Note: Descriptions are shown in the official language in which they were submitted.
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
BETA3-ADRENERGIC RECEPTOR AGONISTS
FIELD OF THE INVENTION
The present invention relates to a3 adrenergic receptor agonists and
uses thereof to treat diseases, conditions and/or disorders modulated by ~3
adrenergic receptor agonists.
BACKGROUND OF THE INVENTION
The disease diabetes mellitus is characterized by metabolic defects in
the production and utilization of carbohydrates that result in the failure to
maintain appropriate blood sugar levels. The results of these defects include,
inter alia, elevated blood glucose or hyperglycemia. Research in the treatment
of diabetes has centered on attempts to normalize fasting and postprandial
blood glucose levels. Current treatments include administration of exogenous
insulin, oral administration of drugs and dietary therapies.
Two major forms of diabetes mellitus are recognized. Type 1 diabetes, or
insulin-dependent diabetes mellitus (IDDM), is the result of an absolute
deficiency of insulin, the hormone that regulates carbohydrate utilization.
Type 2
diabetes, or non-insulin-dependent diabetes mellitus (NIDDM), often occurs
with
normal, or even elevated, levels of insulin and appears to be the result of
the
inability of tissues to respond appropriately to insulin. Most Type 2 diabetic
patients are also obese.
The compounds of the invention effectively lower blood glucose levels
when administered orally to mammals with hyperglycemia or diabetes.
Obesity constitutes a major health risk that leads to mortality and
incidence of Type 2 diabetes mellitus, hypertension, and dyslipidemia. In the
United States, more than 50% of the adult population is overweight, and almost
25% of the population is considered to be obese. The incidence of obesity is
increasing in the United States at a three-percent cumulative annual growth
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-2
rate. While the vast majority of obesity occurs in the United States and
Europe,
the prevalence of obesity is also increasing in Japan. Furthermore, obesity is
a
devastating disease which can also wreak havoc on an individual's mental
health and self-esteem, which can ultimately affect a person's ability to
interact
socially with others. Unfortunately, the precise etiology of obesity is
complex and
poorly understood, and societal stereotypes and presumptions regarding obesity
only tend to exacerbate the psychological effects of the disease. Because of
the
impact of obesity on society in general, much effort has been expended in
efforts to treat obesity, however, success in the long-term treatment and/or
prevention thereof remains elusive.
The compounds, pharmaceutical compositions, and combinations of the
present invention also reduce body weight, or decrease weight gain, when
administered to a mammal, including a human subject. The ability of the
compounds to affect weight gain is due to activation of a3 adrenergic
receptors
that stimulate the metabolism of adipose tissue.
~-Adrenergic agents have been generally classified into ~3~, ~2, and ~3
receptor-specific subtypes. Agonists of ~i-receptors promote the activation of
adenyl cyclase. Activation of ~~ receptors invokes an increase in heart rate
while
activation of ~2 receptors induces smooth muscle tissue relaxation that
produces
a drop in blood pressure and the onset of skeletal muscle tremors. Activation
of
~i3 receptors is known to stimulate lipolysis (e.g., the breakdown of adipose
tissue triglycerides into glycerol and fatty acids) and metabolic rate (energy
expenditure), thereby promoting the loss of fat mass. Accordingly, compounds
that stimulate ~i3 receptors are therefore useful as anti-obesity agents, and
can
be further used to increase the content of lean meat in edible animals. In
addition, compounds that are ~i3 receptor agonists have hypoglycemic activity,
however, the precise mechanism of this effect is presently unknown.
Until recently, (33 adrenergic receptors were believed to be located
predominantly in adipose tissue, however, such ~3 receptors are now known to
be present in such diverse tissues as the intestine, (J. Clin. Invest., 91,
344
(1993)) and the brain (Eur. J. Pharm., 219, 193 (1992)). Stimulation of X33
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-3
receptors has also been demonstrated to induce relaxation of smooth muscle in
the colon, trachea, and bronchi. See, for example, Life Sciences, 44, 1411
(1989), Br. J. Pharm., 112, 55 (1994), and Br. J. Pharmacol., 110, 1311
(1993).
Furthermore, stimulation of (33 receptors has also been found to induce
relaxation of histamine-contracted guinea pig ileum. See, for example, J.
Pharm.
Exp. Ther., 260, 1, 192 (1992).
The ~i3 receptor is also expressed in the human prostate (J. Clin. Invest.,
91, 344 (1993). Because stimulation of the ~3 receptor causes relaxation of
smooth muscles that have been shown to express the ~3 receptor, i.e.
intestinal
smooth muscle, one of ordinary skill in the art would also predict relaxation
of
prostate smooth muscle. Therefore, X33 agonists are useful in the treatment or
prevention of prostate disease.
U.S. Patent No. 5,977,124 discloses certain ~i3 adrenergic receptor
agonists that may be used in the treatment of, inter alia, hypoglycemia and
obesity.
U.S. Patent No. 5,776,983 discloses certain catecholamines as ~i3-
agonists.
U.S. Patent No. 5,030,640 discloses certain a-heterocyclic ethanol amino
alkyl indoles that may be used as growth promoters, bronchodilators, anti-
depressants, and anti-obesity agents.
U.S. Patent No. 5,019,578 discloses certain a-heterocyclic
ethanolamines that may be used as growth promoters.
U.S. Patent No. 4,478,849 discloses pharmaceutical compositions
comprising certain ethanolamine derivatives and methods of using such
compositions in the treatment of obesity and/or hyperglycaemia.
U.S. Patent No. 4,358,455 discloses certain heterocyclic compounds that
may be used for treating glaucoma and cardiovascular disease.
U.S. Patent No. 5,393,779 (EP 516 349 B1 ) discloses certain 2-
hydroxyphenethyl amines that may be used as anti-obesity and hypoglycemic
agents, as well as, other related utilities.
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
U.S. Patent No. 5,153,210 discloses certain heterocyclic compounds that
may be used as anti-obesity and anti-hyperglycaemic agents.
U.S. Patent No. 6,251,925 discloses biaryl compounds that may be used
for the treatment of diseases susceptible to amelioration by administration of
an
atypical beta-adrenoceptor agonist.
U.S. Publication No. 2002-0052392A1 (PCT Publication No. WO
02/32897) discloses certain heterocyclic (33-adrenergic receptor agonists that
may be used in the treatment of intestinal motility disorders, depression,
prostate disease, dyslipidemia, and airway inflammatory disorders, and in
increasing the content of lean meat in edible animals.
SUMMARY OF THE INVENTION
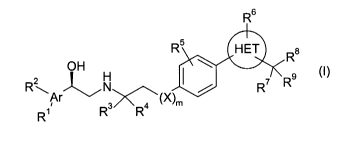
The present invention provides X33-adrenergic receptor agonists of
structural Formula (I)
Rs
R5 HET Ra
OH H I \ ~ Rs
R\Ar v N X / R
Rs ( )m
R'
wherein
Ar is phenyl, a 5- or 6-membere aromatic or non-aromatic heterocyclic
ring having 1 to 4 heteroatoms selected from O, S, or N, a benzene ring fused
to
a (C3-C8)cycloalkyl, a benzene ring fused to a 5- or 6-membered aromatic or
non-aromatic heterocyclic ring having 1 to 3 heteroatoms selected from O, S,
or
N, or a 5- or 6-membered aromatic or non-aromatic heterocyclic ring having 1
to
3 heteroatoms selected from O, S, or N fused to a 5- or 6-membered aromatic
or non-aromatic heterocyclic ring having 1 to 3 heteroatoms selected from O,
S,
or N (preferably, Ar is phenyl or pyridyl, more preferably pyridyl);
R' and R2 are each independently hydrogen, hydroxy, halogen, cyano,
nitro, -NR'aR2a, -NR'aS02R2a, -OR'a, -S02R2a, -CF3, (C3-C$)cycloalkyl, phenyl,
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-5
-NR'aCOR2a, -COR2a, or (C~-C6)alkyl optionally substituted with one or more
substituents selected from the group consisting of hydroxy, nitro, halogen,
and
cyano, where R'a and R2a are each independently hydrogen, (C3-C$)cycloalkyl,
phenyl optionally substituted with 1 to 3 substituents selected from the group
consisting of halo, (C~-C6)alkyl, and (C~-C6)alkoxy, or (C~-C6)alkyl
optionally
substituted with 1 to 3 substituents selected from the group consisting of
hydroxy, fluoro, -C02H, phenyl, and -NR'bR2b, where R'b and R2b are each
independently hydrogen, amino, amino(C~-C6)alkyl, aminoaryl, (C~-C6)alkyl
optionally substituted with one or more substituents selected from the group
consisting of hydroxy, (C~-C6)alkoxy, fluoro, amino, (C~-C6)alkylamino, and
acyl,
(C3-C$)cycloalkyl optionally substituted with one or more substituents
selected
from the group consisting of fluoro, alkyl, (C~-C6)alkoxy, hydroxy, amino,
aminoalkyl-, acyl, and amido, a 3- to 8-membered aromatic or non-aromatic
heterocyclic ring optionally substituted with one or more substituents
selected
from the group consisting of halogen, (C~-C6)alkyl, (C~-C6)alkoxy, hydroxy,
amino, aminoalkyl-, acyl, and amido; or R'b and R2b taken together with the
nitrogen to which they are attached form a 3-to 8-membered aromatic or non-
aromatic heterocyclic ring optionally containing 1 to 2 additional heteroatoms
selected from O, S, or N;
R3 and R4 are each, independently, hydrogen, or (C~-C6)alkyl optionally
substituted with 1 to 3 substituents selected from the group consisting of
hydroxy, (C~-C6)alkoxy, and fluoro;
R5 is hydrogen, (C~-C6)alkyl optionally substituted with 1 to 3 substituents
selected from the group consisting of hydroxy, (C~-C6)alkoxy, and fluoro;
R6 and R' are each independently hydrogen, halogen, or (C~-C6)alkyl
optionally substituted with one or more substituents selected from the group
consisting of hydroxy, (C~-C6)alkoxy, and fluoro;
R8 is -CONR'bRzb, -SOR'b, -SO2R'b, -S02NR'bR2b, -NR'bS02Rzb, or -
CO2R'b (preferably, R$ is is -CONR'bR2b);
R9 is hydrogen, (C~-C6)alkoxy, or (C~-C6)alkyl optionally substituted with
one or more substituents selected from the group consisting of fluoro,
hydroxy,
and (C~-C6)alkoxy;
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-6
X is -O-, -NH-, -NR'a-, -CH2-, -CH2CH2- or -CH20- (preferably, X is -O-);
mis0or1;and
HET is an aromatic heterocyclic ring selected from the group consisting
of imidazole, oxazole, pyrazole, and thiazole (preferably, HET is oxazole or
pyrazole, more preferably oxazole);
a pharmaceutically acceptable salt thereof, a prodrug of the compound or
the salt, or a solvate or hydrate of the compound, the salt or the prodrug.
In a preferred embodiment, compounds of Formula (IA) are provided.
R9
R Rs
N-
R5 ~ O
OH
H
R ~Ar~ N X /
R3 ( )m
R'
(IA)
where R', R2, R3, R4, R5, R6, R', Rs, R9, X and m are as defined above; a
pharmaceutically acceptable salt thereof, a prodrug of the compound or the
salt,
or a solvate or hydrate of the compound, the salt or the prodrug. In preferred
embodiments of the compound of Formula (IA), Ar is pyridyl (more preferably, 3-
pyridyl); R3, R4, R5, and R6 are hydrogen; R' and R9 are each independently
hydrogen, fluoro, or (C~-Cs)alkyl; Rs is -CONR'bR2b (where Rib and R2b are are
each independently selected from hydrogen, (C3-C6)cycloalkyl, or (C~-C6)alkyl
optionally substituted one or more fluoro, or R'b and R2b taken together with
the
nitrogen to which they are attached form a 4- to 6-membered non-aromatic
heterocyclic ring optionally containing one additional heteroatom selected
from
O and N, more preferably R'b and R2b are are each independently selected from
hydrogen or (C~-C6)alkyl, most preferably R'b and R2b are are each
independently selected from hydrogen or methyl); X is -O- and m is 1; a
pharmaceutically acceptable salt thereof, a prodrug of the compound or the
salt,
or a solvate or hydrate of the compound, the salt or the prodrug.
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-7
Preferred compounds of Formula (IA) include: 2-[4-(4-{2-[2(R)-hydroxy-
2-(6-methyl-pyridin-3-yl)-ethylamino]-ethoxy}-phenyl)-oxazol-2-yl)-N,N-
dimethyl-acetamide; 2-(4-{4-[2-(2(R)-hydroxy-2-pyridin-3-yl-ethylamino)-
ethoxy)-
phenyl}-oxazol-2-yl)-N,N-dimethyl-acetamide; N,N-diethyl-2-(4-{4-[2-(2(R)-
hydroxy-2-pyridin-3-yl-ethylamino)-ethoxy]-phenyl}-oxazol-2-yl)-acetamide; 2-
[4-
(4-{2-[2-(6-chloro-pyridin-3-yl)-2(R)-hydroxy-ethylamino]-ethoxy}-phenyl)-
oxazol-
2-yl]-N-ethyl-N-(2,2,2-trifluoro-ethyl)-acetamide; 2-[4-(4-{2-[2-(6-chloro-
pyridin-3-
yl)-2(R)-hydroxy-ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-diisopropyl-
acetamide; 2-[4-(4-{2-[2-(6-chloro-pyridin-3-yl)-2(R)-hydroxy-ethylamino]-
ethoxy}-phenyl)-oxazol-2-yl]-N,N-dimethyl-isobutyramide; 2-(4-{4-[2-(2(R)-
hydroxy-2-pyridin-3-yl-ethylamino)-ethoxy]-phenyl}-oxazol-2-yl)-N,N-dimethyl-
isobutyramide; 2-(4-{4-[2-(2(R)-hydroxy-2-pyridin-3-yl-ethylamino)-ethoxy]-
phenyl}-oxazol-2-yl)-N,N-dimethyl-butyramide; 2-[4-(4-{2-[2(R)-hydroxy-2-(6-
methyl-pyridin-3-yl)-ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-dimethyl-
propionamide; 2-[4-(4-{2-[2(R)-hydroxy-2-(6-methyl-pyridin-3-yl)-ethylamino]-
ethoxy}-phenyl)-oxazol-2-yl]-N,N-dimethyl-butyramide; and 2-[4-(4-{2-[2(R)-
hydroxy-2-(6-methyl-pyridin-3-yl)-ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-
dimethyl-isobutyramide; a pharmaceutically acceptable salt thereof, a prodrug
of
the compound or the salt, or a solvate or hydrate of the compound, the salt or
the prodrug.
More preferred compounds of Formula (IA) include: 2-[4-(4-{2-[2(R)-
hydroxy-2-(6-methyl-pyridin-3-yl)-ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-
dimethyl-acetamide; 2-(4-{4-[2-(2(R)-hydroxy-2-pyridin-3-yl-ethylamino)-
ethoxy]-
phenyl}-oxazol-2-yl)-N,N-dimethyl-acetamide; N,N-diethyl-2-(4-{4-[2-(2(R)-
hydroxy-2-pyridin-3-yl-ethylamino)-ethoxy]-phenyl}-oxazol-2-yl)-acetamide; 2-
[4-
(4-{2-[2(R)-hydroxy-2-(6-methyl-pyridin-3-yl)-ethylamino]-ethoxy}-phenyl)-
oxazol-
2-yl]-N,N-dimethyl-propionamide; 2-[4-(4-{2-[2(R)-hydroxy-2-(6-methyl-pyridin-
3-
yl)-ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-dimethyl-butyramide; and 2-[4-
(4-{2-[2(R)-hydroxy-2-(6-methyl-pyridin-3-yl)-ethylamino]-ethoxy}-phenyl)-
oxazol-
2-yl]-N,N-dimethyl-isobutyramide; a pharmaceutically acceptable salt thereof,
a
prodrug of the compound or the salt, or a solvate or hydrate of the compound,
the salt or the prodrug.
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
_g_
In another preferred embodiment, compounds of Formula (IA-1 ) are
provided.
R9
R Ra
N-
R5 ~ O
OH \ ~(
H ~ Rs
\ N i!~ (X)m /
R3 Ra
R' N
(IA-1 )
wherein R' is hydrogen, hydroxy, halogen, (C~-C6)alkyl, or (C~-
C6)alkoxy (preferably, R' is hydrogen, halogen or (C~-C6)alkyl); R3 and R4 are
hydrogen; R5, R6, R' and R9 are each independently hydrogen or (C~-C6)alkyl
optionally substituted with one or more fluoro substituents (preferably, R5,
R6,
R' and R9 are all hydrogen); R$ is -CONR'bR2b, where R'b and R2b are each
independently selected from hydrogen, (C3-C6)cycloalkyl or (C~-C6)alkyl
optionally substituted one or more fluoro, or R'a and R'b taken together with
the nitrogen to which they are attached form a 4- to 6-membe~ed non-aromatic
heterocyclic ring optionally containing one additional heteroatom selected
from
O or N (preferably, R'a and R'b are each independently hydrogen or (C~-
C6)alkyl, more preferably R'a and R'b are each independently hydrogen or
methyl); X is -O-; and m is 1; a pharmaceutically acceptable salt thereof, a
prodrug of the compound or the salt, or a solvate or hydrate of the compound,
the salt or the prodrug.
Preferred compounds include 2-[4-(4-{2-[2(R)-hydroxy-2-(6-methyl-
pyridin-3-yl)-ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-dimethyl-acetamide;
and 2-[4-(4-{2-[2(R)-hydroxy-2-(6-methyl-pyridin-3-yl)-ethylamino]-ethoxy}-
phenyl)-oxazol-2-yl]-N-methyl-acetamide; or a pharmaceutically acceptable
salt thereof, a solvate or hydrate of the compound or the salt.
In yet another preferred embodiment, compounds of Formula (IB) are
provided.
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
_g_
R9
R \ / Ra
N-N
R
OH ~ \ ~~Rs
R\Ar v N X /
R3 ( )m
R'
(IB)
where R', R2, R3, R4, R5, R', R8, R9, X and m are as defined above; a
pharmaceutically acceptable salt thereof, a prodrug of the compound or the
salt,
or a solvate or hydrate of the compound, the salt or the prodrug. In preferred
embodiments of the compound of Formula (IB), Ar is pyridyl (more preferably, 3-
pyridyl); R3, R4, R5, and Rs are hydrogen; R' and R9 are each independently
hydrogen, fluoro, or (C~-Cs)alkyl; R$ is -CONR'bR2b (where R'b and R2b are are
each independently selected from hydrogen, (C3-Cs)cycloalkyl, or (C~-Cs)alkyl
optionally substituted one or more fluoro, or R'b and R2b taken together with
the
nitrogen to which they are attached form a 4- to 6-membered non-aromatic
heterocyclic ring optionally containing one additional heteroatom selected
from
O and N, more preferably R'b and R2b are are each independently selected from
hydrogen or (C~-Cs)alkyl, most preferably R'b and R2b are are each
independently selected from hydrogen or methyl); X is -O- and m is 1; a
pharmaceutically acceptable salt thereof, a prodrug of the compound or the
salt,
or a solvate or hydrate of the compound, the salt or the prodrug.
Preferred compounds of Formula (IB) include: 2-(3-{4-[2-(2(R)-hydroxy-2-
pyrid in-3-yl-ethylamino)-ethoxy]-phenyl}-pyrazol-1-yl)-N, N-dimethyl-
acetamide;
N-ethyl-2-(3-{4-[2-(2(R)-hydroxy-2-pyridin-3-yl-ethylamino)-ethoxy]-phenyl}-
pyrazol-1-yl)-N-methyl-acetamide; 2-(3-{4-[2-(2(R)-hydroxy-2-pyridin-3-yl-
ethylamino)-ethoxy]-phenyl}-pyrazol-1-yl)-1-morpholin-4-yl-ethanone; 2-(3-{4-
[2-
(2(R)-hydroxy-2-pyridin-3-yl-ethylamino)-ethoxy]-phenyl}-pyrazol-1-yl)-1-
pyrrolidin-1-yl-ethanone; and N-cyclopentyl-2-(3-{4-[2-(2(R)-hydroxy-2-pyridin-
3-
yl-ethylamino)-ethoxy]-phenyl}-pyrazol-1-yl)-acetamide; a pharmaceutically
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-10
acceptable salt thereof, a prodrug of the compound or the salt, or a solvate
or
hydrate of the compound, the salt or the prodrug.
In another aspect of the present invention, a pharmaceutical composition
is provided which comprises (1 ) a compound of the present invention, and (2)
a
pharmaceutically acceptable excipient, diluent, or carrier. The pharmaceutical
composition may further comprise an additional pharmaceutical agent. A
preferred pharmaceutical agent is an anti-obesity agent selected from the
group
consisting of an apolipoprotein-B secretion/microsomal triglyceride transfer
protein (apo-B/MTP) inhibitor, a MCR-4 agonist, a cholecystokinin-A (CCK-A)
agonist, a monoamine reuptake inhibitor (e.g., sibutramine), a sympathomimetic
agent, a cannabinoid receptor antagonist (e.g., rimonabant (SR-141,716A)), a
dopamine agonist (e.g., bromocriptine), a melanocyte-stimulating hormone
receptor analog, a 5HT2c agonist, a melanin concentrating hormone antagonist,
leptin (the OB protein), a leptin analog, a leptin receptor agonist, a galanin
antagonist, a lipase inhibitor (e.g., tetrahydrolipstatin, i.e. orlistat), an
anorectic
agent (e.g., a bombesin agonist), a Neuropeptide-Y antagonist, a thyromimetic
agent, a dehydroepiandrosterone or an analog thereof, a glucocorticoid
receptor
agonist or antagonist, an orexin receptor antagonist, a glucagon-like peptide-
1
receptor agonist, a ciliary neurotrophic factor (e.g., AxokineT""), a human
agouti-
related protein (AGRP), a ghrelin receptor antagonist, a histamine 3 receptor
antagonist or inverse agonist, and a neuromedin U receptor agonist.
In yet another embodiment of the present invention, a method for treating
a disease, condition or disorder modulated by a ~3 adrenergic receptor agonist
in animals that includes the step of administering to an animal in need of
such
treatment a therapeutically effective amount of a compound of the present
invention (or a pharmaceutical composition thereof). Diseases, conditions,
and/or disorders modulated by X33 adrenergic agonists include weight loss
(e.g.,
increased energy expenditure), obesity, diabetes, irritable bowel syndrome,
inflammatory bowel disease, esophagitis, duodenitis, Crohn's disease,
proctitis,
asthma, intestinal motility disorder, ulcer, gastritis, hypercholesterolemia,
cardiovascular disease, urinary incontinence, depression, prostate disease,
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-11
dyslipidemia, fatty liver, and airway inflammatory disorder. Accordingly, the
compounds of the present invention may be used in the manufacture of a
medicament for treating a disease, condition or disorder which is modulated by
a ~3 adrenergic receptor antagonist.
Compounds of the present invention may be administered in combination
with at least one additional pharmaceutical agent described hereinbelow.
Preferred pharmaceutical agents include anti-obesity agents (described above).
The combination therapy may be administered as (a) a single
pharmaceutical composition which comprises a compound of the present
invention, at least one additional pharmaceutical agent described above and a
pharmaceutically acceptable excipient, diluent, or carrier; or (b) two
separate
pharmaceutical compositions comprising (i) a first composition comprising a
compound of the present invention and a pharmaceutically acceptable
excipient, diluent, or carrier, and (ii) a second composition comprising at
least
one additional pharmaceutical agent described above and a pharmaceutically
acceptable excipient, diluent, or carrier. The pharmaceutical compositions
may be administered simultaneously or sequentially and in any order.
In yet another aspect of the present invention, a pharmaceutical kit is
provided for use by a consumer to treat diseases, conditions and/or disorders
modulated by a3 adrenergic receptor agonists in an animal. The kit comprises
a) a suitable dosage form comprising a compound of the present invention; and
b) instructions describing a method of using the dosage form to treat diseases
linked to the modulation of the X33 adrenergic receptor.
In yet another embodiment of the present invention is a pharmaceutical
kit comprising: a) a first dosage form comprising (i) a compound of the
present
invention and (ii) a pharmaceutically acceptable carrier, excipient or
diluent; b) a
second dosage form comprising (i) an additional pharmaceutical agent
described above, and (ii) a pharmaceutically acceptable carrier, excipient or
diluent; and c) a container.
In yet another aspect of the present invention, an intermediate
compound having the Formula (I-a) is provided.
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-12
R$
N
\ ~ O
R'HN~
O Re
(I-a)
wherein
R' is hydrogen or an amino-protecting group;
R5 is hydrogen, (C~-C6)alkyl optionally substituted with 1 to 3 substituents
selected from the group consisting of hydroxy, (C~-C6)alkoxy, and fluoro; and
R8 IS -CONR'bR2b, -SOR'b, -SO2R'b, -S02NR'bR2b, -NR~bS02R2b, Or
-CO2R'b, where R'b and R2b are each independently hydrogen, amino,
amino(C~-C6)alkyl, aminoaryl, (C~-C6)alkyl optionally substituted with one or
more substituents selected from the group consisting of hydroxy, (C~-
C6)alkoxy,
fluoro, amino, (C~-C6)alkylamino, and acyl, (C3-CS)cycloalkyl optionally
substituted with one or more substituents selected from the group consisting
of
fluoro, alkyl, (C~-C6)alkoxy, hydroxy, amino, aminoalkyl-, acyl, and amido, a
3- to
8-membered aromatic or non-aromatic heterocyclic ring optionally substituted
with one or more substituents selected from the group consisting of halogen,
(C1-C6)alkyl, (C~-C6)alkoxy, hydroxy, amino, aminoalkyl-, acyl, and amido; or
R'b
and R2b taken together with the nitrogen to which they are attached form a 3-
to
8-membered aromatic or non-aromatic 3-to 8-membered heterocyclic ring
optionally containing 1 to 2 additional heteroatoms selected from O, S, or N.
Definitions
As used herein, the term "alkyl" refers to a hydrocarbon radical of the
general formula C~H2~+~. The alkane radical may be straight or branched. For
example, the term "(C,-C6)alkyl" refers to a monovalent, straight, or branched
aliphatic group containing 1 to 6 carbon atoms (e.g., methyl, ethyl, n-propyl,
i-
propyl, n-butyl, i-butyl, s-butyl, t-butyl, n-pentyl, 1-methylbutyl, 2-
methylbutyl, 3-
methylbutyl, neopentyl, 3,3-dimethylpropyl, hexyl, 2-methylpentyl, and the
like).
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-13
Unless specified otherwise, the alkane radical may be unsubstituted or
substituted with one or more substituents (generally, one to three
substituents
except in the case of halogen substituents such as perchloro or
perfluoroalkyls)
selected from the group of substituents listed below in the definition for
"substituted." For example, "halo-substituted alkyl" refers to an alkyl group
substituted with one or more halogen atoms (e.g., fluoromethyl,
difluoromethyl,
trifluoromethyl, perfluoroethyl, and the like). Similarly, the alkyl portion
of an
alkoxy, alkylamino, dialkylamino, and alkylthio group has the same definition
as
above.
The term "cycloalkyl" refers to nonaromatic rings that are fully
hydrogenated and may exist as a single ring, bicyclic ring or a spiro-fused
ring.
For example, cycloalkyl includes groups such as cyclopropyl, cyclobutyl,
cyclopentyl, cyclohexyl, norbornyl (bicyclo[2.2.1]heptyl),
bicyclo[2.2.2]octyl, and
the like. Generally, the cycloalkyl ring is a 3 to 8 membered ring. Unless
specified otherwise, the cycloalkyl may be optionally substituted with one of
more substituents (typically, one to three substituents) selected from the
group
of substituents listed below in the definition for "substituted." The
cycloalkyl
group may be attached to the chemical entity or moiety by any one of the
carbon atoms within the carbocyclic ring system. A cycloalkyl fused to a
benzene ring refers to groups such as indanyl.
The term "non-aromatic heterocyclic ring" (also referred to as
"heterocycle") refers to nonaromatic rings that are either partially or fully
hydrogenated and may exist as a single ring, bicyclic ring or a spiro-fused
ring.
Partially saturated or fully saturated heterocyclic rings include groups such
as
epoxy, aziridinyl, tetrahydrofuranyl, dihydrofuranyl, dihydropyridinyl,
pyrrolidinyl,
N-methylpyrrolidinyl, imidazolidinyl, imidazolinyl, piperidinyl, piperazinyl,
pyrazolidinyl, 2H-pyranyl, 4H-pyranyl, 2H-chromenyl, oxazinyl, morpholino,
thiomorpholino, tetrahydrothienyl, tetrahydrothienyl 1,1-dioxide, and the
like.
Generally, the heterocycle is 3 to 8 membered ring containing 1 to 3
heteroatoms selected from oxygen, sulfur and nitrogen. Unless specified
otherwise, the non-aromatic heterocyclic groups may be optionally substituted
with one of more substituents (typically, one to three substituents) selected
from
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-14
the group of substituents listed below in the definition for "substituted." A
heterocyclic ring that is fused to an aryl group includes groups such as 2,3-
dihydrobenzofuranyl, 2,3-dihydroindolyl, 2,3-dihydrobenzothiophenyl, 2,3-
dihydrobenzothiazolyl, etc. The heterocyclic group may be attached to the
chemical entity or moiety by any one of the atoms within the heterocyclic ring
system.
The term "aryl" refers to aromatic moieties having single (e.g., phenyl) or
fused ring system (e.g., naphthalene, anthracene, phenanthrene, etc.). Unless
indicated otherwise, the aryl groups may be unsubstituted or substituted with
one or more substituents (preferably no more than three substituents) selected
from the group of substituents listed below in the definition for
"substituted."
Substituted aryl groups include a chain of aromatic moieties (e.g., biphenyl,
terphenyl, phenylnaphthalyl, etc.) The aryl group may be attached to the
chemical entity or moiety by any one of the carbon atoms within the aromatic
ring system. Preferred aryl substituents are halogens (F, CI, Br or I,
preferably F
or CI), (C~-C4)alkoxy, (C~-C4)alkyl, halo-substituted(C~-C4)alkyl (e.g., CH2F,
CHF2 and CF3) and cyano. An aryl group fused to a cycloalkyl group includes
groups such as indanyl. Similarly, the aryl portion (i.e., aromatic moiety) of
an
aroyl or aroyloxy (i.e., (aryl)-C(O)-O-) has the same definition as above.
The term "aromatic heterocyclic ring" or "heteroaryl" refers to aromatic
moieties containing at least one heteratom (e.g., oxygen, sulfur, nitrogen or
combinations thereof) within the aromatic ring system (e.g., pyrrolyl,
pyridyl,
pyrazolyl, indolyl, indazolyl, thienyl, furanyl, benzofuranyl, oxazolyl,
oxadiazolyl,
imidazolyl, tetrazolyl, triazinyl, pyrimidyl, pyrazinyl, thiazolyl, purinyl,
benzimidazolyl, quinolinyl, isoquinolinyl, benzothiophenyl, benzoxazolyl,
etc.).
The heteroaromatic moiety may consist of a single or fused ring system. A
typical single heteroaryl ring is a 5- to 6-membered ring containing one to
three
heteroatoms selected from oxygen, sulfur and nitrogen and a typical fused
heteroaryl ring system is a 9- to 10-membered ring system containing one to
four heteroatoms selected from oxygen, sulfur and nitrogen. Unless specified
otherwise, the heteroaryl groups may be unsubstituted or substituted with one
or
more substituents (preferably no more than three substituents) selected from
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-15
the group of substituents listed below in the definition for "substituted."
The
heteroaryl group may be attached to the chemical entity or moiety by any one
of
the atoms within the aromatic ring system (e.g., imidazol-1-yl, imidazol-2-yl,
imidazol-4-yl, imidazol-5-yl, pyrid-2-yl, pyrid-3-yl, pyrid-4-yl, pyrid-5-yl,
or pyrid-6-
yl). Similarly, the heteroaryl portion (i.e., heteroaromatic moiety) of a
heteroaroyl
(i.e., (heteroaryl)-C(O)-O-) has the same definition as above.
The term "acyl" refers to alkyl, partially saturated or fully saturated
cycloalkyl, partially saturated or fully saturated heterocycle, aryl, and
heteroaryl
substituted carbonyl groups. For example, acyl includes groups such as (C1-
C6)alkanoyl (e.g., formyl, acetyl, propionyl, butyryl, valeryl, caproyl, t-
butylacetyl,
etc.), (C3-C6)cycloalkylcarbonyl (e.g., cyclopropylcarbonyl,
cyclobutylcarbonyl,
cyclopentylcarbonyl, cyclohexylcarbonyl, etc.), heterocyclic carbonyl (e.g.,
pyrrolidinylcarbonyl, pyrrolid-2-one-5-carbonyl, piperidinylcarbonyl,
piperazinylcarbonyl, tetrahydrofuranylcarbonyl, etc.), aroyl (e.g., benzoyl)
and
heteroaroyl (e.g., thiophenyl-2-carbonyl, thiophenyl-3-carbonyl, furanyl-2-
carbonyl, furanyl-3-carbonyl, 1 H-pyrroyl-2-carbonyl, 1 H-pyrroyl-3-carbonyl,
benzo[b]thiophenyl-2-carbonyl, etc.). In addition, the alkyl, cycloalkyl,
heterocycle, aryl and heteroaryl portion of the acyl group may be any one of
the
groups described in the respective definitions above. Unless indicated
otherwise, the acyl group may be unsubstituted or optionally substituted with
one of more substituents (typically, one to three substituents) selected from
the
group of substituents listed below in the definition for "substituted."
The term "substituted" specifically envisions and allows for one or more
substitutions that are common in the art. However, it is generally understood
by
those skilled in the art that the substituents should be selected so as to not
adversely affect the pharmacological characteristics of the compound or
adversely interfere with the use of the medicament. Those skilled in the art
will
also appreciate that certain substitutions may be inherently unstable and
therefore do not form a part of this invention. Suitable substituents for any
of
the groups defined above include (C~-C6)alkyl, partially or fully saturated
(C3-
C~)cycloalkyl, (C2-C6)alkenyl, aryl, heteroaryl, partially or fully saturated
3- to 6-
membered heterocycle, halo (e.g., chloro, bromo, iodo and fluoro), cyano,
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-16
hydroxy, (C~-C6)alkoxy, aryloxy, sulfhydryl (mercapto), (C~-C6)alkylthio,
arylthio,
amino, mono- or di-(C~-C6)alkyl amino, quaternary ammonium salts, amino(C~-
C6)alkoxy, aminocarboxylate (i.e., -NH-C(O)-O-(C~-C6)alkyl), N-(C~-
C6)alkylaminocarboxylate, hydroxy(C~-C6)alkylamino, amino(C~-C6)alkylthio,
cyanoamino, formamido, acylamino (e.g., acetamido and benzamido), N-(C~-
C6)alkyl-acylamino (e.g., N-methylacetamido), nitro, (C~-C6)carbamyl, keto
(oxy),
acyl, (C~-C6)alkoxycarbonyl, aryloxycarbonyl, (C~-C6)carboxy, glycolyl,
glycyl,
hydrazino, guanyl, sulfamyl, sulfonyl, sulfinyl, thio(C~-C6)carbonyl, thio(C~-
C6)carboxy, and combinations thereof. In the case of substituted combinations,
such as "substituted aryl(C~-C6)alkyl", either the aryl or the alkyl group may
be
substituted, or both the aryl and the alkyl groups may be substituted with one
or
more substituents (typically, one to three substituents except in the case of
perhalo substitutions). An aryl substituted carbocyclic or heterocyclic group
may
be a fused ring (e.g., indanyl, dihydrobenzofuranyl, dihydroindolyl, etc.). A
cycloalkyl substituted carbocyclic or heterocyclic group may be a spiro-fused
ring.
The term "solvate" refers to a molecular complex of a compound of the
present invention with one or more solvent molecules. Such solvent
molecules are those commonly used in the pharmaceutical art, which are
known to be innocuous to the recipient, e.g., water, ethanol, and the like.
The
term "hydrate" refers to the complex where the solvent molecule is water.
The term "protecting group" or "Pg" refers to a substituent that is
commonly employed to block or protect a particular functionality while
reacting
other functional groups on the compound. For example, an "amino-protecting
group" is a substituent attached to an amino group that blocks or protects the
amino functionality in the compound. Suitable amino-protecting groups include
acetyl, trifluoroacetyl, t-butoxycarbonyl (BOC), benzyloxycarbonyl (CBz) and 9-
fluorenylmethylenoxycarbonyl (Fmoc). Similarly, a "hydroxy-protecting group"
refers to a substituent of a hydroxy group that blocks or protects the hydroxy
functionality. Suitable protecting groups include acetyl and silyl. A "carboxy-
protecting group" refers to a substituent of the carboxy group that blocks or
protects the carboxy functionality. Common carboxy-protecting groups include
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-17
-CH2CH2S02Ph, cyanoethyl, 2-(trimethylsilyl)ethyl, 2-(trimethylsilyl)ethoxy-
methyl, 2-(p-toluenesulfonyl)ethyl, 2-(p-nitrophenylsulfenyl)ethyl,
2-(diphenylphosphino)-ethyl, nitroethyl and the like. For a general
description of
protecting groups and their use, see T. W. Greene, Protective Groups in
Organic Synthesis, John Wiley & Sons, New York, 1991.
The phrase "therapeutically effective amount" means an amount of a
compound of the present invention that (i) treats or prevents the particular
disease, condition, or disorder, (ii) attenuates, ameliorates, or eliminates
one or
more symptoms of the particular disease, condition, or disorder, or (iii)
prevents
or delays the onset of one or more symptoms of the particular disease,
condition, or disorder described herein.
The term "animal" refers to humans (male and female), companion
animals (e.g., dogs, cats and horses), food-source animals, zoo animals,
marine
animals, birds and other similar animal species. "Edible animals" refers to
food-
source animals such as cows, pigs, sheep and poultry.
The phrase "pharmaceutically acceptable" indicates that the substance
or composition is compatible chemically and/or toxicologically with the other
ingredients comprising a formulation and/or the animal being treated
therewith.
The phrase "modulated by a a3 adrenergic receptor" or "modulation of a
~i3 adrenergic receptor" refers to the activation or deactivation of ~3
adrenergic
receptors. For example, a X33 adrenergic receptor ligand may act as an
agonist,
partial agonist, inverse agonist, antagonist, partial antagonist, and the
like.
The term "agonist" refers to both full and partial agonists.
The terms "treating", "treat", or "treatment" embrace both preventative,
i.e., prophylactic, and palliative treatment.
The term "compounds of the present invention" (unless specifically
identified otherwise) refer to compounds of Formula (I), (IA), (IA-1 ), and
(IB)
prodrugs thereof, pharmaceutically acceptable salts of the compounds, and/or
prodrugs, and hydrates or solvates of the compounds, salts, and/or prodrugs,
as
well as, all stereoisomers (including diastereoisomers and enantiomers),
tautomers and isotopically labeled compounds.
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-18
DETAILED DESCRIPTION
The present invention provides compounds and pharmaceutical
formulations thereof that are useful in the treatment of diseases, conditions
and/or disorders modulated by X33 adrenergic receptor agonists.
Compounds of the present invention may be synthesized by synthetic
routes that include processes analogous to those well-known in the chemical
arts, particularly in light of the description contained herein. The starting
materials are generally available from commercial sources such as Aldrich
Chemicals (Milwaukee, WI) or are readily prepared using methods well known to
those skilled in the art (e.g., prepared by methods generally described in
Louis
F. Fieser and Mary Fieser, Reagents for Organic Synthesis, v. 1-19, Wiley, New
York (1967-1999 ed.), or Beilsteins Handbuch der organischen Chemie, 4, Aufl.
ed. Springer-Verlag, Berlin, including supplements (also available via the
Beilstein online database)).
For illustrative purposes, the reaction schemes depicted below provide
potential routes for synthesizing the compounds of the present invention as
well
as key intermediates. For a more detailed description of the individual
reaction
steps, see the Examples section below. Those skilled in the art will
appreciate
that other synthetic routes may be used to synthesize the inventive compounds.
Although specific starting materials and reagents are depicted in the schemes
and discussed below, other starting materials and reagents can be easily
substituted to provide a variety of derivatives and/or reaction conditions. In
addition, many of the compounds prepared by the methods described below
can be further modified in light of this disclosure using conventional
chemistry
well known to those skilled in the art.
In the preparation of compounds of the present invention, protection of
remote functionality (e.g., primary or secondary amine) of intermediates may
be
necessary. The need for such protection will vary depending on the nature of
the remote functionality and the conditions of the preparation methods.
Suitable
amino-protecting groups (NH-Pg) include acetyl, trifluoroacetyl, t-
butoxycarbonyl
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-19
(BOC), benzyloxycarbonyl (CBz) and 9-fluorenylmethyleneoxycarbonyl (Fmoc).
The need for such protection is readily determined by one skilled in the art.
For
a general description of protecting groups and their use, see T. W. Greene,
Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
Scheme I illustrates one means for preparing a compound of the present
invention where HET is an oxazole.
O O R8
\ Br ~Rs N=\
HzN \ O
\ v
Me0 Rs
(1 a) Me0 R5 (1 b)
R8 ' Ra
NHPg N
O MsO~ O
I \
PgNH~
O R5 (1d) HO
R (1
Ra ~ Rs
O
~ N-
N~RWA~ O
\ ~ O Rz' (Q) OH ~ \ \
~ 'H
HzN~ I R~~Ar~N~O Rs
O RS Rz
(1e) (~)
Scheme 1
In Scheme 1 above, an a-bromoketone (1a) is cyclocondensed with an
appropriately substituted amide to provide oxazole (1b). The cyclocondensation
is typically conducted at elevated temperature in a polar protic or aprotic
solvent
(e.g., dimethylformamide or N-methylpyrrolidine). The a-bromoketone (1 a)
starting material may be prepared by conventional methods, for example,
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-20
according to those methods disclosed in Scheme 2 below for the conversion of
a protected acetophenone derivative (2b) to a-bromoketone (2c). The
intermediate oxazole (1 b) is then demethylated, preferably with
methanesulfonic
acid/methionine under standard conditions to provide phenol (1 c), which is
then
functionalized with methanesulfonic acid 2-phenoxycarbonylamino-ethyl ester in
the presence of a weak base (e.g., potassium carbonate) in an aprotic solvent
(e.g., dimethylsulfoxide) to afford the protected amine (1d). The protected
amine (1d) is then deprotected, preferably by catalytic hydrogenation in a
polar
protic solvent to provide amine (1e). Coupling of amine (1e) with a
substituted
oxirane derivative (Q) affords an oxazole derivative (a compound of the
present
invention where HET is oxazole). The oxirane intermediates may be prepared
according to methods well-known to those skilled in the art, such as those
described in U.S. Patent Nos. 5,541,197; 5,561,142; 5,705,515; and 6,037,362,
all of which are incorporated herein by reference. Certain oxirane derivatives
are also commercially available.
Alternatively, compounds of the present invention where HET is oxazole
may be prepared according to the procedures outlined in Scheme 2 below.
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-21
0 0 0
HO~NHPg ~ Br
HO Rs ~ P9HN~0 I s ~ PgHN~O I s v
R R
(2a) ~ (2b)
OII
HzN~Ra
Ra Ra
N%1 N
w O w O
HZN~O I s PgHN~O ( s
R (1e) R~ o R (1
\Ar~
z~ (Q)
Ra
N
~O
OH
H
i ~ '
R~Ar~N~O Rs
Rz
Scheme 2
As outlined in Scheme 2 above, 4-hydroxyacetophenone is condensed
with a protected N-(2-hydroxyethyl)-carbamate to form the protected
acetophenone derivative (2a). The condensation may be accomplished
according to methodologies that are well-known to those skilled in the art.
Preferably, the condensation is effected via a Mitsunobu reaction. This
reaction
is typically performed with stirring at room temperature (or at elevated
temperature if required) in the presence of a dehydrating reagent (e.g., a
stoichiometric amount of a diazocarboxyl compound, e.g., 1,1'-(azodicarbonyl)-
dipiperidine (ADDP), and a phosphine, e.g., triphenylphosphine). The
condensation reaction may be carried out in any reaction-inert solvent (e.g.,
tetrahydrofuran, dimethylformamide, a hydrocarbon, or halogenated
hydrocarbon solvent). The protected acetophenone derivative (2a) is then a-
brominated to provide a.-bromoketone (2b). The bromination is performed
according to conventional methods, preferably by the reaction of (2a) with
tetrabutylammonium tribromide (TBA'Br3). Compound (2b) is cyclocondensed
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-22
with an appropriately substituted amide to afford the protected oxazole (1d)
which is then deprotected to afford the amine (1e). Such deprotection may be
accomplished using conventional deprotection methods. For example, when Pg
is a benzyl group, then the benzyl group may be removed by treating with
methanesulfonic acid, or various other deprotecting agents using standard
conditions well-known to those skilled in the art. Preferably, the
deprotection is
performed by hydrogenolysis in the presence of a suitable metal catalyst
(e.g.,
palladium on carbon) in an inert solvent. Amine (1e) is then coupled with an
appropriately substituted oxirane derivative (Q) to provide compounds of the
present invention where HET is an oxazole.
Compounds of the present invention where HET is a pyrazole moiety
may be prepared by the synthetic routes outlined below in Schemes 3 and 4.
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-23
0 0
\ (CH3)zNCH(OEt)z \ _~ N~
PgHN~ ~ PgHN
O Rs \/~O Rs
(2b) (3a)
OEt
O
N~N -N
\ I / I
\
PgHN~
O Rs PgHN
(3c) ~O Rs (3b)
H
R~bRzb
N_N O
I / N.N O
\
PgHN~ ~ ~ \
O Rs (3d) PgHN~O s
R (3e)
R~bRzb ~ R~bRzb
O
-N O R~ ~ ,N O
\ I ~ RZ Ar ~o) \
OH
~H
R 2 Ar NCO Rs (~) HzN~O Rs (3
R
Scheme 3
In Scheme 3 above, the protected amine (2b) is heated with N,N-
dimethylformamide diethyl acetal to afford the protected amine (3a).
Preferably,
the reaction between the protected amine (2b) and N,N-dimethylformamide
diethylacetal is achieved by simply combining both reactants together neat and
heating the resulting mixture for an extended period of time, generally for
about
twenty-four to about forty-eight hours. The resulting product is then
precipitated
by the addition of a non-polar solvent (e.g., hexanes). The subsequent
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-24
cyclocondensation of (3a) with hydrazine hydrate is preferably achieved by
combining the reactants in a polar protic solvent (e.g., ethanol) and heating
the
mixture for about twelve to about twenty-four hours. The resulting pyrazole
(3b)
is then N-alkylated with ethyl bromoacetate, preferably in the presence of a
base
(e.g., sodium ethoxide) in a polar protic solvent (e.g., ethanol) to provide
acetate
(3c). Basic saponification of (3c), preferably with lithium hydroxide in
tetrahydrofuran, affords acid (3d) which is then reacted with an appropriately
substituted amine in the presence of 1-(3-dimethylaminopropyl)-3-
ethylcarbodiimide hydrochloride (EDC) and 1-hydroxybenzotriazole (HOBT) to
form amide (3e). Deprotection of (3e) as described in Scheme 2 above,
followed by coupling with a substituted oxirane derivative (Q), affords a
compound of the present invention where HET is a pyrazole.
Alternatively, compounds of the present invention where HET is pyrazole
may be prepared according to the procedures outlined in Scheme 4 below.
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-25
N-N O ~ Et
\ I / Br~OEt N.N O
\ /
Me0 Rs
(4a) Me0 Rs
(4b)
R~bRzb
H
N.N O
\ I / HNR'bRzb N-N O
\ /
Me0 Rs
(4d) Me0 Rs (4c)
NR~bRzt ~ R~bRzb
N, ~ ~O~NHPg
N-N O
\ I / I /
\ v
HO s PgHN~O s
R (4e) R (3e)
O '
R~~~O NR~bRzb
w ,N~NR~bRzb Rz~ (Q)
OH H ~ I N ~ N_
R~~~N~O ~ Rs \ I /
z
R (') HzN~O I Rs
Scheme 4
In Scheme 4 above, the commercially available pyrazolo-anisole
derivative (4a) is N-alkylated with ethyl bromoacetate to provide acetate
(4b),
which is then saponified with base (e.g., sodium hydroxide) in an aqueous
solvent system (e.g., aqueous tetrahydrofuran) to provide acid (4c). Amide
(4d)
is then prepared by reacting acid (4c) with an appropriately substituted
amine,
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-26
preferably in an inert solvent (e.g., 1,2-dichloroethane) in the presence of
benzotriazole-1-yl-oxy-tris-pyrrolidino-phosphonium hexafluroide (PyBop).
Demethyation of amide (4d), as described above in Scheme 1, affords phenol
(4e) which is subsequently condensed with methanesulfonic acid 2-
phenoxycarbonylamino-ethyl ester to provide the protected amine (3e).
Catalytic deprotection of (3e), preferably with palladium on carbon in a
protic
solvent (e.g., methanol), affords amine (3f7 which is subsequently coupled
with
an appropriately substituted epoxide (Q) to afford a compound of the present
invention where HET is a pyrazole.
In the preceding reaction schematics, the oxazole and pyrazole residues
disclosed contain substitutents that are limited to an R$ moiety. Methods for
preparing other heterocyclic congeners comprising R' and/or R9 moieties is
outlined in Scheme 5 below. One of ordinary skill in the art will appreciate
that
the methylene linking group interposed between the HET and R$ groups of the
intermediate compound (5a) depicted in Scheme 5 contains at least one acidic
hydrogen atom which may be displaced and substituted with an R' and/or R9
group(s). In compound (5a), Pg represents a conventional O-protecting group,
(e.g., methyl, benzyl, tetrahydropyranyl, and the like) and HET is as defined
above. Preferably HET denotes an oxazole, pyrazole, or thiazole heterocyclic
moiety. In the instance where HET is an imidazole, the NH functional group of
such residue should be appropriately protected using conventional protection
schemes described earlier.
R' R'
R
Rg R8 R8
ET Base ET R9-L ET
R~_L
P9_O Rs (5a) P9_O Rs ~5b) P9_O Rs (5C)
Scheme 5
In Scheme 5 above, the active methylene group of compound (5a) is
deprotonated with a suitable base and the resulting anions) treated with an
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-27
appropriate reagent containing a leaving group (e.g., R'-L and/or R9-L,
wherein
R' and R9 are as defined above, except that R' and R9 are neither hydrogen nor
halogen) to afford the functionalized, protected phenol derivative (5b).
Suitable
leaving groups include halogen (preferably bromo, or iodo), triflate, and the
like.
The deprotonation is normally accomplished with a strong base (e.g., lithium
diisopropylamide, sodium hydride, lithium carbonate, lithium
bis(trimethylsilyl)amide, and the like) in a reaction-inert solvent (e.g.,
tetrahydrofuran or ether). Preferably, the deprotonation is accomplished with
lithium bis(trimethylsilyl)amide in tetrahydrofuran. The exact stoichiometric
amounts of base and R'-L and/or R9-L employed will dictate whether compound
(5b) is further functionalized to form compound (5c). Although the preparation
of compound (5c) has been depicted in Scheme 5 as a separate reaction
sequence involving a distinct intermediate (5b), it is generally preferred
that,
when R' and R9 are identical, compound (5c) is generated in a one-pot process.
For a more detailed description of the deprotonation and functionalization
sequence depicted in Scheme 5 see the preparation of Intermediate 1-3a in the
Examples section below.
The protected phenol derivatives) (5b) and/or (5c) may then be
deprotected according to conventional methods well-known to those skilled in
the art, including those methods disclosed above. Functionalization, followed
by
coupling with a substituted oxirane derivative (Q) according to the methods
disclosed above in Schemes 1 through 4 provides a compound of the present
invention.
Exemplification of the coupling reaction between an oxirane derivative
(Q) and an appropriately-substituted amine may be found in Example 1A-1 of
the Examples section below. Alternatively, compounds of the present invention
may be prepared by dehalogenation of a coupled compound of the present
invention where Ar is a 2-chloro-substituted pyridine derivative, an example
of
which is exemplified in Example 1A-2 of the Examples section below.
Conventional methods and/or techniques of separation and purification
known to one of ordinary skill in the art can be used to isolate the compounds
of
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
_28_
the present invention, as well as the various intermediates related thereto.
Such
techniques will be well-known to one of ordinary skill in the art and may
include,
for example, all types of chromatography (high pressure liquid chromatography
(HPLC), column chromatography using common adsorbents such as silica gel,
and thin-layer chromatography), recrystallization, and differential (i.e.,
liquid-
liquid) extraction techniques.
The compounds of the present invention may be isolated and used per
se or in the form of its pharmaceutically acceptable salt, solvate and/or
hydrate.
The term "salts" refers to inorganic and organic salts of a compound of the
present invention. These salts can be prepared in situ during the final
isolation
and purification of a compound, or by separately reacting the compound, N-
oxide, or prodrug with a suitable organic or inorganic acid and isolating the
salt
thus formed. Representative salts include the hydrobromide, hydrochloride,
hydroiodide, sulfate, bisulfate, nitrate, acetate, trifluoroacetate, oxalate,
besylate, palmitiate, pamoate, malonate, stearate, laurate, malate, borate,
benzoate, lactate, phosphate, hexafluorophosphate, benzene sulfonate,
tosylate, formate, citrate, maleate, fumarate, succinate, tartrate,
naphthylate,
mesylate, glucoheptonate, lactobionate, and laurylsulphonate salts, and the
like.
These may include cations based on the alkali and alkaline earth metals, such
as sodium, lithium, potassium, calcium, magnesium, and the like, as well as
non-toxic ammonium, quaternary ammonium, and amine cations including, but
not limited to, ammonium, tetramethylammonium, tetraethylammonium,
methylamine, dimethylamine, trimethylamine, triethylamine, ethylamine, and the
like. See, e.g., Berge, et al., J. Pharm. Sci., 66, 1-19 (1977).
The term "prodrug" means a compound that is transformed in vivo to
yield a compound of Formula (I) or a pharmaceutically acceptable salt, hydrate
or solvate of the compound. The transformation may occur by various
mechanisms, such as through hydrolysis in blood. A discussion of the use of
prodrugs is provided by T. Higuchi and W. Stella, "Pro-drugs as Novel Delivery
Systems," Vol. 14 of the A.C.S. Symposium Series, and in Bioreversible
Carriers
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-29
in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association
and Pergamon Press, 1987.
For example, if a compound of the present invention contains a
carboxylic acid functional group, a prodrug can comprise an ester formed by
the
replacement of the hydrogen atom of the acid group with a group such as (C~-
Ca)alkyl, (C2-C~2)alkanoyloxymethyl, 1-(alkanoyloxy)ethyl having from 4 to 9
carbon atoms, 1-methyl-1-(alkanoyloxy)-ethyl having from 5 to 10 carbon atoms,
alkoxycarbonyloxymethyl having from 3 to 6 carbon atoms, 1-
(alkoxycarbonyloxy)ethyl having from 4 to 7 carbon atoms, 1-methyl-1-
(alkoxycarbonyloxy)ethyl having from 5 to 8 carbon atoms, N-
(alkoxycarbonyl)aminomethyl having from 3 to 9 carbon atoms, 1-(N-
(alkoxycarbonyl)amino)ethyl having from 4 to 10 carbon atoms, 3-phthalidyl, 4-
crotonolactonyl, gamma-butyrolacton-4-yl, di-N,N-(C1-C2)alkylamino(C2-C3)alkyl
(such as ~i-dimethylaminoethyl), carbamoyl-(C~-C2)alkyl, N,N-di(C~-
C2)alkylcarbamoyl-(C~-C2)alkyl and piperidino-, pyrrolidino- or ~morpholino(C2-
C3)alkyl.
Similarly, if a compound of the present invention contains an alcohol
functional group, a prodrug can be formed by the replacement of the hydrogen
atom of the alcohol group with a group such as (C1-C6)alkanoyloxymethyl, 1-
((C~-C6)alkanoyloxy)ethyl, 1-methyl-1-((C~-C6)alkanoyloxy)ethyl, (C~-
C6)alkoxycarbonyloxymethyl, N-(C~-C6)alkoxycarbonylaminomethyl, succinoyl,
(C~-C6)alkanoyl, a-amino(C~-C4)alkanoyl, arylacyl and a-aminoacyl, or a-
aminoacyl-a-aminoacyl, where each a-aminoacyl group is independently
selected from the naturally occurring L-amino acids, P(O)(OH)2, P(O)(O(C~-
C6)alkyl)2 or glycosyl (the radical resulting from the removal of a hydroxyl
group
of the hemiacetal form of a carbohydrate).
If a compound of the present invention incorporates an amine functional
group, a prodrug can be formed by the replacement of a hydrogen atom in the
amine group with a group such as R-carbonyl, RO-carbonyl, NRR'-carbonyl
where R and R' are each independently (C~-C~o)alkyl, (C3-C~)cycloalkyl,
benzyl,
or R-carbonyl is a natural a-aminoacyl or natural a-aminoacyl-natural a-
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-30
aminoacyl, -C(OH)C(O)OY' wherein Y' is H, (C~-C6)alkyl or benzyl, -C(OYo)Y~
wherein Yo is (C~-C4) alkyl and Y~ is (C~-C6)alkyl, carboxy(C~-C6)alkyl,
amino(C~-
C4)alkyl or mono-N- or di-N,N-(C~-C6)alkylaminoalkyl, -C(Y2)Y3 wherein Y2 is H
or methyl and Y3 is mono-N- or di-N,N-(C~-C6)alkylamino, morpholino, piperidin-
1-yl or pyrrolidin-1-yl.
The compounds of the present invention may contain asymmetric or
chiral centers, and, therefore, exist in different stereoisomeric forms. It is
intended that all stereoisomeric forms of the compounds of the present
invention
as well as mixtures thereof, including racemic mixtures, form part of the
present
invention. In addition, the present invention embraces all geometric and
positional isomers. For example, if a compound of the present invention
incorporates a double bond or a fused ring, both the cis- and traps- forms, as
well as mixtures, are embraced within the scope of the invention. Both the
single positional isomers and mixture of positional isomers resulting from the
N-
oxidation of the pyrimidine and pyrazine rings are also within the scope of
the
present invention.
Diastereomeric mixtures can be separated into their individual
diastereoisomers on the basis of their physical chemical differences by
methods
well known to those skilled in the art, such as by chromatography and/or
fractional crystallization. Enantiomers can be separated by converting the
enantiomeric mixture into a diastereomeric mixture by reaction with an
appropriate optically active compound (e.g., chiral auxiliary such as a chiral
alcohol or Mosher's acid chloride), separating the diastereoisomers and
converting (e.g., hydrolyzing) the individual diastereoisomers to the
corresponding pure enantiomers. Also, some of the compounds of the present
invention may be atropisomers (e.g., substituted biaryls) and are considered
as
part of this invention. Enantiomers can also be separated by use of a chiral
HPLC column.
The compounds of the present invention may exist in unsolvated as well
as solvated forms with pharmaceutically acceptable solvents such as water,
ethanol, and the like, and it is intended that the invention embrace both
solvated
and unsolvated forms.
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-31
It is also possible that the compounds of the present invention may exist
in different tautomeric forms, and all such forms are embraced within the
scope
of the invention. For example, all of the tautomeric forms of the imidazole
and
pyrazole moieties are included in the invention. Also, for example, all keto-
enol
and imine-enamine forms of the compounds are included in the invention.
The present invention also embraces isotopically-labeled compounds of
the present invention which are identical to those recited herein, but for the
fact
that one or more atoms are replaced by an atom having an atomic mass or
mass number different from the atomic mass or mass number usually found in
nature. Examples of isotopes that can be incorporated into compounds of the
invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus,
fluorine, iodine, and chlorine, such as 2H, 3H,'3C,'4C,'SN,'80, "O, 3'P, 32P,
35S~ ~sF, X231, and 36CI, respectively.
Certain isotopically-labeled compounds of the present invention (e.g.,
those labeled with 3H and'4C) are useful in compound and/or substrate tissue
distribution assays. Tritiated (i.e., 3H) and carbon-14 (i.e.,'4C) isotopes
are
particularly preferred for their ease of preparation and detectability.
Further,
substitution with heavier isotopes such as deuterium (i.e., 2H) may afford
certain
therapeutic advantages resulting from greater metabolic stability (e.g.,
increased
in vivo half-life or reduced dosage requirements) and hence may be preferred
in
some circumstances. Isotopically labeled compounds of the present invention
can generally be prepared by following procedures analogous to those disclosed
in the Schemes and/or in the Examples hereinbelow, by substituting an
isotopically labeled reagent for a non-isotopically labeled reagent.
Compounds of the present invention are useful for treating diseases,
conditions and/or disorders modulated by (33 adrenergic receptor agonists;
therefore, another embodiment of the present invention is a pharmaceutical
composition comprising a therapeutically effective amount of a compound of the
present invention and a pharmaceutically acceptable excipient, diluent or
carrier.
A typical formulation is prepared by mixing a compound of the present
invention and a carrier, diluent or excipient. Suitable carriers, diluents and
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-32
excipients are well known to those skilled in the art and include materials
such
as carbohydrates, waxes, water soluble and/or swellable polymers, hydrophilic
or hydrophobic materials, gelatin, oils, solvents, water, and the like. The
particular carrier, diluent or excipient used will depend upon the means and
purpose for which the compound of the present invention is being applied.
Solvents are generally selected based on solvents recognized by persons
skilled
in the art as safe (GRAS) to be administered to a mammal. In general, safe
solvents are non-toxic aqueous solvents such as water and other non-toxic
solvents that are soluble or miscible in water. Suitable aqueous solvents
include
water, ethanol, propylene glycol, polyethylene glycols (e.g., PEG400, PEG300),
etc. and mixtures thereof. The formulations may also include one or more
buffers, stabilizing agents, surfactants, wetting agents, lubricating agents,
emulsifiers, suspending agents, preservatives, antioxidants, opaquing agents,
glidants, processing aids, colorants, sweeteners, perfuming agents, flavoring
agents and other known additives to provide an elegant presentation of the
drug
(i.e., a compound of the present invention or pharmaceutical composition
thereof) or aid in the manufacturing of the pharmaceutical product (i.e.,
medicament).
The formulations may be prepared using conventional dissolution and
mixing procedures. For example, the bulk drug substance (i.e., compound of
the present invention or stabilized form of the compound (e.g., complex with a
cyclodextrin derivative or other known complexation agent)) is dissolved in a
suitable solvent in the presence of one or more of the excipients described
above. The compound of the present invention is typically formulated into
pharmaceutical dosage forms to provide an easily controllable dosage of the
drug and to give the patient an elegant and easily handleable product.
The pharmaceutical composition (or formulation) for application may be
packaged in a variety of ways depending upon the method used for
administering the drug. Generally, an article for distribution includes a
container
having deposited therein the pharmaceutical formulation in an appropriate
form.
Suitable containers are well-known to those skilled in the art and include
materials such as bottles (plastic and glass), sachets, ampoules, plastic
bags,
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-33
metal cylinders, and the like. The container may also include a tamper-proof
assemblage to prevent indiscreet access to the contents of the package. In
addition, the container has deposited thereon a label that describes the
contents
of the container. The label may also include appropriate warnings.
The present invention further provides a method of treating diseases,
conditions and/or disorders modulated by ~i3 adrenergic receptor agonists in
an
animal that includes administering to an animal in need of such treatment a
therapeutically effective amount of a compound of the present invention or a
pharmaceutical composition comprising an effective amount of a compound of
the present invention and a pharmaceutically acceptable excipient, diluent, or
carrier.
Investigations of ~i3 adrenergic agonists have indicated that the following
diseases, disorders and/or conditions are modulated by X33 adrenergic
agonists:
weight loss (e.g., increased energy expenditure), obesity, diabetes, irritable
bowel syndrome, inflammatory bowel disease, esophagitis, duodenitis, Crohn's
disease, proctitis, asthma, intestinal motility disorder, ulcer, gastritis,
hypercholesterolemia, cardiovascular disease, urinary incontinence,
depression,
prostate disease, dyslipidemia, fatty liver, and airway inflammatory disorder.
Accordingly, the compounds of the present invention described herein
are useful in treating diseases, conditions, or disorders that are modulated
by (i3
adrenergic receptor agonists. Consequently, the compounds of the present
invention (including the compositions and processes used therein) may be used
in the manufacture of a medicament for the therapeutic applications described
herein.
The compounds of the present invention can be administered to a patient
at dosage levels in the range of from about 0.7 mg to about 7,000 mg per day.
For a normal adult human having a body weight of about 70 kg, a dosage in the
range of from about 0.01 mg to about 100 mg per kilogram body weight is
typically sufficient. However, some variability in the general dosage range
may
be required depending upon the age and weight of the subject being treated,
the
intended route of administration, the particular compound being administered
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-34
and the like. The determination of dosage ranges and optimal dosages for a
particular patient is well within the ability of one of ordinary skill in the
art having
the benefit of the instant disclosure. It is also noted that the compounds of
the
present invention can be used in sustained release, controlled release, and
delayed release formulations, which forms are also well known to one of
ordinary skill in the art.
The compounds of this invention may also be used in conjunction with
other pharmaceutical agents for the treatment of the diseases, conditions
and/or
disorders described herein. Therefore, methods of treatment that include
administering compounds of the present invention in combination with other
pharmaceutical agents are also provided. Suitable pharmaceutical agents that
may be used in combination with the compounds of the present invention
include anti-obesity agents such as apolipoprotein-B secretion/microsomal
triglyceride transfer protein (apo-B/MTP) inhibitors, MCR-4 agonists,
cholecystokinin-A (CCK-A) agonists, monoamine reuptake inhibitors (e.g.,
sibutramine), sympathomimetic agents, cannabinoid receptor antagonists (e.g.,
rimonabant (SR-141,716A)), dopamine agonists (e.g., bromocriptine),
melanocyte-stimulating hormone receptor analogs, 5HT2c agonists, melanin
concentrating hormone antagonists, leptin (the OB protein), leptin analogs,
leptin receptor agonists, galanin antagonists, lipase inhibitors (e.g.,
tetrahydrolipstatin, i.e. orlistat), anorectic agents (e.g., a bombesin
agonist),
Neuropeptide-Y antagonists, thyromimetic agents, dehydroepiandrosterone or
an analog thereof, glucocorticoid receptor agonists or antagonists, orexin
receptor antagonists, glucagon-like peptide-1 receptor agonists, ciliary
neurotrophic factors (e.g., AxokineT"" available from Regeneron
Pharmaceuticals, Inc., Tarrytown, NY and Procter & Gamble Company,
Cincinnati, OH), human agouti-related proteins (AGRP), ghrelin receptor
antagonists, histamine 3 receptor antagonists or inverse agonists, neuromedin
U receptor agonists and the like. Other anti-obesity agents, including the
preferred agents set forth hereinbelow, are well known, or will be readily
apparent in light of the instant disclosure, to one of ordinary skill in the
art.
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-35
Especially preferred are anti-obesity agents selected from the group
consisting of orlistat, sibutramine, bromocriptine, ephedrine, leptin, and
pseudoephedrine. Preferably, compounds of the present invention and
combination therapies are administered in conjunction with exercise and a
sensible diet.
Representative anti-obesity agents for use in the combinations,
pharmaceutical compositions, and methods of the invention can be prepared
using methods known to one of ordinary skill in the art, for example,
sibutramine
can be prepared as described in U.S. Pat. No. 4,929,629; bromocriptine can be
prepared as described in U.S. Pat. Nos. 3,752,814 and 3,752,888; and orlistat
can be prepared as described in U.S. Pat. Nos. 5,274,143; 5,420,305;
5,540,917; and 5,643,874. All of the above recited U.S. patents are
incorporated herein by reference.
Other pharmaceutical agents that may be useful include antihypertensive
agents; antidepressants; insulin and insulin analogs (e.g., LysPro insulin);
GLP-
1 (7-37) (insulinotropin) and GLP-1 (7-36)-NH2; sulfonylureas and analogs
thereof: chlorpropamide, glibenclamide, tolbutamide, tolazamide,
acetohexamide, Glypizide~, glimepiride, repaglinide, meglitinide; biguanides:
metformin, phenformin, buformin; a2-antagonists and imidazolines: midaglizole,
isaglidole, deriglidole, idazoxan, efaroxan, fluparoxan; other insulin
secretagogues: linogliride, A-4166; glitazones: ciglitazone, Actos~
(pioglitazone),
englitazone, troglitazone, darglitazone, Avandia~ (BRL49653); fatty acid
oxidation inhibitors: clomoxir, etomoxir; a-glucosidase inhibitors: acarbose,
miglitol, emiglitate, voglibose, MDL-25,637, camiglibose, MDL-73,945; ~-
agonists: BRL 35135, BRL 37344, RO 16-8714, ICI D7114, CL 316,243;
phosphodiesterase inhibitors: L-386,398; lipid-lowering agents: benfluorex:
fenfluramine; vanadate and vanadium complexes (e.g., Naglivan~) and
peroxovanadium complexes; amylin antagonists; glucagon antagonists;
gluconeogenesis inhibitors; somatostatin analogs; antilipolytic agents:
nicotinic
acid, acipimox, WAG 994, pramlintide (SymIinTM), AC 2993, nateglinide, aldose
reductase inhibitors (e.g., zopolrestat), glycogen phosphorylase inhibitors,
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-36
sorbitol dehydrogenase inhibitors, sodium-hydrogen exchanger type 1 (NHE-1 )
inhibitors and/or cholesterol biosynthesis inhibitors or cholesterol
absorption
inhibitors, especially a HMG-CoA reductase inhibitor, or a HMG-CoA synthase
inhibitor, or a HMG-CoA reductase or synthase gene expression inhibitor, a
CETP inhibitor, a bile acid sequesterant, a fibrate, an ACAT inhibitor, a
squalene
synthetase inhibitor, an anti-oxidant or niacin. The compounds of the present
invention may also be administered in combination with a naturally occurring
compound that acts to lower plasma cholesterol levels. Such naturally
occurring
compounds are commonly called nutraceuticals and include, for example, garlic
extract, Hoodia plant extracts, and niacin.
The dosage of the additional pharmaceutical agent will also be generally
dependent upon a number of factors including the health of the subject being
treated, the extent of treatment desired, the nature and kind of concurrent
therapy, if any, and the frequency of treatment and the nature of the effect
desired. In general, the dosage range of an anti-obesity agent is in the range
of
from about 0.001 mg to about 100 mg per kilogram body weight of the individual
per day, preferably from about 0.1 mg to about 10 mg per kilogram body weight
of the individual per day. However, some variability in the general dosage
range
may also be required depending upon the age and weight of the subject being
treated, the intended route of administration, the particular anti-obesity
agent
being administered and the like. The determination of dosage ranges and
optimal dosages for a particular patient is also well within the ability of
one of
ordinary skill in the art having the benefit of the instant disclosure.
According to the methods of the invention, a compound of the present
invention or a combination of a compound of the present invention and at least
one additional pharmaceutical agent is administered to a subject in need of
such
treatment, preferably in the form of a pharmaceutical composition. In the
combination aspect of the invention, the compound of the present invention and
at least one other pharmaceutical agent may be administered either separately
or in the pharmaceutical composition comprising both. It is generally
preferred
that such administration be oral. However, if the subject being treated is
unable
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-37
to swallow, or oral administration is otherwise impaired or undesirable,
parenteral or transdermal administration may be appropriate.
According to the methods of the invention, when a combination of a compound
of the present invention and at least one other pharmaceutical agent are
administered together, such administration can be sequential in time or
simultaneous with the simultaneous method being generally preferred. For
sequential administration, a compound of the present invention and the
additional pharmaceutical agent can be administered in any order. It is
generally preferred that such administration be oral. It is especially
preferred
that such administration be oral and simultaneous. When a compound of the
present invention and the additional pharmaceutical agent are administered
sequentially, the administration of each can be by the same or by different
methods.
According to the methods of the invention, a compound of the present
invention or a combination of a compound of the present invention and at least
one additional pharmaceutical agent (referred to herein as a "combination") is
preferably administered in the form of a pharmaceutical composition.
Accordingly, a compound of the present invention or a combination can be
administered to a patient separately or together in any conventional oral,
rectal,
transdermal, parenteral, (for example, intravenous, intramuscular, or
subcutaneous) intracisternal, intravaginal, intraperitoneal, intravesical,
local (for
example, powder, ointment or drop), or buccal, or nasal, dosage form.
Compositions suitable for parenteral injection generally include
pharmaceutically acceptable sterile aqueous or nonaqueous solutions,
dispersions, suspensions, or emulsions, and sterile powders for reconstitution
into sterile injectable solutions or dispersions. Examples of suitable aqueous
and nonaqueous carriers, diluents, solvents, or vehicles include water,
ethanol,
polyols (propylene glycol, polyethylene glycol, glycerol, and the like),
suitable
mixtures thereof, vegetable oils (such as olive oil) and injectable organic
esters
such as ethyl oleate. Proper fluidity can be maintained, for example, by the
use
of a coating such as lecithin, by the maintenance of the required particle
size in
the case of dispersions, and by the use of surfactants.
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-38
These compositions may also contain adjuvants such as preserving,
wetting, emulsifying, and dispersing agents. Prevention of microorganism
contamination of the compositions can be accomplished with various
antibacterial and antifungal agents, for example, parabens, chlorobutanol,
phenol, sorbic acid, and the like. It may also be desirable to include
isotonic
agents, for example, sugars, sodium chloride, and the like. Prolonged
absorption of injectable pharmaceutical compositions can be brought about by
the use of agents capable of delaying absorption, for example, aluminum
monostearate and gelatin.
Solid dosage forms for oral administration include capsules, tablets,
powders, and granules. In such solid dosage forms, a compound of the present
invention or a combination is admixed with at least one inert customary
pharmaceutical excipient (or carrier) such as sodium citrate or dicalcium
phosphate or (a) fillers or extenders (e.g., starches, lactose, sucrose,
mannitol,
silicic acid and the like); (b) binders (e.g., carboxymethylcellulose,
alginates,
gelatin, polyvinylpyrrolidone, sucrose, acacia and the like); (c) humectants
(e.g.,
glycerol and the like); (d) disintegrating agents (e.g., agar-agar, calcium
carbonate, potato or tapioca starch, alginic acid, certain complex silicates,
sodium carbonate and the like); (e) solution retarders (e.g., paraffin and the
like); (f) absorption accelerators (e.g., quaternary ammonium compounds and
the like); (g) wetting agents (e.g., cetyl alcohol, glycerol monostearate and
the
like); (h) adsorbents (e.g., kaolin, bentonite and the like); and/or (i)
lubricants
(e.g., talc, calcium stearate, magnesium stearate, solid polyethylene glycols,
sodium lauryl sulfate and the like). In the case of capsules and tablets, the
dosage forms may also comprise buffering agents.
Solid compositions of a similar type may also be used as fillers in soft or
hard filled gelatin capsules using such excipients as lactose or milk sugar,
as
well as high molecular weight polyethylene glycols, and the like.
Solid dosage forms such as tablets, dragees, capsules, and granules can
be prepared with coatings and shells, such as enteric coatings and others well
known in the art. They may also contain opacifying agents, and can also be of
such composition that they release the compound of the present invention
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-39
and/or the additional pharmaceutical agent in a delayed manner. Examples of
embedding compositions that can be used are polymeric substances and
waxes. The drug can also be in micro-encapsulated form, if appropriate, with
one or more of the above-mentioned excipients.
Liquid dosage forms for oral administration include pharmaceutically
acceptable emulsions, solutions, suspensions, syrups, and elixirs. In addition
to
the compound of the present invention or the combination, the liquid dosage
form may contain inert diluents commonly used in the art, such as water or
other
solvents, solubilizing agents and emulsifiers, as for example, ethyl alcohol,
isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl
benzoate, propylene glycol, 1,3-butylene glycol, dimethylformamide, oils
(e.g.,
cottonseed oil, groundnut oil, corn germ oil, olive oil, castor oil, sesame
seed oil
and the like), glycerol, tetrahydrofurfuryl alcohol, polyethylene glycols and
fatty
acid esters of sorbitan, or mixtures of these substances, and the like.
Besides such inert diluents, the composition can also include adjuvants,
such as wetting agents, emulsifying and suspending agents, sweetening,
flavoring, and perfuming agents.
Suspensions, in addition to the compound of the present invention or the
combination, may further comprise suspending agents, e.g., ethoxylated
isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters,
microcrystalline
cellulose, aluminum metahydroxide, bentonite, agar-agar, and tragacanth, or
mixtures of these substances, and the like.
Compositions for rectal or vaginal administration preferably comprise
suppositories, which can be prepared by mixing a compound of the present
invention or a combination with suitable non-irritating excipients or
carriers, such
as cocoa butter, polyethylene glycol or a suppository wax which are solid at
ordinary room temperature but liquid at body temperature and therefore melt in
the rectum or vaginal cavity thereby releasing the active component(s).
Dosage forms for topical administration of the compounds of the present
invention and combinations of the compounds of the present invention with anti-
obesity agents may comprise ointments, powders, sprays and inhalants. The
drugs are admixed under sterile condition with a pharmaceutically acceptable
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-40
carrier, and any preservatives, buffers, or propellants that may be required.
Ophthalmic formulations, eye ointments, powders, and solutions are also
intended to be included within the scope of the present invention.
The following paragraphs describe exemplary formulations, dosages, etc.
useful for non-human animals. The administration of the compounds of the
present invention and combinations of the compounds of the present invention
with anti-obesity agents can be effected orally or non-orally (e.g., by
injection).
An amount of a compound of the present invention or combination of a
compound of the present invention with an anti-obesity agent is administered
such that an effective dose is received. Generally, a daily dose that is
administered orally to an animal is between about 0.01 and about 1,000 mg/kg
of body weight, preferably between about 0.01 and about 300 mg/kg of body
weight.
Conveniently, a compound of the present invention (or combination) can
be carried in the drinking water so that a therapeutic dosage of the compound
is
ingested with the daily water supply. The compound can be directly metered
into drinking water, preferably in the form of a liquid, water-soluble
concentrate
(such as an aqueous solution of a water-soluble salt).
Conveniently, a compound of the present invention (or combination) can
also be added directly to the feed, as such, or in the form of an animal feed
supplement, also referred to as a premix or concentrate. A premix or
concentrate of the compound in a carrier is more commonly employed for the
inclusion of the agent in the feed. Suitable carriers are liquid or solid, as
desired,
such as water, various meals such as alfalfa meal, soybean meal, cottonseed
oil
meal, linseed oil meal, corncob meal and corn meal, molasses, urea, bone meal,
and mineral mixes such as are commonly employed in poultry feeds. A
particularly effective carrier is the respective animal feed itself; that is,
a small
portion of such feed. The carrier facilitates uniform distribution of the
compound
in the finished feed with which the premix is blended. Preferably, the
compound
is thoroughly blended into the premix and, subsequently, the feed. In this
respect, the compound may be dispersed or dissolved in a suitable oily vehicle
such as soybean oil, corn oil, cottonseed oil, and the like, or in a volatile
organic
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-41
solvent and then blended with the carrier. It will be appreciated that the
proportions of compound in the concentrate are capable of wide variation since
the amount of the compound in the finished feed may be adjusted by blending
the appropriate proportion of premix with the feed to obtain a desired level
of
compound.
High potency concentrates may be blended by the feed manufacturer
with proteinaceous carrier such as soybean oil meal and other meals, as
described above, to produce concentrated supplements, which are suitable for
direct feeding to animals. In such instances, the animals are permitted to
consume the usual diet. Alternatively, such concentrated supplements may be
added directly to the feed to produce a nutritionally balanced, finished feed
containing a therapeutically effective level of a compound of the present
invention. The mixtures are thoroughly blended by standard procedures, such
as in a twin shell blender, to ensure homogeneity.
If the supplement is used as a top dressing for the feed, it likewise helps
to ensure uniformity of distribution of the compound across the top of the
dressed feed.
Drinking water and feed effective for increasing lean meat deposition and
for improving lean meat to fat ratio are generally prepared by mixing a
compound of the present invention with a sufficient amount of animal feed to
provide from about 10-3 to about 500 ppm of the compound in the feed or water.
The preferred medicated swine, cattle, sheep and goat feed generally
contain from about 1 to about 400 grams of a compound of the present
invention (or combination) per ton of feed, the optimum amount for these
animals usually being about 50 to about 300 grams per ton of feed.
The preferred poultry and domestic pet feeds usually contain about 1 to
about 400 grams and preferably about 10 to about 400 grams of a compound of
the present invention (or combination) per ton of feed.
For parenteral administration in animals, the compounds of the present
invention (or combination) may be prepared in the form of a paste or a pellet
and administered as an implant, usually under the skin of the head or ear of
the
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-42
animal in which increase in lean meat deposition and improvement in lean meat
to fat ratio is sought.
In general, parenteral administration involves injection of a sufficient
amount of a compound of the present invention (or combination) to provide the
animal with about 0.01 to about 20 mg/kg/day of body weight of the drug. The
preferred dosage for poultry, swine, cattle, sheep, goats and domestic pets is
in
the range of from about 0.05 to about 10 mg/kg/day of body weight of drug.
Paste formulations can be prepared by dispersing the drug in a
pharmaceutically acceptable oil such as peanut oil, sesame oil, corn oil or
the
like.
Pellets containing an effective amount of a compound of the present
invention, pharmaceutical composition, or combination can be prepared by
admixing a compound of the present invention or combination with a diluent
such as carbowax, carnuba wax, and the tike, and a lubricant, such as
magnesium or calcium stearate, can be added to improve the pelleting process.
It is, of course, recognized that more than one pellet may be
administered to an animal to achieve the desired dose level which will provide
the increase in lean meat deposition and improvement in lean meat to fat ratio
desired. Moreover, implants may also be made periodically during the animal
treatment period in order to maintain the proper drug level in the animal's
body.
The present invention has several advantageous veterinary features. For
the pet owner or veterinarian who wishes to increase leanness and/or trim
unwanted fat from pet animals, the instant invention provides the means by
which this may be accomplished. For poultry and swine breeders, utilization of
the method of the present invention yields leaner animals that command higher
sale prices from the meat industry.
Embodiments of the present invention are illustrated by the following
Examples. It is to be understood, however, that the embodiments of the
invention are not limited to the specific details of these Examples, as other
variations thereof will be known, or apparent in light of the instant
disclosure, to
one of ordinary skill in the art.
EXAMPLES
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-43
Preparations of Key Intermediates
Preparation for Intermediate l4-(4-Mefhoxy-phenyl)-oxazol-2-yll-acetic acid
methyl ester ~(1-1a):
OMe
N~O
~ O
Me0
I-1 a
p-Methoxybromoacetophenone (3.0 g, 13 mmol) and methyl malonate
monoamide (23 g, 196 mmol) were combined in a round-bottomed flask and
heated to about 130 C for 90 minutes. The reaction mixture was then allowed to
cool to room temperature, and the resulting orange solid was partitioned
between ethyl acetate and water, and extracted with ethyl acetate. The
combined organic extracts were washed with brine, dried over magnesium
sulfate, and concentrated in vacuo. The resulting crude solid was purified by
column chromatography (5% ethyl acetate/hexanes to 10% ethyl
acetate/hexanes) to afford 2.5 g (77% yield) of title compound (I-1 a) as a
white
solid. LRMS ([M+H]+): 248.3.
Preparation forlntermediate f4-Methoxy-phenyl,)-oxazol-2-yl)-acetic acid (I-
1b):
I-1 b
To a round-bottomed flask containing [4-(4-methoxy-phenyl)-oxazol-2-yl]-
acetic acid methyl ester I-1 a (2.4 g, 9.7 mmol) was added 97 ml each of
tetrahydrofuran, methanol, and 1 N NaOH, sequentially. The resulting solution
was stirred at room temperature for about 3 hours, and was then concentrated
to remove the volatiles in vacuo. The resulting mixture was partitioned
between
water and ethyl acetate, and the pH of the aqueous layer was adjusted to about
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-44
3 with concentrated HCI. The aqueous layer was then extracted with ethyl
acetate, and the combined organic layers were washed with brine, dried over
magnesium sulfate, filtered, and concentrated in vacuo. The crude material was
recrystallized from ethyl acetate to afford the title product (I-1 b) as a
white solid
(1.5 g, 66 % yield).
Preparation for2-(4-~(4-Methoxy-phenyl)-oxazol-2-yll-1-pyrrolidin-1-yl-
ethanone
I-1 c
N ~O
O
v
Me0
I-1 c
In a round-bottomed flask, [4-(4-methoxy-phenyl)-oxazol-2-yl]-acetic acid
I-1 b (500 mg, 2.14 mmol) was combined with pyrrolidine (228 mg, 3.21 mmol),
EDC (615 mg, 3.21 mmol), and hydroxybenzotriazole (433 mg, 3.21 mmol) in 21
ml of dichloromethane. The resulting mixture was stirred overnight,
concentrated
in vacuo to approximately one third of the reaction volume, and loaded
directly
onto a silica gel column for chromatography (50 % EtOAc/hexanes). The
product (I-1c) was obtained as a white solid (670 mg, 109 % yield). LRMS
QM+H]+): 287.2.
Preparation forlntermediate 2-f4-~(Hydroxyphenyl)-oxazol-2-yll-1-pyrrolidin-1-
yl-
ethanone (I-1d):
N ~O
O
HO
I-1 d
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-45
2-[4-(4-Methoxy-phenyl)-oxazol-2-yl]-1-pyrrolidin-1-yl-ethanone I-1c (670
mg, 2.34 mmol) was combined with D,L-methionine (489 mg, 3.28 mmol) in
methanesulfonic acid (10 ml) and the resulting mixture was heated to 60
°C for
about 24 hours. The reaction was then cooled to room temperature, and then
slowly added to stirring saturated aqueous sodium carbonate and ethyl acetate.
The pH was adjusted to about 9, and the phases were separated. The aqueous
phase was extracted with ethyl acetate, and the combined organic layers were
washed with brine, dried over magnesium sulfate, filtered, and concentrated in
vacuo. The resulting crystalline solids were suspended in a small volume of
ethyl acetate, and collected by vacuum filtration to afford 480 mg (75% yield)
of
the desired title product (I-1d). LRMS ([M+H]+): 273.2.
Preparation forlntermediate (2-f4 ~2-~(2-Oxo-2-pyrrolidin-1-yleth~l -oxazol-4-
yll-
phenoxy)-ethyl)-carbamic acid benzyl ester ~(I-1e):
N~O
O
CbzHN~O
I-1 a
In a round-bottomed flask, 2-[4-(4-hydroxy-phenyl)-oxazol-2-yl]-1-
pyrrolidin-1-yl-ethanone I-1d (474 mg, 1.74 mmol) was dissolved in
dimethylsulfoxide (6 ml), and potassium carbonate (powdered, 722 mg, 5.22
mmol) was added in a single portion. Methanesulfonic acid 2-
benzyloxycarbonylamino-ethyl ester (952 mg, 3.48 mmol) was added to the
mixture, and the resulting heterogeneous solution was heated to 70° C
for about
18 hours. The reaction was judged complete by thin-layer chromatography, and
was then cooled to room temperature, and poured into 50 ml of water, and 50
ml of ethyl acetate. The phases were separated and the aqueous phase was
extracted with ethyl acetate. The combined organic extracts were washed with
brine, dried over magnesium sulfate, filtered, and concentrated in vacuo to
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-46
afford a dark yellow oil. This crude material was purified by column
chromatography (2% methanol/dichloromethane) to afford the desired title
product I-1 a (529 mg, 68% yield). LRMS ([M+H]+): 450.1.
Preparation forlntermediate 2-{4-f4-(2 Amino-ethoxy)-phenyll-oxazol-2-ylj-1-
wrrolidin-1-yl-ethanone (I-1t):
N~O
O
H2N ~O
I-1 f
In a hydrogenation bottle, (2-{4-[2-(2-oxo-2-pyrrolidin-1-yl-ethyl)-oxazol-4-
yl]-phenoxy}-ethyl)-carbamic acid benzyl ester I-1 a (529 mg, 1.18 mmol) was
dissolved in methanol (30 ml), and 10 % Pd/C (30 wt %, 160 mg) was added in
one portion. The mixture was hydrogenated under 45 psi of hydrogen for about
2 hours until the reaction was judged complete by thin-layer chromatography.
The mixture was then filtered through a pad of diatomaceous earth, and rinsed
with methanol to remove the catalyst. The filtrate was then concentrated in
vacuo to afford the desired product I-1f (370 mg, 100% yield) as a white
solid.
LRMS ([M+H]+): 316.2.
Preparation for Intermediate Benzyl-f2-~(4-acetyl-phenoxy)-ethyl)-carbamate (1-
0
w w
CbzHN ~O
I-2a
In a round-bottomed flask equipped with a mechanical stirrer, 4-
hydroxyacetophenone I-2a (5.00 g, 36.7 mmol) was dissolved in toluene (122
ml), and triphenylphosphine (14.4 g, 55.1 mmol) and benzyl N-(2-
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-47
hydroxyethyl)carbamate (10.8 g, 55.1 mmol) were then added. The reaction
mixture was cooled to about 0 C, and 1,1'-(azodicarbonyl)dipiperidine (13.9 g,
55.1 mmol) was added in one portion. The mixture was allowed to warm to room
temperature, and after stirring for about 10 minutes, an additional 122 ml of
toluene and 122 m of tetrahydrofuran were added to the thick orange solution.
The mixture was stirred for about 24 hours, and then the solids were filtered
off.
The filtrate was concentrated in vacuo and the resulting solid was purified by
column chromatography (hexanes to 2:1 hexanes/ethyl acetate) to afford 9.68 g
(84% yield) of the desired product (I-2a) as a white solid. LRMS ([M-H]-):
312.2.
Preparation for Intermediate Benzyl-~2-~(4-bromoacetylphenoxy)-ethyll-
carbamate (I-2b):
o
Br
CbzHN ~O
I-2b
Benzyl [2-(4-acetyl-phenoxy)-ethyl]-carbamate I-2a (10.2 g, 32.5 mmol)
was dissolved in dichloromethane (100 ml) and methanol (50 ml), and
tetrabutylammonium tribromide (15.7 g, 32.5 mmol) was added in one portion.
The reaction mixture was stirred for about 16 hours, and was then quenched
with water. The aqueous phase was extracted with ethyl acetate, and then
washed with saturated aqueous sodium bicarbonate and saturated aqueous
sodium bisulfite. The combined organic extracts were dried over magnesium
sulfate, filtered, and concentrated in vacuo, and the resulting crude material
was
purified by column chromatography (hexanes to 2:1 hexanes/ethyl acetate) to
afford a colorless oil which solidified on standing I-2b (11.5 g, 90 % yield).
Preparation for Intermediate ~4 I4-~(2-8enzyloxycarbonylamino-ethoxy -phen~-
oxazol-2-YI)-acetic acid methyl ester ~(I-2c,~:
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-48
O
i
N~-OMe
CbzHN~O I / O
I-2c
Methoxy malonamide (10.6 g, 90.6 mmol) and benzyl [2-(4-bromoacetyl-
phenoxy)-ethyl]-carbamate 1-2b (2.37 g, 6.04 mmol) were combined in a round-
bottomed flask and heated to 130~C for about 90 minutes. The reaction mixture
was then allowed to cool to room temperature, and the resulting orange solid
was partitioned between ethyl acetate and water and extracted with ethyl
acetate. The combined organic extracts were washed with brine, dried over
magnresium sulfate, filtered, and concentrated in vacuo. The resulting crude
solid was purified by column chromatography (30% hexanes/ethyl acetate to
50% hexanes/ethyl acetate) to afford 1.29 g (50% yield) of the title product
as a
white solid (I-2c).
Preparation for Intermediate ~4-(4-(2-Benzyoxycarbonylamino-ethox~~~,phenyll-
oxazol-2-ylj-acetic acid ~(I-2d~:
OH
N~O
O
CbzHN~O
I-2d
To a round-bottomed flask containing {4-[4-(2-benzyloxycarbonylamino-
ethoxy)-phenyl]-oxazol-2-yl}-acetic acid methyl ester I-2c (1.29 g, 3.00 mmol)
was added 10 ml each of tetrahydrofuran, methanol, and 1 N NaOH,
sequentially. The resulting solution was stirred at room temperature for about
5
minutes, and then quenched with 1 N HCI, and extracted with ethyl acetate. The
combined organic layers were washed with brine, dried over magnesium sulfate,
filtered, and concentrated in vacuo. The resulting yellow solid (I-2d) was
determined to be pure by'H NMR and was used directly in the next reaction
(1.20 g, 100% yield).
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-49
Preparation for Intermediate f2-(4-~2-f(Diisopropylcarbamoyl)-methyl)-oxazol-4-
yl~-phenoxy)-ethyl)-carbamic acid benzyl ester (I-2e):
N
N ~O
O
CbzHN ~O
I-2e
In a round-bottomed flask, {4-[4-(2-benzyloxycarbonylamino-ethoxy)-
phenyl]-oxazol-2-yl}-acetic acid I-2d (350 mg, 0.883 mmol) was combined with
diisopropylamine (160 ~I, 1.15 mmol), benzotriazol-1-yl-
oxytripyrrolidinophosphonium hexafluorophosphate (PyBop) (598 mg, 1.15
mmol), and diisopropylethyl amine (230 ~.I, 1.32 mmol) in 1,2-dichloroethane
(4.4 ml). The resulting mixture was stirred overnight, concentrated in vacuo
to
approximately one third of the reaction volume, and loaded directly onto a
silica
gel column for chromatography (50 % hexanes/ethyl acetate). The product was
obtained as a white solid I-2e (219 mg, 52% yield). LRMS ([M+H]+): 480.2.
Preparation for Intermediate 2-l4-(4-Methoxy-phenyl)-oxazol-2-y11-hexanoic
acid
N,N-dimethylamide 1-3a):
1
N-
N- O
~ O
Me0
I-3a
To a stirred solution of 300 mg (1.15 mmol) of 2-[4-(4-methoxy-phenyl)-
oxazol-2-yl]-N,N-dimethyl-acetamide in 4 ml of tetrahydrofuran under nitrogen
at
O~C was added 1.15 ml of a 1.0 M tetrahydrofuran solution of lithium
bis(trimethylsilyl)amide, and the resulting solution was stirred for 30
minutes. To
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-50
this solution was added 0.13 ~I (1.15 mmol) of 1-iodobutane, and the reaction
mixture was allowed to warm to room temperature and stir overnight. After
cooling to O~C, water was added and the mixture was warmed to room
temperature. The mixture was extracted with ethyl acetate and the combined
organic layers were dried over sodium sulfate and concentrated in vacuo. The
resulting material was purified by column chromatography using 2%
acetone/dichloromethane to 5% acetone/dichloromethane as the gradient eluant
to afford the desired title product I-3a (212 mg, 58% yield) as a solid. LRMS
([M+H]+): 317.5.
Preparation for Intermediate {2-(4-~(3-Dimethylamino-acryloyl)-phenoxyl-
eth~il~
carbamic acid benzyl ester ~(I-4a):
0
/ N'
CbzHN~C
I-4a
Benzyl [2-(4-acetyl-phenoxy)-ethyl]-carbamate (28.3 g, 90.3 mmol) and
N,N-dimethylformamide diethyl acetal (62 ml, 361 mmol) were combined in a
round-bottomed flask and heated to 70°C for about 28 hours. The
reaction was
then cooled to room temperature, allowed to stand overnight, and then 25 ml of
hexanes was added to the heterogeneous reaction mixture. The resulting slurry
was filtered, and the solids were dried under vacuum to afford 30.11 g (81.7
mmol, 90% yield) of a mustard-yellow powder (I-4a). LRMS ([M+H]+): 369.3.
Preparation for Intermediate ~2-~4-(9H-Pyrazol-3-yl)-phenoxyj-ethy-carbamic
acid benzyl ester (I-4b):
N-NH
I
CbzHN~C I /
I-4b
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-51
In a round-bottomed flask, {2-[4-(3-dimethylamino-acryloyl)-phenoxy]-
ethyl}-carbamic acid benzyl ester I-4a (12.85 g, 34.87 mmol) was suspended in
ethanol (70 ml), and hydrazine hydrate (3.38 ml, 69.76 mmol) was added
dropwise via syringe. The reaction flask was fitted with a reflux condenser,
and
then heated to 80°C for about 18 hours. The reaction was then allowed
to cool
to room temperature, and the resulting solids were suspended in a minimum
amount of ethanol, and filtered under vacuum. The solids were dried in vacuo
to
yield a colorless solid I-4b (6.55 g), and the filtrate was concentrated and
re-
suspended in ethanol to afford a second crop of solids I-4b (2.27 g, for a
combined yield of 75%). LRMS ([M+H]+): 338.3.
Preparation for Intermediate f3-f4-~(2-Benz~ycarbonylamino-ethoxy)-phenyl)-
wrazol-1-ail)-acetic acid eth I ey ster ~(1-4c~:
,N~ /OEt
~N
O
CbzHN ~O
I-4C
A 500 ml round-bottomed flask was charged with {2-[4-(1 H-pyrazol-3-yl)-
phenoxy]-ethyl}-carbamic acid benzyl ester I-4b (8.39 g, 24.87 mmol) and
ethanol (80 ml). Sodium ethoxide (27.9 ml of a 21 wt % solution in ethanol,
74.6
mmol) was added dropwise via addition funnel over a period of about 5 minutes,
followed by bromoacetic acid ethyl ester (5.51 ml, 49.73 mmol). The resulting
mixture was stirred for about 15 hours, and was then quenched to neutral pH by
the addition of concentrated HCI. The volatiles were removed in vacuo, and 500
ml of diethyl ether were added to form a slurry. The solids were removed by
vacuum filtration to yield 10.6 g of brown-colored solid which was discarded.
The
ether filtrate was then concentrated to an oil (8.1 g) which was purified by
column chromatography (40 % hexanes/ethyl acetate) to afford 3.06 g of the
desired title product I-4c (7.22 mmol, 29% yield). LRMS ([M+H]+): 424.3.
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-52
Preparation for Intermediate ~3-f4-(2-Benzyloxycarbon ylamino-ethoxy -phenyll-
~yrazol-1-~I,~-acetic acid (1-4d):
rN~OH
~'(N
/ O
CbzHN~O
I-4d
In a round-bottomed flask, {3-[4-(2-benzyloxycarbonylamino-ethoxy)-
phenyl]-pyrazol-1-yl}-acetic acid ethyl ester I-4c (1.19 g, 2.81 mmol) was
dissolved in tetrahydrofuran (9.0 ml). Methanol (9 ml) and 1 N LiOH (9 ml)
were
added sequentially, and the mixture was stirred for about 10 minutes. The
reaction was brought to pH 3 with 3 N HCI, and was then diluted with water and
dichloromethane. The aqueous phase was extracted with dichloromethane and
the combined organics were dried over sodium sulfate, filtered, and
concentrated in vacuo. The resulting crude material (1.17 g) was triturated
with
ether and decanted to afford 936 mg (84% yield) of the desired product (I-4d).
LRMS ([M+H]+): 396.3.
Preparation for Intermediate f2 [4-(1-C cly opentylcarbamoylmethyl-1 H-pyrazol-
3-
yl)-phenoxy)-ethyl}-carbamic acid benzyl ester (1-4e):
H
N
INN
O
v
CbzHN~O
I-4e
In a round-bottomed flask, {3-[4-(2-benzyloxycarbonylamino-ethoxy)-
phenyl]-pyrazol-1-yl}-acetic acid I-4d (244 mg, 0.617 mmol) and
diisopropylethyl
amine (322 ~I, 1.85 mmol) were dissolved in dichloromethane (6 ml). EDC~HCI
(178 mg, 0.926 mmol) was added to the solution, followed by
hydroxybenzotriazole hydrate (125 mg, 0.926 mmol) and cyclopentylamine (122
~I, 1.23 mmol). The reaction mixture was stirred for about 48 hours, and was
then diluted with ethyl acetate and saturated aqueous sodium bicarbonate. The
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-53
aqueous phase was extracted with ethyl acetate, and the combined organics
were dried over magnesium sulfate, filtered, and concentrated in vacuo. The
crude material was purified by column chromatography (1.5
methanol/dichloromethane) to afford 290 mg (102% yield) of the desired product
(I-4e). LRMS ([M+H]+): 463.4.
Preparation for Intermediate 2 ~3-(4-~(2-Amino-ethoxy)-phenyl~l-pyrazol-1-YI
cyclopentyl-acetamide~l-4f2:
H
N~N
N
/ O
v
H2N ~O I
I-4f
In a nitrogen-purged round-bottomed flask, {2-[4-(1-
cyclopentylcarbamoylmethyl-1 H-pyrazol-3-yl)-phenoxy]-ethyl}-carbamic acid
benzyl ester I-4e (285 mg, 0.616 mmol) was dissolved in methanol (6.1 ml). To
this solution, 10% Pd/C (100 mg, 30 wt %) and formic acid (2.46 ml, 95 mmol)
were added, and the reaction was stirred overnight. The reaction mixture was
then filtered through a pad of diatomaceous earth, and the filtrate was
concentrated. The resulting material was then dissolved in water, the pH was
adjusted to 12 with 5 N NaOH, and the aqueous phase was extracted with ethyl
acetate. The combined organic layers were dried over sodium sulfate, filtered,
and concentrated in vacuo. The resulting product I-4f (173 mg, 85 % yield) was
carried directly into the next reaction. LRMS ([M+H]+): 329.4.
Preparation for Intermediate 3-Dimethylamino-1~4-methoxy-phenyl)-prop-2-en-
1-one ~(I-5a):
0
N'
I I
Me0
1-5a
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-54
p-Methoxyacetophenone (4.50 g, 30.0 mmol) and N,N-
dimethylformamide diethyl acetal (25.7 ml, 150 mmol) were combined in a
round-bottomed flask and heated to 130 C for about 18 hours. The reaction
was then cooled to room temperature and concentrated in vacuo. Diethyl ether
(30 ml) was added to the reaction mixture and the resulting solids (2.59 g)
were
collected by vacuum filtration. The filtrate was then concentrated to dryness
and
re-suspended in diethyl ether to yield a second crop of solids which was
collected by filtration (1.38 g). The combined solids I-5a (3.97 g, 65% yield)
were
carried directly into the next step.
Preparation for Intermediate (5-(4-Methoxy-phenyl)-pyrazol-1-yl)-acetic acid
ethyl ester (I-5b):
Et02C'~N_N
i
Me0
I-5b
Ethyl hydrazinoacetate hydrochloride (1.91 g, 12.3 mmol) and 3-
dimethylamino-1-(4-methoxy-phenyl)-prop-2-en-1-one I-5a (2.53 g, 12.3 mmol)
were dissolved in ethanol (40 ml). Potassium carbonate (1.70 g, 12.3 mmol) was
added to this solution, and the resulting mixture was heated to 80 °C
for about
16 hours. The reaction was then cooled to room temperature and concentrated
in vacuo. The crude paste was suspended in water (50 ml), and the pH was
adjusted to 9. The aqueous mixture was extracted with ethyl acetate, and the
combined organic extracts were washed with brine, dried over magnesium
sulfate, filtered, and concentrated in vacuo. The material was then purified
by
column chromatography (hexanes to 35% ethyl acetate/hexanes) to afford 2.61
g (81 % yield) of the desired product (I-5b). LRMS ((M+H]+): 261.3.
Preparation for Intermediate~5-(4-Methoxy,phenyl)-pyrazol-1-ail)-acetic acid
(I-
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-55
H02C'1N_N
Me0
I-5c
[5-(4-Methoxy-phenyl)-pyrazol-1-yl]-acetic acid ethyl ester I-5b (2.50 g,
9.60 mmol) was dissolved in tetrahydrofuran (30 ml) and methanol (30 ml). To
this mixture, LiOH (902 mg, 38.4 mmol), and water (30 ml) were added. The
mixture was stirred for about 15 minutes, and was then partitioned between
ethyl acetate and water. The pH was adjusted to 3, and the aqueous phase was
extracted with ethyl acetate. The combined organic extracts were dried over
sodium sulfate, filtered, and concentrated in vacuo to afford the title
compound
(I-5c) as a colorless solid (2.08 g, 93% yield). LRMS ([M+H]+): 233.3.
Preparation for Intermediate 2-(5-(4-Methoxy-,chenyl~~uyrazol-1-yl)-1-
pyrrolidin-1-
yl-ethanone (I-5d):
I-5d
In a round-bottomed flask, [5-(4-methoxy-phenyl)-pyrazol-1-yl]-acetic
acid I-5c was dissolved in dichloromethane (9.5 ml). To this solution, EDC~HCI
(817 mg, 4.26 mmol), diisopropylethyl amine (1.48 ml, 8.52 mmol),
hydroxybenzotriazole hydrate (576 mg, 4.26 mmol), and pyrrolidine (475 ~L,
5.68 mmol) were added sequentially. The reaction was allowed to stir for about
2 days, and was then diluted with dichloromethane and quenched with saturated
aqueous sodium bicarbonate. The aqueous phase was extracted with
dichloromethane, and the combined organics were dried over magnesium
sulfate, filtered, and concentrated in vacuo. The resulting crude material was
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-56
purified by column chromatography (3% methanol/ dichloromethane) to afford
475 mg (59% yield) of the desired product (I-5d). LRMS ([M+H]+): 286.3.
Preparation for Intermediate 2-(5-~(4-Hydroxy-,ahenyl)-pyrazol-1-yl)-1-
pyrrolidin-1-
yl-ethanone (I-5e):
n
I-5e
In a round-bottomed flask, 2-[5-(4-methoxy-phenyl)-pyrazol-1-yl]-1-
pyrrolidin-1-yl-ethanone I-5d (440 mg, 1.54 mmol) was combined with D,L-
methionine (345 mg, 2.31 mmol) in methanesulfonic acid (6.2 ml) and the
resulting mixture was heated to 70~ C for about 22 hours. The reaction was
then
cooled to room temperature, and then slowly added to stirring saturated
aqueous sodium carbonate and ethyl acetate. The pH was adjusted to 9, and
the phases were separated. The aqueous phase was extracted with ethyl
acetate, and the combined organic layers were washed with brine, dried over
magnesium sulfate, filtered, and concentrated in vacuo. The resulting crude
material was purified by column chromatography (dichloromethane to 3%
methanol/ dichloromethane) to afford 280 mg (1.03 mmol, 67% yield) of the
desired product (I-5e) as a colorless solid. LRMS ([M+H]+): 272.2.
Preparation for Intermediate (2-~4-(2-~(2-Oxo-2-pyrrolidin-1-yl-ethyl -
~pyrazol-
3-yll-phenoxy)-ethyl-carbamic acid benzyl ester (I-5~:
CbzH
I-5f
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-57
In a round-bottomed flask, 2-[5-(4-hydroxy-phenyl)-pyrazol-1-yl]-1-
pyrrolidin-1-yl-ethanone I-5e (273 mg, 1.00 mmol) was dissolved in
dimethylsulfoxide (2 ml), and potassium carbonate (powdered, 415 mg, 3.00
mmol) was added in one portion. Methanesulfonic acid 2-
benzyloxycarbonylamino-ethyl ester (547 mg, 2.00 mmol) was then added to the
mixture, and the resulting heterogeneous solution was heated to 70~ C for
about
18 hours. The reaction was judged complete by thin-layer chromatography, and
was then cooled to room temperature, and poured into 20 ml of water. The
aqueous phase was extracted 3 x with dichloromethane, and the combined
organic extracts were washed with brine, dried over magnesium sulfate,
filtered,
and concentrated in vacuo to afford a dark yellow oil. This crude material was
purified by column chromatography (2% methanol/dichloromethane) to afford
the desired product I-5f (390 mg, 87% yield). LRMS ([M+H]+): 449.4.
Preparation for Intermediate 2-~5-~4-(2 Amino-ethoxy)-phenJ~ll-pyrazol-1-yll-1-
pyrrolidin-1-yl-ethanone ~(I-SQL:
H2N
I-5g
In a nitrogen-purged, round-bottomed flask, (2-{4-[2-(2-oxo-2-pyrrolidin-1-
yl-ethyl)-2H-pyrazol-3-yl]-phenoxy}-ethyl)-carbamic acid benzyl ester I-5f
(390
mg, 0.870 mmol) was dissolved in methanol (8.70 ml). To this solution, 10
Pd/C (150 mg, 30 wt %), and formic acid (3.48 ml, 133 mmol) were added, and
the reaction was stirred overnight. The reaction mixture was then filtered
through
a pad of diatomaceous earth, and the filtrate was concentrated. The resulting
material was then dissolved in water and the pH was adjusted to 12 with 5 N
NaOH, and the aqueous phase was extracted with ethyl acetate. The combined
organics were dried over sodium sulfate, filtered, and concentrated in vacuo.
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-58
The resulting product I-5g (225 mg, 72% yield) was carried directly into the
next
reaction. LRMS ([M+H]+): 315.4.
Example 1 illustrates the preparation of compounds of the present
invention where Ar is a pyridyl group.
Example 1
Preparafion for 2-f4-(4-f2-f2-(6-Chloro-~oyridin-3-yl)-2(R)-hydroxy-
ethylaminol-
ethoxy~-phenyl -oxazol-2-yll-1-pyrrolidin-1-yl-ethanone (1-1A):
N~O
O
OH
w NCO
C~ N J
1-1A
In a round-bottomed flask, (R)-2-chloro-5-oxiranyl-pyridine (123 mg, 0.79
mmol) and 2-{4-[4-(2-amino-ethoxy)-phenyl]-oxazol-2-yl}-1-pyrrolidin-1-yl-
ethanone I-1f (370 mg, 1.18 mmol) were dissolved in 20 ml of ethanol, and the
mixture was heated to 80~ C for about 16 hours. The solution was then
concentrated in vacuo to an oil, and the crude material was purified by column
chromatography (dichloromethane to 10 % methanol/dichloromethane) to afford
200 mg (54% yield) of the title product as a white solid. LRMS ([M+H]+):
471.3.
'H NMR: (400 MHz, CD30D): 8 1.91 (m, 2H), 1.99 (m, 2H), 2.85 (m, 2H), 3.03
(m, 2H), 3.45 (m, 2H), 3.61 (m, 2H), 3.95 (s, 2H), 4.10 (m, 2H), 4.84 (m, 1
H),
6.97 (d, 2H, J = 8.8 Hz), 7.42 (d, 1 H, J = 8.0 Hz), 7.64 (d, 2H, J = 8.4 Hz),
7.83
(dd, 1 H, J = 2.8, 8.4 Hz), 8.10 (s, 1 H), 8.37 (d, 1 H, J = 2.8 Hz).
Preparation for 2-(4-f4-~2-L~(R,)-Hydrox ~-~2-pyridin-3-yl-efhylamino)-efhoxyl-
phenyl)-oxazol-2-yl)-1-pyrrolidin-1-yl-ethanone (1-18,x:
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-59
N~O
O
OH \
\ NCO
~J
N
1-1B
In a nitrogen-purged, round-bottomed flask, (R)-2-[4-(4-{2-[2-(6-chloro-
pyridin-3-yl )-2-hyd roxy-ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-1-
pyrrolidin-1-yl-
ethanone 1-1A (200 mg, 0.42 mmol) was dissolved in methanol (15 ml). 10
Pd/C (160 mg, 80 wt %), and ammonium formate (321 mg, 5.1 mmol) were then
added sequentially. The reaction mixture was stirred overnight, and was then
filtered through a pad of diatomaceous earth, and the filter cake rinsed with
ethyl
acetate. The filtrate was concentrated to a white solid, which was taken up in
ethyl acetate and saturated aqueous sodium carbonate, and extracted. The
organic extracts were washed with brine, dried over magnesium sulfate,
filtered,
and concentrated in vacuo to afford a crude solid. This material was suspended
in diethylether and isolated by vacuum filtration to afford a crystalline
solid 1-1 B
(110 mg, 59% yield). This material was then converted to the corresponding
hydrochloride salt. Analytical data for the HCI salt: LRMS ([M+H]+): 437.4. 'H
NMR: (400 MHz, CD30D): 8 1.91 (m, 2H), 2.02 (m, 2H), 3.37-3.29 (m, 2H), 3.45
(m, 2H), 3.54-3.63 (m, 5H), 3.97 (s, 2H), 4.37 (t, 2H, J = 4.8 Hz), 5.38 (dd,
1 H, J
= 3.2, 10.4 Hz), 7.07 (dd, 2H, J = 2.8, 9.6 Hz), 7.69 (dd, 2H, J = 1.8, 6.4
Hz),
8.14 (m, 2H), 8.75 (m, 1 H), 8.86 (d, 1 H, J = 6.0 Hz), 8.99 (m, 1 H).
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-60
Preparation for 2-f414-f2-~2(R)-Hydroxy-2-~(6-methyl-pyridin-3-yl)-ethylaminol-
ethoxy)-phenyl)-oxazol-2-yll-N,N-dimethyl-acetamide (1-1C):
I \
N / NCO \
OH I / N
-/
O N
O
1-1C
In a round-bottomed flask, (R)-2-methyl-5-oxiranyl-pyridine (4.4 g, 0.015 mol)
and 2-{4-[4-(2-amino-ethoxy)-phenyl]-oxazol-2-yl}-N,N-dimethyl-acetamide (2.3
g, 0.017 mol) were dissolved in 16 ml of ethanol, and the mixture was heated
to
60~ C for about 16 hours. A precipitate began forming after ca. 1 hour. The
reaction mixture was then cooled to room temperature and diluted with ethyl
acetate. The resulting mixture was stirred for 1 hour, and the solids were
then
removed by filtration and washed with ethyl acetate. These solids were then
resuspended in ethyl acetate and heated to 70~ C to give a pale yellow
solution.
This solution was then cooled slowly and the resulting solids were isolated to
afford 1.9 g (29 %) of the title product as a pale pink solid. LRMS ([M+H]+):
425Ø 'H NMR: (400 MHz, CD30D): 8 8.42 (d, J=2.49, 1 H), 8.12 (s, 1 H), 7.76
(dd, 1 H), 7.67 (dt, 2H), 7.29 (d, J=8.3, 1 H), 6.99 (dt, 2H), 4.87 (t,
J=6.23, 1 H),
4.16 (m, 2H), 4.03(s, 2H), 3.16 (s, 3H), 3.12(m, 2H), 2.98 (s, 3H), 2.93(d,
J=6.64, 2H), 2.52 (s, 3H)
Table I below lists compounds that were prepared using the general
procedures described above for the preparation of compounds 1-1A, 1-1 B and
1-1C with the appropriate starting materials.
TABLE 1
Example Compound Name LRMS
No. M+H
2-(4-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
ethoxy]-phenyl}-oxazol-2-yl)-1-morpholin-4-yl-471.3
1-1D ethanone h drochloride salt
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-61
Example Compound Name LRMS
No. M+H
2-(4-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
1-1E ethoxy]-phenyl}-oxazol-2-yl)-N,N-dimethyl-437.4
acetamide h drochloride salt
N-Cyclopentyl-2-(4-{4-[2-(2(R)-hydroxy-2-pyridin-3-
1-1F yl-ethylamino)-ethoxy]-phenyl}-oxazol-2-yl)-425.0
acetamide h drochloride salt
2-(4-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
1-1G ethoxy]-phenyl}-oxazol-2-yl)-1-piperidin-1-yl-453.3
ethanone h drochloride salt
N,N-Diethyl-2-(4-{4-[2-(2(R)-hydroxy-2-pyridin-3-yl-
1-1H ethylamino)-ethoxyJ-phenyl}-oxazol-2-yl)-acetamide411.2
h drochloride salt
2-[4-(4-{2-[2-(6-Chloro-pyridin-3-yl)-2(R)-hydroxy-
1-11 ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-1-451.4
mor holin-4- I-ethanone
2-[4-(4-{2-[2-(6-Chloro-pyridin-3-yl)-2(R)-hydroxy-
1-1J ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-451.4
dieth I-acetamide
2-[4-(4-{2-[2-(6-Chloro-pyridin-3-yl)-2(R)-hydroxy-
1-1K ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-1-piperidin-439.4
1- I-ethanone
2-[4-(4-{2-[2-(6-Chloro-pyridin-3-yl)-2(R)-hydroxy-
1-1 L ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N-487.3
c clo ent I-acetamide
2-(4- f 4-[2-(2(R)-Hyd roxy-2-pyrid in-3-yl-ethyla
mino)-
1-1M ethoxy]-phenyl}-oxazol-2-yl)-N-methyl-acetamide473.4
h drochloride salt
N-Ethyl-2-(4-{4-[2-(2(R)-hydroxy-2-pyridin-3-yl-
1-1N ethylamino)-ethoxy]-phenyl}-oxazol-2-yl)-N-methyl-485.4
acetamide h drochloride salt
2-[4-(4-f 2-[2-(6-Chloro-pyridin-3-yl)-2(R)-hydroxy-
1-10 ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N-methyl-485.3
acetamide
2-[4-(4-{2-[2-(6-Chloro-pyridin-3-yl)-2(R)-hydroxy-
1-1P ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N-ethyl-N-397.4
meth I-acetamide
1-Azetid i n-1-yl-2-(4-{4-[2-(2 ( R )-h
yd roxy-2-pyrid i n-3-
1-1Q yl-ethylamino)-ethoxy]-phenyl}-oxazol-2-yl)-425.4
ethanone h drochloride salt
N-Ethyl-2-(4-f4-[2-(2(R)-hydroxy-2-pyridin-3-yl-
1-1R ethylamino)-ethoxy]-phenyl}-oxazol-2-yl)-N-431.4
iso ro I-acetamide h drochloride salt
2-(4-{4-[2-(2 ( R )-H yd roxy-2-pyrid
i n-3-yl-ethyl a m i n o )-
1-1S ethoxy]-phenyl}-oxazol-2-yl)-N-isopropyl-N-methyl-459.2
acetamide h drochloride salt
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-62
Example Compound Name LRMS
No. M+H
2-[4-(4-{2-[2-(6-C h I o ro-pyrid i
n-3-yl )-2 ( R )-h yd roxy-
1-1T ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N-ethyl-N-423.4
2,2,2-trifluoro-eth I -acetamide
2-[4-(4-{2-[2-(6-Chloro-pyridin-3-yl)-2(R)-hydroxy-
1-1U ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-453.5
diiso ro I-acetamide
N-Ethyl-2-(4-{4-[2-(2(R)-hydroxy-2-pyridin-3-yl-
1-1 V ethylamino)-ethoxy]-phenyl}-oxazol-2-yl)-N-(2,2,2-439.4
trifluoro-eth I -acetamide h drochloride
salt
2-(4-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
1-1W ethoxy]-phenyl}-oxazol-2-yl)-N,N-diisopropyl-527.4
acetamide h drochloride salt
2-(4-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
1-1X ethoxy]-phenyl}-oxazol-2-yl)-N-methyl-N-(2,2,2-501.5
trifluoro-eth I -acetamide h drochloride
salt
2-[4-(4-{2-[2-(6-C hloro-pyrid in-3-yl
)-2(R)-hyd roxy-
1-1Y ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-493.2
diiso ro I- ro ionamide
2-(4-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
1-1Z ethoxy]-phenyl}-oxazol-2-yl)-N,N-diisopropyl-467.5
ro ionamide
2-[4-(4-{2-[2-(6-Ch loro-pyrid i n-3-yl
)-2( R)-hyd roxy-
1-2A ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N-methyl-479.3
N- 2,2,2-trifluoro-eth I -acetamide
2-[4-(4-{2-[2-(6-Chloro-pyridin-3-yl)-2(R)-hydroxy-
1-2B ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N-ethyl-N-515.4
2,2,2-trifluoro-eth I - ro ionamide
N-Ethyl-2-[4-(4-{2-[2( R)-hyd roxy-2-(6-methoxy-
1-2C pYridin-3-yl)-ethylamino]-ethoxy}-phenyl)-oxazol-2-481.5
yl]-N-(2,2,2-trifluoro-ethyl)-acetamide
hydrochloride
salt
N-Ethyl-2-(4-{4-[2-(2(R)-hydroxy-2-pyridin-3-yl-
1-2D ethylamino)-ethoxy]-phenyl}-oxazol-2-yl)-N-(2,2,2-513.3
trifluoro-eth I - ro ionamide
N-Ethyl-2-(4-{4-[2-(2(R)-hydroxy-2-pyridin-3-yl-
1-2E ethylamino)-ethoxy]-phenyl}-oxazol-2-yl)-N-(2,2,2-541.3
trifluoro-eth I - ro ionamide h drochloride
salt
N-Ethyl-2-(4-{4-[2-(2(R)-hydroxy-2-pyridin-3-yl-
1-2F ethylamino)-ethoxy]-phenyl}-oxazol-2-yl)-N-(2,2,2-523.3
trifluoro-eth I - ro ionamide h drochloride
salt
N-Ethyl-2-(4-{4-[2-(2(R)-hydroxy-2-pyridin-3-yl-
1-2G ethylamino)-ethoxy]-phenyl}-oxazol-2-yl)-N-(2,2,2-507.3
trifluoro-eth I -acetamide tos late
salt
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-63
Example Compound Name LRMS
No. M+H
2-[4-(4-{2-[2-(6-Chloro-pyrid in-3-yl
)-2( R)-hyd roxy-
1-2H ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-507.3
dimeth I-but ramide h drochloride salt
2-[4-(4-{2-[2-(6-C h I o ro-pyrid i
n-3-yl )-2 ( R )-h yd roxy-
1-21 ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-pentanoic507.3
acid dimeth lamide h drochloride salt
2-[4-(4-{2-[2-(6-Chloro-pyridin-3-yl)-2(R)-hydroxy-
1-2J ethylamino]-ethoxy}-phenyl)-oxazol-2-yl)-hexanoic493.2
acid dimeth lamide h drochloride salt
2-[4-(4-{2-[2-(6-Ch loro-pyrid in-3-yl
)-2( R)-hyd roxy-
1-2K ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-4-methyl-473.1
entanoic acid dimeth lamide h drochloride
salt
2-[4-(4-{2-[2-(6-C h I o ro-pyrid i
n-3-yl )-2 ( R )-h yd roxy-
1-2L ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-487.1
dimeth I- ro ionamide h drochloride
salt
2-(4-{4-[2-(2 ( R )-H yd roxy-2-pyri
d i n-3-yl-ethyl a m i n o )-
1-2M ethoxy]-phenyl}-oxazol-2-yl)-N,N-dimethyl-501.2
but ramide h drochloride salt
2-(4-{4-[2-(2 ( R )-H yd roxy-2-pyri
d i n-3-yl-ethyl a m i n o )-
1-2N ethoxy]-phenyl}-oxazol-2-yl)-pentanoic 501.2
acid
dimeth lamide h drochloride salt
2-(4-{4-[2-(2(R)-Hyd roxy-2-pyrid in-3-yl-ethylamino)-
1-20 ethoxy]-phenyl}-oxazol-2-yl)-hexanoic 549.2
acid
dimeth lamide h drochloride salt
2-(4-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
1-2P ethoxy]-phenyl}-oxazol-2-yl)-4-methyl-pentanoic439.2
acid dimeth lamide h drochloride salt
2-(4-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
1-2Q ethoxy]-phenyl}-oxazol-2-yl)-N,N-dimethyl-453.2
ro ionamide h drochloride salt
2-[4-(4-{2-[2-( 6-C h I o ro-pyrid i
n-3-yl )-2 ( R )-h yd roxy-
1-2R ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-467.3
dimeth I-isobut ramide
2-(4-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
1-2S ethoxy]-phenyl}-oxazol-2-yl)-N,N-dimethyl-467.2
isobut ramide h drochloride salt
2-[4-(4-{2-[2-( 6-C h I o ro-pyrid i
n-3-yl )-2 ( R )-h yd roxy-
1-2T ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N-ethyl-N-425.2
2,2,2-trifluoro-eth I -isobut ramide
N-Ethyl-2-(4-{4-[2-(2(R)-hydroxy-2-pyridin-3-yl-
1-2U ethylamino)-ethoxy]-phenyl}-oxazol-2-yl)-N-(2,2,2-473.1
trifluoro-eth I -isobut ramide h drochloride
salt
2-(4-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
1-2V ethoxy]-phenyl}-oxazol-2-yl)-N,N-dimethyl-439.2
but ramide
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-64
Example Compound Name LRMS
No. M+H
2-(4-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
1-2W ethoxy]-phenyl}-oxazol-2-yl)-pentanoic 555.2
acid
dimeth lamide
2-(4-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
1-2X ethoxy]-phenyl}-oxazol-2-yl)-hexanoic 521.3
acid
dimeth lamide
2-(4-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
1-2Y ethoxy]-phenyl}-oxazol-2-yl)-4-methyl-pentanoic439.2
acid dimeth lamide
2-(4-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
1-2Z ethoxy]-phenyl}-oxazol-2-yl)-N,N-dimethyl-453.2
ro ionamide
2-(4-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
1-3A ethoxy]-phenyl}-oxazol-2-yl)-N,N-dimethyl-467.3
but ramide h drochloride salt
2-(4-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
1-3B ethoxy]-phenyl}-oxazol-2-yl)-pentanoic 467.2
acid
dimeth lamide h drochloride salt
2-(4-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
1-3C ethoxy]-phenyl}-oxazol-2-yl)-hexanoic 425.2
acid
dimeth lamide h drochloride salt
2-(4-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
1-3D ethoxy]-phenyl}-oxazol-2-yl)-4-methyl-pentanoic439.2
acid dimeth lamide h drochloride salt
2-(4-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
1-3E ethoxy]-phenyl}-oxazol-2-yl)-N,N-dimethyl-453.2
ro ionamide h drochloride salt
2-[4-(4-{2-[2(R)-Hydroxy-2-(5-methyl-pyridin-3-yl)-
1-3F ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-467.2
dimeth I- ro ionamide h drochloride
salt
2-[4-(4-{2-[2 ( R)-Hyd roxy-2-(6-methyl-pyrid
i n-3-yl )-
1-3G ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-467.2
dimeth I- ro ionamide h drochloride
salt
2-[4-(4-{2-[2(R)-Hydroxy-2-(6-methyl-pyridin-3-yl)-
1-3H ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-425.1
dimeth I-but ramide h drochloride salt
2-[4-(4-{2-[2(R)-Hydroxy-2-(5-methyl-pyridin-3-yl)-
1-31 ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-439.0
dimeth I-but ramide h drochloride salt
2-[4-(4-{2-[2(R)-Hydroxy-2-(5-methyl-pyridin-3-yl)-
1-3J ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-439.0
dimeth I-isobut ramide h drochloride
salt
2-[4-(4-{2-[2(R)-Hydroxy-2-(6-methyl-pyridin-3-yl)-
1-3K ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N---
dimeth I-isobut ramide h drochloride
salt
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-65
Example Compound Name LRMS
No. M+H
N-Ethyl-2-(4-{4-[2-(2(R)-hydroxy-2-pyridin-3-yl-
1-3L ethylamino)-ethoxy]-phenyl}-oxazol-2-yl)-N-(2,2,2-453.0
trifluoro-eth I -acetamide
2,2-Difluoro-2-[4-(4-{2-[2(R)-hydroxy-2-(6-methyl-
1-3M pyridin-3-yl)-ethylamino]-ethoxy}-phenyl)-oxazol-2-453.3
I -N,N-dimeth I-acetamide
2-[4-(4-{2-[2(R)-Hydroxy-2-(6-methyl-pyridin-3-yl)-
1-3N ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-453.3
dimeth I- ro ionamide
2-[4-(4-{2-[2 ( R )-H yd roxy-2-(6-methyl-pyri
d i n-3-yl )-
1-30 ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-493.2
dimeth I-but ramide
2-(4-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
1-3P ethoxy]-phenyl}-oxazol-2-yl)-N,N-dimethyl-461.4
acetamide
2-[4-(4-{2-[2 ( R )-H yd roxy-2-(6-m
ethyl-pyri d i n-3-yl )-
1-3Q ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-439.3
dimeth I-acetamide h drochloride salt
2-[4-(4-{2-[2(R)-Hydroxy-2-(6-methyl-pyridin-3-yl)-
1-3R ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-453.3
dimeth I-acetamide tos late salt
2-[4-(4-{2-[2(R)-Hydroxy-2-(6-methyl-pyridin-3-yl)-
1-3S ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-411.2
dimeth I-acetamide mes late salt
2-[4-(4-{2-[2(R)-Hydroxy-2-(6-methyl-pyridin-3-yl)-
1-3T ethylaminoJ-ethoxy}-phenyl)-oxazol-2-yl]-N-methyl-425.0
acetamide
Example 2 illustrates the preparation of compounds of the present
invention where Ar is a phenyl group.
Example 2
Preparation forty-{5 f2-~(~4-~2-(2-Azetidin-1-yl-2-oxo-ethyl)-oxazol-4-y11-
phenoxy}-ethylamino'~,(R)-hydroxy-ethyl)-2-chloro-phenyl,~
methanesulfonamide hydrochloride salt ~(2-1A,~
~NJ
N.N II
O I / O
II H OH H ~
O S_N ~ NCO
I~
CI
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-66
2-1 A
In a round-bottomed flask, (R)-N-(2-chloro-5-oxiranyl-phenyl)-
methanesulfonamide (18 mg, 0.073 mmol) and 2-{4-(4-(2-amino-ethoxy)-
phenyl]-oxazol-2-yl)-1-azetidin-1-yl-ethanone (33 mg, 0.11 mmol) were
dissolved in 0.7 mL of ethanol, and the mixture was heated to 80~ C for 12
hours. The solution was then concentrated in vacuo to an oil, and the crude
material was purified by column chromatography (2
methanol/dichloromethane to 7 % methanol/dichloromethane) to afford 16 mg
(40 % yield) of the coupled product as a white solid. This material was
dissolved in dichloromethane and ethyl acetate (1:1 ), and 0.06 mL of 1 N HCI
in ether was added to the solution to afford the HCI salt. This solution was
concentrated to give the title compound as a yellow solid. LRMS ([M+H]+):
349.1. 'H NMR: (400 MHz, CD30D): b 2.33 (m, 2H), 2.98 (s, 3H), 3.19 (m,
1 H), 3.28 (s, 2H), 3.33 (m, 1 H), 3.54 (m, 2H), 4.04 (m, 2H), 4.33 (m, 4H),
5.02
(m, 1 H), 7.05 (d, 2H, J = 8.8 Hz), 7.31 (m, 1 H), 7.50 (d, 1 H, J = 8.4 Hz),
7.64
(d, 1 H, J = 1.6 Hz), 7.69 (d, 2H, J = 8.4 Hz), 8.13 (s, 1 H).
Table II below lists compounds having the following general structure
that were prepared using the general procedures described above for the
preparation of compound 2-1A with the appropriate starting materials.
TABLE II
Example Compound Name LRMS
No. M+H
N-{5-[2-(2-{4-[2-(2-Azetidin-1-yl-2-oxo-ethyl)-
2-1A oxazol-4-yl]-phenoxy}-ethylamino)-1 (R)-hydroxy-549.1
ethyl]-2-chloro-phenyl}-methanesulfonamide
h drochloride salt
2-[4-(4-{2-[2-(3-Chloro-phenyl)-2(R)-hydroxy-
2-1B ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N-472.4
iso ro I-N-meth I-acetamide
2-[4-(4-{2-[2-(3-Chloro-phenyl)-2(R)-hydroxy-
2-1C ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N-ethyl-N-4$6.2
iso ro I-acetamide
2-[4-(4-{2-[2-(3-Chloro-phenyl )-2( R)-hyd
roxy-
2-1 D ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-472.0
dieth I-acetamide
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-67
Example Compound Name LRMS
No. M+H
2-[4-(4-{2-[2-(4-Chloro-3-methanesulfonylamino-
2-1E phenyl)-2(R)-hydroxy-ethylamino]-ethoxy}-phenyl)-581.5
oxazol-2- I -N-eth I-N-iso ro I-acetamide
2-[4-(4-{2-[2-(4-Chloro-3-methanesulfonylamino-
2-1 F phenyl)-2(R)-hydroxy-ethylamino]-ethoxy}-phenyl)-565.5
oxazol-2-yl]-N-isopropyl-N-methyl-acetamide
h drochloride salt
2-[4-(4-{2-[2-(3-Chloro-phenyl)-2(R)-hydroxy-
2-1G ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N-ethyl-N-526.4
2,2,2-trifluoro-eth I -acetamide h drochloride
salt
2-[4-(4-{2-[2-(3-Chloro-phenyl)-2(R)-hydroxy-
2-1H ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-500.4
diiso ro I-acetamide h drochloride slat
2-[4-(4-{2-[2-(4-B a nzyl oxy-3-
2-11 di(methanesulfonyl)amino-phenyl)-2(R)-hydroxy-715.7
ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-
dieth I-acetamide
2-[4-(4-{2-[2-(4-Benzyloxy-3-methanesulfonylamino-
2-1J phenyl)-2(R)-hydroxy-ethylamino]-ethoxy}-phenyl)-637.6
oxazol-2- I -N,N-dieth I-acetamide
N , N-D i eth yl-2-[4-(4-{2-[2 ( R )-h
yd roxy-2-(4-h yd roxy-3-
2-1 K methanesulfonylamino-phenyl)-ethylamino]-ethoxy}-547.5
hen I -oxazol-2- I -acetamide h drochloride
salt
2-[4-(4-{2-[2-(3-Chloro-phenyl)-2(R)-hydroxy-
2-1 L ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N,N-444.2
dimeth I-acetamide
N-Ethyl-2-[4-(4-{2-[2 ( R )-h yd roxy-2-(3-
2-1 M methanesulfonylamino-phenyl)-ethylamino]-ethoxy}-545.6
hen I -oxazol-2- I -N-iso ro I-acetamide
N-Ethyl-2-[4-(4-{2-[2( R)-hyd roxy-2-(4-hyd
roxy-3-
2-1 N methanesulfonylamino-phenyl)-ethylamino]-ethoxy}-561.2
phenyl)-oxazol-2-yl]-N-isopropyl-acetamide
h drochloride salt
2-[4-(4-{2-[2-(3-Chloro-phenyl)-2(R)-hydroxy-
2-10 ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N-methyl-512.2
N- 2,2,2-trifluoro-eth I -acetamide
h drochloride salt
2-[4-(4-{2-[2-(4-Benzyloxy-3-
2-1P di(methanesulfonyl)amino-phenyl)-2(R)-hydroxy-728.3
ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-N-ethyl-N-
iso ro I-acetamide
2-[4-(4-{2-[2-(4-Benzyloxy-3-methanesulfonylamino-
2-1Q phenyl)-2(R)-hydroxy-ethylamino]-ethoxy}-phenyl)-651.3
oxazol-2- I -N-eth I-N-iso ro I-acetamide
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-68
Example Compound Name LRMS
No. M+H
2-[4-(4-{2-[2(R)-Hyd roxy-2-(3-trifluoromethyl-
2-1 R phenyl)-ethylamino]-ethoxy}-phenyl)-oxazol-2-yl]-4-534.3
methyl-pentanoic acid dimethylamide
hydrochloride
salt
Example 3 illustrates the preparation of compounds of the present
invention where HET is a pyrazole.
Example 3
Preparation for 2-f3-(4-~2-f2-(6-Chloro-pyridin-3-yl)-2(R)-hydrox~i-
ethylaminol-
ethoxyj-phenyl)-pyrazol-1-yll-1-pyrrolidin-1 yl-ethanone (3-1A):
~N
N-N II
O
OH ~
NCO
C. N
3-1 A
In a round-bottomed flask, (R)-2-chloro-5-oxiranyl-pyridine (23 mg, 0.15
mmol) and 2-{4-[1-(2-amino-ethoxy)-phenyl]-pyrazol-1-yl}-1-pyrrolidin-1-yl-
ethanone I-4f (71 mg, 0.23 mmol) were dissolved in 1.5 mL of ethanol, and the
mixture was heated to 80~ C for about 16 hours. The solution was then
concentrated in vacuo to an oil, and the crude material was purified by column
chromatography (dichloromethane to 11 % methanol/dichloromethane) to afford
45 mg (63 % yield) of the title product as a white solid. LRMS ([M+H]+):
470Ø
'H NMR: (400 MHz, CD30D): b 1.91 (m, 2H), 2.03 (m, 2H), 2.88 (m, 2H), 3.05
(m, 2H), 3.46 (t, 2H, J = 6.8 Hz), 3.59 (t, 2H, J = 6.7 Hz), 4.12 (m, 2H),
4.86 (m,
1 H), 5.04 (s, 2H), 6.59 (d, 1 H, J = 4.5 Hz), 6.96 (d, 2H, J = 8.0 Hz), 7.43
(d, 1 H,
J = 8.4 Hz), 7.62 (d, 1 H, J = 2.8 Hz), 7.69 (d, 2H, J = 8.0 Hz), 7.85 (dd, 1
H, J =
2.5, 8.3 Hz), 8.35 (d, 1 H, J = 2.2 Hz).
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-69
Table I II below lists compounds that were prepared using the general
procedures described above for the preparation of compounds 3-1 A with the
appropriate starting materials.
TABLE III
Example Compound Name LRMS
No. M+H ''
2-[3-(4-{2-[2-(6-Chloro-pyridin-3-yl)-2(R)-hydroxy-
3-1A ethylamino]-ethoxy}-phenyl)-pyrazol-1-yl]-1-470.0
rrolidin-1- I-ethanone
2-[3-(4-{2-[2-(6-Chloro-pyridin-3-yl)-2(R)-hydroxy-
3-1B ethylamino]-ethoxy}-phenyl)-pyrazol-1-yl]-1-486.0
mor holin-4- I-ethanone
2-(3-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
3-1C ethoxy]-phenyl}-pyrazol-1-yl)-1-pyrrolidin-1-yl-436.3
ethanone h drochloride salt
2-(3-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
3-1D ethoxy]-phenyl}-pyrazol-1-yl)-1-morpholin-4-yl-452.3
ethanone h drochloride salt
2-[3-(4-{2-[2-(6-Chloro-pyridin-3-yl)-2(R)-hydroxy-
3-1E ethylamino]-ethoxy}-phenyl)-pyrazol-1-yl]-1-484.2
i eridin-1- I-ethanone
2-[3-(4-{2-[2-(6-C h I o ro-pyrid i n-3-yl
)-2 ( R )-h yd roxy-
3-1F ethylamino]-ethoxy}-phenyl)-pyrazol-1-yl]-N-484.2
c clo ent I-acetamide
2-[3-(4-{2-[2-(6-Chloro-pyridin-3-yl)-2(R)-hydroxy-
3-1G ethylamino]-ethoxy}-phenyl)-pyrazol-1-yl]-N-ethyl-N-458.2
meth I-acetamide
N-Cyclopentyl-2-(3-{4-[2-(2(R)-hydroxy-2-pyridin-3-
3-1H yl-ethylamino)-ethoxy]-phenyl}-pyrazol-1-yl)-450.4
acetamide h drochloride salt
N-Ethyl-2-(3-{4-[2-(2(R)-hydroxy-2-pyridin-3-yl-
3-11 ethylamino)-ethoxy]-phenyl}-pyrazol-1-yl)-N-methyl-424.4
acetamide h drochloride salt
2-[3-(4-{2-[2-(6-Chloro-pyridin-3-yl)-2(R)-hydroxy-
3-1J ethylamino]-ethoxy}-phenyl)-pyrazol-1-yl]-N,N-444.2
dimeth I-acetamide
2-(3-{4-[2-(2(R)-Hydroxy-2-pyridin-3-yl-ethylamino)-
3-1K ethoxy]-phenyl}-pyrazol-1-yl)-N,N-dimethyl-410.4
acetamide h drochloride salt
BIOLOGICAL ASSAYS
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-70
The utility of the compounds of the present invention, in the practice of
the methods of the instant invention, can be evidenced by activity in at least
one
of the protocols described in detail below.
Assay 1
X33 Receptor Selectivity Over /3~ and ,a2 Adrenergic Receptors
In vitro ~3 receptor agonist activity and selectivity over ~~ and ~2
adrenergic receptors may be determined by measurement of cyclic adenosine
monophosphate (CAMP) accumulation in Chinese hamster ovary cells.
Chinese hamster ovary cells uniquely transfected with the cDNA for the
human ~~, ~i2, or X33 adrenergic receptor are grown to confluence in Ham's F12
media (Gibco BRL, Life Technologies, Inc., Grand Island, NY) containing 10%
fetal bovine serum, 500 mg/ml geneticin, 100 U/ml penicillin, 100 mg/ml
streptomycin, and 250 ng/ml fungizone according to the procedure described in
American Type Culture Catalog of Cell Lines and Hybridomas, Seventh Edition,
1992, p. 36, ATCC CCL 61 CHO-K1. Compounds are prepared as 25 mM stock
solutions in DMSO (0.1 % DMSO final concentration), diluted in Ham's F12
media and added to the cells at 10-'° to 10'5 M along with 10-5 M
isobutylmethylxanthine to inhibit phosphodiesterase activity. The media and
cells are then incubated for sixty minutes at 37~ C. At the end of the
incubation
period, the media is aspirated and the cells lysed in 0.01 N HCI. The cellular
content of cAMP is then determined by radioimmunoassay (RIA) using a kit from
New England Nuclear (Burlington, MA). There is a direct correlation between
the
cellular content of cAMP and the agonism of the ~3~, ~i2, or ~i3 adrenergic
receptor. The non-selective, full (3-adrenergic agonist isoproterenol is
included
as a positive control at 10-5 M.
A range of EC5° values from 13 ~.M to 155 ~M were observed for the
compounds listed in Examples 1, 2 and 3 (Example 1-1A through Example 3-
1 K). As a specific example, the compound of Example 3-1 H had an EC5°
of 88
~M. Example 3-1 H was chosen for illustrative purposes only and does not imply
that the compound of Example 3-1 H is a preferred compound.
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-71
Assay 2
Many G protein-coupled receptors (GPCRs) exhibit at least two agonist
affinity states. High affinity agonist binding to GPCRs requires the
association
or coupling of the receptor with the GDP-bound heterotrimeric G protein
complex. In general, the low affinity agonist binding site is indicative of
the
uncoupled receptor state. The high affinity agonist binding site can be
converted
to the low affinity site by addition of GTP or its analogs. In the absence of
agonist, G proteins display high affinity for GDP. In the presence of agonist,
G
proteins display high affinity for GTP. Thus, when agonist and GTP are added
to
the receptor/G protein complex, GTP displaces GDP and uncouples the
receptor from the G protein. Two affinity states for agonists can be detected
in
radioligand competetion binding assays. A two-site fit is generally observed
for
agonists for many GPCRs and can be calculated using commercially available
software. The high affinity site (K;H) corresponds to the G protein-coupled
state
and, in the case of [i3-adrenergic receptors correlates well with the
functional
EDSO for stimulation of cAMP accumulation.
In order to identify compounds that attenuate the binding of
['251]cyanopindolol (ICYP) to [33 adrenergic receptors, the following
radioligand
binding assay can be used.
Radioligand Binding Assays
ICYP,~3 Adrenergic Receptor Competition Binding Assay
The specific activity of ['251]ICYP is 2000 Ci/mmole. ICYP undergoes
catastrophic decay upon radiolysis. Therefore, the specific activity always
remains at 2000 Ci/mmole, but the concentration will decrease over time. The
final concentration of ICYP is 250 pM. Therefore, a 2.5 nM (10 x) stock needs
to
be made. ['251]CYP can be obtained from New England Nuclear, Boston, MA.
Competitors
Up to four compounds can be tested in thirteen competition curves in a
96 well format. An example for a single compound is outlined below.
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-72-
[Comp 1]
A -10
1,2
B -9.3
1,2
C -9
1,2
D -8.3
1,2
E -8
1,2
F -7.3
1,2
G -7
1,2
H -6.3
1,2
A -6
3,4
B -5
3,4
C -4
3,4
D pindolol
1,3
E TOTAL
3,4
The next compound would begin in F 3,4. Two pairs of totals and non-
specific binding are added to the plates. Wells E 3,4 and G 7,8 are for total
cpm
bound. Wells D 3,4 and H 7,8 are for 100 ~M pindolol to determine non-specific
binding.
To each well in order add: 20 ~,I buffer to "total" wells; 20 ~I 1 mM
pindolol to pindolol wells; 20 wl of each concentration of compound to the
appropriate wells; 20 ~I of 2.5 nM ICYP to all wells; and 160 ~,I membranes
diluted to 15 ~g/160 pl.
Procedure
1. Set up assay for Packard 96 well Unifilter with GF/C filters (Packard;
Meriden, CT) using a 96 well microtiter plate.
2. Incubate 90-120 minutes with shaking at room temperature
3. Using Packard cell harvester (Packard; Meriden, CT), aspirate
samples into processing head. Use a pre-soaked (0.3% PEI) filter.
4. Wash four times with cold wash buffer.
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-73
5. Dry plate, and add 25 pl Microscint (ICN Manufacturers; Costa Mesa,
CA) to each well.
6. Count samples in Wallac beta plate reader (Wallac; Turku, Finland).
Binding Buffer: 50 mM Hepes/10 mM MgCl2, pH 7.4 (prepared from 10 x
stock solution) and 0.2 % BSA (fraction V)
Protease inhibitors (prepared as 100 x stock solution): 100 ~g/ml
bacitracin; 100 ~g/ml benzamidine; 5 ~.g/ml aprotin; and 5 ~g/ml leupeptin.
Wash Buffer: 50 nM Hepes/10 mM MgCl2, pH 7.4, ice cold (prepared
from 10 x stock solution)
Assay 3
Oxygen Consumption
As will be well known to one of ordinary skill in the art, during increased
energy expenditure, animals generally consume increased amounts of oxygen.
In addition, metabolic fuels such as, for example, glucose and fatty acids,
are
oxidized to C02 and H20 with the concomitant evolution of heat, an effect
commonly referred to in the art as thermogenesis. Accordingly, the
measurement of oxygen consumption in animals, including humans and
companion animals, is an indirect measure of thermogenesis, and indirect
calorimetry may be commonly used in animals, e.g., humans, by one of ordinary
skill in the art, to measure such energy expenditures.
The ability of the compounds of Formula (I), the stereoisomers and
prodrugs thereof, and the pharmaceutically acceptable salts of the compounds,
stereoisomers, and prodrugs, to generate a thermogenic response may be
demonstrated according to the following protocol using male Sprague-Dawley
rats (Charles River, Wilmington, MA).
Whole animal oxygen consumption may be measured using an open
circuit, indirect calorimeter (OxymaxTM, Columbus Instruments, Columbus, OH).
The gas sensors are calibrated with nitrogen gas and gas mixture (0.5% carbon
dioxide, 20.5% oxygen, 79% nitrogen; Abco Industrial Supplies, Waterford, CT)
before each experiment. Male Sprague-Dawley rats (300 - 380 g body weight)
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-74
are placed in sealed chambers (43 x 43 x 10 cm) of the calorimeter and the
chambers placed in activity monitors. Air flow rate through the chambers is
set
at 1.6 - 1.7 I/min. The calorimeter software calculates the oxygen consumption
(ml/kg/hour) based on the flow rate of air through the chambers and the
difference in oxygen content at inlet and outlet ports. The activity monitors
have
fifteen infrared light beams spaced one inch apart on each axis; ambulatory
activity is recorded when two consecutive beams are broken (repeated
interruptions of the same beam are not registered) and the results are
recorded
as counts. Basal oxygen consumption and ambulatory activity are measured
every ten minutes for two and one-half to three hours. At the end of the basal
period, the chambers are opened and the test compound (0.01 - 20 mg/kg,
prepared in water, 0.5% methyl cellulose, or other suitable vehicle) or an
equivalent amount of vehicle is administered by oral gavage. Oxygen
consumption and ambulatory activity are measured every ten minutes for an
additional two to six hours post-dosing. Percent change in oxygen consumption
is calculated by averaging the post-dosing values and dividing by basal oxygen
consumption (average of the pre-dosing values except the first hour). Oxygen
consumption values obtained during time periods where ambulatory activity
exceeds 100 counts are excluded from the calculation. Thus, the values
represent % change in resting oxygen consumption.
Assay 4
HVpoc~lycemic Activity
The compounds of the present invention may be tested for hypoglycemic
activity accoprding to the following procedure, and as an aid in determining
dosages when compared to other test compounds and standards.
Five to eight-week old C57 BL/6J-ob/ob mice (Jackson Laboratory, Bar
Harbor, ME) are housed five animals per cage at an ambient temperature of 66
~C under standard animal care practices. After a one week acclimation period,
the animals are weighed and 25 microliters of blood is collected via an
occular
bleed prior to any treatment. The blood sample is immediately diluted 1:5 with
saline containing 2% sodium heparin, in tubes held on ice. Blood samples are
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-75
centrifuged for two minutes to remove red blood cells and the supernatant is
analyzed for glucose concentration using a clinical autoanalyzer (Abbott
Spectrum~ CCx; Abbott Laboratories, Abbott Park, IL). Animals are then
regrouped, in groups of five animals per cage, such that the mean glucose
values of the groups are similar. The mice are then dosed once or twice daily
for
five days with test compound (0.01 - 20 mg/kg), with a positive control such
as
englitazone or ciglitazone (50 mg/kg p.o.) (U.S. Pat. No. 4,467,902; Sohda et
al., Chem. Pharm. Bull., 32, 4460-4465, (1984)), or with vehicle. All
compounds
are administered by oral gavage in a vehicle consisting of 0.5% w/v methyl
cellulose, or with other suitable vehicle. On Day 5, the animals are weighed
again and bled (via the occular route) for blood glucose levels as described
hereinabove. Plasma glucose is then calculated by the equation:
Plasma Glucose (mg/dl) = Sample Value x 5 x 1.67 = 8.35 x Sample
Value, where 5 is the dilution factor and 1.67 is the plasma hematocrit
adjustment (assuming the hematocrit is 40%).
The animals dosed with vehicle maintain substantially unchanged
hyperglycemic glucose levels (e.g. 300 mg/dl), while positive control animals
have depressed glucose levels (e.g. 130 mg/dl). The glucose lowering activity
of
test compounds is expressed in terms of % glucose normalization. For example,
a glucose level which is the same as the positive control is expressed as
100%.
Assay 5
~3, and ~3z Receptor Selectivity
In vivo selectivity for ~i~ and ~3z receptors may be determined by
measurements of heart rate, blood pressure, and plasma potassium
concentration gathered on conscious catheterized rats (male, Sprague-Dawley,
300-400 g body weight). To implant catheters, rats are anesthetized with
pentobarbital (50-60 mg/kg i.p.) and the left carotid artery is cannulated
with
PE50 tubing. The catheter is tunneled subcutaneously, exteriorized at the back
of the neck, filled with a solution of polyvinylpyrrolidone in heparinzied
saline,
flame sealed, and taped. Experiments are performed seven days after surgery.
On the day of the experiment, the catheters are untaped and flushed with
saline.
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-76
After at least thirty minutes, basal values for heart rate and blood pressure
are
measured by attaching the catheter to a pressure transducer, the results
recorded on a Grass Model 7 polygraph (Grass Medical Instruments, Quincy,
MA), and a basal blood sample (0.5 ml) is obtained from the arterial catheter.
After obtaining basal values, the test compound or vehicle is administered by
oral gavage and blood pressure (measure of ~2 activity) and heart rate
(measure
of (3~ activity) measurements are taken at 15, 30, 45, and 60 minutes, and
blood
samples for potassium determination (~i2) are obtained at 30 and 60 minutes.
Isoproterenol, a non-selective ~i-agonist, can be tested as a positive control
at
doses ranging from 0.001 to 1 mg/kg (injected s.c. in saline vehicle). Plasma
potassium is determined by flame spectrophotometry. To determine changes,
basal values are subtracted from the average of the post-dosing values.
Assay 6
Reducing Intestinal Motility
The compounds of Formula (I) have the effect of reducing intestinal
motility and thus have utility in aiding in the treatment of various
gastrointestinal
disorders such as irritable bowel syndrome, peptic ulceration, esophagitis,
gastritis, duodenitis (including that induced by Helicobacter pylon,
intestinal
ulcerations (including inflammatory bowel disease, ulcerative colitis, Crohn's
Disease and proctitis), and gastrointestinal ulcerations. It has been proposed
that the motility of non-sphincteric smooth muscle contraction is mediated by
activity at ~3 adrenergic receptors. The availability of a ~i3 specific
agonist, with
little activity at ~3~ and ~i2 receptors, will assist in the pharmacologic
control of
intestinal motility without concurrent cardiovascular effects.
In vivo activity of the compounds of Formula (I) for the treatment or
prevention of intestinal motility disorders can be determined according to the
following procedures. Eighteen-hour fasted male Sprague-Dawley derived (CD)
rats (175 - 225 g) are dosed with 0.01 - 20 mg/kg p.o. of test compound or
vehicle (distilled water). Thirty minutes after administration of test
compound,
the rats are orally dosed with 0.25 ml of a solution of sodium chromate in
0.9%
saline containing about 20,000 cpm of S~Cr (specific activity 350 mCi/mg Cr).
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-77
Twenty minutes later, the rats are sacrificed, the gastroesophageal, pyloric,
and
ileocecal junctions are then ligated, and the stomachs and small intestines
are
removed. The small intestines are then divided into ten equal lengths, and the
stomach and each length of intestine assayed for radioactivity with a gamma
counter. Gastric emptying rate may then be determined for each rat by
comparing the amount of radioactivity in the intestine relative to the total
in the
intestine plus stomach. In addition, the geometric center of the distribution
of the
radioactive marker is then used as a measure of the overall transit rate
through
the stomach and intestine. The geometric center is calculated by summing the
products of the fractions of 5'Cr in each segment times the segments number:
geometric center = S ((fraction of 5'Cr per segment) x (segment number)). For
these calculations, the stomach is considered segment number 0, and and the
ten intestinal segments as numbers 1 to 10. Thus, a geometric center of 0.0
indicates that the entire load of 5'Cr remains in the stomach. Data from the
two
experiments are pooled, and statistical evaluations are made using Dunnett's
multiple comparison test.
Alternatively, in groups of eight, overnight-fasted male Sprague-Dawley
(CD) rats (175 - 225 g) may be anesthetized with methoxyflurane. A small
abdominal incision is then made, and the pylorus ligated. Immediately after
the
ligation, a solution of the test compound or vehicle (distilled water) is
injected
into the proximal duodenum. The doses of test compound used should be 0.01
20 mg/kg body weight. The incisions are then closed and the rats allowed to
recover from the anesthesia. Two hours after the ligation, the rats are
sacrificed
and the gastric fluid collected and cleared by centrifugation. Total volume of
secretion is determined by weight, and acidity is determined by titration to
pH
7.0 with 0.1 N sodium hydroxide using an automatic titrator. The data from two
experiments are then pooled. A group of rats treated with 10 mg/kg of of the
anti-secretory histamine H2-receptor antagonist cimetidine may be included as
a
positive control. Statistical evaluations can be made using Student's t-test.
In vitro activity for relaxation of contracted ileum from isolated guinea pig
ileum is determined according to the following procedures. Fresh, isolated
segments of guinea pig ileum (about 1.5 cm in length) are mounted in tissue
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-78
baths containing Tyrode's physiological salt solution at about 30~ C and
aerated
continuously with oxygen:carbon dioxide (95%:5%). Tissues are then
equilibrated for 60 - 90 minutes under 4.0 gm tension in order to achieve
stable
baselines. Histamine is then added to the baths and in a cumulative fashion in
concentrations ranging from 1 nM to 10 mM. The maximum tension generated
after each addition of histamine is recorded on a Grass Physiograph (Grass
Medical Instruments, Quincy, MA). The tissues are then washed with several
changes of Tyrode's solution, basal tension is readjusted to 4.0 gm, and a
stable
baseline is then again obtained. Each tissue is then exposed to a single
concentration of a test compound (1 nM - 10mM) or vehicle and, after a thirty
minute equilibration period, the histamine dose response curve is then
repeated.
Results from multiple experiments are standardized (0-100%) to the maximum
response of the control tissues and plotted as percent maximum tension vs. the
log of the histamine concentration in the absence and presence of the test
compound.
Assay 7
Protection Against Gastric Ulceration
Food (but not water) is withheld from female Sprague-Dawley rats
(Charles River, Wilmington, MA) weighing 70 - 120 g. Access is then permitted
to food for ninety minutes. A single dose of test compound is then
administered
p.o. (0.01-20 mg/kg in a dosing volume of 1 ml/100 g), and indomethacin
(Sigma Chemical Co., St. Louis, MO) (60 mg/kg, 1 ml/100 g body weight) is then
injected subcutaneously. Control rats receive the subcutaneous injection of
indomethacin and oral administration of vehicle (0.5% methyl cellulose in
distilled water) for the ~i-adrenoceptor agonist. The animals are then allowed
continued access to food but water is withdrawn. The animals are then
sacrificed by cervical dislocation six hours after dosing with indomethacin.
The
stomach are then removed, opened along the greater curvature and washed in
0.9% saline. An assessment of gastric damage is carried out by an observer
who is unaware of the dosing regimen. A transparent plastic grid divided into
1
mm2 sections is placed over the antrum and the area of macroscopic damage
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
_79_
assessed as the total area of visible lesions in mm2. This value is then
expressed as a percentage of the total antral area.
Assay 8
Anti-Depressant Activity
Male CD1 mice weighing between 20 and 25 g, and Sprague-Dawley rats
weighing between 200 and 250 g are obtained from Charles River, Wilmington,
MA. Test compounds of Formula (I) are dissolved in water. The compounds are
administered to mice in a volume of 10 ml/kg, and to rats in a volume of 2
ml/kg.
Control animals receive the vehicle. Positive test results for the following
parameters indicate anti-depressant activity.
(1 ) Antagonism of Hypothermia Induced by Reserpine
Mice are administered reserpine (2.5 mg/kg i.p. dissolved in 1 % citric
acid). Their rectal temperatures are measured three and one-half hours later.
The mice are then divided into different groups so as to obtain the same mean
rectal temperature in each group. One=half hour later, (i.e., four hours after
reserpine administration), the mice are given the vehicle or test compound.
Rectal temperature is measured again ninety minutes later (i.e., five hours
and
thirty minutes after reserpine administration) (Bourin, et al., The Value of
the
Reserpine Test in Psychopharmacology, Arzneim. Forsch., 33, 1173, (1983)).
(2) Antaqonism of Hypothermia Induced by Apomorphine
One-half hour after the mice are placed in individual cages, their rectal
temperatures are recorded. The animals are allocated so as to obtain the same
mean rectal temperature in each group. Apomorphine (16 mg/kg s.c.) is given
thirty minutes after the test compound or vehicle. Rectal temperature is then
measured again thirty minutes after the apomorphine treatment (Puech, et al.,
Antagonism of Hypothermia and Behavioral Response to Apomorphine; A
Simple, Rapid, and Discriminating Test for Screening Anti-Depressants and
Neuroleptics, Psychopharmacology, 75, 84, (1981 )).
(3) Effect on Learned Helplessness Behavior
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-80
This test is performed essentially as described by Giral, et al., Reversal of
Helpless Behavior in Rats by Putative 5-HT~A Agonists, Biol. Psychiat., 23,
237
(1988). Electric footshocks are delivered to male albino Sprague-Dawley rats
placed in chambers (20 x 10 x 10) with Plexiglass~ walls and covers. The
floors
are made of stainless-steel grids (1.5 cm mesh). A constant-current shock is
delivered as sixty scrambled, randomized inescapable shocks (15 sec. duration,
0.8 mA, every 60+15 sec.) to the grid floor. Control rats are then placed in
identical chambers, but no shock is administered. All preconditioning trials
are
performed on Day 1 between 9 and 11 a.m. Avoidance training is initiated 48
hours (Day 3) after inescapable shock in automated two-way shuttle boxes (60 x
21 x 30 cm) with Plexiglass~ walls and a floor consisting of stainless-steel
rods
spaced 1.0 cm apart in order to evaluate escape deficits. Each shuttle box is
divided into two chambers of equal size by a stainless-steel partition with a
gate
providing access to the adjacent compartment through a 7 x 7 cm space.
Shuttle box sessions are performed for three consecutive days (Days 3, 4, and
5). The animals are placed individually in the shuttle box and allowed to
habituate to the environment for five minutes (for the first session only) and
then
subjected to thirty trials. The intertrial interval should be thirty seconds.
A light
signal, used as a conditioned stimulus, is presented during the first three
seconds of each trial. Crossing the gate into the other compartment of the box
during this "conditioned stimulus only" period (referred to as avoidance
response) allows rats to avoid shocks. A period with conditioned stimulus plus
foot-shock (0.8 mA) may be presented if an avoidance response does not occur.
Crossing the gate into the other compartment during this conditioned stimulus
plus shock period is referred to as an escape response. Absence of escape
response during the three-second duration conditioned stimulus plus shock is
considered to be an escape failure.
The rats (n = 10 per group) are treated randomly according to one of the
following protocols: the control sample, which receives no shock, and is given
only vehicle, or experimental animals with inescapable shock are treated daily
with vehicle or test compound. Animals are treated orally over five
consecutive
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-81
days, i.e. six hours after shock pretreatment on Day 1, and then twice per
day, a
half dose in the morning (30 minutes before shuttle box session) and half a
dose
in the afternoon (except on day 5). Statistical analysis is performed on the
mean
number of escape failures using a two-way analysis of variance (subjects x
sessions) followed by Dunnett's test.
Assay 9
Bronchial Relaxation and Ciliary Motility
In vitro activity of the compounds of Formula (I) for the treatment of
airway inflammatory disorders, such as asthma and obstructive lung disease,
may be determined by measurement of guinea pig bronchial ring relaxation
according to the following procedure.
Guniea pig bronchial rings are obtained from tri-colored guinea pigs of
either sex (250 - 350 g), anesthized with urethane (1.25 g/kg) and suspended
under an initial tension of 2.0 g in Krebs solution at 37~ C gassed with 95%
oxygen:5% carbon dioxide. After about one hour of equilibration, the guinea
pig
bronchial rings are contracted with acetylcholine (10-3 M), relaxed to maximal
relaxation with theophylline (10-3 M), and then allowed to equilibrate for a
further
sixty minutes while they are washed with Krebs solution every fifteen minutes.
Changes in tension are measured isometrically with strain guages and
amplifiers and displayed on a recorder. The composition of the Krebs solution
is
(mM):NaCI 118.0, FCI 5.4, CaCl2, 2.5, KHP04 1.2, MgS04 1.2, NaHC03 25.0,
and glucose 11.7.
To test effects of test compounds on resting tension, cumulative
concentration-response curves are obtained by addition of the test compounds
(10-9 - 10-6 M) every ten to twenty minutes until a plateau is reached. The
relaxant effects of the test compounds are expresed as percentages of the
maximal relaxations induced by theophylline (3 x10-3 M).
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-82
Assay 10
Prostate Disease
Ventral prostates of male Sprague-Dawley rats (300 - 400 g)
anesthetized with diethyl ether are quickly excised and placed in oxygenated
Krebs solution. While maintained at room temperature in this buffer, adherent
fatty and connective tissues are removed. The prostates are then suspended in
ml organ baths containing Krebs solution warmed to 37°C and aerated
with a
mixture of 95% oxygen and 5% carbon dioxide. The composition of the Krebs
solution is 118.4 mM NaCI, 4:7 mM KCI, 1.2 mM MgS04, 2.5 mM CaCl2, 11.1
mM dextrose, 25.0 mM NaHC03 and 1.2 mM KH2P04, dissolved in distilled and
demineralized water. The tissues are attached to isometric force-displacement
transducers and isometric contraction is recorded under a loading tension of
0.5g. Equilibration is undertaken for one or two hours before the addition of
test
compounds. Submaximal contractions are first elicited by repeated
concentrations of 1 x 10-6M phenylephrine until constant responses are
obtained. The control and test compound-treated experiments are performed in
different preparations. A concentration-response curve to cumulate
concentrations of phenylephrine or acetylcholine (10-9 to 10~M) is determined.
For testing compounds, a concentration response curve to phenylephrine or
acetylcholine is determined in the presence of the compounds.
In vitro activity of compounds of Formula (I) can also be determined for
specific efficacy in human prostate as follows.
Prostatic tissue specimens are obtained from patients with symptomatic
BPH, who are undergoing open prostatectomy. Isolated human prostatic tissue
is cut into five to eight strips (3mm wide, 3mm thick and 15mm long in each
strip). The strips are mounted vertically in organ baths containing 20 ml
Krebs-
Henseleit solution of the following composition (mM): NaCI 112, KCI 5.9, MgCl2
1.2, CaCl2 2, NaHC03 25, NaHP04 1.2, glucose 11.5. The medium is
maintained at 37°C and at pH 7.4, and is equilibrated with a gas
mixture
consisting of 95% oxygen and 5% carbon dioxide. A resting tension of 0.5g is
applied and the responses are recorded isometrically through a force-
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-83
displacement transducer. The preparations are equilibrated for ninety minutes
before starting the experiments.
Concentration-response curves for phenylephrine or acetylcholine (10-9 to
10~M) are determined by adding the compound directly to the bathing media in
a cumulative fashion. For testing compounds, the prostate strips are incubated
in the presence of compound (1 or 10NM) for thirty minutes before and then
phenylephrine or acetylcholine are added to the medium in a cumulative fashion
to obtain to the concentration-response curve in the presence of the compound.
Assay 11
Effect on Trigylceride Levels and Dyslipidemia
Compounds of the Formula (I) lower triglyceride levels and cholesterol
levels and raise high density lipoprotein levels and are therefore of use in
combating medical conditions wherein such lowering (and raising) is thought to
be beneficial. Thus, the compounds of Formula (I) can be used in the treatment
of hypertriglyceridaemia, hypercholesterolemia, and conditions of low HDL
(high
density lipoprotein) levels in addition to the treatment of atherosclerotic
disease
such as of coronary, cerebrovascular and peripheral arteries, cardiovascular
disease and related conditions.
Activity of compounds of Formula (I) for dyslipidemia can be determined
according to the following procedure. C57BL/6J ob/ob mice (male, 30-40 g body
weight, Jackson Lab, Bar Harbor, ME), housed 5 mice per cage in an
environmentally controlled room, are dosed once or twice daily for three weeks
with test compound (0.01 - 20 mg/kg, n=15 per group) or vehicle (0.5% w/v
methyl cellulose/distilled water, water, or other suitable vehicle) by oral
gavage.
At the end of the study, twenty-four hours after giving the final dose of
compound, the mice are sacrificed by decapitation and blood collected. Plasma
concentrations of free fatty acids and triglyceride are determined using a
clinical
autoanalyzer (Abbott Spectrum° CCx; Abbott Laboratories, Abbott Park,
IL).
CA 02476316 2004-08-12
WO 03/072572 PCT/IB03/00590
-84
Assay 12
Decrease in Body Fat
Activity of compounds of the present invention for decrease in body fat
can be determined according to the following procedure. C57BL/6J ob/ob mice
(male, 30-40 g body weight, Jackson Lab, Bar Harbor, ME) are housed five mice
per cage in an environmentally controlled room with food (pelleted rodent
chow)
and water available ad libitum. The compound or vehicle (0.5% w/v methyl
cellulose/distilled water, water, or other suitable vehicle) is dosed once or
twice
daily for three weeks (0.01 - 20 mg/kg, n=15 per group) by oral gavage. Body
weight of each mouse is measured daily and food intake per cage determined
by weighing the amount of food left in the trough. At the end of the study,
twenty-four hours after giving the final dose of compound, the mice are
weighed
and then sacrificed by cervical dislocation. The epididymal fat pads from each
mouse are excised and weighed. The fat versus body weight ratio is determined
for each mouse using the absolute body weights and the fat pad weights. A
reduction in fat pad weight is indicative of a reduction in total body fat.