Note: Descriptions are shown in the official language in which they were submitted.
CA 02506082 2010-12-16
25771-1032(S)
1
NOVEL MEDICAMENTS FOR THE TREATMENT OF CHRONIC
OBSTRUCTIVE PULMONARY DISEASE
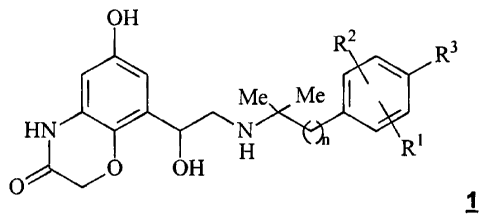
The present invention relates to the use of the compounds of general formula
1
OH 2
R R3
Me Me
HN H n Rt
O O OH
1
wherein the groups R1, R2 and R3 have the meanings provided below,
for preparing a pharmaceutical composition for
the treatment of COPD (chronic obstructive pulmonary disease), as well as
new compounds of general formula 1 and processes for preparing them.
Background to the invention
Betamimetics (1-adrenergic substances) are known from the prior art.
Reference may be made, for example, to the disclosures of US 4,460,581,
which proposes betamimetics for the treatment of a variety of complaints.
For drug treatment of diseases it is often desirable to prepare medicaments
with a longer duration of activity. As a rule, this ensures that the
concentration
of the active substance in the body needed to achieve the therapeutic effect
is
guaranteed for a longer period without the need to re-administer the drug at
frequent intervals. Moreover, giving an active substance at longer time
intervals contributes to the well-being of the patient to a high degree.
It is particularly desirable to prepare a pharmaceutical composition which can
be used therapeutically by administration once a day (single dose). The use of
a drug once a day has the advantage that the patient can become
accustomed relatively quickly to taking the drug regularly at certain times of
the day.
The aim of the present invention is therefore to provide betamimetics which
have a therapeutic benefit in the treatment of COPD and are characterised by
WO 2004/045618 2 PCT/EP2003/012565
CA 02506082 2005-05-13
a longer duration of activity and can thus be used to prepare pharmaceutical
compositions with a longer duration of activity. A particular aim of the
invention is to prepare betamimetics which, by virtue of their long-lasting
effect, can be used to prepare a drug for administration once a day for
treating
COPD. In addition to the above objectives, the present invention also sets out
to provide betamimetics which are not only exceptionally potent but are also
characterised by a high degree of selectivity with respect to the R2-adreno-
receptor.
Detailed description of the invention
Surprisingly it has been found that the abovementioned problems are solved
by compounds of general formula 1.
Accordingly, the present invention relates to compounds of general formula 1
OH 2
R R3
\ Me Me
HN H n R'
~~O OH
O 1
wherein
n denotes I or 2,
R1 denotes hydrogen, C1-C4-alkyl, halogen, OH or -O-C1-C4-alkyl;
R2 denotes hydrogen, Cl-C4-alkyl, halogen, OH or -O-C1-C4-alkyl;
R3 denotes hydrogen, CI-C4-alkyl, OH, halogen, -O-Cl-C4-alkyl,
-0-C1-C4-alkylene-COOH or -O-C1-C4-alkylene-CO-O-Ci-C4-alkyl,
for preparing a pharmaceutical composition for the treatment of COPD.
WO 2004/045618 3 PCT/EP2003/012565
CA 02506082 2005-05-13
It is preferable to use compounds of general formula 1, wherein
n denotes 1 or 2,
R1 denotes hydrogen, halogen or C1-C4-alkyl;
R2 denotes hydrogen, halogen or Cl-C4-alkyl;
R3 denotes hydrogen, Ci-C4-alkyl, OH, halogen, -O-C1-C4-alkyl,
-0-C1-C4-alkylene-000H or -0-C1-C4-alkylene-CO-O-C,-C4-alkyl,
for preparing a pharmaceutical composition for the treatment of COPD.
It is preferable to use compounds of general formula 1 wherein
n denotes 1 or 2;
R' denotes hydrogen, fluorine, chlorine or methyl;
R2 denotes hydrogen, fluorine, chlorine or methyl;
R3 denotes hydrogen, C1-C4-alkyl, OH, fluorine, chlorine, bromine, -0-
C1-C4-alkyl, -O-C1-C4-alkylene-000H, -0-C1-C4-alkylene-CO-O-Cl-C4-alkyl,
for preparing a pharmaceutical composition for the treatment of COPD.
It is particularly preferred to use compounds of general formula 1 wherein
n denotes 1 or 2,
R1 denotes hydrogen, methyl or ethyl ;
R2 denotes hydrogen, methyl or ethyl ;
R3 denotes hydrogen, methyl, ethyl, OH, methoxy, ethoxy, -O-CH2-
000H, -O-CH2-CO-O-methyl or -O-CH2-COOethyl;
for preparing a pharmaceutical composition for the treatment of COPD.
It is particularly preferred to use compounds of general formula I wherein
n denotes 1 or 2,
R1 denotes hydrogen or methyl;
R2 denotes hydrogen or methyl;
R3 denotes hydrogen, methyl, OH, methoxy, -O-CH2-000H or
-0-CH2-COOethyl;
for preparing a pharmaceutical composition for the treatment of COPD.
Also of particular importance according to the invention is the use of
compounds of general formula I wherein
n denotes 1 or 2,
R1 denotes hydrogen or methyl;
R2 denotes hydrogen or methyl;
R3 denotes hydrogen, OH, methoxy or -0-CH2-COON;
WO 2004/045618 4 PCT/EP2003/012565
CA 02506082 2005-05-13
for preparing a pharmaceutical composition for the treatment of COPD.
A preferred aspect of the present invention further relates to the use of
compounds of general formula 1 wherein n = 1 and the groups R1 , R2 and R3
may have the abovementioned meanings, for preparing a pharmaceutical
composition for the treatment of COP D.
Another preferred aspect of the present invention relates to the use of
compounds of general formula 1 wherein n = 1 or 2 , R3 denotes a group
selected from among hydrogen, OH, -O-Ci-C4-alkyl and -O-Cy-C4-alkylene-
COOH and wherein the groups R' and R2 may have the abovementioned
meanings, for preparing a pharmaceutical composition for the treatment of
COPD.
Another preferred aspect of the present invention relates to the use of
compounds of general formula 1 wherein n = 2, R' and R2 hydrogen and the
group R3 may have the abovementioned meanings, for preparing a
pharmaceutical composition for the treatment of COPD.
In the compounds of formula I the groups R' and R2, if they do not represent
hydrogen, may each be arranged in the ortho or meta position relative to the
bond to the benzylic "-CH2" group. If neither of the groups R1 and R2 denotes
hydrogen, it is preferable according to the invention to use those compounds
of formula 1 wherein the two groups R1 and R2 are either in the ortho
configuration or both groups R1 and R2 are in the meta configuration, while
the
use of those compounds wherein both groups R1 and R2 are in the ortho
configuration is particularly important.
In the compounds of formula 1 wherein one of the groups R1 and R2 does not
denote hydrogen, it may be in the ortho or meta configuration with respect to
the bond to the benzylic "-CH2" group. In this case it is particularly
preferred
according to the invention to use those compounds of formula 1 wherein the
group R1 or R2 , which does not denote hydrogen, is in the ortho
configuration.
In another aspect the present invention relates to the abovementioned use of
the compounds of formula I in the form of the individual optical isomers,
mixtures of the individual enantiomers or racemates. It is particularly
preferable to use the compounds of formula I as mentioned above in the form
of the enantiomerically pure compounds, while the use of the R enantiomers
WO 2004/045618 5 PCT/EP2003/012565
CA 02506082 2005-05-13
of the compounds of formula I is of particular importance according to the
invention.
In another aspect the present invention relates to the abovementioned use of
the compounds of formula 1 in the form of the acid addition salts with
pharmacologically acceptable acids as well as optionally in the form of the
solvates and/or hydrates thereof.
The present invention further relates to the use of the abovementioned
compounds of general formula 1 for preparing a pharmaceutical composition
for once-a-day treatment of COPD.
Moreover the present invention relates to a process for the treatment of
COPD, characterised in that one or more of the abovementioned compounds
of general formula 1 are administered in therapeutically effective amounts.
The present invention also relates to processes for treating COPD,
characterised in that one or more of the abovementioned compounds of
general formula 1 are administered once a day in therapeutically effective
amounts.
The compounds of general formula 1 are partly known from the prior art.
Reference is made here to the disclosure of US 4460581. In some cases,
however, the compounds of general formula 1 have not yet been disclosed in
the prior art. Another aspect of the present invention relates to these new
compounds of formula 1 as such.
Accordingly the present invention also relates to compounds of general
formula 1
OH 2
R R3
Me Me
HN H n R
~O OH
O 1
wherein
n denotes 1;
R1 denotes hydrogen, halogen, C1-C4-alkyl or -O-Cl -C4-alkyl;
R2 denotes hydrogen, halogen, C1-C4-alkyl or -O-C1-C4-alkyl;
CA 02506082 2010-12-16
25771-1032(S)
6
R3 denotes C1-C4-alkyl, OH, halogen, -O-C1-C4-alkyl, -O-C,-C4-
alkylene-COON, -O-C1-C4-alkylene-CO-O-C1-C4-alkyl,
with the proviso that if R1 and R2 each represent ortho-methyl, R3 cannot
simultaneously be OR In one embodiment, R3 denotes hydrogen, halogen,
C1-C4-alkyl, -O-C1-C4-alkyl, -O-CH2-COON, -O-CH2-COOmethyl or
-O-CH2-COOethyl.
Preferred compounds of general formula 1 are those wherein
n denotes 1;
R1 denotes hydrogen, fluorine, chlorine, methyl or methoxy;
R2 denotes hydrogen, fluorine, chlorine, methyl or methoxy;
R3 denotes C1-C4-alkyl, OH, fluorine, chlorine, bromine,
-O-C1-C4-alkyl, -O-C1-C4-alkylene-COON, -O-C1-C4-alkylene-CO-O-C1-C4-alkyl,
with the proviso that if R1 and R2 each represent ortho-methyl, R3 cannot
simultaneously be OR
Preferred compounds of general formula 1 are those wherein
n denotes 1;
R1 denotes hydrogen or C1-C4-alkyl;
R2 denotes hydrogen or C1-C4-alkyl;
R3 denotes C1-C4-alkyl, OH, -O-C1-C4-alkyl, -O-C1-C4-alkylene-COON
or -O-C1-C4-alkylene-CO-O-C1-C4-alkyl,
with the proviso that if R1 and R2 each represent ortho-methyl, R3 cannot
simultaneously be OH.
Preferred compounds of general formula 1 are those wherein
n denotes 1;
R' denotes hydrogen, methyl or ethyl;
CA 02506082 2010-12-16
25771-1032(5)
6a
R2 denotes hydrogen, methyl or ethyl;
R3 denotes methyl, ethyl, OH, methoxy, ethoxy, -O-CH2-COON,
-O-CH2-COOmethyl or -O-CH2-COOethyl,
with the proviso that if R' and R2 each represent ortho-methyl, R3 cannot
simultaneously be OH.
Also preferred are the compounds of general formula 1 wherein
n denotes 1;
R1 denotes hydrogen or methyl;
R2 denotes hydrogen or methyl;
R3 denotes methyl, OH, methoxy, -O-CH2-COON or -O-CH2-COOethyl,
WO 2004/045618 7 PCT/EP2003/012565
CA 02506082 2005-05-13
with the proviso that if R1 and R2 each represent ortho-methyl, R3 cannot
simultaneously be OR
Also preferred according to the invention are compounds of general formula 1
wherein
R3 denotes methoxy, ethoxy, -O-CH2-000H, -O-CH2-COOmethyl or
-O-CH2-COOethyl,
and R1, R2 and n may have the above meanings.
The present invention also relates to compounds of general formula 1
wherein
n denotes 1;
R1 denotes halogen, C1-C4-alkyl or -O-C1-C4-alkyl;
R2 denotes halogen, C1-C4-alkyl or -O-C1-C4-alkyl;
R3 denotes halogen, C1-C4-alkyl or -O-C1-C4-alkyl.
The present invention also relates to compounds of general formula 1
wherein
n denotes 1;
R1 denotes fluorine, chlorine, methyl or methoxy;
R2 denotes fluorine, chlorine, methyl or methoxy ;
R3 denotes fluorine, chlorine, methyl or methoxy.
In another preferred aspect the present invention relates to the compounds of
general formula I wherein
n denotes 1;
R1 denotes hydrogen;
R2 denotes hydrogen, fluorine, chlorine or methyl;
R3 denotes methyl, ethyl, iso-propyl, tert.-butyl, OH, fluorine, chlorine,
bromine, methoxy, ethoxy, -O-CH2-COOH, -O-CH2-CH2-COON,
-O-CH2-CH2-CH2-COON, -0-CH2-COOmethyl, -O-CH2-COOethyl,
-O-CH2-CH2-COOmethyl, -O-CH2-CH2-COOethyl, -O-CH2-CH2-
CH2-000methyl and -O-CH2-CH2-CH2-COOethyl.
Also particularly preferred are compounds of general formula I wherein
n denotes 1;
R1 denotes hydrogen;
R2 denotes hydrogen, fluorine, chlorine or methyl;
WO 2004/045618 8 PCT/EP2003/012565
CA 02506082 2005-05-13
R3 denotes OH, fluorine, chlorine, methyl, methoxy, ethoxy or -O-CH2-
000H.
Other particularly preferred compounds of general formula I according to the
invention are those wherein
n denotes 1;
R' denotes hydrogen;
R2 denotes halogen, Cl-C4-alkyl or -O-Cl-C4-alkyl,
preferably fluorine, chlorine, methoxy or methyl;
R3 denotes halogen, C1-C4-alkyl or -O-C1-C4-alkyl,
preferably fluorine, chlorine, methoxy or methyl.
Another preferred aspect of the present invention relates to the compounds of
general formula 1 wherein n = 1 , R1 and R2 denote hydrogen and the group
R3 may have the abovementioned meanings.
Another preferred aspect of the present invention relates to the compounds of
general formula I wherein
n denotes 1;
R1 and R2 denote hydrogen;
R3 denotes methyl, ethyl, iso-propyl, tert.-butyl, OH, fluorine, chlorine,
bromine, methoxy, ethoxy, -O-CH2-COON, -O-C12-CH2-000H, -O-CH2-
CH2-CH2-COON, -O-CH2-COOmethyl, -O-CH2-COOethyl, -0-CH2-CH2-
COOmethyl, -O-CH2-CH2-COOethyl, -O-CH2-CH2-CH2-COOmethyl and -0-
CH2-CH2-CH2-COOethyl.
Particularly preferred are compounds of general formula I wherein
n denotes 1;
R1 and R2 denote hydrogen;
R3 denotes OH, fluorine, chlorine, methoxy, ethoxy,-O-CH2-COOH,
preferably OH, fluorine, chlorine, ethoxy or methoxy.
Particularly preferred are compounds of general formula 1 wherein
n denotes 1;
R' and R2 denote hydrogen;
R3 denotes fluorine, chlorine, methoxy or ethoxy.
The present invention also relates to compounds of general formula I
WO 2004/045618 9 PCT/EP2003/012565
CA 02506082 2005-05-13
wherein
n denotes 1;
R1 denotes hydrogen, halogen, C1-C4-alkyl or -O-Ci-C4-alkyl;
R2 denotes hydrogen, halogen, C1-C4-alkyl or -O-C1-C4-alkyl;
R3 denotes hydrogen.
Also preferred are compounds of general formula 1 wherein
n denotes 1;
R1 denotes hydrogen, fluorine, chlorine, methyl or methoxy;
R2 denotes hydrogen, fluorine, chlorine, methyl or methoxy;
R3 denotes hydrogen.
The present invention also relates to compounds of general formula 1
wherein
n denotes 1;
R1 denotes fluorine, chlorine, methyl or methoxy;
R2 denotes fluorine, chlorine, methyl or methoxy;
R3 denotes hydrogen.
In the compounds of formula 1 , the groups R' and R2, if they do not represent
hydrogen, may each be arranged in the ortho or meta position relative to the
bond to the benzylic "-CH2" group. If neither of the groups R1 and R2 denotes
hydrogen, preferred compounds of formula 1 are those wherein the two
groups R1 and R2 are either in the ortho configuration or both groups R1 and
R2 are in the meta configuration, while the use of those compounds wherein
both groups R1 and R2 are in the ortho configuration is particularly
important.
In the compounds of formula 1 wherein one of the groups R1 and R2 does not
denote hydrogen, it may be in the ortho or meta configuration with respect to
the bond to the benzylic "-CH2" group. In this case particularly preferred
compounds of formula 1 are those wherein the group R' or R2 , which does
not denote hydrogen, is in the ortho configuration.
Also particularly preferred are compounds of general formula 1 which are
selected from among
- 6-hydroxy-8-{1-hydroxy-2-[2-(4-methoxy-phenyl)-1,1-dim ethyl-
ethylam ino]-ethyl}-4H-benzo[1,4]oxazin-3-one;
- 6-hydroxy-8-{1-hydroxy-2-[2-(4-phenoxy-ethyl acetate)-1, 1 -dim ethyl-
ethylamino]-ethyl}-4H-benzo[1,4]oxazin-3-one;
WO 2004/045618 10 PCT/EP2003/012565
CA 02506082 2005-05-13
- 6-hydroxy-8-{1-hydroxy-2-[2-(4-phenoxy-acetic acid)-1,1-dimethyl -
ethyl am ino]-ethyl}-4H-benzo[ 1,4]oxazin-3-one;
- 8-{2-[1,1-dim ethyl-2-(2,4,6-trim ethyl phenyl)-ethylamino]-1-hydroxy-
ethyl}-6-hydroxy-4H-benzo[ 1,4]oxazin-3-one;
- 6-hydroxy-8-{1-hydroxy-2-[2-(4-hydroxy-phenyl)-1,1-dim ethyl-
ethylam ino]-ethyl}-4H-benzo[1,4]oxazin-3-one;
- 6-hydroxy-8-{1-hydroxy-2-[2-(4-isopropyl-phenyl)-1,1 dimethyl-
ethylam ino]-ethyl}-4H-benzo[ 1,4]oxazin-3-one;
- 8-{2-[2-(4-ethyl-phenyl)-1,1-dimethyl-ethylam ino]-1-hydroxy-ethyl}-6-
hydroxy-4H-benzo[1,4]oxazin-3-one;
- 8-{2-[2-(4-fluoro-3-methyl-phenyl)-1,1-dimethyl-ethylamino]-1-hydroxy-
ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one;
- 8-{2-[2-(4-fiuoro-2-methyl-phenyl)-1,1-dimethyl-ethylamino]-1-hydroxy-
ethyl}-6-hydroxy-4H-benzo[ 1,4]oxazin-3-one;
- 8-{2-[2-(2,4-difluoro-phenyl)-1,1-dimethyl-ethylam inoJ-1-hydroxy-ethyl}-6-
hydroxy-4H-benzo[1,4]oxazin-3-one;
- 8-{2-[2-(3, 5-difluoro-phenyl)-1,1-dimethyl-ethylam ino]-1-hydroxy-ethyl}-6-
hydroxy-4H-benzo[1,4]oxazin-3-one;
- 8-{2-[2-(4-ethoxy-phenyl)-1,1-dimethyl-ethylamino]-1-hydroxy-ethyl}-6-
hydroxy-4H-benzo[1,4]oxazin-3-one;
- 8-{2-[2-(3, 5-dimethyl-phenyl)-1,1-dimethyl-ethylamino]-1-hydroxy-ethyl}-
6-hydroxy-4H-benzo[1,4]oxazin-3-one;
- 4-(4-{2-[2-hydroxy-2-(6-hydroxy-3-oxo-3, 4-dihydro-2H-benzo[1, 4]oxazin-
8-yl)-ethylamino]-2-methyl-propyl}-phenoxy)-butyric acid;
- 8-{2-[2-(3, 4-difluoro-phenyl)-1,1-dimethyl-ethylam ino]-1-hydroxy-ethyl}-6-
hydroxy-4H-benzo[1,4]oxazin-3-one;
- 8-{2-[2-(2-chloro-4-fluoro-phenyl)-1,1-dimethyl-ethylamino]-1-hydroxy-
ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one;
- 8-{2-[2-(4-chloro-phenyl)-1,1-dim ethyl-ethylamino]-1-hydroxy-ethyl}-6-
hydroxy-4H-benzo[1,4]oxazin-3-one;
- 8-{2-[2-(4-bromo-phenyl)-1, 1 -dimethyl-ethylam ino]-1-hydroxy-ethyl}-6-
hydroxy-4H-benzo[1,4]oxazin-3-one;
- 8-{2-[2-(4-fiuoro-phenyl)-1,1-dimethyl-ethylamino]-1-hydroxy-ethyl}-6-
hydroxy-4H-benzo[1,4]oxazin-3-one;
- 8-{2-[2-(4-fluoro-3-methoxy-phenyl)-1,1-dimethyl-ethylamino]-1-hydroxy-
ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one;
- 8-{2-[2-(4-fluoro-2,6-dimethyl- phenyl)-1,1-dimethyl-ethylamino]-1-
hydroxy-ethyl}-6-hydroxy-4H-benzo[ 1,4]oxazin-3-one;
WO 2004/045618 11 PCT/EP2003/012565
CA 02506082 2005-05-13
- 8-{2-[2-(4-chloro-2-methyl-phenyl)-1,1-dim ethyl-ethyl amino]-1-hydroxy-
ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one;
- 8-{2-[2-(4-chloro-3-fluoro-phenyl)-1,1-dimethyl-ethylamino]-1-hydroxy-
ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one;
- 8-{2-[2-(4-chloro-2-fluoro-phenyl)-1,1-dimethyl-ethylamino]-1-hydroxy-
ethyl}-6-hydroxy-4H-benzo[ 1, 4]oxazin-3-one;
- 8-{2-[2-(3-chloro-4-flu oro-phenyl)-1,1-dimethyl-ethylamino]-1-hydroxy-
ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one;
- 8-{2-[2-(2,6-difluoro-4-methoxy-phenyl)-1,1-dim ethyl-ethylamino]-1-
hydroxy-ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one;
- 8-{2-[2-(2,5-difluoro-4-methoxy-phenyl)-1,1-dimethyl-ethylamino]-1-
hydroxy-ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one;
- 8-{2-[2-(4-fluoro-3, 5-dim ethyl-phenyl)-1,1-dimethyl-ethylamino]-1-
hydroxy-ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one;
- 8-{2-[2-(3, 5-dichloro-phenyl)-1,1-dimethyl-ethylamino]-1-hydroxy-ethyl}-
6-hydroxy-4H-benzo[1,4]oxazin-3-one;
- 8-{2-[2-(4-chloro-3-methyl-phenyl)-1,1-dim ethyl-ethylamino]-1-hydroxy-
ethyl}-6-hydroxy-4H-benzo[ 1, 4]oxazin-3-one;
- 8-{2-[2-(3,4,5-trifluoro-phenyl)-1,1-dim ethyl-ethylamino]-1-hydroxy-ethyl}-
6-hydroxy-4H-benzo[1,4]oxazin-3-one;
- 8-{2-[2-(3-methyl-phenyl)-1,1-dim ethyl-ethylam ino]-1-hydroxy-ethyl}-6-
hydroxy-4H-benzo[1,4]oxazin-3-one and
- 8-{2-[2-(3,4-dichloro-phenyl)-1,1-dimethyl-ethylamino]-1-hydroxy-ethyl}-
6-hydroxy-4H-benzo[1,4]oxazin-3-one.
In another aspect the present invention relates to the abovementioned new
compounds of formula I in the form of the individual optical isomers, mixtures
of the individual enantiomers or racemates. Particularly preferred are
compounds of formula I in the form of the enantiomerically pure compounds,
while the R-enantiomers of the compounds of formula I are of exceptional
importance according to the invention. Methods of separating racemates into
the respective enantiomers are known in the prior art and may be used
analogously to prepare the enantiomerically pure R- and S-enantiomers of the
compounds of formula 1.
In another aspect the present invention relates to the abovementioned
compounds of formula 1 in the form of the acid addition salts with
WO 2004/045618 12 PCT/EP2003/012565
CA 02506082 2005-05-13
pharmacologically acceptable acids as well as optionally in the form of the
solvates and/or hydrates thereof.
In another aspect the present invention relates to the abovementioned
compounds of formula I for use as pharmaceutical compositions. The
present invention further relates to the use of the abovementioned new
compounds of general formula I for preparing a pharmaceutical composition
for the treatment of COPD. The present invention further relates to the use of
the abovementioned new compounds of general formula 1 for preparing a
pharmaceutical composition for the once-a-day treatment of COPD.
Moreover the present invention relates to a process for the treatment of
COPD, characterised in that one or more of the abovementioned compounds
of general formula 1 are administered in therapeutically effective amounts.
The present invention also relates to processes for treating COPD,
characterised in that one or more of the abovementioned new compounds of
general formula 1 are administered once a day in therapeutically effective
amounts.
By acid addition salts with pharmacologically acceptable acids are meant, for
example, the salts selected from among the hydrochloride, hydrobromide,
hydroiodide, hydrosulphate, hydrophosphate, hydromethanesulphonate,
hydronitrate, hydromaleate, hydroacetate, hydrobenzoate, hydrocitrate,
hydrofumarate, hydrotartrate, hydrooxalate, hydrosuccinate, hydrobenzoate
and hydro-p-toluenesulphonate, preferably the hydrochloride, hydrobromide,
hydrosulphate, hydrophosphate, hydrofumarate and
hydromethanesulphonate.
Of the abovementioned acid addition salts the salts of hydrochloric acid,
methanesulphonic acid, benzoic acid and acetic acid are particularly preferred
according to the invention.
For use according to the invention the compounds of general formula 1 may
optionally be used in the form of their individual optical isomers, mixtures
of
the individual enantiomers or racemates. If the compounds are used in
enantiomerically pure form, the R-enantiomers are preferred.
WO 2004/045618 13 PCT/EP2003/012565
CA 02506082 2005-05-13
Unless otherwise stated, the alkyl groups are straight-chained or branched
alkyl groups having 1 to 4 carbon atoms. The following are mentioned by way
of example: methyl, ethyl, propyl or butyl. In some cases the abbreviations
Me, Et, Prop or Bu are used to denote the groups methyl, ethyl, propyl or
butyl. Unless otherwise stated, the definitions propyl and butyl include all
the
possible isomeric forms of the groups in question. Thus, for example, propyl
includes n-propyl and iso-propyl, butyl includes iso-butyl, sec.butyl and
tert.-
butyl, etc.
Unless otherwise stated, the alkylene groups are branched and unbranched
double-bonded alkyl bridges having 1 to 4 carbon atoms. The following are
mentioned by way of example: methylene, ethylene, n-propylene or n-
butylene.
Unless otherwise stated, the term alkyloxy groups (or -0-alkyl groups)
denotes branched and unbranched alkyl groups having 1 to 4 carbon atoms
which are linked via an oxygen atom. Examples of these include: methyloxy,
ethyloxy, propyloxy or butyloxy. The abbreviations MeO-, EtO-, PropO- or
BuO- are used in some cases to denote the groups methyloxy, ethyloxy,
propyloxy or butyloxy. Unless otherwise stated, the definitions propyloxy and
butyloxy include all possible isomeric forms of the groups in question. Thus,
for example, propyloxy includes n-propyloxy and iso-propyloxy, butyloxy
includes iso-butyloxy, sec.butyloxy and tert.-butyloxy, etc. In some cases,
within the scope of the present invention, the term alkoxy is used instead of
the term alkyloxy. Accordingly, the terms methoxy, ethoxy, propoxy or butoxy
may also be used to denote the groups methyloxy, ethyloxy, propyloxy or
butyloxy.
Halogen within the scope of the present invention denotes fluorine, chlorine,
bromine or iodine. Unless stated otherwise, fluorine, chlorine and bromine
are the preferred halogens.
The compounds according to the invention may be prepared analogously to
methods already known from the prior art. Suitable methods of preparation
are known for example from US 4460581, to the entire contents of which
reference is made at this point.
WO 2004/045618 14 PCT/EP2003/012565
CA 02506082 2005-05-13
The examples of synthesis described below serve to illustrate compounds
known from the prior art, which may surprisingly be used according to the
present invention for the treatment of COPD.
WO 2004/045618 15 PCT/EP2003/012565
CA 02506082 2005-05-13
Example 1: 6-hydroxy-8-{1-hydroxy-2-f2-(4-hydroxy-2,6-dimethyl-phenyl)lr)-1,1
_
dimethyl-ethylaminol-ethyl}-4H-benzof 1,41oxazin-3-one
0
, ~O OH H Me
HN N
Me Me
Me OH
OH
The compound is known from US 4460581.
Example 2: 8-f2-[1,1 -dimethyl-3-phenylpropylam inol-1-hydroxy-ethyl}-6-
hhydroxy-4H-benzof 1,4loxazin-3-one
O~O OH
HN N \ I
Me Me
OH
The compound is known from US 4460581.
The examples of synthesis described below serve to illustrate new
compounds according to the invention more fully. They are intended only as
examples of procedure to illustrate the invention without restricting it to
the
subject matter described hereinafter.
Example 3: 6-hydroxy-841-hydroxy-2-f2-(4-methoxy-phenyl)-1,1-dimethyl-
ethylaminol-ethyl}-4H-benzof 1,41oxazin-3-one
O1 OH
H
HN N
Me Me
OMe
OH
CA 02506082 2010-12-16
25771-1032(S)
16
a) 8-{2-[1 1-dimethyl-2-(4-methoxy-phenyl)-ethylaminol-l -hydroxy-ethyl}-6-
benzyloxy-4H-benzo[1,4]oxazin-3-one
7.5 g of (6-benzyloxy-4H-benzo[1,4]oxazin-3-one)-glyoxal hydrate are added
at 70 C to a solution of 3.6 g of 1,1-dimethyl-2-(4-methoxyphenyl)-ethylamine
in 100 mL ethanol and stirred for 15 minutes. Then I g of sodium borohydride
is added within 30 minutes at 10 to 20 C. The mixture is stirred for one hour,
combined with 10 mL acetone and stirred for a further 30 minutes. The
reaction mixture is diluted with 150 mL ethyl acetate, washed with water,
dried
with sodium sulphate and evaporated down. The residue is dissolved in 50
mL methanol and 100 mL ethyl acetate and acidified with conc. hydrochloric
acid. After the addition of 100 mL diethyl ether the product precipitates out.
The crystals are filtered off, washed and recrystallised in 50 mL ethanol.
Yield: 7 g (68%; hydrochloride); melting point = 232-234 C.
b) 84241,1 -dimethyl-2-(4-methoxy-phenyl)-ethylam inol-1-hydroxy-ethyl}-6-
hydroxy-4H-benzoll ,41oxazin-3-one
6.8 g of the benzyl compound obtained above are hydrogenated in 125 mL
methanol with the addition of 1 g palladium on charcoal (5%) at ambient
temperature and under normal pressure. The catalyst is filtered off and the
filtrate is freed from solvent. After recrystallisation of the residue in 50
mL
acetone and some water, a solid is obtained, which is filtered off and washed.
Yield: 5.0 g (89 %; hydrochloride); melting point = 155-160 C.
The (R)- and (S)-enantiomers of Example 3 may be obtained from the
TM
racemate for example by chiral HPLC (e.g. column: Chirobiotic l , 250 x 22.1
mm made by Messrs Astec). The mobile phase may be methanol with 0.05 %
triethylamine and 0.05% acetic acid. Silica gel with a particle size of 5 pm,
to
which the glycoprotein Teicoplanin is covalently bound, can be used as the
column material.
Retention time (R-enantiomer) = 40.1 min, retention time (S-enantiomer) _
45.9 min. Both enantiomers are obtained according to this method in the form
of their free base.
The R-enantiomer of Example 3 is of exceptional importance according to the
invention.
WO 2004/045618 17 PCT/EP2003/012565
CA 02506082 2005-05-13
Example 4: 6-hydroxy-8-{1-hydroxy-2-f2-(4-phenoxy- ethyl acetate)-1 1-
dimethyl-ethylaminol-ethyl}-4H-benzof 1 4loxazin-3-one
O~O OH
HN N
Me Me O OEthyl
OH O
a) 842-f 1, 1 -dimethyl-2-(4-phenoxy-ethyl acetate)-ethylaminol-l -hydroxy-
ethyl}-6-benzyloxy-4H-benzof 1,41oxazin-3-one
The title compound is obtained analogously to the method described in
Example 3a) from 15 g of (6-benzyloxy-4H-benzo[1,4]oxazin-3-on)-glyoxal
hydrate and 11.8 g 1,1-dimethyl-2-(4-phenoxy-ethyl acetate)-ethylamine
hydrochloride.
Yield: 16.5 g (69%, hydrochloride); melting point = 212-214 C.
b) 842-f 1,1-dimethyl-2-(4-phenoxy-ethyl-acetate)-ethylaminol-l -hydroxy-
ethyl}-6-hydroxy-4H-benzo[1,4loxazin-3-one
8 g of the benzyl alcohol obtained above are dissolved in 100 mL ethanol, 100
mL methanol and 10 mL water and hydrogenated in the presence of 1 g
palladium on charcoal (5%). After the theoretically calculated amount of
hydrogen has been taken up the catalyst is filtered off and the filtrate is
evaporated down. The product which crystallises out when the solvent is
distilled off is suction filtered and washed.
Yield: 5.5 g (81 %; hydrochloride); melting point = 137-140 C.
The (R)- and (S)-enantiomers of this example can be obtained by separating
the racemate analogously to current methods of racemate cleaving known in
the prior art.
WO 2004/045618 18 PCT/EP2003/012565
CA 02506082 2005-05-13
Example 5: 6-hydroxy-8-{1-hydroxy-2-f2-(4-phenoxy-acetic acid)-1 1-
dimethyl-ethylaminol-ethyl}-4H-benzof 1,4loxazin-3-one
O r OH
HN N
Me Me I /
O COOH
OH
11 g of 8-{2-[1,1-dimethyl -2-(4-phenoxy-ethyl-acetate)-ethylamino]-1-hydroxy-
ethyl}-6-benzyloxy-4H-benzo[1,4]oxazin-3-one hydrochloride (Example 4a)
are dissolved in 125 mL methanol and hydrogenated in the presence of 1 g
palladium on charcoal (5%). After the theoretically calculated amount of
hydrogen has been taken up the catalyst is filtered off. 2.6 g sodium
hydroxide
dissolved in 20 mL water are added to the filtrate. The mixture is refluxed
for
30 minutes, the methanol is distilled off and combined with 10 mL water, 20
mL n-butanol and 3.9 mL acetic acid. The solid precipitated is suction
filtered
and washed with diethyl ether.
Yield: 7 g (87%). The hydrochloride is obtained by recrystallisation from 0.5
molar hydrochloric acid. Melting point = 152 C.
The (R)- and (S)-enantiomers of this example can be obtained by separating
the racemate analogously to current methods of racemate cleaving known in
the prior art.
Example 6: 8424 1,1-dim ethyl-2-(2,4,6-trimethylphenyl)-ethylaminol-1-
hydroxy-ethyl}-6-hydroxy-4H-benzof 1,4loxazin-3-one 0- OH Me
HN N
Me Me
Me Me
OH
a) 1-(6-benzyloxy-4H-benzof 1,41oxazin-3-one)-2-f 1,1-dimethyl-2-(2,4,6-
trimethylphenyl)-ethylim inol-ethanone
7.2 g (6-benzyloxy-4H-benzo[1,4]oxazin-3-one)-glyoxal hydrate and 3.6 g 1,1-
dimethyl-2-(2,4,6-trimethylphenyl)-ethylamine are heated to 70 C for one hour
in 100 mL ethanol. After cooling the crystals precipitated are filtered off
and
WO 2004/045618 19 PCT/EP2003/012565
CA 02506082 2005-05-13
washed with ethanol and diethyl ether. Yield: 8.6 g (94%); melting point =
175 C.
b) 842-(1,1-dimethyl-2-(2,4,6-trim ethyl phenyl) -ethylaminol-1-hydroxy-ethyl}-
6-
benzyloxy-4H-benzo[1,41oxazin-3-one
8.6 g of the Schiff base obtained according to method 6a) are dissolved in 100
mL ethanol and 20 mL THE, combined with 0.7 g sodium borohydride within
30 min at 10-20 C and stirred for one hour. After the addition of 10 mL
acetone the mixture is stirred for 30 minutes and then diluted with ethyl
acetate and water. The product which crystallises out on acidification with
conc. hydrochloric acid is filtered off and washed.
Yield: 7.4 g (80%, hydrochloride); melting point = 235 C (decomposition).
c) 8-{2-11,1-dimethyl-2-(2 4 6-trim ethylphenyl)-ethylaminol-l -hydroxy-ethyl}-
6-
hhydroxy-4H-benzo(1,4loxazin-3-one
7.4 g of the benzyl compound obtained in Step b) are hydrogenated in 125 mL
methanol with the addition of 1 g palladium on charcoal (5%) at ambient
temperature under normal pressure. Then the catalyst is filtered off and the
filtrate is evaporated down. The product which crystallises out when acetone
is added is suction filtered and washed with acetone and diethyl ether. Yield:
g (78%, hydrochloride); melting point 160 C (decomposition).
The (R)- and (S)-enantiomers of this example can be obtained by separating
the racemate analogously to current methods of racemate cleaving known in
the prior art.
Example 7: 6-hydroxy-8-{1-hydroxy-2-(2-(4-hydroxy-phenyl)-1 1-dimethyl
ethylaminol-ethyl}-4H-benzo[1,41oxazin-3-one
O~ OH
H
HN N
Me Me
OH
OH
WO 2004/045618 20 PCT/EP2003/012565
CA 02506082 2005-05-13
a) 8-{2-f 1,1-dimethyl-2-(4-hydroxy-phenyl)-ethylaminol-1-hydroxy-ethyl} 6
benzyloxy-4H-benzof 1 4loxazin-3-one
The title compound is prepared from 10 g (6-benzyloxy-4H-benzo[1,4]oxazin-
3-one)-glyoxal hydrate and 4.6 g 1, 1 -dimethyl-2-(4-hydroxy-phenyl)-
ethylamine analogously to the method for Example 3a).
Yield: 9.0 g (64%, hydrochloride); melting point = 255-258 C.
b) 842-11,1 -dimethyl-2-(4-hydroxy-phenyl)-ethylaminol-1-hydroxy-ethyl}-6-
hydroxy-4H-benzoj1,41oxazin-3-one
5.7 g of the coupling product obtained above are hydrogenated in the
presence of 0.6 g palladium on charcoal (5%) in 100 mL methanol. After the
theoretically calculated amount of hydrogen has been taken up the catalyst is
filtered off and the filtrate is freed from solvent. The residue is dissolved
in
ethanol with heating and then combined with diethyl ether. The product
precipitated is suction filtered and recrystallised once in water.
Yield: 3.6 g (72%, hydrochloride); melting point = 159-162 C.
The (R)- and (S)-enantiomers of this example can be obtained by separating
the racemate analogously to current methods of racemate cleaving known in
the prior art.
Example 8: 6-hydroxy-8-{1-hydroxy-2-[2-(4-isopropyl-phenyl)-1 1 dimethyl-
ethylaminol-ethyl}-4H-benzof 1 4loxazin-3-one
o
TO OH
HN N
OH
a) 1-(4-isopropyl-phenyl)-2-methyl-propan-2-ol
The reaction of a Grignard compound, prepared from 20 g (119 mmol)
4-isopropylbenzylchloride, with 11.4 ml (155 mmol) acetone yields the target
compound as a colourless oil.
Yield: 13.0 g (57%); mass spectrometry: [M+H]+ = 193.
WO 2004/045618 21 PCT/EP2003/012565
CA 02506082 2005-05-13
b) N-[2-(4-isopropyl-phenyl)-1,1-dimethyl-ethyll-acetamide
A Ritter reaction is carried out with 10.2 g (53 mmol) 1-(4-isopropyl-phenyl)-
2-
methyl-propan-2-ol in the manner described for Example 9b). The reaction
mixture is poured onto ice water and made alkaline with sodium hydroxide
solution, whereupon a solid is precipitated. This is suction filtered and
dried.
Yield: 9.90 g (80%); mass spectrometry: [M+H]+ = 234.
c) 2-(4-isopropyl-phenyl)-1 1-dimethyl-ethylamine
Reaction of 9.80 g (42 mmol) N-[2-(4-isopropyl-phenyl)-1 ,1-dim ethyl-ethyl]-
acetamide analogously to the method for Example 9c).
Yield: 7.00 g (71 %, hydrochloride); melting point 202-206 C.
d) 6-benzyloxy-8-{1-hydroxy-2-[2-(4-isopropyl-phenyl)-1,1-dimethyl-
ethylaminol-ethyl}-4H-benzo[1,41oxazin-3-one
2.18 g (6.1 mmol) benzyloxy-8-(2-ethoxy-2-hydroxy-acetyl)-4H-
benzo[ 1, 4]oxazin-3-one and 1.1 g (5.8 mmol) 2-(4-isopropyl-phenyl)-1,1-
dimethyl-ethylamine are stirred in 40 mL ethanol at 50-80 C for one hour.
After cooling to ambient temperature 0.24 g (6.3 mmol) sodium borohydride
are added. The mixture is stirred for one hour, diluted with 5 mL acetone and
stirred for a further 30 minutes. The reaction mixture is acidified with
hydrochloric acid, combined with 100 mL water and 80 mL ethyl acetate and
made alkaline with ammonia. The organic phase is separated off, dried with
sodium sulphate and freed from solvent. The residue is dissolved in 20 mL
ethyl acetate and 10 mL water, acidified with conc. hydrochloric acid and
diluted with diethyl ether. After the addition of a crystallisation aid the
solid
precipitate is suction filtered and washed. White solid.
Yield: 1.7 g (52 %, hydrochloride); melting point 220-222 C.
e) 6-hydroxy-8-{1-hydroxy-2-[2-(4-isopropyl-phenyl)-1 1 dimethyl-ethylaminol-
ethyl}-4H-benzo[1,41oxazin-3-one
1.6 g (3.0 mmol) 6-benzyloxy-8-{1-hydroxy-2-[2-(4-isopropyl-phenyl)-1,1-
dimethyl-ethylamino]-ethyl}-4H-benzo[1,4]oxazin-3-one are dissolved in
methanol and hydrogenated with palladium on charcoal as catalyst at normal
pressure and ambient temperature. The catalyst is suction filtered, the
solvent
distilled off and the residue recrystallised from isopropanol. White solid.
Yield: 1.1 g (85%, hydrochloride); melting point 248-250 C; mass
spectrometry: [M+H]+ = 399.
WO 2004/045618 22 PCT/EP2003/012565
CA 02506082 2005-05-13
The (R)- and (S)-enantiomers of this example can be obtained by separating
the racemate analogously to current methods of racemate cleaving known in
the prior art.
Example 9: 8-{2-[2-(4-ethyl-phenyl)-1 1 -dim ethyl-ethylam inol-1-hydroxy-
ethyl}-6-hydroxy-4H-benzof 1,41oxazin-3-one
T OH
HN N
OH
a) 1-(4-ethyl-phenyl)-2-methyl-propan-2-ol
14.8 g (90 mmol) of 1-(4-ethyl-phenyl)-propan-2-one, dissolved in diethyl
ether, are added dropwise to 39 mL of a 3 molar solution of methylmagnesium
bromide in diethyl ether while being cooled with an ice bath in such a way
that
the temperature does not exceed 30 C. After the addition has ended the
reaction mixture is refluxed for 1.5 hours and then hydrolysed with 10%
ammonium chloride solution. After the removal of the organic phase the
aqueous phase is extracted with diethyl ether. The combined ether phases
are washed with water, dried with sodium sulphate and evaporated down. The
oil thus obtained is further reacted directly. Yield: 15.5 g (90%).
b) N-[2-(4-ethyl-phenyl)-1, 1-dim ethyl-ethyll-acetam ide
6.2 mL conc. sulphuric acid are added dropwise to 15.5 g (87 mmol) 1-(4-
ethyl-phenyl)-2-methyl -propan-2-ol in 4.8 mL (91 mmol) acetonitrile and 15
mL glacial acetic acid within 15 minutes, during which time the temperature
rises to 65 C. It is then stirred for one hour, diluted with ice water and
made
alkaline with conc. sodium hydroxide solution. After another 30 minutes'
stirring the solid precipitated is suction filtered and washed with water. The
crude product is dissolved in ethyl acetate, dried with sodium sulphate and
evaporated down. The oil remaining is combined with petroleum ether,
whereupon a solid is precipitated which is filtered off and dried.
Yield: 16.3 g (85%); melting point 90-92 C.
WO 2004/045618 23 PCT/EP2003/012565
CA 02506082 2005-05-13
c) 2-(4-ethyl-phenyl)-1,1-dimethyl-ethylam ine
16.3 g (74 mmol) of N-[2-(4-ethyl-phenyl)-1.,1-dimethyl-ethyl]-acetamide and
8.0 g of potassium hydroxide are refluxed for 15 hours in 60 mL
ethyleneglycol. The reaction mixture is combined with ice water and extracted
three times with diethyl ether. The combined organic phases are washed with
water, dried with sodium sulphate and freed from solvent. To prepare the
hydrochloride the crude product is dissolved in acetonitrile and ethereal
hydrochloric acid and diethyl ether are added successively. The solid
precipitated is suction filtered and dried.
Yield: 11.0 g (69%, hydrochloride); melting point 165-167 C.
d) 6-benzyloxy-8-{2-[2-(4-ethyl-phenyl)-1 1-dimethyl-ethylaminol-1-hydroxy-
ethyl)-4H-benzo[1 4]oxazin-3-one
The target compound is prepared analogously to the method for Example 8d)
from 2.14 g (6.0 mmol) 6-benzyloxy-8-(2-ethoxy-2-hydroxy-acetyl)-4H-
benzo[1,4]oxazin-3-one and 1.0 g (5.6 mmol) 2-(4-ethyl-phenyl)-1,1-dimethyl-
ethylamine. White solid.
Yield: 1.7 g (54%, hydrochloride); melting point 210-214 C.
e) 8-{2-[2-(4-ethyl-phenyl)-1 1 -dimethyl-ethylam inol-1-hydroxy-ethyl}-6
hydroxy-4H-benzo[1 4]oxazin-3-one
The hydrogenolysis of 1.45 g (2.75 mmol) 6-benzyloxy-8-{2-[2-(4-ethyl-
phenyl)-1,1-dimethyl-ethylam ino]-1-hydroxy-ethyl}-4H-benzo[1,4]oxazin-3-one
according to the method for Example 8e) yields the target compound in the
form of a white solid.
Yield: 1.07 g (92%; hydrochloride); melting point 266-269 C; mass
spectrometry: [M+H]+ = 385.
The (R)- and (S)-enantiomers of this example can be obtained by separating
the racemate analogously to current methods of racemate cleaving known in
the prior art.
WO 2004/045618 24 PCT/EP2003/012565
CA 02506082 2005-05-13
Example 10: 842-[2-(4-Fluoro-3-methyl-phenyl)-1 1-dim ethyl-ethylamino]-1-
hydroxy-ethyl}-6-hydroxy-4H-benzo[ 1 4]oxazin-3-one
0"
,:"r O OH
HN yL, N
F
OH
a) 1 -fluoro-2-methyl-4-(2-methyl-propenyl)-benzene
100 mL of a 0.5 molar solution of 4-fluoro-3-methyl-phenylmagnesium
bromide in THE are combined with 4.7 mL (50 mmol) isopropylaldehyde within
30 minutes, while the temperature rises to 45 C. The mixture is stirred for 30
minutes, refluxed for 1 hour and then hydrolysed with 10% ammonium
chloride solution. After separation of the organic phase extraction is carried
out with diethyl ether. The organic phases are combined, dried and
evaporated down. The alcohol thus obtained is dissolved in 100 mL toluene,
combined with 1 g of p-toluenesulphonic acid monohydrate and refluxed for
three hours using the water separator. The reaction mixture is poured onto
water and made alkaline with conc. sodium hydroxide solution. After
separation of the organic phase it is washed with water, dried with sodium
sulphate and freed from solvent. Fractional distillation of the residue yields
the
product in the form of a colourless liquid (boiling point 80-85 C/10 mbar).
Yield: 4.1 g (50%).
b) N-f2-(4-fluoro-3-methyl-phenyl)-1,1-dimethyl-ethyl]-form amide
4.9 mL conc. sulphuric acid are added dropwise at 5-15 C to 1.5 g (31 mmol)
sodium cyanide in 5 mL glacial acetic acid. Then the mixture is combined
with 3.9 g (24 mmol) 1-fluoro-2-methyl -4-(2-methyl-propenyl)-benzene,
dissolved in 10 mL glacial acetic acid, and stirred for 1 hour at 50-60 C. The
reaction mixture is diluted with ice water, made alkaline with conc. sodium
hydroxide solution and extracted with dichloromethane. The organic phase is
dried with sodium sulphate and freed from solvents in vacuo. The slightly
yellow oil thus obtained is further reacted directly. Yield: 4.3 g (87%).
WO 2004/045618 25 PCT/EP2003/012565
CA 02506082 2005-05-13
c) 2-(4-fluoro-3-methyl-phenyl)-1,1-dimethyl-ethylamine
4.3 g (20.6 mmol) N-[2-(4-fluoro-3-methyl-phenyl)-1,1-dimethyl-ethyl]-
formamide, 20 mL conc. hydrochloric acid and 20 mL water are refluxed for 2
hours. The reaction mixture is diluted with water, made alkaline with conc.
sodium hydroxide solution and extracted with dichloromethane. The organic
phases are dried with sodium sulphate and evaporated down. The residue is
dissolved in ethyl acetate, combined with ethereal hydrochloric acid and
cooled. The crystals precipitated are suction filtered and washed with diethyl
ether and dried. White solid.
Yield: 3.9 g (87%, hydrochloride); melting point 196-198 C.
d) 6-benzyloxy-8-{2-f2-(4-fluoro-3-methyl-phenyl)-1,1-dimethyl-ethylaminol-1-
hydroxy-ethyl}-4H-benzof 1,41oxazin-3-one
1.10 g (3.1 mmol) benzyloxy-8-(2-ethoxy-2-hydroxy-acetyl)-4H-
benzo[1,4]oxazin-3-one and 0.50 g (2.8 mmol) 2-(4-fluoro-3-methyl-phenyl)-
1, 1 -dimethyl-ethylamine are reacted and worked up analogously to the
method for Example 8d). White solid.
Yield: 0.75 g (47%, hydrochloride); melting point 228-230 C.
e) 8-{2-f2-(4-fluoro-3-methyl-phenyl)-1 1-dimethyl-ethylaminol-1-hydroxy-
ethyl}-6-hydroxy-4H-benzof 1,41oxazin-3-one
The hydrogenation of 0.70 g (1.4 mmol) 6-benzyloxy-8-{2-[2-(4-fluoro-3-
methyl-phenyl)-1,1-dimethyl-ethylamino]-1-hydroxy-ethyl}-4H-
benzo[1,4]oxazin-3-one yields the target compound as a white solid.
Yield: 0.50 g (87%, hydrochloride); melting point 278-280 C; mass
spectroscopy: [M+H]+ = 389.
The (R)- and (S)-enantiomers of this example can be obtained by separating
the racemate analogously to current methods of racemate cleaving known in
the prior art.
WO 2004/045618 26 PCT/EP2003/012565
CA 02506082 2005-05-13
Example 11: 8-{2-f2-(4-fluoro-2-methyl-phenyl)-1 1-dimethyl-ethylaminol-l -
hydroxy-ethyl}-6-hydroxy-4H-benzof 1,41oxazin-3-one
O OH
HN N
F
OH
a) 1-(4-fluoro-2-methyl-phenyl)-2-methyl-propyl acetate
500 mL of a 0.5 molar solution of 4-fluoro-6-methylphenylmagnesium bromide
and 23.2 mL (260 mmol) isopropylaldehyde are reacted analogously to
Example 10a). After hydrolysis with 10% ammonium chloride solution the
aqueous phase is separated off and extracted with diethyl ether . The
combined organic phases are dried with sodium sulphate and evaporated
down. The alcohol thus obtained is then dissolved in 50 mL acetic anhydride,
combined with 1 mL conc. sulphuric acid and refluxed for three hours. Then
the reaction mixture is poured onto water, stirred for a further hour and made
alkaline. It is extracted with dichloromethane, the organic phases are dried
with sodium sulphate and the solvents are distilled off. Fractional
distillation of
the residue yields the product in the form of a colourless liquid (boiling
point
105-110 C/8 mbar). Yield 29.0 g (52%).
b) N-[2-(4-fluoro-2-methyl-phenyl)-1,1-dimethyl-ethyll-formamide
29.0 g (130 mmol) 1-(4-fluoro-2-methyl-phenyl)-2-methyl-propyl acetate
are reacted and worked up analogously to the method for Example 10b).
Yellow oil. Yield: 27.0 g (99%).
c) 2-(4-fluoro-2-methyl-phenyl)-1,1-dimethyl-ethylam ine
In order to prepare the amine 27.0 g (130 mmol) of N-[2-(4-fluoro-2-methyl-
phenyl)-1,1-dimethyl-ethyl]-forma mide are reacted as described in the method
for Example 10c) reacted. White solid. Yield: 15.5 g (55%, hydrochloride);
melting point 277-280 C.
WO 2004/045618 27 PCT/EP2003/012565
CA 02506082 2005-05-13
d) 6-benzyloxy-8-{2-[2-[4-fluoro-2-methyl-phenyl)-1 1-dimethyl-ethylaminol-1-
hydroxy-ethyl}-4H-benzo[1,41oxazin-3-one
Prepared analogously to the method for Example 8d) from 0.95 g (2.66 mmol)
benzyloxy-8-(2-ethoxy-2-hydroxy-acetyl)-4H-benzo[1,4]oxazin-3-one and 0.43
g (2.37 mmol) 2-(4-fluoro-2-methyl-phenyl)-1,1-dimethyl-ethylamine.
Yield: 0.75 g (55%, hydrochloride); melting point 233-236 C.
e) 8-{2-[2-(4-fluoro-2-methyl-phenyl)-1,1-dimethyl-ethylaminol-1-hydroxy
ethyl }-6-hydroxy-4H-benzo[1,41oxazin-3-one
The debenzylation of 0.70 g (1.36 mmol) 6-benzyloxy-8-{2-[2-[4-fluoro-2-
methyl-phenyl)-1,1-dimethyl-ethylamino]-1-hydroxy-ethyl}-4H-
benzo[1,4]oxazin-3-one yields the target compound in the form of a white
solid. Yield: 0.50 g (87%, hydrochloride); melting point 278-280 C; mass
spectroscopy: [M+H]+ = 389.
The (R)- and (S)-enantiomers of this example can be obtained by separating
the racemate analogously to current methods of racemate cleaving known in
the prior art.
Example 12: 8-{2-[2-(2,4-difluoro-phenyl)-1,1-dimethyl-ethylaminol-1-
hydroxy-ethyl}-6-hydroxy-4H-benzo[1,41oxazin-3-one
0
y0 OH F
HN N
OH
a) 1-(2,4-difluoro-phenyl)-2-methyl propan-2-oi
11.0 mL acetone, diluted with 50 mL diethyl ether, are added dropwise to a
solution of 500 mL of 0.25 molar 2,4-difluorobenzylmagnesium bromide in
diethyl ether within 20 minutes. Then the mixture is refluxed for 1.5 hours
and
then hydrolysed with 10% ammonium chloride solution. The ether phase is
separated off, washed with water, dried with sodium sulphate and evaporated
down. The fractional distillation of the residue yields the alcohol as a
colourless liquid (boiling point 70-73 C/ 2 mmbar). Yield: 20.0 g (86%).
WO 2004/045618 28 PCT/EP2003/012565
CA 02506082 2005-05-13
b) N-f 2-(2 4-difluoro-phenyll-1,1-dimethyl-ethyl)-formamide
Ritter reaction with 20 g (110 mmol) 1-(2,4-difluoro-phenyl)-2-methyl-propan-
2-ol according to the method described for Example 10b). Yellow oil. Yield:
22.0 g (94%).
c) 2-(2 4-difluoro-phenyl)-1,1-dimethyl-ethylam ine
Reaction of 22.0 g (100 mmol) N-[2-(2,4-difluoro-phenyl]-1,1-dimethyl-ethyl]-
formamide analogously to the method for Example 10c).
Yield: 16.0 g (72%, hydrochloride); melting point 201-203 C.
d) 6-benz lox -8 2- 2- 2 4-difluoro- hen I -1 1-dimeth l-eth lamino -1-
hydroxy-ethyl}-4H-benzo( 1,41oxazin-3-one
Reaction of 0.89 g (2.49 mmol) benzyloxy-8-(2-ethoxy-2-hydroxy-acetyl)-4H-
benzo[1,4]oxazin-3-one and 0.40 g (2.16 mmol) 2-(2,4-difluoro-phenyl)-1,1-
dimethyl-ethylamine in the manner described for Example 8d).
Yield: 0.80 g (62%, hydrochloride); melting point 245-247 C.
e) 8-{2-f 2-(2 4-difluoro-phenyl)-1 1-dimethyl-ethylaminol-1-hydroxy-ethyl}-6-
hydroxy-4H-benzo[1 4loxazin-3-one
The hydrogenolysis of 0.70 g (1.35 mmol) 6-benzyloxy-8-{2-[2-(2,4-difluoro-
phenyl)-1,1-dim ethyl-ethylamino]-1-hydroxy-ethyl}-4H-benzo[ 1,4]oxazin-3-one
yields the target compound as a white solid. Yield: 0.48 g (83%,
hydrochloride); melting point 279-280 C; mass spectroscopy: [M+H]+ = 393.
The (R)- and (S)-enantiomers of this example can be obtained by separating
the racemate analogously to current methods of racemate cleaving known in
the prior art.
Example 13: 8-{2-[2-(3, 5-difluoro-phenyl)-1 1-dimethyl-ethylamino)-1-
hydroxy-ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one
o
~O OH
HN N F
OH F
WO 2004/045618 29 PCT/EP2003/012565
CA 02506082 2005-05-13
a) 1-(3,5-difluoro-phenyl)-2-methyl-propan-2-ol
The target compound is obtained by reacting a Grignard compound, prepared
from 25.0 g (121 mmol) 3,5-difluorobenzyl bromide, with 12.6 mL (171 mmol)
acetone. Yellow oil. Yield: 13.5 g (60%).
b) 2-(3,5-difluoro-phenyl)-1,1-dimethyl-ethylamine
The Ritter reaction of 5.5 g (29.5 mmol) 1-(3,5-difluoro-phenyl)-2-methyl-
propan-2-ol and 1.8 g sodium cyanide yields 7.0 g of formamide, which is
treated with hydrochloric acid to cleave the formyl group. Slightly yellow
oil.
Yield: 4.6 g (75%).
c) 6-benzyloxy-8-{2-[2-(3,5-difluoro-phenyl)-1,1-dimethyl-ethylaminol-l-
hydroxy-ethyl}-4H-benzo[1,41oxazin-3-one
Prepared from 1.73 g (4.84 mmol) benzyloxy-8-(2-ethoxy-2-hydroxy-acetyl)-
4H-benzo[1,4]oxazin-3-one and 0.80 g (4.32 mmol) 2-(3,5-difluoro-phenyl)-
1, 1 -dimethyl-ethylamine in the usual way.
Yield: 1.50 g (58 %, hydrochloride); melting point 240-244 C.
d) 8-{2-[2-(3,5-difluoro-phenyl)-1,1-dimethyl-ethylamino]-1-hydroxy-ethyl}-6-
hydroxy-4H-benzo[1,4]oxazin-3-one
Hydrogenolysis of 1.30 g (2.43 mmol) 6-benzyloxy-8-{2-[2-(3,5-difluoro-
phenyl)-1,1-dim ethyl -ethylamino]-1-hydroxy-ethyl}-4H-benzo[1,41oxazin-3-one
yields the target compound as a white solid. Yield: 0.90 g (86%,
hydrochloride); melting point 150-158 C; mass spectroscopy: [M+H]+ = 393.
The (R)- and (S)-enantiomers of this example can be obtained by separating
the racemate analogously to current methods of racemate cleaving known in
the prior art.
Example 14: 8-{2-[2-(4-ethoxy-phenyl)-1,1-dimethyl-ethylaminol-1-hydroxy-
ethyl}-6-hydroxy-4H-benzo[ 1,4]oxazin-3-one
o
~O OH
HN N
OH
WO 2004/045618 30 PCT/EP2003/012565
CA 02506082 2005-05-13
a) benzyl [2-(4-ethoxy-phenyl)-1,1-dim ethyl-ethyl]-carbam inate
15.0 g (50 mmol) benzyl [2-(4-hydroxy-phenyl)-1,1-dimethyl-ethyl]-
carbaminate are stirred with 7.5 mL (92 mmol) ethyl iodide and 21 g (150
mmol) potassium carbonate for 10 hours at 90-100 C. The reaction mixture is
combined with ethyl acetate, washed twice with water and dried with sodium
sulphate. After the solvents have been distilled off a yellow oil remains
(15.0
g, 92%), which is further reacted directly.
b) 2-(4-ethoxy-phenyl)-1 1-dimethyl-ethylamine
A solution of 15.0 g (49 mmol) benzyl [2-(4-ethoxy-phenyl)-1,1-dimethyl-
ethyl]-carbaminate in 100 mL glacial acetic acid is combined with 2 g
palladium on charcoal (10%) and then hydrogenated at 5 bar and 40 to 50 C.
The catalyst is filtered off and the filtrate is freed from solvent. The
residue is
dissolved in a little water, made alkaline with conc. sodium hydroxide
solution
and extracted with ethyl acetate. The organic phase is washed with water,
dried with sodium sulphate and evaporated down. The crude product is
dissolved in acetonitrile and acidified with ethereal hydrochloric acid. The
solid
precipitated after the addition of diethyl ether is suction filtered and
dried.
Yield: 8.8 g (hydrochloride, 84%); melting point 198-200 C.
c) 6-benzyloxy-8-{2-[2-(4-ethoxy-phenyl)-1 1-dim ethyl-ethylam inol-1-hydroxy-
ethyl}-4H-benzo[1,4loxazin-3-one
2.14 g (6.0 mmol) 6-benzyloxy-8-(2-ethoxy-2-hydroxy-acetyl)-4H-
benzo[1,4]oxazin-3-one and 1.0 g (5.2 mmol) 2-(4-ethoxy-phenyl)-1,1-
dimethyl-ethylamine are stirred in 40 mL ethanol for one hour at 50-80 C.
After cooling to ambient temperature 0.23 g (6.0 mmol) sodium borohydride
are added and the mixture is stirred for a further hour. The reaction mixture
is
combined with 5 ml acetone, stirred for 30 minutes, acidified with glacial
acetic acid and evaporated down. The residue is combined with water and
ethyl acetate and made alkaline. The organic phase is separated off, washed
with water, dried with sodium sulphate and freed from solvent in vacuo. The
residue is dissolved again in ethyl acetate and water, combined with conc.
hydrochloric acid and diluted with diethyl ether. The solid precipitated is
suction filtered and washed with diethyl ether. White solid.
Yield: 2.0 g (61 %, hydrochloride); melting point 214-216 C.
WO 2004/045618 31 PCT/EP2003/012565
CA 02506082 2005-05-13
d) 8-{2-f2-(4-ethoxy-phenyl)-1,1-dimethyl-ethylam inol-1-hydroxy-ethyl}-6-
hydroxy-4H-benzof 1,41oxazin-3-one
1.5 g (2.8 mmol) 6-benzyloxy-8-(2-[2-(4-ethoxy-phenyl)-1,1-dimethyl-
ethylamino]-1-hydroxy-ethyl}-4H-benzo[1,4]oxazin-3-one in 80 mL methanol
are hydrogenated with 250 mg palladium on charcoal (10 %) as catalyst at
ambient temperature and normal pressure. The catalyst is suction filtered and
the filtrate is evaporated down. The residue is dissolved in 5 mL ethanol by
heating, seeded and diluted with ethyl acetate. The solid precipitated is
filtered off and washed. White solid.
Yield 1.0 g (83%, hydrochloride); melting point 232-235 C; mass
spectrometry: [M+H]+ = 401.
The (R)- and (S)-enantiomers of this example can be obtained by separating
the racemate analogously to current methods of racemate cleaving known in
the prior art.
Example 15: 8-{2-f2-(3 5-dimethyl-phenyl)-1 1-dimethyl-ethylaminol-1-
hydroxy-ethyl}-6-hydroxy-4H-benzof 1,41oxazin-3-one
T O OH
HN N
OH
a) 1-(3 5-dimethyl-phenyl)-2-methyl-propanol-2-oI
Obtained by reacting ethyl (3,5-dimethyl-phenyl)-acetate with
methylmagnesium bromide.
b) 2-(3 5-dimethyl-phenyl)-1 1-dimethyl-ethylamine
By reacting 6.00 g (34 mmol) 1-(3,5-dimethyl-phenyl)-2-methyl-pro panol-2-oI
and 2.00 g (41 mmol) sodium cyanide in a Ritter reaction 2.40 g of 2-(3,5-
dimethyl-phenyl)-1,1-dimethyl-ethylformamide (35% yield) are obtained. To
liberate the amine the formamide (2.40 g, 11.7 mmol) is treated with
hydrochloric acid. The method and working up are analogous to the method
for Example 10c). Oil. Yield: 1.70 g (82%); mass spectroscopy: [M+H1+ = 178.
W(J 2004/045618 32 PCT/EP2003/012565
CA 02506082 2005-05-13
c) 6-benzyloxy-8-{2-[2-(3,5-dimethyl phenyl)-1,1-dimethyl-ethylaminol-1-
hydroxy-ethyl}-4H-benzof 1,41oxazin-3-one
Prepared analogously to the method for Example 8d) from 1.47 g (4.1 mmol)
benzyloxy-8-(2-ethoxy-2-hydroxy-acetyl)-4H-benzo[ 1, 4]oxazin-3-one and 0.65
g (3.7 mmol) 2-(3,5-dimethyl- phenyl)-1,1-dimethyl-ethylamine.
Yield: 1.1 g (51 %, hydrochloride); melting point 220-222 C.
d) 8-{2-[2-(3,5-dimethyl-phenyl)-1,1-dim ethyl-ethylaminol-1-hydroxy-ethyl}-6-
hydroxy-4H-benzo[1,41oxazin-3-one
The target compound was obtained after hydrogenolysis of 0.90 g (1.71
mmol) 6-benzyloxy-8-{2-[2-(3,5-dimethyl-phenyl)-1,1-dim ethyl-ethylamino]-1-
hydroxy-ethyl}-4H-benzo[1,4]oxazin-3-one and recrystallisation of the crude
product from isopropanol. White solid. Yield: 0.50 g (69%, hydrochloride);
melting point 235-238 C; mass spectroscopy: [M+H]+ = 385.
The (R)- and (S)-enantiomers of this example can be obtained by separating
the racemate analogously to current methods of racemate cleaving known in
the prior art.
Example 16: 4-(4-{2-[2-hydroxy-2-(6-hydroxy-3-oxo-3,4-dihydro-2H-
benzof 1 4loxazin-8-yl)-ethylaminol-2-methyl-propyl}-phenoxy)-butyric acid
0
~O OH
HN N
OH
OH O
a) ethyl 4-[4-(2-amino-2-methyl-propyl)-phenoxyl-butyrate
4.5 g (15.0 mmol) benzyl [2-(4-hydroxy-phenyl)-1,1-dimethyl-ethyl]-
carbaminate, 2.3 mL (16.0 mmol) ethyl 4-bromo-butyrate, 2.3 g (16.6 mmol)
potassium carbonate and 0.3 g (1.8 mmol) potassium iodide in 20 mL
dimethylformamide are heated to 120 C for 13 h. The reaction mixture is
diluted with ethyl acetate and washed successively with water, sodium
hydroxide solution and water. The organic phase is dried with sodium
sulphate and evaporated down. The residue is purified by chromatography
(eluant: cyclohexane/ethyl acetate = 9:1). 5.0 g of a yellow oil are isolated
which is dissolved in 50 mL acetic acid and hydrogenated with 1.0 g palladium
WO 2004/045618 33 PCT/EP2003/012565
CA 02506082 2005-05-13
on charcoal as catalyst at 40 C and 3 bar. The catalyst is filtered off and
the
filtrate is freed from solvent. The residue is dissolved in diethyl ether and
combined with ethereal hydrochloric acid. The solid precipitated is suction
filtered and dried.
Yield: 2.9 g (66% in two stages, hydrochloride); melting point = 103-105 C.
b) ethyl 4-(4-{2-[2-(6-benzyloxy-3-oxo-3,4-dihydro-2H-benzof 1,41oxazin-8-yl)-
2-hydroxy-ethylaminol-2-methyl-propyl}-phenoxy)-butyrate
1.20 g (3.36 mmol) benzyloxy-8-(2-ethoxy-2-hydroxy-acetyl)-4H-
benzo[1,4]oxazin-3-one and 0.90 g (3.22 mmol) ethyl 4-[4-(2-amino-2-methyl-
propyl)-phenoxy]-butyrate are reacted in the manner described for Example
8d). The crude product is dissolved in 10 mL ethyl acetate and 10 mL water
and combined with oxalic acid with stirring. The solution is diluted with
diethyl
ether and the solid precipitated is suction filtered and washed with diethyl
ether. Yield: 1.20 g (54%, oxalate); melting point 223-227 C.
c) 4-(442-[2-(6-benzyloxy-3-oxo-3 4-dihydro-2H-benzo[I,4]oxazin-8-yl)-2-
hydroxy-ethylam ino]-2-methyl-propyl}-phenoxy)-butyric acid
A solution of 1.00 g (1.73 mmol) ethyl 4-(4-{2-[2-(6-benzyloxy-3-oxo-3,4-
dihydro-2H-benzo[ 1, 4]oxazin-8-yl)-2-hydroxy-ethylam ino]-2-m ethyl-propyl}-
phenoxy)-butyrate in 25 mL methanol is combined with 2.5 mL 1 N sodium
hydroxide solution, refluxed for 30 minutes and then neutralised with 1 N
hydrochloric acid. The solution is evaporated down and the residual oil is
dissolved by heating in 5 mL of n-butanol. After the addition of a
crystallisation aid a solid is precipitated out which is suction filtered and
washed with acetone and diethyl ether.
Yield: 0.75 g (79%); melting point 216-218 C.
d) 4-(4-{2-[2-hydroxy-2-(6-hydroxy-3-oxo-3 4-dihydro-2H-benzo[1,41oxazin-8-
yl)-ethylamino)-2-methyl-propyl}-phenoxy)-butyric acid
0.70 g (1.28 mmol) 4-(4-{2-[2-(6-benzyloxy-3-oxo-3,4-dihydro-2H-
benzo[1, 4]oxazin-8-yl)-2-hydroxy-ethylam ino]-2-methyl-propyl}-phenoxy)-
butyric acid are dissolved in 25 mL methanol and 2 mL acetic acid and
hydrogenated in the presence of 150 mg palladium on charcoal (10%) at
ambient temperature and normal pressure. The catalyst is filtered off and the
filtrate is freed from solvent. The product is obtained by crystallisation
from a
methanol/acetone mixture. Yield: 0.40 g (68%); melting point 201-204 C;
mass spectroscopy: [M+H]+ = 459.
WO 2004/045618 34 PCT/EP2003/012565
CA 02506082 2005-05-13
The (R)- and (S)-enantiomers of this example can be obtained by separating
the racemate analogously to current methods of racemate cleaving known in
the prior art.
Example 17: 8-{2-[2-(3,4-difluoro-phenyl)-1 1-dimethyl-ethylaminol-1-
hydroxy-ethyl}-6-hydroxy-4H-benzo[1 4]oxazin-3-one
0
O OH
HN N F
F
OH
a) 1-(3,4-difluoro-phenyl)-2-methyl-propan-2-ol
From 23.0 g (111 mmol) 3,4-difluorobenzyl bromide a Grignard is prepared,
which is then reacted with 11.6 mL (158 mmol) acetone. Slightly yellow oil.
Yield: 9.7 g (47%); Rf value: 0.55 (ethyl acetate/petroleum ether = 1:3).
b) N-[2-(3,4-difluoro-phenyl)-1,1-dimethyl-ethyl]-formamide
The target compound is obtained by a Ritter reaction with 4.0 g (21.5 mmol)
1-(3,4-difluoro-phenyl)-2-methyl-propan-2-ol. Slightly yellow oil.
Yield: 4.0 g (87%); mass spectrometry: [M+H]+ = 214.
c) 2-(3,4-difluoro-phenyl)-1,1-dimethyl-ethylam ine
4.00 g (18.5 mmol) N-[2-(3,4-difluoro-phenyl)-1,1-dimethyl-ethyl]-formamide
are dissolved in ethanol, combined with conc. hydrochloric acid and refluxed
overnight. The reaction solution is poured onto ice water, made alkaline with
sodium hydroxide and extracted with tert-butylmethyl ether. The organic
phases are washed with water, dried with sodium sulphate and evaporated
down. Yellow oil. Yield: 3.2 g (92%); mass spectrometry: [M+H]+ = 186.
d) 842-[2-(3,4-difluoro-phenyl)-1,1-dimethyl-ethylaminol-1-hydroxy-ethyl}-6-
hydroxy-4H-benzo[1,41oxazin-3-one
357 mg (1 mmol) 6-benzyloxy-8-(2-ethoxy-2-hydroxy-acetyl)-4H-
benzo[1,4]oxazin-3-one and 185 mg (1 mmol) 2-(3,4-difluoro-phenyl)-1,1-
dimethyl-ethylamine are stirred for 30 minutes in 5 mL tetrahydrofuran at
ambient temperature. It is cooled to 0 C and under an argon atmosphere 1.5
VV" ~vv+iv t. ola 35 PCT/EP2003/012565
CA 02506082 2005-05-13
mL of a 2 molar solution of lithium borohydride in tetrahydrofuran is added
dropwise. The mixture is stirred for 30 min at ambient temperature, combined
with 10 mL dichloromethane and 3 mL water, stirred for a further hour and
then filtered through Extrelut . The eluate containing the ethanolamine is
freed from solvent. The residue is dissolved in methanol and hydrogenated
with palladium on charcoal (10%) as catalyst at 2.5 bar and ambient
temperature. Then the catalyst is separated off and the crude product is
purified by chromatography. White solid. Yield: 31 mg (6%, trifluoroethyl
acetate); mass spectroscopy: [M+H]+ = 393.
The (R)- and (S)-enantiomers of this example can be obtained by separating
the racemate analogously to current methods of racemate cleaving known in
the prior art.
Example 18: 8-(2-[2-(2-chloro-4-fluoro-phenyl)-1 1-dimethyl-ethylaminol-1-
hydroxy-ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one
0
O OH CI
HN N
F
OH
a) 1-(2-chloro-4-fluoro-phenyl)-2-methyl-propan-2-ol
Prepared from 20 g (97 mmol) methyl (2-chloro-4-fluoro-phenyl)-acetate and
98 mL of a 3 molar solution of methylmagnesium bromide analogously to the
method for Example 8a).
b) N-[2-(2-chloro-4-fluoro-phenyl)-1 1-dimethyl-ethyll-formamide
7.5 g (37 mmol) 1-(2-chloro-4-fluoro-phenyl)-2-methyl-propan-2-ol were
reacted and worked up according to the method described for Example 10b).
The oil thus obtained was chromatographed for further purification on a short
silica gel column (petroleum ether/ethyl acetate = 9:1). Oil.
Yield 7.4 g (87%); mass spectrometry: [M+H]+ = 230/232.
c) 2-(2-chloro-4-fluoro-phenyl)-1 1-dimethyl-ethylamine
Reaction of 7.4 g (32 mmol) N-[2-(2-chloro-4-fluoro-phenyl)-1,1-dimethyl-
ethyl]-formamide as described in the method for Example 17c). Brown oil.
Yield: 5.14 g (79%); mass spectrometry: [M+H]+ = 202/204.
WO 2004/045618 36 PCT/EP2003/012565
CA 02506082 2005-05-13
d) 8-{2-[2-(2-chloro-4-fluoro-phenyl)-l, 1-dimethyl-ethylam inol-1 -hydroxy-
ethyl}-6-hydroxy-4H-benzof 1,41oxazin-3-one
357 mg (1 mmol) of 6-benzyloxy-8-(2-ethoxy-2-hydroxy-acetyl)-4H-
benzo[1,4]oxazin-3-one and 202 mg (1 mmol) 2-(2-chloro-4-fluoro-phenyl)-
1,1-dimethyl-ethylamine are reacted with lithium borohydride analogously to
the method for Example 10d). To debenzylate the ethanolamine thus obtained
it is dissolved in 3 mL of dichloromethane and cooled to -78 C. At this
temperature 2 ml of a 1 molar solution of boron tribromide in dichloromethane
is added and the mixture is slowly allowed to come up to ambient
temperature. The reaction mixture is combined with 10 mL dichloromethane
and 3 mL water and filtered through Extrelut . The eluate is freed from
solvent
and the residue is purified by chromatography. White solid.
Yield: 70 mg (13%, trifluoroethyl acetate); mass spectroscopy: [M+H]+
409/11.
The (R)- and (S)-enantiomers of this example can be obtained by separating
the racemate analogously to current methods of racemate cleaving known in
the prior art.
Example 19: 8-{2-[2-(4-chloro-phenyl)-1 1-dimethyl-ethylaminol-1-hydroxy-
ethyl}-6-hydroxy-4H-benzo[1,41oxazin-3-one
0
TO OH
HN N
~ I / CI
OH
A solution of 300 mg (0.91 mmol) 6-benzyloxy-8-(2,2-dihydroxy-acetyl)-4H-
benzo[1,4]oxazin-3-one and 200 mg (1.09 mmol) 2-(4-chloro-phenyl)-1,1-
dimethyl-ethylamine in 3 mL ethanol was combined with molecular sieve and
stirred for 90 minutes at 80 C. It was allowed to cool to ambient temperature,
35 mg (0.91 mmol) of sodium borohydride were added and the mixture was
stirred for 1 hour. Then the reaction mixture was combined with sodium
hydrogen carbonate solution and extracted with ethyl acetate. The combined
organic phases were freed from solvent and the residue was
chromatographed (eluant: hexane/ethyl acetate/methanol), yielding 305 mg of
WO 2004/045618 37 PCT/EP2003/012565
CA 02506082 2005-05-13
ethanolamine. This was dissolved in 3 mL dichioromethane and cooled to
-78 C under an argon atmosphere. 3 mL of a 1 molar solution of boron
tribromide in dichioromethane were added dropwise and the mixture was
stirred for one hour at -78 C and for 20 minutes at ambient temperature. Then
at -78 C 3 mL of conc. ammonia solution were added dropwise and the
mixture was stirred for 5 minutes. The reaction mixture was combined with
ammonium chloride solution and extracted with ethyl acetate. The combined
organic phases were evaporated down and the residue was further purified by
chromatography (silica gel; eluant: dichioromethane/methanol + 1 %
ammonia). Beige-coloured solid: 93 mg (26%); mass spectrometry: [M+H] " _
391.
The (R)- and (S)-enantiomers of this example can be obtained by separating
the racemate analogously to current methods of racemate cleaving known in
the prior art.
Example 20: 8-{2-[2-(4-bromo-phenyl)-1,1-dimethyl-ethylamino]-1-hydroxy-
ethyl}-6-hydroxy-4H-benzof 1,41oxazin-3-one
0
T OH
HN N
Br
OH
The preparation of the ethanolamine and debenzylation were carried out as
described in Example 19 from 300 mg (0.91 mmol) 6-benzyloxy-8-(2,2-
dihydroxy-acetyl)-4H-benzo[1,4]oxazin-3-one and 250 mg (1.09) mmol) 2-(4-
bromo-phenyl)-1,1-dimethyl-ethylamine. Beige solid.
Yield: 54 mg (14%); mass spectrometry: [M+HJ+ = 435, 437.
The (R)- and (S)-enantiomers of this example can be obtained by separating
the racemate analogously to current methods of racemate cleaving known in
the prior art.
WO 2004/045618 38 PCT/EP2003/012565
CA 02506082 2005-05-13
Example 21: 8-{2-[2-(4-fluoro-phenyl)-1 1-dimethyl-ethylaminol-1-hydroxy-
ethyl}-6-hydroxy-4H-benzo[1 4loxazin-3-one
O
~O OH
HN N
OH
300 mg (0.91 mmol) of 6-benzyloxy-8-(2,2-dihydroxy-acetyl)-4H-
benzo[1,4]oxazin-3-one and 183 mg (1.09 mmol) 2-(4-fluoro-phenyl)-1,1-
dimethyl-ethylamine were dissolved in 3 ml of ethanol. Molecular sieve was
added and the mixture was heated to 80 C for 30 minutes. After cooling to
ambient temperature 35 mg (0.91 mmol) sodium borohydride were added.
The mixture was stirred for 1 hour at ambient temperature, then sodium
hydrogen carbonate solution was added to the reaction mixture and it was
extracted with ethyl acetate. The organic phases were evaporated down and
the residue was chromatographed (eluant: hexane/ethyl acetate/methanol).
The ethanolamine thus obtained (223 mg) was dissolved in methanol to
cleave the benzyl protecting group and hydrogenated with 150 mg palladium
hydroxide as catalyst at ambient temperature and normal pressure. The
catalyst was separated off by filtering through Celite , the filtrate was
freed
from solvent and the residue was chromatographed (silica gel; eluant:
dichloromethane/methanol). Beige solid. Yield: 76 mg (22%); mass
spectrometry: [M+H]+ = 375.
The (R)- and (S)-enantiomers of this example can be obtained by separating
the racemate analogously to current methods of racemate cleaving known in
the prior art.
The following compounds of formula 1 according to the invention may be
obtained analogously to the synthesis examples described above:
Example 22: 8-{2-[2-(4-fluoro-3-methoxy-phenyl)-1,1-dimethyl-ethylamino]-1-
hydroxy-ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one;
Example 23: 8-{2-[2-(4-fluoro-2,6-dimethyl-phenyl)-1,1-dimethyl-ethylamino]-
1-hydroxy-ethyl}-6-hydroxy-4H-benzo[I ,4]oxazin-3-one;
WO 2004/045618 39 PCT/EP2003/012565
CA 02506082 2005-05-13
Example 24: 8-{2-[2-(4-chloro-2-methyl-phenyl)-1,1-dim ethyl-ethylamino]-1-
hydroxy-ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one;
Example 25: 8-{2-[2-(4-chloro-3-fluoro-phenyl)-1,1-dimethyl-ethylamino]-1-
hydroxy-ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one;
Example 26: 8-{2-[2-(4-chloro-2-fluoro-phenyl)-1,1-dimethyl-ethylamino]-1-
hydroxy-ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one;
Example 27: 8-{2-[2-(3-chloro-4-fluoro-phenyl)-1,1-dimethyl-ethylamino]-1-
hydroxy-ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one;
Example 28: 8-{2-[2-(2,6-difluoro-4-methoxy-phenyl)-1,1-dimethyl-
ethylam ino]-1-hydroxy-ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one;
Example 29: 8-{2-[2-(2,5-difluoro-4-methoxy-phenyl)-1,1-dimethyl-
ethylam ino]-1-hydroxy-ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one;
Example 30: 8-{2-[2-(4-fluoro-3,5-dimethyl-phenyl)-1,1-dimethyl-ethylamino]-
1-hydroxy-ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one;
Example 31: 8-{2-[2-(3,5-dichloro-phenyl)-1,1-dimethyl-ethylamino]-1-
hydroxy-ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one;
Example 32: 8-{2-[2-(4-chloro-3-methyl-phenyl)-1,1-dim ethyl-ethylamino]-1-
hydroxy-ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one;
Example 33: 8-{2-[2-(3,4,5-trifluoro-phenyl)-1,1-dim ethyl-ethylam ino]-1-
hydroxy-ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one;
Example 34: 8-{2-[2-(3-methyl-phenyl)-1,1-dimethyl-ethylamino]-1-hydroxy-
ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one;
Example 35: 8-{2-[2-(3,4-dichloro-phenyl)-1,1-dimethyl-ethylamino]-1-
hydroxy-ethyl}-6-hydroxy-4H-benzo[1,4]oxazin-3-one.
WO 2004/045618 40 PCT/EP2003/012565
CA 02506082 2005-05-13
The compounds of general formula I may be used on their own or combined
with other active substances of formula 1 according to the invention. The
compounds of general formula 1 may optionally also be combined with other
pharmacologically active substances. These include, in particular,
anticholinergics, optionally other betamimetics, antiallergic agents, PDE-IV
inhibitors, PAF-antagonists, leukotriene-antagonists and corticosteroids and
combinations of these active substances.
Examples of preferred anticholinergics which may be mentioned include
ipratropium, oxitropium and tiotropium salts. Pharmaceutical combinations
which contain the abovementioned salts, in addition to the compounds of
formula 1 according to the invention, preferably contain those salts of
ipratropium, oxitropium or tiotropium wherein the anion is selected from
among the chloride, bromide, iodide, sulphate, phosphate,
methanesulphonate, nitrate, maleate, acetate, citrate, fumarate, tartrate,
oxalate, succinate, benzoate and p-toluenesulphonate, optionally in the form
of one of the solvates or hydrates thereof.
Within the scope of the present invention, the corticosteroids which may
optionally be used in conjunction with the compounds of formula 1 may be
compounds selected from among flunisolide, beclomethasone, triamcinolone,
budesonide, fluticasone, mometasone, ciclesonide, rofleponide and
dexamethasone. In some cases, within the scope of the present patent
application, the term steroids is used on its own instead of the word
corticosteroids. Any reference to steroids within the scope of the present
invention includes a reference to salts or derivatives which may be formed
from the steroids. Examples of possible salts or derivatives include: sodium
salts, sulphobenzoates, phosphates, isonicotinates, acetates, propionates,
dihydrogen phosphates, palmitates, pivalates or furoates. In some cases the
corticosteroids may also occur in the form of their hydrates.
Within the scope of the present invention, the term dopamine agonists, which
may optionally be used in conjunction with the compounds of formula 1,
denotes compounds selected from among bromocriptine, cabergolin, alpha-
dihydroergocryptine, lisuride, pergolide, pramipexol, roxindol, ropinirol,
talipexol, tergurid and viozan. Any reference to the abovementioned
dopamine agonists also includes, within the scope of the present invention, a
reference to any pharmacologically acceptable acid addition salts and
-------------
WO 2004/045618 41 PCT/EP2003/012565
CA 02506082 2005-05-13
hydrates thereof which may exist. By the physiologically acceptable acid
addition salts thereof which may be formed by the abovementioned dopamine
agonists are meant, for example, pharmaceutically acceptable salts selected
from among the salts of hydrochloric acid, hydrobromic acid, sulphuric acid,
phosphoric acid, methanesulphonic acid, acetic acid, fumaric acid, succinic
acid, lactic acid, citric acid, tartaric acid and malefic acid.
Examples of antiallergic agents which may be used according to the invention
as a combination with the compound of formula 1 include epinastin, cetirizin,
azelastin, fexofenadin, levocabastin, loratadine, mizolastin, ketotifen,
emedastin, dimetinden, clemastine, bamipin, cexchloropheniramine,
pheniramine, doxylamine, chlorophenoxamine, dimenhydrinate,
diphenhydramine, promethazine, ebastin, desloratidine and meclizine. Any
reference to the abovementioned antiallergic agents also includes, within the
scope of the present invention, a reference to any pharmacologically
acceptable acid addition salts thereof which may exist.
Examples of PDE-IV inhibitors which may be used according to the invention
as a combination with the compound of formula I include compounds
selected from among enprofylline, roflumilast, ariflo, Bay-198004, CP-
325,366, BY343, D-4396 (Sch-351591), V-11294A and AWD-12-281. Any
reference to the abovementioned PDE-IV inhibitors also includes, within the
scope of the present invention, a reference to any pharmacologically
acceptable acid addition salts thereof which may exist. By the physiologically
acceptable acid addition salts which may be formed by the abovementioned
PDE-IV inhibitors are meant, for example, pharmaceutically acceptable salts
selected from among the salts of hydrochloric acid, hydrobromic acid,
sulphuric acid, phosphoric acid, methanesulphonic acid, acetic acid, fumaric
acid, succinic acid, lactic acid, citric acid, tartaric acid and maleic acid.
According to the invention, the salts selected from among the acetate,
hydrochloride, hydrobromide, sulphate, phosphate and methanesulphonate
are preferred in this context.
Suitable preparations for administering the compounds of formula 1 include
for example tablets, capsules, suppositories, solutions and powders etc. The
content of the pharmaceutically active compound(s) should be in the range
from 0.05 to 90 wt.-%, preferably 0.1 to 50 wt.-% of the composition as a
whole. Suitable tablets may be obtained, for example, by mixing the active
WO 2004/045618 42 PCT/EP2003/012565
CA 02506082 2005-05-13
substance(s) with known excipients, for example inert diluents such as
calcium carbonate, calcium phosphate or lactose, disintegrants such as corn
starch or alginic acid, binders such as starch or gelatine, lubricants such as
magnesium stearate or talc and/or agents for delaying release, such as
carboxymethyl cellulose, cellulose acetate phthalate, or polyvinyl acetate.
The
tablets may also comprise several layers.
Coated tablets may be prepared accordingly by coating cores produced
analogously to the tablets with substances normally used for tablet coatings,
for example collidone or shellac, gum arabic, talc, titanium dioxide or sugar.
To achieve delayed release or prevent incompatibilities the core may also
consist of a number of layers. Similarly the tablet coating may consist of a
number or layers to achieve delayed release, possibly using the excipients
mentioned above for the tablets.
Syrups or elixirs containing the active substances or combinations thereof
according to the invention may additionally contain a sweetener such as
saccharine, cyclamate, glycerol or sugar and a flavour enhancer, e.g. a
flavouring such as vanillin or orange extract. They may also contain
suspension adjuvants or thickeners such as sodium carboxymethyl cellulose,
wetting agents such as, for example, condensation products of fatty alcohols
with ethylene oxide, or preservatives such as p-hydroxybenzoates.
Solutions are prepared in the usual way, e.g. with the addition of isotonic
agents, preservatives such as p-hydroxybenzoates or stabilisers such as
alkali metal salts of ethylenediaminetetraacetic acid, optionally using
emulsifiers and/or dispersants, while if water is used as diluent, for
example,
organic solvents may optionally be used as solubilisers or dissolving aids,
and
the solutions may be transferred into injection vials or ampoules or infusion
bottles.
Capsules containing one or more active substances or combinations of active
substances may for example be prepared by mixing the active substances
with inert carriers such as lactose or sorbitol and packing them into gelatine
capsules.
WO 2004/045618 43 PCT/EP2003/012565
CA 02506082 2005-05-13
Suitable suppositories may be made for example by mixing with carriers
provided for this purpose, such as neutral fats or polyethyleneglycol or the
derivatives thereof.
Excipients which may be used include, for example, water, pharmaceutically
acceptable organic solvents such as paraffins (e.g. petroleum fractions),
vegetable oils (e.g. groundnut or sesame oil), mono- or polyfunctional
alcohols (e.g. ethanol or glycerol), carriers such as e.g. natural mineral
powders (e.g. kaolins, clays, talc, chalk), synthetic mineral powders (e.g.
highly dispersed silicic acid and silicates), sugars (e.g. cane sugar, lactose
and glucose), emulsifiers (e.g. lignin, spent sulphite liquors,
methylcellulose,
starch and polyvinylpyrrolidone) and lubricants (e.g. magnesium stearate,
talc,
stearic acid and sodium lauryl sulphate).
For oral use the tablets may obviously contain, in addition to the carriers
specified, additives such as sodium citrate, calcium carbonate and dicalcium
phosphate together with various additional substances such as starch,
preferably potato starch, gelatin and the like. Lubricants such as magnesium
stearate, sodium laurylsulphate and talc may also be used to produce the
tablets. In the case of aqueous suspensions the active substances may be
combined with various flavour enhancers or colourings in addition to the
abovementioned excipients.
For administering the compounds of formula 1 for the treatment of COPD it is
particularly preferred according to the invention to use preparations or
pharmaceutical formulations which are suitable for inhalation. Inhalable
preparations include inhalable powders, propellant-containing metered-dose
aerosols or propellant-free inhalable solutions. Within the scope of the
present invention, the term propellant-free inhalable solutions also includes
concentrates or sterile inhalable solutions ready for use. The formulations
which may be used within the scope of the present invention are described in
more detail in the next part of the specification.
The inhalable powders which may be used according to the invention may
contain I either on its own or in admixture with suitable physiologically
acceptable excipients.
WO 2004/045618 44 PCT/EP2003/012565
CA 02506082 2005-05-13
If the active substances I are present in admixture with physiologically
acceptable excipients, the following physiologically acceptable excipients may
be used to prepare these inhalable powders according to the invention:
monosaccharides (e.g. glucose or arabinose), disaccharides (e.g. lactose,
saccharose, maltose), oligo- and polysaccharides (e.g. dextrans),
polyalcohols (e.g. sorbitol, mannitol, xylitol), salts (e.g. sodium chloride,
calcium carbonate) or mixtures of these excipients. Preferably, mono- or
disaccharides are used, while the use of lactose or glucose is preferred,
particularly, but not exclusively, in the form of their hydrates. For the
purposes
of the invention, lactose is the particularly preferred excipient, while
lactose
monohydrate is most particularly preferred.
Within the scope of the inhalable powders according to the invention the
excipients have a maximum average particle size of up to 250 pm, preferably
between 10 and 150 pm, most preferably between 15 and 80 pm. It may
sometimes seem appropriate to add finer excipient fractions with an average
particle size of 1 to 9 pm to the excipient mentioned above. These finer
excipients are also selected from the group of possible excipients listed
hereinbefore. Finally, in order to prepare the inhalable powders according to
the invention, micronised active substance 1, preferably with an average
particle size of 0.5 to 10 pm, more preferably from 1 to 5 m, is added to the
excipient mixture. Processes for producing the inhalable powders according
to the invention by grinding and micronising and finally mixing the
ingredients
together are known from the prior art.
The inhalable powders according to the invention may be administered using
inhalers known from the prior art.
The inhalation aerosols containing propellant gas according to the invention
may contain the compounds 1 dissolved in the propellant gas or in dispersed
form. The compounds 1 may be contained in separate formulations or in a
common formulation, in which the compounds 1 are either both dissolved,
both dispersed or in each case only one component is dissolved and the other
is dispersed. The propellant gases which may be used to prepare the
inhalation aerosols are known from the prior art. Suitable propellant gases
are selected from among hydrocarbons such as n-propane, n-butane or
isobutane and halohydrocarbons such as fluorinated derivatives of methane,
ethane, propane, butane, cyclopropane or cyclobutane. The abovementioned
WO 2004/045618 45 PCT/EP2003/012565
CA 02506082 2005-05-13
propellant gases may be used on their own or mixed together. Particularly
preferred propellant gases are halogenated alkane derivatives selected from
TG134a and TG227 and mixtures thereof.
The propellant-driven inhalation aerosols may also contain other ingredients
such as co-solvents, stabilisers, surfactants, antioxidants, lubricants and pH
adjusters. All these ingredients are known in the art.
The propellant-driven inhalation aerosols according to the invention
mentioned above may be administered using inhalers known in the art (MDIs
= metered dose inhalers).
Moreover, the active substances 1 according to the invention may be
administered in the form of propellant-free inhalable solutions and
suspensions. The solvent used may be an aqueous or alcoholic, preferably
an ethanolic solution. The solvent may be water on its own or a mixture of
water and ethanol. The relative proportion of ethanol compared with water is
not limited but the maximum is preferably up to 70 percent by volume, more
particularly up to 60 percent by volume and most preferably up to 30 percent
by volume. The remainder of the volume is made up of water. The solutions
or suspensions containing I are adjusted to a pH of 2 to 7, preferably 2 to 5,
using suitable acids. The pH may be adjusted using acids selected from
inorganic or organic acids. Examples of particularly suitable inorganic acids
include hydrochloric acid, hydrobromic acid, nitric acid, sulphuric acid
and/or
phosphoric acid. Examples of particularly suitable organic acids include
ascorbic acid, citric acid, malic acid, tartaric acid, maleic acid, succinic
acid,
fumaric acid, acetic acid, formic acid and/or propionic acid etc. Preferred
inorganic acids are hydrochloric and sulphuric acids. It is also possible to
use
the acids which have already formed an acid addition salt with one of the
active substances. Of the organic acids, ascorbic acid, fumaric acid and
citric
acid are preferred. If desired, mixtures of the above acids may be used,
particularly in the case of acids which have other properties in addition to
their
acidifying qualities, e.g. as flavourings, antioxidants or complexing agents,
such as citric acid or ascorbic acid, for example. According to the invention,
it
is particularly preferred to use hydrochloric acid to adjust the pH.
If desired, the addition of editic acid (EDTA) or one of the known salts
thereof,
sodium edetate, as stabiliser or complexing agent may be omitted in these
WO 2004/045618 46 PCT/EP2003/012565
CA 02506082 2005-05-13
formulations. Other embodiments may contain this compound or these
compounds. In a preferred embodiment the content based on sodium edetate
is less than 100 mg/100ml, preferably less than 50mg/100ml, more preferably
less than 20mg/100ml. Generally, inhalable solutions in which the content of
sodium edetate is from 0 to 10mg/100ml are preferred.
Co-solvents and/or other excipients may be added to the propellant-free
inhalable solutions. Preferred co-solvents are those which contain hydroxyl
groups or other polar groups, e.g. alcohols - particularly isopropyl alcohol,
glycols - particularly propyleneglycol, polyethyleneglycol,
polypropyleneglycol,
glycolether, glycerol, polyoxyethylene alcohols and polyoxyethylene fatty acid
esters. The terms excipients and additives in this context denote any
pharmacologically acceptable substance which is not an active substance but
which can be formulated with the active substance or substances in the
physiologically suitable solvent in order to improve the qualitative
properties of
the active substance formulation. Preferably, these substances have no
pharmacological effect or, in connection with the desired therapy, no
appreciable or at least no undesirable pharmacological effect. The excipients
and additives include, for example, surfactants such as soya lecithin, oleic
acid, sorbitan esters, such as polysorbates, polyvinylpyrrolidone, other
stabilisers, complexing agents, antioxidants and/or preservatives which
guarantee or prolong the shelf life of the finished pharmaceutical
formulation,
flavourings, vitamins and/or other additives known in the art. The additives
also include pharmacologically acceptable salts such as sodium chloride as
isotonic agents.
The preferred excipients include antioxidants such as ascorbic acid, for
example, provided that it has not already been used to adjust the pH, vitamin
A, vitamin E, tocopherols and similar vitamins and provitamins occurring in
the
human body.
Preservatives may be used to protect the formulation from contamination with
pathogens. Suitable preservatives are those which are known in the art,
particularly cetyl pyridinium chloride, benzalkonium chloride or benzoic acid
or
benzoates such as sodium benzoate in the concentration known from the
prior art. The preservatives mentioned above are preferably present in
concentrations of up to 50 mg/100 ml, more preferably between 5 and 20
Mg/100 MI.
WO 2004/045618 47 PCT/EP2003/012565
CA 02506082 2005-05-13
Preferred formulations contain, in addition to the solvent water and the
active
substance 1, only benzalkonium chloride and sodium edetate. In another
preferred embodiment, no sodium edetate is present.
The dosage of the compounds according to the invention is naturally highly
dependent on the method of administration and the complaint which is being
treated. When administered by inhalation the compounds of formula 1 are
characterised by a high potency even at doses in the pg range. The
compounds of formula 1 may also be used effectively above the pg range.
The dosage may then be in the gram range, for example.
In another aspect the present invention relates to the above-mentioned
pharmaceutical formulations as such which are characterised in that they
contain a compound of formula 1, particularly the above-mentioned
pharmaceutical formulations which can be administered by inhalation.
The following examples of formulations illustrate the present invention
without
restricting its scope:
Examples of pharmaceutical formulations
A) Tablets per tablet
active substance 1 100 mg
lactose 140 mg
maize starch 240 mg
polyvinylpyrrolidone 15 mg
magnesium stearate 5 mg
500 mg
The finely ground active substance, lactose and some of the maize starch are
mixed together. The mixture is screened, then moistened with a solution of
polyvinylpyrrolidone in water, kneaded, wet granulated and dried. The
granules, the remaining maize starch and the magnesium stearate are
screened and mixed together. The mixture is pressed into tablets of suitable
shape and size.
WO 2004/045618 48 PCT/EP2003/012565
CA 02506082 2005-05-13
B) Tablets per tablet
active substance 1 80 mg
lactose 55 mg
maize starch 190 mg
microcrystalline cellulose 35 mg
polyvinylpyrrolidone 15 mg
sodium carboxymethyl starch 23 mg
magnesium stearate 2 mg
400 mg
The finely ground active substance, some of the corn starch, lactose,
microcrystalline cellulose and polyvinylpyrrolidone are mixed together, the
mixture is screened and worked with the remaining corn starch and water to
form a granulate which is dried and screened. The sodium carboxymethyl
starch and the magnesium stearate are added and mixed in and the mixture is
compressed to form tablets of a suitable size.
C) Ampoule solution
active substance 1 50 mg
sodium chloride 50 mg
water for inj. 5 ml
The active substance is dissolved in water at its own pH or optionally at pH
5.5 to 6.5 and sodium chloride is added to make the solution isotonic. The
resulting solution is filtered to remove pyrogens and the filtrate is
transferred
under aseptic conditions into ampoules which are then sterilised and heat-
sealed. The ampoules contain 5 mg, 25 mg and 50 mg of active substance.
D) Metering aerosol
active substance 1 0.005
sorbitan trioleate 0.1
monofluorotrichloromethane and
TG 134a : TG227 2:1 ad 100
The suspension is transferred into a conventional aerosol container with
metering valve. Preferably 50 pl suspension are released on each actuation.
WO 2004/045618 49 PCT/EP2003/012565
CA 02506082 2005-05-13
The active substance may also be released in higher doses if desired
(e.g. 0.02 wt.-%).
E) Solutions (in mg/100ml)
active substance 1 333.3 mg
benzalkonium chloride 10.0 mg
EDTA 50.0 mg
HCI (1 N) ad pH 3.4
This solution can be prepared in the usual way.
F) Inhalable powder
active substance 1 12 lag
lactose monohydrate ad 25 rig
The inhalable powder is prepared in the usual way by mixing the individual
ingredients.