Note: Descriptions are shown in the official language in which they were submitted.
CA 02488397 1997-07-25
CRYSTAL FORM II OF CLARITHROMYCIN
Technical Field
This invention relates to compounds having therapeutic utility and to methods
for
their preparation. More particularly, the present invention concerns the novel
compound 6-
O-methylerythromycin A crystal Form I and a process for its preparation and
uses of same.
Further, the present invention concerns the novel compound 6-O-
methylerythromycin A
crystal Form II, and compositions and uses thereof.
Ba~ound of the Invention
6-O-methylerythromycin A (Clarithromycin) is a semisynthetic macrolide
antibiotic of
formula
o ' ~'
15 t'i°~.. ~~~.,, E ....,o
0
G
20 6-O-methyl erythromycin A
which exhibits excellent antibacterial activity against gram-positive
bacteria, some gram-
negative bacteria, anaerobic bacteria, Mycoplasma, and Chlamidia. It is stable
under acidic
conditions and is efficacious when administered orally. Clarithromycin is a
useful therapy for
25 infections of the upper respiratory tract in children and adults.
Brief Description of the Drawings
FIGS. la, lb and lc show, respectively, the powder X ray diffraction spectrum,
the
infrared spectrum, and the differential scanning calorimetric (DSC) thermogram
of 6-O-
methylerythromycin A Form I.
CA 02488397 1997-07-25
-2-
FIGS. 2a, 2b and 2c show, respectively, the powder X ray diffraction spectrum,
the
infrared spectrum and the differential scanning calorimetric (DSC) thermogram
of 6-O-
methylerythromycin A Form II.
Summary of the Invention
We have discovered that 6-O-methylerythromycin A can exist in at least two
distinct
crystalline forms, which for the sake of identification are designated "Form
I" and "Form II".
The crystal forms are identified by their infrared spectrum, differential
scanning calorimetric
thermogram and powder x ray diffraction pattern. Form I and Form II crystals
have an
identical spectrum of antibacterial activity, but Form I crystals unexpectedly
have an intrinsic
1o rate of dissolution about three times that of Form II crystals.
Investigations in our laboratory
have revealed that 6-O-methylerythromycin A when recrystallized from ethanol,
tetrahydrofuran, isopropyl acetate, and isopropanol, or mixtures of ethanol,
tetrahydrofuran,
isopropyl acetate, or isopropanol with other common organic solvents results
in exclusive
formation of Form I crystals, not identified hitherto.
15 Drugs currently on the market are formulated from the thermodynamically
more
stable Form II crystals. Therefore, preparation of the current commercial
entity requires
converting the Form I crystals to Form II. Typically this is done by heating
the Form I
crystals under vacuum at temperature of greater than 80°C. Therefore,
the discovery of a
novel form of 6-O-methylerythromycin A which can be prepared without the high
20 temperature treatment results in substantial processing cost savings. In
addition, the
favorable dissolution characteristics of Form I relative to Form II increases
bioavailability of
the antibiotic and provides significant formulation advantages.
The present invention in one embodiment provides, in a process for the
production of
6-O-methylerythromycin A Form I by conversion of erythromycin A to 6-0-
25 methylerythromycin A with intermediate protection and deprotection
reactions and
subsequent crystallization or recrystallization of the 6-O-methylerythromycin
A to 6-O-
methylerythromycin A Form I, that step comprising crystallizing 6-O-
methylerythromycin A
Form I from a syrup or semisolid containing at least one residual solvent from
the
deprotection reaction.
30 In another embodiment, the aforementioned intermediate protection and
deprotection
reactions are carried out on an erythromycin A 9-oxime derivative.
CA 02488397 1997-07-25
-3-
In a further embodiment, in the aforementioned crystallizing step, at least
one solvent
additional to the residual solvent is utilized. Preferred additional solvents
are selected from
one or more or combinations of ethanol, isopropyl acetate, isopropanol,
tetrahydrofuran, a
hydrocarbon of from 5 to 12 carbon atoms, a ketone of from 3 to 12 carbon
atoms, a
carboxylic ester of from 3 to 12 carbon atoms, an ether of from 4 to 10 carbon
atoms,
benzene, benzene substituted with one or more substitutents selected from the
group
consisting of alkyl of from 1 to 4 carbon atoms, alkoxy of from 1 to 4 carbon
atoms, nitro,
and halogen, and a polar aprotic sovlent. A particularly preferred additional
solvent is
ethanol.
1o In a yet further embodiment, there is provided the use of such 6-O-
methylerythromycin A Form I as an antibiotic and also in the manufacture of an
antibiotic
medicament, as well as 6-O-methylerythromycin A Form I when prepared by the
process of
the present invention.
In another aspect of the invention, there is provided 6-O-methylerythromycin A
Form
~5 II produced from the aforesaid 6-O-methylerythromycin A Form I by heating
the latter.
In a further embodiment, there is provided an antibiotic pharmaceutical
composition
comprising the aforesaid 6-O-methylerythromycin A Fonm II together with a
pharmaceutically acceptable Garner. Preferred is a solid pharmaceutical dosage
form for oral
administration comprising such 6-O-methylerythromycin A Form II in combination
with one
20 or more pharmaceutically acceptable excipients, selected from one or more
of fillers, binders,
humectants, disintegrating agents, solution retarding agents, absorption
accelerators, wetting
agents, absorbents and lubricants.
In a further embodiment, there is provided the use of such 6-O-
methylerythromycin A
Form II as an antibiotic and also in the manufacture of an antibiotic
medicament.
25 Detailed Description
6-O-methylerythromycin A is prepared by methylation of the 6-hydroxy group of
erythromycin A. However, in addition to the 6 position, erythromycin A
contains hydroxy
groups at the 11, 12, 2' and 4" positions, and a nitrogen at 3' position, all
of which are
potentially reactive with alkylating agents. Therefore, it is necessary to
protect the various
3o reactive functionalities prior to alkylation of the 6-hydroxy group.
Representative 6-O-
' ' CA 02488397 1997-07-25
-4-
methylerythromycin A preparations are described in U.S. Pat. Nos. 4,331,803,
4,670,549,
4,672,109 and 4,990,602 and European Patent Specification 260 938 B 1.
Following final removal of the protecting groups, the 6-O-methylerythromycin A
may
exist as a solid, a semisolid, or a syrup containing residual solvents from
the deprotection
reactions, inorganic salts, and other impurities. 6-O-methylerythromycin A
Form I may be
crystallized directly from the syrup or semisolid using the solvents described
above.
Alternatively, if the crude reaction product solidifies, the solid may be
recrystallized from
any of the solvents described above. Pure 6-O-methylerythromycin A Form I may
also be
obtained by recrystallizing Form II or mixtures of Form I and Form II from any
of the solvent
systems described above. The term "6-O-methylerythromycin A" as used herein is
meant to
include 6-O-methylerythromycin A Form I or II in any state of purity, or
mixtures thereof.
The term "treating" refers to crystallizing or recrystallizing 6-O-
methylerythromycin A as defined above from any of the solvents described
above.
The term "hydrocarbon" as used herein refers to straight chain or branched
alkanes
15 having the formula C"H2"+2. HY~'ocarbons useful in the solvent mixtures of
the present
invention include hexane, heptane, octane and the like.
The term "alkyl" refers to a monovalent group derived from a straight or
branched
chain saturated hydrocarbon by the removal of a single hydrogen atom. Alkyl
groups are
exemplified by methyl, ethyl, n- and iso-propyl, n-, sec-, iso- and tert-
butyl, and the like.
2o The term "ketone" refers to a solvent of formula RC(O)R' where R and R' are
straight
or branched alkyl. Ketones useful in the solvent mixtures of the present
invention include
acetone, methyl ethyl ketone, 2- , and 3-pentanone, and the like.
The term "carboxylic ester" means a solvent of formula RCOZR' where R and R'
are
straight or branched alkyl. Carboxylic esters useful in the solvent mixtures
of the present
25 invention include methyl acetate, ethyl acetate, isobutyl acetate, and the
like.
The term "ether" means a solvent of formula ROR' where R and R' are straight
or
branched alkyl. Ethers useful in the solvent mixtures of the present invention
include ethyl
ether, diisopropyl ether, methyl tert-butyl ether, and the Like.
CA 02488397 1997-07-25
-5-
The term "polar aprotic" refers to solvents which do not contain hydroxy
groups but
have a relatively high dipole moment. Polar aprotic solvents useful in the
solvent mixtures of
the present invention include acetonitrile, N,N dimethylformamide (DMF),
dimethyl
sulfoxide (DMSO), 1,1- dimethoxyethane (DME), hexamethylphosphoric triamide
(HMPA),
and the like.
By "pharmaceutically acceptable salt" it is meant those salts which are,
within the
scope of sound medical judgment, suitable for use in contact with the tissues
of humans and
lower animals without undue toxicity, irritation, allergic response and the
like, and are
commensurate with a reasonable benefit/risk ratio. Pharmaceutically acceptable
salts are well
1o known in the art. For example, S. M Berge, et al. describe pharmaceutically
acceptable salts
in detail in J. Pharmaceutical Sciences, 1977, 66: 1-19 . The salts can be
prepared in situ
during the final isolation and purification of the compounds of the invention,
or separately by
reacting the free base function with a suitable organic acid. Representative
acid addition salts
include acetate, adipate, alginate, ascorbate, aspartate, benzenesulfonate,
benzoate, bisulfate,
15 borate, butyrate, camphorate, camphersulfonate, citrate,
cyclopentanepropionate, digluconate,
dodecylsulfate, ethanesulfonate, fumarate, glucoheptonate, glycerophosphate,
hemisulfate,
heptonate, hexanoate, hydrobromide, hydrochloride, hydroiodide, 2-hydroxy-
ethanesulfonate,
lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate,
methanesulfonate, 2-
naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palmitate,
pamoate, pectinate,
2o persulfate, 3-phenylpropionate, phosphate, picrate, pivalate, propionate,
stearate, succinate,
sulfate, tartrate, thiocyanate, toluenesulfonate, undecanoate, valerate salts,
and the like.
Representative alkali or alkaline earth metal salts include sodium, lithium,
potassium,
calcium, magnesium, and the like, as well as nontoxic ammonium, quaternary
ammonium,
and amine cations, including, but not limited to ammonium,
tetramethylammonium,
25 tetraethylammonium, methylamine, dimethylamine, trimethylamine,
triethylamine,
ethylamine, and the like.
CA 02488397 1997-07-25
-6-
6-O-methylerythromycin A is prepared from erythromycin A by a variety of
synthetic
routes. In one method, erythromycin A is converted to 2'-O-3'-N
bis(benzyloxycarbonyl)-N
8z0
O a..l ~ OSz
"~,..~.....,
I-lo_ o _ Ocl~
O
O 4-
O
I
demethylerythromycin A (I).
The 6-hydroxy group is then methylated by reaction with an alkylating agent
such as
bromomethane or iodomethane and a base. Removal of the benzoyl groups by
catalytic
t 5 hydrogenation and reductive methylation of the 3' N gives 6-O-
methylerythromycin A. See
U.S. Pat. No. 4,331,803.
An alternative synthetic route involves methylation of 6-O-methylerythromycin
A-9-
oxime. 6-O-methylerythromycin A-9-oxime is prepared by methods well known in
the art
such as reaction of erythromycin A with hydroxylamine hydrochloride in the
presence of
2o base, or by reaction with hydroxylamine in the presence of acid as
described in US Pat. No.
5,274,085. Reaction of the oxime with RX wherein R is allyl or benzyl and X is
halogen
results in formation of 2'-0,3'-N-diallyl or dibenzylerythromycin A-9-O-allyl
or benzyloxime
halide. Methylation of this quaternary salt as described above, followed by
elimination of the
R groups and deoximation gives 6-O-methylerythromycin A. See U.S. Pat. No.
4,670,549.
25 Methylation of 6-O-methylerythromycin A oxime derivatives of formula II,
OH ~ 2'
_,~ _
HO~,,, ~~.~,,
HO~
O
_ O 4~
' ' CA 02488397 1997-07-25
-7-
wherein R is alkyl, alkenyl, substituted or unsubstituted benzyl, oxyalkyl, or
substituted
phenylthioalkyl, RZ is benzoyl, and R3 is methyl or benzoyl, followed by
deprotection,
deoximation, and reductive methylation when R3 is benzoyl gives 6-O-
methylerythromycin
A. See U.S. Pat. No. 4,672,109.
A particularly useful preparation of 6-O-methylerythromycin A involves
methylation
of the
N"'
R ON~
ia.
OR3
.. O
oxime derivative III, wherein R' is alkenyl, substituted or unsubstituted
benzyl, or
alkoxyalkyl, RZ is substituted silyl, and R3 is RZ or H. Removal of the
protecting groups and
deoximation is then accomplished in a single step by treatment with acid to
give 6-O-
methylerythromycin A. See European Patent Specification 260 938 B1 and U.S.
Pat. No.
4,990,602.
A preferred route to 6-O-methylerythromycin A is outlined in Scheme 1.
Erythromycin A, prepared by fermentation of Streptomyces erythreus is oximated
to give
oxime 4 wherein R' is alkoxyalkyl. The group R' may be introduced by reaction
of
erythromycin A with the substituted hydroxylamine R~ONH2, or by reaction of
erythromycin
A with hydroxylamine hydrochloride in the presence of base, or hydroxylamine
in the
presence of acid, followed by reaction with R~X. The two hydroxy groups are
then protected
simultaneously, in which RZ or R3 are the same, or sequentially in which RZ
and R3 are
different. Particularly useful protecting groups are substituted silyl groups
such as
trimethylsilyl, tert-butyldimethylsilyl, tert-butyldiphenylsilyl and the like.
The protecting
groups are then removed and the compound is deoximated to produce 6-O-
methylerythromycin A. The order of deprotection/deoximation is not critical.
When the
3o protecting groups are substituted silyl, deprotection and deoximation can
be accomplished in
a single step by treatment with acid, for example using formic acid or sodium
hydrogen
sulfite. See U.S. Pat. No. 4,990,602.
~ ' CA 02488397 1997-07-25
. -$- _
Scheme 1
O \ ~ R' O \ N''
OH 2' OH 2'
9 ..' 9 ..'
O ~ O
6 ~ HO 6 O
... ''~. ....a
~' Oximation
r.... r.... _
O ~H3 ~_ O ' OCH3
OH O OH
O O 4. O O 4.
Erythromycin A I V
2 \
RIO OH R~ 2' N'
9 ,° O
O
Protection r~._ Methylation
HO~ ~ OCH3
I -O
O ORa
O
O
V
R2 \ ~ O \ N''
R'O 1 2, 9 .,OH 2'
9 ,.OCH3 O O ~... HO O
deprotection
r....
O - OCH3 .... 'I ~O -."
'r O
~OR3 O 4.
O O' \ 4 O
V I 6-0-methylerythromycin A
In accordance with the process aspect of the present invention, 6-O-
methylerythromycin A prepared by any of the methods described above is
suspended in the
desired solvent and heated to about the reflux temperature of the solvent.
Heating is then
continued and the suspension is stirred for an amount of time sufficient to
dissolve most of
the solid, generally about 10 minutes to 2 hours. The suspension is then
filtered hot. If
3o necessary, the filtrate may be heated to at or about the reflux temperature
of the solvent to
form a clear solution. The filtrate is then slowly cooled to ambient
temperature with optional
further cooling in an ice water bath. For purposes of this specification,
ambient temperature is
from about 20 °C to about 25 °C. Crystalline 6-O-
methylerythromycin A is then isolated,
preferably by filtration, and the wet solid is converted to 6-O-
methylerythromycin A Form I
CA 02488397 1997-07-25
-9-
by drying in a vacuum oven at a temperature of between ambient temperature and
about 70
°C, preferably from about 40 to about 50 °C and a pressure of
between about 2 inches of
mercury and atmospheric pressure to remove any remaining solvent.
In accordance with the aspects of this invention wherein 6-O-
methylerythromycin A
is recrystallized from solvent mixtures, 6-O-methylerythromycin A is suspended
in the first
solvent and heated to about the reflux temperature of the solvent. Heating is
then continued
and the suspension is stirred for an amount of time sufficient to dissolve
most of the solid,
generally about 10 minutes to 2 hours. The suspension is then filtered hot.
The filtrate may be
heated to reflux to form a clear solution if necessary. A second solvent is
then added to the
1o hot filtrate and the mixture is cooled slowly to ambient temperature with
optional further
cooling in an ice bath. Representative second solvents include, but are not
limited to, hexane,
heptane, octane, acetone, methyl ethyl ketone, 2-, and 3-pentanone, methyl
acetate, ethyl
acetate, isobutyl acetate, ethyl ether, diisopropyl ether, methyl tert-butyl
ether, acetonitrile,
N,N dimethylfonmamide, dimethyl sulfoxide, l,l-dimethoxyethane,
hexamethylphosphoric
triamide, benzene, toluene, and chlorobenzene. Hydrocarbons of from 5 to 12
carbon atoms
are preferred second solvents. The most preferred second solvent is heptane.
After cooling, 6-
O-methylerythromycin A crystal Form I is isolated by filtration and drying as
described
above. The amount of second solvent added is dependent on the solubility of
the drug in the
first solvent and the second solvent, and can be readily determined by one of
ordinary skill in
2o the art. Typical ratios fall in the range of about 1:10 to about 2:1 parts
by volume of second
solvent. A preferred ratio of first solvent to second solvent is 1:1 parts by
volume.
Preferred solvents for the isolation of 6-O-methylerythromycin A Form I are
ethanol,
isopropyl acetate, tetrahydrofuran, and isopropanol.
The most preferred solvent for the isolation of 6-O-methylerythromycin A Fonm
I is
ethanol.
Pharmaceutical Compositions
The present invention also provides pharmaceutical compositions which comprise
6-
O-methylerythromycin A Fonm I formulated together with one or more non-toxic
pharmaceutically acceptable Garners. The pharmaceutical compositions may be
specially
3o formulated for oral administration in solid or liquid form, for parenteral
injection, or for rectal
administration.
CA 02488397 1997-07-25
-10-
The pharmaceutical compositions of this invention can be administered to
humans
and other animals orally, rectally, parenterally, intracisternally,
intravaginally,
intraperitoneally, topically (as by powders, ointments, or drops), bucally, or
as an oral or
nasal spray. The term "parenteral" administration as used herein refers to
modes of
administration which include intravenous, intramuscular, intraperitoneal,
intrasternal,
subcutaneous and intraarticular injection and infusion.
Pharmaceutical compositions of this invention for parenteral injection
comprise
pharmaceutically acceptable sterile aqueous or nonaqueous solutions,
dispersions,
suspensions or emulsions as well as sterile powders for reconstitution into
sterile injectable
1o solutions or dispersions just prior to use. Examples of suitable aqueous
and nonaqueous
carriers, diluents, solvents or vehicles include water, ethanol, polyols (such
as glycerol,
propylene glycol, polyethylene glycol, and the like), and suitable mixtures
thereof, vegetable
oils (such as olive oil), and injectable organic esters such as ethyl oleate.
Proper fluidity can
be maintained, for example, by the use of coating materials such as lecithin,
by the
15 maintenance of the required particle size in the case of dispersions, and
by the use of
surfactants.
These compositions may also contain adjuvants such as preservatives, wetting
agents,
emulsifying agents, and dispersing agents. Prevention of the action of
microorganisms may
be ensured by the inclusion of various antibacterial and antifungal agents,
for example,
2o paraben, chlorobutanol, phenol sorbic acid, and the like. It may also be
desirable to include
isotonic agents such as sugars, sodium chloride, and the like. Prolonged
absorption of the
injectable pharmaceutical form may be brought about by the inclusion of agents
which delay
absorption such as aluminum monostearate and gelatin.
In some cases, in order to prolong the effect of the drug, it is desirable to
slow the
25 absorption of the drug from subcutaneous or intramuscular injection. This
may be
accomplished by the use of a liquid suspension of crystalline or amorphous
material with
poor water solubility. The rate of absorption of the drug then depends upon
its rate of
dissolution which, in turn, may depend upon crystal size and crystalline form.
Alternatively,
delayed absorption of a parenterally administered drug form is accomplished by
dissolving or
3o suspending the drug in an oil vehicle.
Injectable depot forms are made by forming microencapsule matrices of the drug
in
biodegradable polymers such as polylactide-polyglycolide. Depending upon the
ratio of drug
CA 02488397 1997-07-25
-11-
to polymer and the nature of the particular polymer employed, the rate of drug
release can be
controlled. Examples of other biodegradable polymers include poly(orthoesters)
and
poly(anhydrides). Depot injectable formulations are also prepared by
entrapping the drug in
liposomes or microemulsions which are compatible with body tissues.
The injectable formulations can be sterilized, for example, by filtration
through a
bacterial retaining filter, or by incorporating sterilizing agents in the form
of sterile solid
compositions which can be dissolved or dispersed in sterile water or other
sterile injectable
medium just prior to use.
Solid dosage forms for oral administration include capsules, tablets, pills,
powders,
1o and granules. In such solid dosage forms, the active compound is mixed with
at least one
inert, pharmaceutically acceptable excipient or carrier such as sodium citrate
or dicalcium
phosphate and/or
(a) fillers or extenders such as starches, lactose, sucrose, glucose,
mannitol, and
silicic acid,
15 (b) binders such as, for example, carboxymethylcellulose, alginates,
gelatin,
polyvinylpyrrolidone, sucrose, and acacia,
(c) humectants such as glycerol,
(d) disintegrating agents such as agar-agar, calcium carbonate, potato or
tapioca
starch, alginic acid, certain silicates, and sodium carbonate,
2o (e) solution retarding agents such as paraffin,
(fj absorption accelerators such as quaternary ammonium
compounds,
(g) wetting agents such as, for example, cetyl alcohol
and glycerol monostearate,
(h) absorbents such as kaolin and bentonite clay, and
(i) lubricants such as talc, calcium stearate, magnesium
stearate, solid
25 polyethylene glycols, sodium lauryl sulfate, and mixtures
thereof.
In the case of capsules, tablets and pills, the dosage form may also comprise
buffering agents.
CA 02488397 1997-07-25
-12-
Solid compositions of a similar type may also be employed as fillers in soft
and hard-
filled gelatin capsules using such excipients as lactose or milk sugar as well
as high
molecular weight polyethylene glycols and the like.
The solid dosage forms of tablets, dragees, capsules, pills, and ganules can
be
prepared with coatings and shells such as enteric coatings and other coatings
well known in
the pharmaceutical formulating art. They may optionally contain opacifying
agents and can
also be of a composition that they release the active ingredients) only, or
preferentially, in a
certain part of the intestinal tract, optionally, in a delayed manner.
Examples of embedding
compositions which can be used include polymeric substances and waxes.
The active compounds can also be in micro-encapsulated form, if appropriate,
with
one or more of the above-mentioned excipients.
Liquid dosage forms for oral administration include pharmaceutically
acceptable
emulsions, solutions, suspensions, syrups and elixirs. In addition to the
active compounds, the
liquid dosage forms may contain inert diluents commonly used in the art such
as, for
~ 5 example, water or other solvents, solubilizing agents and emulsifiers such
as ethyl alcohol,
isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl
benzoate, propylene
glycol, 1,3-butylene glycol, dimethyl formamide, oils (in particular,
cottonseed, groundnut,
corn, germ, olive, castor, and sesame oils), glycerol, tetrahydrofurfuryl
alcohol, polyethylene
glycols and fatty acid esters of sorbitan, and mixtures thereof.
2o Besides inert diluents, the oral compositions can also include adjuvants
such as
wetting agents, emulsifying and suspending agents, sweetening, flavoring, and
perfuming
agents.
Suspensions, in addition to the active compounds, may contain suspending
agents as,
for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and
sorbitan esters,
25 microcrystalline cellulose, aluminum metahydroxide, bentonite, agar agar,
and tragacanth,
and mixtures thereof.
Compositions for rectal or vaginal administration are preferably suppositories
which
can be prepared by mixing the compounds of this invention with suitable non-
irritating
excipients or Garners such as cocoa butter, polyethylene glycol or a
suppository wax which
CA 02488397 1997-07-25
-13-
are solid at room temperature but liquid at body temperature and therefore
melt in the rectum
or vaginal cavity and release the active compound.
Compounds of the present invention can also be administered in the form of
liposomes. As is known in the art, liposomes are generally derived from
phospholipids or
other lipid substances. Liposomes are formed by mono- or multi-lamellar
hydrated liquid
crystals that are dispersed in an aqueous medium. Any non-toxic,
physiologically acceptable
and metabolizable lipid capable of forming liposomes can be used. The present
compositions
in liposome form can contain, in addition to a compound of the present
invention, stabilizers,
preservatives, excipients, and the like. The preferred lipids are the
phospholipids and the
phosphatidyl cholines (lecithins), both natural and synthetic.
Methods to form liposomes are known in the art. See, for example, Prescott,
Ed.,
Methods in Cell Biolo~y, Volume XIV, Academic Press, New York, N.Y. (1976), p.
33 et
seq.
Dosage forms for topical administration of a compound of this invention
include
powders, sprays, ointments and inhalants. The active compound is mixed under
sterile
conditions with a pharmaceutically acceptable carrier and any needed
preservatives, buffers,
or propellants which may be required. Opthalmic formulations, eye ointments,
powders and
solutions are also contemplated as being within the scope of this invention.
Actual dosage levels of active ingredients in the pharmaceutical compositions
of this
invention may be varied so as to obtain an amount of the active compounds)
that is effective
to achieve the desired therapeutic response for a particular patient,
composition, and mode of
administration. The selected dosage level will depend upon the activity of the
particular
compound, the route of administration, the severity of the condition being
treated, and the
condition and prior medical history of the patient being treated. However, it
is within the skill
of the art to start doses of the compound at levels lower than required to
achieve the desired
therapeutic effect and to gradually increase the dosage until the desired
effect is achieved.
Generally dosage levels of about 1 to about 1000, more preferably of about 5
to about
200 mg of 6-O-methylerythromycin A Form I per kilogram of body weight per day
are
administered to a mammalian patient. If desired, the effective daily dose may
be divided into
3o multiple doses for purposes of administration, e.g. two to four separate
doses per day.
' ' CA 02488397 1997-07-25
-14-
The following Examples are provided to enable one skilled in the art to
practice the
invention and are merely illustrative of the invention. They should not be
read as limiting the
scope of the invention as defined in the claims.
Example 1
Preparation of 6-O-methyler omycin Form I
6-O-methylerythromycin A was prepared from erythromycin A by oximation of the
C-9 carbonyl, protection of the C-2' and C-4" hydroxy groups, methylation of
the C-6
hydroxy group, deoximation and removal of the protecting groups, and
recrystallization from
ethanol according to the method of U.S. Pat No. 4,990,602. The material
obtained from the
1o recrystallization was dried in a vacuum oven (40-45 °C, 4-8 in. Hg)
to give 6-O-
methylerythromycin A Form I.
6-O-methylerythromycin A Form I is characterized by its infrared spectrum, the
differential scanning calorimetric (DSC) thermogram and the powder x-ray
diffraction
pattern. The differential scanning calorimetric thermogram is obtained by
methods known in
15 the art and is illustrated in Figure lc. In Figure lc, an exothermic
transition at 132.2 °C can be
seen, which is believed to be due to a phase transition. An endothermic peak
at 223.4 °C,
which may be due to melting, can also be seen. Another endothermic peak at
283.3 °C
followed by an exothermic peak at 306.9 °C may be due to decomposition.
After the DSC
scan the color of the sample was black.
2o The powder x-ray diffraction pattern is obtained by the methods known in
the art.
Figure 1 a illustrates the powder x-ray diffraction pattern. The 2-theta angle
positions in the
powder x- ray diffraction pattern of 6-O-methylerythromycin A Form I are
5.16°~0.2,
6.68°t0.2, 10.20°t0.2, 12.28°t0.2, 14.20°~0.2,
15.40°~0.2. 15.72°t0.2, and 16.36°t0.2.
Example 2
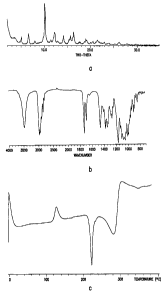
25 Conversion of 6-O-methylerythromycin Form I Crystals to Form II Crystals
6-O-methylerythromycin A Form I crystals (0.40 g), prepared as in Example 1,
were
placed in a vial and heated in the vacuum oven (4-9 in Hg, 100-110 °C)
for 18 hours to give
6-0- methylerythromycin A Form II crystals. 6-O-methylerythromycin A Form II
melts at
223.4 °C. In the differential scanning calorimetric thermogram of 6-O-
methylerythromycin A
3o Form II there can be seen an endothermic peak at 283.3 °C which may
be due to
CA 02488397 1997-07-25
-15-
decomposition. After the DSC scan the color of the sample was black. The 2-
theta angle
positions in the powder x-ray diffraction pattern of 6-O-methylerythromycin A
Form II are
8.52°t0.2, 9.48°~0.2, 10.84°t0.2, 11.48°t0.2,
11.88°t10.2, 12.36°t0.2, 13.72°10.2,
14.12°t0.2, 15.16°t0.2. 16.48°~0.2, 16.92°~0.2,
17.32°~0.2, 18.08°~0.2, 18.40°~0.2,
19.04°t0.2, 19.88°t0.2, and 20.48°t0.2.
Example 3
Isolation of 6-O-methyler hromycin Form I by RecrYstallization
Recrystallization from Tetrahydrofuran
A mixture of 6-O-methyleryyhromycin A (20 g), prepared as described in Example
1,
io in tetrahydrofuran (100 mL) was warmed to reflux and stirred for 15
minutes. The hot
solution was filtered to remove traces of insoluble material and cooled to
ambient
temperature. No crystallization occurred so 10 g of 6-O-methylerythromycin A
was added to
the solution and the suspension was again heated to reflux, hot filtered, and
cooled in an ice
bath. The resulting solid was collected by filtration and dried in the vacuum
oven (40-45 °C,
4-8 in. Hg) to give 6-O- methylerythromycin A Form I (16.74 g).
Recrystallization from Isopropyl Alcohol
A mixture of 6-O-methylerythromycin A (15 g), prepared as described in Example
1,
and isopropyl alcohol (100 mL) was warmed to reflux and heated for 20 minutes.
The hot
solution was filtered to remove traces of insoluble material. The filtrate was
transferred to
another flask along with a 50 mL isopropanol rinse, and the solution was again
heated to
reflux. The clear solution was then cooled slowly to ambient temperature and
left standing for
seven hours. The resulting solid was collected by filtration and dried in the
vacuum oven (40-
45 °C, 4-8 in. Hg) to give 6-O-methylerythromycin A Form I (13.3g).
Recrystallization from Isopropyl Acetate
A mixture of 6-O-methylerythromycin A (10 g), prepared as described in Example
1,
and isopropyl acetate (100 mL) was warmed to 73 °C. The hot solution
was filtered to
remove traces of insoluble material. The clear solution was then cooled slowly
to ambient
temperature. The resulting solid was collected by filtration and dried in the
vacuum oven (40-
45 °C, 4-8 in. Hg) to give 6-O-methylerythromycin A Form I (3.6 g).
CA 02488397 1997-07-25
-16-
Recrystallization from Isopropyl Acetate-Heptane
A mixture of 6-O-methylerythromycin A (10 g), prepared as described in Example
1,
and isopropyl acetate (100 mL) was warmed to reflux. A small amount of
insoluble material
was removed by filtration and the filtrate was transferred to another vessel.
The filter flask
was rinsed with isopropyl acetate (5 mL) and the filtrate and rinse were
combined and heated
to reflux. To the resulting clear solution was added heptane (100 mL) and the
clear solution
was cooled to ambient temperature over 1.5 hours during which time a
precipitate formed.
The solid was collected by filtration and dried overnight in the vacuum oven
(45-50 °C, 4-8
in. Hg) to give 6-O-methylerythromycin A Form I (7.0 g).
1o Recrystallization from Isopropyl Acetate-N,N dimethylformamide
A mixture of 6-O-methylerythromycin A (12 g), prepared as described in Example
1,
and isopropyl acetate (100 mL) was warmed to reflux. A small amount of
insoluble material
was removed by filtration and the filtrate was transferred to another vessel.
The filtrate was
heated to reflux and N,N dimethylformamide (30 mL) was added. The clear
solution was
15 cooled to ambient temperature over 1.5 hours during which time a
precipitate formed. The
solid was collected by filtration and dried overnight in the vacuum oven (49-
50 °C, 4-8 in.
Hg) to give 6-O-methylerythromycin A Form I (6.4 g).
Recrystallization from Tetrahydrofuran-Heptane
To a clear solution of 6-O-methylerythromycin A (10 g), prepared as described
in
2o Example 1, in tetrahydrofuran (75 mL) was added heptane (150 mL). The
resulting cloudy
solution was heated to 71.5 °C at which point it turned clear. The
mixture was cooled to
ambient temperature over 2 hours, and then was cooled in an ice-water bath for
0.5 hours.
The resulting solid was filtered and dried in the vacuum oven (45-50
°C, 3-4 in. Hg) for four
days to give 6-O- methylerythromycin A Form I (0.50 g).
25 Recrystallization from Ethanol-Heptane
A mixture of 6-O-methylerythromycin A (10 g), prepared as described in Example
1,
and ethanol (100 mL) was warmed to reflux. A small amount of insoluble
material was
removed by filtration and the filtrate was transferred to another vessel. The
filter flask was
rinsed with ethanol (20 mL) and the filtrate and rinse were combined and
heated at 78 °C
3o until a clear solution was obtained. To the clear solution was added
heptane (100 mL) and the
CA 02488397 1997-07-25
-17-
clear solution was cooled slowly to ambient temperature and stirred far four
days. The
resulting solid was collected by filtration and dried in the vacuum oven (45-
SO °C, 4-8 in. Hg)
to give 6-O-methylerythromycin A Form I (4.5 g).
Recrystallization from lsopropanol-Heptane
A mixture of 6-O-methylerythromycin A (4.0 g), prepared as described in
Example 1,
and isopropanol (50 mL) was warmed to reflux. Heptane (50 mL) was added and
the solution
was cooled slowly to ambient temperature and then was cooled in an ice-water
bath. The
resulting solids were collected by filtration and dried in the vacuum oven (4-
8 in. Hg) to give
6-O-methylerythromycin A Form I (3.6 g).
1o Example 4
Dissolution Rates of 6-O-methylerythromycin Forms Land II
Dissolution studies were carried out at 60 rpm in 300 mL of 0.05 M phosphate
buffer
at 37°C using a constant surface area (13/32" diameter) drug compact.
Aliquots were
removed periodically and assayed directly by HPLC (Scm x 4.6mm 3~, ODS 2
"Little
15 Champ" (Regis) column; 50:50 acetonitrile-0.05 M pH 4.0 phosphate buffer
mobile phase;
1.0 mL/min flow rate). As shown in Table 1, 6-O-methylerythromycin A Form I
has an
intrinsic rate of dissolution about three times greater than Form II.
Table 1
Intrinsic Dissolution Rates of 6-O-methylerythromycin A forms I and II
Crystal Form Dissolution Rate ~ S.D.
(ICg/min/cm2)
I 636 ~ 2.5
II 203 t 14
The foregoing examples are presented for purposes of illustration and are not
intended
to limit the scope of the invention. Variations and changes which are obvious
to one skilled in
the art are intended to be within the scope and nature of the invention as
defined in the
appended claims.