Note: Descriptions are shown in the official language in which they were submitted.
CA 02475337 2004-08-06
WO 98/33886 PGT/US98/00911
METHODS FOR CULTIVATING CELLS AND PROPAGATING VIRUSES
BACKGROUND OF THE INVENTION
Many established cell lines are available for a
variety of purposes in biotechnology. Some cell lines can be
cultivated as single-cell suspensions, but other cell lines do
not grow well without a support. The growth of a cell line
that requires support is often limited by the surface area
available for the cells to grow on, since many cell lines will
form only a monocellular layer on the surface. In addition,
some cell lines may tend to grow in clumps or aggregates in the
absence of a support, which is an undesirable result when they
are needed as single-cell suspensions, but more especially when
the cells are to be infected with a virus or transformed with a
recombinant vector, since the virus or vector may not gain
access to the cells within the clump or aggregate. Thus, there
can be sever.e problems in scaling up the cultivation of a cell
line, in particular in providing enough surface area for the
cells to grow on and/or avoiding clumping of the cells.
Microcarrier technology has been used to cultivate
cells in culture. For example, Forestell et al. (Biotech.
Bioena. 40: 1039-1044 (1992)) disclosed extended serial
subculture of human diploid fibroblasts on microcarriers using
a medium supplement that reduced the need of the cultured cells
for serum. Furthermore, Ohlson et al. (Cvtotechnoloay 14:67-80
(1994)) disclosed the bead to bead transfer of Chinese hamster
ovary cells using macroporous gelatin microcarriers. Finally,
Hu et al. (Biotech. Bioeng. 27: 1466- 1476 (1985)) disclosed
the serial propagation of mammalian cells on microcarriers
using a selection pH trypsinization technique.
However, in view of the problems noted above, there
is a need for improvements in methods of cultivating cell
lines, i:n methods of producing viruses for clinical uses, and,
in methods of scaling up the production of viruses for larger-
scale commercialization, espec.ially recombinant viruses for
gene therapy. The in.st-.ant invention meets these needs and
CA 02475337 2004-08-06
WO 98/33886 PCT/US98l00911
2
more. -
SUMMARY OF THE INVENTION
One aspect of the invention is a method for
cultivating cells, comprising:
(a) cultivating the cells on a first batch of
microcarriers until the cells are substantially confluent;
(b) detaching the cells from the microcarriers without
removing the microcarriers from suspension;
(c) Adding a second batch of microcarriers; and
(d) cultivating the cells further:
Another aspect of the invention is a method of
detaching cells from a first batch of microcarriers comprises
the following steps:
(a) washing the microcarriers and attached cells to remove
soluble materials;
(b) contacting the microcarriers and washed cells with a
chelating agent;
(c) removing the chelating agent;
(d) trypsinizing the cells for a short period to detach the
cells from the microcarriers; and
(e) neutralizing the trypsin by adding protein,
wherein (a)-(e) are conducted in a single cultivation vessel.
A further aspect of the invention is a method for the ~
separation of cells from microcarriers on which they have been
cultivated but from which they have become detached, comprising
introducing an aqueous suspension of cells and microcarriers
through an inlet into a separation device, the device comprising
(a) an inlet;
(b) a column;
(c) an outlet for the collection of cells and the aqueous
solution; and
(d) a mesh screen;
wherein the microcarriers are retained in suspension by an upward
flow in the separation device and are retained in the separation
device by a mesh screen, and wherein the cells and aqueous
solution are collected through the outlet.
A further aspect oi the invention is a system for
CA 02475337 2004-08-06
WO 9&33986 3 PCTIUS98l00911
separating cells from microcarriers on which the cells have been
cultivated, the system comprising;
(a) a bioreactor in which the cells were cultivated on the
microcarriers;
(b) a flow path from the bioreactor to a separation device;
(c) a separation device comprising
(I) an inlet;
(ii) a column;
(iii) an outlet for the collection of cells and the
aqueous solution; and
(iv) a mesh screen;
wherein the microcarriers are retained in suspension by
the upward flow in the separation device and are retained in the
separation device by a mesh screen, and the cells and aqueous
solution are collected through the outlet; and
(d) a pump, wherein the pump directs the flow of the aqueous
solution from the bioreactor to the outlet.
BRIEF DESCRIPTION OF THE FIGURES
Figure 1 is a side view of a system according to the
present invention used for separating media containing free cells
and virus from microcarriers.
Figure 2 is an exploded view of a separation device
according to the present invention used for separating media
containing free ce'lls and virus from microcarriers.
DETAILED DESCRIPTION OF THE INVENTION
The instant invention addresses the large scale
cultivation of cells for the propagation of viruses, especially
recombinant viruses for gene therapy, vaccine production, and so
on. In particular, the instant invention addresses three aspects
of large scale cultivation; the use of bead-to-bead transfer of
adherent cells to sequentially scale up the number of cells in
culture, including the use of trypsin to dissociate cells from
microcarriers in bioreactors, the use of fluidized bed-like
separation of cells from the beads during harvest, and the use of
microfiltration to disrupt celis so as to liberate virus
particles.
The term "virus" as used herein includes not only
CA 02475337 2004-08-06
wo 9W3M PCT/US98/00911
4
naturally occurring viruses but also recombinant viruses,
attenuated viruses, vaccine strains, and so on. Recombinant
viruses include but are not limited to viral vectors comprising
a heterologous gene. In some embodiments, a helper function(s)
for replication of the viruses is provided by the host cell, a
helper virus, or a helper plasmid. Representative vectors
include but are not limited to those that will infect mammalian
cells, especially human cells, and can be derived from viruses
such as retroviruses, adenoviruses, adeno-associated viruses,
herpes viruses, and avipox viruses. Adenoviral vectors are
preferred. Type 2 and type 5 adenoviral vectors are more
preferred, with type 5 adenoviral vectors being especially
preferred. ACN53 is a recombinant adenovirus type 5 encoding the
human wild-type p53 tumor-suppressor protein and is described, ~.._~
for example, in published PCT international patent application
WO 95/11984.
As used herein, the term "confluent" indicates that the
cells have formed a coherent monocellular layer on the surface
(e.g., of the microcarrier), so that virtually all the available
surface is used. For example, "confluent" has been defined (R.I.
Freshney, Culture of Animal Cells - A Manual of Basic Techniques,
Wiley-Liss, Inc. New York, NY, 1994, p. 363) as the situation
where "all cells are in contact all around their periphery with
other cells and no available substrate is left uncovered". For
purposes of the present invention, the term "substantially
confluent" indicates that the cells are in
general contact on the
surface, even though interstices may remain, such that over about
70%, preferably over about 90%, of the available surface is used.
Here, "available surface" means sufficient surface area to
accommodate a cell. Thus, small interstices between adjacent
cells that cannot accommodate an additional cell do not
constitute "available surface".
The cultivation steps in the methods of the present
invention can be carried out in a bioreactor or fermentor known
,in the art of about 1 to 5000 L equipped with appropriate inlets
for introducing the cells and microcarriers, sterile oxygen,
various media for cultivation, etc.; outlets for removing cells,
microcarriers and media; and means for agitating the culture
medium in the bioreactor, preferably a spin filter (which -also
CA 02475337 2004-08-06
WO 98133856 PCT/US98/00911
functions as an outlet for media). Exemplary media are disclosed
in the art; see, for example, Freshney, Culture of Animal Cells -
A Manual of Basic Techn.iques, Wiley-Liss, Inc. New York, NY,
1994, pp. 82-100. The bioreactor will also have means for
controlling the temperature and preferably means for
electronically monitoring and controlling the functions of the
bioreactor.
Exemplary microcarriers on which the cells are allowed to
grow are known in the art and are preferabiy specially adapted
for the purpose of cell cultivation. General reference is made
to the handbook Microcarrier Cell Culture - Principles & Methods,
published by Pharmacia. However, it should be noted that some
cell lines used in the present invention may not adhere strongly
to the surfaces of microcarriers; it is well within the abili'y
of one of ordinary skill in the art to determine a suitable
combination of cell line, virus (where applicable) , microcarrier
and culture conditions. The microcarrier preferably has a
particle size in the range of about 100 to 250 microns, more
preferably in the range of about 130 to 220 microns, and should
be composed of a non-toxic material. 'I'he median of the sample
size preferably falls in these ranges, such that these size
ranges are preferably those of at least the mi.ddle 90% of the
microcarrier sample. In a preferred embodiment, the microcarrier
consists of substantially spherical microbeads with a median
particle size of about 150 to 200 microns, preferably 170 to 180
microns. The microcarrier surface may be treated to modify cell
adhesion, in particular to enhance cell adhesion yet permit
proliferation and spreading; thus the microcarriers may be
coated, e.g., with collagen_ Preferably, the microcarriers are
slightly denser than the culture medium, so that gentle agitation
will keep them in suspension, whereas simple means such as
sedimentation or centrifugation allows their separation. A
density of 1.03 to 1.045 g/mi when the microcarriers are
equilibrated with a standard solution such as 0.975 NaCl (or with
the culture medium) is suitable. The present inventors have
found that Pharmacia's Cytodex-3microcarriers in general will
meet these requirements, although the partic-ular requirements
that apply for certain cell lines or viriises may require the
selection of a particular Cytodex microcarrier.
* Trade-mark
CA 02475337 2004-08-06
wo 98l3388G PCT/US98/00911
6
The cells may be those of any suitable host cell-line that
is able to replicate itself and in particular support the
replication of the virus of interest. A particularly preferred
cell line is the human embryonic kidney cell line 293 (ATCC
catalog number CRL 1573). These cells do not adhere strongly to
all microcarriers, and are preferably used with Pharmacia's -
CytodexTh'-3 microcarriers, which are collagen-coated for better
cell adhesion. CytodexT"'-3 microcarriers have a median particle
size of about 175 microns with the middle 90% of the sample
having a size of about 140 to 210 microns; the density of such
microcarriers when equilibrated with 0.9% NaCl is 1.04 g/ml. The
cells are preferably cultivated on such a microcarrier in a first
step, and then loosened therefrom and transferred to addit'-onal
microcarriers for a production step.
Stirring can conveniently be effected not only by a paddle
at the bottom of the bioreactor but also by a rotating
spinfilter, which preferably extends downwards from the top of
the bioreactor into the bulk of the medium= The cells and
microcarriers can be kept in suspension in the culture by
rotation of the spinfilter; the spinfilter may also be equipped
with fine orifices that permit the removal of medium without loss
of cells_ The medium can be removed and replaced simultaneously
or alternately; it is frequently convenient to remove a
substantial fraction (e.g., up to about 50%) of the medium and
then replenish it with the appropriate replacement medium while
still removing medium, e.g., through the spinfilter.
Typically, cells are scaled-up from a master working cell
bank vial through various sizes of T-flasks, and, preferably,
finally to bioreactors. A preferred flask is the CELL FACTORYTM
tissue culture flask (CF; NUNC), a specially designed large flask
that conveniently has several internal compartments providing a
large surface area to which the cells can adhere or attach and on
which t-hey can grow. After cultivation until substantially
confluent, the cells can be loosened by trypsinization and
isolated. The trypsinization is effected for a short period
(preferably less than 5 minutes, more preferably about 3
minutes), and the trypsin is then neutralized by the rapid
addition of serum in growth medium_ If desired, the cells can be
centrifuged and the trypsin-containing medium removed before the
CA 02475337 2004-08-06
WO 98/33886 PCTIUS98/00911
7
serum is added. The resulting cell suspension is then typically
fed into a seed production bioreactor (typically 20-30L volume)
for further cultivation, and in some embodiments, to a larger
production bioreactor (typically 150-180L volume).
The ratio of volume of the second (larger) bioreactor to
the seed bioreactor depends upon the degree to which the cell
line is propagated in the first bioreactor, but is typically from
5:1 to 10:1, e.g., in the range of (6-8):1.
Cells are detached from microcarriers by a trypsinization
procedure performed in the cultivation vessel, preferably a
bioreactor, while the microcarriers are suspended. The
spinfilter is utilized to perform medium exchanges to reduce the
serum and calcium levels in the medium, which increases the
efficiency of the trypsinization while maintaining a constant
volume in the bioreactor. Settling steps are avoided which might
cause damage to the cells on the microcarriers. The resultant
cell/microcarrier suspension can then be transferred to a
production bioreactor which is previously charged with culture
media and microcarriers.
After the transfer of cell/microcarrier suspension from
the seed bioreactor, the production bioreactor (for example,
about 200 L) is operated, e.g., at about 37'C and about pH 7.3.
A perfusion of fresh medium during cell propagation can then be
performed in order to maintain the lactate concentration below
about 1.0 g/L. Cells are typically allowed to grow on the
microcarriers for about 4 to 7 days until more than 50oof the
microcarriers are completely confluent. Preferably, a virus
infection process is then initiated. A vial of 40 to 50 ml viral
inoculum, typically containing approximately 1.0x1013 total viral
particles is used to infect the production bioreactor. Virus is
allowed to replicate in the production bioreactor for about 3 to
days until about the time of maximum virus titer. Typically,
more than 90% of cells will have detached from the microcarriers
due to cytopathic effects of the virus. The final recombinant
adenovirus yield from the production bioreactor is typically
about 8.5x109 viral particles/ml.' This gives a total -yield of
viral particles of J.4x1015 from each 160-L batch.
In other embod'i.ments of the invention, the production
bioreactor is inoculated with cells harvested by trypsinization
CA 02475337 2004-08-06
wo 9srMM rcrIos9&4"11
8
and then used directly to inoculate the production bioreactor.
Typically 8 to 12 CELL FACTORYTm tissue culture flasks are
utilized to achieve the overall bioreactor inoculum seeding
density of 0.6 to 1x105 cells/ml. The typical virus yield in
this method ranges from about 1.7 to 2.6x1010 viral particles/ml.
Therefore, this particular method provides a total virus particle
number of about 3 to 4x1015 from each 160 L batch.
In some embodiments of the invention, a fluidized bed-like
process is used to harvest the cells from the bioreactor.
Typically, the bioreactor is harvested after about 90% of the
cells detach from the microcarriers. Without being limited to
any one theory, the cytopathic effect of viral propagation in the
host cells appears to be responsible for the cell detachment. In
other embodiments, uninfected cells can be detached from
microcarriers by the trypsinization method of the instant
invention. After the bioreactor is harvested, the broth
contains cells, microcarriers and medium. Virus is present in
the cells and the medium. Therefore, all of this material is
preferably collected for processing. The speci'Lic gravity
(density) of the microcarriers is similar to that of the cells.
Preferably, the microcarriers are kept freely suspended while
separating the cells from the beads, as processirig steps using
sedimentation causes the cells to settle with the microcarriers,
which results in recovery losses.
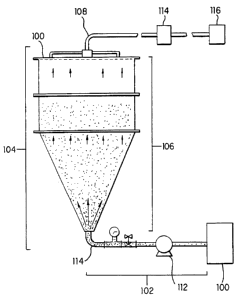
A preferred embodiment of a separation device is provided
e. ~
in Figures 1 and 2. In some embodiments of the invention, the
separation device is provided as part of a system. An exemplary
system is depicted in Figure 2. The system thus comprises a
bioreactor 100 in which the cells are cultivated on
microcarriers; a flow path 102 from the bioreactor to the
separation device 104; the separation device comprising a column
106; an outlet 108 for the collection of cells and the aqueous
solution; and a mesh screen 110. The microcarriers are retained
in suspension by an upward flow in the separation device and are
retained in the separation device by the mesh screen, and the
cells and aqueous solution are collected through the outlet.
Also provided in the system is a pump 112, wherein the pump
directs the flow of the aqueous solution from the bioreactor to
the outlet. In some embodiinencs, a microfilter 114 and an
CA 02475337 2004-08-06
WO 98/33886 9 PCT/US98l00911
ultrafilter 116 may be provided as components of the system.
In the embodiment shown in Figure 1, the separation device
typically comprises a column 106, such as a chromatography
column, having an inlet 114 through which an aqueous suspension
of cells and microcarriers from a bioreactor 100 is introduced
into the separation device 104; and at least one outlet 108 for
the collection of cells and the aqueous solution; and a mesh
screen 110. The microcarriers are retained in suspension in the
column by an upward flow in the separation device and are
retained in the separation device by a mesh screen, and wherein
the cells and aqueous solution are collected through the outlet.
The flow rate in the separation device is about 1 to about 3
cm/min. Typically, an upward flow through the column is
,_generated by pumping an aqueous solution, such as the cell
suspension or a buffer, through the inlet, wherein the inlet is
situated at the bottom of the device and the outlet is situated
at the top of the device.
Figure 2 is an expanded schematic view of a separation
device 200, depicting in more detail an outlet assembly 210, a
mesh screen assembly 212, an inlet 214, and a column 216 having
an upper section 218 and a lower section 220. The lower section
typically comprises about 20 to 50%, more preferably about 30%,
of the volume of the column and contains the inlet. The lower
section is preferably conical, with a preferred angle of about 15
to about 45 degrees.
Thus, the fermentation broth from the bioreactor is pumped
into the base of the column. The flow rate is regulated to
provide an upward flow sufficient to keep the cells and viral
particles suspended in the medium while allowing for the
retention of the microcarriers within the separation device.
Preferably, the flow rate is approximately 1-2 cm/min since the
cells have a specific gravity similar to that of the
microcarriers. The clarified broth containing cells and virus
passes through the mesh screen on the upper end of the fluidized
bed-like column and is collected for microfiltration.
For a 200 L scale device, the lower section of the column
preferably is conical. The cone allows for a gradual reduction
of the linear._velocity of the fermentation broth entering the
cone. The fJ.uid velocity of the inlet line is reduced to achieve
CA 02475337 2004-08-06
WO 98/33886 PCT/US98/00911
a reduced linear flow in a uniform distribution across t-he cross
sectional afea at the upper end of the cone. The walls of the
cone are at an angle which allows the beads that settle on the
walls to move downward to the inlet. In this way, these beads
are resuspended to avoid entrapment of the cells between the
settled beads. The angle of the conical walls is preferably
about 30 degrees. Angles less than 15 degrees provide an
exceptionally long cone and angles greater than approximately 45
degrees may not disperse the inlet feed effectively_ The upper
section of the column functions as a zone in which the beads
settle at a rate greater than the linear flow rate of the
fermentation medium. This section of the column is cviindrical
in shape_ Within this zone a boundary is formed such that the
microcarriers accumulate in the lower region ol' the column_ An
end plate assembly 222 (Figure 2) of the column-functions as a
collection point for the clarified fermentation media containing
cells and virus_ This consists of an end plate 224 fitted with a
mesh screen assembly. This screen, preferably about 50 to 120
mesh, more preferably about 100 mesh, functions as a second point
for removal of the microcarriers.
The above-described embodiment is the pre.-Lerred ernbodiment
used in the examples herein. The column dimensions and screen
mesh may be varied based upon the volume of solution to be
processed, the concentration of beads, the particular
microcarrier used.and the media formulation (e.(,-., speci.fic
gravity of media). A preferred column consists of a bottom cone
custom fabricated out of stainless steel to be attached to two
Phatn'acia''r' KS370 section tubes fitted with a KS370 end assembly to
which the vendor screen was replaced with a stainless steel (ss)
mesh (preferably about 50 to 120 mesh, more preferably about 100
mesh).
After the cells are collected, they are preferably lysed
to liberate additional virus particles. Homogenization or
freeze-thawing may be used to liberate the virus particles_ In a
_preferred embodiment of the invention, microfiltration is used to
simultaneously lyse virus-containing cells and clarify the broth
of cell debris which would otherwise interfere with viral
purification_ For example, the microfiltration can be performed
using a Prostak (Millipore) -ystem with a 0_55 micron,
* Trade-mark
CA 02475337 2004-08-06
WO 98/33886 PCTIUS98/00911
11
hydrophilic or hydrophobic membrane and at a shear rate of 7000
1/sec. The shear rate is generated by the flow of retentate
through the tangential flow channels of the membrane. Therefore,
the cross-flow is used not only to prevent the membrane from
fouling but can also be used to create enough shear for lysing
the cells. The pore size of the filter should be sufficient to
allow passage of virus while retaining cell debris_ Thus,
typically the pore size range is about 0.2-0_65 micron. The
shear rate range is typically about 2000 to 10,000 1/sec, more
preferably about 7000 1/sec.
Tm
Typically, BENZONASE endonuclease (American International
Chemical, Inc.) is added to the clarified broth to digest
cellular nucleic acids, as viral particles can become complexed
with cellular nucleic acids. In a preferred embodiment,
ultrafiltration using a Pellicon system (Millipore) with a 1
million nominal molecular weight cut-off, Pellicon I-regenerated
cellulose membrane is used to concentrate the virus. The
ultrafiltration step accomplishes two functions; the virus is
concentrated for purification and diafiltration is performed to
exchange the buffer so that the virus suspension can be applied
directly to a DEAE column. The eluate from the microfilter
contains the liberated virus and is preferably concentrated e.g.,
by ultrafiltration.
Between each cultivation step, the cells can be loosened
and stripped from the microcarrier by trypsinization, e.g., by
treatment with trypsin. In the present invention it is preferred
to remove the serum used in the cultivation, since the serum
proteins inhibit the trypsin; removal of the serum therefore
allows a smaller amount of trypsin to be used. This is
advantageous since addition of a larger amount may cause
localized high concentrations of trypsin that could damage the
cells. Wit.h regard to the next step, Ca++ ions are removed since
the removal of these ior_s from the cells tends to loosen the
cells and enables one to use less trypsin. Thus, loosening and
stripping cells, especially of the human embryonic kidney cell
line 293, carf conveniently include the following steps:
i) rapi.dly wasiiing the cells to remove serum and other
soluble mater_ial:;;
(ii) removing Ca++ from the washed cells by adding a chelating
* Trade-mark
CA 02475337 2004-08-06
WO 98/33886 PCT/US98/00911
12
agent ;
(iii)rapidly removing the chelating agent;
(iv) rapidly adding trypsin;
(v) trypsinizing the cells for a short period of time
(preferably ranging from about 3 minutes to about 15 minutes);
and
(vi) rapidly neutralizing the trypsin by adding protein.
In step (i) above, the phrase "rapidly washing" means at a
constant bioreactor volume perfusing one volume change of medium
at a rate of about 1-3 liters per minute, more preferably about
2 liters per minute. In step (iii) above, the phrase "rapidly
removing the chelating agent" means at a constant bioreactor
volume perfusing one and a half volume changes of medium at a
rate of about 1-3 liters per minute, more preferably about 2 47
liters per minute. In step (iv) above, the phrase "rapidly
adding trypsin" means adding the appropriate volume of trypsin
solution (typically a 2.5% solution) at a rate of about 1-3
liters per minute, more preferably about 2 liters per minute. In
step (vi) above the phrase "rapidly neutralizing the trypsin by
adding serum" means adding the appropriate volume of serum at a
rate of about 1-3 liters per minute, more preferably about 2
liters per minute.
If the serum is not removed in step (i), then the addition
of the necessary large amounts of trypsin can lead to locally
high concentrations, of trypsin, which can actually damage or even
kill the cells rather than simply loosen them. The removal of
serum in Step (i) and of Ca++ in Step (ii) reduces the amount of
trypsin needed in Steps (iv) and (v). To avoid actually damaging
or even killing the cells, the treatment with the chelating agent
and with the trypsin should preferably be kept short (i.e., long
enough to detach the cells from the microcarriers, but preferably
not longer). Examples of preferred chelating agents include EDTA
(ethylene diamine tetraacetic acid) and EGTA (ethylene-
bis(oxyethylene-nitrilo)tetraacetic acid).
The serum is removed by a process of medium exchange; for
example, the medium can be pumped off through a spinfilter.
Serum-free wash medium is added to replace what was pumped off
and the mixture stirred. Alternatively, the addition of serum-
free wash mediurn can be continuous with the removal of medium
CA 02475337 2004-08-06
WO 98/33886 13 PCT/US98/00911
through the spin-filter. The process is repeated until the serum
concentration has been reduced to a sufficiently low level, e.g.-,
less than about 1.0 to 0.2%, preferably about 0.2%. The
chelating agent, preferably EDTA, is added in serum-free
chelating medium, the mixture again stirred, and the chelating
agent pumped off. Alternatively, the addition of the chelating
agent in serum-free medium can be continuous with the removal of
the medium through the spinfilter.
The trypsin is preferably used in step (v) to provide a
concentration in the bioreactor of from about 0.05 to 0.1o, and
it is allowed to act on the cells for from 5 to 10 minutes, e.g.,
preferably a trypsin concentration of about 0.065% for about 8
minutes. Protein, typically in the form of bovine calf serum, is
preferably added to the bioreactor in a final concentration of
about 10 to 20% to inhibit the trypsin.
Thus the addition of serum in step (vi) not only prepares
the cells for further cultivation but also neutralizes residual
trypsin. The entire sequence of steps (i)-(vi) can take place in
situ in the bioreactor; in some embodiments the suspension of
microcarriers and cells can be transferred to a larger
bioreactor, where further microcarriers are added for the next
step of the cultivation. The cells are allowed to attach to the
microcarriers and then are cultivated further. Once they become
substantially confluent again (e.g., 3-4 days' cultivation at
about 37 C), they can be put through the next stage, which may
for example be harvesting, loosening for a further upstaging, or
inoculation with virus. If the cells have been cultivated simply
for harvest, then they can be harvested at this stage, e.g., by a
repetition of Steps (i) through (vi) above. If they are needed
for a further upstaging, then Steps (i)-(vi) above can be
repeated. If they have been cultivated for propagation of a
virus, the virus can now be inoculated into the medium.
The examples herein serve to illustrate but do not in any
way limit the present invention. The selected vectors and hosts
and other materials, the concentration of reagents, the
temperatures, and the values of other variables are only to
exemplify the application of the present invention and are not to
be considered limitations thereof.
CA 02475337 2004-08-06
WO 98/33886 PCT/US9&%911
14
EXPERIMENTAL EXAMPLES
I. OVERVIEW
A. Cell Inoculum Preparation
Each viral fermentation batch is started from a cell line
propagated from a vial of 293 cell Manufacturers Working Cell
Bank (MWCB). The bioreactors are inoculated with 293 cells (ATCC
catalog number CRL 1573) maintained by propagation in T-flasks
TM
and CELL FACTORY using growth medium (Medium 1), as illustrated
in Table 1 below. Each transfer represents a passage.
Typically, passage numbers of 4 to 30 are employed for
inoculation of a seed bioreactor.
TABLE 1: MEDIA COMPOSITION
Medium Purpose Component Typical Typical
Function Component Component
Range
Medium 1 Cell Cell Growth DMEM Powderl 10-20 g/l
(Growth medium) Growth
Cell Growth Glutamine 0.1-1.0 g/1
Cell Growth Bovine Calf Serum3 5-15%
Buffer Sodium Bicarbonate 2-4 g/1
Medium 2 Prepara- Reduce Ca++ DMEM Powder, 10-20 g/1
(Serum-free wash tion for for tryp- Ca++ free
medium) trypsiniz- sinization,
ation Later wash
EDTA ~
Cell Glutamine 0.1-1.0 g/1
Viability
Buffer Sodium Bicarbonate 2-4 g/l
Medium 3 Prepara- Reduce Ca++ DMEM Powder, 10-20 g/l
(Serum-free tion for for tryp- Ca++ free2
chelating medium) trypsiniz- sinization
ation
Ca++ EDTA4 200-400
Chelation mg/l
Cell Glutamine5 0.1-1.0 g/1
Viability
Buffer Sodium Bicarbonate 2-4 g/1
1- DMEM powder (available from American Biorganics, Catalog no. D2807):
preferably used to provide 4.5 g/L glucose and 0.584 g/L L-glutamine;
no sodium bicarbonate and no HEPES.
2 Ca++-free DMEM powder (available from American Biorganics, Catalog no.
D2807403): preferably used to provide 4.5 g/L glucose and 0.584 g/L L-
glutamin.e; no sodium bicarbonate, no calcium chloride and no HEPES-.
CA 02475337 2004-08-06
wo 9&r&V" 15 rCTrUS9&4"11
3 Bovine calf serum: available from Hyclone, Catalog no. 2151.
4 EDTA stock solution: 186.1 g/L EDTA and 20 g/L NaOH pellets.
Glutamine stock solution: 29.22 g/L.
To prepare for the transfer of cells from flask to flask
during cell expansion, the spent medium is poured off and the
cells in the flask are then washed with phosphate-buffered
saline (PBS). A trypsin solution is added to the cell
monolayer on the flask's surface and the cells are exposed
until they detach from the surface. Trypsin action is then
largely neutralized by adding growth medium (Medium 1, Table 1)
containing serum; complete neutralization. is not necessary,
since residual trypsin will have low activity. The cells can
be recovered by centrifugation and resuspended in fresh growth
medium (Medium 1). Table 2 shows the typical volumes are used.
TABLE 2: TYPICAL VOLUMES USED IN TRANSFER OF CELLS
Flask Surface Volume of Volume of serum- Volume of
volume area 0.05% containing Medium
Trypsin Medium to for Cell Growth
Solution Inactivate
Trypsin
T-75 75 cm2 3 ml 10 ml 30 ml
T-500 500 cm2 25 ml 50 ml 200 ml
CELL 6000 250 ml 500 ml 1500 ml
FACTORYTM cm2
B. Virus Inoculum Preraaration
Virus inocula can be prepared by infecting mature CELL
FACTORY7" tissue culture flasks. In this procedure, 293 cells
are first propagated from T-flasks to CELL FACTORY' tissue
culture flasks. When CELL FACTORY7" tissue culture flask
cultures are mature (typically 80-90% confluent) they are
infected with an inoculum from the manufacturer's working virus
bank (MWVB). The infected CELL FACTORY~ tissue culture flasks
are incubated until the 293 cells detach from their supporting
surface. The cells are collected by centrifugation and
ruptured by.multiple freeze-thaw cycles. After a subsequent
centrifugation, the virus is recovered in the supernatant and
stored as aliquots at -20 C or below. This material is the
"Virus Inoculum" which is used to infect bioreactors.
Optionally, the virus inoculum can also be derived from the
CA 02475337 2004-08-06
WO "MM6 PCT/IIS98/00911
16
bioreactor harvest which is filter-sterilized'(see "Production
Bioreactor Harvest" below).
C. Seed Bioreactor Preparation and Operation
Preferably, a seed bioreactor is used to prepare the 293
cell inoculum for the production bioreactor. The seed
bioreactor is steam-sterilized and charged with a batch of
filter-sterilized growth medium (Medium 1, Table 1, above) for
the free cell suspension process. For the microcarrier
process, however, swelled sterile microcarrier beads (Cytodex 3
or equivalent) are preferably added at this stage.
The seed bioreactor is inoculated with the 293 cells
harvested from CELL FACTORYTM tissue culture flasks. The
operating conditions are set as shown in Table 3. The pH and
dissolved oxygen (DO) are controlled by sparging CO2 and
oxygen, respectively. Extra growth medium can be added to the
bioreactor by perfusion. Cell growth is monitored by
microscopic examination and by measuring the lactate production
and glucose consumption. Typically, when the cell density in
the suspension culture reaches 1 x 106 cells/ml in the seed
bioreactor, it is ready for inoculating the production
bioreactor. However, inoculation requires a few additional
steps for the microcarrier process. Typically, when the cells
on the microcarriers are >50% confluent, the serum and calcium
in the bioreactor medium are washed off using media described
in Table 1. Trypsin is then added rapidly, and when the cell
detachment reaches typical level, serum is added to inactivate
the trypsin. The seed bioreactor contents are now transferred
to the production bioreactor.
Optionally, 293 cells harvested from multiple CELL
FACTORYTM tissue culture flasks can directly be used as
inoculum for a production bioreactor. Typically, 8-12 such
cultures are harvested and pooled to provide an inoculum.
TABLE 3: SEED BIOREACTOR OPERATING CONDITIONS
Variable Recommended Range Typical or preferred
Temperature 34 C-39 C 37 C
pH 6.9-7.5 7.3
Dissolved Oxygen > 10% 30% air saturation
Pressure > 0.05 bar 0.1 bar
CA 02475337 2004-08-06 -
WO 98133886 17 PCT/US98/00911
Aeration overlay > 50 Liters/hour 180 Liters/hour
Agitation > 20 rpm Initially 50-70 rpm,
with
optional stepwise
increases
Spinfilter > 20 rpm Initially 50-70 rpm,
with
optional stepwise
increases
Trypsinization > 25% of > 50% of microcarriers
microcarriers have
have confluent confluent cells
cells
D. Production Bioreactor Preparation and Operation
The viral production process is exemplified in a 200-L
production bioreactor using growth medium (Medium 1, Table 1).
In the suspension culture process, filter-sterilized medium is
batched into the bioreactor. However, in the microcarrier
process, microcarriers are either sterilized in situ in the
production bioreactor or autoclaved externally and charged.
These microcarriers are then conditioned in the growth medium
(Medium 1) prior to inoculation with 293 cells.
The production bioreactor is inoculated with the 293
cells from the seed bioreactor. The operating conditions are
set as shown in Table 4. The pH and dissolved oxygen (DO) are
controlled by sparging CO2 and oxygen, respectively.
Optionally, extra growth medium can be added to the bioreact.or
by perfusion_ Cell growth is monitored by microscopic
examination, and by measurement of lactate production and
glucose consumption. Cells are allowed to grow to
approximately l x 106 cells/ml. The bioreactor is then
inoculated with virus. Preferably, a multiplicity of infection
(MOI) ratio expressed as the total viral particles per cell of
50:1 to 150:1 is used. Viral titer is typically performed
using theResource QT"'HPLC assay_ Virus is allowed to propagate
until the cell viability drops to.about 10%.
The type of virus and its action upon the host cells may
determine whether it is necessary to detach the host cells from
the microcarriers and/or lyse the cells. At about the time of
maximtun virus titer (frequently when the cells start to detach
from the microcarrier and some of which may lyse, so that the
virus starts to escape), the incubation can be stopped and the
CA 02475337 2004-08-06
'~ ..
WO 98133886 PGT/US98/00911
18
cells and virus can be harvested. Indeed, in the microcarrier
process, 80-90% of the cells, sometimes even more than 90%, may
detach from the microcarriers. Without being limited to any
one theory, the cytopathic effect of viral propagation in the
host cells appears to be responsible for cell detachment.
Thus, when adenovirus ACN53 is used with 293 cells, the cells
start to detach from the microcarriers after 3 or 4 days'
cultivation with the virus.
TABLE 4: PRODUCTION BIOREACTOR OPERATING CONDITIONS
Variable Recommended Range Typical or preferred
Temperature 34 C-39 C 37 C
pH 6.8-7.5 7.3
6.8-7.6 7.4 after virus
infection
Dissolved -Oxygen > 10% 30% air saturation
Pressure > 0.05 bar 0.1 bar
Aeration Overlay > 500 Liters/hour 2500 Liters/hour
Agitation > 20 rpm 50-70 rpm
Spinfilter > 20 rpm 50-70 rpm
Virus infection > 25% of > 50% of microcarriers
time microcarriers have
have confluent confluent cells
cells
Harvest time > 50% of > 90% of microcarriers
microcarriers are
are em t empt
E. Production Bioreactor Harvest
1. Ser)aration of Cells from Microcarrier
At harvest, the bioreactor contents have to be handled
differently depending on whether the process uses free
suspension or a microcarrier. In the microcarrier process, a
fluidized bed column is preferably used to separate the
microcarriers from cells and supernatant. An upward flow rate
is maintained so as to retain the microcarriers while the cells
and supernatant pass through. The fluidized bed is washed with
medium or wash buffer to recover most residual cells and virus,
and the washings are combined with the cells and supernatant as
an eluate. The fluidized bed operation is not required for the
free cell-suspension process.
The eluate containing cells and virus is further
processed in that the cells are lysed by high shear to release
virus, and the eluate is then clarified by means of a cr_oss-
CA 02475337 2004-08-06
WO 98/33886 19 PCT/US98100911
flow microfiltration. Typically, 0.65 m Durapore" (Millipore)
or equivalent membranes are used. Towards the end of the
microfiltration, the retentate is washed with wash buffer to
recover the residual virus into the permeate. After
microfiltration, the permeate can be optionally treated with a
nuclease such as BENZONASEz'"' endonuclease.
The permeate from the microfiltration is concentrated by
ultrafiltration (with a typically 1 million molecular weight
cutoff) and a buffer exchange is performed using the wash
buffer. The concentrated and diafiltereci retentate containing
the virus is then passed through a final filter. The resulting
filtrate is stored as "Viral Concentrate" in a freezer at -20 C
or below.
2. Comnosition and Preparation of Media
Table 1 above lists the media used in the preparation of the
viral inoculum and in the fermentation process_ All these
culture media are prepared by first dissolving the dry DMEM,
powder and other reagents in purified water. After dissolving
the drv powders, these media are adjusted to pH 7.2-7.6 with
hydrochloric acid_ The media are then sterilized by passing
through a 0_2 ~tln filter into an appropriate storage container_
The sterile media are refriaerateci below 10 C and discarded one
month after preparation.
Table 5 lists various buffers used in the process.
TABLE 5: BUFFERS USED IN FERMENTATION AND HARVESTING
Step Procedure Component Typical Typical
Function Component Component
Range
Cell inoculum Wash with
preparation in PBS prior to
flasks trypsinization Celi Potassium 0.1-0.3
Electrolyte Chloride g/l
Balance
Cell Potassium 0.1-0.3 gil
Electrolyte Phosphate,
Balance monobasic
Ionic Sodium 6-10 g/l
Strength Chloride
Buffer Sodium 0.~-2 g! i
Phosphate,
dibasic
* Trade-mark
CA 02475337 2004-08-06
WO 98C33886 PCr/irs98/00911
Buffer Sodium 1-3 g/1
Phosphate,
dibasic
he tah drate
Microcarrier Swell micro- Same Same Same ranges
preparation carriers with functions as components as above +
PBS and Tween 80 above + as above + 0.1-1 ml/1
Wetting agent Tween 80
Production Wash fluidized Buffer Tris Base 4-8 g/l
bioreactor bed, microfilter
harvest and ultrafilter
retentate with
Tris Buffer
Ionic Sodium 7-11 g/l
Strength Chloride
Stabilizer Magnesium 0.2-0.6 g/l
Chloride
Stabilizer Sucrose 16-24 g/l
Table 6 summarizes the in-process controls during the
fermentation and harvesting processes.
TABLE 6: IN-PROCESS CONTROLS
Step In-Process Control Recommended Range
T-flasks; CELL Microscopic Normal growth and
FACTORYTM: observation morphology. No
Preparation of contaminants.
inoculum
Seed bioreactor Microscope Normal cell growth
observation, and morphology on
measurement of microcarriers,
lactate and glucose lactate <1.2 g/L at
concentrations, transfer point, no
supernatant cell contaminants. ~
count, sample
sterility check.
Production Microscope Normal cell growth
bioreactor observation, and morphology on
measurement of microcarriers,
lactate and glucose lactate <1.2 g/L
concentrations, prior to infection,
supernatant cell majority of cells
count, sample detach from
sterility check. microcarriers at
harvest, no
contaminants.
Fluidized bed Visual observation of Fluidized bed
fluidized bed, feed volume to be
pressure and feed smaller than total
flow rate. column volume and
feed pressure
should be below-the
leak point of the
R _ column assembly.~
CA 02475337 2004-08-06
wp gg/33886 PCTIUS98/00911
21
Microfiltration Observation and Feed flow rate
control of feed flow, adjusted to achieve
permeate flow, feed, high shear without
retentate and exceeding a
transmembrane transmembrane
pressure, retentate pressure of 0.5
temperature. bar. Temperature
should not exceed
37 C.
Ultrafiltration Observation and Feed pressure
control of feed should not exceed 1
pressure and bar. Temperature
retentate should not exceed
temperature. 37 C.
Final filtration Observation and Feed pressure
control of feed should not exceed 2
ressure bar
II. Bead to Bead Transfer from Seed Bioreactor to
Production Bioreactor
A. 293 Cell Inoculum Preparation for Seed Bioreactor
1. Scale-up from vial to T75 flask
A frozen vial of the 293 cell line containing a total
of 2 x 107 cells was thawed in a 37 C water bath. The cells
were washed with 10 ml of Medium 1. The washed cells were
resuspended in a total volume of 30 ml of Medium 1 and placed
in a 75 cm2 tissue culture flask (T75). The culture was
placed in an incubator at 37 C with a 5% CO2 atmosphere and a
humidity level of 100%. This was passage 1 of the culture.
2. Scale-up from T75 Culture to T500 Flask Using
Trvnsinization
The T75 culture reached a confluency level of 90% in
three days. At this time the T75 culture was trypsinized in
the following manner. The 30 ml of supernatant medium was
removed from the flask. A volume of 10 ml of CNF-PBS
(Dulbecco's phosphate buffered saline without calcium chloride
and without magnesium chloride) was used to wash the culture
surface. The supernatant CMF-PBS was removed from the flask.
Two ml of TE (0.05% crude trypsin with 0.53 mM EDTA-4Na)
solution was added to the flask. The flask was moved so that
the solution covered thP entire culture surface. The cells
detached from the-flask' surface within five miziutes. Ten ml
of Medium 1 was added to the flask immediately after the cells
detached from the suz iat::e . ~ilhe c:ell 5u5pension was
CA 02475337 2004-08-06
wo 9sr33es6 rcr/US9$f00911
22
centrifuged at 1000 rpm for ten minutes at ambient temperature
with the break. The supernatant was removed. The cells were
resuspended in five ml of Medium 1. The cell suspension was
transferred to a sterile bottle with 200 ml of Medium 1. The
200 ml cell suspension was placed in a 500 cm2 tissue culture
flask (T500). The liquid in the T500 was allowed to
equilibrate between chambers before placement in a horizontal
position in the incubator. The culture was placed in an
incubator at 37 C with a 5% CO2 atmosphere and a humidity
level of 100%. This was passage 2 of the culture.
3. Scale-up and Passaaing of T500'Culture Usina
Try-psinization
The -TSO0 culture reached a confluency level of 90% in
four days. On the fourth day, the T500 culture was
trypsinized and scaled-up in the following manner. The
supernatant medium was discarded. The culture surface was
washed with 25 ml of CMF-PBS. A volume of 25 ml of TE was
added to the flask. The flask was moved so that the TE
solution covered all three layers of culture surface. The
cells detached from the flask surface within five minutes.
After the cells detached, 50 ml of Medium 1 was added to the
flask. All of the surfaces were contacted with the medium by
moving the flask. The resultant cell suspension was poured
into a 200 ml conical centrifuge bottle. The cells were
pelleted by centrifugation at 1000 rpm for ten minutes at
ambient temperature with the brake. The supernatant was
discarded. The cells were resuspended in 5 to 15 ml of Medium
1. The cell suspension was placed in 800 ml (200 ml per new
T500 flask) of Medium 1. The cell suspension was mixed. A
volume of 200 ml of the cell suspension was added to each of
four T500 flasks. The liquid level in each flask was allowed
to equilibrate between chambers before placement in a
horizontal position in the incubator. The split ratio for
this passage was 1:4. This was passage 3. The culture was
passaged in this manner for passages 4 through 13. At passage
14, four T500 cultures were trypsinized in the manner given
above. The cell suspensions were pooled and placed in a
bottle containing 1.5 liters of Medium 1. This cell
CA 02475337 2004-08-06
wo 9WAN
23 PCT/U$98M911
suspension was added to a 6000 cm2 CELL FACTORYTM (CF) tissue
culture flask. The liquid level in the CF was allowed to
equilibrate between chambers before placement in a horizontal
position in the incubator.
4. Scale-up and Passaaina of CELL FACTORY7" tissue
culture flask Cultures
The CF culture reached an 80% confluency level in three
days. The trypsinization was performed in the following
manner for passage 15. The-1.5 liters of Medium 1 was drained
from the CF culture. The culture surfaces were washed with
500 (+/- 100) ml of CMF-PBS. After the wash, 250 (+/- 50) ml
of TE solution was added to the CF culture. The CF was moved
so that the TE solution covered each of the.surfaces. After
the cells detached from the surface, 500 ml of Medium 1 was
added to the CF. The CF was moved so that the Medium 1
contacted each of the surfaces. The resultant cell suspension
was aliquotted into four, 250 ml conical centrifuge bottles.
The cells were pelleted by centrifugation at 1000 rpm for ten
minutes with the brake on. The supernatant medium was
discarded from each centrifuge bottle. In each centrifuge
bottle, the cells were resuspended in 5 ml of Medium 1. The
cells suspensions were pooled into one centrifuge bottle. The
three remaining centrifuge bottles were washed with an
additional 5-10 ml of Medium 1 which was added to the pooled
cell suspension. This cell suspension was split equally among
six bottles containing 1.5 liters of Medium 1. Each of the
six 1.5 liter cell suspensions was added to a CF. The liquid
level of each CF was allowed to equilibrate among the chambers
before the CF was placed in a horizontal position in the
incubator. The culture was passaged in the same manner for
passage 16.
Passaging data are provided in Table 7.
TABLE 7: PASSAGING DATA
Passage Culture Time Confluency Source Flask Recipient
(Days) Level at Flask
PassLc e~~_
1 3 not viali T75
a licable
2 3 90= 95~ ~ 1 x T75 1 x T5100
CA 02475337 2004-08-06
WO 98/'3388G PCTIUS9S/00911
24
3 4 90-95% 1 x T500 4 x'T500
4 3 70% 1 x T500 4 x T500
4 90% 1 x T500 6 x T500 -
6 4 85% 1 x T500 8 x T500
7 3 70% 1 x T500 4 x T500
8 4 80% 1 x T500 4 x T500
9 4 95% 1 x T500 4 x T500
3 95% 1 x T500 6 x T500
11 3 70% 1 x T500 6 x T500
12 4 90% 1 x T500 6 x T500
13 4 70% 1 x T500 6 x T500.
14 4 90% 4 x T500 1 CF
3 80% 1 CF 6 x CF
16 5 80% 1 CF 6 x CF
5. Preparation of Cell Inoculum from CELL FACTQRY1'M
tissue culture flask Cultures to the Seed
Bioreactor
The CF cultures reached an 80% confluency level in five
days. Four of the six CF cultures were used to inoculate the
seed bioreactor as follows. The 1.5 liters of Medium 1 was
drained from the CF culture. The culture surfaces were washed
with 500 (+/- 100) ml of CMF-PBS. After the wash, 250 (+/-
50) ml of TE solution was added to the CF culture. The CF was
moved so that the'TE solution covered each of the surfaces.
Immediately after the cells detached from the surface 500 ml
of Medium 1 was added to the CF. The CF was moved so that the
Medium 1 contacted each of the surfaces. The resultant cell
suspension was aliquotted into four, 250 ml conical centrifuge ~
bottles. The cells were pelleted by centrifugation at 1000
rpm for ten minutes at ambient temperature with the brake on.
The supernatant medium was discarded from each centrifuge
bottle. In each centrifuge bottle, the cells were resuspended
in 5 ml of Medium 1. The cells suspensions were pooled into
one centrifuge bottle. The remaining three centrifuge bottles
were washed with an additional 5-10 ml of Medium 1 which was
added to the pooled cell suspension. The cell suspensions
from each of the four centrifuge bottles were then pooled
together, yielding a total volume of 50-100 ml. An additional
volume of Medium 1 was added to bring the total volume to 1000
ml. This was the cell inoculum. The total amount of cells in
the cell inoculum was 2.88 x 109 total cells and 2.84 x 109
CA 02475337 2004-08-06
= ,~
"9WM6 25 PCT/US98/00911
viable cells. The 1000 ml of cell inoculum was transfer-red to
a sterile Erlenmeyer flask and inoculated into the 30 liter
seed bioreactor which contained a total volume of 18 liters of
Medium 1 with 66 g of Cytodex 3 microcarriers.
B. Seed Bioreactor
1. Prebaration of Cytodex 3 Microcarriers for the 30
L Seed Bioreactor
One batch of 66 grams of Cytodex 3 microcarriers was
prepared in the following manner. The 66 grams of Cytodex 3
microcarriers was placed in a five liter, glass, Erlenmeyer
flask. Two liters of CMF-PBS with 0.2 ml of Tween 80 was
added. The microcarriers were allowed to swell at ambient
temperature for five hours and thirty minutes. After this
swelling period, the supernatant CMF-PBS was decanted from the
flask leaving behind the Cytodex 3 microcarrier slurry. The
Cytodex 3 microcarrier slurry was washed with two liters of
CMF-PBS then resuspended in CMF-PBS to a total volume of two
liters. The batch of Cytodex 3 was autoclaved in the five
liter flask at 121 C for three and a half hours on a liquids
cycle. The sterilized Cytodex 3 batch was used the next day
for the 30 L Seed Bioreactor.
On the day of the Cytodex 3 addition to the 30 L Seed
Bioreactor the following actions were performed. The
supernatant CMF-PBS was decanted from the five liter flask.
The microcarrier slurry was washed with two liters of Medium
1. After the wash, Medium 1 was added to the flask to a final
volume of two liters.
2. Prevaration of the 30 L Seed Bioreactor
The 30 L Seed Bioreactor which contained a spinfilter
was cleaned and steam sanitized. The bioreactor was
sterilized for fifty minutes at 121 C. One day prior to 293
cell inoculation, the 30 L Seed Bioreactor was filled with 18
liters_of Medium 1. The two liters containing 66 grams of the.
Cytodex 3'microcarriers with Medium 1 solution was added to
the 30 L Bioreactor. The 30 L Seed Bioreactor operating
conditions are listed in_Table 8.
CA 02475337 2004-08-06
WO 98/'338$6 PGT/US98/00911
26
TABLE 8: 30 L SEED BIOREACTOR OPERATING CONDITIONS
Operating Value Value Range
Condition
g 7.3 7.1-7.4
Temperature 37 C 35-38 C
Dissolved Oxygen 30% 20-140%
Bioreactor Pressure 0.1 bar 0.05-0.4 bar
Aeration Overla 180 lph 100-300 1 h
A itation 70 rpm 10-100 rpm
Spinfilter 70 rpm 10-100 rpm
3. Cultivation of 293 Cells in the 30 L Seed
Bioreactor
The 293 cells were propagated on the Cytodex 3
microcarriers for five days. The actual operating conditions
in the 30 L Seed Bioreactor during this time period are listed
in Table 9.
TABLE 9: OPERATING CONDITIONS IN THE 30 L SEED BIOREACTOR
Operating Actual Range Target
Condition
Tem erature 37-37.1 C 37 C
pH 7.2-7.6 7.3
Dissolved Oxygen 28-100% 30%
Pressure 0.1 bar 0.1 bar
Aeration Overla 200 1 h 200 1 h
~
Agitation 67-94 rpm 67-94 rpm
Spinfilter 69-87 rpm 69-87 rpm
On the fifth day of cultivation, 54% of the
microcarrier population contained greater than 50
cells/microcarrier, 38% contained 1-25 cells, 8% contained no
cells, and 8% were in aggregates of two microcarriers as
determined by examination of a sample under the microscope at
100X magnification. Results are provided in Table 10.
TABLE 10: RESULTS FROM MICROSCOPIC EXAM ON THE FIFTH DAY OF
CULTIVATION OF 293 CEL,LS ON CYTODEX 3 MICROCARRIERS IN THE 30L
BIOREACTOR
-Percentage of Total Microcarrier Population
>50 10-50 1-10 empty
cells/ cells/microcarr cells/microcarr microcarrier
microcarrier ier ier
54% 1.6% 17%
~~f 7~
CA 02475337 2004-08-06
WO 98/33886 27 PCT/US98/00911
B. Bead to Bead Transfer Procedure
1. Preparation of Cytodex 3 microcarriers for the
Production Bioreactor
One batch of 420 grams of Cytodex 3 microcarriers was
prepared in the following manner. The 420 grams of Cytodex 3
microcarriers was placed in a fifty liter carboy. A volume of
21.5 liters of CMF-PBS with 2.0 ml of Tween 80'was added. The
microcarriers were allowed to swell at ambient temperature for
17 hours. After this swelling period, the supernatant CN~-PBS
was removed from the carboy leaving behind the Cytodex 3
microcarrier slurry. The Cytodex 3 microcarrier slurry was
washed with 25 liters of CMF-PBS then resuspended in Cyl-F-PBS
to a total volume of 20 liters.
The 200 L bioreactor which contained a spinf-ilter was
cleaned and steam sanitized. The 20 liter Cytodex 3
microcarrier slurry was transferred to the bioreactor. Five
liters of CMF-PBS was used to wash out the 50 liter carboy and
transferred to the bioreactor. The bioreactor was sterilized
for 50 minutes at 123 C. The bioreactor was maintained at. 4 C
overnight. The next day, 120 liters of Medium I was added to
the 200 L bioreactor. The microcarrier solution was aaitated
at 90 rpm for ten minutes in the bioreactor. The volume was
brought doc%,_ to 55 liters by withdrawing liquid throug'rn the
spinfilter. An additional 110 liters of medium 1 was added to
the 200 L bioreactor. The microcarrier solution was agitated
at 90 rom for ten minutes in the bioreactor. The volume was
brought down to 55 liters by withdrawing liquid through the
spinfilter. Medium 1 was added to the bioreactor to bring the
volume to 125 iiters. The bioreactor's operating conditions
were set according to Table 11.
TABLE 11: BIOREACTOR OPERATING CONDITIONS
Operating Target Actual Range
Condit-ion
Temperature 37-37.1 C 36i-38 C
DH 7.3
Dissolved O en 30% .20-1000
Pressure 0_1 bar 0_05-0_5 bar
Aeration Overla 2500 2500 lph
Aitation 50-90 rr)m 50-90 _ m
Spinfilter Pm 60-100 rprj: _YW
* Trade-mark
CA 02475337 2004-08-06
WO 98J'33886 PCT/US98100911
28
The 200 L production bioreactor was ready to receive
the inoculum from the seed bioreactor.
2. Trygsinization of the Seed Bioreactor Culture
On the fifth day of cultivation of the 293 cells on the
Cytodex 3 microcarriers, the bead to bead transfer procedure
was performed. The serum and calcium levels of the medium in
the culture were reduced by perfusing 22 liters of Medium 2 at
a rate of 2 liters per minute using the spinfilter with a
constant bioreactor volume of 20 liters. Perfusion was
continued with 22 liters of Medium 3 at a perfusion rate of 2
liters per minute with a constant bioreactor volume of 20
liters. This reduced the serum and calcium levels further. .- ~
Medium 3 contained disodium ethylenediaminetetraacetate
dihydrate (EDTA) which chelates divalent cations such as
magnesium and calcium. A third round of perfusion was
performed using 33 liters of Medium 2 which was designed to
further reduce the serum and calcium levels and to reduce the
concentration of EDTA in the medium. At this point, the
medium was withdrawn through the spinfilter to reduce the
total culture volume to 15.5 liters. A volume of 480 ml of a
2.5% trypsin solution was added to the bioreactor in one
minute. By microscopic observation, eight minutes after the
addition of the trypsin solution, 90% of the cells had
'..~
detached from the microcarriers. At this point, four liters
of serum was added to the bioreactor in two and a half minutes
to inhibit the action of trypsin and to protect the cells from
shear during the transfer procedure to the Production
Bioreactor. The trypsinized cells and microcarriers were
transferred to the Production Bioreactor by pressure. The
transfer was achieved in eight minutes. Immediately after the
transfer, five liters of Medium 1 was added to the Seed
Bioreactor as a flush and transferred to the Production
Bioreactor by pressure. Operating conditions of the seed
bioreactor during the bead-bead transfer procedure are
provided in table 12.
CA 02475337 2004-08-06
WO 98J33886 29 PGT/U598100911
TABLE 12: OPERATING CONDITIONS OF THE SEED BIOREACTOR
DURING THE BEAD-BEAD TRANSFER PROCEDURE
Operating Actual Range Target
Condition
Temperature 37 C 37 C
pH 7.2-7.5 '7.3
Dissolved Oxygen 28-55% 30%
Pressure 0.1 bar 0.1 bar
Aeration Overlay 200 1 h 200 1 h
Agitation 89-94 rpm 89-94 rom
S infilter 86-87 rpm 86-87 rpm
C. Cultivation of 293 Cells in the 200 L Production
Bioreactor Before Infection
The 293 cells were propagated on the Cytodex 3
microcarriers for six days. The actual operating conditions
in the 200-L Production Bioreactor during this time period are
listed in Table 13.
Table 13: OPERATING CONDITIONS IN THE 200 L PRODUCTION
BIOREACTOR
Operating Actual Range Target
Condition
Temperature 37-37.8 C 37 C
pH 7.2-7.5 7.3
Dissolved Oxygen 43-160% 30%
Pressure 0.08 bar 0.1 bar
Aeration Overlay 2500 1 h 2500 lph
Agitation 69-74 rpm 69-74 rpm
S infilter 71-80 rpm 71-80 rpm
A total volume of 115 liters of Medium 1 was perfused
from days four through six. The rates were as follows; 24
liters was perfused in one hour on day four, 40 liters was
perfused in one hour on day five, and 50 liters was perfused
in one hour on day six. The oxygen uptake rate measured as
the decrease of the dissolved oxygen level (percent of air
saturation, % DO) per minute (% DO decrease/min) reached
1.65%/min on day six. Results from microscopic exam on the
sixth day of cultivation of 293 cells on Cytodex 3
microcarriers in the 200 L bioreactor are provided in Table
14.
CA 02475337 2004-08-06
WO 9W3886 PCT/U898/00911
TABLE 14: RESULTS FROM MICROSCOPIC EXAM ON THE SIxTH DAY OF
CULTIVATION OF 293 CELLS ON CYTODEX 3 MICROCARRIERS IN THE 200
L BIOREACTOR
Percentage of Total Microcarrier Population
> 50 10-50 1-10 empty
cells/ cells/ cells/ microcarriers
microcarrier microcarrier microcarrier
31% 23% 25% 21%
D. Infection of the 293 Cells in the 200 L Production
Bioreactor -
The bioreactor culture was inoculated with virus on day
6. The viral inoculum had been stored frozen at -80 C. A
volume of 45 ml of the viral inoculum, 2-2, was thawed in a
water bath at 20-25 C. The total amount of virus added to the
tank was 1.1 x 1013 viral
particles as measured by the
Resource Q HPLC assay. The viral inoculum was mixed and
placed in a bottle with one liter of Medium 4(293-1-R07
Dulbecco's modified.Eagle's medium with L-glutamine and sodium
bicarbonate (3.7 g/1)). The viral solution was filtered
through a Gelman Maxi Culture Capsule into a sterile five
liter addition flask. The viral suspension was added to the
200 L Production Bioreactor with sterile connections made via
the tubing welder. Production bioreactor operating conditions
after infection are provided in Table 15.
TABLE 15: PRODUCTION BIOREACTOR OPERATING CONDITIONS AFTER
INFECTION
Operating Actual Range Tar"get
Condition
Temperature 37-37.8 C 37 C
pH 7.08-7.23 7.4
Dissolved Oxygen 20-73% 30%
Pressure 0.08 bar 0.1 bar
Aeration Overlay 2500 1 h 2500 1 h
A itation 69-74 r m 69-74 rpm
S infilter 71-80 rpm 71-80 m
Three days after infection, 89% of the microcarriers
did not have attached cells and the oxygen uptake rate
measured was 0.53%/min. The total infected cell concentration
present in the supernatant broth was 1.0 x 106 cells/ml. The
total volume in the bioreactor was 162 liters. The bioreactor
CA 02475337 2004-08-06
WO 98/33886 31 PCT/US98/00911
was harvested at this time. -
E. Recovery Operations
A volume of 400 liters of the harvest recovery buffer
was prepared and filtered through a Pall Ultipor N66-"(0.2
micron pore size) and aliquotted into sterile vessels in the
following manner. Three aliquots of 210 liters, 130 liters,
and 50 liters were prepared. The volume of 210 liters was
used for the bioreactor wash and the fluidized bed column
operation. The 130 liter aliquot was used during the
microfiltration. The 50 liter aliquot was utilized during the
ultrafiltration process.
F. Separation of Cells from the Microcarriers usinc the
Fluidized Bed Colum.n
The fluidized bed was sanitized using a caustic
solution (0.1 N sodium hvdroxide). A T-fitting was connected
to the harvest port of the bioreactor. On one side of the T-
fitting, a sanitary hose (15.9 mm id) and vaive were connected
to the fluidized bed column. A peristaltic pump was placed on
this line (Watson Marlow Model 604S). The second side of the
T-fitting was connected to a sanitary hose (15.9 mm id) and
valve leading to the buffer tank. The outlet of the fluidized
bed column, through which the broth containing cells and virus
passed, was connected to a tank used as the microfiltration
recirculation vessel.
The broth from the bioreactor was passed through the
fluidized bed column at a target flow rate of 2-3 liters per
minute. The flow rate was controlled with the peristaltic
pump. Agitation was maintained in the bioreaccor. When the
bioreactor volume was less than 100 liters the spin filter was
turned off. When the bioreactor volume was less than 30
liters, the agitator was turned off. After the bioreactor
contents were processed through the fluidized bed column, the
bioreactor was washed with 90 liters of harvest recovery
buffer. This wash material was processed through the
fluidized bed column. At the end of the brocess, the
microcarriers remained in. the f lu-J.dized bed col.u_mr: and were
discarded. Data are provided in Table 16.
* Trade-mark
CA 02475337 2004-08-06
wo 9$/33886 rMosM"11
32
TABT.E 16= DATA FROM A FLUIDIZED BED COLUMN OPERATIQN
Time (minutes) Flow Rate Volume of Comments
(liters per Broth
minute) processed
(liters)
0 9.8 0 Start
2.9 20
60 2.9 160
70 2.9 160 Added 90 liters
of harvest
recovery buffer
to bioreactor
100 2.9 253 Finish
G. Microfiltration of the Microcarrier-clarified broth -
Lvsina the infected cells and BENZONASE'" endonuclease
treatment
The starting material for the microfiltration process
was the broth from the fluidized bed column that was clarified
of the microcarriers and contained cells and virus. During
the microfiltration step, the cells were lysed due to the
shear rate used, the broth was clarified of debris larger than
0.65 microns and the residual nucleic acids from the lysed
cells was digested by BENZONASE'" endonuclease (e.g., 0.5
million units per 200 L batch), an enzymatic preparation.
The microfiltration unit was a Prostak system
(Millipore). It contained a Durapore, 0.65 micron pore size,
hydrophilic, membrane (catalog number SK2P446E0) with a
surface area of 54 square feet. The feed and retentate lines
of the Prostak filter unit were connected to the
microfiltration recirculation vessel which contained the
microcarrier-clarified broth from the fluidized bed column. A
line used to feed the harvest recovery buffer into the
microfiltration recirculation vessel was connected. The
permeate line from the Prostak unit was connected to the
ultrafiltration recirculation vessel. The temperature of the
broth was maintazned in the range of 25-35 C. When the broth
feeding into the Prostak unit was reduced to a volume of 10 to
30 liters in the microfiltration recirculation vessel, 50
liters of the harvest recovery buffer was added to the vessel
and the microfiltration continued. This step was repeated-
CA 02475337 2004-08-06
WO 98133886 33 PCT/Us98/00911
once. The microfiltration continued until the volume in-the
microfiltration recirculation vessel was reduced to 10 to 30
liters. At this time, 0.5 million units of BENZONASE:M
endonuclease was added to the clarified both in the
ultrafiltration recirculation vessel. The contents of the
vessel were mixed well and the broth was held for two hours
before the ultrafiltration was started. Data are provided in
Table 17.
TABLE 17: DATA FROM A MICROFILTRATION OPER.ATION
Time Feed Permeate Inlet Retentat Permeate Broth
flow flowrate pressure e pressure volume Comments
rate (m3/hr) (mbar) pressure (mbar) processed
(m3/hr) (mbar} (liters)
0 9.6 0.17 1190 273 678 2
4 9.6 0.16 1206 278 664 12
20 9.6 0.16 1225 258 571 56
;- ~
36 9.6 0.15 1230 249 551 98
50 9.6 0.16 1225 244 532 138
63 0 176 Stop and
add 50
liters
of
buffer
71 9.6 0.18 1250 268 541 176 Start
a f ter
buffer
addition
81 9.6 0.18 1250 249 532 207
91 0 0 234 Stop and
add 50
liters
of
buffer
96 9.6 0.18 1250 253 522 237 Start
after
buffer
addition
102 9.6 0.18 1264 249 527 258
112 9.7 0.17 2284 249 524 286
118 299 Finish
H. Ultrafiltration of Broth to Concentrate the virus with
Diafiltration to Perform Buffer Excrianae
The starting material for the ultrafiltration process
in the ultrafiltration recirculation vessel was the
BENZONASEr endonuclease-treated, clarified broth from the
microfiltration permeate. .The ultrafiltration unit was a
Pellicon system (Millipore): It contained a 1 million nominal
CA 02475337 2004-08-06
WO 98/33886 PCT/ITS98/00921
34
molecular weight cut-off, Pellicon II-regenerated cellulose
membrane (catalog number P2C01MC05) with a surface area of 40
square feet. The feed and retentate lines of the Pellicon
unit were connected to the ultrafiltration recirculation
vessel. The ultrafiltration permeate line was connected to a
waste vessel. A vessel containing the harvest recovery buffer
(50 mM Tris base, 150 mM sodium chloride, 2 mM magnesium
chloride hexahydrate, and 2% sucrose) was connected to the
ultrafiltration recirculation vessel. When the
ultrafiltration retentate volume reached 5 to 10 liters, an
addition of 15 liters of the harvest recovery buffer was made
and the ultrafiltration was continued. This step was repeated
once. The ultrafiltration was continued until the retentate
volume was -less than 5 to 10 liters. The retentate from the
ultrafiltration contained the concentrated virus. The
retentate was collected from the Pellicon unit. A flush of 3
to 6 liters of the harvest recovery buffer was used to collect
all of the material from Pellicon unit. This flushed material
was added to the ultrafilter retentate broth. This was
filtered through a Millipore, Durapore, 0.45 micron pore size
filter (catalog number, CVHL71PP3) into a sterile bag. The
material in the sterile bag was stored frozen at -80 C. Data
are provided in Table 18.
TABLE 18: DATA FROM hN ULTRAFILTRATION OPERATION
Time Flow rate Inlet Retentate Permeate
(minutes) (liters pressure pressure pressure Comments
per (bar) (bar) (bar)
minute)
0 16.5 0.9 3.1 0.4 Start
16.5 0.9 3.1 0.4
39 17.3 0.9 3.1 0.4
86 17.3 0.9 3.1 0.4 Stop and
added
buffer
93 17.3 0.9 3.0 0.4 Start after
buffer
addition
100 Stop and
added
buffer
107 15.7 0.9 ,3.0 0.4 Start after
buffer
addition
CA 02475337 2004-08-06
WO 98133886 35 PCT/US98/00911
J119 15.7 10_8 3.0 0.4 Flughed
unit -
finish
I. General Comments
All 293 cell cultures in T75, T500 and CELL FACTORY'FM
tissue culture flasks were cultivated in Medium 1 in an
incubator at 37 C, 100% humidity and a 5% CO2 atmosphere. All
open operations were performed aseptically under a biosafety
(laminar flow) hood. Medium fills and additions were
performed through a 0.2 micron pore size, PALL Ultipor N66,
in-line filter installed on the feed port of the bioreactor
which was steam sterilized for 30 minutes at 121 C. All other
additions to the bioreactors were performed each using a
sterile Erienmeyer flask with PHARMED'm tubing that was
aseptically connected between the bioreactor and the addition
flask by a tubing welder. All buffers used in the recovery
.process were filtered through a 0.2 micron pore size, PALL
Ultipor N66, in-line filter (SLK7002NFP) installed on a port
of the receiving vessel. Note that for the microfiltration
operations either a hydrophil.ic or hydrophobic membrane can be
used.
r, .
Modifications and variations of this invention will be
apparent to those skilled in the art. The specific
embodiments described herein are offered by way of example
only, and the invention is not to be construed as limited
thereby.