Note: Descriptions are shown in the official language in which they were submitted.
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
DESCRIPTION
PRODUCTION OF PEPTIDES IN PLANTS
AS VIRAL COAT PROTEIN FUSIONS
This invention was made with United States Government Support under
cooperative
agreement number 70NANB2H3048 awarded by the National Institute of Standards
and
Technology.
TECHNICAL FIELD
The present invention relates to the field of genetically engineered peptide
t0 production in plants, particularly to the use of tobamovirus vectors to
express fusion
proteins.
BACKGROUND ART
Peptides are a diverse class of molecules having a variety of important
chemical and
biological properties. Some examples include; hormones, cytokines,
immunoregulators,
peptide-based enzyme inhibitors, vaccine antigens, adhesions, receptor binding
domains,
enzyme inhibitors and the like. The cost of chemical synthesis limits the
potential
applications of synthetic peptides for many useful purposes such as large
scale therapeutic
drug or vaccine synthesis. There is a need for inexpensive and rapid synthesis
of milligram
and larger quantities of naturally-occurnng polypeptides. Towards this goal
many animal
and bacterial viruses have been successfully used as peptide carriers.
The safe and inexpensive culture of plants provides an improved alternative
host for
the cost-effective production of such peptides. During the last decade,
considerable progress
has been made in expressing foreign genes in plants. Foreign proteins are now
routinely
produced in many plant species for modification of the plant or for production
of proteins
for use after extraction. Animal proteins have been effectively produced in
plants (reviewed
in Krebbers et al., 1992).
Vectors for the genetic manipulation of plants have been derived from several
naturally occurnng plant viruses, including TMV (tobacco mosaic virus). TMV is
the type
member of the tobamovirus group. TMV has straight tubular virions of
approximately 300
by 18 nm with a 4 nm-diameter hollow canal, consisting of approximately 2000
units of a
single capsid protein wound helically around a single RNA molecule. Virion
particles are
95% protein and 5% RNA by weight. The genome of TMV is composed of a single-
stranded RNA of 6395 nucleotides containing five large ORFs. Expression of
each gene is
regulated independently. The virion RNA serves as the messenger RNA (mRNA) for
the 5'
genes, encoding the 126 kDa replicase subunit and the overlapping 183 kDa
replicase
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
2
subunit that is produced by read through of an amber stop codon approximately
5% of the
time. Expression of the internal genes is controlled by different promoters on
the minus-
sense RNA that direct synthesis of 3'-coterminal subgenomic mRNAs which are
produced
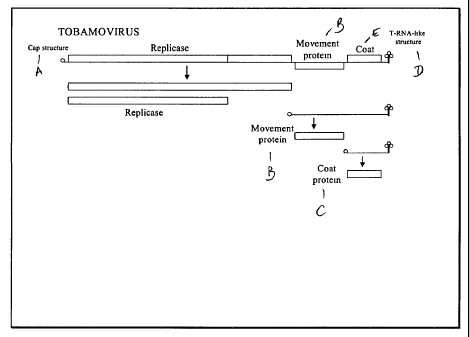
during replication (FIG. 1) Other tobamoviruses have a similar construction
with genomic
RNA of approximately 6.5 kb. The genomic RNA is used as an mRNA and translated
to
produce the replicase protein. These viruses may produce two replicase
proteins, with the
larger protein being produced by translational readthrough of an amber (AUG)
stop codon.
Both viruses produce two smaller coterminal subgenomic RNAs. The coat protein
is
encoded by the 3'-most RNA, and the movement proteins by the larger sgRNA. The
virion
to RNA and sgRNAs are capped. Tobamovirus RNAs are not polyadenylated, but
contain a
tRNA-like structure at the 3' end. Potevirus genomic and sgRNAs are
polyadenylated.. A
detailed description of tobamovirus gene expression and life cycle can be
found, among
other places, in Dawson and Lehto, Advances in Virus Research 38:307-342
(1991).
For production of specific proteins, transient expression of foreign genes in
plants
t5 using virus-based vectors has several advantages. Products of plant viruses
are among the
highest produced proteins in plants. Often a viral gene product is the major
protein produced
in plant cells during virus replication. Many viruses are able to quickly move
from an initial
infection site to almost all cells of the plant. Because of these reasons,
plant viruses have
been developed into efficient transient expression vectors for foreign genes
in plants.
20 Viruses of multicellular plants are relatively small, probably due to the
size limitation in the
pathways that allow viruses to move to adjacent cells in the systemic
infection of entire
plants. Most plant viruses have single-stranded RNA genomes of less than 10
kb.
Genetically altered plant viruses provide one efficient means of transfecting
plants with
genes coding for peptide Garner fusions.
25 Human papillomaviruses (HPVs) are the etiologic agents of many benign and
malignant tumors of stratified squamous epithelium (see recent reviews by
Alani and
Munger, 1998; zur Hausen, 1999; Einstein and Goldberg, 2002). In general,
these tumors
arise from keratinocytes of oral, epidermal, and anogenital sites, although
some tumors (e.g.
adenocarcinoma of the cervix) have a glandular morphology and origin. Not only
do 95-
30 99% of cervical cancers originate from papillomavirus-infected cells (zur
Hausen 1999), but
papillomaviruses also appear to contribute significantly to the development of
oral and
epidermal cancers (Balaram et al., 1995). Malignant conversion of cervical
epithelium
appears to be restricted to a "high risk" subset of papillomaviruses, whose
association with
cancer correlates with the ability of their E6 and E7 proteins to efficiently
inactivate the
35 cellular p53 and pRb tumor suppressor proteins, respectively. A single
"high risk" HPV
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
3
type, HPV-16 is associated, with approximately 60% of cervical carcinomas.
Papillomavirus infection has become a significant public health issue in the
United States,
where at least 17.9% of women are seropositive for HPV-16 infection (Stone et
al., 2002);
this figure does not include rates of infection with other "high risk" HPV
types, and is still
significantly lower than infection rates in developing countries. There is
thus a great need
for development of efficacious and cost-effective vaccines that will prevent
papillomavirus
infection and associated disease.
Papillomavirus are small (55 nm), non-enveloped, double-stranded DNA viruses
with an 8 kb genome enclosed by a T=7 icosahedral capsid (Fields Virology
text). Seven, or
l0 in some viruses eight early genes are involved in such processes as viral
DNA replication
(E1 and E2), RNA transcription (E2), and cell transformation (E5, E6, E7). The
late genes
encode the major capsid protein, L1, and the minor capsid protein, L2. The
viral capsid is
comprised of 72 pentamers, or capsomeres, of LI. Approximately 12 molecules of
the L2
protein are associated with each capsid, probably at the capsid vertices.
Regions of the L2
t5 protein located towards the N-terminus are thought to be displayed on the
surface of
papillomavirus virions, since L2 antibodies can recognize both native virions
and L1:L2
pseudovirions (Roden et al., 1994b; Liu et al., 1997; Kanawa et al., 1998a).
The L2 protein
interacts with the viral DNA and is probably involved in virion assembly (Day
et al., 1998).
Recombinant expression of the L1 protein in eukaryotic cells, e.g. in Sf9
insect cells using
20 baculovirus expression vectors, results in the self assembly of the Ll
protein within the
nuclear compartment into capsid-like structures termed "virus-like particles"
or VLPs. Co-
expression of L2 with L1 in eukaryotic expression systems results in
incorporation of L2
into VLPs. Evidence suggests that L1:L2 VLPs are more stable than VLPs
containing Ll
alone (Kirnbauer et al., 1993). Papillomavirus LI:L2 VLPs can encapsidate
plasmid DNA
25 as well as genomic DNA from other papillomaviruses, and these pseudovirions
have proven
useful for development of surrogate infection assays that have allowed both
antibody-
mediated virus neutralization studies and investigation of the mechanism of
papillomavirus
binding and entry into host cells (Roden et al., 1996; Giroglou et al., 2001;
Kawana et al.,
1998b; 2001b). While L1 VLPs can efficiently bind the cell surface,
pseudovirions
30 containing L1 alone are much less efficient at DNA transfer than L1:L2
particles, implying
that L2 plays a critical role in virus entry (Roden et al., 1997; Unckell et
al., 1997).
Early efforts to express L1 protein-based vaccines showed that denatured
protein
purified from bacteria could not induce virus neutralizing antibodies in
vaccinated animals.
Conformational integrity of L1-based vaccines is critical because host
antibodies recognized
35 native, conformational epitopes on the virion (Ghim et al., 1991; Thompson
et al., 1987).
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
4
In the early to mid 1990's several groups demonstrated that L1 protein
expressed in
eukaryotic expression systems-recombinant baculovirus-transduced insect cells
and
yeast-could assemble into virus-like particles (VLPs) that retain
conformational epitopes
essential for induction of neutralizing antibodies. These purified VLPs were
effective
vaccines and protected rabbits, dogs and cattle from experimental infections
(Suzich et al.,
1995; Breitburd et al., 1995; Kirbauer et al., 1996). These results have been
corroborated in
several studies that show that sera from animals vaccinated with HPV L1 VLPs
neutralize
homologous HPV types in psuedovirus-based cell infection studies, and more
recently that
sera from participants in a HPV16 L1 VLP trial are also neutralizing
(Schiller, 1999; Evans
to et al., 2001; Harro et al., 2001; Pastrana et al., 2001). Recent data show
that small T=1
VLPs and L1 capsomere structures purified from bacteria expressing L1 fusion
proteins
retain many of the conformational epitopes that are required for effective L1
prophylactic
vaccination, and this has been confirmed in the COPV model (Yuan et al. 2001).
Hemorrhagic fever viruses (HFVs) in the viral taxonomic families Filoviridae,
Arenaviridae, Bunyaviridae and Flaviviridae threaten the health of humans and
their
livestock, particularly in developing countries. With the exception of yellow
fever, there
are no widely available, safe and efficacious vaccines that might prevent
infection by any of
the hemorrhagic fever viruses. In the wake of the attacks on the USA in
September 2001,
there is heightened awareness of the theoretical threat that biological
terrorism, or biological
2o warfare to human health. Given that HFVs were known to have been weaponized
by the
former Soviet Union, Russia, and the United States prior to 1969, development
of safe, and
easy-to-administer vaccines against high-priority HFVs would appear prudent
from a
National safety perspective (Borio et al., 2002). Certain of the HFVs, such as
Rift Valley
fever virus (RVFV) and Ebola virus (EBOV), present a threat to health of US
military
personnel deployed in Africa and the Middle East, as well as to travelers to
those areas
(Isaacson 2001 ).
Ideally, a vaccine designed to protect against infection with human
immunodeficiency type 1 (HIV-1) will induce sterilizing immunity against a
broad range of
virus variants. However, generation of broadly-neutralizing antibodies (Nabs)
by
vaccination, let alone natural infection, has proven nearly impossible thus
far. There have
been some notable advances in development of vaccine regimens that are able to
generate
significant levels of protection against development of AIDS in non-human
primate models
(reviewed in 1,2,3,4). These vaccines allow animals to control viral challenge
by strong
priming of virus-specific CD8+ T-cells (cytotoxic T cells, CTLs). However, a
CTL
response alone cannot prevent infection, and mechanisms to induce Nabs that
will neutralize
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
a wide range of isolates remains a vital goal, especially in light of the fact
that viral escape
from vaccine-induced CTL control can sometimes occur (5). The Env spikes on
the surface
of the HIV-1 virion are the primary target for antibody-mediated
neutralization. However,
the Env proteins of HIV-1 are poorly antigenic, and generation of Nabs is
difficult to
s achieve, probably because functionally important domains of the proteins are
obscured by
protein folding and carboydrate chains. Nevertheless, many infected people do
mount a Nab
response that is generally highly specific to the autologous virus, and not
cross-neutralizing.
This is not surprising given the phenomenal sequence and structural variation
that is present
in the Env proteins. However, a rare subset of infected individuals do produce
broadly
neutralizing Abs, which gives hope that induction of sterilizing immunity is
possible.
The envelope proteins of T-cell line-adapted (TCLA) strains of HIV-1 elicit
Nabs
that mostly target linear epitopes in the third variable cysteine loop (V3
loop) of gp120, a
region that is involved in co-receptor binding and hence vital for virus
entry. Subtype C
isolates of HIV-1, which infect more people worldwide than any other subtype,
have
relatively low level of sequence variation in the V3 loop (6,7). However,
neutralization of
subtype C virus by V3 loop Abs is not extremely efficient in vitro, perhaps
reflecting poor
immunogenicity of epitopes in this region (7). There is concern that the V3
loop may be
hidden in the native gp120 structure and not accessible to the immune system,
and therefore
that generation of V3-specific Nabs will be difficult with gp120 subunit
vaccines.
2o However, the V3 loop is vital for viral entry, and so significant levels of
V3 loop-targeted
Nabs should help prevent transmission of HIV-1.
To date, six human monoclonal antibodies (Mabs) have been described that are
capable of neutralizing a broad spectrum of HIV-1 variants in vitro. Three of
these
(IgGb 12; 2612 and 2F5) were described several years ago, and lend insight
into the
domains of the Env proteins that are important in viral entry, and thus for
vaccine design.
Monoclonal antibody "b12" recognizes a conformational epitope in the CD4
binding site of
gp120; 2612 recognizes a discontinuous epitope in the C2-V4 region of gp120
that includes
N-glcyosylation sites, and 2F5 maps to a linear epitope (ELDKWA) in the
membrane-
proximal ectodomain of gp41 (9). Recently, two broadly neutralizing monoclonal
3o antibodies 4E10 and Z13 were shown to recognize a continuous epitope with
core sequence
NWFDIT, just C-terminal to the 2F5 recognition sequence (10,11). This strongly
indicates
that the membrane proximal region of gp41 plays a critical role in virus
entry. Another
recently described monoclonal Fab was selected for binding to gp120-CD4-CCRS
complexes, and also displays a broad neutralization phenotype (12).
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
6
Passive transfer studies have shown that neutralizing Mabs are able to confer
concentration-dependent sterilizing immunity to virus challenge by
intravenous, oral and
vaginal routes in Rhesus macaques. It is encouraging that the mAbs tested
display
significant synergy in their neutralization activity: this will reduce the
minimum antibody
concentration that is required for effective neutralization (reviewed in
13,14). A recent
publication (15) demonstrates that MAb neutralizing activity can also be
generated in vivo:
in mice that expressed the gene for b12 from a recombinant adeno-associated
virus vector.
These studies on neutralizing Mabs have helped to demonstrate that one should
be able to
achieve significant levels of protection against HIV-1 infection and reduced
rates of
1o transmission of virus, if a way is found to induce robust production of
Nabs in vaccinated
animals and is incorporated into a vaccine regimen that includes strong
priming of a CTL
response.
In the light of the disappointing performance of whole Env-based vaccines, and
the
problems associated with poor immunogenicity of Env subunit vaccines, several
studies
have focused on the use of immunogens based on domains of Env proteins that
are
presumed targets for Abs. Data presented by Letvin et al. (8), that showed
that antibodies
induced against the V3 loop could provide partial protection against challenge
with primary
isolate-like SHIV-89.6 in Rhesus macaques. Efforts at generation of
neutralizing antibodies
with immunogens containing the core linear epitope recognized by the 2F5
antibody have
been generally disappointing, with only non-neutralizing antibodies being
produced (16,17).
However, there is one notable exception: recently, Marusic et al. ( 18) showed
that virus-like
particles of the flexuous plant virus potato virus X (PVX) displaying the 2F5
ELDKWA
epitope could induce high levels of HN-1 specific IgG and IgA in mice
immunized with the
recombinant virus-like particles (VLPs). This immunogen was able to induce
production
of human HIV-1 specific neutralizing antibodies (measured by in vitro
inhibition of
syncytium formation) in severe combined immunodeficient mice reconsrituted
with human
periferal blood lymphocytes (hu-PBL-SCID) that had been immunized with human
dendritic cells (DCs) pulsed with the PVX-2F5 VLPs. These authors speculate
that
presentation of the ELDKWAS sequence in a highly repetitive fashion on the
surface of the
PVX virion rendered the sequence highly immunogenic, and thus were able to
generate
Nabs. These results clearly warrant further investigation.
Until the recent discovery of the 4E10/Z3 human Mab, 2F5 was the only human
Mab that appeared to recognize a linear epitope, and so peptides that could
mimic the
neutralizing epitope of b12 and 2612 were not available for testing as
potential
immunogens. However, a linear peptide mimotope of the b12 epitope has recently
been
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
7
discovered using phage peptide display technology (19). This peptide (B2.1)
appears to
bind best to b 12 when presented as a disulphide-linked homodimer on the
surface of the
phage. This phage particle is being optimized for use as an imrnunogen. Scala
et al. (20)
selected epitopes from libraries of peptides displayed on the surface of
filamentous phage
particles with sera from HIV+ patients, both from long term infected non-
progressor donors
and from donors who had progressed to AIDS illness. Five epitopes, presumed to
be
mimotopes of Env-specific neutralizing epitopes, were able to induce
production of
antibodies that neutralized TCLA HIV-1 strains IIIB and NL4-3, as well as the
primary
isolate ADB, but this less strongly than the TCLA strains (20). Subsequently,
these authors
to showed that sera from individuals infected with all group M HIV-1 subgroups
were able to
recognize the phage-displayed mimotopes (21 ). Rhesus monkeys were immunized
with
phage particles displaying the five epitopes that had shown potentially
protective immune
responses in mice, and challenged with pathogenic SHIV-89.6PD. While the
immunized
animals were not protected from SHIV infection, there was evidence of
significant control
of the challenge virus and the monkeys were protected from progression to
AIDS. These
results show similar levels of control to vaccines designed to generate virus-
specific CTLs
and infer that the antibody response was able to control viremia in the
challenged animals.
A recent publication (22) described successful isolation of a number of human
Nabs from
XenoMouse immunized with gp120 derived from a primary Subtype B isolate
(SF162).
2o The authors noted potent neutralizing activity against the autologous virus
isolate, and
reactivity against both R5 and X4 isolates in Subtype B. The Nabs mapped to
novel
epitopes in domains known to possess neutralizing epitopes: V2-, V3- and CD4-
binding
domains of gp120, as well as in the C-terminal region of the V1 loop.
Some non-structural HIV-1 proteins, particularly Tat and Vpr, are found in the
serum of infected individuals, and exert biological function, resulting in
immunodeficiency
and disease. The Tat protein is required for HIV-1 replication and
pathogenesis. It is
produced early in the viral life cycle. In the nucleus of the infected cell,
it interacts with
host factors and the TAR region of the viral RNA to enhance transcript
elongation and to
increase viral gene expression (Jeang et al., 1999). Tat also is also found
extracellularly,
where it has distinct functions that may indirectly promote virus replication
and disease,
either through receptor mediated signal transduction or after internalization
and transport to
the nucleus. Tat suppresses mitogen-, alloantigen- and antigen-induced
lymphocyte
proliferation in vitro by stimulating suppressive levels of alpha interferon
and by inducing
apoptosis in activated lymphocytes. In vivo, it is thought that Tat may alter
immunity by
upregulating IL-10 and reducing IL-12 production, or through its ability to
increase
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
8
chemokine receptor expression (Gallo et al., 2002; Tikhonov et al., 2003).
Antibody
production against Tat has, in some cases, correlated with delayed progression
to AIDS in
HIV-1 infected people (Gallo et al., 2002). Recently, Agwale et al. (2002)
showed that
antibodies induced in mice against a Tat protein subunit vaccine could negate
the immune
suppression activities of Tat in vivo. Subsequently, Tikhonov et al. (2003)
identified linear
epitopes on Tat that were reactive with Tat-neutralizing antibodies produced
in vaccinated
Rhesus macaques. From these data it is clear that antibodies that target the N-
terminus, an
internal basic domain, and the cell-binding domain of Tat (containing the
integrin-binding
motif "RGD") can neutralize the extracellular version of Tat, and reduce the
negative
t 0 impact of Tat on the immune system.
Parvoviruses that are associated with enteric disease in domestic cats, dogs,
mink
and pigs are closely related antigenically, with different isolates diverging
less than 2% in
the sequence of the viral structural proteins. Vaccination with killed or live-
attenuated
parvovirus protects animals against infection by Feline panleukopenia virus
(FPV), canine
parvovirus (CPV), mink enteritis virus (MEV) and porcine parvovirus (PPV).
However,
maternal antibodies neutralize the vaccine, making it ineffective in animals
that have not
been weaned. Subunit vaccines might overcome this limitation, and provide
useful
alternatives to conventional vaccines.
DISCLOSURE OF THE INVENTION
The present invention includes an immunological reagent having a plant viral
protein covalently bound to an epitope peptide having the same linear sequence
as an
immunologically recognized epitope of a human papilloma virus, human
immunodeficiency
virus, ebola virus, rift valley fever virus or parvovirus.
The present invention also includes an immunological reagent having a plant
viral
protein covalently bound to an epitope peptide having the same linear sequence
as an
immunologically recognized epitope of a human papilloma virus, human
immunodeficiency
virus, ebola virus, rift valley fever virus or parvovirus, wherein the epitope
peptide contains
a sequence selected from the group consisting of the peptide sequences of
Table 1, the
peptide sequences of Table 6, the peptide sequences of Table 7, the peptide
sequences of
Table 8, HNTPVYKLDISEATQVE, ATQVEQHHRRTDNDSTA,
GKLGLITNTIAGVAGLI, VQPDGGQPAVRNERAT, MSDGAVQPDGGQPAVRNERA,
MSDGAVQPDGGQPAVRNERAT and KGTMDSGQTKREL.
The invention also includes a vaccine having an immunological reagent having a
plant viral protein covalently bound to an epitope peptide having the same
linear sequence
as an immunologically recognized epitope of a human papilloma virus, human
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
9
immunodeficiency virus, ebola virus, rift valley fever virus or parvovirus,
wherein the
epitope peptide contains a sequence selected from the group consisting of the
peptide
sequences of Table 1, the peptide sequences of Table 6, the peptide sequences
of Table 7,
the peptide sequences of Table 8, HNTPVYKLDISEATQVE, ATQVEQHHRRTDNDSTA,
s GKLGLITNTIAGVAGLI, VQPDGGQPAVRNERAT, MSDGAVQPDGGQPAVRNERA,
MSDGAVQPDGGQPAVRNERAT and KGTMDSGQTKREL, and a pharmaceutically
acceptable Garner or excipient.
The present invention also includes a method for eliciting an immune response
in an
animal by administering a vaccine having an immunological reagent having a
plant viral
l0 protein covalently bound to an epitope peptide having the same linear
sequence as an
immunologically recognized epitope of a human papilloma virus, human
immunodeficiency
virus, ebola virus, rift valley fever virus or parvovirus, wherein the epitope
peptide contains
a sequence selected from the group consisting of the peptide sequences of
Table l, the
peptide sequences of Table 6, the peptide sequences of Table 7, the peptide
sequences of
15 Table 8, HNTPVYKLDISEATQVE, ATQVEQHHRRTDNDSTA,
GKLGLITNTIAGVAGLI, VQPDGGQPAVRNERAT, MSDGAVQPDGGQPAVRNERA,
MSDGAVQPDGGQPAVRNERAT and KGTMDSGQTKREL, and a pharmaceutically
acceptable Garner or excipient to the animal.
'The present invention includes a virus-like particle having a plurality of
assembled
2o protein subunits wherein each protein subunit is a plant viral coat protein
covalently bound
to an epitope peptide having the same linear sequence as an immunologically
recognized
epitope of a human papilloma virus, human immunodeficiency virus, ebola virus,
rift valley
fever virus or parvovirus.
The present invention also includes a virus-like particle having a plurality
of
25 assembled protein subunits wherein each protein subunit is a plant viral
coat protein
covalently bound to an epitope peptide having the same linear sequence as an
immunologically recognized epitope of a human papilloma virus, human
immunodeficiency
virus, ebola virus, rift valley fever virus or parvovirus, wherein the
sequence selected from
the group consisting of the peptide sequences of Table l, the peptide
sequences of Table 6,
3o the peptide sequences of Table 7, the peptide sequences of Table 8,
HNTPVYKLDISEATQVE, ATQVEQHHRRTDNDSTA, GKLGLITNTIAGVAGLI,
VQPDGGQPAVRNERAT, MSDGAVQPDGGQPAVRNERA,
MSDGAVQPDGGQPAVRNERAT and KGTMDSGQTKREL.
The invention includes a vaccine having a virus-like particle having a
plurality of
35 assembled protein subunits wherein each protein subunit is a plant viral
coat protein
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
covalently bound to an epitope peptide having the same linear sequence as an
immunologically recognized epitope of a human papilloma virus, human
immunodeficiency
virus, ebola virus, rift valley fever virus or parvovirus, and a
pharmaceutically acceptable
carrier or excipient.
5 The invention also includes a method for eliciting an immune response in an
animal
including administering the vaccine having a virus-like particle having a
plurality of
assembled protein subunits wherein each protein subunit is a plant viral coat
protein
covalently bound to an epitope peptide having the same linear sequence as an
immunologically recognized epitope of a human papilloma virus, human
immunodeficiency
t0 virus, ebola virus, rift valley fever virus or parvovirus, and a
pharmaceutically acceptable
carrier or excipient to the animal.
The invention includes a plant virus having at least one plant viral coat
protein
covalently bound to an epitope peptide having the same linear sequence as an
immunologically recognized epitope of a human papilloma virus, human
immunodeficiency
virus, ebola virus, rift valley fever virus or parvovirus.
The invention also includes a plant virus having at least one plant viral coat
protein
covalently bound to an epitope peptide having the same linear sequence as an
immunologically recognized epitope of a human papilloma virus, human
immunodeficiency
virus, ebola virus, rift valley fever virus or parvovirus, wherein the
sequence sequence is
selected from the group consisting of the peptide sequences of Table 1, the
peptide
sequences of Table 6, the peptide sequences of Table 7, the peptide sequences
of Table 8,
HNTPVYKLDISEATQVE, ATQVEQHHRRTDNDSTA, GKLGLITNTIAGVAGLI,
VQPDGGQPAVRNER.AT, MSDGAVQPDGGQPAVRNERA,
MSDGAVQPDGGQPAVRNERAT and KGTMDSGQTKREL.
The present invention also includes a vaccine having a plant virus having at
least
one plant viral coat protein covalently bound to an epitope peptide having the
same linear
sequence as an immunologically recognized epitope of a human papilloma virus,
human
immunodeficiency virus, ebola virus, rift valley fever virus or parvovirus,
wherein the
sequence sequence is selected from the group consisting of the peptide
sequences of Table
1, the peptide sequences of Table 6, the peptide sequences of Table 7, the
peptide sequences
of Table 8, HNTPVYKLDISEATQVE, ATQVEQHHRRTDNDSTA,
GKLGLITNTIAGVAGLI, VQPDGGQPAVRNERAT, MSDGAVQPDGGQPAVRNERA,
MSDGAVQPDGGQPAVRNERAT and KGTMDSGQTKREL and a pharmaceutically
acceptable carrier or excipient.
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
11
The invention also includes a method for eliciting an immune response in an
animal
including administering a vaccine having a plant virus having at least one
plant viral coat
protein covalently bound to an epitope peptide having the same linear sequence
as an
immunologically recognized epitope of a human papilloma virus, human
immunodeficiency
virus, ebola virus, rift valley fever virus or parvovirus, wherein the
sequence sequence is
selected from the group consisting of the peptide sequences of Table l, the
peptide
sequences of Table 6, the peptide sequences of Table 7, the peptide sequences
of Table 8,
HNTPVYKLDISEATQVE, ATQVEQHHRRTDNDSTA, GKLGLITNTIAGVAGLI,
VQPDGGQPAVRNERAT, MSDGAVQPDGGQPAVRNERA,
1o MSDGAVQPDGGQPAVRNERAT and KGTMDSGQTKREL and a pharmaceutically
acceptable Garner or excipient to the animal.
The present invention also includes the composition of the sixth paragraph of
this
section or the composition of the tenth paragraph of this section containing a
plurality of
different epitope peptides, each on a separate plant viral coat protein
molecule.
The present invention also includes a method for preparing an antibody against
a
papilloma virus, ebola virus, HIV virus, Rift Valley Fever virus or a
parvovirus including:
exposing an animal to the vaccine described in the third, seventh, or eleventh
paragraph of
this section, recovering cells or body fluids from the animal, and preparing
an antibody
from said cells or body fluids.
The present invention includes the method of the above paragraph wherein the
antibody is neutralizing.
The present invention includes a method for detecting a papilloma virus, ebola
virus,
HIV virus, Rift Valley Fever virus or a parvovirus comprising contacting an
antibody
produced by the method of the 14'h paragraph of this section with a sample
suspecting of
containing a virus, and detecting the presence or absence of antibody binding
to the virus.
The present invention includes a method for inducing an immune response in an
animal against a peptide epitope including: coupling the peptide epitope to a
first carrier
antigen to make a first vaccine composition, coupling the peptide epitope to a
second carrier
antigen, which is different from the first Garner antigen, to make a second
vaccine
composition, immunizing the animal with the first vaccine composition, at a
later time,
immunizing the animal with the second vaccine composition, wherein the immune
response
to the peptide epitope is boosted greater than the boosting of either carrier
antigen.
The present invention also includes the method according to the previous
paragraph
further including: coupling a second peptide epitope to a third carrier
antigen to make a
third vaccine composition, coupling the second peptide epitope to a fourth
carrier antigen,
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
12
which is different from the third carrier antigen but may be the same as
either the first
carrier antigen or the second carrier antigen, to make a fourth vaccine
composition,
immunizing an individual animal with the first vaccine composition and the
third
composition, at a later time, immunizing the same individual animal with the
second
vaccine composition and the fourth composition, wherein the immune responses
to the first
and second peptide epitope are boosted greater than the boosting of the
carrier antigens.
It is still another object of the present invention to provide polynucleotides
encoding
the genomes of the subject recombinant plant viruses.
It is another further object of the present invention to provide the coat
fusion
proteins encoded by the subject recombinant plant viruses.
It is yet another further object of the present invention to provide plant
cells that
have been infected by the recombinant plant viruses of the invention.
Figure 1: Tobamovirus gene map and expression products are diagrammed.
Figure 2: A series of flow charts showing methods used for construction of
recombinant tobamoviruses with useful peptides genetically fused to the coat
protein gene.
Figure 3: An uninfected Glurk plant leaf is shown on the left and a leaf with
lesions
is shown on the right, where each necrotic local lesion indicates a virus
infection event.
Figure 4: SDS PAGE and MALDI-TOF analysis. The vaccine samples were run in
triplicate, with the Markl2 protein molecular weight markers (Invitrogen) in
the fourth lane
2o in every case. The molecular weight marker bands, from top to bottom are
36.5 kDa; 31
kDa; 21.5 kDa and 14.4 kDa. The molecular weight of the upper viral band, as
determined
by MALDI-TOF is indicated in the figure.
Figure S: Western blot analysis of TMV:papillomavirus vaccines. Samples were
loaded as indicated in the coomassie blue stained gel (lower right) and probed
with rabbit
antisera indicated above the blots.
Figure 6: Scatter plot indicating ELISA (IgG) response of all immunized
animals to
the cognate peptide antigen. Sera analyzed here were from bleed 3, post
vaccine 4.
Figure 7: Bar graph showing responses to peptide antigens, pooled data with
error
bars indicating 95% confidence interval. Sera analyzed were from bleed 3, post
vaccine 4.
3o Figure 8: Analysis of serum cross-reactivity between papillomavirus peptide
antigens.
Figure 9: Comparison of IgG antibody response to vaccination with CRPV2.1
vaccines, BEI treated and non-treated (left) and to the HPV6/11 vaccine
(right). Each bar
represents the specific IgG level of an individual mouse.
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
13
Figure 10: shows the results of IgG subtype measurement in sera of animals
vaccinated with the five different papillomavirus L2 vaccines. The immune
response
appears balanced; but, the concentration of IgGI subtype appears to be at
least 3-fold
greater than that of IgG2, perhaps indicating a dominant Th2 response.
Figure 11: ELISA measurement of relative amounts peptide specific IgG after
vaccine 3 (left) and 4 (right).
Figure 12: IgG subtype measurements in sera of Guinea Pigs vaccinated with
TMV:papillomavirus vaccines.
Figure 13: Cross-reactivity of sera of guinea pigs immunized with CRPV- or HPV
6/11 TMV peptide fusions, against HPV 16 L2 peptide capture antigen
(LVEETSFIDAGAP). Each bar indicates the antibody response induced in an
individual
animal. The dashed line indicates the probable level of non-specific cross-
reactive
antibodies that were induced on vaccination with TMV virions carrying the very
distantly
related cottontail rabbit papillomavirus peptide 2.1. Figure 14, below,
illustrates the amino
acid identity between these three peptides.
Figure 14: Shared amino acid identity between the HPV-11 L2 peptide present on
recombinant TMV virion LSB2282; the CRPV 2.1 peptide present on recombinant
TMV
virion LSB2283, and the HPV-16 L2 peptide LVEETSFIDAGAP that was conjugated to
bovine serum albumin and used as the capture antigen in the ELISA.
Figure 15: Solubility of example coat fusion proteins carrying Ebola epitopes.
Photograph of SDS-PAGE gel of crude proteins extracts from plants inoculated
with
infectious transcripts carrying the Ebola epitope-coat protein fusions.
BEST MODE FOR CARRYING OUT THE INVENTION
An "immunologically recognized epitope peptide" generally has at least 8 amino
acids unique to an antigen, or closely related antigens, and is a binding site
for a specific
antibody or T-cell receptor. The antibody and/or cytotoxic T-lymphocyte
containing the T-
cell receptor are induced upon immunization or infection with an antigen
containing this
epitope peptide.
An "epitope peptide" or a "peptide epitope" includes the specific sequences
3o described below chemically bonded to the N-terminal, the C-terminal or an
internal region
of an antigen. The epitope peptide may be longer than the specific sequences
described
below with boardering sequences) having the same sequence as the viral
pathogen's
antigens. The epitope may contain slight amino acid substitutions (preferably
conservative
substitutions) slight deletions in the sequences recited provided that the
epitope peptide
contains a sufficient amount of the sequence to bind to a specific antibody
and/or to elicit a
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
14
specific antibody capable of binding specifically to the natural antigen.
Examples of a
shorter epitope peptide include the 1 N-terminal amino acid in the HPV-16 L1
protein
epitope and Ebola virus epitope GP-1 amino acid number 405.
The term "protein" is intended to also encompass derivitized molecules such as
glycoproteins and lipoproteins as well as lower molecular weight polypeptides.
The terms "binding component", "ligand" or "receptor" may be any of a large
number of different molecules, and the terms are sometimes usable
interchangeably. In the
context of the present invention the receptor is usually an antibody and the
ligand is usually
the pathogenic virus such as a papilloma virus, ebola virus, HIV virus, Rift
Valley Fever
t0 virus or a parvovirus.
The term "bind" includes any physical attachment or close association, which
may
be permanent or temporary. Generally, an interaction of hydrogen bonding,
hydrophobic
forces, van der Waals forces etc. facilitates physical attachment between the
ligand
molecule of interest and the receptor. The "binding" interaction may be brief.
as in the
situation where binding causes a chemical reaction to occur. Reactions
resulting from
contact between the binding component and the analyte are within the
definition of binding
for the purposes of the present invention. Binding is preferably specific.
Specific binding
indicates substantially no strong binding to other antigens. A comparison of
the binding of
different papilloma viruses as shown below emphasizes the nature of the
specific binding.
2o The binding may be reversible, particularly under different conditions.
The tenor "bound to" refers to a tight coupling of the two components
mentioned.
The nature of the binding may be chemical coupling through a linker moiety, as
a fusion
protein produced by expression of a single ORF, physical binding or packaging
such as in a
macromolecular complex. Likewise, all of the components of a cell are "bound
to" the cell.
"Labels" include a large number of directly or indirectly detectable
substances
bound to another compound and are known per se in the immunoassay and
hybridization
assay fields. Examples include radioactive, fluorescent, enzyme,
chemiluminescent, hapten,
a solid phase, spin labels, particles, etc. Labels include indirect labels,
which are detectable
in the presence of another added reagent, such as a receptor bound to a biotin
label and
3o added avidin or streptavidin, labeled or subsequently labeled with labeled
biotin
simultaneously or later.
An "antibody" is a typical receptor and includes fragments of antibodies,
e.g., Fab,
Fab2, recombinant, reassortant, single chain, phage display and other antibody
variations.
The receptor may be directly or indirectly labeled.
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
In situations where a chemical label is not used in an assay, alternative
methods may
be used such as agglutination or precipitation of the ligand/receptor complex,
detecting
molecular weight changes between complexed and uncomplexed ligands and
receptors,
optical changes to a surface and other changes in properties between bound and
unbound
5 ligands or receptors.
The term "biological sample" includes tissues, fluids, solids (preferably
suspendable), extracts and fractions that contain proteins. These protein
samples are from
cellular or fluids originating from an organism. In the present invention, the
host is
generally a mammal, most preferably a human.
to The present invention provides recombinant plant viruses that express
fusion
proteins that are formed by fusions between a plan viral coat protein and
protein of interest.
By infecting plant cells with the recombinant plant viruses of the invention,
relatively large
quantities of the protein of interest may be produced in the form of a fusion
protein. The
fusion protein encoded by the recombinant plant virus may have any of a
variety of forms.
15 The protein of interest may be fused to the amino terminus of the viral
coat protein or the
protein of interest may be fused to the carboxyl terminus of the viral coat
protein. In other
embodiments of the invention, the protein of interest may be fused internally
to a coat
protein. The viral coat fusion protein may have one or more properties of the
protein of
interest. The recombinant coat fusion protein may be used as an antigen for
antibody
development or to induce a protective immune response.
The subject invention provides novel recombinant plant viruses that code for
the
expression of fusion proteins that consist of a fusion between a plant viral
coat protein and a
protein of interest. The recombinant plant viruses of the invention provide
for systemic
expression of the fusion protein, by systemically infecting cells in a plant.
Thus by
employing the recombinant plant viruses of the invention, large quantities of
a protein of
interest may be produced.
The fusion proteins of the invention comprise two portions: (i) a plant viral
coat
protein and (ii) a protein of interest. The plant viral coat protein portion
may be derived
from the same plant viral coat protein that serves a coat protein for the
virus from which the
genome of the expression vector is primarily derived, i.e., the coat protein
is native with
respect to the recombinant viral genome. Alternatively, the coat protein
portion of the fusion
protein may be heterologous, i.e., non-native, with respect to the recombinant
viral genome.
In a preferred embodiment of the invention, the 17.5 KDa coat protein of
tobacco mosaic
virus is used in conjunction with a tobacco mosaic virus derived vector. The
protein of
interest portion of the fusion protein for expression may consist of a peptide
of virtually any
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
16
amino acid sequence, provided that the protein of interest does not
significantly interfere
with (1) the ability to bind to a receptor molecule, including antibodies and
T cell receptors
(2) the ability to bind to the active site of an enzyme (3) the ability to
induce an immune
response, (4) hormonal activity, (5) immunoregulatory activity, and (6) metal
chelating
activity. The protein of interest portion of the subject fusion proteins may
also possess
additional chemical or biological properties that have not been enumerated.
Protein of
interest portions of the subject fusion proteins having the desired properties
may be obtained
by employing all or part of the amino acid residue sequence of a protein known
to have the
desired properties. For example, the amino acid sequence of hepatitis B
surface antigen may
1o be used as a protein of interest portion of a fusion protein invention so
as to produce a
fusion protein that has antigenic properties similar to hepatitis B surface
antigen. Detailed
structural and functional information about many proteins of interest are well
known; this
information may be used by the person of ordinary skill in the art so as to
provide for coat
fusion proteins having the desired properties of the protein of interest. The
protein of
interest portion of the subject fusion proteins may vary in size from one
amino acid residue
to over several hundred amino acid residues, preferably the sequence of
interest portion of
the subject fusion protein is less than 100 amino acid residues in size, more
preferably, the
sequence of interest portion is less than 50 amino acid residues in length. It
will be
appreciated by those of ordinary skill in the art that, in some embodiments of
the invention,
2o the protein of interest portion may need to be longer than 100 amino acid
residues in order
to maintain the desired properties. Likewise, it will be appreciated that a
smaller sequence
containing only the particular epitope or even a fraction of it may be used.
Preferably, the
size of the protein of interest portion of the fusion proteins of the
invention is minimized
(but retains the desired biological/chemical properties), when possible.
While the protein of interest portion of fusion proteins of the invention may
be
derived from any of the variety of proteins, proteins for use as antigens are
particularly
preferred. For example, the fusion protein, or a portion thereof, may be
injected into a
mammal, along with suitable adjutants, so as to produce an immune response
directed
against the protein of interest portion of the fusion protein. The immune
response against
the protein of interest portion of the fusion protein has numerous uses, such
uses include,
protection against infection, and the generation of antibodies useful in
immunoassays.
The location (or locations) in the fusion protein of the invention where the
viral coat
protein portion is joined to the protein of interest is referred to herein as
the fusion joint. A
given fusion protein may have one or two fusion joints. The fusion joint may
be located at
the carboxyl terminus of the coat protein portion of the fusion protein
(joined at the amino
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
17
terminus of the protein of interest portion). The fusion joint may be located
at the amino
terminus of the coat protein portion of the fusion protein (joined to the
carboxyl terminus of
the protein of interest). In other embodiments of the invention, the fusion
protein may have
two fusion joints. In those fusion proteins having two fusion joints, the
protein of interest is
located internal with respect to the carboxyl and amino terminal amino acid
residues of the
coat protein portion of the fusion protein, i.e., an internal fusion protein.
Internal fusion
proteins may comprise an entire plant virus coat protein amino acid residue
sequence (or a
portion thereof) that is "interrupted" by a protein of interest, i.e., the
amino terminal
segment of the coat protein portion is joined at a fusion joint to the amino
terminal amino
to acid residue of the protein of interest and the carboxyl terminal segment
of the coat protein
is joined at a fusion joint to the amino terminal acid residue of the protein
of interest.
When the coat fusion protein for expression is an internal fusion protein, the
fusion
joints may be located at a variety of sites within a coat protein. Suitable
sites for the fusion
joints may be determined either through routine systematic variation of the
fusion joint
locations so as to obtain an internal fusion protein with the desired
properties. Suitable sites
for the fusion jointly may also be determined by analysis of the three
dimensional structure
of the coat protein so as to determine sites for "insertion" of the protein of
interest that do
not significantly interfere with the structural and biological functions of
the coat protein
portion of the fusion protein. Detailed three dimensional structures of plant
viral coat
2o proteins and their orientation in the virus have been determined and are
publicly available to
a person of ordinary skill in the art. For example, a resolution model of the
coat protein of
Cucumber Green Mottle Mosaic Virus (a coat protein bearing strong structural
similarities
to other tobamovirus coat proteins) and the virus can be found in Wang and
Stubbs J. Mol.
Biol. 239:371-384 (1994). Detailed structural information on the virus and
coat protein of
Tobacco Mosaic Virus can be found, among other places in Namba et al, J. Mol.
Biol.
208:307-325 (1989) and Pattanayek and Stubbs J. Mol. Biol. 228:516-528 (1992).
Knowledge of the three dimensional structure of a plant virus particle and the
assembly process of the virus particle permits the person of ordinary skill in
the art to
design various coat protein fusions of the invention, including insertions,
and partial
substitutions. For example, if the protein of interest is of a hydrophilic
nature, it may be
appropriate to fuse the peptide to the TMVCP (Tobacco mosaic tobamovirus coat
protein)
region known to be oriented as a surface loop region. Likewise, alpha helical
segments that
maintain subunit contacts might be substituted for appropriate regions of the
TMVCP
helices or nucleic acid binding domains expressed in the region of the TMVCP
oriented
towards the genome.
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
18
Polynucleotide sequences encoding the subject fusion proteins may comprise a
"leaky" stop codon at a fusion joint. The stop codon may be present as the
codon
immediately adjacent to the fusion joint, or may be located close (e.g.,
within 9 bases) to the
fusion joint. A leaky stop codon may be included in polynucleotides encoding
the subject
coat fusion proteins so as to maintain a desired ratio of fusion protein to
wild type coat
protein. A "leaky" stop codon does not always result in translational
termination and is
periodically translated. The frequency of initiation or termination at a given
start/stop codon
is context dependent. The ribosome scans from the 5'-end of a messenger RNA
for the first
ATG codon. If it is in a non-optimal sequence context, the ribosome will pass,
some
1 o fraction of the time, to the next available start codon and initiate
translation downstream of
the first. Similarly, the first termination codon encountered during
translation will not
function 100% of the time if it is in a particular sequence context.
Consequently, many
naturally occurring proteins are known to exist as a population having
heterogeneous N
and/or C terminal extensions. Thus by including a leaky stop codon at a fusion
joint coding
t 5 region in a recombinant viral vector encoding a coat fusion protein, the
vector may be used
to produce both a fusion protein and a second smaller protein, e.g., the viral
coat protein. A
leaky stop codon may be used at, or proximal to, the fusion joints of fusion
proteins in
which the protein of interest portion is joined to the carboxyl terminus of
the coat protein
region, whereby a single recombinant viral vector may produce both coat fusion
proteins
20 and coat proteins. Additionally, a leaky start codon may be used at or
proximal to the fusion
joints of fusion proteins in which the protein of interest portion is joined
to the amino
terminus of the coat protein region, whereby a similar result is achieved. In
the case of
TMVCP, extensions at the N and C terminus are at the surface of viral
particles and can be
expected to project away from the helical axis. An example of a leaky stop
sequence occurs
25 at the junction of the 126/183 kDa reading frames of TMV and was described
over 15 years
ago (Pelham, H. R. B., 1978). Skuzeski et al. (1991) defined necessary 3'
context
requirements of this region to confer leakiness of termination on a
heterologous protein
marker gene (beta-glucuronidase) as CAR-YYA (C=cytidine, A=adenine,
Y=pyrimidine).
In another embodiment of the invention, the fusion joints on the subject coat
fusion
3o proteins are designed so as to comprise an amino acid sequence that is a
substrate for
protease. By providing a coat fusion protein having such a fusion joint, the
protein of
interest may be conveniently derived from the coat protein fusion by using a
suitable
proteolytic enzyme. The proteolytic enzyme may contact the fusion protein
either in vitro or
m vivo.
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
19
The expression of the subject coat fusion proteins may be driven by any of a
variety
of promoters functional in the genome of the recombinant plant viral vector.
In a preferred
embodiment of the invention, the subject fusion proteins are expressed from
plant viral
subgenomic promoters using vectors as described in U.S. Pat. No. 5,316,931.
Recombinant DNA technologies have allowed the life cycle of numerous plant RNA
viruses to be extended artificially through a DNA phase that facilitates
manipulation of the
viral genome. These techniques may be applied by the person ordinary skill in
the art in
order make and use recombinant plant viruses of the invention. The entire cDNA
of the
TMV genome was cloned and functionally joined to a bacterial promoter in an E.
coli
l0 plasmid (Dawson et al., 1986). Infectious recombinant plant viral RNA
transcripts may also
be produced using other well known techniques, for example, with the
commercially
available RNA polymerases from T7, T3 or SP6. Precise replicas of the virion
RNA can be
produced in vitro with RNA polymerase and dinucleotide cap, m7GpppG. This not
only
allows manipulation of the viral genome for reverse genetics, but it also
allows
manipulation of the virus into a vector to express foreign genes. A method of
producing
plant RNA virus vectors based on manipulating RNA fragments with RNA ligase
has
proved to be impractical and is not widely used (Pelcher, L. E., 1982).
Detailed information
on how to make and use recombinant RNA plant viruses can be found, among other
places
in U.S. Pat. No. 5,316,931 (Donson et al.), which is herein incorporated by
reference. The
invention provides for polynucleotide encoding recombinant RNA plant vectors
for the
expression of the subject fusion proteins. The invention also provides for
polynucleotides
comprising a portion or portions of the subject vectors. The vectors described
in U.S. Pat.
No. 5,316,931 are particularly preferred for expressing the fusion proteins of
the invention.
Figure 2 demonstrates one way used in the present invention for constructing
the
recombinant tobamoviruses used in the present invention. An infectious clone
of TMV
strain U1 called pBSG801 was used as the basic vector for construction of
peptide fusion
constructs, as well as for building other peptide fusion-acceptor vectors. In
some cases, an
NcoI restriction site was required for peptide insertions. A version of
pBSG801 was created
where the NcoI site in the movement protein gene was mutated, without altering
the amino
3o acid sequence of the movement protein. In this construct (pBSG80101Vco),
NcoI is
available as a cloning site. A. shows a method that was used for construction
of peptide
fusion constructs using a PCR-ligation method. PCR primers F
(GGAGTTTGTGTCGGTGTGTATTG)and R (GGAGTTTGTGTCGGTGTGTATTG)
amplify a fragment of the pBSG801 or plasmid that spans the 3' end of the
viral genome to
a point upstream of the native NcoI site within the movement protein open
reading frame.
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
Peptides may be fused to internal positions in the coat protein open reading
frame by
addition of synthetic DNA encoding the a fragment of the peptide of interest
to internal
primers F' and R'. Primers F and R' and R and F' are then used to amplify PCR
products A
and B. Ligation of A and B reconstitutes the peptide of interest in the same
reading frame
5 as the coat protein. The ligated product is digested with NcoI and KpnI The
engineered
coat protein-peptide fusion is then translated in vivo when in vitro-generated
infectious
RNA is used to infect Nicotiana plants. B. Shows part of the plasmid pLSB2268
which
was generated from pBSG801~lVco: an NcoI site (CCATGG) was inserted at the
start of the
coat protein open reading frame to facilitate cloning of N-terminal peptide
fusions by PCR.
10 Synthetic DNA encoding peptides of interest was inserted in frame with the
ATG in the
NcoI site into a primer homologous with the 5' 1 end of the coat protein gene.
The specific
PCR primer was used in PCR reactions with primer R
(GGAGTTTGTGTCGGTGTGTATTG) and resulting PCR product was digested with NcoI
and KpnI and cloned into pLSB2268. An alternative strategy for insertion of
synthetic
15 DNA encoding peptides of interest in different positions of tobamovirus
coat proteins is
shown in C. Three different vectors were created; all were derived from
pBSG80101Vco.
These acceptor vectors, pLSB2268; pLSB2269 and pLSB2109 contain restriction
sites
suitable for accepting double stranded oligonucleotides with sticky ends
compatible with
NcoI (5') and NgoMIV (3'). Complementary single stranded oligonucleotides are
20 synthesized that encode the peptide of interest, such that the sense (top)
strand has the
sequence 5'-CATG(NNN)"G-3' and the antisense (bottom) strand has the sequence
5'-
CCGGC(NNNJ"-3' where (NNN)" denotes a sequence of DNA that encodes amino acids
in
the peptide of interest. The complementary oligonucleotides are annealed in
vitro and the
resulting dsDNA oligonucleotide with overhanging CATG and CCGG ends is ligated
with
acceptor vector that has been digested with NcoI and NgoMIV to create various
coat protein
fusion constructs.
In addition to providing the described viral coat fusion proteins, the
invention also
provides for virus particles that comprise the subject fusion proteins. The
coat of the virus
particles of the invention may consist entirely of coat fusion protein. In
another embodiment
of the virus particles of the invention, the virus particle coat may consist
of a mixture of
coat fusion proteins and non-fusion coat protein, wherein the ratio of the two
proteins may
be varied. As tobamovirus coat proteins may self assemble into virus
particles, the virus
particles of the invention may be assembled either in vivo or in vitro. The
virus particles
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
21
may also be conveniently disassembled using well known techniques so as to
simplify the
purification of the subject fusion proteins, or portions thereof.
The invention also provides for recombinant plant cells comprising the subject
coat
fusion proteins and/or virus particles comprising the subject coat fusion
proteins. These
plant cells may be produced either by infecting plant cells (either in culture
or in whole
plants) with infectious virus particles of the invention or with
polynucleotides encoding the
genomes of the infectious virus particle of the invention. The recombinant
plant cells of the
invention have many uses. Such uses include serving as a source for the fusion
coat proteins
of the invention.
The protein of interest portion of the subject fusion proteins may comprise
many
different amino acid residue sequences, and accordingly may have different
possible
biological/chemical properties however, in a preferred embodiment of the
invention the
protein of interest portion of the fusion protein is useful as a vaccine
antigen. The surface of
TMV particles and other tobamoviruses contain continuous epitopes of high
antigenicity
and segmental mobility thereby making TMV particles especially useful in
producing a
desired immune response. These properties make the virus particles of the
invention
especially useful as carriers in the presentation of foreign epitopes to
mammalian immune
systems.
While the recombinant RNA viruses of the invention may be used to produce
numerous coat fusion proteins for use as vaccine antigens or vaccine antigen
precursors, it is
of particular interest to provide vaccines against viral pathogens of humans,
and domestic
animals. It is of particular interest to provide vaccines against human
papillomavirus
(HPV) types that are implicated in the etiology of cervical cancer, and other
neoplasias,
including but not limited to HPV-16, HPV-18, HPV-31, HPV-33, HPV-35 and HPV-
52.
While not implicated in cervical cancer a vaccine against HPV-6 and HPV-11 is
also
desirable as such viruses cause much disease. It is also of particular
interest to provide
vaccines against hemorrhagic fever-causing viruses such as Rift Valley fever
virus (RVFV)
and Ebola viruse (EBOV), as these pathogens present significant threat to the
US population
if weaponized by terrorists. In addition, it is of interest to provide
vaccines against human
immunodeficiency virus type 1 (HIV-1), and against parvoviruses that are
significant
pathogens of human companion animals (particularly cats and dogs), and
livestock
(especially pigs).
When the fusion proteins of the invention, portions thereof, or viral
particles
comprising the fusion proteins are used in vivo, the proteins are typically
administered in a
composition comprising a pharmaceutical carrier. A pharmaceutical carrier can
be any
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
22
compatible, non-toxic substance suitable for delivery of the desired compounds
to the body.
Sterile water, alcohol, fats, waxes and inert solids may be included in the
carrier.
Pharmaceutically accepted adjuvants (buffering agents, dispersing agent) may
also be
incorporated into the pharmaceutical composition. Additionally, when the
subject fusion
proteins, or portion thereof, are to be used for the generation of an immune
response,
protective or otherwise, formulation for administration may comprise one or
immunological
adjuvants in order to stimulate a desired immune response.
When the fusion proteins of the invention, or portions thereof, are used in
vivo, they
may be administered to a subject, human or animal, in a variety of ways. The
to pharmaceutical compositions may be administered orally or parenterally,
i.e.,
subcutaneously, intramuscularly or intravenously. Thus, this invention
provides
compositions for parenteral administration which comprise a solution of the
fusion protein
(or derivative thereof) or a cocktail thereof dissolved in an acceptable
earner, preferably an
aqueous earner. A variety of aqueous carriers can be used, e.g., water,
buffered water, 0.4%
t5 saline, 0.3% glycerine and the like. These solutions are sterile and
generally free of
particulate matter. These compositions may be sterilized by conventional, well
known
sterilization techniques. The compositions may contain pharmaceutically
acceptable
auxiliary substances as required to approximate physiological conditions such
as pH
adjusting and buffering agents, toxicity adjusting agents and the like, for
example sodium
2o acetate, sodium chloride, potassium chloride, calcium chloride, sodium
lactate, ete. The
concentration of fusion protein (or portion thereof) in these formulations can
vary widely
depending on the specific amino acid sequence of the subject proteins and the
desired
biological activity, e.g., from less than about 0.5%, usually at or at least
about 1% to as
much as 15 or 20% by weight and will be selected primarily based on fluid
volumes,
25 viscosities, ete., in accordance with the particular mode of administration
selected.
Actual methods for preparing parenterally administrable compositions and
adjustments necessary for administration to subjects will be known or apparent
to those
skilled in the art and are described in more detail in, for example,
Remington's
Pharmaceutical Science, current edition, Mack Publishing Company, Easton, Pa.,
which is
3o incorporated herein by reference.
The invention having been described above, may be better understood by
reference
to the following examples. The examples are offered by way of illustration and
are not
intended to be interpreted as limitations on the scope of the invention.
The vaccine compositions of the present invention are used for inducing an
immune
35 response to prevent infection by one or more of the pathogenic viruses.
When the infection
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
23
is of a long duration such as with HPV and HIV, the vaccines may be provided
to help in
clearing the infection or to suppress the infection. Generally, vaccines are
given by
injection or contact with mucosal, buccal, lung, eye or similar tissues.
Transdermal and oral
administration may be used when sufficiently adsorbed and stable, particularly
when
tolerization is desired.
One or more of the vaccines may be used cross-immunize the individual
recipient
against related strains or viruses. Likewise, a single vaccine designed
against one pathogen
may be used against other related ones. For example, a single parvovirus
vaccine
composition may be used to induce an immune response against feline, canine
and porcine
t0 parvoviruses in cats, dogs and pigs respectively due to a very similar
viral antigen common
to each virus. The peptide epitope containing compositions may also be used as
positive
controls for diagnostic, epidemiological and other screening purposes.
The same compositions as used for vaccines may be used to immunize an animal
for
the production of antibodies, antibody-secreting cells (e.g. for monoclonal
antibody
production), T-cell receptors and corresponding T-cells. These materials may
be used for
diagnostic purposes, given by injection to provide passive immunity
prophalactically or to
treat an active infection.
A number of different binding assay formats may be used to detect the
pathogenic
viruses or antibodies to the viruses as a measure of past infection. Both
competitive and
non-competitive assays may be used with direct or indirect labels to one or
more binding
partners. These binding assays, particularly immunoassays are well known in
the art.
EXAMPLE 1: Papillomavirus Vaccines
Antigens are most effectively delivered to the immune system in a repetitive
configuration, like that presented by virus-like particles. For B cell
responses, a crucial
factor for immunogenicity is repetitiveness and order of antigenic
determinants. Many
viruses display a quasicrystalline surface with a regular array of epitopes
which efficiently
crosslink antigen-specific immunoglobulins on the surface of B cells, leading
to B cell
proliferation and production of secreted antibodies (Bachmann et al., 1993;
Fehr et al.,
1998). Triggered B cells can activate helper T cells, leading to long-lived B
cell memory-
3o essential for any vaccine. In part due to these observations, and because
only very low
levels of L2-specific antibodies are detected in vaccinated or infected
animals, only L1 VLP
vaccines have been pursued in clinical trials of prophylactic vaccines.
However, because
VLP and capsomeric L1 vaccines induce mainly type-specific neutralizing
antibodies, a
comprehensive solution to HPV prophylactic vaccination probably requires
vaccination
with L1 from multiple types.
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
24
The dominant virus neutralizing immune response against HPV-16 particles is
directed against a conformational epitope, described by the monoclonal
antibody named V5
(Christensen et a1.,1996). There are, in addition, two linear epitopes in HPV-
16 L1 that
may induce antibodies capable of neutralization of other papillomavirus types;
these two
epitopes (QPLGVGISGHPLLNKLDDTE and ENVPDDLYIKGSGS) bind monoclonal
antibodies I23 and J4, respectively. Unfortunately, the immune response that
is generated
to L1-derived VLP vaccines is a dominant type-specific neutralizing response.
If there were
ways to enhance the recognition of the sub-dominant epitopes that might induce
antibodies
with a broader specificity against other papillomavirus types, this method
could be
1 o incorporated into a vaccine regimen to generate a protective immune
response against
multiple high risk papillomavirus types. The cross-neutralizing epitopes I-23
and J-4 were
displayed on the surface of TMV particles as shown in Table 1. Other peptide
fusion
vaccines are also shown in Table 1.
Table 1: TMV - Papillomavirus Peptide Fusion Vaccines
Construct Virus Name Origin of Peptide Peptide Sequence
Name
LSB2283 TMV:CRPV2.1Cottontail rabbit papillomavirusVGPLDIVPEVADPG
(GPAT) L2
protein GPTL
LSB2288 TMV:CRPV2.2Cottontail rabbit papillomavirusPGGPTLVSLHELPA
(GPAT) L2
protein ETP
LSB2285 TMV:ROPV2.1Rabbit oral papillomavirusVGPLEVIPEAVDPA
(GPAT) L2 protein
GSSI
LSB2280 TMV:ROPV2.2Rabbit oral papillomavirusPAGSSIVPLEEYPAE
(GPAT) L2 protein
IP
LSB2282 TMV:HPV-11 Human papillomavirus LIEESAIINAGAP
(GPAT) L2 type 11 L2 protein
LSB2406
(N-ter)
LSB2278 TMV:HPV-16 Human papillomavirus LVEETSFIDAGAP
(GPAT) L2 type 16 L2 protein
LSB2291
(N-ter)
LSB2281 TMV:HPV-18 Human papillomavirus LIEDSSVVTSGAP
(GPAT) L2 type 18 L2 protein
LSB2297 '
(N-ter)
LSB2284 TMV:HPV-16J4Human papillomavirus GENVPDDLYIKGSG
(GPAT) type 16 Ll protein
LSB2404 S
(N-ter)
LSB2279 TMV:HPV-16I23Human papillomavirus QPLGVGISGHPLLN
(GPAT) type 16 L1 protein
KLDDTE
TMV wild N/A N/A
type
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
Antibodies against the N-terminus of L2 can be neutralizing in pseudoinfection
studies, but paradoxically the neutralizing antibodies do not inhibit virion
binding to the cell
surface (Gaukroger et al., 1996; Roden et al., 1994). It is possible that
domains of L2 that
bind neutralizing antibodies are not accessible in native virions or
pseudovirions, but are
5 exposed at some point during viral entry into cells. Recently Kawana et al.
(2001b)
showed that amino acids 108-126 of HPV16 L2 (a neutralizing domain) could bind
a
proteinaceous receptor, present at higher level on the surface of epithelial
cells than non-
epithelial cells. These data suggest that L2 binds a co-receptor on the cell
surface and that
at least a subset of virus neutralizing antibodies can block L2-mediated virus
entry.
10 Papillomavirus virions and pseudovirions bind a wide variety of cell types
and the N-
termini of L2 proteins of mucosotropic papillomaviruses show high homology. In
the light
of these facts it is tempting to speculate that the binding specificity
between L2 and a
papillomavirus cell surface coreceptor could be a determinant of
papillomavirus tissue
tropism.
~5 The data of Kawana and colleagues show that immunization of mice (Kawana et
al.,
1999; 2001) and humans (Kawana et al., 2003) with the 13 amino acid HPV-16 L2-
derived
peptide (sequence: LVEETSFIDAGAP) could induce antibodies that can neutralize
papillomavirus infection in vitro. Importantly, sera from animals and humans
immunized
with this peptide can neutralize the homologous virus (HPV-16) as well as
related
20 mucosotropic viruses: HPV-11; HPV-6 and HPV52 (Kawana et al., 1999; 2001;
2003).
These results are very significant, since this is the first time that
antibodies from animals
immunized with papillomavirus antigens have shown cross-type neutralization
activity.
Kawana et al. (2003) had to deliver relatively large quantities of peptide -
SOOUg, by the
intranasal route, to induce papillomavirus L2-specific antibodies. The
inventors predicted
25 that display of the peptide as a highly repetitive antigen array, such as
on the surface of
TMV, would enhance the immunogenicity of the peptide.
Genetic fusion of papillomavirus peptides to the coat protein of tobacco
mosaic virus strain
U1
3o Tobacco mosaic virus strain U1 (vulgare) was used as the carrier for
peptide fusions.
All peptides were fused near the carboxy-terminus of the U1 coat protein, at a
position four
residues before the carboxy terminal amino acids (GPAT). DNA sequences
encoding the
papillomavirus epitopes were synthesized in PCR primers and a PCR strategy was
used to
fuse the sequences to the TMV coat protein at a position four amino acids from
the C-
terminus (position . "GPAT") or at the N-terminus, immediately after the
initiating
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
26
methionine ("N-ter"). A synthetic DNA sequence encoding the L2 peptide of
interest was
inserted into the U1 coat protein DNA sequence, by PCR with specific primers
and
fragment ligation. Recombinant TMV clones were sequenced, and clones with DNA
sequences that matched predicted sequences were assigned clone identifiers, as
indicated in
Table 1.
Infection of plants with infectious chimeric TMV:papillomavirus clones
The plasmids described in Table 1 were transcribed in vitro to generate capped
infectious RNA transcripts (mMESSAGE mMACHINE Kit, Ambion, Austin TX).
to Transcription reactions were diluted in FES buffer, and plants were
inoculated by leaf
abrasion. The four rabbit papillomavirus constructs (pLSB2283, pLSB2288,
pLSB2285
and pLSB2280) were inoculated on two leaves of each of 40 to 46 Nicotiana
benthamiana
plants, 24 days post-sowing, and infectious transcripts of pLSB2282 (TMV:HPV-
11L2)
were inoculated on two leaves of each of 40, 27 day-old, Nicotiana excelsiana
plants, a
Large Scale Biology Corporation-proprietary field host for TMV (Fitzmaurice
WP, US
Patent 6,344,597). Wild type TMV U1 was prepared from infected tobacco
(Nicotiana
tabaccum). The recombinant TMV:ROPV2.2 virus induced necrotic symptoms on
infected N. benthamiana plants; the other recombinant viruses induced symptoms
typically
seen in Nicotiana plants infected with TMV coat protein fusions, i.e. leaf
crinkling,
2o bubbling and twisting, and a stunted plant growth habit. The number of
grams of tissue
and DPI for each construct is summarized in Table 2.
Table 2: Record of production of recombinant TMV in Nicotiana plants
Virus Name Plant SpeciesDPI # plantsTissue
weight
TMV:CRPV2.1 Nicotiana 8 60 247 g
Benthamiana
TMV:HPV-11 Excelsiana 11 45 267 g
L2
TMV:ROPV2.2*Nicotiana 10 90 143 g
Benthamiana
TMV wild MD609 15 12 258 g
Type*
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
27
TMV:CRPV2.2 Nicotiana 11 81 269 g
Benthamiana
TMV:ROPV2.1 Nicotiana 10 81 281 g
Benthamiana
* very severe viral symptoms- most infected tissue only was harvested.
** only upper infected tissue was harvested
N. benthamiana plants were used for the rabbit papillomavirus constructs.
Excelsiana plants
were chosen for the HPV construct because if this moved forward to a product
it would
most likely be grown in the field and Excelsiana is a better host for the
field. The control
virus was wild type TMV U1 for which MD609 plants are the host of choice.
Virus is
generally allowed to accumulate for longer time periods in the larger MD
plants prior to
harvest.
t0 Purification of chimeric virus constructs from infected Nicotiana plants
Infected plant material was harvested between 8 and 14 days post-inoculation,
when the
virus accumulation was estimated to be the highest in infected leaf tissues.
Only plant
material (stem and leaves) above the inoculated leaf was harvested. The
harvested tissue
was weighed and chopped into small pieces. The virus was extracted by grinding
the tissue
t 5 in a four liter Waring Blender, for two minutes on high speed in a 1:2
ratio (tissue:buffer) of
0.86M sodium chloride, 0.04% sodium metabisulphite solution that had been
chilled to
10°C. The temperature of the homogenate ("green juice") was measured
and recorded: this
averaged 20.5°C. The homogenate was recovered by squeezing through four
layers of
cheesecloth, and the volume of homogenate measured. Two 0.5 ml samples of the
green
2o juice were collected for analysis by SDS-PAGE, and for bioburden analyses.
The pH of the homogenate was measured and adjusted to pH5.0 with concentrated
phosphoric acid. The green juice was then heated to 47°C, and held at
that temperature for
minutes to coagulate contaminating plant proteins. The homogenate was then
cooled to
15°C in an ice bath. The pH/heat treated homogenate was clarified by
centrifugation at
6,000 x g for 5 minutes. The supernatant (S 1 ) was decanted through two
layers of
Miracloth, and the volume of S 1 recovered was recorded. Two 0.5 ml samples
were
collected for SDS-PAGE, protein assay and bioburden analyses. The pellet (P1)
was
resuspended in distilled water, adjusted to pH 7.4 with NaOH and centrifuged
at 6,000 x g
for S minutes to clarify. The volume of the second supernatant (S2) was
recorded, and
sampled for SDS PAGE to verify that the majority of the virus was in the S1
fraction.
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
28
Recombinant virus was precipitated from S 1 by adding polyethylene glycol
(6000
Da molecular weight) to 4% final concentration. The solution was stirred for
20 minutes,
and then chilled on ice for one hour. Precipitated virus was recovered by
centrifugation at
10,000 x g for 10 minutes. The supernatants were decanted and discarded. The
recombinant virus pellets were resuspended in a modified phosphate buffered
saline
containing 0.86M NaCI, and chilled on ice for 30 minutes. The virus was
centrifuged at
8,000 x g for S minutes to clarify. The supernatants were decanted through
miracloth. Two
0.5 ml samples were collected for SDS PAGE analysis. A second PEG-mediated
virus
precipitation was then performed, as before, and the virus pellets resuspended
in phosphate-
1o buffered saline (PBS), pH 7.4. Insoluble material was pelleted by
centrifugation at 10,000 x
g for 5 minutes and the supernatant was recovered with a serological pipette.
The final
purification step involved freezing and thawing of the virus samples to
precipitate any
remaining plant contaminants: samples were frozen at -20°C for several
hours and then
thawed at room temperature. Insoluble material was eliminated by
centrifugation at 10,000
x g. The additional freeze-thaw purification steps were not carried out for
the
TMV:CRPV2.1 and TMV:HPV11L2 samples.
The virus concentration of each fusion was measured using the BCA protein
assay
with IgG as the standard. Based on the virus concentration determination, a
portion of each
virus preparation was diluted to 0.5 mg/ml (live virus) or 0.55 mg/ml for the
virus
inactivation step.
Virus Inactivation with Binary Ethylenimine
Each recombinant TMV preparation was diluted to 0.55 mg/ml in PBS, pH 7.4 to
account for the slight dilution due to reagent addition. Virus was chemically
inactivated by
treatment with binary ethylenimine (BEI), by addition of a O.1M BEI stock
solution to a
final concentration of SmM BEI. Samples were incubated for 48 hours at
37°C with
constant mixing by rotating tubes end over end in a 37°C incubator.
After 48 hours the BEI
was neutralized by addition of a 3 molar excess of sodium thiosulphate.
3o EXAMPLE 2: Viral Hemorrha~ic Fever Vaccines
Amongst all of the HFVs, RVFV is perhaps the easiest to weaponize: aerosols
are
particularly infectious, and have frequently caused infection in laboratory
personnel (Borio
et al., 2002; Isaacson, 2001). Monoclonal antibody 4D4 has been shown to
inhibit RVFV
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
29
plaque formation in cell culture and to protect mice against lethal challenge
(Keegan and
Collet, 1986; London et al., 1992).
The general method used in Example 1 was repeated with the linear epitope that
binds mAb 4D4 (sequence: KGTMDSGQTKREL) inserted at three different positions
in
the TMV U1 coat protein: N-terminal (between amino acids 1 and 2); in the
surface-located
loop structure (between amino acids 64 and 65) and at the C-terminus, between
amino acids
155 and 156. The genetic constructs were verified by DNA sequencing, and
assigned
LSBC identifiers. Table 5 summarizes the expression and MALDI-TOF
characterization
for these viral fusion constructs.
to
Table 5: RVFV peptide fusions to the TMV U1 coat protein.
ConstructDescriptionSystemic infectionSoluble TheoreticalActual
mass
name in N. benthamianavirions Mass determined
extracted by MALDI-
TOF
LSB24724D4 epitopeYes Yes
at N-terminus
of TMV U1
LSB24714D4 epitopeYes No
in surface
loop of
TMV
U1
LSB24704D4 epitopeYes Yes
at C-terminus
of TMV U1
The general method used in Example 1 was repeated with the three known linear
epitopes from EBOV GP1 that bind monoclonal antibodies that neutralize EBOV
infection
in vitro and in vivo (Wilson et al. 2000). The peptide VYKLDISEA is bound by
Mab 6D8-
1-2; Mab 13F6-1-2 binds the amino acid sequence DEQHHRRTDND and mAb 12B5-1-1-
binds amino acid sequence LITNTIAGV (Wilson et al., 2000). Table 6 summarizes
the
expression and solubility data for these recombinant TMV virions.
2o Table 6: Solubility and confirmation of three Ebola epitopes fused to three
locations
on the TMV U1 coat protein.
Epitope (sequence Position SolubilityPredicted MassMALDI
) Da mass
Da
GP1-393 (VYKLDISEA) N Yes 18639 18641
60's LoopNo - -
Near C Yes 18826 18818
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
GP1-405 (DEQHHRRTDND)N Yes 19197 19199
60's No - -
Loop
Near n.d.' - -
C
GP1-481 (LITNTIAGV) N Yes 18634 18632
60's No - -
Loop
Near Yes 18690 18692
C
' This is the minimal consensus sequence.
2 N: N-terminus, Near C: the insertion site is before the last four amino acid
of the coat
~rotein.
Not determined.
5 4 The extra "D" at the N-terminus was added to the minimal consensus
sequence to balance
the overall charge of the coat protein.
Figure 15 shows an SDS PAGE gel where extracts from plants infected with
infectious transcripts of the various EBOV peptide:TMV fusion constructs were
separated
according to molecular mass. Proteins from leaf tissues of two infected plants
were
to extracted in sodium acetate "N" buffer (pH 5), the pellet was further
extracted in TRIS-Cl
"T" buffer (pH 7.5). To extract total protein, another leaf sample was
extracted in SDS
denaturing "S" buffer (75 mM TRIS (pH 7), 2.5% sodium dodecyl sulfate (SDS),
6%
glycerol, 2.5% beta-mecapthoethanol, and 0.05% bromphenol blue). The protein
molecular
weight marker "M12" is Mark 12 (In vitrogen) spiked with 1.2 mcg of wild type
TMV U1
15 coat protein (CP). The arrow indicates the recombinant product (coat
protein fused to an
Ebola GPI epitope).
EXAMPLE 3: Human Immunodeficien~ virus type 1 jHIV-1) vaccines
The general method used in Example 1 was repeated with the linear epitopes
from
HIV proteins. In Table 7, a list of peptides that have been displayed on the
surface of TMV
2o U 1 and/or U5 virions is displayed.
Table 7 HIV-1 Epitopes Expressed on the surface of TMV
EpitopeEpitope sequence Location Comments and
Name on Env (reference)
2F5 ELDKWAS gp41 membraneLinear epitope.
short
proximal Induction of Nabs
region when
displayed on the
surface
of PVX (18); soluble
on
TMV.
gp41 membraneIdentified as being
2F5 NEQELLELDKWASLWN proximal protected by 2F5
long region
#1 antibod b roteol
'c
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
31
protection assays
(46);
insoluble on N-terminus
of TMV.
gp41 membraneSelected by 2F5
from a
2F5 EQELLELDKWASLW proximal gp160 expression
long region library
#2 11
gp41 membraneSelected by 4E10
from a
4E10 NWFDIT proximal gp160 expression
region library
short 11
gp41 membraneCore 4E10 recognition
4E10 LWNWFDITNWLW proximal site (11), flanked
region by
lon ad'acent 41 se
uence
gp41 membranePeptide binds both
2F5
2F5/4E10LLELDKWASLWNWFDIT proximal and 4E10 neutralizing
region
NWLW antibodies 11
Identified from
phage-
P195 KSSGKLISL gp120 V1 displayed peptide
library
with human HIV-1
antisera 20
Identified from
phage-
P217 CNGRLYCGP gp 120 C2 displayed peptide
library
with human HIV-1
antisera 20
Identified from
phage-
P197 GTKLVCFAA Gp41 displayed peptide
library
with human HIV-1
antisera 20
Identified from
phage-
P287 CAGGLTCSV Undetermineddisplayed peptide
library
with human HIV-1
antisera 20
Identified from
phage-
P335 SGRLYCHESW Undetermineddisplayed peptide
library
with human HIV-1
antisera 20
Selected by b12
Mab
B2.1 HERSYMFSDLENRCI gp 120 CD4 from phage displayed
binding sitepeptide library.
May
require display
as a
homodimer 19
Linear peptide
8.22.2 TTSIRNKMQKEYALFYK gp 120 V2 recognized by Mab
region
isolated from
XenoMouse immunized
with 120 22
Tat
N
terminal
B 1 MEPVDPRLEPWKHPGSQ Tat N terminalPeptide corresponding
to
P HIV-1 subtype B
Tat
rotein
B2 ~ MEPVDPKLEPWKHPGSQ Tat N terminalPeptide corresponding
to
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
32
P HIV-1 subtype B
Tat
rotein
B3 MEPVDPNLEPWKHPGSQ Tat N terminalPeptide corresponding
to
P HIV-1 subtype B
Tat
rotein
C 1 MEPVDPNLEPWKHPGSQ Tat N terminalPeptide corresponding
to
P HIV-1 subtype C
Tat
rotein
C2 MDPVDPSLEPWKHPGSQ Tat N terminalPeptide corresponding
to
P HIV-1 subtype C
Tat
rotein
SA MEPVDPSLEPWNHPGSQ Tat N terminalPeptide corresponding
to
P Tat of HIV-1 subtype
found in Ni eria
Tat
Pe tide
3
B1 PTSQSRGDPTGPKE Tat cellularPeptide corresponding
to
binding domainHIV-1 subtype B
Tat
rotein
B2 PSSQPRGDPTGPKE Tat cellularPeptide corresponding
to
binding domainHIV-1 subtype B
Tat
rotein
B3 PASQSRGDPTGPTE Tat cellularPeptide corresponding
to
binding domainHIV-1 subtype B
Tat
rotein
C 1 PLPRTQGDPTGSEE Tat cellularPeptide corresponding
to
binding domainHIV-1 subtype C
Tat
rotein
C2 PLPQTRGDPTGSKE Tat cellularPeptide corresponding
to
binding domainHIV-1 subtype C
Tat
rotein
SA PLPTTRGNPTGPKE Tat cellularPeptide corresponding
to
binding domainHIV-1 subtype C
Tat
rotein
G 41
LQARILAVE Gp41 Peptide from gp41
of an
HIV-1 sub a B strain
SA LQARVLAVEGQARVLAL Gp41 Peptide from gp41
of an
ER HIV-1 type found
in
Ni eria
SAELD EKNEQDLLALDKWASL Gp41 Peptide from gp41
of an
WN HIV-1 type found
in
Ni eria
V3 Loo
V3MN ADTIGPGRAFYTTK Gp120 V3 Peptide from crown
loop of
the V3 loop of
TCLA
HIV-1 strain MN
V3BaL ADTIGPGRAFYTTG Gp120 V3 Peptide from crown
loop of
the V3 loop of
HIV-1
strain BaL
NigeriaADTIGPGQAFYAGG Gp120 V3 Peptide from crown
~ loop of
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
33
the V3 loop of HIV-1
strain found in Nigeria
The expression, extraction and solubility data for these recombinant viruses
is summarized
in Table 8 below.
THE REST OF THIS PAGE INTENTIONALLY LEFT BLANK
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
34
Table 8.
a
y, c,
LSB# ame pitope Peptide PI Charge n
Sequence Charge _o Plant
'x ~o
@pH5 ~ DPI
@pH7 co Score
a
~~
A
'n
'n
~
a
L1
E"
~
US-
U1-
2405_ N 6L2 LVEETSF)DAGAP 4.41-3.7-5.1
1 1
24051 N 6L2 LVEETSF)DAGAP 4.41-3.7-5.1\ + - + 7 SI Y
1 + 28
24095 N 6L2 LVEETSFIDAGAP 4.41-3.7-5.1
1
24095 N 6L2 LVEETSF1DAGAP 4.41-3.7-5.1\ + + + 7 SI Y
1 + 28
2278UlC 6L2C ~ETSFIDAGAP 4.41-3.7-5.1+ na + + 8 SI Y
1 + 23
2278UlC 6L2C VEETSFmAGAP 4.41-3.7-5.1+ + + + 9 SI,C
1 + 26
24021 N 8L2 LIEDSSVVTSGAP 4.50-2.8-4.1
1
24021 N 8L2 LIEDSSVVTSGAP 4.50-2.8-4.1\ + + - 7 U Y
1 + 28
24165 N 8L2 LIEDSSVVTSGAP 4.50-2.8-4.1
1
24165 N 8L2 LIEDSSVVTSGAP 4.50-2.8-4.1\ + + + 7 SI Y
1 + 28
2270U1C 8L2C DSSVVTSGAP 4.50-2.8-4.1 - N
1
2281UlC 8L2C IEDSSVVTSGAP 4.50-2.8-4.1+ na + + 11 SI Y
1 + 21
2281UlC 8L2C DSSVVTSGAP 4.50-2.8-4.1+ + + + 9 SI,CY
1 + 26
2427UlC E10 IT 4.82-1.0-2.1- - - - 13 U N
- 22
2430UlL E10 TT 4.82-1.0-2.1- - - - 13 N N
+ 22
UlL E10 IT 4.82-1.0-2.1- - - - Y
-
24071 N E10 NWFDTT 4.82-1.0-2.1na- - - .21N,SIN
- 24
24071 N E10 NWFDIT 4.82-1.0-2.1\ - - - 7 U N
+ 28
24115 N E10 SIT 4.82-1.0-2.1na+ + + 21 N,SIY
+ 24
24115 N E10 EDIT 4.82-1.0-2.1\ + + + 7 SI Y
+ 28
24061 N /11L2LIEESAIINAGAP 4.54-2.8-4.1
24061 N /11L2LIEESAIINAGAP 4.54-2.8-4.1\ + + + 7 SI Y
+ 28
2282UlC /11L2CIEESAIINAGAP 4.54-2.8-4.1+ na + + 11 SI Y
6 + 21
2282UlC 6/11L2CIEESAIIT1AGAP 4.54-2.8-4.1+ + + + 9 SI,CY
+ 26
24145 N /11L2CLIEESAIINAGAP 4.54-2.8-4.1
24145 N 6/11L2CLIEESAIINAGAP 4.54-2.8-4.1\ + + - 7 U,NY
- 28
VL2 VGPLDIVPEVADPGGPTL
2417U1N 1 4.38-3.8-5.1 _ Y
.
RPVL2VGPLDIVPEVADPGGPTL '
24191 N 1 4.38-3.8-5.1
.
RPVL2GPLDIVPEVADPGGPTL
2283U1C 1C 4.69-1.8-4.0+ na + + 11 SI Y
. + 21
RPVL2GPLDIVPEVADPGGPTL
2283U1C 1C 4.69-1.8-4.0+ + + + 9 SI,CY
. + 26
RPVL2GPLDIVPEVADPGGPTL
2274U1L 1L 4.50-2.8-4.1- na ~ ~ 8 SI,N
. -. 23 N
RPVL2PGGPTLVSLHELPAETP
2420U1N 2 4.69-1.8-4.0 - Y
.
RPVL2PGGPTLVSLHELPAETP
24105 N 2 4.69-1.8-4.0
.
RPVL2GGPTLVSLHELPAETPY
2288U1C 2C 4.69-1.8-~1.0+ + + + Y
. + 24
RPVL2GGPTLVSLHELPAETPY
2287U1L 2L 4.89-0.9-3.0+ + + + N
. + 24
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
RPVL2GGPTLVSLHELPAETPY
2287U1L.2L 4.89-0.9-3.0- - - - 26 9 N
- N
24611 C7CTL RAHYNIV'I'FAG 5.000.0 -1.9nana + 25 9 Y
na
?
24631 C7CTL RAHYNIVT'FAG 5.000.0 -1.9- + + . 24 14 SI
-
24621 L7CTL RAFIYN1VTFAG 5.651.0 -0.9- + + + 27 13 SI
-
24621 L7CTL R.~~AG 5.651.0 -0.9nana + 25 9 Y
na
?
24611 N7CTL DRAHYNIVZ'FAG 5.000.0 -1.9- + + + 26 13 SI
+
24631 N7CTL DRAHYIVIV'TFAG 5.000.0 -1.9nana + 25 9 Y
na
?
24631 N7CTL DRAHYNIVTFAG 5.000.0 -1.9 ++ + S
na
-
26195 C7CTL SAG 4.55-1.3-2.9- - na - S
? N
7CTLTR.~~AG
2428UlC 5.160.1 -2.0+ + + + 22 13 SI,
+ C
Y
7CTLTAEPDRAHYNIVTF W,
2275U L 5.160.1 _-2.0- na - 23 8 SN
1 - N
-
7CTLTAEPDRAHYNIVT'F
24031 N 5.160.1 _-2.0
7CTLTAEPDRAHYMV'TFCCKCD
22901 CL 5.701.1 _-1.2+ + + + 25 9 Y
+
7CTLTAEPDR?~IiYNIVT~'CCKCD N,M,
2276UlLL 5.701.1 _-1.2- na - 23 8 SI
- N
-
7CTLTAEPDR.AHYNIVT~'CCKC
24001 NL 5.701.1 _-1.2
LDKW LDKWAS
22891 CS 4.87-0.9_-2.1+ + + + 25 9 Y
+
LDKW LDKWAS
2277UlLS 4.87-0.9_-2.1+ + + + 24 Y
+
LDKW ELDKWAS
2013U NS 4.87-0.9_-2.+ na + 21 11 SI
1 i + Y
+
ELDKWAS Mi
LDKW xe
2013UlN 4.87-0.9-2.1nad 24 21 SI
S Up Y
LDKW ELDKWAS
2013UlNS 4.87-0.9_-2.1\ + + + 28 7 SI
+ Y
ELDKWAS Mi
LDKW xe
24135 N 4.87-0.9-2.1nad 24 21 SI
S up N
LDKW ELDKWAS
24135 NS 4.87-0.9_-2.1\ + . - 28 ? U
+ Y
+
2429U1C QELLELDKWASLW 4.59-2.7_-4.1- - - - 22 13 U
- N
QELLELDKWASLW
2271U1L 4.59-2.7_-4.1- na - 23 8 M,
- LSI
- N
24011 N EQELLELDKWASLW 4.59-2.7_-4.1na 24 21 SI
N
24011 N EQELLELDKWASLW 4.59-2.7_-4.1\ + - - 28 7 U
- N
24125 N EQELLELDKWASLW 4.59-2.7_-4.1na 24 21 SI
Y
24125 N EQELLELDKWASLW 4.59-2.7_-4.1\ - - - 28 7 U
- N
3431UlL1I23 PLGVGISGHPLLNKLDDTE4.68-1.9_-4.0- - + - 22 13 R
- N
QPLGVGISGHPLLNKLDD
2418U1N1I23 4.68-1.9_-4.0 Y
2279UlC1I23CPLGVGISGHPLLNKLDDTE4.68-1.9_-4.0+ na + 23 8 SI
+ Y
+
2286UlC1I23CPLGVGISGHPLLNKLDDTE4.68-1.9_-4.0- - - - 24 Y
-
UlL1123LPLGVGISGHPLLNKLDDTE4.87-0.9-3.0
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
36
2404UiN 1J4 GGENVPDDLYIKGSGS4.50-2.8-4.1
-
2284U1C 1J4C GENVPDDLY1KGSGS 4.50-2.8~.1 + na + 21 11 SI Y
+
+
24661 C QARILAVEAGA 4.75-0.9-2.0+ + + + 24 14 SI
+
24661 C QARILAVEAGA 4.75-0.9-2.0+ - na na24 SI
?
24651 L SPMLQARILAVEAGAGPS5.050.1 -1.0- + + + 27 13 SI
-
24651 L SPMLQARILAVEAGAGPS5:050.1 -1.0- - na na24 SI
-
24641 N QARB ALGA 4.75-0.9-2.0- + + + 26 13 SI
-
24641 N QARE-~VEAGA 4.75-0.9-2.0+ - na + 24 SI
+
QARVLAVEGQARVLAL
24671 C N AGA 4.75-0.8-2.0- + + + 25 9 Y
-
QARVLAVEGQARVLAL
24671 C N AGA 4.75-0.8-2.0nana na25 N
na
na
SPMLQARVLAVEGQARV
24681 L N EAGAGPS 5.050.2 -1.0- + + + 25 9 Y
+
SPMLQARVLAVEGQARV
24681 L N EAGAGPS 5.050.2 -1.0- - na - 25 SI
-
QARVLAVEGQARVLALE
24691 N N GA 4.75-0.8-2.0- + + + 25 9 Y
-
QARVLAVEGQARVLALE
24691 N N GA 4.75-0.8-2.0nana na25 N
na
na
OPVL2VGPLEVIPEAVDPAGSSI
2421UlN 1 4.45-3.7-5.0 _ Y
.
OPVL2PAGSSIVPLEEYPAEIP
24221 N 1 4.45-3.7-5.0
.
- OPVL2VGPLEVIPEAVDPAGSSI
24155 N 1 4.15-5.0-6.0
.
OPVL2GPLEVIPEAVDPAGSSI
2285UlC 1C 4.41-3.7-5.1+ na + 21 il SI Y
. +
+
OPVL2GPLEVIPEAVDPAGSSI
2285UlC .1C 4.41-3.7-5.1+ + + + 26 9 SI,CY
- +
OPVL2GPLEVIPEAVDPAGSSI
2272UlL .1L 4.54-2.8-4.1- na - 23 8 N, N
- - SI
-
OPVL2AGSSIVPLEEYPAEIPT
2280U1C .2C 4.45-3.7-5.1+ na + 23 8 SI Y
- +
+
OPVL2AGSSIVPLEEYPAEIPT
2280U1C .2C 4.45-3.7-5.1+ + + + 26 9 SI,CY
- +
OPVL2AGSSIVPLEEYPAEIPT N
2273U L .2L l 4.57-2.7-4.1- na - 23 8 LSI N
-
-
ROPVL2PAGSSIVPLEEYPAEIP
2408U5N _.2N 4.25-5.0-6.0 ~ _ _ _
24701 C VFV GTMDSGQTKREAGA 5.050.2 -1.0+ + + + 25 9 Y
+
24701 C VFV GTMDSGQTKREAGA 5.050.2 -1.0+ + na + 24 S
+
SPMYKGTMDSGQTKREA
24711 L VFV AGPS 7.001.1 0.0 - + + + 25 9 Y
-
SPMYKGTMDSGQTKREA
24711 L VFV AGPS 7.001.1 0.0 - + na - 24 S
-
24721 N VFV GTMDSGQTKREAGA 5.050.2 -1.0- + + - 25 9 N
-
24721 N VFV GTMDSGQTKREAGA 5.050.2 -1.0- + na + 24 S
+
PVDPRLEPWKHPGSQAG
24751 C AT1A 4.52-2.8-4.9+ + + + 24 14 SI
+
PVDPRLEPWKHPGSQAG
24751 C AT1A 4.52-2.8-4.9- + na - 24 SI
?
24741 L AT1A SPMEPVDPRLEPWKI~GS4.62-1.8-3.9- - + - 27 13 1N,
-
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
37
AGAGPS 1
SI
SPMEPVDPRLEPWKHPGS
2474UlLAT1A AGAGPS 4.62-1.8-3.9* + na na24 SI
+
24731 NAT1A PuDPRLEPWKHPGSQAGA4.52-2.8-4.9+ + + + 26 13 SI
+
24731 NAT1A PVDPRLEPWKHPGSQAGA4.52-2.8-4.9+ + na na24 SI
+
PVDPSLEPWNHPGSQAG
24781 CAT1B 4.75-0.8-2.9- + + + 24 14 SI
-
PVDPSLEPV~NHPGSQAG
24781 CAT1B 4.77-0.8-2.9- + na + 27 SI
+
SPMEPVDPSLEPWM-IPGS
24771 LATIB AGAGPS 5.050.2 -1.9- + + + 27 13 SI
+
SPMEPVDPSLEPWNHPGS
24771 LATIB AGAGPS 5.050.2 -1.9- na + 27 SI
na
?
24761 NAT1B PVDPSLEPWNHPGSQAGA4.82-0.8-2.9- + + + 26 13 SI
-
24761 NATIB PuDPSLEPV~NHPGSQAGA4.82-0.8-2.9+ na + 27 SI
na
?
24811 CAT3B SQSRGDPTGPKEAGA 4.77-0.8-2.0+ + + + 24 14 SI
+
24811 CAT3B SQSRGDPTGPKEAGA 4.77-0.8-2.0+ na + 25 SI
na
+
SPMPTSQSRGDPTGPKEA
24801 LAT3B AGPS 5.050.1 -1.0- - + - 27 13 SI
-
SPMPTSQSRGDPTGPKEA
24801 LAT3B AGPS 5.050.1 -1.0+ + na + 25 SI
?
24791 NAT3B QSRGDPTGPKEAGA 4.77-0.8-2.0+ - + - 26 13 SI
+
24791 NAT3B TSQSRGDPTGPKEAGA 4.77-0.8-2.0+ + na na25 SI
+
24841 CAT3N LPTTRGNPTGPKEAGA 4.050.1 -1.0+ + + + 24 14 SI
+
24841 CAT3N LPTTRGNPTGPKEAGA 4.050.1 -1.0+ + na + 23 SI
+
SPMPLPTTRGNPTGPKEA
24831 LAT3N AGPS 6.801.1 0.0 \ \ \ \ 27 13 N
\
SPMPLPTTRGNPTGPKEA
24831 LAT3N AGPS 6.801.1 0.0 - - na - 23 SI
-
2482I NAT3N LPTTRGNPTGPKEAGA 5.050.1 -1.0\ \ \ \ 26 13 N
\
24821 NAT3N LPTTRGNPTGPKEAGA 5.050.1 -1.0nana na23 N
na
na
NYIVICRICRIHIGPGR
UlN3 GTIR AHC 9.678.9 _6.1
24851 C3BAL GPGRAFYTTGAGA 5.020.0 -1.0nana - 25 8 S
na
24871 C3BAL GPGRAFYTTGAGA 5.020.0 -1.0- + + + 24 14 SI
-
SPMIGPGRAFYTTGAGAG
24861 L3BAL S 6.801.0 0.0 - + + + 27 13 SI
-
SPMIGPGRAFYTTGAGAG
24861 L3BAL S 6.801.0 0.0 nana + 25 8 S
na
2485I N3BAL GPGRAFYTTGAGA 5.020.0 -1.0+ + + + 26 13 SI
+
24871 N3BAL GPGRAFYTTGAGA 5.020.0 -1.0nana + 25 8 S
na
24881 C3MN GPGRAFYTZTCAGA 6.801.0 0.0 nana na24 N
na
na
24901 C3MN GPGRAFYTZTCAGA 6.801.0 0.0 - + + + 24 14 SI
-
SPMIGPGRAFYTTTCAGAG IN,
24891 L3MN S 9.302.0 1.0 - - + - 27 13 1SI
-
SPMIGPGRAFYTTICAGAG
24891 L3MN S 9.302.0 1.0 - + na ? 24 S,N
?
24881 N3MN GPGRAFYT'TICAGA 6.801.0 0.0 \ \ \ \ 26 13 N
\
2490I N3MN GPGRAFYTfKAGA 6.801.0 0.0 nana na24 N
na
na
24931 C3NIG GPGQAFYAGGAGA 4.72-1.0-2.0- + + + 24 14 IN
-
24931 C3MG GPGQAFYAGGAGA 4.72-1.0-2.0+ + na + 24 9 SI
-
24921 L3NIG DPMIGPGQAFYAGGAGA5.000.0 -1.0- - + - 27 13 SI
-
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
38
GPS
SPMIGPGQAFYAGGAGAG
24921 L3MG S 5.00 0.0 -1.0 - - na + - 24
9 SI
24911 N3MG GPGQAFYAGGAGA 4.70 -1.0 -2.0 - + + + + 26
13 SI
24911 N3MG GPGQAFYAGGAGA 4.70 -1.0 -2.0 + ,+ na + +
24 9 SI
C~sr 6t= Pr~~ In~rC~non~~~~y
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
39
EXAMPLE 4: Parvo Virus Vaccines
The general method used in Example 1 was repeated with the linear epitopes
from
parvo virus. The N-terminus of FPV, CPV and PPV VP2 contains a major
neutralizing
determinant for the virus; this is a linear epitope, present in the first 23
amino acids of the
protein. Neutralizing antibodies may be induced in animals immunized with
peptides
derived from the first 23 amino acids of VP2 (Langeveld et al., 1995; 2001).
The sequence
of the N-terminus of VP2 follows: MSDGAVQPDGGQPAVRNERATGS.
We designed a synthetic DNA sequences which would encode various portions of
the N-terminal VP2 sequence. The synthetic DNA was synthesized in
complementary
oligonucleotides, and inserted into the coat protein of TMV U1 and TMV U5.
These
sequences of the peptides were denoted Parvol; Parvo2; and Parvo3. The amino
acid
sequences of these peptides are as follows:
Parvol: MSDGAVQPDGGQPAVRNERAT (21 amino acids)
Parvo2: MSDGAVQPDGGQPAVRNERA (20 amino acids)
Parvo3: VQPDGGQPAVRNERAT (16 amino acids)
EXAMPLE 5: Determination of viral infectivity and bacterial bioburden of
recombinant
TMV particles carrying vaccine epitopes
A list of final products with titers diluents, Garner are given in Table 3.
Table 3: Papillomavirus Vaccines Final Volumes and Virus Quantities
irus Name irus
CA IgG olume otal olume usedVials VialsConcentrate
irus
(mg/mL)(mL) (mg) for dilutionsT EI ol. @-20oC
mL reatedmL
MV:CRPV2.
1 15.5 11.2 173.6 3.2 7 S 7
MV:CRPV2.
1 Alt 19.6 7 137.2 37 35
MV:HPV-11
2 8.1 35 983.5 3 60 60 32
MV:ROPV2.
6.4 57 364.8 8 50 0* 6
MV:ROPV2.
F/T 5.3 0 106 9.5 9 9 10
MV wild
a F/T 16 1 336 .5 8 68** 15.5
MV:CRPV2..6 50 30 11.2 50 51 38
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
F/T
MV:ROPV2.
1 F/T 19.3 50 965 .65 50 50 6
4/16/03
*frozen as bulk (46 mL)
** froze 24 mL as bulk
F/T = freeze thaw
Process samples and final product for bacterial bioburden were monitored by
5 aseptically plating 1 U ~1 or 100 p,l samples on bacterial nutrient agar in
a laminar flow hood.
Plates were inverted and incubated at room temperature for four days. The
bacterial colony
counts were recorded after four days. The plates were then transferred to a
33°C incubator
for a further four days, and bacterial colony counts were recorded again.
Bioburden assays
for final fill samples were run in duplicate and the results averaged.
Bioburden decreased
l0 with each sequential processing step from 420 - 3800 colony forming units
(CFU) per ml in
the initial homogenate, to 0 - 130 CFU/ml in the final (concentrated) virus
preparations.
The final dilute vaccines had no detectable bioburden in either the untreated
or BEI-treated
samples.
TMV infectivity was determined using a local lesion host Nicotiana tabacum
var.
15 Xanthi, culrivar "Glurk". This assay is accepted by the United States
Department of
Agriculture as a method for evaluating tobacco mosaic virus infectivity. The
limit of
detection for the Glurk assay is 10 pg/~1. Glurk plants were sown into flats
and transplanted
into 3.5 inch pots at two weeks post sowing. The Glurks were prepared for
inoculation by
numbering the leaves to be inoculated with a lab marker on the upper distal
portion of the
20 leaf. A small amount of silicon carbide (400 mesh) was sprinkled on each
numbered leaf.
One hundred microliters of the sample to be assayed was dispensed onto the
upper surface
of the appropriate leaf and gently spread over the entire surface of the leaf.
Glurk plants
were scored 4 to 6 days post-inoculation by counting the number of local
lesions that had
formed on the leaf surface (see Figure 3). The Glurk local lesion assays were
run in
25 triplicate and the results were averaged. The infectivity of the final
vialed vaccine products
is summarized in Table 4; where the average number of local lesions for the
10'3 dilution
was used to derive the infectivity measurement.
Table 4: Infectivity of TMV:papillomavirus epitope vaccines
Construct Virus Name Number of Local Number of Local
Lesions
Name per ml in the Lesions per ml
Untreated in the
Vaccine Sample BEI-treated Vaccine
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
41
Sample
pLSB2283 TMV:CRPV2.1 2.71 x 10" 0
pLSB2288 TMV:CRPV2.2 1.52 x 10" 0
pLSB2285 TMV:ROPV2.1 4.0 x 10' 0
pLSB2280 TMV:ROPV2.2 Tntc; 1.0 x 10'* 10
pLSB2282 TMV:HPV-11 5.6 x 10' 0
L2
Wild type TMV 1.37 x 10" 0
TMV
* figure derived from 10~' dilution; for all other assays the results from the
10-' dilution
point is depicted.
These results demonstrate that treatment with BEI is an effective means for
inactivation of the infectivity of tobacco mosaic virus vaccines.
All of the vaccine products were analyzed for endotoxin using the Associates
of
Cape Cod gel clot assay. Additional release testing was done on all of the
final vaccine
preparations, which included concentration determination by BCA assay, as well
as amino
acid analysis by post column derivitization, SDS-PAGE for purity assessment
and
concentration, molecular weight determination by MALDI-TOF, tryptic MALDI-TOF
if
t0 required, pH and appearance.
There was no endotoxin detected in any of the BEI-treated samples after
testing
multiple dilutions of the samples. Low levels of endotoxin were present in the
TMV:HPV11L2 (1 EU/dose), TMV:ROPV2.1 (2 EU/dose) and TMV:ROPV2.2 (2
EU/dose) samples, but BEI treatment apparently eliminated the reactive
endotoxin in the
LAL endotoxin assay.
Two microgram samples of final fill vaccines, both untreated and BEI treated
were
run in triplicate on 10-14% Tris-HCI SDS-PAGE gels, and stained with Coomassie
brilliant
blue. Figure 4 shows the results of these analyses.
All vaccines, with the exception of TMV:CRPV2.2, contain >90% fully intact
recombinant coat protein. MALDI-TOF analysis confirms that, in all cases, the
upper band
in the virus preparations contains the full B-cell epitope amino acid sequence
as predicted
from the DNA sequence of the clone. About half of the TMV:CRPV 2.2 vaccine is
fully
intact. MALDI-TOF analysis of tryptic fragments of the TMV:CRPV2.2 product
indicate
that the first 10 amino acids of the 14 amino acid epitope are present in the
smaller (18 096
and 17985) bands.
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
42
Membranes with TMV: papillomavirus vaccine antigens were probed with rabbit
antisera specific for rabbit or human papillomaviruses by Western blot
analysis. The results
are shown in Figure 5: there is some cross-reactivity between ROPV2.1 and
CRPV2.1. The
CRPVL2.2 sera reacts only weakly to the vaccine antigen, but all other sera
react
specifically with the vaccines.
Preliminary immunogenicity testing of the papillomavirus:TMV epitope fusions
was
performed to ensure that appropriate antibody responses could be induced by
immunization
of animals with the vaccines, and to determine what, if any, effect BEI-
inactivation of the
TMV virions would have on the immunogenicity of the recombinant viruses. Four
to five
l0 week old, female BALB/c mice were used to assay immunogenicity of the
vaccines, and to
compare the immunogenicity of BEI-inactivation TMV preparations with untreated
controls. In addition, we immunized a small number of female guinea pigs to
confirm that
the vaccines were immunogenic in more than one species of animal, and also to
generate
antisera that could be used in in vitro virus neutralization studies (to be
performed at
Pennsylvania State University).
Four animals per group received a dose of lOp.g of the TMV vaccine product,
administered subcutaneously. Vaccines were administered every second week, and
a total
of four vaccines were given. All six BEI-inactivated vaccines were
administered, and
untreated (non-BEI inactivated) versions of the TMV, TMV:CRPV2.1 and
TMV:HPV11L2
2o vaccines were given to serve as controls for the BEI-inactivated vaccines.
One further
group received a mixed vaccine series containing Spg each of TMV:CRPV2.1 and
TMV:CRPV2.2 to establish whether an immune response to two different epitopes
could be
induced with a mixed vaccine. No PBS control was used, as each vaccine could
serve as a
control for the others. Animals were bled from the tail vein, after mild
hyperthermia, nine
days after vaccines 2, 3 and 4. ELISA, using peptide-conjugated bovine serum
albumin as
the capture antigen, determined antibody titers. Rabbit polyclonal sera
specific for the
peptide epitopes were provided by Neil Christensen, and served as positive
controls, and
tittering standards on ELISA plates. The rabbit sera used as positive control
were: HPV1/11
NC25 C000840; CRPVL2.1 B0229; CRPVL2.2 B0225; ROPVL2.1 B0219 and ROPVL2.2
B0220. For comparison of ELISA titers with the rabbit sera, a dilution of the
rabbit sera
was chosen, and arbitrarily set to 1. The mouse antibody titers were expressed
as a unit of
the rabbit sera. The subclasses of antibodies of the IgG isotype were measured
with
secondary antibodies specific for mouse IgGI or IgG2.
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
43
Figure 6 shows a scatter plot of antibody responses of all vaccinated animals
to the
peptide antigen; Figure 7 shows the same data in bar graph format, with error
bars
indicating 95% confidence intervals. The X axis standard is normalized to the
various
rabbit positive control sera, where 1 unit is the OD obtained for a 1:1000
dilution. This
gives some indication of the range of responses seen in each group, relative
to the positive
control sera. The responses to different antigens are obviously impossible to
compare, since
the antibody titer in the positive control sera are not standardized to each
other. However,
the data show the variability we observed in immune response, and the
magnitude of the
response relative to the rabbit control sera supplied by Neil Christensen
(Pennsylvania State
1 o University, Hershey PA), at a 1:1000 dilution. On the Y-axis, the
different experimental
groups are listed, with the prefix B- indicating BEI-inactivated samples, and
no prefix
indicating untreated samples. Peptide-BSA conjugates were used as coating
antigens,
except for the TMV samples, where wild type TMV was used. For the mixed
vaccine
(CRPV2.1 + CRPV2.2), CRPV 2.1 peptide was used as the coating antigen when the
label
indicates CRPV2.1 first; and vice-versa.
Figure 8 shows an analysis of the antigen-specificity of sera from vaccinated
animals. Pooled sera were reacted with plates carrying all of the different
peptide antigens.
The antibodies appear very specific, in all cases, with no, or very little
cross-reactivity
between antigens.
The effect of BEI inactivation of TMV peptide vaccines, with untreated samples
was
compared. The data depicted in Figure 9 show that the immune response of
animals
vaccinated with BEI-inactivated TMV:CRPV2.1 and untreated TMV:CRPV2.1 was
qualitatively similar. Likewise, animals vaccinated with BEI-inactivated
TMV:HPV6/11L2
and the untreated version reacted similarly. In Figure 8 we show a comparison
of the IgG
subtype profile in pooled sera from animals vaccinated with all of the
vaccines. Note that
BEI inactivation of the CRPV2.1 and HPV6/11 vaccines seemed to have no major
effect on
the quality of the immune response, as measured by IgG subtype.
An immunogenicity study in guinea pigs was performed in addition to the mouse
study described above. A total of six animals were used in this study; two
animals each
3o received the TMV:CRPV2.1; TMV:HPV6/11; and mixed TMV:CRPV2.1 plus
TMV:CRPV2.2 vaccines. The dose of vaccine was 100 pg, administered every
second
week. A total of four vaccines were given. Each dose was administered
subcutaneously, at
four locations on the animal's back. Animals were bled one week post vaccine 3
(bleed 1)
and one week post vaccine 4 (bleed 2). A terminal bleed was collected nine
days after
vaccine 4.
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
44
The ELISAs were performed in the same way as for the mouse study. Figure 11
shows the antibody titer obtained for each individual animal after vaccine 3
(left) and after
vaccine 4 (right). We note that, as for the mouse vaccinations, the anti
CRPV2.2 peptide
response was very low, and only marginally above background. It is possible
that in this
vaccine, which contained more than 50% cleavage, a new epitope comprising the
part of the
TMV coat protein and part of the first 10 amino acids of the CRPV2.2 peptide
is recognized
and is dominant over the authentic CRPV2.2 epitope. These results argue for
elimination of
the TMV:ROPV2.2 vaccine from subsequent studies.
The titer of the CRPV2.1 and HPV6/11 peptide antibodies was significantly
higher
1o in the Guinea pig sera in comparison with the BALB/c mouse sera. In all
cases, both guinea
pigs responded well to the vaccine; apparently well within a Log of the rabbit
titer. It is
worthwhile noting that the mice received 1/10 of the vaccine dose that the
guinea pigs
received, and that the higher dose could have had some positive effect on the
immune
response observed in the guinea pigs.
The IgG subtype analyses presented in Figure 10 show that the guinea pigs
responded similarly to the mice to the vaccines: with a balanced, but
apparently Th2-
dominant response.
Bleeds 1 and 2 and terminal bleeds from all the guinea pigs, and terminal
bleeds
from highest mouse responder in each group are available for CRPV and HPV6 or
HPV 11
2o neutralization assays.
We investigated whether sera from animals immunized with TMV virions
displaying the HPV6/11 L2 epitope (LIEESAIINAGAP) could recognize the HPV 16
L2
epitope sequence LVEETSFIDAGAP, conjugated to BSA, and used as a coating
antigen on
ELISA plates. Figure 12 shows that, indeed sera from the guinea pigs immunized
with the
TMV virions displaying the HPV6/11 L2 epitope (LIEESAIINAGAP) specifically
recognized the heterologous HPV 16 L2 peptide sequence (LVEETSFIDAGAP). These
data are shown in Figure 13, and indicate that immunization with TMV virions
displaying
the HPV6/11 L2 peptide sequence may function as prophylactic vaccines that can
induce
broadly neutralizing papillomavirus L2-specific antibodies. The homology
between the
3o HPV6/11 L2 epitope, the HPV-16 L2 epitope and the CRPV2.1 epitope are shown
in Figure
14.
EXAMPLE 6: Carner Rotation to Improve Immunolo;~ical Responses to Peptide-
Based
Vaccines
Virus like particle (VLP) -based vaccines can carry specific antigens and to
be
particularly effective in inducing humoral, and sometimes, cellular immune
responses. It is
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
now well established that peptides are most efficiently presented to the
mammalian immune
system in a highly ordered, repetitive, quasicrystalline array as provided by
a VLP structure
(Bachmann et al., 1993; Savelyeva et al., 2001). By their structure, VLPs are
capable of
stimulating proliferation of dendritic cells and other antigen presenting
cells resulting in
5 strong immunological responses thus producing protective immunity and even
breaking
tolerance for self antigens (Savelyeva et al., 2001; Fitchen et al., 1995).
This structural
presentation of antigens appears to be critical for induction of strong Thl or
Th2 responses
(including antigen specific CTL responses and long-lived B cell memory) and
cannot be
replicated by soluble proteins or randomly conjugated carriers, such as KLH
(Storm et al.,
l0 2002; Nicholas et al., 2002). These results have led to a great deal of
interest in VLP
epitope display systems for induction of pathogen-specific antibodies for
protection against
infectious disease, as well as for induction of peptide-specific CTL responses
in
immunotherapy of cancer and chronic infectious diseases. There are many
candidates for
VLP technologies, but hepatitis B core antigen (HBcAg) and papillomaviruses
represent
15 well-established methodologies for recombinant production of VLP-epitope
display.
HBcAg VLPs are produced recombinantly in E. coli systems and are effective
tools for VLP
display (Bachman and Kopf, 2002). Purification of endotoxin-free structures is
a challenge
from such systems. In addition, the rate of successful expression of epitopes
genetically
fused to these structures is highly variable. Some groups have addressed this
issue by
2o resorting to in-vitro methods for conjugating synthetic peptides to VLPs,
but these
methodologies do not necessarily replicate the structural advantages of native
VLP
structures, and are technically challenging and expensive to perform,
especially at large
scale. Preexisting immunity can blunt immune responses to VLP carriers. Da
Silva et al.,
(2001) reported that preexisting neutralizing antibodies to human
papillomavirus L1 virus
25 like particles limit the effectiveness of vaccines that use this carrier
for subsequent
inoculations. It should be noted that significant preexisting immunity exists
in the human
population for these viruses (up to 20% by some estimate).
The tobamovirus family, including TMV, offers the tools for building a robust
epitope display vaccine platform. Each of the 13 tobamovirus species encodes a
coat
3o protein with similar structural folding (Stubbs, 1999). Each coat protein
exhibits surface
exposed N and C termini (extreme end and upstream of terminal GPAT motif) and
a single
surface-exposed loop ("60's loop) that have been shown experimentally to
tolerate insertion
of peptide sequences (Figure 1; see references within 1). Although conserved
in overall
structure, TMV strains U1, U5, cucumber green mild mottle virus (CGMMV), and
ribgrass
35 mosaic virus (RMV) are all immunologically distinct, while TMV U1 and ToMV
are
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
46
immunologically similar (Jaegle and Van Regenmortel, 1985; Gibbs 1999; 1997).
Studies
of mammalian immune responses to tobamoviruses pioneered our understanding of
host
responses to virus structures and have continued for over 60 years (Van
Regenmortel,
1999). Extensive studies by phytopathologists determined that mammals
immunized with
tobamoviruses produce antibodies with little cross-reactivity with other
tobamovirus coat
proteins. This structural conservation, coupled with immunologic distinctness,
provides a
unique opportunity for deriving a platform of vaccine protein scaffolds that
share similar
biochemical and purification properties.
Display of peptides on TMV VLPs may be used for the induction of neutralizing
1o responses to biodefense related pathogens was illustrated by VLP vaccine
candidates
generated against the filovirus pathogen Ebola. Additional biodefense related
epitopes have
been identified for bacterial and viral pathogens and include the Rift Valley
Fever
neutralization epitope KGTMDSGQTKREL bound by protective Mab 4D4 (Keegan and
Collett, 1986; London et al., 1992). We have also made the TMV virions
displaying
peptides specifically binding neutralizing antibodies against the Ebola virus
(Wilson et al.,
2000). The minimal consensus sequence, underlined, represents the common
sequence
found on two adjacent overlapping peptides that were bound by the neutralizing
MAb:
1. Ebola glycoprotein 389-405 HNTPVYKLDISEATQVE
2. Ebola glycoprotein 401-417 ATQVEOHHRRTDNDSTA
zo 3. Ebola glycoprotein 477-493 GKLGLITNTIAGVAGLI
Fusion proteins of these minimal consensus peptides were generated at the N-
terminal, 60's loop, and near the C-terminal of the TMV U1 coat protein using
the general
techniques above.. The solubility of peptides fused to the coat proteins
extracted from N.
benthamiana plants inoculated with infectious transcripts is shown in Table 6
and Figure
15. The virions that remain soluble in aqueous solutions differ in terms of
the absolute yield
of recombinant virus recovered from infected tissues, and the optimal buffer
extraction
conditions necessary for extraction. For example, the epitope GP1-481 fused to
N-terminal
of coat protein has a slightly lower yield compared to the same epitope fused
near the C-
terminus of the TMV U1 coat protein. The majority of the virion with an N-
terminal GP1-
481 fusion is soluble in TRIS-Cl buffer (pH 7.5), whereas the virion carrying
the same
fusion near the C-terminus was soluble at either pH 5.0 or 7.5. As expected,
the negative
control samples did not have any SDS-PAGE band near the expected size of the
coat protein
fusions. The integrity of the fusions was further confirmed by MALDI-TOF mass
spectroscopy (Table 6). Viral constructs with the epitope fused in the 60's
loop caused
necrotic lesions on N. benthamiana plants and often resulted in insoluble
recombinant coat
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
47
protein. Approaches to overcome this problem include testing these constructs
in other
Nicotiana species or changing the amino acid sequences surrounding the epitope
to restore
the native charge of (-3) on the TMV U1 coat protein. From these data, it is
clear that
peptide epitopes bound by antibodies capable of neutralizing Ebola virus and
protecting
mice from infection were readily displayed on the surface of TMV virions.
The cloning vectors for fusing peptides to various tobamovirus coat proteins
were
constructed using unique restriction endonuclease sites, PCR-based genetic
fusions and
insertion cloning procedures. For example, for displaying epitopes on the U1
coat protein,
vectors possess unique NcoI and NgoMIV restriction sites at four locations, N-
terminal, C-
to terminal, C-terminal upstream of the GPATmotif, and within the surface
exposed loop
region. These linearized sites can readily accept any hybridized
oligonucleotides (coding
for epitopes) with the same overhangs. We will use the same strategy to
prepare cloning
vectors for the other three coat proteins. Recombinant virus clones were
transcribed and
capped in-vitro, and the infectious transcripts were inoculated onto plants:
N. benthamiana
or N. excelsiana. Infections of plants were scored visually between S and 10
days post
inoculation.
A low pH buffer (50 mM sodium acetate, 5 mM EDTA, pH 5.0) was very useful for
initial extraction of virus coat protein fusions since many host proteins are
insoluble at this
pH and so coat protein bands are easily visible in extracts run in SDS-PAGE
gels and
2o stained with Coomassie Brilliant Blue. However, several coat fusions were
not soluble
under these conditions. Some of these were selectively solubilized from
insoluble plant
material by resuspension of the material in 50 mM Tris-HCl pH 7.5 buffer
followed by
centrifugation to remove insoluble materials. The virus was purified by
differential
centrifugation followed by precipitation of virus by treatment of supernatants
with 4%
polyethylene glycol in the presence of 0.7M NaCI. Accurate sizing of coat
protein subunits
was possible by MALDI-TOF mass spectrometry, and that this methodology was
very
useful for verification that fusion proteins were intact, and not
proteolytically cleaved.
When further verification of protein identity was required, the band can be
excised from
gels and subject to digestion with trypsin followed by MALDI-TOF for
verification that the
3o predicted Cryptic digest matches the observed pattern of ion masses in the
MALDI-TOF
spectrum. Recombinant fusions that are soluble in either pH 5 or pH 7.5, can
be readily
manufactured for vaccine investigations.
Peptide display vaccines applied with a single carrier can induce a response
primarily to the carrier protein, rather than effectively boosting immune
responses to the
peptide antigen. To discourage such carrier-specific boosting responses, a
carrier rotation
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
48
approach to vaccines was used. In this case, the peptide immunogen, such as
Ebola
neutralizing peptide GP1-393 (VYKLDISEA), was fused to the surface of the coat
protein
of TMV U1 and TMGMV or RMV coat protein. The initial immunization was given
with
the TMV U1-peptide vaccine and the boosting immunization will be given 2-4
weeks later
using the TMGMV or RMV fusion. In this manner, the immune system of the
immunized
individual sees only one consistent linear epitope, and that is for the
peptide immunogen.
This enhances the level of immune response and the specificity of the immune
response
over that available for a vaccine using a single carrier in repeated
immunizations. The
principle is useful for any peptide or protein antigen which is presented with
a non-specific
1o antigen. The booster effect of multiple vaccinations is then directed only
to the specific
peptide immunogen, not to the carrier molecule or portion or the carrier
molecule.
This concept was extended to a multi-peptide immunogen vaccine. In this case,
a
set of peptide immunogens was employed in a vaccine to induce a wider anti-
pathogen
response against a single organism (e.g. Ebola: peptides GP1-393, 405, 481).
In contrast, a
set of peptide immunogens to different organisms can be applied in a single
vaccine to
induce an effective immune response against more than one organism
simultaneously (e.g.
Ebola, GP1-393 and RVFV 4D4 peptide). Each is fused to the surface of the coat
protein
of TMV U1 and TMGMV or RMV coat protein. The initial immunization is given
with the
TMV U1-peptide vaccines and the boosting immunization will be given 2-4 weeks
later
2o using the TMGMV or RMV fusions. In this manner, the immune system of the
immunized
individual sees only the two (or more) consistent linear epitopes that are for
the multi-
pathogen peptide immunogens. This approach enhances the level of immune
response and
the specificity of the immune response over that available for a vaccine using
a single
carrier in repeated immunizations.
In either situation or carrier rotation, the epitope peptide may be fused to
the Garner
antigen or it may be mixed therewith to present or enhance the immune
response. Plural
epitope peptides may be bound to the same or different carrier antigens
simultaneously. In
situations where many immunizations to the same peptide epitope are desired,
such as for
allergy treatments, this method is particularly useful. Also, when one does
not know which
3o peptide epitope is best to use for immunization, to produce neutralizing
antibodies for
example, one may prepare many vaccine preparations without concern for the
Garner
antigen becoming immunodominant.
A murine model for Ebola filovirus is an example of the test systems that may
be
used to for such a rotating carrier approach. Murine test hosts were of the
Balb C or
C57B1/6 mouse strains. Mice were immunized with VLP peptide vaccines (a dose
range (2
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
49
and 10 mcg) fused to TMV U1 (first immunization), TMGMV or RMV (second and/or
third
immunization). Peptides were chosen from the group (peptides GP1-393, 405,
481) and
PBS buffer was used a negative control. Mice were immunized at two week
intervals.
Sera from each mouse, pre-immune and two weeks following each immunization,
were
screened against each the VLP vaccines displaying the cognate peptide on the
surface of
either TMV U1, TMGMV or RMV by ELISA. MAbs that recognize different Ebola
antigens (6D8-1-2, 13F6-1-2 and 12B5-1-1, kindly provided by Dr. Mary Kate
Hart, US
AMRIID) recognized the cognate linear neutralizing epitopes on the different
carriers with
peptides. ELISA assays were completed as described (40). Briefly, Nunc
Maxisorp 96 well
to plates were coated overnight with 5 p.g/ml of target antigen in carbonate
buffer. Targets
included cognate peptide conjugated to BSA, TMV-Ebola peptide fusion, TMV-
RVFV
peptide fusion, and TMV. Plates are washed, blocked, and incubated with a 1:3
serial
dilution of sera from immunized or control mice at a starting dilution of
1:10. Plates were
then washed, and incubated with an anti-mouse-HRP conjugate. Following
secondary
incubation, plates were washed, and developed by standard procedure, and read
on a
Molecular Devices Gemini plate reader at 405 nm. The level of bound antibodies
were
determined by comparing to the known amount of neutralizing MAb.
Sera derived from immunized mice were tested for their ability of these immune
sera
to inhibit or alter Ebola virus plaque formation. Sera showing the most robust
anti-peptide
2o immune responses were used. Neutralization assays were carried out as
described in
Wilson et al., (2). Briefly, fourfold serial dilution of sera was mixed with
100 pfu of
murine-adapted Ebola Zaire at 37°C for 1 hour in the presence or
absence of 5% guinea pig
complement (Accurate Scientific) and used to infect Vero E6 cells. Cells were
overlaid
with agarose and a second overlay with S% neutral red added 6 days later.
Plaques were
counted on the 7th day. Neutralization titers were determined to be the last
dilution of the
sera that reduced the number of plaques by 80% compared with control wells
(sera from
PBS or RVFV peptide immunized mice).
The Ebola peptide immunogens fused to tobamovirus VLP structures can be tested
for efficacy in an Ebola challenge model. Ten mice per treatment will be
evaluated in each
of two experiments. C57BL/6 mice will be vaccinated at two doses at 4 week
intervals and
challenged intraperitoneally with 1000 pfu of mouse-adapted Ebola Zaire virus
(2; 43) one
month after the final immunization. Mice will be observed daily for signs of
illness for 28
days after challenge.
EXAMPLE 7: Carrier Rotation to Improve Immunolo;~ical Responses to HIV
Vaccines
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
Ideally, a vaccine designed to protect against infection with human
immunodeficiency type 1 (HIV-1) will induce sterilizing immunity against a
broad range of
virus variants. However, generation of broadly-neutralizing antibodies (Nabs)
by
vaccination, let alone natural infection, has proven nearly impossible thus
far. There have
5 been some notable advances in development of vaccine regimens that are able
to generate
significant levels of protection against development of AIDS in non-human
primate models
(reviewed in McMichael et al., 2002; Letvin et al., 2002; Robinson 2002;
Letvin 2002).
These vaccines allow animals to control viral challenge by strong priming of
virus-specific
CD8+ T-cells (cytotoxic T cells, CTLs). However, a CTL response alone cannot
prevent
1 o infection, and mechanisms to induce Nabs that will neutralize a wide range
of isolates
remains a vital goal, especially in light of the fact that viral escape from
vaccine-induced
CTL control can sometimes occur (Barouch et al., 2002). The Env spikes on the
surface of
the HIV-1 virion are the primary target for antibody-mediated neutralization.
However, the
Env proteins of HIV-1 are poorly antigenic, and generation of Nabs is
difficult to achieve,
15 probably because functionally important domains of the proteins are
obscured by protein
folding and carboydrate chains. Nevertheless, many infected people do mount a
Nab
response that is generally highly specific to the autologous virus, and not
cross-neutralizing.
This is not surprising given the phenomenal sequence and structural variation
that is present
in the Env proteins. However, a rare subset of infected individuals do produce
broadly
2o neutralizing Abs, which gives hope that induction of sterilizing immunity
is possible.
The envelope proteins of T-cell line-adapted (TCLA) strains of HIV-1 elicit
Nabs
that mostly target linear epitopes in the third variable cysteine loop (V3
loop) of gp120, a
region that is involved in co-receptor binding and hence vital for virus
entry. Subtype C
isolates of HIV-1, which infect more people worldwide than any other subtype,
have
25 relatively low level of sequence variation in the V3 loop (Engelbrecht et
al., 2001; Bures et
al., 2002). However, neutralization of subtype C virus by V3 loop Abs is not
extremely
efficient in vitro, perhaps reflecting poor immunogenicity of epitopes in this
region (Bures
et al., 2002). There is concern that the V3 loop may be hidden in the native
gp120 structure
and not accessible to the immune system, and therefore that generation of V3-
specific Nabs
3o will be difficult with gp120 subunit vaccines. However, the V3 loop is
vital for viral entry,
and so significant levels of V3 loop-targeted Nabs should help prevent
transmission of HIV-
1.
To date, six human monoclonal antibodies (Mabs) have been described that are
capable of neutralizing a broad spectrum of HIV-1 variants in vitro. Three of
these
35 (IgGbl2; 2612 and 2F5) were described several years ago, and lend insight
into the
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
51
domains of the Env proteins that are important in viral entry, and thus for
vaccine design.
Monoclonal antibody "b 12" recognizes a conformational epitope in the CD4
binding site of
gp120; 2612 recognizes a discontinuous epitope in the C2-V4 region of gp120
that includes
N-glcyosylation sites, and 2F5 maps to a linear epitope (ELDKWA) in the
membrane-
s proximal ectodomain of gp41 (D'Souza et al., 1997). Recently, two broadly
neutralizing
monoclonal antibodies 4E10 and Z13 were shown to recognize a continuous
epitope with
core sequence NWFDIT, just C-terminal to the 2F5 recognition sequence
(Stiegler et al.,
2001; Zwick et al., 2001). This strongly indicates that the membrane proximal
region of
gp41 plays a critical role in virus entry. Another recently described
monoclonal Fab was
1o selected for binding to gp120-CD4-CCRS complexes, and also displays a broad
neutralization phenotype (Moulard et al., 2002).
Passive transfer studies have shown that neutralizing Mabs are able to confer
concentration-dependent sterilizing immunity to virus challenge by
intravenous, oral and
vaginal routes in Rhesus macaques. It is encouraging that the mAbs tested
display
15 significant synergy in their neutralization activity: this will reduce the
minimum antibody
concentration that is required for effective neutralization (reviewed in
Mascola, 2002; Xu et
al., 2002). A recent publication (Lewis et al., 2002) demonstrates that MAb
neutralizing
activity can also be generated in vivo: in mice that expressed the gene for
b12 from a
recombinant adeno-associated virus vector. These studies on neutralizing Mabs
have
2o helped to demonstrate that we should be able to achieve significant levels
of protection
against HIV-1 infection and reduced rates of transmission of virus, if a way
is found to
induce robust production of Nabs in vaccinated animals and is incorporated
into a vaccine
regimen that includes strong priming of a CTL response.
In the light of the disappointing performance of whole Env-based vaccines, and
the
25 problems associated with poor immunogenicity of Env subunit vaccines,
several studies
have focused on the use of immunogens based on domains of Env proteins that
are
presumed targets for Abs. Data presented by Letvin et al. (2001 ), that showed
that
antibodies induced against the V3 loop could provide partial protection
against challenge
with primary isolate-like SHIV-89.6 in Rhesus macaques. Efforts at generation
of
30 neutralizing antibodies with immunogens containing the core linear epitope
recognized by
the 2F5 antibody have been generally disappointing, with only non-neutralizing
antibodies
being produced (Ferko et al., 1998; Echart et al., 1996). However, there is
one notable
exception: recently, Marusic et al. (2001) showed that virus-like particles of
the flexuous
plant virus potato virus X (PVX) dispaying the 2F5 ELDKWA epitope could induce
high
35 levels of HIV-1 specific IgG and IgA in mice immunized with the recombinant
virus-like
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
52
particles (VLPs). This immunogen was able to induce production of human HIV-1
specific
neutralizing antibodies (measured by in vitro inhibition of syncytium
formation) in severe
combined immunodeficient mice reconstituted with human periferal blood
lymphocytes
(hu-PBL-SCID) that had been immunized with human dendritic cells (DCs) pulsed
with the
PVX-2F5 VLPs. These authors speculate that presentation of the ELDKWAS
sequence in
a highly repetitive fashion on the surface of the PVX virion rendered the
sequence highly
immunogenic, and thus were able to generate Nabs.
Until the recent discovery of the 4E10/Z3 human Mab, 2F5 was the only human
Mab that appeared to recognize a linear epitope, and so peptides that could
mimic the
l0 neutralizing epitope of b12 and 2612 were not available for testing as
potential
immunogens. However, a linear peptide mimotope of the b 12 epitope has
recently been
discovered using phage peptide display technology (Zwick et al., 2001). This
peptide'
(B2.1) appears to bind best to b12 when presented as a disulphide-linked
homodimer on the
surface of the phage. This phage particle is being optimized for use as an
immunogen.
Scala et al. (1999) selected epitopes from libraries of peptides displayed on
the surface of
filamentous phage particles with sera from HIV+ patients, both from long term
infected non-
progressor donors and from donors who had progressed to AIDS illness. Five
epitopes,
presumed to be mimotopes of Env-specific neutralizing epitopes, were able to
induce
production of antibodies that neutralized TCLA HIV-1 strains IIIB and NL4-3,
as well as
the primary isolate ADB, but this less strongly than the TCLA strains (Scala
et al., 1999).
Subsequently, these authors showed that sera from individuals infected with
all group M
HIV-1 subgroups were able to recognize the phage-displayed mimotopes (Chen et
al.,
2001). Rhesus monkeys were immunized with phage particles displaying the five
epitopes
that had shown potentially protective immune responses in mice, and challenged
with
pathogenic SHIV-89.6PD. While the immunized animals were not protected from
SHIV
infection, there was evidence of significant control of the challenge virus
and the monkeys
were protected from progression to AIDS. These results show similar levels of
control to
vaccines designed to generate virus-specific CTLs and infer that the antibody
response was
able to control viremia in the challenged animals. A recent publication (He et
al., 2002)
3o described successful isolation of a number of human Nabs from XenoMouse
immunized
with gp120 derived from a primary Subtype B isolate (SF162). The authors noted
potent
neutralizing activity against the autologous virus isolate, and reactivity
against both R5 and
X4 isolates in Subtype B. The Nabs mapped to novel epitopes in domains known
to possess
neutralizing epitopes: V2-, V3- and CD4-binding domains of gp120, as well as
in the C-
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
53
terminal region of the V 1 loop. Apparently, several Nabs recognize linear
epitopes that
now warrant further investigation as peptide immunogens.
Some non-structural HIV-1 proteins, particularly Tat and Vpr, are found in the
senzm of infected individuals, and exert biological function, resulting in
immunodeficiency
and disease. The Tat protein is required for HIV-1 replication and
pathogenesis. It is
produced early in the viral life cycle. In the nucleus of the infected cell,
it interacts with
host factors and the TAR region of the viral RNA to enhance transcript
elongation and to
increase viral gene expression (Jeang et al., 1999). Tat also is also found
extracellularly,
where it has distinct functions that may indirectly promote virus replication
and disease,
t o either through receptor mediated signal transduction or after
internalization and transport to
the nucleus. Tat suppresses mitogen-, alloantigen- and antigen-induced
lymphocyte
proliferation in vitro by stimulating suppressive levels of alpha interferon
and by inducing
apoptosis in activated lymphocytes. In vivo, it is thought that Tat may alter
immunity by
upregulating IL-10 and reducing IL-12 production, or through its ability to
increase
chemokine receptor expression (Gallo et al., 2002; Tikhonov et al., 2003).
Antibody
production against Tat has, in some cases, correlated with delayed progression
to AIDS in
HIV-1 infected people (Gallo et al., 2002). Recently, Agwale et al. (2002)
showed that
antibodies induced in mice against a Tat protein subunit vaccine could negate
the immune
suppression activities of Tat in vivo. Subsequently, Tikhonov et al. (2003)
identified linear
epitopes on Tat that were reactive with Tat-neutralizing antibodies produced
in vaccinated
Rhesus macaques. From these data it is clear that antibodies that target the N-
terminus, an
internal basic domain, and the cell-binding domain of Tat (containing the
integrin-binding
motif "RGD") can neutralize the extracellular version of Tat, and reduce the
negative
impact of Tat on the immune system. These linear epitopes are thus interesting
targets for
both prophylactic and therapeutic vaccines against HIV-1 and AIDS.
As in the examples above, peptide epitopes were prepared in TMV coat proteins
and
produced as above. In Table 7, a list of peptides that have been displayed on
the surface of
TMV U1 and/or US virions is displayed. The expression, extraction and
solubility data for
these recombinant viruses is summarized in Table 8.
EXAMPLE 8: Veterinary Parvovirus Vaccines
Parvoviruses that are associated with enteric disease in domestic cats, dogs,
mink
and pigs are closely related antigenically, with different isolates diverging
less than 2% in
the sequence of the viral structural proteins. Vaccination with killed or live-
attenuated
parvovirus protects animals against infection by Feline panleukopenia virus
(FPV), canine
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
54
parvovirus (CPV), mink enteritis virus (MEV) and porcine parvovirus (PPV).
However,
maternal antibodies neutralize the vaccine, making it ineffective in animals
that have not
been weaned. Subunit vaccines might overcome this limitation, and provide
useful
alternatives to conventional vaccines.
s The N-terminus of FPV, CPV and PPV VP2 contains a major neutralizing
determinant for the virus; this is a linear epitope, present in the first 23
amino acids of the
protein. Neutralizing antibodies may be induced in animals immunized with
peptides
derived from the first 23 amino acids of VP2 (Casal et al., 1995; Langeveld et
al., 2001).
The sequence of the N-terminus of VP2 follows: MSDGAVQPDGGQPAVRNERATGS
t0 We designed a synthetic DNA sequences which would encode various portions
of
the N-terminal VP2 sequence. The synthetic DNA was synthesized in
complementary
oligonucleotides, and inserted into the coat protein of TMV U1 and TMV U5, as
depicted in
Figure 2. These sequences of the peptides were denoted Parvol; Parvo2; and
Parvo3. The
amino acid sequences of these peptides are as follows:
15 Parvol: MSDGAVQPDGGQPAVRNERAT (21 amino acids)
Parvo2: MSDGAVQPDGGQPAVRNERA (20 amino acids)
Parvo3: VQPDGGQPAVRNERAT (16 amino acids)
These epitope peptide vaccines are then used as in the examples above
It will be understood that various modifications may be made to the
embodiments disclosed
2o herein. Therefore, the above description should not be construed as
limiting, but merely as
exemplifications of preferred embodiments. Those skilled in the art will
envision other
modifications within the scope and spirit of the claims appended hereto.
All patents and references cited herein are explicitly incorporated by
reference in
their entirety.
25 References:
Agwale SM, Shata MT, Reitz MS Jr., Kalyanaraman VS, Gallo RC, Popovic M, Hone
DM.
(2002) A Tat subunit vaccine confers protective immunity against the immune-
modulating activity of the human immunodeficiency virus type-1 Tat protein in
mice. Proc. Natl. Acad. Sci. USA 99:10037-41.
30 Alani RM, Munger K (1998) Human papillomaviruses and associated
malignancies.
Journal ofClinical Oncology 16:330-337.
Bachmann MF, Kopf M. (2002) Balancing protective immunity and immunopathology.
Curr Opin Immunol. 14:413-9.
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
Bachmann MF, Rohrer UH, Kundig TM, Burki K, Hengartner H, Zinkernagel RM.
(1993)
The influence of antigen organization on B cell responsiveness. Science.
262:1448-
S1.
Balaram P, Nalinakumari KR, Abraham E, Balan A, Hareendran NK, Bernard HU,
Chan
SY. (1995) Human papillomaviruses in 91 oral cancers from Indian betel quid
chewers--high prevalence and multiplicity of infections. Int J Cancer. 61:450-
4.
Barouch DH, Kunstman J, Kuroda MJ, Schmitz JE, Santra S, Peyerl FW, Krivulka
GR,
Beaudry K, Lifton MA, Gorgone DA, Montefiori DC, Lewis MG, Wolinsky SM,
Letvin NL. (2002) Eventual AIDS vaccine failure in a rhesus monkey by viral
to escape from cytotoxic T lymphocytes. Nature. 415:335-9.
Borio L, Inglesby T, Peters CJ, Schmaljohn AL, Hughes JM, Jahrling PB< Ksiazek
T,
Johnson KM, Neyerhoff A, O'Toole T, Ascher MS, Bartlett J, Bremen JG, Eitzen
EM, Hamburg M, Hauer J, Henderson DA, Johnson RT, Kwik G, Layton M,
Lillibridge S, Nabel GJ, Osterholm MT, Perl TM, Russell P, Tonat K, for the
u5 Working Group on Civilian Biodefense. 2002. Hemorrhagic fever viruses as
biological weapons. JAMA 287:2391-2405.
Breitburd F, Kirnbauer R, Hubbert NL, Nonnenmacher B, Trin-Dinh-Desmarquet C,
Orth
G, Schiller JT, Lowy DR. (1995) Immunization with viruslike particles from
cottontail rabbit papillomavirus (CRPV) can protect against experimental CRPV
20 infection. J Virol. 69:3959-63.
Bures R, Morris L, Williamson C, Ramjee G, Deers M, Fiscus SA, Abdool-Karim S,
Montefiori DC. (2002) Regional clustering of shared neutralization
determinants on
primary isolates of Glade C human immunodeficiency virus type 1 from South
Africa. J Virol. 76:2233-44.
25 Casal JI, Langeveld JP, Cortes E, Schaaper WW, van Dijk E, Vela C, Kamstrup
S, Meloen
RH. (1995) Peptide vaccine against canine parvovirus: identification of two
neutralization subsites in the N terminus of VP2 and optimization of the amino
acid
sequence. J Virol. 69:7274-7.
Chen X, Scala G, Quinto I, Liu W, Chun TW, Justement JS, Cohen OJ, vanCott TC,
30 Iwanicki M, Lewis MG, Greenhouse J, Barry T, Venzon D, Fauci AS. (2001)
Protection of rhesus macaques against disease progression from pathogenic SHIV-
89.6PD by vaccination with phage-displayed HIV-1 epitopes. Nat Med. 7:1225-31.
Christensen ND, Dillner J, Eklund C, Carter JJ, Wipf GC, Reed CA, Cladel NM,
Galloway
DA. (1996) Surface conformational and linear epitopes on HPV-16 and HPV-18 L1
35 virus-like particles as defined by monoclonal antibodies. Virology. 223:174-
84.
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
56
Da Silva DM, Pastrana DV, Schiller JT, Kast WM. (2001 ) Effect of preexisting
neutralizing
antibodies on the anti-tumor immune response induced by chimeric human
papillomavirus virus-like particle vaccines. Virology. 290:350-60.
Dawson WO, Beck DL, Knorr DA, Grantham G (1986) cDNA cloning of the complete
genome of tobacco mosaic virus and production of infectious transcripts. Proc.
Natl. Acad. Sci. USA 83:1832-1836.
Day PM, Roden RB, Lowy DR, Schiller JT. (1998) The papillomavirus minor capsid
protein, L2, induces localization of the major capsid protein, L1, and the
viral
transcription/replication protein, E2, to PML oncogenic domains. J Virol.
72:142-50.
to D'Souza MP, Livnat D, Bradac JA, Bridges SH. (1997) Evaluation of
monoclonal
antibodies to human immunodeficiency virus type 1 primary isolates by
neutralization assays: performance criteria for selecting candidate antibodies
for
clinical trials. AIDS Clinical Trials Group Antibody Selection Working Group.
J
Infect Dis. 175:1056-62.
15 Eckhart L, Raffelsberger W, Ferko B, Klima A, Purtscher M, Katinger H,
Ruker F. (1996)
Immunogenic presentation of a conserved gp41 epitope of human immunodeficiency
virus type 1 on recombinant surface antigen of hepatitis B virus. J Gen Virol.
77:2001-8.
Einstein MH, Goldberg GL (2002) Human papillomavirus and cervical neoplasia.
Cancer
2o Investigation 20:1080-1085.
Engelbrecht S, de Villiers T, Sampson CC, zur Megede J, Barnett SW, van
Rensburg EJ.
(2001 ) Genetic analysis of the complete gag and env genes of HIV type 1
subtype C
primary isolates from South Africa. AIDS Res Hum Retroviruses. 17:1533-47.
Evans TG, Bonnez W, Rose RC, Koenig S, Demeter L, Suzich JA, O'Brien D,
Campbell M,
25 White WI, Balsley J, Reichman RC. (2001) A Phase 1 study of a recombinant
viruslike particle vaccine against human papillomavirus type 11 in healthy
adult
volunteers. Jlnfect Dis. 183:1485-93.
Fehr T, Naim HY, Bachmann MF, Ochsenbein AF, Spielhofer P, Bucher E,
Hengartner H,
Billeter MA, Zinkernagel RM. (1998) T-cell independent IgM and enduring
3o protective IgG antibodies induced by chimeric measles viruses. Nat Med.
4:945-8.
Ferko B, Katinger D, Grassauer A, Egorov A, Romanova J, Niebler B, Katinger H,
Muster
T. (1998) Chimeric influenza virus replicating predominantly in the murine
upper
respiratory tract induces local immune responses against human
immunodeficiency
virus type 1 in the genital tract. Jlnfect Dis. 178:1359-68.
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
57
Fitchen J, Beachy RN, Hein MB. (1995) Plant virus expressing hybrid coat
protein with
added murine epitope elicits autoantibody response. Vaccine. 13:1051-7.
Gallo RC, Burny A, Zagury D (2002) Targeting Tat and IFNa as a therapeutic
AIDS
vaccine. DNA Cell Biol. 21: 611-618.
Galloway DA (1996). Surface conformational and linear epitopes on HPV-16 and
HPV-18
L1 virus-like panicles as defined by monoclonal antibodies. Virology 223:174-
184.
Gaukroger JM, Chandrachud LM, O'Neil BW, Grindlay GJ, Knowles G, Campo MS.
(1996) Vaccination of cattle with bovine papillomavirus type 4 L2 elicits the
production of virus-neutralizing antibodies. J Gen Virol. 77:1577-83.
to Ghim S, Christensen ND, Kreider JW, Jenson AB. (1991) Comparison of
neutralization of
BPV-1 infection of C 127 cells and bovine fetal skin xenografts. Int J Cancer.
49:285-9.
Gibbs, A., (1999) Evolution and origins of tobamoviruses. Philosophical
Transactions of
the Royal Society ofLondon Series B-Biological Sciences 354:593-602.
t5 Gibbs, A.J., Tobamovirus Group. CMI/AAB Descriptions of Plant Viruses,
1977. 184: p. 1-
6.
Giroglou T, Florin L, Schafer F, Streeck RE, Sapp M. (2001) Human
papillomavirus
infection requires cell surface heparan sulfate. J Virol. 75:1565-70.
Harro CD, Pang YY, Roden RB, Hildesheim A, Wang Z, Reynolds MJ, Mast TC,
Robinson
2o R, Murphy BR, Karron RA, Dillner J, Schiller JT, Lowy DR. (2001 ) Safety
and
immunogenicity trial in adult volunteers of a human papillomavirus 16 L1 virus-
like
particle vaccine. JNatl Cancer Inst. 93:284-92.
He Y, Honnen WJ, Krachmarov CP, Burkhan M, Kayman SC, Corvalan J, Pinter A.
(2002)
Efficient isolation of novel human monoclonal antibodies with neutralizing
activity
25 against HIV-1 from transgenic mice expressing human Ig loci. Jlmmunol.
169:595-
605.
Isaacson M (2001) Viral hemorrhagic fever hazards for travelers in Africa.
Clinical
Infectious Diseases 33:1707-12.
Jaegle, M. and M.H. Van Regenmonel, (1985) Use of ELISA for measuring the
extent of
30 serological cross-reactivity between plant viruses. J Virol Methodsll:189-
98.
Jeang KT, Xiao H, Rich EA (1999) Multifaceted activities of the HIV
transactivator of
transcription Tat. J. BioL Chem. 274:28837-28840.
Kawana K, Kawana Y, Yoshikawa H, Taketani Y, Yoshiike K, Kanda T. (2001 a)
Nasal
immunization of mice with peptide having a cross-neutralization epitope on
minor
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
58
capsid protein L2 of human papillomavirus type 16 elicit systemic and mucosal
antibodies. Vaccine. 19:1496-502.
Kawana Y, Kawana K, Yoshikawa H, Taketani Y, Yoshiike K, Kanda T. (2001b)
Human
papillomavirus type 16 minor capsid protein 12 N-terminal region containing a
common neutralization epitope binds to the cell surface and enters the
cytoplasm. J
Virol. 75:2331-6.
Kawana K, Matsumoto K, Yoshikawa H, Taketani Y, Kawana T, Yoshiike K, Kanda T.
(1998a) A surface immunodeterminant of human papillomavirus type 16 minor
capsid protein L2. Virology. 245:353-9.
t0 Kawana K, Yoshikawa H, Taketani Y, Yoshiike K, Kanda T. (1998b) In vitro
construction
of pseudovirions of human papillomavirus type 16: incorporation of plasmid DNA
into reassembled L1/L2 capsids. J Virol. 72:10298-300.
Kawana K, Yoshikawa H, Taketani Y, Yoshiike K, Kanda T. (1999) Common
neutralization epitope in minor capsid protein L2 of human papillomavirus
types 16
and 6. J Virol. 73:6188-90.
Kawana K, Yasugi T, Kanda T, Kino N, Oda K, Okada S, Kawana Y, Nei T, Takada
T,
Toyoshima S, Tsuchiya A, Kondo K, Yoshikawa H, Tsutsumi O, Taketani Y (2003)
Vaccine in press.
Keegan K, Collett MS. (1986) Use of bacterial expression cloning to define the
amino acid
2o sequences of antigenic determinants on the G2 glycoprotein of Rift Valley
fever
virus. J Virol. 58:263-70.
Kirnbauer R, Taub J, Greenstone H, Roden R, Durst M, Gissmann L, Lowy DR,
Schiller
JT. (1993) Efficient self assembly of human papillomavirus type 16 L1 and L1-
L2
into virus-like particles. J Virol. 67:6929-36.
Kirnbauer R, Chandrachud LM, O'Neil BW, Wagner ER, Grindlay GJ, Armstrong A,
McGarvie GM, Schiller JT, Lowy DR, Campo MS. (1996) Virus-like particles of
bovine papillomavirus type 4 in prophylactic and therapeutic immunization.
Virology. 219:37-44.
Krebbers E, Bosch D, van der Kerckhove J (1992) Prospects and progress in the
production
of foreign proteins and peptides in plants. Plant Protein Engineering (PR
Shewery
and S Gutteridge, eds) Cambridge University Press, Cambridge, pp316-324.
Langeveld JP, Martinez-Torrecuadrada J, Boshuizen RS, Meloen RH, Ignacio Casal
J.
(2001) Characterisation of a protective linear B cell epitope against feline
parvoviruses. Vaccine. 19:2352-60.
Letvin NL (2002) Strategies for an HIV vaccine. .I. Clin. Invest. 109:15-20.
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
59
Letvin NL, Barouch DH, Montefiori DC. (2002) Prospects for vaccine protection
against
HIV-1 infection and AIDS. Annu Rev Immunol. 20:73-99.
Letvin NL, Robinson S, Rohne D, Axthelm MK, Fanton JW, Bilska M, Palker TJ,
Liao HX,
Haynes BF, Montefiori DC (2001) Vaccine-elicited V3 loop-specific antibodies
in
rhesus monkeys and control of a simian-human immunodeficiency virus expressing
a primary patient human immunodeficiency virus type 1 isolate envelope. J
Virol.
75:4165-75.
Lewis AD, Chen R, Montefiori DC, Johnson PR, Clark KR (2002). Generation of
neutralizing activity against human immunodeficiency virus type 1 in serum by
to antibody transfer. J. Yirol. 76:8769-8775.
Liu WJ, Gissmann L, Sun XY, Kanjanahaluethai A, Muller M, Doorbar J, Zhou J.
(1997)
Sequence close to the N-terminus of L2 protein is displayed on the surface of
bovine
papillomavirus type 1 virions. Virology 227:474-83.
London SD, Schmaljohn AL, Dalrymple JM, Rice CM. (1992) Infectious enveloped
RNA
virus antigenic chimeras. Proc Natl Acad Sci U S A. 89:207-11.
Marusic C, Rizza P, Lattanzi L, Mancini C, Spada M, Belardelli F, Benvenuto E,
Capone I.
(2001 ) Chimeric plant virus particles as immunogens for inducing murine and
human immune responses against human immunodeficiency virus type 1. J Virol.
75:8434-9.
Mascola JR. (2002) Passive transfer studies to elucidate the role of antibody-
mediated
protection against HIV-1. Passive transfer studies to elucidate the role of
antibody-
mediated protection against HIV-1. Vaccine. 20:1922-5.
McMichael A, Mwau M, Hanke T. (2002) Design and tests of an HIV vaccine. Br
Med Bull.
62:87-98.
Moulard M, Phogat SK, Shu Y, Labrijn AF, Xiao X, Binley JM, Zhang MY, Sidorov
IA,
Broder CC, Robinson J, Parren PW, Burton DR, Dimitrov DS. (2002) Broadly
cross-reactive HIV-1-neutralizing human monoclonal Fab selected for binding to
gp 120-CD4-CCRS complexes. Proc Natl Acad Sci U S A. 99:6913-8.
Pastrana DV, Vass WC, Lowy DR, Schiller JT. (2001 ) NHPV 16 VLP vaccine
induces
human antibodies that neutralize divergent variants of HPV16. Virology 279:361-
9.
Pelcher LE, Halasa MC (1982) An RNA plant virus vector or portion thereof, a
method of construction thereof, and a method of producing a gene-derived
product
therefrom. European Patent Application 067,533
Pelham HRB (1978) Leaky UAG stop codon in tobacco mosaic virus RNA. Nature
272:469-471.
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
Robinson HL (2002) New hope for an HIV vaccine. Nature Reviews Immunology
2:239-
250.
Roden RB, Weissinger EM, Henderson DW, Booy F, Kirnbauer R, Mushinski JF, Lowy
DR, Schiller JT (1994) Neutralization of bovine papillomavirus by antibodies
to L1
5 and L2 capsid proteins. J Virol. 68:7570-4
Roden RB, Greenstone HL, Kirnbauer R, Booy FP, Jessie J, Lowy DR, Schiller JT.
(1996)
In vitro generation and type-specific neutralization of a human papillomavirus
type
16 virion pseudotype. J Virol. 70:5875-83.
Savelyeva N, Munday R, Spellerberg MB, Lomonossoff GP, Stevenson FK (2001)
Plant
t 0 viral genes in DNA idiotypic vaccines activate linked CD4+ T-cell mediated
immunity against B-cell malignancies. Nat Biotechnol. 19:760-4.
Scala G, Chen X, Liu W, Telles JN, Cohen OJ, Vaccarezza M, Igarashi T, Fauci
AS. (1999)
Selection of HIV-specific immunogenic epitopes by screening random peptide
libraries with HIV-1-positive sera. Jlmmunol. 162:6155-61.
15 Schiller JT (1999) Papillomavirus-like particle vaccines for cervical
cancer. Mol Med
Today. 5:209-1 S.
Skuzeski JM, Nichols LM, Gesteland RF, Atkins JF (1991) The signal for a leaky
UAF
stop codon in several plant viruses includes that two downstream codons. J.
Mol.
Biol. 365-373.
2o Stiegler G, Kunert R, Purtscher M, Wolbank S, Voglauer R, Steindl F,
Katinger H. (2001 )
A potent cross-Glade neutralizing human monoclonal antibody against a novel
epitope on gp41 of human immunodeficiency virus type 1. AIDS Res Hum
Retroviruses. 17:1757-65.
Stone KM, Karen KL, Sternberg MR, McQuilland GM, Poon AD, Unger ER, Reeves WC
25 (2002) Seroprevalence of human papillomavirus type 16 infection in the
United
States. Journal oflnfectious Diseases 186:1396-1402.
Storni T, Lechner F, Erdmann I, Bachi T, Jegerlehner A, Dumrese T, Kundig TM,
Ruedl C,
Bachmann MF. (2002) Critical role for activation of antigen-presenting cells
in
priming of cytotoxic T cell responses after vaccination with virus-like
particles. J
30 Immunol. 168:2880-6.
Stubbs, G., ( 1999) Tobacco mosaic virus particle structure and the initiation
of disassembly.
Philos Trans R Soc Lond B Biol Sci, 354:551-7.
Suzich JA, Ghim SJ, Palmer-Hill FJ, White WI, Tamura JK, Bell JA, Newsome JA,
Jenson
AB, Schlegel R. (1995) Systemic immunization with papillomavirus L1 protein
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
61
completely prevents the development of viral mucosal papillomas. Proc Natl
Acad
Sci USA. 92:11553-7.
Thompson GH, Roman A. (1987) Expression of human papillomavirus type 6 E1, E2,
Ll
and L2 open reading frames in Escherichia coli. Gene. 56:289-95.
Tikhonov I, Ruckwardt TJ, Hatfield GS, Pauza CD (2003) Tat-neutralizing
antibodies in
vaccinated Macaques. J. Virol. 77:3157-3166.
Unckell F, Streeck RE, Sapp M. (1997) Generation and neutralization of
pseudovirions of
human papillomavirus type 33. J Virol. 71:2934-9.
Van Regenmortel, M.H.V. (1999) The antigenicity of tobacco mosaic virus.
Philos Trans R
l0 Soc Lond B Biol Sci, 354:559-68.
Wilson JA, Hevey M, Bakken R, Guest S, Bray M, Schmaljohn AL, Hart MK. (2000)
Epitopes involved in antibody-mediated protection from Ebola virus. Science.
287:1664-6.
Xu W, Hofmann-Lehmann R, McClure HM, Ruprecht RM. (2002) Passive immunization
with human neutralizing monoclonal antibodies: correlates of protective
immunity
against HIV. Vaccine. 20:1956-60.
Yuan H, Estes PA, Chen Y, Newsome J, Olcese VA, Garcea RL, Schlegel R. (2001 )
Immunization with a pentameric L1 fusion protein protects against
papillomavirus
infection. J Virol. 75:7848-53.
2o zur Hausen H (1999) Papillomaviruses in human cancers. Proc Assoc Am
Physicians.
111:581-7.
Zwick MB, Labrijn AF, Wang M, Spenlehauer C, Saphire EO, Binley JM, Moore JP,
Stiegler G, Katinger H, Burton DR, Parren PW. (2001) Broadly neutralizing
antibodies targeted to the membrane-proximal external region of human
immunodeficiency virus type 1 glycoprotein gp41. J Virol. 75:10892-905.
Zwick MB, Wang M, Poignard P, Stiegler G, Katinger H, Burton DR, Parren PW.
(2001)
Neutralization synergy of human immunodeficiency virus type 1 primary isolates
by
cocktails of broadly neutralizing antibodies. J Virol. 75:12198-208.
3o References for HPV epitope peptides
HPV epl
Kawana K, Yoshikawa H, Taketani Y, et al. Common neutralization epitope in
minor capsid
protein L2 of human papillomavirus types 16 and 6 J VIROL 73 (7): 6188-6190
JUL 1999
HPV ep2
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
62
Konya J, Eklund C, afGeijersstam V, et al. Identification of a cytotoxic T-
lymphocyte
epitope in the human papillomavirus type 16 E2 protein J GEN VIROL 78: 2615-
2620 Part
OCT 1997
5 HPV ep3
Bartholomew JS, Stacey SN, Coles B, et al. Identification Of A Naturally
Processed HLA
A0201-Restricted Viral Peptide From Cells Expressing Human Papillomavirus Type-
16 E6
Oncoprotein Eur J Immunol 24 (12): 3175-3179 Dec 1994
1o HPV ep4
Villada IB, Beneton N, Bony C, et al. Identification in humans of HPV-16 E6
and E7
protein epitopes recognized by cytolytic T lymphocytes in association with HLA-
B 18 and
determination of the HLA-B 18-specific binding motif EUR J IMMUNOL 30 (8):
2281-
2289 AUG 2000
HPV ep5
Azoury-Ziadeh R, Herd K, Fernando GJP, et al.T-Helper epitopes identified
within the E6
transforming protein of cervical cancer-associated human papillomavirus type
16
VIRAL IMMUNOL 12 (4): 297-312 1999
HPV ep6
As for HPV ep5
IiPV ep7
Tindle RW, Fernando GJP, Sterling JC, et al. A Public T-Helper Epitope Of The
E7
Transforming Protein Of Human Papillomavirus-16 Provides Cognate Help For
Several E7
B-Cell Epitopes From Cervical Cancer-Associated Human Papillomavirus Genotypes
P
Natl Acad Sci USA 88 (13): 5887-5891 Jul 1991
3o HPV ep8
Ressing ME, Sette A, Brandt RMP, et al. Human CTL Epitopes Encoded By Human
Papillomavirus Type-16 E6 And E7 Identified Through In-Vivo And In-Vitro
Immunogenicity Studies Of HLA-A-Asterisk-0201-Binding Peptides J Immunol 154
(11):
5934-5943 Jun 1 1995
CA 02497798 2005-02-28
WO 2004/032622 PCT/US2003/027563
63
HPV ep9
As for HPV ep8
HPV epl0
As for HPV ep8
HPV epll
As for HPV ep4
HPV epl2
to Garcia AM, Ortiz-Navarrete VF, Mora-Garcia MD, et al. Identification of
peptides
presented by HLA class I molecules on cervical cancer cells with HPV-18
infection
IMMLJNOL LETT 67 (3): 167-177 APR 15 1999
HPV epl3
t5 As for HPV epl
HPV epl4
Rudolf MP, Man S, Melief CJM, et al. Human T-cell responses to HLA-A-
restricted high
binding affinity peptides of human papillomavirus type 18 proteins E6 and E7
CLIN
20 CANCER RES 7 (3): 788S-7955 Suppl. S MAR 2001
HPV epl5
As for HPV epl4
25 HPV epl6
As for HPV epl4
HPV epl7
AS for HPV epl4
HPV epl8
Hohn H, Pilch H, Gunzel S, et al. Human papillomavirus type 33 E7 peptides
presented by
HLA-DR*0402 to tumor-infiltrating T cells in cervical cancer J VIROL 74 (14):
6632-6636
JUL 2000