Note: Descriptions are shown in the official language in which they were submitted.
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
ANTIDEPRESSANT PIPERIDINE DERIVATIVES OF HETEROCYCLE-FUSED
BENZODIOXANS
Cross-Reference to Related Applications
[0001] This application claims the benefit of U.S. Application Serial No.
60/410,033,
filed September 12, 2002, the disclosure of which is incorporated herein by
reference
in its entirety.
Background of the Invention
[0002] Major depression is a serious health problem affecting more than 5% of
the
population, with a life-time prevalence of 15-20%.
[0003] Selective serotonin reuptake inhibitors have produced success in
treating
depression and related illnesses and have become among the most prescribed
drugs. They nonetheless have a slow onset of action, often taking several
weeks to
produce their full therapeutic effect. Furthermore, they are effective in less
than two-
thirds of patients.
[0004] Serotonin selective reuptake inhibitors (SSRIs) are well known for the
treatment of depression and other conditions. SSRIs work by blocking the
neuronal
reuptake of serotonin, thereby increasing the concentration of serotonin in
the
synaptic space, and thus increasing the activation of postsynaptic serotonin
receptors.
(0005] However, although a single dose of an SSRI can inhibit the neuronal
serotonin transporter which would be expected to increase synaptic serotonin,
long-
term treatment is required before clinical improvement is achieved.
[0006] It has been suggested that the SSRIs increase the serotonin levels in
the
vicinity of the serotonergic cell bodies and that the excess serotonin
activates
somatodendritic autoreceptors, 5HT,A receptors, causing a decrease in
serotonin
release in major forebrain areas. This negative feedback limits the increment
of
synaptic serotonin that can be induced by antidepressants.
-1 -
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
[0007] A 5HT,A antagonist would limit the negative feedback and should improve
the efficacy of the serotonin reuptake mechanism (Perez, V., et al., The
Lancet,
349:1594-1597 (1997)). Such a combination therapy would be expected to speed
up
the effect of the serotonin reuptake inhibitor.
(0008] Thus, it is highly desirable to provide improved compounds which both
inhibit serotonin reuptake and which are antagonists of the SHT~A receptor.
Description of the Invention
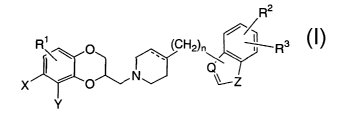
[0009] In accordance with this invention, there is provided a group of novel
compounds of Formula I:
R2
' . CH
R p ~ 2)n ~ R
~\
~~ Z
X ~ ,O
Y
wherein
R', R2 and R3 are, independently, hydrogen, hydroxy, halo, cyano,
carboxamido, carboalkoxy of two to six carbon atoms, trifluoromethyl,
alkyl of 1 to 6 carbon atoms, alkoxy of 1 to 6 carbon atoms, alkanoyl of 2
to 6 carbon atoms, alkanoyloxy of 2 to 6 carbon atoms, amino, mono- or
di-alkylamino in which each alkyl group has 1 to 6 carbon atoms,
alkanamido of 2 to 6 carbon atoms, alkanesulfonyl of 1 to 6 carbon atoms
or alkanesulfonamido of 1 to 6 carbon atoms;
X and Y are, independently, hydrogen, hydroxy, halo, cyano, carboxamido,
carboalkoxy of two to six carbon atoms, trifluoromethyl, alkyl of 1 to 6
carbon atoms, alkoxy of 1 to 6 carbon atoms, alkanoyl of 2 to 6 carbon
atoms, alkanoyloxy of 2 to 6 carbon atoms, amino, mono- or di-alkylamino
in which each alkyl group has 1 to 6 carbon atoms, alkanamido of 2 to 6
carbon atoms, alkanesulfonyl of 1 to 6 carbon atoms or
alkanesulfonamido of 1 to 6 carbon atoms, or X and Y, taken together,
-2-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
form -N=C(R°)-C(RS)=N-, -N=C(R4)-C(R6)=CH-, -N=C(R4)-N=CH-, -
N=C(R4)-O-, -NH-C(R')=N- or -NH-C(R8)=CH-;
R4 and R5 are, independently, hydrogen, halo, amino, mono- or di-alkylamino
in which each alkyl group has 1 to 6 carbon atoms or alkyl of 1 to 6 carbon
atoms;
R6 is hydrogen or alkyl of 1 to 6 carbon atoms;
R' is hydrogen, halo, trifluoromethyl, pentafluoroethyl, amino, mono- or di-
alkylamino in which each alkyl group has 1 to 6 carbon atoms or alkyl of 1
to 6 carbon atoms;
R8 is hydrogen, halo, trifluoromethyl, pentafluoroethyl or alkyl of 1 to 6
carbon
atoms;
the dotted line represents an optional double bond;
Z is oxygen or sulfur;
O is carbon or nitrogen;
n is0orl;
or a pharmaceutically acceptable salt thereof.
[0010] R' is preferably hydrogen, halo, cyano, trifluoromethyl, alkyl of 1 to
6 carbon
atoms or alkoxy of 1 to 6 carbon atoms. More preferably, R' is hydrogen, halo
or
alkoxy of 1 to 6 carbon atoms. In still more preferred embodimants of the
present
invention, R' is hydrogen.
[0011] R2 and R3 are preferably independently selected from hydrogen, hydroxy,
halo, cyano, carboxamido, alkyl of 1 to 6 carbon atoms, or alkoxy of 1 to 6
carbon
atoms. In still more preferred embodiments of the present invention RZ and R3
are
preferably independently selected from hydrogen, cyano or halogen.
[0012] R4 and R5 are preferably independently hydrogen, amino or alkyl of 1 to
6
carbon atoms. More preferably, R4 and R5 are independently hydrogen or alkyl
of 1
to 3 carbon atoms.
[0013] R' and R$ are preferably independently selected from hydrogen,
trifluoromethyl, pentafluoroethyl or alkyl of 1 to 6 carbon atoms. More
preferably, R'
and RB are independently hydrogen, trifluoromethyl or alkyl of 1 to 3 carbon
atoms.
-3-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
[0014] R6 is preferably hydrogen or alkyl of 1 to 3 carbon atoms, Z is
preferably
sulfur, Q is preferably carbon, n is preferably 0 and the dotted line
represents a
double bond.
[0015] In other preferred embodiments of the invention is provided compounds
of
Formula la.
S
n 1 ( R3
N ~~ Ra
Ra
R
la
wherein R','R2, R3, R4 and R6 are as described above.
[0016] In still other preferred embodiments of the invention is provided
compounds
of Formula Ib.
S
3
R~\ O \ I ~ ~~R
i N ~ ~\ R2
HN~ ~O
R~$
Ib
wherein R', Rz, R3 and R8 are as described above.
[0017] This invention relates to both the R and S stereoisomers of the
benzodioxan
methylamines as well as to mixtures of the R and S stereoisomers. Throughout
this
application, the name of the product of this invention, where the absolute
configuration of the compounds of the invention is not indicated, is intended
to
embrace the individual R and S enantiomers as well as mixtures of the two. In
some
-4-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
embodiments of the present invention the S enantiomer is preferred. For
certain of
the compounds of the invention (ie, X and Y form an imidazole), tautomeric
forms
may exist. This application thus encompasses all tautomeric forms of compounds
of
the present invention.
[0018] Where a stereoisomer is preferred, it may, in some embodiments be
provided substantially free of the corresponding enantiomer. Thus, an
enantiomer
substantially free of the corresponding enantiomer refers to a compound which
is
isolated or separated via separation techniques or prepared free of the
corresponding enantiomer. "Substantially free," as used herein, means that the
compound is made up of a significantly greater proportion of one stereoisomer.
In
preferred embodiments the compound is made up of at least about 90% by weight
of
a preferred stereoisomer. In other embodiments of the invention, the compound
is
made up of at least about 99% by weight of a preferred stereoisomer. Preferred
stereoisomers may be isolated from racemic mixtures by any method known to
those
skilled in the art, including high performance liquid chromatography (HPLC)
and the
formation and crystallization of chiral salts or prepared by methods described
herein.
See, for example, Jacques, et al., Enantiomers, Racemates and Resolutions
(Wiley
Interscience, New York, 1981 ); Wilen, S.H., et al., Tetrahedron 33:2725
(1977); Eliel,
E.L. Stereochemistry of Carbon Compounds (McGraw-Hill, NY, 1962); Wilen, S.H.
Tables of Resolving Agents and Optical Resolutions p. 268 (E.L. Eliel, Ed.,
Univ. of
Notre Dame Press, Notre Dame, IN 1972).
[0019] "Alkyl," as used herein, refers to an aliphatic hydrocarbon chain and
includes straight and branched chains such as methyl, ethyl, n-propyl,
isopropyl, n-
butyl, isobutyl, sec-butyl, t-butyl, n-pentyl, isopentyl, neo-pentyl, n-hexyl,
and
isohexyl. Lower alkyl refers to alkyl having 1 to 3 carbon atoms.
[0020] "Alkanamido," as used herein, refers to the group R-C(=O)-NH- where R
is
an alkyl group of 1 to 5 carbon atoms.
[0021] "Alkanoyl," as used herein, refers to the group R-C(=O)- where R is an
alkyl
group of 1 to 5 carbon atoms.
-5-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
[0022] "Alkanoyloxy," as used herein, refers to the group R-C(=O)-O- where R
is an
alkyl group of 1 to 5 carbon atoms.
(0023] "Alkanesulfonamido," as used herein, refers to the group R-S(O)z-NH-
where R is an alkyl group of 1 to 6 carbon atoms.
[0024] "Alkanesulfonyl," as used herein, refers to the group R-S(O)2- where R
is an
alkyl group of 1 to 6 carbon atoms.
[0025] "Alkoxy," as used herein, refers to the group R-O- where R is an alkyl
group
of 1 to 6 carbon atoms.
[0026] "Carboxamido," as used herein, refers to the group NH2-C(=O)- .
[0027] "Carboalkoxy," as used herein, refers to the group R-O-C(=O)- where R
is
an alkyl group of 1 to 5 carbon atoms.
[0028] "Halogen" (or "halo"), as used herein, refers to chlorine, bromine,
fluorine
and iodine.
[0029] Pharmaceutically acceptable salts are those derived from such organic
and
inorganic acids as: acetic, lactic, citric, cinnamic, tartaric, succinic,
fumaric, malefic,
malonic, mandelic, malic, oxalic, propionic, hydrochloric, hydrobromic,
phosphoric,
nitric, sulfuric, glycolic, pyruvic, methanesulfonic, ethanesulfonic,
toluenesulfonic,
salicylic, benzoic, and similarly known acceptable acids.
[0030] Specific examples of compounds of Formula I are:
2-(4-Benzo[b]thiophen-3-yl-3,6-dihydro-2H-pyridin-1-ylmethyl)-8-methyl-2,3-
dihydro-
[1,4]dioxino[2,3-f]quinoline;
2-(4-Benzo[b]thiophen-2-yl-3,6-dihydro-2H-pyridin-1-ylmethyl)-8-methyl-2,3-
dihydro-
[1,4]dioxino[2,3-f]quinoline;
-6-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
2-[4-(5-Fluoro-benzo[b]thiophen-3-yl)-3,6-dihydro-2H-pyridin-1-ylmethyl]-8-
methyl-
2,3-dihydro-[1,4]dioxino[2,3-f]quinoline;
2-[4-(7-Methoxy-benzofuran-3-yl)-3,6-dihydro-2H-pyridin-1-ylmethyl]-8-methyl-
2,3-
dihydro-[1,4]dioxino[2,3-f]quinoline;
2-[4-(5-Fluoro-benzo[b]thiophen-3-yl)-3,6-dihydro-2H-pyridin-1-ylmethyl]-2,3-
dihydro-
[1,4]dioxino[2,3-f]quinoline;
2-(4-Benzo[b]thiophen-3-yl-3,6-dihydro-2H-pyridin-1-ylmethyl)-2,3-dihydro-
[1,4]dioxino[2,3-f]quinoline;
2-(4-Benzo[b]thiophen-3-yl-3,6-dihydro-2H-pyridin-1-ylmethyl)-2,3-dihydro-7H-
[1,4]dioxino[2,3-a]indole;
2-[4-(5-Fluoro-benzo[b]thiophen-3-yl)-3,6-dihydro-2H-pyridin-1-ylmethyl]-2,3-
dihydro-
7H-(1,4]dioxino[2,3-a]indole;
8-(4-Benzo[b]thiophen-3-yl-3,6-dihydro-2H-pyridin-1-ylmethyl)-2-methyl-7,8-
dihydro-
[1,4]dioxino[2,3-g][1,3]benzoxazole;
2-(4-Benzo[b]thiophen-7-yl-3,6-dihydro-2H-pyridin-1-ylmethyl)-8-methyl-2,3-
dihydro-
[1,4]dioxino[2,3-f]quinoline;
2-(4-Benzofuran-2-yl-3,6-dihydro-2H-pyridin-1-ylmethyl)-8-methyl-2,3-dihydro-
[1,4]dioxino[2,3-f]quinoline;
2-(4-Benzofuran-2-yl-piperidin-1-ylmethyl)-8-methyl-2,3-dihydro-
[1,4]dioxino[2,3-
f]quinoline;
2-[4-(5-Chloro-benzo[b]thiophen-3-yl)-3,6-dihydro-2H-pyridin-1-ylmethyl]-8-
methyl-
2,3-dihydro-[1,4]dioxino[2,3-f]quinoline;
2-(4-Benzoxazol-2-yl-piperidin-1-ylmethyl)-8-methyl-2,3-dihydro-
[1,4]dioxino[2,3-
f]quinoline;
_7_
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
[0031] Compounds of the present invention are prepared in accordance with the
following general description and specific examples. Variables used are as
defined
for Formula I, unless otherwise noted. Specifically (Scheme 1 ), the
appropriately
substituted piperidine (2) is combined with a suitably substituted benzodioxan
methyltosylate or bromide (1 ) in a solvent such as dimethyl sulfoxide and
heated to a
temperature of 70-100°C for several hours as illustrated below.
Alternatively, the
appropriately substituted piperidine may be acylated with a suitably
substituted
benzodioxan carboxylic acid chloride, and the resulting amide reduced to the
amine
with a suitable reducing agent such as lithium aluminum hydride or borane/THF.
The
piperidine may also be combined with a suitably substituted benzodioxan
carboxaldehyde in the presence of a reducing agent such as sodium
cyanoborohydride.
R2
R ~ O CH / ~ 3 70-100 °C
2~n ~ ~ R ----
X / O OTs + Q DMSO
HN
Y
1 2
R2
CH / ~ s
R~~ O ( 2O\~ ~ R
'- NJ
X ~ _O
Y I
Scheme 1
[0032] Alternatively (Scheme 2), an appropriately substituted pyridine (3) may
be
alkylated with a suitably substituted benzodioxan methyltosylate or bromide (1
) by
heating the mixture in a high-boiling polar solvent such as dimethyl sulfoxide
to
produce the pyridinium ion (4). The pyridinium ion may be reduced to the
tetrahydropyridine by treatment with a suitable reducing agent such as sodium
borohydride in ethanol or directly to the piperidine by treatment with
hydrogen over a
suitable catalyst such as palladium on carbon.
_g_
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
R2
1
R ~ O H ~ ~ 3 70-100 °C
2)n\~ ~ R
X / O OTs ~ ~ Q DMSO
N / ~Z
Y 1 3
R2
R~\ O ~ C 2)n\~ ~ R NaBH4
Q EtOH
~ O N+ ~ ~Z
X
Y -OTs
R2
1 CH / ~ 3
R\~ O ~ 2)n\~ ~ R
O NJ a~Z
X
Y
Scheme 2
[0033] The benzodioxan methyltosylates and halides (1 ) are known compounds or
they may be prepared from the appropriately substituted salicylaldehydes by
the
method (a) described in Scheme 3 below. The salicylaldehyde (5) is alkylated
with
an epihalohydrin or glycidyl arylsulfonate in the presence of a suitable base.
The
aldehyde moiety is then converted to a phenol by a Baeyer-Villager procedure
and
cyclization to the benzodioxan methanol (7) effected by treatment with a base
such
as potassium carbonate. The alcohol is elaborated to a tosylate (1 ) by
treatment with
p-toluenesulfonyl chloride and a tertiary amine base or to a bromide by
treatment.
Alternatively (b), the substituted salicylaldehyde (8) may be protected with a
suitable
protecting group such as benzyl and the aldehyde (9) converted to a phenol
(10) as
described above. Following elaboration of the phenol to the glycidyl ether (11
) by
treatment with an epihalohydrin or glycidyl arylsulfonate, deprotection and
cyclization
are effected in a single step via a transfer hydrogenation in the presence of
sodium
bicarbonate. The bromide or tosylate is prepared as described above. Or the
benzodioxan methylbromide may be prepared from a suitably substituted guaiacol
_g_
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
(12) by procedure (c) shown above. The guiacol is alkylated with a glycidyl
arylsulfonate or an epihalohydrin as described above. The methyl ether (13) is
then
cleaved by treatment with 48% HBr; this also converts the epoxide to a
bromohydrin
(14). Cyclization directly to the benzodioxan methylbromide (1 ) is effected
by the
Mitsonobu procedure.
-10-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
(a) R~~ OH CIO R1~~ O O,
1. m-CPBA
---
X I ~ H NaH X I ~ H 2. K CO /MeOH
Y o Y ~ z s
6
R~ \ O CBr4/Ph3P R~~ O
~OH or
X ~ ~O TsCI, DMAP, base X ~ _O
Y Y
1 L = Br, OTs
(b) Y Y
X I ~ OH gngr~NaH X I ~ OBn 1. m-CPBA
R1 /~ H R1 /~ H 2. AI203/MeOH
O O
8 9
Y Y
X OBn CI ~ X ~ OBn 1. 1,4-cyclohexadiene
O ~ ~ Pd/C, NaHC03, EtOH
R1 /v 'OH NaH R1 / ~ O~O 2. TsCI, DMAP, base
11
1
R~ ~ o
~OTs
X
Y
1
Y ~ ~I Y
(c) X ~ OCH3 CIO X ~ OCH3 1. 48% HBr (aq)
1 /~pH NaH ' R1 /~O~
R O
12 13
Y
X ~ OH DEAD, Ph3P R ~~ O
1 j I / ~ Br
R O~ Br X O
OH Y
14 1
Scheme 3
-11-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
[0034] The 2,3-dihydro-1,4-dioxino(2,3-f]quinolin-2-ylmethylamines of the
invention
in which R4 is H are alternatively prepared as illustrated in Scheme 4 below.
Specifically, the appropriately substituted nitroguaiacol (15) is alkylated
with allyl
bromide in the presence of a suitable base such as sodium hydride and then
demethylated by a reagent such as sodium hydroxide. The resulting 4-nitro-2-
allyloxyphenol (17) is then alkylated with glycidyl tosylate or an
epihalohydrin in the
presence of a base such as sodium hydride and heated in a high boiling solvent
such
as mesitylene or xylene to effect both rearrangement of the allyl group and
cyclization of the dioxan ring. The resulting primary alcohol (19) is
converted to the
tosylate by reaction with p-toluenesulfonyl chloride in the presence of a
tertiary amine
or pyridine, or alternatively to a halide by reaction with carbon tetrabromide
or carbon
tetrachloride in combination with triphenylphosphine. The allyl side chain is
then
isomerized by treatment with catalytic bis-acetonitrile palladium (II)
chloride in
refluxing methylene chloride or benzene. Allylic oxidation of 20 with selenium
dioxide
in refluxing dioxane/water gives the o-nitrocinnamaldehyde, which upon
reduction
with iron in acetic acid cyclizes to the 2,3-dihydro-1,4-dioxino[2,3-
f]quinoline-2-
methyltosylate (21 ) or halide. Replacement of the tosylate or halide with the
appropriately substituted piperidine in some high boiling solvent such as
dimethyl
sulfoxide gives the title compounds of the invention.
-12-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
R \ \ OCH3 gr~ R ~\ OCH3 NaOH, DMSO/H20
--
02N ~ O-Na+ DMF p2N ~ p~ 80 deg
15 16
R~~ \ OH Ts0~0 R ~ \ C~~O mesitylene,
1 ~5 deg
ON I ~ O~ ~ON
NaH
17 18
R1
1 ) TsCI, pyr
O N 2) (CHaCN)2PdCl2 p2N~~'~O~U i s
CH2CI2 \
. .. 20 I R2
Ri /
p (CH2)n \ ~ Rs
1 ) Se02
2 Fe/AcOH I i ~OTs HN
N p ~Z
-w
21 DMSO
R2
1 CH / ~ 3
R\\ p ( 2)n\ \ R
N I ~ p NJ O~Z
I ,
Scheme 4
[0035] The 2,3-dihydro-1,4-dioxino[2,3-f]quinolin-2-ylmethylamines of the
invention
in which R4 is alkyl may be prepared from the nitro olefin described above in
the
following manner (Scheme 5). The rearranged olefin (20) is treated
sequentially with
ozone and a tertiary amine or with osmium tetroxide and sodium periodate to
give the
o-nitrobenzaldehyde (22). Condensation with the appropriate
-13-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
triphenylphosphorylidene ketone under Wittig conditions gives the o-
nitrocinnamyl
ketone (23), which upon reduction by iron in acetic acid, cyclizes to the
corresponding 2,3-dihydro-1,4-dioxino[2,3-f]quinoline-2-methyltosylate (24).
Replacement of the tosylate with the appropriately substituted piperidine as
above
gives the title compounds of the invention. Substitution of trimethyl
phosphonoacetate for the triphenylphosphorylidene ketone in the Wittig
procedure
above, followed by reduction of the nitro group with tin (II) chloride and
cyclization in
acid gives the compounds of the invention in which R4 is hydroxy. Treatment of
the
hydroxy derivative with an inorganic acid chloride such as phosphoryl chloride
or
bromide gives the compounds of the invention in which R4 is halo. Substitution
of
diethyl cyanomethylphosphonate for the triphenylphosphorylidene ketone in the
Wittig procedure above, followed by reduction of the nitro group with tin (II)
chloride
and cyclization in acid gives the compounds of the invention in which R4 is
amino.
O
R~ R' O Ph3P
1 ) 03, CH2C12 ~ ~ R4
i ~OTs
~OTs 02N O
02N O 2) (i-Pr)2EtN
H O
20 I 22
1
R,
I Fe/AcOH
02N s
23 24
Scheme 5
[0036] Compounds of the invention in which R' is attached to position 6 of the
2,3-
dihydro-1,4-dioxino[2,3-f]quinolin-2-ylmethylamines may be alternatively
prepared by
a variation of the Skraup quinoline synthesis according to Scheme 6 below. The
appropriately substituted benzodioxan methyltosylate (25) is nitrated under
standard
conditions with nitric acid in a solvent such as dichloroethane and the
resulting nitro
compound (26) reduced by treatment with hydrogen in the presence of a catalyst
such as platinum on sulfide carbon. Treatment of the resulting aniline (27)
with
-14-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
acrolein in the presence of hydrogen chloride and an oxidant such as p-
chloranil or
naphthoquinone gives the corresponding 2,3-dihydro-1,4-dioxino[2,3-f]quinoline
(28).
Replacement of the tosylate with the appropriately substituted piperidine as
above
gives the title compounds of the invention.
R1 I W O HN03 R1 ~ O H2, Pt/C-S
~OTs I ~ OTs
O 02N O
25 26
CHO
R' ~ O ~ R1 ~ O
y p ~ w
~OTs -chloranil ' I ~ ~OTs
H2N O HCI/MeOH N O
27 28
Scheme 6
[0037] The 2,3-dihydro-1,4-dioxino[2,3-f]quinazolin-2-ylmethylamines of the
invention are prepared as illustrated below (Scheme 7). The o-
nitrobenzaldehyde
(22) described above is converted to the oxime (29) by treatment with
hydroxylamine
hydrochloride in the presence of a suitable base such as sodium acetate and
the
nitro group reduced to the amine by hydrogenation over palladium on carbon.
Cyclization to the quinazoline N-oxide is effected by treatment at reflux with
the
appropriate ortho ester according to the method of Ostrowski (Heterocycles,
vol. 43,
No. 2, p. 389, 1996). The quinazoline N-oxide may be reduced to the
quinazoline
(30) by a suitable reducing agent such as hydrogen over Raney-nickel.
Alternatively,
an extended period of reflux in the ortho ester gives the reduced quinazoline
directly
via a disproportionation reaction and the 2,3-dihydro-1,4-dioxino[2,3-
f]quinazoline-2-
methyltosylate or halide may be isolated by column chromatography. Replacement
of the tosylate or halide with the appropriately substituted piperidine in
some high
boiling solvent such as dimethyl sulfoxide gives the title compounds of the
invention.
-15-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
R~ R~
\ O 1 ) NH20H, NaOAc ~ \ O
i OTs H N I ~ O OTs
02N O~ 'O 2) H2, Pd/C 2 HN
H 22 , ~ H 29
OH
R
i
4 ~ R \ \ O (CH2)n ~'~ R3
1 ) R C(OR )3 ~ , _ ~OTs
2) H2, ~Ra-Ni ~ ~ O HN J ~Z
R4 N
30 DMSO
R2
/~
R1 O (CH2)n ~~R3
Q
~N~ ~Z
~O
R4 N
Scheme 7
[0038] The 2,3-dihydro-1,4-dioxino[2,3-f]quinazolin-2-ylmethylamines of the
invention may be alternatively prepared from the rearranged olefin described
above
by the method outlined in Scheme 8 below. The nitro olefin (20) is first
reduced to
the aniline by treatment with a suitable reducing agent such as stannous
chloride
dihydrate in refuxing ethyl acetate and the resulting amine acylated with the
appropriate acyl halide or anhydride. The olefin (31 ) is then converted to
the
aldehyde (32) by cleavage with catalytic osmium tetroxide in the presence of
excess
sodium periodate. Cyclization directly to the 2,3-dihydro-1,4-dioxino[2,3-
f]quinazoline-2-methyltosylate (30) or halide is effected by treatment of the
amido
aldehyde (32) with ammonia and replacement of the tosylate or halide with the
appropriately substituted piperidine in some high boiling solvent such as
dimethyl
sulfoxide as described above gives the title compounds of the invention.
-16-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
1
Ri\ O 1 ) SnCl2
O
/~ OTS 4 4 ~ S
02N O 2) R COCI R
Et3N
1 1
Os04, Na104 O R ~ ~ O NH3 R ~ ~ O
/ ~OTs ' I / OTs
R H ~ ~O N ~ -O
H O R4/ 'N
32 30
Scheme 8
(0039] The 2,3-dihydro-1,4-dioxino[2,3-f]quinoxalin-2-ylmethylamines of the
invention are prepared as illustrated in Scheme 9 below. The o-
nitrobenzaldehyde
(22) described above is oxidized to the o-nitrobenzoic acid (33) by a suitable
oxidant
such as chromium trioxide (Jones' oxidation) or sodium chlorite and the acid
converted to the o-nitroaniline (34) with diphenylphosphoryl azide (DPPA) in
the
presence of a tertiary base such as diisopropylethylamine. Reduction of the
resulting
nitroaniline to the diamine (35) with hydrogen and palladium on carbon and
cyclization by treatment with the appropriate dicarbonyl compound (for
example,
glyoxal, 2,3-butanedione, 3,4-hexanedione) gives the 2,3-dihydro-1,4-
dioxino[2,3-
f]quinoxaline-2-methyltosylate (36) or halide. Replacement of the tosylate or
halide
with the appropriately substituted piperidine in some high boiling solvent
such as
dimethyl sulfoxide gives the title compounds of the invention.
-17-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
R1 R'
Cr03, H2S04 ~ ~ O Ph2PON3
OTs I ~ ~OTs
02N ~ ~O acetone 02N ~ O (i-Pr)2EtN
H O 22 O OH 33
O
R1 Ri Ra ll s
H2, Pd/C, HCI ~ ~ O R
O
~OTs I / ~OTs
02N ~ ~O H2N ~ ~O
NH2 NH2 2 HCI
34 35
R
/~
R \ ~ p (CH2)n ~ ERs
~OTs Q
N ~ ~O HN ~Z
~N
R4 36 DMSO R2
R5
/~
R1 p (CH2)n ~~R3
Q
~N~ ~Z
N ~ ~O
~N
R
R5
Scheme 9
[0040] The o-nitrobenzaldehyde (22) used in the chemistry described above may
be alternatively prepared as shown in scheme 10 below. The appropriate mono-
allylated catechol (37) is elaborated with glycidyl tosylate as described
above and
rearranged in refluxing mesitylene. Cyclization to the benzodioxan methanol
(39) is
-18-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
effected by treatment with sodium bicarbonate in ethanol and the alcohol is
converted to the tosylate (40) or halide as described above. After
rearrangement of
the double bond by treatment with catalytic bis-acetonitrile palladium (II)
chloride in
refluxing methylene chloride and cleavage with ozone or osmium
tetroxide/sodium
periodate as described above, the resulting aldehyde (41 ) is regioselectively
nitrated
with a combination of nitric acid and tin (IV) chloride.
1 Ts0 O Ri C>,~O
R \ \ OH ~ ~ \ 1 ) mesitylene,
reflux
O~ .--~ ~ ,
NaH 2) NaHC03, EtOH
37 38
R1 R1
1 ) TsCI, pyr \ ~ O 03, CH2C12
H 2) (CH3CN)2PdCl2 I ~ O OTs Et3N
CH2C12
1 1
R \ w O HN03, SnCl4 R ~ ~ O
O OTs O N I ~ O OTs
2
H O H O
41 22
Scheme 10
[0041] The 7,8-dihydro[1,4]dioxino[2,3-g][1,3]benzoxazol-8-ylmethylamines of
the
invention are prepared as illustrated in Scheme 8 below. The amido olefin (31
)
described in Scheme 8 is cleaved to the corresponding o-amidobenzaldehyde (32)
by treatment with catalytic osmium tetroxide in the presence of sodium
periodate.
The aldehyde is converted to the phenol (42) by treatment with meta-
chloroperoxybenzoic acid in a Baeyer-Villager reaction and cyclization to the
7,8-
dihydro[1,4]dioxino[2,3-g][1,3]benzoxazole (43) is effected by treatment at
reflux with
an appropriate dehydrating agent such as an ortho ester or an acid catalyst
such as
p-toluenesulfonic acid. Replacement of the tosylate or halide with the
appropriately
-19-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
substituted piperidine in some high boiling solvent such as dimethyl sulfoxide
gives
the title compounds of the invention.
1
O R \ ~ O Os04, Na104 O R ~ ~ O
I / ~OTs ~ a~ I i ~OTs
R H ~ 'O R H ~ ~O
:~1 H O 32
1
R \ O R \ O
1 ) m-CPBA O I \ ~ R4C(OR')3
--~ R4 ~ N ~ O OTs I , O OTs
N
2) AI203, MeOH H OH ~O
R2 42 R4 43 R2
(CH2)n
R R ~ \ O (CH2)n\ ~ ~ R
HN~ ~Z I N O
N~ 1' _o
DMSO O
Ra
Scheme 11
[0042] Alternatively (Scheme 12), the nitro olefin (20) may be reduced with
tin (II)
chloride as described in Scheme 8 above and protected with a suitable
protecting
group such as carbobenzoxy (Cbz) before the olefin is cleaved to the aldehyde
(45)
by treatment with osmium tetroxide/sodium periodate and the aldehyde converted
to
a phenol (46) by the Baeyer-Villager procedure. Deprotection by treatment with
hydrogen over palladium on carbon gives the o-aminophenol, (47) which is
cyclized
to the 7,8-dihydro[1,4]dioxino[2,3-g](1,3]benzoxazole (43) by treatment with
the
appropriate ortho ester, carboxylic acid or anhydride. Treatment of the o-
aminophenol
-20-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
with cyanogen bromide or chloride or a suitably substituted carbamoyl chloride
leads
to compounds of the invention in which R4 is amino. Treatment of the o-
aminophenol
with carbonyl diimidazole gives the oxazolone that leads to compounds of the
invention in which R' is halo via treatment with an inorganic anhydride such
as
phosphoryl chloride or bromide. Replacement of the tosylate with the
appropriately
substituted piperidine as above gives the title compounds of the invention.
1 ) SnCl2 P~
02N 2) CbzCl Cbz~N
Et3N H
R1 R'
Os04, Na104 ~ ~ O 1 ) m-CPBA ~ ~ O
Cbz~ ~ , ~OTs Cbz~ , ~OTs
N ~ ~O N ~ ~O
H H O 2) AI203, MeOH H OH
45 46
i
H2, Pd/C R ~ ~ O 4 ~ R \ ~ O
R C(OR )3 ~ , ~OTs
H2N O N OO
OH ~O
R4
4~ 43
Scheme 12
[0043] Compounds of the invention in which R' is hydrogen and R4 is alkyl are
most conveniently prepared according to scheme 13 below. The appropriate
2',3',4'-
trihydroxyacylphenone (48) is regioselectively alkylated with glycidyl
tosylate or an
epihalohydrin in the presence of a base such as sodium carbonate to give the
corresponding 7-acyl-8-hydroxybenzodioxan-2-methanol (49). Following
conversion
of the ketone to the oxime (50) by reaction with hydroxylamine hydrochloride
and
sodium acetate, cyclization to the oxazole (51 ) is effected by treatment with
phosphoryl chloride in the appropriate dimethylalkanoic acid amide. The
resulting
-21 -
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
7,8-dihydro-1,6,9-trioxa-3-aza-cyclopenta[a]naphthalene-8-methanol is
converted to
the tosylate (52) by treatment with p-toluenesulfonyl chloride in pyridine and
combined with the appropriate piperidine as described above to give the title
compounds of the invention.
O
OH TsO~ ~ p NH20H, NaOAc
R4 ~ / ~ R4 I ~ ~OH
OH Na2C03 ( ~ O
O OH O OH
48 49
O POC13 I ~ O
R4 i ~OH 4 i OH
O R CCONMe2 N
~NH OH
HO Ra
50 51
O
TsCI, pyridine
--_ I / ~OTs
N ~ _O
~O
Ra 52
Scheme 13
[0044] The 7,8-dihydro-3H-6,9-dioxa-1,3-diaza-cyclopenta[a]naphthalenes of the
invention are prepared as illustrated in Scheme 14 below. The diamine 35
described
in Scheme 9 is cyclized by treatment at reflux with the appropriate carboxylic
acid to
give the imidazole (53). Refluxing the diamine dihydrochloride in higher
boiling
carboxylic acids occasionally causes replacement of a tosylate group with a
chloride.
Replacement of the tosylate or halide with the appropriately substituted
piperidine in
some high boiling solvent such as dimethyl sulfoxide gives the 7,8-dihydro-3H-
6,9-
dioxa-1,3-diaza-cyclopenta[a]naphthalenes of the invention in which R' is
hydrogen,
perfluoroalkyl or alkyl. Treatment of the diamine described above with
cyanogen
bromide or chloride or a suitably substituted carbamoyl chloride leads to
compounds
-22-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
of the invention in which R' is amino. Treatment of the diamine with carbonyl
diimidazole gives the imidazolone which leads to compounds of the invention in
which R' is halo via treatment with an inorganic anhydride such as phosphoryl
chloride or bromide. Replacement of the tosylate with the appropriately
substituted
piperidine as above gives the title compounds of the invention.
1
R\\ p R' OH R\ \ O
p
H N ~ O OTs / O L
z '~ N
NH2 2 HCI ~NH
R7 L = OTs, CI
35 R2 53 2
R
CH
( 2)n~ ~ ~ R R~ O (CH2)n ~ ~ Rs
HN Q~ ~ \ Q
o NJ
N
DMSO ~NH
R7
Scheme 14
[0045] The 2,3-dihydro-7H-[1,4]dioxino[2,3-a]indoles of the invention are
prepared
as illustrated in Scheme 15 below. Specifically, the primary alcohol (19) from
the
Claisen rearrangement described in Scheme 4 is converted to the tosylate (54)
by
reaction with p-toluenesulfonyl chloride in the presence of a tertiary amine
or
pyridine, or alternatively to a halide by reaction with carbon tetrabromide or
carbon
tetrachloride in combination with triphenylphosphine. The allyl side chain is
then
cleaved to the aldehyde (55) by treatment with ozone at low temperature,
followed by
work-up with a tertiary base such as diisopropylethylamine or triethylamine,
or by
treatment with catalytic osmium tetroxide and sodium periodate. Reduction of
the
nitro group with hydrogen over platinum oxide leads directly to formation of
the indole
(56) in which R8 is hydrogen. Alternatively, the aldehyde may be treated with
an
appropriate alkyl Grignard reagent or with trifluoromethyl trimethylsilane in
the
-23-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
presence of cesium fluoride, then oxidized to a ketone with a suitable oxidant
such as
pyridinium chlorochromate (PCC) or the Swern reagent and reduced with hydrogen
over platinum oxide to give the indoles in which R$ is alkyl or
trifluoromethyl.
Replacement of the tosylate or halide with the appropriately substituted
piperidine in
some high boiling solvent such as dimethyl sulfoxide gives the title compounds
of the
invention.
R1
R \ O TsCI, (i-Pr)2EtN
OH ~ I ~ ~OTs
02N ~O DMAP, CH2C12 02N _O
19 ~ 54
P' R'
03, (i-Pr)2EtN H2, Pt02 \ ~ O
OTs , ~OTs
02N HN~ _O
.... H 56
R2
/ R2
CH
( 2)n R
R1 O (CH2)n ~ ERs
HN J ~Z ~ ~ Q
/ ~N~ ~Z
H N~ ~O
DMSO
H
Scheme 15
[0046] The 2,3-dihydro-7H-[1,4]dioxino[2,3-a]indoles of the invention may
alternatively be prepared from nitroaldehyde 21 by the following procedure
(Scheme
16). The o-nitrobenzaldehyde (22) is condensed with the appropriate
nitroalkane in
the presence of a suitable base catalyst to yield the corresponding o,p-
dinitrostyrene
(57). Reduction of both nitro groups with hydrogen over palladium on carbon is
-24-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
accompanied by cyclization to form the indole (58). Replacement of the
tosylate with
the appropriately substituted piperidine as above gives the title compounds of
the
invention.
i
R ~ ~ O R$CH2N02, KF R \ w O
~OTs ~ ~OTs
02N ~ ~O 02N ~O
N-methylmorpholine ~ R$
H O
22 N02 57
R1
H2, Pd/C \ O
i ~OTs
H N~ 'O
R~$
58
Scheme 16
[0047] The compounds of the invention may be resolved into their enantiomers
by
conventional methods or, preferably, the individual enantiomers may be
prepared
directly by substitution of (2R)-(-)-glycidyl 3-nitrobenzene-sulfonate or
tosylate (for
the S benzodioxan methanamine) or (2S)-(+)-glycidyl 3-nitrobenzene-sulfonate
or
tosylate (for the R enantiomer) in place of epihalohydrin or racemic glycidyl
tosylate
in the procedures above.
[0048] In yet another method, the heterocycle-fused benzodioxans of the
present
invention may be prepared in accordance with Scheme 17. The synthesis of
compound I is comprised of steps that begin with halogenation of 59 where R'
is alkyl
of 1-6 carbon atoms, with reagents such as N-halosuccinimide in acetonitrile
to give
60 (where Hal is halogen such as Br, CI or I). Deprotecting 60 with Lewis
acids such
as boron tribromide, boron trichloride, aluminum trichloride, ferric chloride,
or
trimethylsilyl iodide in a suitable solvent such as methylene chloride, or
with strong
erotic acids such as HBr and HCI gives the salt 61. Free base 61 may be
obtained
-25-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
by neutralization with an Amberlyst A-21 resin slurry in polar solvents such
as
ethanol or methanol.
[0049] Alkylation of 61, either as the free base or as the salt, with benzyl
or
0
substituted benzyl protected glycidyl ethers (y'°R~~, where R" is
benzyl,
substituted benzyl such as 4-bromobenzyl, 3,4-dimethoxybenzyl, 2- or 4-
nitrobenzyl,
or 4-methoxybenzyl) in suitable polar solvents such as DMSO, DMF, or DMA in
the
presence of bases such as sodium carbonate, potassium carbonate, or
triethylamine
gives 62. 62 was then cyclized using palladium catalysts such as
tris(dibenzylideneacetone)dipalladium, tetrakis(triphenylphosphine)palladium,
or
palladium acetate with ligands from the group consisting of (~) BINAP and
separate
enantiomers thereof, (~) Tol-BINAP and separate enantiomers thereof; 1-1'-
bis(diphenylphosphino) ferrocene, 1,3-bis(diphenylphosphino)propane, and 1,2
bis(diphenyl-phosphino)ethane in the presence of bases such as NaH, LiH, KH,
potassium carbonate, sodium carbonate, titanium carbonate, cesium carbonate,
potassium t butoxide or potassium phosphate tribasic in suitable solvent such
as
toluene, or alternatively, with copper catalyst such as copper iodide in the
presence
of bases such NaH, LiH, KH in a suitable solvent such as toluene to afford 63.
-26-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
R~~ OR' N_halosuccinimide R~~ OR' Lewis acid/CH2CI2
X I ~ CH3CN X I ~ Hal or strong erotic acid
Y Y
59 60
OH
R~~ OH O~OR" R~~ O~OR" Pd cat/ligand
or Cu cat
X I ~ Hal ~ I ~ Hal
Y Base, polar solventX Y Base, toluene
61 62
1 1
R \~ O Lewis acid/CH2CI2, R ~\ O
i ~--~,,~OR" or strong erotic acid ( ~ ~~-.,,OOH
X ~ 'O or hydrogenolysis X ~ ~O
Y Y o
63 64
R
R1 UH2)n
\ O Q \ R
Arylsulfonyl chloride ~ , ~..,"OR"' HN~ ~Z
X ~ -O
Base, CH2C12 Y Base, polar solvent
R2
R1 ~CH2)n
\ O Q \ R
X ~ O N~ ~Z
Y I
Scheme 17
[0050] Deprotection of quinoline 63 with Lewis acids such as boron tribromide,
boron trichloride, aluminum trichloride, ferric chloride, trimethylsilyl
iodide in a
suitable solvent such as methylene chloride, or with strong erotic acids such
as HBr
and HCI or under reductive cleavage conditions using Pd catalyst and hydrogen
transfer reagents such as hydrogen, cyclohexene, methyl cyclohexene, or
ammonium formate gives the heterocycle-fused benzodioxanmethanol 64. The
-27-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
hydroxyl moiety of 64 can be activated with an aryl- or alkylsulfonyl chloride
such as
p-toluenesulfonyl chloride, methanesulfonyl chloride, 2-, 3- or 4-
nitrobenzenesulfonyl
chloride, or 2- or 4-bromobenzenesulfonyl chloride in the presence of bases
such as
triethylamine or pyridine in suitable solvents such as methylene chloride,
THF, or
toluene to afford 65 where R"' is a sulfonate such as ~rtoluenesulfonate,
methanesulfonate, 2-, 3-, or 4-nitrobenzenesulfonate, or 2- or 4-
bromobenzenesulfonate. The final coupling of 65 with piperidines appropriate
to the
invention, in the presence of bases such as Hiinig's base
(diisopropylethylamine),
potassium carbonate, or sodium carbonate in polar solvents such as THF,
dioxane,
DMSO, DMF, or DMA affords the compounds of the invention I.
[0051] The phenols, guaiacols, catechols, 2',3',4'-trihydroxyacylphenones and
benzodioxan methyltosylates appropriate to the above chemistry are known
compounds or can be prepared by one schooled in the art. The appropriately
substituted piperidines and tetrahydropyridines are known compounds or can
readily
be prepared by one schooled in the art, for example as illustrated in Scheme
18
below for 4-(7-methoxybenzofuran-3-yl)-1,2,3,6-tetrahydropyridine. The
appropriately substituted benzofuranone (66) is converted to the triflate (67)
by treatment with triflic anhydride and a tertiary base such as triethylamine.
Conversion to the dioxaborolane (68) is effected by treatment with 4,4,5,5-
tetramethyl-1,3,2-dioxaborolane in the presence of a tertiary base and a
suitable
palladium catalyst such as [1,1'-bis(diphenylphosphino)ferrocene]palladium
(II)
chloride. The dioxaborolane (68) is coupled with the appropriately protected
azaheterocyclic triflate under Suzuki conditions involving a palladium
catalyst such as
tetrakis(triphenylphospine)palladium(0) to give (69), which following
deprotection
affords the substituted tetrahydropyridine (70) suitable for production of
certain of the
compounds of the invention. Reduction of the double bond in 70 by
hydrogenation
over a catalyst such as palladium on carbon gives the substituted piperidine,
which is
needed for the production of still other compounds of the invention.
-28-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
O
O
Tf20, Et3N \ OTf HB-O \ B-O
\ \
Et3N
/O 66 /O 67 PdCl2(dppf)2 /O 68
Boc
Boc-N. 'rOTf NH
TFA
CsC03, LiCI, H20
Pd(PPh3)4 ~ O
/O 70
Scheme 18
[0052] The substituted pyridines appropriate to Scheme 2 are known compounds
or may be readily prepared by one schooled in the art by the method
illustrated for 4-
benzo[b]thiophen-7-yl-pyridine in Scheme 19 below. The suitably substituted
bromobenzothiophene or bromobenzofuran (71 ) may be coupled with pyridine-4-
boronic acid under Suzuki conditions involving a palladium catalyst such as
[1,1'-
bis(diphenylphosphino)ferrocene]palladium (II) chloride to give intermediates
(72)
useful for the production of the compounds of the invention.
K3P04, H20
Br N~ ~ B(OH)2
71 \N ~ 72
Pd(dppf)2C12 CH2CI2 I ~ \
S ~ S
Scheme 19
[0053] A protocol similar to that used by Cheetham et al. (Neuropharmacol.
32:737,
1993) was used to determine the affinity of the compounds of the invention for
the
_29_
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
serotonin transporter. The compound's ability to displace 3H-paroxetine from
male
rat frontal cortical membranes was determined using a Tom Tech filtration
device to
separate bound from free 3H-paroxetine and a Wallac 1205 Beta Plate~ counter
to
quantitate bound radioactivity. Ki's thus determined for standard clinical
antidepressants are 1.96 nM for fluoxetine, 14.2 nM for imipramine and 67.6 nM
for
zimelidine. A strong correlation has been found between 3H-paroxetine binding
in rat
frontal cortex and 3H-serotonin uptake inhibition.
[0054] High affinity for the serotonin 5-HT,A receptor was established by
testing the
claimed compound's ability to displace [3H] 8-OHDPAT (dipropylaminotetralin)
from
the 5-HT,A serotonin receptor following a modification of the procedure of
Hall et al.,
J. Neurochem. 44, 1685 (1985) which utilizes CHO cells stably transfected with
human 5-HT,A receptors. The 5-HT,A affinities for the compounds of the
invention
are reported below as Ki's.
[0055] Antagonist activity at 5-HT,A receptors was established by using a 35S-
GTP~yS binding assay similar to that used by Lazareno and Birdsall (Br. J.
Pharmacol.
109: 1120, 1993), in which the test compound's ability to affect the binding
of 35S-
GTPyS to membranes containing cloned human 5-HT,A receptors was determined.
Agonists produce an increase in binding whereas antagonists produce no
increase
but rather reverse the effects of the standard agonist 8-OHDPAT. The test
compound's maximum inhibitory effect is represented as the haX, while its
potency is
defined by the ICSp.
[0056] The results of the three standard experimental test procedures
described in
the preceding three paragraphs were as follows:
5-HT Transporter 5-HT,A Receptor 5-HT,A Function
Affinity Affinity
CompoundKI ~nM) KI nM IC50 (nM) (Ima
Example 9.25 4.89 498.5 (100)
1
Example 78.00 21.59
2
Example 18.50 15.88 436.6 (87.4)
3
Example 70.00 2.66 167.3 (91.0)
4
Example 35.00 23.43 1000.0 (30.0)
Example 8.50 14.53 145.5 (100)
6
-30-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
5-HT Transporter 5-HT,A Receptor 5-HT,A Function
Affinity Affinity
CompoundKI (nM) KI nM ICSp nM Ima
Example 2.48 4.74 25.8 (100)
7
Example 33.00 3.45 146.9 (100)
8
Example 13.00 20.20 1978.0 (50.0)
9
Example 10.00 34.87 391.9 (40.5)
Example 213.00 15.71 15530.0 (70.0)
11
Example 109.00 8.67 589.6 (100)
12
Example 94.00 134.90 2516.0 (100)
13
Example 17.00 19.75 166.8 (100)
14
[0057] Like the antidepressants fluoxetine, paroxetine and sertraline, the
compounds of this invention have the ability to potently block the reuptake of
the
brain neurotransmitter serotonin. They are thus useful for the treatment of
diseases
commonly treated by the administration of serotonin selective reuptake
inhibitor
(SSRI) antidepressants, such as depression (including but not limited to major
depressive disorder, childhood depression and dysthymia), anxiety, panic
disorder,
post-traumatic stress disorder, premenstrual dysphoric disorder (also known as
pre-
menstrual syndrome), attention deficit disorder (with and without
hyperactivity),
obsessive compulsive disorders (including but not limited to
trichotillomania),
obsessive compulsive spectrum disorders (including but not limited to autism),
social
anxiety disorder, generalized anxiety disorder, obesity, eating disorders such
as
anorexia nervosa and bulimia nervosa, vasomotor flushing, cocaine and alcohol
addiction, sexual dysfunction (including but not limited to premature
ejaculation),
incontinence (including, but not limited to fecal incontinence, urge
incontinence,
overflow incontinence, passive incontinence, reflex incontinence, stress
urinary
incontinence urinary exertional incontinence and urinary~incontinence), and
related
illnesses. Moreover, the compounds of this invention have potent affinity for
and
antagonist activity at brain SHT,A serotonin receptors. Recent clinical trials
employing drug mixtures (eg, fluoxetine and pindolol) have demonstrated a more
rapid onset of antidepressant efficacy for a treatment combining SSRI activity
and
5HT,A antagonism (Blier and Bergeron, 1995; F. Artigas et al., 1996; M. B.
Tome et
al., 1997). The compounds of the invention are thus exceedingly interesting
and
useful for treating depressive illnesses.
-31 -
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
[0058] Thus the present invention provides methods of treating, preventing,
inhibiting or alleviating each of the maladies listed above in a mammal,
preferably in
a human, the methods comprising providing a pharmaceutically effective amount
of a
compound of this invention to the mammal in need thereof.
[0059] Also encompassed by the present invention are pharmaceutical
compositions for treating or controlling disease states or conditions of the
central
nervous system comprising at least one compound of Formula I, mixtures
thereof,
and or pharmaceutical salts thereof, and a pharmaceutically acceptable carrier
therefore. Such compositions are prepared in accordance with acceptable
pharmaceutical procedures, such as described in Remington's Pharmaceutical
Sciences, 17th edition, ed. Alfonoso R. Gennaro, Mack Publishing Company,
Easton,
PA (1985). Pharmaceutically acceptable carriers are those that are compatible
with the other ingredients in the formulation and biologically acceptable.
[0060] The compounds of this invention may be administered orally or
parenterally,
neat or in combination with conventional pharmaceutical carriers. Applicable
solid
carriers can include one or more substances that may also act as flavoring
agents,
lubricants, solubilizers, suspending agents, fillers, glidants, compression
aids,
binders or tablet-disintegrating agents or an encapsulating material. In
powders, the
carrier is a finely divided solid that is in admixture with the finely divided
active
ingredient. In tablets, the active ingredient is mixed with a carrier having
the
necessary compression properties in suitable proportions and compacted in the
shape and size desired. The powders and tablets preferably contain up to 99%
of
the active ingredient. Suitable solid carriers include, for example, calcium
phosphate,
magnesium stearate, talc, sugars, lactose, dextrin, starch, gelatin,
cellulose, methyl
cellulose, sodium carboxymethyl cellulose, polyvinylpyrrolidine, low melting
waxes
and ion exchange resins.
[0061] Liquid carriers may be used in preparing solutions, suspensions,
emulsions,
syrups and elixirs. The active ingredient of this invention can be dissolved
or
suspended in a pharmaceutically acceptable liquid carrier such as water, an
organic
-32-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
solvent, a mixture of both or pharmaceutically acceptable oils or fat. The
liquid
carrier can contain other suitable pharmaceutical additives such as
solubilizers,
emulsifiers, buffers, preservatives, sweeteners, flavoring agents, suspending
agents,
thickening agents, colors, viscosity regulators, stabilizers or osmo-
regulators.
Suitable examples of liquid carriers for oral and parenteral administration
include
water (particularly containing additives as above, e.g. cellulose derivatives,
preferably
sodium carboxymethyl cellulose solution), alcohols (including monohydric
alcohols
and polyhydric alcohols e.g. glycols) and their derivatives, and oils (e.g.
fractionated
coconut oil and arachis oil). For parenteral administration the carrier can
also be an
oily ester such as ethyl oleate and isopropyl myristate. Sterile liquid
carriers are used
in sterile liquid form compositions for parenteral administration.
[0062] Liquid pharmaceutical compositions that are sterile solutions or
suspensions
can be administered by, for example, intramuscular, intraperitoneal or
subcutaneous
injection. Sterile solutions can also be administered intravenously. Oral
administration may be either liquid or solid composition form.
[0063] Preferably the pharmaceutical composition is in unit dosage form, e.g.
as
tablets, capsules, powders, solutions, suspensions, emulsions, granules, or
suppositories. In such form, the composition is sub-divided in unit dose
containing
appropriate quantities of the active ingredient; the unit dosage forms can be
packaged compositions, for example packeted powders, vials, ampoules,
prefilled
syringes or sachets containing liquids. The unit dosage form can be, for
example, a
capsule or tablet itself, or it can be the appropriate number of any such
compositions
in package form.
[0064] The amount provided to a patient will vary depending upon what is being
administered, the purpose of the administration, such as prophylaxis or
therapy, and
the state of the patient, the manner of administration, and the like. In
therapeutic
applications, compounds of the present invention are provided to a patient
already
suffering from a disease in an amount sufficient to cure or at least partially
ameliorate
the symptoms of the disease and its complications. An amount adequate to
accomplish this is defined as a "therapeutically effective amount." The dosage
to be
-33-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
used in the treatment of a specific case must be subjectively determined by
the
attending physician. The variables involved include the specific condition and
the
size, age and response pattern of the patient. Generally, a starting dose is
about 5
mg per day with gradual increase in the daily dose to about 150 mg per day, to
provide the desired dosage level in the human.
[0065] Provide, as used herein, means either directly administering a compound
or
composition of the present invention, or administering a prodrug, derivative
or analog
which will form an equivalent amount of the active compound or substance
within the
body.
[0066] The present invention includes prodrugs of compounds of Formula I, la
and
Ib. Prodrug, as used herein, means a compound which is convertible in vivo by
metabolic means (e.g. by hydrolysis) to a compound of Formula I. Various forms
of
prodrugs are known in the art, for example, as discussed in Bundgaard, (ed.),
Design
of Prodrugs, Elsevier (1985); Widder, et al. (ed.), Methods in Enzymology,
vol. 4,
Academic Press (1985); Krogsgaard-Larsen, et al., (ed). "Design and
Application of
Prodrugs, Textbook of Drug Design and Development, Chapter 5, 113-191 (1991 ),
Bundgaard, et al., Journal of Drug Deliver Reviews, 8:1-38(1992), Bundgaard,
J. of
Pharmaceutical Sciences, 77:285 et seq. (1988); and Higuchi and Stella (eds.)
Prodrugs as Novel Drug Delivery Systems, American Chemical Society (1975).
[0067] The following examples illustrate the production of representative
compounds of this invention.
INTERMEDIATE 1
3-Allyloxy-4-methoxynitrobenzene
[0068] 97.5 g (0.51 mole) of the sodium salt of 5-nitroguaiacol was dissolved
in one
liter of DMF and 1.5 equivalents of allyl bromide added. The reaction was
heated to
65°C for two hours, after which time much of the dark color had
discharged and tlc
(1:1 CH2CI2/hexane) indicated loss of starting material. The solvent was
concentrated in vacuum and the residue washed with water. The product was
-34-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
isolated by filtration and dried in a vacuum. This gave 112 g of pale yellow
solid. A
sample recrystallized from methanol, gave m.p. 93-94°C.
INTERMEDIATE 2
2-Allyloxy-4-nitrophenol
[0069] To one liter of dimethyl sulfoxide was added 750 mL of 2 N aqueous
sodium
hydroxide and the mixture was heated to 65°C. The pale yellow solid 3-
allyloxy-4-
methoxynitrobenzene prepared above was added in portions over a 30 minute
period
and then the temperature was raised to 95°C and maintained for 3 hours,
after which
time the starting material had been consumed. The mixture was allowed to cool
and
poured into a mixture of 1 L ice and 1 L 2 N HCI. 73 Grams of crude but
homogeneous (by tlc 1:1 CH2CI2/hexane) desired product was isolated as a light
brown solid by filtration. This material was subsequently dissolved in 1:1
hexane/methylene chloride and filtered through silica gel to give 68 g of pale
yellow
solid, which, when recrystallized from ethyl/acetate/hexane, gave m.p. 61-
62°C. The
aqueous mother liquors from the initial crystallization above were extracted
with 2 L
of ethyl acetate. This was dried over sodium sulfate, filtered and evaporated
to a
dark oil. Column chromatography on silica with 1:1 CH2CI2/hexane gave an
additional 12 g of the title compound as a yellow solid. Elution with 2% MeOH
in
CHCI3 gave 12 g of a dark oil which slowly crystallized in vacuum. This proved
to be
the Claisen product, 3-allyl-4-nitrocatechol.
INTERMEDIATE 3
2-(2-Allyloxy-4-nitrophenoxymethyl)-oxirane
[0070] 20 g (0.50 mole) of 60% NaH/mineral oil was placed in a two liter flask
and
washed with 500 mL of hexane. 1 L of DMF was added, followed by 77 g (0.40
mole) of the 2-allyloxy-4-nitrophenol prepared in the previous step. Addition
of the
phenol was performed in portions under argon. After stirring the mixture for
30
minutes at room temperature under argon, 108 g (0.48 moles) of (R)-glycidyl
tosylate
was added and the mixture heated at 70-75°C under nitrogen overnight.
Upon
cooling, the DMF was removed in vacuum and replaced with one liter of
methylene
-35-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
chloride. This was washed with 500 mL portions of 2 N HCI, saturated sodium
bicarbonate and saturated brine and dried over sodium sulfate. The mixture was
filtered, concentrated to an oil in vacuum and column chromatographed on
silica gel
using 1:1 hexane/methylene chloride as eluant. This gave 43 g of product
contaminated with traces of the two starting materials, followed by 21 g of
pure
product as a pale yellow solid. The impure material was recrystallized from
1.2 L of
10% ethyl acetate/hexane to give 34 g of pure (homogeneous on silica gel tlc
with
1:1 hexane/methylene chloride) (R)-2-(2-allyloxy-4-nitrophenoxymethyl)-oxirane
(m.p.
64°C).
Elemental Analysis for: C12H13N05
Calc'd: C, 57.37; H, 5.21; N, 5.58
Found: C, 57.50; H, 5.21; N, 5.43
INTERMEDIATE 4
(8-AI Iyl-7-nitro-2,3-di hydro-benzo(1,4)dioxi n-2-yl)-methanol
[0071] (R)-2-(2-Allyloxy-4-nitrophenoxymethyl)-oxirane (20 g, 80 mmoles)
prepared
as above was heated at 155°C in mesitylene for 24 hours under nitrogen.
Filtration
of the black solid that formed gave 1.5 g of very polar material. Evaporation
of the
solvent in vacuum followed by column chromatography on silica gel with
methylene
chloride as eluant gave 10 g of recovered starting material and 7.5 g of the
desired
rearranged (S)-(8-allyl-7-nitro-2,3-dihydro-benzo(1,4)dioxin-2-yl)-methanol,
which
slowly crystallized on standing in vacuum (m.p. 67°C). The yield based
on recovered
starting material is 75%.
Elemental Analysis for: C12H13N05
Calc'd: C, 57.37; H, 5.21; N, 5.58
Found: C, 57.26; H, 5.20; N, 5.35
-36-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
INTERMEDIATE 5
Toluene-4-sulfonic acid 8-allyl-7-vitro-2,3-dihydro-benzo(1,4)dioxin-2-
ylmethyl
ester
[0072] 9.55 g (38.0 mmole) of (S)-(8-allyl-7-vitro-2,3-dihydro-
benzo(1,4)dioxin-2-
yl)-methanol was dissolved in 465 mL of pyridine, 29.0 g (152 mmole) of p-
toluenesulfonyl chloride was added and the mixture stirred at room temperature
under nitrogen overnight. Water was then added to quench the excess tosyl
chloride
and the solvent was removed in vacuum and replaced with methylene chloride.
This
solution was washed with 2 N HCI, with saturated sodium bicarbonate, and with
saturated brine, and dried over magnesium sulfate. Filtration, evaporation in
vacuum
and column chromatography on silica gel with 1:1 hexane/methylene chloride as
eluant gave 12.6 g (92%) of toluene-4-sulfonic acid (R)-allyl-7-vitro-2,3-
benzo(1,4)dioxin-2-ylmethyl ester, which slowly crystallized to a tan solid
(m.p. 60-
62°C) upon standing.
Elemental Analysis for: C1 gH 1 gN07S
Calc'd: C, 56.29; H, 4.72; N, 3.45
Found: C, 56.13; H, 4.58; N, 3.44
INTERMEDIATE 6
-Nitro-8-f 1-propenyll-2,3-dihydro-1,4-benzodioxin-2-yl~methyl 4
methylbenzenesulfonate
[0073] To a solution of 10.0 g (24.0 mmole) of (R)-[8-allyl-7-vitro-2,3-
dihydro-1,4-
benzodioxin-2-yl]methyl 4-methylbenzenesulfonate in 700 mL of benzene was
added
1.03 g of bis(acetonitrile)dichloropalladium (II) and the mixture was refluxed
under
nitrogen for 48 hours. The catalyst was then removed by filtration and the
filtrate
concentrated in vacuum to a brown oil. Column chromatography on silica gel
with
methylene chloride as eluant gave 7.2 g of the title compound as a mixture of
E and
Z isomers. A sample of {(2R)-7-vitro-8[(E)-1-propenyl]-2,3-dihydro-1,4-
benzodioxin-
2-yl}methyl 4-methylbenzenesulfonate was obtained as a yellow solid (m.p. 105-
106°C) by evaporation of a pure E isomer-containing fraction.
Elemental Analysis for: C1 gHi gN07S
-37-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
Calc'd: C, 56.29; H, 4.72; N, 3.45
Found: C, 56.12; H, 4.64; N, 3.39
INTERMEDIATE 7
~7-Nitro-8-f3-oxo-1-propenyll-2,3-dihydro-1,4-benzodioxin-2-yllmethyl 4
methvlbenzenesulfonate
[0074] {(2R)-7-nitro-8-(1-propenyl]-2,3-dihydro-1,4-benzodioxin-2-yl}methyl 4-
methyl benzenesulfonate (6.15 g, 15.2 mmole) was dissolved in 180 mL of
dioxane.
Selenium dioxide (4.20 g, 37.9 mmole) was then added, followed by 0.70 mL of
water. The heterogeneous mixture was heated at reflux under nitrogen for 5
hours.
Upon cooling, the reaction was filtered and concentrated in vacuum to yield a
dark
yellow solid. This was dissolved in minimal ethyl acetate and column
chromatographed on silica gel using 30% ethyl acetate in hexane as eluant to
give
5.75 g of the (R)-enantiomer of the title compound as a light yellow solid
(m.p. 138-
140°C).
Elemental Analysis for: C1 gH17NO8S
Calc'd: C, 54.41; H, 4.09; N, 3.34
Found: C, 54.10; H, 3.85; N, 3.31
INTERMEDIATE 8
2,3-Dihydrof1,41dioxinof2,3-flauinolin-2-ylmethyl 4-methylbenzenesulfonate
[0075] To a solution of {(2R)-7-nitro-8-[3-oxo-1-propenyl]-2,3-dihydro-1,4-
benzodioxin-2-yl}methyl 4-methylbenzenesulfonate (3.50 g, 8.35 mmole) in 200
mL
of acetic acid/ethanol (1:1 ) was added 2.35 g (42.1 mmole) of iron powder and
the
mixture was heated at reflux under nitrogen for 8 hours. After the reaction
was
complete, 150 mL of water was added and the mixture filtered through a pad of
celite. The filtrate was neutralized with saturated sodium bicarbonate and
extracted
with ethyl acetate. The extract was dried over magnesium sulfate, filtered and
evaporated in vacuum. The residue was column chromatographed on silica gel
using a gradient elution commencing with 20% ethyl acetate/hexane and ending
with
70% ethyl acetate/hexane to give 1.85 g of the (R)-enantiomer of the title
compound
-38-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
as a yellow oil. 'H-NMR (CDCI3): doublet 8.8 b (1 H); doublet 8.2 b (1 H);
doublet 7.8
8 (2 H); doublet 7.6 8 (1 H); multiplet 7.35 S (1 H); multiplet 7.25 8 (3 H);
multiplet 4.6
8 (1 H); multiplet 4.3-4.4 b (3 H); multiplet 4.2 8 (1 H); singlet 2.4 8 (3
H).
INTERMEDIATE 9
(8-Formyl-7-nitro-2,3-dihydro-1,4-benzodioxin -2-yl)methyl 4
methylbenzenesulfonate
[0076] {(2R)-7-Nitro-8-[1-propenyl]-2,3-dihydro-1,4-benzodioxin-2-yl}methyl 4-
methyl benzenesulfonate (10.5 g, 25.9 mmole) dissolved in 400 mL of methylene
chloride was treated with excess ozone at -78°C. Diisopropylethylamine
(11.5 mL,
66.0 mmole) was then added dropwise over 30 minutes and the mixture allowed to
come to room temperature and stir overnight under a nitrogen atmosphere. The
mixture was then diluted to 600 mL with methylene chloride, washed three times
with
100 mL portions of 2N HCI (aq), twice with 200 mL portions of saturated
aqueous
sodium bicarbonate and with 200 mL of saturated brine. The solution was dried
over
magnesium sulfate, filtered and concentrated in vacuum to a crude brown oil,
which
was column chromatographed on silica gel with 10% hexane/methylene chloride to
give 7.52 g of the (R)-enantiomer of the title compound as a yellow solid. 'H-
NMR
(CDCI3): doublet 7.8 8 (2 H); doublet 7.62 8 (1 H); doublet 7.4 8 (2 H);
doublet 7.0 S
(1 H); multiplet 4.4-4.6 8 (2 H); multiplet 4.2 8 (3 H); singlet 2.4 8 (3 H).
INTERMEDIATE 10
f7-Nitro-8-f(E)-3-oxo-1-butenyll-2,3-dihydro-1,4-benzodioxin-2-yl)methyl 4
methylbenzenesulfonate
[0077] To a solution of 3.00 g (7.37 mmole) of [(2R)-8-formyl-7- nitro-2,3-
dihydro-
1,4-benzodioxin -2-yl]methyl 4-methylbenzenesulfonate in 250 mL of toluene was
added 2.90 g (9.10 mmole) of 1-triphenylphosphorylidene-2-propanone. The
mixture
was stirred at room temperature under nitrogen for 5 hours, during which time
some
product precipitated from solution. The solvent was removed in vacuum and the
crude residue was column chromatographed on silica gel with methylene chloride
as
-39-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
eluant to give 3.0 g of the (R)-enantiomer of the title compound as a yellow
solid.'H-
NMR (CDCI3): doublet 7.8 S (2 H); doublet 7.6 8 (1 H); doublet 7.5 8 (2 H);
doublet
7.4 8 (2 H); doublet 6.95 8 (1 H); doublet 6.6 8 (1 H); multiplet 4.5 8 (1 H);
doublet of
doublets 4.0 8 (1 H); multiplet 4.2 8 (3 H); singlet 2.45 S (3 H); singlet 2.4
8 (3 H).
INTERMEDIATE 11
{8-Methyl-2,3-dihydrofl ,4ldioxinof2,3-flauinolin-2-yl)methyl 4
methylbenzenesulfonate
(0078] To a solution of {(2R)-7-nitro-8-[(E)-3-oxo-1-butenyl]-2,3-dihydro-1,4-
benzodioxin-2-yl}methyl 4-methylbenzenesulfonate (3.40 g, 7.83 mmole) in 200
mL
of acetic acid/ethanol (3:2) was added 2.25 g (40.2 mmole) of iron powder and
the
mixture was heated at reflux under nitrogen for 8 hours. After the reaction
was
complete, 150 mL of water was added and the mixture filtered through a pad of
celite. The filtrate was neutralized with saturated aqueous sodium bicarbonate
and
extracted with ethyl acetate. The extract was dried over magnesium sulfate,
filtered
and evaporated in vacuum. The residue was column chromatographed on silica gel
using a gradient elution commencing with 20% ethyl acetate/hexane and ending
with
70% ethyl acetate/hexane to give 2.5 g of the (R)-enantiomer of the title
compound
as a yellow oil. 'H-NMR (CDCI3): doublet 8.1 b (1 H); doublet 7.6 8 (2 H);
doublet
7.45 8 (1 H); multiplet 7.2 8 (4 H); multiplet 4.6 8 (1 H); multiplet 4.3 8 (3
H); multiplet
4.1 8 (1 H); singlet 2.5 8 (3H); singlet 2.4 8 (3 H).
INTERMEDIATE 12
[7-Nitro-8-(2-oxoethyl)-2,3-dihydro-1,4-benzodioxin-2-yllmethyl 4
methylbenzenesulfonate
[0079] A solution of 4.2 g (10 mmole) of toluene-4-sulfonic acid (2R)-8-allyl-
7-nitro-
2,3-dihydro-benzo(1,4)dioxin-2-ylmethyl ester in 400 mL of methylene chloride
was
cooled in a dry ice/isopropanol bath and saturated with ozone. It was then
purged
with oxygen and 2.6 g (20 mmole) of diisopropylethylamine added. The mixture
was
-40-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
allowed to come to room temperature and stirred under nitrogen for 24 hours.
It was
then washed with 300 mL portions of 2 N HCI (aq), water and saturated brine,
dried
over magnesium sulfate, filtered and concentrated in vacuum to give 3.8 g of
the (R)-
enantiomer of the title compound as a white solid one-quarter hydrate, m.p.
116-
120°C.
Elemental Analysis for: C,SH"NOES ~ 0.25 H20
Calc'd: C, 52.49; H, 4.28; N, 3.40
Found: C, 52.33; H, 3.92; N, 3.36
INTERMEDIATE 13
2,3-Dihydro-7H-f1,41dioxinof2,3-elindol-2-ylmethyl 4-methylbenzenesulfonate
[0080] A mixture of 3.75 g (9.2 mmole) of [(2R)-7-nitro-8-(2-oxoethyl)-2,3-
dihydro-
1,4-benzodioxin-2-yl]methyl 4-methylbenzenesulfonate and 3.0 g of platinum
oxide in
50 mL of ethyl acetate was treated with 45 psi of hydrogen on a Parr
hydrogenation
apparatus for 6 hours. The mixture was then filtered through celite and
concentrated
in vacuum. The residue was column chromatographed on silica gel with first 10%
hexane/methylene chloride, then 1 % methanol/methylene chloride and finally 2%
methanol/methylene chloride to give 1.50 g of the (R)-enantiomer of the title
compound as a white solid one-quarter hydrate, m.p. 145°C.
Elemental Analysis for: C~BH~~NOSS ~ 0.25 H20
Calc'd: C, 59.41; H, 4.85; N, 3.85
Found: C, 59.41; H, 4.57; N, 3.72
INTERMEDIATE 14
1-f5-Hydroxy-3-(hydroxymethyl)-2,3-dihydro-1,4-benzodioxin-6-yll-1-ethanone
[0081] To a solution of 2',3',4'-trihydroxyacetophenone (10.6 g, 63.0 mmole)
in
DMF (75 mL) was added potassium carbonate (17.4 g, 126 mmole). After 5 minutes
(R)-glycidyl tosylate (9.67 g, 42.3 mmole) was added, then the heterogeneous
mixture was heated to 70°C for 3 hours. After removal of the solvent in
vacuum, the
residue was taken into water (800 mL) and was then extracted with ethyl
acetate (4 x
300 mL). The combined organic layers were dried over magnesium sulfate,
filtered
-41 -
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
and evaporate to dryness in vacuum. The crude brown oil thus obtained was
column
chromatographed on silica gel with 40% hexane/ethyl acetate as eluant to give
the
(S)-enantiomer of the title compound as a yellow oil which solidifies upon
standing
(7.5 g, 78%). MS (ESI) m/z 223 (M-H)-.
Elemental Analysis for: C" H,205 ~ 0.10 H20
Calc'd: C, 58.46; H, 5.44
Found: C, 58.02; H, 5.09
INTERMEDIATE 15
1-f 5-Hvdroxy-3-(hydroxymethyl)-2,3-di hydro-1,4-benzodioxi n-6-yll-1-ethanone
oxime
(0082] A solution of hydroxylamine hydrochloride (2.38 g, 34.2 mmole) in 1:1
ethanol/pyridine (100 mL) was added to a solution of 1-[(3S)-5-hydroxy-3-
(hydroxymethyl)-2,3-dihydro-1,4-benzodioxin-6-yl]-1-ethanone (1.92 g, 8.57
mmole)
in ethanol (200 mL). It was then heated to reflux under nitrogen for 5 hours.
Upon
cooling, the solvent was removed and replaced with ethyl acetate. The solution
was
then washed with water (200 mL) and with aqueous 2N HCI (100 mL), dried over
magnesium sulfate, filtered and evaporated in vacuum to give 1.89 g (93%) of
the
(S)-enantiomer of the title compound as a gray solid, m.p. 162 ° C. MS
(ESI) m/z
240 (M+H)+.
Elemental Analysis for: C"H,3N05~ 0.35 H20
Calc'd: C, 53.81; H, 5.62; N, 5.71
Found: C, 53.51; H, 5.30; N, 5.58
INTERMEDIATE 16
[2-Methyl-7,8-dihydrofl ,4ldioxinof2,3-alf 1,3lbenzoxazol-8-yllmethanol
(0083] 3.03 g (12.6 mmole) of 1-[(3S)-5-hydroxy-3-(hydroxymethyl)-2,3-dihydro-
1,4-benzodioxin-6-yl]-1-ethanone oxime was dissolved in a mixture of 1:3 N,N-
dimethylacetamide/acetonitrile (100 mL). The solution was cooled in an
ice/water
bath and a solution of phosphorus oxychloride (1.26 mL, 35 mmole) in 1:3 N,N-
dimethylacetamide/acetonitrile (30 mL) was added. The reaction mixture was
stirred
-42-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
under nitrogen over a period of 48 hours. It was then added to an ice cold,
saturated
solution of sodium acetate, extracted with ethyl acetate, dried over magnesium
sulfate, filtered and evaporated in vacuum. The resulting crude oil was column
chromatographed on silica gel with 60% hexane/ethyl acetate to remove
impurities
and the product eluted with 40% hexane/ethyl acetate. After evaporation of the
solvent in vacuum, 2.08 g (75%) of the (S)-enantiomer of the title compound
was
obtained as a white solid, m.p. 120°C. MS (ESI) m/z 222 (M+H)+.
Elemental Analysis for: C" H" NOQ ~ 0.20 H20
Calc'd: C, 58.77; H, 5.11; N, 6.23
Found: C, 58.93; H, 4.91; N, 6.14
INTERMEDIATE 17
j2-Methyl-7, 8-dihydrof1,41dioxinof2,3-g1f1,31benzoxazol-8-yllmethyl 4
methylbenzenesulfonate
[0084] To a solution of [(8S)-2-methyl-7,8-dihydro[1,4]dioxino[2,3-
g][1,3]benzoxazol-8-yl]methanol (1.80 g, 8.14 mmole) in methylene chloride
(100 mL)
was added p-toluenesulfonyl chloride (3.90 g, 20.4 mmole). The mixture was
cooled
in an ice bath and a solution of diisopropylethylamine ( 3.55 mL, 20.4 mmole)
in
methylene chloride (20 mL) was then added dropwise, followed by 4-
dimethylaminopyridine (0.65 g, 5.30 mmole). The solution was allowed to warm
to
room temperature and was stirred under nitrogen overnight. The reaction was
diluted to 500 mL in volume with methylene chloride, then washed with aqueous
2 N
HCI (200 mL), with saturated aqueous sodium bicarbonate (200 mL), and with
brine
(150 mL), dried over magnesium sulfate, filtered and evaporated in vacuum to a
yellow oil. The crude oil was column chromatographed on silica gel using
methylene
chloride to remove impurities and 3% methanol/methylene chloride to elute the
(R)-
enantiomer of the title compound, which becomes a white solid under vacuum
(2.56
g, 84%), m.p. 123°C. MS (ESI) m/z 376 (M+H)+.
Elemental Anal sis for: C,SH»NO6S ~ 0.20 Hz0
Calc'd: C, 57.04; H, 4.63; N, 3.70
-43-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
Found: C, 56.75; H, 4.62; N, 3.51
INTERMEDIATE 18
5-Bromo-6-methoxy-2-methylauinoline
[0085] A solution of 6-methoxy-2-methylquinoline (177 g, 1.02 mol) in
acetonitrile
(1.77 L) was cooled to 0-3°C followed by portion-wise addition of N-
bromo-
succinimide (200 g, 1.12 mol) over a period of 30 min while maintaining the
same
temperature. The resulted brown slurry was warmed to ambient temperature and
stirred for an additional 6 h. The reaction was then quenched by a 10% NaHS03
solution (211 mL). The reaction mixture was concentrated to a volume of 600 mL
then slowly poured into 0.1 N NaOH (2.5 L). The slurry (pH=9) was stirred at
room
temperature for 1 h then filtered, washed with water (2 x 1 L) and dried in a
vacuum
oven to give 253 g (98.6%) of the title compound as a brown solid. R, = 0.39
(3:7)
EtOAc:heptane; 'H NMR (DMSO) 8 8.30 (d, J=6.5 Hz, 1 H), 7.98 (d, J=6.9 Hz, 1
H),
7.70 (d, J=7.0 Hz, 1 H), 7.47 (d, J=6.5 Hz, 1 H), 4.02 (s, 3H), 2.66 (s, 3H);
Elemental Analysis for: C"H,oNOBr
Calc'd: C 52.40 H 3.97 N 5.56
Found: C 52.13 H 3.94 N 5.61
INTERMEDIATE 19
5-Bromo-2-methyl-6-auinolinol
[0086] A mixture of 5-bromo-2-methyl-6-methoxyquinoline (30 g, 0.12 mol) in
48%
HBr (135 mL) was heated to reflux for 7 h then cooled to 5°C in 1 h to
give a brown
and thick slurry. The slurry was stirred at 0-5°C for 1 h then
filtered, washed with
EtOAc (2 x 50 mL) and dried in a vacuum oven to give 34.9 g (92%) of the
hydrobromide of the title compound as a brown solid. 'H NMR (DMSO) 8 8.26 (d,
J=8.7 Hz, 1 H), 7.85 (d, J=9:1 Hz, 1 H), 7.56 (d, J=9.1 Hz, 1 H), 7.45 (d,
J=8.7 Hz, 1 H),
2.64 (s, 3H). A slurry of the hydrobromide salt of 5-bromo-2-methyl-6-
quinolinol (3.4
g, 10.5 mmol) and Amberlyst A-21 ion-exchange resin (1.7 g, pre-washed with
MeOH
then dried in oven) in MeOH (35 mL) was stirred at room temperature for 3 h.
The
mixture was then filtered and concentrated in vacuo to give 2.5 g (100%) of a
yellow
-44-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
solid. R, = 0.36 (1:1 ) EtOAc:heptane; ' H NMR (DMSO) 8 8.26 (d, J=8.4 Hz, 1
H),
7.82 (d, J=9.3 Hz, 1 H), 7.47 (t, J=9.1 Hz, 2H), 2.66 (s, 3H).
INTERMEDIATE 20
(2S)-1-(Benzyloxy)-3-f(5-bromo-2-methyl-6-auinolinyl)oxyl-2-propanol
[0087] A solution of 5-bromo-2-methyl-6-quinolinol (30.1 g, 126 mmol), (R)-
benzyl
glycidyl ether (24.9 g, 152 mmol) and triethylamine (17.4 g, 172 mmol) in DMA
(200
mL) was heated in a 95-98°C oil bath for 2 days. The solution was
cooled and
poured into water (300 mL) while stirring. The tan precipitate formed was
filtered,
washed with water (100 mL) and dried in a vacuum oven to give 37 g (73%) of
the
title compound as a tan solid. R, = 0.35 (EtOAc); 'H NMR (DMSO) s 8.31 (d,
J=8.8
Hz, 1 H), 7.96 (d, J=9.2 Hz, 1 H), 7.72 (d, J=9.3 Hz, 1 H), 7.74 (d, J=8.7Hz,
1 H), 7.25-
7.36 (m, 5H), 5.28 (d, J=5.1 Hz, 1 H), 4.56 (s, 2H), 4.22-4.29 (m, 2H), 4.08-
4.15 (m,
1 H), 3.61-3.73 (m, 2H), 2.66 (s, 3H); Specific rotation = +6.2 ° (c=1,
CH30H);
Elemental Analysis for: C2oH2oBrN03
Calc'd: C 59.66 H 4.97 N 3.48
Found: C 59.43 H 4.97 N 3.55
INTERMEDIATE 21
(2S)-2f(Benzyloxy)methyl-8-methyl-2,3-dihydrof1,41dioxino X2,3-flauinoline
[0088] To a mixture of (2S)-1-(benzyloxy)-3-[5-bromo-2-methyl-6-
quinolinyl)oxylJ-2-
propanol (100 g, 0.249 mol) and copper (I) iodide (47.4 g, 0.249 mol) in
toluene (2 L),
NaH (10.9 g, 0.45 mol) was added in portions at 30-35°C over 20 min.
The reaction
mixture was kept at 35°C for 30 minutes then heated to 110°C
slowly. After 30
minutes, the reaction was cooled to 60°C, additional NaH (10.9 g, 0.45
mol) was
added. This was warmed to 110°C for an additional 2 hours then cooled
to rt before
dropwise addition of water (200 mL). After stirring for 15 minutes, the
mixture was
filtered through a bed of celite then washed with toluene (3 x 50 mL) and
water (50
mL). The two layers were separated. The organic layer was extracted with water
(100 mL), NH40H (100 mL), 25% NaCI (100 mL) and concentrated in vacuo to give
-45-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
387.6 g of the crude product as a brown syrup. The crude product was carried
through to the debenzylation step before purification.
INTERMEDIATE 22
f(2R)-8-Methyl-2.3-dihydrof1,41dioxinof2,3-flauinotin-2-yllmethanol
[0089] To a solution of (2S)-2((benzyloxy)methyl-8-methyl-2,3-
dihydro[1,4]dioxino
[2,3-f]quinoline (0.16 g, 0.5 mmol) in EtOH (1 mL) was added cyclohexene (0.5
mL)
then 10% Pd/C (0.016 g, 10 mol %). The mixture was heated to reflux under N2
for
18 h then cooled and filtered. The catalyst was rinsed with methanol and the
filtrate
was concentrated in vacuo to afford 0.113 g (98%) of the title alcohol as an
off-white
solid.
'H NMR (CD30D) 8 8.46 (m, 1 H), 7.47 (m, 1 H), 7.38-7.31 (m, 2H), 4.40 (m, 1
H), 4.36
(m, 1 H), 4.18 (m, 1 H), 3.91 (m, 2H), 2.68 (s, 3H).
INTERMEDIATE 23
[(2R)-8-Methyl-2,3-Dihydrof1,41Dioxinof2,3-flQuinolin-2-yllMethyl 4
Bromobenzenesulfonate
[0090] A solution of [(2S)-8-methyl-2,3-dihydro[1,4]dioxino[2,3-f]quinolin-2-
yl]-
methanol (4.0 g, 17.3 mmol), brosyl chloride (4.86 g, 19.0 mmol),
dimethylamino
pyridine (20 mg, 0.16 mmol) and triethylamine (3.62 mL, 25.8 mmol) in toluene
(40
mL) was stirred at 60°C for 6 h. The reaction mixture was cooled to
room temperature
then water (20 mL) was added. After 30 min, the two layers were separated. The
organic layer was extracted with 8% NaHC03 (20 mL) and Hz0 (20 mL), dried over
Na2S04, filtered and concentrated in vacuo. The solid obtained was dissolved
in
isopropyl alcohol (50 mL) and toluene (10 mL) at 80°C, cooled to room
temperature
over 1 h then filtered, washed with (5:1) IPA: toluene (2 x 5 mL) and dried in
a vacuum
oven to give 5.99 g (76.9%) of the title compound as an off-white solid. '3C
NMR
(CDCI3) 8 157.9, 144.3, 138.1, 134.7, 132.9, 129.7, 129.6, 129.0, 122.4,
121.7, 121.3,
118.8, 70.7, 67.6, 64.5, 25.4
-46-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
EXAMPLE 1
2-(4-Benzof blth iophen-3-yl-3,6-di hydro-2H-pyridi n-1-yl methyl)-8-methyl-
2,3
dihydro-f1,41dioxino~2,3-flauinoline
[0091] To a mixture of [(2R)-8-methyl-2,3-dihydro[1,4]dioxino[2,3-f]quinolin-2-
yl]methyl 4-toluenesulfonate (0.87 g, 2.3 mmol) and 4-benzo[b]thiophen-3-yl-
1,2,3,6-
tetrahydro-pyridine (0.48 g, 2.2 mmol) was added 3 mL of dimethylsulfoxide.
The
mixture was stirred at 97°C for 18 hours. The solvent was evaporated
under reduced
pressure. The residue was partitioned between 500 mL each of methylene
chloride
and saturated aqueous sodium bicarbonate. The methylene chloride layer was
washed once with 500 mL of water and dried over anhydrous magnesium sulfate.
Filtration and concentration in vacuum gave 0.91 g of oil. This was
chromatographed
on silica gel with a gradient of ethyl acetate and hexane. The product
fractions were
collected to give 0.059 g of the free base as pure, yellow oil. This was
dissolved in
ethanol and heated. Oxalic acid dihydrate (0.0125 g, 0.0991 mmol) in ethanol
was
added. After the mixture had cooled, filtration gave 0.0619 g of the S
enantiomer of
the title compound as an orange oxalate salt, m.p. 129-133°C.
Elemental Analysis for: C26H24N202S' C2H204' 2/3 H20
Calc'd: C, 63.38; H, 5.19; N, 5.28
Found: C, 63.48; H, 4.97; N, 5.08
EXAMPLE 2
2-(4-Benzof blthiophen-2-yl-3,6-di hydro-2 H-pyridi n-1-yl methyl)-8-methyl-
2,3
dihydro-f1,41dioxinof2,3-flauinoline
[0092] To a mixture of [(2R)-8-methyl-2,3-dihydro[1,4]dioxino[2,3-f]quinolin-2-
yl]methyl 4-toluenesulfonate (0.638 g, 1.66 mmol) and 4-benzo[b]thiophen-2-yl-
1,2,3,6-tetrahydro-pyridine (0.46 g, 2.1 mmol) was added 12 mL of
dimethylsulfoxide.
The mixture was stirred at 90°C for 18 hours. The solvent was
evaporated under
reduced pressure. The residue was partitioned between 500 mL each of methylene
chloride and saturated aqueous sodium bicarbonate. The methylene chloride
layer
was washed with water twice and dried over anhydrous magnesium sulfate.
Evaporation of the solvent gave 0.98 g of oil. This was chromatographed on
silica
-47-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
gel with a gradient of ethyl acetate and hexane. The product fractions were
collected
and concentrated in vacuum to give 0.39 g of the title compound as nearly
pure, light
yellow oil. This was triturated with ethanol to give 0.2563 g of the S
enantiomer of
the title compound as a light yellow solid, m.p. 174-176°C.
Elemental Analysis for: C2gH24N2O2S ~ 1/4 H20
Calc'd: C, 72.11; H, 5.70; N, 6.47
Found: C, 72.01; H, 5.42; N, 6.32
EXAMPLE 3
2-f 4-(5-FI uoro-benzof blthiophen-3-yl)-3,6-dihydro-2H-pyridi n-1-yl methyll-
8
methyl-2,3-dihydro-f1.4ldioxinof2,3-flauinoline
[0093] To a solution of [(2R)-8-methyl-2,3-dihydro[1,4]dioxino[2,3-f]quinolin-
2-
yl]methyl 4-toluenesulfonate (0.50 g, 1.1 mmol), 4-(5-fluoro-benzo[b]thiophen-
3-yl)-
1,2,3,6-tetrahydropyridine (0.35 g, 1.5 mmol), 21 mL of THF and 21 mL of DMF
was
added NaHC03 (0.45 g, 5.4 mmol. The mixture was stirred at reflux for 18
hours.
The solvent was removed in vacuum and the residue partitioned between 500 mL
each of methylene chloride and water. The methylene chloride layer was washed
with water 3 times and dried over anhydrous magnesium sulfate. Filtration and
concentration in vacuum gave 0.62 g of dark oil. This was chromatographed on
silica
gel with a gradient of ethyl acetate and hexane to give 0.12 g of the free
base as a
pure light yellow oil. The oil was dissolved in ethanol and added to a
solution of
oxalic acid dihydrate (0.0400 g, 0.317 mmol) in ethanol. Filtration gave
0.0871 g of
the S enantiomer of the title compound as a yellow oxalate, m.p. 197-201
°C.
Elemental Analysis for: C2gH23FN2O2S ~ C2H204 ~ 2 H20
Calc'd: C, 58.73; H, 5.10; N, 4.89
Found: C, 58.78; H, 4.46; N, 4.63
INTERMEDIATE 24
Trifluoro-methanesulfonic acid 7-methoxy-benzofuran-3-yl ester
[0094] To a cold solution (-20°C) of 3.3 g (20 mmol) 7-methoxy-
benzofuranone in
30 mL methylene chloride was added 8.3 mL (6.0 mmol) of triethylamine. To the
-48-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
cold mixture, a solution of 8.5 g (30 mmol) of triflic anhydride in 20 ml of
methylene
chloride was added dropwise. The temperature was kept at -20°C or 1
hour. The
reaction was then quenched with 100 mL of saturated aqueous sodium bicarbonate
aand extracted with methylene chloride (2 x 150 mL). The combined organic
extracts
were dried over magnesium sulfate and concentrated in vacuum to give 5.6 g of
the
desired product. MS (ES) m/z (relative intensity): 265 (M+H,100);
INTERMEDIATE 25
2-(7-Methoxvbenzo f blfuran-3-y10-4,4,5,5-tetramethyl-f 1,2loxaborolane
[0095] To a mixture of trifluoro-methanesulfonic acid 7-methoxy-benzofuran-3-
yl
ester (0.660 g, 2.23 mmol)) in triethylamine (1 ml) was added first 3.75 mL
(3.75
mmol) of 1 N pinacoleborane in THF followed by 0.10 g of [1,1'-
bis(diphenylphosphino)ferrocene]palladium (II) chloride/methylene chloride 1:1
complex. The reaction was heated at 150°C for 3 minutes in the
microwave. The
solvent was removed under vacuum. The residue was taken up in 300 mL of water
and extracted with ether (2 x 200 mL). The combined organic extracts were
dried
over magnesium sulfate, filtered and the solvent removed in vacuum. The
residue
was filtered through 50 mL of silica gel using 10% ethyl acetate/hexane to
give 0.350
g of the title compound. 1 H NMR (300 MHz, CDCI3); 8 1.36 (s, 12H), 4.01 (s,
3H),
6.81 (d, 1 H), 7.18 (t, 1 H), 7.52 (d, 1 H), 7.95 (s, 1 H).
INTERMEDIATE 26
4-(7-Methoxy-benzofuran-3-yl)-3,6-dihydro-2H-pyridine-1-carboxylic acid tert
butyl ester
[0096] To a solution of 2-(7-methoxybenzo[b]furan-3-yl)-4,4,5,5-tetramethyl-
[1,2]oxaborolane (1.10 g, 4.0 mmol) in dimethoxyethane (1 mL) was added CsC03
(0.650 g, 2.0 mmol), H20 (1 mL), 4-trifluoromethanesulfonyloxy-3,6-dihydro-2H-
pyridine-1-carboxylic acid tert-butyl ester (0.480 g, 1.45 mmol), LiCI (0.10
g, 2.4
mmol) and tetrakis(triphenylphosphine)palladium (0) (0.06 g, 0.05 mmol). The
reaction was heated in the microwave for 5 min at 150°C. The solvent
was removed
in vacuum, the residue taken up in 300 mL of methylene chloride, washed with
200
-49-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
mL portions of saturated aqueous sodium carbonate and 1 N NH40H (aq), dried
over
anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was
filtered through 150 mL of silica gel with 15% ethyl acetate/hexane as eluant
to give
0.50 g of the title compound. 1 H NMR (300 MHz, CDCI3); b 1.5 (s, 9H), 2.51
(t, 2H),
3.66 (t, 2H), 4.04 (s, 3H), 4.12 (dd,2H), 6.24 (br s, 1 H), 6.91 (d, 1 H),
7.17 (t, 1 H), 7.39
(d, 1 H), 7.59 (s, 1 H), MS (ES) m/z (relative intensity): 330 (M+H,100).
INTERMEDIATE 27
4-(7-Methoxy-benzofuran-3-yl)-1,2,3,6-tetrahydro-pyridine
[0097] To a solution of 4-(7-methoxy-benzofuran-3-yl)-3,6-dihydro-2H-pyridine-
1-
carboxylic acid tert-butyl ester (0.50 g, 1.5 mmol) in methylene chloride (10
mL), was
added dropwise a solution of TFA (1 mL) in methylene chloride (5 mL). The
reaction
was stirred at room temperature for one hour, then was diluted with 250 mL of
methylene chloride, washed with 1 N NaOH (100 mL) and with saturated brine,
dried
over magnesium sulfate, filtered and concentrated in vacuum to give 0.30 g of
the
title compound. 1 H NMR (300 MHz, CDCI3); 8 2.5 (t, 2H), 3.19 (t, 2H), 3.64
(dd,2H),
4.01 (s, 3H), 6.30 (br s, 1 H), 7.10 (d, 1 H), 7.21 (t, 1 H), 7.52 (d, 1 H),
7.64 (s, 1 H); MS
(ES) m/z (relative intensity): 230 (M+H,100).
EXAMPLE 4
2-f4-(7-Methoxv-benzofuran-3-yl)-3,6-dihydro-2H-pyridin-1-ylmethyll-8-methyl
2,3-dihydro-f1,4ldioxinof2,3-flquinoline
[0098] A mixture of [(2R)-8-methyl-2,3-dihydro[1,4]dioxino[2,3-f]quinolin-2-
yl]methyl
4-bromobenzenesulfonate ester (0.173 g, 0.38 mmol), 4-(7-methoxybenzofuran-3-
yl)-
1,2,3,6-tetrahydropyridne (0.10 g, 0.44 mmol) and potassium carbonate (0.145
g, 1.0
mmol) in 2 mL of N,N-dimethylformamide was stirred under nitrogen at room
temperature for 2 days and then at 60°C for 6 hours. Water was added
and the
resulting precipitate was filtered, dried and column chromatographed on 100 mL
of
silica gel using first 50% ethyl acetate in hexane and then 75% ethyl
acetate/hexane
as eluant. Combination and concentration of the product fractions gave 0.015 g
of
the S enantiomer of the title compound as a yellow solid.
-50-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
MS (ESI) m/z 443 (M+H)+.
EXAMPLE 5
2-f4-(5-Fluoro-benzofblthiophen-3-yl)-3,6-dihydro-2H-pyridin-1-ylmethyll-2.3
dihydro-f 1,4ldioxinof2,3-flauinoline
[0099] To a solution [(2R)-8-methyl-2,3-dihydro[1,4]dioxino[2,3-f]quinolin-2-
yl]methyl 4-toluenesulfonate (0.51 g, 1.4 mmol), 4-(5-fluoro-benzo[b]thiophen-
3-yl)-
1,2,3,6-tetrahydro-pyridine (0.39 g, 1.7 mmol), 24 mL of THF and 24 mL of DMF
was
added NaHC03 (0.50 g, 5.9 mmol). The mixture was stirred and heated at reflux
for
18 hours. The solvents were evaporated in vacuum and the residue was
partitioned
between 500 mL portions of ethyl acetate and water. The ethyl acetate layer
was
washed with water and dried over anhydrous magnesium sulfate. Filtration and
concentration in vacuum gave 0.731 g of oil. This was chromatographed on
silica gel
with a gradient of ethyl acetate and hexane. Only the fractions clean enough
to use
were combined. They were concentrated to give 0.13 g of the free base as an
oil.
This was dissolved in ethanol. A solution of oxalic acid dihydrate (0.0410 g,
0.325
mmol) in ethanol was added. Filtration gave 0.11 g of the S enantiomer of the
title
compound as a light yellow oxalate, m.p. 183-185°C.
Elemental Analysis for: C25H21 FN202S ~ C2H204 ~ H20
Calc'd: C, 59.99; H, 4.66; N, 5.18
Found: C, 60.05; H, 4.39; N, 4.97
-51 -
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
EXAMPLE 6
2-(4-Benzof blthiophen-3-yl-3.6-dihydro-2H-pyridin-1-ylmethyl)-2,3-dihydro-
f1,41dioxinof2,3-flpuinoline
[0100] To a solution of [(2R)-2,3-dihydro[1,4]dioxino[2,3-f]quinolin-2-
yl]methyl
toluenesulfonate (0.43 g, ~1.2 mmol), 4-benzo[b]thiophen-3-yl-1,2,3,6-
tetrahydro-
pyridine (0.31 g, 1.4 mmol), 21 mL of THF and 21 mL of DMF was added NaHC03
(0.43 g, 5.1 mmol). The mixture was stirred and heated at 60°C for 2
days and then
allowed to stand at room temperature for one day. The solvent was evaporated
in
vacuum. The residue was partitioned between 500 mL each of ethyl acetate and
water. The ethyl acetate layer was washed with water four times and then dried
over
anhydrous magnesium sulfate. Filtration and concentration in vacuum gave 0.24
g of
oil. This was chromatographed on silica gel with a gradient of ethyl acetate
and
hexane to give 0.06 g of the free base as an oil. This was dissolved in
ethanol and
added to a solution of oxalic acid dehydrate (0.021 g, 0.17 mmol) in ethanol.
Filtration
gave 0.042 g of the S-enantiomer of the title compound as a yellow oxalate,
m.p.
178-180°C.
Elemental Analysis for: C25H22N202S' C2H204' 1.6 H20
Calc'd: C, 60.80; H, 5.14; N, 5.25
Found: C, 60.59; H, 4.79; N, 5.00
EXAMPLE 7
2-(4-Benzof blth iophen-3-yl-3,6-dihydro-2H-pyridi n-1-yl methyl)-2,3-di hydro-
7H
f 1,4ldioxinof2,3-elindole
[0101] To a solution of [(2R)-7,8-dihydro-3H-6,9-dioxa-3-aza-
cyclopenta[a]naphthalen-8-yl]methyl toluene-4-sulfonate (0.6 g, 2 mmol), ), 4-
benzo[b]thiophen-3-yl-1,2,3,6-tetrahydro-pyridine ( 0.04 g, 0.2 mmol), 30 mL
of THF
and 25 mL of DMF was added NaHC03 ( 0.6 g, 7 mmol). The mixture was stirred
and heated at 70°C for 1 day and then allowed to stand at room
temperature for 3
days. The solvent was evaporated in vacuum. The residue was partitioned
between
500 mL each of ethyl acetate and water. The ethyl acetate layer was washed
with
water four times and dried over anhydrous magnesium sulfate. Filtration and
-52-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
concentration in vacuum gave 0.86 g of oil. This was chromatographed on silica
gel
with a gradient of ethyl acetate and hexane to give 0.25 g of the free base as
a very
light tan oil. This was dissolved in ethanol. A solution of oxalic acid
dihydrate
(0.0851 g, 0.675 mmol) in ethanol was added. Filtration gave 0.1436 g of the S
enantiomer of the title compound as a light cream color amorphous oxalate.
Elemental Analysis for: C24H22N202S ~ C2H204
Calc'd: C, 63.36; H, 4.91; N, 5.68
Found: C, 63.22; H, 4.86; N, 5.50
EXAMPLE 8
2-f4-(5-Fluoro-benzofblthiophen-3-yl)-3,6-dihydro-2H-pyridin-1-ylmethyll-2,3
dihydro-7H-f 1,4ldioxino f2,3-elindole
[0102] To a mixture of [(2R)-7,8-dihydro-3H-6,9-dioxa-3-aza-
cyclopenta[a]naphthalen-8-yl]methyl 4-toluenesulfonate (0.67 g, 1.9 mmol), 4-
(5-
fluoro-benzo[b]thiophen-3-yl)-1,2,3,6-tetrahydro-pyridine (0.48 g, 2.1 mmol)
and
Na2C03 (0.80 g, 7.5 mmol) was added 21 mL of dimethylsulfoxide. The mixture
was stirred heated at 70°C for 18 hours. TLC on silica gel showed much
tosylate
was unreacted. Stirring and heating at 80°C was continued for 18 hours.
The
solvent was evaporated at reduced pressure. The residue was partitioned
between
500 mL portions of ethyl acetate and water. The ethyl acetate layer was washed
five
times with water and dried over anhydrous magnesium sulfate. Filtration and
concentration in vacuum gave 0.86 g of dark oil. This was chromatographed on
silica
gel with 40% ethyl acetate in hexane to give 0.29 g of the free base as a
light tan oil.
This was dissolved in ethanol and added to a solution of oxalic acid dihydrate
(0.0978 g, 0.776). Filtration gave 0.1049 g of the S enantiomer of the title
compound
as a light gray amorphous oxalate.
Elemental Analysis for: C24H21 FN2 02S ~ C2H204 ~ 0.2 H20
Calc'd: C, 60.74; H, 4.59; N, 5.45
Found: C, 60.72; H, 4.34; N, 5.26
-53-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
EXAMPLE 9
8-(4-Benzof blthiophen-3-yl-3,6-di hydro-2H-pyridin-1-yl methyl)-2-methyl-7,8
dihydro-f 1,4ldioxinof 2,3-alf 1,3lbenzoxazole
[0103] To a mixture of [(2R)-2-methyl-7,8-dihydro-1,6,9-trioxa-3-aza-
cyclopenta[a]naphthalen-8-yl]methyl 4-toluenesulfonate (0.3081 g, 0.8207 mmol)
and
4-benzo[b]thiophen-3-yl-1,2,3,6-tetrahydro-pyridine (0.54 g, 2.5 mmol) was
added 10
mL of dimethylsulfoxide. The solution was stirred and heated at 85°C
for 4.5 hours.
The solvent was evaporated under reduced pressure. The residue was partitioned
between 500 mL each of ethyl acetate and saturated aqueous sodium bicarbonate.
The ethyl acetate layer was washed four times with water. Drying over
magnesium
sulfate, filtration and evaporation of the solvent gave 0.55 g of oil. This
was eluted
from silica gel with a gradient of hexane and ethyl acetate to give 0.09 g of
the free
base as an oil. This was dissolved in EtOH and added to a solution of oxalic
acid
dehydrate 0.0304 g, 0.241 mmol) in ethanol. Filtration gave 0.0948 g of the S
enantiomer of the title compound as a fine white oxalate, m.p. 126-
129°C.
Elemental Analysis for: C24H22N203S' C2H204' 0.5 H20
Calc'd: C, 60.34; H, 4.87; N, 5.41
Found: C, 60.36; H, 4.99; N, 5.26
INTERMEDIATE 28
4-Benzofblthiophen-7-yl-pyridine
[0104] To 7-bromo-benzo[b]thiophene (5.28 g, 24.7 mmol) was added pyridine-4-
boronic acid (2.734 g, 22.24 mmol), K3P04 (12.0 g, 56.5 mmol), 37.5 mL of 1,4-
dioxane and 3.8 mL of water. The mixture was placed under vacuum for several
minutes and flushed with nitrogen. This was repeated 5 times. Pd(dppf)CI2
CH2CI2 (0.909 g, 1.11 mmol), PdCl2 (0.1994 g, 1.124 mmol) and 1,1 "-
bis(diphenylphosphino)ferrocene (0.6234 g, 1.124 mmol) were purged in the same
way using high vacuum. The catalyst was added to the reaction flask, which was
purged again 3 times. The mixture was stirred at 80 °C under nitrogen.
After 1 day
TLC showed much starting material remained. Additional K3P04 (2.3 g, 10.8
mmol)
-54-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
was added after purging. Stirring at 80 °C under nitrogen was continued
for 12 to 14
hours and then at room temperature till the next day. The mixture was
partitioned
between water and ethyl acetate, filtered through Celite and the layers
separated.
The organic layer was concentrated in vacuum. The residue was redissolved in
ethyl
acetate, washed with water and dried over magnesium sulfate. Concentration in
vacuum gave 6.06 g of dark oil. This was eluted from silica gel with a
gradient of
hexane and ethyl acetate to give 2.43 g of the title compound as a tan oil
that
crystallizes slowly, (m.p. 71-72°C).
Elemental Analysis for: Cl3HgNS ~ 1/3 H20
Calc'd: C, 71.86; H, 4.48; N, 6.45
Found: C, 71.93; H, 4.37; N, 5.66
INTERMEDIATE 25
S-4-Benzof blthiophen-7-yl-1-(8-methyl-2,3-dihydro-f 1,4ldioxinof2,3-
flauinolin-2
ylmethyl)-pyridinium 4-bromo-benzenesulfonate
[0105] To a mixture of [(2R)-8-methyl-2,3-dihydro[1,4]dioxino[2,3-f]quinolin-2-
yl]methyl 4-bromobenzenesulfonate (1.00 g, 2.22 mmol) and 4-benzo[b]thiophen-7-
yl-pyridine (0.58 g, 2.7 mmol) was added 4 mL of benzene. The mixture was
stirred
at 75 °C for 18 hours, after which time most of the solvent had
evaporated. TLC on
silica gel showed much of the starting material remained. The reaction mixture
stood
at room temperature and open to the atmosphere for several days. The residual
tar
was triturated with acetone at 52°C to give a solid. The solid was
broken up and the
volume was reduced by evaporation to approximately 20 mL. Filtration gave
0.6581
g of the title compound as a gray solid, (dec.> 175 °C).
Elemental Analysis for: C32H25BrN205S2 ~ 2 H20
Calc'd: C, 55.09; H, 3.61; N, 4.02
Found: C, 55.13; H, 3.88; N, 3.77
-55-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
EXAMPLE 10
2-(4-Benzof blthiophen-7-yl-3,6-dihydro-2H-pyridin-1-ylmethyl)-8-methyl-2,3
dihydro-f1,41dioxinof2,3-flguinoline
[0106] To a stirring suspension of S-4-benzo[b]thiophen-7-yl-1-(8-methyl-2,3-
dihydro-[1,4]dioxino[2,3-fJquinolin-2-ylmethyl)-pyridinium 4-bromo-
benzenesulfonate
((0.5060 g, 0.7648 mmol) in 4.5 mL of EtOH cooled in an ice-bath was added
sodium
borohydride (0.045 g, 1.2 mmol). This was stirred until it had warmed almost
to room
temperature. TLC on silica gel showed product and maybe some starting
material.
A slight excess of sodium borohydride was added to the mixture stirring at 0
°C. The
reaction was allowed to stir and warm to room temperature overnight. The
solvent
was evaporated at reduced pressure. The residue was partitioned between ethyl
acetate and water. The ethyl acetate layer was washed with water twice and
dried
over anhydrous magnesium sulfate. Filtration and concentration in vacuum gave
0.31 g of oil. This was chromatographed on silica gel with a gradient of ethyl
acetate
and hexane to give 0.1307 g of the free base as an oil. This was dissolved in
EtOH
and added to a solution of oxalic acid dihydrate (0.0426 g, 0.338 mmol) in
ethanol.
Filtration gave 0.1237 g of the S enantiomer of the title compound as a white
solid
oxalate, m.p. 183-185°C.
Elemental Analysis for: C26H24N2O2S ~ C2H204' 1/3 H20
Calc'd: C, 64.11; H, 5.12; N, 5.34
Found: C, 64.01; H, 5.05; N, 5.28
EXAMPLE 11
2-(4-Benzofuran-2-yl-3,6-dihydro-2H-pyridin-1-ylmethyl)-8-methyl-2,3-dihydro
[1,4ldioxinof2,3-flguinoline
[0107] A mixture of [(2R)-8-methyl-2,3-dihydro[1,4]dioxino[2,3-f]quinolin-2-
yl]methyl
4-bromobenzenesulfonate ester (0.18 g, 0.40 mmol), 4-benzofuran-2-yl-1,2,3,6-
tetrahydropyridine (0.15 g, 0.75 mmol) and potassium carbonate (0.21 g, 1.5
mmol)
in 3 mL of N,N-dimethylformamide was stirred under nitrogen at 60 °C
for 24 hours.
The mixture was partitioned between 250 mL each of water and ethyl acetate.
The
-56-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
organic fraction was dried over magnesium sulfate, filtered and concentrated
in
vacuum. The residue was column chromatographed on 50 mL of silica gel using
first
25% ethyl acetate in hexane and then 50% ethyl acetate in hexane as eluant.
Combination and concentration of the product fractions gave 0.025 g of the S
enantiomer of the title compound as a pale yellow solid, m.p. 149-
150°C.
Elemental Analysis for: C26H24N203 ~ 0.5 H20
Calc'd: C, 74.09; H, 5.98; N, 6.65
Found: C, 73.96; H, 5.89; N, 6.43
EXAMPLE 12
2-(4-Benzofuran-2-yl-aiperidin-1-yl methyl)-8-methyl-2,3-di hydro
f1,41dioxinof2,3-flauinoline
[0108] A mixture of [(2R)-8-methyl-2,3-dihydro[1,4]dioxino[2,3-f]quinolin-2-
yl]methyl
4-bromobenzenesulfonate ester (0.18 g, 0.40 mmol), 4-benzofuran-2-yl-
piperidine
(0.103 g, 0.51 mmol) and potassium carbonate (0.21 g, 1.5 mmol) in 3 mL of N,N-
dimethylformamide was stirred under nitrogen at room temperature for 3 days
and
then at 60 °C for 6 hours. The mixture was partitioned between 250 mL
each of
water and ethyl acetate. The organic fraction was dried over magnesium
sulfate,
filtered and concentrated in vacuum. The residue was column chromatographed on
50 mL of silica gel using first 25% ethyl acetate in hexane and then 50% ethyl
acetate in hexane as eluant. Combination and concentration of the product
fractions
gave 0.060 g of the S enantiomer of the title compound as a pale yellow solid,
m.p.
103-104°C.
Elemental Analysis for: C26H26N203 ~ 0.25 H20
Calc'd: C, 74.53; H, 6.37; N, 6.69
Found: C, 74.52; H, 6.49; N, 6.63
INTERMEDIATE 26
4-(5-Chloro-benzofblthiophen-3-yl)-pyridine
[0109] To 3-bromo-5-chloro-benzo[b]thiophene (9.80 g, 39.6 mmol) was added
pyridine-4-boronic acid (4.445 g, 36.2 mmol), K3P04 (19.5 g, 91.9 mmol), 61 mL
of
-57-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
1,4-dioxane and 6 mL of water. The mixture was placed under house vacuum for
several minutes and flushed with nitrogen. This was repeated 5 times.
Pd(dppf)CI2
CH2C12 (2.71 g, 3.31 mmol), PdCl2 (0.0563 g, 0.318 mmol) and 1,1 "-
bis(diphenylphosphino)ferrocene (0.1747 g, 0.3151 mmol) were purged in the
same
way. The catalyst was added to the reaction flask which was purged with
nitrogen 3
more times. The mixture was stirred (stir bar) at 80°C for 22 hours.
TLC showed
little change. The mixture was mechanically stirred at 80°C for 4
hours. The mixture
was partitioned between water and ethyl acetate, filtered through , Celite and
the
layers were separated. The organic layer was evaporated. The residue was
dissolved in ethyl acetate, washed with water twice. Saturated brine was added
the
second time to separate the layers more quickly. The organic solution was
dried
over magnesium sulfate. Filtration and contration in vacuum gave 11.35 g of
black
oil. This was eluted from silica gel with a gradient of hexane and ethyl
acetate to give
3.86 g of recovered starting material and 4.28 g of the title compound as
light brown
crystals, m.p. 90-91°C.
Elemental Analysis for: C13H8CINS
Calc'd: C, 63.54; H, 3.28; N, 5.70
Found: C, 63.19; H, 3.26; N, 5.46
EXAMPLE 13
2-f4-(5-Chloro-benzof blthiophen-3-yl)-3,6-dihydro-2H-pyridin-1-ylmethyll-8-
methyl-2,3-dihydro-f1,41dioxinof2,3-flguinoline ,
[0110] To a mixture of [(2R)-8-methyl-2,3-dihydro[1,4]dioxino[2,3-f]quinolin-2-
yl]methyl 4-bromobenzenesulfonate ester (0.92 g, 2.0 mmol) and 4-(5-chloro-
benzo[b]thiophen-3-yl)-pyridine (0.50 g, 2.0 mmol) was added 8 mL of acetone.
The
mixture was refluxed for 4 hours, stirred at room temperature overnight and
refluxed
for 5 hours more. The solvent slowly evaporated and was replenished as needed.
After standing at room temperature overnight some crystals had formed. To this
was
added 15 mL of methylethylketone. The mixture was refluxed overnight. TLC on
silical gel showed starting material and no obvious product. The solvent was
evaporated. The residue was stirred at 130°C overnight. The thick
mixture had
solidified. It was broken up and crushed. To this was added 12 mL of ethanol.
This
-58-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
heterogeneous mixture was stirred in an ice-bath. Sodium borohydride (0.12 g,
3.2
mmol) was added initially. A slight excess was added to insure consumption of
the
pyridinium salt. The reaction was allowed to stir and warm to room temperature
overnight. The solvent was evaporated at reduced pressure. The residue was
partitioned between ethyl acetate and water. The ethyl acetate layer was
washed
with water 3 times and dried over anhydrous magnesium sulfate. Filtration and
concentration in vacuum gave 0:84 g of dark oil. This was chromatographed on
silica
gel with a gradient of ethyl acetate and hexane to give 0.29 g of the free
base as an
oil. This was dissolved in ethanol and added to a solution of oxalic acid
dihydrate
(0.0811 g, 0.643 mmol) in ethanol. Filtration gave 0.2659 g of the S
enantiomer of
the title compound as white oxalate, m.p. 203-207 °C.
Elemental Analysis for: C2gH23CIN202S ~ C2H204 ~ 2/3 H20
Calc'd: C, 59.52; H, 4.70; N, 4.96
Found: C, 59.59; H, 4.40; N, 4.74
G~rennm c ~d
2-(4-Benzoxazol-2-yl-piperidin-1-ylmethyl)-8-methyl-2,3-dihydro
j1,4ldioxinof2,3-flduinoline
[0111] A mixture of [(2R)-8-methyl-2,3-dihydro[1,4]dioxino[2,3-f]quinolin-2-
yl]methyl
4-bromobenzenesulfonate ester (0.18 g, 0.40 mmol), 2-piperidin-4-yl-
benzoxazole
(0.121 g, 0.59 mmol) and potassium carbonate (0.21 g, 1.5 mmol) in 3 mL of N,N-
dimethylformamide was stirred under nitrogen at room temperature for 3 days
and
then at 60 °C for 6 hours. The mixture was partitioned between 250 mL
each of
water and ethyl acetate. The organic fraction was dried over magnesium
sulfate,
filtered and concentrated in vacuum. The residue was column chromatographed on
50 mL of silica gel using first 50% ethyl acetate in hexane and then 75% ethyl
acetate/hexane as eluant. Combination and concentration of the product
fractions
gave 0.040 g of the S enantiomer of the title compound as a dark beige solid,
m.p.
128-130°C.
Elemental Analysis for: C25H25N3~3' 0.25 H20
Calc'd: C, 71.49; H, 6.12; N, 10.00
Found: C, 71.60; H, 6.06; N, 10.19
-59-
CA 02498532 2005-03-10
WO 2004/024733 PCT/US2003/028523
[0112] When ranges are used herein for physical properties, such as molecular
weight, or chemical properties, such as chemical formulae, all combinations
and
subcombinations of ranges specific embodiments therein are intended to be
included.
[0113] The disclosures of each patent, patent application, and publication
cited or
described in this document are hereby incorporated herein by reference, in
their
entirety.
(0114] Those skilled in the art will appreciate that numerous changes and
modifications can be made to the preferred embodiments of the invention and
that
such changes and modifications can be made without departing from the spirit
of the
invention. It is, therefore, intended that the appended claims cover all such
equivalent variations as fall within the true spirit and scope of the
invention.
-60-