Note: Descriptions are shown in the official language in which they were submitted.
CA 02504076 2013-04-18
DEGRADABLE ELASTOMERIC NETWORK
Field of the Invention
This invention relates to biodegradable/biocompatible elastomeric materials.
Such
materials are suitable for use as implantable medical devices. In particular,
this invention
relates to photo-cross-linked biodegradable/biocompatible elastomeric
materials suitable for
use as implantable drug delivery devices.
Background of the Invention
Biodegradable and/or biocompatible polymeric materials are widely used in the
manufacture of implantable medical devices, including drug delivery depots.
Elastomeric
polymers are advantageously used in such applications because they are less
likely to
produce tissue irritation at the implant site and, for setting elastomers,
they maintain their
geometric dimensions during release and degradation. Cured elastomers can be
prepared
using heat or photo-irradiation to form covalent linkages between polymer
chains (see, for
example, U.S. Patent No. 6,984,393). However, for drug delivery devices
involving the
entrapment of temperature-sensitive drugs such as peptides or proteins, a
thermo-setting
elastomer is unsuitable.
Many peptide and protein drugs, e.g. cytokines, are effective at very low
concentrations, have very short biological half-lives, act in a paracrine
fashion, require long-
term delivery and are readily degraded when administered by conventional
routes. For
these reasons considerable effort has been devoted to the development of
formulations for
prolonged localized delivery, most of which have focused on the use of
biodegradable
polymers as delivery vehicles (Amkraut et al., Adv. Drug Delivery Rev. 1990,
4:255-276;
Gombotz et al., Bioconjugate Chem. (1995) 6:332-351; Sinha et al., J. Control.
Rel. (2003)
90:261-280; Schwendeman et al., Peptide, protein, and vaccine delivery from
implantable
polymeric systems. In: Controlled Drug Delivery: Challenges and Strategies,
ed.: Park, K.,
ACS: Washington, D.C., (1997)). In particular, the development of
biodegradable
microparticle formulations has received much attention.
Typically, in such delivery systems the drug is incorporated as a solid
particle
dispersed throughout the polymer matrix. The drug is released by dissolution
and diffusion
of surface resident particles and any particles in contact with those at the
surface.
Subsequent release for biodegradable systems then proceeds through the
creation of
- 1 -
CA 02504076 2005-04-14
micropores within the device as the polymer begins to hydrolyze. For low drug
loadings,
only a fraction of the drug can be released by diffusion, and so the majority
of the drug is
released through the creation of pores by polymer degradation. This generally
results in a
biphasic release pattern, with release by diffusion occurring first and
reaching a plateau, and
erosion-controlled release occurring after a lag period. Thus, for drugs that
should be
released at low concentrations but within a reasonable time frame, use of a
hydrophobic
polymer matrix is a poor choice, as drug release rates are controlled by the
interconnectedness of the particles within the matrix (Gombotz et al.,
Bioconjugate Chem.
(1995) 6:332-351).
One way to increase the amount of drug released in the diffusional phase is by
including physiologically innocuous, water soluble excipients in the delivery
device. Such
excipients increase the porosity of the device by dissolving to generate pores
and may also
enhance polymer degradation by increasing water absorption into the device.
The
incorporated drug is released by diffusion through the pores. The inclusion of
water soluble
excipients may also eliminate the biphasic release pattern. However, a
combination of
enhanced total fraction released and a sustained constant release rate is not
possible with
this approach, because the release rate increases as the porosity of the
device increases.
Other approaches have included the use of block thermoplastic copolymers,
containing a water-soluble polymer block (e.g., poly(ethylene glycol)) and a
hydrophobic
polymer block, typically poly(D,L-lactide). Using these block copolymers, the
protein is
loaded into the polymer device by dissolving the polymer in a suitable organic
solvent and
then using processes such as emulsification, and solvent casting (Kissel et
al., J. ControL
Rel. (1996) 39:315-326; Bezemer et al., J. Control. Rel. (2000) 64:179-192).
This approach
has been demonstrated to be capable of generating constant protein release
rates.
However, this approach often results in a significant initial burst release of
drug, and/or
denaturation of the drug during device fabrication.
Summary of the Invention
In a first aspect, the invention provides adegradable delivery system for
delivering an
agent, comprising: a degradable cross-linked network of: (i) a hydrophobic,
hydrolysable
amorphous star polymer; and a hydrophilic, biocompatible polymer; and an agent
distributed within the network.
- 2 -
CA 02504076 2005-04-14
The star polymer may comprise at least one monomer, said at least one monomer
capable of forming a degradable linkage to another monomer. The at least one
monomer
may be selected from the group consisting of lactones, carbonates, and cyclic
amides, and
combinations thereof. The at least one monomer may be selected from
valerolactone,
caprolactone, dioxepanone, lactide, glycolide, trimethylene carbonate, and 0-
benzyl-L-
serine.
In certain embodiments, the polymers may further comprise one or more cross-
linkable groups on the polymer chain termini.
The cross-linking may be initiated thermally or by irradiation. The delivery
system
may further comprise a photo-cross-linkable group selected from acrylate,
coumarin,
thymine, cinnamates, diacrylate, oligoacrylate, methacrylate, dimethacrylate,
and
oligomethacrylate.
The cross-linked network may be formed through action of an initiator.
In certain embodiments of the delivery system, the polymer chain termini may
contain a carbon-carbon double bond capable of cross-linking and polymerizing
polymers.
In certain embodiments, the initiator may be a free radical initiator selected
from
acetophenone derivatives, camphorquinone, Irgacure (1-hydroxy-cyclohexyl-
phenyl-
ketone, 144-(2-hydroxyethoxy)-pheny1]-2-hydroxy-2-methy1-1-propane-1-one, 2,2-
dimethoxy-1,2-diphenylethan-1-one, or 2-methyl-144-(methylthio) phenyI]-2-(4-
morpho-
linyI)-1-propanone, 2,2-dimethy1-2-phenylacetaphenone, 2-methoxy-2-
phenylacetaphenone), Darocur (144-(2-hydroxyethoxy)-pheny1]-2-hydroxy-2-
methy1-1-
propane-1-one or 2,4,6-trimethylbenzoyl-diphenyl-phosphineoxide), eosin dye,
potassium
persulfate, with or without tetraamethyl ethylenediamine; benzoylperoxide,
with or without
triethanolamine; and ammonium persulfate with sodium bisulfite.
In some embodiments of the delivery system, the star polymer has a glass
transition
temperature (TO below room temperature. The star polymer may comprise star-
poly(c-
caprolactone-co-D,L-lactide).
In some embodiments of the delivery system, the hydrophilic polymer may be
selected from poly(ethylene glycol), poly(ethylene oxide), poly(vinyl
alcohol),
poly(vinylpyrrolidone), poly(ethyloxazoline), poly(ethylene oxide)-co-
poly(propylene oxide)
block copolymers, polysaccharides, carbohydrates such as hyalyuronic acid,
chitosan,
dextran, heparan sulfate, heparin, alginate, and proteins such as gelatin,
collagen, albumin,
ovalbumin, and polyamino acids.
- 3 -
CA 02504076 2005-04-14
In some embodiments of the delivery system, the hydrophilic polymer may
comprise
poly(ethylene glycol) diacrylate.
The hydrophobic polymer may form greater than 70% by weight of the total
polymer
mass, and the rate of agent release increases as the content of hydrophobic
polymer
decreases.
In some embodiments, the agent may be a drug, a peptide, or a protein. In
other
embodiments, delivery system may be a medical device, may be adapted for
implant in a
subject, and may be biodegradable.
In a second aspect, there is provided a method of preparing a degradable
delivery
system for delivering an agent, comprising: combining a hydrophobic,
hydrolysable
amorphous star polymer and a hydrophilic, biocompatible polymer to create a
mixture;
adding an agent to the mixture; and subjecting the mixture to photo-
irradiation to create a
degradable cross-linked solid network.
In some embodiments, the mixture may be disposed in a mold prior to photo-
irradiation. In some embodiments, the star co-polymer may comprise at least
one monomer,
said at least one monomer capable of forming a biodegradable linkage to
another monomer.
In some embodiments, the monomer may be capable of undergoing polymerization
through a ring-opening reaction or a condensation reaction.
In some embodiments, the at least one monomer may be selected from the group
consisting of lactones, carbonates, and cyclic amides, including
valerolactone, caprolactone,
dioxepanone, lactide, glycolide, trimethylene carbonate, and 0-benzyl-L-
serine.
In some embodiments, the star polymer may further comprise one or more photo-
cross-linkable groups on the polymer chain termini, wherein the photo-cross-
linkable group
may be selected from acrylate, coumarin, thymine, cinnamate, diacrylate,
oligoacrylate,
methacrylate, dimethacrylate, and oligomethacrylate.
In some embodiments, the cross-linked network may be formed through action of
an
initiator.
In some embodiments, the termini of the polymers may contain a carbon-carbon
double bond capable of cross-linking and polymerizing polymers.
In some embodiments, the initiator may absorb photons to form a free radical
which
reacts with an allyl group of the photo-cross-linkable group. The initiator
may be selected
from acetophenone derivatives, camphorquinone, Irgacure (1-hydroxy-cyclohexyl-
phenyl-
ketone, 144-(2-hydroxyethoxy)-phenyl]-2-hydroxy-2-methyl-1-propane-1-one, 2,2-
dimethoxy-1,2-diphenylethan-1-one, or 2-methyl-144-(methylthio) phenyI]-2-(4-
morpho-
- 4 -
CA 02504076 2005-04-14
linyI)-1-propanone, 2,2-dimethy1-2-phenylacetaphenone, 2-methoxy-2-
phenylacetaphenone), Darocur (144-(2-hydroxyethoxy)-pheny1]-2-hydroxy-2-
methy1-1-
propane-1-one or 2,4,6-trimethylbenzoyl-diphenyl-phosphineoxide), and eosin
dye.
In a preferred embodiment, the star polymer may comprise star-poly(E-
caprolactone-
co-D,L-lactide).
In some embodiments, the star polymer may be end-functionalized with a vinyl
monomer.
In some embodiments, the hydrophilic polymer may be selected from
poly(ethylene
glycol), poly(ethylene oxide), poly(vinyl alcohol), poly(vinylpyrrolidone),
poly(ethyloxazoline),
poly(ethylene oxide)-co-poly(propylene oxide) block copolymers,
polysaccharides,
carbohydrates such as hyalyuronic acid, chitosan, dextran, heparan sulfate,
heparin,
alginate, and proteins such as gelatin, collagen, albumin, ovalbumin, and
polyamino acids.
In a preferred embodiment, the hydrophilic polymer may comprise poly(ethylene
glycol) diacrylate.
In some embodiments, the hydrophobic polymer may form greater than 70% by
weight of the total polymer mass.
In some embodiments, the agent may be a drug, a peptide, or a protein.
In a third aspect of the invention there is provided a method of delivering a
drug to a
subject, comprising: providing the drug in a delivery system comprising a
cross-linked
network of a hydrophobic, hydrolysable amorphous star polymer and a
hydrophilic,
biocompatible polymer; and disposing the delivery system in the subject.
In some embodiments, wherein the drug may be a peptide or a protein.
In a fourth aspect of the invention there is provided a degradable elastomer,
comprising: a degradable cross-linked network of: (i) a hydrophobic,
hydrolysable
amorphous star polymer; and (ii) a hydrophilic, biocompatible polymer.
In some embodiments, the elastomer may be biodegradable.
In some embodiments, the cross-linking may be photo-cross-linking.
In some embodiments, pores of the elastomer may be connected.
Brief Description of the Drawings
Preferred embodiments of the invention will now be described, by way of
example,
with reference to the drawings, wherein:
Figure 1 is a plot showing the influence of weight percent of poly(ethylene
glycol)
diacrylate (PEGD) incorporated into networks on vitamin B12 release from
cylinders
- 5
CA 02504076 2005-04-14
prepared, using acrylated star co-polymer (ASCP) 1000 (see the Example for
details). The
cylinders had a diameter of 3.5 mm and the vitamin B12 particle size was < 100
gm. The
solid lines represent linear regressions to the data over the region
indicated.
Figure 2 is a plot showing the influence of cylinder diameter on vitamin B12
release.
The cylinders were prepared using ASCP 1000 and contained 10% PEGD. The
vitamin B12
particle size was < 100 gm. The solid lines represent linear regressions to
the data over the
region indicated.
Figure 3 is a plot showing the effect of ASCP molecular weight on vitamin B12
release. The PEGD content was 10%, the cylinder diameter was 1.8 mm, and the
vitamin
B12 particle size was < 100 gm.
Figure 4 is a plot showing the effect of vitamin B12 particle size on release
from 1.8
mm cylinders prepared using ASCP 2700 containing 10% PEGD.
Figure 5 is a plot showing the volume change of vitamin B12 loaded cylinders
with
release time. The data is expressed as the volume at time t, Vt, divided by
the initial
volume, Vo. (A) Cylinders prepared using ASCP 1000 containing 10 w/w% PEGD.
(B)
Cylinders prepared using 10 w/w% PEGD and varying ASCP molecular weight. The
initial
cylinder diameter was 1.8 mm and the vitamin B12 particle size was < 100 gm.
Figure 6 is a plot showing in vitro mass loss with time for cylindrical
networks
prepared with ASCP 1000 and varying amounts of PEGD. The data is expressed as
mass
at time t, mt, divided by the initial mass, mo.
Detailed Description of Preferred Embodiments
In accordance with a broad aspect of the invention, there is provided a
degradable,
preferably biodegradable, and/or biocompatible photo-cross-linked elastomeric
polymer.
The elastomer is useful in applications such as, for example, biomaterials and
biomedical
devices, where it can be used in treatment of human and non-human subjects,
and in
applications such as tissue engineering. Elastomers of the invention can be
formed into
films, rods, screws, needles, stents, catheters, or other structures with or
without
incorporated fibres; implantable drug delivery systems, in which a
pharmaceutical agent is
disposed in the elastomer; film coatings for pills; coatings on biomedical
devices such as
needles, stents, and catheters; as well as other applications such as rubber
tougheners for
ceramic devices. The elastomer may be formed into devices such as scaffolds
for tissue
engineering and tissue restoration of soft tissue, connective tissue, and
bone, in vitro and in
- 6 -
CA 02504076 2005-04-14
vivo. Also, the elastomer may be provided as a coating on devices such as
scaffolds for
tissue engineering and tissue restoration of soft tissue, connective tissue,
and bone, in vitro
and in vivo.
Other applications of the elastomers of the invention include applications
where
delivery of an agent encapsulated in, or loaded into, a
biodegradable/biocompatible polymer
is required, or would be beneficial. For example, in agriculture, an elastomer
of the
invention can be loaded with one or more agents such as a fertilizer or
pesticide.
Application of the loaded elastomer to a crop results in sustained delivery of
the one or more
agents. Such delivery helps to avoid over-fertilizing of crops, and reduces or
eliminates the
need for repeated applications of such agents.
Depending on the properties of the agent loaded into the elastomer, and the
desired
delivery rate of the agent, an excipient, as described below, can be used
together with such
agent. Also depending on the properties of the agent loaded into the
elastomer, it may be
desirable to protect the agent by treating the agent during or prior to
loading into the
elastomer. For example, when the agent is a drug, the drug may be co-
lyophilized with a
protecting agent prior to loading.
As used herein, the term "degradable" is intended to refer to a substance that
can be
chemically degraded or decomposed by natural effectors, for example, via
weather or
biological processes (i.e., biodegradable) such as physiological temperature,
pH, and/or
enzyme activity. For example, degradation may occur by hydrolysis, which can
occur
chemically and/or in a biological system. Biological processes can take place
within an
organism or outside of an organism.
As used herein, the term "biocompatible" is intended to refer to a substance
having
substantially no known toxicity to or adverse affects on biological processes.
The substance
can be a compound in its original state or one or more components or products
of the
compound as the compound biodegrades.
In accordance one aspect of the invention, there is provided a biodegradable/
biocompatible elastomeric polymer. The elastomer comprises a degradable photo-
cross-
linked network of a hydrophobic, hydrolysable amorphous star co-polymer and a
hydrophilic,
biocompatible polymer. In embodiments where the elastomer is used to deliver
an agent,
such as a drug, the drug is distributed either as drug particles throughout or
is dissolved
within the elastomer. The star polymer and the hydrophilic polymer are
modified such that
they contain one or more photo-cross-linkable groups on the polymer chain
termini.
- 7 -
CA 02504076 2005-04-14
It.should be noted that thermal polymerization initiator systems may be used
instead
of photo-cross-linking. Such systems that are unstable at 37 C and would
initiate free
radical polymerization at physiological temperatures include, for example,
potassium
persulfate, with or without tetraamethyl ethylenediamine; benzoylperoxide,
with or without
triethanolamine; and ammonium persulfate with sodium bisulfite. However, photo-
cross-
linking is the preferred method of cross-linking, because it can be
accomplished very rapidly,
with minimal heat generation (Sawhney et al., Macromolecules (1993) 26:581-
587), and
therefore may not lead to degradation of an agent, such as a peptide drug, to
be entrapped.
Suitable star polymers may be prepared from any monomer capable of forming a
biodegradable linkage to another monomer and capable of undergoing
polymerization
through a condensation reaction, or preferably through a ring-opening
reaction. Preferably,
the monomer or monomers are chosen so as to form an amorphous star polymer.
Such
monomers include, for example, lactones, carbonates, cyclic amides (e.g.,
polyester amides,
polyamides), and combinations thereof. Examples of such monomers are
valerolactone,
caprolactone, dioxepanone, lactide, glycolide, trimethylene carbonate, and 0-
benzyl-L-
serine.
A suitable cross-linkable group may be any group with an accessible carbon-
carbon
double bond that can undergo free radical polymerization. Examples of cross-
linkable
groups are coumarin, thymine, cinnamates, acrylates, including, for example
diacrylates,
oligoacrylates, methacrylates, dimethacrylates, and oligomethacrylates. Cross-
linkable
groups may be substituted or unsubstituted. Preferred cross-linkable groups
are acrylates
which cross-link faster than methacrylates. The photo-cross-linking reaction
may be initiated
by a compound which absorbs photons to form a free radical which reacts with
the allyl
group of the photo-cross-linkable group. Examples of such an initiator are
acetophenone
derivatives (2,2-dimethy1-2-phenylacetaphenone, 2-methoxy-2-
phenylacetaphenone),
camphorquinone, Irgacure (1-hydroxy-cyclohexyl-phenyl-ketone, 144-(2-
Hydroxyethoxy)-
pheny1]-2-hydroxy-2-methy1-1-propane-1-one, 2,2-dimethoxy-1,2-diphenylethan-1-
one, or 2-
methy1-144-(methylthio) pheny11-2-(4-morpho-liny1)-1-propanone), Darocur
(14442-
Hydroxyethoxy)-pheny1]-2-hydroxy-2-methy1-1-propane-lone or 2,4,6-
trimethylbenzoyl-
diphenyl-phosphineoxide), and eosin dye. The wavelength (e.g., visible,
ultraviolet (UV))
and intensity of light used for the photo-cross-linking reaction depend on the
specific initiator
used.
The hydrophilic, biocompatible polymer may be crystalline, non-crystalline, or
semi-
crystalline and may be selected from poly(ethylene glycol), poly(ethylene
oxide), poly(vinyl
- 8 -
CA 02504076 2005-04-14
alcohol),,poly(vinylpyrrolidone), poly(ethyloxazoline), poly(ethylene oxide)-
co-poly(propylene
oxide) block copolymers, polysaccharides or carbohydrates such as hyaluronic
acid,
chitosan, dextran, heparan sulfate, heparin, alginate, proteins such as
gelatin, collagen,
albumin, ovalbumin, or polyamino acids.
In a preferred embodiment, the hydrophobic star polymer is star-poly(e-
caprolactone-
co-D,L-lactide) that has been end-functionalized with a vinyl monomer, and the
hydrophilic
polymer is comprised of poly(ethylene glycol) that has been end-functionalized
with a vinyl
monomer.
The elastomer, or delivery device comprising the elastomer, may be prepared by
first
dissolving the star co-polymer and the hydrophilic polymer in a suitable
solvent. In
embodiments where the elastomer is for a delivering an agent such as a drug,
the drug, in
particulate form, is then added to the solution to create a suspension. In
some
embodiments, the solvent may not be necessary. A photo-initiator is then
added, and mixed
throughout the suspension. To obtain an elastomer device of desired shape
(e.g.,
cylindrical), the suspension may be poured into a suitable mold and
immediately subjected
to photo-irradiation. A cross-linked, solid network is formed that entraps the
solid drug
particles. Residual solvent is removed by evaporation.
The rate of drug release is controlled by the weight ratio of hydrophobic
polymer
present. As the hydrophobic polymer content decreases, the release rate
increases. A
long-term continuous release of the entrapped agent, such as a peptide or
protein drug, is
achieved when the hydrophobic polymer forms greater than 70% by weight of the
total
polymer mass.
Requirements for the formation of a useful elastomer using a star polymer as a
prepolymer are that the prepolymer has a glass transition temperature (TO
below
physiological temperature (e.g., 37 C), and preferably below room temperature,
and is
amorphous. Glass transition temperature is the temperature at which a polymer
undergoes
a phase transition from a glassy state to a rubbery state upon heating. It is
the temperature
where the molecules of a polymeric solid begin to move relative to one
another, yielding a
substance that behaves like a rubber, rather than a brittle glass.
Thus, star polymers in which at least one monomer has a very low glass
transition
temperature are the most suitable. An example of a monomer suitable for use in
accordance with the invention is E-caprolactone (T9 = -60 C). Such monomer
can be used
to prepare a star polymer, such as a star co-polymer, with another monomer
such as D,L-
lactide, even though the glass transition temperature of D,L-lactide is 68 C.
- 9 -
CA 02504076 2005-04-14
,
In preparing a star polymer from one or more species of monomers, an initiator
is
used. The initiator can be any polyol, such as, for example, glycerol,
pentaerythritol, and
xylitol.
As noted above, a star polymer in accordance with the invention can comprise
one
or more species monomer. In general, the properties (e.g., physical properties
such as
strength, Young's modulus, etc., and degradation kinetics) of the elastomer
are determined
to a large extent by the composition of the star polymer, and, where two or
more monomers
are employed, by the molar ratios of the monomers. For example, where an
elastomer
having more rapid biodegradation kinetics is desired, a monomer that either
biodegrades
more rapidly, and/or is more hydrophilic, should be chosen for incorporation
into the star
polymer. Thus, in the case of a polymer of E-caprolactone and D,L-lactide, the
relative
proportions of E-caprolactone and D,L-lactide should be controlled so as to
produce a
polymer that is amorphous. However, increasing the D,L-lactide content
increases the
biodegradation rate of the elastomer. It will be appreciated that, in
accordance with the
invention, an elastomer having a desired set of physical properties, including
biodegradation
rate, can be prepared by designing a star polymer with a specific
architecture, and
controlling the amount of cross-linking agent used. Moreover, such an
elastomer is easily
reproduced.
In embodiments where elastomers of the invention are loaded with a
pharmaceutical
agent and used for implantable delivery devices for the agent, the delivery
rate of the agent
will be a function of the hydrophobic star polymer content. For example, we
have found that
when the content of the hydrophobic star polymer is less than about 70 w/10/0
of the
elastomer, and the elastomer is loaded with an agent having a molecular weight
of about
1355 g/mol (i.e., the average weight of many peptide drugs), substantially all
of the agent is
delivered from the elastomer in about four days (see the below example).
However, when
the content of the hydrophobic star polymer is about 90 w/w% of the elastomer,
and the
elastomer is loaded with the same agent, delivery of the agent is slower and
more linear
over the delivery period. Thus, it is preferred that the content of the
hydrophobic star
polymer is at least about 70 % by weight of the total polymer mass, more
preferably about
70 % to about 90 %. Of course, the delivery rate will also depend on the
molecular weight
of the agent, with larger agents having slower delivery.
Advantages of the photo-cross-linked elastomer include:
1. The biodegradable elastomer can be prepared at room or
physiologic temperature
and thus may be prepared in vivo.
-10-
CA 02504076 2005-04-14
2. The low temperature during preparation avoids thermal denaturation of
peptide and
protein drugs.
3. The prepolymer is a star polymer which has a reduced viscosity, which
allows for
easier insertion into molds for part manufacture, and thus may be processed at
lower
temperatures than linear counterparts.
4. The prepolymer is amorphous (non-crystalline) and produces an amorphous
elastomer which degrades at a more homogeneous rate than would a thermoplastic
elastomer which relies on crystalline blocks of homopolymer sections of the
backbone to
provide cross-links (amorphous regions degrade first, then the crystalline
regions which
degrade more slowly).
5. Because of its homogeneous degradation rate, the elastomer maintains its
physical
properties for a longer time period (provides a linear decrease in strength
with respect to
mass loss during degradation).
Further, in embodiments where the elastomer is provided as a drug delivery
device,
the elastomer advantageously provides the combination of low drug loading,
minimal burst
effect, nearly constant release, and 100 % drug entrapment. None of the known
prior drug
delivery systems provides this combination of advantages.
As noted above, the biodegradable/biocompatible photo-cross-linked elastomer
of
the invention is particularly well suited for drug delivery devices, such as
controlled release
devices. Advantages of such an elastomeric device surgically implanted in a
subject
include: administration of a drug at a desired location, with sustained slow
release with
minimal burst effect and depot effect, so that the total dosage administered
to a subject can
be reduced, and the potential for systemic side effects is reduced; further
surgery to retrieve
the delivery device is avoided because the device is biodegradable and
biocompatible; and
the elastomer may protect the drug from degradation until it is released.
Lipophilic drugs, (for example, but not limited to bupivacaine, benzocaine,
lidocaine,
camptothecin, paclitaxel, etoposide, vincristine, vinblastine, vitamin D,
tacrolimus,
hydrocortisone, nitroglycerin, fentanyl, estradiol, testosterone, cortisone
and other
corticosteroids), hydrophilic drugs (for example, but not limited to
pilocarpine nitrate, aspirin,
ibuprofen, potassium chloride, ascorbic acid), and peptide and protein drugs
(for example,
but not limited to cytokines such as interferons, interleukins, granulocyte
macrophage colony
stimulating factor, insulin, erythropoeitin, human growth hormone, epidermal
growth factor,
vascular endothelial growth factor, basic fibroblast growth factor), and
combinations thereof,
may be loaded into a delivery device using an elastomer of the invention.
- 11 -
, _________________________________
CA 02504076 2005-04-14
In some embodiments an excipient is included in addition to a drug or drugs.
Excipients, which may be bulking agents or osmotagens, are physiologically
inert, and may
enhance delivery or increase the rate of delivery of a drug by generating
osmotic pressure
within the elastomer. The mechanism of osmotically controlled release is as
follows: Upon
immersion into an aqueous medium, drug release begins as water vapor
penetrates the
polymer matrix until it reaches a polymer encapsulated particle, hereafter
referred to as a
capsule. The water phase-separates and dissolves the solid drug at the
polymer/drug
interface, forming a saturated solution of drug and excipient particles. Under
the reduced
water activity gradient, water is drawn into the capsule, causing it to swell.
If the osmotic
pressure is great enough, the polymer capsule wall ruptures. Due to the
relaxation process
of the elastomer, the capsule wall slowly collapses and the solution of drug
and excipient
particles is forced out through the rupture. This rupture and collapse process
results in the
drug being released at an almost constant rate. Osmotic drug delivery from
monolithic
polymer devices has been described (Michaels et al., U.S. Patent No.
4,117,256; Di Colo,
Biomaterials. 13(12):850-856, 1992; Amsden et al., J. Controlled Rel. 30:45-
56, 1994) using
non-biodegradable polymers such as poly(ethylene-vinylacetate) and silicone.
The delivery of peptide and protein drugs may be problematic due to the
sensitivity of
such drugs to environmental conditions associated with the delivery system
employed.
Various means of achieving localized delivery of protein drugs have been
investigated and
include the use of liposomes, polymer gels, and biodegradable microspheres.
Problems
with some of these prior delivery systems include inability to maintain
protein stability,
relatively short drug release durations, inefficient drug loadings, and
unsustained and/or
incontrollable release rates. The latter may be manifested as a large amount
of peptide or
protein drug released immediately upon immersion of the delivery device into
an aqueous
medium. This burst effect can be deleterious to the patient if the drug is
potent. Such prior
delivery systems may subject proteins to conditions leading to aggregation,
denaturation
and adsorption at interfaces, deamidation, isomerization, cleavage, oxidation,
thiol disulfide
exchange, and p elimination in aqueous solutions. The major factors affecting
these
changes are mechanical forces such as shear, the presence of surfactants,
buffers, ionic
strength, the presence of oxidizers such as ions, radicals and peroxide,
light, pH,
temperature, and material surface interactions. Protein denaturation may
result in a loss of
potency and the conformation changes in the protein molecule may make the
protein
immunogenic.
-12-
CA 02504076 2005-04-14
For example, polymeric microspheres have been developed that are capable of
delivering a virtually constant amount of an encapsulated protein (Takada et
al., J. Control.
Rel. (1994) 32, 79-85; Sah et al., Journal of Applied Polymer Science (1995)
58, 197-206;
Mehta et al., J. Control. Rel. (1996) 41:249-257). This approach has been
investigated for
local and systemic protein and peptide delivery (Sabel et al., Annals of
Surgical Oncology
(2004) 11:147-156; Mullerad et al., Cancer Investigation (2003) 21:720-728;
Egilmez et al.,
Cancer Research (2000) 60:3832-3837; Jiang et al., Pharmaceutical Research
(2003)
20:452-459). These formulations generally consist of poly(lactide-co-
glycolide) (PLG),
throughout which the protein is distributed as solid particles. The protein is
released in three
phases: an initial burst; diffusion controlled release; and polymer erosion
controlled release.
The initial burst is due to surface resident protein particles, while the
diffusion controlled
release is a result of dissolved protein diffusing through the water-filled
pores and channels
within the microspheres. To obtain a constant release rate from PLG
microspheres, the
diffusion phase must overlap with the erosion release phase such that new
pores or
channels are created during drug release. Polymeric microspheres have one or
more of the
following advantages of not only providing a constant release, but of being
easily injected to
the target site, providing a long term release duration, consisting of proven
biocompatible
materials, having a reasonable shelf-life and degrading to completely bio-
resorbable
compounds.
However, due to the need for the overlapping polymer erosion phase, a
significant
problem with polymeric microspheres as a delivery system is maintenance of
protein and
peptide stability (van de Weert et al., Pharm. Res. (2000) 17:1159-1167). When
polymers
such as PLG degrade, they liberate acidic oligomers and monomers. The presence
of these
acids has been found to decrease the local pH at the surface of the polymer
and in the
pores and channels of the device (Mader et al., Biomaterials (1996) 17:457-
461; Fu et al.,
Pharm. Res. (2000) 17:100-106). In fact, the pH at the centre of a PLG
microsphere has
been determined to be as low as from 1.5 (Fu et al., Pharm. Res. (2000) 17:100-
106) to 1.8
(Shenderova et al., Pharm. Res. (2997) 14:1406-1414). At this pH, many
proteins undergo
backbone cleavage and deactivation. This reduction in the pH of the inner
environment of
the microspheres has been linked to inactivation and denaturation of other
proteins within
PLG microspheres (Park et al., J. Control. Rel. (1995) 33:211-222; Johnson et
al., J.
Control. Rel. (1991) 17:61-67; Takahata et al., J. Control. Rel. (1998) 50:237-
246; Zambaux
et al., J. Control. Rel. (1999) 60:179-188; Tabata et al., Pharm. Res. (1993)
10:487-496;
Aubert-Pouessel et al., Pharm. Res. (2002) 19:1046-1051). Attempts to overcome
this pH
-13-
CA 02504076 2005-04-14
issue have included the incorporation of basic salts into the matrix (Zhu et
al., Nature
Biotechnology (2000) 18:52-57). However, a recent paper, wherein the micro-
environmental
pH of different size distributions of PLG microspheres was mapped, has
demonstrated that
the inclusion of a basic excipient does not prevent the internal pH of the
microspheres from
dropping significantly over a 3 week period (Li et al., J. Control. Rel.
(2005) 101:163-173).
Moreover, protein-loaded microspheres that have been used in the studies to
date have
been prepared using techniques such as double emulsification that typically
result in protein
denaturation (van de Weert et al., Pharm. Res. (2000) 17:1159-1167).
The invention is particularly advantageous for the delivery of peptide and
protein
drugs, as the above-noted problems associated with environmental conditions
are avoided.
The protein delivery device of the invention overcomes such problems by
providing a
polymeric delivery system capable of long-term, relatively constant protein
delivery from a
biodegradable and biocompatible elastomer device. The elastomer minimizes or
avoids
acidic degradation of a protein incorporated therein, because the elastomer
and its
degradation products are not acidic and are biocompatible. That is, the
poly(caprolactone)
homopolymer used in the elastomer of the invention degrades more slowly and
produces
fewer acidic degradation products per molecular weight than do other
biodegradable
polymers, such as poly(lactide-co-glycolide). These properties provide a more
suitable pH
environment for protein stability within the polymer. Thus, the protein
released is more likely
to be bioactive and non-immunogenic. Continuous release from the elastomer is
achieved
by employing an osmotic mechanism and a balance of polymer physical properties
with
polymer degradation. Aggregation of the protein within the delivery device is
minimized or
avoided by incorporating the protein as a solid lyophilized with appropriate
agents. Use of
lyophilization agents provides a driving force for an osmotic drug delivery
mechanism. Use
of the photo-cross-linked elastomer of the invention allows the device to be
fabricated at,
e.g., room temperature, thereby avoiding heat which can denature a protein.
Further, the invention substantially reduces or eliminates the burst effect
discussed
above, due to the rapid setting of the polymer network. The rapid setting is
achieved by
photo-cross-linking during the manufacturing process, which prevents migration
of the
peptide or protein drug particles to the polymer surface. Others have
attempted to reduce
the burst effect by encapsulating the drug in a blend of ,a hydrophilic
polymer with a
hydrophobic polymer (Yeh et al., J. Control. Rel. (1995) 37:1-9; Jiang et al.,
Pharm. Res.
(2001) 18:878-885). In that approach, the presence of the hydrophilic polymer
reduced the
formation of protein crystals at the device surface. However, a combination of
reduced burst
- 14 -
CA 02504076 2005-04-14
effect, nearly constant release, low initial drug loading in the device,
complete drug
entrapment and enhanced total drug released was not demonstrated.
Wu et al. (Journal of Biomaterials Science-Polymer Edition (2003) 14:777-802)
used
a photo-cross-linkable star polymer combined with poly(ethylene glycol)
diacrylate to
produce a cross-linked network containing a model protein drug. In that study
a star
polymer (star-poly(c-caprolactone)) was used, rather than a star co-polymer as
in the
invention, resulting in a highly crystalline (38 % crystallinity) polymer
network, which in turn
resulted in very long polymer degradation times (over a period of years) and
very slow
release of drug. Additionally, a large protein, bovine serum albumin, was
incorporated in a
co-solvent for both the polymers and the protein. This resulted in a
significant protein
release burst effect during the initial stage of release (13 to 45 % within
the first 24 hours).
A disadvantage of this approach is potential denaturation of the protein
during the free
radical cross-linking reaction to prepare the delivery device. Finally, a
combination of low
initial burst and constant release was not achieved with the formulations of
Wu et al. (2003).
The principle of osmotic drug delivery has previously been demonstrated in a
delivery system capable of delivering a variety of proteins at the same,
almost constant
release rate (Amsden et at., J. Control. Rel. (1995) 33:99-105). The proteins
were released
at the same rate because the driving force for release was the same in each
case : the
osmotic pressure generated by an inorganic salt. However, use of such salt
should
preferably be avoided because of its destabilizing effect on a protein and the
potential for
tissue irritation. The necessary polymer properties for this release mechanism
are a radial
extension ratio of greater than 1.05, a water permeation coefficient of
between le and
--12
lu g cm/cm2sec cm Hg, a degradation time of greater than 1 month, and
minor tissue
irritation and inflammation upon implantation. In the previous work, non-
degradable
polymers such as silicone and poly(ethylene-co-vinyl acetate) were used. With
such
polymers a device geometry having a constant cross-sectional area is required
in order to
provide a constant release rate, because the osmotic rupturing mechanism
proceeds in a
serial manner from the surface to the interior of the device. As one moves
from the exterior
of the device, usually cylindrical in shape, to the interior, fewer and fewer
drug capsules
exist within each rupturing layer. This reduction in the number of capsules
produces a
declining release rate with time.
However, this problem is overcome by the biodegradable elastomers of the
invention. Due to their biodegradable nature, their mechanical properties
change with time.
This property produces a drug-loaded device exhibiting a constant release
rate. Although
-15-
CA 02504076 2013-04-18
the mass of drug per cross-sectional area of the device is difficult to
manipulate, the time
required to produce a rupture of the elastomer is more easily manipulated.
This latter
parameter is determined by the extension ratio and Young's modulus of the
polymer. Thus,
according to the invention, the elastomer can be tailored such that its
Young's modulus
decreases with time while the extension ratio remains essentially constant
during the
release period without significant polymer degradation, such that the time
required to rupture
the polymer decreases with time. So long as this decrease keeps pace with the
decrease in
the mass of drug per cross-sectional area of the device, a constant release
rate is achieved.
In one embodiment, an osmotic excipient is used in the protein delivery
device. The
excipient reduces protein aggregation and enhances osmotic protein delivery.
Examples of
suitable excipients include, but are not limited to, polyols (e.g., trehalose,
polyethylene
glycol, glycerin, mannitol) and small, neutral amino acids, and combinations
thereof. Polyols
are preferable because they can generate significant osmotic pressures and are
highly
effective at preventing protein aggregation. They accomplish this by re-
ordering the water
around the protein molecule, exerting pressure to reduce the surface contact
between the
protein and the solvent. This pressure forces hydrophobic portions of the
protein to become
further removed from the solvent, thus decreasing the likelihood of a
hydrophobic-
hydrophobic interaction leading to aggregation. Thus, in accordance with the
invention, the
protein is combined with an excipient by, for example, lyophilization. The
ratio of excipient
to protein can range from 1:1 to 99:1, depending on the specific conditions. A
suspension of
the protein/excipient is added to the photo-cross-linkable polymer of the
invention prior to
cross-linking, and is contained with in the elastomer upon cross-linking.
The invention is further described in the following non-limiting Example.
Example. Delivery of vitamin B12 as a drug analog
In this study, we examined an amorphous hydrophobic star co-polymer co-cross-
linked with a hydrophilic polymer (poly(ethylene glycol) diacrylate) to yield
networks having
less than 30% poly(ethylene glycol) diacrylate, and incorporated a low
molecular weight
drug analog as solid particles during the free radical cross-linking reaction.
Vitamin B12 was
used as the drug analog because it has a molecular weight (1355 g/mol) similar
to that of
many peptide drugs, and is readily detectable due to its red color. The
loading of vitamin
- 16-
CA 02504076 2005-04-14
B12 was.kept to 1 w/w%, and means of modulating its release from the
cylindrical matrix
were investigated.
MATERIALS AND METHODS
D,L-lactide (99%) was obtained from Purac (The Netherlands) and used as
received,
while E-caprolactone was obtained from Lancaster (Canada), dried over CaH2
(Aldrich,
Canada) and distilled under vacuum in a nitrogen atmosphere. Other chemicals
used were
stannous 2-ethylhexanoate, glycerol, acryloyl chloride, triethylamine, 4000
g/mol
poly(ethylene glycol) diacrylate (PEGD), 4-dimethylaminopyridine, and 2,2-
dimethoxy-2-
phenyl-acetophenone, which were all obtained from Aldrich, Canada. Other
chemicals used
included dichloromethane and ethyl acetate obtained from Fisher, Canada.
Polymer synthesis
The photo-cross-linkable star-poly(E-caprolactone-co-D,L-lactide) was prepared
as
described previously (Aoyagi et al., J. Control. Re/. 1994, 32:87-96; Amsden
et al.,
Biomacromolecules 2004, 5:2479-2486). Briefly, 50:50 molar ratio co-polymers
were
prepared of molecular weights of 1000, 2700 and 3900 g/mol by melt ring-
opening
polymerization of E-caprolactone and D,L-lactide at 140 C for 24 hours
initiated by glycerol
and catalyzed by stannous 2-ethylhexanoate. This process yielded a 3-armed
star co-
polymer terminated in hydroxyl groups. The star co-polymer termini were
esterified using
acryloyl chloride in anhydrous dichloromethane containing triethylamine as an
HCI
scavenger and 4-dimethylaminopyridine as a catalyst, at room temperature under
nitrogen
for 48 hours. Purification yielded an acrylated star co-polymer (ASCP) having
a degree of
acrylation greater than 85% (Amsden et at., Biomacromolecules 2004, 5:2479-
2486).
Device preparation
Vitamin B12 as received was ground and sieved into less than 100 pm or less
than
25 pm fractions. Vitamin B12 loaded cylinders were prepared by first
dispersing the vitamin
B12 particles in a solution of ASCP dissolved in different amounts of ethyl
acetate. In this
solution was also dissolved varying amounts of PEGD and 0.015 mg 2,2-dimethoxy-
2-
phenyl-acetophenone (UV photo-initiator) per gram star co-polymer. The vitamin
B12
particles were suspended by agitation using a vortexer, and the suspension
quickly poured
-17-
CA 02504076 2005-04-14
into sealed glass tubing. The tube was placed into a holder and rotated
horizontally at 40
rpm under a long-wave Black-Ray AP UV lamp at an irradiation intensity of 10
mW/cm2 for 5
minutes. One end of the tube was then opened to allow for solvent evaporation.
Cylinders
of length 1 cm were cut from these master cylinders and used in subsequent
release
experiments.
Polymer characterization
Thermal properties of the polymers were measured using a Seiko 220U
differential
scanning calorimeter (DSC) calibrated with indium and gallium standards. 10 mg
samples
were subjected to a heating-cooling-heating cycle from ambient to 100 C to
¨100 C and
back to 100 C at a rate of 10 C/min. All measurements were taken from the
second
heating cycle. The molecular weights of the ASCP were measured using a Waters
Breeze
GPC system connected to a Precision Detectors PD 2000 DLS light scattering
detector
supplied with a Waters 410 Differential Refractometer. The mobile phase
consisted of THF
at a flow rate of 2 ml/min with the system at 30 C. The concentration of the
polymers used
for the GPC measurements were 5 mg/ml and the injection volume was 50 pl. The
column
configuration consisted of an HP guard column attached to a Phenogel linear
(2) 5 p GPC
column. The incremental refractive index (dn/dc) was determined using a Wyatt
Optilab
refractometer at 30 C and found to be 0.064. Sol contents were measured using
dichloromethane extraction at 40 C on a Soxhlet apparatus. Fourier transform
infra-red
spectroscopy (FTIR) of the ASCP, the PEGD, cross-linked ASCP, cross-linked
PEGD and
co-cross-linked ASCP and PEGD was performed by forming a thin film of the
polymers
directly onto the surface of a KBr crystal. The spectra were collected on a
Nicolet XX IR
spectrometer.
Release studies
The vitamin B12 loaded cylinders were placed in 2 ml polypropylene vials
containing
1 ml pH 7.4 phosphate buffered saline (per 100 m1:0.16 sodium bisphosphate,
0,758 g
sodium phosphate, 0,44 g sodium chloride). The vials were placed on a rotary
shaker
maintained at 37 C in an incubator oven. At each sampling period, the 0.5 ml
of release
medium was removed and replaced with fresh buffer. Vitamin B12 concentration
in the
release media was measured at 381 nm using a Spectromax microplate
spectrophotometer.
-18-
CA 02504076 2005-04-14
For every formulation examined the release of 3 cylinders was measured and
averaged.
The error bars shown in the Figures represent one standard deviation about the
mean of
this average.
Network degradation studies
Vitamin B12-free cylinders were prepared in the same fashion as described
above.
The initial mass and dimensions of the cylinders were recorded. The cylinders
were
immersed in 4 ml pH 7.4 phosphate buffered saline maintained at 37 C in 5 ml
glass vials.
The buffer was replaced weekly. At given time points, the cylinders were
removed, wiped
dry with Kim Wipes, their dimensions recorded using calipers, and weighed.
Three cylinders
were also then dried in a vacuum oven for 48 hours in the presence of
dessicant, and
weighed dry.
Statistics
Unless otherwise stated, all experiments were performed in triplicate, with
the data
points in the figures representing the average, and the error bars one
standard deviation
from the average.
RESULTS
As vitamin B12 absorbs within the UV region, it was important to determine
whether
the cross-linking conditions affected the vitamin B12. The vitamin B12 was
therefore
suspended in ethyl acetate in the presence of the photo-initiator, and in a
non-acrylated
polymer solution also containing the photo-initiator, and subjected to 10
mW/cm2 long-wave
UV irradiation for 5 minutes. The vitamin B12 was then filtered from solution,
dried, and
dissolved in varying concentrations and their absorbance measured and compared
to that of
solutions prepared from the as-received vitamin B12. The results indicated
that there was
no significant change in the absorbance of the vitamin B12 due to this
procedure.
In the following discussion, "ASCP" refers to acrylated star co-polymer, while
the
number following refers to the molecular weight of the polymer. For example,
ASCP 1000
refers to the star co-polymer of molecular weight 1000 g/mol. The thermal
characteristics
(heat flow as a function of temperature) and thermal properties (glass
transition temperature
Tg, onset of melting point Tim and latent heat of fusion AH) of the networks
prepared from
these prepolymers and of a network prepared using just PEGD were determined.
-19-
CA 02504076 2005-04-14
The PEGD network did not exhibit a glass transition temperature over the range
of
temperatures examined; however it did possess a distinct melting endotherm
that began at
34 C. The networks prepared without PEGD were amorphous elastomers with glass
transition temperatures well below physiologic temperature. The Tg decreased
as the ASCP
prepolymer molecular weight increased, ranging from 4 C for networks prepared
using
ASCP 1000 to ¨8 C for those prepared using ASCP 3900. As the weight fraction
of PEGD
incorporated into the networks increased, the Tg decreased, and a small
melting endotherm
appeared. The latent heat of fusion of the melting endotherm increased, and
the onset
temperature of melting approached that of PEGD as the PEGD content increased.
From
these data, it can be inferred that at low PEGD concentrations, a homogeneous
co-polymer
network is formed, wherein the ASCP and the PEGD are co-cross-linked together.
As the
PEGD concentration in the network increases, regions of solely PEGD are formed
within the
polymer matrix.
FTIR spectrum analysis showed that the double bonds were completely consumed
during the cross-linking reaction. The C=C stretch at 1635 cm-1, which was
visible in the
uncross-linked ASCP prepolymer and PEGD, disappeared upon exposure to UV
irradiation.
This was supported by the very low sol contents of the networks formed (values
ranged
between 2 1 % sol for 100 A, PEGD and 8 2 % sol for ASCP 1000 with 20
PEGD).
The influence of mass percent PEGD incorporated into the matrix, the diameter
of
the cylinder, ASCP molecular weight, and particle size of the solid vitamin
B12 entrapped
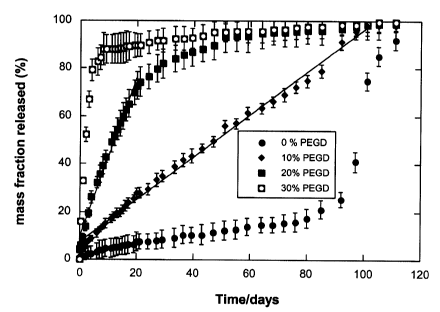
within the cylinder on vitamin B12 release were all examined. Figure 1
illustrates the effect
of increasing the mass percent of PEGD in the matrix on vitamin B12 release
for matrices
prepared using ASCP 1000. The cylinder diameter in this case was 3.5 mm and
the vitamin
B12 particle size in the cylinders was less than 100 pm. Without any PEGD
incorporated
into the polymer matrix, vitamin B12 release proceeded very slowly, with less
than 20 % of
the initially loaded amount released over 80 days. By day 96, the release
began to
accelerate and nearly complete release was obtained by day 111. This release
pattern is
typical of degradation-controlled release from hydrolytically degradable
polymers. As the
content of PEGD incorporated into the polymer matrix increased, the release
rate of vitamin
B12 increased. Cylinders containing 30 w/w% PEGD released approximately 90% of
the
vitamin B12 within 10 days, while those containing 20 w/w% PEGD reached 90%
released
by day 45, and those containing 10 w/w% PEGD reached 90% released by 92 days.
There
was little to no burst effect observed regardless of the weight percent of
PEGD in the
cylinders. Moreover, for a portion of the release period, the release of
vitamin B12 from the
- 20 -
CA 02504076 2005-04-14
cylinders containing 10 w/w% and 20 w/w% PEGD could be approximated as zero
order.
For example, a linear regression of the data from day 1 to day 100 for the
cylinders
containing 10 w/w% PEGD resulted in a correlation coefficient (R2) of 0.995,
while a linear
regression of the data from day 1 to day 20 for cylinders containing 20 w/w%
PEGD resulted
in a correlation coefficient of 0.981.
When the cylinder diameter was decreased to 1.8 mm, all other parameters kept
constant, the release of vitamin B12 increased (Figure 2). Again, a period of
release was
observed that could be approximated as constant, however, the duration of
constant release
decreased. Linear regression of the data from day 1 to day 35 resulted in a
correlation
coefficient in this case of 0.993. The rate of release, determined from the
slope of the linear
portion of the release, was roughly double (0.017 mass fraction released/day)
for the 1.8
mm diameter cylinders compared to that for the 3.5 mm diameter cylinders
(0.0089 mass
fraction released/day).
The influence of the molecular weight of the ASCP prepolymer on vitamin B12
release from cylinders containing 10 w/w% PEGD can be seen in Figure 3. The
vitamin B12
particle size was less than 100 pm and the cylinder diameter was 1.8 mm. The
release
rates are statistically equivalent for the cylinders fabricated with ASCP 1000
and ASCP
2700. For cylinders made with ASCP 3900, the initial release rate is the same
as for those
made with ASCP 1000 and ASCP 2700 up until day 10, after which release becomes
much
slower although it continues to be approximately constant.
To determine the influence of solid vitamin B12 particle size entrapped within
the
matrix on its release, cylinders were prepared using ASCP 2700 containing 10
w/w% PEGD.
The cylinders had a diameter of 1.8 mm. The results can be seen in Figure 4.
There was
no statistical difference in the release pattern of vitamin B12 with respect
to its initial particle
size in the cylinder.
The degradation rate of the networks were determined in vitro and are
displayed in
terms of the volumetric change and dry mass change with time in Figures 5 and
6,
respectively. For cylinders prepared with ASCP 1000, the network swelled to an
initial
maximum within 7 days, and the maximum obtained increased with increasing w/w%
PEGD
in the network (Figure 5A). The initial degree of swelling was small, ranging
from roughly 7
v/v% for the 10 w/w% PEGD networks to 14 v/v(Yo for the 30 w/w% PEGD networks.
The
volume of the cylinders remained constant at this initial maximum until day
135. After this
time, the cylinders began to swell markedly. The swelling behavior of networks
prepared
using varying ASCP molecular weight and 10 w/w% PEGD are shown in Figure 5B.
Again,
-21 -
CA 02504076 2005-04-14
maximal swelling is obtained within 7 days, with the ASCP 1000 and ASCP 2700
reaching
essentially the same swelling extent, while the networks containing ASCP 3900
swelled the
least.
The mass loss of the ASCP 1000 networks, on the other hand, decreased in a
continual, and apparently constant, manner (Figure 6). The rate of mass loss
was the same
regardless of the PEGD content of the cylinders, with the exception of the
cylinders
containing no PEGD. These cylinders lost mass at the same rate as those
containing
PEGD up until day 49, and then began to degrade more quickly than those
containing
PEGD. Thus, it would appear that network degradation does not play a dominant
role in
determining the rate of vitamin B12 release, and that the presence of the PEGD
in the
matrix modulates the degradation of the elastomer.
DISCUSSION
The work presented indicates that a near-linear release period can be achieved
through the co-cross-linking of an amorphous hydrophobic polymer with a
hydrophilic
polymer to entrap solid drug particles in a cylindrical geometry. The drug
loading achieved
is low (i.e. less than 5 v/v /0). The release rate is independent of the
entrapped drug analog
particle size, and of the molecular weight of the hydrophobic polymer, at
least when it is less
than that of the hydrophilic polymer. Furthermore, there is little to no burst
effect. The
method of manufacture of the delivery system results in 100% drug entrapment
efficiency,
and can be adapted to geometries other than cylindrical.
The mechanism of release has not been clearly elucidated, but possibilities
can be
inferred from the data presented. The cylinders swell to an essentially
constant volume
within the first week, which is maintained during the entire release period.
This swelling is
driven by the PEGD content of the matrix. There is only a small mass loss
during the
release period. For example, a mass loss of only approximately 15% occurred
over the 100
days of nearly constant release for the cylinders prepared with ASCP 1000 and
10
w/w%PEGD, and a mass loss of only approximately 8% over the 20 days of nearly
constant
release for the cylinders prepared with ASCP 1000 and 20 w/w% PEGD. Thus, the
degradation of the polymer, to generate a greater matrix porosity and thus an
increase in
solute diffusivity within the matrix, would seem to play only a minor role in
the release
kinetics. It has been suggested by van Dijkhuizen-Radersma et al.
(Biomaterials (2002)
23:1527-1536) who examined vitamin B12 release from poly(ethylene
glycol)/poly(butylenes
terephthalate) multiblock co-polymers, that a nearly constant release period
is a result of a
- 22-
CA 02504076 2013-04-18
_
,
vitamin B12 solubility limitation within the swollen matrix. However, if
dissolution of vitamin
B12 within the matrix was rate-limiting, then decreasing the particle size of
the vitamin B12
should have had an influence on the release rate, which was not observed in
this work.
Another possibility is that the release is driven by the osmotic pressure
generated by the
polymer enveloped vitamin B12 particles within the matrix. This release
mechanism has
been shown to be capable of generating constant release from cylindrical
devices (Amsden
et al. J. Control. Rel. (2003) 93:249-258; Gu et al., J. Control. Rel. (2005)
102:607-617;
Schirrer et al., J. Mater. Sci. (1992) 27:3424-3434). In this situation, water
is drawn into the
polymer matrix due to the osmotic activity of the solute, and the pressure
generated creates
microcracks within the matrix through which the dissolved solute is forced
out. In the
present situation, the PEGD incorporated may act to enhance the rate of water
uptake while
at the same time providing aqueous pathways for the movement of the solute to
the surface.
At present, the release mechanism is not clear, and may be due to a
combination of all the
mechanisms discussed.
- 23 -