Note: Descriptions are shown in the official language in which they were submitted.
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
METHODS OF DETECTING TARGET MOLECULES AND MOLECULAR INTERACTIONS
Field of the invention
The invention relates to methods and kits for
detecting target molecules or the interactions between
target molecules. In particular, the invention
relates to methods based on the use of "two-component
conjugate" systems comprising a catalytic label and a
' substrate label.
Background to the invention
Detection of a target biological molecule in a
sample requires sensitive techniques that are capable
of discriminating between specific recognition events
and non-specific recognition events that might
otherwise lead to false positive results.
Techniques for detecting many different types of
biological molecules, including proteins, nucleic
acids, viruses, bacteria and carbohydrates, are known
in the art. High specificity of detection is often
achieved by using multiple binding entities to bind to
the target molecule that is to be detected.
Known techniques for the detection of nucleic
acids include, for example, the Polymerase Chain
Reaction (PCR). In this technique two short nucleic
acid primers recognise the target nucleic acid.
Detection of the target nucleic acid is only achieved
when both primers are bound to, and linked through,
the same target molecule. Non-specific interactions
of the primers with other molecules are not detected
unless both primers bind to and are linked by this
non-specific interaction. The conditions of the
reaction are such that the latter is highly unlikely.
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
_ 2 -
Other molecular amplification methods known in the art
include nucleic acid sequence-based amplification
(NASBA) (Compton, 1991) and 3SR (Fahy et al., 1991).
Proteins may be detected by well known techniques
such as Western Blotting. This technique is very
useful but is limited in sensitivity and specificity,
due to the lack of an amplification step, and the fact
that there is a single entity binding to the target.
Target molecules can, however, be detected in a
very sensitive and specific manner through the Dual
Phage approach, as disclosed in WO 99/63348. In the
Dual Phage assay two phage are needed in order to
generate a signal and the two phage must be brought
together or linked through the target molecule. This
is most easily achieved by linking the phage to
ligands such as antibodies that are specific for the
target molecule. In this method it is possible to
detect a wide range of target molecules including
nucleic acids, proteins and simple or complex
molecules.
In another approach described in US Patent Nos.
US 5,35,602 and US 5,665,539 two target-specific
antibodies are both linked to the same piece of
nucleic acid, such that the nucleic acid forms a
bridge. After binding to the target this nucleic acid
bridge is specifically cleaved and then re-associated.
The presence of an intact nucleic acid bridge (i.e.
cleaved and re-associated) is shown by the use of PCR
and two primers that recognise the reformed nucleic
acid, because the nucleic acid bridge contains two PCR
primer binding sites. This approach enhanoes the
specificity of the assay because the nucleic acid is
more likely to reform after cleavage if both antibody
molecules are bound to the target and are thus in
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 3 -
close spatial proximity. The nucleic acid bridge is
unlikely to re-form if only one antibody is bound non-
specifically to a molecule other than the intended
target. A disadvantage of this approach is the
problem of ensuring that all of the nucleic acid
bridge molecules are cleaved in the absence of target
antigen. In addition, the method is complex and
involves a number of steps that could involve DNA
restriction enzymes, DNA polymerases and DNA ligation
enzymes.
WO 01/61037 relates to assays for detection of
analytes in solution using so-called proximity probes.
Said proximity probes consist of a binding moiety and
a nucleic acid. Upon binding of the proximity probes
to the analyte the nucleic acids are brought into
close proximity and can thus be ligated and then
detected, usually by amplification. This technique
has the disadvantage that ligation can be an
inefficient reaction and the lipase has to be added to
the reaction in a high enough concentration to allow
efficient ligation of the proximity probes.
Sensitive methods are also needed for use in
monitoring molecular interactions. Drug discovery and
proteomics are just two of the areas of technology
that rely on the monitoring of such interactions. A
variety of methods are currently in use, which are
well known in the art, such as the Scintillation
Proximity Assay (SPA) (Bosworth and Towers, 1989) and
various Yeast Hybrid methodologies (Ma and Ptashne,
1988; Fields and Sternglanz, 1994).
The Dual Phage method can also be applied to the
monitoring of molecular interactions. In this case,
the molecules whose interaction is to be studied each
have a ligand-binding site that can bind one phage
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 4 -
type either directly or indirectly. The interaction
of the molecules is thus able to be monitored through
the linking of the two phage types. If the molecules
interact the two phage types are brought together but
if they do not interact the two phage types remain
separate. This approach can be applied to proteomics
and drug discovery.
Other approaches which can be used to detect
interactions between biological molecules include
fluorescent Resonance Energy Transfer (FRET), a
distance-dependent excited state interaction in which
emission of one fluorophore is coupled to the
excitation of another. FRET relies on a fluorescence
donor which transfers a quantum, or exciton, of energy
to a fluorescence acceptor, thus raising an electron
in the acceptor to a higher energy state as the
photo-excited electron in the donor returns to the
ground state. The resonance interaction between donor
and acceptor fluorophores occurs over distances that
are greater than interatomic. For successful FRET the
fluorescent emission spectrum of the donor must
overlap the absorption spectrum of the acceptor, and
the donor and acceptor transition dipole orientations
must be approximately parallel. The probability that
energy transfer will occur depends on the sixth power
of the distance between the fluorophores.
When the donor and acceptor are different, FRET
can be detected by the appearance of fluorescence of
the acceptor or by quenching of donor fluorescence.
If the donor and acceptor are the same, FRET can be
detected by the resulting fluorescent depolarization.
Energy transfer can be detected by measuring emission
from the acceptor fluorophore when excited at the
donor fluorophore's wavelength. This wavelength does
not produce an emission from the acceptor unless FRET
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 5 -
has occurred. Alternatively, FRET can be detected by
measuring the quenching effect that the aoceptor has
on donor emission at its excitation wavelength.
US3996345, US42~1968 and US199559 describe
immunoassays that employ fluorescer-quencher
chromophoric pairs. Here, either one or both of the
chromophoric pair are bonded to antibodies. Depending
on the particular ligand of interest, various reagent
combinations can be employed, where the amount of
quenching is directly related to the amount of ligand
present in the assay medium.
US 6,251,581 discloses methods for determining an
analyte in a medium suspected of containing the
analyte. One method comprises treating a medium
suspected of containing an analyte under conditions
such that the analyte, if present, causes a
photosensitizer and a chemiluminescent compound to
come into close proximity. The photosensitizer
generates singlet oxygen and activates the
chemiluminescent compound when in close proximity.
The activated chemiluminescent compound subsequently
produces light. The amount of light produced is
related to the amount of analyte in the medium.
Description of the invention
The present invention seeks to provide improved
methods of detecting target molecules and of
monitoring molecular interactions.
In accordance with the first aspect of the
invention, hereinafter referred to as the "target
detection method" there is provided a method of
detecting a target molecule in a sample comprising the
steps of;
(a) contacting a sample with two or more
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 6 -
binding entities specific for the target
molecule, including a first binding entity
labelled with a catalytic label and a second,
separate binding entity labelled with a substrate
label, characterised in that the catalytic label
is capable of acting directly on the substrate
label to generate a detectable change in the
substrate label;
(b) incubating under conditions which permit the
binding entities to bind to the target molecule, '
thus bringing the catalytic and substrate labels
into close proximity;
(c) allowing the catalytic label to act directly
on the substrate label when bound in close
proximity, thereby producing a detectable change
in the substrate label;
(d) detecting the change in the substrate label.
In accordance with a second aspect of the
invention, referred to herein as the "interaction
method", there is provided a method of detecting
interactions between two or more interacting molecules
comprising the steps of:
(a) incubating the interacting molecules such
that they can interact, thereby bringing into
close proximity a catalytic label attached to one
of the interacting molecules and a substrate
label attached a separate interacting molecule,
characterised in that the catalytic label is
capable of acting directly on the substrate label
to generate a detectable change in the substrate
label;
(b) allowing the catalytic label to act directly
on the substrate label when bound in close
proximity, thereby producing a detectable change
in the substrate label;
(c) detecting the change in the substrate label.
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
Both methods rely on specific binding
interactions into order to bring into close proximity
a "catalytic label" and a "substrate label". The
catalytic label is capable of acting directly on the
substrate label to generate a detectable change in the
substrate label. In this context "detectable change"
means a change in structure and/or activity of the
substrate label, caused by the action of the catalytic
label, which change in structure and/or activity can
be directly or indirectly detected. The "detectable
change" in the substrate label is preferably
irreversible under the conditions used to detect the
change.
In the most preferred embodiment the "detectable
change" is caused by direct action of the catalytic
label on the substrate label, meaning that the change
in structure and/or activity is brought about by
direct contact or physical interaction between the
catalytic and substrate labels. The term "direct
action" excludes changes in structure and/or activity
of the substrate label which are brought about solely
though the action of a diffusable intermediate,
without any requirement for a direct contact or
physical interaction between the catalytic and
substrate labels.
The catalytic and substrate labels are attached
to "separate" binding entities/interacting molecules,
meaning that no part of the catalytic label is
attached to the same binding entity/interacting
molecule as any part of the substrate label. The
catalytic and substrate labels are brought into close
proximity by specific binding of binding entities to a
target molecule (in the target detection method) or
specific binding of interacting molecules (interaction
method), thus facilitating direct action of the
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
_ g _
catalytic label on the substrate label. The close
juxtaposition of the catalytic and substrate labels as
a result of specific binding events results in a rate
enhancement that provides an improved signal-to-noise
ratio over the background due to random interactions
between un-bound catalytic and substrate labels in
free solution.
The target detection and interaction methods
according to the invention are types of biological
binding assays and may be carried out in accordance
with standard principles known in the art for these
types of assays. For example, it is common to include
intermediate washing steps between addition of
reagents to remove excess/unbound reagents, as
illustrated in the accompanying examples.
Preferred features of catalytic and substrate labels
Action of the catalytic label on the substrate
label results in a detectable change in structure
and/or activity of the substrate label.
Changes in the structure of the substrate label
may include, for example, physical cleavage of the
label.
Changes in activity of the substrate label may
include, for example, a change in a detectable
property such as fluorescence, luminescence, etc, or a
change in enzymatic activity, electrochemical activity
or redox properties. The change in activity may be
accompanied by, or caused by, a change in structure of
the substrate label.
The "change" in activity of the substrate label
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 9 -
may be a change in activity resulting from a discrete
switch of the substrate label from an "inactive" to an
"active" state (or vice versa) as a result of a
discrete action of the catalytic label on the
substrate label (an example being a cleavage of the
substrate label). However, in other embodiments the
"change" in activity may be the appearance of an
activity which requires continued action of the
catalytic label on the substrate label. An example of
such a "change" is increased transcriptional activity
resulting from interaction between an RNA polymerase
catalytic label and a nucleic acid substrate label
bearing a promoter specific for the RNA polymerase.
Preferred catalytic labels include, but are not
limited to, enzymes.
In one embodiment of the invention the action of
the catalytic label on the substrate label may
generate a fluorophore, which can subsequently be
detected. For example, the substrate label may be
formed of a fluorescent moiety linked to a quencher
moiety which quenches the fluorescence of the
fluorescent moiety by via a linkage which is cleaved
by the action of the catalytic label. Cleavage of the
linkage releases the quencher moiety and thus
increases fluorescence from the fluorescent moiety.
This increase in fluorescence may be detected using a
suitable measuring instrument. In this embodiment the
catalytic label may be an enzyme and the "linkage" may
be a molecular structure which is cleaved by the
action of the enzyme. For example, the enzyme may be
a protease and the linlcage a peptide which is cleaved
by the action of the enzyme. The precise nature of
the linkage is not important, except to the extent
that it must enable the fluorescent and quencher
moieties to interact when "linked", such that the
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 10 -
quencher moiety quenches fluorescence from the
fluorescent moiety.
In a further embodiment the action of the
catalytic label on the substrate label may quench a
fluorophore.
In a still further embodiment the action of the
catalytic label on the substrate label may create an
electrochemically active species or group that can
subsequently be detected. For example the catalytic
label may be the enzyme alkaline phosphatase and the
substrate label may be a phenol phosphate moiety
coupled directly to a binding moiety. In this case the
enzyme, when in close proximity to the substrate,
catalyses the removal of the phosphate group,
generating an electrochemically active group which may
be detected by a redox reaction at an electrode surface.
In a still further embodiment the substrate label
may be an inactive molecule which becomes activated by
the action of the catalytic label. Example of such
labels include, for example, enzyme precursors,
zymogens or pro-enzymes.
Many enzymes are synthesized as inactive
precursors, called zymogens or pro-enzymes, which are
subsequently activated by cleavage of one or a few
specific peptide bonds. This proteolytic activation
is an irreversible process. For example, trypsinogen,
chymotrypsinogen, proelastase and procarboxypeptidase
are all inactive precursors, of the digestive enzymes
trypsin, chymotrypsin, elastase and carboxypeptidase,
respectively. All of these pro-enzymes become
activated by the action of trypsin, which hydrolyses
peptide bonds in the pro-enzymes. Therefore, in a
preferred embodiment of the invention the catalytic
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 11 -
label may be trypsin, or a protease of equivalent
proteolytic activity but differing specificity, and
the substrate label may be one of trypsinogen,
chymotrypsinogen, proelastase and procarboxypeptidase.
These examples are merely illustrative and are not
intended to be limiting to the invention.
Other preferred combinations of enzyme catalytic
labels and enzyme precursor substrate labels include
enteropeptidase/trypsinogen (cleaved to generate
trypsin), thrombin/fibrinogen (cleaved to generate
(fibrin).
In such systems wherein the catalytic label
cleaves an enzyme precursor substrate label to
generate an active enzyme, the "change" brought about
by action of the catalytic label on the substrate
label (i.e. enzyme activation) may be conveniently
detected by a suitable assay specific for the enzyme
activity generated by cleavage of the substrate label.
It is important that this assay is capable of
distinguishing between enzymatic activity generated as
a result of cleavage of the substrate label and the
enzymic activity of the catalytic label.
Other preferred examples of catalytic labels
which may be used in accordance with the invention
include phosphatases and kinases. Such catalytic
labels will, respectively, de-phosphorylate or
phosphorylate a suitable substrate label.
Dephosphorylation or phosphorylation of the substrate
label may, in turn, lead to a detectable change in
structure and/or activity of the substrate label.
In certain embodiments the substrate label which
is phosphorylated/dephosphorylated by the
kinase/phosphatase may be a protein, however the
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 12 -
invention is not limited to only proteins. Any
suitable substrate label which can be phosphorylated
or dephosphorylated, wherein such phosphorylation or
dephosphorylation is accompanied by a detectable
change in structure and/or activity, may be used
within the scope of the invention.
In a still further embodiment of the invention
the substrate label may be a nucleic acid and the
catalytic label may be a species, typically an enzyme,
capable of catalysing a detectable change in the
structure and/or activity of the nucleic acid.
Examples of suitable catalytic labels include, for
example, recombinases, ligases, transposases, DNA
polymerases, reverse transcriptases or RNA
polymerases.
In this embodiment, changes in the "structure" of
a nucleic acid label are taken to include changes in
sequence which are detectable, for example with the
use of suitable sequence-specific probes and/or
nucleic acid amplification techniques. There are many
suitable techniques known to those skilled in the art
which may be used to detect and differentiate specific
nucleic acid sequences.
Changes in the "activity" of a nucleic acid label
may include, as discussed previously, a change in
transcriptional activity, such as may occur if the
catalytic label is an RNA polymerase and the substrate
label contains a promoter specific for the said
polymerase. Preferred RNA polymerase/promoter systems
for use in this embodiment of the invention include
(but are not limited to) the bacteriophage RNA
polymerases, especially T7, T3 and SP6 polymerases,
and their cognate promoters. These polymerases are
well known in the art and are routinely used for in
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 13 -
vitro transcription. Increases in transcriptional
activity brought about by the action of an RNA
polymerase catalytic label on a substrate label
containing an appropriate promoter may be detected by
directly or indirectly detecting the resulting
transcripts. In a preferred embodiment, the substrate
label may be a reporter gene expression construct
comprising a suitable promoter driving expression of a
reporter gene. The increase in transcriptional
activity may then be monitored by detecting expression
of the reporter gene.
In one embodiment of the invention, the substrate
label may be formed of two or more component parts, at
least one of which is attached to a separate binding
entity/interacting molecule to the catalytic label.
There are Various ways in which this can be achieved.
For example, the component parts of the substrate
label may be all attached to a single binding
entity/interacting molecule and the catalytic label
may be attached to a separate binding
entity/interacting molecule.
In a further arrangement, the catalytic label and
component parts of the substrate label may each be
attached to separate binding entities/interacting
molecules. Such a system has the advantage that three
species must be brought into close proximity for the
interaction between catalytic and substrate labels to
take place. Thus, in a "target detection method"
wherein the catalytic label and components of the
substrate label are each attached to separate binding
entities, at least three separate binding events of
binding entities to a common target must take place in
order to enable the interaction between catalytic and
substrate labels. In the "interaction method",
wherein at least three separate interacting molecules
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 14 -
are respectively labelled with a catalytic label and
components of the substrate label, then at least three
separate species of interacting molecules must come
together, in order to enable the interaction between
catalytic and substrate labels.
In a still further embodiment, the catalytic
label and at least one component part of the substrate
label may each be attached to separate binding
entities/interacting molecules and a further component
part of the substrate label may be present in free
solution, preferably in an excess amount. In this
system there will still be a "proximity effect"
between the catalytic label and the component of the
substrate label which is attached to a binding
entity/interacting molecule, as these two labels will
only be brought into proximity by binding of labelled
binding entities to their targets) or interaction of
labelled interacting molecules.
An example of a substrate label formed of two
component parts is a substrate label formed of two
separate nucleic acid tags, which are capable of
interacting, in the presence of a suitable catalytic
label, to generate at least one nucleic acid tag
having novel sequence. This type of label is
particularly useful in an assay system wherein one
nucleic acid tag is attached to a binding
entity/interacting molecule and another nucleic acid
tag is present in free solution in excess.
In the most preferred embodiment of the system
wherein substrate label formed of two separate nucleic
acid tags, the tags are capable of interacting via
recombination to generate at least one tag of novel
sequence. In this embodiment the catalytic label
comprises an enzyme that catalyses recombination
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 15 -
between the nucleic acid tags.
"Recombination" is defined herein to include any
exchange of nucleic acid sequence or deletion or
insertion of sequences between the nucleic acid tags
in order to generate at least one novel sequence that
is capable of being detected. Examples include site-
specific recombination events (e.g. requiring a
specific recombinase) and transposition events (e. g.
requiring a specific transposase).
Site-specific recombination events are non-
homologous recombination events, in so far as they
generally do not require extensive homology between
nucleic acid tags. In most cases site-specific
recombination requires the presence of short
recombination site sequences (generally a few tens of
base-pairs). Many site-specific recombination systems
require the presence of identical recombination site
sequences. However, in other systems the
recombination sites may share little or no sequence
homology, as is the case with the integration sites
attP and attB, derived respectively from bacteriophage
lambda and the E. coli chromosome.
In a preferred embodiment, the "substrate label"
is comprised of two nucleic acid tags, each containing
a site-specific recombination sequence recognised by a
particular site-specific recombinase enzyme, and the
"catalytic label" is the appropriate site-specific
recombinase enzyme.
Suitable site-specific recombination systems
which may be used include the Cre/loxP system, wherein
the nucleic acid tags making up the substrate label
contain loxP sites, and the catalytic label is Cre
recombinase. Another suitable system is the
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 16 -
bacteriophage lambda integration system, wherein the
nucleic acid tags contain attP and attB recognition
sequences or attL and attR sequences, allowing
recombination catalysed by an enzyme label which
recognises these sites. Recombination between attB
and attP sites or between attL and attR sequences is
catalysed by the lambda phage enzyme integrase, and
requires a host accessory factor IHF. The lambda
phage recombination system is well known in the art
l0 and the enzymes required for recombination are
available commercially (e.g. as components of the
GatewayTM cloning system supplied by Invitrogen).
These particular recombination systems are listed by
way of example only and it is not intended to limit
the invention to the use of these specific systems.
Other site-specific recombination systems known in the
art such as, for example the Flp/FRT system, may also
be used.
In a particular embodiment of the "recombinase"
system, the recombinase enzyme may actually be
attached to one of the nucleic acid tags making up the
substrate label. For example, the two nucleic acid
tags making up the recombinase substrate may be
attached to separate binding entities/interacting
molecules and the recombinase enzyme may be attached
to one of the nucleic acid tags. Upon binding of the
binding entities to their target/interaction between
the interacting molecules the two nucleic acid tags
plus the recombinase will be brought into close
proximity, thus enabling the recombination reaction to
take place. For the majority of applications it will,
however, be preferable to have the recombinase and
nucleic acid tags attached to separate components.
In a still further embodiment, recombination may
depend upon a transposition event and rely upon the
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 17 -
use of a transposase as the catalytic label. A
suitable example of such a system depends upon Tn5
transposase that recognizes Mosaic Ends recognition
sequences. However, it is not intended to limit the
invention to the use of this specific system, and
other transposition systems known in the art may be
used.
Systems which rely on the use of a transposase as
the catalytic label in order to generate a "detectable
change" by transposition of a nucleic acid sequence
between two nucleic acid tags making up the substrate
label may or may not require the presence of specific
sequences in both the nucleic acid tags in order to
allow transposition to take place. The requirements
for successful transposition with any particular
transposase enzyme/transposable element will generally
be appreciated by those skilled in the art.
A further preferred embodiment of a system
wherein the substrate label is composed of two nucleic
acid tags is based on the use of a lipase as the
catalytic label. The lipase is capable of ligating
together two separate nucleic acid tags, which
together make up the substrate label, in order to form
a detectable ligation product having novel sequence.
In this system it is possible to attach one of the
nucleic acids to a binding entity/interacting molecule
and add the second tag in free solution in an excess
amount.
The nucleic acid tags used in this embodiment may
be formed of double-stranded RNA, enabling ligation by
T4 DNA lipase. The tags may have complementary
"sticky" ends or blunt ends. In other embodiments,
the tags may be formed of single-stranded RNA, which
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 18 -
can be joined by T4 RNA ligase.
Nucleic acid tags having novel sequence, such as
may be generated by the action of recombinases,
transposases or ligases on "substrate" nucleic acid
tags may be detected using any suitable technique
known in the art.
Most preferably, detection of the novel sequence
will involve an amplification reaction, for example
PCR, NASBA, 3SR or any other amplification technique
known in the art. Amplification is achieved with the
use of amplification primers specific for the novel
sequence. In order to provide specificity for the
novel tag sequence primer binding sites corresponding
to a region of completely novel sequence may be
selected, or else a novel combination of primer
binding sites, not present in the original tags, may
be chosen.
The skilled reader will appreciate that the novel
sequence may also include sequences other than primer
binding sites which are required for detection of the
novel sequence, for example RNA Polymerase binding
sites or promoter sequences required for isothermal
amplification technologies, such as NASBA or 3SR.
In a preferred embodiment detection of the novel
sequence is carried out by amplification with "real-
time" detection of the products of the amplification
reaction. This can be achieved using any
amplification technique which allows for continuous
monitoring of the formation of the amplification
product.
A number of techniques for real-time detection of
the products of an amplification reaction are known in
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 19 -
the art. Many of these produce a fluorescent read-out
that can be continuously monitored, specific examples
being molecular beacons and fluorescent resonance
energy transfer probes. Real-time quantification of
PCR reactions can be accomplished using the TaqMan~
system (Applied Biosystems).
In a most preferred embodiment the method is
carried out in real-time, meaning that specific
binding of the binding entities to the target molecule
(or specific binding of interacting molecules), action
of the catalytic label on the nucleic acid substrate
label and detection of the product of the interaction
are carried out simultaneously in a single reaction
step. Real-time detection requires that the binding
step, action of the catalytic label on the substrate
label, and detection of the product of the interaction
can all be carried out under a single set of reaction
conditions, without the need for intermediate washing
~0 steps. In this embodiment real-time detection of the
novel sequence will preferably be carried out using an
isothermal amplification reaction, for example NASBA
or 3SR, in order to avoid changes of temperature which
might adversely affect the binding of the binding
entities to the target molecule/interaction between
interacting molecules.
In embodiments wherein the substrate label is
comprised of nucleic acid tags, the term "nucleic acid
tags" includes any natural nucleic acid and natural or
synthetic analogues that can be acted upon by a
catalytic label to generate novel sequence, for
example by recombination. Suitable nucleic acid tags
include tags composed of double or single-stranded
DNA, double or single-stranded RNA. Tags which are
partially double-stranded and partially single-
stranded are also contemplated. It is also
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 20 -
contemplated to use single-stranded tags in
combination with double-stranded tags, i.e. one
component labelled with a single-stranded tag and
another component labelled with a double-stranded tag
capable of interacting with the single-stranded tag.
If the catalytic label catalyses a recombination, then
the nucleic acid tags may be composed of any nucleic
acid which is capable of participating in the
recombination reaction, suitable examples including
linear or circular double-stranded DNA (dsDNA) or
double-stranded RNA (dsRNA) or mixtures thereof. Most
preferably the nucleic acid tags will comprise dsDNA.
The term "nucleic acid" encompasses synthetic
analogues which form a substrate for the catalytic
label in an analogous manner to natural nucleic acids,
for example nucleic acid analogues incorporating non-
natural or derivatized bases, or nucleic acid
analogues having a modified backbone. In particular,
the term "double-stranded DNA" or "dsDNA" is to be
interpreted as encompassing dsDNA containing non-
natural bases.
The precise sequence of the nucleic acid tags is
not material to the invention, except to the extent
that certain sequences may be required to enable the
"action" of the catalytic label on the nucleic acid
tags. For example, specific sequences are required to
permit site-specific recombination. Two nucleic acid
tags making up a "substrate label'° will most usually
be of different sequence, so that an interaction event
between the nucleic acid tags leads to production of
at least one novel sequence that can be detected.
However, it is not excluded to use tags of identical
sequence, provided that the tags are able to interact
to generate novel sequence. Most preferably, the
action of the catalytic label on the substrate label
will lead to the production of two separate nucleic
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- ~1 -
acid tags, each having novel sequence. This has the
advantage that the two tags of novel sequence form
independently verifiable products.
In a further embodiment of the invention, the
catalytic label may be formed of two or more component
parts, at least one of which is attached to a separate
binding entity/interacting molecule to the substrate
label. Examples of such catalytic labels may include,
for example enzyme/co-enzymes, multi subunit enzymes,
etc.
The substrate label and component parts of the
catalytic label may each be attached to separate
binding entities/interacting molecules, or the
component parts of the catalytic label may all be
attached to a single binding entity/interacting
molecule and the substrate label may be attached to a
separate binding entity/interacting molecule.
In a still further embodiment, the substrate
label and catalytic label may each. be formed of two or
more component parts, wherein component parts of the
substrate and catalytic labels are attached to
separate binding entities/interacting molecules. In
this embodiment all component parts of the substrate
label may be attached to a single binding
entity/interacting molecule or the component parts may
be attached to a number of separate binding
entities/interacting molecules. Similarly, all
component parts of the catalytic label may be attached
to a single binding entity/interacting molecule or the
component parts may be attached to a number of
separate binding entities/interacting molecules. The
only limitation is that (component parts of) the
substrate and catalytic labels must not be attached to
the same binding entity/interacting molecule.
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
-
It is generally preferred to start with the
catalytic label in an inactivated state and then
activate the catalytic label only after binding of the
binding entities to the target molecule (or
interaction between interacting molecules) has taken
place to position the catalytic and substrate labels
in close proximity. Activation of the catalytic label
may, for example, be achieved by changing the
composition, pH or temperature of the reaction medium.
This "external activation" step increases the
sensitivity of the method by minimising background
resulting from non-specific interactions between the
catalytic label and substrate label in free solution.
This may be important in applications wherein the
"proximity effect" which results from specific binding
of binding entities to their target molecule or
specific interactions between interacting molecules in
order to bring the catalytic and substrate labels into
close proximity does not by itself provide sufficient
signal-to-noise enhancement over background non-
specific interactions.
In a particular embodiment, step (b) of the
target detection method and the equivalent step (a) of
the interaction method may be carried out under
conditions which do not permit the catalytic label to
act on the substrate label. These "conditions" may
be, for example, the lack of a key component required
for the action of the catalytic label on the substrate
label. If the substrate label is formed of two
component parts, one of which is to be added in free
solution, then the "conditions" could be the absence
of this component. After binding of the binding
entities to the target molecule (or interaction
between interacting molecules) has taken place to
position the catalytic and substrate labels in close
proximity, the "conditions" may be changed to permit
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 23 -
the catalytic label to act on the substrate label.
This change could be, for example, addition of the
missing component of the substrate label under
conditions which permit subsequent reaction between
the catalytic label and components of the substrate
label. A "change" in reaction medium may easily be
achieved with the use of an intermediate washing step
or by simple addition of a missing reaction component
(both of which are in accordance with standard
principles of biological binding assays).
In embodiments wherein the substrate label is
formed of two (or more) separate nucleic acid tags
which are to be brought together in an interaction
requiring hybridisation between the two tags, it may
be convenient to carry out step (b) of the target
detection method and the equivalent step (a) of the
interaction method under conditions which do not allow
hybridisation between the tags and then to change the
reaction conditions to permit hybridisation.
In a preferred embodiment the catalytic label and
substrate label are directly attached to the binding
entities or interacting molecules. Direct linkage may
be achieved via a covalent linkage.
Techniques are known in the art for direct
covalent linkage of protein components to other
proteins, or to nucleic acids or carbohydrates, see
for example the techniques described in manuals such
as Bioconjugation; M Aslam and A Dent, eds. Macmillan
Reference Ztd 1998.
For example, amine-derivatized nucleic acid tags
may be coupled to protein binding entities/interacting
molecules using any one of a number of chemical cross-
linking compounds.
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 24 -
It is also within the scope of the invention for
the catalytic or substrate labels to be attached
indirectly to the binding entities/interacting
molecules. For example, "indirect" attachment may be
achieved through linker molecules. Suitable linker
molecules include components of biological binding
pairs which bind with high affinity, for example
biotin/streptavidin or biotin/avidin.
For most applications of the target detection
method, the catalytic and substrate labels will be
attached to the binding entities/interacting molecules
at the start of the reaction, at least before the
binding of the binding entities to the target
molecule. Most preferably, the binding entities will
be supplied pre-labelled with catalytic and substrate
labels, or else the labels will be attached in a
separate reagent labelling step. However, the
possibility of attaching catalytic and/or substrate
labels to the binding entities during the detection
reaction itself, i.e. following binding of the binding
entities to the target, is not excluded.
Preferred features of tarcLet detection method
The "target detection method" may be used to
detect essentially any target molecule for which it is
desired to develop a specific target detection method.
The target molecule may comprise a single
molecule, a multimer, aggregate or molecular
collection or complex. A multimer will generally
comprise a number of repeats of a single molecule
linked together through covalent or non-covalent
interactions. A complex will generally consist of
different molecules interacting through covalent or
non-covalent interactions.
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 25 -
In the context of this invention "binding
entities" are defined as any molecule that can bind
specifically to a target molecule. Binding entities
include, for example, antibodies, lectins, receptors,
transcription factors, cofactors and nucleic acids,
and fragments thereof which retain target-specific
binding activity (e.g. Fab fragments). This list is
merely illustrative and is not intended to be limiting
to the invention.
The binding entities may bind different regions
of a single target molecule. Thus, the catalytic and
substrate labels will be brought into close proximity
when the binding entities bind to their respective
regions of the target molecule.
If the target molecule is a multimer or
aggregate, then the binding entities may bind to
equivalent binding sites on the monomeric components
of the multimer or units making up the aggregate.
The "target detection method" of the invention
may be adapted for the detection of essentially any
"target molecule" for which suitable "binding
entities" of the required specificity are available.
The "sample" to be tested using the method may be
essentially any material which permits the specific
binding reactions that are essential to the operation
of the target detection method.
The "target detection method" is of use in all
areas of technology where it is desirable to provide
specific detection of target molecules, in particular
target biological molecules such as proteins, nucleic
acids, carbohydrates, etc. One important area of
application of the target detection method, though not
intended to be limiting, is in the field of clinical
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 26 -
diagnostics. Typically the "sample" will be a sample
of biological fluid, e.g. whole blood, serum, plasma,
urine etc, taken from a human patient. Other
important applications may include the field of
environmental testing and monitoring.
Preferred features of interaction method
The "interaction method" may be used in
essentially any field of technology where it is
desired to monitor interactions between molecules, and
particularly interactions between biological
molecules.
In a preferred embodiment, the interaction method
may be used in proteomics in order to investigate
molecular interactions. For example a first
interacting molecule may be labeled with either the
catalytic or the substrate label, and a library of
molecules which may potentially interact with the
first interacting molecule may then each be labeled
with the other label type. If an interaction occurs
between the first interacting molecule and a component
from the library of molecules, this brings the
catalytic and substrate labels into close proximity,
thus allowing interaction to generate a change in
structure and/or activity of the substrate label,
which can be detected in order to identify interacting
partners.
A further application is in the field of drug
discovery. For example, the interaction method may be
used to study interactions between particular
combinations of molecules and to identify potential
inhibitors or enhancers of molecular interactions.
Potential inhibitors of a given interaction could be
identified by screening for the ability to reduce the
signal detected following interaction of catalytic and
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
_ 27 _
substrate labels brought into close proximity by
interaction between the interacting molecules.
The "interacting molecules" may be essentially
any combination of interacting molecules which it is
desired to study. These may be, for example, subunits
of a multi-subunit complex, a pair of monomers,making
up a dimer, a ligand and receptor, an enzyme and
substrate or inhibitor, etc.
The "interaction method" differs from the target
detection method only in that the catalytic and
substrate labels are attached to the interacting
molecules which it is desired to evaluate, rather than
l5 to binding entities capable of binding to a target
molecule. The interaction method may therefore
incorporate analogous features to those described
above in connection with the target detection method,
as would be apparent to the skilled reader.
It is particularly preferred to carry out the
interaction method in real-time. The ability to
monitor molecular interactions in real-time provides
significant advantages, particularly in the field of
drug discovery.
ReacLent kits and reaaent labelling kits
The invention also relates to reagent kits
suitable for use in carrying out the target detection
method or the interaction method of the invention.
Reagent kits suitable for use in carrying out the
"target detection method" may comprise a first binding
entity labelled with a catalytic label and a second
binding entity labelled with a substrate label,
characterised in that the catalytic label is capable
of acting directly on the substrate label to generate
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
_ 28 _
a detectable change in the substrate label.
Reagent kits suitable for use in carrying out the
"interaction method" may comprise a first interacting
molecule labelled with a catalytic label and a second
interacting molecule labelled with a substrate label,
characterised in that the catalytic label is capable
of acting directly on the substrate label to generate
a detectable change in the substrate label.
The reagent kits may incorporate any of the
preferred features mentioned in connection with the
target detection and interaction methods. Preferred
combinations of catalytic and substrate labels are as
listed above in the description of the target
detection and interaction methods.
Reagent kits may further include supplies of
suitable reaction buffers) and also reagents required
for detection of the "detectable change" brought about
by action of the catalytic label on the substrate
label. For example, where the "change" in activity of
the substrate label is a change in enzymatic activity,
the kit may include assay reagents for use in
measuring this enzymatic activity. Where the "change"
is generation of a nucleic acid tag of novel sequence,
such as may be detected by nucleic acid amplification,
the kit may also include reagents required for the
amplification reaction, for example: primer sets,
amplification enzymes, probes for detection of the
amplification product (including probes labelled with
fluorescent or other revealing labels), positive
control amplification templates, reaction buffers etc.
The invention still further provides a reagent
labelling kit comprising a catalytic label and a
substrate label, characterised in that the catalytic
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 29 -
label is capable of acting directly on the substrate
label to generate a detectable change in the substrate
label, and means for attaching the catalytic label and
substrate label to interacting molecules or to binding
entities.
In one embodiment the means for attaching the
tags to interacting molecules or binding entities may
be a chemical reagent capable of cross-linking the
catalytic label and/or the substrate label to a
binding entity or interacting molecule.
In a further embodiment the "means for attaching
the tags" may be an indirect linkage. Preferred types
of indirect linkage are provided by components of a
biological binding pair, for example biotin/avidin or
biotin/streptavidin. In this embodiment the catalytic
label and/or substrate label is conjugated with one
half of the biological binding pair, enabling linkage
to a binding entity or interacting molecule conjugated
to the other half of the biological binding pair. The
kit may contain a supply of pre-conjugated catalytic
and substrate labels, or may include tags which have
not yet been conjugated together with means for
conjugating the catalytic or substrate labels with
half of the binding pair. The kit will further
include either binding entity or interacting molecule
pre-conjugated with the other half of the binding
pair, or else means for conjugating a binding entity
or interacting molecule of choice to the other half of
the binding pair.
The means for attaching half of the biological
binding pair to a binding entity or interacting
molecule may (depending on the nature of the binding
pair) be a chemical cross-linking reagent. However,
it may comprise an expression vector which can be used
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 30 -
to express the binding entity or interacting protein
as a fusion protein, either as a direct fusion with
the other half of the binding pair or as a fusion with
a polypeptide tag which enables attachment of the
other half of the binding pair. By way of example,
vectors for the expression of biotinylated fusion
proteins are known in the art and are commercially
available (for example the Pinpoint vector system from
Promega, Madison, V~1I, USA). These vectors allow
expression of proteins as fusions with a biotinylation
domain of the biotin carboxylase carrier protein. The
fusion proteins can be biotinylated in E. coli host
cells in an ATP-dependent enzymic reaction. Thus, the
reagent labelling kit may contain a supply of such a
vector, which enables expression of biotinylated
binding entities/interacting molecules proteins, plus
streptavidin conjugated catalytic and/or substrate
labels.
The invention will be further understood with
reference to the following non-limiting experimental
examples, together with the accompanying drawings, in
which:
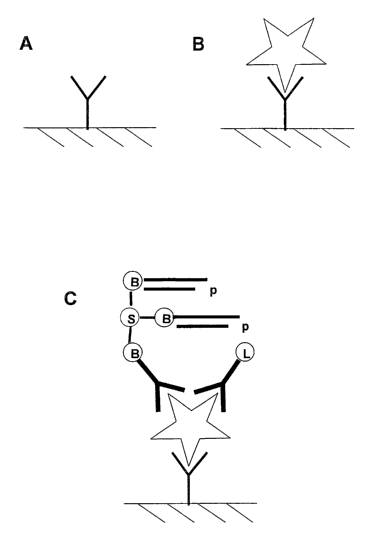
Figure 1 is a schematic illustration of a "target
detection" assay according to the invention.
Figure 2 is a schematic illustration of a further
"target detection" assay according to the invention.
Example 1 Demonstration of detectable DNA
modification by close proximity approach of a binding
entity modified with a DNA substrate and a bindinct
entitw modified with an enzyme
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 31 -
This experiment illustrates that DNA substrates on one
binding entity can be modified in a detectable manner
by an DNA-specific enzyme on another binding entity
under conditions such that the binding entitles are
brought into close proximity by binding to a target
molecule (see Figure 1). In this example a double
stranded piece of DNA with a single-stranded overhang
is linked to a polyclonal anti-hepatitis C core
antibody through a biotin-streptavidin bridge. Another
antibody with specificity for the core is labelled
with T4 DNA ligase. In the assay the target antigen,
Hepatitis C virus core protein, is first
immunocaptured to a solid phase surface and then
incubated with the labelled antibodies under
conditions which allow antibody binding but which do
not allow the enzyme on one antibody to modify the DNA
on the other. After binding of the antibody conjugates
and removal of excess conjugate by washing a DNA
oligomer with a complementary single-strand sequence
to that in the antibody/DNA conjugate is added in
buffer conditions that allow hybridization. If the two
antibodies labelled with the DNA and the ligase
respectively are in close proximity then the
hybridised DNA strands can be ligated to form a stable
double stranded DNA molecule which can then be
detected by nucleic acid amplification methods, such
as PCR.
a) Synthesis of the DNA-anti-hepatitis C core antibody
conjugate
1. Two overlapping oligonucleotides were
constructed:
32251; 5'CGGGCCTCTT GCGGGATATC GTCCATTCCG ACAGCATCGC
CAGTCACTAT GGCG3'
322AS1~ 5'ATAGTGACTG GCGATGCTGT CGGAATGGAC GATATCCCGC
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 32 _
AAGAGGCCCG3'
32251 was synthesized with a biotin at the 5'end to
allow conjugation to streptavidin whereas 322AS1 was
synthesized with a phosphate at the 5' end to allow
subsequent ligation.
2. To form double-stranded DNA with. a 4 by
single-stranded phosphate terminated sequence
extension the oligos were mixed at a 50pmo1/~1
concentration in lOmM Tris pH 8.0, 1mM EDTA, 150mM
NaCl and incubated at 37°C for 1 hour.
3. The double-stranded DNA product was complexed
with streptavidin to form a conjugate with a 1:1
streptavidin/DNA molar ratio. The reaction was
incubated at room temperature (22°C) for 1 hour to
allow the complex formation.
4. This double-stranded DNA-streptavidin complex was
further complexed to anti-hepatitis C core antibody by
adding biotinylated antibody (biotinylated according
to standard biotinylation protocols) at a 1:1
streptavidin/antibody molar ratio. The reaction was
incubated at room temperature (22°C) for 1 hour to
allow the final conjugate formation. In this example
the conjugate was then used in the assay procedure
without prior removal of any free streptavidin or
antibody.
b) Synthesis of the ligase-anti-hepatitis C core
antibody conjugate
1. T4 DNA ligase was obtained from Sigma (Poole, UK)
and linked to the anti-hepatitis C core antibody using
the heterobifunctional reagent SMCC (Pierce Co)
following the supplier's recommended standard chemical
conjugation procedures. A molar ration of 2:1 ligase
to antibody was used in the coupling procedure. In
this example the conjugate was used in the assay
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 33 -
procedure without prior removal of any free ligase or
antibody.
c) Synthesis of the double-stranded DNA used in step 6
below.
1. Two overlapping oligonucleotides were
constructed:
i. 32252; TGCTGCTAGC GCTATATGCG TTGATGCAAT
TTCTATGCGC ACCCGTTCTC GGAGCA3'
ii. 322AS2; 5'TGCTCCGAGA ACGGGTGCGC ATAGAAATTG
CATCAACGCA TATAGCGCTA GCAGCACGCC3'
32252 was synthesized with a phosphate at the 5' end
to allow subsequent ligation.
2. To form double-stranded DNA with a 4 by
single-stranded phosphate terminated sequence
extension the oligos were mixed at a 50pmol/~1
concentration in lOmM Tris pH 8.0, 1mM EDTA, 150mM
NaCl and incubated at 37°C for 1 hour.
d) Detection of HCV recombinant core antigen
1. Microwells in a 96 well microplate were coated
with anti-hepatitis C core antibody at 10 ug/m1 in 50
mM carbonate buffer pH 9.0 (see A in Figure 1).
2. Serial dilutions of recombinant core antigen were
prepared in PBS buffer containing 0.10 (v/v) Tween 20,
1mM EDTA. 100u1 of each dilution was incubated in an
antibody coated microplate well for 60 min to allow
capture (see B in Figure 1).
3. Wells were washed x3 with PBS buffer pH 7.5, O.lo
(v/v) Tween 20, 1mM EDTA.
4. 10 ng of each antibody conjugate in 1001 lOmM
sodium phosphate buffer, O.lo Tween 20, 100mM NaCl,
1mM EDTA pH 7.5 was added and incubated for 30 min to
allow the conjugates to bind to the captured antigen
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 34 -
(see C in Figure 1).
5. Wells were washed x3 in the same buffer and then
x2 in buffer without EDTA and Tween 20.
6. Then 100.1 lOmM sodium phosphate buffer pH 7.5,
100mM NaCl, 1mM ATP, lOmM MgCl2 pH 7.5 containing long
double-stranded DNA with a 4 by complementary overhang
to that of the conjugate DNA was added and incubated
60 min at 37°C to allow hybridization and ligation to
take place (see D and E in Figure 1).
7. After ligation, the generation of newly
covalently-linked double stranded DNA was investigated
using a 'hot-start' quantitative PCR on a Light Cycler
using standard conditions. The primers . 322PCRAS
5'ACGGGTGCGC ATAGAAATTG CATC3' and 322PCRS
5'GCGGGATATC GTCCATTCCG ACAG3' were used, which
amplify across the junction at which the two DNA
fragments have been ligated. The amount of DNA
amplified in the PCR and the time/cycle of PCR at
which the PCR becomes positive is related to the
number of ligated DNA fragments present in the
reaction, which in turn is related to the number of
antibody conjugates brought into close proximity and
is therefore a measure of the number of HCV core
antigen molecules captured.
e) Results
The time and number of cycles at which the PCR became
positive was related to the amount of antigen captured
in the well. As little as 0.1 fg of HCV core antigen
target could be detected by this method.
f) Discussion
This experiment shows that antibody conjugates
labelled with DNA substrate and a DNA-dependent enzyme
that modifies the DNA in a detectable manner can be
used to detect antigen in a sensitive and specific
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 35 -
manner.
Example 2-Demonstration of detectable DNA modification
by close proximity binding of a binding entity
modified with two DNA substrates and a binding entity
modified with an enzyme.
This experiment illustrates that two DNA substrates on
one binding entity can be modified in a detectable
manner by a DNA-specific enzyme on another binding
entity under such conditions such that the binding
entities are brought into close proximity by binding
to a target molecule (see Figure 2). In this example
two double stranded DNA oligomers with single-stranded
sequence extensions are linked to a target-specific
antibody (a polyclonal anti-hepatitis C core antibody)
through a biotin-streptavidin bridge. Another target
specific antibody is labelled with T4 DNA ligase. In
the assay the target antigen is immunocaptured to a
solid phase and then incubated with the labelled
antibodies under conditions which allow antibody
binding but do not allow the enzyme on one antibody to
modify the DNA on the other. After binding of the
antibody conjugates and removal of excess conjugate by
washing, the buffer conditions are then changed to
allow hybridisation. If the two antibody conjugates
(conjugated to the two DNA oligomers and the ligase
respectively) are in close proximity the two DNA
strands can be ligated to yield a stable double-
stranded DNA product which can then be detected by
nucleic acid amplification methods such as PCR.
a) Formation of the DNA-anti-hepatitis C core antibody
conjugate
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 36 -
1. Two overlapping oligonucleotides were
constructed:
32251; 5'CGGGCCTCTT GCGGGATATC GTCCATTCCG ACAGCATCGC
CAGTCACTAT GGCG3'
322AS1; 5'ATAGTGACTG GCGATGCTGT CGGAATGGAC GATATCCCGC
AAGAGGCCCG3'
32251 was synthesized with a biotin at the 5'end to
allow conjugation to streptavidin whereas 322AS1 was
synthesized with a phosphate at the 5' end to allow
subsequent ligation.
2. To form double-stranded DNA with a 4 by
single-stranded phosphate terminated sequence
extension the oligos were mixed at a 50pmo1/ul
concentration in lOmM Tris pH 8.0, 1mM EDTA, 150mM
NaCl and incubated at 37°C for 1 hour.
3. Two further overlapping oligonucleotides were
constructed:
32252; TGCTGCTAGC GCTATATGCG TTGATGCAAT TTCTATGCGC
ACCCGTTCTC GGAGCA3'
322AS2; 5'TGCTCCGAGA ACGGGTGCGC ATAGAAATTG CATCAACGCA
TATAGCGCTA GCAGCACGCC3'
32252 was synthesized with a phosphate at the 5' end
to allow subsequent ligation.
4. To form double-stranded DNA with a 4 by
single-stranded phosphate terminated sequence
extension the oligos were mixed at a 50pmo1/ul
concentration in lOmM Tris pH 8.0, 1mM EDTA, 150mM
NaCl and incubated at 37°C for 1 hour.
5. These double-stranded DNA products were linked to
streptavidin by mixing the two DNAs and the
streptavidin at equimolar concentration. The reaction
was incubated at room temperature for 1 hour to allow
the complex formation to take place.
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 37 -
6. This double-stranded DNA-streptavidin complex was
complexed to anti-hepatitis C core antibody by adding
biotinylated antibody (biotinylated according to
standard biotinylation protocols) at a 1:1 molar ratio
relative to the streptavidin. The reaction was
incubated at room temperature for 1 hour to allow the
complex formation to take place. In this example the
conjugate was used in the assay without removal of any
free streptavidin or antibody.
b) Formation of the ligase-anti-hepatitis C core
antibody conjugate
1. T4 DNA ligase was obtained from Sigma (Poole, UK)
and linked to the anti-hepatitis C core antibody using
the heterobifunctional reagent SMCC (Pierce Co)
following the supplier's recommended standard chemical
conjugation procedures. A molar ration of 2:1 ligase
to antibody was used in the coupling procedure. In
this example the conjugate was used in the assay
procedure without prior removal of any free ligase or
antibody.
c) Detection of HCV recombinant core antigen
1. Microwells in a 96 well microplate were coated
with anti-hepatitis C core antibody at 10 ~g/ml in 50
mM carbonate buffer pH 9.0 (see A in Figure 2). Serial
dilutions of recombinant core antigen were made in PBS
buffer pH 7.2, 0.1o (v/v) Tween 20, 1mM EDTA and 1001
of each dilution incubated in a coated well for 60 min
to allow capture (see B in Figure 2).
2. Wells were washed x3 with PBS buffer pH 7.2 0.10
(v/v) Tween 20, 1mM EDTA.
3. 10 ng of each antibody conjugate in 100u1 IOmM
sodium phosphate buffer, 0.1o Tween20, 100mM NaCl, 1mM
EDTA pH 7.5 was added and incubated for 30 min to
allow the antibody conjugates to bind to the antigen
CA 02487954 2004-11-29
WO 03/102590 PCT/GB03/02383
- 38 -
(see C in Figure 2).
4. Wells were washed x3 in the same buffer and then
x2 in buffer without EDTA and Tween 20.
5. Then 100u1 lOmM sodium phosphate, 100mM NaCl, 1mM
ATP, lOmM MgCl2pH 7.5 was added and incubated 60 min
at 37°C to allow hybridization, ligation and the
formation of covalently linked DNA fragments (see D in
Figure 2).
6. After ligation, the generation of
covalently-linked double stranded DNA was investigated
using a 'hot-start' quantitative PCR on a Zight Cycler
using standard conditions. The primers . 322PCRAS
5'ACGGGTGCGC ATAGAAATTG CATC3' and 322PCRS
5'GCGGGATATC GTCCATTCCG ACAG3' were used which amplify
across the junction at which the two DNA fragments
join. The amount of DNA amplified in the PCR and the
time/cycle of PCR at which the reaction becomes
positive is related to the number of ligated DNA
fragments present in the reaction, which in turn is
related to the number of antibody conjugates brought
into close proximity and is therefore a measure of the
number of HCV core antigen molecules captured.
d) Results
The time and number of cycles at which the PCR became
positive was related to the amount of antigen captured
in the well. As little as 0.2 fg of antigen target
could be detected by this method.
e) Discussion
This experiment shows that antibodies labelled,
respectively, with two DNA substrates and a
DNA-dependent enzyme that modifies the DNA in a
detectable process can be used to detect antigen in a
sensitive and specific manner.