Note: Descriptions are shown in the official language in which they were submitted.
CA 02478970 2004-08-24
FIELD OF THE INVENTION
The present invention relates generally to devices and methods for quantifying
or
identifying components of interest in a biological system, such as in an
animal.
BACKGROUND OF THE INVENTION
Presently, if one wants to accurately assess the concentrations of chemicals
or
drugs inside a living animal a sample of the blood or tissue to be studied is
removed from
the animal and taken to an analytical laboratory to have the chemicals of
interest extracted
and quantified. Typically a first step is a pre-treatment ofthe sample to
convert it to a
form more suitable for chemical extraction. In the case of blood this may be
by the
removal of blood cells and/or some blood components by the preparation of
serum or
plasma. In the case of a tissue sample this may be by many processes including
freezing,
grinding, homogenizing, enzyme treatment leg. protease or cellulase) or
hydrolysis.
Subsequently chemicals of interest are extracted and concentrated from the
processed
sample. For example serum samples may be subjected to liquid-liquid
extraction, solid
phase extraction or protein precipitation followed by drying and
reconstitution in an
injection solvent. A portion of the injection solvent is introduced to an
analytical
instrument for chromatographic separation and quantification of the
components. This
method produces accurate results with high specificity for the compound of
interest, but is
time consuming and labour intensive. Also, because of the large number of
steps in the
process there is a significant chance of errors in sample preparation
impacting the results.
This method has good sensitivity and selectivity and accuracy for the target
compounds
but is limited in that the chemical balance the chemicals exist in inside the
animal is
disrupted during sampling. In many cases this disruption reduces the value of
the results
obtained, and in some cases makes this technique inappropriate for the
analysis. Where
the blood volume removed is a high proportion of the total blood volume of the
animal, as
is commonly the case when mice are used, the death of the animal results. This
means that
a different animal must be used for each data point and each repeat, By
eliminating the
need for a blood draw in this case, fewer animals would be required for
testing and a
significant improvement in inter-animal variation in the results would be
achieved.
-2-
CA 02478970 2004-08-24
Alternatively biosensors have been developed for some applications in analysis
of
chemical concentrations inside animals. In this case a device consisting of a
specific
sensing element with associated transducer is implanted and produces a signal
collected by
an electronic data logger that is proportional to the chemicals to which the
sensor
responds. The main limitations of this type of device are that they normally
respond to a
spectrum of chemicals rather than having specificity for only one chemical. Of
the
spectrum of chemicals to which the sensor responds, some produce a greater and
some a
lesser response. Sensors are also susceptible to interferences where another
chemical
present in a system interferes with the response produced by the target
chemicals. For
these reasons biosensors are normally limited in terms of accuracy and
precision. Finally
biosensors are typically not as sensitive to low chemical concentrations as
state of the art
stand alone detectors such as mass spectrometers that are used in the above
mentioned
conventional analysis techniques and in solid phase microextraction. A
strength of this
technology is that the chemical balance in the system under study is not
disturbed.
The in vivo procedure described here is a significant departure from
conventional
'sampling' techniques, where a portion of the system under study is removed
from its
natural environment and the compounds of interest extracted and analyzed in a
laboratory
environment. There are two main motivations for exploring these types of
configurations.
The first is the desire to study chemical processes in association with the
normal
biochemical milieu of a living system, and the second is the lack of
availability or
impracticality frequently associated with size of removing suitable samples
for study from
the living system. Newer approaches that extend the applicability of
conventional SPME
technology, where an externally coated extraction phase on a micro fibre is
used, seem to
be logical targets for the development of such tools. As with any
microextraction, because
compounds of interest are not exhaustively removed from the investigated
system,
conditions can be devised where only a small proportion of the total compounds
and none
of the matrix are removed, thus avoiding a disturbance of the normal balance
of chemical
components. This could have a benefit in the non-destructive analysis of very
small tissue
sites or samples. Finally because extracted chemicals are separated
chromatographically
and quantified by highly sensitive analytical instruments, high accuracy,
sensitivity and
selectivity are achieved.
-3-
CA 02478970 2004-08-24
With the current commercially available SPME devices a stationary extraction
polymer is coated onto a fused silica fibre. The coated portion of the fibre
is typically 1
cm long and coatings have various thicknesses. The fibre is mounted into a
stainless steel
support tube and housed in a syringe-like device for ease of use. Extractions
are
performed by exposing the extraction polymer to a sample for a pre-determined
time to
allow sample components to come into equilibrium with the extraction phase.
After
extraction the fibre is removed to an analytical instrument (typically a gas
or liquid
chromatography where extracted components are desorbed and analysed. The
amount of a
component extracted is proportional to its concentration in the sample ( J.
Pawliszyn
"Method and Device for Solid Phase Microextraction and Desorption", U.S. Pat.
5,691,
206.).
To date commercial SPME devices have been used in some applications of direct
analysis of living systems. For example they have been applied for the
analysis of
airborne pheromones and semiochemicals used in chemical communications by
insects
(Moneti, G.; Dani, F.R.; Pieraccini, G.T.S. Rapid Gommun. Mass Spectrom. 1997,
11,
857-862.), (Frerot, B.; Malosse, C.; Cain, A.H. J. High Resolut. Chromatogr.
1997, 20,
340-342.) and frogs (Smith, B.P.; Zini, C.A.; Pawliszyn, J.; Tyler, M.J.;
Hayasaka, Y.;
Williams, B.; Caramao, E.B. Chemi.stry and Ecology 2000, 17, 215-225.)
respectively. In
these cases the living animals were non-invasively monitored over time by
assessing the
chemical concentrations in the air around the animal, providing a convenient
means to
study complicated dynamic processes without interference.
The current commercial devices do, however, have some limitations for in vivo
analysis inside a living animal. Firstly, the application to chemical analysis
inside animals
requires greater robustness in both the extraction phase and the supporting
fibre core. In
addition, most of the extraction phases currently available are better suited
for more
volatile and less polar compounds. Only one phase is suitable for liquid
chromatography
(LC) applications (carbowax/templated resin). Analytes of interest that
typically circulate
in living systems are less volatile and more polar and require LC analysis, so
new or
modified extraction phases are indicated. The overall dimension ofthe current
device is
typically too large for direct in vivo analysis and for direct interfacing to
microanalytical
systems, the time required for the LC extraction phase to come into
equilibrium with
-4-
CA 02478970 2004-08-24
chemicals in a sample is relatively long (typically 1 hr or more in a well-
stirred sample)
and analysis is sensitive to degree of convection in the sample. Also the
present SPME
devices cannot be conveniently coupled to positioning devices necessary for in-
vivo
investigation at a well-defined part of the living system.
It is, therefore, desirable to provide a method and a device that allows
minimally
invasive sampling, quantification or analysis of a biological system
SUMMARY OF THE INVENTION
It is an object ofthe present invention to obviate or mitigate at least one
disadvantage of previous devices and methods for evaluating components of
interest in
biological systems.
The invention provides a method for measuring or identifying one or more
component of interest in an animal or animal tissue, said method comprising
the steps of
positioning a fibre within said animal or tissue, said fibre being at least
partially coated
with an extraction phase for adsorbing said one or more component of interest
from said
animal or tissue, said extraction phase being positioned within said animal or
tissue;
adsorbing said one or more component of interest onto the extraction phase for
a pre-
determined period of time; removing the fibre trom said animal or tissue; and
desorbing
said one or more component of interest from the extraction phase into an
analytical
instrument for measurement or identification.
The invention further provides a device for adsorbing one or more component of
interest from an animal or animal tissue, said device comprising: one or more
fibre having
an at least partially coated end, said end being at least partially coated
with an extraction
phase for absorbing one or more component of interest; and a positioning
device for
guiding the at least partially coated end of said fibre into position within
the animal or
animal tissue.
Additionally, the invention provides a method of measuring or identifying one
or
more component of interest in liquid samples arranged in a plurality of wells
in a
multiwell plate, said method comprising the following steps: simultaneously
submerging a
distal end of a plurality of fibres within said plurality of wells,
respectively, the distal end
of each fibre being at least partially coated with an extraction phase for
adsorbing the
-5-
CA 02478970 2004-08-24
component of interest from the liquid sample; adsorbing the component of
interest onto
the extraction phase for a pre-determined period of time; removing the fibres
simultaneously from the wells; and positioning the extraction phase into an
analytical
instrument for desorption, and measurement or identification of the component
of interest.
Other aspects and features of the present invention will become apparent to
those
ordinarily skilled in the art upon review of the following description of
specific
embodiments of the invention in conjunction with the accompanying figures.
BRIEF DESCRIPTION OF THE DRAWINGS
Embodiments of the present invention will now be described, by way of example
only, with reference to the attached Figures, wherein:
Figure 1 shows a general schematic of device design according to an embodiment
of the invention.
Figure 2 illustrates the use of a medical catheter to position device
accurately
within a vein.
Figure 3 shows a schematic of the polypyrrole polymerization reaction.
Figure 4 is a schematic of a catheter with multiple coated fibres.
Figure 5 shows a schematic of housing and device for soft tissue sampling.
Figure 6 illustrates operation of housing and device for soft tissue sampling.
Figure 7 shows a schematic of use of soft tissue sampling housing to position
device for sampling with x-y-z stage.
Figure 8 illustrates a device according to an embodiment of the invention with
hollow fibre with inner coated surface with catheter positioning device.
Figure 9 illustrates a device according to an embodiment of the invention with
hollow fibre sealed at one end with flexible extraction phase.
Figure 10 shows a chromatogram comparing diazepam extraction from fibre with
polypyrrole only versus fibre with anti-diazepam antibody entrapped in
polypyrrole.
Figure 11 illustrates selective extraction of diazepam using anti-diazepam
antibodies immobilized on surface.
Figure 12 illustrates calibration in whole blood used to calibrate device
response.
-6-
CA 02478970 2004-08-24
Figure 13 illustrates an example chromatograph obtained after LC/MS/MS
quantification of device extraction from plasma.
Figure 14 shows a cartridge holding a fibre.
Figure 15 is a schematic of batch process for parallel extraction and multiple
MALDI desorption with positioning devices at both laser source and desorption
ends of
fibres.
Figure 16 is a schematic of the inventive device used with nanospray nebulizer
and ESI MS.
Figure 17 shows a schematic of fibre/MALDI-IMS system according to the
invention.
Figure 18 illustrates an exemplary mass spectrum obtained from a fibre/MALDI-
IMS system according to the invention
Figure 19 is a schematic of a fibre/MALDI source.
Figure 20 shows extraction response versus time for standard devices.
Figure 21 shows extraction response versus time for pre-conditioned devices.
Figure 22 provides a comparison of calibration in buffer and plasma,
demonstration of linear response limit.
Figure 23 illustrates an exemplary pharmacokinetic profile of diazepam.
Figure 24 illustrates an exemplary pharmacokinetic profile of diazepam, with
an
expanded y-axis.
Figure 25 illustrates and exemplary pharmacokinetic profile of nordiazepam.
Figure 26 illustrates an exemplary pharmacokinetic profile of oxazepam.
Figure 27 shows an ion mobility spectrum obtained by matrix spray method at
0.05 mg/mL.
DETAILED DESCRIPTION
The invention relates to a method and micro-device based on coated fibre,
optionally in combination with a positioning device, or separation and
detection
technologies particularly useful for in vivo studies of compounds of interest
(identities and
concentrations) in animals, or parts of animals.
CA 02478970 2004-08-24
The method for measuring or identifying one or more component of interest in
an
animal or animal tissue comprises the steps of positioning a fibre within the
animal or
tissue, wherein the fibre is at least partially coated with an extraction
phase for adsorbing
the component of interest from the animal or tissue. The extraction phase is
positioned
within said animal or tissue, and thus the component of interest is adsorbed
onto the
extraction phase for a pre-determined period of time. Following this, the
fibre is removed
from the animal or tissue; and the component of interest is desorbed from the
extraction
phase into an analytical instrument for measurement or identification. The
method of the
invention may be used in pharmacokinetic studies, wherein observation of
analyte levels
in a biological system over time is desirable to conduct with little or no
blood or biological
fluid removal from the system. In the case where blood samples would normally
be drawn
periodically for pharmacokinetic studies, the invention advantageously allows
similar
observations without removal of blood volume from the subject.
The extraction phase may specifically adsorb the one or more component of
interest, and is preferably located at a terminal end (or "distal" end) of the
fibre.
The period of time for which the fibre is positioned within the animal or
animal
tissue can be any acceptable time allowing adsorption of a detectable amount
of the
component of interest. For example, this time may be equivalent to
equilibration time for
a component of interest, or it can be less than equilibration time for a
component of
interest.
The component of interest can be any desirable component. For example, it may
be a bacteria, viruses, sub-cellular components, biopolymers, DNA, proteins,
drugs, drug
metabolites, hormones, vitamins, environmental contaminants, chemicals, or
cells. Any
component capable of detection can be selected.
The animal or animal tissue can be is selected from the group consisting of
single
cell animals, live eggs, mice, rats, rabbits, dogs, sheep, pigs, monkeys and
humans. As
discussed further herein, an embodiment of the invention requires only
samples, and is not
necessarily conducted in an animal or in animal tissue. The animal tissue
could be, for
example, isolated cells and organs.
The fibre may be positioned within a blood vessel, and this embodiment would
allow analysis of a component of interest adsorbed from blood flowing through
said blood
_g_
CA 02478970 2004-08-24
vessel. Optionally, the step of positioning said fibre comprises guiding the
fibre into
position within the blood vessel using a catheter. Other areas in an animal in
which the
fibre may be positioned include a) muscle, brain, soft tissue, or organ of
said animal; and
the component of interest is adsorbed from interstitial fluid or intracellular
fluid; b) an
inner part of spine, scull or bone; and the component of interest can be
adsorbed from the
bone, inner fluids including spinal fluid, bone marrow or brain fluid; or c) a
cell of an
animal, and an adsorbed component is extracted from the inner cellular fluid
or sub-
cellular component of a single cell of an animal. Of course, the invention is
not limited to
these examples.
During positioning, the fibre may be disposed within a housing having a sealed
penetrating end. In this case, the method may include the step of opening the
penetrating
end once the fibre is positioned as desired within the animal, exposing the
extraction phase
within said animal.
Alternatively, the ftbre may be inactive during said positioning followed by
activating the extraction phase using change of electrical potential or
optical means to
allow adsorption of said component of interest. An example of this could be if
the fibre is
made of a metal which can be activated to attract certain components. Other
possibilities
for electrical activation of the fibre are within the scope of the invention.
The invention may use one fibre, or a plurality of fibres arranged as an array
or
bundle. As used herein, discussion of a fibre in the singular does not
preclude the use of
more than one fibre, or a bundle of fibres. In the case where a plurality of
fibres are used,
they may be disposed in a single position within the animal, or they may be
disposed in
more than one position within said animal, so as to obtain readings from
multiple locations
simultaneously. The fibre may be one or more optical fibres, such as a bundle
of optical
fibres.
In one embodiment of the invention, the extraction phase may additionally
comprise a strongly bound calibrant which is retained in the extraction phase
during the
step of adsorbing. Alternatively, a weakly bound calibrant can be used which
is released
from the extraction phase during the step of adsorbing according to convection
conditions
and diffusion coeffficient. The amount of the weakly bound calibrant remaining
after the
_g_
CA 02478970 2004-08-24
pre-determined period of time can be observed. This can also be used to
deliver a desired
compound to the animal or animal tissue.
In another embodiment, a strongly bound reagent may be added to the extraction
phase prior to extraction. This reagent may be a strongly bound reagent which
reacts with
the component of interest. An example of such a strongly bound reagent is one
that labels
the component of interest with a fluorescence tag. Another example is a
reagent such as
an enzyme, in which case the component of interest may be a substrate for that
enzyme.
Such an enzyme may be one that digests a protein directly onto the fibre, for
example
trypsin or a trypsin cofactor. Further, the reagent may be added to the
extraction phase
after the step of adsorbing, in which case the reagent subsequently reacts
with the
component of interest.
The reagent can be added to the extraction phase by spraying or dipping the
reagent onto the extraction phase.
The method of the invention may be one in which a polymerase chain reaction
(PCR) is conducted directly on the extraction phase. In such an embodiment,
the
components of interest are DNA or DNA fragments, the fibre is subject to
periodic cycles
of alternating cooling and heating, the reagent comprises polymerase and
nucleic acids,
and the method results in a polymerase chain reaction (PCR) on the extraction
phase.
The reagent may comprise an ionization matrix utilized in matrix assisted
laser
desorption and ionization (MALDI). MALDI analysis of the extraction phase can
be
conducted with any embodiment of the invention amenable to such a method of
measurement or compound identification. Any number of analytical instruments
may be
used with the invention, such as a spectrometer such as a time of flight
instrument mass
spectrometer (TOFMS) or an ion mobility spectrometer. After desorbing the
component of
interest from the extraction phase, measurement or identification of the
component may
occur in an analytical instrument such as a gas chromatograph, a liquid
chromatograph, a
capillary electrophoresis instrument, a capillary electrochromatography
instrument and a
microfluidic device.
The invention may include positioning of the fibre in an analytical instrument
after
the step of adsorbing. This could, for example involve laser irradiation of
the fibre to
desorb the component of interest from the extraction phase into the analytical
instrument.
-10-
CA 02478970 2004-08-24
In such a case, the fibre can be irradiated in a region not coated with the
extraction phase,
so as to desorb the component.
The invention may allow introduction of the fibre directly into a mass
spectrometer
prior to the step of desorbing. The fibre may be introduced into a mass
spectrometer by
insertion into a small solvent volume in a nanospray needle, followed by the
step of
desorbing, and electrospray of a desorbed component of interest.
After removing the fibre from the animal or tissue, the fibre may be exposed
to a
high voltage resulting in field desorption of the component of interest
directly from the
extraction phase into the mass spectrometer.
Separation of components of interest from the extraction phase may occur
directly
in a separation capillary or channel of the analytical instrument. The step of
desorbing
may be conducted in a small bore cartridge filled with a desorption solvent
following by
automated measurement or identification of a component of interest in the
analytical
instrument. In such a case, the fibre may be placed in the small bore
cartridge
immediately following the step of removing the fibre from the animal or
tissue, and the
cartridge can either be analysed immediately or sealed and transported or
stored prior to
automated measurement or identification.
The invention also relates to a device for adsorbing one or more component of
interest from an animal or animal tissue. The device comprises one or more
fibre having
an at least partially coated end. The end is at least partially coated with an
extraction
phase for absorbing one or more component of interest. The device also
includes a
positioning device for guiding the at least partially coated end of the fibre
into position
within the animal or animal tissue.
Optionally, the fibre diameter can be of millimeter to nanometer dimensions,
and
formed of any acceptable material that would be amenable for use in the
intended
application. Such materials may include fused silica, plastic, carbon or metal
wire. The
fibre may be a plurality of optical fibres formed from fused silica.
Optionally, the fibre may be a hollow tubing having the extraction phase
coated on
an inside surface of the tubing. In this instance, the tubing may be in
communication with
a pump capable of draw up or ejecting a sample from the tubing. The pump may
be of any
acceptable type known for use with tubing. Alternatively, the fibre may be a
hollow
CA 02478970 2004-08-24
tubing having the extraction phase coated on an outside surface thereof. In
this case, the
tubing could be sealed at one end and have a pump in communication with the
tubing to
blow fluid, such as a gas or liquid, into the tubing. This would allow
expansion of the
tubing as desired, which could increase the surface area of the extraction
phase as
required.
The device of the invention may additionally comprise a sheath surrounding the
fibre for protection and easy handling.
The extraction phase is advantageously biocompatible, as necessary.
Optionally,
the fibre may be additionally at least partially coated with a biocompatible
protection
layer, which can surround the extraction phase. Such a biocompatible
protection layer
may comprise polypyrrole or derivatised cellulose, or any such polymer as
would provide
protection.
The extraction phase itself may comprises any composition capable of binding a
component of interest. It may, for example be a polymeric composition such as
IS substituted or unsubstituted poly (dimethylsiloxane), polyacrylate, poly
(ethylene glycol)
or polypyrrole. Alternatively, the extraction phase may have a bioaffinity
agent on its
surface, such as a selective cavity, a molecular recognition moiety, a
molecularly
imprinted polymer, or an immobilized antibody. The extraction phase may
contain any of
these in combination.
The extraction phase can, alternatively be an extraction and ionization matrix
for
MALDI-TOFMS analysis, and may contain a calibrant molecule, as discussed
above.
The fibre may be contained in a housing closed at one end, for opening and
exposing the fibre when appropriately positioned within the animal or animal
tissue. Such
a housing may be a sealed leaf structure, or any other such openable sealant.
The positioning device itself may a catheter, for those applications where the
fibre
is guided into a blood vessel, such as a vein, or other tubular biological
structures, as
discussed in more detail below. Further, the position device may be an x-y-z
micro
positioning stage, for those applications wherein a tissue can be positioned
on such a
stage, and its movement finely controlled. The positioning device comprises an
automated
system, which may be rendered attachable to the animal or animal tissue. The
positioning
device may additionally be used to position said fibre within an analytical
instrument for
-12-
CA 02478970 2004-08-24
desorption of the component of interest from the extraction phase. The
positioning device
can optionally be used to place the fibre directly inside a separation
capillary or channel,
and could be used to couple the fibre to a Laser beam facilitating desorption
of a
component of interest from the extraction phase. The positioning device may be
used to
facilitate desorption of a component of interest into an analytical
instrument.
In the case where a plurality of fibres are used, these fibres may have the
same or a
different extraction phase coated thereon, so that more than one component of
interest can
be detected. More than one extraction phase can be combined on a fibre, so
that a variety
of components of interest can be detected.
The device may additionally comprise an agitator to cause movement of the
coated
end of a fibre, for example axial or horizontal movement of the fibre. In the
case where
the fibre comprises hollow tubing having the extraction phase coated on an
inside surface
of the tubing, the agitator may force the tubing to draw up a sample into the
tubing. This
can be effected by mechanical means or by creating a pressure differential
forcing the
tubing to draw up a sample into the tubing. The agitator may comprise an
inflatable
balloon.
The invention further relates to a method of measuring or identifying one or
more
component of interest in liquid samples arranged in a plurality of wells in a
multiwell
plate. This involves simultaneously submerging a distal end of a plurality of
fibres within
the plurality of wells, respectively, the distal end of each fibre being at
least partially
coated with an extraction phase for adsorbing the component of interest from
the liquid
sample. Following this, the component of interest is adsorbed onto the
extraction phase
for a pre-determined period oftime. The fibres are then simultaneously removed
from the
wells, and are positioned in an analytical instrument for desorption, and
measurement or
identification of the component of interest from the extraction phase. Such an
analytical
instrument may be any of the ones noted above, such as a MALDI analytical
instrument or
a multichannel micromachined microfuidic device.
The inventive device for measuring or identifying one or more component of
interest from liquid samples arranged in a plurality of wells in a multiwell
plate, for use
with the method described herein comprises a plurality of fibres, each having
an at least
partially coated distal end, said end being at least partially coated with an
extraction phase
-13-
CA 02478970 2004-08-24
for absorbing the component of interest. A positioning device is used for
guiding the
coated distal end of said fibres into a submerged position within the
plurality of wells of
the multiwell plate, for removing said fibres from said wells, and for
positioning said
fibres into an analytical instrument.
According to one embodiment, a small sterile device containing a small
diameter
fibre with an associated extraction phase coated thereon is used. The
extraction phase has
affinity for one or more compounds of interest. After exposure of the
extraction phase in
vivo, the device may be removed for quantitative or qualitative analysis in an
analytical
instrument.
A device for in vivo study of chemical concentrations consists of a fibre or
wire
and associated extraction phase. The fibre or wire may be made of fused
silica, metal,
carbon, graphite or a polymeric material. The device may or may not have an
attached or
removable handle. The device may have an associated or removable housing such
as an
outer needle sheath to provide access for the device to the tissue under
study. Preferably
the device is introduced to the tissue under study via a standard medical
positioning device
such as a catheter or microdialysis cannula. After extraction the housing may
be retained
if it is used in association with desorption, or discarded if it is not so-
needed. Where a
medical device is used to provide access to the tissue under study, multiple
devices may be
used with a single catheter for instance, obviating the need to puncture the
skin or other
tissues separately for each extraction.
A process of carrying out in vivo solid phase microextraction uses a fibre
with
associated extraction phase, which may or may not have an associated housing.
In any
case a means is provided to position the device in the tissue for the desired
extraction. For
extraction the device is left in contact with the tissue under study for a
sufficient period of
time to allow equilibration with the chemicals in the tissue, insensitivity to
convective
forces and/or maximal sensitivity. It is likely the device could be used to
monitor
chemical concentrations in humans or experimental animals such as rats, mice,
dogs,
sheep or rabbits. Subsequent to sampling the device is placed in an
appropriate analytical
instrument or desorption device so that at least one chemical component
extracted is
desorbed for quantification.
- 14-
CA 02478970 2004-08-24
The device and process described are used to monitor chemical concentrations
in
vivo in a living animal, without causing a disruption in the dynamic balance
in the animal
systems. Some specific benefits can be described. Because no blood need be
drawn for
the analysis, animals are less stressed. This would allow for more data points
to be
collected for pharmacokinetic profiles, allowing for better data on which to
make drug
design decisions. It would also allow or for sampling of blood or tissue drug
concentrations at multiple sites in an animal, to better assess the effects of
differing
metabolic processes in different locations in animals. Where more data points
are
collected from one animal, a reduction in inter-animal variation in the
results arises. This
variation can often obscure real pharmacokinetic trends and so by eliminating
it, better
pharmacokinetic data can be collected. Conventional sampling where a specific
sample of
blood/tissue is removed from an animal causes a disruption in the normal
chemical
balance of the animal. Each successive sample enhances the impact on the
normal
dynamics ofthe animal. With sampling according to the invention, where only a
1 S negligible portion of the analytes of interest are removed, the normal
chemical balance
remains unperturbed, thus eliminating the effect of sampling itself on the
results. Genetic
variation in drug metabolism within a population gives rise to differing
pharmacokinetics
for the same drug among individuals. The device and process described would be
beneficial both in monitoring the effect of genetic variation on metabolism of
existing
drugs, and for directing the design of novel drugs to take advantage of
variable genetic
profiles for tailored drug design.
Calibration of the device may be achieved in several ways. Where equilibrium
extraction is achieved, calibration by comparison to matched in vitro samples
is simple
and effective. Under non-equilibrium extraction or where it is not possible to
match in
vitro samples to the in vivo system, calibration may be achieved by pre-
loading the fibre
with a suitable calibrant. Direct quantification based on analyte physico
chemical
properties is also possible using spectroscopic analysis of the analytes
directly from the
fibre.
The device accomplishes both sampling and sample preparation during in vivo
analyte extraction. Sample preparation may be limited to isolation from sample
matrix
and concentration in the extraction phase. It may also include additional
processing on the
-15-
CA 02478970 2004-08-24
fibre. Examples of this are derivatization of analyte to a form with higher
sensitivity in
detection through either a modification of product polarity or fluorescent
tagging,
amplification of analyte copy number in the case of DNA analysis to improve
signal
intensity, and protein or enzymatic digestion in the case of general
biomolecules leg.
proteins) to convert them to a form more amenable to instrumental analysis
leg. peptide
fragments). In all cases the goal of this on-fibre processing is to enhance
detectionlquantification of the target analytes.
In the conventional SPME device the overriding goal in device design was
optimizing the affinity of the analyte for the extraction phase on the fibre,
to maximize
analytical sensitivity. In the case of in vivo analysis the issue of coating
biocompatibility
is equally important. Device design must take into account both
biocompatibility and
affinity in the extraction phase.
Because of the simplicity inherent in both the device design and the process,
multiplexing in both sampling and analysis is much more practical that it has
been for
conventional analyses. Fibres may be grouped together in bundles, with fibres
having
either the same or different coatings, allowing for both sampling and
quantification from
many fibres at once, rather than one at a time.
Another advantage of the device and process is that quantification is
performed
separately from sampling, using conventional high sensitivity instrumental
analysis. This
allows better sensitivity and selectivity than are achievable where the
detection is coupled
directly to the sampling/sample preparation as is the case for biosensors. An
interface is
used to couple the fibre to the analytical instrument. This may be as simple
as the off line
desorption of analytes into solvent filled wells in a multi-well plate, to a
more
sophisticated dedicated interface for thermal, field, solvent or laser
desorption. In the case
of a dedicated interface for solvent desorption, small internal diameter
coupled with
efficient solvent flow enhance desorption kinetics so that analytes may be
removed from
the fibre as quickly as possible.
Although the discussion thus far has focused on using a device without
compounds
of interest initially loaded into the extraction phase, to investigate
chemical concentrations
in a living system, the device described is equally suited to the delivery ofa
precise
amount of a chemical compound to a precisely targeted tissue. If a device is
first loaded
-16-
CA 02478970 2004-08-24
with a pre-determined amount of compound of interest, it can be accurately
positioned at
the site of interest, where compounds will move out of the device according to
kinetic
and/or thermodynamic principles and thus supply the chemical to the tissue.
This would
be of value in targeted drug dosing where only a specific tissue is exposed to
a drug
compound.
Figure 1, part A illustrates an extraction device 1 consists essentially of an
extraction phase 4 coated on a fibre or wire 2 to be used with a positioning
device to
accurately locate the device in a tissue. The entire device is sterilizable by
one or more of
the conventional means of sterilization, such as autoclave, ethylene oxide, UV
or gamma
irradiation. The uncoated end of the wire may or may not include a handle 8 to
facilitate
positioning of the device. The length of the wire is variable 7 depending on
the application
requirements. The extraction phase 4 could be a polymeric layer prepared on
the wire
surface, particulate adsorptive or absorptive material glued or otherwise
affixed to the wire
surface, or immobilized biorecognition agents such as antibodies nucleotides
or protein
receptors. When constructed of the stainless steel wire described below the
extraction
device is quite flexible. It will follow curves in a vein or catheter and
normally resume a
straight configuration when removed. The device is useful for the application
of
monitoring concentrations of drugs and their metabolites in blood or other
tissues, either in
single point monitoring or in multiple point (time course) monitoring.
Figure 1, part B illustrates standard medical catheter is shown in schematic
form
having a catheter body 10 and a sealing septum 12 (PRN). PRN is the commonly
used
term for an i.v. adapter to seal a catheter, incorporating a piercable septum,
marketed by
Beckton Dickinson. In the text that follows applications are described that
use such a
catheter for intra venous (i.v.) sampling. In practice, catheters are
available for accessing
other vessels as well, so applications are not limited to i.v. ones. For
instance arteries,
vessels within organs or capillaries may also be accessed using similar
devices.
Figure 1, part C, illustrates an embodiment comprising the extraction device
alone
with no support rod and no handle may be introduced to a blood vessel through
a
previously placed medical catheter 10 with attached PRN 12. The end of the
extraction
device with the extraction phase 4 may be contained in a sterile hypodermic
needle that is
used to pierce the PRN and provide access to the catheter. The extraction
device is pushed
_ 17_
CA 02478970 2004-08-24
partly into the catheter by means of the support wire 2 and the hypodermic
needle is
withdrawn. In this case the PRN provides a seal around the device to prevent
blood loss.
The extraction device 1 is then pushed into the catheter and blood vessel by
an appropriate
amount so that the extraction phase is exposed to the flowing blood. The
catheter is then
flushed with saline to prevent clotting in the catheter. After the required
time for the
extraction of drugs and metabolites the hypodermic needle is once again used
to pierce the
PRN to provide a port for removal of the extraction device. The extraction
device is then
removed from the housing, rinsed and packaged for transport for analysis. The
coated
fibre can be placed inside a micro-syringe as described in US Patent No.
5,691,206, for
easier handling with a catheter or other positioning device.
Figure 2 shows the use of a medical catheter 10 passing through the skin 20
and
vein wall 18 to position the extraction device 9 with PRN 12 inside a vein 22
with blood
flow 16 past the exposed extraction phase 4. In this position the extraction
device has been
fully depressed through catheter so that the extraction phase is fully exposed
to flowing
blood outside of catheter. PRN 12 is still accessible to allow for flushing to
ensure
patency of the catheter.
Figure 3 illustrates a schematic of the polypyrrole polymerization reaction.
As an
example of an extraction phase, polypyrrole may be deposited onto the surface
of a fine
metal wire by electrolytic oxidation under conditions of controlled potential
The polymer
can be prepared on thin stainless steel wire as described below. The resulting
polymer can
then serve as the extraction phase to extract pre-concentrate drug compounds
directly from
blood flowing in a vessel. An exemplary preparation of coating a stainless
steel wire with
polypyrrole is provided in Example 1.
MEDICAL SAMPLING DEVICE
In use it may be desirable to provide a housing or sheath to allow access to
the
tissue site of interest. The housing is also important to ensure correct
positioning of the
device at a specific location in the tissue or site under study. This may be
by puncture of
the skin and/or blood vessel followed by positioning of the phase at a
specific site for
analysis, incorporation of multiple fibres and agitation means. The housing
may also be
-18-
CA 02478970 2004-08-24
required to provide a seal to prevent blood from escaping past the device
during sampling.
The nature of the associated housing will be dependent on the site to be
sampled.
Figures 4 to 9 provide schematic illustration of options for the devices and
described herein.
Figure 4 shows modifications to the device and a housing for mufti-fibre
sampling
using a commercial catheter. Fibres 24, 26 and 28 are coated by coating 30, 32
and 34
respectively, which can be the same type of coating to increase capacity of
the device, or
preferentially each fibre having different highly selective coatings, such as
antibodies
designed to recognized only defined components of interests in a living
animal.
The device may also be used for sampling from an unpressurized medical port
such
as a microdialysis cannula. Because such a port is not pressurized, there is
no need for a
seal to prevent fluid from flushing past the device during sampling, which
obviates the
need for an additional sheath or specialized housing during sampling. The
device has
significant advantages over conventional microdialysis sampling because it is
not
necessary to either add or remove fluid from the tissue to sample. In
conventional
microdialysis analysis a portion of the fluid that diffuses into the cannula
from the
surrounding tissue may be removed for analysis. Alternatively synthetic fluid
is pumped
into the cannula and then to an analytical instrument for semi-continuous
monitoring. In
both instances the fluid balance of the tissue is disrupted during sampling,
by reduction in
volume in the first instance and by dilution in the second. Analysis using the
device
according to the invention would not disrupt the biochemical balance in the
tissue as it
does not cause such an imbalance.
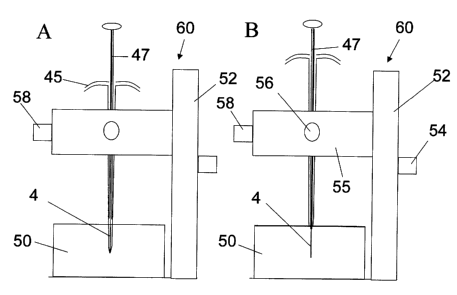
Figure 5 shows a modified housing in part A and extraction device in part B
appropriate for sampling directly from soft tissue. When the device is in the
retracted
position the housing as seen in Figure 5, part A, is constructed of a rigid
tube 40 with a
handle 46 and has a sealed tip 44 for penetrating soft tissue. The tip is
constructed from
two or more leaves separated from each other part way up the housing by a cut
or slot 42
and are normally held together by spring action to seal the tip. Figure 5,
part B shows the
extraction device supported in a thick tubing 6 for opening up the leaves of
the needle end
to allow exposure of extraction phase for sampling.
-19-
CA 02478970 2004-08-24
Figure 6 shows a schematic of the use of the extraction device and housing for
soft
tissue sampling. Figure 6, part A, shows the extraction device 47 within the
housing 45 in
retracted position. Figure 6, part B, shows the extraction device 47 in the
housing 45 in
exposed position. The supporting wire 6 moves with extraction device to force
open the
leaves at tip of needle to allow extraction phase on wire to pass through.
Figure 7 illustrates the housing 45 and extraction device mounted in the x-y-z
positioning device 60 consisting ofthe "z" vertical positioning stage 52 with
high
resolution dial 54 and the x-y stage 55 with appropriate dials 56 and 58
allowing precise
positioning of the extraction phase 4 within the sample 50. This positioning
system is
typically with microscope to monitor insertion and sampling process. The
housing is first
used to prepare a channel for the device at the required position for sampling
(Figure 7,
part A). The housing is then withdrawn slightly while the extraction device 47
is held
still. In this way the extraction phase of the device comes into contact with
the tissue
surrounding the channel prepared by the housing, thus avoiding a plug of
tissue from
traveling into the housing, and avoiding having the extraction device itself
have to bore the
channel in the tissue. In this case the device is used to monitor the
concentrations of
chemicals in the interstital or intracellular fluids in the tissues, as it
would not sample
chemical that is bound to tissue proteins or membranes. This would be
preferred to tissue
biopsy both in terms of the simplified sampling and reduced tissue damage.
Figure 8 shows the catheter with the hollow fibre 3$ coated on the inside wall
surface at the lower portion 70 of the fibre. The schematic cross sectional
view shows the
two layer coating 66 ad 64 on the inner fibre surface 62. The outer coating 66
is chosen to
be biocompatible to eliminate absorption of proteins, while the inner coating
64 is the
extraction phase facilitating removal of well defined components from sample
introduced
to the inner fibre via channel 68. The sample is drawn into the hollow fibre
by using the
device 72 generating pressure differential, such as syringe or metering pump
connected to
the hollow fibre. The action of drawing and ejecting sample produces agitation
and
therefore accelerate the extraction rate. The tubing is mounted in catheter,
but can also be
mounted in a positioning device illustrated in Figure ?.
Figure 9 shows the catheter with the hollow fibre 38 and stretchable coating
sealing one end that can be blown out forming a small balloon structure 75
using the
-20-
CA 02478970 2004-08-24
pressurized gas delivery device 74, such as small compressor or cylinder with
carbon
dioxide and micro-regulator connected to the free end of the hollow fibre. The
material of
the coating or its modified surface 76 can be designed to extract compounds
from sample.
The expended coating has higher surface area resulting in extraction rate
enhancement. In
S addition repeated expansion and retraction of the coating cause induction of
the
convection currents and further increase in the extraction rate.
MINIATURIZATION
While the device described is quite small (127 pm diameter), further
miniaturization would be beneficial, particularly for the study of single
cells. As probe size
is reduced, the effect of the size of the theoretical boundary layer around
the extraction
phase on the rate of extraction is diminished, as is the case with
microelectrodes (Heinze,
J. Angew. Chem. Int. Ed. Engl. 1993, 32, 1268-1288.). In practical terms, this
means the
degree of convection in the sample has less effect on the rate of extraction.
This is
important for sampling of any system where static extraction must be
conducted, as it
would in single cells, or where degree of agitation is variable as it is for
intravenous
sampling. In addition, the dimension of the extraction phase also impacts
extraction
equilibration. Thinner extraction phases equilibrate faster and are less
dependent on
sample convection. Devices with overall dimensions in the range of 1-10 pm
would be
suitable for monitoring the interior of single cells while devices in the sub-
micron range
would be useful for monitoring organelles within cells. There are currently no
feasible
means to accurately assess chemical concentrations occurring within cells. All
currently
available methods either require that the cell is killed (eg. cell lysis
followed by CE of
cytosolic components in microchannels), which may produce an erroneous result,
or suffer
from poor accuracy (fluorescence tagging of specific compounds). The main
strength of
the coated fibre technology is that it can monitor cellular process in a non-
disruptive
manner. Only a negligible portion of the chemical is removed, allowing
cellular processes
to continue unperturbed. Commercially available micropositioning devices using
x-y-z
stage coupled to microscope can be used to position coated end of the fibre in
the well
defined part of the investigated system.
-21
CA 02478970 2004-08-24
Technology that has been developed for genetic manipulation of cells uses fine
capillaries to sample and introduce genetic material in cells, controlled by
micromanipulators and monitored by stereomicroscopes. Cells are maintained in
isotonic
environments during the manipulations, typically by being contained in dishes
or vials
filled with suitable buffers. Similar instruments could be employed for
positioning and
sampling cells with fibre probes.
PORTABLE AUTOMATED SAMPLING
Because the device and process described simplifies sampling and sample
preparation significantly, it provides the opportunity for automated sampling
of tissue
concentrations without the need for continual human involvement. In on-line
microdialysis sampling an animal being monitored is tethered to a stationary
support and
tubing conducting fluid to and from the microdialysis cannula and analytical
instrument
(CE or LC) is included in the tether. In the embodiment, an animal being
monitored does
not need to be tethered, but rather can carry a device for automatically
moving probes in
and out of a catheter, cannula or other sampling port at prescribed times.
After sampling
the device would hold the probes for retrieval and quantification at a later
time. This
embodiment would have similar advantages to the microdialysis system in terms
of
reduced human intervention and hence reduced sampling errors, with the
additional
advantage that animals in a study would be less restricted and stressed, and
experiencing a
more normal environment. This would reduce stress impacts on the integrity of
the
results.
STRATEGY OF SINGLE USE DEVICES
Up to now SPME devices have been designed to be re-used numerous times.
While it is possible to re-use the polypyrrole coated fibre (wire) device
described above, it
is advantageous that this device be employed as a single-use device.
Particularly in
implementations where the device is exposed to blood, it would not be
practical to clean
the device and associated housing sufficiently for re-use. The goal of
manufacture should
be to minimize cost so that users find it cost-effective to dispose of the
device after use.
-22-
CA 02478970 2004-08-24
COATING STRATEGIES
There are a number of additional coating strategies that would be desirable in
the
design of these devices, under certain circumstances. These would extend the
usefulness
of the devices for the purposes described and allow them to be applied for
additional
purposes.
Improved biocompatibility in the extraction phase would be beneficial to
extend
either the time period the phase can be in contact with tissues, or increase
the number of
samplings that can be made from one site. This can be achieved in two
different ways.
Either new phase with better biocompatibility could be selected or a
biocompatible outer
layer could be used in conjunction with an inner extraction phase having lower
biocompatibil ity.
Polypyrrole itself has good biocompatibility. It has been used for several
years in
biosensor devices without any evidence of toxicity, immunogenesis (initiation
of an
immune response) or thrombogenesis (initiation of clotting response). It is an
example of
I S an extraction phase that is suitable for exposing directly to the
investigated system. If it is
desirable to use a less biocompatible extraction phase the device could be
rendered
biocompatible by coating the extraction phase with an outer biocompatible
layer such as
derivatized cellulose. Analytes of interest would diffuse freely through this
outer layer
and be extracted by the extraction phase on the inner layer. This may be
useful if more
traditional extraction phases such as poly (dimethylsiloxane), polyacrylate or
poly
(ethylene glycol) are of interest for extractions.
Biorecognition entities that either comprise the extraction phase or are
immobilized in another phase having low extraction affinity could provide both
higher
selectivity and higher sensitivity in these analyses. Higher affinity would
provide higher
sensitivity and more easily allow for shorter probe residence times. Higher
selectivity
would allow for reduced disturbance of the system under study, further
enhancement of
sensitivity and reduced concern for competition in extraction. This would
permit the
quantitative analysis of one compound present at low concentration when a
competing
compound is present at high concentration.
- 23 -
CA 02478970 2004-08-24
Biorecognition in the extraction phase may be accomplished by entrapment of
antibodies or another molecules capable of biorecognition in an inert
biocompatible
extraction phase. This is demonstrated this in the use of polypyrrole to
entrap antibodies
specific for diazepam.
Figure 10 shows a chromatogram comparing extraction of a sample containing
diazepam, with a device with polypyrrole only, versus a device with entrapped
anti-
diazepam antibody. In this case the analyte affinity to the antibody is much
higher than it
is to the polypyrrole. Alternatively antibodies, nucleic acids or other
molecules may be
covalently attached to the fibre using typical immobilization strategies or
they may be
electrostatically immobilized by means similar to the immobilization of
nucleic acid to
nitrocellulose used in current blotting technologies. For covalent
immobilization either
random or oriented strategies may be used in one application or another.
Figure 11 shows a schematic of the oriented immobilization of antibody 172 on
a
surface 170, and attraction of antigen 174, to form an antibody-antigen
complex 176. The
diazepam may be liberated from the complex for quantification by temporary or
permanent denaturation of the antibody protein.
If a probe with very high selectivity was developed, it could potentially
extract
only the compound of interest, which would eliminate the need for
chromatography in the
analysis. Direct introduction to a mass spectrometer for quantification would
further
simplify the analytical process. Such entities may include antibodies or
antibody
fragments, proteins, protein subunits or peptide sequences, DNA, RNA or
polynucleotides
or the antigens or substrates that bind with any of these. Such biorecognition
entities may
be immobilized by adsorption, electrostatically, covalently or by entrapment
within
another matrix. Covalent immobilization may be by either random or oriented
means.
Biorecognition may also be achieved by using molecular imprinted polymers. In
this case a polymer is prepared in the presence of the analyte of interest.
The polymer
contains functional groups that interact electrostatically with the analyte.
After
polymerization the analyte is removed and cavities remain in the polymer, with
appropriate functional groups located inside. When used for extraction,
analyte freely
soluble in the sample is attracted to the cavities and held there by
electrostatic forces.
These polymers are seen by some as synthetic antibodies due to their high
selectivity for
-24-
CA 02478970 2004-08-24
the analyte of interest. Such polymers provide enhance selectivity when used
as extraction
phases in devices according to the invention.
It is also possible to prepare a coating that can have its extraction
efficiency gated
or activated just prior to extraction. This would allow for the pre-
positioning of the device
in a specific site, and then activate the extraction phase just as extraction
is to start.
Because polypyrrole is a conducting polymer, this may be accomplished by
applying a
small charge to the fibre. This is useful for the extraction of ionic
compounds through
controlling of the oxidation state of the polymer. Alternatively this may be
accomplished
using the device shown in Figure 8 for soft tissue sampling. The device could
first be
positioned in the desired location, but the exposure of the fibre to the
tissue could be
delayed until the proper time to initiate sampling.
USE OF INDICATOR COMPOUND
A common and valuable tool in bioanalytical analysis is the monitoring of the
appearance or disappearance of an indicator compound that is specific for a
biochemical
pathway. This is used for instance to monitor for the presence of specific
cells or bacteria,
or for the presence of free enzymes. In the typical chemical reaction a
substrate (S) is
transformed into a product (P) by interacting with a single enzyme or an
enzyme system
with associated cofactors. Enzymes may or may not be transformed in this
process. The
indicator may be the substrate, in which case its disappearance is monitored,
or it may be
the product, in which case its appearance is monitored. The amount of
indicator formed in
a specific time is correlated to both the amount and activity of the target
enzyme present.
If the indicator has an affinity for the extraction phase, enzyme activities
andlor metabolic
rates may be monitored in situ. The substrate may be either loaded onto the
fibre or
placed into a cell suspension or enzyme solution. When the fibre is placed
into the
solution, indicator will become immobilized in the fibre, and can be
subsequently
quantified by an analytical instrument.
- 25 -
CA 02478970 2004-08-24
PRE-LOADING OF FIBRE WITH CALIBRANT
For conventional SPME analysis, a common difficulty is in devising accurate
means of quantification. For in vitro analysis quantification is often
achieved by adding a
known amount of standard to the sample, and then performing the analysis. This
is
referred to as calibration by internal standard or standard addition. The
amount of the
standard recovered is assumed to be correlated with the amount of unknown
analyte
recovered and the ratio is calculated in order to determine the original
concentration of
unknown. For in vivo and in situ analysis it is typically not practical to add
a standard to
the system under analysis. Until now the most practical means of calibration
is by
t0 preparing a series of synthetic standards that match the sample as closely
as possible, and
comparing the results from the standards analysis with that of the unknown.
This
approach was described above for the calibration of polypyrrole devices in the
in vivo
pharmacokinetic study with reference to Figure 12. In this case whole dog
blood was
obtained from a commercial supplier and samples were prepared with various
drug
concentrations. Upon analysis a calibration curve is constructed and this
curve is used to
interpolate unknown detector responses to estimate unknown drug
concentrations. While
the method is conceptually simple, it is not always highly accurate as it
cannot
accommodate the impact of slight changes in the in vivo site for impact on the
results.
As an alternative to conventional internal standard calibration, a standard
may be
loaded onto the fibre (extraction phase) prior to analysis and the loss of
standard from the
fibre is monitored instrumentally. Where the kinetics of absorption of the
internal
standard analyte to the fibre is equivalent to the kinetics of desorption
(binding is
reversible), absorption and desorption are controlled by diffusion in the
sample and the
rate of loss of standard from the fibre will be correlated with uptake of
analyte by the
fibre. The amount of analyte lost may be correlated with the amount absorbed,
and
consequently with sample concentration of unknown also. Using this strategy
variation in
sample convection may be controlled for by referencing unknown analyte to the
amount of
calibrant lost from the fibre. Alternatively, where the convection conditions
and hence
rate of mass transfer and are known or controlled, the use of an irreversibly
bound
calibrant on the fibre may be used. The fibre would first be exposed to a
matrix-matched
-26-
CA 02478970 2004-08-24
standard with a known concentration of analyte. The fibre would subsequently
be exposed
to the unknown sample. The ratio of unknown to standard extracted by the fibre
would
accurately reflect the ratio of unknown to standard sample concentrations. (G.
Xiong, Y.
Chen and J. Pawliszyn "On-site calibration method based on stepwise solid-
phase
microextraction", ,I. Chromatogr. in press).
Pre-loading of compound onto the fibre may also be used for calibrated
delivery of
compound to a precise tissue region. Where the compound pre-loaded has low to
moderate affinity for the fibre, compound will partition out of the fibre and
into the
surrounding tissue during exposure. This may be used as a means of dosing only
one
targeted tissue region with a drug or other compound of interest, avoiding
dosing of the
whole animal as is commonly the case in therapeutic drug regimens. Tissue
dosage
control may be attained by precisely controlling the exposure time. Dosage may
then be
confirmed by desorbing remaining analytes into an analytical instrument to
quantify the
amount remaining, allowing the calculation of the amount delivered.
An exemplary use of calibrant is discussed below with reference to Example 4.
USE OF MULTIPLE FIBRES
The development of multiple fibre coating strategies has several benefits. In
addition to providing more flexibility in selecting devices for a particular
application,
multiple devices could be used in parallel to provide for a more complete
profiling of the
types and amounts of compounds present in a sample. This may be accomplished
either
by exposing multiple fibres to a sample in parallel, or by preparing one fibre
with multiple
sorbents.
FIBRES FOR CONDUCTING MICRO-CHEMICAL REACTIONS
On-fibre reaction can significantly enhance the detection of components of
' interests. For example on-fibre fluorescence labeling has been has improved
detection
limits for detection of toxins at trace level (A. Namera, A. So, J. Pawliszyn
''Analysis of
Anatoxin-a in Aqueous Samples by Solid - Phase Microextraction Coupled to High
-
-27-
CA 02478970 2004-08-24
Performance Liquid Chromatography with Fluorescence Detection and On - Fiber"
Derivatization J. Chromatogr. 963, 295-302 (2002)). Two of the most important
chemical
reactions for molecular characterization in genomics and proteomics research
are DNA
amplification and enzymatic protein digestion. Both processes are enzymatic
reactions
that are conducted in vitro, with the products either being carried on to a
further
processing step or analysed directly.
In DNA amplification a small number of DNA or polynucleotide fragments are
amplified by the enzyme DNA polymerase. Through the action of the enzyme and
suitable substrates, the copy number of DNA fragments can be increased
exponentially in
just a few hours. The process is characterized by high fidelity so that the
end product is a
very pure solution of identical DNA fragments. Typically the amount of DNA
originally
present is insufficient for further processing andlor analytical
characterization whereas the
concentration in the final product is sufficient. The product may either be
characterized
for nucleotide content and sequence or used for the preparation of peptides or
proteins
coded by the DNA sequence.
For enzymatic protein digestion a protein sample is digested by enzymes that
cleave the polypeptide chains at specific sites. The resulting polypeptide
fragments may
be characterized for molecular weight or peptide content and sequence.
Typically the
intact protein is too large for direct characterization and so a protein is
characterized by a
'fingerprint' analysis of the pattern of polypeptide fragments produced by one
or more
enzymatic cleavages. Alternatively the poiypeptides may be sequenced and the
sequence
of the original protein reconstructed. This allows for example, that the DNA
sequence
coding for the protein may be determined either for the purpose of identifying
its location
in the genome or for development of an expression system to produce the
protein in
quantity.
With the continued miniaturization of genomic and proteomic analyses through
the
use of micromachined or pTAS devices, there is a need to miniaturize the
sample
preparation and introduction steps that come up front. These types of
miniaturized
analyses are increasingly important in the fields of genomics and proteomics
where
sample sizes are small due to the high cost of these samples. Also the
miniaturization
allows for parallelization and higher throughput in analysis to more
efficiently process the
-28-
CA 02478970 2004-08-24
very large number of samples made possible by the completion of the human
genome
project. A porous polymer attached to a fine fibre or wire makes an ideal
medium in
which to conduct these enzymatic reactions in miniature scale, with the added
advantage
that when the reaction is complete, the device is also suitable for
introduction of the
reaction products directly to a microanalytical system.
INTERFACES
As described above one of the strengths of the device and process described is
that
once sampling and sample preparation (pre-concentration and elimination of
matrix) have
been completed, the device of the instant invention is ideally suited for
directly
introducing the extracted analytes to an instrument for separation and
quantification.
Conventional SPME devices are interfacing to GC or LC equipment for
quantification of amount of compound extracted. In the case of GC equipment
the fibre is
exposed in the heated injection sleeve similarly to the way a conventional
syringe injection
is conducted. Analytes for GC analysis are necessarily volatile at the
temperatures
normally used in a GC injector and are efficiently desorbed in the hot carrier
gas flowing
through the injection sleeve and into the separation column. Compounds
analysed by LC
are typically non volatile and/or thermally unstable and so heat cannot be
used for
desorption . For LC desorption a dedicated interface is required to first
remove analytes
from the fibre and transfer them to a solvent. A portion or all of this
solvent is then
injected into the instrument for analysis. In the commercial interface the
fibre is desorbed
in a solvent filled chamber in a valve connected to the instrument inlet.
After desorption
the valve is switched in line with the pressurized solvent flow of the
instrument and the
entire volume of the desorption solution with dissolved analytes is introduced
to the
instrument.
MODIFICATION FOR EFFICIENT LC QUANTIFICATION
The technique has been limited by the relatively large volume of the
commercial
desorption interface ( 100 p.L). Because of the phase thickness of the
commercial SPME
-29-
CA 02478970 2004-08-24
devices for LC (ca. 50 wL) this large volume is required. If desorption volume
is reduced
a significant proportion of the analytes are not removed from the fibre.
Carryovers in the
range of 20% are common (depending on the specific analyte and desorption
solvent used)
as the volume of desorption solvent is reduced below 50 pL. These volumes,
however, are
too large for typical LC applications as injection volumes are in the 10-20 pL
range,
particularly for LC/MS applications. Large injection volumes in these analyses
typically
produce unacceptably broad chromatographic peaks and poor resolution. When
only a
small portion of the total desorption solvent is injected, inferior
sensitivity results. One
strength of device of the instant invention is the ability to introduce all of
the extracted
analyte to the instrument for quantification. This allows for maximal
sensitivity. Fibres
with significantly reduced phase thicknesses, such as the polypyrrole coated
wire
described for the pharmacokinetic analyses, may be efficiently desorbed in 10-
20 pL of
desorption solvent. The entire desorption volume may then be injected for
quantification.
The result is sharp, symmetrical peaks as are shown in Figure 13, which may be
accurately integrated and produce good chromatographic resolution.
The foregoing described the use of static desorption, but dynamic desorption
of
analytes is also of interest in certain applications. This is achieved by
passing desorption
solvent over the fibre during desorption. Because the fibre is continuously
exposed to
fresh desorption solvent, quantitative desorption is theoretically possible.
The rate of
desorption is governed by the rate of solvent flow over the fibre. Faster flow
results in
faster desorption. To achieve the fastest desorption possible and to avoid
ending up with
an overly large solvent injection plug, it is necessary that the inner
diameter of the
desorption chamber is as small as possible. When volumetric flow is constant,
faster
linear flow is achieved in a smaller diameter chamber. This results in a
shorter desorption
time and hence a minimized total desorption volume.
AUTOMATION OF LC QUANTIFICATION
While the reduced volume HPLC interface used to date allows for efficient
transfer
of analytes from the fibre to the instrument, the process is only partially
automated. To
date the introduction and removal of the probe wire tolfrom the interface must
be
performed manually for each injection.
-30-
CA 02478970 2004-08-24
Figure 14 illustrates a micro-cartridge 77, which contains coated piece of
fibre in
its small cavity 79 and sealed with plugs 78. The cavity 79 can be filled with
desorption
solvent. After extraction the coated piece of fibre containing the coating 4
is placed in a
cavity 79 ofthe cartridge 77 for protection during storage and transport.
Determination of
extracted components can be performed in an automated instrument adopted for
use with
cartridges.
INTERFACE FOR CE, USE OF ELECTROKINETIC STACKING
As discussed above, the device of the instant invention provides an ideal
means for
interfacing sampling and sample preparation to microanalytical instruments,
particularly
when devices much smaller than the commercially available SPME devices are
employed.
In capillary electrophoresis and related technologies, analytes are separated
in a capillary
typically 50 pm in diameter. This is too small for conventional syringe
injection.
Injection is typically by hydrodynamic or electrokinetic means. With
hydrodynamic
injection a sample is placed in the buffer reservoir associated with one end
of the capillary.
That end is then lifted above the opposite end by a prescribed amount for a
prescribed
time. The volume of sample entering the capillary may be calculated from the
time, the
elevation difference, the capillary diameter and the solution viscosity. The
sample
solution is then exchanged for running buffer solution prior to applying the
separation
voltage. While simple, the technique suffers from inaccuracies in injection
volume and
poor reproducibility from one analysis to the next. With electrokinetic
injection a sample
is again placed in the buffer reservoir associated with one end ofthe
capillary. An
injection voltage is applied across the reservoir and capillary and analytes
in solution
move into the capillary by electromotive force. Once sufficient material has
been injected
the voltage is removed and the sample solution is again exchanged for running
buffer
solution prior to applying the separation voltage. This method suffers from
inaccuracy in
injections due to the variation in electrophoretic mobility between analytes.
This results in
different amounts of the different compounds present being injected. A small
diameter
fibre with extracted analytes may be introduced directly inside a CE
separation capillary
filled with running buffer (Whang, C.W., Pawliszyn, J. Anal. Commun., 1998,
35, 353-
356). This allows for accurate, quantitative introduction of analytes for
separation.
-31 -
CA 02478970 2004-08-24
As an improvement to this technique for CE analysis, by carefully matching the
outer diameter of the fibre and the inner diameter of the separation
capillary, a stacking of
analytes occurs prior to separation results. This allows for much superior
resolution.
Electrophoretic velocity is inversely proportional to the cross-sectional area
inside the
separation capillary. When this area is reduced, increased velocity results
because of an
increase in electric field gradient. When a fibre is introduced inside a CE
capillary, the
space between the fibre and the capillary wall has a much smaller cross-
sectional area than
the space after the fibre where only buffer is present in the capillary. When
a fibre is
present and separation voltage applied, the analytes move out of the fibre and
along the
restricted channel quite quickly. When they reach the area of open capillary
mobility drops
significantly and the analytes are concentrated in a narrow band. During
separation a
higher resolution is achieved than would otherwise be possible.
Figure 15 illustrates x-y-z positioning device for use with a fibre bundle.
Individual fibres may be positioned precisely in the separation capillary
prior to
desorption. In this case the extraction phase would be coated on more than
just the very
tip of the fibre, as is shown in Figure 15 (132) and desorption would be
accomplished by
applying an appropriate electric potential rather than by laser pulsing.
DIRECT INTRODUCTION TO MS THROUGH NANOSPRAY NEBULIZER
In some instances it is not necessary to chromatographically separate
extracted
analytes prior to quantification. This is the case where the fibre has very
high selectivity
such that only the analyte of interest is extracted with no interfering
substances. It is also
true where mass spectrometry is used for detectionlquantification and
components are
separated by mass rather than by time prior to quantification. For MS
applications it is
possible to place the fibre directly into a nebulizer needle in an
electrospray ionization
source.
Figure 16 describes this process schematically. Solvent flowing through the
nebulizer 150 efficiently desorbs analytes from the fibre 164 prior to being
nebulized and
sprayed in a plume 156 in a mass spectrometer atmospheric pressure ionization
source
160. Ionization is then accomplished by standard ESI with MS detection, ie.
droplets in
-32-
CA 02478970 2004-08-24
the plume 156 are dried and reduced in size by hot gas flow 152 until ions 154
form in the
vicinity of the orifice 166. These then pass into the mass analyzer 162 in the
instrument.
APPLICATION TO MALDI ANALYSIS
Matrix-assisted laser desorptionlionization (MALDI) is a technique for
ionization
of molecules using a laser as the energy source. As a very soft ionization
method, MALDI
yields primarily the singly charged protonated molecule which are then
conveniently
quantified by either ion mobility spectrometry or time of flight mass
spectrometry. This
feature has made MALDI a widespread ionization tool for high molecular weight,
nonvolatile and thermally labile analytes. MALDI has enabled the routine
determination
of large bimolecular such as peptides and proteins (PE. Jackson, PF. Scholl,
and JD.
Groopman, Molecular Medicine Today, 2000, 6, 271.)
The embodiment of the invention wherein the inventive fibre device is coupled
to
MALDI advantageously allows a combination of sample extraction with the
ionization
procedure on the very tip of a fused silica optical fibre for bimolecular
analysis. The
sample end of the fibre was coated for the extraction of peptides and/or
proteins in a
matrix solution. In the case of enkephalin and substance P the matrix used was
alpha-
cyano-4-hydroxy cinnaminic acid. The optical fibre thus served as the sample
extraction
surface, the support for the sample plus matrix, and the optical pipe to
transfer the laser
energy from the laser to the sample. Laser energy was transferred through the
other end of
the optic fibre to ionize and desorb the biomolecules for subsequent analysis.
This fibre /
MALDI combination was coupled with an ion mobility spectrometer and a tandem
quadrupole/time-of flight mass spectrometer (in separate experiments) for the
detection of
the MALDI signal.
Figure 17 shows a schematic of the fibre /MALDI-IMS interface and instrument.
This consists of a laser source 96 and focusing lens 80, which focuses the
laser light onto
the uncoated end of the fibre, held in an x-y-z positioning array 84. The
array movement
may be manual or automated. The fibre 86 transmits light from the source to
the x-y-z
positionable inlet 88 of the mass analyser 90, which in this case was an ion
mobility
spectrometer. In the inlet 88 the coated end of the fibre 86 is held in place
by two silicone
septa 94 and a section of support tubing 92. Only the very tip 98 of the fibre
is coated with
- 33 -
CA 02478970 2004-08-24
extraction phase. A photosensitive diode 82 is positioned at the laser source
84 to sense
the desorption laser pulse and initiate data collection 100.
Figure 18 shows the ion mobility mass spectrum of enkephalin and substance P
were obtained using this system.
One advantage of the MALDI/IMS interface is that the MALDI source is operated
at ambient pressure instead of high vacuum, as it is in conventional MALDI/TOF
mass
spectrometery. Also, loss of sample delivered to the drift tube is negligible
at ambient
pressure and it has been reported recently that atmospheric pressure MALDI
produces a
generally uniform ion cloud at atmospheric pressure. The ionization process is
even softer
than that of the conventional high vacuum MALDI and is capable of producing
protonated
molecular ions for small proteins. This is convenient for the MALDI analysis
of
macromolecules because of the relative absence of metastable fragmentation and
discrimination in the ionization process compared to conventional vacuum
MALDI. The
most promising advantage of this ambient interface is the possibility of
interchangeably
using the same instrument for both electrospray and MALDI sample introduction.
Figure 19 shows a schematic of the laser desorption interface an ion
formation. In
this case two time of flight mass spectrometers (TOF) are used, one to sample
positive
ions 110 and the other to sample negative ions 112. A laser pulse initiates
desorption from
the extraction phase 114 and polarized plates 116 accelerate the appropriate
ions into the
appropriate mass analyzer.
Though MALDI has enabled the routine determination of large bimolecules such
as peptides and proteins, it has always been great interest to develop
quantitative MALDI
analysis. For quantitative work with conventional MALDI analysis, the laser
beam is
scanned cross the sample area on the target plate, and each sample spot is
irradiated with
multiple laser shots until a striking decrease in signal detection is observed
which indicates
the removal of most of the sample loaded on this particular spot. Therefore,
tens to
hundreds of laser shots must be fired to finish the scanning process, and the
final spectrum
is typically a sum or an average of all the spectra obtained from each laser
shot. This
sampling process will lead to the unavoidable poor shot-to-shot and spot-to-
spot sample
reproducibility, and has been considered as the fundamental limitation for
method
quantification in MALDI analysis.
-34-
CA 02478970 2004-08-24
The combination of the inventive device with MALDI has technically solved the
above problem as it combines sample extraction with the ionization procedure
on the tip of
a fused silica optical fibre. The optical fibre thus served as the sample
extraction surface,
the support for the sample plus matrix, and the optical pipe to transfer the
laser energy
from the laser to the sample. Since the sample was loaded directly on the
fibre tip, so the
sample size was identical to that of the laser irradiance area and there
existed no spot-to-
spot desorption difference. In addition, due to the multiple reflections
inside the fibre, the
primary laser profile is converted into a homogeneous intensity profile at the
sample end
fibre surface. This means that laser emission is homogeneous through the fibre
tip surface.
The method was developed as to accomplish all sample desorption that was
extracted on
fibre tip with a single laser shot. As long as this situation could be
satisfied, the spot-to-
spot and shot-to-shot spectral disparity would also be minimized. In this way
it
dramatically improved the quantification aspect of MALDI as well as saved
large amount
of analytical time and analyte consumed. To explore the quantitative aspects
of the
i 5 fibre/MALDI method TOAB was selected as the analyte compound and al I
experiments
were performed in the matrix DHB. The fibre/MALDI-IMS system described in
Figure
17 was used for quantification.
In the extraction step of the previous experiments, the tip of the fibreIMALD1
fibre
was dipped into the solution containing both sample and matrix. For this pre-
mixed
extraction method, the analyte to matrix ratio was pre-optimized and fixed for
the best
performance and this is almost impossible for the detection of analyte in real
samples of
unknown concentrations. Meanwhile, due to the very small capacity of the
extraction
phase, there exists a competition between the analyte and matrix that causes a
further
limitation for the amount of anaiyte that can be extracted. A more practical
way is to load
the matrix in a second step after sample extraction. Spray method with a
nebulizer is an
ideal candidate for this purpose as it forms very fine solution drops smaller
than 100 nm.
After sample extraction, matrix solution is loaded with a nebulizer. The fog
like matrix
drops would help to form more uniform cocrystalline on the fibre tip surface.
The amount
of matrix loaded on the fibre or matrix to analyte ratio could be easily
adjusted by varying
the concentration of the matrix solution and the spray time.
- 35 -
CA 02478970 2004-08-24
Example 2 describes use of a MALD1/IMS interface which is associated with
reduced noise. Reduced noise, though convenient, is not necessary. Thus,
careful
alignment of the laser with the sample surface is optional, as the fibre
itself can
accomplish this. As an alternative however, it would still be feasible to
conduct a
conventional MALDI analysis where the laser is directed at the surface of the
fibre. This
would allow devices to be constructed from non-light conducting fibres, and
would
eliminate the need to optically couple the device and the laser prior to
analysis.
MULTIPLEXING FOR PARALLEL EXTRACTION AND QUANTIFICATION
i0 The inventive device described lends itself to parallelization in both the
sampling
and quantification steps, due to both its cylindrical geometry and
simplification ofthe
analytical process.
Figure 15 illustrates that parallel sampling could be accomplished by bundling
multiple fibres, with the same or different coatings, to either probe multiple
samples at
15 once or to probe a single sample for multiple analytes. The bundle of
fibres could also be
used to provide efficient stirring during extraction. The extraction can be
from mufti-well
autosampler plate, each well containing a different sample is extracted by a
single fibre
facilitating highly parallel determinations. The bundled extraction device
could be
employed for quantification by the MALDI process described above. The bundle
could be
20 multiplexed to a light source, and each individual probe irradiated in
sequence by targeting
the source at each individual fibre in succession. Simultaneously the sample
end of each
fibre would be positioned at the instrument for analysis. As shown in Figure
15, a laser
source 120 is irradiated in sequence onto each fibre in a fibre bundle 130 by
means of a
positioning device 122. The sample ends of the fibres in the bundle are
directed into an
25 extractionldesorption mesh 126. In this case only the tips of the fibres
are coated with
extraction phase 132 as this is the surface that is irradiated by the laser
light. The sample
end is positionable by means of a second positioning device 124. As each fibre
is ready to
be desorbed, it is positioned by the positioning device 124 at the sampling
orifice of the
mass analyzer 128 and the laser 120 is fired to intimate desorption.
-36-
CA 02478970 2004-08-24
Alternatively the probe bundle could be desorbed simultaneously into
individual
solvent desorption wells, with quantification by LC/MS.
The combination of fiber MALDI analysis with multiwell plates may also involve
a positioning device to allow proper placement of the distal end of each
coated fibre within
a small opening of each well, so as to submerge the extraction phase. This
approach
requires design of a relatively small and accurate positioning device, to
accommodate the
large number of wells in a single high density multiwell plate. The technology
now allows
for over 1,000 wells to reside on one plate. Other introduction techniques may
be used to
introduce a sample or fibre into a well, specifically by using micromachined
microfluidic
systems where many microfluidic channels can be placed in one microfluid
device to
accommodate each fibre. This can be performed in combination with nanospray
introduction to MS, where all fibres are desorbed in parallel in a
microstructure, and
subsequently each desorbed solution is introduced to MS in sequence.
Example 1
Preparation of Po~pyrrole Coating on Stainless Steel Wires
and Use in a Biological System
Stainless steel wires (grade T-304V, 0.005") were from Small Parts Inc. (Miami
Lakes FL). Lithium perchlorate (95%) and pyrrole (98%) were from Sigma/Aldrich
(Mississauga, ON). Pyrrole was used as received for one month after opening,
was stored
refrigerated and the bottle was layered with nitrogen after each use.
Polypyrrole (PPY)
films were deposited onto the supporting electrode surface (stainless steel
wire) by anodic
oxidation of the pyrrole monomer in the presence of an aqueous electrolyte
solution
(counter ion). A potentiostat/galvanostat (Model 2?3, EG&G Princeton Applied
Research)
was used for the electrodeposition. The last 15 mm of the wires were coated
potentiostatically at 0.8 V for 20 minutes. The placement of a silicon septum
15 mm from
the end of the wire allowed for accurate control of coating length. The
coating solution
used was pyrrole (0.1 M) and lithium perchlorate (0.1 M) in water and was
prepared fresh
daily. Coating was performed in a custom designed 50 mL flow-through glass
compartment. Coating solution was pumped through the compartment continuously
to
allow for one complete change of solution during each deposition (50 mL/20
min.). The
-37-
CA 02478970 2004-08-24
stainless steel wires were cut into 10.7 cm sections with a razor blade and 2-
4 cm at the
end to be coated was etched with 400 grit silicon carbide polishing paper.
Wires were
then sonicated in acetone until required to prevent accumulation of oxides or
other
contaminants on the wire surface. Immediately before use the wires were rinsed
briefly
with water and were installed as the working electrode. The counter electrode
consisted of
a ca. 10 cm section of platinum wire (0.75 mm OD) formed into a coil of about
1.5 cm
diameter. The stainless steel wire was placed into the coating solution in the
centre of this
coil. A calomel reference electrode was used. The polypyrrole coating
thickness was
estimated to be < 10 wm thick. Prepared probes were then placed into vials
with sufficient
buffer to cover the extraction phase and autoclaved for sterilization.
Wires prepared as described above were characterized in a series of in vitro
experiments. Benzodiazepine standards (1 mg/mL in methanol) were purchased
from
Cerilliant (Austin Texas). These were diluted in methanol to prepare mixtures
of various
concentrations for use in sample preparation and instrument calibration.
Samples were
prepared from buffer, dog plasma or dog blood and spiked with an appropriate
amount of
the analytes of interest. The device was placed directly into the sample
contained in an
appropriate polypropylene sample vial, for a certain period of time. After
extraction the
probe was rinsed briefly with a stream of water and either analysed
immediately or
allowed to dry prior to analysis. Drugs were stable in the extraction phase
when stored dry
at room temperature for at least 24 hours.
Figure 20 and Figure 21 show two alternatives for device response that may be
achieved by this method. In Figure 20 it can be seen that a fast initial
equilibrium
between the sample and native polypyrrole coated wires. After a longer period
of time
additional analyte is extracted as the polymer swells and exposes additional
sites for
extraction.
Figure 21 shows that the polymer was preconditioned with methanol to provide a
swelled polymer prior to extraction. The result the elimination of the initial
lag time seen
in Figure 20 and an immediate increase in amount extracted with maximal
extraction seen
after 30 minutes when the analyte has diffused throughout the bulk of the
polymer to
access the additional sites exposed during swelling. This provides for
additional
sensitivity at the expense of a slower response time.
-38-
CA 02478970 2004-08-24
Figure 22 shows the result of in vitro extraction calibrations from buffer and
plasma. Because the device will only extract unbound drug and because the
drugs under
study are ca. 90% bound to protein, the plasma concentrations tested were 1 Ox
higher than
the buffer concentrations. In buffer I 00% of drug is free and 0% is bound to
protein as no
protein is present. In plasma, it is expected that 10% or less of the drug
will be free.
Figure 22 demonstrates that the linear range attained is similar in buffer and
plasma,
based on free drug concentration. The figure also demonstrates that polymer
extraction
reaches maximal capacity in a solution with 100-200 ng/mI, free drug.
After extraction (either in vivo or in vitro) the compounds on the device are
desorbed in a small volume (10-20 pL) of desorption solvent, 75% methanol in
this case.
Maximal desorption is seen in as little as 20 sec. All or a portion of the
desorption solvent
is injected to an analytical instrument for analysis. This may be accomplished
either on-
line in a dedicated injection interface that takes the place of the regular
injection port on a
LC, or off line in a small desorption chamber, followed by standard syringe
injection of
the desorption solvent by a commercial autosampler.
Figure 12 presents the results of a calibration from whole blood treated with
an
anticoagulant. The figure demonstrates good linearity in extraction over the
range of total
(bound plus free) drug shown.
Figure 13 shows a chromatogram obtained after extraction of drugs spiked at
100
nglmL from dog plasma, demonstrating the good chromatographic peak shape
obtainable
by the method. In this case the injection volume was ca. I 1 pL, using the
desorption
solvent described above.
Figures 23 to 26 show the results ofthe use ofthe device by the catheter
sampling
method described above, for a pharmacokinetic study in dogs. In this case dogs
were
dosed with diazepam at time 0:00. Multiple samplings were performed from a
catheter
over the ensuing 12 hours. Calibration was by comparison to results from an
external
calibration in whole blood similar to that shown in Figure 12. Also shown is a
comparison to results obtained by multiple blood draws over the same time
period, with
conventional sample preparation and analysis as described in the description
of the prior
art. These results demonstrate that the device is useful for the application
described and
-39-
CA 02478970 2004-08-24
that the method described produces results in good agreement with devices and
methods
using invasive prior art sampling techniques.
Table 1 shows the limits of detection achieved in buffer and whole blood for a
"probe" formed according to the invention. As can be seen from these data, the
device
and method allow an extremely sensitive detection of the analytes of interest,
in this case:
diazepam, nordiazepam and oxazepam.
Table 1- Limits of Detection Achieved in Buffer and Whole Blood
detection limit linear
SPME probe calibration from whole blood
Diazepam 1-1000 ng/mL 7.1 215 0.999
Nordiazepam 1-1000 ng/mL 3.1 328 0.994
SPME probe calibration from buffer
Diazepam 10-100 ng/mL 0.43 306 0.999
Nordiazepam 10-100 ng/mL 0.24 281 0.998
to
Example 2
MAtLDI Analysis
In this Example, a medical aerosol compressor was used as the matrix sprayer,
and
mglmL matrix DHB solution was deposited into the nebulizer vial. After analyte
extraction the fibre tip was placed 1.5 cm above the nebulizer vial, and by
turning on the
compressor very fine drops of the matrix DHB solution were formed and attached
to the
fibre tip. The 800pm fibre was tested with the spray method for a 0.05 mglmL
TOAB
sample solution. The times for matrix application were set at 45 seconds and
30 seconds,
respectively, considering the lower analyte concentration. Two 3 minute air-
dry times
were applied before and after the spray of matrix.
-40-
CA 02478970 2004-08-24
Figure 27 shows an IMS spectrum from this analysis. The limit of detection was
found to be 0.02 mg/mL with S/N ~ 2. This level is 10 times lower than the
previous 0.2
mg/mL established by the 400pm fibre using the pre-mixed method. The
sensitivity has
been increased dramatically. This great improvement was attributed to the
larger surface
area as well as the spray method.
In the work described above the laser pulse was shot down the core of the
fibre. In
addition to the advantages associated with reduced noise as described above,
this is
convenient as it is not necessary to carefully align the laser with the sample
surface. The
fibre itself accomplishes this. As an alternative however, it would still be
feasible to
conduct more of a conventional MALDI analysis where the laser is directed at
the surface
of the fibre. This would allow probes to be constructed from non-light
conducting fibres,
and would eliminate the need to optically couple the probe and the laser prior
to analysis.
Example 3
On-Fi er and In-Needle Laboratory
In micromachined devices, controlling flow is not simple since it requires
pumps
or electroosmontic flow means. In addition it is quite difficult to mix
analytes in the small
channels. A more efficient approach is to do sample processing on the surface
or in thin
layers adjacent to the surface. The structures chosen in this Example are the
outer surfaces
of fibers. Alternatively, the inside surface of a tube fiber could be used.
This Example
makes use of a small fiber to demonstrate a convenient sampling method to
collect
analytes from small objects. In this work capillary electrophoresis with
fluorescence
detection has been used to facilitate detection of small amounts of analytes
extracted by
the fiber.
Chemicals and materialx 4-fluoro-7-nitro-2,I,3-benzoxadiazole (NBD-F) was
purchased from Fluka (Sigma-Aldrich Canada Ltd., Oakville, Ontario). Brij35~
and all
amino acids (glycine, L-pheny(alanine, L-proline, L-glutamate and L-aspartate)
were
obtained from Sigma-Aldrich Canada Ltd. (Oakville, Ontario, Canada). Sodium
borate
was from Fisher Scientific (Nepean, Ontario, Canada). All of the solvents used
were
HPLC grade, filtered and degassed and all the aqueous samples were prepared
with
deionized water (NANOpure, Ultrapure water system). A manual SPME assembly and
-41 -
CA 02478970 2004-08-24
replaceable extraction fibers, coated with Carbowax-temprated resin (CW-TPR,
50 um)
were purchased from Supelco (Canada).
Instrument. The high voltage power supply for the CE system was from Spellman
High Voltage Electronics Cooperation, Plainview, NY, USA. The CE separation
capillary
and silica fibers were purchased from Polymicro Technologies LLC, Phoenix, AZ,
USA.
The fundamental components of the laser induced fluorescence detection (LIF)
are
the laser, focusing lens, objective lens, interference filter and
photomultiplier tube. An
Argon ion (Ar+) laser (~ 5 mW) was the excitation source. It provided an
excitation
wavelength of488 nm (its maximum). The microscope objective lens (10x) and the
low
pass filter (530 nm) as an interference filter were from Melles Griot
(Toronto, ON,
Canada). The photomultiplier tube (PMT) and its socket including a high
voltage power
supply were purchased from Hamamatsu (R928 and C6271, Bridgewater, NJ, USA).
In
the design, the optical chopper and lock-in amplifier were used to enhance the
signal
indirectly. The optical chopper (SR-540) and lock-in amplifier (SR-510) were
from
Stanford Research systems (Sunnyvale, CA, USA). The analog output signal from
the
lock-in amplifier was read by a plug-in data acquisition card (Star 4.5,
Varian), which
recorded at 20 Hz followed by the digitalization of the analog signal.
CE system set-up. The CE system was composed of a high voltage power supply
and a separation capillary with an effective length of 45 cm (75 um LD. and
385 um
O.D.). The running buffer was 20 mM sodium borate, 10 mM Brij 35~ and 2.5
methanol. The capillary was conditioned with O.l M sodium hydroxide (NaOH),
water
and running buffer for 15 minutes each. Between runs, the capillary was
reconditioned
with 0.1 M NaOH for 4 minutes followed by running buffer for 2 minutes. The
running
voltage was 12 kV. The injection was done hydrodynamically by raising the
capillary
inlet by 5 cm for 5 seconds.
In-solution derivatization reaction. The reaction solution was prepared by
mixing
10 ul of each amino acid (0.01 mM in water), 10 ul of 1 mg/ml of NBD-F and 60
ul of
IOmM borate buffer at pH 8. This mixture was vortexed for 30 seconds and held
in a 60°
C water bath for various periods of time (2, 5, 10, 30 and 60 minutes). The
reaction
solution was diluted to a final volume of 800 ul with running buffer and
stored in an ice
bath while waiting to be analyzed.
- 42 -
CA 02478970 2004-08-24
On--feber derivatization reaction. A fiber was first cleaned by soaking it in
ethanol.
It was then dipped into a vial containing NBD-F (2-3 mglml in ethanol) for 10
min with
magnetic stirring at 1000 rpm. After that it was transferred to a i-ml Teflon
centrifuge
tube containing 200 ul of amino acids (0.1 mM) in sodium borate buffer (50 mM,
pH 6.0)
and dipped in the sample for 20 sec. A 4-ml amber vial containing 1 ml of
triethylamine
(TEA) was maintained in a 60°C water bath. The headspace ofthis vial
was basic. When
the fiber with extracted analytes was exposed to it, the derivatization
reaction took place
(15 min).
Whole grape sampling with fiberlCE: interface and off column desorption. The
fiber/CE interface with off column desorption was described previously. The
desorption
solvent (2 ul) was placed on the surface of the SPME fiber coating. This small
droplet
was manually rolled around the surface of the fiber coating. Finally this
droplet was
placed on top of a section of quartz tubing and it slipped down to the other
end of the
tubing where the capillary inlet was fixed. The capillary inlet contacted with
this droplet
for approximately 2 s. Since this quartz tubing was positioned 10 cm above the
buffer
vials, the droplet was hydrodynamically injected. The capillary
electrophoresis was
subsequently started. With such an interface, a commercial carbowax/TPR SPME
fiber
from Supelco could be used.
FiberlCE interface: on-column desorption. On-column desorption has been
described previously for a fiber/CE interface. The SPME micro fibers were made
by
attaching a 2-cm long silica fiber (100 um diameter) to a 10-cm polyimide
coated silica
capillary (100 um ID x 365 um OD) with epoxy glue. The unit was air-dried
overnight.
The micro fibers were further etched to approximately 50 um diameter with 50%
HF.
These fibers were finally housed in stainless steel tubes and were sent to
Restek
Corporation, Bellefonte, Pennsylvania for coating with carbowax.
Results and Discussion: Separation of amino acid derivatives. The critical
micellar concentration (CMC) of Brij 35~ is 0.9 mM. The CE running buffer used
in
these experiments had Brij 35~ of 10 mM. Brij 35~ not only forms micelles to
improve
the separation resolution, but also enhances the fluorescence intensity of the
amino acid
derivatives. Some studies have shown that Brij 35~ will enhance the
florescence signal of
such derivatives by at least three times. 1'he methanol (2.5%) in the running
buffer
- 43 -
CA 02478970 2004-08-24
functioned as organic modifier. It helped to increase the solubility of the
solutes and
enlarged the migration time window. As a result, a better resolution was
achieved. Under
these conditions, a mixture of five amino acid derivatives (phenylalanine,
proline, glycine,
glutamate and aspartic acid) was analyzed within 20 minutes.
On-fiber derivatization reaction of amino acids. The on-fiber derivatization
of
amino acids with NBD-F was first established with fibre/HPLC/fluorescence
detector
system described. On-fiber derivatization and a CE/LIF detection system has
been
described in the experimental section. The separation of amino acids
derivatives was
established. NBD-F reacts with to amines and nucleophilies under a mild basic
condition.
The peaks observed of sp-a represented the side products of the reaction of
NBD-F and
TEA. The peaks of sp-b observed are representative of the side products of the
reaction of
NBD-F with the aqueous buffer components and the sample matrix. NBD-OH is the
major side product formed in the reaction of NBD-F with aqueous solution.
Glycine could
not be analyzed under these conditions because the glycine derivative co-
eluted with one
of the side-product peaks, sp-b. Glycine has an average migration time of 6.47
min
(RSD=1.37%). Using a similar procedure, the fibre/CE/LIF detection system was
used for
the amino acid analysis. In this study, the fibre/CE with off column
desorption was used.
Whole grape sampling: Off-column desorption fibrelCE interface. To
demonstrate the application of this technology to the direct analysis of small
living objects
with on fiber derivatization coupled to a CE/LIF detection system, whole
grapes were used
as the samples. With a NBD-F doped fiber, the sampling procedure and
derivatization
reaction took 20 seconds and 20 minutes, respectively. The resulting
electropherograms
from this method with green grape (G) and red seedless grape (R) illustrate a
glutamic
derivative found at 7.05 min for the green grape sample and 7.01 min for the
red seedless
grape sample. These migration times corresponded to the L-glutamate standard
7.04 min.
In the fibre/CE experiments, most of the peaks were saturated such that
phenylalanine and
proline derivatives were hidden in the saturated signal. For further
identification of amino
acids in the sample, the juice from the grapes was analyzed. The glutamate was
also
found in the grape juice sample in the form of glutamic acid.
On-column desorption fibrelCE interface. With the off column desorption,
fibers
with carbowax / TPR coating were used for sampling, while the on-column
desorption was
-44-
CA 02478970 2004-08-24
used with microfiber having thinner coating. These microfibers had a diameter
between
75 - 50 um, and so could be used to sample smaller living objects. These
microfibers
were coated with carbowax. Electron microscopy was used to visualize the
fibers and the
fiber coating was found to be about 10 nm.
The feasibility of using these fibers for the on-fiber labeling.reaction
coupled to
CE/LIF detection system was tested. First, the blank NBD-F solution was
extracted with
the fiber and desorbed on-column. The doping of NBD-F was successful. After
the
sampling of amino acid standard and reaction in the TEA headspace, no product
was
detected.
Example 4
Instrument and Method Calibration using Fibers
Loaded with Calibration Compound
One of the main advantages of the invention is that it allows very convenient
introduction of extracted components onto analytical instrumentation, such as
gas
chromatography, liquid chromatography, supercritical fluid chromatography,
capillary
electrophoresis, micro-channel devices and even directly to mass spectrometry
and
detection instruments. This feature can be further explored for delivering
calibration
standards to analytical instrumentation. Currently standards are delivered to
the
instrument by injecting the solvent mixture containing appropriate calibration
compounds.
However, presence of solvent frequently interferes with calibrarion procedure.
Therefore,
it would be to user benefit to eliminate solvents from the calibration
procedure. It can be
accomplished by desorbing standard loaded fiber into appropriate instrument.
The loading of the fiber can be accomplished by exposing sorbent-coated fiber
(extraction phase coated) to the source of the standard. The standard is then
adsorbed onto
the fiber coating. Another approach is to immobilize chemical standards via
chemical
reaction onto the fiber, which then is released to the instrument under
conditions of
increased temperature, light, chemical potential, mobile phase, etc. The
second approach
ensures stability of the calibrant, but as this Example demonstrates the first
approach can
also be very effective.
Two calibration methods were used. The first approach used solid sorbent
coated
fibres. One of the methods is to deliver the calibration compound by utilizing
SPME fibre
- 45 -
CA 02478970 2004-08-24
to the analytical instrument. The standard is first extracted from the
standard mixture
using strong sorbent followed by introduction ofthe standards loaded fibre
into analytical
system requiring calibration. In this approach liquid injection is avoided and
thus solvent
interference to the determination of trace VOC (volatile components) is
eliminated.
Satisfactory calibration curves were obtained for the very volatile compounds
namely
methanol, acetone, dichloromethane and chloroform when a 75-um carboxenTM/
polydimethylsiloxane (CX/PDMS) fiber/coating was used. The standard gases or
gas
mixtures of VOCs were prepared using the NIST traceable certified permeation
tubes
combined with gas chambers or by microwave-assisted evaporation. "Stepwise" is
the
approach to the second calibration method developed during this work for on-
site
calibration of fibres. In this approach the CX/PDMS coated fibre was loaded
with
standard followed by exposure to the investigated system and then introduction
into GC
system for analysis. The accumulation time of analytes can be controlled equal
to or
different from that of the standard, and the response factors for the analytes
can be
adjusted accordingly. A good reproducibility of the response factors for BTEX
was
obtained with the stepwise method. Satisfactory results were obtained by using
this
method in quantitative analysis of BTEX in the gas station air. The
introduction of
standard via the stepwise procedure makes the technique more useful in field
applications.
This approach in some respects resembles standard addition, but also external
calibration.
It can be used to detect leaks, contaminations and losses from the time of
standard loading
onto the fibre to introduction to analytical instrument.
Preparation of standard gases or gas mixtures. Up to now several methods have
been developed for preparation of standard gas. Two methods were employed in
this work
to prepare the required standard gases or gas mixtures.
Preparation of gas mixture of BTEX using NIST permeation tubes. The
standard gas mixture of BTEX (benzene, toluene, ethylbenzene,p-xylene and o-
xylene)
was generated with the NIST traceable certified permeation tubes (Kin-Tech
Laboratories,
La Marque, TX) and the gas chambers build in our laboratory. It was a flow-
through
system with which a constant concentration of standard gas (or gas mixture)
can be
gained. The temperature was controlled at 50 °C and the air flow rate
was at 300 ml/min.
The gas mixture was sampled from the gas chamber.
-46-
CA 02478970 2004-08-24
Microwave-assisted generation of gas standards of VOCs . A domestic
microwave oven (I OOOW, Model MW5490W, Samsung, Korea) and I-L gas sampling
bulbs (Supelco, Bellefonte, PA) were used for preparation of standard gases
and gas
mixtures of the investigated VOCs with different concentrations. The inner
walls of glass
bulbs were deactivated by silanization and the bulbs were cleaned with
flushing nitrogen
before use. For preparation of standard gases or gas mixtures of BTEX, 1,3-
dichlorobenzene, 1,1,2-trichloroethane and tetrachloroethylene, a clean piece
of glass wool
(ca: I 0 mg) was set inside the sampling port of the bulb each time and was
moistened with
deionized water ( 15 ~L). Water was used to absorb microwave energy and then
to prompt
the evaporation of the compounds that are poor absorbers of microwave. For
preparation
of standard gas mixtures of acetone, methanol, dichloromethane and chloroform,
no glass
wool and water were needed. The port of the glass bulb was sealed with a
Teflon-faced
silicon rubber septum through which a certain volume of liquid of target
compound (or
mixture of several compounds) was injected onto the glass wool. Finally, the
bulb was
placed into the microwave oven to receive microwave radiation for 90 s. The
microwave
output was always set to the maximum power level. After cooling the Supelco
bulb to
room temperature, analysis of the standard gas was performed through the
sampling port
of the bulb where a septum is located.
The device and the "stepwise" procedure. Fiber coatings and conventional
samplers used were provided by Supelco (Bellefonte, PA). The coatings utilized
included
75-~m Carboxenr~~''lPolydimethylsiloxane (CX/PDMS), 85-pm Polyacrylate (PA),
100-
pm Polydimethylsiloxane (PDMS) and 65-um polydimethylsiloxane/divinylbenzene
(PDMSJDVB).
The stepwise procedure was conducted as follows: first, the ftber was exposed
to
tetrachloroethylene standard gas in the bulb, then the fiber was withdrawn
into the needle
after 2-min extraction and a Thermogreen Septum (LB-2, Supelco) was used to
seal the tip
of the needle. When using the field SPME sampler, no separate sealing septum
is needed.
The tetrachloroethylene loaded fiber was then exposed to BTEX standard gas
mixture in
the chamber or to real air sample for a few minutes. Finally, the fiber was
transferred to
the GC injector to desorb the standard and analytes at the same time.
-47-
CA 02478970 2004-08-24
GC=FID analysis of analytes. A Varian model 3500 GC equipped with a flame
ionization detector (FID) was employed for sample analysis. A SPB-5 capillary
column
(30 m x 0.25 mm x 1 pm) from Supelco (Bellefonte, PA) was used and hydrogen as
carrier gas at 30 psi. The column was programmed as follows: 35 °C
initial, held for 1
min, ramp to 135 °C at 10 °C /min and held for 1 min. The
detector was maintained at 280
°C. For the PA, PDMS and PDMS/DVB fibers, the injector was controlled
at 250 °C and
desorption time was 1 min, while CXlPDMS fiber was desorbed for 2 min at 300
°C.
Comparison of introduction of YOCs standards into GC system by syringe
injection of standard solution and by standards-loaded fiber. Several very
volatile
compounds, namely acetone, chloroform, dichloromethane and methanol, were
selected
for investigation. A standard solution was prepared using methanol as the
solvent and the
concentration of acetone, dichloromethane and chloroform was 10 pg/ml for each
compound. The GC-FID chromatogram obtained by injecting 0.1 p.l of the
standard
solution into the GC system illustrated that the solvent peak was too large to
be well
separated from peaks of other compounds and thus made it difficult to
accurately
determine those trace components.
On a contrary, there was no big solvent peak appearing in the chromatograms
obtained by injecting a standards-loaded fiber into the GC system and an ideal
separation
and identification of the VOCs were therefore achieved. The analysis of the
standard gas
mixture was conducted far 3 min using a 75-pm CX/PDMS fiber and the
concentration of
acetone, chloroform, dichloromethane and methanol in the standard gas mixture
was 50.5
pg/L for each compound. Due to the avoidance of solvent injection, it became
easy to get
satisfactory chromatograms for the micro amount of VOC standards, even for the
very
volatile compounds that possess quite short retention times.
In addition, it is difficult to obtain a calibration curve by directly
injecting pure
liquid of individual VOC or liquid mixture of VOCs into GC system to avoid
introduction
of plenty of solvent due to the difficulty of accurate injection of a very
small volume («
0.1 pl) of liquid standards into the GC system to match the quantitative
ranges of trace
analysis.
_ ,~8 _
CA 02478970 2004-08-24
Calibration curves obtained with this technique for GC analysis of some
voes Two different fibers were used to extract the standard gas mixtures of
two different
groups of VOCs. The very volatile compounds, including acetone, chloroform,
dichloromethane and methanol, were extracted with a 75-um CXIPDMS fiber, which
has a
high affinity towards to VOCs as described above. BTEX were extracted with a
100-p.m
PDMS fiber. It is known that the PDMS fiber extract target compounds by
absorption
while CX/PDMS fiber works by adsorption. By introduction of the VOCs standards
into
GC system with fibers, satisfactory calibration curves regarding the
concentration-
response relationship for SPME-GC-FID analysis of the mentioned VOCs were
obtained
and shown in Figure 2. The related calibration equations were listed below:
Methanol: A = 767.69 C+ 3564, RZ = 0.9937 (1-a)
Acetone: A = 3234.8 C +22693, R2 = 0.9952 ( 1-b)
Dichloromethane: A = 1004.6 C+ 7271.5, Rz = 0.9965 (1-c)
Chloroform: A = 980.46 C + 719.5, R' = 0.9993 ( 1-d)
Benzene: A = 106.18 C- 1069.5, RZ = 0.995 (2-a)
Toluene: A = 326.42 C-4320.8, R2 = 0.9993 (2-b)
Ethylbenzene: A = 711.53 C- 6136.8 , RZ = 0.9958 (2-c)
p-Xylene: A = 868.43 C- 10704 , R' = 0.9994 (2-d)
o-Xylene: A = 995.98 C- 9588, l, RZ = 0.9972 (2-e)
where A is the chromatographic peak area (counts) and C is the concentration
of VOCs
standard gas (Pg/L).
The experimental results demonstrated that the investigated fibers are
efficient
for introducing VOCs standards into GC system without solvent injection for
getting
calibration curves and fibre-GC is a highly feasible method for quantitative
analysis of
VOCs, even for the very volatile compounds.
Moreover, it is also possible to establish the "mass:response" calibration
curves for
GC analysis of VOCs by introducing standards with SPME fibers. When absorption-
type
fibres are employed to extract analytes, there is a direct relationship
between the initial
analyte concentration in the sample (C'~) and the amount of the analyte
extracted by the
fibre at equilibrium (n) according to Equation 3-a:
-49-
CA 02478970 2004-08-24
Kfs Vs VJCO
n = (3-a)
VS + Kfs Vf
where Kfs is the fibre/sample partition coefficient, Vfis the fibre coating
volume and Vs the
sample volume. For adsorption-type SPME process, the amount of analyte A
extracted by
the fibre at equilibrium (n) also grows with the increase of the initial
analyte concentration
in the sample (CoA) before saturated adsorption reached:
KAC°AVrVf(Cf mao- CAA )
n=CfA =- ~ (
ys + KA Vf Cf max- CAA
where KA is the adsorption equilibrium constant of analyte A, CfA is the
concentration of
analyte A on the fibre at steady state, Cf,"~ is the concentration of active
sites on the
surface (corresponding to the maximum achievable analyte concentration on the
surface),
VS and Vfare the volumes of the sample and the fibre coating, respectively.
Stability of VOCs on extraction phase coatings after exposing the coatings to
zero air. Analytes present in sample by either absorption or adsorption,
collect or enrich
the target compounds from samples onto the coatings. However, similar to other
extraction
procedures, enrichment is followed by an opposite procedure, the release of
extracted
compounds from the coating phase. Therefore, when a coating loaded with some
VOCs is
exposed to pure air, a part of extracts will transfer to the air and then the
extracts tend to
reach an equilibrium distribution between the coating and air phases. The
release of the
extracts from the coating phase depends on a lot of factors, mainly the
compounds' and
coatings' properties, the temperature of the environment and the differences
of the
compounds' concentrations in the coating phase and in the sample or
environment. It was
shown in Table 2 that the remains of several VOCs (BTEX were included) on the
85-pm
PA, 100-pm PDMS and 65-p,m PDMS/DVB coatings ranged from 0 to 91.5% after
exposing the coatings to zero air for 1 min at room temperature. However, the
75-pm
CX/PDMS coating could store the extracts as much as 89.9%-97.2% even through a
6-min
exposure to zero air under the same conditions. Actually, no obvious losses
could be found
for most of the extracts on the 75-pm CX/PDMS coating when exposure time was
controlled within 4 min. Thus it is possible to allow a "stepwise" procedure
conducted
with the CX/PDMS coating-that is, this coating can be used to extract a
compound in
-50-
CA 02478970 2004-08-24
first step and then transferred to extract other compounds while the compound
extracted
previously still remains on the coating.
Selection of internal standard for BTEX analysis with stepwise Procedure. 1,3-
dichlorobenzene, 1,1,2-trichloroethane and tetrachloroethylene were tested,
respectively,
as internal standards for BTEX analysis when a 75-p,m CX/PDMS coating was
used. The
CX/PDMS coating has a strong affinity towards these compounds and their
storage on the
CX/PDMS coating was close to that of BTEX. Considering their chromatographic
behaviors, tetrachloroethylene is the best internal standard for BTEX analysis
since it can
be well separated from BTEX and its peak is located in the central position of
the
chromatogram. Further investigation demonstrated that, under the selected
conditions, the
loading of tetrachloroethylene on the fiber did not affect the BTEX, and in
turn, the BTEX
did not affect the storage of tetrachloroethylene on the fiber either.
Tetrachloroethylene as
the internal standard for BTEX analysis has another advantage-that is, its
background is
generally not present in main BTEX sources like petroleum. However,
tetrachloroethylene
also has its drawbacks: it is a halogenated compound and the GC-FID response
for it is not
as sensitive as for BTEX. The problem involved in determination sensitivity
for
tetrachloroethylene can be solved by using selective detectors like MSD or
FPD.
Response factors for BTEX when tetrachloroethylene was used as internal
standard For chromatographic analysis, the response factor (F~ can be defined
as the
following form:
Cx Cs
where Ax and As are the peak areas of analyte X and internal standard, while
Cx and Cs the
concentrations of analyte Xand standard after they have been mixed together.
For the use
of tetrachloroethylene as internal standard for BTEX analysis following the
stepwise
procedure described above, the standard is not mixed with analytes before they
were
extracted onto the SPME fiber. In such a case Cs stands for
tetrachloroethylene's
concentration of the standard gas and Cx is individual BTEX's concentration in
the
sample.
For the stepwise GC/FID analysis of BTEX, the response factors were measured.
The time for of BTEX was 2 min, equal to that for the standard. It can be seen
that the
-51 -
CA 02478970 2004-08-24
response factors highly coincided for duplicate tests in almost all cases.
This reflects that
the stepwise procedure is a feasible and practicable method to introduce
internal standard
for GC analysis of BTEX when the CXlPDMS coating is used.
Effect of extraction time on response factors for stepwise procedure. It
should
be noticed that the response factors discussed above are not only related to
the sensitivity
of GC-FID determination to individual BTEX but also depend on the SPME
efficiency for
them. Since the standard and BTEX were not extracted at the same time during
the
stepwise procedure, the time for BTEX can be controlled equal to or different
from that
for the standard. Obviously, time control will significantly affect the
response factors. This
is one of special features of the stepwise procedure distinguishing with the
conventional
way to use internal standards. In the conventional way, the extraction of
analytes and
internal standard is conducted simultaneously. It was found that the response
factors
varied linearly with the time for SPME of BTEX in the range of I-S min when
SPME time
for tetrachloroethylene (standard) was constantly controlled as 2 min. The
linear equations
1 S obtained were as follows:
Benzene: F= 4.265t +1.301, RZ = 0.9979; (5-a)
Toluene: F= 5.776t + 0.402, RZ = 0.9981; (5-b)
Ethylbenzene: F= 4.663t- 0.031, RZ = 0.9996; (5-c)
p-Xylene: F= 4.623t- 0.247, RZ = 0.9993; (S-d)
o-Xylene: F= 4.767t - 0.703, RZ = 0.9963, (5-e)
where F is response factor and t is time in minute for BTEX.
The concentrations of both standard and analytes were within the linear ranges
when the internal standard was used. GC-FID is known to have a linear response
to VOCs
during a very wide range and so is the inventive procedure with the CX/PDMS
coating.
The excellent linearity of the response factors for BTEX varying with the
extraction time
also reflected that the compounds' concentrations studied were located within
the linear
range.
Field application-analysis of BTEX in the air of a gas station. The VOCs
(BTEX were the interests) were sampled from a gas station that is 5-min walk
to our
laboratory, using the home-made field sampler with a 75-p,m CX/PDMS coating.
It was a
clear day and the temperature was ca. 24 °C when the sampling was
conducted. The glass
-52-
CA 02478970 2004-08-24
bulb holding the standard gas of tetrachloroethylene (8. I ~g/L) was carried
to the field and
analysis of tetrachloroethylene was performed prior to BTEX. The sampling time
was 2
minutes for the standard and 4 minutes for the gas station air. As soon as the
sampling
was finished, the sampler was delivered to the laboratory and then the fiber
was
immediately introduced to GC-FID. The identification of individual BTEX was
based on
their retention times as well as GC-MS analysis using a Hewlett-Packard 6890
GC
equipped with a 5973 MSD (Agilent Tech., USA). Tetrachloroethylene was not
found in
the gas station air itself. The separation of standard and BTEX from other
components of
the extracts was very good, only one peak might contain m- and p- xylenes,
which
couldn't be separated from each other under the selected chromatographic
conditions.
Finally, using the peak areas obtained (As and Ax), the standard concentration
(Cs) and the
response factors (F? given by Equations 5-a -5-e, the concentrations of BTEX
in the air
were calculated according to Equation 4.
Conclusion. For getting calibration curves in GC analysis of VOCs in air,
fiber
I S was successfully used to introduce VOCs standards into GC system without
solvent
injection. The avoidance of solvent injection with the inventive technique
made it easy to
obtain satisfactory chromatograms for micro amount of VOC standards, even for
the very
volatile compounds that possess quite short retention times. Moreover, a
stepwise method
was developed to introduce internal standard for GC analysis of BTEX in field
application. The CX/PDMS was proved to be the only suitable coating to fit
this method
due to its extraordinary affinity towards VOCs. Tetrachloroethylene was
selected as the
internal standard for reasons such as its proper retention time compared with
those of
BTEX for GC analysis, similar behaviors to BTEX on the CX/PDMS coating and
very
low background in main BTEX contamination sources in the environments. Using
the
developed method, analytical results can be calibrated without a necessity to
spike
standard material into samples, hence it makes the inventive procedure even
more
advantageous in field applications. However, since the standard is not
directly added into
the sample and the analysis of standard and analytes is conducted stepwise,
this method
may not meet the need of calibration of the air matrix's effects on the
analysis of BTEX.
This approach is also suitable to detected problems with fiber storage in
filed devices, such
as leaks, which will result in analyte and standard losses. Further
development of the
- 53 -
CA 02478970 2004-08-24
technology can include chemical immobilization of compounds, which will
facilitate
production of certified standards.
Example S
Electrophoresis in Non-Uniform Channel Modulated bInsert
Electrophoretic behavior of analyte in capillary consisted of two parts with
different cross-section was investigated. The modulation of the separation
path was
achieved by inserting into the capillary a cylindrical fiber at different
depth. The sample
loaded at the end of lower cross-section and the appropriate zone, at it was
demonstrated,
spatially narrowed in the wide capillary part according to the electric field
strength ratio in
the two parts of the capillary. Additionally, the low conductive sample buffer
can enhance
the further signal narrowness and increase the total probe amount, introduced
into the
capillary by electroinjection. The applications of this concentration
technique includes
focusing after desorption from SPME fibers into the electrophoretic separation
channel
I S prior separation or prior to direct detection using for example UV-
Visible, Fluorescence,
electrochemical, NMR or mass spectrometry detection. Also, focusing of
analytes present
in a buffer is possible by inserting different shapes inserts prior to
separation or direct
detection. Periodic insertion of the insert into the channel will modulate the
concentration
of analyte, which facilitate separation and monitoring of the system connected
to the
separation channel. The modulation input could be random and the signal can be
then
analyzed by multiplex data processing techniques, such as cross-correlation.
Modulation
of the diameter of the channel can be also accomplished by applying external
pressure or
electrical pulses, which will also result in focusing without need for
movement of the
insert in and out of the channel. The results described below indicate that
the focusing
occur in a cross channel configuration since the channel cross-section
diameter increasing
substantially in the area where two channels meet. This can be used
effectively to
facilitate concentration of analyte prior to separation in the second channel,
or in two-
dimensional separation when the separation in the first channel is followed by
focusing at
the interface between the two channels prior to second dimension separation in
the second
channel.
-54-
CA 02478970 2004-08-24
Introduction. Non-constant form of a separation channel in electrophoresis is
a
way of providing the variance of some parameters (electric field strength,
temperature,
pH) which can play an important role for the process concerned. Smooth form
changing is
required should one need to obtain the appropriate smooth function. By varying
a cross-
section of electrophoretic camera one obtains an electric field gradient, this
gradient can
be used both in combination with some other force applied, what is used in so-
called
"gradient focusing techniques " or by itself provided the current density drop
and the
chamber design are appropriate to form sufficient temperature difference. The
latter effect
was used in IEF in thermogradients caused by internal Joule heating.
A separation channel, composed of few different parts but each one of constant
form, can be used rather for sample introducing, detecting, multi step
analysis
development in microarray etc. The results described in this paper are also
important for
the methodologies where the sample application procedure is connected with a
long object
inserting (e.g. microfiber) into the separation capillary.
Experimental: Apparatus. The whole-column imaging detection (WCID) of UV
absorbance was conducted in the iCE280 CIEF instrument (Convergent Bioscience
Ltd.,
Toronto, Canada) with a fixed wavelength of 280 nm. A short fused-silica
capillary (5.5
cm long) with an ID. of 100 um, internally coated with fluorocarbon (J&W
Scientific,
Folsom, CA), was assembled in a cartridge format (Convergent Bioscience Ltd.)
The
entire process of capillary conditioning, sample injection, data collection,
and processing
was implemented by a PC computer, and the electropherogram was recorded as
absorbance versus the distance to the anode.
Materials and Chemicals. Optical fiber with a 50 and 61.5 um core
(FHP050055065 & FVP60072082) was purchased from Polymicro Technologies
Inc.(Phoenix, AZ). pI-markers and buffer chemicals were obtained from Bio-Rad.
Water
was purified using an ultrapure water system (Barnstead/ Thermolyne, Dubuque,
IA) and
was used for all solutions.
Procedures. The fiber was inserted into the capillary at different distances
and the
capillary was filled with running buffer (Phosphate 5-100mM, BioRad). Then,
the sample
was injected electokinetically, with the injection time being specially
selected to achieve
complete replenish of the first the first capillary part (containing an
inserted microfiber).
-55-
CA 02478970 2004-08-24
After, the electrode reservoirs were washed and the desired buffer was placed
and the
electrophoretic run performed.
Results and Discussion. The initial zone width is an important matter in CZE.
For
the case the sample concentration is insufficient to provide sensitive
detection, a number
of on-line preconcentation procedures is developed. The simplest
electrophoresis-based
techniques are connected with a special conductivity profile creation allowing
us to
achieve a higher electric field strength value in the sample zone place,
although
concentration mechanism may be different (e.g., CE- or ITF-based). The similar
effect of
electric field enhancing can be obtained due to stepwise cross-section change.
In these experiments, by inserting the cylindrical microfiber the cross-
section of
the separation channel was modulated. Sample was injected electrokinetically
at SOOv, the
duration of voltage pulse was controlled to achieve complete filling of the
first part of the
capillary (up to the end of microfiber).
The initial starting zone was rather wide (taking into account the "dead"
volume-
around one half of the capillary). Then it was effectively compressed, in the
same
proportion as one could expect starting from cross section difference. In the
case of two
co-axial cylinders the cross-section ratio (R=S2/S1) is: R=D2/(D2-d2), where D
is the
diameter of the capillary and d is the one of the microfiber inserted. By the
assumptions of
constant conductivity, the electric field increase in the narrow part (E1/E2)
is defined by
S2/Sl, and the initial zone length should be narrowed in the same proportion,
approximately.
The effect of observed can be combined with methods traditionally used for
sample preconcentration. With using low conductivity buffer it was possible to
achieve an
essential concentration sample increase in the plug introduced, although the
peak width
change was less evident.
This effect described above does not provide by itself any concentration
increase in
the introduced probe, since the volume of the sample zone should remain
constant and the
sample plug narrowing is due to it form change. But this simple and clearly
visible effect
still opens a lot of important applications to start with to start the
separation from "initially
wide" zone when it is necessary. For example, working with the inventive
technique, one
can insert a microfiber into the capillary and obtain a rather wide starting
zone, with
-56-
CA 02478970 2004-08-24
electric field application the initial zone can effectively be narrowed at the
end of
microfiber. The latter effect, obviously, depends on the relative size of
microfiber inserted,
and to achieve high zone narrowing the (D-d) difference should be small
enough.
Solid phase microextraction and direct desorption of fluorescent labelled
analytes
into the separation channel was observed. The process is monitored by the
fluorescence
whole column imaging detection. The excitation light is delivered to the
separation
channel using the fiber. This work successfully demonstrates the stacking
process that
occurs in CE coupling interface with LIF imaging detection. Based on the
enhancement in
fluorescence intensity, concentration efficiency can be approximated to be as
high as a 10-
fold. Higher concentration efficiency could be expected with further
optimization of
configuration of the interface and the experimental conditions used, such as,
dimensions of
separation capillary and fibre, buffer concentration and applied voltage. The
stacking
effect generated by such an interface is beneficial to separation efficiency
and detection
sensitivity of CE separation.
The above-described embodiments of the present invention are intended to be
examples only. Alterations, modifications and variations may be effected to
the particular
embodiments by those of skill in the art without departing from the scope of
the invention,
which is defined solely by the claims appended hereto.
-57-