Note: Descriptions are shown in the official language in which they were submitted.
CA 02489452 2008-07-02'
30725-332
-1-
PHENYLAMINOPYRIMIDINES AND THEIR USE AS RHO-KINASE INHIBITORS
The invention relates to phenylaminopyrimidines, to a process for their
preparation
and to their use for preparing pharmaceuticals for the treatment and/or
prophylaxis of
diseases in humans and animals, in particular cardiovascular diseases.
An increase in the intracellular calcium concentration is one of the main
factors
triggering the contraction of the vascular musculature (Somlyo, A.P. and
Himpens,
B., FASEB J. 1989, 3, 2266-2276). This is effected primarily by agonists, such
as, for
example, phenylephrine or thromboxane A2 which, after stimulation of the
phosphatidylinositol cascade, cause the release of calcium from the
sarcoplasmatic
reticulum. The elevated intracellular calcium activates the MLC kinase (myosin
light-chain kinase) which phosphorylates the MLC subunits of the myosin
molecule
(Kamm, K.H. and Stull, J.T., Annu. Rev. Pharmacol. Toxicol. 1985, 25, 593-
603).
MLC phosphorylation induces the contraction of smooth muscles, MLC
dephosphorylation after reduction of the intracellular calcium concentration
results in
the relaxation of the vessel.
t
In addition to the calcium-dependent MLC phosphorylation, there is a further
central,
but calcium-independent regulation mechanism of the vascular tone. This is the
Rho/Rho kinase signal path (Noda, -M. et al., FEBS Lett. 1995, 367, 246-250;
Uehata,
M. et al., Nature 1997, 389, 990-994; Fukata, Y. et al., Trends in
Pharmacological
Sciences 2001, 22, 32-39). The binding of agonists such as, for example,
phenylephrine or thromboxane A2 to their receptors results in the activation
of the
small G-proteins Rho which then interact with and activate Rho kinase. The
activated
Rho kinase inhibits myosin phosphatase following phosphorylation of a subunit
of
the enzyme. At the same time, Rho kinase phosphorylates MLC at the position
which
is also phosphorylated by MLC kinase. Inhibition of myosin phosphatase and
phosphorylation of MLC induces the vascular musculature to contract. In
contrast,
inhibition of Rho kinase leads to a relaxation of the vessels. Accordingly,
inhibitors
of Rho kinase lower the blood pressure and increase coronary perfusion.
CA 02489452 2011-03-11
30725-332
-2-
Compounds of a similar structure are known for other indications or other
mechanisms of action. Thus, for example, US 3 478 030 and US 3 432 493
describe
substituted aminopyrimidines capable of increasing coronary perfusion but
acting as
carboanhydrase inhibitors (J. Chem. Inf. Comp. Sciences 2002, 42, 94-102).
Other
pyrimidine derivatives have been described as anti-cancer and anti-HIV agents
(Debi, M.; Indian J. Exp. Biol. 1997, 35, 1208-1213) or as cdk2 inhibitors
(WO-A 01/64654).
It is an object of the present invention to provide pharmaceuticals for
treating
disorders, in particular cardiovascular disorders.
This object is achieved by the compounds of the formula (I), which act as Rho
kinase
inhibitors.
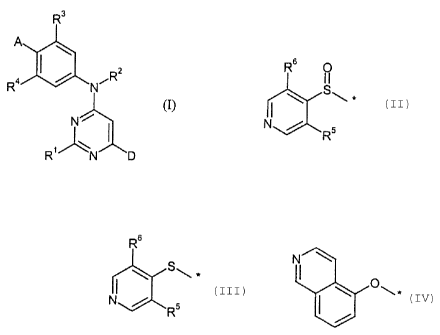
The present invention provides a compound of the formula (I)
R3
2
A 6N
R4 CR
(I),
N
/\ I
R' N D
in which
R' represents amino or hydroxyl,
R2 represents hydrogen, (Ci-C6)-alkyl or (C3-Cg)-cycloalkyl,
R3 and R4 independently of one another represent cyano, hydrogen, fluorine or
chlorine,
Le A 36 136 CA 02489452 2004-12-14
-3-
A represents a radical
R6 O R6
NI
~~ Ste, ~~ Ste.
N or Rs N R5
in which
R5 and R6 independently of one another represent hydrogen, fluorine or
chlorine,
D (1) represents a radical selected from the group consisting of
phenyl, which for its part is substituted by (CI-C4)-alkyl-
carbonylamino, hydroxymethyl, cyano, (C I -C4)-alkoxymethyl or 1,2-
dioxymethylene,
quinoline, isoquinoline, indole or 6-membered heteroaryl having 2 or
3 nitrogen atoms, where the rings are in each case attached via a
carbon atom,
pyridylmethyl, 2-oxo-2H-pyridin-1-yl, 4-oxo-4H-pyridin-l-yl, which
for their part may be substituted by fluorine, chlorine or (CI-C4)-alkyl,
and
pyridyl, which for its part is substituted by fluorine, chlorine or
(CI-C4)-alkyl,
or
(2) represents a radical *-OR7,
Le A 36 136 CA 02489452 2004-12-14
-4-
in which
R7 represents phenyl which may be substituted by
trifluoromethyl, trifluoromethoxy, nitro, cyano, *-NR 8R9,
fluorine, chlorine or 1,2-dioxymethylene or by (Ci-C4)-alkyl
or (C1-C4)-alkoxy, which for their part may be substituted by
hydroxyl and/or *-NR8R9,
3- to 7-membered heterocyclyl having a nitrogen atom which
may be substituted by hydrogen, (C1-C4)-alkyl or (C3-C8)-
cycloalkyl,
5- or 6-membered heteroaryl having up to three nitrogen
atoms,
(C1-C6)-alkyl or (C3-C7)-cycloalkyl which for their part may
be substituted by hydroxyl or *-NR8R9,
thienyl, furyl, pyridylmethyl, naphthyl or benzyl,
in which
R8 and R9 independently of one another represent hydrogen or (C1-
C4)-alkyl which for its part may be substituted by hydroxyl or
amino, or
R8 and R9 together with the nitrogen atom to which they are attached
form a 5- to 7-membered heterocycle which may have an
additional oxygen atom or a group N-H or N-(C1-C4)-alkyl in
the ring,
or
Le A 36 136 CA 02489452 2004-12-14
-5-
(3) represents a radical *-NR1OR11,
in which
R10 represents hydrogen or (C1-C4)-alkyl and
R11 represents amino-substituted (C3-C8)-cycloalkyl or a radical
*-(CH2)x-phenyl, where phenyl may be substituted up to four
times independently of one another by fluorine, chlorine or
(C1-C4)-alkyl, or represents *-(CH2)Y E,
in which
x represents 1, 2 or 3,
y represents 0, 1, 2 or 3 and
E represents pyrrolidine or piperidine, which for their part may
be substituted by (Ci-C4)-alkyl, or represents pyridyl which
may be substituted up to four times independently of one
another by fluorine, chlorine or (C1-C4)-alkyl,
or
R10 and R11 together with the nitrogen atom to which they are attached
form a 5- or 6-membered heterocycle which is substituted by
*-NR 12R13, 1,1-dioxyethylene, (C 1-C4)-alkoxy, hydroxyl- or
(C i -C4)-alkoxy-substituted (C 1-C4)-alkyl, (C 1-C4)-
alkoxycarbonyl or 5- or 6-membered heterocyclyl having one
or two heteroatoms N and/or 0, which for its part may be
substituted by (C i -C4)-alkyl,
in which
Le A 36 136 CA 02489452 2004-12-14
-6-
R12 and R13 independently of one another represent hydrogen, (C1-
C6)-alkyl, (C1-C4)-alkoxycarbonyl, (C3-Cg)-cycloalkyl or (C1-
C4)-alkanoyl or
R12 and R13 together with the nitrogen atom to which they are attached
form a 5- or 6-membered heterocycle,
or
R10 and R11 together with the nitrogen atom to which they are attached
form a 7- to 12-membered bicyclic heterocycle which is fused
or spirocyclic and may have one or two further heteroatoms
from the group consisting of N and 0 in the ring and which
may be substituted by (C1-C4)-alkyl, (C1-C4)-alkoxycarbonyl,
(C1-C4)-alkanoyl or benzyl,
or
R10 and R11 together with the nitrogen atom to which they are attached
form a radical
Ris
*
T--~ 14 or *
_ N \--/ N-R -N SO 2
in which
R14 represents (C2-C6)-alkenyl, (C1-C4)-alkoxycarbonyl or
*-(CH2)Z-G,
in which
z represents 0 or I and
= CA 02489452 2011-03-11
30725-332
-7-
G represents (C3-C8)-cycloalkyl, pyridyl, optionally
(C1-C4)-alkyl- or (Ci-C4)-alkoxyl-substituted phenyl,
tetrahydrofuran or 1,3-dioxolane,
and
R15. represents hydrogen or (C1-C4)-alkyl,
or a salt, hydrate, hydrate of the salt or solvate thereof.
The application also provides processes for preparing a compound as described
herein,
compositions comprising a compound as described herein, oral uses of a
compound as
described herein for the preparation of pharmaceuticals, and uses of the
medicaments as
described herein.
Depending on their structure, the compounds according to the invention can
exist in
stereoisomeric forms (enantiomers, diastereomers). Accordingly, the invention
relates to the enantiomers or diastereomers and to their respective mixtures.
The
stereoisomerically uniform components can be isolated in a known manner from
such mixtures of enantiomers and/or diastereomers.
Depending on the structure of the compounds, the invention also relates to
tautomers
of the compounds.
In the context of the invention, preferred salts are physiologically
acceptable salts of
the compounds according to the invention.
CA 02489452 2011-03-11
30725-332
-7a-
Physiologically acceptable salts of the, compounds (I) include acid addition
salts of
mineral acids, carboxylic acids and sulphonc acids, for example salts of
hydrochloric acid, hydrobromic acid, sulphuric acid, phosphoric acid,
methanesulphonic acid, ethanesulphonic acid, toluenesulphonic acid,
benzenesulphonic acid, naphthalenedisulphonic acid, acetic acid, propionic
acid,
lactic acid, tartaric acid, malic acid, citric acid, fumaric acid, maleic acid
and benzoic
acid.
Physiologically acceptable salts of the compounds (I) also include salts of
customary
bases, such as, by way of example and by way of preference, alkali metal salts
(for
example sodium salts and potassium salts), alkaline earth metal salts (for
example
Le A 36 136 CA 02489452 2004-12-14
-8-
calcium salts and magnesium salts) and ammonium salts, derived from ammonia or
organic amines having 1 to 16 carbon atoms, such as, by way of example and by
way
of preference, ethylamine, driethylamine, triethylamine,
ethyldiisopropylamine,
monoethanolamine, diethanolamine, triethanolamine, dicyclohexylamine,
dimethylaminoethanol, procaine, dibenzylamine, N-methylmorpholine,
dihydroabietylamine, arginine, lysine, ethylenediamine and methylpiperi dine.
In the context of the invention, solvates are those forms of the compounds
which, in
solid or liquid state, form a complex by coordination with solvent molecules.
Hydrates are a specific form of solvates where the coordination is with water.
In the context of the present invention, the substituents are as defined
below, unless
specified otherwise:
Alkyl per se and "alk" and "alkyl" in alkoxy, alkanoyl, alkylcarbonylamino,
alkoxycarbonyl and alkoxymethyl represent a linear or branched alkyl radical
having
generally 1 to 6, preferably 1 to 4, particularly preferably 1 to 3, carbon
atoms, by
way of example and by way of preference methyl, ethyl, n-propyl, isopropyl,
tert-
butyl, n-pentyl and n-hexyl.
By way of example and by way of preference, alkoxy represents methoxy, ethoxy,
n-propoxy, isopropoxy, tert-butoxy, n-pentoxy and n-hexoxy.
By way of example and by way of preference, alkanoyl represents acetyl and
propanoyl.
By way of example and by way of preference, alkoxycarbonyl represents
methoxycarbonyl, ethoxycarbonyl, n-propoxycarbonyl, isopropoxycarbonyl, tert-
butoxycarbonyl, n-pentoxycarbonyl and n-hexoxycarbonyl.
By way of example and by way of preference, alkoxycarbonylamino represents
methoxycarbonylamino, ethoxycarbonylamin, n-propoxycarbonylamino, isopropoxy-
Le A 36 136 CA 02489452 2004-12-14
-9-
carbonylamino, tert-butoxycarbonylamino, n-pentoxycarbonylamino and n-hexoxy-
carbonylamino.
Alkenyl represents a linear or branched alkenyl radical having generally 1 to
6
carbon atoms. Preference is given to a straight-chain or branched alkenyl
radical
having 2 to 4, particularly preferably 2 or 3, carbon atoms. The following
radicals
may be mentioned by way of example and by way of preference, vinyl, allyl, n-
prop-
1-en- l -yl and n-but-2-en- l -yl.
Cycloalkyl represents a cycloalkyl group having generally 3 to 8, preferably 5
to 7,
carbon atoms, by way of example and by way of preference cyclopropyl,
cyclobutyl,
cyclopentyl, cyclohexyl and cycloheptyl.
Heteroarvl represents an aromatic mono- or bicyclic radical having generally 5
to 10,
preferably 5 or 6, ring atoms and up to 5, preferably up to 4, heteroatoms
from the
group consisting of S, 0 and N, by way of example and by way of preference
thienyl,
furyl, pyrrolyl, thiazolyl, oxazolyl, imidazolyl, pyridyl, pyrimidyl,
pyridazinyl,
indolyl, indazolyl, benzofuranyl, benzothiophenyl, quinolinyl, isoquinolinyl.
Heterocyclyl represents a mono- or polycyclic, preferably mono- or bicyclic,
non-
aromatic heterocyclic radical having generally 4 to 12, preferably 5 to 8,
ring atoms
and up to 3, preferably up to 2, heteroatoms and/or hetero groups from the
group
consisting of N, 0, S, SO, SO2. The heterocyclyl radicals can be saturated or
partially
unsaturated. Preference is given to 5- to 8-membered monocyclic saturated
heterocyclyl radicals having up to two heteroatoms from the group consisting
of 0,
N and S, such as, by way of example and by way of preference, tetrahydrofuran-
2-yl,
pyrrolidin-2-yl, pyrrolidin-3-yl, pyrrolinyl, piperidinyl, morpholinyl,
perhydroazepinyl.
A symbol * at a bond denotes the point of attachment in the molecule.
If radicals in the compounds according to the invention are substituted, the
radicals
can be mono- or polysubstituted by identical or different substituents unless
Le A 36 136 CA 02489452 2004-12-14
-10-
otherwise specified. A substitution by up to three identical or different
substituents is
preferred. Very particular preference is given to substitution with one
substituent.
Preference is given to compounds of the formula (I)
in which
R' represents amino,
RZ represents hydrogen,
R3 and R4 independently of one another represent hydrogen, fluorine or
chlorine,
A represents a radical
Rs
N
~ Ste.
or I Off'
N
R s
in which
R5 and R6 represent hydrogen,
D (1) represents a radical selected from the group consisting of
phenyl which is substituted by (C1-C4)-alkylcarbonylamino,
hydroxymethyl, (C1-C4)-alkoxymethyl or 1,2-dioxymethylene,
quinoline, indole or 6-membered heteroaryl having 2 or 3 nitrogen
atoms, where the rings are in each case attached via a carbon atom,
pyridylmethyl, which may be substituted by (C1-C4)-alkyl,
Le A 36 136 CA 02489452 2004-12-14
-11-
and
pyridyl, which is substituted by (C1-C4)-alkyl,
or
(2) represents a radical *-OR7,
in which
R7 represents phenyl, which may be substituted by fluorine,
chlorine, (C1-C4)-alkyl, (C I -C4)-alkoxy or 1,2-
dioxymethylene,
(Ci-C6)-alkyl or (C3-C8)-cycloalkyl, which for their part may
be substituted by hydroxyl or *-NR8R9,
naphthyl or benzyl,
in which
R8 and R9 independently of one another represent hydrogen or (C1-
C4)-alkyl or
R8 and R9 together with the nitrogen atom to which they are attached
form a 5- to 7-membered heterocycle which may have an
additional oxygen atom or a group N-H or N-(Ci-C4)-alkyl in
the ring,
or
(3) represents a radical *-NR1OR",
Le A 36 136 CA 02489452 2004-12-14
-12-
in which
R10 represents hydrogen or (C1-C4)-alkyl and
R" represents amino-substituted (C3-C8)-cycloalkyl or a radical
*-(CH2)X-phenyl, where phenyl may be substituted up to four
times independently of one another by fluorine, chlorine or
(C1-C4)-alkyl, or represents *-(CH2)y E,
in which
x represents 1 or 2,
y represents 0, 1 or 2 and
E represents pyrrolidine or piperidine, which for their
part may be substituted by (C1-C4)-alkyl, or represents
pyridyl which may be substituted up to four times
independently of one another by fluorine, chlorine or
(C 1-C4)-alkyl,
or
R10 and R11 together with the nitrogen atom to which they are
attached form a 5- or 6-membered heterocycle which is
substituted by *-NR '2R13, 1,1-dioxymethylene, (C1-
C4)-alkoxymethyl or by 5- or 6-membered heterocyclyl
having one or two heteroatoms N and/or 0, which for
its part may be substituted by (C1-C4)-alkyl,
in which
Le A. 36 136 CA 02489452 2004-12-14
-13-
R12 and R13 independently of one another represent hydrogen,
(CI-C6)alkyl, (C3-Cg)-cycloalkyl or (CI-C4)-alkanoyl or
R12 and R13 together with the nitrogen atom to which they are
attached form a 5- or 6-membered heterocycle,
or
R10 and RI1 together with the nitrogen atom to which they are
attached form an 8- to 10-membered bicyclic
heterocycle which is fused or spirocyclic and may have
one or two further heteroatoms from the group
consisting of N and 0 in the ring and which may be
substituted by (CI-C4)-alkyl, (CI-C4)-alkoxycarbonyl,
alkanoyl or benzyl,
or
R10 and R" together with the nitrogen atom to which they are
attached form a radical
R15
is
-N N-R
in which
R14 represents (C3-C8)-cycloalkyl, (C2-C6)-alkenyl, (CI-
C4)-alkoxycarbonyl or tetrahydrofuran-2-ylmethyl,
and
R15 represents hydrogen or (CI-C4)-alkyl,
Le A 36 136 CA 02489452 2004-12-14
-14-
and their salts, hydrates, hydrates of the salts and solvates.
Particular preference is given to compounds of the formula (I)
in which
R' represents amino,
R2 represents hydrogen,
R3 and R4 independently of one another represent hydrogen, fluorine or
chlorine,
A represents a radical
R
N
or \\.
N RS
in which
R5 and R6 represent hydrogen,
D (1) represents a radical which is selected from the group consisting of
quinoline, indole, pyrazine, pyridazine and triazine, where the rings
are in each case attached via a carbon atom,
or
(2) represents a radical *-OR7
Le A 36 136 CA 02489452 2004-12-14
-15-
in which
R7 represents phenyl which may be substituted by fluorine,
chlorine, (C1-C4)-alkyl, (C 1 -C4)-alkoxy or 1,2-
dioxymethylene,
(C1-C6)-alkyl or (C3-C8)-cycloalkyl which for their part may be
substituted by hydroxyl or *-NR8R9,
in which
R8 and R9 independently of one another represent hydrogen or
(C1-C4)-alkyl or
R8 and R9 together with the nitrogen atom to which they are attached
form a morpholine or piperazine ring, where the second
nitrogen atom of the piperazine ring may be substituted by
(C 1-C4)-alkyl,
or
(3) represents a radical *-NR10R11,
in which
R10 represents hydrogen or (C1-C4)-alkyl and
R11 represents amino-substituted (C3-C8)-cycloalkyl or a radical
*-(CHZ)y-E,
in which
y represents 0 or 1 and
Le A 36 136 CA 02489452 2004-12-14
-16-
E represents pyrrolidine or pyridyl, which for their part may be
substituted by (C 1-C4)-alkyl,
or
R10 and R11 together with the nitrogen atom to which they are attached
form a pyrrolidine or piperidine ring which may be substituted
by *-NR12R13, 1, 1 -dioxym ethylene, (C1-C4)-alkoxymethyl or
5- or 6-membered heterocyclyl having one or two heteroatoms
N and/or 0, which for its part may be substituted by (C1-C4)-
alkyl,
in which
R12 and R13 independently of one another represent hydrogen, (C1-C6)-
alkyl, (C3-C8)-cycloalkyl or (C1-C4)-alkanoyl or
R12 and R13 together with the nitrogen atom to which they are attached
form a 5- or 6-membered heterocycle,
or
R10 and R11 together with the nitrogen atom to which they are attached
form an 8- to 10-membered bicyclic heterocycle which is
fused or spirocyclic and may have one or two further
heteroatoms from the group consisting of N and 0 and which
may be substituted by (C1-C4)-alkyl, (C1-C4)-alkoxycarbonyl,
(C 1-C4)-alkanoyl or benzyl,
and their salts, hydrates, hydrates of the salts and solvates.
Very particular preference is given to combinations of two or more of the
preferred
ranges mentioned above.
Le A 36 136 CA 02489452 2004-12-14
-17-
The present invention also provides a process for preparing the compounds of
the
formula (I), which process is characterized in that either
[A] compounds of the formula (II)
R3
A
2
R4 N,R
N R' N CI
in which
A, R', R2, R3 and R4 are as defined above,
are reacted with compounds of the formula (III)
D-X' (III)
in which
D is as defined above and
x' represents hydrogen or *-B(OH)2
or
[B] compounds of the formula (IV)
Le A 36 136 CA 02489452 2004-12-14
-18-
CI
N---
I tom,
H2N N D
in which
D is as defined above
are reacted with compounds of the formula (V)
R3
A ~
i 2
R4 / NCR
H
in which
A, R2, R3 and R4 are as defined above
to give compounds of the formula (I).
If X1 represents hydrogen, the reaction in process step [A] is carried out in
inert
solvents or neat, if appropriate in the presence of a base, preferably in a
temperature
range of from 20 C to the reflux of the solvents or in the melt at atmospheric
pressure.
Inert solvents are, for example, alcohols, such as methanol, ethanol,
propanol,
isopropanol or butanol, N-alkylated carboxamides, such as dimethylformamide or
dimethylacetamide, alkyl sulphoxides, such as dimethyl sulphoxide, or other
solvents, such as acetonitrile or pyridine, preferably ethanol or
dimethylformamide.
Le A 36 136 CA 02489452 2004-12-14
-19-
Bases are, for example, alkali metal hydroxides, such as sodium hydroxide or
potassium hydroxide, or alkali metal carbonates, such as caesium carbonate,
sodium
carbonate or potassium carbonate, or amides, such as lithium diisopropylamide,
or
other bases, such as DBU, triethylamine or diisopropylethylamine, preferably
diisopropylethylamine or triethylamine.
If X' represents *-B(OH)2, the conversion into compounds of the formula (I) in
process step [A] is generally carried out in inert solvents in the presence of
a
transition metal catalyst and in the presence of a base, preferably in the
temperature
range of from 70 C to 110 C at atmospheric pressure.
Inert solvents are, for example, ethers, such as dioxane, tetrahydrofuran or
1,2-
dimethoxyethane, hydrocarbons, such as benzene, xylene or toluene, nitrated
aromatic compounds, such as nitrobenzene, optionally N-alkylated carboxamides,
such as dimethylformamide and dimethylacetamide, alkyl sulphoxides, such as
dimethyl sulphoxide, or cyclic lactams, such as N-methylpyrrolidone. The
solvents
are, if appropriate, used with addition of ethanol. Preference is given to
solvents from
the group consisting of dimethylformamide, 1,2-dimethoxyethane and
toluene/ethanol.
Preferred transition metal catalysts are palladium(0) or palladium(II)
compounds, in
particular bis(diphenylphosphaneferrocenyl)palladium(II) chloride,
dichlorobis(tri-
phenylphosphine)palladium or tetrakis(triphenylphosphine)palladium(0).
Preferred bases are potassium tert-butoxide, or alkali metal hydroxides or
salts, such
as potassium acetate, sodium hydroxide, sodium bicarbonate, sodium carbonate
or
potassium carbonate, if appropriate in the form of their aqueous solutions.
In process step [B], the conversion into compounds of the formula (I) is
carried out
in concentrated hydrochloric acid, preferably in a temperature range of from
70 C to
110 C at atmospheric pressure. In this reaction, the amino group at the
pyrimidine
may be hydrolyzed to the hydroxyl group.
Le A 36 136 CA 02489452 2004-12-14
-20-
To prepare compounds of the formula (II) for process step [A], compounds of
the
formula (V) are reacted with the compound of the formula (VI)
Cl
N
H2N N CI
under the reaction conditions described for process step [B].
In this reaction, the amino group at the pyrimidine may be hydrolyzed to the
corresponding hydroxyl group.
To prepare compounds of the formula (IV) for process step [B], compounds of
the
formula (VII)
OH
N
(VII),
H2N N D
in which
D is as defined above
are reacted with phosphoryl chloride in N,N-dimethylaniline, preferably in a
temperature range of from 70 C to 110 C at atmospheric pressure.
In another process variant, to prepare the compounds of the formula (IV),
compounds of the formula (VI) are reacted with compounds of the formula (III)
under the reaction conditions described for process step [A].
To prepare compounds of the formula (VII), compounds of the formula (VIII)
Le A 36 136 CA 02489452 2004-12-14
-21-
O O
s
X'-~ O "kA D (VIII),
in which
D is as defined above and
X2 represents alkyl, preferably methyl or ethyl,
are reacted with the compound of the formula (IX)
NH
H 2 N1NH 2 (IX).
The reaction of the compounds of the formula (VIII) and (IX) is initially
carried out
using concentrated hydrochloric acid in ethanol, preferably in a temperature
range of
from 50 C to the reflux of the solvents at atmospheric pressure, and then with
aqueous sodium hydroxide solution, preferably in a temperature range of from
50 C
to the reflux of the solvents at atmospheric pressure.
To prepare compounds of the formula (Va) for process step [B] in which R2
represents (C,-C6)-alkyl or (C3-C8)-cycloalkyl, compounds of the formula (Vb)
in
which R2 represents hydrogen
are reacted with compounds of the formula (X)
R2-X4 (X)
in which
Le A 36 136 CA 02489452 2004-12-14
-22-
R2 represents (Ci-C6)-alkyl or (C3-C8)-cycloalkyl and
X4 represents halogen, preferably bromine or chlorine.
The reaction is generally carried out in inert solvents, if appropriate in the
presence
of a base, preferably in a temperature range of from room temperature to the
reflux
of the solvents at atmospheric pressure.
Inert solvents are, for example, halogenated hydrocarbons, such as methylene
chloride, trichloromethane or 1,2-dichloroethane, ethers, such as dioxane,
tetrahydrofuran or 1,2-dimethoxyethane, or other solvents, such as acetone,
dimethylformamide, dimethylacetamide, 2-butanone or acetonitrile, preferably
tetrahydrofuran, methylene chloride, acetone 2-butanone, acetonitrile,
dimethylformamide or 1,2-dimethoxyethane.
Bases are, for example, alkali metal carbonates, such as caesium carbonate,
sodium
carbonate or potassium carbonate, or sodium methoxide or potassium methoxide,
or
sodium ethoxide or potassium ethoxide, or potassium tert-butoxide, or amides,
such
as sodium amide, lithium bis(trimethylsilyl)amide or lithium diisopropylamide,
or
organometallic compounds, such as butyllithium or phenyllithium, or other
bases,
such as sodium hydride or DBU, preferably potassium tert-butoxide, caesium
carbonate, DBU, sodium hydride, potassium carbonate or sodium carbonate.
To prepare the compounds of the formula (Vb) for process step [B] in which R2
represents hydrogen, compounds of the formul;a (XI)
R3
A
(XI),
R / N+'O
I-
0
in which
Le A 36 136 CA 02489452 2004-12-14
-23-
A, R3 and R4 are as defined above,
are reacted with reducing agents.
The reaction is generally carried out in inert solvents, if appropriate in the
presence
of hydrazine hydrate, preferably in a temperature range of from room
temperature to
the reflux of the solvents at from atmospheric pressure to 3 bar.
Reducing agents are for example, palladium on carbon and hydrogen, platinum
oxide
on carbon and hydrogen, tin dichloride or titanium trichloride, preferably
palladium
on carbon and hydrogen in the presence of hydrazine hydrate or platinum oxide
on
carbon and hydrogen.
Inert solvents are, for example, ethers, such as diethyl ether, methyl tert-
butyl ether,
1,2-dimethoxyethane, dioxane, tetrahydrofuran, glycol dimethyl ether or
diethylene
glycol dimethyl ether, alcohols, such as methanol, ethanol, n-propanol,
isopropanol,
n-butanol, tert-butanol or 2-ethylhexanol, hydrocarbons, such as benzene,
xylene,
toluene, hexane, cyclohexane or mineral oil fractions, or other solvents, such
as
dimethylformamide, dimethylacetamide, acetonitrile or pyridine; preferred
solvents
are ethanol, n-butanol or 2-ethylhexanol.
To prepare the compounds of the formula (XI), compounds of the formula (XII)
R3
XS
R4 N+=.O
O (XI),
in which
R3 and R4 are as defined above and
Le A 36 136 CA 02489452 2004-12-14
-24-
X5 represents halogen, preferably fluorine or chlorine,
are reacted with compounds of the formula (XIII)
A-H (XIII)
in which
A is as defined above.
The reaction is generally carried out in inert solvents, if appropriate in the
presence
of a base, preferably in a temperature range of from room temperature to the
reflux
of the solvents at atmospheric pressure.
Inert solvents are, for example, halogenated hydrocarbons, such as methylene
chloride, trichloromethane or 1,2-dichloroethane, ethers, such as dioxane,
tetrahydrofuran or 1,2-dimethoxyethane, or other solvents, such as acetone,
dimethylformamide, dimethylacetamide, 2-butanone or acetonitrile, preferably
acetonitrile, dimethylformamide or 1,2-dimethoxyethane.
Bases are, for example, alkali metal carbonates, such as caesium carbonate,
sodium
carbonate or potassium carbonate, or sodium methoxide or potassium methoxide,
or
sodium ethoxide or potassium ethoxide, or potassium tert-butoxide, or amides,
such
as sodium amide, lithium bis(trimethylsilyl)amide or lithium diisopropylamide,
or
organometallic compounds, such as butyllithium or phenyllithium, or other
bases,
such as sodium hydride or DBU, preferably potassium tert-butoxide, caesium
carbonate, potassium carbonate or sodium carbonate.
The compounds of the formulae (III), (VI), (VIII), (IX), (X), (XII) and (XIII)
are
known per se to the person skilled in the art or can be prepared by customary
processes known from the literature.
Le A 36 136 CA 02489452 2004-12-14
-25-
The compounds of the formula (I) can be derivatized further, for example by
reaction
with oxidizing agents.
The preparation -of the compounds according to the invention can be
illustrated by
the reaction schemes below.
[A] 3
3
CI K2CO3, DMF A
A
R N02 R4
NO2
(XII) (X111) (XI)
3 3
hydrazine hydrate A R 4 A
ethanol, Pd/C 2
R4 NH2 (X) R4 NR
H
(Vb) (Va)
Cl 3
A \
H zN N C l R'4 D -'
(I)
(VI)/ (III)
HP N Cl
(II)
Le A 36 136 CA 02489452 2004-12-14
-26-
CBI
O O
Et0 D OH
(VIII)
N
+
NH H2N N D
(VII) POCI3
CI
H 2 N NH2
(IX) N
H z N--"- ~'N D
CI
(IV)
N + D-X' 3
H2N N CI A
(VI) (III) R4 NH2
(V)
(I)
The compounds according to the invention have an unforeseeable useful spectrum
of
pharmacological and pharmacokinetic action. Accordingly, they are suitable for
use
as pharmaceuticals for the treatment and/or prophylaxis of diseases in humans
and
animals.
The pharmaceutic activity of the compounds of the formula (I) according to the
invention can be explained by their action as Rho kinase inhibitors.
Owing to their pharmacological properties, the compounds of the formula (I)
according to the invention can be used on their own or in combination with
other
active compounds for the treatment and/or prevention of disorders, in
particular
cardiovascular disorders.
Le A 36 136 CA 02489452 2004-12-14
-27-
The compounds of the formula (I) are suitable for the prophylaxis and/or
treatment of
cardiovascular disorders such as, for example, hypertension and cardiac
insufficiency, stable and unstable angina pectoris, peripheral and
cardiovascular
disorders, of arrhythmias, of thromboembolic disorders and ischaemias, such as
myocardial infarction, stroke, transitory and ischaemic attacks, obstruction
of
peripheral circulation, prevention of restenoses, such as, for example, after
thrombolysis therapies, percutaneous transluminal angioplasties (PTA),
percutaneous
transluminal coronary angioplasties (PTCA), bypass, and for the prophylaxis
and/or
treatment of arteriosclerosis, asthmatic disorders and diseases of the
urogenital
system, such as, for example, prostate hypertrophy, erectile dysfunction,
female
sexual dysfunction, osteoporosis, gastroparesis and incontinence.
The present invention also relates to the use of the compounds of the formula
(I) for
preparing pharmaceuticals for the prophylaxis and/or treatment of the
syndromes
mentioned above.
The present invention furthermore relates to a method for the prophylaxis
and/or
treatment of the syndromes mentioned above using the compounds of the formula
(I).
The present invention furthermore provides pharmaceuticals which comprise at
least
one compound according to the invention, preferably together with one or more
pharmacologically acceptable auxiliaries or carriers, and their use for the
purposes
mentioned above.
The active compound can act systemically and/or locally. For this purpose, it
can be
administered in a suitable manner, such as, for example, orally, parenterally,
pulmonarily, nasally, sublingually, lingually, buccally, rectally,
transdermally,
conjunctivally, otically, as stents or as an implant.
For these administration routes, the active compound can be administered in
suitable
administration forms.
Le A 36 136 CA 02489452 2004-12-14
-28-
For oral administration, known forms releasing the active compound rapidly
and/or
in modified form are suitable, such as, for example, tablets (non-coated and
coated
tablets, for example enterically coated tablets or film-coated tablets),
capsules, sugar-
coated tablets, granules, pellets, powders, emulsions, suspensions, solutions
and
aerosols.
The parenteral administration can take place with circumvention of an
absorbtion
step (intravenous, intraarterial, intracardiac, intraspinal or intralumbar) or
with
involvement of an absorption (intramuscular, subcutaneous, intracutaneous,
percutaneous or intraperitoneal). For parenteral administration, suitable
administration forms are, inter alia, injection and infusion preparations in
the form of
solutions, suspensions, emulsions, lyophilizates and sterile powders.
For the other administration routes, for example, inhalation pharmaceutical
forms
(inter alia powder inhalers, nebulizers), nasal drops/solutions, sprays;
tablets or
capsules to be applied lingually, sublingually or buccally, suppositories, ear
and eye
preparations, gynocapsules, aqueous suspensions (lotions, shake lotions),
lipophilic
suspensions, ointments, creams, milk, pastes, dusting powder or implants are
suitable.
The active compounds can be converted into the administration forms mentioned
in a
known manner. This takes place using inert non-toxic, pharmaceutically
suitable
auxiliaries. These include, inter alia, carriers (for example microcrystalline
celluose),
solvents (for example liquid polyethylene glycols), emulsifiers (for example
sodium
dodecylsulphate), dispersants (for example polyvinylpyrrolidone), synthetic
and
natural biopolymers (for example albumin), stabilizers (for example
antioxidants
such as ascorbic acid), colorants (for example inorganic pigments such as iron
oxides) or taste and/or odour corrigents.
In general, it has been found to be advantageous both in human and in
veterinary
medicine to administer the active compound according to the invention in total
amounts of from about 0.01 to about 700, preferably 0.01 to 100, mg/kg of body
weight per 24 hours, if appropriate in the form of a plurality of individual
doses, to
Le A 36 136 CA 02489452 2004-12-14
-29-
obtain the desired results. An individual dose contains the active compound
according to the invention preferably in amounts of from about 0.1 to about
80, in
particular 0.1 to 30, mg/kg of body weight.
In spite of this, it may be necessary, if appropriate, to deviate from the
amounts
mentioned, namely depending on the body weight, the route of application, the
individual response to the active compound, the type of preparation and the
time or
interval at which administration takes place. Thus, in some cases it may be
sufficient
to use less than the abovementioned minimum amount, whereas in other cases the
upper limit mentioned has to be exceeded. In the case of the administration of
relatively large amounts, it may advisable to divide these into several
individual
administrations over the course of the day.
The percentages in the tests and examples below are, unless indicated
otherwise,
percentages by weight; parts are parts by weight. Solvent ratios, dilution
ratios and
concentrations of liquid/liquid solutions are in each case based on the
volume.
Le A 36 136 CA 02489452 2004-12-14
-30-
A. Examples
Abbreviations
TLC thin-layer chromatography
DCI direct chemical ionization (in MS)
DCM dichloromethane
DIEA N,N-diisopropylethylamine
DMSO dimethyl sulphoxide
DMF N,N-dimethylformamide
EA ethyl acetate
El electron impact ionization (in MS)
ESI electrospray ionization (in MS)
M.P. melting point
sat. saturated
h hour
HPLC high-pressure, high-performance liquid chromatography
conc. concentrated
LC-MS liquid-chromatography-coupled mass spectroscopy
LDA lithium diisopropylamide
MPLC medium-pressure, medium-performance liquid chromato-
graphy
MS mass spectroscopy
NMR nuclear magnetic resonance spectroscopy
RP-HPLC reverse phase HPLC
RT room temperature
Rf retention index (in TLC)
Rt retention time (in HPLC)
THE tetrahydrofuran
CA 02489452 2010-09-09
30725-332
-31-
HPLC, LCMS and GCMS methods:
Method 1 (HPLC):
Instrument: HP 1100 with DAD detection; column: Kromasil RP-18, 60 mm x 2 nun,
3.5 m; mobile phase: A=5 ml of HC1O4/l of H2O, B=acetonitrile; gradient: 0
min
2% B, 0.5 min 2% B, 4.5 min 90% B, 6.5 min 90% B; flow rate: 0.75 mllmin;
temp.:
30 C; detection UV 210 rim.
Method 2 (HPLC):
Instrument: HP l 100 with DAD detection; column: Kromasil RP-18, 60 mm x 2 mm,
3.5 m; mobile phase: A-5 ml of HC1O4/1 of H2O, B=acetonitrile; gradient: 0
min
2% B, 0.5 min 2% B, 4.5 min 90% B, 9 min 90% B; flow rate: 0.75 ml/min; temp.:
30 C; detection UV 210 nm.
Method 3 (HPLC):
Instrument: Finnigan MAT 900S, TSP: P4000,AS3000,UV3000HR; column:
Symmetry C 18, 150 mm x 2.1 mm, 5.0 m; mobile phase C: water, mobile phase B:
water + 0.3 g of 35% strength HCI, mobile phase A: acetonitrile; gradient: 0.0
min
2% A -~ 2.5 min 95% A -4 5 min 95% A; oven: 70 C; flow rate: 1.2 mllmin; UV
detection: 210 nm.
Method 4 (LCMS):
Instrument: Micromass Quattro LCZ, HP I 100; column: Symmetry C 18, 50 mm x
2.1 mm, 3.5 m; mobile phase A: acetonitrile + 0.1% formic acid, mobile phase
B:
water + 0.1% formic acid; gradient: 0.0 min 10% A --* 4.0 min 90% A - 6.0- min
90% A; oven: 40 C; flow rate: 0.5 mllmin; UV detection: 208-400 nm.
Method 5 (LCMS):
Instrument: Micromass Platform LCZ, HP1100; column: Symmetry C18, 50 mm x
2.1 mm, 3.5 m; mobile phase A: acetonitrile + 0.1% formic acid, mobile phase
B:
water + 0.1% formic acid; gradient: 0.0 min 10% A -a 4.0 min 90% A --> 6.0 min
90% A; oven 40 C; flow rate: 0.5 mllmin; UV detection: 208-400 nm.
*Trade-mark
CA 02489452 2010-09-09
30725-332
-32-
Method 6 (LCMS):
Instrument MS: Micromass ZQ; instrument HPLC: Waters Alliance 2790; column:
Symmetry C 18, 50 mm x 2.1 mm, 3.5 m; mobile phase B: acetonitrile + 0.05%
formic acid, mobile phase A: water + 0.05% formic acid; gradient: 0.0 min 10%
B ->
3.5 min 90% B --* 5.5 min 90% B; oven: 50 C; flow rate: 0.8 mUmin; UV
detection:
210 nm.
Method 7 (LCMS):
Instrument: Micromass Platform LCZ, HP 1100; column: Symmetry C18, 50 mm x
2.1 mm, 3.5 gm; mobile phase A: water + 0.05% formic acid, mobile phase B:
acetonitrile + 0.05% formic acid; gradient: 0.0 min 90% A --a 4.0 min 10% A -p
6.0 min 10% A; oven: 40 C; flow rate: 0.5 ml/min; UV detection: 208-400 rim.
Method 8 (LCMS):
Instrument: Micromass Quattro LCZ, HPI 100; column: Symmetry C18, 50 mm x
2.1 mm, 3.5 m; mobile phase A: water + 0.05% formic acid, mobile phase B:
acetonitrile + 0.05% formic acid; gradient: 0.0 min 90% A --> 4.0 min 10% A -4
6.0
min 10% A; oven: 40 C; flow rate: 0.5 ml/min; UV detection: 208-400 nm.
Method 9 (GCMS):
Column: HP-5 30 in x 320 m x 0.25 pm (thickness of the film); carrier gas:
helium;
temperature gradient: 14 C/min up to 300 C, then I min const. 300 C; flow
rate:
1.5 ml/min; initial temperature: 60 C; starting time: 2 min; front injector
temp.:
250 C.
Method 10 (HPLC):
Instrument: Waters Alliance 2790 LC; column: Symmetry C18, 50 mm x 2.1,
3.5 m; mobile phase A: water + 0.1% formic acid, mobile phase B: acetonitrile
+
0.1% formic acid; gradient: 0.0 min 5% B -4 5.0 min 10% B -4 6.0 min 10% B;
temperature: 50 C; flow rate: 1.0 ml/min; UV detection: 210 nm.
*Trade-mark
Le A 36 136 CA 02489452 2004-12-14
- 33 -
Method 11 (chiral HPLC):
Column: chiral stationary phase, based on the optically active monomer
N-methacrylacyl-L-leucine-dicyclopropylmethylamide; mobile phase A: isohexane,
mobile phase B: ethyl acetate; gradient: A:B - 20:80; flow rate: 15 ml/min.
Le A 36 136 CA 02489452 2004-12-14
-34-
Starting materials
Example I
4-Chloro-6-(1 H-indol-5-yl)-2-pyrimidinamine
N
H N
2
WNN~I-I
H
380 mg (2.33 mmol) of 2-amino-4,6-dichloropyrimidine are suspended in 20 ml of
toluene and 10 ml of ethanol, and 80 mg (0.07 mmol) of tetrakis(triphenylphos-
phine)palladium(0) are added. After 20 minutes of stirring at room
temperature,
450 mg (2.80 mmol) of 5-indoleboronic acid and 3.90 ml of a 2M sodium
carbonate
solution are added. The mixture is stirred at 120 C for 20 hours. After
cooling, the
reaction solution is neutralized using iN hydrochloric acid and extracted
three times
with in each case 50 ml of ethyl acetate. The organic phase is dried over
sodium
sulphate, filtered off and concentrated under reduced pressure. The crude
product is
purified chromatographically on silica gel 60 (mobile phase: cyclohexane --~
cyclohexane: ethyl acetate 1:1).
This gives 32 mg (4% of theory) of product.
'H-NMR (400 MHz, DMSO-d6): b = 6.47 (s, 1H), 7.33 (s, 1H), 7.50 (dd, 2H), 7.98
(s, 1 H), 11.13 (s, 1 H)
MS (ESIpos): m/z = 245 (M+H)+
HPLC (method 1): Rt = 4.12
The example listed in the table below can be prepared analogously to the
procedure
described for Example I using the appropriate starting materials.
Le A 36 136 CA 02489452 2004-12-14
-35-
Example Structure - Analytical data
'H-NMR (300 MHz, DMSO-d6): 8 = 7.24
ci (br.s, 2H), 7.44 (s, IH), 7.61 (dd, IH), 8.12
N \ (d, 1H), 8.45 (dt, 2H), 8.77 (d, 1H), 8.98 (dd,
II H2N N \ \ IH)
N MS (ESIpos): m'z = 257 (M+H)+
HPLC (method 1): Rt = 3.54 min
Example III
Ethyl 3-oxo-3-(4-pyridinyl)propanoate
rCH3
O O
0
N
25 g (203 mmol) of isonicotinic acid, 35.12 g (243.7 mmol) of 2,2-dimethyl-1,3-
dioxolane-4,6-dione and 49.6 g (406 mmol) of 4-dimethylaminopyridine are
initially
charged in 300 ml of dichloromethane and cooled to 0 C. A IN solution of 46.1
g
(223.4 mmol) of 1,3-dicyclohexylcarbodiimide in dichloromethane is added
dropwise. The mixture is stirred at room temperature for 2 hours. The
resulting
precipitate is filtered off and washed with dichloromethane. The filtrate is
concentrated under reduced pressure. The residue is dissolved in 1200 ml of
ethanol,
a solution of 96.6 g (507.7 mmol) of p-toluenesulphonic acid monohydrate in
300 ml
of ethanol is added and the mixture is stirred under reflux for one hour.
After
cooling, the ethanol is removed under reduced pressure. The residue is taken
up in
1000 ml of ethyl acetate and 900 ml of water and dissolved by heating. The
organic
phase is separated off, washed with 600 ml of saturated sodium bicarbonate
solution
Le A 36 136 CA 02489452 2004-12-14
-36-
and saturated sodium chloride solution and dried over sodium sulphate. The
solution
is concentrated under reduced pressure. The crude product is filtered through
a silica
gel frit using dichloromethane:methanol 10:1. Since the aqueous phase still
contains
some product, it is extracted with dichloromethane and the extract is dried
over
sodium sulphate and concentrated under reduced pressure. The crude product is
filtered through a silica gel frit using dichloromethane:methanol 10:1.
This gives a total of 25.9 g (42% of theory) of product.
'H-NMR (300 MHz, DMSO-d6): S = 1.17 (t, 3H), 4.12 (q, 2H), 4.25 (s, 2H), 7.82
(dd, 2H), 8.83 (dd, 2H)
LC-MS (method 3): Rt = 2.40 min
MS (ESIpos): m/z = 194 (M+H)+
Example IV
2-Amino-6-(4-pyridinyl)-4-pyrimidinol
OH
N
N I
H 2 N
iN
25 g (81.52 mmol) of the compound from Example III and 13.22 g (73.37 mmol) of
guanidinium carbonate are dissolved in 250 ml of ethanol, concentrated
hydrochloric
acid is added and the mixture is stirred under reflux overnight. After
cooling, the
precipitate is filtered off with suction, washed with ethanol and dried under
high
vacuum. 250 ml of IN sodium hydroxide solution are added to the solid, and the
mixture is stirred under reflux for 2 hours. After cooling, the mixture is
acidified
using concentrated acetic acid and the precipitated product is filtered off
with suction
and washed with diethyl ether.
Drying under high vacuum gives 12.52 g (82% of theory) of product.
'H-NMR (300 MHz, DMSO-d6): 8 = 6.23 (s, 1H), 6.89 (br.s, 2H), 7.86 (dd, 2H),
8.64 (dd, 2H)
Le A 36 136 CA 02489452 2004-12-14
-37-
LC-MS (method 4): Rt = 0.30 min
MS (ESIpos): m/z = 189 (M+H)+
Example V
4-Chloro-6-(4-pyridinyl)-2-pyrimidinamine
CI
N
\N NHZ
N /
10.4 g (55.26 mmol) of the compound from Example IV are dissolved in 28.33 ml
(303.95 mmol) of phosphoryl chloride. 0.88 g (7.18 mmol) of N,N-
dimethylaniline
are slowly added dropwise, and the mixture is stirred at 100 C for one hour.
The
reaction solution is then stirred at room temperature for another 2 hours. The
phosphoryl chloride is removed under reduced pressure using a rotary
evaporator.
Water:dichloromethane 9:1 is added to the residue, and the mixture is boiled
for
5 minutes. The mixture is then neutralized using saturated sodium bicarbonate
solution and the product is filtered off with suction and dried under high
vacuum.
'H-NMR (300 MHz, DMSO-d6): 6 = 7.31 (br.s, 2H), 7.38 (s, 1H), 8.00 (dd, 2H),
8.74 (dd, 2H)
LC-MS (method 4): Rt = 1.08 min
MS (ESIpos): m/z = 207 (M+H)+
Example VI
1-Chloro-2,3-difluoro-5-nitrobenzene
F
F
C I N IIZZ~
1O
I_
0
The compound can be obtained by oxidizing 3-chloro-4,5-difluoroaniline,
described
in JP 05059067, with hydrogen peroxide in trifluoroacetic acid according to a
Le A 36 136 CA 02489452 2004-12-14
-38-
process described in Heaton, A. et al., J. Fluorine Chem. 1997, 81 (2), 133-
138 and
Krapcho, A.P. et al., J. Org. Chem. 1990, 55 (21), 5662-5664 for the
preparation of
analogous derivatives.
Example VII
4-[(2-Fluoro-4-nitrophenyl)sulphanyl]pyni dine
F
N ItLNv.O
I-
O
21 g (188.9 mmol) of 4-mercaptopyridine, 30.05 g (188.9 mmol) of 3,4-difluoro-
nitrobenzene and 60.05 g (434.5 mmol) of potassium carbonate are dissolved in
dimethylformamide, and the mixture is stirred at 40 C for 3 hours. The
reaction
solution is then diluted with 500 ml of ethyl acetate and 300 ml of water. The
aqueous phase is extracted five times with in each case 100 ml of ethyl
acetate. The
combined organic phases are washed with 200 ml of saturated sodium chloride
solution, dried over sodium sulphate and concentrated under reduced pressure
using
a rotary evaporator. The residue is purified by MPLC (mobile phase: ethyl
acetate:cyclohexane 1:1).
This gives 37.3 g (79% of theory) of product.
'H-NMR (300 MHz, DMSO-d6): 6 = 7.28 (dd, 2H), 7.79 (t, 1H), 8.15 (dd, 1H),
8.30
(dd, I H), 8.50 (dd, 2H)
LC-MS (method 4): Rt = 2.68 min
MS (ESIpos): m/z = 251 (M+H)+
The examples listed in the table below can be prepared from the appropriate
mercapto or hydroxy heterocycles and their corresponding 4-fluoro- and 4-
chloro-
nitrobenzene derivatives, analogously to the procedure described in Example
VII.
Le A 36 136 CA 02489452 2004-12-14
-39-
Example Structure Analytical data
H-NMR (200 MHz, DMSO-d6): S =
F I "0- 7.14 (t, 1H), 7.55 (d, IH), 7.74 (t, 1H),
VIII O7.86 (d, 1H), 8.08 (t, 2H), 8.43 (dd, 1H),
N j j 8.58 (d, 11-1), 9.46 (s, 1H)
HPLC (method 1): R, = 3.80 min
H-NMR (300 MHz, DMSO-d6): 6 =
S F 7.15 (dd, 2H), 8.37 (dd, IH), 8.41- 8.45
IX (m, 3H)
N Cl NO2 HPLC (method 1 ): R, = 3.77 min
MS (Clpos): m/z = 302 (M+NH4)+
H-NMR (200 MHz, DMSO-d6): 6 =
S CI 7.07 (dd, 2H), 8.41 (dd, 2H), 8.54 (s,
X I 2H)
N CI NO2 HPLC (method 1): R, = 3.92 min
MS (ESIpos): m/z = 301 (M+H)+
Example XI
3 -Fluoro-4 -(4-pyridinylsulphanyl) aniline
F
S
N \
NH2
37 g (147.9 mmol) of the compound from Example VII are dissolved in 1000 ml of
ethanol, and 143.86 ml (2.95 mol) of hydrazine hydrate and 4 g of palladium on
carbon are added. The reaction mixture is stirred under reflux overnight.
After
cooling, the mixture is filtered off with suction through silica gel, and the
filter cake
is washed with ethanol. The filtrate is concentrated under reduced pressure
using a
rotary evaporator. The residue is suspended in diethyl ether and filtered off
with
suction. The precipitate is then suspended in water and filtered off with
suction. The
product is washed two more times with a little water.
Le A 36 136 CA 02489452 2004-12-14
-40-
Drying under high vacuum gives 27.3 g (84% of theory) of product.
'H-NMR (300 MHz, DMSO-d6): b = 6.02 (br.s, 2H), 6.49-6.54 (m, 2H), 6.93 (dd,
2H), 7.23 (t, 1H), 8.32 (dd, 2H)
LC-MS (method 4): Rt 0.96 min
MS (ESIpos): m/z = 221 (M+H)+
The example listed in the table below can be prepared analogously to the
procedure
described for Example XI from the appropriate starting materials.
Example Structure Analytical data
'H-NMR (200 MHz, DMSO-d6): S
F . I NHZ = 5.46 (br.s, 2H), 6.46 (d, 1H), 6.55
(dd, 1H), 6.85 (d, 1H), 7.04 (t, IH),
XII 7.54 (t, 1H), 7.77 (d, 1H), 8.10 (d,
N IH), 8.58 (d, 1H), 9.35 (s, 1H)
LC-MS (method 4): R, = 1.95 min
MS (ESIpos): m/z = 255 (M+H)+
Example XIII
3 -Chloro- 5 -fluoro-4-(4-pyri dinylsulphanyl) aniline
F
S
N Cl NH 2
3.19 g (11.205 mmol) of the compound from Example IX are dissolved in 200 ml
of
ethanol. 638 mg (2.81 mmol) of platinum(IV) oxide are then added, and the
mixture
is stirred at RT and atmospheric pressure under an atmosphere of hydrogen for
2 hours. For work-up, the reaction solution is filtered off with suction
through
Le A 36 136 CA 02489452 2004-12-14
-41-
kieselguhr and washed thoroughly with ethanol. The filtrate is concentrated
under
reduced pressure.
This gives 2.755 g (81% of theory) of product.
'H-NMR (200 MHz, DMSO-d6): 6 = 6.37 (s, 2H), 6.49 (dd, 1H), 6.72-6.74 (m, 1H),
6.93 (dd, 2H), 8.34 (dd, 2H)
HPLC (method 1): Rt = 3.68 min
MS (ESIpos): m/z = 255 (M+H)+
The example listed in the table below can be prepared analogously to the
procedure
described for Example XIII from the appropriate starting materials.
Example Structure Analytical data
'H-NMR (200 MHz, DMSO-d6): S =
S CI 6.32 (s, 2H), 6.83 (s, 2H), 6.90 (dd,
XIV N i 2H), 8.33 (dd, 2H).
CI NH 2 HPLC (method 1): R, = 3.80 min
MS (ESIpos): m/z = 270.9 (M+H)+
Example XV
3-Fluoro-N-methyl-4-(4-pyridinylsulphanyl)aniline
F
N / NCH3
H
440.5 mg (2 mmol) of the compound from Example XI are dissolved in 2 ml of
methanol, and 2 ml of a 21 % strength sodium ethoxide solution are added. 84
mg
(2.8 mmol) of paraformaldehyde are added, and the mixture is stirred at room
temperature overnight. 75.7 mg (2 mmol) of sodium borohydride are added to the
Le A 36 136 CA 02489452 2004-12-14
-42-
reaction mixture, which is then heated under reflux for 1.5 hours. After
cooling, the
mixture is carefully hydrolysed using 1M potassium hydroxide solution. Once
the
reaction has stopped, the resulting solid is filtered off with suction and
washed with a
large quantity of water. The solid residue is dissolved in diethyl ether,
dried over
sodium sulphate and concentrated under reduced pressure using a rotary
evaporator.
This gives 387 mg (83% of theory) of product.
'H-NMR (200 MHz, DMSO-do): 6 = 2.73 (d, 3H), 6.45-6.52 (m, 2H), 6.59-6.69 (m,
1H), 6.92 (dd, 2H), 7.29 (t, 1H), 8.32 (dd, 2H)
LC-MS (method 4): Rt = 2.35 min
MS (ESIpos): m/z = 235 (M+H)+
Example XVI
N-(2-Amino-6-chloro-4-pyrimidinyl)-N-[3-fluoro-4-(4-
pyridinylsulphanyl)phenyl]amine
F
I \ S ~ \
N I NH
N
CI N NH 2
2.98 g (18.16 mmol) of 2-amino-4,6-dichloropyrimidine are suspended in 300 ml
of
water, and 4 g (18.16 mmol) of the compound from Example XI and 1.82 ml of
concentrated hydrochloric acid are then added. The mixture is stirred at 100 C
overnight. For work-up, the reaction solution is allowed to cool and made
alkaline
using saturated sodium bicarbonate solution. The precipitated product is
filtered off
with suction and then dried in a vacuum drying cabinet at 40 C.
This gives 6.04 g (74% of theory) of product.
Le A 36 136 CA 02489452 2004-12-14
-43 -
'H-NMR (200 MHz, DMSO-d6): 6 = 6.09 (s, 1H), 6.99 (dd, 4H), 7.41 (dd, 1H),
7.56
(dd, 1H), 8.27 (dd, 1H), 8.36 (dd, 2H), 9.89 (s, 1H)
HPLC (method 1): R, = 3.69 min
MS (ESIpos): m/z = 348 (M+H)+
The examples listed in the table below can be prepared analogously to the
procedure
described above for Example XVI from the appropriate starting materials.
Example Structure Analytical data
F
0--i LC-MS (method 3): Rt = 1.95 min
XVII NH MS (ESIpos): m/z = 382 (M+H)+
N
H2NN CI
F 'H-NMR (200 MHz, DMSO-d6): 6 =
S 6.08 (s, 1H), 7.00 (d, 2H), 7.09 (s,
N CI I NH 2H), 7.74 (s, I H), 8.15 (dd, IH), 8.37
XVIII
N (dd, 2H), 10.00 (s, 1H)
N HPLC (method 1): R, = 3.81 min
MS (ESIpos): m/z = 382 (M+H)+
CI 'H-NMR (200 MHz, DMSO-d6): 8 =
% 6.34 (s, 1H), 7.69 (d, 2H), 8.36 (s,
g~,
XIX CINH 2H), 8.71 (d, 2H), 10.72 (s, 1H).
HPLC (method 1): R, = 3.8 min
ct N NH2 MS (ESIpos): m/z = 398 (M+H)+
Le A 36 136 CA 02489452 2004-12-14
-44-
Example XX
-Benzyl- l -oxa-5-azaspiro [4.2]heptane
0
N -
The compound can be prepared from N-benzyl-3-pyrrolidinone by a procedure of
5 E.J. Corey et al. according to US 4,508,724.
Example XXI
1-Benzyl-3-hydroxy-3-(2-hydroxyethylaminomethyl)pyrrolidine
OH
HN
OH
QjN
32.7 g (0.17 mol) of Example XX are added dropwise to 31 g (0.52 mol) of
ethanolamine in 250 ml of water, and the mixture is stirred at room
temperature
overnight. The mixture is extracted with diethyl ether, the aqueous phase is
concentrated and the residue is distilled under high vacuum.
This gives 42.1 g (96% of theory) of product.
Boiling point: 180-190 C/0.1 mbar
Example XXII
7-Benzyl- l -oxa-4,7-diazaspiro[5.4]decane
Le A 36 136 CA 02489452 2004-12-14
-45-
rNH
0
N
85 g (340 mmol) of Example XXI are dissolved in a mixture of 280 ml of
concentrated sulphuric acid and 140 ml of water, and the mixture is heated at
180 C
overnight. The mixture is made alkaline using 45% strength aqueous sodium
hydroxide solution, precipitated salts are dissolved in water and the mixture
is
extracted five times with in each case 200 ml of chloroform. The organic
phases are
dried over potassium carbonate, the drying agent is separated off and the
solution is
concentrated.
The residue is distilled under high vacuum.
This gives 60 g (76% of theory) of product.
Boiling point: 125 C/0.08 mbar
Example XXIII
tert-Butyl 7-benzyl- l -oxa-4,7-diazaspiro[5.4] decane-4-carboxylate
H3C CH3
CH3
rN O
0
N
2 g of sodium hydroxide pellets in 25 ml of water are added to 10.3 g (47
mmol) of
Example XXII in 30 ml of tert-butanol, and 11 g (50 mmol) of di-tert-butyl
pyrocarbonate are added dropwise. The mixture is stirred at room temperature
overnight, 50 ml of water are added, the mixture is extracted three times with
Le A 36 136 CA 02489452 2004-12-14
-46-
chloroform, the extract is dried over potassium carbonate, the drying agent is
filtered
off with suction, the filtrate is concentrated and the residue is distilled
under high
vacuum.
This gives 13.8 g (88% of theory) of product.
Boiling point: 160 C/0.3 mbar
Example XXIV
tert-Butyl 1 -oxa-4,7-diazaspiro [5.4] decane-4-carboxylate
H 3 C CH
O-<
CH3
~N O
NH
0
13.7 g (41 mmol) of Example XXIII are dissolved, 3 g of 10% palladium on
carbon
are added and the mixture is hydrogenated at 100 C and 100 bar. The catalyst
is
filtered off with suction, the filtrate is concentrated and the residue is
distilled under
high vacuum.
This gives 7.6 g (75% of theory) of product.
Boiling point: 113 C/0.07 mbar
Example XXV
9-Methyl-6-oxa-2,9-diazaspiro[4.5]decane
0
NH
The compound can be prepared from Example XXII by reductive alkylation
according to the method described for the preparation of Example XV, followed
by
Le A 36 136 CA 02489452 2004-12-14
-47-
hydrogenolytic cleavage of the benzyl group according to the method described
for
the preparation of Example XXIV.
Example XXVI
3,7-Dibenzyl-3,7-diazabicyclo[3.3.0]octane-2,4-dione
\ N O
/ I
N
O
18.7 g (0.1 mol) of N-benzylmaleimide, 25 g (0.15 mol) of N-benzylglycine and
5 g
(0.157 mol) of paraformaldehyde are heated under reflux in 500 ml of toluene
until
the evolution of CO2 has ended. The mixture is concentrated and the crude
product is
used without further purification.
Example XXVII
3-Benzyl-3,7-diazabicyclo[3.3.0]octane-2,4-dione
HN O /
N \
O
150 g (0.4 mol) of the crude product from Example XXVI in 750 ml of ethanol
are
hydrogenated on 15 g of 10% palladium on carbon at 100 C and 100 bar. The
catalyst is filtered off and the solution is concentrated and filtered through
1.2 kg of
silica gel using dichloromethane.
This gives 38 g (42% of theory) of product.
Rf value: 0.35 (mobile phase: dichloromethane 96%/methanol 4%).
Example XXVIII
3-Benzyl-3,7-diazabicyclo[3.3.0] octane
Le A 36 136 CA 02489452 2004-12-14
-48-
H N
N ~ I
16 g (0.42 mol) of lithium aluminium hydride are initially charged in 350 ml
of
absolute tetrahydrofuran, and 38 g (0.165 mol) of Example XXVII in 150 ml of
absolute tetrahydrofuran are added dropwise. The mixture is heated under
reflux for
another five hours, 16 ml each of water, 16% strength aqueous potassium
hydroxide
solution and again water are added dropwise and the inorganic salts are
filtered off
with suction and decocted twice with tetrahydrofuran. The filtrates are
concentrated
and distilled under membrane-pump vacuum.
This gives 31.4 g (94% of theory) of product.
Boiling point: 140 C/4 mbar
Example XXIX
tert-Butyl 7-benzyl-3,7-diazabicyclo[3.3.0]octane-3-carboxylate
H3 C \ CH3
CH 3
O N
6 g (29.7 mmol) of Example XXVIII are initially charged, 1.4 g (35 mmol) of
sodium hydroxide in 30 ml of water are added and 8 g (36.7 mmol) of tert-butyl
pyrocarbonate are then added dropwise. The mixture is stirred at room
temperature
overnight and then extracted with chloroform, the extract is dried over
potassium
carbonate, the drying agent is filtered off, the filtrate is concentrated and
the residue
is distilled.
This gives 7.6 g (75% of theory) of product.
Boiling point: 153-156 C/0.2 mbar
Le A 36 136 CA 02489452 2004-12-14
-49-
Example XXX
tert-Butyl 3,7-diazabicyclo[3.3.0]octane-3-carboxylate
H 3 C /CH3
CH3
0 N
NH
7.6 g (25.3 mmol) of Example XXIX in 100 ml of ethanol are hydrogenated on 1.5
g
of 10% palladium/carbon at 100 C and 100 bar. The catalyst is filtered off,
the
filtrate is concentrated and the residue is distilled.
This gives 3.6 g (76% of theory) of product.
Boiling point: 92 C/0.08 mbar
Example XXXI
3,7-Diazabicyclo[3.3.0] octane
HN
NH
The compound can be prepared from Example XXX by removing the tert-
butoxycarbonyl group with hydrogen chloride (4 M in dioxane) or with
trifluoroacetic acid/dichloromethane (1:1).
Example XXXII
(4aR,7aS)-4-Benzyloctahydropyrrolo[3,4-b] [ 1,4]oxazine
H
N
N H
~-O H
The preparation of the compound is described in US 6 004 956.
Le A 36 136 CA 02489452 2004-12-14
-50-
Example XXXIII
-Methyl octahydrop yrro to [3 ,4-b]pyrrole
H
N
The preparation of the compound is described in DE-A 4 032 560.
5
Example XXXIV
[S,S]-8-Benzyl-2,8-diazabicyclo[4.3.0]nonane
H H
N
CN
3 g (20 mmol) of D-(-)-tartaric acid are dissolved in 10 ml of
dimethylformamide by
heating at 80 C, and a solution of 2.16 g (10 mmol) of cis-8-benzyl-2,8-
diazabicyclo[4.3.0]nonane in 3 ml of dimethylformamide is added. The mixture
is
stirred at 0 C for 1 hour and then filtered off with suction, and the product
is washed
with dimethylformamide and methoxyethanol. (Yield: 1.93 g)
Melting point: 146-151 VC
[a]D24 = -19.3 (c=1, H2O)
One recrystallization from methoxyethanol gives diastereomerically pure [S,S]-
8-
benzyl-2,8-diazabicyclo[4.3.0]nonane D-tartrate.
Melting point: 148-154 C
[a]D24 = -22.7 (c=1, H2O)
40 g of the salt are dissolved in 250 ml of water, and 32 g of 45% strength
aqueous
sodium hydroxide solution are added. The oil that has separated off is taken
up in
150 ml of tert-butyl methyl ether and the aqueous phase is re-extracted with
150 ml
Le A 36 136 CA 02489452 2004-12-14
-51-
of tert-butyl methyl ether. The organic phases are combined and, after drying
over
sodium sulphate, concentrated. The residue is then distilled under reduced
pressure.
This gives 18.5 g of product.
Boiling point: 107-109 C/0.1 mbar
[a]D24 = 17.3 (undiluted)
Example XXXV
[S,S]-2,8-Diazabicyclo[4.3.0]nonane
H H
CICNH
H
28.4 g (0.131 mol) of [S,S]-8-benzyl-2,8-diazabicyclo[4.3.0]nonane in 190 ml
of
methanol are hydrogenated over 5.8 g of palladium on carbon (5%) at 90 C and
90 bar for 5 hours. The catalyst is then filtered off with suction and washed
with
methanol and the filtrate is concentrated under reduced pressure. The residue
is
distilled without fractionation.
This gives 15 g (91 % of theory) of product.
Boiling point: 44-59 C/0.18 mbar
[UID24 = -2.29 (undiluted)
ee> 99% (determined by gas chromatography after derivatization with Mosher's
reagent)
Example XXXVI
[R,R]-8-Benzyl-2,8-diazabicyclo[4.3.0]nonane
H H
N
H
Le A 36 136 CA 02489452 2004-12-14
-52-
At 80 C, 75 g (0.5 mol) of L-(+)-tartaric acid are dissolved in 250 ml of
dimethylformamide, and 54.1 g (0.25 mol) of cis-8-benzyl-2,8-
diazabicyclo[4.3.0]nonane are added dropwise as a solution in 75 ml of
dimethylformamide. The mixture is slowly cooled to 20 C, and the crystal
suspension is stirred for another 1 hour. The crystals ([R,R]-8-benzyl-2,8-
diazabicyclo[4.3.0]nonane L-tartrate) are filtered off with suction. (The
filtrate can
be processed further to give [S,S]-8-benzyl-2,8-diazabicyclo[4.3.0]nonane).
These
crystals are washed with dimethylformamide and methoxyethanol (crude yield:
49.2 g) and recrystallized from 300 ml of methoxyethanol. This gives 45.6 g of
enantiomerically pure [R,R]-8-benzyl-2,8-diazabicyclo[4.3.0]nonane L-tartrate
(enantiomeric purity determined by gas chromatography after derivatization
with
menthyl chloroformate).
Melting point: 121-124 C
[X]D24 = +22.3 (c = 1, H2O)
The resulting salt is now converted into the free base. To this end, 44.5 g
are
dissolved in 280 ml of water, and 35.6 g of 45% strength aqueous sodium
hydroxide
solution are added. The precipitated oil is taken up in 170 ml of tert-butyl
methyl
ether and the aqueous phase is re-extracted with 170 ml of tert-butyl methyl
ether.
The organic phases are combined and, after drying over sodium sulphate,
concentrated. The residue is then distilled under reduced pressure.
Boiling point: 107-111 C/0.04 mbar
[a]D24 = -17.5 (undiluted)
Example XXXVII
[R,R]-2,8-Diazabicyclo[4.3.0]nonane
H H
CJNH
H
19.4 g (0.09 mol) of [R,R]-8-benzyl-2,8-diazabicyclo[4.3.0]nonane in 130 ml of
methanol are hydrogenated over 3.96 g of palladium on carbon (5%) at 90 C and
Le A 36 136 CA 02489452 2004-12-14
-53-
90 bar for 5 hours. The catalyst is then filtered off with suction and washed
with
methanol and the filtrate is concentrated under reduced pressure. The residue
is
distilled without fractionation.
This gives 9.61 g (85% of theory) of product.
Boiling point: 45-58 C/0.08 mbar
[a]D24 = +2.30 (undiluted)
Example XXXVIII
N-[2-Amino-6-(4-pyridinyl)-4-pyrimidinyl]-N-[3-fluoro-4-(4-pyridinylsulphanyl)-
phenyl]amine
F
N
INH
N
H2N N
N
29.6 ml of water are added to 4.32 g (20.9 mmol) of Example V and 4.61 g
(20.9 mmol) of Example XI. 9.25 ml of concentrated hydrochloric acid are added
to
the mixture, which is then stirred at 100 C overnight. After cooling to room
temperature, the mixture is diluted with methanol and neutralized with
saturated
aqueous sodium bicarbonate solution. The mixture is absorbed on silica gel and
purified by column chromatography on silica gel 60 using
dichloromethane/methanol
95:5 -* methanol.
Yield: 3.87 g (47% of theory)
'H-NMR (300 MHz, DMSO-d6): 6 = 6.65 (s, 1H), 6.71 (s, 2H), 7.02 (dd, 2H), 7.47-
7.58 (m, 2H), 7.87 (dd, 2H), 8.30-8.41 (m, 3H), 8.72 (d, 2H), 9.90 (s, 1H)
LC-MS (method 1): R1 = 3.34 min
MS (ESlpos): m/z = 391 (M+H)+
Le A 36 136 CA 02489452 2004-12-14
-54-
Working Examples
Example 1
N- [2-Amino-6-(6-quinolinyl)-4-pyrimidinyl]-N- [3 -fluoro-4-(4-
pyridinylsulphanyl)-
phenyl] amine
F
S ItLNH
N
\NNH2
N
1.50 g (5.84 mmol) of the compound from Example II are, together with 1.29 g
(5.84 mmol) of the compound from Example XI, initially charged in 70 ml of
water,
and 10 drops of concentrated hydrochloric acid (37% strength) are added. The
suspension is stirred at 100 C overnight. After cooling, the reaction mixture
is made
alkaline using concentrated sodium bicarbonate solution and evaporated to
dryness
under reduced pressure.
The residue is purified by preparative HPLC and then by flash chromatography
on
silica gel using the mobile phase dichloromethane/methanol 30/1.
This gives 330 mg (12% of theory) of product.
'H-NMR (300 MHz, DMSO-d6): S = 6.66 (br.s, 2H), 6.74 (s, 1H), 7.01 (d, 2H),
7.53
(d, 1H), 7.56-7.62 (m, 2H), 8.13 (d, 1H), 8.32-8.38 (m, 4H), 8.50 (d, 1H),
8.58 (d,
I H), 8.95 (dd, I H), 9.87 (s, 1H)
HPLC (method 1): Rt = 4.22 min
The example listed in the table below can be prepared analogously to the
procedure
described for Example I from the appropriate starting materials.
Le A 36 136 CA 02489452 2004-12-14
-55-
Example Structure Analytical data
'H-NMR (300 MHz, DMSO-d,): 6
= 6.44 (br.s, 2H), 6.54 (br.s, 1H),
i y s 6.60 (s, IH), 6.99 (d, 1H), 7.01 (s,
f "" 1H), 7.40 (d, 1H), 7.45-7.54 (m,
2
4H), 7.74 (d, 1H), 8.23 (s, 1H), 8.36
-COO (d, 3H), 9.68 (s, 1H)
HPLC (method 1): Rt = 3.80 min
Example 3
N-[2-Amino-6-(6-quinolinyl)-4-pyrimidinyl]-N-[3-fluoro-4-(5-isoquinolinyloxy)-
phenyl]amine
N F
b"'NH
N
CI
I \ \N NH2
N
110 mg (0.29 mmol) of the compound from Example XVII are suspended in 12 ml of
a solvent mixture of toluene and ethanol in the ratio 2:1. 10 mg (0.01 mmol)
of
tetrakis(triphenylphosphine)palladium(0) are added, and the mixture is stirred
at
room temperature for 20 minutes. 60 mg (0.35 mmol) of 6-quinolineboronic acid
and
1 ml of a 2M sodium carbonate solution are then added to the suspension, which
is
then stirred under reflux overnight. After cooling, the reaction mixture is
concentrated under reduced pressure. The residue is purified
chromatographically on
silica gel 60 (mobile phase: dichloromethane/methanol 30:1 --* 10:1).
This gives 82 mg (60% of theory) of product.
Le A 36 136 CA 02489452 2004-12-14
-56-
'H-NMR (300 MHz, DMSO-d6): 6 = 6.57 (br.s, 2H), 6.70 (s, 1H), 6.99 (d, 1H),
7.92
(t, IH), 7.41-7.47 (m, 1H), 7.55-7.63 (m, 2H), 7.85 (d, 1H), 8.13 (t, 2H),
8.28-8.37
(m, 2H), 8.50 (br.d, 1H), 8.58-8.62 (m, 2H), 8.95 (dd, 1H), 9.39 (s, 1H), 9.66
(br.s,
I H)
HPLC (method 1): Rt = 3.49 min.
MS (ESIpos): m/z = 475 (M+H)+
The examples listed in the table below can be prepared analogously to the
procedure
described for Example 3 from the appropriate starting materials.
Example Structure Analytical data
tH-NMR (300 MHz, DMSO-d6): 6 =
6.84 (s,1 H), 7.01 (s, I H), 7.18 (s, I H),
F 7.30 (d, 1H), 7.41 (t,1H), 7.58 (br.d,
rot 7.80 (t, 1H), 8.12 (d, 1H), 8.29 (d,
L NH
4 1H), 8.44 (d, 1H), 8.70 (d, 1H) 9.30 (s,
HzN" _N f N 2H), 9.41 (s, 1H), 9.75 (s, 1H), 11.25
N' (br.s, 1 H)
LC-MS (method 5): R, = 2.42 min
MS (ESlpos): m/z = 426 (M+H)+
Le A 36 136 CA 02489452 2004-12-14
-57-
Example Structure Analytical data
LC-MS (method 5): R, = 2.73 min
H MS (ESlpos): m/z = 449 (M+H)+
GV
'H-NMR (300 MHz, DMSO-d6): S =
F 6.68 (s, 1H), 7.25 (d, IH), 7.40 (t, 1H),
0
I 7.53-7.66 (m, 4H), 7.67-7.76 (m, IH),
NH
N 7.76 (t, 1H), 8.08 (d, IH), 8.28-8.39
6 H2N" 'N I \ I (m, 4H), 8.68 (d, 1H), 9.68 (s, 1H),
HNYCH, 10.34 (s, I H), 11.21 (s, 1H)
o LC-MS (method 5): Rt = 2.53 min
MS (ESlpos): m/z = 481 (M+H)+
'H-NMR (300 MHz, DMSO-d6): S =
F 4.62 (s, 2H), 6.86 (s, 1H), 7.46 (d,
6
N NH 2H), 7.58 (d, 2H), 7.69-7.77 (m, 2H),
7 7.88 (d, 2H), 8.41 (br.d, IH), 8.56 (d,
H NI(2H), 11.51 (s, 1H)
ON LC-MS (method 3): R, = 1.84 min
MS (ESIpos): m/z = 420 (M+H)+
'H-NMR (300 MHz, DMSO-d6): S =
F 2.07 (br.s, 2H), 3.13 (s, 3H), 3.20
YS (br.s, 2H), 3.52 (br.s, 2H), 4.63 (br.s,
8 I NH 1H), 5.17 (s, IH), 6.03 (br.s, 2H), 6.97
N
I o (d, 2H), 7.33-7.49 (m, 2H), 8.25 (dd,
HZN~N
I > 1H), 8.34 (d, 2H), 9.29 (s, 1H)
LC-MS (method 3): R, = 2.33 min
MS (ESIpos): m/z = 434 (M+H)+
Example 9
N- {2-Amino-6-[3-(dimethylamino)-1-pyrrolidinyl]-4-pyrimidinyl}-N-[3-fluoro-4-
(4-
5 pyridinylsulphanyl)phenyl]amine
Le A 36 136 CA 02489452 2004-12-14
-58-
F
S
NH
N
9NH2
H3C-rN\
CH3
600 mg (1.73 mmol) of the compound from Example XVI are dissolved in 40 ml of
ethanol, and 788 mg (6.9 mmol) of 3-(dimethylamino)pyrrolidine and 3 ml
(17.25 mmol) of N,N-diisopropylethylamine are added. The mixture is stirred at
80 C overnight. After cooling, the reaction solution is purified by MPLC
(mobile
phase: dichloromethane/methanol 5:1 + 1% concentrated ammonia solution).
This gives 475 mg (58% of theory) of product. (Mixture of enantiomers)
'H-NMR (300 MHz, DMSO-d6): S = 1.69-1.83 (m, 1H), 2.06-2.15 (m, 1H), 2.20 (s,
6H), 2.69-2.80 (m, 1H), 3.05 (t, 1H), 3.43-3.70 (br.m, 2H), 4.05 (q, 1H), 5.16
(s, 1H),
5.95 (br.s, 2H), 6.97 (d, 2H), 7.36-7.47 (m, 2H), 8.21 (dd, 1H), 8.34 (dd,
2H), 9.23 (s,
1H)
LC-MS (method 7): Rt = 0.44 min
MS (ESIpos): m/z = 426 (M+H)+
The two enantiomers below are obtained form Example 9 by separating the
enantiomers using chiral HPLC (Method 11).
Le A 36 136 CA 02489452 2004-12-14
-59-
Example Structure Analytical data
F
S
N I NH
Chiral HPLC (method 11):
10~aN NH Rt = 6.79 min
H3C-N,
CHI
(-)-enantiomer
F
S
N~ ( NH
Chiral HPLC (method 11):
N
11 //~N \N"NH Rt = 5.77 min
2
HNC-N
CHI
(+)-enantiomer
The examples listed in the table below can be prepared analogously to the
procedure
described for Example 9 from the corresponding starting materials.
Le A 36 136 CA 02489452 2004-12-14
-60-
Example Structure Analytical data
F 'H-NMR (300 MHz, DMSO-d6): S =
s-,( 1.42 (s, 9H), 2.07 (m, 2H), 2.74 (s,
NH 3H), 3.20 (m, 2H), 3.52 (m, 2H), 4.63
N (q, 1H), 5.17 (s, I H), 6.03 (br.s, 2H),
12 N -NNHZ
6.97 (d, 2H), 7.33-7.49 (m, 2H), 8.25
H3 C-N0 (dd, 1H), 8.34 (d, 2H), 9.29 (s, IH)
H'C
ko LC-MS (method 6): R, = 1.79 min
H3C CH3
+
MS (ESIpos): m/z = 512 (M+H)
'H-NMR (300 MHz, DMSO-d6): b =
F 1.39 (s, 9H), 2.92 (m, 2H), 3.16 (m,
4H), 3.52 (m, 4H), 5.16 (s, 1H), 5.95
NH (br.s, 2H), 6.97 (d, 2H), 7.36-7.47 (m,
13
H3C(CH,~N NNH, 2H), 8.20 (dd, 1H), 8.34 (d, 2H), 9.25
H,C O~N (s, 1 H)
O
LC-MS (method 7): Rt = 3.04 min
MS (ESIpos): m/z = 524 (M+H)+
'H-NMR (200 MHz, CDC13): 8 =
1.57-1.83 (m, 1H), 2.43-2.56 (m, IH),
F c,,;,a, 2.68-2.99 (m, 1H), 3.30-3.52 (m, 3H),
N s 3.57-3.98 (m, 6H), 4.14 (br.s, 1H),
NH
4.66 (br.s,IH), 5.12 (br.s, I H), 5.30 (s,
14 H N N, NH, 1H), 6.52 (s, I H), 6.94 (d, 1H), 7.07
C N
\--o H (dd, 1H), 7.28-7.48 (m, 6H), 7.60 (dd,
1H), 8.35 (d, 2H)
LC-MS (method 7): Rt = 2.90 min
MS (ESIpos): m/z = 530 (M+H)+
Le A 36 136 CA 02489452 2004-12-14
-61-
Example Structure Analytical data
'H-NMR (400 MHz, DMSO-d6):
S =
F 1.70-1.90 (m, 1H), 1.78 (s, 3H),
2.68-3.01 (m, 1H), 3.16-3.61 (m,
N ! NH overlaps with H2O signal, - 3H)),
i N 4.18-4.32 (m, 1H), 5.15 (s, 1H), 5.98-
(N N NHZ 6.10 (br.s., 2H), 6.98 (d, 2H), 7.31-
HN\J~~ 7.49 (m, 2H), 8.14 (d, IH), 8.28 (dd,
H 0 1H), 8.38 (dd, 2H), 9.29 (s, 1H)
LC-MS (method 7): R, = 2.58 min
MS (ESIpos): m/z = 440 (M+H)+
Example 16
N-[ l-(2-Amino-6- ([3 -fluoro-4-(4-pyridinylsulphanyl)phenyl]amino}-4-
pyrimidinyl)-
5 3-pyrrolidinyl]-N-methylamine
F
' ~ S
N
NH
N
9NH2
H3CJH
8 ml of 4M hydrochloric acid in dioxane are added to 46 mg (0.09 mmol) of the
compound from Example 12, and the mixture is stirred at room temperature for
4 hours. The mixture is then concentrated under reduced pressure. A little
10 concentrated ammonia solution is added to the residue, the mixture is
concentrated
Le A 36 136 CA 02489452 2004-12-14
-62-
using a rotary evaporator and the residue is then suspended in 2 ml of water.
The
precipitate is filtered off with suction and washed with 2 ml of
dichloromethane.
This gives 23 mg (62% of theory) of product.
'H-NMR (300 MHz, DMSO-d6): S = 1.40 (s, 3H), 2.60 (s, 1H), 3.51-3.90 (m, 6H),
5.29 (s, 1H), 7.00 (d, 1H), 7.16 (s, 1H), 7.33-7.52 (m, 3H), 8.18 (br.d, 1H),
8.36 (d,
1H)
LC-MS (method 1): Rt = 0.68 min
MS (ESIpos): m/z = 412 (M+H)+
Example 17
N-[2-Amino-6-(4-cyclopentyl- l -piperazinyl)-4-pyrimidinyl]-N-[3-fluoro-4-(4-
pyridinylsulphanyl)phenyl] amine
F
1 ~ s
N NH
N
rN \N NH2
<yNj
300 mg (0.86 mmol) of the compound from Example XVI are suspended in 8 ml of
2-ethyl-l-hexanol, and 266 mg (1.73 mmol) of 1-cyclopentylpiperazine and 0.75
ml
(4.31 mmol) of N,N-diisopropylethylamine are added. The mixture is stirred at
150 C overnight. After cooling, the reaction solution is purified by MPLC
(mobile
phase: dichloromethane/methanol 10:1 + 1% concentrated ammonia solution).
216 mg (52% of theory) of product are obtained.
'H-NMR (400 MHz, DMSO-d6): S = 1.28-1.41 (m, 2H), 1.44-1.55 (m, 2H), 1.57-
1.68 (m, 2H), 1.70-1.85 (m, 2H), 2.43 (br.s, 5H), 3.41 (br.s, 4H), 5.39 (s,
1H), 6.04
Le A 36 136 CA 02489452 2004-12-14
- 63 -
(br.s, 2H), 6.97 (d, 2H), 7.36 (dd, 1H), 7.45 (t, 1H), 8.23 (dd, 1H), 8.34 (d,
2H), 9.28
(s, 1H)
LC-MS (method. 7): Rt = 2.38 min
MS (ESIpos): m/z = 466 (M+H)+
The examples in the table below can be prepared analogously to the procedure
described for Example 17 from the corresponding starting materials.
Example Structure Analytical data
'H-NMR (400 MHz, DMSO-d6): 8
= 1.21-1.74 (m, 5H), 2.09-2.33 (m,
F Chiral 1H), 2.72-2.93 (m, 2H), 3.06-3.26
I as (m, 3H), 3.42-3.65 (m, 1H), 5.12 (s,
18 N NH
I H), 5.76 (s, I H), 5.95 (br.s, I H),
t~H "NJ``NH 6.98 (d, 2H), 7.33-7.49 (m, 2H),
Z 8.24 (dd, 1H), 8.34 (d, 2H), 9.25 (s,
I H)
LC-MS (method 6): Rt = 0.34 min
MS (ESIpos): mlz = 438 (M+H)'
F
5
N NH LC-MS (method 7): Rt = 0.41 min
19 N MS (EIneg): rn/z = 422 (M-)
N NNH2
HN~
Le A 36 136 CA 02489452 2004-12-14
-64-
Example Structure Analytical data
'H-NMR (200 MHz, DMSO-d6): S
F Chiral 1.23-1.75 (m, 5H), 2.21 (br.s,
\ S 2H), 2.84 (d, 1H), 3.63-3.22 (m,
I
N NH 4H), 4.11 (q, IH), 5.11 (s, 1H), 5.94
20 (br.s, 2H), 6.97 (d, 2H), 7.32-7.49
N-"% N N NH2 (m, 2H), 8.29 (dd, 1H), 8.34 (d,
2H), 9.23 (s, 1H)
LC-MS (method 7): Rt = 0.37 min
MS (ESIpos): m/z = 438 (M+H)+
'H-NMR (400 MHz, DMSO-d6): 6
= 1.03- 1.22 (m, 4H), 1.23 (s, 2H),
F chiral 1.65 (br.s, 2H), 1.87 (t, 2H), 3.16
i \ \
N I (s, 2H), 5.26 (s, 1H), 5.86 (s, 2H),
21 H 6.35 (d, 1H), 6.97 (d, 2H), 7.36 (d,
IH), 7.43 (t, 1H , 8.17 (d, IH), 8.35
~N N NHZ )
H
NHZ (d, 2H), 9.15 (s, 1H)
MS (ESIpos): m/z = 426 (M+H)+
LC/MS (method 1): R, = 3.53 min
'H-NIvIR (300 MHz, DMSO-d6): 6
= 1.36- 1.47 (m, 1H), 1.52- 1.75
F
(m, 3H), 2.22- 2.38 (m, 1H), 2.57
chiral
N ` (t, 1H), 2.84- 2.93 (m, 1H), 3.17 (s,
Ct NH 3H), 4.06 (br.s, 1H), 5.14 (s, IH),
22 nP .. j 6.00 (s, 2H), 6.97 (d, 2H), 7.74 (s,
N NH2 1H), 8.12 (dd, 1H), 8.36 (d, 2H),
9.42 (s, I H)
MS (ESlpos): m/z = 472 (M+H)+
LCIMS (method 1): R, = 3.53 min
Le A 36 136 CA 02489452 2004-12-14
-65-
Example Structure Analytical data
'H-NMR (400 MHz, DMSO-d6): 6
= 1.34-1.66 (m, 5H), 2.04-2.29 (m,
F chiral 2H), 2.80-2.83 (rn, 1H), 3.17 (d,
I 3H), 4.09 (q, IH), 5.07 (s, IH), 5.75
I NH
23 (s, 1 H), 5.82 (s, 2H), 6.93 (d, I H),
I
",IN N\,,- 7.19 (br.d, IH), 7.56 (t, IH), 7.82
(d, 1H), 8.10 (d, IH), 8.17 (dd, 1H ,
H )
8.59 (d, 1H), 8.92 (s, 1H), 9.36 (s,
IH)
MS (ESIpos): m/z = 472 (M+H)+
'H-NMR (400 MHz, DMSO-d6): 8
= 1.82- 2.10 (m,5H), 3.29-3.30 (m,
F 4H), 3.41-3.48 (m, IH), 3.54- 3.65
(m, 2H), 4.15 (br.s, 1H), 5.29 (s,
NH 1H), 7.02 (d, 2H), 7.34 (dd, IH),
24
7.42 (t, 1H), 7.93 (dd, 1H), 8.25 (d,
H2N N N
2H)
H MS (ESlpos): m/z = 413 (M+H)+
HPLC (method 1): Rt= 3.48 min
Le A 36 136 CA 02489452 2004-12-14
-66-
Example Structure Analytical data
F
Is
1 \ \
NH
N
HZNN N
o MS (CIpos): m/z = 454 (M+H)'
25 H HPLC (method 1): Rt = 3.39 min
from Example XVI and
Example XXIV; during the
chromatographic purification
with addition of acid, the Boc
group is cleaved off
F
MS (ESIpos): m/z = 468 (M+H)T
26 HPLC (method 1): Rt = 3.41 min
H=fJ N N~ )
J
N
H,C
'H-NMR (300 MHz, DMSO-d6):5
=0.92- 1.03 (m, 2H), 1.13- 1.30 (m,
5H), 1.64- 1.80 (m, 6H), 2.99 (d,
F
6H), 4.22 (d, 2H), 5.53 (s, 1H), 6.42
27 b'NH (br.s, 1H), 7.22 (d, 2H), 7.40 (dd,
H NJN N~ 1H), 7.52 (t, 1H), 8.19 (d, 1H9,
2 ON,_~ 8.45 (d, 1H), 9.31
(br.s, 1H), 9.57
(s, 1 H)
MS (ESIpos): m/z = 494 (v1+H)+
HPLC (method 1): Rt=3.77 min
Le A 36 136 CA 02489452 2004-12-14
67-
Example Structure Analytical data
'H-NMR (300 IvfHz, DMSO-d6): 6
= 2.50 (s, 3H), 3.20 (br.s, 4H), 3.64
F (br.s, 4H), 5.61 (s, 1H), 6.89 (d,
N ' tL NH 3H), 7.06 (d, 3H), 7.19 (d, d, 2H),
28 N 7.38 (dd, 1H), 7.58 (t, 1H), 8.07
HZN~N N~
ON (br.s, IH), 8.44 (d, 2H), 9.85 (br.s,
1 H)
MS (ESIpos): m/z = 488 (M+H)+
HPLC (method 1):.Rt= 3.81 min
'H-NMR (400MHz, MeOH-d4): 5 =
F 1.35 (t, 3H), 3.19 (t, 4H), 3.75 (s,
N s 4H), 3.98 (quart., 2H), 4.89 (s,1H),
I NH
N- 6.85 (d, 2H), 6.99 (d, 2H), 7.43 (d,
29 HZNN ~N 3H), 7.65 (t, 1H), 7.84 (br.s, IH),
o'er 8.43 (d, 2H)
MS (ESIpos): m/z = 518 (M+H)+
HPLC (method 1): R,=3.82 min
'H-NMR (400 MHz, MeOH-d4): 6
= 1.29-1.41 (m, 2H), 2.03 (br.s,
F
s 1H), 2.32 (br.s, IH), 2.67 (s, 2H),
I
30 NH 2.95 (s, 5H), 3.61-3.68 (m, 2H),
4.67 (s, IH), 7.14 (d, 3H), 7.38 (dd,
H2N N N
/__I N 1H), 7.52 (t, 2H), 7.93 (d, IH), 8.32
(dd, 3H)
HPLC (method 1): Rt= 3.35 min
Le A 36 136 CA 02489452 2004-12-14
-68-
Example Structure Analytical data
1H-NMR (200 MHz, DMSO-d6): S
S F = 3.17 (br.s, 4H), 3.97 (br.s, 4H),
g-, NH 5.61 (s, 1H), 7.29 (d, 3H), 7.39 (dd,
31 ~-:I' 1H), 7.54 (t, 1H), 8.15 (br.d, 1H),
H2N N N'~ 8.48 (d, 3H), 9.61 (br.s, 1H)
l~s, 0
0 MS (ESIpos): m/z 447 (M+H)+
HPLC (method 1): Rt=3.38 min
1H-NMR (200 MHz, DMSO-d6): S
F = 4.47 (d, 3H), 5.41 (s, 1 H), 7.21
(d, 3H), 7.32 (d, 4H), 7.43- 7.48 (m,
HN
I H), 7.61 (t, I H), 7.64- 7.86 (m,
32 N'
H N~N I NH 1H), 8.00- 8.16 (m, IH), 8.24-8.33
2
(m, 1H), 8.46 (d, 2H), 10.17 (s, 1H)
MS (ESIpos): m/z = 419 (M+H)+
HPLC (method 1): R, = 3.85 min
1H-NMR (200 MHz, DMSO-d6): S
F
s ' = 4.43- 4.50 (m, 3H), 5.39 (s, 1H),
HN 7.15- 7.29 (m, 5H9, 7.32- 7.44 (m,
33 4H), 7.52-7.66 (m, 2H), 8.44 (dd,
H2N N NH 2H), 10.14 (br.s, 1 H)
F MS (ESIpos): m/z = 437 (M+H)+
HPLC (method 1): R,=3.89 min
Le A 36 136 CA 02489452 2004-12-14
-69-
Example Structure Analytical data
'H-NMR (200 MHz, DMSO-d6): 6
F = 1.22 (d, IH), 1.26 (d, 1H), 3.52
~
HN I I 'N (d, H), 5.51 (s, 1H), 7.38 (d, 3H),
NN 7.64 (t,2H), 7.72-7.78 (m, 2H), 8.19
34 HZN N NH (d, 21-1), 8.53 (d, 2H), 8.66- 8.73 (m,
3H), 10.26 (s, 1H)
" MS (ESIpos): m/z = 434 (M+H)+
HPLC (method 1): R,=3.34 min
F
I \ S I \
HN / i N
35 MS (ESIpos): m/z = 433 (M+H)+
d H NN NH CH LC/MS (method 4): R,= 2.90 min
x
I \
'H-NMR (200 NIHz, DMSO-d6): 6
= 1.04-1.10 (m, I H), 1.44-1.66 (m,
C1 6H), 2.47-2.62 (m, 1H), 2.69-2.84
s
ri I (m, 2H), 2.96-3.13 (m, 4H), 5.06 (s,
CI NH
36 N 1H), 6.88 (d, 2H), 7.91 (s, 2H), 8.23
I
HZN~N N H N
H7(d, 2H), 8.83 (br.s, 1H), 9.55 (br.s,
U I H)
MS (ESIpos): m/z = 488 (M+H)+
HPLC (method 1): Rt=3.61 min
.............
Le A 36 136 CA 02489452 2004-12-14
-70-
Example Structure Analytical data
'H-NMR (400 MHz, DMSO-d6): 6
ci = 1.22-1.28 (m, 1H), 2.15-2.26 (m,
s
N 111), 2.84 (s, 6H), 3.17 (s, 1H), 3.82
CI NH
37 N (m, 3H), 3.92-4.02 (m, 2H) 5.26 (s,
'
~ I N 1H), 7.07 (d, 2H9, 8.09 (s, 2H),
HzN N 1~
~J\ 8.42 (d, 2H), 9.71 (br.s, 1H), 10.05
N-CH3
H3c (br.s, 1 H)
HPLC (method 1): Rt=3.57 min
N MS (ESlpos): m/z = 419 (M+H)'
38N
s YY LC-MS (method 4): R, = 2.30 min
F N
Example 39
N-(2-Amino-4,5'-bipyrimidin-6-yl)-N-[3-fluoro-4-(4-pyridinylsulphanyl)phenyl]-
amine
F
N a,,,, l NH
N
N
H 2 N N'-
N J
100 mg (0.253 mmol) of the compound from Example XI are initially charged in
ml of dimethylformamide, and 350 mg (2.53 mmol) of potassium carbonate are
added. In an argon countercurrent, 52.13 mg (2.53 mmol) of 5-(4,4,5,5-
tetramethyl-
10 1,3,2-dioxaborolan-2-yl)pyrimidine and 20.7 mg (0.025 mmol) of 1,1'-
bis(diphenyl-
phosphine)ferrocene-dichloropalladium(II) complex are added with dichloro-
methane. After a short period of time, the colour of the reaction solution
turns to
............ .
Le A 36 136 CA 02489452 2004-12-14
-71-
black. The mixture is stirred at 120 C overnight. After cooling, the reaction
mixture
is diluted with about 30 ml of ethyl acetate and extracted with water. The
aqueous
phase is re-extracted twice with 20 ml of ethyl acetate. The organic phase is
washed
once with saturated sodium chloride solution, dried over sodium sulphate and
concentrated under reduced pressure. The dark-brown residue is purified on
silica gel
60 (mobile phase: dichloromethane/methanol 20:1). Since it was not possible to
isolate a clean product, the product was purified once more by preparative
HPLC.
This gives 9 mg (8% of theory) of product.
'H-NMR (400 MHz, DMSO-d6): 6 = 6.66 (s, 1H), 7.33 (d, 2H), 7.55-7.65 (m, 2H),
8.40 (dd, 1H), 8.50 (br.s, 2H), 9.25 (s, 2H), 9.32 (s, 1H), 10.26 (br.s 1H).
MS (ESIpos): m/z = 392 (M+H)+
HPLC (method 1): RI = 3.53 min
Example 40
N- {2-Amino-6z[4-(2-pyri dinyl)-1-piperazinyl]-4-pyrimidinyl } -N-[3-fluoro-4-
(4-
pyridinylsulphanyl)phenyl] amine
N
F / NY rNJ
S \ I NYiN
I
NH2
6N
34.78 mg (0.1 mmol) of the compound from Example XVI are dissolved in 0.5 ml
of
dimethylformamide, and 32.6 mg (0.2 mmol) of 1-(2-pyridinyl)piperazine and
25.8 mg (0.2 mmol) of N,N-diisopropylethylamine are added. The mixture is
stirred
at 120 C overnight. For work-up, the reaction solution is purified initially
by LC-MS
and then once more by UV/HPLC.
13.1 mg (20% of theory) of product are obtained.
LC-MS (method 4): Rt = 1.30 min
MS (ESIpos): m/z = 475 (M+H)+
CA 02489452 2004-12-14
Le A 36 136
-72-
The examples listed in the table below can be prepared analogously to the
procedure
described for Example 40 using Example XVI and the appropriate starting
materials.
Example Structure Analytical data
LC-MS (method 4): Rt = 2.80 min
41 MS (ESlpos): m/z = 518 (M+H)+
Hh ~
F LC-MS (method 4): Rt = 0.70 min
42 N 'N MS (ESIpos): m/z = 484 (M+H)+
`
NHi
7LC-MS (method 4): Rt = 0.70 min
43~~ MS (ESIpos): m/z = 482 (M+H)r
rN~o ' LC-MS (method 4): Rt = 2.40 min
44 FlYyY" MS (ESIpos): m/z = 456 (M+H)+
LC-MS (method 4): Rt = 2.40 min
F~N~
45 -~J NhyNN MS (ESlpos): m/z = 455 (M+H)+
NH,
i
LC-MS (method 4): Rt = 2.20 min
~N
46 F Y MS (ESIpos): m/z = 494 (M+H)+
Y
N ,
Le A 36 136 CA 02489452 2004-12-14
-73-
Structure Analytical data
LC-MS (method 4):R1=3.10 min
47 MS (ESIpos): m/z = 4.88 (M+H)+
LC-MS (method 4): Rt = 0.50 min
F N J
48 MS (ESIpos): m/z = 426 (M+H)+
N r-NLC-MS (method 4): Rt = 1.30 min
49 MS (ESIpos): m/z = 466 (M+H)+
LC-MS (method 4): Rt = 0.50 min
50 MS (ESIpos): m/z = 466 (M+H)+
LC-MS (method 4): Rt = 1.90 min
51 MS (ESIpos): m/z = 488 (M+H)+
õ H LC-MS (method 4): Rt = 2.30 min
52 Y N MS (ESIpos): m/z = 528 (M+H)+
NMI
Example 53
N-[2-Amino-6-(4-methoxyphenoxy)-4-pyrimidinyl]-N-[3-fluoro-4-(4-pyridinyl-
sulphanyl)phenyl]amine
Le A 36 136 CA 02489452 2004-12-14
-74-
N
Y F
S
I(tLNH
/ CH3
O
H 2 N N
30 mg (0.086 mmol) of the compound from Example XVI, 37.31 mg (0.345 mmol)
of p-cresol and 9.679 mg (0.173 mmol) of solid potassium hydroxide are mixed
thoroughly and, as a melt, stirred under argon at 150 C for 3 hours. For work-
up, the
melt is purified by column chromatography on silica gel 60 using
dichloromethane/methanol 95:5.
This gives 15 mg (41% of theory) of product.
LC-MS (method): Rt = 2.40 min
MS (ESIpos): m/z 420 (M+H)+
The examples listed in the table below can be prepared analogously to the
procedure
described for Example 53 from the appropriate starting materials.
Le A 36 136 CA 02489452 2004-12-14
-75-
Example Structure Analytical data
F
S
/
g~,
\ NH _
LC-MS (method 10): Rt 2.20 min
54 MS (ESIpos): m/z = 406 (M+H)+
HZN' N\O
F
S
ri LCMIS (method 10): Rt = 2.27 min
NH
55 MS(ESIpos): m/z = 412 (M+H)+
ell
H2N N O`
F LCMS (method 10): R( =2.56 min
s
ri MS(ESIpos): m/z = 440 (M+H)+
NH
56
N `1J~
H2N' N O
cl
B. Assessment of the physiological activity
The inhibition of the enzyme is investigated in an in vitro assay with
recombinant
Rho kinase II. The vessel-relaxing action is determined using phenylephrin-
induced
contractions of isolated rings of the saphenous artery of rabbits. The
suitability of the
compounds according to the invention for treating cardiovascular disorders can
be
demonstrated by examining the hypotensive effect on anaesthetized rats.
Inhibition of recombinant Rho kinase II (ROKa)
CA 02489452 2010-09-09
-76-
The activity of Rho kinase is determined by the uptake of 33P phosphate into a
substrate peptide. To this end, commercially available Rho kinase II (Upstate
Biotechnology) is pre-incubated at 37 C in the presence of the S6 phosphate-
acceptor peptide with the test substances or a solvent control for 10 min. The
kinase
reaction is then started by addition of 33P-labelled ATP. After 20 min at 37
C, the
reaction is stopped by addition of H3PO4. Aliquots are pipetted onto filters
and the
filters are washed and then covered with scintillator. The radioactivity of
the 33P-
labelled peptides bound to the filter is measured in a Micro-Beta counter. The
IC50
value corresponds to the concentration of a test substance at which the Rho-
kinase-
catalysed uptake of 33P into the peptide is inhibited by 50%, compared to a
solvent
control. The experimental data are summarized in the table below.
Example No. IC5o (nM
1 7
11
21 25
22 6
56 70
Vessel-relaxing action in vitro
Individual 3-mm-wide rings of the isolated saphenous artery of rabbits are
introduced
into 5 ml organ baths with Krebs-Henseleit solution (37 C, gassed with
carbogen).
The vessel tone is monitored isometrically and registered. Contractions are
induced
by addition of 3x10"8 g of phenylephrin/ml, which is washed out again after 4
min.
After a number of control cycles, the rings are pre-incubated with the
substance to be
examined, with the dosage being increased for each further cycle, and the
subsequent
contraction is compared to the intensity of the last control contraction. The
concentration required to reduce the intensity of the control value by 50%
(IC50) is
calculated. The experimental data are summarized in the table below.
*Trade-mark
Le A 36 136 CA 02489452 2004-12-14
-77-
Example No. IC50 (nM)
1 760
20 1700
21 870
22 900
Measurement of blood pressure in anaesthetized rats
Male Wistar rats of a body weight of 300 - 350 g are anaesthetized with
thiopental
(100 mg/kg i.p.). Following tracheotomy, a catheter is introduced into the
femoral
artery to measure the blood pressure. The substances to be tested are
administered as
solutions, either orally via a stomach tube or intravenously via the femoral
vein.
C. Working examples for pharmaceutical compositions
The compounds according to the invention can be converted into pharmaceutical
preparations as follows:
Tablet:
Composition:
100 mg of the compound from Example 1, 50 mg of lactose (monohydrate), 50 mg
of
maize starch (native), 10 mg of polyvinylpyrrolidone (PVP 25) (from BASF,
Ludwigshafen, Germany) and 2 mg of magnesium stearate.
Tablet weight 212 mg. Diameter 8 mm, spherical radius 12 mm.
Preparation:
The mixture of active compound, lactose and starch is granulated with a 5%
strength
solution (w/w) of the PVP in water. After drying, the granules are mixed for 5
min
with the magnesium stearate. This mixture is compacted in a conventional
tablet
press (dimensions of the tablet: see above). The standard value used for
compacting
is a compaction force of 15 kN.
CA 02489452 2010-09-09
-78-
Suspension for oral administration:
Composition:
1000 mg of the compound from Example 1, 1000 mg of ethanol (96%), 400 mg of
Rhodigel (xanthan gum from FMC, Pennsylvania, USA) and 99 g of water.
A single dose of 100 mg of the compound according to the invention corresponds
to
ml of oral suspension.
Preparation:
10 The Rhodigel is suspended in ethanol and the active compound is added to
the
suspension. The water is added with stirring. The mixture is stirred for about
6 h until
the Rhodigel is completely swollen.
*Trade-mark