Note: Descriptions are shown in the official language in which they were submitted.
CA 02494322 2005-02-01
WO 2004/013204 PCT/US2003/023034
POLYMER EXTRACTION METHODS
Technical Field
The invention relates to polymer extraction methods.
Background
A polyhydroxyalkanoate ("PHA") can be extracted from biomass having cells
that contain the PHA. Generally, this process involves combining the biomass
with a
solvent for the PHA, followed by heating and agitation. Typically, this
provides a
system including two phases, with one phase being a solution that contains the
solvent
and the PHA, and the other phase containing residual biomass with cells
containing a
reduced amount of the PHA. Usually, the two phases are separated, and the PHA
is
then removed from the solvent.
Summ
In general, the invention relates to polymer extraction methods.
In one aspect, the invention features a method of separating a polymer from a
biomass containing the polymer. The method includes contacting the biomass
with a
solvent system to provide a residual biomass and a solution. The solvent
system
includes a solvent for the polymer and a precipitant for the polymer, and the
solution
includes the polymer, the solvent for the polymer and the precipitant for the
polymer.
The method also includes applying a centrifugal force to the solution and
residual
biomass to separate at least some of the solution from the residual biomass.
In another aspect, the invention features a method of separating a polymer
from biomass containing the polymer. The method includes contacting the
biomass
with a solvent system to provide a residual biomass and a solution including
the
polymer and the solvent system, and separating at least some of the solution
from the
residual biomass. The method also includes adding a precipitant for the
polymer to
the solution to remove at least some of the polymer from the solvent system.
In a further aspect, the invention features a method of separating a polymer
from biomass containing the polymer. The method includes contacting the
biomass
with a solvent system to provide a residual biomass and a solution that
includes the
polymer and the solvent system. The solution has a polymer concentration of at
least
1
CA 02494322 2005-02-01
WO 2004/013204 PCT/US2003/023034
about two percent and a viscosity of at most about 100 centipoise. The method
also
includes separating at least some of the solution from the residual biomass.
In one aspect, the invention features a method of separating a polymer from
biomass containing the polymer. The method includes contacting the biomass
with a
solvent system to provide a residual biomass and a solution. The solvent
system
includes a solvent for the polymer, and the solution includes the polymer and
the
solvent for the polymer. The solvent for the polymer may have a boiling point
greater
than 100 C. The method also includes separating the polymer from the residual
biomass.
In another aspect, the invention features a method of separating a polymer
from biomass containing the polymer. The method includes contacting the
biomass
with a volume of a solvent system to provide a residual biomass and a solution
including the polymer and the solvent for the polymer, and separating at least
some of
the solution from the residual biomass. The method also includes adding a
volume of
a precipitant for the polymer to the separated solution to remove at least
some of the
polymer from the solution. The volume of the precipitant added is less than
about two
parts relative to the volume of the solvent system.
In a further aspect; the invention features a method of separating a polymer
from a biomass containing the polymer and biomass impurities. The method
includes
contacting the biomass with a precipitant for the polymer to remove at least
some of
the biomass impurities from the biomass containing the polymer and the biomass
impurities, thereby providing a purified biomass containing the polymer. The
method
also includes contacting the purified biomass with a solvent system to provide
a
residual biomass and a solution including the polymer and the solvent for the
polymer.
In another aspect, the invention features a method of separating a polymer
from the biomass containing the polymer and biomass impurities. The method
includes pre-treating the biomass chemically to remove at least some of the
biomass
impurities from the biomass containing the biomass and the impurities, thereby
providing a purified biomass containing the polymer. The chemical treatments
include manipulation of pH, temperature and contact time with or without the
presence of additional chemicals such as surfactants, detergents, enzymes or
similar
2
CA 02494322 2005-02-01
WO 2004/013204 PCT/US2003/023034
materials that can aid removal of the biomass impurities. The method also
includes
contacting the purified biomass with a solvent system to provide a residual
biomass
and a solution including the polymer and the solvent for the polymer.
In one aspect, the invention features a method of separating a polymer from a
biomass containing the polymer. The method includes contacting the biomass
with a
solvent system under countercurrent flow conditions.
In another aspect, the invention features a method of separating a polymer
from a biomass containing the polymer. The method includes contacting the
biomass
with a solvent system using a one-stage process that forms a PHA phase and a
residual biomass phase. The ratio of volume of the solvent system present in
the PHA
phase to volume of the solvent system contacted with the biomass is at least
about 0.8.
In a further aspect, the invention features a method of separating a polymer
from a biomass containing the polymer. The method includes contacting the
biomass
with a solvent system using a one-stage process that forms a PHA phase and a
residual biomass phase. The ratio of volume of the solvent system present in
the
residual biomass phase to volume of the solvent system contacted with the
biomass is
at most about 0.2.
In certain embodiments, the methods can extract polymer (e.g., PHA) from
biomass in relatively high yield. In some embodiments, a relatively high yield
of
polymer (e.g., PHA) can be extracted from biomass without using multiple
stages
(e.g., with a one-stage process).
In some embodiments, the methods can extract relatively pure polymer (e.g.,
PHA).
In certain embodiments, the methods can use solvent(s) and/or precipitant(s)
in a relatively efficient manner. For example, a relatively high percentage of
the
solvent(s) and/or precipitant(s) used in the methods can be recovered (e.g.,
for re-use).
In some embodiments, the methods can have a reduced environmental impact.
In certain embodiments, the methods can extract the polymer at relatively high
space velocity (e.g. at high throughput with overall low residence time in
process
equipment).
In certain embodiments, the methods can result in a relatively small amount of
undesirable reaction side products (e.g., organic acids). This can, for
example,
3
CA 02494322 2010-09-16
51668-21
decrease the likelihood of corrosion or other undesirable damage to systems
used
in the methods and/or extend the useful lifetime of such systems.
In some embodiments, the methods can provide relatively high
volumetric throughput (e.g., by using a one-stage process).
In certain embodiments, the methods can provide relatively high
solvent recovery.
In certain embodiments, the process can be performed with
one-stage device (e.g., a countercurrent centrifugal contacter).
In some embodiments, a relatively low viscosity residual biomass is
formed (e.g., using countercurrent conditions), which can enhance subsequent
processing such as stripping of residual solvent and concentration of the
solids
content (e.g. by evaporation, filtration or drying).
According to one aspect of the present invention, there is provided a
method of separating a polymer from a biomass containing the polymer, the
method comprising: contacting the biomass with a solvent system, including a
solvent for the polymer and a precipitant for the polymer, to provide a
residual
biomass and a solution that includes the polymer, the solvent for the polymer
and
the precipitant for the polymer; and applying a centrifugal force to the
solution and
residual biomass to separate at least some of the solution from the residual
biomass; wherein: the solvent system comprises a solvent for the polymer that
is a
ketone, ester or alcohol; the polymer is a polyhydroxyalkanoate ("PHA"); and
the
biomass comprises a slurry in water.
According to another aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer has
a
logK value relative to water at 100 C of at least 1.5.
According to still another aspect of the present invention, there is
provided the method described herein, wherein the solution comprises at most
25% by volume of the precipitant for the polymer.
4
CA 02494322 2010-09-16
51668-21
According to yet another aspect of the present invention, there is
provided the method described herein, further comprising removing at least
some
of the polymer from the solution.
According to a further aspect of the present invention, there is
provided the method described herein, wherein removing at least some of the
polymer from the solution includes adding a second precipitant for the polymer
to
the solution.
According to yet a further aspect of the present invention, there is
provided the method described herein, wherein the first and second
precipitants
for the polymer are the same.
According to still a further aspect of the present invention, there is
provided the method described herein, further comprising evaporating a portion
of
the solution before removing at least some of the polymer from the solution.
According to another aspect of the present invention, there is provided
the method described herein, further comprising, after applying the
centrifugal force
to the solution, adding a volume of a second precipitant for the polymer to
remove
at least some of the polymer from the solution, wherein the volume of the
second
precipitant is less than two parts relative to the volume of the solvent
system.
According to yet another aspect of the present invention, there is
provided the method described herein, wherein the precipitant for the polymer
dissolves less than 0.2% by weight of the polymer at room temperature.
According to another aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer and
the
precipitant for the polymer have a relative volatility of at least two at an
equimolar
bubble point for the solvent for the polymer and the precipitant for the
polymer.
According to still another aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer and
the precipitant for the polymer do not form an azeotrope.
4a
CA 02494322 2010-09-16
51668-21
According to yet another aspect of the present invention, there is
provided the method described herein, wherein the precipitant for the polymer
comprises an alkane.
According to a further aspect of the present invention, there is
provided the method described herein, wherein the solution has a polymer
concentration of at least two weight percent.
According to yet a further aspect of the present invention, there is
provided the method described herein, wherein the solution has a viscosity of
at
most 100 centipoise.
According to still a further aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer has
a
density of less than 0.95 kilograms per liter.
According to another aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer is a
non-halogenated solvent.
According to yet another aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer has
a
solubility in water of less than one percent by weight.
According to another aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer has
a
boiling point greater than 100 C.
According to still another aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer is
methyl isobutyl ketone (MIBK), butyl acetate, cyclohexanone or a combination
thereof.
According to yet another aspect of the present invention, there is
provided the method described herein, wherein the biomass containing the
polymer
is of microbial origin and has a polymer content of at least 50 weight
percent.
4b
CA 02494322 2010-09-16
51668-21
According to a further aspect of the present invention, there is
provided the method described herein, wherein the biomass containing the
polymer
is of plant origin and has a polymer content of less than 50 weight percent.
According to yet a further aspect of the present invention, there is
provided the method described herein, wherein the solvent system is contacted
with the biomass under countercurrent flow conditions.
According to still a further aspect of the present invention, there is
provided the method described herein, wherein the ratio of volume of the
solvent
system present to volume of the solvent system contacted with the biomass is
at
least 0.8.
According to another aspect of the present invention, there is provided
the method described herein, wherein the method is a one-stage method.
According to yet another aspect of the present invention, there is
provided the method described herein, wherein the method is a multi-stage
method.
According to another aspect of the present invention, there is
provided the method described herein, wherein the countercurrent flow
conditions
include a pressure of at least 65 psig.
According to a further aspect of the present invention, there is
provided a method of separating a polymer from biomass containing the polymer,
the method comprising: contacting the biomass with a solvent system to provide
a
residual biomass and a solution including the polymer and the solvent system;
adding a precipitant for the polymer to the solution; and separating at least
some
of the solution from the residual biomass after adding the precipitant for the
polymer; wherein: the solvent system comprises a solvent for the polymer that
is a
ketone, ester or alcohol; the polymer is a polyhydroxyalkanoate ("PHA"); and
the
biomass comprises a slurry in water.
According to yet a further aspect of the present invention, there is
provided the method described herein, wherein the precipitant for the polymer
dissolves less than 0.2% by weight of the polymer at room temperature.
4c
CA 02494322 2010-09-16
51668-21
According to still a further aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer and
the
precipitant for the polymer have a relative volatility of at least two at an
equimolar
bubble point for the solvent for the polymer and the precipitant for the
polymer.
According to another aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer and
the precipitant for the polymer do not form an azeotrope.
According to yet another aspect of the present invention, there is
provided the method described herein, wherein the precipitant for the polymer
comprises an alkane.
According to another aspect of the present invention, there is
provided the method described herein, wherein the solution has a polymer
concentration of at least two weight percent.
According to still another aspect of the present invention, there is
provided the method described herein, wherein the solution has a viscosity of
at
most 100 centipoise.
According to yet another aspect of the present invention, there is
provided the method described herein, further comprising evaporating a portion
of
the solution before adding the precipitant for the polymer to the solution to
remove
at least some of the polymer from the solvent system.
According to a further aspect of the present invention, there is
provided the method described herein, further comprising, after separating,
adding
a volume of a second precipitant for the polymer to remove at least some of
the
polymer from the solution, wherein the volume of the second precipitant is
less
than two parts relative to the volume of the solvent system.
According to yet a further aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer that
has a density of less than 0.95 kilograms per liter.
4d
CA 02494322 2010-09-16
51668-21
According to still a further aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer is a
non-halogenated solvent.
According to another aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer has
a
boiling point greater than 100 C.
According to yet another aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer is
methyl isobutyl ketone (MIBK), butyl acetate, cyclohexanone or a combination
thereof.
According to another aspect of the present invention, there is provided
the method described herein, wherein the biomass containing the polymer is of
microbial origin and has a polymer content of at least 50 weight percent.
According to still another aspect of the present invention, there is
provided the method described herein, wherein the biomass containing the
polymer
is of plant origin and has a polymer content of less than 50 weight percent.
According to yet another aspect of the present invention, there is
provided the method described herein, wherein separating at least some of the
solution from the residual biomass includes applying a centrifugal force to
the
solution and the residual biomass.
According to a further aspect of the present invention, there is
provided the method described herein, wherein the solvent system is contacted
with the biomass under countercurrent flow conditions.
According to yet a further aspect of the present invention, there is
provided a method of separating a polymer from biomass containing the polymer,
the
method comprising: contacting the biomass with a solvent system to provide a
residual biomass and a solution including the polymer and the solvent system,
the
solution having a polymer concentration of at least two weight percent and a
viscosity
of at most 100 centipoise; and separating at least some of the solution from
the
4e
CA 02494322 2010-09-16
51668-21
residual biomass; wherein: the solvent system comprises at least one solvent
for the
polymer that is a ketone, ester or alcohol; the polymer is a
polyhydroxyalkanoate
("PHA"); and the biomass comprises a slurry in water.
According to still a further aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer that
has a density of less than 0.95 kilograms per liter.
According to another aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer is a
non-halogenated solvent.
According to yet another aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer has
a
solubility in water of less than one percent by weight.
According to another aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer has
a
boiling point greater than 100 C.
According to still another aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer is
methyl isobutyl ketone (MIBK), butyl acetate, cyclohexanone or a combination
thereof.
According to yet another aspect of the present invention, there is
provided the method described herein, wherein the solvent system further
comprises a precipitant for the polymer that dissolves less than 0.2 wt.% of
the
polymer at room temperature.
According to a further aspect of the present invention, there is provided
the method described herein, wherein the solvent system further comprises a
precipitant for the polymer, the solvent for the polymer and the precipitant
for the
polymer having a relative volatility of at least two at an equimolar bubble
point for the
solvent for the polymer and the precipitant for the polymer.
4f
CA 02494322 2010-09-16
51668-21
According to yet a further aspect of the present invention, there is
provided the method described herein, wherein the solvent system further
comprises a precipitant for the polymer, and the solvent for the polymer and
the
precipitant for the polymer do not form an azeotrope.
According to still a further aspect of the present invention, there is
provided the method described herein, wherein the solvent system further
comprises a precipitant for the polymer that comprises an alkane.
According to another aspect of the present invention, there is provided
the method described herein, wherein the biomass containing the polymer is of
microbial origin and has a polymer content of at least 50 weight percent.
According to yet another aspect of the present invention, there is
provided the method described herein, wherein the biomass containing the
polymer
is of plant origin and has a polymer content of less than 50 weight percent.
According to another aspect of the present invention, there is
provided the method described herein, further comprising removing at least
some
of the polymer from the solution.
According to still another aspect of the present invention, there is
provided the method described herein, wherein removing at least some of the
polymer from the solution includes adding a precipitant for the polymer to the
solution.
According to yet another aspect of the present invention, there is
provided the method described herein, further comprising evaporating a portion
of
the solution before removing at least some of the polymer from the solution.
According to a further aspect of the present invention, there is
provided the method described herein, wherein separating at least some of the
solution from the residual biomass includes applying a centrifugal force to
the
solution and the residual biomass.
4g
CA 02494322 2010-09-16
51668-21
According to yet a further aspect of the present invention, there is
provided the method described herein, wherein the solvent system is contacted
with the biomass under countercurrent flow conditions.
According to still a further aspect of the present invention, there is
provided a method of separating a polymer from a biomass containing the
polymer
and biomass impurities, the method comprising: contacting the biomass with at
least one alkane to remove at least some of the biomass impurities from the
biomass containing the polymer and the biomass impurities, thereby providing a
purified biomass containing the polymer; and contacting the purified biomass
with
a solvent system comprising a solvent for the polymer to provide a residual
biomass and a solution including the polymer and the solvent for the polymer;
wherein: the solvent system comprises at least one solvent that is methyl
isobutyl
ketone ( MIBK), butyl acetate, cyclo-hexanone or a combination thereof; and
the
polymer is a polyhydroxyalkanoate.
According to another aspect of the present invention, there is provided
the method described herein, further comprising separating at least some of
the
solution from the residual biomass, and adding a precipitant for the polymer
to the
solution to remove at least some of the polymer from the solution.
According to yet another aspect of the present invention, there is
provided the method described herein, wherein the solvent system comprises a
solvent for the polymer that has a density of less than 0.95 kilograms per
liter.
According to another aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer
comprises a non-halogenated solvent.
According to still another aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer has
a
solubility in water of less than one weight percent.
According to yet another aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer that
has a boiling point greater than 100 C.
4h
CA 02494322 2010-09-16
51668-21
According to a further aspect of the present invention, there is
provided the method described herein, wherein the solvent system comprises a
precipitant for the polymer that dissolves less than 0.2% by weight of the
polymer
at room temperature.
According to yet a further aspect of the present invention, there is
provided the method described herein, wherein the solvent system further
comprises a precipitant for the polymer, and the solvent for the polymer and
the
precipitant for the polymer have a relative volatility of at least two at an
equimolar
bubble point for the solvent for the polymer and the precipitant for the
polymer.
According to still a further aspect of the present invention, there is
provided the method described herein, wherein the solvent system further
comprises a precipitant for the polymer, and the solvent for the polymer and
the
precipitant for the polymer do not form an azeotrope.
According to another aspect of the present invention, there is
provided the method described herein, wherein the alkane is hexane, heptane or
an isoalkane.
According to yet another aspect of the present invention, there is
provided the method described herein, wherein the solution has a polymer
concentration of at least two weight percent.
According to another aspect of the present invention, there is
provided the method described herein, wherein the solution has a viscosity of
at
most 100 centipoise.
According to still another aspect of the present invention, there is
provided the method described herein, wherein the biomass containing the
polymer
is of microbial origin and has a polymer content of at least 50 weight
percent.
According to yet another aspect of the present invention, there is
provided the method described herein, wherein the biomass containing the
polymer
is of plant origin and has a polymer content of less than 50 weight percent.
4i
CA 02494322 2010-09-16
51668-21
According to a further aspect of the present invention, there is
provided the method described herein, wherein separating at least some of the
solution from the residual biomass includes applying a centrifugal force to
the
solution and the residual biomass.
According to yet a further aspect of the present invention, there is
provided the method described herein, further comprising evaporating a portion
of
the solution before adding a precipitant for the polymer to the solution to
remove
at least some of the polymer from the solution.
According to still a further aspect of the present invention, there is
provided the method described herein, wherein the at least one alkane is
contacted with the biomass under countercurrent flow conditions.
According to another aspect of the present invention, there is provided
the method described herein, wherein the method is a one-stage method.
According to yet another aspect of the present invention, there is
provided the method described herein, wherein the method is a multi-stage
method.
According to another aspect of the present invention, there is
provided the method described herein, wherein the countercurrent conditions
include a pressure of at least 65 psig.
According to still another aspect of the present invention, there is
provided a method of separating a polymer from a biomass containing the
polymer,
the method comprising: contacting the biomass with a solvent for the polymer,
to
provide a residual biomass and a solution that includes the polymer, and the
solvent
for the polymer; and applying a centrifugal force to the solution and residual
biomass to separate at least some of the solution from the residual biomass;
wherein: the polymer is a polyhydroxyalkanoate; the solvent for the polymer is
3-methyl-2-pentanone, 3-methyl-2-butanone, 2-pentanone, diisobutyl ketone,
2-hexanone, 3-pentanone, 2-methyl-3-heptanone, 3-heptanone, 2-octanone,
5-methyl-3-heptanone, 5-methyl-2-hexanone, heptanone, or cyclopentanone; and
the biomass comprises a slurry in water.
4j
CA 02494322 2010-09-16
51668-21
According to yet another aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer is
3-methyl-2-pentanone, 3-methyl-2-butanone, 2-pentanone, diisobutyl ketone,
2-hexanone, 3-pentanone, 2-methyl-3-heptanone, 3-heptanone, 2-octanone,
5-methyl-3-heptanone, 5-methyl-2-hexanone, or heptanone.
According to a further aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer is
2-pentanone, 2-hexanone, 3-pentanone, 2-methyl-3-heptanone, 3-heptanone,
2-octanone, 5-methyl-3-heptanone, 5-methyl-2-hexanone, or heptanone.
According to yet a further aspect of the present invention, there is
provided the method described herein, wherein the solvent for the polymer is
2-pentanone, 2-hexanone, or 3-pentanone.
According to still a further aspect of the present invention, there is
provided the method described herein further comprising contacting the biomass
with a precipitant for the polymer.
According to another aspect of the present invention, there is provided
the method described herein, wherein the precipitant comprises at least one
alkane.
According to yet another aspect of the present invention, there is
provided the method described herein, wherein the solution has a viscosity of
at
most 100 centipoise.
According to another aspect of the present invention, there is provided
the method described herein, wherein the biomass containing the polymer is of
microbial origin and has a polymer content of at least 50 weight percent.
According to still another aspect of the present invention, there is
provided the method described herein, wherein the biomass containing the
polymer
is of plant origin and has a polymer content of less than 50 weight percent.
According to yet another aspect of the present invention, there is
provided the method described herein, further comprising removing at least
some
of the polymer from the solution.
4k
CA 02494322 2010-09-16
51668-21
According to a further aspect of the present invention, there is
provided the method described herein, wherein removing at least some of the
polymer from the solution includes adding an additional precipitant for the
polymer
to the solution.
According to yet a further aspect of the present invention, there is
provided the method described herein, further comprising, after applying the
centrifugal force to the solution, adding a volume of an additional
precipitant for
the polymer to remove at least some of the polymer from the solution, wherein
the
volume of the additional precipitant is less than two parts relative to the
volume of
the solvent system.
According to still a further aspect of the present invention, there is
provided the method described herein, wherein the biomass is a dried biomass.
Features, objects and advantages of the invention are in the
description, drawings and claims.
Brief Description of the Drawing
Fig. I is a flow diagram of an embodiment of a method of extracting
PHA from a biomass with cells containing PHA;
Fig. 2 is a flow diagram of a portion of an embodiment of a method
of extracting PHA from a biomass with cells containing PHA; and
Fig. 3 is a graph showing viscosity and polymer content from
Example Ill.
Detailed Description
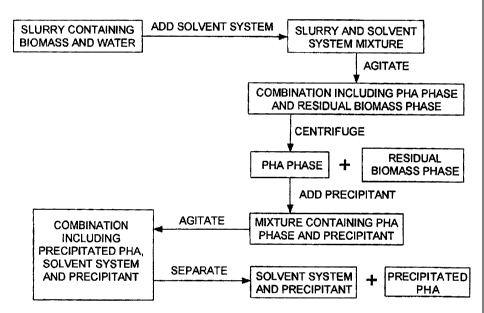
Fig. 1 is a flow diagram of an embodiment of a process for extracting
a PHA from biomass having cells that contain the PHA. A slurry is provided
that
contains the biomass and water. A solvent system is added to the slurry to
form a
mixture that contains the slurry and the solvent system. The mixture is
agitated
(e.g., stirred) to provide a combination that includes two phases. One phase
is
formed of a solution containing the PHA and the solvent system with trace
41
CA 02494322 2010-09-16
51668-21
amounts of biomass ("the PHA phase"). The second phase is formed of residual
biomass having cells with reduced polymer content, water and a carry over
portion
of the solvent system ("the residual biomass phase"). The two phases included
in
the combination are separated using an appropriate device that exploits
centrifugal force to facilitate the separation (e.g. disc centrifuge, bowl
centrifuge,
decanter centrifuge, hydroclone, countercurrent
4m
CA 02494322 2005-02-01
WO 2004/013204 PCT/US2003/023034
centrifugal contacter). Optionally, one or more solvents can be added to the
device
that exploits centrifugal force to facilitate the separation. A precipitant
for the PHA is
added to the PHA phase to form a mixture that contains the PHA phase and the
precipitant. The mixture is agitated (e.g., stirred) to form a combination
that contains
precipitated PHA, the solvent system and the precipitant. In certain
embodiments, the
solvent system and the precipitant are miscible which results in the
combination
(precipitated PHA, solvent system and precipitant) having two phases (e.g.,
one phase
containing the precipitated PHA, and one phase containing the solvent system
and
precipitant). The combination (precipitated PHA, solvent system and
precipitant) is
separated (e.g., by filtration or using centrifugal force) to provide the
isolated,
extracted PHA.
The process in Fig. 1 can be referred to as a one-stage process. In general, a
one-stage process is a process in which only one centrifugation step is used
during
separation of the polymer (e.g., PHA) from the biomass. In general, a multi-
stage
process refers to a process in which more than one centrifugation step is used
during
separation of the polymer (e.g., PHA) from the biomass (see additional
discussion
below). For example, the residual biomass formed in the process in Fig. 1 can
be
treated and ultimately centrifuged, thereby creating a two-stage process (see,
for
example, Fig. 2 and discussion below).
In some embodiments, the process results in a relatively high yield of the
PHA. For example, in some embodiments a ratio of the dry weight of extracted
PHA
to the dry weight of the PHA initially contained in the biomass is at least
about 0.9
(e.g., at least about 0.95, at least about 0.97, at least about 0.98). In
certain
embodiments, a relatively high yield of PHA can be achieved without using a
multi-
stage process (e.g., with a one-stage process).
In certain embodiments, the process can be performed with relatively large
amount of the solvent being transferred to the PHA phase. For example, in some
embodiments a ratio of the volume of solvent recovered in the PHA phase to the
volume of solvent contacted with the biomass is at least about 0.8 (e.g.,
0.85, at least
about 0.9, at least about 0.95, at least about 0.98, at least about 0.99). In
some
embodiments, a relatively large amount of solvent can be transferred to the
PHA
5
CA 02494322 2005-02-01
WO 2004/013204 PCT/US2003/023034
phase using, for example, countercurrent conditions during separation of the
polymer
(e.g., PHA) from the biomass.
In certain embodiments, the process can be performed with a relatively small
amount of the solvent being transferred to the residual biomass phase. For
example,
in some embodiments a ratio of the volume of solvent recovered in the residual
biomass phase to the volume of solvent contacted with the biomass is at most
about
0.2 (e.g., at most about 0.15, at most about 0.1, at most about 0.05, at most
about 0.02,
at most about 0.01). In some embodiments, a relatively small amount of the
solvent is
transferred to the residual biomass phase using, for example, countercurrent
conditions during separation of the polymer (e.g., PHA) from the biomass.
The slurry can be provided in any desired manner. Typically, the slurry is
provided by forming a fermentation broth containing water and the biomass, and
removing a portion of the water from the fermentation broth. The water can be
removed, for example, by filtration (e.g., microfiltration, membrane
filtration) and/or
by decanting and/or by using centrifugal force . In certain embodiments,
biomass
impurities, such as cell wall and cell membrane impurities, can be removed
during the
process of providing the slurry. Such impurities can include proteins, lipids
(e.g.,
triglycerides, phospholipids, and lipoproteins) and lipopolysaccharides.
The PHA content of the biomass (e.g., PHA content of the dry biomass,
20, inclusive of its polymer content, on a weight percent basis) can be varied
as desired.
As an example, in embodiments in which the biomass is of microbial origin, the
biomass can have a PHA content of at least about 50 weight percent (e.g., at
least
about 60 weight percent, at least about 70 weight percent, at least about 80
weight
percent). As another example, in embodiments in which the biomass is of plant
origin, the biomass can have a PHA content of less than about 50 weight
percent (e.g.,
less than about 40 weight percent, less than about 30 weight percent, less
than about
20 weight percent).
In some embodiments, the slurry has a solids content (e.g., dry biomass,
inclusive of its PHA content, weight relative to total wet weight of slurry)
of from
about 25 weight percent to about 40 weight percent (e.g., from about 25 weight
percent to about 35 weight percent).
6
CA 02494322 2010-04-14
51668-21
The biomass can be formed of one or more of a variety of entities. Such
entities include, for example, microbial strains for producing PHAs (e.g.,
Alcaligenes
eutrophus (renamed as Ralstonia eutropha), Alcaligenes latus, Azotobacter,
Aeromonas, Comamonas, Pseudomonads), genetically engineered organisms for
producing PHAs (e.g., Pseudomonas, Ralstonia, Escherichia coli, Klebsiella),
yeasts
for producing PHAs, and plant systems for producing PHAs. Such entities are
disclosed, for example, in Lee, Biotechnology & Bioengineering 49:1-14 (1996);
Braunegg et al., (1998), J. Biotechnology 65: 127-161; Madison and Huisman,
1999;
and Snell and Peoples 2002, Metabolic Engineering 4: 29-40.
In embodiments in which the biomass contains microbial cells, the size of the
microbial cells contained in the biomass can also be varied as desired. In
general, the
microbial cells (e.g., bacterial cells) have at least one dimension with a
size of at least
about 0.2 micron (e.g., at least about 0.5 micron, at least about one micron,
at least
about two micronsõ at least about three microns, at least about four microns,
at least
about five microns). In certain embodiments, using relatively large microbial
cells
(e.g., relatively large bacterial cells) in the biomass can be advantageous
because it
can facilitate the separation of the biomass to form the biomass slurry.
In general, a PHA is formed by polymerization (e.g., enzymatic
polymerization) of one or more monomer units. Examples of such monomer units
include, for example, 3-hydroxybutyrate, 3-hydroxypropionate, 3-
hydroxyvalerate, 3-
hydroxyhexanoate, 3-hydroxyheptanoate, 3-hydroxyoctanoate, 3-hydroxynonaoate,
3-
hydroxydecanoate, 3-hydroxydodecanoate, 3-hydroxydodecenoate, 3-
hydroxytetradecanoate, 3-hydroxyhexadecanoate, 3-hydroxyoctadecanoate, 4-
hydroxybutyrate, 4-hydroxyvalerate, 5-hydroxyvalerate, and 6-hydroxyhexanoate.
In some embodiments, the PHA has at least one monomer unit with the
chemical formula -OCR1R2(CR3R4)õCO-. n is zero or an integer (e.g., one, two ,
three, four, five, six, seven, eight, nine, 10, 11, 12, 13, 14, 15, etc.).
Each of R1, R2,
R3 and R4 is a hydrogen atom, a saturated hydrocarbon radical, an unsaturated
hydrocarbon radical, a substituted radical (e.g., a substituted hydrocarbon
radical) or
an unsubstituted radical (e.g., an unsubstituted hydrocarbon radical).
Examples of
substituted radicals include halo-substituted radicals (e.g., halo substituted
7
CA 02494322 2005-02-01
WO 2004/013204 PCT/US2003/023034
hydrocarbon radicals), hydroxy-substituted radicals (e.g., hydroxy-substituted
hydrocarbon radicals), halogen radicals, nitrogen-substituted radicals (e.g.,
nitrogen-
substituted hydrocarbon radicals) and oxygen-substituted radicals (e.g.,
oxygen-
substituted hydrocarbon radicals). Substituted radicals include, for example,
substituted, saturated hydrocarbon radicals and substituted, unsaturated
hydrocarbon
radicals. Ri is the same as or different from each of R2, R3 and R4. R2 is the
same as
or different from each of R1, R3 and R4. R3 is the same as or different from
each of
R2, Rl and R4, and R4 is the same as or different from each of R2, R3 and Rt.
In some embodiments, the PHA is a copolymer that contains two or more
different monomer units. Examples of such copolymers include poly-3-
hydroxybutyrate-co-3-hydroxypropionate, poly-3-hydroxybutyrate-co-3-
hydroxyvalerate, poly-3-hydroxybutyrate-co-3-hydroxyhexanoate, poly-3-
hydroxybutyrate-co-4-hydroxybutyrate, poly-3-hydroxybutyrate-co-4-
hydroxyvalerate, poly-3-hydroxybutyrate-co-6-hydroxyhexanoate, poly 3-
hydroxybutyrate-co-3-hydroxyheptanoate, poly-3-hydroxybutyrate-co-3-
hydroxyoctanoate, poly-3-hydroxybutyrate-co-3-hydroxydecanoate, poly-3-
hydroxybutyrate-co-3-hydroxydodecanotate, poly-3-hydroxybutyrate-co-3-
hydroxyoctanoate -co-3-hydroxydecanoate, poly-3-hydroxydecanoate-co-3-
hydroxyoctanoate, and poly-3-hydroxybutyrate-co-3-hydroxyoctadecanoate.
In certain embodiments, the PHA is a homopolymer. Examples of such
homopolymers include poly-4-hydroxybutyrate, poly-3-hydroxypropionate, poly-3-
hydroxybutyrate, poly-3-hydroxyhexanoate, poly-3-hydroxyheptanoate, poly-3-
hydroxyoctanoate, poly-3-hydroxydecanoate and poly-3-hydroxydodecanoate.
The PHA can have a polystyrene equivalent weight average molecular weight
of at least about 500 (e.g., at least about 10,000, at least about 50,000)
and/or less than
about 2,000,000 (e.g., less than about 1,000,000, less than about 800,000).
In general, the amount of solvent system added to the slurry can be varied as
desired. In certain embodiments, an amount of solvent system is added to the
slurry
so that, after centrifugation, the PHA phase has a PHA solids content of less
than
about 10 weight percent (e.g., less than about eight weight percent, less than
about six
weight percent, less than about five weight percent, less than about four
weight
percent, less than about three weight percent).
8
CA 02494322 2005-02-01
WO 2004/013204 PCT/US2003/023034
The solvent system includes one or more solvents for the PHA, and can
optionally include one or more precipitants for the PHA. Without wishing to be
bound by theory, it is believed that including a precipitant for the PHA in
the solvent
system can reduce the viscosity of the solution containing the polymer and the
solvent
system and/or enhance the selectivity of the process in extracting the desired
PHA.
In general, a solvent for a given polymer is capable of dissolving the polymer
to form a substantially uniform solution at the molecular or ionic size level.
In
general, a precipitant for a given polymer is capable of inducing the
precipitation of
the polymer and/or weakening the solvent power of a solvent for the polymer.
The choice of solvent(s) and/or precipitant(s) generally depends on the given
PHA to be purified. Without wishing to be bound by theory, it is believed that
an
appropriate solvent for a given polymer can be selected by substantially
matching
appropriate solvation parameters (e.g., dispersive forces, hydrogen bonding
forces
and/or polarity) of the given polymer and solvent. Such solvation parameters
are
disclosed, for example, in Hansen, Solubility Parameters - A User's Handbook,
CRC
Press, NY, NY (2000).
In certain embodiments in which the PHA is a poly-3-hydroxybutyrate
copolymer (e.g., poly-3-hydroxybutyrate-co-3-hydroxypropionate, poly-3-
hydroxybutyrate-co-3-hydroxyvalerate, poly-3-hydroxybutyrate-co-3-
hydroxyhexanoate and/or poly-3-hydroxybutyrate-co-4-hydroxybutyrate, poly-3-
hydroxybutyrate-co-3-hydroxyoctanoate-co-3-hydroxydecanote-co-3-
hydroxydodecanote), where the majority of the monomer units are 3-
hydroxybutyrate
(e.g., at least about 50% of the monomer units are 3-hydroxybutyrate, at least
about
60% of the monomer units are 3-hydroxybutyrate), the solvent(s) maybe selected
from ketones, esters and/or alcohols with at least four carbon atoms, and the
precipitant(s) may be selected from alkanes, methanol and ethanol.
In some embodiments in which the PHA is poly-3-hydroxyoctanoate, the
solvent(s) may be selected from ketones, esters, alcohols with at least four
carbon
atoms or alkanes (e.g., hexane).
In general, the ketones can be cyclic or acyclic, straight-chained or
branched,
and/or substituted or unsubstituted. Examples of acyclic ketones and cyclic
ketones
include methyl isobutyl ketone ("MIBK"), 3-methyl-2-pentanone (butyl methyl
9
CA 02494322 2005-02-01
WO 2004/013204 PCT/US2003/023034
ketone), 4-methyl-2-pentanone (methyl isobutyl ketone), 3-methyl-2-butanone
(methyl isopropyl ketone), 2-pentanone (methyl n-propyl ketone), diisobutyl
ketone,
2-hexanone (methyl n-butyl ketone), 3-pentanone (diethyl ketone), 2-methyl-3-
heptanone (butyl isopropyl ketone), 3-heptanone (ethyl n-butyl ketone), 2-
octanone
(methyl n-hexyl ketone), 5-methyl-3-heptanone (ethyl amyl ketone), 5-methyl-2-
hexanone (methyl iso-amyl ketone), heptanone (pentyl methyl ketone), cyclo-
pentanone, cyclo-hexanone.
In general, the esters can be cyclic or acyclic, straight-chained or branched,
and/or substituted or unsubstituted. Examples of acyclic esters and cyclic
esters
include ethyl acetate, propyl acetate, butyl acetate, amyl acetate, butyl iso-
butyrate,
methyl n-butyrate, butyl propionate, butyl butyrate, methyl valerate, ethyl
valerate,
methyl caproate, ethyl butyrate, ethyl acetate, gamma-butyrolactone, gamma-
valerolactone.
In general, the alcohols having at least four carbon atoms can be cyclic or
acyclic, straight-chained or branched, and/or substituted or unsubstituted.
Examples
of such cyclic alcohols and acyclic alcohols include methyl- l-butanol, ethyl-
l-
butanol, 3-methyl-l-butanol (amyl alcohol), 2-methyl-l-pentanol, 2-methyl-2-
butanol
(amyl alcohol), 3-methyl-2-pentanol (methyl iso-butyl carbinol), methyl-2-
pentanol,
4-methyl-2-pentanol, butyl alcohol, pentyl alcohol, hexyl alcohol, heptyl
alcohol,
cyclo-hexanol, methyl-cyclo-hexanol and fusel oil (a mixture of higher
alcohols,
which is often a by-product of alcohol distillation, and typically is
predominantly
amyl alcohol (methyl butanol)).
In general, the alkanes can be cyclic or acyclic, straight-chained or
branched,
and/or substituted or unsubstituted. In some embodiments, the alkanes include
straight-chain alkanes and have five or more carbon atoms (e.g., heptane,
hexane,
octane, nonane, dodecane). In certain embodiments the alkanes include
isoalkanes
(e.g. methyl heptane, methyl octane, dimethyl heptane). In certain
embodiments,
Soltrol' 100 (a mixture of C9-C 11 isoalkanes, commercially available from
Chevron
Phillips Chemical Company located in Houston, TX) can be used.
Generally, the amount of solvent present in the solvent system can be varied
as
desired. In certain embodiments, the solvent system has at least about five
parts (e.g.,
at least about 10 parts, at least about 15 parts) solvent per part PHA and/or
less than
CA 02494322 2010-04-14
51668-21
about 50 parts (e.g., less than about 30 parts, less than about 25 parts)
solvent per part
PHA.
In some embodiments, a solvent for the PHA is non-halogenated. Using a
non-halogenated solvent can be advantageous because this can reduce the
negative
environmental impact of the solvent, reduce the health risks associated with
using the
solvent, and/or reduce the costs associated with storing, handling and/or
disposing the
solvent.
In certain embodiments, a solvent for the PHA can have a relatively low
density. For example, a solvent for the PHA can have a density of less than
about
0.95 kilograms per liter (e.g., less than about 0.9 kilograms per liter, less
than about
0.8 kilograms per liter, less than about 0.7 kilograms per liter) at 20 C.
Without
wishing to be bound by theory, it is believed that using a relatively low
density
solvent can enhance the quality of the separation of the PHA phase from the
residual
biomass phase.
In some embodiments, a solvent for the PHA has a relatively low solubility in
water. For example, a solvent for the PHA can have a solubility in water of
less than
about one percent (e.g., less than about 0.5 percent, less than about 0.2
percent) at
C. A solvent with a relatively low solubility in water can be desirable
because
such a solvent is less likely to intermix with water. This can enhance the
ease of
20 providing two separate phases during the process, thereby reducing the cost
and/or
complexity of the process.
In certain embodiments, a solvent for the PHA is substantially non-
hydrolyzable. For example, the solvent can be at most as hydrolyzable as ethyl
acetate. Using a substantially non-hydrolyzable solvent can reduce the
likelihood of
undesirable side product formation (e.g., chemically reactive species, such as
organic
acids). This can reduce the amount and/or rate of, for example, corrosion of
portions
(e.g., plumbing) of the system in which the PHA extraction is performed.
In some embodiments, a solvent for the PHA is relatively easily stripped from
water. For example, the solvent can have a logK value relative to water at 100
C of at
least about 1.5 (e.g., at least about 1.8, at least about two, at least about
2.2) as
determined according to Hwang et at., Ind. Eng. Chem. Res., Vol. 31, No. 7,
pp.
1753-1767 (1992). Using a solvent that is
11
CA 02494322 2005-02-01
WO 2004/013204 PCT/US2003/023034
readily stripped from water can be desirable because such a solvent can be
more
readily recovered and recycled relative to other solvents that are not as
readily
stripped from water.
In certain embodiments, a solvent for the PHA has a boiling point greater than
100 C.
In certain embodiments, an appropriate solvent is non-halogenated, has
relatively low (e.g., less than ethyl acetate) water solubility, and
relatively low
reactivity from the perspective of hydrolysis and/or from the perspective of
reactivity
towards the polymer.
In some embodiments, the solubility of the PHA in the precipitant is less than
about 0.2 percent (e.g., less than about 0.1 percent) of the PHA at 20 C.
In certain embodiments, a relatively small volume of precipitant is added to
the PHA phase relative to the volume of solvent system added to the slurry.
For
example, the ratio of the volume of precipitant added to the PHA phase to the
volume
of solvent system added to the slurry is less than about 0.2 (e.g., less than
about 0.1,
less than about 0.07, less than about 0.05).
In embodiments in which the solvent system contains one or more solvents for
PHA and one or more precipitants for PHA, the solvent(s) and the
precipitant(s) can
have a relative volatility of at least about two (e.g., at least about three,
at least about
four) at the equimolar bubble point of the solvent(s) and the precipitant(s)
at
atmospheric pressure.
In some embodiments in which the solvent system contains one or more
solvents for the PHA and one or more precipitants for the PHA, the solvent(s)
and the
precipitant(s) do not form an azeotrope. Using solvent(s) and precipitant(s)
that do
not form an azeotrope can be desirable because it can be easier to separate
and recover
the solvent and precipitant for re-use relative to a solvent and precipitant
that form an
azeotrope.
In certain embodiments in which the solvent system contains a solvent for the
PHA and a precipitant for the PHA, the solution formed of the PHA and the
solvent
system contains less than about 25 volume percent (e.g., less than about 20
volume
percent, less than about 15 volume percent, less than about 10 volume percent)
of the
precipitant.
12
CA 02494322 2005-02-01
WO 2004/013204 PCT/US2003/023034
In general, the mixture containing the solvent system and the slurry is heated
to enhance the interaction of the solvent system with the PHA, thereby
allowing the
PHA to be removed from the biomass.
In general, the temperature of the solvent system and slurry mixture during
agitation can be varied as desired. In some embodiments, the temperature is
less than
about 160 C (e.g., less than about 125 C, less than about 95 C, less than
about 65 C)
and/or at least about 20 C. In certain embodiments, the temperature is from
ambient
temperature to about 95 C (e.g., from about 40 C to about 80 C, from about 60
C to
about 70 C). In certain embodiments the pressure can be regulated to greater
than
atmospheric pressure to facilitate extraction at elevated temperature (e.g.
greater than
1 atmosphere, up to 20 atmosphere).
Generally, the shear force used when agitating the solvent system and slurry
mixture can be varied as desired. In certain embodiments, the solvent system
and
slurry mixture is agitated by stirring so that the dissolution time is
reduced. In some
embodiments, to assist dissolution, a high shear impeller and agitator (e.g.
flat blade
impeller such as the 6 bladed Rushton turbine) can be used at tip speeds of,
for
example, about five meters per second or more (e.g., to about 10 meters per
second).
In certain embodiments a high speed disperser having a low profile blade can
be used
at tip speeds of, for examples, about 10 meter per second or more (e.g. about
15 meter
per second or more, about 20 meter per second to about 25 meter per second),
Typically, the high speed dispersers have a blade with a low profile bladed or
saw
tooth edge to generate high shear at enhanced tip speeds. In certain
embodiments, a
rotor/stator system is used that generates relatively high shear (e.g., at tip
speeds up to
about 50 meters per second) in the gap between a high speed rotor that spins
within a
slotted stator. In general the geometry of the rotor and stator can be varied
to suit
particular applications and many designs are commercially available.
In general, the solvent system and slurry mixture is agitated until a
centrifuged
sample of the mixture has a PHA phase with a desired PHA solids content. In
some
embodiments, the solvent system and slurry mixture is agitated for less than
about
three hours (e.g., less than about two hours) and/or at least about one minute
(e.g., at
least about 10 minutes, at least about 30 minutes).
13
CA 02494322 2010-04-14
51668-21
In certain embodiments, the PHA phase contains less than about 0.5 weight
percent (e.g., less than about 0.25 weight percent, less than about 0.1 weight
percent)
biomass relative to the amount of dissolved PHA in the PHA phase.
In some embodiments, the biomass phase contains less than about 25 weight
percent (e.g., less than about 20 weight percent, less than about 15 weight
percent) of
the solvent that was initially present in the solvent system and or at least
about one
weight percent (e.g., at least about five weight percent, at least about 10
weight
percent) of the solvent that was initially present in the solvent system.
In some embodiments, the PHA phase has a relatively low viscosity. For
example, this phase can have a viscosity of less than about 100 centipoise
(e.g., less
than about 75 centipoise, less than about 50 centipoise, less than about 40
centipoise,
less than about 30 centipoise). Without wishing to be bound by theory, it is
believed
that preparing the PHA phase such that it has a relatively low viscosity can
result in a
relatively good separation of the PHA phase from the residual biomass phase.
In
particular, it is believed that the rate of separation of the phases during
centrifugation
is inversely proportional to the viscosity of the PHA phase so that, for a
given
centrifugation time, decreasing the viscosity of the PHA phase results in an
improved
separation of the phases relative to certain systems in which the PHA phase
has a
higher viscosity.
In certain embodiments, the PHA phase has a relatively high polymer
concentration. For example, the PHA phase can have a polymer concentration of
at
least about two percent (e.g., at least about 2.5 percent, at least about
three percent, at
least about 3.5 percent, at least about four percent, at least about 4.5
percent, at least
about five percent).
Various types of devices can be used that exploit centrifugal force. As an
example, in some embodiments centrifugation is performed using a disc stack
(e.g., a
model SC-6, available from Westfalia Separator US, Inc., located in Northvale,
NJ).
In certain embodiments centrifugation is performed using a decanter (e.g., a
model
CA-220, available from Westfalia Separator US, Inc., located in Northvale,
NJ). In
some embodiments, a hydroclone can be used.
In certain embodiments a countercurrent centrifugal contacter (e.g., a
TM
Podbielniak centrifugal contacter, a Luwesta centrifugal contacter, Taylor-
Couette
14
CA 02494322 2010-04-14
51668-21
centrifugal contacter) can be used. In general, a countercurrent centrifugal
contacter
is used by having two (or possibly more) fluid streams contact each other. One
stream (the solvent stream) begins as a fluid stream that is relatively rich
in solvent.
Another stream (the biomass stream) begins as a fluid stream that is
relatively rich in
PHA. The two streams contact each other under countercurrent conditions such
that a
portion of the solvent stream that is richest in solvent contacts a portion of
the
biomass stream that is poorest in PHA (to enhance, e.g., optimize, the
recovery of
PHA from the biomass stream), and/or such that a portion of the biomass stream
that
is richest in PHA contacts a portion of the solvent stream that is most laden
with PHA
(to enhance, e.g., optimize, the concentration of PHA in the solvent stream).
In
certain embodiments, this is achieved by flowing the solvent stream reverse to
the
biomass stream (reverse flow conditions). Countercurrent centrifugal
contacters are
available from, for example, B&P Process Equipment (Saginaw, MI) and
Quadronics.
Examples of commercially available countercurrent centrifugal contacters
include the
TM
Podbielniak A-1 countercurrent centrifugal contacter (B&P Process Equipment)
and
TM
the Podbielniak B-10 countercurrent centrifugal contacter (B&P Process
Equipment).
In general, the conditions (e.g., force, time) used for centrifugation can be
varied as desired.
In some embodiments in which a disc stack is used, centrifugation can be
performed using at least about 5,000 RCF (Relative Centrifugal Force) (e.g.,
at least
about 6,000 RCF, at least about 7,000 RCF, at least about 8,000 RCF) and/or
less than
about 15,000 RCF (e.g., less than about 12,000 RCF, less than about 10,000
RCF). In
certain embodiments in which a decanter is used, centrifugation can be
performed
using at least about 1,000 RCF (e.g., at least about 1,500 RCF, at least about
2,000
RCF, at least about 2,500 RCF) and/or less than about 5,000 RCF (e.g., less
than
about 4,000 RCF, less than about 3,500 RCF). In certain embodiments in which a
countercurrent centrifugal contacter is used, centrifugation can be performed
using at
least about 1,000 RCF (e.g., at least about 1,500 RCF, at least about 2,000
RCF, at
least about 2,500 RCF) and/or less than about 5,000 RCF (e.g., less than about
4,000
RCF, less than about 3,500 RCF).
In some embodiments in which a disc stack is used, centrifugation can be
performed for less than about one hour (e.g., less than about 30 minutes, less
than
CA 02494322 2005-02-01
WO 2004/013204 PCT/US2003/023034
about 10 minutes, less than about five minutes, less than about one minute)
and/or at
least about 10 seconds (e.g., at least about 20 seconds, at least about 30
seconds). In
certain embodiments in which a decanter is used, centrifugation can be
performed for
less than about one hour (e.g., less than about 30 minutes, less than about 10
minutes,
less than about five minutes, less than about one minute) and/or at least
about 10
seconds (e.g., at least about 20 seconds, at least about 30 seconds). In
certain
embodiments in which a countercurrent centrifugal contacter is used,
centrifugation
can be performed for less than about one hour (e.g., less than about 30
minutes, less
than about 10 minutes, less than about five minutes, less than about one
minute)
and/or at least about 10 seconds (e.g., at least about 20 seconds, at least
about 30
seconds).
After centrifugation, a precipitant for the PHA. is added to the separated PHA
phase to form a mixture. In embodiments in which the solvent system contains
one or
more precipitants for the PHA, the precipitant added to the separated PHA
phase may
be the same as or different from the precipitant(s) contained in the solvent
system.
In general, the amount of the precipitant added to the separated PHA phase
can be varied as desired. In some embodiments, the amount of precipitant added
to
the separated PHA phase is at least about 0.1 part (e.g., at least about 0.25
part, at
least about 0.5 part) precipitant by volume relevant to the volume of solvent
in the
PHA phase and/or less than about two parts (e.g., less than about 1.5 parts,
less than
about one part, less than about 0.75 part) precipitant by volume relevant to
the volume
of solvent in the PHA phase.
The PHA phase/precipitant mixture is agitated to enhance the interaction of
the PHA with the precipitant for the PHA. This allows the PHA to precipitate
from
the mixture, resulting in a combination formed of precipitated PHA and a
mixture
containing the solvent system and the added precipitant for the PHA. In
general,
agitation of the PHA phase/precipitant mixture is performed at room
temperature, but
other temperatures can be used if desired. In some embodiments, the PHA
phase/precipitant mixture is mixed using high shear devices such as high shear
impellers (e.g., a six-bladed Rushton turbine), high speed dispersers and
rotor/stator
high shear in-line or in-tank mixers. The shear rates are determined by the
tip speeds
of the various devices and can be varied between, for example, from about five
meters
16
CA 02494322 2005-02-01
WO 2004/013204 PCT/US2003/023034
per second to about 50 meters per second (e.g., from about 10 meters per
second to
about 25 meters per second). Without wishing to be bound by theory, it is
believed
that the high shear mixing can, under certain conditions, improve the quality
of the
precipitated polymer.
The precipitated PHA is then separated from the remaining liquid (e.g.,
solvent system and precipitant). This separation can be performed by, for
example,
filtration or centrifugation (e.g., using a basket centrifuge, using a vacuum
belt filter).
Typically, the precipitated PHA is then washed to assist removing undesired
impurities, such as remaining solvent and/or precipitant. In some embodiments,
the
polymer can be washed with solvent (e.g., relatively freshly prepared
solvent), such
as, for example, a mixture of the PHA solvent and the PHA precipitant (e.g.,
with any
ratio between 0-100%). Usually, the composition for washing is selected to
reduce
(e.g., minimize) the re-dissolution of the polymer and/or to enhance (e.g.,
maximize)
removal of impurities. In certain embodiments, the appropriate ratio can be
dependent on the particular polymer composition and/or can be determined by
standard experimentation (washing efficiency). In some embodiments this
washing
step can be conducted at elevated temperature and appropriate residence time
to
further facilitate the washing and removal of impurities
Typically, the washed, precipitated PHA is dried (e.g., at a temperature of
from about 40 C to about 100 C). Drying can be performed under vacuum (e.g.,
to
assist in facilitating recovery of the residual solvent). In certain
embodiments it may
be desirable to directly extrude the precipitated polymer still containing
solvent in, for
example, a devolatilizing extruder. Such extrusion can be performed, for
example, at
a temperature close to the polymer melting point, and the solvent can be
recovered
directly from the extruder. Water can optionally be injected under pressure
into the
devolatilizing extruder (e.g., to generate steam in-situ to facilitate
efficient stripping
and removal of traces of residual solvent). A gas stream (e.g. air, CO2 or
steam) can
optionally be injected into the extruder (e.g., to facilitate solvent
removal). Extrusion
can consolidate drying and product formation (e.g. pelletizing) operations
into a
single unit with, for example, capital and process operating cost savings.
The remaining liquid (solvent system and precipitant) can be further processed
so that the components of the liquid (solvent(s) and/or precipitant(s)) can be
re-used.
17
CA 02494322 2005-02-01
WO 2004/013204 PCT/US2003/023034
For example, the liquid can be distilled to separate solvent from precipitant.
In some
embodiments, the separated solvent and/or precipitant can be re-used in the
process
described above (e.g., as a solvent in the solvent system, as a precipitant in
the solvent
system, as a precipitant added to the PHA phase). In certain embodiments, the
separated solvent and/or precipitant can be re-used in the process described
in Fig. 2
(see discussion below) (e.g., as a solvent in the solvent system, as a
precipitant in the
solvent system, as a precipitant added to the PHA phase).
In certain embodiments, the process (or portions of the process) can be
performed in a continuous and/or an in-line manner. As an example, the process
can
involve an in-line rotor/stator process for dissolution, and/or an in-line
rotor/stator
process for precipitation of the PHA and/or an in-line devolatilizing extruder
(e.g. a
Werner and Pfleiderer ZSK extruder supplied by Coperion Corporation of Ramsey,
NJ) for removing the solvent and forming PHA solids (e.g. pellets).
In some embodiments, the process uses the solvent in a relatively efficient
manner. For example, at least about 90 volume percent (e.g., at least about 95
volume
percent, at least about 97 volume percent, at least about 98 volume percent)
of the
solvent initially used in the solvent is recovered for re-use.
In certain embodiments, the process uses the precipitant in a relatively
efficient manner. For example, at least about 90 volume percent (e.g., at
least about
95 volume percent, at least about 97 volume percent, at least about 98 volume
percent) of the combined amount of precipitant initially used in the solvent
and added
to the PHA phase is recovered for re-use.
Fig. 2 is a flow diagram showing an embodiment of a second stage of a two-
stage process that can be used to enhance the efficiency of PHA extraction by
extracting at least a portion of the PHA present in the residual biomass phase
(Fig. 1).
As shown in Fig. 2, a solvent system is added to the biomass phase to provide
a
mixture containing the biomass phase and the solvent system. The mixture is
agitated
(e.g., using the conditions described above with respect to agitation of the
slurry and
solvent system mixture) to provide a combination including a PHA phase
(containing
predominantly solvent system and PHA) and a biomass phase (containing
predominantly biomass, water and carry-over solvent system). The PHA phase and
biomass phase are separated using centrifugation (e.g., using the conditions
described
18
CA 02494322 2005-02-01
WO 2004/013204 PCT/US2003/023034
above with respect to centrifuging the PHA phase and biomass phase). The PHA
phase can be treated as described above (e.g., by adding a precipitant for the
PHA,
agitating, separating, washing, drying), or the PHA phase can be added to the
slurry
and solvent mixture described above. The components of the solvent system
(e.g.,
solvent(s) and/or precipitant(s)) can be stripped from the remaining biomass
phase
using standard techniques. The residual solvent contained in the biomass can
be
recovered through a variety of means such as steam stripping in a suitable
column, a
desolventizing drier (e.g. desolvantizer toaster used commonly in recovering
residual
solvent from soybean meal after oil extraction) or direct drying with solvent
recovery
(e.g. vacuum drier, fluid bed drier with inert gas circulation and solvent
condensation). In some embodiments, the biomass containing the solvent can be
co-
dried with a compatible animal feed material (e.g., gluten feed, distiller dry
grain, oil
seed meal) in a drier that is suitably rated to handle and recover and/or
safely
eliminate (e.g. adsorption or incineration) the residual solvent. In the
overall process
in Fig. 2, the first stage is shown in Fig. 1, and the second stage is shown
in Fig. 2.In
certain embodiments, the residual biomass can be used as a nutrient for
fermentation
(e.g. ethanol fermentation using Saccharomyces), optionally after removing the
residual solvent as described above. In some embodiments, the biomass can be
hydrolyzed (e.g. by exposure to acidic conditions at elevated temperature,
treatment
with protease enzymes, lytic enzymes) to improve its nutrient profile for
fermentation.
While certain methods for extracting a PHA from biomass have been
described, other embodiments are also possible.
As an example, dry biomass can be used. In some embodiments, the dry
biomass can be combined with water to provide a slurry.
As another example, a precipitant for the PHA can be added to the slurry
before adding the solvent system. In some embodiments, the amount of
precipitant
added is at least about 0.5 volumes (e.g., from about 0.5 volumes to about two
volumes) relative to the slurry.
Adding precipitant before adding the solvent system can result in the
formation of a relatively pure isolated, extracted PHA (e.g., a purity of at
least about
99%, a purity of at least about 99.5%, at purity of at least about 99.9%). The
polymer
purity can be determined by gas chromatography (GC) analysis (e.g., with a
Hewlett
19
CA 02494322 2005-02-01
WO 2004/013204 PCT/US2003/023034
Packard 5890 Series II GC equipped with Supelco 24044 SBPTM-1 column of 30m x
0.32 mm ID with 0.25 m film) following butanolysis of the polymer sample
under
acidic conditions to form butyl esters of the PHA monomeric units as well as
the butyl
esters of the lipids and phospholipids fatty acid residues. Suitable standards
of the
fatty acids and hydroxy acids (e.g. palmitic acid, stearic acid, oleic acid,
linoleic acid
and 3-hydroxy butyric acid) are used to calibrate and standardize and quantify
the
chromatographic response. This can be used to quantify both the polymer
content as
well as the impurity content. Inorganic impurities can be quantified by
ashing.
Without wishing to be bound by theory, it is believed that adding a
precipitant
for the PHA to the slurry prior to adding the solvent system can assist in
removing
biomass impurities present in the biomass (e.g., phospholipids, neutral
lipids,
lipoproteins). This can be particularly advantageous if the PHA solids content
in the
biomass is relatively high (e.g., a PHA solids content of at least about 65%,
at least
about 75%).
As a further example, the biomass and/or the slurry can be chemically pre-
treated for example with relatively mild caustic conditions (e.g., a pH of
from about
8.5 to 10, from about 8.5 to about 9, from about 9 to about 9.5, from about
9.5 to
about 10) followed by neutralization before adding the solvent system. This
can
result in the formation of a relatively pure isolated, extracted PHA (e.g., a
purity of at
least about 99%, at least about 99.5%). The caustic conditions can be prepared
using
one or more relatively basic materials, such as, for example, potassium
hydroxide,
sodium hydroxide and/or ammonium hydroxide.
As another example, the temperature can be elevated (e.g. any temperature
between room temperature and about 95 C) and other chemicals such as
surfactants,
detergents and/or enzymes added during the chemical pre-treatment step to
further
facilitate the formation of a relatively pure isolated, extracted PHA (e.g., a
purity of at
least about 99%, at least about 99.5%).
Without wishing to be bound by theory, it is believed that a chemical
treatment (e.g., a relatively mild caustic treatment) of the slurry prior to
adding the
solvent system can assist in removing biomass impurities present in the
biomass (e.g.,
lipids, phospholipids, lipoproteins). This can be particularly advantageous if
the PHA
CA 02494322 2005-02-01
WO 2004/013204 PCT/US2003/023034
solids content in the biomass is relatively high (e.g., a PHA solids content
of at least
about 65%, at least about 75%).
As another example, the methods can include concentration (e.g., evaporation)
of the PHA phase after separation of this phase from the residual biomass
phase but
before addition of the precipitant for the PHA to the PHA phase. This can
reduce the
volume of solution, thereby reducing precipitant.
As a further example, in some embodiments the processes can be performed
without adding a precipitant for the PHA to the PHA phase.
Moreover, the solvent system can be formed and then contacted with the
biomass, or the biomass can be contacted with fewer than all the components of
the
solvent system, followed by subsequent addition of the remaining portion of
the
solvent system (e.g., in series or all at once). For example, in embodiments
in which
the solvent system includes a solvent for the PHA and a precipitant for the
PHA, the
slurry can be contacted with the solvent, followed by addition of the
precipitant, or
vice-versa. Alternatively, the solvent and precipitant can be combined to form
the
solvent system, followed by contacting the biomass.
Furthermore, while the extraction of a single PHA from a biomass has been
described, the processes could be used to extract multiple PHAs (e.g., two,
three, four,
five, six) from a biomass. Such processes could involve the use of multiple
solvents,
precipitants and/or solvent systems.
In addition, while solvent systems containing a single solvent for the PHA and
optionally a single precipitant for the PHA have been described, multiple
solvents for
the PHA (e.g., two, three, four, five, six) and/or multiple precipitants for
the PHA
(e.g., two, three, four, five, six) can be used.
As another example, in some embodiments the slurry/solvent system mixture
can be agitated without heating. Alternatively the slurry/solvent system
mixture can
be agitated under pressure with heating.
As a further example, the methods can include distilling the solvent
system/precipitant mixture formed (e.g., distilled) to separate the components
(e.g.,
solvent for the PHA, precipitant for the PHA) so that one or more of the
components
can be re-used.
21
CA 02494322 2010-04-14
51668-21
The following examples are illustrative and not intended to be limiting. In
the
examples, the chemicals were from Aldrich Chemical Co. Inc. (Milwaukee, WI),
the
overhead stirrer was an Ika -werke Eurostar power control-vise overhead
stirrer (Ika
TM
Work Inc., Wilmington, NC), and the centrifuge was a Sorvall RC 5B plus
centrifuge.
Example I
A batch of Escherichia coli biomass slurry containing 70% polymer on a dry
basis with a composition of polyhydroxybutyrate co 4-hydroxybutyrate with 25%
4-
hydroxybutyrate was split three ways and treated as follows:
a. Spray dried and 30 g of dry biomass collected.
b. Spray dried, 30 g of biomass collected and re-wetted with deionized (DI)
water to 100 g.
c. 100 g of original slurry containing 30 g of dry biomass without
modification.
Each batch was extracted with 400 ml butyl acetate at room temperature with
overhead stirring at 500 rpm for 2 hours. The resulting slurry was centrifuged
at 5000
g for 20 minutes and the PHA phase recovered. The PHA content was determined
by
precipitation of the PHA from the PHA phase using hexane as precipitant
followed by
drying overnight under vacuum of one millimeter Hg and 40 C. The recovered
polymer represented 32% dissolution of the starting polymer (approach 1), 43%
of the
starting polymer (approach 2), and greater than 97% dissolution of the
starting
polymer (approach 3).
Example II
In a side by side test broth from an Escherichia coli fermentation containing
cells with one dimension exceeding 2 microns was compared to Ralstonia
eutropha
containing cells with a maximum dimension of 0.5 micron. The time to obtain a
clear
TM
supernatant was determined at 12000 rpm in an Eppendorf 5415C micro-
centrifuge,
using 1.5 mL centrifuge tubes filled with 1 mL of broth. In the case of the E.
coli
broth clear supernatant was obtained in less than 1 minute of centrifugation
time
while the Ralstonia eutropha required more than 5 minutes centrifugation for
similar
clarity.
22
CA 02494322 2010-04-14
51668-21
Example III
A polymer solution containing 5% polymer by weight (expressed relative to
the total solution weight) was prepared by dissolving of Escherichia coli
biomass
slurry containing 70% polymer on a dry basis with a composition of
polyhydroxybutyrate co 4-hydroxybutyrate with 25% 4-hydroxybutyrate in butyl
acetate (Aldrich Chemical Co. Inc., Milwaukee, WI) using the procedure of
Example
Ic. The viscosity of the resulting solution was measured as 365 centiPoise
(cP) using
TM
a Brookfield LVF Viscometer (Brookfield Engineering Laboratories Inc.,
Stoughton,
MA). For solutions with viscosity less than 100 cP, a No. 1 spindle was used,
and, for
solutions with viscosity greater than 100 eP, a No. 2 spindle was used. The
solution
was further diluted. with additional butyl acetate to 4% and 3% polymer by
weight
total solution. The resulting viscosities were found to be 150 cP and 40 cP,
respectively.
Some of the 5% polymer solution in butyl acetate prepared above was
subsequently diluted using hexane (Aldrich Chemical Co. Inc., Milwaukee, WI)
to
prepare a 4.5%, 4.3%, 4.1% and 3.9% solution by weight. The viscosities of
these
solutions were measured as described above and determined to be 215 cP, 37.5
cP, 5
cP and 27.5 cP, respectively.
The viscosities upon dilution of a 5% polymer solution in butyl acetate with
additional butyl acetate (PHA solvent) compared to dilution with hexane (PHA
precipitant at room. temperature) are depicted in Fig. 3. Dilution with the
precipitant
has a non-linear and desirable impact on reducing viscosity. The increased
viscosity
observed with hexane dilution to 3.9% by weight polymer in solution coincided
with
polymer precipitation from solution at that level of hexane addition.
Example IV
A recombinant E. coli was used to produce poly 3-hydroxybutyrate-co-4-
hydroxybutyrate (30% 4-hydroxybutyrate on a molar basis) in a fed-batch
fermentation, using glucose as the major carbon source. At the completion of
the
fermentation the E. coli cells had expanded in size to greater than 2 microns
in at least
one dimension. The biomass accumulated 70% polymer on a dry weight basis. The
biomass was subsequently harvested using centrifugation to produce a wet
biomass
pellet, substantially free of dissolved impurities.
23
CA 02494322 2010-04-14
51668-21
100 g of the wet biomass pellet (48% dry solids) containing 70% poly 3-
hydroxybutyrate co 4-hydroxybutyrate on a dry basis was charged with 500 ml of
ethyl acetate and agitated with a overhead stirrer at room temperature for 1
hour. The
polymer composition was 30% 4-hydroxybutyrate on a molar basis. The mixing
time
was terminated after the viscosity increased to the extent that further
stirring was not
effective in mixing the material. A total of 350 ml of the slurry was
collected and
TM
centrifuged at 5000 g for a total of 20 minutes (Sorvall RC 5B plus
centrifuge, Kendro
Laboratory Products, Newtown CT). The theoretical amount of ethyl acetate that
should be recoverable from the 350 mL of slurry was 300 mL according to mass
balance calculations.
The PHA content of the PHA phase was about 5.3%. 220 milliliters of the
PHA phase was recovered by decantation after centrifugation, constituting
about 73%
by volume of the total recoverable ethyl acetate of slurry prior to
centrifugation.
Example V
The preceding example was repeated, except butyl acetate (Aldrich Chemical
Co. Inc., Milwaukee, WI) was used rather than ethyl acetate. The polymer in
solution
was about 4.3%. There was the appearance of an emulsified layer at the
interface
after centrifugation. 250 milliliters of the PHA phase was recovered by
decantation
after centrifugation, constituting about 83% of the total recoverable butyl
acetate
present in the slurry before centrifugation.
Example VI
The preceding example was repeated, except M1BK (Aldrich Chemical Co.
Inc., Milwaukee, WI) was used rather than butyl acetate. The polymer in
solution was
about 4.2%. 290 milliliters of the PHA phase was recovered by decantation
after
centrifugation, constituting about 97% of the total recoverable MIBK present
in the
slurry before centrifugation.
Example VII
100 g of wet E. coli biomass paste with 28% dry solids containing 75% Poly
3-hydroxybutyrate co 4-hydroxybutyrate with 35% 4-hydroxybutyrate on a dry
solids
basis was contacted with 200 g of hexane (Aldrich Chemical Co. Inc.,
Milwaukee,
WI) and extracted for 2 hours with overhead stirring (Ika -werke Eurostar
power
control-visc overhead stirrer, Ika Work Inc., Wilmington, NC) at room
temperature.
24
CA 02494322 2010-04-14
51668-21
The hexane supernatant was separated by centrifugation at 3,500 g for 20
minutes,
and the solid pellet recovered after decanting the hexane supernatant. The
pellet was
subsequently extracted using 425 g of MIBK (Aldrich Chemical Co. Inc.,
Milwaukee,
WI) at room temperature with overhead stirring (IkaO-werke Eurostar power
control-
visc overhead stirrer, Ika Work Inc., Wilmington, NC) for 3 hours. The
supernatant
(solution of polymer in MIRK) was separated by centrifugation at 3,500 g for
20
minutes and the polymer precipitated by addition of 355 g of hexane. The
precipitated polymer was recovered by filtration using a funnel lined with
fluted filter
paper (VWR Scientific Products, West Chester, PA) and dried overnight at 45 C
TM
under vacuum of 1 mm Hg under vacuum in a Biichi rotavap to yield 13 gram of
dried
polymer. The dried polymer was subjected to hot film pressing at 180 C. A
suitable
amount of PHA (typically 0.5 gram) is placed between two PET sheets separated
by
shims to form a film of 100 micron thickness. The film assembly (i.e. two
sheets,
shims and PHA) was placed between the heated (180 C) blocks of the press
(Carver
Hydraulic Press model #3912, Carver Inc., Wabash, IN) and a load of 10 tons
was
applied for 30 seconds. The film was then cooled between aluminum blocks and
then
inspected for color and clarity. This yielded a substantially clear film with
substantially no fuming or objectionable odors at the operating temperature of
180 C
during the press cycle.
Example VIII
The preceding example was repeated, except that heptane (Aldrich Chemical
Co. Inc., Milwaukee, WI) was used rather than hexane. The processed yielded a
substantially clear film with substantially no fuming or objectionable odors.
Example IX
The preceding example was repeated, except that Soltrol 100 (a mixture of
C9-C11 isoalkanes, commercially available from Chevron Phillips Chemical
Company
located in Houston, TX) was used instead of hexane. The process yielded a
substantially clear film with substantially no fuming or objectionable odors.
Example X
100 g of wet E. coli biomass paste with 28% dry solids containing 75% Poly
3-hydroxybutyrate co 4-hydroxybutyrate with 35% 4-hydroxybutyrate on a dry
solids
basis was treated with an effective 0.02 N of NaOH (Aldrich Chemical Co. Inc.,
CA 02494322 2010-04-14
51668-21
Milwaukee, WI) at 65 C for 20 minutes and thereafter rapidly cooled to room
temperature over 5 minutes. The resulting slurry was neutralized to pH of 7
using
85% phosphoric acid (Aldrich Chemical Co. Inc., Milwaukee, WI) and then
centrifuged (3,500 g) for 20 minutes, and washed with two volumes of DI water.
The
supernatant was discarded and the paste extracted using 425 g of MIBK at room
temperature with overhead stirring (Ika -werke Eurostar power control-visc
overhead
stirrer, Ika Work Inc., Wilmington, NC) for three hours. The supernatant
(solution of
polymer in MIBK) was separated by centrifugation at 3,500 g for 20 minutes and
the
polymer precipitated by addition of 355 g of hexane. The precipitated polymer
was
TM
recovered by filtration and dried under vacuum in a Biichi B-171 rotavap (65
C and 1
mm Hg vacuum for 8 hours) to yield 12 gram of dried polymer. The dried polymer
was subjected to hot film pressing at 180 C. This yielded a film with only
very slight
discoloration/opaqueness.
Example XI
The preceding example was repeated, but without the steps of treating with
NaOH at 65 C for 20 minutes and rapid cooling. The PHA thus recovered yielded
a
film with strong yellow discoloration and opaqueness during the hot film
press. There
was also evidence of thermal degradation during the test as evidenced by
fuming
during the film test pressure cycle (180 C and pressure of 10 tons of 30
seconds
duration).
Example XII
The following is an example of a one-stage process using a countercurrent
centrifugal contacter.
11 kg of biomass paste containing 26% E. coli dry solids was contacted with
38.6 kg methyl isobutyl ketone (4-methyl 2-pentanone or MIBK) for three hours
at
C in a dissolution tank equipped with an agitator with marine impeller to
maintain
a homogeneous mixture. The biomass contained 71% by weight of poly-3-
hydroxybutyrate co 4-hydroxybutyrate (22% molar 4-hydroxybutyrate) on a dry
basis.
After three hours the supernatant solution of MIBK and PHA obtained by
centrifuging
30 a sample from the dissolution tank contained 4.1 % PHA by weight
representing
91.2% dissolution.
26
CA 02494322 2010-04-14
51668-21
TM
The mixture of cell paste and MIRK was fed to an A-1 pilot scale Podbielniak
extractor (B&P Process Equipment, Saginaw, MI) as the heavy liquid in (HLI) at
a
rate of 635 ml/min. At the same time fresh M1BK was fed as the light liquid in
(LLI)
to effect countercurrent washing and extraction of the cell paste within the
TM
Podbielniak contactor. The LLI was fed at a rate of 175 ml/min to maintain a
feed
ratio of HLI:LLI of 3.6:1. A total of 49.6 kg of HLI and 12.8 kg of LLI was
fed over
a 90 minute period. A total of 8.9 kg of residual cell paste was collected as
the heavy
liquid out (HLO) and 53.6 kg of PHA solution in MIRK was collected as the
light
liquid out (LLO) over the course of the 90 minute period. The LLO contained
3.75%
by weight PHA in solution as determined by drying a sample of this material. A
total
of 2.0 kg PHA was recovered in the LLO compared to the 2.04 kg PHA contained
in
the HLI cell paste feed (98.4% overall recovery).
Mass balance measurements indicated that more than 98% of the total MIBK
contained in the combined HLI and LLI was recovered in the clarified PHA in
MIBK
solution (LLO). Laboratory centrifugation indicated that a very clear
interface was
formed after 1 minute of centrifugation at 3000 g. The absence of any
interfacial
accumulation was also confirmed by the LLO remaining clear for the duration of
the
90 minute trial.
TM
The improved PHA recovery of the Podbielniak extractor (98.4%) compared
to that achieved with a single stage of dissolution (91.2%) confirms the
efficacy of
countercurrent contracting with fresh solvent to improve PHA recovery. The
residual
biomass paste viscosity is also reduced dramatically by countercurrent
contacting as a
result of the nearly complete removal of the PHA
Example XIII
The following is an example of PHA extraction using cyclo-hexanone.
90 g of wet.E. coli biomass paste with 28% dry solids containing 80% poly 3-
hydroxybutyrate co 4-hydroxybutyrate (PHA) with 12% 4-hydroxybutyrate on a dry
solids basis was added to 400 g of cyclo-hexanone (Aldrich Chemical Co., Inc.,
Milwaukee, WI) at 90 C. The solution was homogenized for 5 minutes using a
hand-held homogenizer equipped with a single slotted rotor stator combination
at
30,000 rpm (Virtis, Gardiner, NY) and then agitated for 30 minutes using an
overhead
stirrer (Ika -werke Eurostar power control-vise overhead stirrer, Ilea Work
Inc.,
27
CA 02494322 2010-04-14
51668-21
Wilmington, NC). The temperature was controlled at 90 5 C during the solvent
contacting step. The biomass paste/cyclo-hexanone mixture was then centrifuged
at
3000g for 5 minutes to separate the supernatant (solution of polymer in cyclo-
TM
hexanone) by decanting from the residual biomass paste pellet using a Sorvall
RC 5B
plus centrifuge (Kendro Laboratory Products, Newtown, CT).
The supernatant was then re-heated in a beaker to 80 5 C and an equal
volume of heptane (held at room temperature) was slowly added to the solution
over
the course of 5 minutes while mixing vigorously with an overhead stirrer (Ikao-
werke
Eurostar power control-vise overhead stirrer, Ilca Work Inc., Wilmington, NC)
to
precipitate the polymer while maintaining the temperature between 70 C and 80
C.
The precipitated polymer was recovered by filtration using a funnel lined with
fluted
filter paper (VWR Scientific Products, West Chester, PA) and air dried
overnight in a
chemical fume hood to yield 16 g of white polymer granules (80% overall
recovery).
A film was prepared by placing approximately 0.5 g of polymer between two
PET sheets separated by shims to form a film of 100 micron thickness. The film
assembly (i.e. two sheets, shims and PHA) was placed between the heated (180
C)
blocks of the press (Carver Hydraulic Press model #3912, Carver Inc., Wabash,
IN)
and a load of 10 tons was applied for 30 seconds. The film was then cooled
between
aluminum blocks and inspected for color and clarity. This yielded a
substantially
clear film with substantially no fuming or objectionable odors at the
operating
temperature of 180 C during the press cycle.
Example XW
The following is an example of PHBH extraction.
Wet cell paste of Ralstonia eutropha (27% biomass solids on a weight basis in
water) containing approximately 65% poly 3-hydroxybutyrate co 3-
hydroxyhexanoate
(PHBH) on a dry biomass basis with a composition of 5-7% hydroxyhexanoate on a
molar basis (Kichise et. al., (1999), Intl. J. Biol. Macromol. 25: 69-77) was
prepared
using a genetically engineered Ralstonia strain prepared as described in
(Kichise et.
al., (1999), Intl. J. Biol. Macromol. 25: 69-77) and the fermentation process
described
by Naylor in US patent No. 5,871,980 using fructose and lauric acid as carbon
sources. This biomass was added to a suitable quantity of MIBK targeting a 5%
solution (w/w) of PHBH in the solvent. The solution was homogenized for 5
minutes
28
CA 02494322 2010-04-14
51668-21
using a hand-held homogenizer equipped with a single slotted rotor stator
combination at 30,000 rpm (Virtis, Gardiner, NY) and then agitated for 30
minutes
using an overhead stirrer (Ika -werke Eurostar power control-vise overhead
stirrer,
Ika Work Inc., Wilmington, NC). The temperature was controlled at 801 5 C
during
the solvent contacting step. The resulting biomass/solvent mixture was
separated by
TM
centrifugation using a Sorvall RC 5B plus centrifuge (Kendro Laboratory
Products,
Newtown, CT). The biomass paste/cyclo-hexanone mixture was then centrifuged at
3000g for 5 minutes to separate the supernatant (solution of polymer in cyclo-
hexanone) by decanting from the residual biomass paste pellet.
The supernatant was added to a beaker and an equal volume of heptane was
slowly added to the solution over the course of 5 minutes while mixing
vigorously
with an overhead stirrer (Ika -werke Eurostar power control-vise overhead
stirrer, Ika
Work Inc., Wilmington, NC) to precipitate the polymer. A white crystalline
polymer
powder was recovered after overnight drying in a chemical fume hood.
Example XV
The following is an example of PHBX extraction.
Wet cell paste of genetically engineered Pseudomonas sp was prepared and
grown on glucose as described by Matsusakai et al., (1999, Biomacromolecules
1: 17-
22) containing approximately 50% poly 3-hydroxybutyrate co 3-hydroxyoctanoate
co
3-hydroxydecanoate co3-hydroxydodecanoate co 3-hydroxydodecenoate (PHBX) on
a dry biomass basis with a composition of 92% 3-hydroxybutyrate, 1% 3-
hydroxyoctanoate, 3% 3-hydroxydecanoate, 3% 3-hydroxydodecanoate and 1% 3-
hydroxydodecenoate on a molar basis. This biomass was added to a suitable
quantity
of MIBK targeting a 5% solution (w/w) of PHBX in the solvent. The solution was
homogenized for 5 minutes using a hand-held homogenizer equipped with a single
slotted rotor stator combination at 30,000 rpm (Virtis, Gardiner, NY) and then
agitated for 30 minutes using an overhead stirrer (lka -werke Eurostar power
control-
vise overhead stirrer, Ika Work Inc., Wilmington, NC). The temperature was
controlled at 80 5 C during the solvent contacting step. The resulting
TM
biomass/solvent mixture was separated by centrifugation using a Sorvall RC 5B
plus
centrifuge (Kendro.Laboratory Products, Newtown, CT). The biomass paste/cyclo-
hexanone mixture was then centrifuged at 3000g for 5 minutes to separate the
29
CA 02494322 2005-02-01
WO 2004/013204 PCT/US2003/023034
supernatant (solution of polymer in cyclo-hexanone) by decanting from the
residual
biomass paste pellet.
The supernatant was added to a beaker and an equal volume of heptane was
slowly added to the solution over the course of 5 minutes while mixing
vigorously
5D with an overhead stirrer (Ilea -werke Eurostar power control-visc overhead
stirrer, Ika
Work Inc., Wilmington, NC) to precipitate the polymer. A white crystalline
polymer
powder was recovered after overnight drying in a chemical fume hood.
Other embodiments are in the claims.