Note: Descriptions are shown in the official language in which they were submitted.
CA 02477159 2004-08-20
HIGHLY HOMOGENEOUS MOLECULAR MARKERS FOR
ELECTROPHORESIS
BACKGROUND OF THE INVENTION
Field of the Invention
The present invention is in the fields of molecular biology and protein
biochemistry. The invention relates to marker molecules for identifying
physical properties of molecular species separated by the use of
electrophoretic systems. The invention further relates to methods for
preparing and using marker molecules.
Background Art
Gel electrophoresis is a common procedure for the separation of
biological molecules, such as deoxyribonucleic acid (DNA), ribonucleic acid
(RNA), polypeptides and proteins. A common method of electrophoresis of
proteins involves equilibrating the sample with a negatively-charged
surfactant
such as sodium dodecylsulfate (SDS) before electrophoresis. This causes all
the proteins to have a net negative charge and thus migrate toward the anode.
Nucleic acids are charged without further change. In gel electrophoresis, the
molecules are separated into bands according to the rates at which an imposed
electric field causes them to migrate through a medium.
A commonly used variant of this technique consists of an aqueous gel
enclosed in a glass tube or sandwiched as a slab between glass or plastic
plates. The gel has an open molecular network structure, defining pores that
are saturated with an electrically conductive buffered solution of a salt.
These
pores through the gel are large enough to admit passage of the migrating
macromolecules.
The gel is placed in a chamber in contact with buffer solutions which
make electrical contact between the gel and the cathode or anode of an
electrical power supply. A sample containing the macromolecules and a
CA 02477159 2004-08-20
-2-
tracking dye is placed on top of the gel. An electric potential is applied to
the
gel causing the sample macromolecules and tracking dye to migrate toward
one of the electrodes depending on the charge on the macromolecule. The
electrophoresis is halted just before the tracking dye reaches the end of the
gel.
The locations of the bands of separated macromolecules are then determined.
By comparing the distance moved by particular bands in comparison to the
tracking dye and macromolecules of known mobility, the mobility of other
macromolecules can be determined. The size of the macromolecule can then
be calculated or macromolecules of different sizes can be separated in the
gel.
Isoelectric focusing (IEF) is an electrophoresis method based on the
migration of a molecular species in a pH gradient to its isoelectric point
(pI).
The pH gradient is established by subjecting an ampholyte solution containing
a large number of different-pI species to an electric field, usually in a
cross-
linked matrix such as a gel. Analytes added to the ampholyte-containing
medium will migrate to their isoelectric points along the pH gradient when an
electrical potential difference is applied across the gel.
For complex samples, multidimensional electrophoresis methods have
been employed to better separate species that co-migrate when only a single
electrophoresis dimension is used. Common among these is two dimensional
electrophoresis or 2D-E. For 2D-E analysis of proteins, for example, the
sample is usually fractionated first by IEF in a tube or strip gel to exploit
the
unique dependence of each protein's net charge on pH. Next, the gel
containing the proteins separated by pI is extruded from the tube in the case
of
a tube gel, equilibrated with SDS and laid horizontally along one edge of a
slab gel, typically a cross-linked polyacrylamide gel containing SDS. Other
methods for IEF fractionation allow pieces or strips of gel supported on non-
conductive backing to be laid directly onto the slab of gel. Electrophoresis
is
then performed in the second dimension, perpendicular to the first, and the
proteins separate on the basis of molecular weight. This process is referred
to
as SDS polyacrylamide gel electrophoresis or SDS-PAGE. The rate of
migration of macromolecules through the SDS-PAGE gel depends upon four
CA 02477159 2004-08-20
-3-
principle factors: the porosity of the gel; the size and shape of the
macromolecule; the field strength; and the charge density of the
macromolecule. It is critical to an effective electrophoresis system that
these
four factors be precisely controlled and reproducible from gel to gel and from
sample to sample. However, maintaining uniformity between gels is difficult
because each of these factors is sensitive to many variables in the chemistry
of
the gel and the other reagents in the system as well as the characteristics of
the
macromolecules. Thus, proteins having similar net charges, which are not
separated well in the first dimension (IEF), will separate according to
variations of the other principle factors in the second dimension (SDS-PAGE).
Since these two separation methods depend on independent properties, the
overall resolution is approximately the product of the resolution in each
dimension.
Essential to the practice of many of these electrophoretic techniques,
including 2D-E and SDS-PAGE, are molecular marker standards, i.e. standard
protein molecules with known molecular weights and pIs. Molecular markers
are used as benchmarks in electrophoresis systems for comparison of physical
properties with the unknown samples of interest. Although there are numerous
applications for molecular markers, some particular examples include:
conventional two-dimensional gel electrophoresis using broad pH range
immobilized pH gradient (IPG) strips, overlapping two-dimensional gel
electrophoresis using narrow pH range IPG strips, stand-alone SDS-PAGE,
IEF gels with carrier ampholytes, capillary electrophoresis, electrokinetic
chromatography. Many other forms of gel electrophoresis are well known to
those of skill in the art.
Thus, it is desirable to have reliable standard markers with well-
defined properties with which to compare an unknown sample. This is
particularly true in high-resolution systems such as 2D-E. Unfortunately,
commercially available 2D-E standards (BioRad, Hercules, CA, Catalogue
No. 161-0320; Sigma, St. Louis, MO, Catalogue No. 60653; Pharmacia,
Uppsala, Sweden, Catalogue Nos. 17-0471-Ol and 17-0582-O1) consist mainly
CA 02477159 2004-08-20
-4-
of unstained natural proteins that are only available in a limited range of
pIs
and molecular weights. These commercial markers randomly distribute on
two-dimensional gels and cannot be distinguished from the analyte.
Furthermore, manipulation of pI and molecular weight of proteins using
various agents generates a heterogeneous mixture of products that do not
migrate in a sharp zone under electrophoretic conditions. This is particularly
a
problem when using conventional techniques to make proteins visibly
detectable by attaching chromophoric groups. In the current state of the art,
proteins are labeled by treating the protein with a reactive agent which may
be
a chromophoric group or other label. Since the protein has multiple
potentially reactive sites such as NH2 or -SH groups, and since complete
reaction of all sites is never achieved, the labeling reaction results in a
mixture
of products. A single population of markers may have varying numbers of
labels depending on how many active sites are available. This heterogeneous
mixture of molecules will vary in pIs and molecular weights and will produce
smeared or diffused bands or spots under electrophoretic conditions. Lack of
precision for molecular markers will have a negative effect on all separation
techniques, especially those involving isoelectric focusing. The smearing or
blurred appearance of the markers during visualization of the results will
lead
to ambiguous or unreliable representation of the experimental data.
Consequently, there is an unmet need for highly homogeneous visible
molecular markers that are compatible with commercially available separation
techniques, especially techniques that separate proteins on the basis of
charge
andlor molecular weight.
SUMMARY OF THE INVENTION
The present invention is directed to methods for preparing
homogeneous visible, preferably colored marker molecules with known pIs
CA 02477159 2004-08-20
-S-
and molecular weights. The invention is further directed to methods of
altering the pI and molecular weight of proteins or nucleic acids in a
consistent, reproducible fashion using organic molecules or peptides. Marker
molecules of the present invention will generally separate to give narrow,
sharp bands or spots under electrophoretic conditions. The present invention
is also directed to methods of preparing marker molecules of the present
invention and methods for using these molecules.
In one embodiment, the present invention relates to marker molecule
compositions comprising same pI and same molecular weight marker
molecules. In another embodiment, the present invention relates to marker
molecule compositions comprising same pI and different molecular weight
marker molecules. In yet another embodiment, the present invention relates to
marker molecule compositions comprising different pI and different molecular
weight marker molecules. In a further embodiment, the present invention
relates to marker molecule compositions comprising different pI and same
molecular weight.
In another embodiment, the present invention relates to a marker
molecule comprising: a molecular weight from about 200 daltons to about
2,000 daltons, from about 300 daltons to about 2,500 daltons, from about
3,000 daltons to about 250,000 daltons, an isoelectric point (pI) from about 2
to about 12, and at least one or more labeling molecules. Such labeling
molecules may include chromophores, fluorophores, or ultraviolet light (UV)
absorbing groups. Labeling may also be achieved by introducing natural
amino acids containing UV absorbing moieties such as the aromatic groups in
tryptophan and tyrosine (Shimura, K. et al., Electrophoresis 21:603-610
(2000)). In another embodiment, the present invention relates to a marker
molecule of the formula:
Segment A-L-,Segment B
wherein,
Segment A is a labeled molecule (e.g., natural or synthetic, including,
without
limitation, organic molecules, polypeptide, polynucleotides, macromolecule
CA 02477159 2004-08-20
-6-
such as carbohydrates, small molecules, oligopeptides, natural or non-natural
amino acids), preferably labeled with one or more chromophores,
fluorophores, or UV absorbing groups;
L is a linker or a bond;
Segment B is a protein (e.g., native, recombinant or synthetic protein) or
nucleic acid (e.g., DNA or RNA).
In a further embodiment, the present invention relates to marker
molecule compositions comprising a collection of two or more (e.g., one, two,
three, four, five, six, seven, eight, nine, ten, fifteen, twenty, etc.) marker
molecules of the present invention wherein the marker molecules differ in
molecular weight and/or isoelectric point (pI).
In another embodiment, the present invention relates to marker
molecules wherein the labeling molecules are selected from the group
consisting of chromophores, fluorophores, and W absorbing groups.
In a further embodiment, the present invention relates to the use of
marker molecules of the present invention in gel electrophoresis systems (eg.,
two-dimensional gel electrophoresis systems).
In another embodiment, the present invention relates to methods of
separating one or more proteins present in a sample by gel electrophoresis,
comprising adding the marker molecule composition of the present invention
to the sample containing one or more proteins, applying the sample to an
electrophoresis gel, and subjecting the electrophoresis gel to an electric
field.
In a further embodiment, the present invention relates to methods
further comprising detecting one or more marker molecules and comparing the
position of one or more marker molecules to the position of the one or more
proteins after subjecting the gel to an electric field. In yet another
embodiment, the present invention relates to methods of separating one or
more proteins present in a sample by using two-dimensional gel
electrophoresis.
1n yet another embodiment, the present invention relates to methods of
separating one or more molecules present in a sample, comprising adding the
CA 02477159 2004-08-20
marker molecule composition of the present invention to the sample
containing one or more molecules, applying the sample to a matrix, and
separating the one or more molecules.
In another embodiment, the present invention relates to a method of
preparing marker molecule comprising:
(a) labeling a molecule (e.g., a polypeptide of known
molecular weight); and
(b) ligating the molecule with a protein or nucleic acid
(e.g., a protein or nucleic acid of known molecular
weight), wherein the molecule or protein (or nucleic
acid) contains an a-thioester and the other contains a
thiol-containing moiety.
In yet another embodiment, the present invention relates to a method of
preparing marker molecule compositions further comprising:
(c) repeating (a)-(b) one or more times to obtain a number
of labeled marker molecules of different molecular
weights and pIs; and
(d) combining the labeled marker molecules having
different molecular weights and pIs.
In one embodiment, the number of labels attached to the marker
molecule is known. In a further embodiment, the number of labels is at least
one and will generally be one or more (e.g., one, two, three, four, five,
etc.).
Labels such as charged chromophoric groups may alter the pI of the final
marker molecule. Chromophores with a sulfonic acid group (pKa of 1.5) will
shift the pI of the marker molecule to acidic pH or chromophores with amino
groups will shift the pI to basic pH. Therefore, the pI may be manipulated and
as a result, marker molecules of known pI may be prepared. In yet another
embodiment, the collection of marker molecules is at least more than one,
preferably at least two or more (e.g., two, three, four, five, etc.).
In a further embodiment, the present invention relates to a method of
preparing a marker molecule comprising:
CA 02477159 2004-08-20
_8_
(a) labeling a molecule, preferably a molecule of known
molecular weight, comprising an amino-terminal
cysteine residue; and
(b) ligating the molecule with a protein or nucleic acid of
known molecular weight and comprising an Ca-
thioester.
In yet another embodiment, the present invention relates to a method of
preparing a marker molecule composition further comprising:
(c) repeating (a)-(b) one or more times to obtain a number
of labeled marker molecules of different weights and
pIs; and
(d) combining the labeled marker molecules of different
weights and pIs.
In a further embodiment, the present invention relates to a method of
labeling a marker molecule comprising:
(a) attaching a first amino acid to a solid phase;
(b) coupling said first amino acid to a second amino acid
protected by blocking groups resulting in a chain of
amino acids, wherein said blocking groups are removed
before the addition of amino acids;
(c) extending the length of the chain by solid phase
synthesis with additional amino acids, wherein said
chain comprises at least one labeled amino acid,
resulting in a labeled oligopeptide;
(d) releasing the labeled oligopeptide from the solid phase;
and
(e) ligating the labeled oligopeptide with a protein of
known molecular weight.
In one embodiment, one, two or more (e.g., two, three, four, five, etc.)
additional amino acids are modified with a label. Preferably, the blocking
CA 02477159 2004-08-20
-9-
groups are selected from the group consisting tert-butyloxycarbonyl (BOC), 9-
fluorenylmethoxycarbonyl (FMOC) and their derivatives thereof.
In yet another embodiment, the present invention relates to a method of
characterizing one or more proteins, comprising:
(a) electrophoresing one or more proteins (e.g., one, two,
three, four, five, six, eight, ten, etc.) in a gel with at
least one (e.g., one, two, three, four, five, six, eight, ten,
etc.) marker molecule of the present invention;
(b) comparing the migration of the one or more proteins
with the migration of the at least one marker molecule
of the present invention; and
(c) optionally, determining the isoelectric point (pI) and/or
molecular weight of the one or more proteins.
In a further embodiment, the present invention relates to a method of
characterizing one or more molecules, comprising:
(a) separating one or more molecules (e.g., one, two, three,
four, five, six, eight, ten, etc.) in a matrix with at least
one (e.g., one, two, three, four, five, six, eight, ten, etc.)
marker molecule of the present invention;
(b) comparing the migration of the one or more molecules
with the migration of the at least one marker molecule
of the present invention; and
(c) optionally, determining the isoelectric point (pI) and/or
molecular weight of the one or more molecules.
In yet another embodiment, the present invention relates to a method of
characterizing one or more molecules, comprising:
(a) electrophoresing one or more molecules (e.g., one, two,
three, four, five, six, eight, ten, etc.) in a matrix with at
least one (e.g., one, two, three, four, five, six, eight, ten,
etc.) marker molecule of the present invention;
CA 02477159 2004-08-20
-10-
(b) comparing the migration of the one or more molecules
with the migration of the at least one marker molecule
of the present invention; and
(c) optionally, determining the isoelectric point (pn and/or
S molecular weight of the one or more molecules.
In one embodiment, two-dimensional gel electrophoresis may be used
to analyze one or more proteins to determine their molecular weights and/or
pIs. In another embodiment, the marker molecule may contain at least one
(e.g., one, two, three, four, five, etc.) labeled protein, preferably at least
two
(e.g., two, three, four, five, etc.) labeled proteins of the present
invention.
In another embodiment, the present invention relates to a peptide
having the formula:
Cysr-Yri Z
where,
Y is one or more amino acid selected from the group consisting of alanine,
arginine, aspartic acid, asparagine, cysteine, glutamic acid, glutamine,
glycine,
histidine, iso-leucine, leucine, lysine, methionine, phenylalanine, proline,
serine, threonine, tryptophan, tyrosine and valine or
any non-natural amino acid with appropriate functionality, without limitation,
traps-4-hydroxyproline, 3-hydroxyproline, cis-4-fluoro-L-proline,
dimethylarginine, and homocysteine; wherein at least one amino acid is
labeled with a chromophore, fluorophore, or UV absorbing group, in many
instances at least two (e.g., two, three, four, five, etc.) amino acids are
labeled;
Z is a C-terminal amino acid (the Ca-carboxyl group may be modified to have
an amide function) or non-natural amino acid; and
n=1-100 covalently linked amino acid(s). In one embodiment, Y may be a
non-natural amino acid which is not one of the twenty amino acids commonly
found in proteins. Further, as one skilled in the art would recognize, Y can
be
composed of different amino acids (e.g., amino acids listed above). In another
embodiment, Z may be any amino acid listed above including non-natural
amino acids listed above.
CA 02477159 2004-08-20
- 11 -
In another embodiment, the present invention is directed to a method
of ligating nucleic acids to oligopeptides. For example, incorporation of a
thiol-containing group (e.g., 1-amino-2-mercaptoethyl) into one terminus of
the nucleic acid (e.g., nucleic acid-CH(NHZ)-CHZ-SH) and subsequent ligation
with an oligopeptide containing Ca-thioester forms nucleic acid-oligopeptide
conjugate. This method may be used, for example, for the construction of
nucleic acid markers. Ligation of nucleic acid-CH(NHZ)-CHZ-SH with a
labeled macromolecule or a labeled small organic molecule containing Ca-
thioester may be used to form a labeled nucleic acid.
Kits serve to expedite the performance of, for example, methods of the
invention by providing multiple components and reagents packed together.
Further, reagents of these kits can be supplied in pre-measured units so as to
increase precision and reliability of the methods. Kits of the present
invention
will generally comprise a carton such as a box; one or more containers such as
boxes, tubes, ampules, jars, or bags; one or more (e.g., one, two, three,
etc.)
pre-casted gels and the like; one or more (e.g., one, two, three, etc.)
buffers;
and instructions for use of kit components.
In another embodiment, the present invention relates to marker
molecule kits comprising a carrier having in close confinement therein at
least
one (e.g., one, two, three, four, five, etc.) container where the first
container
comprises at least one (e.g., one, two, three, four, five, six, seven, eight,
nine,
ten, fifteen, twenty, etc.) marker molecule of the present invention. In yet
another embodiment, the marker molecule kit of the present invention further
comprises instructions for use of kit components. In a further embodiment,
the marker molecule kit of the present invention further comprises one or more
(e.g., one, two, three, etc.) pre-casted electrophoresis gels.
In another embodiment, the present invention relates to a marker
molecule of the formula I:
Segment A-L~egment B
wherein,
Segment A is a labeled molecule;
CA 02477159 2004-08-20
-12-
L is a linker or a bond; and
Segment B is a protein which contains no deamidation sites. In another
embodiment, the present invention relates to marker molecules wherein
Segment B is a protein which contains no arginine or glutamine residues. In
yet another embodiment, the present invention relates to marker molecules
wherein the protein comprises an amino acid sequence selected from the group
consisting of:
(a) the amino acid sequences shown in SEQ ID
NO:11;
(b) the amino acid sequences shown in SEQ ID
N0:12;
(c) the amino acid sequences shown in SEQ ID
N0:13;
(d) the amino acid sequences shown in SEQ ID
N0:14;
(e) the amino acid sequences shown in SEQ ID
NO:15;
(f) the amino acid sequences shown in SEQ ID
N0:16;
(g) the amino acid sequences shown in SEQ ID
N0:17; and
(h) the amino acid sequences shown in SEQ ID
N0:18.
In another embodiment, the present invention relates to a marker
molecule of formula I:
Segment A-L~egment B
wherein,
Segment A is a labeled molecule;
L is a linker or a bond; and
Segment B is modified naturally occurnng protein which contains a reduced
number of post-translational modification sites. In yet another embodiment,
the present invention relates to a marker molecule wherein the post-
translational sites are selected from the group consisting of
(a)deamidation sites;
(b)glycosylation
sites; and
(c)phosphorylation
sites.
CA 02477159 2004-08-20
-13-
In yet another embodiment, the present invention relates to marker molecules
wherein in which contains none of one or more amino acid selected from the
group consisting o~
(a) asparagine;
(b) glutamine;
(c) proline;
(d) serine;
(e) threonine;
(f) tyrosine; and
(g) aspartic acid.
Other embodiments of the invention will be apparent to one of
ordinary skill in light of what is known in the art, the following drawings
and
description of the invention, and the claims.
BRIEF DESCRIPTION OF THE FIGURES
FIG. 1 depicts a scheme showing solid phase synthesis of a peptide to
be used as Segment A of the marker molecules of the invention. In this
example, a resin linker is present which contains a thioester-linked glycine.
Further, a Na-Fmoc-NE-TMR-Lysine is used as a building block amino acid
that is labeled with tetramethylrhodamine (TMR). The N-terminal amino acid
is an iminobiotin labeled glycine. The labeled peptide is released from the
solid phase by treatment with benzylthiol (Ph-CHZ-SH) and the product
peptide is purified by reverse phase HPLC (RP-HPLC).
FIG. 2 depicts a scheme showing a ligation of Segment A (TMR- and
biotin-labeled peptide) to a protein containing N-terminal cysteine (Segment
B). Upon transthioesterification of the thioester with the cysteine thiol, a
SAN
acyl shift takes place to generate a ligated product with the two segments,
now
connected by an amide bond; resulting in the generation of a final product
which is a labeled protein of known molecular weight and pI.
CA 02477159 2004-08-20
-14-
FIGS. 3A and 3B depict schemes showing preparation of a TMR-
labeled protein by coupling an organic thioester labeled with a fluorescent
dye
such as tetramethylrhodamine (Segment A) to a protein with N-terminal
cysteine (Segment B). FIG. 3A depicts a scheme for forming a labeled protein
by acylating triethylenetetramine (TREN, available from Aldrich, Milwaukee,
WI, Catalogue No. 90462) with 3.5 equiv. of an activated ester of
carboxytetrarhodamine (TMR), available from Molecular Probes, OR
(Catalogue No. e-6123), to form (TMR)3-TREN 5. Acylation of Na-Fmoc-
Lysine with 2-iminobiotin-N-hydroxysuccinimide ester (Biotin-NS ester)
yields NE-Fmoc-Na -biotin-Lysine 6. Deblocking of the a-amino group of 6
followed by acylation with bromoacetyl chloride forms N~ bromoacetamido-
N°'-biotinyl-Lysine 8. The carbodiimide coupling of 8 with a-
toluenethiol
results in 9. The alkylation of 5 with the thioester 9 in the presence of
sodium
iodide generates the quaternary ammonium salt 10 (Segment A) that upon
coupling with Segment B under the same conditions described above affords
11 (chromophore to protein ratio = 3). FIG. 3B depicts a scheme for forming
a TMR-labeled protein by first preparing a thiol benzyl ester (13).
Deprotection of the amino group of 13 in the presence of trifluoroacetic acid,
14, followed by coupling to N-hydroxy succinimidyl ester of TMR generates
the benzyl thioester derivative of N-TMR-8-heptanoic acid 15. The reaction
of the thioester 15 (Segment A) with recombinant protein with N-terminal
cysteine (Segment B) forms TMR-protein 16 (chromophore to protein ratio =
1) that can be purified by dialysis.
FIG. 4 shows solid phase synthesis of a peptide labeled with TMR
(Segment A). The resin linker is a thioester-linked histidine and N°'-
Fmoc-s
TMR-Lysine is the building block amino acid labeled with TMR. In this
scheme, the N-terminal amino acid is cysteine. After treatment with
trifluoroacetic acid (TFA), the resulting product is an oligopeptide labeled
with the chromophore, TMR, and tagged with the metal affinity binding
(histidine)6 sequence.
CA 02477159 2004-08-20
-15-
FIG. 5 depicts a scheme showing the labeling of a protein via in vitro
chemical ligation. In this method, a recombinant protein with C-terminal
thioester (Segment B) ligates to a TMR-labeled, polyhistidine-tagged peptide
(Segment A) with N-terminal cysteine in the presence of toluene thiol,
benzylthiol and thiophenol. The reaction results in a product of known
molecular weight and pI.
FIG. 6 depicts a scheme showing site-specific modification of a protein
that contains an N-terminal threonine or cysteine. The amino and hydroxyl
groups on adjacent carbons of an N-terminal amino acid can be readily
oxidized to form a protein with N-terminal aldehyde (17, Segment B).
Coupling of Segment B to 19 (Segment A) results in a visibly colored protein
(21) with known molecular weight and pI.
FIG. 7 depicts a scheme showing solid phase synthesis of a peptide
with N-terminal cysteine (Segment A) using Fmoc-PAL-PEG-PS resin or any
amide resin as described by Schnolzer, M. et al., Intl. J. Peptide Protein
Research 40:180 (1990).
FIG. 8 depicts a scheme illustrating labeling of a protein via in vitro
chemical ligation. In this method a recombinant protein, MBP-95aa (a 95
amino acid segment of Maltose Binding Protein) with a C-terminal thioester
(Segment B) ligates to a TMR-labeled peptide with N-terminal cysteine.
FIG. 9 depicts a scheme illustrating in vitro chemical ligation using a
peptide without N-terminal cysteine. The N°'-(1-phenyl-2-mercaptoethyl)
auxiliary is coupled to the oligopeptde N-terminus using solid phase peptide
synthesis. Upon ligation, the auxiliary group is removed under mild
conditions.
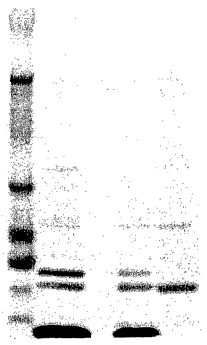
FIG. 10 is a photograph of a NU-PAGE~ 4-12% Bis-Tris gel
characterizing MBP-110aa-(TMR)2. Lane 1 is the Multimark (Invitrogen
Corporation, Carlsbad, CA) protein marker. Lane 2 is reaction mixture
containing MBP-110aa-(TMR)Z (highest molecular weight), MBP-95aa,
unreacted Cys-Leu-Lys(TMR)-Asp-Ala-Leu-Asp-Ala-Leu-Asp-Ala-Leu-
Lys(TMR)-Asp-Ala-amide (lowest band) (SEQ ID N0:3). Lane 3 is blank.
CA 02477159 2004-08-20
- 16-
Lane 4 is reaction mixture containing MBP-110aa-(TMR)Z (highest molecular
weight), MBP-95aa, unreacted Cys-Leu-Lys(TMR)-Asp-Ala-Leu-Asp-Ala-
Leu-Asp-Ala-Leu-Lys(TMR)-Asp-Ala-amide (SEQ ID N0:3). Lane 5 is
MBP-95 aa.
DETAILED DESCRIPTION OF THE INVENTION
Generally, when proteins are modified by the addition of specific
labels to produce marker molecules for gel electrophoresis systems, the
proteins are typically linked to the labels in a manner which results in the
production of a mixture of products. These product mixtures typically contain
molecules having various pIs and molecular weights and often smear under
electrophoretic conditions. Further, the molecules lack the precision or
uniformity required for molecular markers especially when such markers are
to be separated by their isoelectric points. Therefore, methods for preparing
marker molecules should result in the incorporation of a chromophore or other
detectable group (e.g., a visibly colored molecule) in the marker molecules in
such a way as to direct the label onto a single site (e.g., at one amino acid)
or
at a small number of locations (e.g., one, two, three, four, or five
locations)
rather than randomly.
The present invention relates to a marker molecule comprising:
Segment A-L~egment B
wherein,
Segment A is a labeled molecule (e.g., natural or synthetic, including,
without
limitation, organic molecules, polypeptide, polynucleotides, macromolecule
such as carbohydrates, small molecules, oligopeptides, natural or non-natural
amino acids), preferably labeled with one or more chromophores,
fluorophores, or UV absorbing groups;
L is a linker or a bond;
Segment B is a protein (e.g., native, recombinant or synthetic protein) or
nucleic acid (e.g., DNA or RNA, polynucleotide). For example, Segment B
CA 02477159 2004-08-20
-17-
may be a protein of known molecular weight (e.g., a protein having a
molecular weight from about 200 daltons to about 2,000 daltons, from about
300 daltons to about 2,500 daltons, from about 1,000 daltons to about 250,000
daltons, from about 2,000 daltons to about 250,000 daltons, from about 3,000
daltons to about 250,000 daltons, from 1,000 daltons to about 200,000 daltons,
from about 2,000 daltons to about 200,000 daltons, from about 3,000 daltons
to about 200,000 daltons, from about 4,000 daltons to about 150,000 daltons,
from about 6,000 daltons to about 100,000 daltons, from about 2,000 daltons
to about 50,000 daltons, from about 3,000 daltons to about 50,000 daltons,
from about 8,000 daltons to about 50,000 daltons); and wherein the marker
molecule has a known pI from about 0 to about 14, from about 2 to about 12,
from about 3 to about 11, from about 4 to about 10, from about 5 to about 9,
from about 6 to about 8. Segment A may be linked to Segment B in either
orientation.
In one embodiment, Segment A may comprise 1-100 covalently linked
amino acids (e.g., 1, 2, 3, 4, 5, 6, 10, 30, 50, 75, 100, etc. covalently
linked
amino acids or 10-30, 5-50, 15-40, 20-50, 30-60, 40-70, 50-80, 60-90, 70-100,
etc. covalently linked amino acids), most preferably, 15 covalently linked
amino acids. In a further embodiment, one, two or more (two, three, four,
five, etc.) of the amino acids in Segment A are labeled. In another
embodiment, one or more amino acids in Segment A are from tyrosine or
tryptophan. In yet another embodiment, the labeled amino acid is a lysine. In
yet another embodiment, the polypeptide or polynucleotide is labeled with
carboxytetramethylrhodamine (TMR).
In another embodiment, Segment B may comprise from about 100
nucleotides (nt) to about 1,000 nt, from about 200 nt to about 2,000 nt, from
about 300 nt to about 3,000 nt, from about 1,000 nt to about 5,000 nt, from
about 3,000 nt to about 10,000 nt, from about 5,000 nt to about 20,000 nt,
from about 6,000 nt to about 30,000 nt, from about 10,000 nt to about 50,000
nt, from about 20,000 nt to about 100,000 nt, from about 50,000 nt to about
200,000 nt, from about 70,000 nt to about 250,000 nt.
CA 02477159 2004-08-20
-18-
The invention further provides marker molecules having a molecular
weight from about 300 daltons to about 3,000 daltons, from about 500 daltons
to about 4,000 daltons, from about 1,000 daltons to about 5,000 daltons, from
about 3,000 daltons to about 8,000 daltons, from about 5,000 daltons to about
12,000 daltons, from about 10,000 daltons to about 15,000 daltons, from about
12,000 daltons to about 18,000 daltons, from about 15,000 daltons to about
25,000 daltons, from about 20,000 daltons to about 30,000 daltons, from about
25,000 daltons to about 40,000 daltons, from about 30,000 daltons to about
50,000 daltons, from about 40,000 daltons to about 60,000 daltons, from about
50,000 daltons to about 80,000 daltons, from about 60,000 daltons to about
90,000 daltons, from about 75,000 daltons to about 110,000 daltons, from
about 90,000 daltons to about 140,000 daltons, from about 110,000 daltons to
about 160,000 daltons, from about 130,000 daltons to about 180,000 daltons,
from about 140,000 daltons to about 200,000 daltons, from about 180,000
daltons to about 220,000 daltons, or from about 200,000 daltons to about
250,000 daltons.
The invention further provides marker molecules having a pI from
about 0.5 to about 2, from about 1 to about 3, from about 2 to about 4, from
about 3 to about 5, from about 4 to about 6, from about 5 to about 7, from
about 6 to about 8, from about 7 to about 9, from about 8 to about 10, from
about 9 to about 11, from about 10 to about 12, from about 11 to about 13,
from about 12 to about 13.5, from about 2 to about 6, from about 3 to about 7,
from about 5 to about 9, from about 6 to about 10, from about 8 to about 12,
or
from about 9 to about 13.
In another embodiment, the present invention relates to a marker
molecule of wherein Segment A comprises a labeled organic molecule, L is a
linker bond, and Segment B is a peptide, protein or polynucleotide, wherein
Segment A can form bond L in only in one position of Segment B.
In a further embodiment, the present invention relates to a marker
molecule wherein Segment A comprises a thioester and Segment B contains a
single 1-amino-2-mercaptoethyl group. In yet another embodiment, the
CA 02477159 2004-08-20
-19-
present invention relates to Segment A comprising a labeled polypeptide
thioester or a labeled organic thioester. In a further embodiment, the present
invention relates to Segment B comprising a protein, peptide or polynucleotide
containing a 1-amino-2-mercaptoethyl group. In yet another embodiment, the
present invention relates to the 1-amino-2-mercaptoethyl group in the protein
or peptide comprising the N-terminal amino acid cysteine. In another
embodiment, the present invention relates to the 1-amino-2-mercaptoethyl
group in the polynucleotide comprising a single modified base. In yet another
embodiment, the present invention relates to the peptide or protein comprising
a recombinant protein constructed to have an N-terminal cysteine. In further
embodiment, the present invention relates to the polynucleotide prepared with
a single modified base by an enzymatic reaction. In another embodiment, the
present invention relates to the marker molecule wherein Segment A
comprises a single 1-amino-2-mercaptoethyl group and Segment B comprises
a thioester. In another embodiment, the present invention relates to Segment
A comprising a labeled polypeptide having the amino acid cysteine as the N-
terminal amino group. In another embodiment, the present invention relates to
Segment A comprising an organic molecule containing a 1-amino-2-
mercaptoethyl group. In another embodiment, the present invention relates to
Segment A comprising a cysteinyl carboxy ester or amide. In another
embodiment, the present invention relates to Segment A constructed by
automated peptide synthesis. In another embodiment, the present invention
relates to the marker molecule wherein Segment A comprises an aldehyde
reactive group and Segment B contains an aldehyde formed from oxidation of
an N-terminal serine or threonine of a polypeptide or protein. In another
embodiment, the present invention relates to marker molecule wherein
Segment A comprises a labeled hydrazone. In another embodiment, the
present invention relates to the marker molecule wherein L is a hydrazide
bond.
In another embodiment, the present invention relates to a method of
preparing a marker composition, the method comprising labeling an organic
CA 02477159 2004-08-20
-20-
molecule and ligating it to a single position in a peptide, protein or
polynucleotide. In another embodiment, the present invention relates to a
method of labeling a marker molecule, comprising: ligating a first labeling
molecule to a single position on a second molecule consisting of a protein,
peptide or polynucleotide. In another embodiment, the present invention
relates to a method of modifying the isoelectric point of a marker molecule
comprising: ligating a first labeling molecule containing acidic or basic
ionizable groups to a second molecule consisting of a protein, peptide or
polynucleotide.
As used herein, the term "known pI," when applied to marker
molecules and their composition, means that the pI is theoretically calculated
using the polynomial equations described in Sillero, A. et al., Analytical
Biochem. 179:319-325 (1989) and Ribeiro, J. et al., Comput. Biol. Med.
20:235-242 (1990), which are incorporated herein by reference, or determined
empirically.
In a further embodiment, the linker comprises a peptide bond or one of
the following bifunctional linkers, without limitation:
- (CHZ )9 - NH-,
wherein q is 2-10;
O O
II II
-(CH~9 NH-C-(CH~x C
wherein q = 2-S,
x = 2-12; and
O
I I
- (CH~~ C-
CA 02477159 2004-08-20
-21 -
wherein y = 1-3.
In one embodiment, Segment A may be preferably and specifically
labeled with chromophores, fluorophores, or LTV absorbing groups such as 5-
carboxyfluoresceine (FAM), fluorescein, fluorescein isothiocyanate, 2'7'-
dimethoxy-4'S'-dichloro-6-carboxyfluorescein (JOE), rhodamine, N,N,N',N'-
tetramethyl-6-carboxyrhodamine (TAMRA), tetramethyl rhodamine or
carboxytetramethylrhodamine (TMR). In a further embodiment, Segment A
may comprise a capture or binding tag such as biotin, fluoroscein,
digoxigenin, polyhistidine or derivatives thereof. In another embodiment,
Segment A may be used to modify the pI of Segment B by the presence of one
or more acidic amino acids such as aspartate and glutamate or one or more
basic amino acids such as lysine, arginine and histidine. In another
embodiment, the addition of charged chromophoric groups or chromophores
with a sulfonic acid also affect the pI. In a further embodiment, Segment A
may be used to introduce reactive sites for covalent attachment of proteins.
In another embodiment, the present invention relates to the use of a
labeled thioester wherein the labeled thioester may be a single amino acid
thioester such as N-tetramethylrhodamine amide glycyl thioester to attach as a
labeled Segment A to a protein, polypeptide, or polynucleotide having a 1-
amino-2-mercaptoethyl group.
In a further embodiment, the present invention relates to the use of a
labeled 1-amino-2-mercaptoethyl group to attach a label to a protein or
polypeptide having a C-terminal thioester group.
In yet a further embodiment, the present invention relates to the use of
labeled hydrazides and other aldehyde reactive groups as Segment A to attach
a label to a protein or polypeptide having an oxidized (or oxidizable) N-
terminal serine or threonine group.
Proteins may be modified so as to eliminate or introduce functional
groups which may be targeted by selective reagents. For example, if a protein
has no naturally occurring cysteines in its primary sequence, and nucleic acid
CA 02477159 2004-08-20
-22-
(e.g., DNA) clone encoding the protein is available, mutagenesis may be
undertaken to introduce one or more cysteines. Procedures for such
modifications are well known in the art (Ausubel, F.M. et al., in Current
Protocols in Molecular Biology, John Wiley and Sons, Chapter 8 (1995)).
Briefly, in one example, the wild type nucleic acid encoding the protein to be
modified is incorporated in a single stranded bacteriophage vector containing
random uracil bases. The single stranded nucleic acid is hybridized with a
complementary synthetic oligonucleotide sequence incorporating a codon at
the site of modification encoding the new amino acid desired to be in that
position. The new double stranded sequence is extended with T4 DNA
polymerase and the resulting phage used to transform E. coli bacteria. The
expressed protein may then be isolated by standard techniques well known to
those of ordinary skill in the art.
Such procedures may be used not only to incorporate amino acids of
interest, but also to replace amino acids and to eliminate reaction sites. For
example, one may reduce the number of cysteine groups in a wild type protein
so that there are few sites available for modification. Cysteine groups are
particularly useful because of the large number of reagents available to
selectively react with the sulfhydryl sidechain. Examples include maleimidyl
or iodoacetamidyl derivatives of chromophoric compounds or other labels that
are commercially available (e.g., eosin-5-maleimide, item E-118 from
Molecular Probes, Inc., Bothell, WA; Oregon Green iodoacetamide, item 0-
6010 also from Molecular Probes).
Other groups may also be selectively modified. For example, oxalyl
groups on a labeling reagent will selectively react with the amidino group of
arginine. So proteins may be cloned so as to add or delete arginines as
described for cysteine. Such modified proteins may then be selectively
labeled. As another example, N-hydroxysuccinimidyl esters will react with
lysine groups on the protein. N-hydroxysuccinimidyl esters are also widely
available commercially and include, for example, carboxyfluorescein-N-
hydroxysuccinimidyl ester (available from Research Organics, Cleveland, OH,
CA 02477159 2004-08-20
- 23 -
as item 1048C). Lysines may be selectively added or eliminated as desired
using standard cloning techniques. Use of lysine or arginine as sites for
modification is less attractive than cysteine, because there are generally
more
of these basic amino acids and their elimination often results in changes in
the
solubility characteristics and pI of the recombinant protein.
Nucleic acids may also be modified using the techniques described
herein. For example, it is well known that modified bases such as biotin-16-
dUTP, biotin-11-dUTP and biotin-14-dATP, among others, may be
incorporated as labels by the action of polymerases when such building blocks
are added to the typical nucleotide triphosphate mix used for in vitro
synthesis
of DNA (Ausubel, F.M. et al., in Current Protocols in Molecular Biology,
John Wiley and Sons, 3.18.3 (1995)). Bases modified to contain 1-amino-2-
mercaptoethyl groups may be prepared and incorporated by enzymatic action
into DNA to form Segment B. Such labeling results in a nonspecific
incorporation of the modified base into sites of the DNA. However, this group
is reactive with molecules or macromolecules as Segment A bearing a
thioester such as shown in FIGS. 1, 3A and 3B, so the reactive group could be
used to attach labels to the nucleic acid after enzymatic synthesis. Molecules
with a thioester may include polypeptides as well as smaller molecules.
As an example, N6-(6-aminohexyl)ATP is commercially available
(Invitrogen Corporation). This compound may be readily ligated to a blocked
cysteine activated with carbodiimide to form the 6-aminohexyl
cysteinylamide. Once unblocked, this compound may be used in enzymatic
synthesis of oligonucleotides as describe above. The resulting 1-amino-2-
mercaptoethyl group is reactive with thioesters and allows the facile
incorporation of labels and even the attachment of oligopeptides and proteins
bearing a thioester group. Many other structural analogs of purine and
pyrimidine bases may be modified in this manner, and as an example
attachment to the N4 position of CTP or the N2 position of guanine. Modified
bases that are suitable for preparation of nucleotide triphosphates
incorporating 1-amino-2-mercaptoethyl groups such as, without limitation,
CA 02477159 2004-08-20
-24-
04-Triazolyl-dT-CE (CE is (3-cyanoethyl), 06-Phenyl-dI-CE, and 04-
Triazolyl-dU-CE are also available from Glen Research, Sterling, VA, and
from TriLink Biotechnologies, San Diego, CA.
Another method of incorporating modified bases into a nucleic acid to
S form Segment B is to append it to the end of a nucleic acid chain. Terminal
nucleotide transferase (Invitrogen Corporation) is a well known enzyme that
may be used to append oligonucleotides to the 3' end of DNA (Flickinger, J. et
al., Nucleic Acids Res., 20:9 (1992)). This enzyme is used to incorporate
biotinylated oligonucleotides and will readily incorporated bases modified
with less bulky side groups such as 1-amino-2-mercaptoethyl groups capable
of forming amide bonds with thioesters.
Yet another method of incorporation of labels into RNA employs
guanylyltransferase (Invitrogen Corporation) which appends GMP onto the 5'
terminus of an RNA transcript which has a diphosphate or triphosphate group
at the 5' terminus. Use of a modified guanylyltriphosphate will give a base
bearing a 1-amino-2-mercaptoethyl group that allows the incorporation of
thioester-ligatable functions into RNA (Melton, D.A. et al., Nucleic Acids
Res.
12:18 (1984)). Guanylyl transferase possesses GTP exchange properties so
capped mRNA may be labeled with a thioester reactive base by incubating the
capped mRNA with the enzyme and 1-amino-2-mercaptoethyl-modified GTP.
In particular embodiments, the present invention provides different
chemical ligation strategies, further described below, to prepare homogeneous
molecular marker compositions for gel electrophoresis systems.
As used herein, the term "isolated," when applied to marker molecules,
means that the molecules are separated from substantially all of the
surrounding contaminants. "Surrounding contaminants" include molecules
(e.g., amino acids, uncoupled Segment A, uncoupled Segment B, side
products, etc.) associated with the production of the marker molecules but
does not include molecules or agents associated with the isolation process or
which confer particular properties upon either the marker molecules or
compositions which contain the marker molecules. Examples of molecules
CA 02477159 2004-08-20
-25-
which are typically not considered to be surrounding contaminants include
water, salts, buffers, and reagents used in processes such as HPLC (e.g.,
acetonitrile). Thus, marker molecules which have been separated from
unreacted molecules associated with marker molecule production by reverse
S phase HPLC (RP-HPLC), for example, are considered isolated even if present
in a solution which contains 10% purification reagents such as organic
solvents and buffers (e.g., acetonitrile and 10 mM Tris-HCl). This is the case
even when the marker molecules are present in solutions at a concentration of,
for example, 75 ~g/ml. Further, the term "isolated" means that marker
molecules being isolated are at least 90% pure, with respect to the amount of
contaminants. In other words, the marker molecules which are isolated are
separated from at least 90% of the surrounding contaminants.
The invention further includes isolated marker molecules, as well as
compositions comprising one or more (e.g., one, two, three, four, five, six,
eight, ten, twelve, twenty, fifty, etc.) isolated marker molecules, methods
for
preparing isolated marker molecules, methods for preparing compositions
comprising isolated marker molecules, methods for using isolated marker
molecules, and methods for using compositions comprising one or more (e.g.,
one, two, three, four, five, six, eight, ten, twelve, twenty, fifty, etc.)
isolated
marker molecules. The invention also includes compositions comprising one
or more isolated marker molecules.
Marker molecules of the invention may be isolated and/or purified by
any number of methods. Examples of such methods include HPLC (e.g.,
reverse phase HPLC), fast protein liquid chromatography (FPLC), cellulose
acetate electrophoresis (CAE), isoelectric fractionation, column
chromatography (e.g., affinity chromatography, molecular sieve
chromatography, ion exchange chromatography, etc.), capillary zone
electrophoresis, dialysis, isoelectric focusing, and field-flow fractionation.
One example of an apparatus which may be used to isolate and/or
purify marker molecules of the invention is the Hoefer Isoprime isoelectric
CA 02477159 2004-08-20
-26-
purification unit of Amersham Pharmacia Biotech Inc. (Piscataway, NJ 08855)
(Catalog No. 80-6081-90).
Chemical ligation involves a chemoselective reaction between
synthetic unprotected oligopeptides, polynucleotides, organic compounds,
macromolecules or small molecules, termed Segment A, with another
unprotected protein (e.g., synthetic, recombinant or native 'proteins) or
modified nucleic acid of known mass and charge, termed Segment B. The
ligation reaction is site-specific and allows only a single specific coupling
reaction between one site on one segment and one site of another segment, in
the presence of other potentially reactive groups. Chemical ligation is useful
for joining, for example, two segments which are both polypeptides. Peptides
may be made by stepwise solid phase peptide synthesis and may have either
an N-terminal cysteine (or N°'-(1-phenyl-2-mercaptoethyl)) or C-
terminal
thioester depending on the ligation strategy. Incorporation of chromophoric,
acidic, and basic groups into the peptide chain may be achieved by using
amino acids labeled with such groups during peptide synthesis.
Chemical ligation of proteins has the following advantages in the
present invention:
- It is site-specific and allows only a single specific coupling
reaction between the Ca of one segment (e.g., Segment A or
Segment B) and Na of another segment (e.g., Segment A or
Segment B), in the presence of other reactive groups.
- It generates only one product.
- The resulting product has a known pI and a known molecular
weight. These parameters can be determined theoretically and
experimentally.
- It allows protein labeling using chromophores and fluorophores
in a consistent, reproducible fashion.
It allows nucleic acid labeling using chromophores and
fluorophores in a consistent, reproducible fashion.
CA 02477159 2004-08-20
-27-
- It can be used to alter the pI of proteins. The incorporation of
charged amino acid residues, or of charged chromophoric
groups into Segment A, will alter the pI of the final protein
product. For example, the guanidino group of arginine (pKa
S >12) will shift the pI of the product to basic pH, whereas,
chromophores with a sulfonic acid group (pKa of 1.5) will shift
the pI of the product to acidic pH. Other charged
chromophores or charged amino acids will have similar effects.
- It allows manipulation of the molecular weight of proteins. For
example, a 30-residue oligopeptide (Segment A) increases the
molecular weight of the protein (Segment B) by approximately
3.0 daltons (kD), depending on the amino acid sequence, upon
ligation.
- It allows incorporation of tags into proteins. Addition of tags
such as biotin, fluorescein, digoxigenin, polyhistidine to the
synthetic peptide followed by ligation of the peptide to the
protein generates a tagged protein. This tagging strategy may
be used to facilitate purification.
- It allows ligation of polynucleotides to labeled oligopeptides in
a consistent, reproducible fashion.
In the present invention, depending upon the N-terminal amino acid or
the C-terminal carboxylate of the protein (Segment B), ligation strategies
such
as Native Chemical Ligation, in vitro chemical ligation or site-specific
modification may be employed for attaching Segment A to Segment B.
In particular aspects, the present invention provides for: 1) synthesis of
segments A and B, 2) ligation of Segment A to Segment B to form molecular
markers, and/or 3) use of the molecular marker as molecular weight and
isoelectric point markers.
Proteins with low numbers of post-translational modification sites or
comprising low numbers of post-translational modifications are useful for
preparing marker molecules suitable for use in methods and compositions of
CA 02477159 2004-08-20
-28-
the invention and may be incorporated into such markers, for example, as
Segment A and/or Segment B. For purposes of illustration, when a protein is
deamidated at an asparagine or glutamine residue, in most instances, the
molecular weight of the protein, and hence a marker molecule which
S comprises this protein, will change relatively little. However, the change
in
the isoelectric point of the protein will typically be relatively pronounced.
In a
situation where a marker molecule contains, for example, ten potential
deamidation sites, a composition comprising this marker may contain a
considerable number of different molecular species which vary based on
whether deamidation has occurred at one or more of these ten sites. In other
words, since deamidation may occur in individual marker molecules at one or
more of these deamidation sites and deamidation at each site results in a
change in charge, deamidation leads to the formation of a heterogeneous
population of marker molecules.
Other post-translational modifications may substantially change both
the charge and molecular weight of a protein. Ubiquitin, for example, is a
seventy-six amino acid residue protein which is highly conserved among
eukaryotes. Further, proteins are typically ubiquinated at lysine residues,
and
proteins may be poly-ubiquinated. Due to the size of ubiquitin, substantial
changes in the molecular weight, as well as the charge, of a protein can occur
upon ubiquination.
In one aspect, the present invention is directed to marker molecules,
and methods for preparing such markers, which are relatively homogenous
with respect to differences in post-translational modifications. As explained
below, there are several ways to generate marker molecules which are
relatively homogenous with respect to differences in post-translational
modifications. For example, markers may be formed using proteins which
comprise no, few, fewer or a reduced number of post-translational
modification sites. In addition, markers may be formed using proteins which
are produced and/or stored in such a manner as to result in no, few, fewer or
a
reduced number of post-translational modifications. The second option noted
CA 02477159 2004-08-20
-29-
above may result, for example, from the use of proteins which (1) comprise
no, few, fewer or a reduced number of post-translational modification sites,
(2) are produced in systems which do not result in the introduction of post
translational modifications (e.g., the proteins may be synthetically
produced),
or (3) a combination of (1) and (2).
Eukaryotic and/or prokaryotic expression systems which are
genetically altered, mutated, or modified so as to produce proteins which
contain no, few, fewer or a reduced number of post-translational modifications
or post-translational modification sites may be used to produce proteins which
are used to prepare marker molecules of the invention. In particular
instances,
these expression systems may also comprise cells genetically programmed or
altered to block regions of protein which are subject to post-translational
modifications. In other particular instances, genetic alterations, mutations,
and/or modifications can be designed or selected such that one or more post-
translational modification system is either ( 1 ) rendered non-functional or
(2) inducibly or constitutively repressed.
As used herein, the term "few," when used in reference to post-
translational modification sites or post-translational modifications refers to
proteins wherein less than SO%, less than 40%, less than 30%, less than 20%,
less than 10%, less than 5%, less than 2%, or less than 1% of the individual
amino acids which make up the protein are either (1) potential post-
translational modification sites or (2) are post-translationally modified. As
one skilled in the art would recognize, proteins may be subject to a
considerable number of types of post-translational modifications. Thus, the
term "few" may be used in reference to a single type of post-translational
modification site or post-translational modification or multiple types (e.g.,
two, three, four, five, six, seven, ten, etc.) of post-translational
modification
sites or post-translational modifications.
As used herein, the term "fewer," when used in reference to post-
translational modification sites or post-translational modifications refers to
proteins which exhibit less than an expected average number of one or more
CA 02477159 2004-08-20
-30-
different types of post-translational modification sites or post-translational
modifications than would be expected for an average protein from the
particular organism from which the protein was derived. For example, if the
average ratio of potential deamidation sites (e.g., sites in proteins where
glutamine or asparagine residues are found) in proteins from a particular
organism is 1:15 (i.e., one deamidation sites/15 amino acid residues), then a
protein from that organism which has the ratio of potential deamidation sites
of 1:20 is said to have "fewer" deamidation sites. Thus, the term "fewer" is
relative and requires reference to the organism from which the protein is
derived. Further, the term "fewer" will typically be used to describe native
proteins which are selected for use in marker molecules of the invention due
to
their amino acid sequences. Proteins used to prepare marker molecules of the
invention may have any number of fewer post-translational modification sites
or post translational modifications. In particular instances, proteins used to
prepare marker molecules of the invention will have at least 5%, at least 10%,
at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least
40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at
least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least
95%,
at least 97%, or at least 99% fewer of one or more post-translational
modification sites or post translational modifications as compared to average
proteins of the particular organism in which the protein is naturally found.
As used herein, the term "reduced number," when used in reference to
post-translational modification sites or post-translational modifications
refers
to proteins that have been altered to decrease the number of post-
translational
modification sites or post-translational modifications. One example of a
situation where the number of post-translational modification sites is reduced
is where a protein which contains three glutamine and one asparagine residue
is modified to remove two of the glutamine residues. Thus, the number of
deamidation sites is reduced by two. As above from the term "few", the term
"reduced" may be used in reference to a single type of post-translational
modification site or post-translational modification or multiple types (e.g.,
CA 02477159 2004-08-20
-31-
two, three, four, five, six, seven, ten, etc.) of post-translational
modification
sites or post-translational modifications. In particular instances, proteins
used
to prepare marker molecules of the invention will exhibit a reduction in the
number of one or more post-translational modification sites or post
translational modifications of at least 5%, at least 10%, at least 15%, at
least
20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at
least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least
75%,
at least 80%, at least 85%, at least 90%, at least 95%, at least 97%, or at
least
99%.
Known post-translational modifications include, but are not limited to,
acetylation, acylation, ADP-ribosylation, amidation, carbamylation, covalent
attachment of flavin, covalent attachment of a heme moiety, covalent
attachment of a nucleotide or nucleotide derivative, covalent attachment of a
lipid or lipid derivative, covalent attachment of phosphotidylinositol, cross-
linking, cyclization, deamidation, disulfide bond formation, demethylation,
formation of covalent crosslinks, formation of cystine, formation of
pyroglutamate, formylation, gamma carboxylation, glycosylation, GPI anchor
formation, hydroxylation, iodination, methylation, myristoylation, oxidation,
proteolytic processing, phosphorylation, prenylation, racemization,
selenoylation, sulfation, transfer-RNA mediated addition of amino acids to
proteins such as arginylation, and ubiquitination. Such post-translational
modifications are well known to those of skill in the art. Several
particularly
common modifications, glycosylation, lipid attachment, sulfation, gamma-
carboxylation of glutamic acid residues, hydroxylation and ADP-ribosylation,
for instance, are described in most basic texts, such as Proteins--Structure
and
Molecular Properties, 2nd Ed., T. E. Creighton, W. H. Freeman and Company,
New York (1993). Many detailed reviews are available on this subject, such
as by Wold, F., Posttranslational Covalent Modification of Proteins, B. C.
Johnson, Ed., Academic Press, New York 1-12 (1983); Seifter et al. (Methods
of Enzymology 182:626-646 (1990)) and Rattan et al. (Ann. New York Acad.
Sciences 663:48-62 (1992)). In a particular aspect, proteins used to prepare
CA 02477159 2004-08-20
-32-
marker molecules of the present invention may contain no, few, fewer or a
reduced number of post-translational modifications or sites for such post-
translational modifications listed above, as well as other post-translational
modifications. Thus proteins used to prepare marker molecules of the
invention, as well as the marker molecules themselves, may comprise no, few,
fewer or a reduced number of post-translational modifications or post-
translational modification sites for one or more of the post-translational
modification processes referred to above, or other post-translational
modification processes.
A number of post-translational modifications occur in regions of
proteins wherein more than one amino acid residue is required for
modification recognition. In other words, while post-translational
modifications generally occur at a single amino acid residue within a protein,
intramolecular disulfide bonds being an exception, post-translational
1 S modifications often require a particular recognition sequence comprising
more
than one amino acid residue or a particular local conformation (e.g.,
secondary
structure). The invention thus includes marker molecules which comprise
proteins which contain no, few, fewer or a reduced number of such recognition
sites, as well as methods for making and using such markers and compositions
comprising such markers. Such marker molecules may be prepared, for
example, using proteins where a single amino acid comprising a
post-translational modification recognition region is altered or missing or
where local conformation required for one or more particular
post-translational modifications is lacking or is disrupted.
As noted above, proteins which may be used to prepare marker
molecules of the invention include proteins which comprise no, few, fewer or
a reduced number of post-translational modifications or post-translational
modification sites. Examples of amino acids (e.g., L-amino acids) which are
subject to post-translational modifications include the following: alanine,
asparagine, aspartic acid, glutamine, glutamic acid, glycine, histidine,
phenylalanine, proline, methionine, cysteine, lysine, tyrosine, serine, and
CA 02477159 2004-08-20
-33-
threonine. The invention thus includes marker molecules which contain no,
few, fewer or a reduced number of one or more (e.g., one, two, three, four,
five, etc.) of the amino acid residues referred to above, as well as methods
for
making and using such markers and compositions comprising such markers.
S These proteins may comprise, for example, (1) repeating sequences of amino
acid residues or repeating sets of two or more (e.g., two, three, four, five,
six,
etc.) amino acid residues, (2) amino acid sequences which are essentially
random, or (3) predefined amino acid sequences which are not based on any
particular order of the amino acids.
Amino acid residues which, with certain exceptions, are not subjected
to post-translational modifications include glycine, methionine, tryptophan,
alanine, valine, leucine, and isoleucine. Thus, the invention includes marker
molecules in which Segment A and/or Segment B is a protein wherein at least
40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 85%, at
least 90%, at least 95% of the amino acid residues present are one or more
(e.g., one, two, three, four, five six, or seven) of these amino acid residues
set
out directly above.
Examples of repeating sequences of amino acid residues or repeating
sets of two or more (e.g., two, three, four, five, six, etc.) amino acid
residues
which, under particular circumstances, may be subject to no, few, fewer or a
reduced number of post-translational modifications include proteins
comprising poly-glycine (e.g., a protein comprising 25, 30, 40, or 50 glycine
residues), repeating sets of glycine and alanine residues (e.g., a protein
comprising 10, 15, 25, 30, 40, or 50 sets of Gly-Ala residues), repeating sets
of
glycine, alanine, and valine residues (e.g., a protein comprising 10, 15, 25,
30,
40, or 50 sets of Gly-Ala-Val residues), etc.
Amino acid residues such as glycine and alanine are subject to
post-translational modifications such as GPI-anchoring (see Nalivaeva and
Turner, Proteomics 1:35-747 (2001)). Thus, when these amino acid residues
are present in a protein and post-translational modification at these residues
is
CA 02477159 2004-08-20
-34-
not desired, the protein may be produced in a manner such that GPI-anchoring
does not occur.
The invention also includes marker molecules wherein Segment A
and/or Segment B comprises a protein which does not contain any one, two,
three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen,
fourteen,
or fifteen of the following amino acids: alanine, arginine, asparagine,
aspartic
acid, glutamine, glutamic acid, glycine, histidine, phenylalanine, leucine,
isoleucine, proline, methionine, cysteine, lysine, tryptophan, tyrosine,
serine,
valine, and threonine. These proteins will typically also have other
characteristics referred to herein (e.g., will be of a particular molecular
weight).
Marker molecules of the invention will typically have a predefined
molecular weight and isoelectric point. Further, in order to function as a
member of a set of markers which vary in both molecular weight and
isoelectric point, typically it will be necessary to include amino acids which
may be subject to particular post-translational modifications. As noted
elsewhere herein, whether and/or when post-translational modification occurs,
as well as the number of post-translational modifications present, can be
regulated by the selection of the protein production method and/or the
conformation of the protein where the amino acid residue which functions as
the post-translational modification site is located. For example, ubiquination
is believed to occur exclusively in eukaryotic cells. Thus, when a
ubiquination
site is present in a protein, ubiquination can be prevented by either
synthetically producing the protein or by expressing the protein in a
prokaryotic cell. Similar methods can be used, for example, to produce
proteins which contain other post-translational modification sites (e.g.,
glycosylation sites) which are not post-translational modified at those sites
(e.g., not glycosylated).
One example of a post-translational modification which is not typically
directed by biological processes is deamidation. Thus, a purified protein, for
example, which contains amino acid residues which can undergo deamidation
CA 02477159 2004-08-20
- 35 -
(e.g., asparagine (N) and glutamine (Q) residues) will often undergo
deamidation during storage. When a protein, or marker molecule which
contains such a protein, contains more than one asparagine and/or glutamine
residues and these amino acid residues undergo deamidation, the result is a
heterogeneous population of molecules in which the degree of heterogeneity
varies with the number of amino acid residues in each protein which are
capable of undergoing deamidation and the differences between the individual
molecules in which these amino acid residues have undergone deamidation.
In other words, if all of the amino acid residues present in molecules which
are
capable of undergoing deamidation have either not undergone deamidation or
have undergone deamidation, then a homogenous population of molecules will
be present. However, if partial deamidation has occurred, the heterogeneity of
the population will be determined by the degree to which deamidation of the
individual amino acid residues in the molecules has occurred. Similar
1 S considerations apply to other forms of post-translational modifications
(e.g.,
glycosylation, ubiquination, etc.).
In particular aspects, the marker molecules of the present invention, as
well as proteins used to prepare marker molecules of the invention, may have
no, few, fewer, or a reduced number of asparagine and/or glutamine residues
within their sequences. The invention further includes methods for preparing
and using such markers and compositions comprising such markers.
The invention further includes methods for selecting proteins to be
used as Segment A and/or Segment B of marker molecules. These proteins
may be selected by any method including visual review of (1) nucleotide
sequences of coding regions or putative coding regions to identify nucleic
acid
molecules which encode proteins that contain no, few, fewer, or a reduced
number of one or more post-translational modification sites or (2) amino acid
sequences of proteins to identify proteins which contain no, few, fewer, or a
reduced number of one or more post-translational modification sites. In
addition, a computer program can be used to conduct the above review and
identify proteins or nucleic acid molecules which encode such proteins
CA 02477159 2004-08-20
-36-
suitable for preparing marker molecules. One example of a computer program
which would identify proteins suitable for preparing marker molecules is a
program which reviews amino acid sequence data by first identifying amino
acid sequences data which correspond to individual full-length proteins or
fragments thereof and then reviews the data to identify full-length proteins
or
fragments thereof which contain no, few, fewer, or a reduced number of one or
more post-translational modification sites. Thus, the invention provides, in
part, methods for selecting proteins for use in preparing marker molecules of
the invention, as well as marker molecules prepared using proteins selected by
these methods.
In one particular instance, the genome sequence for Eschericia coli
strain K12, which is accessible through the National Library of Medicine
GenBank database (www.ncbi.nlm.nih.gov) (see, e.g., sequence m
NC 000913) was searched using a computer program for protein sequences
containing no asparagine or glutamine residues, which are potential sites of
deamidation that can lead protein heterogeneity in a protein population. A
short PERL script (Practical Extraction Report Language) was used to identify
(1) the longest amino acid sequences in the genome containing neither of these
amino acids and (2) individual proteins which contained neither of these
amino acids. Alternatively, amino acid sequences expressed in the 4,639,221
by DNA may be searched manually (e.g., by visual inspection) for sequences
which contain no, few, fewer, or a reduced number of sites susceptible to one
or more post-translational modifications.
A number of proteins of Eschericia coli strain K12 proteins which
contain no deamidation sites have been identified and may be suitable for
preparing marker molecules of the invention. Examples of such proteins
include the following:
MHTGSTTLPDFFAGMSDDFTPPIFAGYCRDDSHELRFRLYALL (SEQ ID
NO:11);
MKAIFVLKGWWRTS (SEQ m N0:12);
CA 02477159 2004-08-20
-37-
MSFMVSEEVTVKEGGPRMIVTGYSSGMVECRWYDGYGVKREAFHET
ELVPGEGSRSAEEV (SEQ ID N0:13);
MKHIPFFFAFFFTFP (SEQ ID N0:14);
MTISDIIEIIVVCALIFFPLGYLARHSLRRIItDTLRLFFAKPRYVKPAGTL
RRTEKARATKK (SEQ ID N0:15);
MTALLRVISLVVISVVVIIIPPCGAALGRGKA (SEQ ID N0:16);
ALLWLTGSLWGRDWSFVKIAIPLMILFLPLSLSFCRDLDLLALGDARA
TTLGVSVPHTRFWALLLAVAMTSTGVAACGPISFIGLVVPHMMRSITG
GRHRRLLPVSALTGALLLVVADLLARIIHPPLELPVGVLTAIIGAPWFV
WLLVRMR (SEQ ID N0:17); and
GASLGEMIKEEMGPVPGTIALFGCFLIMIIILAVLALIVVKALAESPWGV
FTVCSTVPIALFMGIYMRFIRPGRVGEVSVIGIVLLVASIYFGGVIAHDP
YWGPALTFKDTTITFALIGYAFVSALLPVWLILAPRDYLATFLKIGVIV
GLALGIVVL (SEQ ID N0:18).
1 S In addition, when Segment A and/or Segment B is a protein,
post-translational modifications may occur at either the carboxyl terminus or
amino terminus. Examples of post-translational modifications which occur at
such termini include formylation, acetylation, pyroglutamate formation,
GPI-anchoring, amidation, and polyglycylation. Alternatively, the amino
and/or carboxyl termini may be blocked to prevent post-translational
modifications or the proteins may be produced under conditions in which
post-translational modification does not occur. Further, when Segment A
and/or Segment B is a protein, these proteins may be produced under
conditions in which one or both termini will not be post-translationally
modified.
The invention further includes marker molecules which contain a
particular numbers of sites (e.g., one, two, three, four, five, six, seven,
eight,
nine, ten, etc. sites) for the direct or indirect attachment of labels. These
attachments sites may comprise charged or uncharged chemical groups.
Further, the attachment of labels to these sites may or may not result in the
alteration to the charge of the attachment site chemical group. For instance,
CA 02477159 2004-08-20
-38-
Segment B may be a protein which contains one, two, three, four, five, six,
seven, eight, nine, ten, etc. sites for the direct or indirect attachments of
labels.
As a specific example, a protein which is about 159 amino acids in length
(e.g., the putative carbon starvation protein of E. coli) and contains two
cysteine residues may be contacted with a label (e.g., a dye) under conditions
which allow for direct (e.g., without the presence of an intervening linker)
covalently attachment to the cysteine residues. Attachment of the dye to the
cysteine residues, which are normally uncharged at neutral pH, will typically
result in the formation of a heterogeneous population of molecules. More
specifically, assuming both cysteine residues are accessible to the label but
the
labeling process does not go to completion, four groups of molecules will be
present in the population. These groups will be composed of the following
molecules: (1) molecules which contain no label, (2) molecules wherein the
label is attached to the first cysteine residue, (3) molecules wherein the
label is
attached to the second cysteine residue, and (4) molecules wherein the label
is
attached to both cysteine residues. When the label is used to detect the
presence of the marker molecules, the first group of molecules will not be
detected. Further, the second and third groups of molecules will generally
migrate virtually identically with respect to both molecular weight and
isoelectric point. Finally, the fourth group will typically migrate
differently
and be distinguishable from the molecules in the second and third groups after
separation by isoelectric point. Thus, when the above mixture, for example,
(1) is separated on a two dimensional gel electrophoresis system in which
molecular weight separation is used in the first dimensional and isoelectric
point separation is used in the second dimension and (2) the label is used to
detect the marker molecules, then two separate spots can be detected. In such
an instance, the isoelectric points of the marker molecules will often be
sufficiently close that when a relatively low resolution isoelectric focusing
gel
is used, either one spot or two spots which are located very close together
will
typically be detected but when a relatively high resolution isoelectric
focusing
gel is used two spots will be detected. Further, the isoelectric points of
both
CA 02477159 2004-08-20
-39-
marker molecules can be determined and/or calculated. Thus, the
modification of a single protein by the direct or indirect attachment of a
label
can be used to generate a heterogeneous population of marker molecules in
which a limited number of molecular species are present and the properties of
these species, with respect their ability to function as marker molecules
(e.g.,
molecular weight, isoelectric point, etc.), can be determined and/or
calculated.
Thus, the invention includes, in part, methods for preparing marker
molecules in which attachment of a label and/or post-translational
modifications are used to generate populations of molecules wherein
properties of the individual molecules present in the populations, with
respect
their ability to function as marker molecules (e.g., molecular weight,
isoelectric point, etc.), can be determined and/or calculated. For example,
such marker molecules may be prepared by providing a Segment A and/or
Segment B molecule which has more than one (e.g., two, three, four, five,
etc.)
site (e.g., a site for label attachment, a post-translational modification
site,
etc.) which can be modified either by the attachment of a label or other
molecule. As described above, when the process used to modify the Segment
A and/or Segment B molecules does not go to completion, the number of
species present in the mixed population will be determined by the number of
modification sites present. The invention thus includes marker molecule
populations which comprise two, three, four, five, six, seven, eight, nine,
ten,
etc. different marker molecules generated by the attachment of one or more
label or other molecule to single starting molecule (e.g., a protein). The
invention further includes methods for using such marker molecule
populations and marker molecules, as well as the marker molecule populations
and the marker molecules themselves and compositions comprising such
marker molecule populations and marker molecules. These marker molecule
populations are homogeneous in the sense that they are composed of a defined
set of molecules, each of which has characteristics which can be readily
determined either empirically or by calculation.
CA 02477159 2004-08-20
-40-
Further, when molecules are ligated to charged groups in a Segment B
molecule, this ligation can be used to modulate the overall charge of the
product (e.g., the Segment B molecule and/or the marker molecule). This
charge modulation can occur, for example, in two different ways, as well as a
S combination of these two ways. First, if the group on the Segment B where
attachment occurs is charged (e.g., positively or negatively charged), then
the
charge of the group may be altered (e.g., neutralized). Second, the molecule
which is ligated to the Segment B molecule may contain its own charged
groups and the addition of these charged groups to the product may confer a
new isoelectric point upon the product. In many instances, charge
modification by such methods will result in changes in isoelectric point of
less
than 1.0 pH unit, less than 0.8 pH units, less than 0.6 pH units, less than
0.4
pH units, or less than 0.2 pH units.
Native Chemical Ligation
Native Chemical Ligation involves ligation of a macromolecule or
small molecule containing a thioester (Segment A) with a protein (e.g., a
native, recombinant or synthetic protein) having an N-terminal cysteine or an
Na-(1-phenyl-2-mercaptoethyl) group (Segment B). Recombinant proteins
with desired termini may be produced in prokaryotic expression systems so
that they have preferably no, few, fewer, or reduced numbers post-
translational modifications. Marker molecules of the invention may be
generated using native chemical ligation.
Native proteins are suitable as long as they have appropriate termini
and have no, few, fewer, or a reduced number of sites susceptible to post
translational modification. Coupling of an auxiliary group, such as 1-phenyl
2-mercaptoethyl, to an N-terminal amino group is done post-transcriptionally
when all active side chains are blocked.
Peptides suitable as Segment A, may be prepared by solid phase
synthesis methods such as a highly optimized stepwise solid phase peptide
CA 02477159 2004-08-20
-41
synthesis (Kent, S.B.H., et al. U.S. Patent 6,184,344 B1; Dawson, P.E., et
al.,
Science 266:776-779 (1994); Lu, W., et al., J. Am. Chem. Soc. 118:8518-8523
(1996); Tolbert, T.J., et al., J. Am. Chem. Soc 122 (23):5421-5428 (2000); and
Swinen, D. et al., Org. Lett. 2:2439-2442 (2000)).
Solid phase chemical synthesis is a technique for the systematic
construction of a polypeptide from individual amino acids. Blocked amino
acids (e.g., with a-amino groups) such as the following may be used in solid
phase chemical synthesis: Alanine, Arginine, Aspartic Acid, Asparagine,
Cysteine, Glutamic Acid, Glutamine, Glycine, Histidine, Iso-leucine, Leucine,
Lysine, Methionine, Phenylalanine, Proline, Serine, Threonine, Tryptophan,
Tyrosine and Valine. Amino acids other than the twenty amino acids
commonly found in native proteins may also be incorporated into proteins by
solid phase synthesis and may be used to prepare markers molecules of the
invention. Examples of such non-natural amino acids include traps-4-
hydroxyproline, 3-hydroxyproline, cis-4-fluoro-L-proline, dimethylarginine,
homocysteine, the enantiomeric and racemic forms of 2-methylvaline, 2-
methylalanine, (2-i-propyl)-(3-alanine, phenylglycine, 4-methylphenylglycine,
4-isopropylphenylglycine, 3-bromophenylglycine, 4-bromophenylglycine, 4-
chlorophenylglycine, 4-methoxyphenylglycine, 4-ethoxyphenylglycine, 4-
hydroxyphenylglycine, 3-hydroxyphenylglycine, 3,4-dihydroxyphenylglycine,
3,5-dihydroxyphenylglycine, 2,5-dihydrophenylglycine, 2-
fluorophenylglycine, 3-fluorophenylglycine, 4-fluorophenylglycine, 2,3-
difluorophenylglycine, 2,4-difluorophenylglycine, 2,5-difluorophenylglycine,
2,6-difluorophenylglycine, 3,4-difluorophenylglycine, 3,5-
difluorophenylglycine, 2-(trifluoromethyl)phenylglycine, 3-
(trifluoromethyl)phenylglycine, 4-(trifluoromethyl)phenylglycine, 2-(2-
thienyl)glycine, 2-(3-thienyl)glycine, 2-(2-furyl)glycine, 3-pyridylglycine, 4-
fluorophenylalanine, 4-chlorophenylalanine, 2-bromophenylalanine, 3-
bromophenylalanine, 4-bromophenylalanine, 2-naphthylalanine, 3-(2-
quinoyl)alanine, 3-(9-anthracenyl)alanine, 2-amino-3-phenylbutanoic acid, 3-
chlorophenylalanine, 3-(2-thienyl)alanine, 3-(3-thienyl)alanine, 3-
CA 02477159 2004-08-20
-42-
phenylserine, 3-(2-pyridyl)serine, 3-(3-pyridyl)serine, 3-(4-pyridyl)serine, 3-
(2-thienyl)serine, 3-(2-furyl)serine, 3-(2-thiazolyl)alanine, 3-(4-
thiazolyl)alanine, 3-(1,2,4-triazol-1-yl)-alanine, 3-(1,2,4-triazol-3-yl)-
alanine,
hexafluorovaline, 4,4,4-trifluorovaline, 3-fluorovaline, 5,5,5-
trifluoroleucine,
2-amino-4,4,4-trifluorobutyric acid, 3-chloroalanine, 3-fluoroalanine, 2-
amino-3-flurobutyric acid, 3-fluoronorleucine, 4,4,4-trifluorothreonine, L-
allylglycine, tert-Leucine, propargylglycine, vinylglycine, S-methylcysteine,
cyclopentylglycine, cyclohexylglycine, 3-hydroxynorvaline, 4-azaleucine, 3-
hydroxyleucine, 2-amino-3-hydroxy-3-methylbutanoic acid, 4-thiaisoleucine,
acivicin, ibotenic acid, quisqalic acid, 2-indanylglycine, 2-aminoisobutyric
acid, 2-cyclobutyl-2-phenylglycine, 2-isopropyl-2-phenylglycine, 2-
methylvaline, 2,2-diphenylglycine, 1-amino-1-cyclopropanecarboxylic acid, 1-
amino-1-cyclopentanecarboxylic acid, 1-amino-1-cyclohexanecarboxylic acid,
3-amino-4,4,4-trifluorobutyric acid, 3-phenylisoserine, 3-amino-2-hydroxy-5-
methylhexanoic acid, 3-amino-2-hydroxy-4-phenylbutyric acid, 3-amino-3-(4-
bromophenyl)propionic acid, 3-amino-3-(4-chlorophenyl)propionic acid, 3-
amino-3-(4-methoxyphenyl)propionic acid, 3-amino-3-(4-
fluorophenyl)propionic acid, 3-amino-3-(2-fluorophenyl)propionic acid, 3-
amino-3-(4-nitrophenyl)propionic acid, and 3-amino-3-(1-naphthyl)propionic
acid. Thus, the invention includes marker molecules which contain one or
more amino acids other than the twenty amino acids commonly found in
proteins.
In solid phase chemical synthesis of peptides, amino acids are
covalently linked one at a time to a polypeptide chain in a C-terminal to N-
terminal direction. The C-terminal amino acid is generally coupled to a solid
support, such as a cross-linked polystyrene resin or other suitable insoluble
support. Typically, amino acids are systematically added, first to a resin
linker, and then to the previously added amino acid. Each amino acid added to
the growing chain must be chemically blocked at its a-amino group to prevent
addition of numerous amino acids to the chain in a single cycle. Common
blocking agents include tert-butyloxycarbonyl (BOC), 9-
CA 02477159 2004-08-20
- 43 -
fluorenylmethoxycarbonyl (FMOC), acetamidomethyl, acetyl, adamantyloxy,
benzoyl, benzyl, benzyloxy, benzyloxycarbonyl, benzyloxymethyl, 2-
Bromobenzyloxycarbonyl, t-butoxy, t-butoxymethyl, t-butyl, t-butylthio, 2-
chlorobenzyloxycarbonyl, cyclohexyloxy, 2,6-dichlorobenzyl, 4,4'-
dimethoxybenzhydryl, 1-(4,4-dimethyl-2,6-dioxocyclohexylidene)ethyl, 2,4-
dinitrophenyl, formyl, mesitylene-2-sulphonyl, 4-methoxybenzyl, 4-methoxy-
2,3,6-trimethyl-benzenesulphonyl, 4-methoxytrityl, 4-methyltrityl, 3-nitro-2-
pyridinesulphenyl, 2,2,5,7,8-pentamethylchroman-6-sulphonyl, tasyl,
trifluoroacetyl, trimethylacetamidomethyl, trityl, xanthyl and others known to
those of ordinary skill in the art. Such blocked amino acids are available
from
Sigma, St. Louis, MO. Thus, each cycle of amino acid addition typically
requires a deblocking step followed by an amino acid coupling step.
Following the systematic coupling of select amino acids to form a polypeptide
chain, the peptide may be released from the resin linker by the addition of an
agent such as a-toluenethiol, or other suitable solvent. Further, the peptide
may be recovered by purification techniques such as reverse phase, high-
pressure liquid chromatography (RP-HPLC), affinity chromatography, or
isoelectric fractionation.
In one example of the preparation of a suitable Segment A, the first
amino acid is a glycine attached by thioesterification to a polystyrene bead
and
protected by an FMOC group. The building block amino acid is Na Fmoc-NE
TMR-Lysine, which is also blocked by FMOC, and can be obtained from
many vendors, including Molecular Probes, (Eugene, OR, Catalogue No. F-
11830). The blocking group is present to prevent unwanted reactions during
the synthesis of the peptide. Extension of the peptide takes place by first
removing the blocking group with an agent such as trifluoroacetic acid (TFA),
and then allowing the newly free amino group to form a peptide bond with the
next building block amino acid. Following extension of the resin linker, an N-
terminal glycine may be added and labeled with iminobiotin, for recovery of
the peptide, by treating the peptide with 2-iminobiotin-N-hydroxysuccinimide
ester (available from Calbiochem-Novabiochem, San Diego, CA, Catalogue
CA 02477159 2004-08-20
-44-
No. 401778) in 0.1 M sodium phosphate as described by Greg T. Hermanson
(in Bioconjugate Techniques, Academic Press, San Diego, CA, p. 159 (1996)).
After cleavage with a-toluenethiol, the crude thioester peptide may be
purified
by a process such as RP-HPLC (FIG. 1). Synthesis of Segment A by the
above sequential and tightly controlled approach results in a homogeneous
population of specifically labeled peptides. The methods of the present
invention, such as those described above, may be used to sequentially
introduce a predetermined number of charged and/or chromophoric groups
into a sequence of amino acids to form a Segment A with a C-terminal
thioester and may be readily carried out by one of ordinary skill in the art.
In another embodiment, Segment A may have the formula:
Cys~Yp Z
where,
Y is one or more amino acid selected from the group consisting of alanine,
1 S arginine, aspartic acid, asparagine, cysteine, glutamic acid, glutamine,
glycine,
histidine, iso-leucine, leucine, lysine, methionine, phenylalanine, proline,
serine, threonine, tryptophan, tyrosine and valine or non-natural amino acids
such as traps-4-hydroxyproline, 3-hydroxyproline, cis-4-fluoro-L-proline,
dimethylarginine, and homocysteine,
wherein at least one amino acid is labeled with a chromophore, fluorophore, or
a UV absorbing group, preferably at least two amino acids are labeled;
Z is a C-terminal amino acid (Ca-carboxyl group may be modified to have an
amide function); and
n=1-100 covalently linked amino acid, (e.g., 1, 2, 3, 4, S, 6, 10, 30, 50, 75,
100, etc. covalently linked amino acids or 10-30, 5-50, 15-40, 20-50, 30-60,
40-70, 50-80, 60-90, 70-100 covalently linked amino acids) and/or 14
covalently linked amino acids. In another embodiment, Z may be any amino
acid listed above including non-natural amino acids such as those set out
herein. In another embodiment, the peptide is prepared via chemical
synthesis, preferably solid phase chemical synthesis. In a further embodiment,
the amino acid is labeled specifically with carboxytetramethylrhodamine
CA 02477159 2004-08-20
-45-
(TMR). In yet a further embodiment, the labeled amino acid is lysine. In
another embodiment, the N-terminal cysteine-labeled peptide may be ligated
with a protein with known molecular weight having an a-thioester. Ligation
occurs via Native Chemical Ligation or in vitro chemical ligation. In a
further
embodiment, the resulting product of the ligation reaction is a protein marker
of known molecular weight and pI.
In a further embodiment, the present invention relates to a polypeptide,
protein and marker molecules of the present invention further comprising a tag
molecule. In another embodiment, the tag molecule is selected from the group
consisting of biotin, fluorescein, digoxigenin, polyhistidine and their
derivatives thereof. Tag molecules may be used to facilitate protein
purification using ligands capable of binding to the tag such as avidin (binds
to
biotin), antibodies (binds to fluorescein or digoxigenin), lectin (binds to
sugars), or chelated metal ions (bind to polyhistidine). In another
embodiment,
1 S the polyhistidine comprises from two through ten contiguous histidine
residues (e.g., two, three, four, five, six, seven, eight, nine, or ten
contiguous
histidine residues). The tag may also be a peptide tag comprising an amino
acid sequence having the formula:
R1-(His-X)n-R2
wherein (His-X)" represents a metal chelating peptide and n represents a
number between two through ten (e.g., two, three, four, five, six, seven,
eight,
nine, or ten), and X is an amino acid selected from the group consisting of
alanine, arginine, aspartic acid, asparagine, cysteine, glutamic acid,
glutamine,
glycine, histidine, iso-leucine, leucine, lysine, methionine, phenylalanine,
proline, serine, threonine, tryptophan, tyrosine and valine. Further, Rz is a
polypeptide which is covalently linked to the metal chelating peptide and Rl
is
either a hydrogen or one or more (e.g., one, two, three, four, five, six,
seven,
eight, nine, ten, twenty, thirty, fifty, sixty, etc.) amino acid residues.
Tags of
this nature are described in U.S. Patent No. 5,594,115, the entire disclosure
of
which is incorporated herein by reference.
CA 02477159 2004-08-20
-46-
Segment B may be any N-terminal cysteine-containing protein (e.g.,
synthetic, recombinant or native), preferably of known molecular weight and
pI. A recombinant protein with N-terminal cysteine may be prepared using any
one of a number of E. coli expression vectors such as, but not limited to,
pBAD/Thio-TOPO~ (Invitrogen Corporation), pET (Invitrogen Corporation),
pTWIN (New England Biolabs), pTYB (New England Biolabs), and others
that are known in the art.
Ligation of Segment A to Segment B: The ligation reaction may be
carned out according to the optimized protocol of Kent in U.S. Patent
6,184,344 B1, the entire disclosure of which is incorporated herein by
reference (FIG. 2).
The first step is a chemoselective reaction of the N-terminal cysteine of
Segment B with the C-terminal thioester of Segment A (1.5 equivalents), for
example, in 6M guanidine hydrochloride HCI, pH 7.5 in the presence of 1%
toluenethiol and 5% thiophenol. Segment A's a-carbonyl thioester undergoes
nucleophilic attack by the cysteine residue at Segment B's N-terminus,
resulting in a thioester intermediate. The resulting thioester-linked
intermediate undergoes spontaneous intramolecular acyl transfer to the nearby
amine and forms a peptide bond (FIG. 2). The reaction is allowed to proceed
to completion, e.g. in 24 hours, and the resulting product is purified, e.g.
by
affinity chromatography.
In another embodiment, Segment A may be a TMR-labeled organic
thioester (see FIG. 3A). Acylation of triethylenetetramine (TREN, available
from Aldrich, Milwaukee, WI, Catalogue No. 90462) with 3.5 equiv. of an
activated ester of carboxytetramethylrhodamine (TMR), available from
Molecular Probes, OR (Catalogue No. e-6123), forms (TMR)3-TREN 5.
Acylation of N"-Fmoc-Lysine with 2-iminobiotin-N-hydroxysuccinimide ester
(Biotin-NS ester) yields N~ Fmoc-Na -biotin-Lysine 6, see FIGS. 3A and 3B.
Deblocking of the a-amino group of 6 followed by acylation with bromoacetyl
chloride forms N~ bromoacetamido-Na biotinyl-Lysine 8. The carbodiimide
CA 02477159 2004-08-20
-47-
coupling of 8 with a-toluenethiol results in 9. The alkylation of 5 with the
thioester 9 in the presence of sodium iodide generates the quaternary
ammonium salt 10 (Segment A) that upon coupling with Segment B under the
same conditions described above affords 11 (chromophore to protein ratio =
3).
In a further embodiment, Segment A may be a synthetic organic
molecule that is labeled with a chromophore with a high extinction coefficient
such as tetramethylrhodamine (TMR) as shown in FIG. 3B. In the reaction of
N-Boc-8-heptanoic acid 12 with a-toluenethiol in the presence of 1-[(3-
dimethylamino)propyl]-3-ethyl carbodiimide, methyl iodide and
dimethylaminopyridine (DMAP, available from Aldrich, Milwaukee, WI,
Catalogue No. 33,245-3) yields the corresponding thiobenzyl ester (FIG. 3B).
Deprotection of the amino group of 13 in the presence of TFA and subsequent
coupling of 14 to N-hydroxy succinimidyle ester of TMR generates the benzyl
thioester derivative of N-TMR-8-heptanoic acid 15. The reaction of the
thioester 15 (Segment A) with recombinant protein with N-terminal cysteine
(Segment B) forms TMR-protein 16 (chromophore to protein ratio = 1) that
can be purified by dialysis.
In another embodiment, Segment B may have the formula:
Cysteine-oligonucleotide
Coupling of N°'-(6-aminohexyl)ATP to N-a-t-Boc-S-trityl-L-cysteine
in the
presence of a water soluble carbodiimide such as EDC forms N-a-t-Boc-S-
trityl-6-aminohexylcysteinylamide. Deblocking of N-a-t-Boc-S-trityl-6-
aminohexylcysteinylamide in the presence of trifluoroacetic acid and
triisopropylsilane forms cysteine-ATP that can be added to an oligonecleotide
chain enzymatically to generate cysteine-oligonucleotide (Segment B).
Ligation of an oligopeptide with C°'-thioester labeled with
chromophores,
fluorophores, and LJV absorbing groups to the cysteine-oligonucleotide
segment in the presence of thiophenol and toluenethiol forms a labeled
oligopeptide-oligonucleotide.
CA 02477159 2004-08-20
-48-
In vitro chemical ligation
This method may involve ligation of Segment A, which is a labeled
molecule with N°'-cysteine or N°' -(1-phenyl-2-mercaptoethyl) or
small
organic molecule which is labeled and contains 1-amino-2-mercaptoethyl
moiety on a cysteine residue residue which is labeled through its carboxyl
group to a recombinant protein with a C-terminal thioester (Evans, Jr., T.C.,
et
al., J. Biol. Chem. 274:18359-8363 (1999)). However, the present invention is
not limited to molecules with an N-terminal cysteines (Low, D.W., et al.,
Proc. Nat. Acad. Sci. U.S.A. 98:6554-6655 (2001)). Thus, a molecule which
does not contain an N-terminal cysteine may be modified to form Na linked
removable moiety (Carne, L. et al., J. Amer. Chem. Soc. 118:5891-5896
(1996)). In a specific embodiment, any synthetic peptide with a thiol-
containing removable auxiliary moiety, such as 1-phenyl-2-mercaptoethyl,
appended to the N-terminus, may be used as Segment A. Following the
peptide bond formation, the auxiliary group can be removed in the presence of
appropriate deblocking reagents. See FIG. 9. In another embodiment, any
labeled organic molecule which contains 1-amino-2-mercaptoethyl group
maybe be used as Segment A. In a specific embodiment, a labeled cysteine
can be used as Segment A.
Segment B may be a protein (e.g., native, recombinant or synthetic
protein) or a nucleic acid with a C-terminal thioester. In a further
embodiment,
the commercially available pTWINI expression plasmid such as IMPACT
(New England Biolabs) with two modified mini inteins, Ssp DnaB and Mxe
GyrA, may be employed to express Mxe GyrA intein genetically fused to the
C-terminus of the protein of interest. Following affinity purification of the
fusion protein (for example, via a chitin binding domain (CBD) placed
downstream of Mxe GyrA), the target protein may be released simultaneously
forming a thioester by treatment with an external thiol such as ethane thiol,
n-
butane thiol, or 2-mercaptoethanesulfonic acid (MESNA). Inteins and their
use are described in U.S. Patent No. 5,834,247, the entire disclosure of which
CA 02477159 2004-08-20
-49-
is incorporated herein by reference. The IMPACT vectors have been used to
express Maltose Binding Protein (MBP), McrB, T4 DNA ligase, Bst DNA
polymerase Large Fragment, Bam HI, Bgl II, CDK2, CamK II and E. coli
RNA polymerases with C-terminal thioester, as well as altered forms of these
proteins.
Ligation of Segment A to Segment B: The feasibility of in vitro
chemical ligation to make visibly colored protein markers was first explored
in
a series of model reactions. A recombinant fragment corresponding to amino
acids 1-92 of the 404 amino acid-long E. coli maltose binding protein (MBP)
was genetically fused to the intein-CBD. The gene was modified at the DNA
level to append the sequence Met-Arg-Met at the C-terminus. This addition
was carried out to improve in vitro cleavage of the target protein (MBP-95aa)
from intein as well as to enhance the ligation reaction. Exposure of the
immobilized intein-fusion construct to MESNA has been shown to induce
cleavage, and this was confirmed in the present system. The target protein was
eluted as MBP-95aa-CO-S-CH2-CHZ-S03Na and was characterized by mass
spectroscopy (MS) and SDS gel. It was then evaluated whether the
immobilized construct could be chemically ligated to a short synthetic peptide
labeled with a chromophore (Cys-Lys(fluorescein)-Lys-Arg-Lys(fluorescein)-
Lys-His-His-His-His-His-His) (SEQ 117 NO:1) containing an N-terminal
cysteine. Overnight exposure of the chitin beads to 1.0 mM of the peptide and
mM of MESNA at 4 °C generated MBP-107aa-(fluorescein)2 which was
characterized by mass spectrometry. MBP-95aa (10.6 kD, pI 5.12) was
treated with Cys-Leu-Lys(TMR)-Asp-Ala-Leu-Asp-Ala-Leu-Asp-Ala-Leu-
25 Lys(TMR)-Asp-Ala-amide (SEQ m N0:3) in the presence of
tributylphosphine, toluene thiol and thiophenol at room temperature, 37
°C
and 50 °C (FIG. 8). The product was purified by RP- HPLC and
characterized
by MALDI/MS (13.0 kD, pI 4.75). In vitro chemical ligation using
recombinant proteins has been reported (Muir, T.W. et al., Proc. Natl. Acad.
30 Sci. USA 95:6704-6710 (1998)).
CA 02477159 2004-08-20
-50-
Site-specific modification
Site-specific modification may involve conjugation of peptides or
organic molecules to proteins with N-terminal serine or threonine. This
method is described in Geoghegan, K.F. and Stroh, J.G., Bioconjugate Chem.
3:138-146 (1992).
A further embodiment, depicted in FIG. 6, provides for the conjugation
of peptides or organic molecules to proteins with N-terminal serine or
threonine. The hydroxy group of these N-terminal amino acids is oxidized in
the presence of periodate (available from Aldrich, Milwaukee, WI) to form an
aldehyde, 17 (Segment B). Segment A is prepared from an oligopeptide or a
synthetic organic molecule, such as 8-aminocaprylic acid, 7-aminoheptanoic
acid and 6-aminohexanoic acid with a carboxyl function (18). Esterification
of Ca of the peptide or carboxyl group of the organic molecule and
subsequent exposure to hydrazine forms hydrazide 19. Coupling of Segment
A with Segment B, e.g., using Geoghegan protocol (Geoghegan K.F. and
Stroh, J.G., Bioconjugate Chem. 3:138-146 (1992)), forms the corresponding
hydrazone 20 that can be reduced in the presence of sodium
cyanoborohydride, to generate a more stable product, 21. Chromophoric
labels can be introduced into Segment A during synthesis; therefore, the
resulting product will be.visibly colored. This procedure is less preferred
than
using either native peptide ligation or in vitro chemical ligation procedures
because it requires the use of an oxidant to create the reactive group at the
N-
terminus that may damage the protein of Segment B.
The marker molecules and marker molecule compositions of the
present invention may be used as standards in any system commonly used to
separate macromolecules, e.g. by size, pI, or other physical or chemical
property. The marker molecules and marker molecule compositions may be
added to a matrix and exposed to an electromagnetic field which results in
movement of the molecular markers through the matrix. Examples of such
matrixes include, without limitation, agarose, cross-linked polyacrylamide
CA 02477159 2004-08-20
-51-
gels, cross-linked dextran, DEAE-cellulose, DEAF-Sephadex, DEAF
Sephacel and the like. The matrices may be in any form or shape, size or
porosity. The shapes include slabs, blocks, tubes, columns, membranes and
the like. The matrices may contain a number of additives which include,
without limitation, denaturant, and buffers. In another embodiment, the
marker molecules and marker molecule compositions may be used as markers
in capillary electrophoresis. In another embodiment, the marker molecules
and marker molecule compositions are used as standards when separating
macromolecules by any other method including column chromatography,
density gradient centrifugation, ion-exchange chromatography, size exclusion
chromatography, thin layer chromatography, liquid chromatography, and the
like.
In particular, marker molecules of the present invention may be used in
gel electrophoresis systems such as those described below. A considerable
number of gel electrophoresis separation systems are known in the art.
Further, these systems operate to separate molecules by a variety of
properties
associated with the molecules being separated. Further, multiple separation
principles may be combined to separate molecules (1) in a single gel
electrophoresis system or (2) in different gels electrophoresis systems. In
other words, molecules may be separated from each other in a
one-dimensional gel system which separates molecules based on one or more
(e.g., one, two, three, four, five, six, etc.) properties or the same
molecules
may be separated from each other using a two-dimensional gel, wherein each
phase of the separation process separates molecules based on one or more
(e.g., one, two, three, four, five, six, etc.) properties. Typically, when a
two-
dimensional gel system is used, molecules are separated in each of the two
dimensions based on at least one different property (e.g., charge in the first
dimension and molecular weight in the second dimension). Marker molecules
of the present invention may be employed in one-dimensional and
two-dimensional gel electrophoresis systems.
CA 02477159 2004-08-20
-52-
As noted above, gel electrophoresis systems may separate molecules
based on a variety of properties. Examples of these properties including
molecular weight, isoelectric point, and the ability of the molecules to bind
detergents (e.g., non-ionic detergents), as well as combinations of these
properties. Further, examples of gel electrophoresis systems in which marker
molecules of the invention may be employed include SDS-polyacrylamide gel
electrophoresis (SDS-PAGE), acid-urea gel electrophoresis, acid-urea gel
electrophoresis conducted in the presence of one or more detergents (e.g., one
or more non-ionic detergent such as TRITON X-100TM, sodium deoxycholate,
NONIDET P-40TM, etc.), and isoelectric focusing. Markers molecules of the
invention may be used, for example, with electrophoretic systems such as
one-dimensional gel electrophoresis systems, two-dimensional gel
electrophoresis systems, capillary electrophoresis systems, and electrokinetic
chromatography systems, as well as other gel electrophoresis systems.
In one aspect, the invention includes marker molecules of uniform
molecule weight, as well as compositions containing one or more (e.g., one,
two, three, four, five, six, eight, ten, twelve, twenty, fifty, etc.) marker
molecules which differ in molecular weight. These marker molecules are
particularly suited for use with gel electrophoresis systems which separate
molecules on the basis of molecular weight. Examples of gel electrophoresis
systems which separate molecules mainly on the basis of molecular weight
include SDS-PAGE systems (Laemmli, U.K., Nature 227:680-685 (1970)).
In another aspect, the invention includes marker molecules of uniform
isoelectric point, as well as compositions containing one or more (e.g., one,
two, three, four, five, six, eight, ten, twelve, twenty, fifty, etc.) marker
molecules which differ in isoelectric point. These marker molecules are
particularly suited for use with gel electrophoresis systems which separate
molecules on the basis of isoelectric point (e.g., isoelectric focusing
systems).
It will be understood by one of ordinary skill in the relevant arts that
other suitable modifications and adaptations to the methods and applications
described herein are readily apparent from the description of the invention
CA 02477159 2004-08-20
-53-
contained herein in view of information known to the ordinarily skilled
artisan, and may be made without departing from the scope of the invention or
any embodiment thereof. Having now described the present invention in
detail, the same will be more clearly understood by reference to the following
examples, which are included herewith for purposes of illustration only and
are not intended to be limiting of the invention.
EXAMPLES
EXAMPLE 1
Reaction of Cys-Ser-Thr-Met-Met-Ser-Arg-Ser-His-Lys-Thr-Arg-Ser-His-
His-Val-OH (SEQ ID N0:2) with TMR-thioester 15 using Native Chemical
Ligation
The model peptide, Cys-Ser-Thr-Met-Met-Ser-Arg-Ser-His-Lys-Thr-
Arg-Ser-His-His-Val-OH (SEQ >D N0:2), was prepared by optimized
stepwise solid phase peptide synthesis. The thioester 15 was prepared as
outlined in FIG. 3B. To a 1 mL solution of 6.0 M guanidine hydrochloride
buffered at pH 7.3 with 0.1 M sodium phosphate containing 5.0 mg (2.65 x
10-36 mmol) of the peptide was added 3.0 mg (1.5 x 10-3 mmol) of TMR-
thioester 15 dissolved in 20 p.L of acetonitrile. To this was added 10 pL (1%,
v/v) toluenethiol and 30 ~L (3%, v/v) thiophenol and stirred at room
temperature under Argon overnight. Mass spectroscopy data and SDS gel
electrophoresis showed that the product, TMR-labeled peptide was formed.
EXAMPLE 2
Cloning of Maltose Binding Protein-95aa (MBP-95aa) Gene into
pTWINl Vector
TOPO Cloning of MBP-95aa Gene: Two restriction sites, Spel and
Ndel, were introduced on either side of MBP-95aa gene. The PCR amplified
gene was purified and TOPO-cloned into pCR-TOPO vector. The pCR-
CA 02477159 2004-08-20
-54-
TOPOMBP-95aa gene was transformed into TOP10 competent cells and grew
on LB/AMP plate overnight. Ten colonies were taken and used to inoculate
ten 2-mL LB/AMP cultures (one colony/tube) and grown at 37 °C
overnight.
The DNA from each culture was isolated using S.N.A.P.TM (Simple Nucleic
Acid Prep) Miniprep kit (Invitrogen Corporation, Carlsbad, CA) and analyzed
by DNA sequencing.
Restriction Digestion and Ligation: The pCR-TOPOMBP-95aa was
digested simultaneously with SpeI and NdeI at 37 °C overnight. The
pTWINI
vector was digested with the same enzymes. Both reaction mixtures were
purified on a 1.2% agarose gel. The insertion of MBP-95aa gene into
pTWINl plasmid was conducted at 14 °C for 3-1/2 hours.
Transformation: TOP10 cells were transformed with the above
ligation mixture and plated on LB/AMP/Xgal along with control experiments.
Several 2-mL LB/AMP cultures were inoculated with different colonies (one
colony/tube) and grew at 37 °C overnight. pTWINIMBP-95aa was isolated
by S.N.A.P. Miniprep.
Screening for Insert: To confirm the insertion, pTWINIMBP-95aa was
digested with SpeI and NdeI enzymes. This reaction resulted in two
fragments: the insert, 250-300 by and the backbone, 7000 bp.
Cell Culture and Fusion Protein Expression
BL21/BAD cells were transformed with pTWINIMBP-95aa and were
plated on LB/AMP and grew at 37 °C overnight. A 2-mL LB/CAR (200 p,g
carbenicillin/mL LB) culture was inoculated with one colony and grew at 37
°C overnight. 1 liter LB/CAR medium containing 0.01% glucose was
inoculated with the above culture and grew at 30 °C. Mid-log phase
cells
were induced with 0.1 mM isopropyl-1-(3-D-galactopyranoside (IPTG) and
0.1% arabinose at 30 °C for 2-1/2 hours.
CA 02477159 2004-08-20
-SS-
Cell Harvest
The cells from the induced culture were spun down at 5000 X g for 1 S
minutes at 4 °C and the supernatant was discarded. At this stage, the
cell
pellets were stored at -80 °C.
S
Affinity Purification and On-column Cleavage
Preparation of crude cell extract
A 2.0 g pellet was resuspended in 100 mL of ice-cold lysis buffer (25
mM Tris pH 8.0, 800 mM KCI, 0.1 mM EDTA, 0.5% Triton X-100, 1.0 mM
PMSF) and was split into two portions. Each portion was sonicated for 1 min
X 4. Combined lysate was clarified by centrifugation at 12000 X g for 30
minutes at 4 °C.
Preparation of chitin column
A column packed with 15 mL of chitin beads (bed volume) was
prepared and equilibrated with 100 mL of column buffer (20 mM Tris, pH 8.5,
500 mM NaCI, 0.1 mM EDTA, 0.1% Triton X-100.
Loading the clarified cell lysate
The clarified cell lysate was loaded onto the chitin column at a flow
rate of 0.5 mL/min. The flow-through was collected and loaded onto the same
column at a flow rate of 1.0-2.0 mL/min.
Washing the chitin column
The column was washed with 500 mL of column buffer at a flow rate
of 2.0 mL/min.
All traces of crude extract were washed off the sides of the column.
CA 02477159 2004-08-20
-56-
Induction of on-column cleavage
The column was loaded with 50 mL of MESNA buffer (200 mM
mercaptoethane sulfonic acid in the column buffer), flushed quickly until the
buffer is slightly above the chitin beads. The flow was stopped and the
column was slowly rocked at room temperature overnight.
Elusion of the target protein
Following on-column cleavage of the intein, MESNA derivative of
MBP-95aa was released as a-thioester and eluted using column buffer. All
fractions were analyzed by SDS-PAGE. Combined fractions were
concentrated using Millipore Ultrafree - 15 Centrifugal Filter Device Biomax
- SK to yield 5.6 mg of the desired protein.
EXAMPLE 3
Synthesis of Peptides
A peptide suitable as a "Segment A" and having the following amino
acid sequence: Cys-Leu-Lys(TMR)-Asp-Ala-Leu-Asp-Ala-Leu-Asp-Ala-Leu-
Lys(TMR)-Asp-Ala-amide (SEQ ID N0:3), was prepared by highly optimized
stepwise solid phase peptide synthesis. In a 30-mL reaction vessel fitted with
a glass frit 909 mg (0.2 mmol) of Fmoc-PAL-PEG-PS resin (Applied
Biosystems, 0.22 meq.) was soaked in 10 ml of 20% of piperidine/DMF
solution containing 0.05 M HOBt for 5 minutes. The liquid was drained, and
the same procedure was repeated 2 more times. The resin was washed with 10
ml of DMF six times. In another reaction vessel, the carboxyl group of Fmoc-
Ala (249.0 mg, 0.8 mmol) was activated with of 303.0 mg (0.8 mmol) O-
benzotriazol-1-yl-N,N,N',N'-tetramethyluronium hexafluorophosphate
(HBTU) in the presence of 30.0 mg (0.2 mmol) of 1-hydroxybenzotriazole
(HOBT) and 280.0 ~.L (1.6 mmol) of N,N-diisopropylethylamine (DIEA) in
10 ml of DMF. The mixture was stirred for 3 minutes at room temperature,
CA 02477159 2004-08-20
-57-
added to the resin and stirred at room temperature for 1.5 hours. The mixture
was washed with DMF several times. The activation and coupling of the
second amino acid, Fmoc-Asp(O-t-Bu), was done under the same conditions
described for Fmoc-Ala. The third amino acid, Fmoc-Lys(TMR) was
purchased as N-hydroxysuccinimido ester (Molecular Probes). It did not
require further activation and was added to the reaction mixture (250 mg 0.32
mmol), protected from light and left at room temperature overnight.
Following Fmoc-Lys(TMR) coupling, the mixture was transferred into
Applied Biosystems Pioneer Peptide Synthesizer vessel. A peptide having the
amino acid sequence: Asp-Ala-Leu-Asp-Ala-Leu-Asp-Ala-Leu (SEQ ID
N0:4), was then assembled onto the Lys(TMR)-Asp-Ala-resin. The synthesis
protocol for the synthesizer was: 5 min deprotection step with piperidine/DMF
(1:4, v/v) containing 0.05M HOBt, 1 hr coupling time with Fmoc-amino
acid/HBTU/HOBTlDIEA (4:4:1:8). After the synthesis was done on the
synthesizer, the reaction mixture containing Asp-Ala-Leu-Asp-Ala-Leu-Asp-
Ala-Leu-Lys(TMR)-Asp-Ala-resin (SEQ ID N0:5) was transferred into the
manual reaction vessel, and the rest of the sequence Cys-Leu-Lys(TMR)) was
coupled stepwise and manually as described before (FIG. 8).
Deblocking: A reaction mixture containing 1.364 g of Cys-Leu-
Lys(TMR)-Asp-Ala-Leu-Asp-Ala-Leu-Asp-Ala-Leu-Lys(TMR)-Asp-Ala-
resin (SEQ ID N0:3) was added with 300 p,L of scavenger mixture
(thioanisole 10 ml/triisopropylsiline 4 ml/phenol 600 mg), 200 ~,1 of
mercaptopropionic acid (MPA) and 10 ml of 95% TFA/5% HZO was left at
room temperature for 3 hours with occasional stirnng. A 100 ml of tert-butyl
methyl ether (MTBE)/hexane (1:1) was added to the reaction mixture and
centrifuged. The supernatant was decanted, and the residue was washed with
50 ml of MTBE/hexane (1:1) and centrifuged again. The solid was separated
by decantation, extracted with 50 ml of 50% of acetonitrile in H20 and
lyophilized. The crude mixture was purified on preparative C-18 RP-HPLC to
yield 198 mg of pure peptide that was MS analyzed by MS (Found 2397.67,
Calc. 2398.71).
CA 02477159 2004-08-20
-58-
The following peptides were prepared:
Cys-Asp-Asp-Lys(TMR)-Asp-Asp-Asp-Asp-Leu-Ala-Asp-Asp-Asp-
Lys(TMR)-Asp-amide (SEQ ID N0:6)
Cys-Asp-Lys(TMR)-Asp-Ala-Asp-Asp-Leu-Ala-Asp-Leu-Asp-Lys(TMR)-
Asp-Ala-amide (SEQ ID N0:7)
Cys-Gly-Lys(TMR)-Ser-Gly-Ser-Gly-Lys-Ser-Gly-Lys-Gly-Lys(TMR)-Ser-
Gly-amide (SEQ ID N0:8)
Cys-Ala-Lys(TMR)-Leu-Lys-Ala-Lys-Ala-Lys-Leu-Ala-Lys-Lys(TMR)-Leu-
Ala-amide (SEQ ID N0:9)
Cys-Lys-Lys(TMR)-Lys-Ala-Lys-Leu-Lys-Ala-Lys-Lys-Lys-Lys-Lys(TMR)-
Ala-amide (SEQ ID NO:10)
Ligation of Cys-Leu-Lys(TMR)-Asp-Ala-Leu-Asp-Ala-Leu-Asp-Ala-
Leu-Lys(TMR)-Asp-Ala-amide (Segment A) (SEQ ID N0:3) to MBP-95aa
(Segment B): A mixture of MBP-95aa (0.4 x 10-6 mmol, 4.0 mg) and Cys-
Leu-Lys(TMR)-Asp-Ala-Leu-Asp-Ala-Leu-Asp-Ala-Leu-Lys(TMR)-Asp-
Ala-amide (0.4 x 10-5 mmol, 8.9 mg) (SEQ ID N0:3) was stirred in 6.0 M
guanidine hydrochloride buffered at pH 7.3 with 0.1 M sodium phosphate in
the presence of SmM tri-butylphosphine (25 p.L of 200 mM solution in 1-
methyl-2-pyrrolidinone) and 20 mM mercaptoethanol. To this was added 3%
(v/v) thiophenol as a catalyst and stirred at room temperature for 96 hours.
Every 24 hours, 25 pL of 200 mM solution of tributylphosphine was added to
the reaction mixture. The reaction mixture was monitored by SDS gel
electrophoresis and it went to 60% completion. The desired product, MBP-
110aa-(TMR)2, was purified on preparative RP HPLC and characterized by
SDS-gel and MALDI-MS (Found 13061.1, Calc. 13037.01; pI value 4.75).
MBP-110aa-(TMR)z, pI 4.75 was tested on NuPAGE Bis-Tris, 4-12%
(Invitrogen Corporation) and 16% Tricine gel (Invitrogen Corporation) using
MultiMark (Invitrogen Corporation) as protein marker; gel shown in FIG. 10.
The ligation of Cys-Asp-Asp-Lys(TMR)-Asp-Asp-Asp-Asp-Leu-Ala-
Asp-Asp-Asp-Lys(TMR)-Asp-amide (SEQ ID N0:6) to MBP-95aa, results in
a marker molecule, MBP(110a)-(TMR)2; calculated pI 4.3. The ligation of
CA 02477159 2004-08-20
-59-
Cys-Asp-Lys(TMR)-Asp-Ala-Asp-Asp-Leu-Ala-Asp-Leu-Asp-Lys(TMR)-
Asp-Ala-amide (SEQ ID N0:7) to MBP-95aa results in a marker molecule,
MBP(110a)-(TMR)Z; calculated pI 4.5. The ligation of Cys-Gly-Lys(TMR)-
Ser-Gly-Ser-Gly-Lys-Ser-Gly-Lys-Gly-Lys(TMR)-Ser-Gly-amide (SEQ ID
N0:8) to MBP-95aa results in a marker molecule, MBP(110a)-(TMR)2;
calculated pI 6.5. The ligation of Cys-Ala-Lys(TMR)-Leu-Lys-Ala-Lys-Ala-
Lys-Leu-Ala-Lys-Lys(TMR)-Leu-Ala-amide (SEQ ID N0:9) to MBP-95aa
results in a marker molecule, MBP(110a)-(TMR)Z; calculated pI 7.4. The
ligation of Cys-Lys-Lys(TMR)-Lys-Ala-Lys-Leu-Lys-Ala-Lys-Lys-Lys-Lys-
Lys(TMR)-Ala-amide (SEQ ID NO:10) to MBP-95aa results in MBP(110a)-
(TMR)2; calculated pI 9.5.
Having now fully described the present invention in some detail by
way of illustration and example for purposes of clarity of understanding, it
will be obvious to one of ordinary skill in the art that the same can be
performed by modifying or changing the invention within a wide and
equivalent range of conditions, formulations, and other parameters without
affecting the scope of the invention or any specific embodiment thereof, and
that such modifications of changes are intended to be encompassed within the
scope of the appended claims.
All publications and patents mentioned in this specification are
indicative of the level of skill of those skilled in the art to which this
invention
pertains, and are herein incorporated by reference to the same extent as if
each
individual publication or patent was specifically and individually indicated
to
be incorporated by reference.
CA 02477159 2004-08-20
1/8
SEQUENCE LISTING
<110> Tadayoni-Rebek, Mitra
Amshey, Joseph W.
Rooney, Regina
<120> Highly Homogeneous Molecular Markers for Electrophoresis
<130> 0942.530PC03
<150> US 60/357,634
<151> 2002-02-20
<160> 18
<170> PatentIn version 3.2
<210> 1
<211> 12
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<220>
<221> MOD_RES
<222> (2) .(2)
<223> Modified with fluorescein
<220>
<221> MOD_RES
<222> (5) .(5)
<223> Modified with fluorescein
<400> 1
Cys Lys Lys Arg Lys Lys His His His His His His
1 5 10
<210> 2
<211> 16
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic Peptide
CA 02477159 2004-08-20
2/8
<400> 2
Cys Ser Thr Met Met Ser Arg Ser His Lys Thr Arg Ser His His Val
1 5 10 15
<210> 3
<211> 15
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<220>
<221> MOD_RES
<222> (3) . (3)
<223> Modified with TMR
<220>
<221> MOD_RES
<222> (13) .. (13)
<223> Modified with TMR
<220>
<221> MOD_RES
<222> (15)..(15)
<223> AMIDATION
<400> 3
Cys Leu Lys Asp Ala Leu Asp Ala Leu Asp Ala Leu Lys Asp Ala
1 5 10 15
<210> 4
<211> 9
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<400> 4
Asp Ala Leu Asp Ala Leu Asp Ala Leu
1 5
<210> 5
<211> 12
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
CA 02477159 2004-08-20
3/8
<220>
<221> MOD_RES
<222> (10)..(10)
<223> Modified with TMR
<400> 5
Asp Ala Leu Asp Ala Leu Asp Ala Leu Lys Asp Ala
1 5 10
<210> 6
<211> 15
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<220>
<221> MOD_RES
<222> (4) .(4)
<223> Modified with TMR
<220>
<221> MOD_RES
<222> (14)..(14)
<223> Modified with TMR
<220>
<221> MOD_RES
<222> (15)..(15)
<223> AMIDATION
<400> 6
Cys Asp Asp Lys Asp Asp Asp Asp Leu Ala Asp Asp Asp Lys Asp
1 5 10 15
<210> 7
<211> 15
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<220>
<221> MOD_RES
<222> (15)..(15)
<223> AMIDATION
<220>
CA 02477159 2004-08-20
4/8
<221> MOD_RES
<222> (3) .(3)
<223> Modified with TMR
<220>
<221> MOD_RES
<222> (13)..(13)
<223> Modified with TMR
<400> 7
Cys Asp Lys Asp Ala Asp Asp Leu Ala Asp Leu Asp Lys Asp Ala
1 5 10 15
<210> 8
<211> 15
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<220>
<221> MOD_RES
<222> (3) .(3)
<223> Modified with TMR
<220>
<221> MOD_RES
<222> (13)..(13)
<223> Modified with TMR
<220>
<221> MOD_RES
<222> (15)..(15)
<223> AMIDATION
<400> 8
Cys Gly Lys Ser Gly Ser Gly Lys Ser Gly Lys Gly Lys Ser Gly
1 5 10 15
<210> 9
<211> 15
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<220>
<221> MOD_RES
<222> (3) . (3)
CA 02477159 2004-08-20
5/8
<223> Modified with TMR
<220>
<221> MOD_RES
<222> (13) . . (13)
<223> Modified with TMR
<220>
<221> MOD_RES
<222> (15)..(15)
<223> AMIDATION
<400> 9
Cys Ala Lys Leu Lys Ala Lys Ala Lys Leu Ala Lys Lys Leu Ala
1 5 10 15
<210> 10
<211> 15
<212> PRT
<213> Artificial Sequence
<220>
<223> Synthetic peptide
<220>
<221> MOD_RES
<222> (3) . (3)
<223> Modified with TMR
<220>
<221> MOD_RES
<222> (14)..(14)
<223> Modified with TMR
<220>
<221> MOD_RES
<222> (15)..(15)
<223> AMIDATION
<400> 10
Cys Lys Lys Lys Ala Lys Leu Lys Ala Lys Lys Lys Lys Lys Ala
1 5 10 15
<210> 11
<211> 43
<212> PRT
<213> Escherichia coli
<400> 11
Met His Thr Gly Ser Thr Thr Leu Pro Asp Phe Phe Ala Gly Met Ser
1 5 10 15
CA 02477159 2004-08-20
6/8
Asp Asp Phe Thr Pro Pro Ile Phe Ala Gly Tyr Cys Arg Asp Asp Ser
20 25 30
His Glu Leu Arg Phe Arg Leu Tyr Ala Leu Leu
35 40
<210> 12
<211> 14
<212> PRT
<213> Escherichia coli
<400> 12
Met Lys Ala Ile Phe Val Leu Lys Gly Trp Trp Arg Thr Ser
1 5 10
<210> 13
<211> 60
<212> PRT
<213> Escherichia coli
<400> 13
Met Ser Phe Met Val Ser Glu Glu Val Thr Val Lys Glu Gly Gly Pro
1 5 10 15
Arg Met Ile Val Thr Gly Tyr Ser Ser Gly Met Val Glu Cys Arg Trp
20 25 30
Tyr Asp Gly Tyr Gly Val Lys Arg Glu Ala Phe His Glu Thr Glu Leu
35 40 45
Val Pro Gly Glu Gly Ser Arg Ser Ala Glu Glu Val
50 55 60
<210> 14
<211> 15
<212> PRT
<213> Escherichia coli
<400> 14
Met Lys His Ile Pro Phe Phe Phe Ala Phe Phe Phe Thr Phe Pro
1 5 10 15
<210> 15
<211> 62
<212> PRT
<213> Escherichia coli
<400> 15
Met Thr Ile Ser Asp Ile Ile Glu Ile Ile Val Val Cys Ala Leu Ile
CA 02477159 2004-08-20
7/8
1 5 10 15
Phe Phe Pro Leu Gly Tyr Leu Ala Arg His Ser Leu Arg Arg Ile Arg
20 25 30
Asp Thr Leu Arg Leu Phe Phe Ala Lys Pro Arg Tyr Val Lys Pro Ala
35 40 45
Gly Thr Leu Arg Arg Thr Glu Lys Ala Arg Ala Thr Lys Lys
50 55 60
<210> 16
<211> 32
<212> PRT
<213> Escherichia coli
<400> 16
Met Thr Ala Leu Leu Arg Val Ile Ser Leu Val Val Ile Ser Val Val
1 5 10 15
Val Ile Ile Ile Pro Pro Cys Gly Ala Ala Leu Gly Arg Gly Lys Ala
20 25 30
<210> 17
<211> 152
<212> PRT
<213> Escherichia coli
<400> 17
Ala Leu Leu Trp Leu Thr Gly Ser Leu Trp Gly Arg Asp Trp Ser Phe
1 5 10 15
Val Lys Ile Ala Ile Pro Leu Met Ile Leu Phe Leu Pro Leu Ser Leu
20 25 30
Ser Phe Cys Arg Asp Leu Asp Leu Leu Ala Leu Gly Asp Ala Arg Ala
35 40 45
Thr Thr Leu Gly Val Ser Val Pro His Thr Arg Phe Trp Ala Leu Leu
50 55 60
Leu Ala Val Ala Met Thr Ser Thr Gly Val Ala Ala Cys Gly Pro Ile
65 70 75 80
Ser Phe Ile Gly Leu Val Val Pro His Met Met Arg Ser Ile Thr Gly
85 90 95
Gly Arg His Arg Arg Leu Leu Pro Val Ser Ala Leu Thr Gly Ala Leu
CA 02477159 2004-08-20
8/8
100 105 110
Leu Leu Val Val Ala Asp Leu Leu Ala Arg Ile Ile His Pro Pro Leu
115 120 125
Glu Leu Pro Val Gly Val Leu Thr Ala Ile Ile Gly Ala Pro Trp Phe
130 135 140
Val Trp Leu Leu Val Arg Met Arg
145 150
<210> 18
<211> 158
<212> PRT
<213> Escherichia coli
<400> 18
Gly Ala Ser Leu Gly Glu Met Ile Lys Glu Glu Met Gly Pro Val Pro
1 5 10 15
Gly Thr Ile Ala Leu Phe Gly Cys Phe Leu Ile Met Ile Ile Ile Leu
20 25 30
Ala Val Leu Ala Leu Ile Val Val Lys Ala Leu Ala Glu Ser Pro Trp
35 40 45
Gly Val Phe Thr Val Cys Ser Thr Val Pro Ile Ala Leu Phe Met Gly
50 55 60
Ile Tyr Met Arg Phe Ile Arg Pro Gly Arg Val Gly Glu Val Ser Val
65 70 75 80
Ile Gly Ile Val Leu Leu Val Ala Ser Ile Tyr Phe Gly Gly Val Ile
85 90 95
Ala His Asp Pro Tyr Trp Gly Pro Ala Leu Thr Phe Lys Asp Thr Thr
100 105 110
Ile Thr Phe Ala Leu Ile Gly Tyr Ala Phe Val Ser Ala Leu Leu Pro
115 120 125
Val Trp Leu Ile Leu Ala Pro Arg Asp Tyr Leu Ala Thr Phe Leu Lys
130 135 140
Ile Gly Val Ile Val Gly Leu Ala Leu Gly Ile Val Val Leu
145 150 155