Note: Descriptions are shown in the official language in which they were submitted.
CA 02485739 2000-O1-13
-1-
Process for Preparing 1-(6-methylpyridin-3-yl)-2-[(4-
(methylsulfonyl)phenyl~ Ethanone
This application is a division of Canadian Patent
Application Serial No. 2,359,958. The claims of the
present application are directed to a process for the
production of 1-(6-methylpyridin-3-yl)-2-((4-
(methylsulfonyl)phenyl]ethanone, as well as to an N,N-
dialkylamino-(6-methyl-3-pyridyl)acetonitrile, in which
the alkyl radicals are C1-C4 and are identical or
different. However, for a ready understanding of the
overall invention, including all features which are
inextricably bound up in one and the same inventive
concept, the teachings of those features claimed in
Canadian Patent Application Serial No. 2,359,958 are all
retained herein.
Description
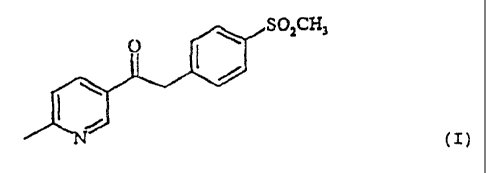
The invention comprises a novel process for preparing
1-(6-methylpyridin-3-yl)-2-[(4-(rnethylsulfonyl)phenyl]-
ethanone of the formula
OZCH3
(I)
1- (6-Methylpyridin-3-yl) -2- [ (4- (methylsulfonyl) phenyl] -
ethanone is an important intermediate for preparing so-
called COX-2 inhibitors, which are pharmaceutically active
compounds having analgesic and anti-inflammatory action
(R. S. Friesen et al., Bioorganic & Medicinal Chemistry
Letters 8 (1998) 2777-2782; WO 98/03484). 1-(6-
Methylpyridin-3-yl)-2-[(4-(methylsulfonyl)phenyl]ethanone
of the formula (I) is a novel compound which is not known
from the literature.
CA 02485739 2000-O1-13
-la-
It is an object of the invention to provide a novel
intermediate for the production of COX-2 inhibitors and to
provide an industrially feasible process for preparing the
intermediate of the formula (I).
As an aspect of the invention which is claimed in
Canadian Patent Application Serial No. 2,359,958, there is
provided a process for preparing 1-(6-methylpyridin-3-yl)-
2- [ (4- (methylsulfonyl)phenyl] ethanone of the formula
SOiCH3
(I)
wherein 2-methyl-5-vinylpyridine is initially converted,
by reaction with ozone and subsequent reduction, into 2-
methylpyridine-5-carbaldehyde or into 1-hydroxy-(6-
methylpyridin-3-yl)methanesulfonic acid or acid salt of
the formula
30
/ SOC M~
(II)
in which M is an alkali metal, which is then reacted with
a dialkylamine and a cyano compound to give a N,N-
dialkylamino-(6-methyl-3-pyridyl)acetonitrile of the
formula
CA 02485739 2000-O1-13
-lb-
R~
~T
jV~~,R2
~~ (III)
in which R1 and Rz are identical or different and are each
C1_4-alkyl, and the acetonitrile (III) is finally reacted,
in the presence of a base, with a 4-(methylsulfonyl)benzyl
halide to give 1-(6-methylpyridin-3-yl)-2-[(4-
(methylsulfonyl)phenyl]-ethanone of the formula (I).
As another aspect of the invention, which is claimed
in Canadian Patent Application Serial No. 2,359,958, there
is provided a 1-hydroxy-(6-methylpyridin-3-
yl)methanesulfonic acid salt of the formula
SOGM~~~
3
(II)
in which M is an alkali metal.
A further aspect of the invention claimed in Canadian
Patent Application Serial No. 2,359,958, provides a
process for preparing a 1-hydroxy-(6-methylpyridin-3-
yl)methanesulfonic acid salt as defined herein, wherein 2-
methyl-5-vinylpyridine is converted, by reaction with
ozone and subsequent reduction with an alkali metal
hydrogen sulfite, into the end product of the formula
(II) .
CA 02485739 2000-O1-13
-1C-
Yet another aspect of the invention claimed in
Canadian Patent Application Serial No. 2,359,958, provides
a process for preparing a N,N-dialkylamino-(6-methyl-3-
pyridyl)acetonitrile of the formula (III)
T
r'yRz
C1j (III)
in which R1 and RZ are identical or different and are each
C1_4-alkyl, wherein 2-methylpyridine-5-carbaldehyde or 1-
hydroxy-(6-methylpyridin-3-yl)methanesulfonic acid salt of
the formula (II)
so~ M°
(II)
is reacted with a dialkylamine and a e:yano compound to
give the end product of the formula (III).
The invention also provides, as claimed in Canadian
Patent Application Serial No. 2,359,958, a process for
preparing 1-(6-methylpyridin-3-yl)-2-[(4-
(methylsulfonyl)phenyl]ethanone of the formula
35
S 42C~-i3
(I)
CA 02485739 2000-O1-13
-1d-
wherein in a first step a), 2-methyl-5-ethylpyridine is
converted at a temperature of from 500°C to 700°C in the
presence of a catalyst into 2-methyl-5-vinylpyridine,
in a second step b), the 2-methyl-5-vi.nylpyrdine is
converted by reaction with ozone and subsequent reduction
into 2-methylpyridine-5-carbaldehyde,
in a third step c), 2-methylpyridine-S-carbaldehyde is
converted, using a dialkylamine and a cyano compound, into
the corresponding N,N-dialkylamino-(6-methyl-3-
pyridyl)acetonitrile, and finally,
35 in a last step d), the N,N-dialkylamino-(6-methyl-3-
pyridyl)acetonitrile is reacted in the presence of a base
with a 4-(methylsulfonyl)benzyl halide to give 1-(6-
methylpyridin-3-yl)-2-[(4-(methylsulfonyl)phenyl]ethanone.
As a further aspect, the invention provides a process
for preparing 1-(6-Methylpyridin-3-yl)-2-[(4-
(methylsulfonyl)phenyl]-ethanone of the formula
S02CH3
(I)
wherein N,N-Dialkylamina-(6-methyl-3-pyridyl)acetonitrile
in which the alkyl radicals are Cl_C4 alkyl and are
identical or different, is reacted in the presence of a
base with a 4-(Methylsulfonyl)benzyl halogenide to give 1-
(6-Methylpyridin-3-yl)-2-[(4-(methylsulfonyl)phenyl]-
CA 02485739 2000-O1-13
-le-
ethanone, wherein the base is an alkali alkoholate and is
used in the presence of an organic solvent at a
temperature of 15°C to 25°C.
Finally, the invention provides a N,N-dialkylamino-
(6-methyl-3-pyridyl)acetonitrile of formula
~f
1 '1
(III)
wherein R1 and R2 are the same or different and represent
Cl_4-alkyl .
The process according to the invEntion comprises four
steps, where in the first step a) 2-methyl-5-ethylpyridine
is converted at from 500°C to 700°C ire the presence of a
catalyst into 2-methyl-5-vinylpyridine, in the second step
b) the 2-methyl-5-vinylpyridine is converted with ozone
and subsequent reductive work-up into 2-methylpyridin-5-
carbaldehyde, .in the third step c) the 2-methylpyridin-5-
carbaldehyde is converted with a dialkylamine and a CN
CA 02485739 2000-O1-13
- 2 -
compound into the corresponding N,N-dialkylamino-~6-
methyl-3-pyridyl)acetonitrile
and f finally in the last step d) the N, N, -dialkylamino-
(6-methyl-3-pyridyl)acetonitrile is reacted in the
presence of a base with a 4-(methylsulfonyl)benzyl
halide to give 1-(6-methylpyridin-3-yl)-2-j(4-
(methylsulfonyl)phenyl]ethanone to give the end
product.
A considerable advantage of the process
according to the invention consists in the fact that
industrially available 2-methyl-5-ethylpyridine can be
used as starting material.
Step a:
The dehydration of 2-methyl-5-ethylpyridine to
give 2-methyl-5-vinylpyridine is .known from the
literature (for example A. Nenz et al., Hydrocarbon
Processing, 47 (11), 1958, 139-144; lJS-A 2,769,773) .
The reaction proceeds at from 500°C to 700°C in the
presence of a large number of different catalysts.
In general, catalysts based on silica, silica gel, iron
oxide, zinc oxide, chromium oxide, copper chroriite,
magnesium oxide, potassium oxide, alumina or boron
phosphate, alone or as a mixture, if appropriate
applied to a support, are employed.
Good results can be obtained inter alia with a zinc
oxide catalyst applied to pumice as support.
It is furthermore advantageous for the reaction to
dilute the 2-methylpyridine with steam or an inert gas,
but preferably with steam.
The 2-methyl-5-vinylpyridine can be purified in
a simple manner, for example by removal of the aqueous
phase' and subsequent steam distillation or vakuum
distillation, such that it is suitable for the
3S subsequent step b).
Step b:
The reaction with ozone is advantageously
carried out in the presence of a mineral acid at a
CA 02485739 2000-O1-13
temperature of from -20°C to 0°G, preferably at a
temperature of from -1S°C to -5°C. Suitable mineral
acids are sulfuric acid or phosphoric acid, and in
particular sulfuric acid. Suitable reaction media are
water and/or a polar solvent. As a polar solvent
C1_6 alcohols can be used such as methanol, ethanol,
propanol, butanol, pentanol or hexanol. Mixtures of a
lower alcohol, such as methanol or ethanol, with water
have been found to be useful.
The ozone complex which is formed as an intermediate is
worked up reductively, preferably with an alkali metal
hydrogen sulphite, to obtain the 2-methyl-5-
carbaldehyde.
Suitable alkali metal hydrogen sulphites are sodium or
15. potassium hydrogen sulphite. However, it is also
possible to choose other known reducing agents, such
as, for example, dimethyl sulphide, thiourea or
trimethyl phosphite, or hydrogen in the presence of a
suitable catalyst.
In the case of the preferred reductive work-up
with alkali metal hydrogen sulphite, the reaction is
carried out in essentially the same medium as used for
the ozonization, generally at a temperature of from
-20°C to 20°C, preferably from -10°C to 0°C.
Depending on the further work-up steps, the
2-methylpyridine-5-carbaldehyde or an adduct of alkali
metal hydrogen sulphite with the 2-methylpyridine-5-
carbaldehyde can be formed.
If it is desired to isolate the 2-methyl-5
carbaldehyde, it is possible to selectively extract the
reaction mixture at a pH of about 4 to 5 with a
suitable organic solvent, such as, for example, with
ethyl acetate. Alternatively, but preferred, an adduct
of alkali metal hydrogen sulphite with the
2-methylpyridine-5-carbaldehyde may be formed
initially, which is then cleaved at a pH of about 10
into the 2-methyl-5-carbaldehyde.
Particularly preferably, however, the adduct of
alkali metal hydrogen sulphite with the 2-methyl-
CA 02485739 2000-O1-13
pyridine-S-carbaldehyde is employed immediately for
further reaction 'in step c) . Thus, it is possible to
circumvent isolation of the relatively unstable
2-methylpyridine-5-carbaidehyde in an elegant manner.
The adduct of alkali metal hydrogen sulphite
with the 2-methyipyridine-5-carbaldehyde is novel and
not known from the literature and accordingly also part
of the subject-matter of the invention. The adducts
have the general formula
O~I
.''~ $ ~°SUUM~? (
s
in which M is an alkali metal, and are referred to as
1-hydroxy-(6-methylpyridin-3-yl)methanesulfonic acid
salts. The alkali metal M, is advantageously Na or K.
Step c:
The reaction .of the 2-rnethylpyridine-5-
carbaldehyde or the adduct of alkali metal hydrogen
sulphite with the 2-methylpyridi,ne-5-carbaldehyde is
2o carried out accordirAg to the principle of the Strecker
synthesis using a CN compound and a dialkylamine to
give the corresponding' N,N-dialkylamino-(6-methyl-3-
pyridyl)acetonitriie.
An aqueous HCN solution or an aqueous solution of an
alkali metal cyanide may serve as CN compound here. A
suitable dialkylamine is a C~_4-dialkylamine, in
particular dimethylamine or diethylamine.
The reaction temperature is advantageously in
the range of from 0°C to 30°C.
It may be advantageous to add a water-
immiscible solvent, such as, for example, toluene or
t-butyl methyl ether. Work-up and isolation of the
corresponding N,N-dialkylamino-(6-methyl-3-pyridyl)-
acetonitrile can then be carried out by simple phase
CA 02485739 2000-O1-13
- 5 -
separation. The N,N-dialkylamino-(6-methyl-3-pyridyl)-
acetonitriles of the general formula
N(Alk)i
(III)
i
N
in which Alk is an al kyl group of from 1 to 4 carbon
atoms are novel compounds which are not known from the
literature, and they therefore also form part of the
subject-matter of the invention.
Alkyl is specifically methyl, ethyl, n-propyl,
isopropyl, n-butyl, isobutyl or tent-butyl. The
preferred meaning of Alk is methyl or ethyl.
Step d: .
The conversion of the N,N-dialkyla~ino-(5
methyl-3-pyridyl) acetonitrile by reaction with the 4
(methylsulfonyl)benzyl halide to give the end product
of the formula I is carried out in the presence of a
base. A suitable 4-(methylsulfonyl)benzyl halide is
4-(a~ethylsulfonyl)benzyi chloride.
The base used can be an aqueous alkali metal
hydroxide solution, preferably an aqueous sodium
hydroxide solution, where in this case the presence of
a customary phase-transfer catalyst is useful. Suitable
phase-transfer catalysts are, for example,
tetraalkylammonium halides, such as, for example,
tetra-n-butylammonium chloride or tetra-n-butylammonium
bromide. The reaction temperature is in the range of
from ~0°C to 70°C. It may be advantageous to add a
water-immiscible solvent, such as, for example,
toluene, methylene chloride or t-butyl methyl ether.
Alternatively and preferably; the base used is
an alkali metal ~alkoxide. Suitable alkali metal
alkoxides are, for example, sodium tart-butoxide,
potassium tart-butoxide or sodium tart-pentoxide, and
preferably potassium tart-butoxide. Recommended
CA 02485739 2000-O1-13
- 6 -
solvents are ethers, such as, for example,
tetrahydrofuran. The reaction temperature in this
variant is generally from 15°C to 25°C.
The 1-(6-methylpyridin-3-yl)-2-[(4-(methyl
sulfonyl)phenyl]ethanone can be isolated in a manner
known to the person skilled in the art, for example by
acidifying the reaction mixture, followed b_y extraction
with, for example, toluene. Further purification can be
carried out by recrystallization, for example in
acetonitrile.
Example 1
Preparation of 2-methyl-5-vinylpyriciine
Pumice having a particle size of from 6 to 8 mrn
is moistened with water and mixed with 25% of its dry
weight of zinc oxide in powder form, filled moist into
the reactor (length of the tube 750 mm, diameter of the
tube 60 mm) and left in a stream of nitrogen at from
650°C to 700°C for 24 h.
76 ml/h of 2-methyl-5-ethylpyridine togethe r with
87 m1/h of steam were passed over: the abovementioned
catalyst at from 670°C to 680°C and 665 mbar. At the
end of the reactor, a product stream consisting of
40.6% by weight of 2--methyl-5-vinylpyridine and 56.3%
of 2-methyl-5-ethylpyridine was taken off. Based on
reacted 2-methyl-5-ethylpyridine, a yield of 93.0% was
achieved.
To prepare pure 2-methyl-5-vinyipyridine, the product
mixture was subsequently su'.bjected to steam
distillation (266 mbar, overhead temperature 59°C-60°C)
or to vacuum distillatian (20 mbar, temperature 90 °C) .
Example 2
Preparation of 2-methylpyridine-5-carbaldehyde
11.92 g of 2-methyl-5-vinylpyridine (content
85%, 85 mmol)., 50 ml of methanol and 10 ml of water
were initially charged. Concentrated sulphuric acid
19.81 g, 98 mmol) was metered in such that the
temperature did not exceed 20°C;. The solution was
CA 02485739 2000-O1-13
_ 7 _
cooled from -12°C and an ozone/oxygen mixture (about 50
03 in OZ, SO 1/h) was introduced until the 2-methyl-5-
vinylpyridine had reacted completely. Water (50 ml) and
40% aqueous NaHS03 solution (22.7 g, 85 mrnol) were
carefully metered in. The reaction mixture was warmed
to 20°C and neutralized using 30% NaOH (about 32 g,
0.24 mol). Methanol was distilled off at 30-40°C, and
then, to form the bisulphate adduct, another 22.7 g of
40% NaH503 solution were added. The mixture was stirred
for 30 min, after which the pH was readjusted to
neutral, and the neutral impurities were subsequently
extracted using 35 ml of t-butyl methyl ether. The
aqueous phase was adjusted to pH 10 using 30% NaOH, and
26.5 g of Na2C03 (0.25 mol) were added. The liberated
aldehyde was extracted using 2 x 80 ml of t-butyl
methyl ether. Concentration of the solvent gave 9 g of
2-methylpyridine-5-carbaldehyde as a slightly yellowish
oil.
1H-NMR (CDC13) : 2.66 (s, 3H) ;
7.35 (d, J =~ 8 Hz, 1H)
8.07 (dd, J = 8 Hz and 2.1 Hz, 1H);
8.96 (d, J = 2.1 Hz, 1H);
10.08 (s, 1H).
;3C-NMR (CDC13) : 24.98 (CH3)
;
123.72 (C-5);
129.32 (C-3);
135.88 (C-4);
151.8? (C-2);
164.87 (C-6);
190.51 (C=0).
Example 3a.
Preparation of N,N-diethylamino-(6-methylpyridin-3-
yl)acetonitrile (from 2-methylpyridine-5-carbaldehyde)
At from 10°C to 15°C, 73.2 g (1.25 eq.) of
diethylamine and 100.3 g (1.15 eq.) of a 25% HCN
solution were added simultaneously over a period of one
CA 02485739 2000-O1-13
hour to a mixture of 98.3 g (1.0 eq.) of
2-methylpyridine-S-carbaldehyde in 200 ml of water and
200 ml of toluene which was stirred efficiently. The
reaction mixture was stirred at 30°C for 3 h.
The phases were then separated and the aqueous phase
was extracted with 2 x 100 ml of toluene. The organic
phases were combined and the toluene was then removed,
giving the title product in the form of a yellowish oil
and in a yield of 172.3 g (90.10).
1H-NMR (CDC13) : 8.65 (1H, s)
;
7.?5 (1H, d)
;
7 . 20 ( d)
1H, ;
5.00 (1H, S)
i
2. 68 (2H, m)
;
2.59 (13H;s);
2.50 (2H, m);
1. 10 ( t
6H, )
.
1H-NMR ( D6-DMSO) : 8 . ( s ) ;
5 d 1H,
7.70 (1H, d);
7.32 (1H, d) ;
5.45 (1H, s);
2.58 (2H, m);
2.50 (3H, S);
2.40 (2H, m);
1.02 (6H, t).
Example 3b.
Preparation of N,N-diethylaminc~-(6--methylpyridin-3
yl)acetonitrile (via the adduct of 2-methylpyridine-5
carbaldehyde ~rith sodium hydrogen sulphite)
The ozonolysis was carried out as in Example 2,
starting from 23.84 g of 2-methyl-5-vinylpyridine
(83.10 GC, 106.2 mmol). After the impurities had been
extracted at neutral pH, the aqueous phase was cooled
to 15°C and diethylamine (21.94 g, 0.3 mol) and then
9.8 g of NaCN (0.2 mol) were added (in each case
addition over a period of 10 min). The solution was
CA 02485739 2000-O1-13
_ g _
stirred at 15°C for 4.5 h, and the product was
subsequently extracted with 3 x 85 ml of toluene. The
combined extracts were concentrated. Obtained: 37.4 g
of N,N-diethylamino-(6-methylpyridin-3-yl)acetonitrile
S as an orange oil. Content: 83.7% {GC, % by weight),
0.34% of aldehyde). Yield: 92.?% based on 2-methyl-5-
vinylpyridine.
1H-NMR (CDC13) : 1.08 (t, 6H) ;
2:50 (m, 2H);
2. 58 (s~,3H) ;
2. 65 (m, 2H) ;
5.00 (s, 1H);
? . 18 (d, J = 8 Hz, 1H) ;
7.74 (dd,J = 8 Hz, 2 Hz, 1H)
;
8. 66 (d, J = 2 Hz, 2H) .
Example 3c.
Preparation and characterization of the adduct of
2-methylpyridine-5-carbaldehyde tait:h sodium hydrogen
sulphite
After the addition of bisulphite, the 1H- and
13C-NMR of a sample were measured. The NMR signals of
the aldehyde had disappeared completely, and the
following signals were observed instead:
1H-NMR (DMSO-ds): 1.96 (s, 3H);
5.01 (s, 1H};
6.85 (d, J = 8 Hz, 1H);
7. 45 (dd, J = 8 and 2 Hz, 1H) ;
7. 93 (d, J = 2 Hz, 1H) .
isC-NMR {DMSO-d6): 20.23 (GH3};
81.78 (CH);
124.14 (C-5);
130.02 (C-3);
138.76 (C-4);
143.08 (C-2);
156.04 (C-6) .
CA 02485739 2000-O1-13
- 10 -
Example 4a.
Preparation of 1-(6-methylpyridin-3-yl)-2-j(4-(methyl-
aultonpl)pbsayl,ethanons (aqueous ~ao$ as baae)
41.07 g (89.1, 1.00 eq.) of N,N-diethylamino-
(6-methylpyridin-3-yl)acetonitrile, 30 ml of toluene
and 10.0 g of Celite were initially charged. ?2 g
(5 eq.) of a 50o aqueous NaOH solution were then added
over a period of 15 minutes such that the temperature
could be maintained at 20°C. The reaction mixture was
heated to 45°C. With vigorous stirring, a first portion
of 0.32 g of tetra-n-butylammonium bromide was added.
Immediately after that, a solution of 0.32 g of tetra-
n-butylammonium bromide and 44.52 g (1.2 eq.) ~ of
4-(methylsulphonyl)benzyl chloride in 200 ml of toluene
was added over a period of 1.5 h. After half had been
added, a third portion of 0.32 g of tetra-n-
butylammonium bromide was added, and stirring was
continued at 45°C for 6 h.
The reaction mixture was then. warmed to room
temperature, and 100 ml of water and 100 ml of toluene
were then added. The mixture was filtered, the residue
was washed with 25 ml of toluene and the phases were
then separated. The aqueous phase was extracted' with
2 x 50 ml of toluene. The combined organic phases were
then extracted with 380 ml of 1N HCl. Neutralization
with 29.6 g of 50% aqueous NaOH solution to pH 4.5
resulted in the title product crystallizing out. The
suspension was filtered and the product was washed with
2 x 100 ml of water and 2 x 80 mi of isopropanol/water
1:1 and subsequently dried at 20°C/20 mbar.
This gave 40.19 g (?6.4%) of~the title product having a
content of 99 . 0 0 .
M.p. 182°C-183°C
1H-NMR (CDCi~) : 9.15 (1H, s) ;
8.18 (1H, d):
7. 92 (2H, d) ;
CA 02485739 2000-O1-13
- 11 -
7.47 (2H,d);
7.30 (1H,d);
4.39 (2H,s);
3.04 (3H,s);
2. (3H,s)
63 .
_...-.. ..._ ._.. .......... , _._ ......_ _...~.-._ .....-.,."",.. . .. . .~x
. > .~:..r.:aa , s"..anv.,~.....".~".r,.... ..."....,.w-......., ..-
..........,__.....~_~..._...._.. .._._.._...__.. ....._.~._...
........____.__.,.."""..
,. s°r~m-r.;.w. n:~~,::..,.-°-.-.: 'crrr"~.aac.. ~n=:..
. CA 02485739 2000-O1-13
- 12 -
Example 4b.
Preparation of 1-(6-methylpyridin-3-yl)-2-j(4-(methyl-
suifonyl)phenyl]ethanone ~alkouide, anhydrous)
At 20°C, 48.16 g (84.5, 1.00 eq.) of N,N
diethylamino-(6-methylpyridin-3-yl)acetonitrile in
20 ml of tetrahydrofuran were added over a period of 30
minutes to a suspension of 38.58 g (1.7 eq.) of
potassium t-butoxide in 60 ml of tetrahydrofuran.~
Immediately afterwards, 42.59 g (1.03 eq.) of 4
(methylsulphonyl)benzyl chloride in 60 ml of
tetrahydrofuran were added at from 20°C to 25°C over a
period of 1.5 h.
The reaction mixture was stirred at. 20°C for 0.5 h and
then diluted with 100 ml of water, and adjusted to pH 2
by addition of 180 ml of 2N HCl over a period of one
hour. After a further 0.5 h at 20°C, the mixture was
adjusted to pH 3 using 10 g of a 30~ aqueous NaOH
solution. The suspension was stirred at 20°C for one
hour and then filtered, and the product was washed with
2 x 150 ml of water and 2 x 100 ml of water/isopropanol
1:1. Drying at 20°C/20 mbar gave 53.72 g (92~) of the
title product having a content of 99.1°x.
M.p. 182°C-183°C
1H-NMR (CDC13) : 9.15 (1H,s)
;
8.18 (1H,d)
?.92 (2H,d);
7 . 47 (2H;d)
7.30 (1H,d);
4. 39 (2H,s)
;
3.04 (3H,s)
;
2.63 (3H,s)
.