid burst and the long-term
accumulation of PGE2. Thus, different GPCR and IL-1 pathways for intracellular
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-132-
signal transduction synergistically interact to bring about either rapid or
slower, long-
term regulation of PGE2 responses.
The synergism between IL-1 and calcium-regulatory GPCRs in synoviocytes
that produce the rapid PGE2 burst may in part be explained by the rapid
augmentation of arachidonic acid release, a measure of cPLA2 activation in
many
cell types. In addition to inducing COX-2 expression, IL-1 increases
expression of
cPLA2 (Hulkower et al., 1994). These two proteins act together to provide free
arachidonic acid substrate for COX-2. The upregulation of the key eicosonoid
metabolizing enzymes induced by IL-l, combined with the ability of the GPCR
ligands to activate arachidonate release, would therefore be predicted to
increase
overall substrate flux through prostanoid synthesis. cPLA2 is the only known
PLA2
that exhibits functional properties indicative of receptor regulation and is
likely to be
involved in eicosonoid production and intracellular signaling. Since cPLA2 is
activated by increasing calcium concentrations for full activity and
bradykinin B2
and histamine H1 receptor activation is coupled to mobilization of
intracellular
calcium, this is likely the predominant factor regulating the rapid agonist-
stimulated
burst in PGE2 production. Finally, the very rapid and transient increase in
cytoplasmic calcium triggered by B2 or Hl receptor activation is similar to
the
kinetics known for cPLA2 activation, arachidonic acid release, and the
observed
PGE2 burst.
STUDY 3
Inhibition of PGE2 burst formation by clooxygenase inhibitors.
The actions of ketoprofen, a cyclooxygenase inhibitor, to attenuate PGE2
formation were determined by co-incubation with IL-1 during prolonged exposure
(16 hr); and by brief pre-incubation prior to a subsequent GPCR agonist
challenge
interval, as shown in FIGURE 8. Addition of specified concentrations of
ketoprofen
during overnight priming with IL-1 abolishes PGE2 formation, with IC50 = 4.5 +
0.8
nM determined by nonlinear regression analysis (mean SEM, n=4 synoviocyte
cell
lines). Similar determinations (data not shown) were performed with the
cyclooxygenase inhibitors etodolac (IC50 = 15.2 + 4.6 nM, n=4), ketorolac (2.2
+ 0.4
nM, n=4), and indomethacin (3.2 + 1.5 nM, n=2).
FIGURE, 8 also shows the ketoprofen concentration-dependent inhibition of
the agonist-elicited PGE2 burst in response to a challenge by 100 M histamine
(IC50
= 3.4 + 0.2 nM, n=3) or 1 gM bradykinin (IC50 = 9.5 + 2.0 nM, n=3) in
synoviocytes
primed overnight with IL-1 (10 U/ml). These values are comparable to those
observed for ketoprofen inhibition during overnight IL-1 induction of PGE2.
This
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-133-
result demonstrates that the onset of inhibition by the COX inhibitor occurs
within
the 10 min pretreatment interval prior to GPCR agonist addition, consistent
with a
direct, reversible inhibition of the COX activity and not due a mechanism
linked to
changes in the expression levels of the prostanoid regulatory enzymes. This
immediate inhibitory effect also provides a basis for the immediate
effectiveness of
this drug when delivered locally to the intra-articular in an irrigation
solution during
arthroscopic surgery.
STUDY 4
Induction of IL-6 production by IL-1 and GPCR agonists and inhibition by
ketoprofen.
The kinetics of induction of interleukin-6 in response to stimulation with IL-
1
are described. Synoviocyte cultures were exposed to the indicated treatments
with
IL-1 plus either histamine to activate signaling through inositol
trisphosphate
(InsP3)/protein kinase C pathway or isproterenol to activate increases in
intracellular
CAMP. Production of PGE2, IL-6, and IL-8 were measured in the culture
supernatants following 1, 2, 4, 6, and 24 hr treatments. In this experiment,
each
treatment interval was performed in a separate culture. In the above treatment
regime, production of IL-6 was robustly increased by IL-1 following 24-hr
exposure,
but no IL-6 was detected within the initial 6 hr interval. IL-6 production in
response
to IL-1 was not augmented further by addition of histamine, and histamine
alone
failed to stimulate IL-6 production. IL-1 also produced a significant
elevation of IL-
8 (2000 pg/ml), which was first measurable at 6 hr of treatment. IL-8
production was
sustained and greatly increased at 24-hr exposure to IL-1.
The effect of ketoprofen on the induction of cytokine production by IL-1 and
GPCR agonists was examined. The protocol also. tested the effects of IL-1
concentration dependence on the IL-6 steady state induction. Synoviocyte
cultures
were exposed to indicated concentrations of IL-1 and GPCR agonists. Culture
supernatants were collected and replaced with fresh media aliquots containing
the
same agonist additions at 8-hr intervals. PGE2, IL-6, and IL-8 in the
supernatants
were assayed as described.
Data for IL-6 production are shown in FIGURE 9, which shows IL-6
production at 16 hr (corresponding to treatment interval from 9-16 hr) in the
presence
of indicated concentrations of IL-1 plus added ligand. Addition of histamine
or
isoproterenol does not enhance IL-6 production compared to IL-1 alone. At 1.0
pg/ml IL-1, ketoprofen causes a partial (<50%) inhibition of IL-1-elicited IL-
6
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
20541PCT
-134-
production. Furthermore, ketoprofen inhibited IL-6 production in the histamine
or
isoproterenol/IL-1 co-stimulated samples.
The synoviocyte cell culture model system was used to characterize the
synergistic interactions between IL-1 and non-cytokine inflammatory mediators
that
are important in modulating the destruction of joint tissue, including damage
that
occurs as a consequence of tissue injury during arthroscopic surgery. The
results can
be summarized as follows: (1) IL-1 induces large increases in PGE2, IL-6, and
IL-8
in cultured synoviocytes, whereas quiescent cultures do not produce detectable
quantities of these mediators, (2) the induction of PGE2 occurs most rapidly
and
results in release of PGE2 to the culture supernatant at 4 hr, followed by IL-
8 at 6 hr,
and IL-6 at longer intervals, and (3) all three mediators remain elevated in
the culture
supernatant following 24 hr IL-1 exposure.
In contrast to their actions on PGE2 production, the GPCR agonists do not
enhance IL-1 induction of IL-6 or IL-8 and also do not increase IL-6 and IL-8
release
following priming with IL-1. IL-1 induction of IL-6 and IL-8 appears to be
reinforced by the concomitant induction of PGE2 since ketoprofen reduces the
production of these cytokines in response to IL-1. This result indicates that
ketoprofen could provide a therapeutic chondroprotective effect when delivered
to
the joint during surgical procedures.
Taken together, these results demonstrate interactions between specific
G-coupled receptor signaling pathways and the activation of synoviocytes by
pro-
inflammatory' stimulation with IL-1. A similar mechanism is expected to be
operative in chondrocytes. These interactions provide a means of integrating
and
modulating pro-inflammatory responses ofsynoviocytes and chondrocytes
depending
on inputs from other autocoid or neurotransmitter receptor systems within the
joint.
These findings underscore the rationale and potential clinical benefit of,
therapeutic.
interventions which target inhibition of G-protein coupled receptors that
mediate
signaling through calcium mobilization, phosphoinositide hydrolysis ' and PKC
activation and are coupled to increases in. production of PGE2 in arthroscopic
surgery. These receptors on synoviocytes and chondrocytes include histamine
H1,
bradykinin, Substance P, 5HT2, and the purinergic P2Y receptors.
While the preferred embodiment of the invention has been illustrated and
described, it will be appreciated that various changes to the disclosed
solutions and
methods can be made therein without departing from the spirit and scope of the
invention. For example, alternate chondroprotective agents, antibodies and
delivery'
vehicles may be discovered that may augment or replace the disclosed agents,
CA 02474645 2004-07-28
WO 03/063799 PCT/US03/03175
2U541FU I
-135-
targeting antibodies and delivery vehicles in accordance with the disclosure
contained herein. It is therefore intended that the scope of letters patent
granted
hereon be limited only by the definitions of the appended claims.