Note: Descriptions are shown in the official language in which they were submitted.
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
"Orthopoxvirus vectors, genes and products thereof"
Field of the invention
The invention relates to a viral protein that is a novel inhibitor of
intracellular
signalling mediated by the immunologically important interleukin-1 / Toll-like
receptor (IL-lRITLR) superfamily. The invention also relates to the mechanism
whereby the inhibitor functions, and the use of the inhibitor, or information
derived
from its mechanism of action, in designing peptides or small molecule
inhibitors for
use in IL-1R/TLR related diseases and conditions. The invention also relates
to a
recombinant vaccinia virus (VV) as a vaccine candidate for the prevention of
smallpox or other infectious diseases, or for the prevention or treatment of
cancer.
Background
Members of the IL-1R/TLR superfamily are key mediators in innate and adaptive
immunity (Akira, S., Takeda, K. & Kaisho, T. Nature Immuhol. 2, 675-680
(2001)).
The superfamily is defined by the presence of a cytosolic motif termed the
Toll/IL-1
receptor (TIR) domain. The family includes receptors for the proinflammatory
cytokines IL-1 and IL-18 as well as the TLR members, which participate in the
recognition of pathogens by responding to pathogen associated molecular
patterns
(PAMPs) and activating signalling pathways leading to altered gene expression
(Bowie, A. & O'Neill, L.A.J. J. Leuk. Biol. 67, 508-514 (2000)). The TLRs were
discovered on the basis of their amino acid similarity to Toll, a Drosophila
protein
involved in mediating antifungal defence (Lemaitre, B., et al. Cell 86, 973-
983
(1996)). Ten mammalian TLRs have been identified to date. TLR4, the first TLR
to
be discovered, is essential for the response to lipopolysaccharide (LPS)
(Poltorak, A.
et al. Sciehce 282, 2085-2088 (1998); Qureshi, S.T, et al. J. Exp. Med. 189,
615-625
(1999)). TLRS recognises and responds to bacterial flagellin (a 55kD monomer
of
bacterial flagella) (Hayashi, F. et al. Nature 410, 1099-1103 (2001)), while
TLR9 is
required for the recognition of unmethylated CpG motifs which are present in
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
2
bacterial DNA (Hemmi, H. et al. Nature 408, 740-745 (2000)). TLR2 recognises
diverse bacteria and their products, including bacterial lipoproteins,
peptidoglycan
and other Gram-positive molecular patterns, but only when present as a
heterodimer
in combination with another TLR, such as TLR1 or TLR6 (Brightbill, H.D. et al.
Science 285, 732-736 (1999); Aliprantis, A. et al. Science 285, 736-739
(1999);
Underhill, D. et al. Nature 401, 811-815 (1999); Takeuchi, O. et al. Immunity
11,
443-451 (1999); Ozinsky, A. et al. Proc. Natl. Acad. Sci. USA 97, 13766-13771
(2000); Takeuchi, O. et al. Int. Immuhol. 13, 933-940 (2001)).
TLRs have also been implicated in sensing viral infections. TLR4 has been
shown to
be necessary for the cytokine-stimulating ability of F protein from
respiratory
syncytial virus (RSV) and also for murine retrovirus activation of B cells
(Kurt-
Jones, E. A. et al. Nature Immuhol. 1, 398-401 (2000); Rassa, J.C. et al.
Proc. Natl.
Acad. Sci. USA 99, 2281-2286 (2002)). TLR3 meanwhile was identified as a
receptor activated in response to poly(I:C), a synthetic double-stranded RNA
(dsRNA) mimic of viral dsRNA. Poly(I:C) activation of cells via TLR3 led to
the
activation of the transcription factor NFxB and the production of type I
interferons,
which are important in anti-viral innate immunity (Alexopoulou, L. et al.
Nature
413, 696-712 (2001)). Further, imidazoquinoline compounds known to have potent
anti-viral properties, such as R-848, activated immune cells via TLR7 (Hemmi,
H. et
al. Nature Immu~ol. 3, 196-200 (2002)).
Since these receptors all contain the signalling TIR domain, stimulation of
all the
family members with the appropriate ligands leads to activation of NFxB and
also
the mitogen-activated protein kinases (MAPKs), p38, c-Jun N terminal kinase
(JNK) and p42144. NFxB is a homo- or hetero- dimer of members of the Rel
family of transcriptional activators that is involved in the inducible
expression of a
wide variety of important cellular genes. The activation of NFxB by IL-1, IL-
18,
TLR2, TLR7 and TLR9 is absolutely dependent on the cytoplasmic TIR domain-
containing protein MyD88 (Hemmi, H. et al. Nature Immuhol. 3, 196-200 (2002);
Adachi, O. et al. Immunity 9, 143-150 (1998); Takeuchi, O. et al. J. Immuhol.
164,
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
3
554-557 (2000); Schnare, M. et al. Curr. Biol. 10, 1139-1142 (2000)), which is
recruited to receptor TIR domains (Medzhitov, R. et al. Mol. Cell 2, 253-258
(1998); Wesche, H. et al. Immunity 7, 837-847 (1997); Muzio, M. et al. Science
278, 1612-1615 (1997)). Further, the induction of IL-6 by flagellin via TLRS
was
completely dependent on MyD88 (Hayashi, F. et al. Nature 410, 1099-1103
(2001)). TLR4 activates NFxB, by a MyD88-dependent pathway, although an
alternative MyD88-independent pathway also exists (Kawai, T. et al. Immunity
11,
115-122 (1999)). Thus MyD88 is a crucial adaptor molecule for the entire IL-
1R/TLR superfamily, with the exception of TLR3, where NFxB activation is
MyD88-independent (Alexopoulou, L. et al. Nature 413, 696-712 (2001)).
The MyD88 dependent pathway is involved in TNF induction by LPS in dendritic
cells whereas the MyD88 independent pathway leads to the upregulation of
costimulatory molecules required for dendritic cell maturation, and induction
of
genes dependent on the transcription factor Interferon Regulatory Factor 3
(IRF3)
(Kaisho, T. et al. J. Immunol. 166, 5688-5694 (2001)). An important example of
such a gene is Interferon- (IFN ) . For TLR4 and TLR2, another TIR adapter
molecule, MyD88Adaptor-Like (Mal, also known as TIRAP) is involved in the
MyD88 dependent pathway (Fitzgerald, K. A. et al. Nature 413, 78-83 (2001);
Horng, T., Burton, G. M. & Medzhitov, R. Nature Immunol. 2, 835-841 (2001);
Yamamoto, M. et al Nature 420, 324-329 (2002); Horng, T. et al Nature 420, 329-
333 (2002)).
Activation of NFxB by the MyD88 dependent pathway can proceed via
recruitment by MyD88 of IL-1 receptor-associated kinase (IRAK) and/or IRAK2,
while Mal functions via the recruitment of IRAK2 (Fitzgerald, K. A. et al.
Nature
413, 78-83 (2001)). IRAK or IRAK2 activation in turn leads to recruitment of
tumour necrosis factor receptor-associated factor 6 (TRAF6). TRAF6 is required
for the ubiquitination and activation of the kinase TAK-1, which, in complex
with
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
TAB1, phosphorylates IxB kinase (IKK) leading to NFxB activation (Wang, C. et
al. Nature 412, 346-351 (2001)).
Other signalling pathways apart from NFxB are also important in TLR-mediated
gene induction. Clearly the MAP kinase pathways are also important, leading as
they do to the activation of transcription factors, as well as to post-
transcriptional
events that enhance gene induction. All IL-1R/TLR activators tested have been
shown to activate one or all of the three MAP kinase pathways, extracellular
regulated kinase (ERK), p38 and JNK. In response to IL-1R/TLR activators, the
MAP kinases can be triggered through MyD88 (shown for IL-18, TLR2, TLR9), but
also, in the case of LPS and poly(I:C), through MyD88-independent pathways
(Adachi, O. et al. Immunity 9, 143-150 (1998); Takeuchi, O. et al. .J.
Immu~zol. 164,
554-557 (2000); Hacker, H. et al. J. Exp. Med. 192, 595-600 (2000); Kawai, T.
et al.
Immunity 11, 115-122 (1999); Alexopoulou, L. et al. Nature 413, 696-712
(2001)).
A role for all three MAP kinase pathways in the induction of different genes
by LPS
has been clearly demonstrated (reviewed in Guha, M. & Mackman, N. Cellular
Sig~allihg 13, 85-94 (2001)).
Another important transcription factor activated by some TLR3 and TLR4 is
IRF3,
which has importance in IFN-dependent anti-viral defense (Servant, M.J.,
Grandvaux, N. & Hiscott, J. Biochem. Pharmacol. 64, 985-992 (2002)). Recently
another TIR adapter termed TICAM-1 or TRIF has been discovered (Yamamoto, M.
et al J. Immunol. 169, 6668-6672 (2002); Oshiumi, H. et al Nature Immuuol. 4,
161-
167 (2003)). It has been shown that for TLR4, TRIF mediates the MyD88-
independent pathway to IRF3, while for TLR3, TRIF mediates both NF B and IRF3
activation (Hoebe, K. et al Nature doi:10.1038/nature01889 (2003); Yamamoto,
M.
et al Science doi:10.11261science.1087262 (2003)).
Any novel method to inhibit IL-1R/TLR superfamily signalling would have
important therapeutic application.
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
Statements of Invention
According to the invention there is provided an orthopoxvirus vector, such as
vaccinia, Wherein the A46R protein from vaccinia, or a closely related protein
from
5 any orthopoxvirus is not expressed or is expressed but is non-functional.
In one embodiment of the invention part or all of the nucleotide sequence
encoding
A46R is deleted from the viral genome.
In another embodiment of the invention the nucleotide sequence encoding A46R
is
inactivated by mutation or the insertion of foreign DNA.
The nucleotide sequence encoding A46R may be changed.
In one embodiment of the invention the A46R gene comprises amino acid SEQ ID
No. 1.
In another embodiment the vector comprises DNA sequences encoding one or more
heterologous polypeptides.
The orthopoxvirus vector of the invention has enhanced irnmunogenicity andlor
safety compared to the wild type orthopoxvirus.
The invention also provides a medicament comprising an orthopoxvirus vector of
the
invention.
In another aspect the invention provides a vaccine comprising an orthopoxvirus
vector of the invention.
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
6
In another aspect the invention provides a recombinant orthopoxvirus incapable
of
expressing a native A46R protein. A vaccine may comprise such a recombinant
virus.
In a further aspect the invention provides a method of attenuating an
orthopoxvirus
vector such as vaccinia virus, comprising the steps of:
(a) deleting part or all of the nucleotide sequence encoding A46R
from the viral genome; and/or
(b) inactivating one or more of said nucleotide sequence by mutating
said nucleotide sequence or by inserting foreign DNA; and/or
(c) changing said nucleotide sequence to alter the function of a
protein product encoded by said nucleotide sequence.
In one embodiment the invention provides a method of inhibiting IL1R/TLR
superfamily signalling comprising administering an effective amount of
vaccinia
A46R protein, or a closely related protein from any orthopoxvirus or a
functional
peptide, peptidometic, fragment or derivative thereof, or a DNA vector capable
of
expressing such a protein or fragment thereof.
In another embodiment the invention provides a method of modulating anti-viral
immunity in a host comprising administering a vaccinia virus vector of the
invention
or a functional peptide, peptidometic, fragment or derivative thereof.
The invention also provides an immunogen comprising a vaccinia virus vector or
a
recombinant virus vector of the invention.
In another aspect the invention provides use of a vaccinia virus A46R protein,
or a
closely related protein from any orthopoxvirus, or a functional peptide,
peptidometic,
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
fragment or derivative thereof, or a DNA vector expressing any of the above in
the
modulation and/or inhibition of IL-1R/TLR superfamily-signalling.
The use may be in the modulation and/or inhibition of IL-1R/TLR superfamily-
induced NFxB activation.
The use may be in the modulation and/or inhibition of IL-1RJTLR superfamily-
induced MAP kinase activation.
The use may be in the modulation and/or inhibition of TLR induced IRF3
activation.
In one aspect the vaccinia virus A46R protein, or a closely related protein
from any
orthopoxvirus, inhibits Toll-like receptor proteins.
The use as may be in the modulation and/or inhibition of NF-xB activity or MAP
kinase activation by interaction of A46R with MyD88.
The use may be in the modulation and/or inhibition of NF-xB activity or MAP
kinase activation by interaction of A46R with Mal.
In one aspect the vaccinia virus A46R protein inhibits MyD88- andlor Mal-
dependent signalling.
The use rnay be in the modulation and/or inhibition of IRF3 or NF-xB activity
by
interaction of A46R with TRIF.
In one aspect of the invention the vaccinia virus A46R protein inhibits TRIF-
dependent signalling.
The invention also provides a peptide derived from, and/or a small molecule
inhibitor designed based on vaccinia virus A46R protein.
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
g
The invention further provides a method of screening compounds that modulate
the
IL-1R/TLR-induced NF-xB or MAP kinase pathway comprising measuring the
effect of a test compound on the interaction of vaccinia virus A46R protein or
a
functional peptide, peptidometic, fragment or derivative thereof with MyD88.
The invention also provides a method of screening compounds that modulate the
IL-
1R/TLR-induced NF-xB or MAP kinase pathway comprising measuring the effect of
a test compound on the interaction of vaccinia virus A46R protein or a
functional
peptide, peptidometic, fragment or derivative thereof with Mal.
The invention further provides a method of screening compounds that modulate
the
IL-1RITLR-induced NF-xB, IRF3 or MAP kinase pathway comprising measuring
the effect of a test compound on the interaction of vaccinia virus A46R
protein or a
functional peptide, peptidometic, fragment or derivative thereof with TRIF.
The invention also provides a method of identifying signalling pathways that
require
MyD88 and/or Mal andlor TRIF, comprising measuring their sensitivity to A46R.
In another aspect the invention provides use of a functional peptide,
peptidometic, or
fragment derived from vaccinia virus A46R protein, or a small molecule
inhibitor
designed based on A46R protein or a DNA vector capable of expressing such a
protein or fragment in the treatment and/or prophylaxis of IL-1R/TLR
superfamily-
induced NF-xB, IRF3 or MAP kinase related diseases or conditions.
Preferably the NF-xB related disease or condition is selected from any one or
more
of a chronic inflammatory disease, allograft rejection, tissue damage during
insult
and injury, septic shock and cardiac inflammation, autoimmune disease, cystic
fibrosis or any disease involving the blocking of Thl responses. The chronic
inflammatory disease may include any one or more of rheumatoid arthritis,
asthma or
inflammatory bowel disease. The autoimmune disease may include systemic lupus
erythematosus.
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
9
The use may be in the treatment and/or prophylaxis of inflammatory disease,
infectious disease or cancer.
One aspect of the invention also provides use of a viral protein derived from
an
A46R-like protein or a functional peptide, gene, or peptidometic thereof in
the
treatment andlor prophylaxis of inflammatory disease.
The A46R-like protein may be derived from an orthopoxvirus.
The term functional peptide, peptidometic, fragment or derivative as used
herein are
understood to include any molecule or macromolecule consisting of a portion of
the
A46R protein, or designed using sequence or structural information from A46R.
The term non-functional is understood to mean not functioning in the normal
way
compared to how the wild-type A52R protein would function.
The term 'closely related' is understood to mean 'greater than 50% amino acid
identity'.
The invention is in the field of poxviruses. The family name is poxvirus, the
subfamily name is chordopoxvirinae (infect vertebrates) and the genus is
orthopoxvirus which includes species of virus some of which have A46R related
proteins. The best known species of this genus are vaccinia, variola,
camelpox,
cowpox, monkeypox and ectromelia (which infects mice).
The invention relates to any orthopoxvirus vector in which the A46R protein is
deleted/modified.
The invention further relates to the use of a DNA vector expressing A46R
protein.
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
Brief description of the drawings
The invention will be more clearly understood from the following description
thereof
given by way of example only with reference to the accompanying drawings in
5 which:-
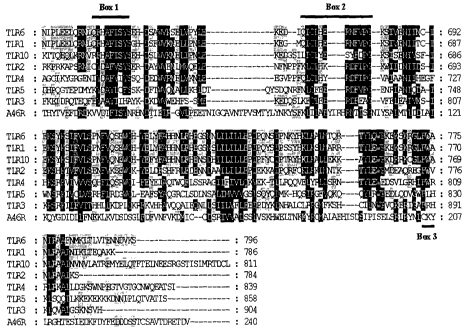
Fig. 1 is a sequence alignment showing the sequence similarity between
A46R and TIR domains from IL-1R/TLR signalling proteins;
10 Figs. 2a to d are graphs showing the inhibition by A46R of the activation
of
NFxB and MAP kinases by IL-1 in human 293 cells;
Figs. 3a to d are graphs showing the inhibition by A46R of the activation of
NFxB and MAP kinases by TLR4 in human 293 cells;
Fig. 4 is a graph showing the inhibition by A46R of the activation of NFxB
by
TLR agonists LPS (TLR4), R-848 (TLR7/8) and flagellin (TLRS) in a murine
macrophage cell line, RAW264.7;
Fig. 5 is an immunoblot showing the ectopic expression of A46R in 293T
cells;
Figs. 6a and b are imrnuno-blots showing association of A46R with MyD88,
by co-immunoprecipitation and GST pull-down.
Figs. 7a and b are immuno-blots showing association of A46R with Mal, by
co-immunoprecipitation and GST pull-down.
Figs. 8a to a are graphs showing the inhibition by A46R of the activation of
NFxB and MAP kinases by MyD88 and Mal;
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
lI
Figs. 9a and b are bar graphs showing the inhibition by A46R of TLR3-
induced Interferon-Stimulated Response Element (ISRE) and Interferon-(3
(IFN(3) promoter;
Figs. 10a and b are immuno-blots showing association of A46R with TRIF,
by co-immunoprecipitation and GST pull-down.
Figs. l la and b are graphs showing the inhibition by A46R of the activation
of NFxB and IFN(3 promoter by TRIF;
Figs. 12 a to c are graphs showing the inhibition by A46R of TLR4-mediated
TRIF-dependent pathways. Fig 12c also shows inhibition of ISRE by the
TLR activators LPS, poly(I:C), R-848 and flagellin;
Figs. 13a and b are immuno-blots showing the phase during infection at
which the A46R protein is expressed, and lack of expression of A46R in cells
infected with the VV deletion mutant lacking the A46R gene;
Figs. 13c and d are graphs showing that deletion of A46R does not affect the
growth or replication of VV in cell culture; and '
Fig. 14 is a graph showing that deletion of A46R from the vaccinia virus
genome attenuates the virus, as measured by weight loss in a murine
intranasal model of infection.
Detailed description
Poxviruses are a family of complex DNA viruses that include variola virus, the
causative agent of smallpox, and the antigenically related virus used to
eradicate this
disease, vaccinia virus (VV). Orthopoxvirus such as VV display unique
strategies
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
12
for the evasion of host immune responses such as the ability to produce
secreted
decoy receptors for cytokines such as IL-1, TNF, and the interferons IFNcx(3
and
IFN~y. Often these inhibitors of host immune function display sequence
similarity to
host proteins.
The present invention concerns a VV protein A46R, which is known to be an
intracellular inhibitor of signalling via IL-1 (Bowie, A. et al. Proc. Natl
Acad. Sci.
USA 97, 10162-10167 (2000)). Using PROFILESEARCH (Genetics Computer Group,
Madison, WI) to search sequence databases for novel proteins containing TIR
domains, we identified A46R (Bowie, A. et al. P~oc. Natl Acad. Sci. USA 97,
10162-10167 (2000)) as a VV protein with a putative TIR domain. The name A46R
is based on the standard VV nomenclature of the Copenhagen strain (Goebel, S.
J. et
al. Virology 179, 247-266 (1990)). A46R was cloned from the laboratory VV
strain
Western Reserve (WR), where it was previously called SalF9R (Smith, G. L.,
Chan,
Y. S. & Howard, S. T. J. Ge~c. Vz~-ol. 72, 1349-1376 (1991)). A46R displays a
high
degree of sequence conservation between different strains of VV, including WR,
Copenhagen, Modified Virus Ankara and Tian Tian, while many other
orthopoxviruses also have a closely related version of A46R, namely variola
major,
variola minor, camelpox, monkeypox and cowpox. This high degree of
conservation
could reflect an important role for A46R in viral virulence.
Fig. 1 shows an alignment of A46R with other TIR domains from TLRs. Within the
TIR domain there are three regions of important sequence conservation, which
have
been termed Box 1, 2 and 3 (Bowie, A. & O'Neill, L.A.J. J. Leuk. Biol. 67, 508-
514
(2000)). Box 1 is particularly strong in A46R, the sequence DTFISY being as
closely related to the Box 1 consensus of other proven family members.
The crystal structures of the TIR domains for TLR1 and TLR2 have been
determined
and show that the domain adopts a three-Iayer a(3a sandwich conformation,
similar
to the bacterial protein CheY. Threading the A46R amino acid sequence through
secondary structure prediction programmes revealed that A46R could also fold
in
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
13
such a manner. Therefore the A46R protein does appear to contain a bona fide
TIR
domain.
In the present invention it was found that, in addition to blocking IL-1
mediated
. NFxB activation A46R inhibits numerous signalling pathways activated by IL-
1,
including c-Jun N terminal kinase (JNK) and extracellular-regulated kinase
(ERK)
MAP kinase activation (Fig. 2a to d). Further, A46R is also shown to inhibit
NFxB
activation by multiple TLRs including TLR4 (Fig. 3a), TLR7l8 (Fig. 4) and TLRS
(Fig. 4). The fact that A46R could associate with both MyD88 (Fig. 6) and Mal
(Fig.
7) provides a rationale for these inhibitory effects. In addition A46R was
able to
block MyD88-independent pathways (Figs 10 and 13) by associating with TRIF
(Fig. 11). A46R was shown to be expressed early on in cells infected with VV
(Fig.
14). Furthermore, a deletion mutant VV lacking the A46R gene was shown to be
attenuated compared to wild type and revenant controls in vivo (Fig. 15),
indicating
the importance of A46R in viral virulence.
There is intense interest in the IL-1R/TLR family at present, given its
emerging
central importance in the innate immune response to diverse pathogens (Akira,
S.,
Takeda, K. & Kaisho, T. Nature Immuhol. 2, 675-680 (2001)). During the course
of
viral infection the body mounts several lines of host defence involving
constituents
of the IL-1R/TLR superfamily. The cytokines IL-1 and IL-18 are key regulators
of
the innate and adaptive immune response to viral infection. In particular IL-1
is
antagonized by the production of a soluble IL-1 binding protein (B15R) by VV
(Alcami, A. & Smith, G.L. Cell 71, 153-167 (1992). IL-18 is a potent inducer
of
IFN- , and administration of IL-18 has been shown to elicit antiviral effects
in VV-
infected mice (Tanaka-Kataoka, M. et al. Cytokihe 11, 593-599 (1999)). Recent
work has suggested that TLR3, TLR4 and TLR7 are crucial mediators of an innate
immune response to viral infection (Kurt-Jones, E. A. et al. Nature Immu~ol.
1, 398-
401 (2000); Rassa, J.C., Meyers, J.L., Zhang, Y., Kudaravalli, R. & Ross, S.
Proc.
Natl. Acad. Sci. USA 99, 2281-2286 (2002), Alexopoulou, L., Czopik-Holt, A.,
Medzhitov, R. & Flavell, R. Natzsre 413, 696-712 (2001) and Hernrni, H. et al.
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
14
Nature Immunol. 3, 196-200 (2002)). Furthermore, TLR2 and TLR9 have also been
implicated in responding to some viruses (Lund, J. et al J. Exp. Mea: 198, 513-
520
(2003); Compton, T. et al J. Virol. 77, 4588-4596 (2003). It is possible that
other
TLRs also have a role in responding to viral infection. The TLR family is
therefore
important in anti-viral host defense. Viral mechanisms to antagonise this
family
would have valuable therapeutic potential.
In the present invention the VV protein A46R has been found to be an IL-1R/TLR
inhibitor. Deletion of A46R from VV causes the virus to be attenuated in a
murine
model of infection (Fig. 15). These results further support the emerging role
of
TLRs in the host response to viral infection.
A46R is very similar to the TIR domain and has wide-ranging effects on IL-
1R/TLR
signalling. The inhibitory data suggests that A46R blocks IL-1R/TLR signalling
close to the receptors, before the NFxB and MAP kinase pathways bifurcate,
probably by disrupting TIR-dependent interactions necessary for signalling.
One
important target for A46R is likely to be MyD88, since A46R can be co-
immunoprecipitated with MyD88 (Fig. 6a), while GST-A46R can pull MyD88 out of
a cell lysate (Fig. 6b), probably due to an association between the A46R and
MyD88
TIR domains. This is the first demonstration of a viral protein targeting
MyD88. The
fact that A46R can also target Mal (as assessed by co-immunoprecipitation and
GST
pull-down, (Fig. 7) is consistent with the fact that TLR4 inhibition by A46R
is
particularly potent (compare Fig. 3 to Fig. 2).
In addition to targeting MyD88- and Mal-dependent signalling, A46R was also
found to block MyD88-independent TLR signalling events, such as those
emanating
from TLR3. Fig. 9 shows that both Interferon-Stimulated Response Element
(ISRE)
and Interferon-(3 (IFN(3) promoter induction by TLR3 was potently blocked by
A46R. This indicated that A46R might also target the adapter TRIF, since this
has
been shown to be responsible for most if not all TLR3-dependent. Fig. 10 shows
that
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
A46R could co-immunoprecipitate TRIF and also that GST-A46R was capable of
pulling down TRIF.
Thus A46R, by nature of its similarity to the TIR domain, can target three key
5 adapters involved in signalling by the IL-1/TLR superfamily, thus allowing
A46R to
antagonise a huge array of signalling pathways. The targeting of TRIF is
particularly
interesting, since it has recently been shown that TRIF controls the IFN-
dependent,
and thus anti-viral arm, of TLR3 and TLR4 signalling. When mice in which the
TRIF gene was mutated were infected with cytomegalovirus, no IFN a/(3 was
10 detected in the serum, although a robust IFN response was evident in
control mice
(Hoebe, K. et al Natu~~e doi:10.1038/nature01889 (2003). Further, in
macrophages
in which TRIF is disrupted, vaccinia virus was able to replicate to a higher
titre,
compared to in control cells (Hoebe, K. et al Nature doi:10.1038/nature01889
(2003)). This demonstrates a direct role for TRIF in containing viral
infections, and
15 hence makes it an important target for viral immune evasion strategies.
From the profile of expression of A46R in infected cells it was shown that the
protein is expressed quite early in infection, compared to D8L, a known late
expressed VV protein (Fig. 13a). This would be consistent with a role for A46R
in
suppressing IL-1R/TLR signalling in infected cells, since these receptors are
generally involved in the triggering of host immune responses in the early
phases of
infection.
The present invention relates to a recombinant vaccinia virus in which the
gene
sequence of A46R is deleted. Fig. 13b confirms that the mutant virus does not
express the A46R protein, while a revertant virus (in which the A46R gene is
inserted into the virus deletion mutant lacking A46R) does. Deletion of A46R
did
not affect the replication of the virus in cell culture (Fig. 13c and d).
However, the
absence of A46R led to an attenuation of the virus, in that when mice were
infected
intranasally, the deletion mutant caused a reduction in the weight loss
induced in the
animals, compared to wild type and revertant virus (Fig. 14).
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
16
Live vaccinia virus is currently used as the vaccine to immunise against and
eradicate smallpox. However there is a need to develop more effective and
safer
smallpox vaccines due to the threat of bioterrorism. It is possible to
engineer
recombinant vaccinia viruses in which vaccinia genes are deleted or altered.
Deletion or alteration of vaccinia virus genes involved in modulating the host
immune response can alter the immunogenicicty and safety of a vaccinia virus
for
use as a vaccine against smallpox or other orthopoxviruses, or for the
development
of recombinant vaccinia viruses as vaccines against other infectious diseases
and
cancer. Such recombinant vaccinia viruses can be engineered in which genes
derived
from other organisms are inserted (Market, M. & Smith, G.L. J. Geh. V'irol.
67,
2067-2082 (1986)). The recombinant viruses retain their infectivity and
express any
inserted genes during the normal replicative cycle of the virus. Immunisation
of
animals with recombinant viruses containing foreign genes has resulted in
specific
immune responses against the proteins) expressed by the vaccinia virus,
including
those proteins) expressed by the foreign genes) and in several cases has
conferred
protection against the pathogenic organism from which the foreign gene was
derived.
Recombinant vaccinia viruses have, therefore, potential application as new
live
vaccines in human or veterinary medicine.
The present invention also relates to a vaccinia virus wherein 93.5 % of the
nucleotide sequence encoding A46R is deleted. Alteration or deletion of A46R
from
the vaccinia genome may increase virus safety and immunogenicity. Such a virus
or
a derivative virus expressing one or more foreign antigens may have
application as
an improved vaccine against smallpox or other orthopoxvirses, or for the
application
of recombinant vaccinia viruses as vaccines against other infectious diseases
and
cancer.
The examples presented are illustrative only and various changes and
modifications
within the scope of the present invention will be apparent to those skilled in
the art.
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
17
Examples
Cell Culture. HEK 293, HEK 293T and RAW 264.7 cells were cultured in
Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS),
supplemented with 100 units/ml penicillin, 100 mg/ml streptomycin, and 2 mM L-
glutamine.
Expression Plasmids. The chimeric TLR receptor CD4-TLR4, composed of the
extracellular domain of CD4 fused to the transmembrane domain and cytosolic
tail
of TLR4 was a gift from R. Medzhitov, (Yale University, New Haven, CT). AUl-
MyD88 expression vector was a gift from M. Muzio (Muzio, M., Ni, J., Feng, P.
&
Dixit, V.M. Science 278, 1612-1615 (1997)). The mammalian expression vector
pRKS was kindly provided by Tularik Inc. (San Francisco, CA). Flag-TRIF was
from S.Akira (Research Institute for Microbial Diseases, Osaka University,
Japan).
A46R, Flag-A46R and HA-Mal expression plasmids have been previously described
(Bowie, A. et al. P~oc. NatZ Acad. Sci. USA 97, 10162-10167 (2000);
Fitzgerald, K.
A. et al. Nature 413, 78-83 (2001)).
The name A46R is based on the standard VV nomenclature of the Copenhagen
strain
(Goebel, S.J et al, Vi~~alogy 179, 247-266 (1990)). A46R was cloned from the
laboratory VV strain WR where it was previously called SalF9R (Smith, G.L et
al .T.
Gen. Virol, 72 1349-1376 (1991); Goebel, S.J et al, Virology 179, 247-266
(1990))
into the mammalian expression vector ARKS. Any other suitable mammalian
expression vector such as pcDNA3.1 (available from Invitrogen) or pEF-BOS
(Mizushima et al Nucleic Acids Res. 18, 5322 (1990)) for example may also be
used.
The A46R ORF was cloned by PCR amplification from WR DNA with primers
incorporating restriction sites for EcaRI upstream and HihdIII downstream of
the
ORF. The primers used were 5' - CGTGAATTCCGAGAAT~GCGTTTGA (sense)
and 5' -CGGAAGCTTTTATACATCCGTTTCCT (antisense). The restriction sites
and start and stop codons are underlined. The resulting EcoRI-HindIII fragment
was
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
18
ligated into the multiple cloning site of the mammalian expression vector
pRKS. For
irnmunoblot analysis, an epitope-tagged A46R expression vector was
constructed,
employing the same strategy, except that the 8-amino acid Flag coding
sequences
was inserted into the antisense primer 5' of the stop codon.
Antibodies. Polyclonal antibodies were raised against a purified, bacterially
expressed glutathione S-transferase (GST) fusion of A46R, encoded by a plasmid
synthesised by inserting full length A46R downstream of GST in the bacterial
expression vector GEX4T2. Other antibodies used were anti-AU1 monoclonal
antibody (BabCO), anti-HA polyclonal antibody (Y-11, Santa Cruz Biotechnology)
and anti-flag M2 monoclonal antibody (Sigma)
Lueiferase reporter gene assays. HEK 293 cells (2 x 104 cells per' well) or
RAW
264.7 cells (4 x 104 cells per well) were seeded into 96-well plates and
transfected
the next day with expression vector, and reporter plasmids. GeneJuiceTM
(Novagen)
was used for transient transfections, according to the manufacturer's
instructions.
For experiments involving NFxB, ISRE or IFN(3 promoter, 60 ng of xB-luciferase
reporter gene, ISRE-luciferase reporter gene (Stratagene) or INF(3 promoter
luciferase reporter (a gift from Prof. Taniguchi, University of Tokyo)
respectively
were used as previously described (Fitzgerald, K. A. et al. Nature 413, 78-83
(2001)). For MAP kinase reporter assays the Stratagene Pathdetect SystemTM was
used whereby c-jun (2 ng) or Elkl (5 ng) Gal4 fusion vectors were used in
combination with 80 ng pFR-luciferase reporter to measure JNK and ERK
activation
respectively (Fitzgerald, K. A. et al. Nature 413, 78-83 (2001)). For p65 or
IRF3
transactivation assay, a p65-Gal4 or IRF3-Gal4 fusion vector was used in
combination with pFR-luciferase reporter (Jefferies, C., et al. Mol. Cell.
Biol. 21,
4544-4552 (2001)). In all cases 40 ng of l2ehilla-luciferase internal control
(Promega) was used. The total amount of DNA per transfection was kept constant
at
200 ng by addition of pcDNA3.l (Stratagene). After 24 h cells were harvested
into
passive lysis buffer (Promega) and reporter gene activity was measured in a
luminometer. Data are expressed as mean fold induction ~ s.d. relative to
control
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
19
levels, for a representative experiment from a minimum of three separate
experiments, each performed in triplicate.
Immunoprecipitation and GST-Pulldown Assays. HEK 293T cells were seeded
into 100 mm dishes (1.5 x 106) 24 hrs prior to transfection. Transfections
were
carried out using GeneJuiceTM (Novagen) according to manufacturers
instructions.
Four g of each construct was transfected. Where only one construct was
expressed
the total amount of DNA (8 g) was kept constant by supplementation with vector
DNA. Cells were harvested 24 hrs post transfection in 7501 o f lysis buffer
(50 mM
HEPES, pH 7.5, 100 mM NaCI, 1 mM EDTA, 10% glycerol, 0.5% NP40 containing
1 mM PMSF and protease inhibitor cocktail (1/100) (Sigma), and 1 mM sodium
orthovanadate). For immunoprecipitation the indicated antibodies were
precoupled
to either protein A sepharose or protein G sepharose (anti-AUl) for 1 hr at
4°C,
washed, and then incubated with the cell lysates for 2 hrs at 4°C. The
immune
complexes were washed twice with lysis buffer and once with lysis buffer
without
NP40 and glycerol. Associated proteins were eluted from the beads by boiling
in 35
1 of 3x SPB (final concentrations in sample: 62.5 mM Tris, 2% (wlv) SDS, 10%
v/v
glycerol, 0.1% (w/v) bromophenol blue)). The immune complexes were analyzed by
SDS PAGE. 30 1 of the immune complex was immunoblotted for co-precipitating
protein and the remaining 5 1 was blotted directly for the protein directly
recognised
by the immunoprecipitating antibody. For immunoblotting, primary antibodies
were
detected using horseradish peroxidase conjugated secondary antibodies,
followed by
enhanced chemiluminescence (Amersham). For GST-pulldown experiments, a
similar transfection protocol was employed, except that cells were transfected
with 8
p,g of plasmid expressing a TIR adapter. GST-pulldown assays were performed
using recombinant GST-A46R fusion protein (prepared and purified using
standard
techniques) coupled to GSH-sepharose. Lysates prepared as described above were
incubated for two hours with GST-A46R, washed three times as above, and
subjected to SDS-PAGE and immunoblotting.
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
Plaque Assays. Aliquots from vaccinia virus stocks (WR strain) were frozen and
thawed 3 times, sonicated and serial dilutions were made in 2.5% FBS DMEM.
Three of these dilutions were inoculated in duplicate onto confluent
monolayers of
BSC-1 cells in 6-well plates (0.5 ml of each dilution per well). After
infection for 90
5 min at 37 C, the inoculum was aspirated, cells were overlaid with 2 ml of
1.5%
carboxymethylcellulose (CMC) in 2.5% FBS DMEM and incubated for 2 days at
37 C. The semi-solid overlay was aspirated, cells were washed briefly with PBS
and
stained with 0.1% (w/v) crystal violet in 15% ethanol. After rinsing with
water, the
plate was air-dried and the number of plaques was determined.
Example 1 - A46R inhibition of multiple signals induced by the IL-1R/TLR
superfamily
(i) A4bR inhibits multiple IL-1-dependent signals
A46R has been shown to block IL-1 induced NFkB activation, while not affecting
TNF (Bowie, A. et al. Proc. Natl Acad. Sci. USA 97, 10162-10167 (2000)).To
determine what other signals activated by IL-1 would also be blocked by A46R
HEK
293 cells (2 x 104 cells per well) were transfected with a xB-luciferase
reporter gene
and Renilla-luciferase internal control as described above. Six hours prior to
harvesting, cells were stimulated with 100 nglml IL-1. Cells were harvested 24
h
after transfection, and the reporter gene activity was measured. Data is
expressed as
mean fold induction ~ s.d. relative to control levels, for a representative
experiment
from a minimum of three separate experiments, each performed in triplicate.
Fig. 2a
shows that as the amount of expression vector encoding .A46R transfected into
cells
increased, there was a modest dose-dependent inhibition of IL-1 induced NFxB
activation, as previously shown (Bowie, A. et al. P~~oc. Natl Acad. Sci. USA
97,
10162-10167 (2000)). A46R was found to block other signals induced by IL-1. A
single dose of A46R cDNA transfected into cells was capable of blocking the
ability
of the transactivating subunit of NFxB, p65 to activate a reporter gene (Fig.
2b), JNK
activation (Fig.2c), and ERK activation (Fig.2d), respectively.
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
21
(ii) A46R inhibits multiple TLR4-dependent signals
As A46R has a putative TIR domain we determined whether other TIR-dependent
signals were sensitive to inhibition by examining the TLR4 pathway. Chimeric
versions of the TLRs, comprising the murine CD4 extracellular domain fused to
the
cytoplasmic domain of a given human TLR have proved useful in probing TLR
signalling pathways (Hayashi, F. et al. Nature 410, 1099-1103 (2001); Ozinsky,
A.
et al. Proc. Natl. Acad. Sca. USA 97, 13766-13771 (2000); Medzhitov, R.,
Preston-
Hurlburt, P. & Janeway, C. A. Jr. Nature 388, 394-397 (1997)). The
extracellular
domain of CD4 promotes homodimerisation of the molecules. Chimeras composed
of the extracellular domain of CD4 fused to the intracellular domain of TLR4
are
constitutively active, in that overexpression of CD4-TLR4 induces NF B
activation
and gene induction (Medzhitov, R., Preston-Hurlburt, P. & Janeway, C. A. Jr.
Nature
388, 394-397 (1997)). In the present invention HEK 293 cells (2 x 104 cells
per
well) were transfected with a Renilla-luciferase internal control and either
xB-
luciferase construct (Fig. 3a), or plasmids encoding Gal4 fused to p65 (for
p65, (b)),
Elkl (for ERK1/2, (c)) or CHOP (for p38, (d)) in the presence or absence of 50
ng
CD4-TLR4, together with increasing amounts of A46R cDNA. Cells were harvested
24 hours after transfection, and the reporter gene activity was measured. Data
are
expressed as mean fold induction ~ s.d. relative to control levels, for a
representative
experiment from a minimum of three separate experiments, each performed in
triplicate. Cells transfected with empty vector were used as control.
NFxB activation was measured as shown in Fig. 3a. Overexpression of CD4-TLR4
in HEK293 cells led to induction of the NFxB-dependent reporter gene (Fig. 3a)
which was inhibited dose-dependently by A46R. The effect of A46R on TLR4
appears to be more potent than its effects on IL-1 (compare Fig. 2a and Fig.
3a).
A46R was found to block other signals induced by CD4-TLR4. A single dose of
A46R cDNA transfected into cells was capable of blocking the ability of the
transactivating subunit of NFxB, p65, to activate a reporter gene (Fig. 3b),
ERK
activation (Fig. 3c), and p38 activation (Fig. 3d), respectively.
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
22
(iii) A46R inhibits TLR ligand-induced NFxB activation in murine
macrophages
Murine macrophage RAW264.7 cells were transfected with an NKxB luciferase
construct and a Renilla-luciferase internal control, together with empty
vector (EV)
or 100 ng cDNA encoding A46R. Cells were stimulated for 6 hours with the TLR
agonists 1 M R-848 (TLR7), 1000 nglml LPS (TLR4) or 250 nglml flagellin
(TLRS) before harvesting. 24h after transfection cells were harvested, and the
reporter gene activity was measured: Data are expressed as mean fold induction
~
s.d. relative to control levels, single experiment, performed in triplicate.
A46R inhibited NKxB activation induced by R-848, LPS, and flagellin. As in the
case of CD4-TLR4 in 293 cells (Fig. 3a), TLR4 (LPS) activation was again
particularly sensitive to A46R inhibition. R-848 induced NKxB activation was
also very strongly inhibited by A46R.
These results show that A46R is capable of blocking multiple signals emanating
from the IL-1R and TLRs. The fact that A46R has a putative TIR domain suggests
that it might block these TIR-dependent pathways by disrupting the TIR
receptor
interactions necessary for signalling. Thus A46R probably acts close to the
receptors, before signal bifurcation.
Example 2 - A46R associates with MyD88 and Mal, and can block signals
induced by MyD88 and Mal overexpression
(i) Association of A46R with MyD88.
The activation of NFxB by IL-1R/TLR family members is mediated by a common
set of signalling molecules. The ability of A46R to inhibit NFxB activation
induced
by IL-1 TLR4, TLRS and TLR7 suggested that its effects may be due to its
interaction with a molecule whose function is critical to signalling by all
these
receptors. MyD88 was a likely target, given its role in these pathways, and
the fact
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
23
that it has a TIR domain. Therefore the ability of A46R to interact with MyD88
was
examined.
Firstly, dose-dependent expression of A46R was demonstrated in HEK 293T cells,
HEK 293T cells were seeded at 1x105 cells/ml in 6 well plates 24 h prior to
transfection. Transfections were carried out using GeneJuiceTM (Novagen)
according
to manufacturers instructions. Increasing amounts of a plasmid vector encoding
A46R was transfected, as indicated in Fig. 5. The total amount of DNA (2 g) w
a s
kept constant by supplementation with vector DNA. Cells were harvested 24 h
post
transfection and resolved by SDS-PAGE. The blot was probed with an antibody
specific for A46R and Fig. 5 shows that a clear dose-dependent pattern of
expression
was observed, at the correct molecular mass for A46R (near 26kD).
In order to test the ability of A46R to associate with MyD88, HEK 293T cells
were
transfected with plasmids encoding A46R and AU1-MyD88, and co-
immunoprecipitation perFormed, as described above. The results are shown in
Fig.
6a, where lanes 1-3 correspond to lysates directly blotted for expression of
MyD88,
lanes 4-6 correspond to lysates immunoprecipitated with anti-A46R antibody and
blotted for the presence of MyD88, while lanes 7-9 correspond to
immunoprecipitation using anti-AUl antibody directed towards AU1-MyD88.
Therefore, the appearance of a band in lane 6 that is not detected in lanes 4
or 5 is
indicative of an interaction between A46R and MyD88. This is clearly evident
in
Fig. 6a, demonstrating that A46R immunoprecipitates in complex with MyD88.
Association between A46R and MyD88 was also shown by GST-pulldown, where
GST-A46R could pull MyD88 out of a cell lysate (Fig. 6b). Lane 1 corresponds
to
lysate directly blotted for expression of MyD88, lane 2 corresponds to lysate
incubated with GST and lane 3 corresponds to lysate incubated with GST-A46R.
(ii) Association of A46R with Mal
The ability of A46R to associate with Mal, another TIR domain containing
adaptor
molecule, that has a specific role in TLR2 and TLR4 signalling was also
tested.
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
24
A46R was expressed in HEK 293T cells along with Flag-tagged Mal. To isolate
complexes, immunoprecipitations were carried out using antibodies directed
against
Flag or A46R. The results are shown in Fig. 7a, where lanes 1-3 correspond to
lysates directly blotted for expression of A46R, lanes 4-6 correspond to
lysates
immunoprecipitated with Flag antibody and blotted for the presence of the
A46R,
while lanes 7-9 correspond to immunoprecipitation using A46R antibody.
Therefore, the appearance of a band in lane 6 that is not detected in lanes 4
or 5 is
indicative of an interaction between A46R and Mal. Similar to MyD88, Mal could
also be found associated with A46R, since Mal immunoprecipitated in complex
with
A46R (Fig. 7a, lane 6). This result was confirmed by GST-pulldown, where GST-
A46R pulled HA-Mal out of a cell lysate (Fig. 7b, lane 3).
(iii) Inhibition by A46R of signals induced by either MyD88 or Mal
overexpression
The effect of A46R on signals induced by ectopic expression of MyD88 or Mal
was
determined in order to test more directly the implications of A46R associating
with
both MyD88 and Mal. HEK 293 cells (2 x 104 cells per well) were transfected
with
different amounts of cDNAs encoding either MyD88 or Mal, in the presence or
absence of A46R, together with a Renilla-luciferase internal control and
either xB-
luciferase construct (Fig. 8a), or plasmids encoding Gal4 fused to p65 (for
p65, (Fig.
8b)), Elkl (for ERKll2, (Fig. 8c)), c-JUN (for JNK, (Fig. 8d)) or CHOP (for
p38,
(Fig. 8e)). Cells were harvested 2.4 hours after transfection, and the
reporter gene
activity was measured. Data are expressed as the mean fold induction ~ s.d.
relative
to control levels, for a representative experiment from a minimum of three
separate
experiments, each performed in triplicate. Cells transfected with empty vector
were
used as control. The amounts of cDNA used were 2 ng Mal or 5 ng MyD88 for
NF B reporter gene assay (a), 50 ng MyD88 or Mal for p65 transactivation assay
(b), 25 ng MyD88 or Mal for JNK assay (d), and 10 ng MyD88 or 25 ng Mal for
ERK (c) and p38 (e) assays. These amounts of cDNA were determined as being
optimal for activation of the different reporters in previous experiments.
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
Consistent with the targeting of MyD88 and Mal by A46R, both MyD88- and Mal
mediated NF B , p65 and ERK signals are inhibited very potently by A46R (Fig.
8a
to c). However, A46R inhibits MyD88 mediated JNK and p38 signals only
slightly,
when compared to the very strong inhibition of Mal mediated JNK and p38
signals
5 by A46R (Fig. 8d and e).
Thus the ability of A46R to antagonise multiple signals induced by IL-1 and
TLRs is
likely due to its ability to associate with MyD88 and Mal. The functional
consequences of these associations is that A46R can inhibit signals emanating
from
10 these adaptors.
Example 3 - A46R blocks MyD88-independent pathways by associating with
TRIF.
15 A46R was also capable of antagonising TLR signalling pathways known to be
independent of MyD88 and Mal. One important example of such a pathway is the
activation of ISRE promoter elements and subsequent induction of IFN(3 by
poly(I:C) via TLR3. In order to test the effect of A46R on this pathway, HEK
293
cells (2 x 10~ cells per well) were transfected with 0.5 ng TLR3 in the
presence or
20 absence of A46R, with an ISRE-luciferase construct (Fig. 9a), or an IFN(3
promoter
reporter plasmid (b) and Renilla-luciferase internal control. Cells were
harvested at
24 hours, 6 hours after stimulation with 25 ~,g/ml poly(I:C), and the reporter
gene
activity was measured. Data are expressed as mean fold induction ~ s.d.
relative to
control levels, for a representative experiment from a minimum of three
separate
25 experiments, each performed in triplicate. Fig. 9 shows that A46R potently
blocked
both ISRE (a) and IFN(3 promoter (b) activation, suggesting that A46R can also
target the MyD88-independent, IRF3-dependent pathway. This pathway has
recently
been shown to be controlled by the TIR adapter TRIF (Hoebe, K. et al Nature
doi:10.1038/nature01889 (2003); Yamamoto, M. et al Science
doi:10.1126/science.1087262 (2003)). The same immunoprecipitation and GST-
pulldown approach used for MyD88 and Mal was employed to test whether A46R
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
26
could associate with TRIF, in addition to MyD88 and Mal. Fig. 10 shows that
A46R
was indeed able to associate with TRIF in both assays (Fig, 10a, lane 6 and
Fig. lOb,
lane 3).
Thus A46R can target TRIF-dependent signalling by association with this TIR
adaptor. This was confirmed in other experiments where activation of either
NFxB
or induction of the IFN(3 promoter by ectopic expression of TRIF was blocked
by
A46R (Fig. lla and b respectively). Furthermore, TLR4-induced ISRE or IRF3,
which are also dependent on TRIF, were also potently inhibited (Fig. 12a and
b).
Finally, Fig. 12c shows that TLR ligand induced ISRE induction in murine
macrophages by LPS, and other TLR agonists, is also sensitive to A46R
inhibition.
Overall these results show that by targeting three different TIR adapters,
A46R can
inhibit multiple IL-1 and TLR induced signals.
Example 4 - Comparison of a VV deletion mutant lacking A46R gene with wild
type and revertant viruses.
A mutant virus lacking the A46R gene was constructed in order to determine
what
contribution A46R might make to VV virulence. A revertant virus, in which the
A46R gene was re-inserted into the mutant, was also constructed.
(i) Expression of A46R in cells infected with wild type virus, and
characterisation of VV deletion mutant lacking A46R
The expression profile of A46R in cells infected with wild-type virus was
determined. BSC-1 cells were mock infected or infected with VV WR at 10
pfu/cell
in the absence or presence of 40 mg/ml cytosine arabinoside (an inhibitor of
DNA
synthesis). At various times p.i., cells were harvested, and extracts were
prepared,
separated by SDS-PAGE and analysed by immunoblotting. Blots were detected with
anti-A46R antibody (diluted 1:1000) and anti-D8L antibody (diluted 1:1000).
D8L
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
27
is known to be expressed late during VV infection. Bound IgG was detected with
HRP-conjugates goat anti-rabbit IgG antibody (diluted 1:2500) and ECL reagents
(Amersham) and blots were exposed to X-GMAT film.
Fig 13a shows that in comparison to DBL, which was only detected at 24 h p.i.,
A46R could be seen after just 6 h. The level of protein continued to
accumulate up
to 24 h. This profile of expression of A46R in infected cells shows that the
protein is
expressed quite early in infection, and rapidly accumulates, compared to DBL,
a
known late expressed VV protein. This is consistent with a role for A46R in
suppressing IL-1R/TLR signalling in infected cells, since these receptors are
generally involved in the triggering of host immune responses in the early
phases of
infection.
The role of A46R in the VV life cycle was investigated by the construction of
a
deletion mutant lacking the A46R gene and by the comparison with wild type and
revertant controls. A VV mutant lacking 93.5 % of the A46R gene (v A46R) Was
constructed by transient dominant selection (Falkner, F.G. & Moss, B. (1991)
,l.
Tirol. 64, 3108-3111). A plaque purified wild type virus (vWT-A46R) and a
revertant virus (vA46R-RV) in which the A46R gene was reinserted at its
natural
locus were also isolated. Fig. 13b confirms that no A46R was expressed in
cells
infected with the v A46R virus, while those infected with vWT-A46R and vA46R-
RV did express the protein. BSC-1 cells were infected with vWT-A46R, vA46 R
and vA46R-RV at 10 pfu/cell. Cells were harvested 8 h p.i. and extracts were
prepared, separated by SDS-PAGE and analysed by immunoblotting as before.
The loss of the A46R gene did not affect the replication of the virus in cell
culture, as
shown in Fig. 13c and d. Fig. 13c is a single step growth analysis of
recombinant
viruses. BSC-1 cells were infected with the indicated viruses at 10 pfu/cell.
After 24
h, the virus present in the clarified supernatants and cells were determined
by plaque
assay in duplicate. Results show the mean of duplicate experiments. As can be
seen,
there is no difference between the viral titre for vWT-A46R, vA4 6 R and vA46R-
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
28
RV. Fig. 13d shows multi-step growth curves for the recombinant viruses. BSC-1
cells were infected with the indicated viruses at 0.01 pfulcell. At various
times post-
infection, virus was harvested by scraping cells into the culture
supernatants.
Samples were frozen and thawed 3 times and sonicated. Total virus levels were
determined by plaque assay on BSC-1 cells. Results show the mean of duplicate
experiments. vWT-A46R, v A 46R and vA46R-RV display the same growth
characteristics.
The deletion of A46R therefore does not affect the growth or replication of VV
in
cell culture.
(ii) Deletion of A46R gene from VV attenuates the virus
The virulence of the virus was examined in a mouse intranasal model. Groups of
five female, 6-week old Balb/c mice were anaesthetized and inoculated with 104
pfu
of VV in 20 ~.1 of phosphate-buffered saline (PBS). A control group was mock
infected with PBS. Each day the weights of the animals was measured as
described
previously (Alcami, A. & Smith, G.L. (1992) Fell 71, 153-167). Data are
presented
as the mean weight of each group of animals compared to the mean weight of the
same group on day 0. As can be seen in Fig. 14, the deletion mutant caused
reduced
weight loss in mice, compared to wild-type and revertant viruses.
Thus the A46R protein contributes to virus virulence and this is likely to be
due to
the inhibition of IL-1R/TLR signalling.
These results demonstrate that A46R from VV is able to inhibit IL-1R/TLR-
induced
intracellular signalling, by associating with a TIR adapter-containing
complexes.
The ability of A46R to disrupt TLR signalling has relevance to VV virulence,
since
deletion of A46R attenuates the virus.
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
29
In this specification some references have been included which Were published
after
the priority date of the application. These are included for the reader's
assistance
only.
The invention is not limited to the embodiments hereinbefore described which
may
be varied in detail.
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
SEQUENCE LISTING
<110> The Provost, Fellows & Scholars of the College of the Ho
ly and
Undivided Trinity of Queen Elizabeth, near Dublin
<120> Orthopoxvirus vectors, genes and products thereof
<130> TRII~104 . C
<160> 1
<170> PatentIn version 3.1
<210> 1
<211> 240
<212> PRT
<213> Vaccinia virus-A46R
<400> 1
Met Ala Phe Asp Ile Ser Va1 Asn Ala Ser Lys Thr Ile Asn Ala Leu
1 5 10 15
Va1 Tyr Phe Ser Thr Gln Gln Asn Lys Leu Val Ile Arg Asn Glu Val
20 25 30
Asn Asp Thr His Tyr Thr Val Glu Phe Asp Arg Asp Lys Val Va1 Asp
35 40 45
Thr Phe Ile Ser Tyr Asn Arg His Asn Asp Thr Ile Glu Ile Arg G1y
50 55 60
Val Leu Pro Glu Glu Thr Asn Ile Gly Cys Ala Val Asn Thr Pro Val
65 70 75 80
Ser Met Thr Tyr Leu Tyr Asn Lys Tyr Ser Phe Lys Leu Ile Leu Ala
Page 1
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
85 90 95
Glu Tyr Ile Arg His Arg Asn Thr Ile Ser Gly Asn Ile Tyr Ser Ala
100 105 110
Leu Met Thr Leu Asp Asp Leu A1a Ile Lys Gln Tyr Gly Asp Ile Asp
115 120 125
Leu Leu Phe Asn Glu Lys Leu Lys Val Asp Ser Asp Ser Gly Leu Phe
130 135 140
Asp Phe Val Asn Phe Val Lys Asp Met Ile Cys Cys Asp Ser Arg Ile
145 150 155 160
Val Val Ala Leu Ser Ser Leu Val Ser Lys His Trp Glu Leu Thr Asn
165 170 175
Lys Lys Tyr Arg Cys Met Ala Leu Ala Glu His Ile Ser Asp Ser Ile
180 185 190
Pro Ile Ser Glu Leu Ser Arg Leu Arg Tyr Asn Leu Cys Lys Tyr Leu
195 200 205
Arg Gly His Thr G1u Ser Ile Glu Asp Lys Phe Asp Tyr Phe Glu Asp
210 215 220
Page 2
CA 02499871 2005-03-22
WO 2004/031225 PCT/IE2003/000131
Asp Asp Ser Ser Thr Cys Sex Ala Val Thr Asp Arg Glu Thr Asp Val
225 230 235 240
Page 3