Note: Descriptions are shown in the official language in which they were submitted.
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
CONTROLLED RELEASE DOSAGE FORMS
CROSS-REFERENCE TO RELATED APPLICATIONS
This application is a continuation-in-part application of U.S. application
Serial
Number 10/291619, filed on November 12, 2002 and claims the benefit of
provisional
application Serial Number 60/342,442, filed December 24, 2001, and provisional
application Serial Number 60/361,821, filed March 4, 2002, both of which are
incorporated herein by reference.
FIELD OF THE INVENTION
The present invention relates to oral pharmaceutical dosage forms and more
particularly to controlled release forms and forms designed to mask the taste
of the active
ingredient.
BACKGROUND OF THE INVENTION
Tailoring drug delivery to the needs of therapy is a current goal in the
development
of drug delivery systems. The delivery profile rnay be desired to be one of
immediate
release within the oral cavity (the so-called "immediate dissolve" or "fast
dissolve" systems),
immediate release in the stomach or in the intestine, controlled slow release
of the drug in
the gastrointestinal (G)] tract, concomitant release of more than one drug at
the same or at
different rates, and many combinations of the above. There are systems that
exist to provide
drug delivery profiles that approximate the above requirements, but in each
category there is
room for improvement.
Immediate dissolve systems for immediate delivery of drugs in the oral cavity
have
been developed by R. P. Scherer Corporation in the form of a freeze dried
tablet that
readily dissolves on the tongue called Zydis~ and by Cima labs, Inc. in the
form of the
OraSolv~ system. These systems dissolve quickly in the mouth and are useful
for cases
where the delivery of the drug is needed immediately and in cases where the
patient has
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
difficulty swallowing tablets. Both of these systems suffer from being
relatively fragile and
very sensitive to moisture. They are therefore difficult to handle with the
moisture of the
fingers damaging the integrity of the delivery system ("melts on the hands and
not in the
mouth" to paraphrase an old advertisement).
In the world of controlled release drug delivery systems there have been
certain
axioms upon which much development has been based. One such axiom is that
'flatter is
better' i.e. the flatter the delivery curve is vs. time the better the system
will behave. It is
therefore considered desirable to have delivery systems that give essentially
a zero order
release profile. The amount of drug released is not dependent on the amount
left within the
delivery system and remains constant over the entire delivery profile.
Tailoring the drug
delivery to the needs of the therapy is another axiom of delivery improvement.
One can
conceive of therapies that need a sudden burst of drug after several hours of
constant
delivery or a change in the rate of drug delivery after several hours.
A swelling hydrogel tablet delivery system or an eroding tablet delivery
system,
gives drug delivery that tapers off with time. In the eroding system, the
surface that provides
drug delivery is shrinking with time so the rate falls off proportionally. If
the drug is delivered
by diffusion through a non eroding hydrogel the rate falls off as drug
depletion changes the
force of the chemical gradient. These systems do not offer the opportunity to
carefully tailor
the drug release rates.
Zero order delivery has been achieved with the "Oros" osmotic pumps as is
documented in many patents held by the Alza company (e.g. US Patent 3,995,631
to
Higuchi, T. et. al., US Patent 3,977,404 to Theeuwes,F. and many other
patents).The
"Oros" system is based on osmotic pressure pushing the drug out of an almost
microscopic
orifice. The zero order profile is achieved due to the constant, small, cross
section of the
orifice being the rate determining step in the drug release. The "Oros" system
has proven
itself in several products but has limitations. It is most useful for soluble
drugs with insoluble
drugs having limited applicability. The technology of manufacture is somewhat
complicated with the need of a laser drilled hole in the semipermeable
coating. The drug
release through an almost microscopic hole can also lead to several drawbacks.
Clogging of
2
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
the hole may limit drug release and the streaming of a concentrated solution
of drug from the
delivery system to the intestinal lumen can cause damage to the intestinal
wall (see Laidler,
P.; Maslin, S. C.; and Gihome, R. W. Pathol Res Pract 1985 180 (1) 74-76) .
Delays of
the start of drug release can be achieved by coating the system (such as with
an enteric
coating) but the small orifice may be clogged by the coating and give erratic
results in
opening (if at all). The "Oros" system is best suited for a simple zero order
delivery profile.
Complicated patterns can be achieved with the "Oros" such as described in US
Patent
5,156,850 to Wong, P. S. et. al. and in PCT WO 9823263 to Hamel, L. G. et. al.
with
concomitant complication of the manufacture and of the system, and without
solving the
drawbacks of the almost microscopic hole.
Zero order delivery profiles have been achieved with clever manipulation of
the
geometric surface of drug delivery as embodied in the "Geomatrix" delivery
systems. ( US
Patents 4,839,177 to Colombo, P. et. al. and 5,422,123 to Conte, U. et. al.
and assigned
to Jagotech AG and many other patents). These systems achieve a zero order
profile by
1 S sandwiching the drug delivery layer between two layers that are
impermeable. Only the drug
delivery layer is eroded and the cross-section of the eroding layer is
constant. Again here,
there are several drawbacks. The manufacture of the system requires special
equipment to
produce two and three layer tablets. The system does not easily lend itself to
changing the
rate of delivery during the release profile. The amount of drug available in
the tablet is
somewhat limited since only one of the layers is used for drug delivery. The
zero order
profile may not be followed up to 100% of drug release due to tablet breakup
once most of
the central layer has eroded.
In view of the foregoing, it would be highly desirable to have a versatile
solid
dosage form that enables controlled release of an active ingredient
approaching zero order
release. Accordingly, one obj ect of the present invention is to provide a
solid dosage form
that can release a drug according to a predetermined release profile.
3
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
SUMMARY OF THE INVENTION
The present invention provides controlled release pharmaceutical dosage forms
in
which a core tablet is sheathed in an annular body of compressed powder or
granular
material.
The drug layer may be recessed from the opening of the annular body on one or
both sides. The drug layer is recessed from the surface so that any contact,
whether with
hands or with the mucosa, is with the walls of the annular body. The annular
body is
preferably made of non ulcerative and non sensitive pharmaceutical ingredients
such as
hydroxypropyl cellulose, hydroxypropyl rnethylcellulose, microcrystalline
cellulose, starch,
lactose, sugars, polyvinyl pyrrolidone, calcium phosphate and any other
regular tablet
excipients.
The controlled release pharmaceutical dosage forms of the invention release
the
active ingredient from the core tablet into the environment of the dosage form
at a rate in the
range of from 3% per hour to 12% per hour.
: The present invention further provides a pharmaceutical dosage form wherein
the
pharmaceutical dosage form is adapted for extended or zero-order release of
active drug
material.
The present invention further provides a pharmaceutical dosage form wherein
the
pharmaceutical dosage form is adapted for immediate release of active drug
material.
The present invention further provides a pharmaceutical dosage form wherein
the
pharmaceutical dosage form is adapted for sublingual administration.
The present invention further provides a pharmaceutical dosage form wherein
the
pharmaceutical dosage form is adapted so as to mask the taste of the active
material.
The present invention further provides a method of independently controlling
the
rate of release of coactive ingredients in a single dosage form.
The present invention further provides a pharmaceutical dosage form for co-
administration of coactive ingredients in a single dosage form.
4
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
BRIEF DESCRIPTION OF THE FIGURES
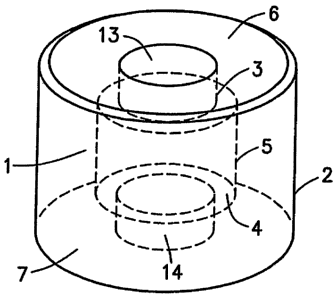
FIG. 1 shows sectional perspective, side and top down views of a solid dosage
form with a recessed core tablet of active ingredient in a compressed annular
body of
powder or granular material in accordance with the invention.
FIG. 2 is a perspective view of a single station tableting press shown with
the
toolset installed.
FIG. 3 is a sectional side view of the columnar punch and punch assembly.
FIGs. 4a-4e are sectional side views depicting stages in a cycle of operation
from
delivery of powder or granular material to ej ection of a finished tablet at a
tableting station
equipped with a toolset in accordance with the invention.
FIG. 5 is a plot of the average rate of alendronate excretion in urine of
humans who
had taken a dosage form in accordance with the present invention containing 70
mg
monosodium alendronate and a prior art 70 mg monosodium alendronate dosage
form.
FIG. 6 is a plot of the rate of release of oxybutynin from a dosage form in
accordance with the invention, wherein the rate of release is maintained
between 3% h'I and
12% h'I for seven hours or more.
FIG. 7 is a plot of the rate of release of oxybutynin from a dosage form in
accordance with the invention. The proportion of hydrogel in the core tablet
is increased
relative to the dosage form that produced FIG. 6 resulting in a decreased
maximum rate of
release and an extended release between 3% and 12% per hour for about twelve
hours.
FIG. ~ is a plot of the rate of release of oxybutynin from a dosage form in
accordance with the invention. The proportion of release-inhibiting hydrogel
in the annular
body was increased relative to the dosage form that produced FIG. 7. The
maximum rate
of release was further reduced to less than 7% h''.
FIG. 9 is a plot of the rate of release of carbidopa from the core tablet and
of
levodopa from the annular body of a dosage form in accordance with the present
invention.
The core tablet is cylindrically shaped and annular having a 2.5 mm diameter
hole
therethrough.
5
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
FIG. 10 is a plot of the rate of release of carbidopa from the core tablet and
of
levodopa from the annular body of a dosage form in accordance with the present
invention.
The core tablet of this dosage form has a 4.6 mm hole, larger than that in the
dosage form
that produced Fig. 9, resulting in greater surface area and a more rapid rate
of release of
carbidopa.
FIG. 11 is a plot of the rate of release of carbidopa from the core tablet and
of
levodopa from the annular body of a dosage form in accordance with the present
invention.
The dosage form that produced this figure had an oval core tablet with a 3 mm
hole
therethrough which resulted in a release similar to the cylindrical core table
with a 2.5 mm
hole (FIG. 9).
DESCRIPTION OF THE PREFERRED EMBODIMENTS
The present invention provides a novel solid dosage form, as well as tooling
and a
process for producing the novel dosage form. Preferred embodiments of the
invention are
well suited for the controlled release of drugs, especially extended release
approaching
zero-order, and for taste masking of unpleasant tasting drugs.
The novel dosage form comprises a core tablet containing an active
pharmaceutical
ingredient sheathed in an annular body (also called a mantle in this
disclosure) comprised of
compressed powder or granular material. The core tablet has first and second
opposed
surfaces and a circumferential surface. "Sheathed" means that the annular body
encircles
the core tablet and is in contact with the core tablet about its
circumferential surface, but
leaves opposed surfaces of the core tablet substantially exposed. The core
tablet contains
at least one active pharmaceutical ingredient, but otherwise its formulation
is not critical to
the invention. The core tablet can be formulated for any desired release
profile, such as
immediate release, delayed release, burst or pulsed release, sustained or zero
order release.
The annular body can be formulated to achieve any desired purpose, such as
gastric
retention, ease of swallowing, taste masking and control of the rate of drug
release from the
core tablet. The annular body also can contain or be coated with a co-active
ingredient.
6
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
The terms "drug" and "active pharmaceutical ingredient" broadly include any
biologically, physiologically, or pharmacologically active the agent. Active
pharmaceutical
ingredients that can be administered in the compressed dosage form of the
present invention
include adrenergic receptor agonists and antagonists; muscarinic receptor
agonists and
antagonists; anticholinesterase agents; neuromuscular blocking agents;
ganglionic blocking
and stimulating agents; sympathomirnetic drugs; serotonin receptor agonists
and antagonists;
central nervous system active drugs such as psychotropic drugs, antipsychotic
drugs,
antianxiety drugs, antidepressents, antimanic drugs, anesthetics, hypnotics,
sedatives,
hallucinogenic drugs and antihallucinogenic drugs; antiepileptic drugs;
antimigraine drugs;
drugs for treatment of Parkinson's, Alzheimer's and Huntington's disease;
analgesics;
antitussive agents; antihistaminic drugs; HI, HZ, and H3 receptor antagonists;
bradykinin
receptor antagonists; antipyretic agents; antiinflammatory agents; NSA)J7s;
diuretics;
inhibitors of Na+-Cl- symport; vasopressin receptor agonists and antagonists;
ACE
inhibitors; angiotensin II receptor antagonists; renin inhibitors; calcium
channel blockers; (3-
adrenergic receptor antagonists; antiplatelet agents; antithrombic agents;
antihypertensive
agents; vasodilators; phosphodiesterase inhibitors; antiarrhythmic drugs; HMG
CoA
reductase inhibitors; H~, I~+-ATPase inhibitors; prostaglandins and
prostaglandin analogs;
laxatives; antidiarrheal agents; antiemetic agents; prokinetic agents;
antiparasitic agents such
as antimalarial agents, antibacterial agents, drugs for treatment of protozoal
infections and
antihelinintic drugs; antimicrobial drugs such as sulfonamides, quinolones, [3-
lactam
antibiotics, aminoglycosides, tetracyclines, chloramphenicol and erythromycin;
drugs for
treatment of tuberculosis, drugs for treatment of leprosy; antifungal agents;
antiviral agents;
antineoplastic agents; immunomodulators; hematopoietic agents; growth factors;
vitamins;
minerals; anticoagulants; hormones and hormone antagonists such as antithyroid
drugs,
estrogens, progestins, androgens, adrenocortical steroids and adrenocortical
steroid
inhibitors; insulin; hypoglycemic agents; calcium resorption inhibitors;
glucocorticoids;
retinoids and heavy-metal antagonists.
The annular body can be formed of any powdered or granular pharmaceutically
acceptable excipients and can itself include a pharmaceutically active
ingredient. In
7
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
particular, it may be mentioned that diluents, binders, disintegrants,
glidants, lubricants,
flavorants, colorants and the like can be included in the annular body.
Powdering and
granulation with conventional excipients and the techniques for forming
compressed bodies
therefrom with given characteristics in terms of friability, hardness and
freedom from
capping is well within the knowledge of those skilled in the art of tableting.
Preferred excipients for forming the annular body include hydroxypropyl
cellulose
(e.g., Klucel'z''t), hydroxypropyl methylcellulose (e.g. Methocel~),
microcrystalline
cellulose (e.g., Avicel~), starch, lactose, sugars, polyvinylpyrrolidone
(e.g., Kollidon~,
PlasdoneTM) and calcium phosphate.
In an especially preferred compressed dosage form illustrated in FIG. 1, core
tablet
1 containing the active pharmaceutical ingredient is recessed in the annular
body 2, which is
composed of non-ulcerative pharmaceutical excipients. The "recessed" tablet is
especially
well suited for oral delivery of ulcerative drugs. It reduces the incidence of
pill esophagitis
and contact gastritis by localizing the ulcerative drug in a core tablet that
is shielded from
contact with the mucosa lining the gastrointestinal tract. The drug is
shielded because the
core tablet is recessed. Recessing the core tablet does not significantly
alter the release
profile of the core tablet because a sizable portion of the surface of the
core tablet is in fluid
communication with the environment. In contrast, in coated or encapsulated
dosage forms,
the coating or capsule must be breached by gastric fluid before the drug is
released. 1n the
present invention, the outer contour of the dosage form protects the mucosa
lining the
gastrointestinal tract without interrupting fluid communication between the
core tablet and
the environment.
Exemplary of drugs that can be advantageously delivered using the preferred
recessed dosage form of this invention are monosodium alendronate monohydrate,
monosodium alendronate trihydrate, sodium etidronate, sodium risedronate,
pamidronate,
aspirin, ibuprofen, naproxen, fenoprofen, ketoprofen, oxaprozin, flubiprofen,
indomethacin,
sulindac, etodolac, mefenamic acid, meclofenamate sodium, tolmetin, ketorolac,
diclofenac,
piroxicam, meloxicam, tenoxicam, phenylbutazone, oxyphenbutazone, oxybutynin,
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
alendronate, carbidopa, levodopa, tizanidine, sumatriptan, pharmaceutically
acceptable
salts, hydrates, isomers, esters and ethers thereof, and mixtures thereof.
Both the core tablet and the annular body may be formed into any suitable
shape.
Specific shapes can be achieved by use of specifically designed punches.
Preferably the
core tablet and the annular body are cylindrical in shape. The core tablet and
the annular
body may be the same or different in shape. The exposed surfaces of the core
tablet may
be of any suitable shape. Preferably, the exposed surfaces of the core tablet
are circular or
oval.
Turning again to FIG. l, core tablet 1 has opposed first and second surfaces 3
and
4 and an outer circumferential surface 5 extending between the opposed
surfaces. Core
tablet 1 is preferably cylindrical or disk shaped for ease of manufacture, but
need not be so.
In a dosage form for administration to humans, the maximum distance across
either of the
opposed surfaces 3 or 4 is preferably from about 2 mm to about 12 mm, more
preferably
from about 4 mm to about 7 mm, most preferably about S mm. Opposed surfaces 3
and 4
can be flat, concave or convex and are preferably flat for bearing modest
axial compression
forces exerted by flat pressing surfaces during formation of the annular body
about the core
tablet.
In outer contour, annular body 2 is preferably cylindrically shaped, but it
can have
any cross section, such as oval, elliptical or oblong. The outer diameter is
preferably of
from about 5 mm to about 15 mm, more preferably of from about 7 mm to about 12
mm,
most preferably about 9 mm. The inner diameter can be any size up to about 2
mm less
than the outer diameter. A narrow inner diameter less than 2 mm may slow
release of the
drug if an excipient in the annular body swells upon contact with gastric
fluid. However, in
some embodiments, a lower limit 0.5 mm may still be useful. Preferably, the
inner diameter
is 3 mm or greater.
Annular body 2 has opposed first and second annular faces 6 and 7, an outer
circumferential surface 8 extending between the annular faces from their outer
edges, and an
inner circumferential surface 9 extending between the annular surfaces from
their inner
edges, thus defining an annulus.
9
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
As best seen in side view (FIG. 1B), inner circumferential surface 9 of
annular body
2 consists of three longitudinal (axial) segments. First and second segments
10 and 11 are
terminal and do not contact the sides of the core tablet. They are separated
by an internal
third segment 12 that contacts the outer circumferential surface 5 of core
tablet 1. Opposed
surfaces 3 and 4 of the core tablet are therefore recessed from annular faces
6 and 7 of the
annular body. Opposed surfaces 3 and 4 are preferably recessed from about 0.5
mm to
about 4 mm, more preferably about 1.5 mm relative to the annular faces 6 and 7
of the
annular body (said recessed distance corresponding to the length of the
corresponding
terminal segment). The recess depth of surfaces 3 and 4 can be the same or it
can be
different.
By recessing the drug-containing core tablet, any contact between the dosage
form
and the gastrointestinal mucosa occurs with a surface of the annular body
formed of non-
ulcerative excipients, and optionally one or more non-ulcerative co-active
ingredient, rather
than with the solid ulcerative active ingredient. However, one or both of
opposed surfaces
3 and 4 can be flush with annular faces 6 and 7 of the annular body without
deleterious
effect when the dosage form of the present invention is used to administer non-
ulcerative
drugs.
To better apprehend the preferred recessed dosage form embodiment of the
invention, it is useful to conceive of surface 3 of the core tablet and first
longitudinal segment
10 as defining a first void 13. Likewise, surface 4 of the core tablet and
second longitudinal
segment 11 define a second void 14. Voids 13 and 14 fill with gastric fluid
when the
dosage form is immersed in gastric fluid after reaching the stomach. Gastric
fluid passes
through the voids to contact the core tablet and the drug leaves through the
voids after it is
dissolved. Voids 13 and 14 are preferably from about 0.5 mm to about 10 mm,
more
preferably from about 3 mm to about 6 mm and most preferably about 4.5 mm in
width
(measured parallel to first or second opposed surfaces). Drug release,
therefore, does not
occur by an osmotic mechanism such as occurs with pierced dosage forms made
using the
apparatus of U.S. Patent No. 5,071,607. Rather, in a large still fluid
environment, drug
concentration drops off roughly isotropically and exponentially by diffusion.
In contrast,
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
osmotic release of the drug product would produce a streaming flow that can
cause locally
high concentrations of the drug and osmotic agents at considerable distance
from the tablet.
Osmotic streams highly concentrated in an ulcerative drug are potentially
irntating to the
mucosa, just like the solid drug, particularly if the tablet is lodged in a
fold in the
gastrointestinal wall.
Opposed surfaces 3 and 4 of the core tablet are preferably substantially
exposed,
i. e. are not substantially covered by the annular body. "Substantially
exposed" means that
less than about 50°10 of each of the opposed surfaces is concealed or
hidden from visual
inspection by the annular body. A portion of opposed surfaces 3 and 4 can be
concealed
by the annular body because of differences between the diameter and shape of
the core
tablet and the diameter and shape of certain pressing portions of the tooling
used to
compress the annular body, as will become apparent from consideration of the
description
of the tooling aspect of the invention. Such differences may result in inner
segment 12 being
offset from terminal segments 10 and 11, which, themselves, can have different
longitudinal
cross sections, e.g. have different diameters, as depicted in FIG 1.
Alternatively, the cross
section of the annulus defined by inner circumferential surface 9 can be
uniform throughout
its length. Although a portion of opposed surfaces 3 and 4 can be concealed by
the annular
body that is not necessarily the case.
Further, the invention contemplates that the rate of release of the drug is
determined
by the formulation and shape of the core tablet, not by diffusion of the drug
through the
annular body which contributes to the versatility of the dosage form for
different release
profiles.
In one embodiment, the pharmaceutical dosage form is an extended release
dosage
form. Active drug material is delivered via the exposed axial surfaces of the
core tablet.
The exposed axial surfaces retain a constant cross-section during delivery of
the active
material, thus producing a zero-order release profile. For extended release
applications, the
core tablet can be formulated to be of an eroding or diffusive nature.
An extended release core tablet preferably contains a hydrogel such as
hydroxypropyl methylcellulose, hydroxypropyl cellulose, ethylcellulose and the
like.
11
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
Optionally, the core tablet also contains a more rapidly dissolving substance
like
compressible sucrose to open pores in the hydrogel matrix and thereby modulate
the
hydrogel's grip on the active ingredient. In a zero order extended release
dosage form
wherein the active ingredient is contained in the core tablet, the annular
body will be
formulated to be yet slower dissolving than the core tablet so that the
surface area of the
core tablet will remain constant. Mixtures of about 1 part high molecular
weight'
polyethylene glycol (PEG) and 3-5 parts ethyl cellulose will retain their
shape and rigidity in
water for the time that it takes for most conventional eroding or swelling
hydrogel matrices
to completely release the drug. An especially preferred composition of the
annular body of
an extended release dosage form in accordance with this invention comprises
about 15-25
parts PEG 4000, about 70-80 parts ethylcellulose and about 5 parts
polyvinylpyrrolidone.
The rate of release of active material from the core tablet of extended
release dosage forms
is less than about 15% by weight per hour. Preferably the rate of release is
from about 3%
per hour to about 12% by weight per hour. Extended release dosage forms are
adapted for.
the release of active material over a period of at least about 4 hours, more
preferably at
least about 7 hours, and most preferably at least about 10 hours. The rate of
release of
active ingredient is measured in a United States Pharmacopeia standard
apparatus II
solution tester in an aqueous solution buffered at 6.8 at 37°C with a
stirring rate of 50
revolutions per minute.
Dosage forms in accordance with this invention also are adaptable for
immediate
release and have unique advantages when used for immediate release. The
annular body or
sheathing layer provides protection for the immediate release core tablet
while being
handled by the patient or caregiver. The core drug layer is recessed from the
surface so
that any contact is with the walls of the annular body. While the core tablet
may be fragile,
ones hands would contact only the non fragile annular body. The core tablet
can be
formulated to be of a "fast dissolve" nature without the drawbacks of the
current "fast
dissolve" systems. The drug can be released by dissolution into the saliva as
the "fast
dissolve" form is held in the mouth for a few minutes. The outer annular body
can be
formulated to dissolve too but at a slower rate so that it not be as sensitive
to moisture or
12
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
alternately could be swallowed (by those that can swallow a tablet) or
expectorated.
Dissolution of the released drug is preferably carried out in less than about
5 minutes, more
preferably in less than about 2 minutes. The rate of dissolution of active
ingredient is
measured in a United States Pharmacopeia standard apparatus III dissolution
unit at 37°C
or a United States Pharmacopeia standard apparatus II dissolution unit at
37°C with stirring
at SO revolutions per minute. The dosage form can be formulated so as to be
suitable for
rapid dissolution in the oral cavity without co-administration of liquid.
As a consequence of the protection afforded by the annular body, many active
ingredients can be used in a greater proportion in the core tablet formulation
than they could
be in conventional tablets. Thus, a core tablet can contain a very high
concentration of
active drug material without thereby producing a dosage form that is too
delicate to be
handled. An immediate release core tablet preferably contains a
superdistintegrant. Other
preferred excipients for an immediate release formulation include sodium
saccharin,
microcrystalline cellulose, lactose and menthol.
One immediate release core tablet formulation that has been found to compress
well in the tooling of the invention contains 5 parts active ingredient, 20
parts crospovidone,
74 parts MicrocelLac~, 1 part lubricant and 0.4 parts menthol.
When the core tablet is formulated for immediate release, the annular body can
be
formulated differently than the annular body of an extended release
formulation because it
does not need to remain rigid for as long a time. The annular body will
generally be
formulated to dissolve more slowly than the core tablet, however. As further
illustrated in
Example 3, an annular body can be made by modifying an immediate release core
formulation by reducing the proportion of superdisintegrant, and optionally
substituting a
dissolving, but non-swelling excipient, like compressible sugar.
Immediate release dosage forms in accordance with the invention are useful for
administering active pharmaceutical ingredients that have an unpleasant taste,
like
sumatriptan succinate.One method of achieving taste masking includes recessing
the
surface of the core tablet within the annular body, thus avoiding contact
between the tongue
and the core tablet. Immediate release dosage forms in accordance with the
invention also
13
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
are useful for sublingual and buccal administration of drugs. It is often
desirable that a
sublingually administered drug be released from the dosage form as rapidly as
possible.
Buccal administration can also be via immediate release dosage forms. To
achieve rapid
release, such dosage forms can be formulation with a high proportion of the
active
ingredient. However, a high proportion of active ingredient will, in many
cases, make the
tablet fragile. As previously discussed in another context, the annular body
protects fragile
core tablets in the dosage forms of this invention, making them well adapted
for sublingual
and buccal administration of drugs. Preferred drugs for sublingual and buccal
administration
in the dosage forms of the present invention axe tizanidine, nitroglycerin,
isosorbide dinitrate,
isosorbide mononitrate, vaccines, ergotamine and other anti-migraine
compounds,
lorazepam and other tranquilizers, vitamin B 12 and folic acid, and mixtures
thereof. A
dosage form of tizanidine is further illustrated in Example 3.
The rate of release of active material from the core tablet of immediate
release or
sublingual dosage forms is greater less than about 90% in 30 minutes.
Preferably the rate of
release is greater than about 85% in 15 minutes. The rate of release of active
ingredient is
measured in a United States Pharmacopeia standard apparatus III dissolution
unit at 37 °C
or a United States Pharmacopeia standard apparatus II dissolution unit at
37°C with stirring
at 50 revolutions per minute.
The core tablet also can be a bilayer tablet with each layer containing the
same or
different drugs and each layer releasing the drug at the same or at different
rates. One of the
layers could be an immediate release layer and the other a slow release layer,
or both can
be slow release layers. The inner tablet can be formulated to be a three layer
tablet with the
central layer being a drug to be delivered after a delay. The two outer layers
can be delay
layers or drug delivery layers with the same or different drugs and with the
same or different
release profiles. The middle layer can contain again the same or different
drugs compared
to the outer layers and can be of a controlled release or an immediate release
nature. Thus,
one can have controlled release of two drugs each at its desired release rate
and a delayed
release or delayed pulse of a third drug. The currently described invention
thus gives a very
14
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
wide range of drug delivery capabilities not addressed by conventional dosage
forms and
improves upon the performance of other known delivery systems.
Dosage forms in accordance with the invention also can be formulated to
deliver
two drugs by locating one of the drugs in the core tablet and the other in the
annular body.
Such an arrangement enables the release rate of each active ingredient to be
controlled
independently by formulation adjustments to the portion of the dosage form,
i.e. core tablet
or annular ring, that contains the drug that is being released either too
slowly or too quickly.
In addition, the shape of one of the portions can be changed without adjusting
the
formulation. For instance, the powder or granular material may be pressed
around the core
tablet into a body having an oval cross-section rather than a circular cross-
section to
achieve a faster rate of release (resulting from increased surface area). In
addition, the core
tablet may have a hole extending from one axial face to the other in order to
increase the
surface and thereby increase the release rate. The release rate can be further
controlled
through changes to the diameter of the hole, as further illustrated in Example
4.
Preferred drug combinations for use with the invention include
levodopa/carbidopa,
acetaminophen/caffeine, acetaminophen/codeine, acetaminophen/antihistamines,
vitamin and
mineral combinations and combinations of antibiotics. The combination of
levodopa/carbidopa is especially preferred. In Example 5, especially preferred
levodopa/carbidopa dosage forms are illustrated wherein the levodopa is
dispersed in a
hydrogel matrix in the annular body and carbidopa is direct compressed with a
direct
compression excipient rnix and a superdistintegrant in the core tablet.
The rate of release of levodopa material from the core tablet of a combination
levodopa/carbidopa drug dosage forms is less than about 35% by weight per
hour.
Preferably the rate of release is from about 3% per hour to about 30% by
weight per hour,
more preferably from about 6% per hour to about 30% per hour.
Levodopa/carbidopa
combination dosage forms are adapted for the release of active material over a
period of at
least about 2 hours, more preferably at least about 3 hours. The rate of
release of active
ingredient is measured in a United States Pharmacopeia standard apparatus II
solution
tester in O.1N HCl at 37°C with a stirring rate of 50 revolutions per
minute.
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
The solid dosage forms with a drug-containing core tablet sheathed in a
compressed
annular body of non-ulcerative excipients can be produced using a novel
toolset that
constitutes a second aspect of the invention.
The toolset can be used in conjunction with conventional tablet presses such
as
rotary presses and reciprocating presses or with presses that have been
specially designed
and manufactured. Examples of commercially available rotary presses are the
Manesty
Express 25, the Kilian RUD or RTS series and comparable equipment. Examples of
commercially available reciprocating presses are the Manesty F3 and comparable
equipment made by Stokes, Kilian and I~ey Industries.
The principle elements of the toolset are a columnar punch and a punch
assembly
comprising an annular punch having an annulus (or bore), a core rod slidably
engageable
within the annulus of the annular punch, wherein the core rod is capable of
movement
between a retracted position and an extended position, the core rod being
biased in the
extended position. The columnar punch and punch assembly are sized and shaped
to fit into
the die bore of a rotary or reciprocating tablet machine.
The toolset is well adapted for use with conventional single station tablet
presses in
which opposing upper and lower punches cooperatively compress a powder or
granular
material within a die. Referring to FIG. 2, single station presses are
provided with a
horizontal die table 15 having an aperture for receiving a die 16 and
associated gripping
means for locking the die into position. Dies for such presses customarily
have opposed flat
surfaces with a centrally located bore 17 having a highly polished wall
surface extending
from surface to surface and a circumferential locking groove 18 for engaging
the gripping
means. The bore serves as a receptacle for receiving powder or granular
material to be
compressed when the lower punch is partially inserted. The rims of the bore
are
customarily chamfered to help guide the punches into the bore. The bore's
cross section
determines the size and shape of the finished tablet in cross section. The
quantity of material
and pressure of compression determine the tablet's height. The bore can be
cylindrical, but
also can be any other shape.
16
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
In operation, the bore is filled with material and the upper punch is inserted
into the
bore and pressed against the material under high pressure thereby compressing
the powder
or granulated material into a tablet between the pressing, or contact,
surfaces of the
punches.
Together, the wall of the bore and the contact surfaces of the upper and lower
punches define a mold that determines the size and surface contours of the
final product.
The final product can have any external contour by selection of appropriate
bore shape and
contact face contour.
After compression, the upper punch is withdrawn and the lower punch is
advanced
to ej ect the tablet.
The upper and lower punches are advanced and withdrawn by independently
actuated upper and lower reciprocating rams 19 and 20. Customarily, single
punch
presses are also provided with a stationary mounting point 21 below the die
table coaxial
with the aperture.
A toolset of this invention adapted for use in. a single station press
comprises a
columnar punch and a punch assembly comprising a collar, core rod and annular
punch.
Refernng now to FIG. 3, columnar punch 22 can be of a conventional columnar
shape and is provided with locking means, such as locking flat 23 to secure it
to the upper
reciprocating ram 19 of the tablet press.
Columnar punch 22 includes a contact face 24. Contact face 24 can have any
desired contour, e.g. standard concave, deep concave, extra deep concave,
modified ball
or flat. Preferably, the contour of contact face 24 is flat with a beveled
edge.
A columnar punch for use in producing a dosage form of the present invention
having a recessed core also has a protrusion 25 centrally located on the
contact face 24, as
illustrated. Preferably, the height of protrusion 25 is from about 0.5 mm to
about 4 rnm,
more preferably about 1.5 mm. The shape of the protrusion is preferably
cylindrical or
tapered cylindrical but can also be oval, ellipsoid, oblong or any other shape
desired. The
protrusion is preferably cylindrical and has a flat raised surface 26.
Protrusion 25 preferably
has a diameter of from about 3 mm to about 7 mm, more preferably about 4.5 mm.
In
17
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
other embodiments, particularly suited to use when non-ulcerative active
pharmaceutical
ingredients are to be administered, protrusion 25 is absent.
Punch assembly 27 comprises collar 28, core rod 29 slidably engaged with
collar
28 and annular punch 30 slidably engageable with core rod 29.
Collar 28 is provided with mounting means, such as external threads 31 around
its
circumference for mounting to stationary mounting point 21 located below the
die table. As
illustrated, the distal end 32 of collar 28 relative to the die table when
installed, has a
gripping section (shown with optional hexagonal cross section) for gripping by
a wrench for
mounting to stationary mounting point 21. At the proximal end 33 of the collar
28 relative
to the die table when installed, the annulus is dimensioned to receive and
guide the core rod
29.
Away from the proximal end of the collar, the diameter of the annulus is
substantially
greater than that of the core rod to provide a housing 34 for a biasing means
such as spring
35. The coils of spring 35 encircle the core rod. Although a coil spring 35 is
a preferred
biasing means, biasing can be accomplished by other means, such as a stack of
Belleville
washers or an elastic insert.
Spring 35 or other biasing means engages retaining ring 36 mated to core rod
29.
Retaining ring 36 can be mated to the core rod by clamping engagement with a
circumferential groove 37 in the rod. The retaining ring can be a conventional
C-clip which
engages the groove, or it can be a clamp or any other structure against which
the biasing
means can exert a biasing force and which is restrained from movement relative
to core rod
29 in a direction parallel to the long axis of the core rod.
As illustrated, an annular locking bolt 38 engages internal threads 39 at the
distal
end of collar 32. The bore 40 through locking bolt 38 is dimensioned to
receive and, in
conjunction with the annulus at the proximal portion of the collar, to
restrain motion of core
rod 29 to axial movement. Locking bolt 38 also retains and can compress the
biasing
means. Core rod 29 is biased in the direction of the die table when the collar
is installed on
stationary mounting point 21 and is retained in slidable engagement with
collar 28 by
retaining ring 36 and locking bolt 38. The height of rod tip 41 is adjusted by
advancing or
18
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
retracting collar 28 relative to stationary mounting point 21, e.g. by
rotating the collar when
in threaded engagement with the stationary mounting point.
Core rod 29 can vary in diameter along its length. A preferred diameter of rod
tip
41 is from about 0.5 mm to about 10 mm, more preferably about 4.5 mm. However,
for
rigidity, the core rod should be thicker, preferably from about 4 mm to about
12 mm
throughout most of its length, more preferably about 9 mm. The rod can taper
gradually
from a narrow diameter at the tip to a larger shank diameter or it can change
abruptly at a
shoulder 42.
The core rod can be of two-piece construction. For instance, the core rod tip
41
could be adapted to attach to the core rod by providing external threads at
its lower end
and a socket with internal threads at the upper end of the core rod, or vice
versa. A two-
piece construction allows the core rod tip to be replaced if it is damaged or
if a core rod tip
of a different shape is desired. The core rod tip can have any desired
diameter or shape.
Punch assembly 27 further comprises annular punch 30. Annular punch 30 is
provided with means for attaching to lower reciprocating ram 20, such as
locking flat 43.
The bore 44 through annular punch 30 is dimensioned to receive and surround
core rod 29
while permitting axial movement of annular punch 30 independent of the core
rod. The bore
through annular punch 30 can vary in diameter along the length of the punch
providing an
annular flange 45 for engagement with shoulder 42 on the core rod. Engagement
of flange
45 with shoulder 42 prevents annular punch 30 and collar 28 from abutting each
other
during handling and installation. Annular punch contact surface 46 presses
against the
powder or granular material during compression. Contact face 46 can have any
desired
contour, e.g. standard concave, deep concave, extra deep concave, modified
ball or flat.
Preferably contact face 46 is flat with a beveled edge for ease of ej ection
of the finished
tablet.
The columnar punch, annular punch, core rod and collar are preferably made of
metal, more preferably steel, most preferably stainless steel.
In the final dosage form with recessed core tablet, the depth of first void 13
(FIG.
1 ) is determined by the height of protrusion 25 . The depth of second void 14
is determined
19
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
by the fill depth, strength of the bias on the core rod, the compressibility
of the material and
the thickness of the core tablet. These parameters can be adjusted by routine
experimentation to control the depth of second void 14, which is suitably
commensurate
with the depth of first void 13.
In a second dosage form embodiment, either one or both of opposed surfaces 3
and 4 of the core tablet are flush with the annular faces 6 and 7 of the
annular body 2. This
alternative embodiment can be produced by using a columnar punch as previously
described but lacking a protrusion 25. Surface 3 will generally be flush with
annulax face 6
if the columnar punch has a flat contact face. Whether the opposed surface 4
is flush with
annular face 7 will depend on the fill depth, compressibility of the powder or
granular
material and thickness of the core tablet, which factors can be adjusted by
routine
experimentation to yield a dosage form with surface 4 recessed the desired
distance relative
to annular face 7.
To further illustrate the invention and the operation of the toolset, a cycle
of
operation will now be described. The cycle of operation is embodied in a
process that
constitutes a third aspect of the invention.
The cycle of operation is first illustrated on a single station press. The
cycle begins
with the first action that occurs after ejection of the tablet formed in a
previous cycle.
Referring now to FIG. 4a, feed shoe 47 moves laterally over the die bore while
the annular
punch 30 is in an advanced position such that contact surface 46 is
substantially flush with
the top surface of the die. In so doing, the feed shoe sweeps a finished
tablet from atop the
annulax punch toward a chute leading to a receptacle where the tablets are
collected.
Annular punch 30 is retracted while the tip 41 of core rod 29 remains flush
with the die
surface (FIG. 4b). Retraction of the annular punch causes an annular cavity to
form into
which particles of the powder or granular material are fed from the feed shoe
by gravity
and/or pressure differential. Once the cavity is filled, the feed shoe is
shifted away from the
die bore.
Pre-compressed core tablet 1 is positioned atop the core rod using any
conventional apparatus for producing tablets with a compressed coating such as
that of a
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
I~ilian RUD press (FIG. 4c). The positioning means forms no part of the
invention and has
been omitted for clarity.
Columnar punch 22 is advanced by upper reciprocating ram 19 (FIG. 4d). As
columnar punch 22 approaches the bore, the raised surface 26 of protrusion 25
presses
upon core tablet 1. As columnar punch 22 enters bore 17, core tablet 1 is
pushed into the
bore by the protrusion against the biasing force exerted on core rod 29.
Continued
movement of columnar punch 22 into the die bore compresses the powder or
granular
material into an annular body around the core tablet. Strong compressive
forces can be
exerted on the powder or granular material without breaking the core tablet
because the
core tablet travels into the bore before the powder or granular material is
fully compressed.
Those skilled in the art may also appreciate that protrusion 25 could be
replaced
with a core rod in the columnar punch that is biased toward an extended
position so that the
tip of the rod would press against core tablet 1 during compression. Such a
core rod for
the columnar punch would not necessarily be attached to a stationary mounting
point on the
press. It would be biased with greater force than core rod 29 so that pressure
exerted by
the columnar punch would push the core tablet into the bore against the
resistence of the
core rod.
After the powder or granular material is compressed, the columnar punch is
withdrawn. Either concurrently or subsequently, annular punch 30 is advanced
by lower
reciprocating ram 20 to a position such that contact face 46 is substantially
flush with the
upper surface of the die to elevate the finished tablet above the die where it
can be swept
from the die table in a subsequent cycle of operation (FIG. 4e). Meanwhile,
the core rod is
biased back to its original position flush with the die surface.
The toolset is well adapted for use in a rotary tablet press. The cross-
sectional
dimension and shape of the columnar punch, and the dimensions and shape of the
protrusion
(if present) are the same as in a punch adapted for use in a reciprocating
tablet press. The
other dimensions of the toolset are generally dictated by the dimensions and
layout of a
particular tableting press. These dimensions can be readily determined by
those skilled in
the art. The cross-sectional dimensions and shape of the annular punch and of
the core rod
21
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
are the same as in a punch adapted for use in a reciprocating tablet press,
again with other
dimensions being dictated by the dimensions and layout of a particular
tableting press.
These dimensions can be readily determined by those skilled in the art. In
addition, the
punches include conventional bearing surfaces at the end distal to their
contact surfaces for
engaging the cams and rollers that control their motion along the axis of the
die bore, such as
those shown in the patents that are incorporated by reference below.
In an annular punch for use in a rotary machine, the core rod biasing means
preferably is housed in the annular punch and includes a means for adjusting
the degree of
extension of the core rod and/or the bias, such as a set screw or similar
device.
Conventional rotary tablet presses are well known in the art. Some rotary
presses
and improvements related thereto are described in U.S. Patents Nos. 5,462,427,
5,234,646, 5,256,046 and 5,635,223, which are incorporated herein by reference
in their
entirety. Rotary presses have a moving die table that rotates around a
vertical axis.
Mounted above and below the die table are upper and lower punch carriers that
rotate
synchronously with the die table. The punch carriers can be generally drum
shaped bodies
of about the same diameter as the die table or they can have arms that extend
outward from
a lesser diameter ring. The punch Garners are provided with a plurality of
vertical holes or
slots at regular intervals around their circumference or through the ends of
the arms. When
the press is in operation, punches are inserted into each slot with their
contact faces pointing
toward the die table. Each punch has a bearing means at the end opposite the
contact face.
The bearing means engage stationary cams and rollers which control the
vertical motion of
each punch during a cycle of operation. The cams and rollers are arranged such
that in a
cycle of operation, a powder ox granular material is fed into a die while the
lower punch is
inserted into the die. Pressure is applied to the powder or granular material
to produce a
compressed body. After compression, one or more of the punches is removed from
the die
and the dosage form is released. Rotary presses are especially suited for high
volume
production because they typically contain numerous punch and die sets
operating
simultaneously.
22
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
A cycle of operation using the toolset of this invention adapted for use in a
rotary
press will now be described. As the die table rotates, one of the dies passes
under a fill
shoe or force feeder. While the die is passing underneath the shoe or feeder,
the annular
punch is withdrawn by the cam. The core rod remains in an extended position,
up to the
upper die face. The annular space left by withdrawal of the annular punch is
filled with
powder or granulate. At the next station, a core tablet is inserted onto the
tip of the core
rod by conventional means, such as those used in "press coat" machines like
the Kilian
RUD. The core tablet can be positioned atop the core rod by any method. On
further
rotation, the die comes to the compression station where the columnar punch
with, or
without, its protrusion moves downward and pushes the core tablet into the bed
of powder
or granular material. The force of the columnar punch retracts the core rod
against the bias
and the powder or granular material is compressed into an annular shape around
the core
tablet. In the dosage form product, one recess is defined by the height of the
protrusion and
the other recess is defined by a combination of the factors such as the
strength of the bias,
the fill depth, the compactability of the.powder or granular material and the
thickness of the
core tablet. After the powder is compressed, the die rotates further to where
the columnar
punch is withdrawn.from the die. Either concurrently or subsequently, the
annular punch is
raised until it reaches the die face. The core rod rises concurrently to the
die face due to the
bias. The tablet is swept out of the die by an ejection element and is
collected.
While reference has been made to "upper" and "lower" elements in the
description
of the toolset and process for making solid dosage form according to the
invention, the
spatial relationships of the elements are determined by the design and
construction of the
press in which they are used. Use of the terms "upper" and "lower" is not
intended to limit
the invention to a vertical arrangement of the elements.
Having thus described the present invention with reference to certain
preferred
embodiments, the invention will now be further illustrated by the following
example.
23
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
EXAMPLES
Example 1
Immediate Release Monosodium Alendronate Tablets
This example summarizes a study designed to determine the rate and extent of
absorption of alendronate sodium in human subjects upon administration of a
solid
pharmaceutical dosage form of the present invention ("protected tablet")
Materials and Methods
Protected tablets were made as follows.
Tablet Core: 85.4 g of alendronate trihydrate (TEVA Assia Ltd.) and 2.6 g of
xylitol (Danisco Sweeteners ~Y) were granulated with 20 g water in a Diosna
(model P1/6)
granulator for 3 min. The granulate was dried at..40°C for one hour in
a fluidized bed dryer
and milled through a 0.8 mm screen. The granulate was blended with 11 g
crospovidone
NF (BASF Pharma ) for five minutes. One gram magnesium stearate NFBP
(Mallinkrodt
Inc.) was added and the granulate was further blended for an additional 0.5
minutes. The
blend was compressed using a Manesty F3 single punch tablet machine fitted
with a 5 mm
flat beveled punch. The tablet weight was 94.9 mg ~ 1.0% RSD. The hardness of
the
core tablets was 3 - 6 kP.
Protected Tablets: A mixture of 94 grams compressible sucrose (Nu-TabTM, DMV
International ) and S grams microcrystalline cellulose (AvicelTM pH102, FMC
International)
were blended for five minutes. ~ne gram magnesium stearate (NF/EP, Mallinkrodt
Inc.)
was added and the mixture was blended for another half a minute.
A Manesty f3 single punch tableting machine was fitted with a spring-biased
columnar punch and punch assembly constructed in accordance with the present
invention.
The core rod was designed for a 5 mm round core tablet and the die and punches
for the
outer tablet were designed to produce a round, 9 mm diameter, flat beveled
solid
24
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
pharmaceutical dosage form. The upper punch had a protrusion of diameter 4.5
mm and
1.2 mm height. The tablet press was operated and the protected tablets were
produced.
The tablet weight was 474 mg ~ 0.62% RSD and the hardness of the protected
tablets
was 12 -15 kP. The alendronate trihydrate content, expressed as alendronic
acid was
66.8 mg ~ 1.38% RSD (82.4 mg alendronate trihydrate being equivalent to 70 mg
alendronic acid).
The drug-containing inner tablet was recessed from the surface of the annular
body
by about 1 mm.
Pharmacokinetic Studv
A clinical trial involving twelve (12) human volunteers was conducted to
demonstrate the pharmacokinetics of a solid dosage form of the present
invention containing
70 mg alendronate. Its pharmacokinetics was compared to that of a commercial
70 mg
FosalanTM tablet of the prior art (Merck, Sharpe & Dohme).,
Method
The study was a randomized, open-label, 2-treatment, 2 period, 2 sequence
crossover design under fasting conditions. Twelve (12) healthy adult male
volunteers, 18-
55 years of age were the subjects in the study.
The study was divided into first and second study periods, each of 36 hours
duration, with a 14 day "wash-out" period between the study periods. All
subjects who
completed both study periods were included in the analysis. Subjects were
randomly
assigned to two groups. One group was administered alendronate via the
protected tablet
in the first period and administered control Fosalan in the second period. The
order of
administration to the second group was reversed.
In both periods, alendronate was administered in the fasted state. A
standardized
meal was provided 4 hours after administration. Snacks were provided on a
standardized
schedule that was the same for all subjects in both study periods. Water was
provided ad
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
libitum. In addition, subjects were encouraged to drink at least 200 ml of
water at regular
intervals during each study period.
The bioavailability of alendronate was determined by measuring the cumulative
levels of alendronate excreted in the urine over a 36 hour period following
oral ingestion of
the test and control tablets (hereafter "Aeo_36~~). An initial (t = 0) urine
sample was taken
immediately after administration. Urine samples were taken at 11 regularly
scheduled points
in time over the 36 hour test period. All urine samples were analyzed for
alendronate using
a validated HPLC-FLR assay.
Results
The main pharmacokinetic parameters obtained from the analyses of urine
samples
are collected in Table 1.
Table 1: Pharmacokinetic Parameters
Administration Administration via Fosalan
via (control)
Protected
Tablet
Parameter Mean ~ SD CV (%) Mean ~ SD CV (%)
Ae_36 (fig) 113.6 77.2 67.9 102.6 36.8 36.8
R",a,~ (~g/h) 37.9 19.9 51.5 31.7 11.8 38.3
T",~ (h) 1.4 0.9 --- 1.4 0.9 _
A comparison of the pharmacokinetic parameters of the dosage form in
accordance
with this invention with the pharmacokinetic parameters of the prior art
dosage form is
provided in Table 2.
Table 2. Comparison of Pharmacokinetics of the Protected Tablet to Prior Art
Ae°_3s (mg) R",~ (m~)
Geometric Mean of Ratio 0.99 1.12
90% Geometric C. I. 75.31 % to 128.79% 93.98% to 135.01
Intra-subject C.V. 37.48% 24.85%
26
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
By reference to Tables 1 and 2, and FIG. 5, one can see that alendronate
administered via the solid dosage form of the present invention gives
essentially the same
pharmacokinetic results as administration via Fosalan. The total amount of the
alendronate
excreted into urine over 36 hours is essentially the same for both treatments
with the
maximum rates of excretion (parallel to Cm~ in a pharmacokinetic study of
plasma levels of
drug) also close.
The profile of excretion into urine was similar for all subjects and in both
treatments.
The majority of the subjects had their maximum rate of excretion (R,r,ax)
between one and
two hours. For five of the subj ects, the RmaX occurred earlier than 1 hour
after
administration when they took Fosalan. Four of the subj ects experienced a Rm~
in less than
an hour when they took the protected tablet. One of the subjects had an RmaX
in the third
hour when he took Fosalan while two of the subjects had a RmaX in the third
hour when they
took the protected tablet.
The total amount of excreted alendronate ranged frdm 36.9 p,g to 158.6 ~,g
when
15~ Fosalan was administered and from 30.1p,g to 284.4 pg when the solid oral
dosage form of
the present invention was administered. In only two subjects was there a
greater than two
fold difference between the total amount of excreted alendronate between the
two
treatments. Another subject excreted a very low amount of alendronate
regardless of how
the alendronate was administered.
The bioavailability of alendronate administered via the novel solid dosage
form of
the present invention is equivalent to that of alendronate administered by
dosage forms of
the prior art. However, the dosage form of the prior art does not provide any
protection
against contact of the alendronate with the mucous membranes of the
esophageous and
stomach while the bioequivalent novel dosage form of the present invention
affords such
protection.
27
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
Drug Release Profile
Dissolution was measured in a USP apparatus BI dissolution unit (Hanson B-3)
unit
at 37°C. The alendronate content of samples taken at 5, 10, 15 and 30
minutes was
determined by HPLC on an anion column using refractive index detection. The
results of the
dissolution are reported in Table 3.
Table 3
Time (m) Cumulative Percent
Release
5 48
10 70
85
30 98
The outer mantle took more than one hour to dissolve.
15 The tablets were tested in a human pharmacokinetic study and shown to be
bioequivalent to commercially available alendronate (70 mg).
Example 2
Extended Release Zero Order Release Ox~t~in Tablets
The annular coated tablet is uniquely fit for extended controlled release,
particularly
when one needs to approximate zero order release over an extended period of
time. The
drug is delivered through the exposed axial faces of the delivery system.
These faces retain a
constant cross-section during drug delivery, thereby aiding in the achieving
of a constant rate
of drug release.
28
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
A. Inner tablet
Oxybutynin (50 g), was mixed with anhydrous lactose (50 g) in a Zanchetta
RotolabTM one pot granulator. The granulation solution, 5% w/w
hydroxypropylcellulose
(KlucelTM LF, 21 ml), was added with stirnng at 500 rpm until thorough mixing
was
achieved. The granulate was dried in the one pot granulator at 45-SO°C
with gas stripping
for a time of about 20 minutes. The granulate was milled in a Quadro ComilTM
milling
machine using a screen size of 1143 Vim.
The oxybutynin granulate (27.6 g), was mixed with
hydroxypropylmethylcellulose,
(HPMC, MethocelTM K15M, 19 g), and compressible sucrose (Nu-TabTM, 52.4 g).
Magnesium stearate, 1 g, was added with mixing. The blend was compressed into
tablets
on a Manesty f3 single punch tablet machine using 6 mm flat beveled punches to
produce
tablets weighing about 110 mg and having a hardness of 4 Kp.
B. Non Dissolving Outer Mantle on Cylindrical Surfaces
Polyethylene glycol (PEG 4000) was milled and passed through a 500 ~,m screen.
The milled PEG 4000 (24 g), was mixed with polyvinylpyrrolidone (PovidoneTM,
PVP K_
30, 5 g), and ethylcellulose (EthocelTM 7 cps, 71 g), for 3 minutes. Magnesium
stearate (1
g), was added and the blend mixed for another 0.5 minutes. The inner cores,
produced
above, were pressed within the outer mantle using this blend and a 9 mm outer
cylinder
spring loaded core rod tooling previously described. The core rod diameter was
4.5 rnm.
The upper punch had a protrusion of 5 mm diameter tapering to 4.5 mm at its
upper surface
with a height of 1.2 mm. The final product, an annular ring coated tablet with
recessed
exposed axial faces, had an outer diameter of 9 mm, a total weight of 350 mg
and contained
15 mg oxybutynin (Formulation A).
29
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
C. Dnx~ Release Profile
The drug release profile of oxybutynin from the delivery system of Example 1
was
tested in an USP apparatus II dissolution tester using 900 ml of phosphate
buffer pH = 6.8
at 37° C, 50 rpm. The oxybutynin content of the samples were determined
by an HPLC
method with UV detection. The results are reported in Table 4, below, and
presented
graphically in FIG. 6.
Table 4
Time (h) Cumulative Percent
Release
1 1.7
2 4.9
4 20.0
6 41.8
8 58.3
10 75.1
14 79.0
16 79.1
18 79.5
D. Control of the Release by Changes in the Inner Tablet Formulation
The above procedure for the preparation of the inner tablet was repeated,
using 30
g of MethocelTM Kl SM and 41.4 g of Nu-TabTM, thus raising the content of the
gel forming
HPMC and lowering the content of the dissolving sucrose (Formulation B). The
results of
the dissolution experiment are reported in Table 5, below, and depicted in
FIG. 7.
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
Table 5
Time (h) Cumulative Percent
Release
1 0.8
2 3.4
4 11.8
6 29.1
8 47.5
59.8
12 68.8
10 14 76.2
16 79.8
18 82.0
A significant slowing of drug release in the first ten hours was observed.
E. Control of Release by Changes in the Formulation of the Outer Mantle
The procedure for the preparation of Formulation B was repeated, with the
outer
mantle containing 14 g of PEG 4000 and 81 g of EthocelTM (Formulation C). The
results of
the dissolution experiment are shown in Table 6, below, and depicted
graphically in FIG. 8.
31
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
Table 6
Time (h) Cumulative Percent
Release
1 0.6
2 1.2
4 7.6
6 20.5
8 30.5
39.6
12 46.1
10 14 51.5
16 55.5
l~ 5~.0
Again, significant changes in the rate of drug release were observed,
demonstrating
that changes in the formulation of the inner core tablet or the outer annular
body can
determine the rate of release of active drug material.
Example 3
Fast Dissolving Tizanidine Tablets for Sublingual delivery
Sublingual tablets were formed into an inner core of a fast disintegrating
formulation
containing tizanidine (2 mg) and an outer annular ring of protective
excipients.
A. Inner Tablet
The inner cores were made by mixing tizanidine hydrochloride (4.5 parts) and
crospovidone (20 parts), for 2 minutes. Sodium saccharin (0.5 parts),
MicrocelLac100TM
(73.6 parts), and menthol (0.4 parts) were added and the mixing continued for
3 more
32
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
minutes. Magnesium stearate (1 part) was added and the mixing continued for a
half a
minute. The mixture was compressed on a Manesty f3 tablet press fitted with a
five mm flat
beveled punch. The tablets formed were of 5 mm diameter, weighed 45 mg each,
were
about 2 mm thick and had a hardness of 1- 3.5 Kp.
B. Dissolving Outer Mantle
The outer annular ring was made by mixing Nu-TabTM (48.5 parts),
MicrocelLac100TM (a 25:75 mixture of microcrystalline cellulose and lactose
commercially
available for direct compression, 45 parts), sodium saccharin (0.5 parts) and
of
crospovidone (5 parts) for 5 minutes. Magnesium stearate (1 part) was added
and the
mixing continued for a half a minute. The mixture was compressed on a Manesty
f3 tablet
press fitted with the spring loaded core rod tooling as previously described.
The entire
tablet weight was 290 mg, the outer diameter was 9 mm, the tablet height about
4.5 mm and
the hardness 5 - 9 Kp.
C. Drug Release Profile
The tablets were tested for total disintegration of the inner tablet in 3 ml
water within
4 minutes and at least 85% dissolution of the tizanidine in 450 ml water at
37°C and 50 rpm
in a USP apparatus II dissolution system in 15 minutes. The outer mantle
dissolves after
about 15 minutes.
Example 4
Release of Two Dru;~s at Different Rates
The annular body and core tablet can be formulated to contain different drugs
and
to release the drugs with totally different release profiles. The rates of
release can be
controlled by the formulation of the core tablet and annular ring and by their
geometries. In
33
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
this case,we have formulated a carbidopa immediate release profile in the core
tablet with
controlled release of levodopa from the annular body while using an oval
tablet as the
annular ring around either a cylindrical tablet or an inner oval tablet. The
inner cores, both
cylindrical and oval, were themselves hollow with a cylindrical hole in each
of them.
A. Inner Tablets
Carbidopa (160 g) was mixed with pre sieved (500 ~m screen) xylitol (40 g) in
a
Diosna pl/6 granulator. Water (45 ml) was added as the granulation solution.
The mixture
was granulated for 5 minutes at 500 rpm and further massed at 800 rpm for 1.5
minutes.
The granulate was air dried at room temperature overnight and then milled,
while still wet,
through a 1.6 mm screen. The milled granulate was dried in a fluidized bed for
30 minutes
at 40°C and then milled through a 0.8 mm screen. This granulate, 56.3
g, was mixed with
crospovidone (10 g) and MicrocelLac100TM (32.7 g) for three minutes. Magnesium
stearate (1 g) was added to the blend which was further mixed for 0.5 minutes.
The blend
was compressed in a Manesty f3 single punch tableting machine using three
different core
rod punches to make hollow cylinders of the following dimensions:
Formulation D: cylindrical outer diameter 7.5 mm inner diameter 2.5 mm
Formulation E: cylindrical outer diameter 7.0 mm inner diameter 4.6 mm
Formulation F: oval outer diameters 12 x 6 mrn, inner diameter 3 mm.
Each tablet contained 54 mg carbidopa.
B. Drug Containing, Non Dissolving, Oval Outer Mantle
Levodopa (150 g) was mixed with xylitol (75 g) and hydroxypropylcellulose
(KlucelTM LF, 25 g) at 500 rpm for 5 minutes. Ethanol (SO ml) was added slowly
and the
granulate was formed at 500 rpm over 1.5 minutes. The granulate was air dried
overnight at
room temperature and milled through a 0.8 mm screen.
34
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
The levodopa granulate (44.4 g) was mixed with ethylcellulose (EthocelTM 7
cps, 30
g) and Cellactose 80TM (25:75 mixture of powdered cellulose: lactose for
direct
compression, 24.6 g) for three minutes. Magnesium stearate (1 g) was added and
the blend
mixed for another 0.5 minutes.
The previously formed inner tablets, Formulations D, E and F were compressed
in
an oval shaped mantle core on their radial surfaces using an oval shaped
spring loaded core
rod punch as previously described, with dimensions 17.6 x 8.8 mm with an
internal core rod
of 5 mm diameter and an upper punch with a protrusion of 5 mm diameter
tapering to 4.5
mm at its height of 1.8 mm. The total weight of each tablet was 750 rng and
each contained
200 mg of levodopa.
C. Dru~Release Profile
Dissolution was carried out in O.1N HCl (900 ml) at 37°C in a USP
Apparatus II
dissolution tester at 50 rpm and the levodopa and carbidopa concentrations of
each sample
were determined by HPLC. The results of the dissolution experiments are
provided in
Tables 7, 8 and 9 and depicted in FIGS. 9, 10, and 11.
Table 7
Dissolution Results for Formulation D
Cumulative Percent Release
Time (h) Levodopa (%) Carbidopa(%)
0.5 21 71
1 33 87
2 50 105
3 62
4 70
6 81
8 94
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
Table 8
Dissolution Results for Formulation E
Cumulative Percent Release
Time (h) Levodopa (%) Carbidopa(%)
0.5 27 102
1 43
2 63
3 76
4 85
6 94
8 101
Table 9
Dissolution Results for Formulation F
Cumulative Percent Release
Time (h) Levodopa (%) Carbidopa(%)
0.5 26 72
1 40 95
2 61 103
3 72
4 88
6 93
8 99
Thus, two drugs with totally different release profiles can be delivered with
independent control of the rate of release of each drug. It should be noted
that this control
can be achieved by shaping and sizing the core tablet, e.g. by providing it
with hole of
predetermined size or shape, without necessitating a change in formulation.
36
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
Example 5
Annular Coated Tablet for Taste Masking
A. Inner Tablets
Surnatriptan succinate (70 parts), is granulated in water (20 parts) with
microcrystalline cellulose (AvicelTM PH 101, 80 parts). The granulate is dried
in a fluidized
bed drier for 30 minutes at 40 -50°C and subsequently milled through a
O.g mm screen.
The granulate (75 parts) is mixed with anhydrous lactose (9 parts),
microcrystalline cellulose
(AvicelTM PH101, 10 parts) and croscarmellose sodium (AC-DI-SOLTM, 5 parts)
for 3
minutes. Magnesium stearate (1 g) is added and the blend mixed for another 0.5
minutes.
Tablets are pressed on a Manesty f3 single punch tableting machine using a 6
mm flat
beveled punch. The tablet weight is 100 mg and contains the equivalent of 25
mg
sumatriptan.
B. Dissolving: Outer Mantle
A mixture of compressible sucrose (Nu-TabTM, 94 g), microcrystalline cellulose
(AviceITMPH102, 5 g), and of menthol (1 g) are blended for five minutes.
Magnesium
stearate (1 g) is added and the mixture is blended for another half minute.
Tablets are formed using the inner cores described in Example 4, above, and a
9
mm outer cylinder spring loaded core rod tool previously described. The
tablets obtained
are cylindrical tablets of 9 mm outer diameter with the axial faces uncoated
and recessed
from the surface. The tablet weigh a total of 475 mg.
37
CA 02477701 2004-08-26
WO 03/075893 PCT/US03/06591
C. Drug Release Profile
The release profile of the tablets is measured in a USP Apparatus lI
Dissolution
tester in 900 ml water at 37° C and 50 rpm. The tablets are expected to
provide a drug
release of greater than 80% in 30 minutes.
Having thus described the invention with reference to certain preferred
embodiments, other embodiments will be apparent from this description to those
skilled in
the art to which the invention pertains. It is intended that the specification
is considered
exemplary only, with the scope and spirit of the invention being indicated by
the claims
which follow.
38