Note: Descriptions are shown in the official language in which they were submitted.
CA 02498218 2005-03-08
-1-
DESCRIPTION
CATALYST FOR A FUEL CELL, METHOD OF MANUFACTURING
THE SAME, AND FUEL CELL
TECHNICAL FIELD
The present invention relates to catalysts
for a fuel cell, methods of manufacturing the same,
and fuel cells, and particularly to a catalyst
having Pt or the like attached on the surface of a
conductive carrier.
An oxygen-hydrogen cell is a typical
example of the fuel cell. This utilizes the reverse
reaction of electrolysis of water. By externally
supplying oxygen as the active material of a cathode
and hydrogen as the active material of an anode,
electrical energy can be extracted. Other active
materials of the anode are methanol, ethanol, and
methane.
BACKGROUND ART
Conventionally, fuel cells having large
capacity for spaceships, small-scale power stations,
and automobiles have been developed. Recently,
however, there has been a growing demand for fuel
cells as batteries for portable information
processing apparatuses such as portable terminals,
cellular phones, and notebook PCs.
Fuel cells include direct methanol fuel
cells that use methanol as fuel and directly obtain
H+ from the methanol and indirect methanol fuel
cells that decompose methanol into hydrogen and then
obtain H+ from the hydrogen. The indirect-type fuel
cells require reactions at high temperatures in
order to decompose methanol, and therefore, are not
suitable for portable terminals. The direct-type
fuel cells have a merit in that it is possible to
CA 02498218 2005-03-08
-2-
cause reactions to proceed at room temperature.
In the direct methanol fuel cells, at a
cathode and an anode, the following reactions occur
at the catalyst surfaces of the respective
electrodes:
Anode (fuel electrode) : CH3OH + H2O -+ CO2
+ 6H+ + 6e-
Cathode (air electrode) : 3/202 + 6H+ + 6e-
3H20-
Accordingly, the overall reaction is:
Overall reaction: CH3OH + 3/202 -3 2H2O +
CO2.
That is, electrons obtained in this reaction, for
instance, six moles of electrons, can be utilized as
electrical energy.
Conventionally, a variety of catalysts
have been studied in order to increase the speed of
the above-described reaction. Of these, catalysts
that have a variety of metals, principally platinum,
carried on carbon particles or carbon substrate are
used, which are specifically catalysts that have
metal particulates having electrocatalytic activity,
such as Pt particulates or particulates of a Pt
alloy of, for instance, Pt and Ru, carried on
conductive carbon particles. Rate of reaction on a
catalyst surface relates directly to the amount of
current, and contributes to power generation
efficiency. Accordingly, catalysts having high
rates of reaction, that is, catalysts having a great
surface area per unit mass (specific surface area),
are desired.
According to the conventional method of
preparing catalysts, for instance, carbon particles
are dispersed in an aqueous solution including a Pt
compound. Then, an alkaline aqueous solution is
dropped to reduce the Pt compound, and the carbon
CA 02498218 2005-03-08
-3-
particles are caused to carry precipitated Pt
particulates.
However, according to this method, it is
possible to cause the catalytic Pt particulates to
adhere to the carbon particles, but the amount is
small. Accordingly, there is a problem in that the
catalysis is insufficient, thus resulting in an
insufficient rate of reaction in fuel cells.
In order to improve catalysis, it is
desirable to cover the entire surface of the carbon
particles while maintaining the size of the Pt
particulates. However, a prolonged period of
reduction to increase the amount of precipitated Pt
particulates causes a problem in that adjacent Pt
particulates on the surface of the carbon particles
are coupled by newly precipitated Pt to increase
particle size, thus having the opposite effect of
reducing surface area to decrease catalysis.
DISCLOSURE OF THE INVENTION
Accordingly, it is a general object of the
present invention to provide a novel and useful
catalyst for a fuel cell, a method of manufacturing
the same, and a fuel cell in which the above-
described disadvantages are eliminated.
A more specific object of the present
invention is to provide a catalyst for a fuel cell
that is highly active with a high rate of reaction,
a method of manufacturing the same, and a fuel cell
using the same.
According to one aspect of the present
invention, a catalyst for a fuel cell is provided
that includes a conductive carrier, and a catalyst
layer formed to cover the conductive carrier and
formed of Pt, Ru, or a Pt-based alloy.
According to the present invention, a
catalytic Pt, Ru, or Pt-alloy catalyst layer is
CA 02498218 2005-03-08
-4-
formed like a layer so as to cover the surface of a
conductive carrier. Accordingly, compared with the
conventional case of forming catalyst particulates,
the surface area per conductive carrier mass and
that per catalyst mass can be increased. Further,
since the catalyst is layered, it is possible for
the intermediate of a reactant adsorbed to the
catalyst surface to move on the surface of the
catalyst layer more easily than in the case of the
catalyst particulates, thus increasing activity. As
a result, it is possible to increase the rate of
reaction.
Additionally, metal particulates formed of
Pt, Ru, or a Pt-based alloy may be dispersed on the
surface of the catalyst layer. This makes it
possible to further increase the surface area per
conductive carrier mass and that per catalyst mass.
The conductive carrier may be conductive
carbon particles. The Pt-based alloy may employ Pt
as a principal component and include a Pt group
element other than Pt. The catalyst layer may be
0.5 nm to 20 nm in thickness.
According to another aspect of the present
invention, a method of manufacturing a catalyst for
a fuel cell is provided that includes the steps of
reducing a compound of a Pt group element in a
mixture in a gel or highly viscous state, the
mixture including a solution including the compound
of the Pt group element and a conductive carrier;
and forming a catalyst layer on a surface of the
conductive carrier by burning, the catalyst layer
being formed of the Pt group element.
According to the present invention, a Pt
group element compound is reduced in a gel or highly
viscous state. Therefore, the reduced Pt group
element is prevented from making a Brownian motion,
thus being prevented from growing into particulates.
CA 02498218 2005-03-08
By decomposing and evaporating a gel or the like by
burning in this state, a catalyst layer formed of
the Pt group element is formed on the surface of a
conductive carrier. Accordingly, the catalyst can
be higher in activity and rate of reaction as
described above.
The step of causing particulates of a Pt
group element to precipitate on the surface of the
catalyst layer may be further included. The Pt
group element compound may be a Pt compound; be a Ru
compound; or employ a Pt compound as a principal
component and include a compound of a Pt group
element other than Pt. Further, viscosity may fall
within the range of 10 cps to 1x104 cps in the
highly viscous state.
According to another aspect of the present
invention, a fuel cell is provided that includes a
solid electrolyte membrane and a fuel electrode and
an air electrode having the solid electrolyte
membrane sandwiched therebetween, wherein the fuel
electrode and the air electrode each include a
collector and a catalyst layer, and one of the
catalyst layers of the fuel electrode and the air
electrode includes a catalyst, the catalyst having a
conductive carrier and a catalyst layer formed to
cover the conductive carrier and formed of Pt, Ru,
or a Pt-based alloy.
According to the present invention, it is
possible to increase the rate of reaction of
oxidation and reduction reaction in the fuel
electrode and the air electrode because of a
catalyst having a catalytic Pt, Ru, or Pt-alloy
catalyst layer formed like a layer so as to cover
the surface of a conductive carrier. As a result, a
fuel cell having high power generation efficiency is
realized.
CA 02498218 2010-12-07
27879-185
-5a-
In one embodiment, the invention relates to a
catalyst for a fuel cell, comprising: a conductive carrier;
and a catalyst layer, wherein: the catalyst layer is formed
continuously over an entire surface of the conductive
carrier to fully cover the conductive carrier; and the
catalyst layer comprises Pt, Ru or a Pt-based alloy.
In a further embodiment, the invention relates to
a fuel cell, comprising: a solid electrolyte membrane; and a
fuel electrode and an air electrode having the solid
electrolyte membrane sandwiched therebetween, wherein: the
fuel electrode and the air electrode each include a
collector and a layer of a catalyst which is in contact with
the solid electrolyte membrane; and the catalyst of one of
the layers of the catalyst of the fuel electrode and the air
electrode includes a conductive carrier and a catalyst layer
formed continuously over an entire surface of the conductive
carrier to fully cover the conductive carrier, and the
catalyst layer comprises Pt, Ru, or a Pt-based alloy.
CA 02498218 2005-03-08
-6-
BRIEF DESCRIPTION OF THE DRAWINGS
FIG. 1A is a cross-sectional view of a
catalyst prepared by the conventional reduction
method;
FIG. 1B is a cross-sectional view of a
catalyst prepared by reduction of a longer period
than FIG. 1A;
FIG. 2 is a cross-sectional view of a
catalyst for a fuel cell according to the present
invention;
FIG. 3 is a flowchart illustrating a
process of manufacturing a catalyst for a fuel cell
according to a first embodiment of the present
invention;
FIG. 4 is a flowchart illustrating a
process of manufacturing a catalyst for a fuel cell
according to a second embodiment of the present
invention;
FIG. 5 is a flowchart illustrating a
process of manufacturing a catalyst for a fuel cell
according to a third embodiment of the present
invention;
FIG. 6 is a cross-sectional view of the
catalyst for a fuel cell according to the third
embodiment;
FIG. 7 is a diagram illustrating a fuel
cell according to a fourth embodiment of the present
invention; and
FIG. 8 is a diagram illustrating the power
generation efficiencies of fuel cells employing the
catalysts for a fuel cell of example implementations
and a comparative example.
BEST MODE FOR CARRYING OUT THE INVENTION
A description is given below of a catalyst
for a fuel cell according to an embodiment of the
present invention.
CA 02498218 2009-01-13
27879-185
-7-
The inventor of the present invention
found out, through various experiments to increase
the activity of a Pt catalyst carried by carbon
particles according to the conventional reduction
method, that it is difficult to improve activity by
a catalyst preparation method according to the
conventional reduction method. That is, observation
of a cross section of a Pt catalyst 10 prepared by the
conventional reduction method illustrated in FIG. 1A
with an HRTEM (High Resolution Transmission Electron
Microscope) shows that Pt particulates 12 only
adhere discretely to the surface of a carbon
particle 11, which is a conductive carrier, and do
not adhere enough to cover the surface of the carbon
particle 11. Accordingly, the mass of the Pt
particulates 12 adhering to the carbon particle 11
is small. That is, the specific surface area of the
Pt particulates 12 is also small. In a catalyst 15
illustrated in FIG. 1B, prepared by prolonging a
period of reduction in the conventional reduction
method, the mass of Pt particulates 16 adhering to
the carbon particle 11 is increased. However, since
the Pt particulates 16 are increased in particle
size, there is no increase in the specific surface
area. Accordingly, it is inferred that activity is
not increased compared with the catalyst illustrated
in FIG. 1A.
It is inferred that this is because when a
certain amount of Pt particulates adhere to a carbon
particle so as to increase the entire surface area
of the Pt particulates, the Pt particulates become
unstable in terms of energy, so that the Pt
particulates adhere to each other to grow as a unit
so as to be stable in terms of energy.
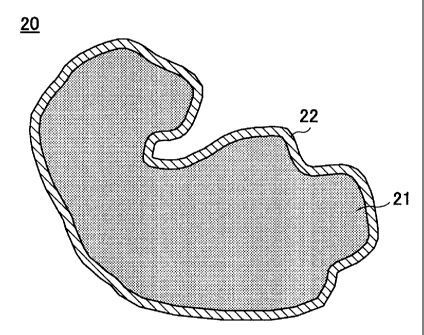
FIG. 2 is a cross-sectional view of a
catalyst for a fuel cell according to an embodiment
of the present invention. Referring to FIG. 2, a
CA 02498218 2005-03-08
-8-
catalyst 20 for a fuel cell is made up of a
conductive carrier 21 and a catalyst layer 22 formed
on the surface of the conductive carrier 21. The
catalyst layer 22 is made of, for instance, Pt, and
the conductive carrier 21 is made of, for instance,
a conductive carbon particle. The Pt layer is thus
formed to be thin on the surface of the conductive
carbon particle. Accordingly, there is an increase
in the surface area and activity compared with a
catalyst according to the conventional reduction
method. Further, the catalyst layer 22 is formed
continuously on the surface of the conductive
carrier. Accordingly, it is inferred that it is
easy for a reactant or the intermediate of the
reactant adsorbed to the surface of the catalyst
layer 22 to move on the surface of the catalyst
layer 22 so that activity is further increased.
Accordingly, the rate of reaction is further
increased.
An electroconductive material such as a
carbon particle or porous Ni with a great surface
area is employed as the conductive carrier 21. The
BET value of the conductive carrier 21 is preferably
within the range of 100 m2/g to 2000 m2/g. The
surface area of the catalyst layer 22 of Pt or the
like cannot be secured sufficiently with values less
than 100 m2/g. With values greater than 2000 m2/g,
the conductive carrier 21 is so small as to make it
difficult to disperse in a solution in below-
described manufacturing methods. The specific
resistance of the conductive carrier 21 is
preferably 10-'Q-cm to 102 0-cm in serving as a
medium that conducts electrons and protons generated
by the catalyst oxidizing methanol or the like. For
instance, in the case of a carbon particle,
KETJENBLACK EC-600J (name of a Ketjenblack
International Corporation product) is employed.
CA 02498218 2005-03-08
-9-
The catalyst layer 22 may be not only Pt
but also a Ru or Pt alloy, for instance, an alloy
composed principally of Pt with another Pt group
element added thereto, such as PtRu or PtRh. PtRu
or the like can reduce poisoning with respect to
carbon monoxide generated in a fuel cell. According
to measurement of an HRTEM photograph of a cross
section, the catalyst layer 22 is preferably formed
to be 0.5 nm to 20 nm in thickness. The catalyst
layer 22 cannot sufficiently cover the surface of
the conductive carrier 21 if it is thinner than 0.5
nm. On the other hand, if it is thicker than 20 nm,
the specific surface area is reduced.
With respect to the specific surface area
of the catalyst 20 for a fuel cell of this
configuration according to this embodiment of the
present invention, the specific surface area
according to pulse CO adsorption is preferable
within the range of 200 m2/g to 5000 m2/g. The rate
of reaction in a fuel cell is low if it is less than
200 m2/g, and the stability of the catalyst for a
fuel cell over time is reduced if it is greater than
5000 m2/g.
A description is given below of methods of
manufacturing a catalyst for a fuel cell according
to embodiments of the present invention.
The manufacturing methods according to the
present invention are characterized in that a Pt
group element compound solution including a Pt group
element acid or salt is reduced in a gel or highly
viscous state; a precipitated catalyst is prevented
from growing into catalyst particles by confining
the precipitated catalyst in a three-dimensional
network structure of a gel or highly viscous
material to restrict Brownian motion; and a catalyst
layer is formed in a layered manner on the surface
of a conductive carrier by burning. Compared with
CA 02498218 2005-03-08
-10-
the conventional reduction method in a solution, the
specific surface area per catalyst mass and that per
conductive carrier mass can be increased, so that
the rate of reaction as a catalyst can be increased.
A specific description is given below of the
manufacturing methods.
[First Embodiment]
This embodiment is a case of manufacturing
a catalyst for a fuel cell by reducing a Pt group
element compound in a gel state.
FIG. 3 is a flowchart illustrating a
process of manufacturing a catalyst for a fuel cell
according to this embodiment. A description is
given below, with reference to FIG. 3, of the
manufacturing process.
First, a solution of a gel material and a
Pt group element compound is prepared (SlOl).
Specifically, the gel material and a predetermined
amount of the Pt group element compound are mixed
with water and completely dissolved by heating.
For instance, a monomer, a dimmer, an
oligomer, or a polymer is employable as the gel
material. The gel material may be any of those
producing gel by a crosslinking reaction with a
below-described gelatinizer. That is, the gel
material may be any of those becoming an organic
polymer by a crosslinking reaction or be a polymer
itself forming a three-dimensional network structure
with a low molecular weight material.
The Pt group element compound is an acid
or salt of a Pt group element such as Pt, Ru, or Rh.
For instance, a hexachloroplatinic acid (H2PtCl6)
platinum chloride (PtC14), ruthenium chloride
(RuCl3), or rhodium chloride (RhC13) is employable
as a Pt, Ru, or Rh acid or salt. These compounds
may also be employed in combination.
Next, a gelatinizer serving as a
CA 02498218 2005-03-08
-11-
crosslinking agent is added to the obtained solution,
to which a conductive carrier is further added. The
conductive carrier is dispersed in the solution
while degassing is performed under reduced pressure
(S102). It is preferable to use a homogenizer or an
ultrasonic disperser in order to disperse the
conductive carrier.
Specifically, a crosslinking agent
suitable for the above-described gel material is
selected. For instance, bis-acrylamide or a
diacrylate monomer is employable with respect to the
gel material of acrylamide. These crosslinking
agents have two or more reactive bonding parts,
which bond to the reactive bonding parts of the gel
material to cause crosslinkage to be generated
between the principal chains of the polymer, thereby
forming a three-dimensional network structure.
The above-described material is employed
for the conductive carrier. A dispersing agent
promoting dispersion of carbon may be employed as
required.
Next, this solution is subjected to
bubbling with nitrogen so as to reduce the oxygen
concentration of the solution. While removing
oxygen, which inhibits the reaction of a reducing
agent described below, the solution is heated to
approximately 90 C for approximately 1 hour using a
hot plate so as to be gelatinized (S103).
Specifically, heating temperature is set to 50 C to
200 C, and heating time is set to 0.1 hours to 5
hours. The obtained gel is preferably a hard gel
substantially like agar in terms of prevention of
growth of catalyst particles.
Next, the gel is crushed into 5 mm pieces
using a rotary mixer, and is introduced into an
aqueous solution including a reducing agent. After
being heated approximately at 80 C for 2 hours, the
CA 02498218 2005-03-08
-12-
solution is left at rest at room temperature (S104).
Formaldehyde or hydroquinone may be employed as the
reducing agent. Specifically, in the case of, for
instance, formaldehyde, the concentration of the
reducing agent is preferably 0.1% to 100, and more
preferably 1% to 3%, in terms of the rate of
reaction. Further, heating temperature is set to
50 C to 100 C, and heating time is set to 0.5 hours
to 10 hours. The rest period after heating is
preferably 8 hours to 15 hours in terms of uniform
formation of a catalyst layer.
Next, after discarding the reducing agent
and washing the gel with water, the gel is heated to
approximately 150 C in the atmosphere to be dried
(S106).
Next, the gel is burned at 650 C for 2
hours in the atmosphere using an oven (S107). The
network structure forming the gel is decomposed and
gasified, so that a catalyst incorporated in the
network structure is formed like a layer on the
conductive carrier.
According to the manufacturing method of
this embodiment, the Pt group element compound is
reduced in a gel state. Accordingly, the Pt group
element precipitated by reduction, whose Brownian
motion is restricted by the three-dimensional
network structure of the gel, is prevented from
growing into particulates. Accordingly, as a result
of decomposition and evaporation of the three-
dimensional network structure by burning, a layer-
like catalyst layer is formed on the surface of the
conductive carrier. As a result, it is possible to
increase the specific surface area of the catalyst
per catalyst mass and that per conductive carrier,
and thus to increase activity.
Alternative gel materials are animal
proteins such as casein, gelatin, and collagen;
CA 02498218 2005-03-08
-13-
plant proteins such as wheat-derived protein and
soybean-derived protein; cellulose such as wood pulp
cellulose; plant seed-derived mucilage such as gum
guaiac and locust beam gum; seaweed-derived mucilage
such as agar and carrageenan; plant leaf mucilage
such as gum Arabic and tragacanth gum; plant fruit
mucilage such as pectin; plant rhizome mucilage such
as mannan; microbe-produced mucilage such as
pullulan, xanthan gum, and dextran; cellulose
derivatives such as methyl cellulose, ethyl
cellulose, hydroxypropylcellulose,
carboxymethycellulose, carboxymethylethylcellulose,
methyl cellulose, celluloseacetate phthalate, and
hydroxypropylmethylcellulose phthalate; and starch
derivatives such as soluble starch and carboxymethyl
starch. Methods of forming a gel state using these
gel materials do not require the above-described
crosslinking agent. The heating temperature and the
temperature after heating of step 103 are set in
accordance with the gelatinization conditions of the
gel material, such as gelatinization temperature.
For instance, gelatin and agar are gelatinized when
being cooled to or below the gelatinization
temperature, and pectin is gelatinized when the
gelatinization temperature is reached or exceeded.
These gel materials are also employable as a below-
described thickener depending on heating temperature
and blending quantity. Polyvinyl acetal and a gel
body formed of a polyion complex are employable as
alternative gel materials.
[Second Embodiment]
This embodiment is a case of manufacturing
a catalyst for a fuel cell by reducing a Pt group
element compound in a highly viscous state.
FIG. 4 is a flowchart illustrating a
process of manufacturing a catalyst for a fuel cell
according to this embodiment. A description is
CA 02498218 2005-03-08
-14-
given below, with reference to FIG. 4, of the
manufacturing process.
First, a solution of a thickener and a Pt
group element compound is prepared (S201).
Specifically, a predetermined amount of the Pt group
element compound is gradually added to an aqueous
solution in which the thickener is dissolved. While
being heated to approximately 60 C, the solution is
mixed with water, thereby heating and completely
dissolving the Pt group element compound.
The thickener may be any of the following
polyoxyalkylene compounds and acrylic water viscous
agent polymers, and mixtures of two or more of them.
The polyoxyalkylene compounds are polyethyleneglycol,
polyoxyethyleneoxide, alkyleneoxide (such as
ethyleneoxide or propyleneoxide)-modified polyhydric
alcohols, and polyoxyethylene oxypropyleneglycol (a
block or random copolymer of ethyleneoxide and
propyleneoxide). The acrylic water viscous agent
polymers are polyacrylamide, polymethacrylamide,
polyacrylate or salts thereof, polymethacrylate or
salts thereof, 2-alkyl-2-acrylamide propane
sulfonate or salts thereof such as 2-alkyl-2-
acrylamide propane sodium sulfonate,
(meth)acryloyloxyalkyltrialkyl tetraammonium such as
methacryloyloxyethyltrimethylammonium chloride, and
(meth)acryloyloxyalkyldialkyl amine salt such as a
tri- or tetra-salt of diethylaminoethylmethacrylate.
Further, as the Pt group element compound, the same
acid or salt as in the first embodiment may be
employed.
Next, a conductive carrier is added to the
obtained solution, and the conductive carrier is
dispersed in the solution while degassing is
performed under reduced pressure (S202). A
homogenizer or an ultrasonic disperser may be
employed in order to disperse the conductive carrier.
CA 02498218 2005-03-08
-15-
The same conductive carrier as in the first
embodiment may be employed as the conductive carrier.
Next, an aqueous solution including a
reducing agent is gradually added to this solution.
After being stirred for approximately 2 hours at
approximately 80 C, the solution is left at rest at
room temperature (S203). The reducing agent is the
same as in the first embodiment. The heating
temperature here is set to 50 C to 95 C, and the
heating time is set to 0.1 hours to 5 hours. The
rest period after heating is preferably 8 hours to
hours in terms of uniform growth of a catalyst
layer. At the time of heating, a viscosity at, for
instance, 80 C is preferably 10-1x104 cps according
15 to a B-type viscometer. Further, viscosity at room
temperature after slow cooling is preferably 100 cps
to lxlO5 cps according to a B-type viscometer.
Next, this aqueous solution after
completion of the reduction reaction is concentrated
and dried and hardened using a rotary evaporator,
and is further heated to approximately 150 C so as
to be dried completely (S204).
Next, this dry material is subjected to
burning for approximately 2 hours at approximately
650 C in the atmosphere using an oven (S205). A
material forming the state of high viscosity is
decomposed and evaporated, so that an incorporated
catalyst is formed like a layer on the conductive
carrier. Specifically, burning temperature is set
to 500 C to 800 C, and burning time is set to 1 hour
to 5 hours. At burning temperatures higher than
800 C, the surface of the catalyst layer is oxidized
to become as if poisoned, thus resulting in a
decrease in catalysis. At temperatures lower than
500 C, it is not possible to decompose and evaporate
the highly viscous material sufficiently.
According to the manufacturing method of
CA 02498218 2005-03-08
-16-
this embodiment, the Pt group element compound is
reduced in the state of high viscosity. Accordingly,
the Pt group element precipitated by reduction,
whose Brownian motion is restricted by the high
viscosity of the solution, is prevented from growing
into particulates. Accordingly, as a result of
decomposition and evaporation of the material
realizing high viscosity by burning, a layer-like
catalyst layer is formed on the surface of the
conductive carrier. As a result, it is possible to
increase the specific surface area of the catalyst
per catalyst mass and that per conductive carrier,
and thus to increase activity.
[Third Embodiment]
This embodiment is a case of manufacturing
catalysts for a fuel cell by further having
catalytic Pt group element particulates precipitated
and carried on the surface of the catalysts for fuel
cells obtained by the first and second embodiments.
FIG. 5 is a flowchart illustrating a
process of manufacturing a catalyst for a fuel cell
according to this embodiment. A description is
given below, with reference to FIG. 5, of the
manufacturing process.
First, a Pt group element compound is
prepared, a catalyst obtained by the first or second
embodiment is added thereto, and dispersion is
performed using a homogenizer (S301).
Next, a reducing agent is gradually added
to this mixture solution, which is heated
approximately at 80 C for 2 hours and is left at
rest at room temperature (S302).
After subjecting to centrifugal separation
and washing with water, this precipitation is heated
for 2 hours at 300 C in a N2 atmosphere so as to
form a catalyst (S303).
FIG. 6 is a cross-sectional view of a
CA 02498218 2009-01-13
27879-185
-17-
catalyst for a fuel cell according to this
embodiment. Referring to FIG. 6, catalyst
particulates 26 formed of a Pt group element are
precipitated on and adhere to the catalyst layer
obtained by the first or second embodiment. These
catalyst particulates make it possible to increase
the specific surface area of the catalyst per
catalyst mass and that per conductive carrier and
thus to increase activity.
According to this embodiment, as described
above, catalyst particulates are further formed on
the catalyst layer formed on the surface of the
conductive carrier, the catalyst layer being
obtained by the first or second embodiment. These
catalyst particulates make it possible to increase
the specific surface area of the catalyst per
catalyst mass and that per conductive carrier and
thus to increase activity.
[Fourth Embodiment]
FIG. 7 is a diagram showing a fuel cell
according to this embodiment. Referring to FIG. 7,
the fuel cell of this embodiment is made up
principally of a solid electrolyte membrane 31, a
fuel electrode 32 and an air electrode 33 on both
sides of the solid electrolyte membrane 31, a case
34 housing these, an external circuit 35 to which a
load is connected for extracting power from the fuel
cell, etc.
The solid electrolyte membrane 31 is
formed of a proton-conductive polymer material, and
for instance, Nafion N-115 (trade-mark) of DuPont
is employable.
The fuel electrode 32 and the air
electrode 33 are each formed of a collector 36 and a
catalyst layer 38 applied on a carbon paper 37. The
catalyst layer 38 is configured to come into contact
with the solid electrolyte membrane 31. The
CA 02498218 2005-03-08
-18-
catalyst for a fuel cell of any of the first through
third embodiments is employed as the catalyst layer
38. Approximately 2 g of a catalyst for a fuel cell
obtained by any of the first through third
embodiments is kneaded with 20 g of 5% by mass of a
Nafion solution to be formed like a paste, which is
applied on a carbon paper by doctor blade coating or
bar coating with a thickness being set to
approximately 50 m to 300 m.
The collectors 36A and 36B, each formed of
a mesh of a highly corrosion-resistant alloy such as
stainless steel, collect electrons generated in the
catalyst layer 38A of the fuel electrode 32 through
the carbon paper 37A, or supply electrons flowing
from the external circuit 35 evenly to the catalyst
layer 38B.
A methanol aqueous solution is supplied to
the fuel electrode 32 side, so that the reaction of
CH3OH + H2O -+ C02 + 6H+ + 6e- occurs on the catalyst
surface of the catalyst layer 38A. The generated
protons are conducted through the solid electrolyte
membrane 31 to reach the air electrode 33, and the
electrons flow through the load connected to the
external circuit 35 to reach the air electrode 33.
oxygen in the air is supplied to the air electrode
33 side, so that the reaction of 3/202 + 6H+ + 6e- -*
3H20 occurs on the catalyst surface of the catalyst
layer 38B. As a result, water is generated from the
oxygen, protons, and electrons.
The fuel cell of this embodiment is
characterized by the catalyst of the catalyst layer.
The catalyst covers the surface of a carbon particle,
which is a carrier, in a layered manner. Therefore,
the specific surface area of the catalyst of the
catalyst layer with respect to mass is great, so
that it is highly probable that a reactant comes
into contact with the catalyst, that is, the rate of
CA 02498218 2005-03-08
-19-
reaction is high. As a result, power generation
efficiency is improved.
A description is given below of example
implementations according to the present invention
and a comparative example that is not according to
the present invention.
[First Example Implementation]
200 ml of a 30% aqueous solution of
acrylamide and 200 ml of a 2% aqueous solution of
bis-acrylamide were mixed. Further, 1.5 g of
hexachloroplatinic acid was added, and was dissolved
completely by heating to 60 C. Next, after adding
40 ml of a 10% aqueous solution of potassium
persulfate, 0.6 g of KETJENBLACK EC-600J was
introduced as conductive carrier carbon particles.
Degassing was performed under reduced pressure, and
stirring was performed. While being subjected to
bubbling with nitrogen gas so as to reduce the
concentration of oxygen in the solution, this
solution was heated for 1 hour at 90 C using a hot
plate. As a result, a gel was obtained.
Next, the gel was crushed into pieces of
several mm and introduced into 1000 ml of a 3.5%
aqueous solution of formaldehyde. After being
heated for 2 hours at 80 C, it was left at rest for
10 hours at room temperature. Then, the
formaldehyde aqueous solution was discharged, and
the gel was lightly washed with water. The gel was
dried by heating for 3 hours at 150 C in the
atmosphere. The gel was further subjected to
burning for 2 hours at 650 C in the atmosphere. As
a result, the Pt catalyst of this example
implementation was obtained.
An HRTEM observation of a cross section of
the Pt catalyst of this example implementation
showed that the Pt catalyst layer was 2 nm in
thickness, and the specific surface area according
CA 02498218 2005-03-08
-20-
to pulse CO adsorption was 1200 m2/g.
[Second Example Implementation]
1.5 g of hexachloroplatinic acid was
gradually added to and was dissolved completely in
500 ml of a 10% aqueous solution of
polyvinylpyrrolidone (K-90) by heating to 60 C.
Next, 0.6 g of KETJENBLACK EC-600J was introduced as
carrier carbon. Stirring was performed while
performing degassing under reduced pressure. 1000
ml of a 3.5% aqueous solution of formaldehyde was
gradually added to this solution. It was heated for
2 hours at 80 C while being stirred, and thereafter
was left at rest for 10 hours at room temperature.
The viscosity at the time of heating at 80 C was
2500 cps.
Next, this mixture aqueous solution was
concentrated and dried and hardened using a rotary
evaporator, and was further heated for 3 hours at
150 C so as to be dried completely. Further, this
hardened material was subjected to burning for 2
hours at 650 C in the atmosphere. As a result, the
Pt catalyst of this example implementation was
obtained.
An HRTEM observation of a cross section of
the Pt catalyst of this example implementation
showed that the Pt catalyst layer was 3 nm in
thickness, and the specific surface area according
to pulse CO adsorption was 1100 m2/g.
[Third Example Implementation]
After dispersing 1 g of the Pt catalyst of
the first example implementation in 100 ml of a 1%
aqueous solution of hexachloroplatinic acid, 200 ml
of a 3.5% aqueous solution of formaldehyde was
gradually added thereto. After being retained for 2
hours at 80 C, it was left for 10 hours at room
temperature. After being subjected to centrifugal
separation and washing with water, an obtained
CA 02498218 2005-03-08
-21-
precipitation was heated for 2 hours at 300 C in an
oven of a N2 atmosphere. As a result, the Pt
catalyst of the third example implementation was
obtained.
An HRTEM observation of a cross section of
the Pt catalyst of this example implementation
showed that the Pt catalyst layer was 5 nm in
thickness, and the specific surface area according
to pulse CO adsorption was 1800 m2/g.
[Fourth Example Implementation]
After dispersing 1 g of the Pt catalyst of
the second example implementation in 100 ml of a 1%
aqueous solution of hexachloroplatinic acid, 200 ml
of a 3.5% aqueous solution of formaldehyde was
gradually added thereto. After being retained for 2
hours at 80 C, it was left for 10 hours at room
temperature. After being subjected to centrifugal
separation and washing with water, an obtained
precipitation was heated for 2 hours at 300 C in an
oven of a N2 atmosphere. As result, the Pt catalyst
of the fourth example implementation was obtained.
An HRTEM observation of a cross section of
the Pt catalyst of this example implementation
showed that the Pt catalyst layer was 6 nm in
thickness, and the specific surface area according
to pulse CO adsorption was 1700 m2/g.
[Fifth Example Implementation]
200 ml of a 20% aqueous solution of 2-
acrylamide-2-methylpropane sulfonate and 200 ml of a
2% aqueous solution of bis-acrylamide were mixed.
Further, 1.5 g of hexachloroplatinic acid was added,
and was dissolved completely by heating to 60 C.
Next, after adding 40 ml of a 10% aqueous solution
of potassium persulfate, 0.6 g of KETJENBLACK EC-
600J was introduced as carrier carbon. Degassing
was performed under reduced pressure, and stirring
was performed. The subsequent process was performed
CA 02498218 2005-03-08
-22-
in the same manner as in the first example
implementation. As a result, the Pt catalyst of
this example implementation was obtained.
[Sixth Example Implementation]
200 ml of a 20% aqueous solution of 2-
hydroxyethylmethacrylate and 200 ml of a 2% aqueous
solution of 4,4'-bisphenol A-diacrylate were mixed.
Further, 1.5 g of hexachloroplatinic acid was added,
and was dissolved completely by heating to 60 C.
Next, after adding 40 ml of a 10% aqueous solution
of potassium persulfate, 0.6 g of KETJENBLACK EC-
600J was introduced as carrier carbon. Degassing
was performed under reduced pressure, and stirring
was performed. The subsequent process was performed
in the same manner as in the first example
implementation. As a result, the Pt catalyst of
this example implementation was obtained.
[Seventh Example Implementation]
1.5 g of hexachloroplatinic acid was added
to 400 ml of a 15% aqueous solution of quaternary-
stilbazolium-group-introduced polyvinyl alcohol, and
was dissolved completely by heating to 60 C. Next,
after adding 40 ml of a 10% aqueous solution of
potassium persulfate, 0.6 g of KETJENBLACK EC-600J
was introduced as carrier carbon. Degassing was
performed under reduced pressure, and stirring was
performed. The subsequent process was performed in
the same manner as in the first example
implementation. As a result, the Pt catalyst of
this example implementation was obtained.
[Eighth Example Implementation]
1.5 g of hexachloroplatinic acid was added
to 200 ml of a 20% aqueous solution of polystyrene
sodium sulfonate, and was dissolved completely by
heating to 60 C. Next, 0.6 g of KETJENBLACK EC-600J
was introduced as carrier carbon. Degassing was
performed under reduced pressure, and stirring was
CA 02498218 2005-03-08
-23-
performed. 200 ml of a 20% aqueous solution of
poly(4-ethylvinyl pyridine) was added to this
solution, and was mixed therewith by stirring with a
stirrer, so that the solution was gelatinized in 10
minutes. The subsequent process was performed in
the same manner as in the first example
implementation. As a result, the Pt catalyst of
this example implementation was obtained.
[Ninth Example Implementation]
200 ml of a 20% aqueous solution of sodium
acrylate and 200 ml of a 2% aqueous solution of
4,4'-bisphenol A-diacrylate were mixed. Further,
1.5 g of hexachloroplatinic acid was added, and was
dissolved completely by heating to 60 C. Next,
after adding 40 ml of a 10% aqueous solution of
potassium persulfate, 0.6 g of KETJENBLACK EC-600J
was introduced as carrier carbon. Degassing was
performed under reduced pressure, and stirring was
performed. The subsequent process was performed in
the same manner as in the first example
implementation. As a result, the Pt catalyst of
this example implementation was obtained.
[Tenth Example Implementation]
200 ml of a 20% aqueous solution of
vinylpyrrolidone and 200 ml of a 2% aqueous solution
of 4,4'-bisphenol A-diacrylate were mixed. Further,
1.5 g of hexachloroplatinic acid was added, and was
dissolved completely by heating to 60 C. Next,
after adding 40 ml of a 10% aqueous solution of
potassium persulfate, 0.6 g of KETJENBLACK EC-600J
was introduced as carrier carbon. Degassing was
performed under reduced pressure, and stirring was
performed. The subsequent process was performed in
the same manner as in the first example
implementation. As a result, the Pt catalyst of
this example implementation was obtained.
[11th Example Implementation]
CA 02498218 2005-03-08
-24-
1.5 g of hexachloroplatinic acid was
gradually added to and was dissolved completely in
an 8% aqueous solution of gelatin by heating to 90 C.
Next, 0.6 g of KETJENBLACK EC-600J was introduced as
carrier carbon. Degassing was performed under
reduced pressure, and stirring was performed. After
being cooled down slowly to room temperature, this
solution was cooled for three hours at 4 C. As a
result, a gel was obtained. The subsequent process
was performed in the same manner as in the first
example implementation. As a result, the Pt
catalyst of this example implementation was obtained.
[12th Example Implementation]
1.5 g of hexachloroplatinic acid was
gradually added to and was dissolved completely in a
5% aqueous solution of agar by heating to 90 C.
Next, 0.6 g of KETJENBLACK EC-600J was introduced as
carrier carbon. Degassing was performed under
reduced pressure, and stirring was performed. After
being cooled down slowly to room temperature, this
solution was cooled for three hours at 4 C. As a
result, a gel was obtained. The subsequent process
was performed in the same manner as in the first
example implementation. As a result, the Pt
catalyst of this example implementation was obtained.
[13th Example Implementation]
200 ml of a 20% aqueous solution of
carboxymethycellulose and 200 ml of a 2% aqueous
solution of oligo (ethyleneoxide) acrylate 4,4'-
bisphenol A-diacrylate were mixed. Further, 1.5 g
of hexachloroplatinic acid was added, and was
dissolved completely by heating to 60 C. Next,
after adding 40 ml of a 10% aqueous solution of
potassium persulfate, 0.6 g of KETJENBLACK EC-600J
was introduced as carrier carbon. Degassing was
performed under reduced pressure, and stirring was
performed. The subsequent process was performed in
CA 02498218 2005-03-08
-25-
the same manner as in the first example
implementation. As a result, the Pt catalyst of
this example implementation was obtained.
[14th Example Implementation]
200 ml of a 20% aqueous solution of
polyethyleneoxide-acrylate and 200 ml of a 2%
aqueous solution of oligo (ethyleneoxide) acrylate
4,4'-bisphenol A-diacrylate were mixed. Further,
1.5 g of hexachloroplatinic acid was added, and was
dissolved completely by heating to 60 C. Next,
after adding 40 ml of a 10% aqueous solution of
potassium persulfate, 0.6 g of KETJENBLACK EC-600J
was introduced as carrier carbon. Degassing was
performed under reduced pressure, and stirring was
performed. The subsequent process was performed in
the same manner as in the first example
implementation. As a result, the Pt catalyst of
this example implementation was obtained.
[15th Example Implementation]
1.5 g of hexachloroplatinic acid was
gradually added to and was dissolved completely in
500 ml of a 10% aqueous solution of pectin by
heating to 60 C. Next, 0.6 g of KETJENBLACK EC-600J
was introduced as carrier carbon. Degassing was
performed under reduced pressure, and stirring was
performed. The subsequent process was performed in
the same manner as in the second example
implementation. As a result, the Pt catalyst of
this example implementation was obtained.
[16th Example Implementation]
1.5 g of hexachloroplatinic acid was
gradually added to and was dissolved completely in
500 ml of a 10% aqueous solution of
polyethyleneglycol (5000 molecular weight) by
heating to 60 C. Next, 0.6 g of KETJENBLACK EC-600J
was introduced as carrier carbon. Degassing was
performed under reduced pressure, and stirring was
CA 02498218 2005-03-08
-26-
performed. The subsequent process was performed in
the same manner as in the second example
implementation. As a result, the Pt catalyst of
this example implementation was obtained.
[17th Example Implementation]
1.5 g of hexachloroplatinic acid was
gradually added to and was dissolved completely in
500 ml of a 10% aqueous solution of polyacrylamide
(2000 molecular weight) by heating to 60 C. Next,
0.6 g of KETJENBLACK EC-600J was introduced as
carrier carbon. Degassing was performed under
reduced pressure, and stirring was performed. The
subsequent process was performed in the same manner
as in the second example implementation. As a
result, the Pt catalyst of this example
implementation was obtained.
[18th Example Implementation]
200 ml of a 30% aqueous solution of
acrylamide and 200 ml of a 2% aqueous solution of
bis-acrylamide were mixed. Further, 0.3 g of
ruthenium trichloride was added, and was dissolved
completely by heating to 60 C. Next, after adding
40 ml of a 10% aqueous solution of potassium
persulfate, 0.6 g of KETJENBLACK EC-600J was
introduced as carrier carbon. Degassing was
performed under reduced pressure, and stirring was
performed. The subsequent process was performed in
the same manner as in the first example
implementation and thereafter in the third example
implementation. As a result, the Pt catalyst of
this example implementation formed of Ru and Pt was
obtained.
[19th Example Implementation]
200 ml of a 30% aqueous solution of
acrylamide and 200 ml of a 2% aqueous solution of
bis-acrylamide were mixed. Further, 1.00 g of
hexachloroplatinic acid and 0.25 g of ruthenium
- - ----------
CA 02498218 2005-03-08
-27-
trichloride were added so that the Pt-Ru molar ratio
was 2:1, and were dissolved completely by heating to
60 C. Next, after adding 40 ml of a 10% aqueous
solution of potassium persulfate, 0.6 g of
KETJENBLACK EC-600J was introduced as carrier carbon.
Degassing was performed under reduced pressure, and
stirring was performed. The subsequent process was
performed in the same manner as in the first example
implementation. As a result, the Pt-Ru alloy
catalyst of this example implementation was obtained.
[Comparative Example]
1.5 g of hexachloroplatinic acid was added
to and was dissolved completely in 400 ml of water
by heating to 60 C. Next, 0.6 g of KETJENBLACK EC-
600J was introduced as conductive carrier carbon
particles. Degassing was performed under reduced
pressure, and stirring was performed. This solution
was subjected to bubbling with nitrogen gas so as to
reduce the concentration of oxygen in the solution.
Next, 1000 ml of a 3.5% aqueous solution
of formaldehyde was gradually added to this mixture.
After being heated for 2 hours at 80 C, the mixture
was left at rest for 10 hours at room temperature.
Next, the formaldehyde aqueous solution was
discharged, washing was performed lightly with water,
and suction and filtration were performed. As a
result, the Pt catalyst of this comparative example
was obtained.
[Evaluation]
Fuel cells were formed using the catalysts
of the above-described first through 19th example
implementations and comparative example. 20g of a
5% by mass solution of Nafion was added to 2 g of
each catalyst. They were kneaded to be formed like
a paste. Next, it was applied to carbon paper (200
cm2 in area) by doctor blade coating to be 60 m in
thickness. After moisture was evaporated, it was
CA 02498218 2005-03-08
-28-
adhered to one side of a polymer solid electrolyte
membrane (Nafion N-115 of DuPont [127 [tm in
thickness]). An electrode for an air electrode
formed in the same manner was adhered to the other
side. A stainless steel mesh was pressed and
attached to each electrode as a collector. These
were contained in an acrylic case. A 10% by mass
aqueous solution of methanol was supplied to the
fuel electrode side at a rate of 30 ml/min., and air
was supplied to the air electrode at a rate of 50
ml/min.
A load was connected to each of these fuel
cells to measure power generation efficiency.
FIG. 8 is a diagram showing the power
generation efficiency of each of the example
implementations and comparative example. The power
generation efficiency is expressed by power per fuel
cell electrode surface area (W/cm2).
Referring to FIG. 8, the power generation
efficiencies of the first and second example
implementations were improved to be 1.55-1.65 times
that of the conventional Pt catalyst of the
comparative example. The power generation
efficiencies of the third and fourth example
implementations, in which catalyst particulates were
further precipitated and attached with respect to
the first and second example implementations, were
improved to be 1.90-1.95 times that of the
comparative example.
A detailed description is given above of
preferred example implementations according to the
present invention. However, the present invention
is not limited to the specific embodiments, and
variations and modifications may be made within the
scope of the present invention recited in CLAIMS.
For instance, the 18th example
implementation may be combined suitably with the
CA 02498218 2005-03-08
-29-
first through 17th example implementations except
the third and fourth example implementations.
Further, the first, second and fifth through 18th
example implementations may be combined with the
third and fourth example implementations. Further,
the noble metals of a catalyst layer or catalyst
particles may be suitably combined.
INDUSTRIAL APPLICABILITY
According to the present invention, by
forming a catalyst layer on the surface of a
conductive carrier, a catalyst for a fuel cell
having high activity and a high rate of reaction
with fuel, a method of manufacturing the same, and a
fuel cell employing the catalyst for a fuel cell can
be provided.