Note: Descriptions are shown in the official language in which they were submitted.
---. j ~t r~ .... t_,:,.~~ ° $~. ;.a, ... ~.~.~~ ~°~~ 1
CA 02497609 2005-03-07
Solvay Pharmaceuticals GmbH
30173 Hannover
Non-peptidic BRS-3 agonists
Description
The present invention relates to novel, non-peptidic
compounds which exhibit a selective agonistic action on the
bombesin receptor of subtype 3 (BRS-3), and to pharmaceutical
preparations containing these compounds and also processes
for the preparation of these compounds.
Bombesin (Bn) is a peptide consisting of 14 amino acids
which was originally isolated from amphibians: The two
peptides neuromedin B (NMB) and the "gastrin-releasing
peptide" (GRP) which have been identified in mammals
represent structurally similar peptides. These bombesin-like
peptides are the naturally endogenous ligands of the
corresponding bombesin receptors, the "neuromedin B receptor"
(NMB-R, BB1) and the "gastrin-releasing peptide receptor"
(GRP-R, BB2). The bombesin receptors belong to the group of
the G-coupled receptors with 7 transmembrane domains.
Owing to the homology of its amino acid sequence, the
bombesin receptor of subtype 3 (BRS-3 or BB3) is assigned to
this family of bombesin receptors [cf. Fathi et a1. (1993) J.
Biol. Chem.. 268:5979-84; cited below as "Fathi et a1."]. The
natural ligand of BRS-3 is hitherto unknown. The expression
of BRS-3 was demonstrated in various regions of the brain
[cf. Yamada et a1. (1999) Physiol. Behav.. 66:863-7], in
secondary spermatocytes [cf. Fathi et al.], in pancreatic
islet cells [cf. Fleischmann et a1. (2000) Lab. Invest.
80:1807-17] and in the uterine tissue of pregnant animals
[cf. Gorbulev et a1. (1992) Eur. J. Biochem. 208:405-10].
Furthermore, BRS-3 was identified in different human cancer
cell lines (e. g. lung [cf. Fathi et a1.], breast [cf.
CA 02497609 2005-03-07
2
Gorbulev et a1. (1994) FEBS Lett. 340:260-4], prostate [cf.
Sun et a1. (2000) Prostate. 42:295-303] or ovary [cf. Sun et
a1. (2000) Regul. Pept. 90:77-84]).
Genetically altered mice in which the BRS-3 gene had
been knocked out ("BRS-3 Knockout Mice") exhibited a clinical
picture which comprised obesity, hyperphagia and also
hypertension and diabetes [cf. Okhi-Hamazaki et a1. (1997)
Nature 390:165-9]. According to this, BRS-3 appears to be an
essential participant in the regulation of glucose metabolism
and lipometabolism, in maintaining the energy status and in
controlling blood pressure, and also in influencing eating
behaviour. It can therefore be assumed of BRS-3-agonistic
compounds that they are suitable in particular for the
prophylaxis and/or treatment of pathological conditions such
as obesity (= adiposity), diabetes, hyperinsulinism,
cardiovascular diseases, eating disorders (hyperphagia,
anorexia, bulimia) and/or metabolic syndrome (= syndrome X).
Syndrome X manifests itself above all by Type II diabetes
mellitus and/or reduced glucose tolerance, arterial
hypertension, lipometabolism disorders, obesity and also
coronary heart disease.
Furthermore, it is known that the activation of BRS-3
can have a neuro-protective action [cf. WO 01/68120]. Also
BRS-3 appears to be connected to taste perception [cf. Yamada
et a1. (1999) Physiol. Behav. 66:863-7], influencing of
social behaviour [cf. Yamada et al. (2000) Physiol. Behav.
68:555-61] and certain emotional behaviours [cf. Yamada et
a1. (2002) Mol. Psychiatry. 7:113-7]. It can therefore
likewise be assumed that BRS-3-modulatory compounds may be
suitable for the prophylaxis and/or treatment of psychic
clinical pictures such as depression or anxiety states, taste
perception disorders and/or degenerative diseases of the
central nervous system, for example Parkinson's or
Alzheimer's.
Some synthetic peptidic ligands are already known which
bind with a certain affinity to BRS-3 and exert an agonistic
action thereon, namely the BRS-3 selective octapeptide [D-
3
CA 02497609 2005-03-07
Phe6, Phel3]Bn(6-13) propylamide [cf. Wu et a1. (1996) Mol.
Pharmacol. 50:1355-63] and also the less-selective
nonapeptide [D-Tyr&, (3-A1a11, Phel3, N1e14] Bn ( 6-14 ) [cf . Mantey
et a1. (1997) J. Biol. Chem. 272:26062-71] and its
derivatives [cf. Pradhan et a1. (1998) J. Pharmacol..
343:275-87; Mantey et al. (2001) J. Biol. Chem. 276:9219-29].
Low-molecular, non-peptidic bombesin-analogous compounds
are furthermore already known from WO 98107718, but these are
selective antagonists of the other two subtypes of the
bombesin receptor family (NMB-R and GRP-R). Low-molecular,
non-peptidic compounds which have a selective agonistic
effect with high affinity to BRS-3 on the other hand have not
been described hitherto.
It is therefore the object of the present invention to
make available novel, low-molecular, non-peptidic compounds
which have a selective agonistic effect with high affinity to
BRS-3.
It has now surprisingly been discovered that the low-
molecular and non-peptidic novel compounds according to the
invention are selective BRS-3 agonists and are thus suitable
for the prophylaxis and/or treatment of clinical pictures
which can be influenced beneficially by stimulating the
BRS-3. Owing to their action profile, the compounds according
to the invention appear to be suitable in particular for the
prophylaxis andlor treatment of obesity (= adiposity),
diabetes, hyperinsulinism, cardiovascular diseases, eating
disorders (hyperphagia, anorexia, bulimia) and/or syndrome X.
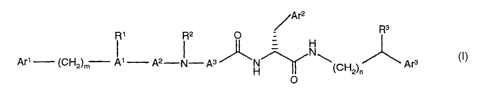
The subject of the invention is novel compounds of the
general formula I,
/Ar2
R3
N
1 2- - 3 ~ 3
Ar (CHZ)m A A N A H~ (CHZ)"
O
wherein
4
Al is CH or, if A2 does not stand for a bond and at the
same time A3 does not stand for NH, also nitrogen,
A2 is a bond, C~_2-alkylene or, if A1 stands for CH and R2
stands for hydrogen, also carbonyl,
A3 is methylene which is optionally substituted by C1_4-
alkyl or C1_q-alkyl carbonylamide or, if R2 is hydrogen
or together with R1 stands for a bond, is also NH,
R~- is hydrogen or, if A2 stands for carbonyl, also amino,
and
R2 is hydrogen, or
R1 and R2 together form C1_2-alkylene or, if A2 is a bond, R1
and R2 may also together stand for a bond,
R3 is hydrogen or methyl,
Arl is phenyl which is optionally substituted 1 to 2 times
by halogen or Cl_4-alkyl or by C1_2-alkylenedioxy bonded
to two adjacent ring carbon atoms; pyridyl, furyl,
indolyl or tetrahydroisoquinolyl,
Ar2 is furyl, benzofuranyl, thienyl, benzothiophenyl,
pyrrolyl or indolyl,
Ar3 is phenyl which is optionally substituted 1 to 2 times
by halogen, or pyridyl,
m is 0 or 1 and
n is 0 or 1,
and also optionally their physiologically compatible acid
addition salts. Further subjects of the invention are
pharmaceutical preparations containing the compounds of
Formula I and also a process for the preparation of such
compounds.
Where substituents C1_g-alkyl are contained in the
compounds of Formula I, this may be straight-chain or
branched. Where substituents contain halogen, this may be in
particular fluorine, chlorine or bromine. Chlorine is
preferred.
Where A3 is substituted by C1_q-alkyl, methyl is
preferred. Where A3 is substituted by Cl_4 alkyl
carbonylamide, n-propylamide is preferred.
R3 preferably stands for hydrogen.
CA 02497609 2005-03-07
5
CA 02497609 2005-03-07
Arl is preferably phenyl which is optionally substituted
once by halogen; pyridyl, furyl, in particular 2-furyl, or
indolyl, in particular 2-indolyl.
Ar2 preferably stands for benzothiophenyl or for
indolyl. Indolyl, in particular 3-indolyl, is preferred.
Ar3 preferably stands for phenyl, in particular non-
substituted phenyl.
n is preferably 1.
Preferred compounds of Formula I are the compounds of
the general formula Ia given below,
~Ar2o'
Rio /~
O _:
N~ N
Ar' (CH2}m = H~ ~' Ia
O - O
Ra
wherein Arl and m have the above meanings, Rlo1 is hydrogen
or amino, R~ is hydrogen, C1_4-alkyl or Cl_q-alkyl
carbonylamide and Ar2o1 is benzothiophenyl or indolyl, of the
general formula Ib,
~Arzo~
O
H - H
Are (CHZ}m N - N
Ib
R4 O
wherein R~, Arl, Ar2o1 and m have the above meanings, of the
general formula Ic,
~Arzo,
O _
N~ ~ N
Art (CHz)m~ H H~ IC
O O
CA 02497609 2005-03-07
6
wherein Ar', Ar2o1 and m have the above meanings, of the
general formula Id,
,Ar2°'
H O - H
Art (CH2)rt, N~ ~ N
N"N ~ Id
H H
wherein Arl, Ar2o1 and m have the above meanings, of the
general formula Ie,
,Arz°,
O _/_
=Nw ~ N
Ie
Ar (CHz)m
O
CA 02497609 2005-03-07
wherein Arl, Ar2o1 and m have the above meanings, and of the
general formula If,
~Ar2o~
Ar' (CH2}m A~~ O =
IN ~ N \
N ~ ~ If
H O
wherein A1, Ark-, Ar2o1 and m have the above meanings .
The compounds of Formula I represent non-peptidic
compounds, which however contain peptide bonds. The compounds
of Formula I can therefore be regarded as non-natural
polypeptides and be constructed partially or completely in a
manner known for polypeptide synthesis, far example by
conventional solid- or liquid-phase synthesis techniques with
suitable amino and carboxyl building blocks, preferably
sequentially. Where additionally also other organic-chemical
synthesis methods are used for constructing the compounds of
Formula I, known conventional organic-chemical synthesis
methods may be used.
Thus the compounds of Formula I and their acid addition
salts may for example be prepared in that
a) for the preparation of a compound of the general formula
Ig,
Ar2
Rio O ~ Rs
H - H
N\ ~ N\ ~ s
Ar' (CH2}m A3 H~ (CHZ}~ Ar Ig
O O
CA 02497609 2005-03-07
8
wherein A3, Rlol, R3, Arl, Ar2, Ar3, m and n have the above
meanings, a compound of Formula II,
R "'
OH II
Ar"° (CHz)m
O
wherein m has the above meaning, Arllo has the meaning
given above for Arl, any reactive groups being protected
by protective groups, and 8111 has the meaning given above
for Rlol, any amino group being protected by a protective
group, is reacted with a compound of the general formula
III,
~Arz~ o
R3
O __
~ H
H N-A3'o/' \ N\ /~ s
z H~ (C~..~2)~ Ar III
O
wherein R3, Ar3 and n have the above meanings, Ar2lo has
the meaning given above for Ar2, any reactive groups being
protected by protective groups, and A310 has the meaning
given above for A3, any reactive nitrogen atoms being
protected by protective groups, or
b) for the preparation of a compound of the general formula
Ih
/Arz
R~ Rz O _ Ra
Z~N\AaoWN N\
Ar (CHz)m A A H~ (CHz)" Ar3 Ih
O
wherein A1, A2, R1, R2, R3, Arl, Ar2, Ar3, m and n have the
above meanings and A3o1 has the meaning given above for A3
with the exception of NH, a compound of the general
formula IV,
9
CA 02497609 2005-03-07
Rllo 8201
Ariio (CH2)rt, A1 A2-N-A311 OH
IV
wherein Al, A2, ArllO and m have the above meanings, A311
has the meaning given above for A3ol~ any reactive
nitrogen atoms being protected by protective groups, 8110
has the meaning given above for R1, any amino groups being
protected by a protective group, and R2o1 has the meaning
given above for R2 with the exception of hydrogen, or
represents an amino protective group, is reacted with a
compound of the general formula V,
Arzio
_ Rs
- H
N
HN
2 ~ (CH2)~ Ar V
O
wherein R3, Ar210, Ar3 and n have the above meanings, or
c) for the preparation of a compound of the general formula
Ii,
/Ar2
O - Rs
~ H
N
Ari-(CH2)m A1-A2o~ N-N~N~ ~ ~Ar3 Ii
H H H 11 ~C~"'j2)n
O
wherein A1, R3, Arl, Ar2, Ar3, m and n have the above
meanings and A2o1 has the meaning given above for A2 with
the exception of carbonyl, a compound of Formula V is
reacted with a carbonyl-group synthesis equivalent and
with a compound of the general formula VI,
CA 02497609 2005-03-07
Rtoa
Ar~jo (CHz)m A1 /~zo~--.- ~ -NHz VI
SG
wherein Al, A2ol, Arllo and m have the above meanings, Rlo4
stands for hydrogen or, if Al is nitrogen, may also stand
for a nitrogen protective group, and SG stands for a
protective group suitable in peptide chemistry, or
d) for the preparation of a compound of the general formula
Ij,
/Arz
O _ R3
H ~
Ar' CH '~N\A3'o~N N\
( 2)m H~ (CHz)" IJ
O
wherein A310~ R3~ Arl~ Ar2, Ar3, m and n have the above
meanings, a compound of Formula III is reacted with a
compound of the general formula VIII,
Ar~~o (CHz)m CHO VIII
wherein Arllo and m have the above meanings,
. and any protective groups are each subsequently cleaved off
again, and a resulting compound of Formula I if desired is
converted into its acid addition salt or an acid addition
salt is converted into a free compound of Formula I.
According to process variant a), a compound of Formula
Ig can be prepared by reacting a carboxylic acid derivative
of Formula II with a primary amine of Formula III and
subsequently cleaving off possibly present protective groups
again. The reaction can be carried out in the manner known in
peptide chemistry as a reaction in the liquid phase or
alternatively as a solid-phase reaction, for example in the
manner of a "Merrifield" solid-phase peptide synthesis. Where
11
CA 02497609 2005-03-07
synthesis is performed in the solid phase, preferably a
resin-bound compound of Formula III is reacted in a polar
aprotic solvent such as N-methylpyrrolidinone (= NMP) with a
compound of Formula III and also with compounds suitable as
coupling reagents, in particular N-hydroxybenzotriazole
(= HOBT), 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium
tetrafluoroborate (= TBTU), N-hydroxy-9-azabenzotriazole
(= HOAt) and/or 2-(1H-9-azabenzotriazol-1-yl)-1,1,3,3-
tetramethyluronium hexafluorophosphate (= HATU) and also in
the presence of a non-nucleophilic organic base, in
particular diisopropylethylamine (= DIPEA). A suitable resin
for the solid-phase synthesis is in particular 2-(4-formyl-3-,
methoxyphenoxy)ethyl resin (= FMPE resin, cf. e.g.
A. Floersheimer et al., Pept. 1990, Proc. Eur. Pept. Symp.,
21st (1991), Meeting Date 1990, E. Giralt et a1. (eds.)
ESCOM: Leiden, 1991; 131). The resin can be loaded with the
compound intended for the further reaction each time in known
manner (see below).
Compounds of Formula II are known per se or may be
prepared in known manner from known compounds (cf. e.g.
R. Gretler et a1. (1978) Helv. Chim. Acta 61(5):1730-1755).
Thus for example compounds of Formula II wherein Arllo
represents optionally protected 2-indolyl may be obtained in
known manner by reductive reaction of nitrophenyl
acetoacetate derivatives with titanium trichloride (cf. e.g.
C.J. Moody et a1. (1990) J. Chem. Soc. Perkin Trans. 1:673-
679; A. Mai et a1. (1999) J. Med. Chem. 42:619-627).
Protective groups which are used in the context of the
present invention may each be introduced in known manner and
usually selectively and independently of each other and
cleaved off again. Suitable protective groups for peptide
synthesis are known, far example, from J. A. W. McOmie,
"Protective Groups in Organic Chemistry", Plenum Press 1971,
or T. W. Green and P. G. M. Wuts, "Protective Groups in
Organic Synthesis", Wiley and Sons 1999. Where substituents
8111 are protected by suitable protective groups, in
particular protective groups known from peptide chemistry are
CA 02497609 2005-03-07
12
suitable. Preferably the tent. butylcarbonyloxy (= Boc) or
the (9H-fluoren-9-ylmethoxy)carbonyl (= Fmoc) protective
group is suitable.
Compounds of Formula III can for example be prepared by
reacting a compound of Formula V with a compound of the
general formula X,
O
SG-N- A3'o~OH X
H
wherein A310 has the above meaning and SG has the above
meaning and is preferably the Fmoc protective group, and
subsequently cleaving off the protective group SG again in
known manner. The reaction may be carried out in the manner
set forth above for the reaction of a compound of Formula II
with a compound of Formula III, with the compound of Formula
V preferably being resin-bound. Where reactive nitrogen atoms
present in the group A3lo are protected by suitable
protective groups, in particular the triphenylmethyl
(= trityl, Trt) protective group is suitable for this.
Compounds of Formula X are known per se or can be prepared in
known manner from known compounds.
Compounds of Formula V can be prepared in that a
compound of the general formula XI,
Arz,o
OH
SG-H ~ XI
O
wherein Ar2lo and SG have the above meanings, is reacted with
a compound of the general formula XII,
13
CA 02497609 2005-03-07
R3
HzN\ / \Ar3
(CHz)~ XII
wherein R3, Ar3 and n have the above meanings, and a
protective group SG is subsequently cleaved off again. The
reaction may for example be carried out in the manner set
forth above for the reaction of a compound of Formula II with
a compound of Formula III, as solid-phase synthesis, with the
compound of Formula XII preferably being resin-bound. Where
FMPE resin is used, the resin may be loaded with a compound
of Formula XII in known manner in the manner of a reductive
amination (cf. B. Dorner et al., Pept. 1998, Proc. Eur. Pept.
Symp, 25th (1999), Meeting Date 1998; S. Bajusz et al.
(eds.), Akademiai Kiado: Budapest, 1999; 90). Compounds of
Formula XI are known per se or can be prepared in known
manner from known compounds. Where substituents Ar2lo in
compounds of Formula XI are protected by protective groups,
in particular protective groups known from peptide chemistry
are suitable. Preferably the Boc protective group is
suitable. Compounds of Formula XII are known per se or can be
prepared in known manner from known compounds.
In one embodiment of process variant a), a compound of
the formula Ik,
Arz
Rio, O ~ Rs
H - H
Nw ~ N\ ~ s
Are (CHz)m H H~ (CHz)~ Ar Ik
O O
wherein Rlol~ R3~ Arl, Ar2, Ar3, m and n have the above
meanings, can be prepared in that a carboxylic acid
derivative of Formula II is reacted with a hydrazine
derivative of Formula IIIa,
CA 02497609 2005-03-07
14
~p,rz~o
R3
O _-
HZN~ ~ ~N~
H H 1I ~OH2)n Ar3 IIIa
O
wherein R3, Ar2lo, Ar3 and n have the above meanings, and
possibly present protective groups are subsequently cleaved
off again. The reaction may for example be carried out in the
manner set forth above for the reaction of a compound of
Formula II with a compound of Formula III, as solid-phase
synthesis. Compounds of Formula IIIa can be prepared by
reacting a compound of Formula V with known 5-(9H-fluoren-9-
ylmethoxy)-3H-[1,3,4]oxadiazol-2-one and subsequently
cleaving off undesirable protective groups. The reaction may
for example be carried out in the manner set forth above for
the reaction of a compound of Formula II with a compound of
Formula III, as solid-phase synthesis, with dichloromethane
in particular possibly being used as solvent.
According to process variant b), a compound of Formula
Ih can be prepared by reacting a carboxylic acid derivative
of Formula IV with a primary amine of Formula V and
subsequently cleaving off possibly present protective groups
again. The reaction may for example be carried out in the
manner set forth above for the reaction of a compound of
Formula II with a compound of Formula III, as solid-phase
synthesis or alternatively in liquid phase. Where the
reaction is carried out in solid phase, preferably the
compound of Formula V is resin-bound. Where the reaction is
carried out in liquid phase, it is possible to work in a
polar aprotic solvent such as dimethyl formamide (= DMF) and
in the presence of compounds suitable as coupling reagents
given under process variant a), and in the presence of a non-
nucleophilic organic base, in particular sym. collidine.
Where R2ol represents an amino protective group, this may
preferably be the Fmoc protective group. Where substituents
Rllo are protected by suitable protective groups, the
15
CA 02497609 2005-03-07
protective groups given above as being suitable for
substituents 8111 are suitable. Compounds of Formula IV can
be prepared by reacting a compound of the general formula
XIII,
R»o R2
Ar"° (CH2)m A~ AZ-NH XIII
wherein A1, A2, R110~ R2~ Arllo and m have the above meanings,
with a compound of the general formula XIV,
O
X- p~s~i / \O- SGT XIV
wherein A311 has the above meaning, X stands for a cleavable
leaving group and SG1 stands for a carboxylic acid protective
group, subsequently cleaving off a carboxylic acid protective
group SG1 again in known manner and if necessary introducing
a protective group into substituents R2. The reaction may be
carried out in an aromatic solvent such as toluene at
temperatures between -20°C and room temperature (= RT),
preferably at 0°C. In particular halogen, preferably chlorine
or bromine, is used as leaving group X in compounds of
Formula XIV. A suitable carboxyl protective group SG1 is in
particular lower alkyl, preferably ethyl or tert. butyl.
Compounds of Formula XIII are known per se or can be prepared
in known manner from known compounds. Compounds of Formula
XIV are known per se or can be prepared in known manner from
known compounds.
According to process variant c), a compound of Formula
Ii can be prepared by reacting a primary amine of Formula V
with a carbonyl-group synthesis equivalent and with a
hydrazine derivative of Formula VI and subsequently cleaving
off possibly present protective groups again. The reaction
may preferably be carried out at room temperature in the
liquid phase, in particular in a dipolar aprotic solvent such
CA 02497609 2005-03-07
16
as dichloromethane. Expediently, operation is in the presence
of an organic non-nucleophilic base which is soluble in the
solvent, such as 4-dimethylaminopyridine (= DMAP). Suitable
carbonyl-group synthesis equivalents are preferably
dipentafluorophenyl carbonate or alternatively phosgene, bis-
(trichloromethyl)carbonate (= triphosgene), trichloromethyl
chloroformate (= diphosgene) or carbonyl diimidazole.
Compounds of Formula VI are known per se, or can be prepared
in known manner from known compounds. Thus for example a
compound of the general formula VIa,
Ar"°-(CH2)m CHZ N-NH2 VIa
SG
wherein Arllo, m and SG have the above meanings, can be
prepared in known manner by reductive amination from a
corresponding aldehyde of the general formula VIII and a
corresponding amine of the general formula XVII,
HZN-H- SGZ XVII
wherein SG2 represents a protective group known in peptide
chemistry, preferably the Boc protective group, subsequent
introduction of a protective group SG and finally cleavage of
the protective group SG2. The reaction may be carried out in
a dipolar aprotic solvent such as tetrahydrofuran (= THF) and
preferably at room temperature. The reduction of a
corresponding imine compound obtained as intermediate product
can be carried out in a dipolar-aprotic solvent such as THF
and at temperatures between -20°C and room temperature,
preferably at 0°C. Suitable reducing agents are complex
borohydrides such as NaCNBH3. The compounds of Formulae VIII
and XVII are known per se or can be prepared from known
compounds in known manner.
17
CA 02497609 2005-03-07
According to process variant d), a compound of Formula
Ij can be prepared by reacting an amino compound of Formula
III with an aldehyde of Formula VIII and subsequently
cleaving off possibly present protective groups again. The
reaction can be carried out in the manner set forth above for
the reaction of compounds of Formula XVI with compounds of
Formula XVII, the resulting imine in this case however not
being reduced.
The resulting compounds of Formula I may in each case be
isolated from the reaction mixture and purified in known
manner. Acid addition salts may be converted into the free
bases in conventional manner, and these may if desired be
converted in known manner into physiologically compatible
acid addition salts.
Physiologically compatible salts of compounds of Formula
I are their salts with inorganic acids, for example sulphuric
acid, phosphoric acids or hydrohalic acids, preferably
hydrochloric acid, or with organic acids, for example lower
aliphatic monocarboxylic, dicarboxylic or tricarboxylic acids
such as malefic acid, fumaric acid, lactic acid, tartaric
acid, citric acid, or with sulphonic acids, for example lower
alkanesulphonic acids such as methanesulphonic acid or
benzenesulphonic acids optionally substituted in the benzene
ring by halogen or lower alkyl, such as p-toluenesulphonic
acid.
The compounds of Formula I may in addition to the carbon
atom bearing the -CH2-Ar2 radical also contain further chiral
centres, namely the carbon atom bearing the substituent R3,
the carbon atom, substituted by Cl_4 alkyl or by C1_q alkyl
carbonylamide, of the methylene group A3 and/or the carbon
atom of the CH group A1, where Rl is amino. The compounds of
Formula I may thus be present in several stereoisomeric
forms. The present invention comprises both the mixtures of
optical isomers and the isomerically pure compounds of
Formula I. Isomerically pure compounds of Formula I are
preferred, in particular the compounds of Formula I, wherein
the carbon atom, substituted by C1_q-alkyl or by C1_q alkyl
CA 02497609 2005-03-07
18
carbonylamide, of the methylene group A3 is in the S
configuration. Where mixtures of optical isomers of the
starting compound are used in the synthesis of the compounds
of Formula I, the compounds of Formula I are also obtained in
the form of mixtures of optical isomers. Departing from
stereochemically uniform forms of the starting compound,
stereochemically uniform compounds of Formula I can also be
obtained. The stereochemically uniform compounds of Formula I
can also be obtained from the mixtures of optical isomers in
known manner, for example by chromatographic separation on
chiral separating materials or by reaction with suitable
optically active acids, for example tartaric acid or
10-camphorsulphonic acid, and subsequent separation into
their optically active antipodes by fractional
crystallisation of the diastereomeric salts obtained.
The novel compounds of Formula I and their
physiologically compatible acid addition salts are
distinguished by a high affinity to the bombesin receptor of
subtype 3 which is selective in comparison to the further
known bombesin receptor subtypes, NMB-R and GRP-R, on which
they act as agonists. It should therefore be expected of the
compounds of Formula I that they are suitable for the
prophylaxis and/or treatment of clinical pictures which can
be beneficially influenced by stimulation of the BRS-3. In
particular, the compounds according to the invention appear
to be suitable for the prophylaxis and/or treatment of
obesity (= adiposity), diabetes, hyperinsulinism,
cardiovascular diseases, eating disorders (hyperphagia,
anorexia, bulimia) and/or syndrome X.
Description of the pharmacological test method:
The BRS-3-agonistic effects of the test substances can for
example be demonstrated in vitro in a pharmacological
standard test operating in accordance with the FLIPR method
("Fluorometric Imaging Plate Reader").
19
CA 02497609 2005-03-07
For this, first of all CHO cells (_ "Chinese hamster ovary
cells") were transfected in known manner with an expression
vector for the subtype 3 of the human bombesin receptor, i.e.
BRS-3.
The cDNA of the human BRS-3 (nucleotide sequence under
GenBank Accession No. L08893) was excised from the plasmid
vector pGEM4 (from Promega, USA) using the restriction
endonuclease EcoRI and was subcloned into the expression
vector pcDNA3.1(-) (from Invitrogen, USA). CHO-K1 cells,
which were already stably transfected with the expression
vector RD-HGA16, which bears the cDNA sequence of the human
Gal6 protein (nucleotide sequence under GenBank Accession
No. M63904), were placed in sample plates with 24 sample
wells ("24-well plate") and incubated overnight under sterile
conditions in an air-humidified incubator at 37°C and 5o COz
in F-12 medium plus Glutamax-I (from GibcoBRL, cat. No.
31765), to which loo-strength foetal calf serum (inactivated
at 56°C for 1 h, from GibcoBRL), 25 ug/ml gentamicin (from
GibcoBRL) and 0.2 mg/ml hygromycin B (from GibcoBRL) had been
added. The next day, the cells were transfected with the BRS-
3 expression vector by adding, using the "Effectene
Transfection Reagent" (from Qiagen), 12 ul of a solution
containing 0.3 ug/ul DNA of the expression vector per sample
well. One day after transfection, the culture medium was
replaced by selection medium. For this, the transfected
cells, which each simultaneously express BRS-3 and the human
Gal6 protein, were cultivated under sterile conditions at
37°C and 5o COZ in F-12 medium plus Glutamax-I (from
GibcoBRL, cat. No. 31765), to which loo-strength foetal calf
serum (inactivated at 56°C for 1 h, from GibcoBRL), 25 ug/ml
gentamicin (from GibcoBRL), 0.2 mg/ml hygromycin B (from
GibcoBRL) and 0.5 mg/ml geneticin (from GibcoBRL) had been
added. To optimise the cell test, the cells with the highest
receptor expression rate were selected. For this, the
transfected cells were diluted 1:30,000 with the selection
medium described above and were placed in sample plates with
96 sample wells ("96-well plate"). The cells were incubated
CA 02497609 2005-03-07
overnight at 37°C and 5o CO2, then those sample wells which
contained only an individual cell were selected. These cells
were first started in sample plates with 24 sample wells
("24-well plate") and then cultivated in Costar plastic
flasks (first 25 ml and then 225 ml). The BRS-3 receptor
expression of the respective individual cell clone was
estimated by determination of the ECSO value of the synthetic
nonapeptide [ D-Phe6, (3-A1a11, Phel3, Nlel9 ] Bn ( 6-14 ) as ligand ( for
performance of the test see below). The transfected cells
were stored at -80°C in aliquots of 1.8 ml medium each with
loo dimethyl sulphoxide (= DMSO) (cell concentration 1 x 106
cells/ml). For cultivation, a frozen aliquot was heated to
37°C, transferred into a Costar plastic flask (225 ml) and
diluted with 50 ml of the selection medium described above.
The medium was first changed once after 30 minutes'
incubation. On each following first to third day, the medium
was removed, the adherent cells (40 - 95o confluence) were
washed with PBS Dulbecco's (from GibcoBRL) and detached from
the bottom of the flask by a 2-minute treatment with trypsin-
EDTA solution (from GibcoBRL) at 37°C. If the cells were to
be cultivated further, they were transferred into a new
plastic flask with fresh medium. If experiments were to be
carried out with the cells, the cells were transferred into
Costar sample plates with 96 sample wells, a clear baseplate
and cover ("Costar 96-well assay plates", from Corning), once
the cell concentration had been set to 1.2 x 109 cells/ml.
BRS-3 is coupled via G-proteins to the Ca2+-signal
transduction path of the CHO cell. If an agonist binds to the
receptor, the phospholipase C is activated via the G-protein,
and then in turn catalyses the synthesis of water-soluble
inositol phosphates. These water-soluble inositol phosphates
cause Ca2+ to be released, which is stored in the endoplasmic
reticulum. The transient increase in the cytosolic Ca2+
concentration was measured in what is called the FLIPR
experiment. To this end, the cells were laden with a Ca2+-
binding, fluorescent dye, Fluo4 (from Molecular Probes). This
intracellular dye binds the cytosolic Ca2+ ions released
21
CA 02497609 2005-03-07
after activation and in so doing intensifies its fluorescent
intensity. The change in fluorescent intensity is
proportional to the change in the intracellular Ca2+
concentration and is a measurement of the activation of the
cell by the corresponding agonists. Below the maximum
fluorescence response, the degree of activation is dependent
on the concentration of the compounds used. The change in
fluorescence due to activation of BRS-3 was determined for
each substance to be tested at different substance
concentrations. The maximum fluorescence response upon
activation of the BRS-3 with the synthetic nonapeptide
[ D-Phe6, (3-Alan, Phel3, N1e14 ] Bn ( 6-14 ) served as reference value
for 1000 activation [cf. Mantey et a1. (1997) J. Biol. Chem.
272: 26062-26071]. The concentration of the compound at which
50o activation occurred was determined as EC5p value and
served as a measure of the effectiveness of the respective
test compound as BRS-3 agonist.
The transfected CHO cells were cultivated for 18 to 24 hours
(= h) in the "Costar 96-well assay plates" (from Corning)
until they were confluent. A 250 mM stock solution of
probenecid was freshly prepared each day. For this, 710 mg
probenecid (from Sigma # P8761) was dissolved in 5 ml 1 N
NaOH and then diluted to 10 ml with HBSS medium without
phenol red (GibcoBRL), which contained 20 mM HEPES (from PAA
Laboratories). A 2 mM stock solution of the fluorescent
calcium-ion indicator dye Fluo4 was prepared by dissolving
1 mg Fluo4 in 440 ~1 DMSO and was stored at -20°C.
Furthermore, a 20o-strength (w/v) solution of Pluronic F-127
(from Sigma) in DMSO was used. Immediately before use, a 22
ul aliquot of the Fluo4 stock solution was thawed. The
loading medium was always freshly prepared by mixing 42 ml
HBSS medium without phenol red (GibcoBRL), which contained
60 mM HEPES (from PAA Laboratories), with 420 ul of the
probenecid stock solution and 22 ul of each of Fluo4 stock
solution and Pluronic F-127 solution. The cells were each
incubated per sample well with 100 ul fresh loading medium
for 45 - 60 min at 37°C and 5o CO2. Then the cells were
CA 02497609 2005-03-07
22
washed three times with 100 ul HBSS medium with 20 mM HEPES
and 2.5 mM probenecid each time. Following the final washing
step, 100 ~l volume remained on the cells in each of the 96
sample wells.
In each case 10 mM stock solutions in DMSO were prepared of
the compounds of Formula I, of which dilution series with
HBSS medium with 20 mM HEPES were loaded into microtitration
plates with 96 sample wells ("96-well plates", from Greiner)
. The maximum concentration used in the measurements was
usually 33 uM, but in some cases also only 1 uM. The
solutions were diluted 1:2, 1:3, 1:4 or 1:10 on 8 or 16
different sample wells, according to the respective compound.
Each microtitration plate contained as a reference a dilution
series of the nonapeptide [D-Phe6, (3-Alan, Phel3, N1e14] Bn ( 6-
14).
The FLIPR apparatus (from Molecular Devices) was programmed
to measure the background fluorescence over a period of 30
seconds (= sec.) at 6-second intervals. After transferring
50 ul in each case from each sample well of the
microtitration plate into the corresponding sample well of
the cell plate, the change in fluorescence over a period of
100 seconds (= sec.) was plotted at 1-second intervals, and
at 6-second intervals during the final 42 sec.
The changes in fluorescence of the reference compound as a
function of the concentration were plotted, and the peptide
concentration of the nonapeptide at which the maximum change
in fluorescence had already been observed was determined
(usually 16 ~M). The value of the maximum change in
fluorescence per sample well was exported to the Excel
spreadsheet program (from Microsoft) and standardised using
the maximum value of the change in fluorescence for the
corresponding reference compound, which was adopted as 1000
value. The curves for the gradient of the relative change in
fluorescence dependent on the concentration of the compound
to be investigated and the corresponding ECSp value were
calculated using the Graphpad Prism program (Version 3.00,
from Graphpad Software).
23
CA 02497609 2005-03-07
In the pharmacological FLIPR test described above, all the
example compounds given below exhibited EC5p values (in nM)
which were less than or equal to 2600. The compounds of
Examples 13 to 34 exhibited EC5p values which were less than
or equal to 710. The ECSp values determined in,the FLIPR
experiment described above are listed in Table 1 below for
individual compounds of Formula I. The example numbers given
in Table 1 relate to the preparation examples below.
CA 02497609 2005-03-07
24
Table 1; Agonistic activity of the test substances on BRS-3
Example No. EC ~ [nM]
2 2.1
3 4.0
4 1.4
6. 0
6 1.5
7 30
I 15 57
19 32
i
21 21
22 2. 9
23 21
24 17
26 25
27 3.1
28 0.19
i
30 2.2
The compounds of Formula I may be administered in
conventional pharmaceutical preparations. The doses to be
used may vary individually and will naturally vary according
to the type of condition to be treated and the substance
used. In general, however, medicinal forms with an active
substance content of 0.1 to 300 mg per individual dose are
suitable for administration to humans and larger mammals.
The compounds of Formula I may be contained according to
the invention, together with conventional pharmaceutical
auxiliaries and/or excipients, in solid or liquid
pharmaceutical preparations. Examples of solid preparations
are preparations which can be administered orally, such as
tablets, coated tablets, capsules, powders or granules, or
alternatively suppositories. These preparations may contain
conventional pharmaceutical inorganic and/or organic
excipients, such as talcum, lactose or starch, in addition to
25
CA 02497609 2005-03-07
conventional pharmaceutical auxiliaries, for example
lubricants or tablet disintegrating agents. Liquid
preparations such as suspensions or emulsions of the active
substances may contain the usual diluents such as water, oils
and/or suspension agents such as polyethylene glycols and the
like. Other auxiliaries may additionally be added, such as
preservatives, taste correctives and the like.
The active substances may be mixed and formulated with
the pharmaceutical auxiliaries and/or excipients in known
manner. For the preparation of solid medicament forms, the
active substances may for example be mixed with the
auxiliaries and/or excipients in conventional manner and may
be wet or dry granulated. The granules or powder may be
poured directly into capsules or be pressed into tablet cores
in conventional manner. These may be coated in known manner
if desired.
T.he following examples are intended to explain the
invention further, without limiting its scope.
Example 1:
IV1- [ ( 1R) -2- ( 1H-3-indolyl) -1- (phenethylcarbamoyl) -ethyl] - (2S) -
2-{[(1R)-1-amino-2-phenethyl]-carboxamido}-pentane diamide;
(H-D-Phe-Gln-D-Trp-phenylethylamide)
H
N
H OII H
H2N~N~N~N w
O H O
O NH2
CA 02497609 2005-03-07
26
A) 100 mg FMPE resin (maximum capacity 0.54 mmol/g) was
allowed to swell in 1 ml dichloroethane for 10 min. 0.5 ml
trimethyl orthoformate (= TMOF), 68 ~1 2-phenylethylamine
and 114 mg NaBH(OAc)3 were added to this receiving
solution, the resulting mixture was treated for 10 min.
with ultrasound and then shaken overnight at RT. Then the
resin was washed in succession three times for three min.
with 3 ml dichloromethane each time and three times for
three min. with 3 ml NMP each time. Then a solution of 56
mg Fmoc-D-Trp(Boc)-OH, 41 mg HATU, 14.6 mg HOAt and 143 ~1
sym. collidine in 1 ml NMP was added to the washed resin.
The resin was shaken in this solution for 5 h at RT,
washed three times for three min. with 3 ml NMP each time
and the resin was again treated overnight with a solution
of 56 mg Fmoc-D-Trp(Boc)-OH, 41 mg HATU, 14.6 mg HOAt and
143 ul sym. collidine in 1 ml NMP. Finally the resin was
washed three times for three min. with 1 ml
dichloromethane each time and was dried in an oil pump
vacuum. 129 mg of an FMPE resin laden with Fmoc-D-
Trp(Boc)-phenylethylamide [loading 0.366 mmol/g;
corresponding to 30 mg (0.047 mmol) free Fmoc-D-Trp(Boc)-
phenylethylamide], which was used directly without
cleavage of the intermediate product for the reaction
below.
B) The entire amount of the laden FMPE resin obtained above
[laden with 0.366 mmo1/g Fmoc-D-Trp(Boc)-phenylethylamide,
corresponding to 30 mg (0.047 mmol) of the free compound]
was allowed to swell for 10 min. in 5 ml NMP. Then the
FMPE resin was treated for 15 min. with 5 ml of a freshly
prepared 20o-strength (v/v) solution of piperidine in NMP,
was washed five times for three min. with 5 ml NMP each
time and the FMPE resin was finally treated again for 15
min. with 5 ml of a freshly prepared 20o-strength (v/v)
solution of piperidine in NMP. Finally, the resin was
washed five times for three min, with 5 ml NMP each time.
The resulting FMPE resin laden with D-Trp(Boc)-
27
CA 02497609 2005-03-07
phenylethylamide was used directly for the reaction below
without isolating the intermediate product.
C) The entire amount of the laden FMPE resin obtained above
[at assumed 1000 conversion laden with 0.366 mmol/g
D-Trp(Boc)-phenylethylamide, corresponding to 19.2 mg
(0.047 mmol) of the free compound] was washed five times
for three min. with 5 ml NMP each time. Then a solution of
57.4 mg Fmoc-Gln(Trt)-OH, 12.7 mg HOBTXH20 and 30 mg TBTU
in 2 ml NMP was added to the laden FMPE resin. Finally 46
ul DIPEA was added to the resulting receiving solution and
the mixture was shaken for 45 min. Once the FMPE resin had
been washed five times for three min. with 5 ml NMP each
time, the coupling step described above was repeated.
Finally it was washed another five times for three min.
with 5 ml NMP each time. An FMPE resin laden with Fmoc-
Gln(Trt)-D-Trp(Boc)-phenylethylamide was obtained, which
was used directly for the reaction below without isolating
the intermediate product.
D) The entire amount of the laden FMPE resin obtained above
[at assumed 1000 conversion laden with 0.366 mmol/g Fmoc-
Gln(Trt)-D-Trp(Boc)-phenylethylamide, corresponding to
47 mg (0.047 mmol) of the free compound] was treated to
cleave off the Fmoc protective group as described above
under B). An FMPE resin laden with Gln(Trt)-D-Trp(Boc)-
phenylethylamide was obtained, which was used directly for
the reaction below without isolating the intermediate
product.
E) The entire amount of the laden FMPE resin obtained above
[at assumed 1000 conversion laden with 0.366 mmol/g
Gln(Trt)-D-Trp(Boc)-phenylethylamide, corresponding to
36.6 mg (0.047 mmol) of the free compound] was washed five
times for three min. with 5 ml NMP each time. Then a
solution of 36.4 mg Fmoc-Phe-OH, 12.7 mg HOBTXH20 and
30 mg TBTU in 2 ml NMP was added to the laden FMPE resin.
Finally 46 ul DIPEA was added to the resulting receiving
solution and the mixture was shaken for 45 min. Once the
FMPE resin had been washed five times for three min. with
CA 02497609 2005-03-07
28
ml NMP each time, the coupling step described above was
repeated. Finally it was washed another five times for
three min. with 5 ml NMP each time. An FMPE resin laden
with Fmoc-Phe-Gln(Trt)-D-Trp(Boc)-phenylethylamide was
obtained, which was used directly for the reaction below
without isolating the intermediate product.
F) The entire amount of the laden FMPE resin obtained above
[at assumed 1000 conversion laden with 0.366 mmol/g Fmoc-
Phe-Gln(Trt)-D-Trp(Boc)-phenylethylamide, corresponding to
54 m,g (0.047 mmol) of the free compound] was treated to
cleave off the Fmoc protective group as described above
under B). An FMPE resin laden with Phe-Gln(Trt)-D-
Trp(Boc)-phenylethylamide was obtained, which was used
directly for the reaction below without isolating the
intermediate product.
G) The entire amount of the laden FMPE resin obtained above
[at assumed 1000 conversion laden with 0.366 mmol/g Phe-
Gln(Trt)-D-Trp(Boc)-phenylethylamide, corresponding to
43.5 mg (0.047 mmol) of the free compound] was washed
three times for ten min. with 3 ml dichloromethane each
time. Then the laden FMPE resin was treated three times
for 30 min. with 2 ml each time of a mixture of
trifluoroacetic acid (= TFA)/triisopropylsilane
(= TIPS)/H20 (18:1:1 v/v/v) and the FMPE resin was
filtered off. Then it was again washed three times for
three min. with 3 ml dichloromethane each time and the
FMPE resin was filtered off. The combined filtrates were
evaporated in a water pump vacuum with nitrogen-cooled
receiving solution. The remaining residue was taken up in
DMSO and purified by reversed-phase HPLC [HPLC system from
Amersham Pharmacia Biotech Akta Basic 100F; pump system P-
900 and detector UV-900; column ODS-A C1$ from Omnicrom
YMC (250 mm x 20 mm, 10 um, flow rate: 8 ml/min); elution
with linear gradient (30 min.) of water (solvent A) in
acetonitrile (solvent B) and 0.10 (v/v) TFA]. Freeze-
drying of the purified fractions yielded 19.2 mg of the
title compound as colourless powder.
29
CA 02497609 2005-03-07
HPLC-MS (ESI) m/z 276.1 (32), 308.1 (90), 583.3 (72)
[m+H]+, 605.4 (100) [m+Na]+, 893.6 (13), 1165.2 (10)
[2m+H]+, 1187.2 (40) [2m+Na]+.
Example 2:
Nl-phenethyl-(2R)-2-[(1S)-1-(benzylcarboxamido)-ethyl]-
carboxamido-3-(1H-3-indolyl)-propanamide
H
N
H O~[ H
w N~N~N w
O H IOI
A) 100 mg of an FMPE resin laden with Fmoc-D-Trp(Boc)-
phenylethylamide [for preparation see Example lA); loading
0.354 mmol/g; corresponding to 22.3 mg (0.035 mmol) free
Fmoc-D-Trp(Boc)-phenylethylamide] was reacted in a manner
corresponding to Example 1B. The resulting resin-bound
Trp(Boc)-phenylethylamide was used directly for the
reaction below without isolation or purification.
B) The entire amount of the FMPE resin laden with D-Trp(Boc)-
phenylethylamide obtained above [at assumed 1000
conversion laden with 0.354 mmol/g D-Trp(Boc)-
phenylethylamide, corresponding to 14.4 mg (0.035 mmol) of
the free compound] was washed five times for three min.
with 5 ml NMP each time. Then a solution of 22 mg Fmoc-
Ala-OH, 9.5 mg HOBTxH20 and 22.5 mg TBTU in 2 ml NMP was
added to the laden FMPE resin. Finally 34 ul DIPEA was
added and the resulting mixture was shaken for 45 min.
Once the FMPE resin had been washed five times for three
min. with 5 ml NMP each time, the coupling step described
above was repeated. Finally it was washed another five
times for three min. with 5 ml NMP each time. An FMPE
resin laden with Fmoc-Ala-D-Trp(Boc)-phenylethylamide was
CA 02497609 2005-03-07
obtained, which was used directly for the reaction below
without isolation.
C) The entire amount of the laden FMPE resin obtained above
[at assumed 1000 conversion laden with 0.354 mmol/g Fmoc-
Ala-D-Trp(Boc)-N-phenylethylamide, corresponding to
24.5 mg (0.035 mmol) of the free compound] was treated to
cleave off the Fmoc protective group as described above
under Example 1B). An FMPE resin laden with Ala-D-
Trp(Boc)-phenylethylamide was obtained, which was used
directly for the reaction below without isolation.
D) The entire amount of the laden FMPE resin obtained above
[at assumed 1000 conversion laden with 0.354 mmolJg Ala-D-
Trp(Boc)-phenylethylamide, corresponding to 16.7 mg
(0.035 mmol) of the free compound] was washed five times
for three min. with 5 ml NMP each time. Then a solution of
9.6 mg phenylacetic acid, 9.5 mg HOBTxH20 and 22.5 mg TBTU
in 2 ml NMP was added to the laden FMPE resin. Finally 34
ul DIPEA was added and the resulting mixture was shaken
for 45 min. Once the FMPE resin had been washed five times
for three min. with 5 ml NMP each time, the coupling step
described above was repeated. Finally it was washed
another five times for three min. with 5 ml NMP each time.
An FMPE resin laden with phenyl acetate-Ala-D-Trp(Boc)-
phenylethylamide was obtained, which was used directly for
the reaction below without isolation.
E) The entire amount of the laden FMPE resin obtained above
[at assumed 1000 conversion laden with 0.354 mmol/g phenyl
acetate-Ala-D-Trp(Boc)-N-phenylethylamide, corresponding
to 20.9 mg (0.035 mmol) of the free compound] was treated
to cleave off the FMPE resin and remove the Boc protective
group as described above in Example 1G). Purification of
the resulting crude product by means of HPLC and
subsequent freeze-drying yielded 12.6 mg (0.025 mmol) of
the title compound as colourless powder with a melting
point of 205-207°C.
1H-NIA (500 MHz, DMSO-d6, 300 K) 8 = 10.78 (s, 1H, NH-CH-
C) , 8.24 (d, JHH = 6. 8 Hz, 1H, NH-CH-CH3) , 8. 18 (d, J =
31
CA 02497609 2005-03-07
8.4 Hz, 1H, NH-CH-CH2), 8.00 (t, J = 5.5 Hz, 1H, NH-CH2-
CH2), 7.57 (d, J = 7.9 Hz, 1H, arom), 6.95-7.32 (m, 14H,
arom), 4.38-4.43 (m, 1H, NH-CH-CH2), 4.21-4.25 (m, 1H, NH-
CH-CH3 ) , 3 . 4 5 ( s, 2H, CO-CH2 ) , 3 . 19-3 . 2 4 (m, 2H, NH-CH2-
CH2 ) , 3 . 11 (dd, J = 14 . 7 Hz, J = 4 . 6 Hz, 1H, NH-CH-CH2 ) ,
2. 84 (dd, J = 14. 6 Hz, J = 9. 6 Hz, 1H, NH-CH-CH2) , 2. 61
(t, J = 7. 6 Hz, 2H, NH-CH2-CH2) , 1. O1 (d, J = 7. 0 Hz, 3H,
C H3 ) .
HPLC-MS (ESI) m/z 159.1 (40), 291.2 (35), 308.1 (100),
497.2 (80) [M+H]+, 519.4 (55) [M+Na]+, 764.5 (20) , 993.1
(10) [2M+H]+, 1015.2 (100) [2M+Na]+.
Example 3:
N1-phenethyl-(2R)-2-{1-[(4-chlorobenzyl)-amino]-
ethylcarboxamido}-3-(1H-3-indolyl)-propanamide
H
N
CI , O
N N
N
H O
A) A solution of 8.35 g ethyl 2-bromopropionate in 18 ml
toluene was added dropwise to a solution of 7.08 g
4-chlorobenzylamine and 5.06 g triethylamine in 38 ml
toluene with stirring and ice cooling over a period of
4 h, and the reaction mixture was then stirred for 4 days.
Then the organic phase was extracted with 300 ml water and
then dried over Na2SOq. The solvent was evaporated in a
water pump vacuum and the resulting residue was purified
by means of flash chromatography at a pressure of 1-1.2
bar (stationary phase: Silicagel 60, grain size 0.040-
0.063 mm, mobile phase: ethyl acetate/hexane 1:1). After
concentration of the solvent in a water pump vacuum and
drying of the residue in an oil pump vacuum, 4.25 g N-(4-
chlorobenzyl)-alanine ethyl ester was obtained as yellow
oil.
CA 02497609 2005-03-07
32
1H-NMR (250 MHz, DMSO-d6, 300 K) 8 = 7.31-7.38 (m, 4H,
atom) , 4. 09 (q, J = 7 . 1 Hz, 2H, CH2-CH3) , 3. 65 (q, J =
13.9 Hz, NH-CH2-C6H4C1), 3.20-3.24 (m, 1H, NH-CH), 2.51
(bs, 1H, NH) , 1 . 17-1 . 22 (m, 6H, CH2-CH3 and CH-CH3 ) .
B) 6.2 ml 1 N NaOH and 12 ml methanol were added to 1.0 g of
the N-(4-chlorobenzyl)-alanine ethyl ester obtained above
and the mixture was stirred for 30 min. It was neutralised
with 1 N HCl and the solvent was evaporated in a water
pump vacuum. The resulting residue was taken up in a
mixture of 15 ml saturated aqueous NaHC03 solution and
4 ml dioxane. A solution of 1.1 g FmocCl in 8 ml dioxane
was added dropwise to this receiving solution over a
period of 15 min. with ice cooling. The reaction mixture
was stirred for 30 min. with ice cooling and then
overnight at RT. Then 20 ml water was added to the
reaction mixture, the aqueous phase was separated off and
extracted with 100 ml diethyl ether. The aqueous phase was
set to pH 1 by addition of concentrated hydrochloric acid
and then extracted again three times with 100 ml ethyl
acetate each time. The combined organic phases were dried
over Na2SOq and the solvent was evaporated in a water pump
vacuum. The resulting residue was purified by column
chromatography (stationary phase: Silicagel 60, grain size
0.040-0.063 mm, mobile phase: ethyl acetate/hexane/acetic
acid 1:1:1). After evaporating the solvent in a water pump
vacuum and drying the residue in an oil pump vacuum, 1.3 g
2-{4-chlorobenzyl-[(9H-fluoren-9-ylmethoxy)-carbonyl]-
amino}-propanoic acid was obtained as colourless oil
(2.98 mmol). The ratio of cis/trans-isomers was not
determined.
HPI~C-MS (ESI) m/z 179.1 (95), 436.0 (75) [M+H]+, 458.2
(40) [M+Na]+, 893.0 (50) [2M+Na]+, 909.2 (100) [2M+K]+.
C) 0.26 g phenylethylamine, 1.13 g Fmoc-D-Trp(Boc)-OH, 0.43 g
HOBT and 1.03 g TBTU were dissolved in 15 ml DMF. 1.19 g
DIPEA was added dropwise to this receiving solution over a
period of 5 min. Then the reaction mixture was stirred for
one hour, the solvent was evaporated in a water pump
33
CA 02497609 2005-03-07
vacuum and the resulting residue was purified by means of
flash chromatography at a pressure of 1-1.2 bar
(stationary phase: Silicagel 60, grain size 0.040-0.063
mm, mobile phase: ethyl acetate/hexane 1:1). After
evaporating the solvent in a water pump vacuum and drying
the residue in an oil pump vacuum, 1.33 g N1-phenethyl-
(2R)-2-(9H-fluoren-9-ylmethoxy)-carboxamido-3-[1-(tert-
butoxycarbonyl)-3-indolyl]-propanamide (= Fmoc-D-Trp(Boc)-
phenylethylamide) was obtained as colourless, waxy solid
with a melting point of 140°C.
HPLC-MS (ESI) m/z 308.2 (20), 530.3 (40), 630.3 (40)
[M+H]+, 652.4 (10) [M+Na]+, 1259.5 (100) [2M+H]+, 1281.5
(30) [2M+Na]+.
D) 1.0 g of the Fmoc-D-Trp(Boc)-phenylethylamide obtained
above was dissolved in 6 ml of a 20o-strength (v/v)
solution of piperidine in DMF. The reaction mixture was
stirred for 30 min. and the solvent was then evaporated
off in a water pump vacuum with nitrogen-cooled receiving
solution. The remaining residue was separated off from the
resulting Fmoc-piperidine complex by means of flash
chromatography at a pressure of 1-1.2 bar (stationary
phase: Silicagel 60, grain size 0.040-0.063 mm, mobile
phase: ethyl acetate/hexane 2:1) and then eluted (mobile
phase: chloroform/methanol 15:1). Concentrating the
solvent in a water pump vacuum and drying the residue in
an oil pump vacuum yielded 0.63 g tert-Butyl-3-[(2R)-2-
amino-2-(phenethylcarbamoyl)-ethyl]-1H-1-indole
carboxylate (= D-Trp(Boc)-phenylethylamide) as yellow oil.
MS (ESI) m/z 159.1 (20), 291.2 (75), 308.1 (95), 352.1
(70), 408.1 (100) [M+H]+, 430.1 (35) [M+Na]+, 815.2 (15)
[2M+H]+, 837.1 (20) [2M+Na]+.
E) 0.32 g 2-(4-chlorobenzyl-[(9H-fluoren-9-ylmethoxy)-
carbonyl]-amino}-propanoic acid (0.736 mmol, for
preparation see above under B), 0.30 g tert-butyl-3-[(2R)-
2-amino-2-(phenethylcarbamoyl)ethyl]-1H-1-indole
carboxylate, for preparation see above under D), 0.42 g
HATU and 0.15 g HOAt were dissolved in 7 ml DMF. Then 1.33
CA 02497609 2005-03-07
34
g sym. collidine was added dropwise over a period of 10
min. The reaction mixture was stirred for 1 h and then the
solvent was evaporated in a water pump vacuum with
nitrogen cooling. The remaining residue was purified by
means of flash chromatography at a pressure of 1-1.2 bar
(stationary phase: Silicagel 60, grain size 0.040-
0.063 mm, mobile phase: ethyl acetate/hexane 1:1).
Concentrating the solvent in a water pump vacuum and
drying the residue in an oil pump vacuum yielded 0.54 g
N1-phenethyl-(2R)-2-{1-[N-(4-chlorobenzyl)-N-(9H-fluoren-
9-ylmethoxycarbonyl)-amino]-ethylcarboxamido}-3-[1-(tert-
butoxycarbonyl)-3-indolyl]-propanamide as colourless foam.
MS (ESI) m/z 179.2 (10) , 769.4 (10) , 825.4 (100) [m+H]+,
1651.6 (55) [2m+H]+.
F) The entire amount (0. 54 g) of the IVl-phenethyl- (2R) -2-{ 1-
[N-(4-chlorobenzyl)-N-(9H-fluoren-9-ylmethoxycarbonyl)-
amino]-ethylcarboxamido}-3-[1-(tert-butoxycarbonyl)-3-
indolyl]-propanamide obtained above (0.654 mmol) was
dissolved in 2.5 ml dichloromethane. 0.25 ml
triisopropylsilane was added to this receiving solution
and the resulting mixture was cooled to 0°C. Then 2.5 ml
TFA was added dropwise to the mixture over a period of
min. and the mixture was stirred for 1 h at 0°C. The
solvent was evaporated in a water pump vacuum with
nitrogen-cooled receiving solution. The remaining residue
was taken up in a mixture of 20 ml DMSO, 2.5 ml water and
2.5 ml acetic acid and stirred overnight at RT. Then the
solvent was evaporated in a water pump vacuum and the N1-
phenethyl-(2R)-2-{1-[N-(4-chlorobenzyl)-N-(9H-fluoren-9-
ylmethoxycarbonyl)-amino]-ethylcarboxamido}-3-[1H-3-
indolyl]-propanamide remaining as residue was used
directly for the reaction given below without further
purification or characterisation.
G) The entire amount of the Fmoc-protected propanamide
obtained above (0.654 mmol at 1000 conversion) was
dissolved in 10 ml of a 20o-strength (v/v) solution of
piperidine in DMF and stirred for 30 min. The solvent was
35
CA 02497609 2005-03-07
evaporated in a water pump vacuum with nitrogen-cooled
receiving solution and the resulting residue was purified
by means of flash chromatography at a pressure of 1-
1.2 bar (stationary phase: Silicagel 60, grain size 0.040-
0.063 mm, mobile phase: ethyl acetate). After freeze-
drying the purified fractions, 320 mg of the title
compound (0.637 mmol) was obtained as colourless powder
with a melting point of 107-110°C. The ratio of the two
isomers to one another was 1:1.24.
1H-NMFt (500 MHz, DMSO-d6, 300 K) 8 = 10.85 and 10.83 (s,
1H, NH-CH-C), 9.2 (m, 1H, NH-CH-CH3), 8.72 and 8.77 (d, J
- 8.5 Hz, 1H, NH-CH-CH2), 8.28 and 8.34 (t, J = 5.5 Hz,
1H, NH-CH2-CH2), 7.68 and 7.63 (d, J = 7.7 Hz, 1H, arom),
6.98-7.48 (m, 13H, arom), 4.61-4.69 (m, 1H, NH-CH), 3.98-
4.02 (m, 1H, NH-CH2-C6HQC1), 3.75 (m, 1H, NH-CH-CH3), 3.60-
3.67 and 3.39-3.43 (m, 1H, NH-CH2-C6HqC1), 3.34-3.38 (m,
1H, NH-CH2-CH2), 3.24-3.29 (m, 1H, NH-CH2-CH2), 3.05-3.08
(m, 1H, NH-CH-CH2), 2.85-2.92 (m, 1H, NH-CH-CH2), 2.67-
2.71 (m, 2H, NH-CH2-CHZ), 1.33 and 1.10 (d, J = 6.9 Hz,
3H, CH3) .
HPLC-MS (ESI) m/z 291.2 (30), 308.1 (100), 503.2 (35)
[M+H]+, 525.4 (15) [M+Na]+, 1027.1 (20) [2M+Na]+.
Example 4:
N1-phenethyl-(2R)-2-{N'-[2-(3-pyridyl)-ethanoyl]-hydrazino}-
carboxamido-3-(1H-3-indolyl)-propanamide (181)
H
N
O
N ~ N.N~N~N
I , O H H O I ,
A) 10.0 g Boc-hydrazine was dissolved in 200 ml dry
dichloromethane and 12.95 ml DIPEA (75.6 mmol) and the
CA 02497609 2005-03-07
36
solution was then cooled to 0°C . A solution of 19.6 g
FmocCl in 100 ml dry dichloromethane was added dropwise to
this receiving solution over a period of 30 min. Then the
reaction mixture was stirred overnight at RT. Following
this, the organic phase was extracted with 200 ml water,
dried over Na2S04 and concentrated in a water pump vacuum
to a volume of 100 ml. Then 100 ml trifluoroacetic acid
was added with ice cooling and the mixture was stirred for
1.5 h. 300 ml saturated aqueous Na2C03 solution was added
to the mixture, the mixture was filtered and the organic
phase separated off was dried over Na2SOq. Evaporating the
solvent in a water pump vacuum and drying the resulting
residue in an oil pump vacuum yielded 18.02 g N-[(9H-
fluoren-9-ylmethoxy)-carbonyl]-hydrazine (70.8 mmol) as
colourless solid with a melting point of 150-153°C.
1H-NMR (250 MHz, DMSO-d6, 300 K) 8 = 10.10 (bs, 1H, NH),
9.60 (bs, 1H, NH), 7.89 (d, J = 7.6 Hz, 2H, atom), 7.70
(d, J = 7.3 Hz, 2H, atom), 7.30-7.45 (m, 4H, atom), 4.48
(d, J = 6. 6 Hz, 2H, CO-CH2) , 4.27 (t, J = 6.7 Hz, 1H, CO-
CH2-CH) .
B) A suspension of 1.49 g of the N-[(9H-fluoren-9-ylmethoxy)-
carbonyl]-hydrazine obtained above (5.78 mmol), 60 ml
dichloromethane and 60 ml saturated aqueous NaHC03
solution was stirred vigorously for 5 min. at 0°C and then
left to stand for 5 min. at this temperature. Then 7.95 ml
of a 1.89 M phosgene solution in toluene was added to the
bottom organic phase using a syringe. Once addition was
complete, the reaction mixture was stirred vigorously for
a further 10 min. Then 20 ml water and 20 ml
dichloromethane were added to the reaction mixture and the
phases were quickly separated. The aqueous phase was
extracted with 50 ml dichloromethane and the combined
organic phases were dried over Na2S04. After evaporating
off the solvent in a water pump vacuum and drying the
residue in an oil pump vacuum, 1.35 g 5-(9H-fluoren-9-
ylmethoxy)-3H-[1,3,4]oxadiazol-2-one (4.82 mmol) was
obtained as colourless solid with a melting point of
37
CA 02497609 2005-03-07
125°C.
1H-NMR (250 MHz, CDC13, 300 K) 8 = 8.72 (bs, 1H, NH), 7.77
(d, J = 7.5 Hz, 2H, arom), 7.59 (d, J = 7.4 Hz, 2H, arom),
7.28-7.45 (m, 4H, arom), 4.49 (d, J = 7.8 Hz, 2H, C H2-CH),
4.32-4.41 (m, 1H, CH2-CH).
C) 100 mg of an FMPE resin laden with Fmoc-D-Trp(Boc)-
phenylethylamide (for preparation see Example lA); loading
0.354 mmol/g; corresponding to 22.3 mg (0.035 mmol) free
Fmoc-D-Trp(Boc)-phenylethylamide) was allowed to swell for
min. in 5 ml NMP and then treated twice, each time for
min., with 5 ml each time of a freshly prepared 20%-
strength (v/v) solution of piperidine in NMP. Following
this, the resin was washed five times for three min. each
time with 5 ml NMP each time, and once again five times
each time for three min. with 5 ml dichloromethane each
time. Then the resin was left to stand for 30 min. in 5 ml
dry dichloromethane. After separation of the solvent by
filtration, a solution of 30.5 mg of the 5-(9H-fluoren-9-
ylmethoxy)-3H-[1,3,4]oxadiazol-2-one obtained above under
B) in 1 ml dry dichloromethane was added to the FMPE resin
laden with D-Trp(Boc)-phenylethylamide and the mixture was
shaken for 90 min. Finally, the resin was washed five
times for three min. with 5 ml dichloromethane each time
and then another five times for three min. with 5 ml NMP
each time. An FMPE resin laden with Fmoc-hydrazine-
carbonyl-D-Trp(Boc)-phenylethylamide was obtained, which
was used directly for the reaction below without isolating
the intermediate product.
D) The entire amount of the laden FMPE resin obtained above
[at assumed 1000 conversion laden with 0.354 mmol/g Fmoc-
hydrazine-carbonyl-D-Trp(Boc)-phenylethylamide,
corresponding to 24 mg (0.035 mmol) of the free compound]
was treated to cleave off the Fmoc protective group as
described in Example 1B). An FMPE resin laden with
hydrazine-carbonyl-D-Trp(Boc)-phenylethylamide was
obtained, which was used directly for the reaction below
CA 02497609 2005-03-07
38
without cleaving off and isolating the intermediate
product.
E) The entire amount of the laden FMPE resin obtained above
[at assumed 1000 conversion laden with 0.354 mmol/g
hydrazine-carbonyl-D-Trp(Boc)-phenylethylamide,
corresponding to 16.5 mg (0.035 mmol) of the free
compound] was washed five times for three min. with 5 m1
NMP each time. Then a solution of 12 mg 3-pyridylacetic
acid (0.07 mmol), 9.5 mg HOBTXH20 (0.07 mmol) and 22.5 mg
TBTU (0.07 mmol) in 2 ml NMP was added to the laden FMPE
resin. Finally 34 ~1 DIPEA (0.2 mmol) was added to the
resulting receiving solution and the mixture was shaken
for 45 min. Once the FMPE resin had been washed five times
for three min. with 5 ml NMP each time, the coupling step
described above was repeated. Finally it was washed
another five times for three min. with 5 ml NMP each time.
An FMPE resin laden with 3-pyridyl acetate-hydrazine-
carbonyl-D-Trp(Boc)-phenylethylamide was obtained, which
was used directly for the reaction below without isolating
the intermediate product.
F) The entire amount of the laden FMPE resin obtained above
[at assumed 1000 conversion laden with 0.354 mmol/g
3-pyridyl acetate-hydrazine-carbonyl-D-Trp(Boc)-
phenylethylamide, corresponding to 20.5 mg (0.035 mmol) of
the free compound] was treated to cleave off the FMPE
resin and remove the Boc protective group as described
under Example 1G). After HPLC purification and freeze-
drying, 4.5 mg (0.0093 mmol) of the title compound was
obtained as colourless powder with a melting point of 116-
120°C.
1H-NMR (500 MHz, DMSO-d6, 300 K) 8 = 10.79 (s, 1H, NH),
9.89 (s, 1H, NH), 8.63 (s, 1H, arom), 8.61 (d, J = 5.0 Hz,
1H, arom), 8.02-8.04 (m, 2H, NH and arom), 7.96 (bs, 1H,
NH-CH2-CH2), 7.63 (t, J = 5.5 Hz, 1H, arom), 7.51 (d, J =
7.9 Hz, 1H, arom), 7.30 (d, J = 8.2 Hz, 1H, arom), 7.25
(t, J = 7.6 Hz, 2H, arom), 6.99-7.18 (m, 5H, arom), 6.95
(t, J = 8.0 Hz, 1H, arom), 6.44 (d, J = 8.1 Hz, 1H, NH-
39
CH), 4.32-4.36 (m, 1H, NH-CH), 3.59 (s, 2H, CO-CH2-C5H4N),
3.22-3.26 (m, 1H, NH-CH2-CH2), 3.15-3.19 (m, 1H, NH-CH2-
CH2) , 3.01 (dd, J = 14. 4 Hz, J = 5. 6 Hz, 1H, NH-CH-CH2) ,
2. 91 (dd, J = 14 . 6 Hz, J = 7. 4 Hz, 1H, NH-CH-CH2) , 2. 58
(t, J = 7.5 Hz, 2H, NH-CH2-CH2) .
HPLC-MS (ESI) m/z 152.1 (40), 185.2 (30), 334.3 (30),
485.3 (100) [M+H]+, 507.3 (70) [M+Na]+, 523.3 (10) [M+Na]+,
969.3 (20) [2M+H]+, 991.4 (50) [2M+Na]+, 1007.5 (20)
[2M+K] +.
Example 5:
N1-phenethyl-(2R)-2-[N'-(4-chlorobenzyl)-hydrazino]-
carboxamido-3-(1H-3-indolyl)-propanamide (185)
H
N
\ I
CI
H C H
N.N~N~N
H H ~ I ,
A) 2.12 g 4-chlorobenzaldehyde in 5 ml THF was added dropwise
to a solution of 1.98 g tert-butyl carbazate (Boc-
hydrazine) in 15 ml THF with constant stirring at room
temperature over a period of 10 min. After 3 hours, the
solvent was evaporated in a water pump vacuum and the
resulting residue was purified by means of flash
chromatography at a pressure of 1-1.2 bar (stationary
phase: Silicagel 60, grain size 0.040-0.063 mm, mobile
phase: ethyl acetate/hexane 1:5). After evaporating the
solvent in a water pump vacuum and drying the residue in
an oil pump vacuum, 3.64 g tert-butyl N'-(4-
chlorophenylmethylene)-hydrazine-carboxylate was obtained
as colourless solid with a melting point of 170-171°C.
MS (EI) m/z 41.2 (20), 57.2 (100), 154.0 (10), 181.0 (5),
197.9 (20), 253.9 (5) [M]+.
CA 02497609 2005-03-07
CA 02497609 2005-03-07
B) 0.55 g NaCNBH3 was added to a suspension of 1.5 g of the
tent-butyl N'-(4-chlorophenylmethylene)-hydrazine-
carboxylate obtained above in 25 ml dry THF with ice
cooling and under argon protective gas atmosphere. 10 ml
acetic acid was added dropwise to this mixture over a
period of 10 min. The resulting clear solution was stirred
overnight at RT. Then 60 ml water and 60 ml ethyl acetate
were added and the pH value of the aqueous phase was set
to 8 with NaHC03. The organic phase was separated off and
washed in succession with 50 ml saturated aqueous NaHC03
solution and with 50 ml saturated aqueous common salt
solution. The organic phase was dried over Na2SOq and the
solvent was evaporated in a water pump vacuum. 40 ml
methanol and 20 ml 1N NaOH were added to the remaining
colourless residue in succession and the resulting mixture
was first stirred for 1 hour at RT and then heated to
boiling under reflux cooling for 1 hour. The mixture
cooled to RT was extracted three times with diethyl ether,
the combined ether phases were dried over Na2SOq and the
solvent was evaporated in a water pump vacuum. The
remaining yellow oil was purified by means of flash
chromatography at a pressure of 1-1.2 bar (stationary
phase: Silicagel 60, grain size 0.040-0.063 mm, mobile
phase: ethyl acetate/hexane 1:4). Evaporating the solvent
in a water pump vacuum and drying the residue in an oil
pump vacuum yielded 1.05 g N-(tert-butoxycarbonyl)-N'-(4-
chlorobenzyl)-hydrazine as colourless solid with a melting
point of 77-82°C.
1H-NMR (250 MHz, DMSO-d6, 300 K) 8 = 8.23 (bs, 1H, NH-CO),
7.35 (m, 4H, arom), 4.84 (bs, 1H, NH-CH2), 3.85 (s, 2H,
NH-CH2) , 1. 37 (s, 9H, CH3) .
C) 0.8 g of the N-(tert-butoxycarbonyl)-N'-(4-chlorobenzyl)-
hydrazine obtained above was suspended with ice cooling in
a mixture of 4 ml dioxane and 16 ml loo-strength aqueous
NaHC03 solution. Then a solution of 0.89 g FmocCl in 10 ml
dioxane was added over a period of 10 min. and the
reaction mixture was then stirred overnight at RT. 50 ml
41
CA 02497609 2005-03-07
water was added and the aqueous phase was extracted three
times with 100 ml diethyl ether each time. The organic
phases which had been separated off were combined, dried
over Na2SOq and the solvent was finally evaporated in a
water pump vacuum. The residue was purified by means of
flash chromatography at a pressure of 1-1.2 bar
(stationary phase: Silicagel 60, grain size 0.040-0.063
mm, mobile phase: ethyl acetate/hexane 1:2). Evaporating
the solvent in a water pump vacuum and drying the residue
in an oil pump vacuum yielded 1.37 g N-(4-chlorobenzyl)-N-
[(9H-fluoren-9-ylmethoxy)-carbonyl]-N'-(tert-
butoxycarbonyl)-hydrazine as colourless solid with a
melting point of 53-55°C.
1H-NMR (250 MHz, DMSO-d6, 300 K) 8 = 9. 62 (s, 1H, NH) ,
7.89 (d, J = 7.3 Hz, 2H, arom), 7.74 (d, J = 6.7 Hz, 1H,
arom), 7.56 (m, 1H, arom), 7.27-7.43 (m, 7H, arom), 6.99
(m, 1H, arom), 4.2-5.52 (m, 5H, N-CH2 and CO-CH2-CH), 1.43
(s, 9H, CH3) .
D) 0.1 ml triisopropylsilane was added to a solution of
0.30 g of the N-(4-chlorobenzyl)-N-[(9H-fluoren-9-
ylmethoxy)-carbonyl]-N'-(tert-butoxycarbonyl)-hydrazine
obtained above under C) in 2.5 ml dichloromethane and the
mixture was cooled to 0°C. Then 2.5 ml TFA was added
dropwise over a period of 5 min. and the solution was then
stirred for 30 min. The solvent was evaporated in a water
pump vacuum with nitrogen cooling and the residue was
taken up again in a solution of 77 mg DMAP (0.63 mmol) in
ml dry dichloromethane. This mixture was added dropwise
and with stirring over a period of 20 min. to a solution
of 0.25 g dipentafluorophenyl carbonate (0.63 mmol) in 20
ml dry dichloromethane. After complete addition, a
solution of 0.26 g tert-butyl-3-[(2R)-2-amino-2-
(phenethylcarbamoyl)-ethyl]-1H-1-indole carboxylate (for
preparation see Example 3D)), 77 mg DMAP (0.63 mmol) and
10 ml dry dichloromethane was added to the receiving
solution thus obtained, with stirring. It was stirred for
30 min, at RT, the solvent was evaporated in a water pump
CA 02497609 2005-03-07
42
vacuum and the residue was taken up in 4 ml
dichloromethane. Then 0.1 ml triisopropylsilane was added
and the mixture cooled to 0°C. Then 4 ml TFA was added
dropwise over a period of 5 min. and the mixture was then
stirred for 30 min. The solvent was removed in an oil pump
vacuum and 10 ml of a 20o-strength (v/v) solution of
piperidine in DMF was added to the dried residue for 30
min. at RT. The solvent was evaporated in a water pump
vacuum with nitrogen-cooled receiving solution, the
remaining residue was taken up in DMSO and this was
purified by reversed-phase HPLC [(HPLC system Amersham
Pharmacia Biotech Akta Basic 100F; pump system P-900 and
detector UV-900; column ODS-A C18 from Omnicrom YMC (250
mm x 20 mm, 10 um, flow rate: 8 ml/min); elution with
linear gradient (30 min.) of water (solvent A) in
acetonitrile (solvent B) and 0.10 (v/v) TFA]. Freeze-
drying the purified fractions yielded 26.8 mg of the title
compound as colourless powder with a melting point of 75-
80°C.
1H-NMFt (500 MHz, ACN-d3, 300 K) 8 = 9.74 (bs, 1H, NH) ,
7 . 91 (d, J = 7.7 Hz, 1H, arom) , 7. 98 (d, J = 8. 1 Hz, 1H,
arom), 7.58-7.83 (m, 12H, arom), 6.99 (bs, 1H, NH-CH),
6.89 (bs, 1H, NH-CH2-CH2), 4.87 (q, J = 6.9 Hz, NH-CH),
4. 34 (bs, 2H, NH-CH2-C6HqCl) , 3. 87-3. 94 (m, 1H, NH-CH2-
CH2), 3.76-3.82 (m, 1H, NH-CH2-CH2), 3.66 (d, J = 6.2 Hz,
2H, NH-CH-CH2) , 3. 17 (t, J = 7. 3 Hz, 2H, NH-CH2-CH2) .
HPI~C-MS (ESI) m/z 490.1 (70) [M+H]+, 512.3 (50) [m+Na]+,
754.8 (100), 978.9 (25) [2M+H]+, 1001.0 (90) [2M+Na]+,
1063.1 (20) .
Example 6:
N1-phenethyl-(2R)-2-[N'-(furan-2-ylmethylene)-hydrazino]-
carboxamido-3-(1H-3-indolyl)-propanamide (189)
43
CA 02497609 2005-03-07
H
N
/ O
~~N~N~N N w
H H
A) 500 mg tert butyl-3-[(2R)-2-amino-2-(phenethylcarbamoyl)-
ethyl]-1H-1-indole carboxylate (for preparation see
Example 3D)) and 515 mg freshly prepared 5-(9H-fluoren-9-
ylmethoxy)-3H-[1,3,4]oxadiazol-2-one (for preparation see
Example 4B)) were dissolved in 20 ml dry DMF and stirred
for 75 min. at RT. The solvent was then evaporated in a
water pump vacuum with nitrogen-cooled receiving solution
and the residue was purified by means of column
chromatography (stationary phase: Silicagel 60, grain size
0.040-0.063 mm, mobile phase: chloroform/methanol, 20:1).
The solvent was again evaporated in a water pump vacuum
and the residue was dried in an oil pump vacuum.
0. 58 g N1-phenethyl- (2R) -2-{ [N' - ( 9H-fluoren-9-ylmethoxy) -
carbonyl]-hydrazino}-carboxamido-3-[1-tert-
butoxycarbonyl)-3-indolyl]-propanamide (= Fmoc-hydrazine-
carbonyl-D-Trp(Boc)-phenylethylamide) was obtained as
colourless solid with a melting point of 135-137°C.
HPI~C-MS (ESI) m/z 179.2 (10), 334.3 (10), 378.2 (10),
588.4 (10) , 632.3 (25) , 654.4 (35) , 688.3 (80) [M+H]+,
710.4 (100) [M+Na]+, 1375.5 (40) [2M+H]+, 1397.5 (25)
[2M+Na]+.
B) 179 mg of the Fmoc-hydrazine-carbonyl-D-Trp(Boc)-
phenylethylamide obtained above was dissolved in 2 ml of a
20o-strength (v/v) solution of piperidine in DMF and
stirred for 30 min. at RT. Then the solvent was evaporated
in a water pump vacuum with nitrogen-cooled receiving
solution and the residue was taken up in 10 ml THF. 25 mg
furan-2-carbaldehyde was added to this receiving solution
and the mixture was stirred for 24 hours at RT. Then a
further 50 mg furan-2-carbaldehyde was added and the
CA 02497609 2005-03-07
44
mixture was stirred for another 24 hours. The solvent was
evaporated in a water pump vacuum and the residue was
purified by means of flash chromatography at a pressure of
1-1.2 bar (stationary phase: Silicagel 60, grain size
0.040-0.063 mm, mobile phase: ethyl acetate/hexane 1:1).
After evaporating the solvent in a water pump vacuum and
drying the residue in an oil pump vacuum, 100 mg
N1-phenethyl-(2R)-2-[N'-(furan-2-ylmethylene)-hydrazino]-
carboxamido-3-[1-(tert-butoxycarbonyl)-3-indolyl]-
propanamide was obtained as colourless, crystalline solid.
MS (ESI) m/z 510.3 (15), 544.3 (55) [M+H]+, 566.3 (50)
[M+Na]+, 835.2 (25) [(3M+K+H)/2]2+, 1087.4 (45) [2M+H]+,
1109.5 (100) [2M+Na]+, 1630.3 (5) [3M+H]+, 1652.2 (20)
[3M+Na]+.
C) 100 mg of the N1-phenethyl-(2R)-2-[N'-(furan-2-
ylmethylene)-hydrazino]-carboxamido-3-[1-(tert-
butoxycarbonyl)-3-indolyl]-propanamide obtained above was
dissolved in 3 ml dichloromethane. 0.1 ml
triisopropylsilane was added thereto and the mixture was
then cooled to 0°C. Then 3 ml TFA was added dropwise over
a period of 5 min. and the reaction mixture was stirred
for 1 hour. Then the solvent was evaporated in a water
pump vacuum, the remaining residue was taken up in a
mixture of 8 ml DMSO, 1 ml water and 1 ml acetic acid, and
was stirred overnight at RT. Then the solvent was
evaporated to dryness in a water pump vacuum with
nitrogen-cooled receiving solution and the residue was
taken up in DMSO. Reversed-phase HPLC [HPLC system
Amersham Pharmacia Biotech Akta Basic 100F; pump system P-
900 and detector UV-900; column OI?S-A C18 from Omnicrom
YMC (250 mm x 20 mm, 10 um, flow rate: 8 ml/min); elution
with linear gradient (30 min.) of water (solvent A) in
acetonitrile (solvent B) and 0.10 (v/v) TFA and freeze-
drying of the purified fractions yielded 46 mg of the
title compound as colourless powder with a melting point
of 100-101°C.
1H-NMFt (500 MHz, DMSO-d6, 300 K) 8 = 10.82 (s, 1H, NH-GH-
45
C) , 10. 41 (s, 1H, N-NH) , 8 . 09 (t, J = 5. 5 Hz, 1H, NH-CH2) ,
7.75 (s, 1H, arom), 7.73 (s, 1H, arom), 7.55 (d, J = 7.9
Hz, 1H, arom), 7.30 (d, J = 8.1 Hz, 1H, arom), 7.23-7.26
(m, 2H, arom), 7.14-7.17 (m, 3H, arom), 7.07 (s, 1H,
arom), 7.03 (t, J = 7.4 Hz, 1H, arom), 6.93 (t, J = 7.5
Hz, 1H, arom), 6.74 (d, J = 3.2 Hz, 1H, arom), 6.58-6.60
(m, 2H, NH-CH-CH2 and arom), 4.45 (q, J = 6.8 Hz, 1H, NH-
CH) , 3.24-3. 31 (m, 1H, NH-CH2) , 3. 17-3.24 (m, 1H, NH-CH2) ,
3.02-3.10 (m, 2H, NH-CH-CH2), 2.62 (t, J = 7.4 Hz, 2H, NH-
CH2-CH2 ) .
HPLC-MS (ESI) m/z 444.2 (30) [M+H]+, 466.3 (65) [M+Na]+,
685.1 (90) , 909.2 (100) [2M+Na]+, 1352.1 (15) [3M+Na]+.
Example 7:
N1-phenethyl-(2R)-2-[(4-benzylpiperidino)-methyl]-
carboxamido-3-(1H-3-indolyl)-propanamide
H
N
II H
N~ N
H
A) 3.0 g 4-benzylpiperidine and 1.73 g triethylamine was
added dropwise to 13 ml toluene with stirring and ice
cooling. A solution of 2.86 g ethyl bromoacetate in 6.2 ml
toluene was added dropwise to this receiving solution over
a period of 4 h. Following this the reaction mixture was
stirred for 4 days at RT. Then the organic phase was
extracted with 100 ml water and then dried over Na2SOq.
The solvent was evaporated in a water pump vacuum and the
residue was dried in an oil pump vacuum. 4.02 g ethyl (4-
benzyl-piperidin-1-yl) acetate was obtained as colourless
oil.
HPI~C-MS (ESI) m/z 188.2 (100), 234.2 (45), 262.2 (100)
[M+H]+.
CA 02497609 2005-03-07
CA 02497609 2005-03-07
46
B) 1.0 g of the ethyl (4-benzyl-piperidin-1-yl) acetate
obtained above was added to a receiving solution
consisting of 5.755 ml 1N aqueous NaOH and 11.5 ml
methanol, It was stirred overnight at RT, then neutralised
with conc. hydrochloric acid and the solvent was
evaporated in a water pump Vacuum. The residue was
purified by means of flash chromatography at a pressure of
1-1.2 bar (stationary phase: Silicagel 60, grain size
0.040-0.063 mm, mobile phase: methanol/chloroform 1:1).
After evaporating the solvent in a water pump vacuum and
drying the residue in an oil pump vacuum, 0.89 g
(4-benzyl-piperidin-1-yl)-acetic acid was obtained as a
colourless solid with a melting point of 250-252°C.
GC=MS (EI) m/z 44.1 (10), 91.1 (15), 188.1 (100), 233.0
(5) [M]+.
C) 86 mg of the (4-benzyl-piperidin-1-yl)-acetic acid
obtained above under B), 150 mg tent butyl-3-[(2R)-2-
amino-2-(phenethylcarbamoyl)-ethyl]-1H-1-indole
carboxylate (for preparation see Example 3D)), 75 mg HOBT
and 177 mg TBTU were dissolved in 2.6 ml DMF. 0.20 g DIPEA
was added dropwise to this receiving solution over a
period of 5 min. Then the reaction mixture was stirred for
23 h, the solvent was evaporated in a water pump Vacuum
with nitrogen-cooled receiving solution and the remaining
residue was purified by means of flash chromatography at a
pressure of 1-1.2 bar (stationary phase: Silicagel 60,
grain size 0.040-0.063 mm, mobile phase:
chloroform/methanol, 10:1). 180 mg N1-phenethyl-(2R)-2-[(-
benzylpiperidino)-methyl]-carboxamido-3-[1-(tert-
butoxycarbonyl)-3-indolyl]-propanamide was obtained as
yellow oil.
HPLC-MS (ESI) m/z 188.1 (20), 567.3 (70), 623.3 (100)
[M+H]+, 645.2 (25) [M+Na]+, 1245.2 (10) [2M+H]+, 1267.3
(40) [2M+Na]+.
D) 180 mg of the Nl-phenethyl-(2R)-2-[(4-benzylpiperidino)-
methyl]-carboxamido-3-[1-(tert-butoxycarbonyl)-3-indolyl]-
propanamide obtained above was dissolved in 3 ml
47
CA 02497609 2005-03-07
dichloromethane. 0.1 ml triisopropylsilane was added
thereto and the solution was cooled to 0°C. Then 3 ml TFA
was added dropwise over a period of 5 min. and the
reaction mixture was stirred for 1 hour. Following this
the solvent was evaporated in a water pump vacuum with
nitrogen-cooled receiving solution. The remaining residue
was taken up in a mixture of 8 ml DMSO, 1 ml water and 1
ml acetic acid and stirred overnight. Then the solvent was
evaporated to dryness in a water pump vacuum with
nitrogen-cooled receiving solution. The residue was taken
up in DMSO and purified by reversed-phase HPLC [HPLC
system from Amersham Pharmacia Biotech Akta Basic 100F;
pump system P-900 and detector UV-900; column ODS-A Cls
from Omnicrom YMC (250 mm x 30 mm, 10 um, flow rate: 25
ml/min); elution with linear gradient (30 min.) of water
(solvent A) in acetonitrile (solvent B) and O.lo (v/v)
TFA]. Freeze-drying the purified fractions yielded 109 mg
of the title compound as colourless powder with a melting
point of 73-75°C.
1H-NMR (500 MHz, DMSO-d6, 300 K) 8 = 10.84 (s, 1H, NH-CH-
C) , 8. 81 (d, J = 8. 3 Hz, 1H, NH-CH) , 8.25 (t, J = 5.2 Hz,
NH-CH2-CH2) , 7. 61 (d, J = 7.7 Hz, 1H, arom) , 7.07-7. 33 (m,
12H, arom), 7.02 (t, J = 7.3 Hz, 1H, arom), 6.96 (t, J =
6.9 Hz, 1H, arom), 4.60 (q, J = 6.9 Hz, 1H, NH-CH), 3.81
(d, J = 15. 2 Hz, 1H, N-CH2-CO) , 3. 67 (d, J = 13. 1 Hz, 1H,
N-CH2-CO), 3.21-3.34 (m, 3H, NH-CH2-CH2 and N-CH2-CH2-CH),
3. 06 (dd, J = 14 . 4 Hz, J = 4. 9 Hz, 1H, NH-CH-CH2) , 2 . 97-
2. 93 (m, 3H, NH-CH-CH2 and N-CH2-CH2-CH) , 2. 59-2. 67 (m,
3H, NH-CH2-CH2 and N-CH2-CH2-CH), 2.46-2.48 (m, 2H, CH-
CH2-C6H5) , 1. 57-1. 70 (m, 3H, N-CH2-CH2-CH and CH) , 1. 32-
1.46 (m, 2H, NCH2-CH2-CH).
HPLC-MS (ESI) m/z 188.1 (70), 523.3 (100) [M+H]+, 803.7
(20), 1045.1 (20) [2M+H]+, 1067.3 (40) [2M+Na]+.
The compounds of Formula I listed below in Table 2 can also
be prepared according to the preparation processes described
CA 02497609 2005-03-07
48
above or analogously to these preparation processes. Table 2
contains the following abbreviations:
bo: bond
dm: dioxolanylmethyl
Ind: indolyl
Phe: phenyl
Py: pyridyl
rac.: racemic
THI: tetrahydroisoquinolyl
CA 02497609 2005-03-07
49
N ~ 01 ~-IO
N N N ('7
61 N M r-iN
0 0 ~ c~
p o ,-~ ~-I ~ ,~ ,--i r-, ~I ~-I
-I o ,~ 0 0 0
I
'
A, W W N W W f.~ W !~ W 0.~
I
'C3 '~ "O 'C~'O
N !~ ~ ~ ~ ~ ~ S~ !~ f~
Z.1 H H H H H H H H H H
I I I I I I I i I I
(~ (~ (~ (~ c'~ c~ ~'7 (~ n'1('~
N N
H ~ ~ '~ ~ H
a~ x U s~ x
U
.s~ ,~ E-~rti~ H ~ E-~
(>~
pa pa W !~ t~ I ~-1U I U I
~-I
rl I N 1 c-I
x x x c~~ v~ ~ x x x x x
I I
x x x x x x x x x x
H N N N N N
~a rx z z z z z x x x x x
H i I I I I
o x x x x x x x x
~ ~ ~ ~ Z ~ Z ~ x
I I I I , 1 I I x
w x o x o x o x o x o x o x o x o U
p , U U U U U U U U U U U U U U U U x x
~, I I I I I I I I --U U
~ .-, I I
CIlU~
x ~ x v x v x v x '~x '~x '~x ~-
O U U U U U U U U
O
U
N O O O O O O O O O O
_ _ _ _
U U U U U U U U U U
Czi 1 I I I I I 1 I 1 I
~ x ~ x ~ x ~ x ~ x x x x x x
. ~; v U ~ U ~ U ~ U ~ U U U U U U
N I I 1 I I I I I I I
00 01 O ~-I N ('~1 ~' tn lOI
' W -I v--I ri r-i c~ r-I r-Ir-i
W
z
CA 02497609 2005-03-07
V7 ~ ~ I~~Tc--ict'~ l9COM 01OW T' ct'N N O
,~ N N N N N N N N N N r-1N N N N N I
, M
. r-I OJl0tnO1~ alOlM d ~ Ol (~ l9l~~'V'
N
aj
~
,"t',V' 61c'')Lf~CO~ LO~ CDCOo-iC'7 N Ln~'~ N
L~ ~'LfW ~'Wit'~'V'~'ct'Ll~LW .I Wt''d'~'Lf~
t'
('~r '--I~ ,-Ir-If--irl o-Ir-Ir-ir-1v-ir-i r-1c--Iw-1,---W
-4
O O O O O O v-IO riO O O O O O O r-1
M N ~ ~ ~ N ~ N N N ~ ~ ~ ~ (U~ N
.~.~-,.~.r.-S~
P-i W f-~OaW W fI~W Cl.~O-iW W W W t~CIPa
'~ ~ ~ ~ ~ ~ ~ 'Z.~~ '
N S~ ~ ~ S~~ ~ ~ ~ ~ ~ ~ S~
~1 H H H H H -. H H H H H H H H H H H
I I I I I H I i A I I I I I I I f
I
C'7 M c'7C'~M C''7('7c'7l'~C'~l~c~'7(~'7(~l('~('~(''~
N N
I ~'~ ~ ~' ~ ~ ~ H
'~ p'' a'
N N N ~ N N x N ~ N N
i.l I a'H ,~~ ' .C,~~ ~ ~ H H ~ ~ s~
.~ ~
~t~ ~,.~I 0.iU ~.,~U 0.. . U I I . 4-a. W
Pa r Wr0.i ~ W C~
<,.~ N I I ~- N N
V' cr
x x x x x x x x x x x x x x x x x
N I
x x x x x x x x x x x x x x x
0
r,i U
x x x x x x x x x x x x x x x
1
x x x x x
U U U I I I I U U I I 1 I l I I 1
U U U x x x x U U x x x x x x x x
' ~ Z Z Z Z Z Z
I I I U U U U I I U
I I I I i I 1 I I I I
_ _ I
I
I
I _O O__OO O O O O O O _O_O O O O O
'N
'~.~.CZ~ .~-~ ~ .~2.~
I
U U U U U U U I
I I 1 I I I I I I I I I I I i I 1
x x x x x x x x x x x x x x x x z
U U U U U U U U U U U U U U U U I
I I I I 1 I 1 I I I 1 I 1 I 1 I
O CO 61O r-IN (''7~'Ilkl0l~COOl O r-IN M ~1'
?C ~ ~ N N N N N N N N N N t''7c~t'r7t'~'7C'7
7.a
CA 02497609 2005-03-07
51
Example I:
Capsules containing M-phenethyl-(2R)-2-{1-[(4-chlorobenzyl)-
amino]-ethylcarboxamido}-3-(1H-3-indolyl)-propanamide:
Capsules with the following composition per capsule were
produced:
N1-phenethyl-(2R)-2-{1-[(4-chlorobenzyl)-amino]-
ethylcarboxamido}-3-(1H-3-indolyl)-propanamide 20 mg
Corn starch 60 mg
Lactose 300 mg
Ethyl acetate q.s.
The active substance, the corn starch and the lactose were
processed into a homogenous pasty mixture using ethyl
acetate. The paste was ground and the resulting granules were
placed on a suitable tray and dried at 45°C in order to
remove the solvent. The dried granules were passed through a
crusher and mixed in a mixer with the further following
auxiliaries:
Talcum 5 mg
Magnesium stearate 5 mg
Corn starch 9 mg
and then poured into 400 mg capsules (= capsule size 0).
CA 02497609 2005-03-07
52
Claims
1. Compounds of the general formula I,
/Arz
R3
R' Rz O _=
H
Ar' CrH ~ 1 2- ~ - 3 ~ N\ ~ 3
( z)m A N A H~ (Cf-Iz)n Ar j
O
wherein
Al is CH or, if A2 does not stand for a bond and at the
same time A3 does not stand for NH, also nitrogen,
A2 is a bond, Cl_2-alkylene or, if Al stands for CH and R2
stands for hydrogen, also carbonyl,
A3 is methylene which is optionally substituted by C1_q-
alkyl or Cl_q-alkyl carbonylamide or, if R2 is hydrogen
or together with Rl stands for a bond, is also NH,
R1 is hydrogen or, if A2 stands for carbonyl, also amino,
and
R2 is hydrogen, or
Rl and R2 together form Cl_2-alkylene or, if A2 is a bond, R1
and R2 may also together stand for a bond,
R3 is hydrogen or methyl,
Ar1 is phenyl which is optionally substituted 1 to 2 times
by halogen or Cl_q-alkyl or by C1_2-alkylenedioxy bonded
to two adjacent ring carbon atoms; pyridyl, furyl,
indolyl or tetrahydroisoquinolyl,
Ar2 is furyl, benzofuranyl, thienyl, benzothiophenyl,
pyrrolyl or indolyl,
Ar3 is phenyl which is optionally substituted 1 to 2 times
by halogen, or pyridyl,
m is 0 or 1 and
n is 0 or 1,
CA 02497609 2005-03-07
. 53
and also optionally their physiologically compatible acid
addition salts.
2. Compounds of Formula I according to Claim 1, wherein
n stands for 1 and R3 is hydrogen.
3. Compounds of Formula I according to one of the
preceding claims, wherein Ar2 stands for indolyl or for
benzothiophenyl.
4. Compounds of the general formula Ia,
/Ar2o1
810 /1
O _
N~ N
Ar1 (CHZ)m = H~ ~ Ia
O = O
Ra
wherein
Rlo1 is hydrogen or amino,
R4 is hydrogen, C1-4-alkyl or C1_q-alkyl carbonylamide,
Arl is phenyl which is optionally substituted 1 to 2 times
by halogen or C1-4-alkyl or by C1-2-alkylenedioxy bonded
to two adjacent ring carbon atoms; pyridyl, furyl,
indolyl or tetrahydroisoquinolyl,
Ar2olis benzothiophenyl or indolyl and
m is 0 or 1
and also optionally their physiologically compatible
acid addition salts.
CA 02497609 2005-03-07
54
5. Compounds of the general formula Ib,
/Arzoi
O /_
H - H
Ar' (CHz)m~N N~N
H ~ Ib
0 /
wherein
R4 is hydrogen, C1_q-alkyl or C1_4-alkyl carbonylamide,
Arl is phenyl which is optionally substituted 1 to 2 times
by halogen or C1_q-alkyl or by C1_2-alkylenedioxy bonded
to two adjacent ring carbon atoms; pyridyl, furyl,
indolyl or tetrahydroisoquinolyl,
Ar2olis benzothiophenyl or indolyl and
m is 0 or 1
and also optionally their physiologically compatible acid
addition salts.
6. Compounds of the general formula Ic,
~Arzo,
O /_
N~ ~ N
Ar' (CHz)m~ H H~ ~ ~ IC
wherein
Arl is phenyl which is optionally substituted 1 to 2 times
by halogen or C1_q-alkyl or by Cl-2-alkylenedioxy bonded
to two adjacent ring carbon atoms; pyridyl, furyl,
indolyl or tetrahydroisoquinolyl,
Ar2olis benzothiophenyl or indolyl and
m is 0 or 1
CA 02497609 2005-03-07
and also optionally their physiologically compatible acid
addition salts.
7. Compounds of the general formula Id,
/Af201
H O - H
Ar1 (CHz)m~NWN~N N ~ Id
H H
wherein
Arl is phenyl which is optionally substituted 1 to 2 times
by halogen or C1_4-alkyl or by C1_2-alkylenedioxy bonded
to two adjacent ring carbon atoms; pyridyl, furyl,
indolyl or tetrahydroisoquinolyl,
Ar2olis benzothiophenyl or indolyl and
m is 0 or 1
and also optionally their physiologically compatible
acid addition salts.
8. Compounds of the general formula Ie,
~Arzo1
O _=
~N~ ~ N
Ar1 (CH2)m H H~ Ie
O /
wherein
Arl is phenyl which is optionally substituted 1 to 2 times
by halogen or C1_q-alkyl or by C1_2-alkylenedioxy bonded
to two adjacent ring carbon atoms; pyridyl, furyl,
indolyl or tetrahydroisoquinolyl,
Ar2olis benzothiophenyl or indolyl and
CA 02497609 2005-03-07
56
m is 0 or 1
and also optionally their physiologically compatible acid
addition salts.
9. Compounds of the general formula If,
~,qr2o~
Ar' /_
(CHZ)m A O
H
~N~N N \
H ~ ~ If
0
wherein
A1 is CH or nitrogen,
Arl is phenyl which is optionally substituted 1 to 2 times
by halogen or Cl_4-alkyl or by Cl-2-alkylenedioxy bonded
to two adjacent ring carbon atoms; pyridyl, furyl,
indolyl or tetrahydroisoquinolyl,
Ar201is benzothiophenyl or indolyl and
m is 0 or 1
and also optionally their physiologically compatible acid
addition salts.
10. Compounds of Formula I according to Claim 1, for use
as medicaments.
11. Medicament, containing a pharmacologically active
quantity of a compound of Formula I according to Claim 1 and
additionally conventional pharmaceutical auxiliaries and/or
excipients.
12. The use of compounds of Formula I according to Claim
1 for the preparation of a medicament for the treatment
and/or prophylaxis of obesity, diabetes, hyperinsulinism,
cardiovascular diseases, eating disorders and/or syndrome X.
CA 02497609 2005-03-07
57
13. A process for the preparation of compounds of the
general formula I,
/Arz
3
Rz O _ R
H
N
Ar' CH A' Az- ~ 3 ~N~ ~ s
z)m N A H II ~CHz)n Ar j
O
wherein
A1 is CH or, if A2 does not stand for a bond and at the
same time A3 does not stand for NH, also nitrogen,
A2 is a bond, Cl_2-alkylene or, if Al stands for CH and R2
stands for hydrogen, also carbonyl,
A3 is methylene which is optionally substituted by Cl_q-
alkyl or C1_4-alkyl carbonylamide or, if R2 is hydrogen
or together with Rl stands for a bond, is also NH,
R1 is hydrogen or, if A2 stands for carbonyl, also amino,
and
R2 is hydrogen, or
R1 and R2 together form C1_2-alkylene or, if A2 is a bond, R1
and R2 may also together stand for a bond,
R3 is hydrogen or methyl,
Arl is phenyl which is optionally substituted 1 to 2 times
by halogen or C1_4-alkyl or by C1_2-alkylenedioxy bonded
to two adjacent ring carbon atoms; pyridyl, furyl,
indolyl or tetrahydroisoquinolyl,
Ar2 is furyl, benzofuranyl, thienyl, benzothiophenyl,
pyrrolyl or indolyl,
Ar3 is phenyl which is optionally substituted 1 to 2 times
by halogen, or pyridyl,
m is 0 or 1 and
n is 0 or 1,
CA 02497609 2005-03-07
58
and also optionally their physiologically compatible acid
addition salts, characterised in that
a) for the preparation of a compound of the general formula
Ig
Arz
Rio O ~ Rs
H - H
N N
1 \ 3 ~ ~ \ ~ 3
AC (CHz)m A H II (CHz)r, Ar Ig
O O
wherein A3, R3, Arl, Ar2, Ar3, m and n have the above
meanings and Rlol is hydrogen or amino, a compound of the
general formula II,
OH Ij
Ar~~o
(CHz)m
O
wherein m has the above meaning, Ar110 has the meaning
given above for Ark, any reactive groups being protected
by protective groups, and 8111 has the meaning given above
for 8101, any amino group being protected by a protective
group, is reacted with a compound of the general formula
III,
~,4rz, o
R3
O _-
H
H N-A3io~N N\ ~ s
z H ~ (CHz)n Ar III
O
wherein R3, Ar3 and n have the above meanings, Ar2lo has
the meaning given above for Ar2, any reactive groups being
protected by protective groups, and A3lo has the meaning
given above for A3, any reactive nitrogen atoms being
protected by protective groups, or
CA 02497609 2005-03-07
59
b) for the preparation of a compound of the general formula
Ih
/Ar2
R1 R2 O _/ Rs
1 ~ 1 2~N~A3ol~N N\
Ar3 Ih
Ar (CH2)m A A H~ (CH2)~
O
wherein Al, A2, Rl, R2, R3, Arl, Ar2, Ar3, m and n have the
above meanings and A3o1 has the meaning given above for A3
with the exception of NH, a compound of the general
formula IV,
110 ~ 201 O
Arllo (CH2)m A1 A2-N- Asl1 / 'Ohi IV
wherein A1, A2, Arllo and m have the above meanings, A311
has the meaning given above for A3ol, any reactive
nitrogen atoms being protected by protective groups, Rllo
has the meaning given above for Rl, any amino group being
protected by a protective group, and R2o1 has the meaning
given above for R2 with the exception of hydrogen, or an
amino protective group, is reacted with a compound of the
general formula V,
Ar210
_ R3
- H
H N N ~ ~ Ar3
2 ~ (CH2)~ V
O
CA 02497609 2005-03-07
wherein R3, Ar2lo, Ar3 and n have the above meanings, or
c) for the preparation of a compound of the general formula
Ii,
/Ar2
Rs
H
N ~
Are-(CHZ)m /~~-Azo, N-N N '~- ~ i \Ar3 Ii
H H H II (CHz)~
O
wherein Al, R3, Arl, Ar2, Ar3, m and n have the above
meanings and A201 has the meaning given above for A2 with
the exception of carbonyl, a compound of Formula V is
reacted with a carbonyl-group synthesis equivalent and
with a compound of the general formula VI,
Rioa
Ar~~o (CHZ)m A1 Azo~- ~ -NHZ VI
SG
wherein Al, A2ol~ Arllo and m have the above meanings,
stands for hydrogen or, if Al is nitrogen, may also stand
for a nitrogen protective group, and SG stands for a
protective group suitable in peptide chemistry, or
d) for the preparation of a compound of the general formula
Ij,
/Ar2
O
H ~
Ar'- CH ~N~'A3'o~N N~ / \Ar3
( 2)m H~ (Cf-i2)n h1
0
CA 02497609 2005-03-07
61
wherein A310 ~ R3 ~ Arl ~ Ar2, Ar3, m and n have the above
meanings, a compound of Formula III is reacted with a
compound of the general formula VIII,
Ar"° (CHz)m CHO
VIII
wherein Arllo and m have the above meanings,
and any protective groups are each subsequently cleaved off
again, and a resulting compound of Formula I if desired is
converted into its acid addition salt or an acid addition
salt is converted into a free compound of Formula I.