Note: Descriptions are shown in the official language in which they were submitted.
CA 02500763 2005-03-30
WO 2004/033416 PCT/EP2003/010501
1
Description
Carboxyalkoxy-substituted acyl-carboxyphenylurea derivatives, method for their
production and their use as medicaments
s
The invention relates to carboxyalkoxy-substituted acyl-carboxyphenylurea
derivatives and to their physiologically tolerated salts and physiologically
functional
derivatives.
io Acylphenylurea derivatives have already been described as antitumor agents
and
as antidiabetics in the prior art (EP 0 193 249 and WO 01/94300).
The invention was based on the object of providing compounds which display a
therapeutically utilizable blood glucose-lowering effect. In particular, the
object was
is to provide compounds with an improved effect in comparison with the
compounds
from WO 01/94300.
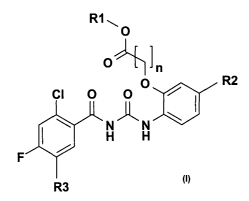
The invention therefore relates to compounds of the formula I,
R1 ~
O
Jn
O
CI O O
N~N
H H
F ~
2o R3
in which
CA 02500763 2005-03-30
2
R1 is H, (C~-C6)-alkyl, (Co-C6)-alkyl-phenyl, where the phenyl ring may be
substituted up to twice by F, CI, CN, OH, (C~-C6)-alkyl, O-(C~-Cs)-
alkyl, CF3, OCF3, COOH, COO(C~-C6)-alkyl or CONH2;
R2 is H, (C~-C6)-alkyl, O-(C~-C6)-alkyl, CO-(C~-C6)-alkyl, COO-(C~-C6)-
alkyl, (Co-C6)-alkylene-COOH;
R3 is H, F, CI, Br, OH, CF3, N02, CN, OCF3, O-(C~-C6)-alkyl, (C~-C6)-
alkyl;
to
n is 1, 2, 3, 4, 5, 6, 7, 8;
and the physiologically tolerated salts thereof.
Preference is given to compounds of the formula I in which
R1 is H, (C~-C6)-alkyl;
2o R2 is H, (C~-C6)-alkyl, O-(C~-C6)-alkyl, CO-(C~-C6)-alkyl, COO-(C~-C6)-
alkyl, (Co-C6)-alkylene-COOH;
R3 is H, F, CI, Br, OH, CF3, N02, CN, OCF3, O-(C~-Cs)-alkyl, (C~-Cs)-
alkyl;
n is 1, 2, 3, 4, 5, 6, 7, 8;
and the physiologically tolerated salts thereof.
Particularly preference is given to compounds of the formula I in which
CA 02500763 2005-03-30
3
R1 is H, (C~-C6)-alkyl;
R2 is H, COO-(C~-C6)-alkyl, -COOH;
s R3 is H, F;
n is 1, 2, 3, 4;
and the physiologically tolerated salts thereof.
io
The invention relates to compounds of the formula I in the form of their
racemates,
racemic mixtures and pure enantiomers, and to their diastereomers and mixtures
thereof.
is The alkyl radicals in the substituents R1, R2 and R3 may be both straight-
chain
and branched.
Pharmaceutically acceptable salts are, because their solubility in water is
greater
than that of the initial or basic compounds, particularly suitable for medical
2o applications. These salts must have a pharmaceutically acceptable anion or
cation. Suitable pharmaceutically acceptable acid addition salts of the
compounds
of the invention are salts of inorganic acids such as hydrochloric acid,
hydrobromic, phosphoric, metaphosphoric, nitric and sulfuric acid, and of
organic
acids such as, for example, acetic acid, benzenesulfonic, benzoic, citric,
2s ethanesulfonic, fumaric, gluconic, glycolic, isethionic, lactic,
lactobionic, malefic,
malic, methanesulfonic, succinic, p-toluenesulfonic and tartaric acid.
Suitable
pharmaceutically acceptable basic salts are ammonium salts, alkali metal salts
(such as sodium and potassium salts), alkaline earth metal salts (such as
magnesium and calcium salts), and trometamol (2-amino-2-hydroxymethyl-1,3-
3o propanediol), diethanolamine, lysine or ethylenediamine.
Salts with a pharmaceutically unacceptable anion such as, for example,
trifluoroacetate likewise belong within the framework of the invention as
useful
CA 02500763 2005-03-30
4
intermediates for the preparation or purification of pharmaceutically
acceptable
salts andlor for use in nontherapeutic, for example in vitro, applications.
The term "physiologically functional derivative" used herein refers to any
s physiologically tolerated derivative of a compound of the formula I of the
invention,
for example an ester, which on administration to a mammal such as, for
example,
a human is able to form (directly or indirectly) a compound of the formula I
or an
active metabolite thereof.
io Physiologically functional derivatives include prodrugs of the compounds of
the
invention, as described, for example, in H. Okada et al., Chem. Pharm. Bull.
1994,
42, 57-61. Such prodrugs can be metabolized in vivo to a compound of the
invention. These prodrugs may themselves be active or not.
~s The compounds of the invention may also exist in various polymorphous
forms, for
example as amorphous and crystalline polymorphous forms. All polymorphous
forms of the compounds of the invention belong within the framework of the
invention and are a further aspect of the invention.
2o All references to "compound(s) of formula I" hereinafter refer to
compounds) of
the formula I as described above, and their salts, solvates and
physiologically
functional derivatives as described herein.
The compounds) of formula (I) may also be administered in combination with
2s other active ingredients.
The amount of a compound of formula I necessary to achieve the desired
biological effect depends on a number of factors, for example the specific
compound chosen, the intended use, the mode of administration and the clinical
3o condition of the patient. The daily dose is generally in the range from 0.3
mg to
100 mg (typically from 3 mg to 50 mg) per day and per kilogram of bodyweight,
for
example 3-10 mg/kg/day. An intravenous dose may be, for example, in the range
from 0.3 mg to 1.0 mg/kg, which can suitably be administered as infusion of 10
ng
CA 02500763 2005-03-30
to 100 ng per kilogram and per minute. Suitable infusion solutions for these
purposes may contain, for example, from 0.1 ng to 10 mg, typically from 1 ng
to
mg, per milliliter. Single doses may contain, for example, from 1 mg to 10 g
of
the active ingredient. Thus, ampoules for injections may contain, for example,
from
s 1 mg to 100 mg, and single-dose formulations which can be administered
orally,
such as, for example, capsules or tablets, may contain, for example, from 1.0
to
1000 mg, typically from 10 to 600 mg. For the therapy of the abovementioned
conditions, the compounds of formula I may be used as the compound itself, but
they are preferably in the form of a pharmaceutical composition with an
acceptable
io carrier. The carrier must, of course, be acceptable in the sense that it is
compatible with the other ingredients of the composition and is not harmful
for the
patient's health. The carrier may be a solid or a liquid or both and is
preferably
formulated with the compound as a single dose, for example as a tablet, which
may contain from 0.05% to 95% by weight of the active ingredient. Other
i$ pharmaceutically active substances may likewise be present, including other
compounds of formula I. The pharmaceutical compositions of the invention can
be
produced by one of the known pharmaceutical methods, which essentially consist
of mixing the ingredients with pharmacologically acceptable carriers andlor
excipients.
Pharmaceutical compositions of the invention are those suitable for oral,
rectal,
topical, peroral (for example sublingual) and parenteral (for example
subcutaneous, intramuscular, intradermal or intravenous) administration,
although
the most suitable mode of administration depends in each individual case on
the
2s nature and severity of the condition to be treated and on the nature of the
compound of formula I used in each case. Coated formulations and coated slow-
release formulations also belong within the framework of the invention.
Preference
is given to acid- and gastric juice-resistant formulations. Suitable coatings
resistant
to gastric juice comprise cellulose acetate phthalate, polyvinyl acetate
phthalate,
3o hydroxypropylmethylcellulose phthalate and anionic polymers of methacrylic
acid
and methyl methacrylate.
CA 02500763 2005-03-30
6
Suitable pharmaceutical compounds for oral administration may be in the form
of
separate units such as, for example, capsules, wafers, suckable tablets or
tablets,
each of which contain a defined amount of the compound of formula I; as
powders
or granules, as solution or suspension in an aqueous or nonaqueous liquid; or
as
s an oil-in-water or water-in-oil emulsion. These compositions may, as already
mentioned, be prepared by any suitable pharmaceutical method which includes a
step in which the active ingredient and the carrier (which may consist of one
or
more additional ingredients) are brought into contact. The compositions are
generally produced by uniform and homogeneous mixing of the active ingredient
io with a liquid and/or finely divided solid carrier, after which the product
is shaped if
necessary. Thus, for example, a tablet can be produced by compressing or
molding a powder or granules of the compound, where appropriate with one or
more additional ingredients. Compressed tablets can be produced by tableting
the
compound in free-flowing form such as, for example, a powder or granules,
where
is appropriate mixed with a binder, glidant, inert diluent and/or one or more
surface-
active/dispersing agents) in a suitable machine. Molded tablets can be
produced
by molding the compound, which is in powder form and is moistened with an
inert
liquid diluent, in a suitable machine.
2o Pharmaceutical compositions which are suitable for peroral (sublingual)
administration comprise suckable tablets which contain a compound of formula I
with a flavoring, normally sucrose and gum arabic or tragacanth, and pastilles
which comprise the compound in an inert base such as gelatin and glycerol or
sucrose and gum arabic.
2s
Pharmaceutical compositions suitable for parenteral administration comprise
preferably sterile aqueous preparations of a compound of formula I, which are
preferably isotonic with the blood of the intended recipient. These
preparations are
preferably administered intravenously, although administration may also take
3o place by subcutaneous, intramuscular or intradermal injection. These
preparations
can preferably be produced by mixing the compound with water and making the
resulting solution sterile and isotonic with blood. Injectable compositions of
the
invention generally contain from 0.1 to 5% by weight of the active compound.
CA 02500763 2005-03-30
Pharmaceutical compositions suitable for rectal administration are preferably
in the
form of single-dose suppositories. These can be produced by mixing a compound
of the formula I with one or more conventional solid carriers, for example
cocoa
s butter, and shaping the resulting mixture.
Pharmaceutical compositions suitable for topical use on the skin are
preferably in
the form of ointment, cream, lotion, paste, spray, aerosol or oil. Carriers
which can
be used are petrolatum, lanolin, polyethylene glycols, alcohols and
combinations
io of two or more of these substances. The active ingredient is generally
present in a
concentration of from 0.1 to 15% by weight of the composition, for example
from
0.5 to 2%.
Transdermal administration is also possible. Pharmaceutical compositions
suitable
is for transdermal uses can be in the form of single plasters which are
suitable for
long-term close contact with the patient's epidermis. Such plasters suitably
contain
the active ingredient in an aqueous solution which is buffered where
appropriate,
dissolved and/or dispersed in an adhesive or dispersed in a polymer. A
suitable
active ingredient concentration is about 1 % to 35%, preferably about 3% to
15%. A
2o particular possibility is for the active ingredient to be released by
electrotransport
or iontophoresis as described, for example, in Pharmaceutical Research, 2(6):
318
(1986).
Further active ingredients suitable for combination products are:
2s all antidiabetics mentioned in the Rote biste 2001, chapter 12. They may be
combined with the compounds of the formula I of the invention in particular
for a
synergistic improvement of the effect. Administration of the active ingredient
combination may take place either by separate administration of the active
ingredients to the patient or in the form of combination products in which a
plurality
30 of active ingredients are present in one pharmaceutical preparation. Most
of the
active ingredients listed below are disclosed in the USP Dictionary of USAN
and
International Drug Names, US Pharmacopeia, Rockviile 2001.
CA 02500763 2005-03-30
g
Antidiabetics include insulin and insulin derivatives such as, for example,
Lantus~
(see www.lantus.com) or HMR 1964, fast-acting insulins (see US 6,221,633),
GLP-1 derivatives such as, for example, those disclosed in WO 98108871 of Novo
Nordisk AIS, and orally effective hypoglycemic active ingredients.
s The orally effective hypoglycemic active ingredients include, preferably,
sulfonylureas, biguanides, meglitinides, oxadiazolidinediones,
thiazolidinediones,
glucosidase inhibitors, glucagon antagonists, GLP-1 agonists, potassium
channel
openers such as, for example, those disclosed in WO 97126265 and WO 99/03861
of Novo Nordisk AIS, insulin sensitizers, inhibitors of liver enzymes involved
in the
to stimulation of gluconeogenesis and/or glycogenolysis, modulators of glucose
uptake, compounds which alter lipid metabolism, such as antihyperlipidemic
active
ingredients and antilipidemic active ingredients, compounds which reduce food
intake, PPAR and PXR agonists and active ingredients which act on the ATP-
dependent potassium channel of the beta cells.
In one embodiment of the invention, the compounds of the formula I are
administered in combination with an HMG-CoA reductase inhibitor such as
simvastatin, fluvastatin, pravastatin, lovastatin, atorvastatin, cerivastatin,
rosuvastatin.
In one embodiment of the invention, the compounds of the formula I are
administered in combination with a cholesterol absorption inhibitor such as,
for
example, ezetimibe, tiqueside, pamaqueside.
2s In one embodiment of the invention, the compounds of the formula ! are
administered in combination with a PPAR gamma agonist, such as, for example,
rosiglitazone, pioglitazone, JTT-501, GI 262570.
In one embodiment of the invention, the compounds of the formula I are
3o administered in combination with a PPAR alpha agonist, such as, for
example,
GW 9578, GW 7647.
CA 02500763 2005-03-30
9
In one embodiment of the invention, the compounds of the formula I are
administered in combination with a mixed PPAR alpha/gamma agonist, such as,
for example, GW 1536, AVE 8042, AVE 8134, AVE 0847, or as described in
PCTIUS00/11833, PCT/US00/11490, DE10142734.4.
In one embodiment of the invention, the compounds of the formula I are
administered in combination with a fibrate such as, for example, fenofibrate,
clofibrate, bezafibrate.
io In one embodiment of the invention, the compounds of the formula I are
administered in combination with an MTP inhibitor such as, for example,
implitapide, BMS-201038, R-103757.
In one embodiment of the invention, the compounds of the formula I are
is administered in combination with a bile acid absorption inhibitor (see, for
example,
US 6,245,744 or US 6,221,897), such as, for example, HMR 1741.
In one embodiment of the invention, the compounds of the formula I are
administered in combination with a CETP inhibitor, such as, for example, JTT-
705.
In one embodiment of the invention, the compounds of the formula I are
administered in combination with a polymeric bile acid adsorbent such as, for
example, cholestyramine, colesevelam.
2s In one embodiment of the invention, the compounds of the formula I are
administered in combination with an LDL receptor inducer (see US 6,342,512),
such as, for example, HMR1171, HMR1586.
In one embodiment of the invention, the compounds of the formula I are
3o administered in combination with an ACAT inhibitor, such as, for example,
avasimibe.
CA 02500763 2005-03-30
to
In one embodiment of the invention, the compounds of the formula I are
administered in combination with an antioxidant, such as, for example, OPC-
14117.
s In one embodiment of the invention, the compounds of the formula I are
administered in combination with a lipoprotein lipase inhibitor, such as, for
example, NO-1886.
In one embodiment of the invention, the compounds of the formula I are
io administered in combination with an ATP-citrate lyase inhibitor, such as,
for
example, SB-204990.
In one embodiment of the invention, the compounds of the formula I are
administered in combination with a squalene synthetase inhibitor, such as, for
is example, BMS-188494.
In one embodiment of the invention, the compounds of the formula I are
administered in combination with a lipoprotein(a) antagonist, such as, for
example,
CI-1027 or nicotinic acid.
In one embodiment of the invention, the compounds of the formula I are
administered in combination with a lipase inhibitor, such as, for example,
orlistat.
In one embodiment of the invention, the compounds of the formula I are
2s administered in combination with insulin.
In one embodiment, the compounds of the formula I are administered in
combination with a sulfonylurea such as, for example, tolbutamide,
glibenclamide,
glipizide or glimepiride.
In one embodiment, the compounds of the formula I are administered in
3o combination with a biguanide, such as, for example, metformin.
In one further embodiment, the compounds of the formula I are administered in
combination with a meglitinide, such as, for example, repaglinide.
CA 02500763 2005-03-30
11
In one embodiment, the compounds of the formula I are administered in
combination with a thiazolidinedione, such as, for example, troglitazone,
ciglitazone, pioglitazone, rosiglitazone or the compounds disclosed in WO
97141097 of Dr. Reddy's Research Foundation, in particular 5-[[4-[(3,4-dihydro-
3-
s methyl-4-oxo-2-quinazolinylmethoxy]phenyl]methyl]-2,4-thiazolidinedione.
In one embodiment, the compounds of the formula I are administered in
combination with an a-glucosidase inhibitor, such as, for example, miglitol or
acarbose.
In one embodiment, the compounds of the formula I are administered in
io combination with an active ingredient which acts on the ATP-dependent
potassium
channel of the beta cells, such as, for example, tolbutamide, glibenclamide,
glipizide, glimepiride or repaglinide.
In one embodiment, the compounds of the formula I are administered in
combination with more than one of the aforementioned compounds, e.g. in
is combination with a sulfonylurea and metformin, with a sulfonylurea and
acarbose,
repaglinide and metformin, insulin and a sulfonylurea, insulin and metformin,
insulin and troglitazone, insulin and lovastatin, etc.
In a further embodiment, the compounds of the formula I are administered in
2o combination with CART modulators (see "Cocaine-amphetamine-regulated
transcript influences energy metabolism, anxiety and gastric emptying in mice"
Asakawa, A, et al., in: Hormone and Metabolic Research (2001 ), 33(9), 554-
558),
NPY antagonists, e.g. naphthalene-1-sulfonic acid {4-[(4-aminoquinazolin-2-
ylamino)methyl] cyclohexylmethyl}amide hydrochloride (CGP 71683A)), MC4
2s agonists (e.g. 1-amino-1,2,3,4-tetrahydronaphthalene-2-carboxylic acid [2-
(3a-
benzyl-2-methyl-3-oxo-2,3,3a,4,6,7-hexahydropyrazolo[4,3-c]pyridin-5-yl)-1-(4-
chlorophenyl)-2-oxoethyl]-amide; (V110 01191752)) , orexin antagonists (e.g. 1-
(2-
methylbenzoxazol-6-yl)-3-[1,5]naphthyridin-4-ylurea hydrochloride (SB-334867-
A)), H3 agonists (3-cyclohexyl-1-(4,4-dimethyl-1,4,6,7-tetrahydroimidazo[4,5-
3o c]pyridin-5-yl)propan-1-one oxalic acid salt (WO 00/63208)); TNF agonists,
CRF
antagonists (e.g. [2-methyl-9-(2,4,6-trimethylphenyl)-9H-1,3,9-triazafluoren-4-
yl]dipropylamine (WO 00/66585)), CRF BP antagonists (e.g. urocortin),
urocortin
agonists, ~3 agonists (e.g. 1-(4-chloro-3-methanesulfonylmethylphenyl)-2-[2-
(2,3-
CA 02500763 2005-03-30
12
dimethyl-1 H-indol-6-yloxy)ethylamino]-ethanol hydrochloride (V110 01183451
)),
MSH (melanocyte-stimulating hormone) agonists, CCK-A agonists (e.g. f2-[4-(4-
chloro-2,5-dimethoxyphenyl)-5-(2-cyclohexylethyl)thiazol-2-ylcarbamoyl]-5,7-
dimethylindol-1-yl)acetic acid trifluoroacetic acid salt (WO 99/15525)),
serotonin
reuptake inhibitors (e.g. dexfenfluramine), mixed serotoninergic and
noradrenergic
compounds (e.g. WO 00/71549), 5HT agonists e.g. 1-(3-ethylbenzofuran-7-
yl)piperazine oxalic acid salt (WO 01/09111), bombesin agonists, galanin
antagonists, growth hormone (e.g. human growth hormone), growth hormone-
releasing compounds (6-benzyloxy-1-(2-diisopropylaminoethylcarbamoyl)-3,4-
to dihydro-1 H-isoquinoline-2-carboxylic acid tertiary butyl ester (WO
01/85695)), TRH
agonists (see, for example, EP 0 462 884), uncoupling protein 2 or 3
modulators,
leptin agonists (see, for example, Lee, Daniel W.; Leinung, Matthew C.;
Rozhavskaya-Arena, Marina; Grasso, Patricia. Leptin agonists as a potential
approach to the treatment of obesity. Drugs of the Future (2001 ), 26(9), 873-
Is 881 ), DA agonists (bromocriptine, Doprexin), lipase/amylase inhibitors
(e.g. WO
00/40569), PPAR modulators (e.g. WO 00/78312), RXR modulators or TR-[i
agonists.
In one embodiment of the invention, the other active ingredient is leptin;
see, for
2o example, "Perspectives in the therapeutic use of leptin", Salvador, Javier;
Gomez-
Ambrosi, Javier; Fruhbeck, Gema, Expert Opinion on Pharmacotherapy (2001 ),
2(10), 1615-1622.
In one embodiment, the other active ingredient is dexamphetamine or
2s amphetamine.
In one embodiment, the other active ingredient is fenfluramine or
dexfenfluramine.
In another embodiment, the other active ingredient is sibutramine.
In one embodiment, the other active ingredient is mazindol or phentermine.
3o In one embodiment, the compounds of the formula I are administered in
combination with bulking agents, preferably insoluble bulking agents (see, for
example, carob/Caromax~ (Zunft H J; et al., Carob pulp preparation for
treatment
of hypercholesterolemia, ADVANCES IN THERAPY (2001 Sep-Oct), 18(5), 230-
CA 02500763 2005-03-30
13
6.) Caromax is a carob-containing product from Nutrinova, Nutrition
Specialties 8~
Food Ingredients GmbH, Industriepark Hochst, 65926 Frankfurt/Main)).
Combination with Caromax~ is possible in one preparation or by separate
administration of compounds of the formula I and Caromax~. Caromax~ can in
this
s connection also be administered in the form of food products such as, for
example,
in bakery products or muesli bars.
It will be appreciated that every suitable combination of the compounds of the
invention with one or more of the aforementioned compounds and optionally one
to or more other pharmacologically active substances is regarded as falling
within the
protection conferred by the present invention.
CA 02500763 2005-03-30
14
CH_ \
~N / OiCHs
NJ
OPC-14117
JTf-705 CI \
O
',./ w/ ~/ O O
CI
~~ O OH
SB-204990 HO
/ N (. \ O CH3
H \\ ~O~"/
/ P~O~CH3
NO-1886 p OH
H3C OH O CH3
v v -p v v
H3C CH3
.O CI-1027
O I_
BMS-1 f
O
CH3
O
-- N O
O
CH3
O \
\ I / ~ ~ 612625
i N O O H
JTT-501
CA 02500763 2005-03-30
1$
The examples detailed below serve to illustrate the invention without,
however,
restricting it. The measured solidification or decomposition points have not
been
corrected and generally depend on the heating rate
s Table 1: Examples
R,i ~ O
O Jn
CI O O O
'H H
F
X
Example X R1 R2 n m.p. MS
No. [C]
1 H H H 1 180-181 ok
2 H H COOH 1 279-281 ok
3 F H H 1 179-180 ok
4 F Me COOMe 1 268 ok
F H COOH 1 294-297 ok
6 F H H 3 168 ok
7 F Me H 3 228 ok
8 F Me COOMe 3 209 ok
9 F H COOH 3 194 ok
F Me COOH 3 227 ok
11 H H H 4 197 ok
12 H H COOH 4 253-254 ok
13 F H H 4 164 ok
14 F H COOH 4 293-296 ok
F Me COOH 4 220 ok
io
* The statement "MS is ok" means that a mass spectrum was recorded and the
molecular peak (molecular mass + H+) was detected therein
The compounds of the formula I are distinguished by beneficial effects on
lipid and
is carbohydrate metabolism, they lower in particular the blood glucose level
and they
CA 02500763 2005-03-30
16
are suitable for the treatment of type 2 diabetes, of insulin resistance, of
dyslipidemias and of the metabolic syndromelsyndrome X. The compounds are
additionally suitable for the prophylaxis and treatment of arteriosclerotic
manifestations. The compounds can be employed alone or in combination with
s other blood glucose-lowering active ingredients. The compounds of the
formula I
are also outstandingly suitable because of their pharmacological properties
(see
J. L. Treadway, P. Mendys, D. J. Hoover, Exp. Opin. Invest. Drugs 2001, 10(3),
439-454) as cardioprotective medicaments for the prophylaxis of infarction and
the
treatment of infarction, and for the treatment of angina pectoris, where they
also
to preventively inhibit or greatly reduce the pathophysiological processes
associated
with the development of ischemia-induced damage, in particular in the
triggering of
ischemia-induced cardiac arrhythmias. The compounds can be employed alone or
in combination with other cardioprotective or antiarrhythmic active
ingredients.
The compounds of the formula I are also because of their pharmacological
is properties of suitable for the treatment oncoses. It was possible to show
in various
investigations of this that there is a direct connection between the glycogen
level
and various parameters of tumor growth and tumor development (see M. Rousset,
E. Dussaulx, G. Chevalier, A. Zweibaum, J. Natl. Cancer Inst. 1980, 65(5),
885-889; K. Yano, S. Ohoshima, Y. Shimizu, T. Moriguchi, H. Katayama, Cancer
2o Letters 1996, 110(1,2), 29-34; L. Skwarski, Z. Namiot, J. Stasiewicz, A.
Kemona,
M. Kralisz, J. Gorski, Cancer Letters 1998, 127(1,2), 123-128; S. Takahashi,
A. Satomi, K. Yano, H. Kawase, T. Tanimizu, Y. Tuji, S. Murakami, R. Hirayama,
J. Gastroenterology 1999, 34(4), 474-480). It is thus also possible to reduce
tumor
growth through manipulation of the amount of glucose released. The compounds
2s of the formula I can for this purpose be employed where appropriate also in
conjunction with other antitumor medicaments.
CA 02500763 2005-03-30
17
The efficacy of the compounds was tested as follows:
Analysis of test substances for their inhibitory potency on glycogenolysis in
primary cultures of rat hepatocytes
s
Hepatocytes were isolated from the livers of fed rats (Sprague-Dawley or
Wistar,
bodyweight 220-240 g) by means of a standard 2-stage perfusion first with
calcium-free buffer solution followed by perfusion with collagenase-containing
solution to break up the tissue assembly (Seglen et al, 1979). The cells were
io incubated in an incubator with 90% humidity at 37°C and an
atmosphere with 5%
COZ in air. The cultivation normally took place with Williams' medium E
supplemented with 20 mM glucose, 0.1 mM fructose, 1 NM dexamethasone and
100 nM insulin. Glycogenolysis was induced by changing the culture medium to
prewarmed carbogen-gassed Krebs-Henseleit bicarbonate Hepes (20 mM) buffer,
is pH 7.4, supplemented with 100 nM glucagon (time 0 min). The test substances
were normally added at time 0 min as 100 x stock solution in DMSO. The final
DMSO concentration was not higher than 1 % (v/v). The amount of glucose in the
culture supernatant was determined after addition of glucagon in the presence
and
absence of test substance by removing small aliquots of the cell culture
2o supernatant at times 0, 30, 60 and 90 min. The glucose concentration (mM)
was
determined by enzymatic methods in a biochemical analysis laboratory of
Aventis
Pharma Deutschland. The glucose production rate was found by linear regression
of the glucose concentration at times 30, 60 and 90 min using the following
formula:
Glucose production rate (mM/h) = mM glucose (90 min) - mM glucose (30 min).
The percent inhibition was calculated using the following formula:
Percentage inhibition = 100 x 1 _ glucose production rate in the presence of
test substance
glucose production rate in the absence of test substance
CA 02500763 2005-03-30
I8
ICSO values (IC5o: the concentration (NM) of test substance which brings about
a
reduction of 50% in the glucose production rate) was estimated by standard
curve
fitting methods using the percent inhibitions obtained at relevant
concentrations of
test substance.
Table 2: Biological activity
io
Example IC50 on hep.
No.
1 1.161
2 1.46
3 0.776
4 1.191
0.407
6 0.134
7 0.226
8 0.069
9 < 0.1
0.106
11 0.156
12 0.34
13 0.113
14 0.312
r 0.032
It is evident from the table that the compounds of the formula I inhibit
glycogenolysis in rat hepatocytes and thus bring about a reduction in the
blood
glucose level.
is
Two compounds from WO 01/94300 were tested as comparative examples.
Table 3: Biological activity of the comparative examples
CA 02500763 2005-03-30
19
. . Structure MW IC~ on hep.
Example no.105 from WO 01!94300 491_73 17.524
a
~o
I ~"
/ a
FI/
Example no.116 from WO 01194300 . 45728 13.522
F~O
ai
y
hRJ"O
p I
~a
It is evident from the table that the compounds of the formula I have a
considerably better effect than the comparative examples from WO 01/94300. The
s effect of the compounds of the invention is 11 to 548 times greater.
The preparation of some examples is described in detail below, and the other
compounds of the formula I were obtained analogously:
Experimental part:
to
Example 1:
2-[3-(2-Chloro-4-fluorobenzoyl)ureido]phenoxyacetic acid
is a) tert-Butyl2-nitrophenoxyacetate
2.8 g (14.4 mmol) of tert-butyl bromoacetate and 2.6 g (7.9 mmol) of cesium
carbonate are added to a solution of 1.0 g (7.2 mmol) of 2-nitrophenol in 20
ml of
acetone. The suspension is heated to reflux for 48 hours. Then 50 ml of water
are
added, and the mixture is extracted twice with 50 ml of ethyl acetate each
time.
2o The combined organic phases are washed with water, dried over Na2S04 and
concentrated in a rotary evaporator. The product is employed without
purification
in the next step. Crude yield: 2.4 g
CA 02500763 2005-03-30
b) tert-Butyl 2-aminophenoxyacetate
0.5 g of crude material from a) are dissolved in methanol and, after addition
of
s 0.3 g of Raney Ni, hydrogenated with hydrogen at room temperature. The
reaction
is monitored by LC-MS.
After reduction is complete, the mixture is filtered with suction through
Celite and
washed with methanol. The filtrate is concentrated in a rotary evaporator, and
the
product is employed without purification in step d). Crude yield: 0.34 g
io
c) 2-Chloro-4-fluorobenzoyl isocyanate
2-Chloro-4-fluorobenzamide was dissolved in dichloromethane and, after
addition
1.5 eq. of oxalyl chloride, heated to reflux for 16 hours, The reaction
mixture was
~s concentrated under high vacuum and employed without further purification in
stage d).
d) tert-Butyl 2-[3-(2-chloro-4-tluorobenzoyl)ureido]phenoxyacetate
2o A solution of 230 mg (1.2 mmol) of 2-chloro-4-fluorobenzoyl isocyanate in 5
ml of
acetonitrile is added at room temperature to a solution of 129 mg (0.6 mmol)
of
tert-butyl 2-aminophenoxyacetate in 5 ml of dry acetonitrile under a
protective gas
atmosphere. The mixture is heated to reflux for 4 hours and cooled to room
temperature. The solvent is distilled out in a rotary evaporator, and the
resulting
2s residue is employed without further purification in the next step. Crude
yield:
0.33 g.
e) tert-Butyl 2-[3-(2-chloro-4-fluorobenzoyl)ureido]phenoxyacetate
0.33 g of crude material from stage d) are taken up in 10 ml of methylene
chloride
and, after addition of 10 ml of trifluoroacetic acid, stirred at room
temperature for
two hours. The solution is then concentrated after addition of 3 ml of toluene
twice
in a rotary evaporator, and the residue obtained in this way is purified in a
CA 02500763 2005-03-30
21
preparative HPLC system (column: Waters Xterra TMMS C~8, 5 Nm, 30 x 100 mm,
mobile phases: A: H20 + 0.2% trifluoroacetic acid, B: acetonitrile, gradient:
2.5 minutes 90 % A/10% B to 17.5 minutes 10% A/90% B). 88 mg (0.23 mmol) of
the desired product are obtained.
s Melting point: 180-181 °C
Example 3 was prepared in accordance with this method.
io Example 2:
3-Carboxymethoxy-4-[3-(2-chloro-4-fluorobenzoyl)ureido]benzoic acid
a) Methyl3-methoxycarbonylmethoxy-4-nitrobenzoate
0.77 g (5.1 mmol) of methyl bromoacetate and 0.91 g (2.8 mriiol) of cesium
is carbonate are added to a solution of 0.50 g (2.5 mmol) of methyl 3-hydroxy-
4-nitrobenzoate in 10 ml of acetone. The suspension is heated to reflux for 48
hours. Then 50 ml of water are added, and the mixture is extracted twice with
50 ml of ethyl acetate each time. The combined organic phases are washed with
water, dried over Na2S04 and concentrated in a rotary evaporator. Crude yield:
20 0.64 g
b) 3-Carboxymethoxy-4-[3-(2-chloro-4-fluorobenzoyl)ureido]benzoic acid
The crude product from step a) is converted in analogy to examples 1 b) to 1
d) into
methyl 3-methoxycarbonylmethoxy-4-[3-(2-chloro-4-fluorobenzoyl)ureido]-
2s benzoate, of which 50 mg (0.11 mmol) are dissolved in 5 ml of THF, and 1 ml
of
water is added. To this are added 27 mg (1.1 mmol) of LiOH, and the reaction
is
stirred at room temperature until the precursor has reacted completely. The
reaction is monitored by LC-MS. After completion of the reaction, the mixture
is
acidified with dilute hydrochloric acid and extracted twice with 20 ml of
ethyl
3o acetate each time. The combined organic phases are dried over Na2S04,
filtered
and concentrated in a rotary evaporator, and the residue obtained in this way
is
purified in a preparative HPLC system (column: Waters Xterra TMMS C,a, 5 Nm,
30 x 100 mm, mobile phases: A: HZO + 0.2% trifluoroacetic acid, B:
acetonitrile,
CA 02500763 2005-03-30
22
gradient: 2.5 minutes 90% A110% B to 17.5 minutes 10% A/90% B). 28 mg
(0.068 mmol) of the desired product are obtained.
Melting point: 279-281 °C
s Examples 4-9 and 11-14 were prepared in analogy to this method.
Example 10:
4-[3-(2-Chloro-4,5-difluorobenzoyl)ureido]-3-(3-methoxycarbonylpropoxy)benzoic
io acid
a) Benzyl3-hydroxy-4-nitrobenzoate
2.0 g (11 mmol) of 3-hydroxy-4-nitrobenzoic acid are suspended in 25 ml of
toluene, and 1.8 g (16 mmol) of benzyl alcohol and 0.2 g (1.1 mmol) of
is p-toluenesulfonic acid are added. The reaction is heated to reflux using a
water
trap until no further water separates out. After the reaction is complete, the
solution is evaporated in a rotary evaporator and the crude product is
purified by
chromatography. Yield 1.6 g (5.9 mmol).
2o b) 4-[3-(2-Chloro-4,5-difluorobenzoyl)ureido]-3-(3-methoxycarbonylpropoxy)-
benzoic acid
The benzyl 3-hydroxy-4-nitrobenzoate from step a) was alkylated in analogy to
method 2a) to form benzyl 3-(3-methoxycarbonylpropoxy)-4-nitrobenzoate.
450 mg (1.2 mmol) of the benzyl ester are dissolved in 20 ml of methanol and,
2s after addition of 13 mg (0.1 mmol) of palladium on activated carbon (10%),
hydrogenated at room temperature. The reaction is monitored by LC-MS.
After the reduction is complete, the mixture is filtered with suction through
Celite
and washed with methanol. The filtrate is concentrated in a rotary evaporator,
and
the product is reacted without previous purification in analogy to method 1 d)
to
3o give 4-(3-(2-chloro-4,5-difluorobenzoyl)ureido]-3-(3-
methoxycarbonylpropoxy)-
benzoic acid. The final purification takes place by preparative HPLC (column:
Waters Xterra T""MS Cog, 5 Nm, 30 x 100 mm, mobile phases: A: H20 + 0.2%
CA 02500763 2005-03-30
23
trifluoroacetic acid, B: acetonitrile, gradient: 2.5 minutes 90% A/10% B to
17.5
minutes 10% A190% B). Yield: 0.51 mg (0.11 mmol)
Melting point: 227°C
Example 15 was prepared in analogy to this method.