Note: Descriptions are shown in the official language in which they were submitted.
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-1-
QUINOLINE DERIVATIVES
Backctround of the Invention
The present invention relates to novel quinoline derivatives, to intermediates
used in
their preparation, to pharmaceutical compositions containing them and to their
medicinal use.
The compounds of the present invention are agonists of serotonin 7 (5HT7)
receptors. They
are useful in treating CNS disorders, including depression and disorders that
can be treated
by modulating circadian rhythms. Examples of such disorders and conditions are
seasonal
affective disorder, bipolar disorder, jet lag, sleep disorders such as
circadian sleep rhythm
disorder, sleep deprivation, REM sleep disorders, hypersomnia, parasomnias,
sleep-wake cycle
disorders, narcolepsy, sleep disorders associated with blindness, sleep
disorders associated
with obesity, and sleep disorders associated with shift work or irregular work
schedules;
nocturnal enuresis, and restless leg syndrome.
Serotonin 7 receptors are present in the suprachiasmatic nucleus (SCN), the
brain
region that contains the biological clocks, and their activation leads to a
resetting of the clocks
as a function of dose and timing of treatment. Such a mechanistic link is
evident in numerous
paradigms - in in vitro electrophysiological studies of SCN neuronal activity,
and in light induced
changes in wheel running behavior and nighttime melatonin suppression - in
each case
activation of 5HT7 receptors having the potential to modulate both clock
function and the clock
resetting ability of light. Full agonists and partial agonists of the 5HT7
receptor therefore offer a
wide range of clinically useful therapeutics.
Glennon's article "Serotonin Receptors: Clinical Implications", Neuroscience
and
Behavioral Reviews. 14, 35-47 (1990), refers to the pharmacological effects
associated with
serotonin receptors including appetite suppression, thermoregulation,
cardiovascularihypotensive effects, sleep, psychosis, anxiety, depression,
nausea, emesis,
Alzheimer's disease, Parkinson's disease and Huntington's disease.
Summary of the Invention
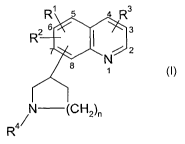
The present invention relates to compounds of the Formula
R1 5 4 R3
R2 6 ~ 7'~ 3
~C / 2
~1
/N (CH2)n
R4
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
_2_
wherein R', R~ and R3 are independently selected from hydrogen, halo, (C~-
Cs)alkyl optionally
substituted with from one to three halo (i.e., chloro, fluoro, bromo or iodo)
atoms; and (C~-
C6)alkoxy optionally substituted with from one to three halo atoms;
R4 is hydrogen or (C~-C3) alkyl; and
n is one or two;
and to the pharmaceutically acceptable salts thereof.
Compounds of Formula I and their pharmaceutically acceptable salts (also
referred to
collectively herein as "the active compounds of this invention") are potent
agonists of 5HT7
receptors.
As used herein, the non-quinoline ring refers to the ring containing the
nitrogen to
which R4 is attached, i.e.,
n
/N-(CH2)n
R4
In one embodiment, the present invention provides compounds of Formula I
wherein
n is 1. In another embodiment, the present invention provides compounds of
Formula I
wherein either R~ and R2 are both hydrogen or one of R~ and R~ is hydrogen and
the other is
attached at position 5. In another embodiment, n is 1, and either R~ and R2
are both
hydrogen or one of R~ and R~ is hydrogen and the other is attached at position
5.
In another embodiment, the invention provides compounds of Formula I wherein
the
non-quinoline ring is attached at position 7 or 8.
In another embodiment, compounds of Formula I are provided wherein n is 1, and
either R' and RZ are both hydrogen or one of R~ and R2 is hydrogen and the
other is attached
at position 5, and the non-quinoline ring is attached at position 7.
In another embodiment, the present provides compounds of Formula I having the
Formula:
R R~
R3 R3
Or
R
Ra
wherein R4, R' and R3 are defined as above.
Examples of preferred compounds of the Formula I of the invention are:
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-3-
R and S - (3-Ethyl-7-methyl-8-piperidin-3-yl-quinoline);
R, S - (3-Ethyl-7-methyl-8-piperidin-3-yl-quinoline);
R and S - (3,6-Dimethyl-8-piperidin-3-yl-qui,noline);
R, S - (3,6-Dimethyl-8-piperidin-3-yl-quinoline);
R and S - (3,7-Dimethyl-8-piperidin-3-yl-quinoline);
R, S - (3,7-Dimethyl-8-piperidin-3-yl-quinoline);
R and S - (3,5-Dimethyl-8-piperidin-3-yl-quinoline);
R and S - (3,5-Dimethyl-8-piperidin-3-yl-quinoline);
R and S - (6-Chloro-3-methyl-8-piperidin-3-yl-quinoline);
R, S - (6-Chloro-3-methyl-8-piperidin-3-yl-quinoline);
R and S - (3-Ethyl-8-piperidin-3-yl-quinoline);
R, S - (3-Ethyl-8-piperidin-3-yl-quinoline);
R and S - (4-Methyl-8-piperidin-3-yl-quinoline);
R, S - (4-Methyl-8-piperidin-3-yl-quinoline);
R and S - (3-Methyl-8-piperidin-3-yl-quinoline);
R, S - (3-Methyl-8-piperidin-3-yl-quinoline);
R and S - (3-Ethyl-8-piperidin-3-yl-quinoline);
R, S - (3-Ethyl-8-piperidin-3-yl-quinoline);
R and S - (Ethyl-7-piperidin-3-yl-quinoline);
R, S - (Ethyl-7-piperidin-3-yl-quinoline);
R and S - [3-Methyl-8-(1-methyl-piperidin-3-yl)-quinoline]; and
R, S - [3-Methyl-8-(1-methyl-piperidin-3-yl)-quinoline];
and pharmaceutically acceptable salts thereof.
Other examples of specific compounds of the Formula I of the invention are:
3-Ethyl-7-methyl-8-(1-methyl-piperidin-3-yl,)-quinoline;
3-Ethyl-8-methyl-8-(1-ethyl-piperidin-3-yl)-7-methyl-quinoline;
3,6-Dimethyl-8-(1-methyl-piperidin-3-yl)-quinoline;
8-(1-Ethyl-piperidin-3-yl)-3,6-dimethyl-quinoline;
3,7-Dimethyl-8-(1-methyl-piperidin-3-yl)-quinoline;
8-(1-Ethyl-piperidin-3-yl)-3,7-dimethyl-quinoline;
3,5-Dimethyl-8-(1-methyl-piperidin-3-yl)-quinoline;
8-(1-Ethyl-7-piperidin-3-yl)-3,5-dimethyl-quinoline;
6-Chloro-3-methyl-8-(1-methyl-piperidin-3-yl)-quinoline;
6-Chloro-8-(1-ethyl-piperidin-3-yl)-3-methyl-quinoline;
3-Ethyl-8-(1-methyl-piperidin-3-yl)-quinoline;
3-Ethyl-8-(1-ethyl-piperidin-3-yl)-quinoline;
4-Methyl-8-(1-methyl-piperidin-3-yl)-quinoline;
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-4-
8-(1-Ethyl-piperidin-3-yl)-4-methyl-quinoline;
3-Methyl-8-(1-methyl-piperidin-3-yl)-quinoline;
8-(1-Ethyl-piperidin-3-yl)-3-methyl-quinoline;
3-Ethyl-8-(1-methyl-pyrrolidin-3-yl)-quinoline;
3-Ethyl-8-(1-ethyl-pyrrolidin-3-yl)-quinoline;
3-Ethyl-7-(1-methyl-piperidin-3-yl)-quinoline;
3-Ethyl-7-(1-ethyl-piperidin-3-yl)-quinoline;
3-Ethyl-7-pyrrolidin-3-yl)-quinoline;
3-Ethyl-7-(1-methyl-pyrrolidin-3-yl)-quinoline; and
3-Ethyl-7-(1-ethyl-pyrrolidin-3-yl)-quinoline;
and pharmaceutically acceptable salts thereof.
The present invention also provides a pharmaceutical composition comprising a
therapeutically effective amount of a compound of Formula I, or a
pharmaceutically
acceptable salt thereof, and a pharmaceutically acceptable carrier.
The present invention also provides a method for treating a disorder or
condition that
can be treated by modulating serotonergic neurotransmission in a mammal,
comprising
administering to a mammal requiring such treatment a serotonin 7 receptor
agonizing
effective amount of a compound of Formula I or a pharmaceutically acceptable
salt thereof.
The present invention also provides a pharmaceutical composition for treating
a
condition or disorder that can be treated by modulating serotonergic
neurotransmission in a
mammal, comprising:
a) a pharmaceutically acceptable carrier;
b) an amount of a first compound of Formula I or a pharmaceutically acceptable
salt thereof; and
c) an amount of a second compound selected from the group consisting of a
5HT reuptake inhibitor, a 5HT7 receptor antagonist or a NK1 receptor
antagonist or a
pharmaceutically acceptable salt thereof;
wherein the amounts of (b) and (c) are together effective in treating such
disorder or
condition.
The present invention also provides a method for treating a disorder or
condition that
can be treated by modulating serotonergic neurotransmission in a mammal,
comprising
administering to a mammal requiring such treatment:
a) an amount of a compound of Formula I or a pharmaceutically acceptable salt
thereof; and
b) an amount of a second compound selected from the group consisting of 5HT
reuptake inhibitor, a 5HT7 receptor antagonist and an NI<1 receptor antagonist
or
pharmaceutically acceptable salt thereof;
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-5-
wherein the amounts of (a) and (b) are together effective in treating such
disorder or
condition.
The present invention also provides a method for treating a disorder or
condition
selected from depression, anxiety, avoidant personality disorder, premature
ejaculation,
eating disorders, migraine, premenstrual syndrome, premenstrual dysphoric
disorder,
seasonal affective disorder, bipolar disorder, jet lag, sleep disorder,
nocturnal enuresis, and
restless leg syndrome in a mammal, comprising administering to a mammal in
need of such
treatment an amount of a compound of Formula I, or a pharmaceutically
acceptable salt
thereof, which amount is (a) effective in treating such disorder or condition,
or (b) effective in
agonizing 5HT7 receptors.
In different embodiments of the methods described in the preceding paragraphs,
the
sleep disorder is circadian sleep rhythm disorder, sleep deprivation, REM
sleep disorder,
hypersomnia, parasomnia, sleep-wake cycle disorder, sleep disorder associated
with blindness,
steep disorder associated with obesity, narcolepsy or sleep disorder
associated with shift work
or irregular work schedules.
The present invention also provides a method of treating a disorder or
condition
selected from depression, anxiety, avoidant personality disorder, premature
ejaculation,
eating disorders, migraine, premenstrual syndrome, premenstrual dysphoric
disorder,
seasonal affective disorder, bipolar disorder, jet lag, sleep disorder,
nocturnal enuresis, and
restless leg syndrome in a mammal, comprising administering to a mammal
requiring such
treatment: (a) and amount of a first compound of Formula I or pharmaceutically
acceptable salt
thereof; and (b) an amount of a second compound selected from the group
consisting of a 5HT7
receptor antagonist, a NK1, receptor antagonist and an a 5HT7 receptor
antagonist or
pharmaceutically acceptable salts of said second compound; wherein the amounts
of (a) and
(b) are together effective in treating such disorder or condition.
In different embodiments of the method described in the preceding paragraph,
the
sleep disorder is circadian sleep rhythm disorder, sleep deprivation, REM
sleep disorder,
hypersomnia, parasomnia, sleep-wake cycle disorders, sleep disorder associated
with
blindness, sleep disorder associated with obesity, narcolepsy, or sleep
disorder associated with
shift work or irregular work schedules.
The present invention also provides compounds of the Formula
R2 R~ s
R
~~ J
Br v 'N
XI I
and compounds of the Formula
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-6-
R2 R~ s
R
~~ J
~N
Br
wherein for each of the above two Formulae R', RZ and R3 are independently
selected from
hydrogen, halo, (C~-C6)alkyl optionally substituted with from one to three
halo atoms; and (C~-
C6)alkoxy optionally substituted with from one to three halo atoms. Compounds
of these two
Formulae are useful as intermediates for synthesizing compounds of Formula I.
The present invention also provides a method for synthesizing a compound of
the
Formula
R2 R~ a
R
~~ J
Br v N
XI I
wherein R~, RZ and R3 are independently selected from hydrogen, halo, (C~-
C6)alkyl optionally
substituted with from one to three halo atoms; and (C~-C6)alkoxy optionally
substituted with
from one to three halo atoms;
which method comprises reacting a compound of the Formula
R2 R~
Br NH2
X
wherein R~ and R~ are as recited above,
with a compound of the Formula
R3 C-CHO
CH
XI
wherein R3 is as recited above,
or with a compound
R3 O H
wherein R3 is as recited above,
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-7-
wherein said reaction is in the presence of an aqueous acid and 3-
nitrobenzenesulfonic acid or a salt thereof, and wherein said reaction is at a
temperature of
from about 100°C to about 140°C.
The present invention also provides a method for synthesizing a compound of
the
Formula
R2 R~ a
R
~~ J
~N
Br
wherein R', R~ and R~ are independently selected from hydrogen, halo, (C~-
C6)alkyl optionally
substituted with from one to three halo atoms; and (C~-C6)alkoxy optionally
substituted with
from one to three halo atoms;
which method comprises reacting a compound of the Formula
R2 R~
NH2
r
wherein R' and RZ are as recited above,
with a compound of the Formula
R3 C-CHO
CH
XI
wherein R3 is as recited above,
or, preferably, with a compound
R3 OH
wherein R3 is as recited above,
wherein said reaction is in the presence of an aqueous acid and 3
nitrobenzenesulfonic acid or a salt thereof, and wherein said reaction is at a
temperature of
from about 100°C to about 140°C.
In either of the above-described synthetic methods, the aqueous acid is in one
embodiment sulfuric acid.
Compounds of Formula I may contain chiral centers and therefore may exist in
different enantiomeric and diastereomeric forms. This invention relates to all
optical isomers
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
_g_
and all stereoisomers of compounds of the Formula I, both as racemic mixtures
and as
individual enantiomers and diastereoisomers of such compounds, and mixtures
thereof, and
to all pharmaceutical compositions and methods of treatment defined above that
contain or
employ them, respectively. Individual isomers can be obtained by known
methods, such as
optical resolution, optically selective reaction, or chromatographic
separation in the
preparation of the final product or its intermediate. Individual enantiomers
of the compounds
of Formula I may have advantages, as compared with the racemic mixtures of
these
compounds, in the treatment of various disorders or conditions.
Insofar as the compounds of Formula I of this invention are basic compounds,
they
are capable of forming a wide variety of different salts with various
inorganic and organic
acids which are used to prepare the pharmaceutically acceptable acid addition
salts of the
aforementioned base compounds of this invention are those which form non-toxic
acid
addition salts, i.e., salts containing pharmaceutically acceptable anions,
such as the
hydrochloride, hydrobromide, hydroiodide, nitrate, sulfate or bisulfate,
phosphate or acid
phosphate, acetate, lactate, citrate or acid citrate, tartrate or bi-tartrate,
succinate, maleate,
fumarate, gluconate, saccharate, benzoate, methanesulfonate, ethanesulfonate,
benzenesulfonate, p-toluenesulfonate and pamoate (i.e., 1,1'-methylene-bis-(2-
hydroxy-3-
naphthoate))salts.
The present invention also includes isotopically labelled compounds, which are
identical to those recited in Formula I, but for the fact that one or more
atoms are replaced by
an atom having an atomic mass or mass number different from the atomic mass or
mass
number usually found in nature. Examples of isotopes that can be incorporated
into
compounds of the present invention include isotopes of hydrogen, carbon,
nitrogen, oxygen,
phosphorous, sulfur, fluorine and chlorine, such as ZH, 3H,'3C,
"C,'4C,'SN,'$O, "O, 3'P, 32P,
35S, '$F, and 36CI, respectively. Compounds of the present invention, prodrugs
thereof, and
pharmaceutically acceptable salts of said compounds or of said prodrugs which
contain the
aforementioned isotopes and/or other isotopes of other atoms are within the
scope of this
invention. Certain isotopically labelled compounds of the present invention,
for example
those into which radioactive isotopes such as 3H and'4C are incorporated, are
useful in drug
and/or substrate tissue distribution assays. Tritiated, i.e., 3H, and carbon-
14, i.e., '4C,
isotopes are particularly preferred for their ease of preparation and
detectability. Further,
substitution with heavier isotopes such as deuterium, i.e., ~H, can afford
certain therapeutic
advantages resulting from greater metabolic stability, for example increased
in vivo half-life or
reduced dosage requirements and, hence, may be preferred in some
circumstances.
Isotopically labelled compounds of Formula I of this invention and prodrugs
thereof can
generally be prepared by carrying out the procedures disclosed in the Schemes
and/or in the
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
_g_
Examples below, by substituting a readily available isotopically labelled
reagent for a non-
isotopically labelled reagent.
"Serotonin" and "5HT7" are used interchangeably herein, unless otherwise
indicated.
"Serotonin 7 agonists are useful for the treatment of depression.
As used herein, the term "depression" includes major depressive disorder;
single
episode or recurrent major depressive episodes; recurrent depression;
dysthymia; cyclothymia;
depressive disorders not otherwise specified; seasonal affective disorder; and
bipolar
disorders, for example, bipolar I disorder, bipolar II disorder and bipolar
disorder not otherwise
specified.
Other mood disorders encompassed within the term "depression", as used herein,
include dysthymic disorder with early or late onset and with or without
atypical features;
dementia of the Alzheimer's type, with early or late onset, with depressed
mood; vascular
dementia with depressed mood; mood disorders induced by alcohol, amphetamines,
cocaine,
hallucinogens, inhalants, opioids, phencyclidine, sedatives, hypnotics,
anxiolytics or other
substances; schizoaffective disorder of the depressed type; and adjustment
disorder with
depressed mood.
Encompassed within the term "depression", as used herein, are: depression in
cancer
patients, depression in Parkinson's patients, postmyocardial infarction
depression,
subsyndromal symptomatic depression, depression in infertile women, pediatric
depression,
child abuse induced depression, and post partum depression.
Major depression is characterized by feelings of intense sadness and despair,
mental
slowing and loss of concentration, pessimistic worry, agitation, and self-
deprecation. Physical
changes also occur, especially in severe or "melancholic" depression. These
include
insomnia or hypersomnia, anorexia and weight loss (or sometimes overeating),
decreased
energy and libido, and disruption of normal circadian rhythms of activity,
body temperature,
and many endocrine functions.
The Serotonin 7 agonists of Formula I of the invention are also useful for the
treatment
of anxiety. As used herein, the term "anxiety" includes anxiety disorders,
such as panic
disorder with or without agoraphobia, agoraphobia without history of panic
disorder, specific
phobias, for example, specific animal phobias, social phobias, obsessive-
compulsive
disorder, stress disorders including post-traumatic stress disorder and acute
stress disorder,
and generalized anxiety disorders.
"Generalized anxiety" is typically defined as an extended period (e.g., at
least six
months) of excessive anxiety or worry with symptoms on most days of that
period. The
anxiety and worry is difficult to control and may be accompanied by
restlessness, being easily
fatigued, difficulty concentrating, irritability, muscle tension, and
disturbed sleep.
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-10-
"Panic disorder" is defined as the presence of recurrent panic attacks
followed by at
least one month of persistent concern about having another panic attack. A
"panic attack" is
a discrete period in which there is a sudden onset of intense apprehension,
fearfulness or
terror. During a panic attack, the individual may experience a variety of
symptoms including
palpitations, sweating, trembling, shortness of breath, chest pain, nausea and
dizziness.
Panic disorder may occur with or without agoraphobia.
"Phobias" includes agoraphobia, specific phobias and social phobias.
"Agoraphobia"
is characterized by an anxiety about being in places or situations from which
escape might be
difficult or embarrassing or in which help may not be available in the event
of a panic attack.
Agoraphobia may occur without history of a panic attack. A "specific phobia"
is characterized
by clinically significant anxiety provoked by a feared object or situation.
Specific phobias
include the following subtypes: animal type, cued by animals or insects;
natural environment
type, cued by objects in the natural environment, for example storms, heights
or water; blood-
injection-injury type, cued by the sight of blood or an injury or by seeing or
receiving an
injection or other invasive medical procedure; situational type, cued by a
specific situation
such as public transportation, tunnels, bridges, elevators, flying, driving or
enclosed spaces;
and other type, where fear is cued by other stimuli. Specific phobias may also
be referred to
as simple phobias. A "social phobia" is characterized by clinically
significant anxiety provoked
by exposure to certain types of social or performance circumstances. Social
phobia may also
be referred to as social anxiety disorder.
Other anxiety disorders encompassed within the term "anxiety" include anxiety
disorders induced by alcohol, amphetamines, caffeine, cannabis, cocaine,
hallucinogens,
inhalants, phencyclidine, sedatives, hypnotics, anxiolytics and other
substances, and
adjustment disorders with anxiety or with mixed anxiety and depression.
Anxiety may be present with or without other disorders, such as depression in
mixed
anxiety and depressive disorders. The compositions of the present invention
are therefore
useful in the treatment of anxiety with or without accompanying depression.
The term "alkyl", as used herein, unless otherwise indicated, includes
saturated
monovalent hydrocarbon radicals having straight, branched or cyclic moieties
or combinations
thereof. Examples of "alkyl" groups include, but are not limited to, methyl,
ethyl, propyl,
isopropyl, butyl, iso- sec- and tert-butyl, pentyl, hexyl, heptyl, 3-
ethylbutyl, cyclopropyl,
cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, norbornyl, and the like.
The term "alkoxy", as used herein, unless otherwise indicated, means "alkyl-O-
",
wherein "alkyl" is as defined above. Examples of "alkoxy" groups include, but
are not limited
to, methoxy, ethoxy, propoxy, butoxy and pentoxy.
The term "alkenyl", as used herein,.unless otherwise indicated, includes
unsaturated
hydrocarbon radicals having one or more double bonds connecting two carbon
atoms,
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-11-
wherein said hydrocarbon radical may have straight, branched or cyclic
moieties or
combinations thereof. Examples of "alkenyl" groups include, but are not
limited to, ethenyl,
propenyl, butenyl, pentenyl, and dimethylpentyl, and include E and Z forms
where applicable.
The term "aryl", as used herein, unless otherwise indicated, includes an
aromatic ring
system with no heteroatoms, which can be either unsubstituted or substituted
with one, two or
three substituents selected from the group consisting of halo, (C~-C4)alkyl
optionally
substituted with from one to three fluorine atoms and (C~-CQ)alkoxy optionally
substituted with
from one to three fluorine atoms.
The term "heteroaryl", as used herein, unless otherwise indicated, includes an
aromatic heterocycle containing five or six ring members, of which from 1 to 4
can be
heteroatoms selected, independently, from N, S and O, and which rings can be
unsubstituted,
monosubstituted or disubstituted with substituents selected, independently,
from the group
consisting of halo, (C~-C4)alkyl, and (C~-C4)alkoxy, optionally substituted
with from one to
three fluorine atoms;
The term "one or more substituents", as used herein, refers to a number of
substituents that equals from one to the maximum number of substituents
possible based on
the number of available bonding sites.
The terms "halo" and "halogen", as used herein, unless otherwise indicated,
include,
fluoro, chloro, bromo and iodo.
The term "treating", as used herein, refers to reversing, alleviating,
inhibiting the
progress of, or preventing the disorder or condition to which such term
applies, or preventing
one or more symptoms of such condition or disorder. The term "treatment", as
used herein,
refers to the act of treating, as "treating" is defined immediately above.
"Modulating serotonergic neurotransmission," as used herein, refers to
increasing or
improving, or decreasing or retarding the neuronal process whereby serotonin
is released by
a pre-synaptic cell upon excitation and crosses the synapse to stimulate or
inhibit the post
synaptic cell.
Unless indicated to the contrary, when used herein the term "active compounds"
and
"active agents" are synonymous and are therefore interchangeable. This term
refers to the
compounds of Formula I or its pharmaceutically acceptable salts thereof either
alone or in
combination with one or more of the compounds selected from the group
consisting of 5HTID
receptor antagonists, NK1 receptor antagonists, 5HT7 receptor antagonists or
pharmaceutically acceptable salts of any of the compounds identified herein.
Detailed Description of the Invention
Compounds of Formula I may be prepared according to the following reaction
schemes and discussion. Unless otherwise indicated, R', R2, R3' R4 and n, and
structural
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-12-
Formulae I through IX in the reaction schemes and discussion that follow are
as defined
above.
SCHEME 1
R2 R~
I R3 II CHO
Br / NH2 CH2
X XI
S03Na+
/I
H2S04/H20/heat
N 02
R2 R~
R
~~ J
Br N
XII
H3C~ B~CH3
Pd(PPh3)2CI2
Na2C03, heat ~ /
\ N
R2 R~
I \ R3
\ \
_N
i
N
XIII
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-13-
SCHEME 1 CONTIUNED
R2 R1 R1 R~
\ 3 I-I+B(C2Hs)sH ~ \~R3
~R
THF \ N
N~ XIV
XIII H
di-t-butyl N(Et)3
dicarbonate I CHZCI2
R3
R3
chiral column
preparative
chromatography
~\
p OC(CH3)s /
_ p / OC(CH3)s
separate enantiomers of
(racemic)
HCI
methanol
CH2CI2 R1
R2 ~ \
R3
v ~N
N
H ~ HCI
Separate enantiomers of
IA
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-14-
SCHEME 2
2 R2
/ R H3C~ B~CH3
R
R \~I \ \
~N02 I ~ _NOa
Br ~ N
THF I
XVI
Pd(PPh3)2CI2
Na2C03, heat XVII
' 2 H
R Pt02 2
R~ / I CH30H
\ NH 50 psi
2
Li+ R2
NH -BCH H ~ / I
( 2 5)3 R
XIX THF \ NH2
O
l
R3 CHO \ N
H2SO4 XVIII
- H20
Na+ / S03 R3
\ I R2 \
. II J
N02 R~ Y ~N
NH
IB
(racemic)
CA 02505873
2005-05-11
WO 2004/043929 PCT/IB2003/004903
-15-
SCHEME 2
CONTINUED
3 R3
R 2
R
R~
\
R~ N
~ ~N di-tert-butyl-
R dicarbonate,
triethylamine
N
O
t
Bu
NH CH2C12 I I
O
IB
(racemic)
(racemic)
chiral
column
preparative
chromatography
R3
Rs~ R2
2 \
R
\ i
~ ~
R~ ~ R~ N
i HCI
\
~N
methanol I
CH2CI2 N II
O t
BU
NH HCI O
Separate enantiomers Separate
of enantiomers
of
IB
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-16-
SCHEME 3
R~ Na2C03
Pd(PPh3)2CI2 R2
R' I THF/H~O
w
NHS
R~ _NH2
Br H3C.~B~CH3
\ \
XXI
i ~ ~H
N~
XVIII
Na+ S03
H2S04
H20 N02
R3\ /CHO
~CH
R3
Li+ -B(CH2H5)sH
R
IC XXII
(racemic)
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-17-
SCHEME 3 CONTINUED
R2 R3
R
IC
(racemic)
R2 R3 R2 Rs
1 ~ \ \
R
\ N
R1 ~ ~N
N-C-O-tB a t
~N-C-O-t-Bu
O IO
Separate enantiomers of XXI
XXI (racemic)
R2 R3
1
R
\ N
1
NH ~ HCI
Separate enantiomers of
IC
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-18-
SCHEME 4
R1 R1
\ \ 3 n-BuLi ~ I \ Rs
~R THF \ N
N -77°C
Br HO
IV(A) O N-C-Ot-Bu
II XXIII
N-C-O-tBu
1 ) H2S04
2) basic work -up
1 R1
3
i R Pt0 CH OH \ ~ I ~ Rs
\ N z/ s N
H2
50 psi H
NH ' NH
ID
(racemic) XXIV
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-19-
SCHEME 5
R~
H-C-R4
CH3OH
Na+ -B[O(C=O)CH3]3H R
R4-CH3' H a
N-R
n-1,2
ID'(n=1)
I D (n=1 )
IE (n=2) IE' (n=2)
Scheme 1 illustrates the synthesis of compounds of the Formula I wherein R4 is
hydrogen, n is two, and the saturated nitrogen containing ring is piperidin-3-
yl and is attached
to position "7" of the quinoline nucleus. Referring to Scheme 1, a compound of
Formula X is
reacted with a compound of the Formula XI and 3-nitrobenzenesufonic acid or
salt thereof,
such as the Group IA salt, e.g., sodium salt thereof in aqueous acid, e.g.,
sulfuric acid, at a
temperature from about 100°C, to about 140°C, preferably at
110°C, to form the
corresponding quinoline derivative of Formula XII. An alcohol, R3-OH, may be
used instead
of the reactant XI. Reaction of the resulting compound of Formula XII with
palladium
triphenylphosphine dichloride and diethyl (3-pyridyl) borane in the presence
of sodium
carbonate or other inorganic basesuch as potassium carbonate, calcium
carbonate, or cesium
carbonatein an organic solvent such as, tetrahydrofuran (THF), 1,4,-dioxane or
1,2,-
dichloroethane, and the like, preferably THF, at a temperature ranging from
about 80°C to
about 120°C, preferably at about 90°C, yields the corresponding
compound of Formula XIII.
Reduction of the pyridine derivative of Formula XIII using lithium
triethylborohydride in
tetrahydrofuran (THF) yields the corresponding piperidine derivative of
Formula XIV. This
reaction is typically carried out at a temperature from about 0°C to
about 70°C, preferably at
about room temperature. Alternate reducing agents and solvents can be used.
These are
well known to those of ordinary skill in the art (e.g., lithium tri-
isobutylborohydride, lithium
triphenyl borohydride, and the like.). Lithium triphenyl borohydride in THF is
preferred.
The piperidine derivative of Formula XIV is then converted into the
corresponding
ester of Formula XV by reacting it with di-t-butyldicarbonate in the presence
of a tertiary
amine base such as triethylamine, 4-methyl morpholine, or DBU (1,8 -
diazabicyclo[5.4Ø ]
under - 7 -ene preferably triethylamine. Suitable solvents for this reaction
include chloro
alkanes (e.g., methylene chloride), 1,4-dioxane, THF and 1,2,-dichloroethane.
Methylene
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-20-
chloride is preferred. The reaction temperature can range from about 0
°C to about 50°C, and
is preferably about 25°C.
Chiral column chromatography can be used to separate the enantiomers that
comprise the racemic compound of Formula XV. Each enantiomer ester can then be
deprotected using methods well known to those of skilled in the art, for
example, by hydrolysis
with a strong acid such as hydrochloric acid, sulfuric acid, acetic acid or
trifluoroacetic acid, in
a solvent such as methanol, methylene chloride, dioxane, ethyl ether, or ethyl
acetate, to form
the corresponding enantiomeric acid salt of a compound of the Formula IA.
Preferably,
hydrochloric acid is used. This reaction is typically carried out at a
temperature from about
0°C to about 70°C, and is preferably carried out at about room
temperature.
Scheme 1 can also be used to prepare compounds identical to those of Formula
IA
but for the piperidine ring is being attached to the quinoline nucleus at
position "8". This can
be accomplished by replacing the starting material of Formula X with the
analogous
compound wherein the bromo and amino groups are ortho to each other. In
synthesizing
intermediates analogous to intermediate XII having the bromine in position "8"
according to
Scheme 1, use of an alcohol R3-OH instead of a compound of formula XI is
preferred.
Scheme 2 illustrates the synthesis of compounds of the Formula I wherein n is
2, R4
is hydrogen, R3 is attached to position "4" of the quinoline nucleus, and the
saturated nitrogen
containing ring of Formula I is a piperidin-3-yl ring that is attached to
position "8" of such
nucleus. Referring to Scheme 2, a compound of~the Formula XVI is reacted with
palladium
triphenylphosphine dichloride and [diethyl (3-pyridyl) borane] in the presence
of an inorganic
base, e.g., metal carbonates such as sodium carbonate potassium carbonate,
calcium
carbonateor cesium carbonate, and the like, preferably sodium carbonate, in an
organicsolvent such as THF, 1,4,-dioxaneor 1,2,-dichloroethane, preferably
THF, at a
temperature from about 80 °C to about 120°C, preferably at about
90°C, to form the
corresponding compound of Formula XVII. Reduction of the nitrobenzene
derivative of
Formula XVII yields the corresponding aniline derivative of Formula XVIII.
This reduction can
be accomplished using methods well known to those of skill in the art, e.g.,
reaction with
hydrogen gas at a pressure of 50 psi, in a methanol solvent, in the presence
of a platinum
oxide catalyst. This reaction is typically conducted at a temperature from
about zero to about
40°C, and is preferably conducted at about room temperature.
The resulting pyridine derivative of Formula XVIII is then reduced to form the
corresponding piperidine derivative of Formula XIX using the methods described
above, in the
description of the reactions in Scheme I, for reducing compounds of the
Formula XIII. , The
desired compound of Formula IB can be prepared by reacting the compound of
Formula XIX
with a compound of the Formula R3(C=CH)CHO and 3-nitrobenzenesulfonic acid
sodium salt
or ferric chloride hexahydrate and zinc chloride in an organic solvent such as
ethanol, n-
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-21-
propanol, isopropanoland an acid, e.g., inorganic acid, such as sulfuric acid
or hydrochloric
acid or in an aqueous inorganic acid, such as, or aqueous sulfuric acid, and
more preferably
aqueous hydrochloric acid, at a temperature from about 60°C, to about
100°C, preferably at
about 60 °C. The racemic compound of Formula IB can be separated into
its enantiomers as
illustrated in Scheme 2 and described above for the preparation of the
enantiomers of
compounds of the Formula IA.
Scheme 3 illustrates the preparation of compounds of the Formula I wherein n
is 2, R4
is hydrogen, R3 is attached to position "3",of the quinoline nucleus, and the
saturated nitrogen
containing ring of Formula I is a piperidin-3-yl ring that is attached to
position "8" of such
nucleus. Referring to Scheme 3, the compound of Formula XXI is reacted with
palladium
triphenylphosphine dichloride and [diethyl (3-pyridyl) borane] in the presence
of sodium
carbonate or another inorganic base such as potassium carbonate, calcium
carbonate or
cesium carbonatein an organic solvent such as tetrahydrofuran (THF), 1,4,-
dioxaneor 1,2,-
dichloroethane, preferably THF, at a temperature from about 80 °C to
about 120°C, preferably
at about 90°C, to form the corresponding compound of Formula XVIII.
The compound of Formula XVIII is then reacted with a compound of the Formula
R3(C=CH)CHO and sodium [3-nitrobenzenesulfonic acid or salt thereof,
especially metal salts
thereof, such as the sodium salt or ferric chloride hexahydrate and zinc
chloride] in an organic
solvent such as an alcohol of 1-6 carbon atoms, e.g., ethanol, n-propanol,
isopropanol, and
an acid, e.g., inorganic acid, e.g., aqueous inorganic acid, such as
hydrochloric acid, sulfuric
acid, preferably aqueous hydrochloric acid and more preferably aqueous,
sulfuric acid, at a
temperature from about 100°C, to about 140°C, preferably at
about 110 °C, to form the
racemic pyridine substituted quinoline derivative of Formula XXII. Reduction
of the pyridine
substituted quinoline derivative of Formula XXII using lithium
triethylborohydride in
tetrahydrofuran (THF) yields the corresponding piperidine substituted
quinoline derivative of
Formula IC. This reaction is typically carried out at a temperature from about
0°C to about
70°C, preferably at about 25°C. Alternate reducing agents and
solvents can be used. These
are well known to those of ordinary skill in the art.
The racemic compound of Formula IC can be separated into its enantiomers as
illustrated in Schemes 1, 2 and 3 and described above for the preparation of
the enantiomers
of compounds of the Formula IA.
Scheme 4 illustrates the syntheses of compounds of the Formula I wherein n is
1, R4
and RZ are hydrogen, and R' is attached to position "5" of the quinoline
nucleus. Referring to
Scheme 4, the compound of Formula IVA is reacted with [3-oxo-pyrrolidine-1-
carboxylic acid
tent-butyl ester] and n-butyl lithium in an organic solvent such as
tetrahydrofuran (THF),
diethyl ether or 1,4-dioxane, preferably THF, at a temperature from about -
77°C to about
-100°C, preferably at about -77°C, to form the corresponding
compound of Formula XXIII.
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
_22_
The addition of acid thereto followed by a basic workup hydrolyzed the amide
to form the
pyrrolene XXIV. The resulting compound of Formula XXIV can be reduced to form
the
corresponding racemic compound of Formula ID. This reduction can be
accomplished using
methods well known to those of ordinary skill in the art, e.g., reaction with
hydrogen gas at a
pressure of 50 psi, in a solvent such as acetic acid, methanol or ethanol, in
the presence of a
platinum oxide catalyst. Acetic acid is preferred for reduction of the free
base pyrrolene to the
free base pyrrolidine, while methanol or ethanol is preferred for reducing the
hydrochloride
salt of the pyrrolene to the hydrochloride salt of the pyrrolidine. This
reaction is typically
conducted at a temperature from about zero to about 40°C, and is
preferably conducted at
about room temperature.
Scheme 4 can also be used to prepare compounds identical to those of Formula
ID
but for the fact that the pyrrolidine ring is attached to the quinoline
nucleus at position "7".
This can be accomplished by replacing the starting material of Formula IVA
with the
analogous compound wherein the bromo group is attached to the quinoline ring
at position
"7".
Scheme 5 illustrates the formation of compounds of the Formula I wherein R4 is
other
than hydrogen from the corresponding compounds of the Formula I wherein R4 is
hydrogen.
Referring to Scheme 5, the compound of Formula IE or 1 F is reacted with a
compound of the
Formula HC(=O)R4 and an alkylating agent such as sodium triacetoxyborohydride,
sodium
cyanoborhydride and the like in an organic solvent especially an alcohol such
as ethanol, or
methanol, preferably methanol, at a temperature from about 0°C to about
30°C, preferably
about 25°C, to form the corresponding compound of Formula IE' or IF',
respectively. The
procedure illustrated in Scheme 5 can be used generally to convert compounds
of the
Formula I wherein R4 is other than hydrogen into the corresponding compounds
wherein R4 is
hydrogen.
The racemic compounds of Formula ID, ID', IE and IE" can be separated into its
enantiomers as illustrated in Schemes 1, 2 and 3 and described above for the
preparation of
the enantiomers of compounds of the Formula IA.
Unless indicated otherwise, the pressure of each of the above reactions is not
critical.
Generally, the reactions will be conducted at a pressure of about one to about
three
atmospheres, preferably at ambient pressure (about one atmosphere).
The compounds of Formula I that are basic in nature are capable of forming a
wide
variety of different salts with various inorganic and organic acids. Although
such salts must
be pharmaceutically acceptable for administration to animals, it is often
desirable in practice
to initially isolate a compound of the Formula I from the reaction mixture as
a
pharmaceutically unacceptable salt and then simply convert the latter back to
the free base
compound by treatment with an alkaline reagent, and subsequently convert the
free base to a
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-23-
pharmaceutically acceptable acid addition salt. The acid addition salts of the
base
compounds of this invention are readily prepared by treating the base compound
with a
substantially equivalent amount of the chosen mineral or organic acid in an
aqueous solvent
medium or in a suitable organic solvent such as methanol or ethanol. Upon
careful
evaporation of the solvent, the desired solid salt is obtained.
The acids which are used to prepare the pharmaceutically acceptable acid
addition
salts of the base compounds of this invention are those which form non-toxic
acid addition
salts, i.e., salts containing pharmacologically acceptable anions, such as
hydrochloride,
hydrobromide, hydroiodide, nitrate, sulfate or bisulfate, phosphate or acid
phosphate, acetate,
lactate, citrate or acid citrate, tartrate or bitartrate, succinate, maleate,
fumarate, gluconate,
saccharate, benzoate, methanesulfonate and pamoate [i.e., 1,1'-methylene-bis-
(2-hydroxy-3-
naphthoate)] salts.
It will be appreciated that when using any of the combination methods of the
present
invention, referred to above, whichever components (a) and (b) that are
utilized, i.e.,
whichever combination of a compound of Formula I or pharmaceutically
acceptable salt
thereof and 5HTID receptor antagonist or salt, NK1 receptor antagonist or salt
or sertonin
reuptake inhibitor or salt, the combination will be administered to a patient
within a reasonable
period of time. The compounds may be in the same pharmaceutically acceptable
carrier and
therefore administered simultaneously. They may be in separate pharmaceutical
carriers
such as conventional oral dosage forms that are taken simultaneously. The term
combination, as used above, also refers to the case where the pharmaceutically
active
compounds are provided in separate dosage forms and are administered
sequentially.
Therefore, by way of example, the NK1 receptor antagonist may be administered
as a tablet
and then, within a reasonable period of time, the compound of the Formula I
may be
administered either as an oral dosage form such as a tablet or a fast-
dissolving oral dosage
form. By a °'fast dissolving oral formulation" is meant, an oral
delivery form which when
placed on the tongue of a patient, dissolves within about seconds.
Examples of serotonin reuptake inhibitors that can be used in the methods and
compositions of this invention are sertraline, fluoxetine and paroxetine.
Sertraline, (1 S-cis)-4-
(3,4-dichlorophenyl)-1,2,3,4-tetrahydro-N-methyl-1-naphthalenamine, has the
chemical
formula C~~H~~NCIZ and the following structural formula
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-24-
NHCH3
HCI
CI
Its synthesis is described in United States Patent 4,536,518, assigned to
Pfizer Inc.,
the contents of which are incorporated by reference. Sertraline hydrochloride
is useful as an
antidepressant and anorectic agent, and is also useful in the treatment of
depression,
chemical dependencies, anxiety obsessive compulsive disorders, phobias, panic
disorder,
post traumatic stress disorder, and premature ejaculation.
Examples of NK-1 receptor antagonists that may be used in the methods and
pharmaceutical compositions of this invention are compounds of the Formula
X3
X2 IX
Q/H X~
wherein X' is hydrogen, (C~-Coo) alkoxy optionally substituted with from one
to three
fluorine atoms or (C~-Coo) alkyl optionally substituted with from one to three
fluorine atoms;
Xa and X~ are independently selected from hydrogen, halo, nitro, (C~-C~o)
alkyl
optionally substituted with from one to three fluorine atoms, (C~-Cep) alkoxy
optionally
substituted with from one to three fluorine atoms, trifluoromethyl, hydroxy,
phenyl, cyano,
amino, (C~-C6)-alkylamino, di-(C~-C6)alkylamino, -C(=O)-NH-(C~-C6)alkyl, (C~-
C6) alkyl-C(=O)-
NH-(C~-Cs) alkyl, hydroxy(C~-C4)alkyl, (C~-CQ)alkoxy(C~-C4)alkyl, -NHC(=O)H
and -NHC(=O)-
(C~-C6) alkyl; and Q is a group of the Formula
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-25-
R2
(CH2)
R Rs
N
II RIJ III
R4
R''
IV V
R
R°
Rw
VI
OR
R$
Rs (CH2)~
(CH2)~
Rs
(CH2)y~ N
R'
R~~~CH2)m
Rio
~6
VII
VIII
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-26-
wherein R' is a radical selected from furyl, thienyl, pyridyl, indolyl,
biphenyl and
phenyl optionally substituted with one or two substituents independently
selected from halo,
(C~-Cep) alkyl optionally substituted with from one to three fluorine atoms,
(C~-Coo) alkoxy
optionally substituted with from one to three fluorine atoms, carboxy,
benzyloxycarbonyl and
(C~-C3) alkoxy-carbonyl;
R~3 is selected from (C3-C4) branched alkyl, (C5-C6) branched alkenyl, (C5-C~)
cycloalkyl,
and the radicals named in the definition of R';
R2 is hydrogen or (C,-C6) alkyl;
R3 is phenyl, biphenyl, naphthyl, pyridyl, benzhydryl, thienyl or furyl, and
R3 may
optionally be substituted with from one to three substituents independently
selected from halo,
(C~-Coo) alkyl optionally substituted with from one to three fluorine atoms
and (C~-Cep) alkoxy
optionally substituted with from one to three fluorine atoms;
Y is (CH2)i wherein I is an integer from one to three, or Y is a group of the
Formula
<J)
Z is oxygen, sulfur, amino, (C~-C3)alkylamino or (CH~)~ wherein n is zero, one
or two;
o is two or three;
p is zero or one;
x is an integer from zero to four;
y is an integer from zero to four;
z is an integer from one to six, and the ring in Formula VIII containing
(CHZ)z may contain
from zero to three double bonds, and one of the carbons of said (CH~)Z may
optionally be
replaced by oxygen, sulphur or nitrogen;
R4 is furyl, thienyl, pyridyl, indolyl, biphenyl, or phenyl optionally
substituted with one or
two substituents independently selected from halo, (C~-Coo) alkyl optionally
substituted with from
one to three fluorine atoms, (C~-Cio) alkoxy optionally substituted with from
one to three fluorine
atoms, carboxy, (C~-C3) alkoxy-carbonyl and benzyloxycarbonyl;
R5 is thienyl, biphenyl or phenyl optionally substituted with one or two
substituents
independently selected from halo, (Ci-Coo) alkyl optionally substituted with
from one to three
fluorine atoms and (Ci-Cio) alkoxy optionally substituted with from one to
three fluorine atoms;
X is (CH2)q wherein q is an integer from 1 to 6, and wherein any one of the
carbon-
carbon single bonds in said (CHZ)q may optionally be replaced by a carbon-
carbon double bond,
and wherein any one of the carbon atoms of said (CH~)q may optionally be
substituted with R~,
and wherein any one of the carbon atoms of said (CH2)q may optionally be
substituted with R9;
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-27-
m is an integer from 0 to 8, and any one of the carbon-carbon single bonds of
(CH2)m
may optionally be replaced by a carbon-carbon double bond or a carbon-carbon
triple bond, and
any one of the carbon atoms of said (CH~)m may optionally be substituted with
R";
R6 is a radical selected from hydrogen, (C~-C6) straight or branched alkyl,
(C3-C~)
cycloalkyl wherein one of the carbon atoms may optionally be replaced by
nitrogen, oxygen or
sulfur; aryl selected from biphenyl, phenyl, indanyl and naphthyl; heteroaryl
selected from thienyl,
furyl, pyridyl, thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, triazolyl,
tetrazolyl and quinolyl; phenyl
(C~-C6) alkyl, benzhydryl and benzyl, wherein each of said aryl and heteroaryl
groups and the
phenyl moieties of said benzyl, phenyl (CZ-C6) alkyl and benzhydryl may
optionally be substituted
with one or more substituents independently selected from halo, nitro, (C~-
Cep) alkyl optionally
substituted with from one to three fluorine atoms, (C~-C~°) alkoxy
optionally substituted with from
one to three fluorine atoms, amino, hydroxy-(C~-C6)alkyl, (C~-C6)alkoxy-(C~-
C6)alkyl, (C~-C6)-
alkylamino, (C~-C6)alkyl-O-C(=O)-, (C~-C6) alkyl-O-C(=O)- (C~-C6)alkyl, (C~-
C6)alkyl-C(=O)-O-,
(C~-C6)alkyl-C(=O)- (C~-C6)alkyl-O-, (Ci-C6)alkyl-C(=O)-, (C~-C6)alkyl-C(=O)-
(C~-C6)alkyl-, di-(Ci-
C6)alkylamino, -C(=O)NH-(Ci-C6)alkyl,(C~-Cs)-alkyl-C(=O)-NH-(C~-C6)alkyl, -
NHC(=O)H and -
NHC(=O)-(C~-C6) alkyl; and wherein one of the phenyl moieties of said
benzhydryl may optionally
be replaced by naphthyl, thienyl, furyl or pyridyl;
R7 is hydrogen, phenyl or (C~-C~)alkyl; ,
or R6 and R', together with the carbon to which they are attached, form a
saturated
carbocyclic ring having from 3 to 7 carbon atoms wherein one of said carbon
atoms may
optionally be replaced by oxygen, nitrogen or sulfur;
R$ and R9 are each independently selected from hydrogen, hydroxy, halo, amino,
oxo
(=O), nitrite, hydroxy-(C1-C6)alkyl, (C~-Cs)alkoxy-(C~-C6)alkyl, (C~-
C6)alkylamino, di-(C~-
C6)alkylamino, (C~-C~)alkoxy, (C~-C6)alkyl-O-C(=O)-, (Ci-C6)alkyl-O-C(=O)-(C~-
C6)alkyl, (C~-
C6)alkyl-C(=O)-O-, (C~-C6)alkyl-C(=O)-(C~-C6)alkyl-O-, (C~-C6)alkyl-C(=O)-,
(C~-C6)alkyl-C(=O)-
(C~-C6)alkyl-, and the radicals set forth in the definition of R6;
R'° is NHCR'~, NHCHZR'Z, NHSO~R'2 or one of the radicals set forth in
any of the
definitions of R6, R8 and R9;
R" is oximino (=NOH) or one of the radicals set forth in any of the
definitions of Rs, Ra
and R9; and
R'Z is (C~-C6)alkyl, hydrogen, phenyl(C~-C6)alkyl or phenyl optionally
substituted with
(C~-C6) alkyl; and
with the proviso that (a) when m is 0, R" is absent, (b) neither R8, R9,
R'° nor R" can
form, together with the carbon to which it is attached, a ring with R', (c)
when Q is a group of the
Formula VIII, R$ and R9 cannot be attached to the same carbon atom, (d) when
Ra and R9 are
attached to the same carbon atom, then either each of R$ and R9 is
independently selected from
hydrogen, fluoro, (C~-C6) alkyl, hydroxy-(C~-C6)alkyl and (C~-C6)alkoxy-(C~-
C6)alkyl, or R$ and R9,
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
_28_
together with the carbon to which they are attached, form a (C3-C6) saturated
carbocyclic ring
that forms a spiro compound with the nitrogen-containing ring to which they
are attached, (e)
when neither X~, X~ nor X3 is a fluorinated alkoxy group, at least one of R~,
R3, R4, R5, R6, R' and
R'3 is an aryl group substituted with a fluorinated alkoxy group;
and the pharmaceutically acceptable salts thereof.
Additional examples include the following compounds (hereinafter referred to,
collectively, as "the Group A compounds"):
(2S,3S)-3-(6-methoxy-3-trifluoromethyl-1,3-dihydroisobenzofuran-5-
yl)methylamino-2-
phenylpiperidine;
(2S,3S)-3-(6-methoxy-1-methyl-1-trifluoromethylisochroman-7-yl)methylamino-2-
phenylpiperidine;
(2S,3S)-3-(6-methoxy-3-methyl-3-trifluoromethyl-1,3-dihydroisobenzofuran-5-
yl)methylamino-2-phenylpiperidine;
(2S,3S)-3-(6-methoxy-3-phenyl-3-trifluoromethyl-1,3-dihydroisobenzofuran-5-
yl)methylamino-2-phenylpiperidine;
(2S,3S)-3-[1-(6-methoxy-3-methyl-3-trifluoromethyl-1,3-dihydroisobenzofuran-5-
yl)ethylamino]-2-phenylpiperidine;
(2S,3S)-3-[(1 R)-6-methoxy-1-methyl-1-trifluoromethylisochroman-7-
yl]methylamino-2-
phenylpiperidine;
(2S,3S)-3-[(3R)-6-methoxy-3-methyl-3-trifluoromethyl-1,3-dihydroisobenzofuran-
5-
yl)methylamino-2-phenylpiperidine;
(2S,3S)-N-(5-ethyl-2-methoxyphenyl)methyl-2-diphenylmethyl-1-azabi-
cyclo[2.2.2]-
octan-3-amine;
(2S,3S)-N-(5-isopropyl-2-methoxyphenyl)methyl-2-d i-phenylmethyl-1-
azabicyclo[2.2.2]-octan-3-amine;
(2S,3S)-N-(5-sec-butyl-2-methoxyphenyl)-methyl-2-diphenylmethyl-1-
azabicyclo[2.2.2]-octan-3-amine;
(2S,3S)-N-(5-tert-butyl-2-methoxyphenyl)-methyl-2-diphenylmethyl-1-
azabicyclo[2.2.2]-octan-3-amine; and
(2S,3S)-N-(5-methyl-2-methoxyphenyl)methyl-2-diphenylmethyl-1-
azabicyclo[2.2.2]-
octan-3-amine;
and pharmaceutically acceptable salts thereof.
Preferred methods of this invention include the above combination methods
wherein the
an NI<1 receptor antagonist that is employed in such method is a compound of
the Formula IX
wherein R', R4, R5 and R' are phenyl, R~ is hydrogen, R3 is phenyl optionally
substituted with
chlorine, fluorine, (C~-C6) alkyl optionally substituted with from one to
three fluorine atoms or (C~-
Cs) alkoxy optionally substituted with from one to three fluorine atoms, m is
0 and n is 3 or 4.
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
_29_
More specific preferred methods of this invention include the above
combination
methods wherein the NK1 receptor antagonist is a compound of the Formula IX
selected from:
(2S,3S)-3-(5-tert-butyl-2-methoxybenzyl)amino-2-(3-
trifluoromethoxyphenyl)piperidine;
(2S,3S)-3-(2-isopropoxy-5-trifluoromethoxybenzyl)amino-2-phenyl-piperidine;
(2S,3S)-3-(2-ethoxy-5-trifluoromethoxybenzyl)amino-2-phenyl-piperidine;
(2S,3S)-3-(2-methoxy-5-trifluoromethoxybenzyl)-amino-2-phenylpiperidine;
(2S,3S)-3(-5-tent-butyl-2-trifluoromethoxybenzyl)amino-2-phenylpiperidine;
2-(diphenylmethyl)-N-(2-methoxy-5-trifluoromethoxy-phenyl)methyl-1-
azabicyclo[2.2.2]octan-3-amine;
(2S,3S)-3-[5-chloro-2-(2,2,2-trifluoroethoxy)-benzyl]amino-2-phenylpiperidine;
(2S,3S)-3-(5-tert-butyl-2-trifluoromethoxybenzyl)amino-2-phenylpiperidine;
(2S,3S)-3-(2-isopropoxy-5-trifluoromethoxybenzyl)amino-2-phenylpiperidine;
(2S,3S)-3-(2-difluoromethoxy-5-trifluoromethoxybenzyl)-amino-2-
phenylpiperidine;
(2S,3S)-2-phenyl-3-[2-(2,2,2-trifluoroethoxybenzyl)-aminopiperidine; or
(2S,3S)-2-phenyl-3-(2-trifluoromethoxybenzyl)]aminopiperidine;
or a pharmaceutically acceptable salt thereof.
Other NK1 receptor antagonists useful in the present invention are selected
from:
3-[N-(2-methoxy-5-trifluoromethoxybenzyl)-amino]-5,5-dimethyl-2-
phenylpyrrolidine;
3-[N-(2-methoxy-5-trifluoromethoxy-benzyl)amino]-4,5-dimethyl-2-
phenylpyrrolidine;
3-(2-cyclopropyloxy-5-trifluoromethoxybenzyl)amino-2-phenylpiperidine;
3-(2-cyclopropylmethoxy-5-trifluoromethoxybenzyl)amino-2-phenylpiperidine;
3-(2-difluoromethoxy-5-phenylbenzyl)amino-2-phenylpiperidine;
3-(5-cyclopropylmethoxy-2-difluoromethoxybenzyl)amino-2-phenylpiperidine;
3-(2-methoxybenzyl)amino-2-(3-trifluoromethoxyphenyl)-piperidine;
3-(2-methoxy-5-trifluoromethoxybenzyl)amino-2-(3-tri-
fluoromethoxyphenyl)piperidine;
2-phenyl-3-(5-n-propyl-2-trifluoromethoxybenzyl) amino-piperidine;
3-(5-isopropyl-2-trifluoromethoxybenzyl)amino-2-phenylpiperidine;
3-(5-ethyl-2-trifluoromethoxybenzyl)amino-2-phenyl-piperidine;
3-(5-sec-butyl-2-trifluoromethoxybenzyl)amino-2-phenyl-piperidine;
3-(5-difluoromethoxy-2-methoxybenzyl)amino-2-phenyl-piperidine;
3-(2-methoxy-5-trifluoromethoxybenzyl)amino-2-phenylpyrrolidine;
3-(2-methoxy-5-trifluoromethoxybenzyl)amino-2-phenylhomopiperidine;
2-benzhydryl-3-(2-methoxy-5-trifluoromethoxy-benzyl)aminopyrrolidine;
2-benzhydryl-3-(2-methoxy-5-trifluoromethoxy-benzyl)aminohomopiperidine;
3-[2,5-bis-(2,2,2-trifluoroethoxy)benzyl]amino-2-phenylpiperidine;
2-phenyl-3-(3-trifluoromethoxybenzyl)aminopiperidine;
2-benzhydryl-3-(2-methoxy-5-trifluoromethoxybenzyl)-aminopiperidine;
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-30-
1-(5,6-difluorohexyl)-3-(2-methoxy-5-trifluoromethoxy-benzyl)amino-2-
phenylpiperidine;
1-(6-hydroxyhexyl)-3-(2-methoxy-5-trifluoromethoxy-benzyl)amino-2-
phenylpiperidine;
3-phenyl-4-(2-methoxy-5-trifluoromethoxybenzyl)amino-2-
azabicyclo[3.3.0]octane;
4-benzhydryl-5-(2-methoxy-5-trifluoromethoxybenzyl)-amino-3-
azabicyclo[4.1.0]heptane;
4-(2-methoxy-5-trifluoromethoxybenzyl)amino-3-phenyl-2-
azabicyclo[4.4.0]decane;
2-phenyl-3-(2-methoxy-5-trifluoromethoxybenzyl)-aminoquinuclidine;
8-benzhydryl-N-(2-methoxy-5-trifluoromethoxybenzyl)-9-
azatricyclo[4.3.1.04'9]decan-7-
amine;
9-benzhydryl-N-(2-methoxy-5-trifluoromethoxybenzyl)-10-
azatricyclo[4.4.1.05~'°]undecan-
8-amine;
9-benzhydryl-N-(2-methoxy-5-trifluoromethoxybenzyl)-3-this-10-azatricyclo-
[4.4.1.05~~°]undecan-8-amine;
8-benzhydryl-N-(2-methoxy-5-trifluoromethoxybenzyl)-9-
azatricyclo[4.3.1.04'9]decan-7-
amine;
5,6-pentamethylene-2-benzhydryl-3-(2-methoxy-5-trifluoromethoxybenzyl)amino-
quinuclidine;
5,6-trimethylene-2-benzhydryl-3-(2-methoxy-5-trifluoromethoxybenzyl)amino-
quinuclidine;
9-benzhydryl-N-((2-methoxy-5-trifluoromethoxyphenyl)-methyl)-3-oxa-10-
azatricyclo-
[4.4.1.05~~°]undecan-3-amine;
8-benzhydryl-N-((2-methoxy-5-trifluoromethoxyphenyl)-methyl)-7-azatricyclo-
[4.4.1.05~'o]undecan-9-amine; and
2-benzhydryl-N-((2-methoxy-5-trifluoromethoxyphenyl)-methyl)-1-azabicyclo-
[3.2.2]nonan-3-amine;
and pharmaceutically acceptable salts thereof.
Other more specific embodiments of the present invention relate to the above
combination methods wherein the NK1 receptor antagonist that is employed in
such methods is
a compound of the Formula IX wherein o is two or three and each of Ri and R~3
is phenyl or
substituted phenyl.
Other more specific embodiments of the present invention relate to the above
combination methods wherein the NK1 receptor antagonist that is employed in
such methods is
a compound of the Formula IX wherein Q is a group of the Formula III, R~ is
hydrogen and R3 is
phenyl or substituted phenyl.
Other more specific embodiments of the present invention relate to the above
combination methods wherein the NI<1 receptor antagonist that is employed in
such methods is
a compound of the Formula IX wherein Q is a group of the Formula IV wherein I
is one or finro
and each of R4 and R5 is phenyl or substituted phenyl.
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-31-
Other more specific embodiments of the present invention relate to the above
combination methods wherein the NK1 receptor antagonist that is employed in
such methods is
a compound of the Formula IX wherein Q is a group of the Formula V wherein n
is zero or one
and each of R4 and R5 is phenyl or substituted phenyl.
Other more specific embodiments of the present invention relate to the above
combination methods wherein the NK1 receptor antagonist that is employed in
such methods is
a compound of the Formula IX wherein Q is a group of the Formula VI wherein p
is one and each
of R4 and R5 are phenyl, or substituted phenyl.
Other more specific embodiments of the present invention relate to the above
combination methods wherein the NK1 receptor antagonist that is employed in
such methods is
a compound of the Formula IX wherein Q is a group of the Formula VII wherein q
is two, three or
four, m is zero and Rs is phenyl or substituted phenyl.
Other more specific embodiments of the present invention relate to the above
combination methods wherein the NK1 receptor antagonist that is employed in
such methods is
selected from:
(2S,3S)-3-(6-methoxy-1-methyl-1-trifluoromethylisochroman-7-yl)methylamino-2-
phenylpiperidine;
(2S,3S)-3-[(1 R)-6-methoxy-1-methyl-1-trifluoromethylisochroman-7-
yl]methylamino-2-
phenylpiperidine;
(2S,3S)-N-(5-isopropyl-2-methoxyphenyl)methyl-2-di-phenylmethyl-1-
azabicyclo[2.2.2]-octan-3-amine; and
(2S,3S)-N-(5-tert-butyl-2-methoxyphenyl)-methyl-2-diphenylmethyl-1-
azabicyclo[2.2.2]-octan-3-amine;
and their pharmaceutically acceptable salts.
Examples of 5HT1 D antagonists that can be used in the pharmaceutical
compositions
and methods of this invention are the following:
3-(4-chlorophenyl)-5-[2-(4-methylpiperazin-1-yl)-benzylidene]-imidazolidine-
2,4-dione;
3-(4-chlorobenzyl)-5-[2-(4-methylpiperazin-1-yl)-benzylidene]-imidazolidine-
2,4-dione;
3-(4-chlorobenzyl)-5-[2-(4-methylpiperazin-1-yl)-benzylidene]-thiazolidine-2,4-
dione;
4-benzyl-2-[2-(4-methylpiperazin-1-yl)-benzylidene]-thiomorpholin-3-one;
4-(3,4-dichlorobenzyl)-2-[2-(4-methylpiperazin-1-yl)-benzylidene]-
thiomorpholin-3-one;
3-(4-chlorophenyl)-5-[2-(4-methylpiperazin-1-yl)-benzylidene]-thiazolidine-2,4-
dione;
3-(4-trifluoromethylphenyl)-5-[2-(4-methylpiperazin-1-yl)-benzylidene]-
thiazolidine-2,4-
dione;
2-[2-(4-methylpiperazin-1-yl)-benzylidene]-4-(4-trifluoromethylphenyl)-
thiomorpholin-3-
one;
2-[2-(4-methylpiperazin-1-yl)-benzylidene]-thiomorpholin-3-one;
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-32-
4-(3,4-dichlorophenyl)-2-[2-fluoro-6-(4-methylpiperazin-1-yl)-benzylidene]-
thiomorpholin-3-one;
4-(3,4-dichlorophenyl)-2-[2-(4-methylpiperazin-1-yl)-benzylidene]-morpholin-3-
one;
4-(3,4-dichlorophenyl)-2-[2-(4-methylpiperazin-1-yl)-benzylidene]-
thiomorpholin-3-one;
4-(3,4-dichlorophenyl)-2-[2-(4-methylpiperazin-1-yl)-benzyl]-thiomorpholin-3-
one;
4-methyl-2-[2-(4-methylpiperazin-1-yl)-benzylidene]-thiomorpholin-3-one; and
4-(3,4-dichlorophenyl)-2-(2-piperazin-1-ylbenzylidene)-thiomorpholin-3-one.
and pharmaceutically acceptable salts thereof.
Other specific NK1 receptor antagonists useful in the present invention
include:
5-[2-(4-methylpiperazin-1-yl)-benzylidene]-thiazolidine-2,4-dione;
2-[2,4-dibromo-6-(4-methylpiperazin-1-yl)-benzylidene]-4-(3,4-dichlorophenyl)-
thiomorpholin-3-one;
4-(4-chlorophenyl)-2-[2-(4-methylpiperazin-1-yl)-benzylidene]-
[1,4]oxazepan-3-one;
4-(4-chlorophenyl)-2-[2-(4-methylpiperazin-1-yl)-benzylidene]-
[1,4,5]oxadiazepan-3-one;
4-(4-chlorophenyl)-2-[2-(4-methylpiperazin-1-yl)-benzylidene]-
[1,4]thiazepan-3-one;
4-(3,4-dichlorophenyl)-2-{2-[(2-dimethylaminoethyl)-methyl-amino]-benzylidene}-
thiomorpholin-3-one;
one;
dione;
one;
3-one;
4-(3,4-dichlorophenyl)-2-[2-(1-methylpiperidin-4-yl)-benzylidene]-
thiomorpholin-3-one;
4-(3,4-dichlorophenyl)-2-[2-(1,4-dimethylpiperidin-4-yl)-benzylidene]-
thiomorpholin-3-
4-(3,4-dichlorophenyl)-2-[2-(4-methylpiperazin-1-yl)-benzylidene]-
thiomorpholine-3,5-
4-(3,4-dichlorophenyl)-2-[2-(2-dimethylaminoethoxy)-benzylidene]-thiomorpholin-
3-one;
4-(3,4-dichlorophenyl)-2-[2-(4-isopropylpiperazin-1-yl)-benzylidene]-
thiomorpholin-3-
4-(3,4-dichlorophenyl)-2-[2-(1-methylpyrrolidin-3-ylmethyl)-benzylidene]-
thiomorpholin-
4-(3,4-dichlorophenyl)-2-{2-[methyl-(1-methylpyrrolidin-2-ylmethyl)-amino]-
benzylidene}-thiomorpholin-3-one;
4-(3,4-dichlorophenyl)-2-[2-(1-methylpyrrolidin-2-ylmethoxy)-benzylidene]-
thiomorpholin-3-one;
4-(3,4-dichlorophenyl)-2-{2-[2-(1-methylpyrrolidin-2-yl)-ethyl]-benzylidene}-
thiomorpholin-3-one;
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-33-
1-(3,4-dichlorophenyl)-4-methyl-3-[2-(4-methylpiperazin-1-yl)-benzylidene]-
piperazin-2-
one;
4-methyl-3-[2-(4-methylpiperazin-1-yl)-benzylidene]-1-(4-
trifluoromethylphenyl)-
piperazin-2-one;
1-(4-chlorophenyl)-4-methyl-3-[2-(4-methylpiperazin-1-yl)-benzylidene]-
piperazin-2-one;
2-[2-(4-methylpiperazin-1-yl)-benzylidene]-4-(4-trifluoromethylphenyl)-
morpholin-3-one;
2-[4-fluoro-2-(4-methylpiperazin-1-yl)-benzylidene]-4-(4-
trifluoromethylphenyl)-
thiomorpholin-3-one;
2-[5-fluoro-2-(4-methylpiperazin-1-yl)-benzylidene]-4-(4-
trifluoromethylphenyl)-
thiomorpholin-3-one;
2-{1-[2-(4-methylpiperazin-1-yl)-phenyl]-ethylidene}-4-(4-
trifluoromethylphenyl)-
thiomorpholin-3-one;
2-[2-(4-methylpiperazin-1-yl)-benzyl]-4-(4-trifluoromethylphenyl)-
thiomorpholin-3-one;
4-(4-chlorophenyl)-6-methyl-2-[2-(4-methylpiperazin-1-yl)-benzylidene]-
thiomorpholin-3-
one;
3-(4-chlorophenyl)-2,2-dimethyl-5-[2-(4-methylpiperazin-1-yl)-benzylidene]-
thiazolidin-4-
one;
4-(4-chlorophenyl)-2-[2-(4-methylpiperazin-1-yl)-benzylidene]-[1,4]oxazepan-3-
one;
4-(4-chlorophenyl)-2-[2-(4-methylpiperazin-1-yl)-benzylidene]-4H-[1,4]thiazin-
3-one;
1-(4-chlorophenyl)-4,6,6-trimethyl-3-[2-(4-methylpiperazin-1-yl)-benzylidene]-
piperazin-
2-one;
1-(4-chlorophenyl)-4-methyl-3-[2-(4-methylpiperazin-1-yl)-benzylidene]-
piperazin-2-one;
4-(4-chlorophenyl)-2-[2-(4-methylpiperazin-1-yl)-benzylidene]-morpholin-3-one;
3-(4-chlorophenyl)-5-[2-(4-methylpiperazin-1-yl)-benzylidene]-oxazolidin-4-
one;
3-(4-chlorophenyl)-2,2-dimethyl-5-[2-(4-methylpiperazin-1-yl)-benzylidene]-
imidazolidin-
4-one;
and pharmaceutically acceptable salts thereof.
The following references refer to quinuclidine, piperidine, ethylene diamine,
pyrrolidine and azanorbornane derivatives and related compounds that exhibit
activity as NK1
receptor antagonists and that can be used, in combination with the 5HT7
receptor partial
agonists of the Formula I, in the pharmaceutical compositions and methods of
this invention,
and to methods of preparing the same: United States Patent 5,162,339, which
issued on
November 11, 1992; United States Patent 5,232,929, which issued on August 3,
1993; World
Patent Application WO 92/20676, published November 26, 1992; World Patent
Application
WO 93/00331, published January 7, 1993; World Patent Application WO 92/21677,
published
December 10, 1992; World Patent Application WO 93/00330, published January 7,
1993;
World Patent Application WO 93/06099, published April 1, 1993; World Patent
Application
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-34-
WO 93/10073, published May 27, 1993; World Patent Application WO 92/06079,
published
April 16, 1992; World Patent Application WO 92/12151, published July 23, 1992;
World Patent
Application WO 92/15585, published September 17, 1992; World Patent
Application WO
93/10073, published May 27, 1993; World Patent Application WO 93/19064,
published
September 30, 1993; World Patent Application WO 94/08997, published April 28,
1994; World
Patent Application WO 94/04496, published March 3, 1994; World Patent
Application WO
95/07908, published March 3, 1995; World Patent Application WO 94/20500,
published
September 15, 1994; World Patent Application WO 94/13663, published June 23,
1994;
World Patent Application WO 95/16679, published June 22, 1995; World Patent
Application
WO 97/08144, published March 6, 1997; World Patent Application WO 97/03066,
published
January 30, 1997; World Patent Application WO 99/25714, published May 27,
1999; United
States Patent Application 988,653, filed December 10, 1992; United States
Patent Application
026,382, filed March 4, 1993; United States Patent Application 123,306, filed
September 17,
1993, and United States Patent Application 072,629, filed June 4, 1993. All of
the foregoing
World Patent Applications designate the United States. The foregoing patents
and patent
applications are incorporated herein by reference in their entirety.
NK-1 receptor antagonists of the Formula IX can be prepared as described in
the
following patents and patent applications, all of which are referred to above
and incorporated
herein by reference in their entirety: WO 93/00331, WO 92/21677, WO 92/15585,
WO
92/01688, WO 93/06099, WO 91/18899, United States Patent 5,162,339, and United
States
Patent 5,232,929.
Other NK1 receptor antagonists that can be used, together with the 5HT7
agonists of
the Formula I, for the treatment of anxiety or depression in accordance with
the methods and
pharmaceutical compositions of the present invention are those compounds and
pharmaceutically acceptable salts described in the following references:
European Patent
Application EP 499,313, published August 19, 1992; European Patent Application
EP 520,555,
published December 30, 1992; European Patent Application EP 522,808, published
January 13,
1993, European Patent Application EP 528,495, published February 24, 1993, PCT
Patent
Application WO 93/14084, published July 22, 1993, PCT Patent Application WO
93/01169,
published January 21, 1993, PCT Patent Application WO 93/01165, published
January 21,
1993, PCT Patent Application WO 93/01159, published January 21, 1993, PCT
Patent
Application WO 92/20661, published November 26, 1992, European Patent
Application EP
517,589, published December 12, 1992, European Patent Application EP 428,434,
published
May 22, 1991, and European Patent Application EP 360,390, published March 28,
1990. All of
the foregoing World Patent Applications designate the United States. The
foregoing patents
and patent applications are incorporated herein by reference in their
entirety.
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-35-
For any of the therapeutic methods or pharmaceutical compositions of the
present
invention, the appropriate dose regimen, the amount of each dose of an active
agent
administered, and the specific intervals between doses of each active agent
will depend upon
the subject being treated, the specific active agent being administered and
the nature and
severity of the specific disorder or condition being treated. In general, the
active compounds of
this invention, when used as a single active agent or in combination with
another active agent,
will be administered to an adult human in an amount from about 0.05 to about
1500 mg per day,
in single or divided doses, preferably from about 5 to about 200 mg/day. Such
compounds may
be administered on a regimen of up to 6 times per day, preferably 1 to 4 times
per day,
especially 2 times per day and most especially once daily. Variations may
nevertheless occur
depending upon the species of animal being treated and its individual response
to said
medicament, as well as on the type of pharmaceutical formulation chosen and
the time period
and interval at which such administration is carried out. In some instances,
dosage levels below
the lower limit of the aforesaid range may be more than adequate, while in
other cases still
larger doses may be employed without causing any harmful side effect, provided
that such
larger doses are first divided into several small doses for administration
throughout the day.
A proposed daily dose of a 5HT reuptake inhibitor, preferably sertraline, in
the
combination methods and compositions of this invention, for oral, parenteral
or buccal
administration to the average adult human for the treatment of the conditions
referred to
above, is from about 0.1 mg to about 2000 mg, preferably from about 1 mg to
about 200 mg
of the 5HT reuptake inhibitor per unit dose, which could be administered, for
example, 1 to 4
times per day.
A proposed daily dose of a 5HT1 D receptor antagonist in the combination
methods
and compositions of this invention, for oral, parenteral, rectal or buccal
administration to the
average adult human for the treatment of the conditions referred to above, is
from about 0.01
mg to about 2000 mg, preferably from about 0.1 mg to about 200 mg of the 5HT1
D receptor
antagonist per unit dose, which could be administered, for example, 1 to 4
times per day.
A proposed daily dose of an NK1 receptor antagonist in the combination methods
and
compositions, for oral, parenteral or buccal administration to the average
adult human for the
treatment of the conditions referred to above, is from about 0.1 mg to about
2000 mg,
preferably from about 1 mg to about 200 mg of the NK1 receptor antagonist per
unit dose
which could be administered, for example, 1 to 4 times per day.
The 5HT7 receptor agonists, the NK1 receptor antagonists, the serotonin
reuptake
inhibitors and the 5HT1 D receptor antagonists, and their pharmaceutically
acceptable salts, that
are employed in the pharmaceutical compositions and methods of this invention
are hereinafter
also referred to as "therapeutic agents". The therapeutic agents can be
administered via either
the oral, buccal, nasal or parenteral route. Compositions containing both a
5HT7 receptor
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-36-
agonist and an NI<1 receptor antagonist, a 5HT1 D receptor antagonist or a
serotonin reuptake
inhibitor, will generally be administered orally or parenterally daily, in
single or divided doses, so
that the total amount of each active agent administered falls within the above
guidelines.
The therapeutic agents may be administered alone or in combination with
pharmaceutically acceptable carriers or diluents by either of the routes
previously indicated, and
such administration may be carried out in single or multiple doses. More
particularly, the
therapeutic agents of this invention can be administered in a wide variety of
different dosage
forms, i.e., they may be combined with various pharmaceutically acceptable
inert carriers in the
form of tablets, capsules, lozenges, troches, hard candies, suppositories,
aqueous suspensions,
injectable solutions, elixirs, syrups, and the like. Such carriers include
solid diluents or fillers,
sterile aqueous media and various non-toxic organic solvents, etc. Moreover,
oral
pharmaceutical compositions can be suitably sweetened and/or flavored. In
general, the
therapeutic agents of this invention, when administered separately (i.e., not
in the same
pharmaceutical composition) are present in such dosage forms at concentration
levels ranging
from about 5.0% to about 70% by weight.
For oral administration, the pharmaceutical compositions may take the form of,
for
example, tablets or capsules prepared by conventional means with
pharmaceutically
acceptable excipients such as binding agents (e.g., pregelatinized maize
starch,
polyvinylpyrrolidone or hydroxypropyl methylcellulose); fillers (e.g.,
lactose, microcrystalline
cellulose or calcium phosphate); lubricants (e.g., magnesium stearate, talc or
silica);
disintegrants (e.g., potato starch or sodium starch glycolate); or wetting
agents (e.g., sodium
lauryl sulphate). The tablets may be coated by methods well known in the art.
Liquid
preparations for oral administration may take the form of, for example,
solutions, syrups or
suspensions, or they may be presented as a dry product for constitution with
water or other
suitable vehicle before use. Such liquid preparations may be prepared by
conventional
means with pharmaceutically acceptable additives such as suspending agents
(e.g., sorbitol
syrup, methyl cellulose or hydrogenated edible fats); emulsifying agents
(e.g., lecithin or
acacia); non-aqueous vehicles (e.g., almond oil, oily esters or ethyl
alcohol); and
preservatives (e.g., methyl or propyl p-hydroxybenzoates or sorbic acid).
For preparing solid compositions such as tablets, the principal active
ingredient is
mixed with a pharmaceutical carrier, e.g., conventional tableting ingredients
such as corn
starch, lactose, sucrose, sorbitol, talc, stearic acid, magnesium stearate,
dicalcium phosphate
or gums, and other pharmaceutical diluents, e.g., water, to form a solid
preformulation
composition containing a homogeneous mixture of a therapeutic agent, or a non-
toxic
pharmaceutically acceptable salt thereof. When referring to these
preformulation
compositions as homogeneous, it is meant that the therapeutic agent is
dispersed evenly
throughout the composition so that the composition may be readily subdivided
into equally
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-37-
effective unit dosage forms such as tablets, pills and capsules. This solid
preformulation
composition is then subdivided into unit dosage forms of the type described
above containing,
typically, from 0.05 to about 500 mg of each of the therapeutic agents
contained in the
composition. The tablets or pills of the composition can be coated or
otherwise compounded
to provide a dosage form affording the advantage of prolonged action. For
example, the
tablet or pill can comprise an inner dosage and an outer dosage component, the
latter being
in the form of an envelope over the former. The two components can be
separated by an
enteric layer which serves to resist disintegration in the stomach and permits
the inner
component to pass intact into the duodenum or to be delayed in release. A
variety of
materials can be used for such enteric layers or coatings, such materials
including a number
of polymeric acids and mixtures of polymeric acids with such materials as
shellac acetyl
alcohol and cellulose acetate.
For buccal administration, the composition may take the form of tablets or
lozenges
formulated in conventional manner.
The therapeutic agents may be formulated for parenteral administration by
injection,
including using conventional catheterization techniques or infusion.
Formulations for injection
may be presented in unit dosage form, e.g., in ampules or in multi-dose
containers, with an
added preservative. The compositions may take such forms as suspensions,
solutions or
emulsions in oily or aqueous vehicles, and may contain formulating agents such
as
suspending, stabilizing and/or dispersing agents. Solutions of a therapeutic
agent in either
sesame or peanut oil or in aqueous propylene glycol may be employed. The
aqueous solutions
should be suitably buffered if necessary and the liquid diluent first rendered
isotonic. These
aqueous solutions are suitable for intravenous injection purposes. The oily
solutions are
suitable for intraarticular, intramuscular and subcutaneous injection
purposes. The preparation
of all these solutions under sterile conditions is readily accomplished by
standard
pharmaceutical techniques well known to those skilled in the art.
Alternatively, the active
ingredient may be in powder form for reconstitution with a suitable vehicle,
e.g., sterile
pyrogen-free water, before use.
For intranasal administration or administration by inhalation, the active
compounds of
the invention are conveniently delivered in the form of a solution or
suspension from a pump
spray container that is squeezed or pumped by the patient or as an aerosol
spray
presentation from a pressurized container or a nebulizer, with the use of a
suitable propellant,
e.g., dichlorodifluoromethane, trichlorofluoromethane,
dichlorotetrafluoroethane, carbon
dioxide or other suitable gas. In the case of a pressurized aerosol, the
dosage unit may be
determined by providing a valve to deliver a metered amount. The pressurized
container or
nebulizer may contain a solution or suspension of the active compound.
Capsules and
cartridges (made, for example, from gelatin) for use in an inhaler or
insufflator may be
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-38-
formulated containing a powder mix of a compound of the invention and a
suitable powder
base such as lactose or starch.
Aerosol formulations of the active compounds of this invention for treatment
of the
conditions referred to above in the average adult human are preferably
arranged so that each
metered dose or "puff' of aerosol contains 20 pg to 1000 pg of active
compound. The overall
daily dose with an aerosol will be within the range 100 pg to 10 mg.
Administration may be
several times daily, for example 2, 3, 4 or 8 times, giving for example, 1, 2
or 3 doses each
time.
The compounds of Formula I may advantageously be used in conjunction with one
or
more other therapeutic agents, for instance, different antidepressant agents
such as tricyclic
antidepressants (e.g., amitriptyline, dothiepin, doxepin, trimipramine,
butripyline,
clomipramine, desipramine, imipramine, iprindole, lofepramine, nortriptyline
or protriptyline),
or monoamine oxidase inhibitors (e.g., isocarboxazid, phenelzine or
tranylcyclopramine),
and/or with antiparkinsonian agents such as dopaminergic antiparkinsonian
agents (e.g.,
levodopa, preferably in combination with a peripheral decarboxylase inhibitor
e.g.,
benserazide or carbidopa, or with a dopamine agonist e.g., bromocriptine,
lysuride or
pergolide). It is to be understood that the present invention covers the use
of a compound of
general Formula I or a physiologically acceptable salt or solvate thereof in
combination with
one or more other therapeutic agents.
The affinities of the active compounds for 5HT7 receptors can be determined
using
standard radioligand binding assays as described in the literature. The 5HT7
affinity can be
measured using the following procedure.
~H-5-CARBOXAMIDOTRYPTAMINE (3H-5-CT) BINDING TO RAT 5HT7
RECEPTORS EXPRESSED IN HEK-293 CELLS:
Materials:
HEK-293 cells expressing the rat 5-HT7 receptor
Brinkman Polytron Tissue Homogenizer
Phosphate Buffered Saline (GIBCO)
Capped Centrifuge Tubes
Centrifuge
50mMTrisHCIBuffer, pH7.7 (SigmaT-4378)
EDTA (Sigma E-4884)
MgS04 (Sigma M-7506)
CaCh (MCBCX156)
pargyline (SigmaP-8013)
ascorbicacid (Calbiochem1831)
5-HTcreatinine sulfate complex (Sigma H-7752)
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-39-
3H-5CT (Amersham TRK.1038)
12 x 75 mm boroscilicate glass tubes
96 well V-bottom polypropylene plates (NUNC - 442587)
Skatron 96 Well Harvester
Whatman GF/B Glass Fiber Filters (Brandel FP-105) presoaked in 0.3%
polyethylenimine (Sigma - P-3143)
Betaplate scintillation counter (Wallac/LKB)
Tissue Preparation:
HEK-293 cells expressing 5HT7 receptors are grown according to standard cell
culture techniques. Cells are harvested by removing the media, rinsing the
flasks out with
phosphate buffered saline (PBS) and then allowed to sit for 2-3 minutes with
PBS containing
2.5 mM EDTA. Cells are dislodged and poured into a RcappableS centrifuge tube.
Flasks are
rinsed with PBS and added to a centrifuge tube. The cells are centrifuged for
ten minutes at
40,000 x g (20,000 rpm in a Sorvall SS34 rotor). The supernatant is discarded
and at this
point the remaining pellet is weighed and can be stored frozen (-20 degrees C)
until used in
the binding assay. Pellets (fresh or frozen) are homogenized in 50 mM Tris HCI
buffer (pH
7.4 at 4 degrees C) using a Polytron homogenizer (setting 15,000 rpm) for ten
seconds in a
biologcial hood certified for use with human tissues. The homogenate is
centrifuged for ten
minutes at 40,000 x g. The supernatant is discarded and the pellet resuspended
with the
Polytron in a fresh ice-cold 50 mM Tris HCI (pH 7.4 at 4 degrees) buffer and
centrifuged
again. The final pellet is resuspended in assay buffer (50 mM Tris HCI buffer
(pH 7.7 at 25
degrees) containing 0.5 mM EDTA, 10 mM MgS04, 2 mM CaCl2) for a final tissue
concentration of 5-15 mg wet weight of original pellet per mL buffer (2X final
concentration).
Receptor Bindinct
Incubation is initiated by the addition of tissue to V-bottom polypropylene
plates (in
triplicate). Incubation is at 25 degrees C for 2 hours.
Each tube receives:
100 uL tissue suspension (5-15mg/mL original wet weight), 50 uL 3H-5-CT** (0.4
nM
final concentration), and 50 uL drug or buffer
**3H-5-CT is made up in assay buffer that contains 40 uM pargyline & 0.4%
ascorbic
acid (for final concentrations of 10 uM pargyline & 0.1 % ascorbic acid).
Nonspecific binding is determined using 1 uM 5-HT creatinine sulfate.
Incubation is
ended by rapid filtration under vacuum through fire-treated Whatman GF/B glass
fiber filters
(presoaked in 0.3% PEI for two hours and dried) using a 96 well Skatron
Harvester (3 sec
prewet; 20 seconds wash; 15 seconds dry). Filters are put into LKB sample bags
with 10 mL
BetaScint. Radioactivity is quantified by liquid scintillation counting using
a BetaPlate counter
(LKB).
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-40-
The percent inhibition of specific binding is calculated for each
concentration of test
compound. An ICSO value (the concentration which inhibits 50% of the specific
binding) is
determined by linear regression of the concentration-response data (log
concentration vs.
logit percent values). Ki values are calculated according to Cheng & Prusoff:
Ki = ICSO/(1 +
(L/Kd)), where L is the concentration of the radioligand used in the
experiment and the Kd
value is the dissociation constant for the radioligand determined in separate
saturation
experiments. The binding activities to 5HT7 receptors of approximately 40
compounds of the
invention that were assayed as described above ranged from about 3.5 nM to
about 5 pM.
For example the title compound of Example 8, below, showed a Ki of about 7.6
nM, and the
title compound of Example 10, below, showed a Ki of about 500 nM.
The following assay can be used to evaluate the functional activity of
compounds
at 5HT7 receptors:
5-HT7 RECEPTOR MEDIATED ADENYLATE CYCLASE ACTIVITY:
Materials:
1.5 mL siliconized polypropylene microfuge tubes (Costar 3207)
12 x 75 mm boroscilicate glass tubes
Heated water bath
Glass-Teflon Homogenizer
Centrifuge
HEK-293 cells expressing 5-HT7 receptors
32P-ATP (30 Ci/mmol: NEG-003 - New England Nuclear)
3H-CAMP (30 Ci/mmol: NET-275 - New England Nuclear)
Methods:
Cells are grown according to standard cell culture techniques. Cells are
harvested by
replacing the media with phosphate-buffered saline containing 2.5 mM EDTA. The
cells are
homogenized using a hand-held glass-teflon homogenizer. The homogenate is
centrifuged at
35,000 x g for 10 minutes at 4 degrees C. The pellet is resuspended in 100 mM
HEPES
buffer containing 1 mM EGTA (pH 7.5) to a final protein concentration of 40
microgram
protein per tube.
The "Reaction Mix" is prepared so that the following agents will be at these
final
concentrations in tube: 4.OmM MgClz, 0.5m MATP, 1.Om McAMP, 0.5mM IBMX, 10mM,
phosphocreatine, 0.31 mg/mL creatine phosphokinase, and 100uM GTP0.5-1
microcuries a-
[3zP]_ATP per tube.
Incubation is initiated by the addition of tissue to siliconized microfuge
tubes (in
triplicate). Incubation is at 3TC for 15 minutes.
Each tube receives:
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-41-
20uL tissue, 20uL drug or buffer (at 5X final concentration), 20 uL 100 nM
agonist or
buffer (at 5X final concentration), and 40 uL "Reaction Mix".
Incubation is terminated by the addition of 100 uL 2% SDS, 1.3 mM cAMP, 45 mM
ATP solution containing 40,000 dpm [~H]-CAMP to monitor the recovery of cAMP
from the
columns. The separation of [3aP]-ATP and [32P]-cAMP is accomplished using the
method of
Salomon et al., Analytical Biochemistry 58: 541-548, 1974, which is
incorporated herein by
reference in its entirety. Radioactivity is quantified by liquid scintillation
counting.
The maximal effect of agonists is defined in terms of the maximal effect of
serotonin
(5-HT). Antagonists are evaluated by their ability to inhibit 5HT-stimulated
adenylate cyclase
activity. ICSO values are converted to apparent Ki values by the following
equation: ICSO/ (1 +
([agonist]/ECSO of agonist)).
Activity of a combination of active compounds to produce an antidepressant
effect
and related pharmacological properties can be determined by methods (1 )-(4)
below, which
are described in Koe, B. et al., Journal of Pharmacology and Experimental
Therapeutics, 226
(3), 686-700 (1983), which is incorporated herein by reference in its
entirety. Specifically,
activity can be determined by studying (1 ) their ability to affect the
efforts of mice to escape
from a swim-tank (Porsolt mouse "behavior despair" test), (2) their ability to
potentiate 5HT -
induced behavioral symptoms in mice in vivo, (3) their ability to antagonize
the serotonin-
depleting activity of p-chloroamphetamine hydrochloride in rat brain in vivo,
and (4) their
ability to block the uptake of serotonin, norepinephrine and dopamine by
synaptosomal rat
brain cells in vitro. The ability of the active combination to counteract
reserpine hypothermia
in mice in vivo can be determined according to the methods described in U.S.
Pat.
No. 4,029,731, which is incorporated herein by reference in its entirety.
The following Examples illustrate the present invention. It is to be
understood,
however, that the invention, as fully described herein and as recited in the
claims, is not
intended to be limited by the details of the following Examples.
The following Examples illustrate the preparation of the compounds of the
present
invention. Melting points are uncorrected. NMR data are reported in parts per
million and are
referenced to the deuterium lock signal from the sample solvent
(deuteriochloroform unless
otherwise specified). Specific rotations were measured at room temperature
using the
sodium D line (589 nm). Commercial reagents were utilized without further
purification. THF
refers to tetrahydrofuran. DMF refers to N,N-dimethylformamide. Chromatography
refers to
column chromatography performed using 47-61 micron mesh silica gel and
executed under
nitrogen pressure (flash chromatography) conditions. Room or ambient
temperature refers to
20-25°C. All non-aqueous reactions were run under a nitrogen atmosphere
for convenience
and to maximize yields. Concentration at reduced pressure means that a rotary
evaporator
was used.
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-42-
Examples
EXAMPLE 1
Step 1
8-Bromo-3-ethyl-auinoline
ENANTIOMERIC (BOTH ENANTIOMERS) AND RACEMIC 3-ETHYL-8-PIPERIDIN-3-YL-
QUINOLINE
To a well-stirred mixture consisting of 2-bromo-aniline (5.4 g, 31.4 mmol),
sodium 3-
nitrobenzene sulfonate (4.25 g, 18.9 mmol), concentrated sulfuric acid (8.5 g,
177 mmol), and
water (3.20 ml) heated to 100°C, 2-ethyl acrolein (5.0 ml, 51.06 mmol)
was added. After
maintaining the reaction temperature at 100°C for 1 hour, the
temperature was increased to
110°C. An additional portion of 2-ethyl acrolein (1.0 ml, 10.2 mmol)
was added, and the
reaction was stirred at 110°C for 1 hour. The reaction temperature was
then elevated to
120°C prior to addition of another 1.0 ml (10.2 mmol) portion of 2-
ethyl acrolein. After heating
the reaction at 120 for 1 hour, the temperature was elevated to 130 prior to
addition of 1.0 ml
(10.2 mmol) of 2-ethyl acrolein. Finally, the reaction temperature was raised
to 140°C and
maintained at that temperature for 2 hours following addition of a final
portion (1.3 ml,
13.3mmol) of 2-ethyl acrolein. The cooled reaction was quenched with ice (60
g), and the pH
of the resulting mixture was adjusted to 14 by addition of 6 N aqueous sodium
hydroxide.
The reaction mixture was then extracted with three 100 ml portions of
methylene chloride.
. The combined organic extracts were dried (anhydrous sodium sulfate) and
concentrated in
vacuo yielding an amber oil. Flash chromatography of the entire sample (silica
gel, 47-61
micron mesh; elution with methylene chloride) afforded the title compound
(3.70 g, 50% yield)
as an amber oil.
MS m/z 236, 237, 238, 239 (M+1 ).
'H NMR (400 MHz, CDCI3) 8 8.9 (1 H, m), 7.97 (1 H, m), 7.91 (1 H, m), 7.74 (1
H, m),
7.36 (1 H, m), 2.86 (2H, q, J = 7.5 Hz), 1.34 (3H, t, J = 7.5 Hz) ppm.
Ste~2
3-Ethyl-8-pyridin-3-yl-auinoline
To a well-stirred mixture consisting of the title compound from the previous
step (2.36
g, 10.0 mmol), diethyl (3-pyridyl) borane (1.67 g, 11.0 mmol), and bis
(triphenylphosphine)
palladium (II) chloride (913 mg, 1.3 mmol) in tetrahydrofuran (40 ml), an
aqueous solution of
sodium carbonate (4.24 g, 40 mmol in 20 ml water) is added, and the resulting
reaction
mixture is heated at reflux for 4 hours. Water (50 ml) was added to the well-
stirred mixture.
The aqueous phase of the biphasic reaction mixture is separated and extracted
with three 50
ml portions of ethyl acetate. The solvent of the organic phase of the reaction
mixture is
removed in vacuo, and the residue is extracted with two 50 ml portions of
ethyl acetate. The
combined organic extracts are dried (anhydrous sodium sulfate) and
concentrated in vacuo,
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-43-
yielding a viscous syrup. Flash chromatography of the entire sample (silica
gel, 47-61 micron
mesh; elution with ethyl acetate) yielded a pure portion of the title compound
(830 mg, 35.4%
yield) as a viscous amber syrup and a less pure (judged to be approximately
75% pure by
NMR inspection) second portion of the title compound (700 mg), also an amber
syrup.
MS mlz 234 (M+1 ).
'H NMR (400 MHz, CDCI3) 8 8.93 (1 H, m), 8.80 (1 H, m), 8.63 (1 H, m), 8.10 (1
H, m),
7.96 (1 H, m), 7.81 (1 H, m), 7.66 (1 H, m), 7.60 (1 H, m), 7.42 (1 H, m),
2.84 (2H, q, J = 7.5 Hz),
1.34 (3H, t, J = 7.5 Hz) ppm.
Step 3
3-Eth~yridin-3-yl-auinoline
To a solution of the title compound from the previous step (830 mg, 3.53 mmol)
in
anhydrous tetrahydrofuran (5.0 ml), 28.4 ml (28.4 mmol) of 1.0 M lithium
triethylborohydride in
tetrahydrofuran was added, and the resulting reaction mixture was stirred at
ambient
temperature for 18 hours. The reaction was quenched by cautious dropwise
addition of water
(50 ml). Solvents were removed in vacuo, affording a viscous oil which was
extracted with
three 25 ml portions of methylene chloride. The organic extract was dried
(anhydrous sodium
sulfate) and concentrated in vacuo to afford a viscous yellow syrup. The just-
described
procedure was repeated utilizing, respectively, 817 mg (3.49 mmol) and 27.9 ml
(27.9 mmol)
of the previous step title compound and 1.0 N triethylborohydride in
tetrahydrofuran. The
crude reaction products after work-up from both reactions (i.e., the viscous
yellow syrups)
were combined. Flash chromatography of the entire sample (silica gel, 47-61
micron mesh;
elution with methylene chloride/methanol/concentrated aqueous ammonium
hydroxide =
90:9:1 in volume) afforded the title compound (480 mg) as a viscous yellow
oil.
MS m/z 240 (M+ 1 ).
'H NMR (400 MHz, CDCI3) 8 8.78 (1 H, m), 7.87 (1 H, m), 7.58 (1 H, m), 7.42-
7.50 (2H,
overlapping multiplets), 4.07 (1 H, m), 3.32 (1 H, m), 3.16 (1 H, m), 2.80
(2H, q, J = 7.5 Hz),
2.73-2.64 (2H, m), 2.2-2.0 (1 H, m), 1.66-1.87 (3H, m), 1.32 (3H, t, J = 7.5
Hz) ppm.
'3C NMR (125 MHz, CDCI3) & 150.9, 145.1, 143.3, 136.7, 134.1, 128.7, 126.6,
125.7,
125.3, 54.0, 47.1, 38.1, 31.6, 28.0, 26.4, 15.5 ppm.
Separation of the enantiomers of the racemic title compound
Step 4
Racemic 3-(3-Ethyl-auinolin-8-yl)-piperidine-1-carboxylic acid tert-butyl
ester
To a well-stirred solution of the racemic title compound from the previous
step (5.40
g, 23.9 mmol) in methylene chloride (50 ml) containing triethylamine (6.7 ml,
47.8 mmol), di
tert-butyl dicarbonate (7.80 g, 35.8 mmol) was added, and the resulting
reaction mixture was
stirred at ambient temperature for 5 hours. Saturated aqueous sodium
bicarbonate (50 ml)
was added with efficient stirring. The mixture was then extracted with two 20
ml portions of
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-44-
methylene chloride. The organic extracts were combined, washed with an equal
volume of
brine, dried (anhydrous sodium sulfate), and finally, concentrated in vacuo,
affording a
viscous syrup. Flash chromatography of the entire sample utilizing the Biotage
Flash 401 iT"~
silica gel flash chromatography module (silica gel 32-63 micron mesh prepacked
cartridges
supplied by the manufacturer: Biotage Division of the Dyax Corporation,
Charlottesville, VA),
eluting with methylene chloride/methanol = 99.5:0.5 in volume afforded the
title compound
(4.24 g, 52% yield) as a colorless solid.
MS m/z 340 (M+1 ).
'H NMR (400 MHz, CDCI3) 8 8.80 (1 H, m), 7.88 (1 H, m), 7.62 (1 H, m), 7.50-
7.43 (2H,
overlapping multiplets), 4.32 (1 H, m), 4.18 (1 H, m), 4.10 (1 H, m), 2.92 (1
H, m), 2.85-2.76
(overlapping 1 H, m and 2H, q centered at 2.80, J = 7.5 Hz), 2.12 (1 H, m),
1.82-1.71 (3H, m),
1.44 (s, 9H), 1.33 (3H, t, J = 7.5 Hz) ppm.
Step 5
Enantiomeric 3-(3-Ethyl-auinolin-8-y,-piperidine-1-carboxylic acid tert-butyl
ester (both
enantiomers)
Separation of the enantiomers of the Step 4 title compound:
Utilizing a Waters Prep LC 2000T"~ Preparative Chromatography System (Waters
ChiracelT"' OD 10 cmx 50 cm) preparative column; mobile phase: heptane/ethanol
= 98:2 in
volume with 0.025% diethyl amine modifier; a flow rate of 225 ml/minute; 4.08
g of the title
compound from the previous step dissolved in 10 ml of methylene
chloride/methanol = 4:1 in
volume; injecting 204 mg of compound in the methylene chloride/methanol
solution at a time;
with approximate retention times for the enantiomers of 20 and 28 minutes) the
enantiomers
of the title compound from Step 4 above were isolated as yellow oils. Mass
spectra and iH
NMR spectra of both enantiomers were identical in all respects to those of the
Step 4
racemic compound. The entire 1.5 g sample of the more rapidly eluting
enantiomer was
further purified by flash chromatography utilizing the aforedescribed Biotage
Flash 401 iT"~
silica gel chromatography module (32-63 micron mesh factory packed cartridges;
eluting with
hexanes/ethyl acetate = 8:2 in volume afforded 1.34 g of purified enantiomer
as a colorless
syrup.
MS m/z 340 (M+1 ).
'H NMR (400 MHz, CDCI3) 8
Step 6
Enantiomeric 3-Ethyl-8-piperidin-3-yl-auinoline (both enantiomers)
Dissolution of either purified compound from the previous step with an ethyl
acetate/hydrogen chloride saturated solution (0.25 ml of hydrogen chloride
saturated ethyl
acetate per 10 mg of tert-butyloxycarbonyl functionalized substrate; 4 hours
reaction time at
ambient temperature) yielded the corresponding deprotected enantiomer title
compound of
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-45-
Step 5 as a mono-hydrochloric acid salt in quantitative yield. The free base
of either
enantiomeric title compound hydrocliloric acid salt was obtained in
quantitative yield as a
colorless amorphous solid by dissolution of the salt form into a vigorously
stirred (pH 10)
aqueous sodium hydroxide/ethyl acetate biphasic mixture, separation and
(anhydrous sodium
sulfate drying) of the organic extract, followed by solvent removal in vacuo.
The mass spectra
and'H NMR spectra of the enantiomeric free base compounds are identical in all
respect to
those of the previously described (Step 3 title compound) racemic counterpart.
EXAMPLE 2
ENANTIOMERIC (BOTH ENANTIOMERS) AND RACEMIC 3-ETHYL-7-METHYL-8-
PIPERIDIN-3-YL-QUINOLINE
Step 1
3-(2-Methyl-6-nitro-phenyl)-pyridine
To a solution of 2-bromo-3-nitrotoluene 5.0 g (23 mmol) in tetrahydrofuran
(180 ml);
diethyl-3-pyridyl borane (3.89 g, 26 mmol), bis-triphenylphosphine palladium
(II) chloride (2.42
g, 3.45 mmol), and a solution of sodium carbonate (12.19 g, 115 mmol) in water
(60 ml) were
sequentially added. The resulting well-stirred reaction mixture was then
heated at 75°C for 18
hours. The separated organic layer was diluted with ethyl acetate (200 ml) and
extracted with
an equal volume of water. The organic extract was then dried (anhydrous sodium
sulfate)
and concentrated in vacuo to afford a brown oil (9.4 g). Flash chromatography
of the entire
sample (silica gel, 47-61 micron mesh; elution with ethyl acetate/hexanes =
1:1 in volume)
afforded the title compound (2.40 g, 48% yield) as an amber oil.
'HNMR (400 MHz, CDCI3) 8 8.66 (m, 1 H), 8.47 (m, 1 H), 7.80 (m, 1 H), 7.61 (m,
1 H),
7.55 (m, 1 H), 7.42 (m, 2H), 2.11 (5, 3H) ppm.
Step 2
3-Methyl-2-pyridin-3-yl-phen lamine
The title compound from the previous step (2.40 g, 12 mmol) dissolved in
ethanol (50
ml) was hydrogenated (40 psi; 275 mg platinum oxide catalyst) for 3 hours. The
catalyst was
filtered and the solvent was removed in vacuo yielding an amber oil (1.4 g).
Flash
chromatography of the entire sample (silica gel, 41-67 micron mesh; elution
with methylene
chloride/methanol = 96:4 in volume) afforded the title compound (1.40 g, 69%
yield) as am
amber oil. TLC Rf (silica gel plates; elution with methylene chloride/methanol
= 96:4 in
volume; UV detection): 0.35.
'HNMR (450 MHz, CDCI3) 8 8.62 (m, 1 H), 8.50 (m, 1 H), 7.61 (m, 1 H), 7.40 (m,
1 H),
7.04 (m, 1 H), 6.70 (m, 1 H), 6.60 (m, 1 H), 3.25 (br s, 2H), 2.00 (s, 3H)
ppm.
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-46-
Step 3
3-Ethyl-7-methyl-8-pyridin-3-yl-cl uinol ine
A reaction mixture prepared by combining the title compound of the previous
step
(800 mg, 4.3 mmol), concentrated sulfuric acid (660 pl, 12 mmol), and sodium
meta
nitrobenzene sulfonate (544 mg, 24 mmol) in water (450 ~I) was well stirred
and heated to
100°C while 2-ethyl acrolein (1.26 ml, 13 mmol) was added dropwise over
4 minutes. The
reaction mixture was heated at 100°C; then at 120°C for two
hours. The reaction was then
cooled to 100°C, and an additional 1.26 ml (13 mmol) of 2-ethyl
acrolein was added dropwise
over several minutes. After further heating at 120°C for 2 hours, water
(10 ml) was added
and the solution was made basic (pH 12) with sodium hydroxide. The solution
was then
extracted with three 25 ml portions of methylene chloride. The combined
organic extracts
were dried (anhydrous sodium sulfate), and concentrated in vacuo to afford an
oil (2.36 g).
Flash chromatography of the entire sample (silica gel, 41-67 micron mesh;
elution with
methylene chloridelmethanol = 97:3 in volume) afforded the title compound as a
colorless oil
(567 mg, 53% yield). TLC Rf (silica gel plates, elution with methylene
chloride/methanol =
97:3 in volume; UV detection): 0.31.
MS m/z 249 (M+1 ).
'3C NMR (125 MHz, CDCI3) 8 152.0, 151.2, 148.1, 145.8, 138.4, 136.9, 135.7,
135.1,
133.4, 129.6, 127.4, 126.8, 123.2, 26.3, 21.2, 15.4 ppm.
Step 4
Racemic 3-Ethyl-7-methyl-8-piperidin-3-yl-auinoline
To a solution of the title compound from the previous step (567 mg, 2.3 mmol)
in
anhydrous tetrahydrofuran (20 ml), a solution of lithium triethylborohydride
(1.0 M in
anhydrous tetrahydrofuran; 8.1 ml, 8.1 mmol; Aldrich Chemical Company) was
added
dropwise over several minutes. After stirring at ambient temperature for 3
hours, an
additional 4.05 ml (4.05 mmol) of 1.0 M triethylborohydride in anhydrous
tetrahydrofuran was
added dropwise. After 3 additional hours of stirring at ambient temperature,
the reaction was
quenched by dropwise addition of methanol. Saturated aqueous sodium carbonate
was
added, and the resulting mixture was extracted with three 25 ml portions of
methylene
chloride. The combined organic extracts were dried (anhydrous sodium sulfate)
and
concentrated in vacuo, yielding an oil (670 mg). Flash chromatography of the
entire sample
(silica gel, 47-61 micron mesh; elution with methylene
chloride/methanol/concentrated
aqueous ammonium hydroxide = 79:20:1 in volume) afforded the title compound
(87 mg, 15%
yield) as a colorless oil.
TLC Rf (silica gel plates; elution with methylene
chloride/methanol/concentrated
aqueous ammonium hydroxide = 58.75:40:1.25 in volume; UV detection): 0.14.
MS m/z 255 (M+1 ).
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-47-
'H NMR (400 MHz, CDCI3) S 8.71 (m, 1 H), 7.77 (m, 1 H), 7.46 (m, 1 H), 7.26
(m, 1 H),
4.2 (m, 1 H), 3.5 (m, 2H), 2.9 (m, 2H), 2.75 (q, 2H, J = 7), 2.52 (s, 3H),
1.70 (m, 4H), 1.30 (t,
3H, J = 7) ppm.
Step 5
Enantiomeric (Both Enantiomers)
Utilizing analogously the procedure of Step 4/Example1, the racemic title
compound
of the previous step of this example was converted to the corresponding
racemic nitrogen
substituted tert-butoxycarbonyl compound, the separated/purified enantiomers
of which were
then isolated by the methodology of Step 5/Example1. Finally, by the procedure
of Step
6/Example1, the enantiomers of the title compound of the previous step of this
example were
prepared in both mono-hydrochloride and free base form.
EXAMPLE 3
ENANTIOMERIC (BOTH ENANTIOMERS) AND RACEMIC 3.6-DIMETHYL-8-PIPERIDIN-3-
YL-QUINOLINE
Step 1
4-Methyl-2-pyridin3-yl-phenylamine
To a mixture consisting of a tetrahydrofuran (125 ml) solution of 2-bromo-4-
methylaniline (2.67 ml, 21 mmol), diethyl-3-pyridyl borane (3.08 g, 24 mmol),
and bis
(triphenylphosphine) palladium (II) chloride (2.21 g ,0.32 mmol), an aqueous
solution sodium
carbonate (11.13 g 10.5 mmol in 44 ml of water) was added. The well-stirred
reaction mixture
was heated at 75°C for 18 hours. The upper layer of the cooled biphasic
mixture was
separated, dried (anhydrous sodium sulfate), and then filtered through celite.
Solvent
removal in vacuo yielded an oil (6.5g). Flash chromatography of the entire
sample (silica gel;
elution with methylene chloride/methanol = 95:5 in volume) afforded the title
compound
(1.85g, 48% yield) as a colorless amorphous solid. TLC Rf (silica gel plates;
elution with
methylene chloride/methanol = 95:5; UV detection) :0.53.
MS m/z 185 (M+1 ).
'3C NMR (125 MHz, CDCI~) 8 150.2, 148.5, 141.5, 136.8, 135.6, 131.2, 130.1,
128.4,
124.0, 123.7, 116.3, 20.6 ppm.
Step 2
3 6-Dimethyl-8-pyridin-3-yl-auinoline
To a solid sample of the title compound from the previous step (1.85 g, 10
mmol)
concentrated sulfuric acid (18 M, 27.5 mmol 1.52 ml) was slowly added,
followed by addition
of sodium meta-nitrobenzene sulfonate (1.26 g, 56 mmol) and water (1.05 ml).
The well-
stirred mixture was heated to 100°C while 2-methyl acrolein (4.97 ml,
60 mmol) was added
dropwise over 5 minutes. After stirring at 100°C for 1/2 hour, the
reaction temperature was
elevated to 140°C with continued stirring for 3 hours. After quenching
with ice, the reaction
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-48-
mixture was made basic (pH 12) by addition of 50% aqueous sodium hydroxide.
The mixture
was then extracted with three 30 ml portions of methylene chloride. The
combined organic
extracts were dried (anhydrous sodium sulfate) and the solvent was removed in
vacuo to
afford an amber oil. Flash chromatography of the entire sample (silica gel; 41-
67 micron
mesh; elution with methylene chloride/methanol = 96:4 in volume) afforded the
title compound
(650 mg, 28% yield) as an amorphous solid. TLC Rf (silica gel plates; elution
with methylene
chloride/methanol = 96:4 in volume; UV detection) :0.24.
MS m/z 235 (M+1 ).
~3C NMR (125 MHz, CDCI3) 8 151.8, 1,51.0, 148.4, 138.4, 136.7, 136.3, 135.5,
134.5,
131.7, 130.9, 130.1, 128.9, 126.9, 123.0, 21.8, 18.8 ppm.
Step 3
3 6-Dimethyl-8-piperidin-3-yl-auinoline
To a solution of the title compound from the previous step (650 mg, 28 mmol)
in
anhydrous tetrahydrofuran (20 ml), a 1.0 M solution of lithium
triethylborohydride (9.70 ml, 9.7
mmol); Aldrich Chemical Co.) was added over several minutes. After stirring
the reaction
mixture for 2 hours at ambient temperature, an additional 2.8 ml (2.8 mmol) of
1.0 M lithium
triethylborohydride in tetrahydrofuran was added; and ambient temperature
stirring was
continued for an additional 1 hour. The reaction was quenched by slow,
cautious addition of
methanol. Saturated aqueous sodium carbonate (15 ml) and methylene chloride
were added,
and the resulting mixture was extracted with three 25 ml portions of methylene
chloride. The
combined organic extracts were dried (anhydrous sodium sulfate) and
concentrated in vacuo
to afford an oil (600 mg). Flash chromatography of the entire sample (silica
gel, 41-67 micron
mesh; initial elution with methylene chloride/methanol/concentrated aqueous
ammonia =
84:15:1 in volume, followed by elution with methylene
chloride/methanol/concentrated
aqueous ammonia = 73.75:25:1.25 in volume) afforded the title compound (100
mg, 15%
yield) as a colorless oil. TLC Rf (silica gel plates; elution with methylene
chloride/methanol/concentrated aqueous ammonium hydroxide = 82:15:1 in volume;
UV
detection) :0.25.
MS mlz 241 (M+1 ).
~3C NMR (125 MHz, CDCI3) b 150.6, 143.3, 142.6, 136.2, 134.7, 130.4, 128.6,
127.7,
124.5, 53.6, 46.9, 37.8, 31.5, 27.7, 22.0, 18.8 ppm.
Step 4
Enantiomeric (Both Enantiomers)
Utilizing analogously the procedure of Step 4/Example 1, the racemic title
compound
of the previous step of this example was converted to the corresponding
racemic nitrogen
substituted tert-butoxycarbonyl compound, the separated/purified enantiomers
of which were
then isolated by the methodology of Step 5/Example 1. Finally, by the
procedure of Step
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-49-
6/Example1, the enantiomers of the title compound of the previous step of this
example were
prepared in both mono-hydrochloride and free base form.
EXAMPLE 4
ENANTIOMERIC (BOTH ENANTIOMERS) AND RACEMIC 3.7-DIMETHYL-8-PIPERIDIN-3-
YL-QUINOLINE
Step 1
3-(2-Methyl-6-nitro-~henyl)-pyridine
To a mixture consisting of 2-bromo-3-nitrotoluene (5.0 g, 23 mmol) in
tetrahydrofuran
(180 ml), diethyl-3-pyridyl borane (3.89 g, 26 mmol), and bis
(triphenylphosphine) palladium
(II) chloride (2.42 g, 3.45 mmol), a solution of sodium carbonate (12.19 g,
115 mmol) in water
was added. The well-stirred reaction mixture was heated at 75°C for 18
hours. The organic
and aqueous layers were separated, and the aqueous phase was extracted with
ethyl acetate
(100 ml). The combined organic extracts were dried (anhydrous sodium sulfate)
and
concentrated in vacuo to afford an oil (9.6 g). Flash chromatography of the
entire sample
(silica gel, 41-67 micron mesh; elution with ethyl acetate/hexanes = 1:1 in
volume) afforded
the title compound as a light yellow oil (1.87 g, 38% yield). TLC Rf (silica
gel plates; elution
with ethyl acetate/hexanes = 1:1 in volume; UV detection) :0.50.
MS m/z 215 (M+1 ).
~3C NMR (125 MHz, CDCI3) 8 171.4, 148.1, 148.0, 139.6, 137.2, 134.7, 133.0,
131.7,
129.2, 123.9, 122.0, 21.0 ppm
Step 2
3-Methyl-2-pyridin-3-yl-phen lamine
The title compound from the previous step (1.87 g, 8.7 mmol) dissolved in
ethanol (40
ml) was hydrogenated (40 psi; 200 mg platinum oxide catalyst) for 4 hours. The
catalyst was
removed by filtration through celite. The filtrate was concentrated in vacuo
to afford an amber
oil (1.2 g). Flash chromatography of the entire sample (silica gel, 41-67
micron mesh; elution
with methylene chloride/methanol = 96:4 in volume) afforded the title compound
(1.17 g, 74%
yield) as a tacky solid. TLC Rf (silica gel plates; elution with methylene
chloride/methanol =
96:4 in volume; UV detection) :0.38.
MS m/z 185 (M+1 ).
~3C NMR (125 MHz, CDCI3) 8 151.1, 148.8, 144.4, 138.2, 137.4, 134.3, 129.1,
124.2,
123.8, 120.4, 113.3, 20.9 ppm.
Step 3
3,7-Dimethyl-8-pyridin-3-yl-cpuinoline
To a solid sample of the title compound from the previous step (1.17 g, 6.4
mmol),
concentrated sulfuric acid (18 M, 17.6 mmol 980 pl) was slowly added, followed
by addition of
sodium meta-nitrobenzene sulfonate (800 mg, 3.6 mmol) and water (680 ~,I). The
well-stirred
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-50-
mixture was heated at 100°C while 2-methyl acrolein (1.59 ml, 19.2
mmol) was added
dropwise over 5 minutes. After stirring at 100°C for 1/2 hour, an
addition 1.59 ml (19.2 mmol)
portion of 2-methyl acrolein was added dropwise; and the well-stirred reaction
mixture was
then heated at 140°C for 3 hours. Thin layer chromatography (TLC)
inspection of a reaction
aliquot revealed incomplete reaction. The reaction mixture temperature was
lowered to
100°C, and a final 1.59 (19.2 mmol) portion of 2-methyl acrolein was
added, with subsequent
heating at 140°C for 2 more hours to complete reaction. The reaction
mixture was poured
into ice (50 g) made basic (pH 10) by addition of 50% aqueous sodium
hydroxide, and then
extracted with three 50 ml portions of methylene chloride. The combined
organic extracts
were dried (anhydrous sodium sulfate) and concentrated in vacuo to afford an
oil (3.2 g). .
Flash chromatography of the entire sample (silica gel, 47-61 micron mesh;
elution with
methylene chloride/methanol = 96:4 in volume) afforded the title compound (328
mg, 22%
yield) as an amber oil. TLC Rf (silica gel plates; elution with methylene
chloride/methanol =
96:4 in volume, UV detection) :0.34.
~H NMR (400 MHz, CDCI3) s 8.6 (m, 3H), 7.88 (m, 1 H), 7.64 (m, 2H), 7.40 (m,
2H),
2.42 (s, 3H), 2.38 (s, 3H) ppm.
Step 4
Racemic 3,7-Dimethyl-8-piperidin-3-yl-quinoline
To a solution of the title compound from the previous step (328 mg, 1.4 mmol)
in
anhydrous tetrahydrofuran (10 ml), a 1.OM solution of lithium
triethylborohydride in
tetrahydrofuran (4.90 ml, 4.9 mmol) was added dropwise. The reaction mixture
was stirred at
ambient temperature for 3 hours. After dropwise addition of a second portion
of 1.0 M lithium
triethylborohydride in tetrahydrofuran (1.40 ml, 1.4 mmol), ambient
temperature stirring was
continued for an additional 1.5 hours. The reaction was quenched by cautious
dropwise
addition of methanol (1 ml). Saturated aqueous sodium carbonate and methylene
chloride
were added to the well-stirred mixture, which was then extracted with three 30
ml portions of
methylene chloride. The combined organic extracts were dried (anhydrous sodium
sulfate)
and concentrated in vacuo to afford a yellow oil (470 mg). Flash
chromatography of the entire
sample (silica gel, 47-61 micron mesh; initial elution with methylene
chloridelmethanollconcentrated aqueous ammonium hydroxide = 84:15:1 in volume,
followed
by elution with methylene chloride/methanol/ concentrated aqueous ammonium
hydroxide =
59:40:1 in volume) afforded the title compound (45 mg, 13% yield) as an
amorphous foam.
TLC Rf (silica gel plates; elution with methylene chloride/methanol/
concentrated aqueous
ammonium hydroxide = 84:15:1 in volume, UV detection) :0.28.
MS m/z 241 (M+1 ).
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-51-
H NMR (400 MHz, CDC13) b 8.65 (m, 1 H), 7.76 (m, 1 H), 7.45 (m, 1 H), 7.26 (m,
1 H),
4:32 m, 1 H), 3.22 (m, 1 H), 3.08 (m, 1 H), 2.92 (m, 2H), 2.75 (s, 3H), 2.42
(m, 4H), 1.80 (m,
3H) ppm.
Steih 5
Enantiomeric (Both Enantiomers)
Utilizing analogously the procedure of Step 4/Example 1, the racemic title
compound
of the previous step of this example was converted to the corresponding
racemic nitrogen
substituted tent-butoxycarbonyl compound, the separated/purified enantiomers
of which were
then isolated by the methodology of Step 5/Example 1. Finally, by the
procedure of Step
6/Example 1, the enantiomers of the title compound of the previous step of
this example were
prepared in both mono-hydrochloride and free base form.
EXAMPLE 5
ENANTIOMERIC (BOTH ENANTIOMERS) AND RACEMIC 3.5-DIMETHYL-8-PIPERIDIN-3-
YL-QUINOLINE
, Step 1
3-(4-Methyl-2-nitro-phenyl)-pyridine
To a well-stirred mixture consisting of 4-bromo-3-nitrotoluene (4.0 g, 18.5
mmol) in
tetrahydrofuran (145 ml), diethyl-3-pyridylborane (3.12 g, 21 mmol), and bis
(triphenylphosphine) palladium (II) chloride (1.94 g, 2.8 mmol), a solution of
sodium carbonate
(9.8 g, 92.5 mmol) in water (50 ml) was added. The reaction mixture was heated
to 75°C for
18 hours. The organic layer of the biphasic mixture was dried (anhydrous
sodium sulfate),
and the solvent was removed in vacuo to afford an oil (7.0 g). Flash
chromatography of the
entire sample (silica gel, 47-61 micron mesh; elution with ethyl
acetate/hexanes = 1:1 in
volume) afforded the title compound (2.58 g, 65% yield) as a light yellow
foam. TLC Rf (silica
gel plates; elution with ethyl acetate/hexanes = 1:1 in volume, UV detection)
:0.51.
MS m/z 215 (M+1 ).
~3C NMR (125 MHz, CDCI3) 8 148.4, 147.9, 140.3, 136.4, 134.3, 133.9, 132.1,
130.1,
125.2, 123.6, 21.1 ppm.
Ste .~~2
5-Methyl-2-pyridin-3- I-rLphenylamine
The title compound from the previous step (2.58 g, 12 mmol) dissolved in
ethanol (65
ml) was hydrogenated (40 psi; 275 mg platinum oxide catalyst) for 3 hours. The
catalyst was
removed by filtration through celite, and the filtrate was concentrated in
vacuo to afford an
amber oil (2.27 g). Flash chromatography of the entire sample (silica gel, 47-
61 micron mesh;
elution with methylene chloride/methanol = 95:5 in volume) afforded the title
compound (1.6
g, 73% yield) as a yellow oil. TLC Rf (silica gel plates; elution with
methylene
chloride/methanol = 95:5 in volume, UV detection) :0.59.
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-52-
MS m/z 185 (M+1 ).
'3C NMR (125 MHz, CDCI3) 8 150.2, 148.2, 143.8, 139.7, 135.6, 130.7, 123.8,
121.2,
120.2, 116.7, 21.4 ppm.
Step 3
3 5-Dimethyl-8-pyridin-3-yl-auinoline
To a solid sample of the title compound from the previous step (1.60 g, 8.7
mmol),
concentrated sulfuric acid (18 M, 1.32 ml, 23.9 mmol) was slowly added,
followed by addition
of sodium meta-nitrobenzene sulfonate (1.10 g, 4.9 mmol) and water (1.0 ml).
The well-
stirred mixture was heated at 100°C while 2-methyl acrolein (4.31 ml,
52 mmol) was added
dropwise over a 5 minute period. After 1/2 hour heating at 100°C, the
reaction was heated for
6 hours at 140°C. The mixture was diluted with water (50 ml) and the pH
was adjusted to 10
with 50% aqueous sodium hydroxide. Three successive extractions were made with
40 ml
portions of methylene chloride. The combined organic extracts were dried
(anhydrous
sodium sulfate) and concentrated in vacuo to afford an oil 1.06 g. Flash
chromatography of
the entire sample (silica gel, 47-61 micron mesh; elution with methylene
chloride/methanol =
97:3 in volume) afforded the title compound as a colorless oil (220 mg, 10.8%
yield). TLC Rf
(silica gel plates; elution with methylene chloride/methanol = 97:3 in volume,
UV detection)
:0.20.
MS m/z 235 (M+1 ).
Step 4
Racemic 3 5-Dimethyl-8-piperidin-3-yl-auinoline
To a well-stirred solution of the title compound from the previous step (220
mg, 0.94
mmol) in tetrahydrofuran (7.5 ml), a 1.0 M solution of lithium
triethylborohydride in
tetrahydrofuran (3.30 ml, 3.3 mmol) was added dropwise over several minutes.
The reaction
was stirred at ambient temperature for 4 hours, and quenched by cautious
dropwise addition
of methanol. Methylene chloride (25 ml) and aqueous sodium carbonate (25 ml)
were added
to the well-stirred mixture, which was then extracted with two 30 ml portions
of methylene
chloride. The combined organic extracts were dried (anhydrous sodium sulfate)
and
concentrated in vacuo to afford an orange oil (440 mg). Flash chromatography
of the entire
sample (silica gel, initial elution with methylene
chloride/methanol/concentrated aqueous
ammonium hydroxide - 84:15:1 in volume followed by elution with methylene
chloride/methanol/concentrated aqueous ammonium hydroxide = 73.75:25:1.25 in
volume)
afforded the title compound (17 mg, 7.5% yield) as a colorless oil. TLC Rf
(silica gel plates;
elution with methylene chloride/methanollconcentrated aqueous ammonium
hydroxide =
73.75:25:1.25 in volume, UV detection) :0.33.
MS m/z 241 (M+1 ).
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-53-
'3C NMR (125 MHz, CDCI3) 8 150.9, 144.8, 140.8, 132.1, 131.8, 130.0, 127.7,
127.0,
125.0, 53.4, 46.7, 37.7, 31.3, 27.4, 19.1, 18.8 ppm.
Step 5
Enantiomeric (Both Enantiomers)
Utilizing analogously the procedure of Step 4/Example 1, the racemic title
compound
of the previous step of this example was converted to the corresponding
racemic nitrogen
substituted tent-butoxycarbonyl compound, the separated/purified enantiomers
of which were
then isolated by the methodology of Step 5/Example 1. Finally, by the
procedure of Step
6/Example1, the enantiomers of the title compound of the previous step of this
example were
prepared in both mono-hydrochloride and free base form.
EXAMPLE 6
ENANTIOMERIC (BOTH ENANTIOMERS) AND RACEMIC 6-CHLORO-3-METHYL-8
PIPERIDIN-3-YL-QUINOLINE
Step 1
6-Chloro-3-methyl-8-pyridin-3-yl-auinoline
To a well-stirred mixture consisting of 2-bromo-4-chloroaniline (5.0 g, 24
mmol) in
tetrahydrofuran (180 ml), diethyl-3-pyridyl borane (4.07 g, 28 mmol), and bis
(triphenylphosophine) palladium (II) chloride (2.53 g, 3.6 mmol), a solution
of sodium
carbonate (12.72 g, 120 mmol) in water (60 ml) was added. ' The reaction was
then heated at
75°C for 18 hours. The layers of the biphasic mixture were separated,
and the aqueous
phase was extracted with an equal volume of ethyl acetate. The combined
original reaction
organic phase and ethyl acetate extract were dried and concentrated in vacuo
to afford an oil
(9.4 g). Flash chromatography of the entire sample (silica gel; initial
elution with ethyl
acetate/hexanes = 8:2 in volume followed by elution with pure hexane) afforded
the title
compound as a colorless oil (3.64 g, 74% yield). TLC Rf (silica gel plates;
elution with ethyl
acetate, UV detection) :0.46.
Step 2
6-Chloro-3-methyl-8-pyridin-3-yl-auinoline
To a well-stirred mixture consisting of the title compound from the previous
step (3.64
g, 17.8 mmol), sodium 3-nitrobenzene sulfonate (2.33 g, 10 mmol), and water
(1.79 ml), 2.72
ml (49 mmol) of concentrated sulfuric acid (18 M) was cautiously added. The
reaction mixture
was heated to 100°C, and 2-methyl acrolein (4.39 ml, 53 mol) was added.
The reaction was
stirred at 100°C for 20 minutes, and then stirred at 140°C for 2
hours. After lowering the
reaction temperature back to 100°C, another 4.39 ml (53 mmol) portion
of 2-methyl acrolein
was added dropwise. The reaction temperature was again elevated to
140°C and maintained
at that temperature for 1.5 hours. An equal volume of ice was used to quench
the reaction;
and the resulting mixture was made basic (pH = 12) by addition of 50% aqueous
sodium
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-54-
hydroxide. The mixture was then extracted with 75 ml of methylene chloride.
The organic
extract was dried (anhydrous sodium sulfate) and concentrated in vacuo,
yielding a dark oil.
Flash chromatography of the entire sample (silica gel, 47-61 micron mesh;
elution with initially
ethyl acetate/hexanes = 8:2 in volume, steadily increasing the polarity of the
eluting solvent,
finally to pure ethyl acetate) afforded the title compound as a colorless oil
(343 mg, 7.6%
yield). TLC Rf (silica gel plates; elution with ethyl acetate/hexanes = 8:2 in
volume, UV
detection) :0.35.
MS m/z 255 (M+1 ).
Step 3
Racemic 6-Chloro-3-methyl-8-piperidin-3- rLquinoline
To a well-stirred solution of the title compound of the previous step (150 mg,
0.59
mmol) in anhydrous tetrahydrofuran (5 ml) a 1.0 M solution of lithium
triethylborohydride in
tetrahydrofuran (2.1 ml, 2.1 mmol) was added, and the resulting reaction
mixture was stirred
at ambient temperature for 4 hours, prior to quenching by cautious addition of
200 gl of
methanol. Saturated aqueous sodium carbonate (10 ml) and methylene chloride
were added,
with vigorous stirring. The mixture was then extracted with three 15 ml
portions of methylene
chloride. The combined organic extracts were dried (anhydrous sodium sulfate)
and
concentrated in vacuo, yielding a (230 mg) yellow oil. Flash chromatography of
the entire
sample (silica gel, 47-61 micron mesh; elution with initially methylene
chloride/methanol/concentrated aqueous ammonium hydroxide = 97.25:2.50:0.25 in
volume,
steadily increasing the polarity of the eluting system to a final methylene
chloride/methanol/concentrated aqueous ammonium hydroxide = 89:10:1 in volume)
afforded
the title compound (17 mg, 11 % yield) as a colorless oil. TLC Rf (silica gel
plates; elution with
methylene chloride/methanol/aqueous concentrated ammonium hydroxide = 89:10:1
in
volume, UV detection) :0.39.
MS mlz 261 (M+1 ).
'3C NMR (175 MHz, CDCI3) s 151.6, 145.4, 143.2, 134.4, 132.4, 131.6, 129.2,
126.4,
124.1, 53.5, 46.8, 37.9, 31.3, 27.5, 18.8 ppm.
Step 4
Enantiomeric (Both Enantiomers)
Utilizing analogously the procedure of Step 4/Example 1, the racemic title
compound
of the previous step of this example was converted to the corresponding
racemic nitrogen
substituted tent-butoxycarbonyl compound, the separated/purified enantiomers
of which were
then isolated by the methodology of Step 5/Example 1. Finally, by the
procedure of Step
6/Example 1, the enantiomers of the title compound of the previous step of
this example were
prepared in both mono-hydrochloride and free base form.
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-55-
EXAMPLE 7
ENANTIOMERfC~BOTH ENANTIOMERS) AND RACEMIC 4-METHYL-8-PIPERIDIN-3-YL
QUINOLINE
Step 1
3-(2-Nitro-phenyl)-pyridine
To a mixture consisting of 1-bromo-2-nitrobenzene (2.12 g, 8.7 mmol), diethyl
(3-
pyridyl) borane (1.47 g, 10.0 mmol), and bis (triphenylphosphine) palladium
(II) chloride (913
mg, 1.3 mmol) in tetrahydrofuran (40 ml), sodium carbonate (4.24 g, 40.0 mmol)
was added,
and the resulting reaction mixture was heated at reflux for 4 hours. Water (40
ml) was added
to the cooled reaction mixture which was then extracted with three 25 ml
portions of ethyl
acetate. The combined organic extracts were dried (anhydrous sodium sulfate)
and
concentrated in vacuo to afford a tacky residue. Flash chromatography of the
entire sample
(silica gel, 47-61 micron mesh; elution initially with methylene chloride and
finally with
methylene chiorideimethanol = 98:2 in volume) afforded the title compound as a
viscous
amber oil (713 mg, 41 % yield). Subsequent eluent contained less pure product
which was
further purified by a similar flash chromatography, eluting with methylene
chlorideimethanol =
99:1 in volume, thus affording an additional 488 mg (28% yield) of the
purified title compound,
again as a viscous amber oil.
MS m/z 200 (M+ 1 ).
'H NMR (400 MHz, CDCI3) 8 8.65 (1 H, M), 8.58 (1 H, m), 7.99 (1 H, m), 7.60-
7.73 (2H,
overlapping multiplets), 7.56 (1 H, m), 7.43 (1 H, m), 7.36 (1 H, m) ppm.
Step 2
2-Pyridin-3-YI-phen lay mine
A solution of the title compound from the previous step (16.3 g, 81 mmol) in
methanol
(300 ml) was hydrogenated (50 psi; 1.65 g of platinum oxide catalyst) for 3.5
hours. The
catalyst was filtered, and the filtrate was concentrated in vacuo to afford a
viscous amber oil.
Flash chromatography of the entire sample (silica gel, 47-61 micron mesh;
elution with ethyl
acetate) afforded the title compound 13.5 g, (94.6% yield) as an amber oil.
MS m/z 171 (M+ 1 ).
' H NMR (400 MHz, CDCI3) 8 8.71 (1 H, m), 8.58 (1 H, m), 7.80 (1 H, m), 7.37
(1 H, m),
7.19 (1 H, m), 7.10 (1 H, m), 6.84 (1 H, m), 6.78 (1 H, m), 3.70 (2H, broad s)
ppm.
' Std
Racemic 2-Piperidin-3-y~henylamine
To a well-stirred solution of the title compound from the previous step (1.83
g, 10.8
mmol) in tetrahydrofuran (5.0 ml), 37.8 ml (37.8 mmol) of 1 M lithium
triethylborohydride in
tetrahydrofuran was added dropwise over a 15 minute period. The reaction
mixture was then
stirred at ambient temperature for 18 hours. The reaction was then quenched by
cautious
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-56-
dropwise addition of water (100 ml). Solvents were removed in vacuo, yielding
a viscous oil.
Flash chromatography of the entire sample (silica gel, 47-61 micron mesh;
elution with
methylene chloride/methanol/concentrated aqueous ammonium hydroxide = 90:9:1
in
volume) afforded (after appropriate combining of chromatography column
fractions of similar
purity as per thin layer chromatography inspection) a 470 mg viscous syrup
sample (which
solidified on standing, shown to be predominantly title compound product by
NMR inspection)
and a considerably less pure sample of the desire product (600 mg, a viscous
oil). Trituration
of the 470 mg sample with methylene chloride (2 ml) afforded a 100 mg sample
of the title
compound (a colorless amorphous solid, isolated by suction filtration; no
detectable impurities
by NMR inspection). A second flash chromatography of the entire aforedescribed
600 mg
impure sample (same chromatography conditions) yielded an additional 160 mg of
the
purified title compound product (colorless amorphous solid; 260 mg, 14%
yield).
MS m/z 177 (M+ 1 ).
'H NMR (400 MHz, CDCI3) 8 9.40 (1 H, m), 9.33 (1 H, m), 9.10 (1 H, m), 9.05 (1
H, M),
5.60 (2H. m), 5.0-5.34 (3H, overlapping multiplets), 4.30 (2H, m), 4.12 (2H,
m) ppm.
Step 4
Racemic 4-Methyl-8-piperidin-3-yl-auinoline
To a well-stirred slurry of the title compound from the previous step (254 mg,
1.49
mmol) in ethanol, 0.12 ml of 12 N hydrochloric acid was added, affording a
clear solution.
Ferric chloride hexahydrate (557 mg, 2.06 mmol) and zinc chloride (24 mg,
10.18 mmol) were
added, and the reaction mixture was heated to 60°C. Methyl vinyl ketone
(0.016 ml, 0.19
mmol) was added, and the reaction temperature was maintained at 60°C
for 1 hour, during
which time, additional 0.016 ml portions of methyl vinyl ketone were added at
10 minute
intervals. The reaction was then refluxed for 2 hours. Volatiles were removed
in vacuo,
yielded a viscous syrup. The residual syrup was made basic by thorough
trituration with 10
ml of 3 N aqueous sodium hydroxide. The resulting mixture was extracted with
three 10 ml
portions of methylene chloride. The combined organic extracts were, in turn,
extracted with
an equal volume of brine, dried (anhydrous sodium sulfate), and concentrated
in vacuo,
yielding a viscous oil. Several repetitive flash chromatography procedures
utilizing the entire
crude product sample (silica gel, 47-61 micron mesh; eluting in the initial
procedure with
methylene chloride/methanol = 98:2 in volume, and in the repeated
chromatography with
100% methylene chloride) yielded the purified product as a colorless oil (131
mg, 39% yield).
MS m/z 227 (M+1 ).
~H NMR (400 MHz, CDCI3) 8 8.76 (1 H, m), 7.85 (1 H, m), 7.58-7.48 (2H,
overlapping
multiplets), 7.22 (1 H, m), 4.14 (1 H, m), 3.34 (1 H, m), 3.19 (1 H, m), 2.68
(3H, s), 2.60-2.80
(2H, m), 2.1 (1 H, m), 1.90-1.6 (3H, m) ppm.
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-57-
Separation of Enantiomers of the Title Compound
Step 5
Racemic 3-(4-Methyl-4uinolin-8-yl-pi!peridine-1-carboxylic acid tent-butyl
ester
Utilizing the method of Example 1, Step 6, the entire 131 mg (0.58 mmol) of
the
racemic free base title compound from the previous step was converted into the
corresponding N-tert-butyloxycarbonyl functionalized title compound of this
step (yielding 120
mg, 63.4% yield, as a colorless oil).
MS m/z ( M+1 ).
'H NMR (400 MHz, CDCI3) 8 8.78 (1 H, m), 7.87 (1 H, m), 7.46-7.60 (2H,
overlapping
multiplets), 7.23 (1 H, m), 4.34 (1 H, m), 4.20 (1 H, m), 4.15 (1 H, m), 2.91
(1 H, m), 2.81 (1 H, m),
2.67 (3H, s), 2.12 (1 H, m), 1.76 (3H, m), 1.44 (9H, s) ppm.
Enantiomeric 3-(4-Methyl-c~uinolin-8-yl)-piperidine-1-carboxylic acid tent-
butyl ester
both enantiomers)
Step 6
Separation of the enantiomers of the Step 5 title compound
Utilizing the Waters Prep LC2000T"" Preparative Chromatography System
described
in Example 1 (ChiracalT"" OD 2.1 cm x 25 cm preparative column; mobile phase:
heptane/ethanol = 98:2 in volume with 0.025% diethyl amine as modifier; a flow
rate of 10
ml/minute; 134 mg of the title compound from the previous step dissolved in
methylene
chloride/methanol/mobile phase solution = 1:1:1 in volume; injecting 10 mg of
dissolved
compound at a time; with approximate retention times of 25 and 35 minutes) the
enantiomers
of the title compound from Step 5 above were isolated as colorless oils (31 mg
of the foster
eluting enantiomer and 17 mg of the slower eluting enantiomer were obtained).
The'H NMR
spectra of both enantiomers were identical in all respects to that of the
racemic title
compound of Step 5, this example.
Step 7
Enantiomeric 4-Methyl-8-piperidin-3-yl-auinoline (both enantiomers)
The entire 31 mg and 17 mg samples respectively of the faster and slower
eluting
enantiomeric title compounds prepared in the previous step were dissolved in
0.5 ml of
chloroform. A hydrogen chloride saturated diethyl ether solution (1 ml) was
added to each.
Both reaction mixtures were stirred for 18 hours at ambient temperature.
Solvent removal in
vacuo afforded 15 mg and 9.5 mg respectively of the title compound enantiomers
derived
from the faster and slower elution Step 6 enantiomeric compounds as colorless
amorphous
solids. NMR obtained with mono-hydrochloride salt of more rapidly eluted
enantiomer:
'H NMR (400 MHz, CDCI3) ~ 9.09 (1 H, m), 8.46 (1 H, m), 8.18 (1 H, m), 7.96-
8.09 (2H,
overlapping multiplets), 4.21 (1 H, m), 3.65 (1 H, m), 3.57 (1 H, m), 3.46 (1
H, m), 3.18 (1 H, m),
3.07 (3H, s), 2.08-2.33 (3H, m), 1.93-2.05 (1 H, m) ppm.
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-58-
EXAMPLE 8
ENANTIOMERIC (BOTH ENANTIOMERS) AND RACEMIC 3-METHYL-8-PIPERIDIN-3-YL
QUINOLINE
Step 1
3-Methyl-8-pyridin-3-yl-auinoline
A well-stirred mixture consisting of Example 7, Step 2 (3.20 g, 18.8 mr~iol),
2.51 g
(11.1 mmol) of sodium 3-nitrobenzene sulfonate, 5.1 g (51.9 mmol) of
concentrated sulfuric
acid, and water (1.89 ml) was heated to 100°C. 2-Methyl acrolein (1.0
ml~ 12.1 mmol) was
added, and the reaction temperature was maintained at 100°C for 1 hour.
The reaction
temperature was then elevated to 110°C, an additional 1 ml (12.1 mmol)
portion of 2-methyl
acrolein was added, and the 110°C reaction temperature was maintained
for 1 hour.
Subsequently, the above described sequence of elevating the reaction
temperature by 10°C
increments, followed by an addition of 1.0 ml of 2-methyl acrolein and an hour
of heating at
the newly established temperature was repeated three more times (with reaction
temperatures of 120°C, 130°C, and finally, 140°C). The
reaction temperature was then
lowered to and maintained at 90°C, affording an acidic aqueous phase
and a pliable tacky
gum. The acidic layer was carefully siphoned off, and the residual gum was
thoroughly
triturated/pulped with several 25 ml portions of 1 N hydrochloric acid. The
combined acidic
aqueous extracts were made basic (pH = 14) by addition of 50% aqueous sodium
hydroxide
and, in turn, extracted with two 50 ml portions of methylene chloride. The
organic extract was
dried (anhydrous sodium sulfate) and concentrated in vacuo to afford an amber
syrup. Flash
chromatography of the entire sample (silica gel, 47-61 micron mesh; elution
with ethyl
acetate) yielded the title compound (790 mg, 19.1 % yield) as a viscous
colorless syrup.
MS m/z 221 (M+1 ).
~ H NMR (400 MHz, CDCI3) 8 8.91 (1 H, m), 8.77 (1 H, m), 8.63 (1 H, m), 8.07
(1 H, m),
7.97 (1 H. m), 7.66 (1 H, m), 7.60 (1 H, m), 7.40 (1 H, m), 2.52 (3H, s) ppm.
Step 2
Racemic 3-Methyl-8-piperidin-3-yl-auinoline
To a solution of the title compound from the previous step (590 mg, 2.68 mmol)
in
tetrahydrofuran (8.0 ml), a 1 M solution of lithium triethylborohydride in
tetrahydrofuran (10.72
ml, 10.72 mmol) was added, and the reaction was stirred at ambient temperature
for 7 hours.
An additional 5.36 ml (5.36 mmol) portion of 1 M lithium triethylborohydride
in tetrahydrofuran
was added, and the reaction mixture was stirred at ambient temperature for 18
hours prior to
quenching by cautious dropwise addition of water (50 ml). Solvents were then
removed in
vacuo, and the residue was extracted with three 20 ml portions of methylene
chloride. The
organic extract was dried (anhydrous sodium sulfate) and concentrated in
vacuo, affording an
amber oil. Flash chromatography of the entire sample (silica gel, 47-61 micron
mesh; elution
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-59-
with methylene chloride/methanol/concentrated aqueous ammonium hydroxide =
90:9:1 in
volume) yielded the title compound (302 = mg, 49.8% yield) as a light orange
foam.
MS m/z 227 (M+1 ).
~H NMR (400 MHz, CDCI3) 8 8.76 (1H, m), 7.88 (1H, m), 7.58 (1H, m), 7.40-7.54
(2H,
overlapping multiplets), 4.09 (1 H, m), 3.33 (1 H, m), 3.17 (1 H, M), 2.60-
2.80 (2H, m), 2.50 (3H,
s), 2.09 (1 H, m), 1.56-1.93 (3H, m) ppm.
Separation of the enantiomers of the racemic title compound
Step 3
Enantiomeric 3-(3-Methyl-auinolin-8-yl-piperidine-1-carboxylic acid tert-butyl
ester
To a well-stirred solution of the racemic title compound from the previous
step (302
mg, 1.33 mmol) in methylene chloride (20 ml) containing 0.56 ml (4.0 mmol) of
triethylamine,
436 mg (2.0 mmol) of di-tert-butyl dicarbonate was added, and the resulting
reaction mixture
was stirred for 48 hours at ambient temperature. Aqueous saturated sodium
bicarbonate (20
ml) was added with efficient stirring. The mixture was then extracted with two
20 ml portions
of methylene chloride. The combined organic extracts were, in turn, extracted
with an equal
volume of brine, dried (anhydrous sodium sulfate), and finally, concentrated
in vacuo,
affording a viscous syrup. Flash chromatography of the entire sample (silica
gel, 47-61
micron mesh; elution with methylene chloride/methanol = 99:1 in volume)
affording the title
compound as a colorless oil.
MS m/z 326 (M+1 ).
~ H NMR (400 MHz, CDCI3) 8 8.77 (1 H, m), 7.87 (1 H, m), 7.40-750 (2H,
overlapping
multiplets), 7.60 (1 H, m), 4.34 (1 H, m), 4.21 (1 H, m), 4.09 (1 H, m), 2.92
(1 H, m), 2.80 (1 H, m),
2.50 (3H, s), 2.20 (1 H, m), 1.78 (3H, m), 1.44 (9H, s) ppm.
Step 4
Separation of the enantiomers of the Stela 2 title compound
Utilizing the Waters Prep LC2000T"~ Preparative Chromatography System
described
in Example 1 (ChiracalT"" OD 10 cmx x 50 cm preparative column; mobile phase:
hexanes/ethanol = 98:2 in volume with 0.025% diethyl amine as modifier; a flow
rate of 225
ml/minute; 247 mg of the racemic title compound from the previous step
dissolved in
methylene chloride/methanol = 1:1 in volume; injecting the entire 247 mg
sample of dissolved
compound as a single load; with approximate retention times of 25 and 40
minutes) the
enantiomers were separated. The process yielded 119 mg of the faster eluting
enantiomer.
The'N HMR spectra for the enantiomers are identical to those obtained with the
racemic title
compound of Step 3, above.
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-60-
Step 5
Enantiomeric 3-Methyl-8-piperidin-3-yl-auinoline (both enantiomers)
A 90 mg sample of the faster eluting title compound enantiomer isolated in the
previous step was dissolved in 1 ml of methanol. A saturated hydrogen chloride
solution in
diethyl ether was added, and the reaction mixture was stirred at ambient
temperature for 18
hours. Solvents and excess hydrochloric acid were removed in vacuo, affording
a colorless
glass. Trituration with ethyl acetate (10 ml) yielded the enantiomeric title
compound as an
amorphous solid mono-hydrochloride salt (61 mg).
Mono-hydrochloride salt:
~H NMR (400 MHz, CDCI3) 8 9.15 (1 H, m), 9.09 (1 H, m), 8.21 (1 H, m), 8.12 (1
H, m),
7.96 (1 H, m), 4.24 (1 H, m), 3.66 (1 H, m), 3.57 (1 H, m), 3.46 (1 H, m),
3.20 (1 H, m), 2.75 (3H,
s), 2.08-2.34 (3H, m), 1.90-2.08 (1 H, m) ppm.
The free base of the enantiomeric title compounds of this step was prepared by
the
method of Example 1, Step 6. The mass spectra and ~H NMR spectra of the
enantiomeric
title compound free bases are identical in all respects to the racemic title
compound free base
of Step 2, this Example.
EXAMPLE 9
ENANTIOMERIC (BOTH ENANTIOMERS) AND RACEMIC 3-ETHYL-8-PYRROLIDIN-3-YL-
QUINOLINE
Step 1
3-(3-Ethyl-auinolin-8-yl)-3-hydroxy-pyrrolidine-1-carboxylic acid tert-butyl
ester
To a well-stirred solution of the title compound of Example 1, Step 1 (2.10 g,
8.9
mmol) in 30 ml of anhydrous tetrahydrofuran chilled to and maintained at -
77°C, a 2.5 M
solution of n-butyl lithium in hexanes (3.60 ml, 8.9 mmol; Aldrich Chemical
Co.) is added
dropwise over a 10 minute period. The reaction was stirred at -77°C for
15 minutes betore
adding a solution of 3-oxo-pyrrolidine-1-carboxylic acid tert-butyl ester
(1.65 g, 8.9 mmol) in
anhydrous tetrahydrofuran (10 ml). The reaction mixture was allowed to warm to
ambient
temperature and stir at that temperature for 3 hours before quenching by
cautious dropwise
addition (with cooling) of saturated aqueous sodium bicarbonate (50 ml total).
The resulting
mixture was thoroughly extracted with three 20 ml of ethyl acetate. The
combined organic
extracts were, in turn, extracted with an equal volume portion of brine, dried
(anhydrous
sodium sulfate) and, finally, concentrated in vacuo, yielding a viscous syrup.
Flash
chromatography of the entire sample (silica gel, 47-61 micron mesh; elution
with methylene
chloride/methanol = 98:2 in volume) afforded the title compound (352 mg, 11.5%
yield) as a
viscous yellow oil.
MS m/z 343 (M+1 ).
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-61-
~ H NMR (400 MHz, CDC13) 8 8.70 (1 H, m), 7.96 (1 H, m), 7.70 (1 H, m), 7.53
(1 H, m),
7.44 (1 H. m), 4.14 (1 H, m), 3.93 (1 H, m), 3.60-3.76 (2H, m), 3.48-3.60 (1
H, m), 2.83 (2H, m),
2.42 (2H, m), 1.45 and 1.43 (9H, two singlets), 1.34 (3H, m) ppm.
Step 2
8-(2 5-Dihydro-1 H-pyrrol-3-yl)-3-ethyl-auinoline
The title compound from the previous step (350 mg, 1.02 mmol) was dissolved in
concentrated sulfuric acid. The solution was heated at 100°C for 6
hours, and then stirred at
ambient temperature for 48 hours. The reaction mixture was chilled to ice bath
temperature,
cautiously diluted with water (dropwise addition, 25 ml), and the made basic
(pH = 14) by
addition of 50% aqueous sodium hydroxide. The mixture was then extracted with
two 20 ml
portions of methylene chloride. The combined organic extracts were, in turn,
extracted with
an equal volume portions of water and then brine, dried (anhydrous sodium
sulfate), and
concentrated in vacuo, yielding a viscous syrup. Flash chromatography of the
entire sample
(silica gel, 47-61 micron mesh; elution with methylene
chloride/methanol/concentrated
aqueous ammonium hydroxide = 90:9:1 in volume) afforded the title compound (67
mg,
29.4% yield) as a viscous light yellow syrup.
MS m/z 225 (M+1 ).
'H NMR (400 MHz, CDCI3) 8 8.79 (1 H, m), 7.88 (1 H. m), 7.66 (1 H, M), 7.53 (1
H, m),
7.44 (1 H, m), 6.85 (1 H, M0, 4.46 (1 H, m), 4.10 (1 H, m), 2.82 (2H, q, J =
7.5 Hz), 1.33 (3H, t, J
= 7.5 Hz) ppm. .
The entire 67 mg sample was converted to the mono-hydrochloride salt by
dissolution
in 3 ml of ethyl acetate, followed by addition of 0.5 ml of saturated
hydrochloric acid in ethyl
acetate. The hydrochloride salt immediately precipitated as a pale yellow
amorphous solid,
which was isolated in quantitative yield by removal of solvent and excess
hydrochloric acid in
vacuo. The hydrochloride salt was utilized in the next preparative procedure
(Step 3).
Step 3
_Racemic 3 Ethyl-8-pyrrolidin-3-yl-auinoline (semi-~hurified~ purification:
Steps 4/5 below)
The title compound from the previous step (in hydrochloride salt form, 75 mg,
0.29
mmol) was dissolved in methanol (5.0 ml), and hydrogenated (50 psi, 10 mg of
platinum oxide
catalyst). The catalyst was filtered and the filtrate was concentrated in
vacuo yielding a light
amber residue. The residue was dissolved in 5 ml of water. The solution was
extracted with
10 ml of ethyl acetate, the organic extract then being discarded. The aqueous
solution was
made basic (pH 14) by addition of 50% aqueous sodium hydroxide, and then
extracted twice
with 10 ml portions of methylene chloride. The combined methylene chloride
extracts were, in
turn, extracted with an equal volume portion of brine, dried (anhydrous sodium
sulfate), and
finally concentrated in vacuo to afford a colorless syrup. Flash
chromatography of the entire
sample (silica gel, 47-61 micron mesh; elution with methylene
chloride/methanol/aqueous
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-62-
concentrated ammonium hydroxide = 90:9:1 in volume) afforded the title
compound (30 mg,
46.1 % yield) as a pale yellow amorphous foam.
Final purification of the title compound product
Final purification of the just described 30 mg sample of title compound was
accomplished by tent-butyloxycarbonyl acylation at the pyrrolidine nitrogen
(to facilitate a
further chromatographic purification) followed by acid catalyzed removal of
the tert
butyloxycarbonyl substituent. This procedure afforded the final purified title
compound (after
basic work-up) in the free base form.
Step 4
Racemic 3-(3-Ethyl-auinolin-8-yl)-pyrrolidine-1-carboxylic acid tert-butyl
ester
To a well-stirred solution of the 15 mg (0.07 mmol) sample of semi-purified
title
compound product from the previous step and triethylamine (0.02 ml, 0.14 mmol)
in
methylene chloride, di-tert-butyl carbonate (21.8 mg, 0.10 mmol) was added.
The reaction
was then stirred at ambient temperature for 48 hours. Aqueous saturated sodium
bicarbonate
(5 ml) was added with efficient stirring. The mixture was then extracted with
two 3 ml portions
of methylene chloride. The organic extracts were combined and, in turn,
extracted with an
equal volume of brine, dried (anhydrous sodium sulfate). Concentration in
vacuo afforded a
viscous syrup. Flash chromatography of the entire sample (silica gel, 47-61
micron mesh;
initially eluting with methylene chloride/methanol = 99.75:0.25 in volume)
steadily increasing
the polarity of the eluting system finally to methylene chloride/methanol =
99:1 in volume)
afforded the title compound (15 mg, 69.7% yield) as a colorless oil.
MS m/z 327 (M+1 ).
' H NMR (400 MHz, CDCI3) 8 8.78 (1 H, m), 7.88 (1 H, m), 7.63 (1 H, m), 7.40-
7.54 (2H,
overlapping multiplets), 4.63 (1 H, m), 3.96 (1 H, m), 3.50 (2H, m), 3.33 (1
H, m), 2.82 (2H, q, J
= 7.5 Hz), 2.34 (1 H, m), 2.12 (1 H, m), 1.44 and 1.47 (9H, two singlets),
1.33 (3H, t, J = 7.5
Hz) ppm.
Step 5
Racemic 3-Ethyl-8-pyrrolidin-3-yl-quinoline
To a solution of 15 mg (0.05 mmol) of the title compound from the previous
step in
methylene chloride/methanol = 9:2 in volume, a 1.0 ml saturated anhydrous
hydrogen
chloride/diethyl ether solution was added, and the resulting reaction mixture
was stirred at
ambient temperature for 18 hours. Solvents were removed in vacuo, and the
residue was
extracted into water. The aqueous extract was, in turn, extracted with an
equal volume of
ethyl acetate. Finally, the separated aqueous phase was made basic (pH = 14)
by addition of
50% aqueous sodium hydroxide, and then extracted with three 5 ml portions of
ethyl acetate.
The combined organic extracts were dried (anhydrous sodium sulfate) and
concentrated in
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-63-
vacuo to afford the purified title compound (free base form, 10 mg, 96% yield)
as a colorless
amorphous solid.
MS m/z 227 (M+1 ).
'H NMR (400 MHz, CDCI3) 8 8.76 (1 H, m), 7.89 (1 H, m), 7.60 (1 H, m), 7.53 (1
H, m),
7.44 (1 H, m), 4.38 (1 H, m), 3.46 (1 H, m), 3.27 (1 H, m), 3.12 (1 H, m),
2.97 (1 H, m), 2.82
(2H. q, J = 7.5 Hz), 2.32 (1 H, m), 2.01 (1 H, m), 1.33 (3H, t, J = 7.5 Hz)
ppm.
Step 6
Enantiomeric (Both Enantiomers) 3-Ethyl-8-pyridin-3-yl-auinoline
Utilizing analogously the procedure of Step 4/Example 1, the racemic title
compound
of the previous step of this example was converted to the corresponding
racemic nitrogen
substituted tert-butoxycarbonyl compound, the separated/purified enantiomers
of which were
then isolated by the methodology of Step 5/Example 1. Finally, by the
procedure of Step
6/Example 1, the enantiomers of the title compound of the previous step of
this example were
prepared in both mono-hydrochloride and free base form.
EXAMPLE 10
ENANTIOMERIC (BOTH ENANTIOMERS) AND RACEMIC 3-ETHYL-7-PIPERIDIN-3-YL
QUINOLINE
Step 1
7-Bromo-3-ethyl-auinoline
To a well-stirred mixture consisting of 3-bromo-aniline (5.40 g, 31.4 mmol),
sodium 3-
nitro-benzene sulfonate (4.25 g, 18.9 mmol), 8.5 g (177 mmol) of concentrated
sulfuric acid,
and 3.2 ml of water heated to 100°C, 2-ethyl acrolein (5.0 ml, 51 mmol)
was added. After
maintaining the reaction temperature at 100°C for 1 hour, the
temperature was elevated to
110°C. An additional portion of 2-ethyl acrolein (1.0 ml, 10.2 mmol)
was added, and the
reaction was stirred at 110°C for 1 hour. The reaction temperature was
then elevated to
120°C prior to addition of another 1.0 ml (10.2 mmol) portion of 2-
ethyl acrolein. After heating
the reaction at 120°C for 1 hour, the temperature was increased to
130°C prior to addition of
1.0 ml ( 10.2 mmol) of 2-ethyl acrolein. Finally, the reaction temperature was
raised to 140°C
and maintained at that temperature for 2 hours after addition of a final
portion (1.3 ml, 13.2
mol) of 2-ethyl acrolein. The cooled reaction was quenched with ice (50 g),
and the pH of the
resulting mixture was adjusted to by addition of 6 N aqueous sodium hydroxide.
The mixture
was then extracted with two 30 ml portions of methylene chloride. The combined
organic
extracts were dried (anhydrous sodium sulfate) and concentrated in vacuo,
affording an
amber oil. Flash chromatography of the entire sample utilizing the Biotage
Flash 401 iTM silica
gel flash chromatography module and manufacturer's prepacked silica gel
cartridges
described in Example 1, Step 4, and eluting with methylene chloride, afforded
the title
compound (1.46 g, 19.7% yield) as a viscous light amber syrup which solid on
standing.
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-64-
MS m/z 236, 237, 238, 239 (M+ 1 ).
H NMR (400 MHz, CDCI3) 8 8.77 (1 H, m), 8.23 (1 H, m), 7.87 (1 H, m), 7.54-
7.63 (2H,
overlapping multiplets), 2.80 (2H, q, J = 7.5 Hz), 1.33 (3H, t, J = 7.5 Hz)
ppm.
Step 2
3-Ethyl-7-pyridin-3-yl-e~uinoline
To a well-stirred mixture consisting of the title compound of Step 1, this
Example
(1.40 g, 5.93 mmol), diethyl(3-pyridyl)borane (0.96 g, 6.53 mmol) and
bis(triphenylphosphine)
palladium (II) chloride (458 mg, 0.65 mmol) in tetrahydrofuran (15 ml), a 7.5
ml aqueous
solution of sodium carbonate (2.51 g, 23.7 mmol) was added. The reaction
mixture was then
stirred 'at 90°C for 5 hours, and then at ambient temperature for 18
hours. The aqueous
phase of the biphasic reaction mixture is separated and extracted with an
equal volume of
ethyl acetate. The solvent of the organic phase of the reaction mixture was
removed in vacuo
and the residue is extracted with ethyl acetate (25 ml). The combined organic
extracts are
dried (anhydrous sodium sulfate) and concentrated in vacuo, yielding a dark
viscous oil.
Flash chromatography of the entire sample (silica gel, 47-61 micron mesh;
elution with ethyl
acetate) afford the title compound (807 mg, 58% yield) as a viscous yellow
syrup.
~H NMR (400 MHz, CDCI3) 8 8.93 (1 H, m), 8.82 (1 H, m), 8.62 (1 H, m), 8.27 (1
H, m),
8.01 (1 H, m), 7.94 (1 H, m), 7.86 (1 H, m), 7.74 (1 H, m), 7.36-7.49 (2H,
overlapping
multiplets), 2.84 (2H, q, J = 7.5 Hz), 1.35 (3H, t, J = 7.5 Hz) ppm.
Step 3
Racemic 3-Ethyl-7-piperidin-3-yl-quinoline
To a well-stirred solution of the title compound of the previous step (780 mg,
3.33
mmol) in anhydrous tetrahydrofuran (8 ml), 27.0 ml of a 1 M solution of
lithium triethyl
borohydride (27 mmol) in tetrahydrofuran was added, and the resulting reaction
mixture was
stirred at ambient temperature for 18 hours. The reaction was quenched by
cautious
dropwise addition of water (50 ml). Solvents were removed in vacuo, affording
a viscous oil
which was extracted with three 20 ml portions of methylene chloride. The
combined organic
extracts dried (anhydrous sodium sulfate) and concentrated in vacuo, yielding
a viscous
syrup. Flash chromatography of the entire sample (silica gel, 47-61 micron
mesh; elution with
methylene chloride/methanol/concentrated aqueous ammonium hydroxide = 90:9:1
in
volume) afford the title compound (390 mg, 48.8% yield) as a yellow gum.
MS m/z 241 (M+1 ).
'H NMR (400 MHz, CDCI3) 8 8.73 (1H, m), 7.86 (2H, overlapping multiplets),
7.66
(1 H, m), 7.37 (1 H, m), 3.27 (1 H, m), 3.15 (1 H, m), 2.88 (1 H, m), 2.7-2.9
(2H, overlapping
multiplets), 2.66 (1 H, m), 2.78 (2H, q, J = 7.5 Hz), 1.83 (1 H, m), 1.60-1.82
(2H, overlapping
multiplets), 1.31 (3H, t, J = 7.5 Hz) ppm.
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-65-
Separation of the enantiomers of the racemic title compound
Step 4
Racemic 3-(3-Ethyl-auinolin-7-yl)-piperidine-1-carboxylic acid tert-butyl
ester
A reaction mixture consisting of the free base title compound from the
previous step
(390 mg, 1.63 mmol), triethylamine (0.45 ml, 3.25 mmol), and di-tert-butyl
dicarbonate (530
mg, 2.44 mmol) in methylene chloride (15 ml) was stirred at ambient
temperature for 18
hours. Saturated aqueous sodium bicarbonate (20 ml) was added with efficient
stirring. The
mixture was then extracted with two 10 ml portions of methylene chloride. The
combing
organic extracts were, in turn, extracted with an equal volume portion of
brine, dried
(anhydrous sodium sulfate), and concentrated in vacuo, yielding a viscous
syrup. Flash
chromatography of the entire sample (silica gel, 47-61 micron mesh; elution
with
hexanes/ethyl acetate = 75:25 in volume) afforded the title compound (180 mg,
32% yield) as
a colorless oil.
MS m/z 341 (M+1 ).
Step 5
Enantiomeric 3-(3-Ethyl-auinolin-7-yl)-piperidine-1-carboxylic acid tert-butyl
ester (both
enantiomers)
Utilizing the method of Example 1, Step 5, the enantiomers of the racemic
title
compound of Step 4 of this Example were separated.
Step 6
Enantiomeric 3-Ethyl-7-piperidin-3-yl-auinoline (both enantiomers)
Utilizing the method of Example 1, Step 6, the enantiomers of the previous
Step of
this Example were used to prepare the title compound enantiomers of this step
in both mono-
hydrochloride and free base forms.
EXAMPLE 11
ENANTIOMERIC (BOTH ENANTIOMERS) AND RACEMIC 3-METHYL-8-(1-METHYL
PIPERIDIN-3-YL)QUINOLINE
Step 1
Racemic 3-Methyl-8-(1-methyl-piperidin-3-yl)auinoline
To a well-stirred solution of the title compound of Example 8, Step 2 (30 mg,
0.133
mmol) in 1.0 ml of methanol, 0.10 ml of 37% formaldehyde in 'methanol (1.2
mmol of
formaldehyde) and 100 mg (0.47 mmol) of sodium triacetoxyborohydride were
sequentially
added, and the resulting reaction mixture was stirred at ambient temperature
for 6 hours. The
solvent was removed in vacuo, and the resulting residue was extracted into 10
ml of
methylene chloride. The organic extract was, in turn, extracted with an equal
volume portion
of aqueous saturated sodium bicarbonate, and then with an equal volume portion
of brine.
After drying (anhydrous magnesium sulfate), the methylene chloride was removed
in vacuo,
CA 02505873 2005-05-11
WO 2004/043929 PCT/IB2003/004903
-66-
yielding a light amber solid (40 mg). Flash chromatography of the entire
sample (silica gel,
47-61 micron mesh; elution with methylene chloride/methanol/concentrated
aqueous
ammonium hydroxide = 90:9:1 in volume) afforded the title compound (19 mg, 60%
yield) as
a colorless amorphous solid.
MS m/z 241 (M+).
'H NMR (400 MHz, CDCI3) 8 8.76 (1 H, m), 7.86 (1 H, m), 7.56 (1 H, m), 7.48 (1
H, m),
7.43 (1 H, m), 4.29 (1 H, m), 3.12 (1 H, m), 2.97 (1 H, m), 2.49 (3H, s), 2.33
(3H, s), 1.80-2.13
(5H, overlapping multiplets), 1.64 (1 H, m) ppm.
Step 2
Enantiomeric (Both Enantiomers)3-Methyl-8-(1-methyl-piperidin-3-yl)auinoline
Utilizing the general methodology for enantiomer separation described in Step
5/Example 1, the enantiomers of the racemic title compound of the previous
step were
isolated in free base form. The mono-hydrochloride salts of the enantiomers
were prepared
by the procedure of Step 2/Example 9.