Note: Descriptions are shown in the official language in which they were submitted.
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-1-
MIXED LINEAGE HINASE MODULATORS
BACKGROUND OF THE INVENTION
Mixed lineage kinases (MLKs) are a family of serine/threonine protein kinases
that function in a cascade of phosphorylation reactions to control the
activity of specific
mitogen activated protein kinases (MAPKs). Gallo and Johnson, Nature Reviews;
3: 663-
664 (2002). MLKs act as MAPK-kinase kinases (MAPKKKs) to activate, for
example,
the stress activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) and p38
kinase
pathways, through phosphorylation of MAPK kinases (MKKs). Bloem et al., J.
Mol. Cell
Cardiol.; 33: 1739-1750, (2001). SAPK/JNK and p38 kinase are specifc MAPKs
that,
like other eukaryotic MAPKs, are activated in response to a multitude of
stimuli including
exposure to inflammatory cytokines, hormones, and growth factors as well as
cellular
stresses such as heat shock, inhibition of protein glycosylation, and exposure
to
ultraviolet irradiation. Fanger et al., Curr. Opin. Genet. Dev.; 7(1): 67-74
(1997).
Three subfamilies of MLKs have been previously identified and grouped on the
basis of domain arrangements and sequence homology within their catalytic
domains.
The MLKs (MLKl-MLK4) contain an amino-terminal SRC homology domain (SH3),
followed sequentially by a catalytic kinase domain, a leucine zipper region,
and a
2 0 Cdc42/Rac-interactive binding domain (CRIB Motif). MLKl-MLK4 share
approximately 75% homology within their catalytic domains and approximately
65%
homology from the SH3 domain to the CRIB motif. However, the carboxy terminus
of
each of these kinases diverge; indicating these regions may serve different
regulatory
functions. Nature Reviews; 3: 663-664 (2002). The duel leucine zipper bearing
kinases
2 5 (DLKs) represent the second family of MLKs and are characterized by a
kinase domain
followed by two leucine zipper motifs, separated by a 31 amino acid spacer.
The catalytic
domains of the two DLKs (DLK and leucine zipper kinase (LZK)) share
approximately
87% sequence homology but again diverge in their carboxy terminus. The final
subfamily
of MLK is represented by zipper sterile oc o~motif kinase (ZAK). ZAK shares
homology
3 0 with the other MLKs through the leucine zipper domain, but again diverges
from the
others at the carboxy terminus. Nature Reviews; 3: 663-664 (2002). While all
of the
MLKs have been shown to activate the c-Jun N-terminal kinase pathway, some
have also
been shown to activate the p38 kinase pathway as well. In addition, other
MAPKKKs
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-2-
have been identified which activate the JNK and p38 kinase pathways, including
MEK
kinase; apoptosis inducing kinase 1 (ASKl); and transforming growth factor
beta
(TGF(3)-activated kinase (TAK1). Nature Reviews; 3663-664 (2002).
Congestive heart failure (CHF) is a complex disorder with several etiologies
including hypertension, myocardial injury, and hemodynamic overload. One of
the
adaptive responses of the heart to these stresses is hypertrophy of the
cardiac myocyte,
characterized by altered gene transcriptional regulation, increased protein
synthesis, and
increased organization of the myofibril. This hypertrophy, in turn, may lead
to
remodeling of the heart and subsequent failure. J. Mol. Cell Cardiol; 33: 1739-
1750,
(2001). Evidence suggests that activation of signal transduction pathways,
including
MAPK and stress activated protein kinase (SAPK/JNK) pathways contribute to
hypertrophic changes in the heart. (See generally: Sugden et al., J. Mol.
Med.; 76: 725-
746, (1998); Force et al., Gene Expression; 7: 337-348, (1999); Ramirez et
al., J. Biol.
Chem.; 272: 14057-14061, (1997); Bogoyevitch; Cardiovasc. Res., 45: 826-842,
(2000);
Hines et al.; J. Mol. Cell. Cardiol., 30: 485-494, (1998) and Clerk et al.;
Am. J. Cardiol.,
83: 64H-69H, (1999)).
Recently, a novel MAPKKK, designated as MLK-7, was identified from a
database mining effort of a cDNA library from human failed heart tissue. The
cDNA
encodes for a 55 kDa. protein with serine/threonine kinase activity when
expressed and
purified from insect cells. J. Mol. Cell Cardiol; 33: 1739-1750, (2001) In
addition,
MLK-7 activates the SAPK/JNKl pathway in rat neonatal cardiac myocytes and
modulates fetal expression of marker genes for cardiac hypertrophy.
Specifically, MLK-7
increased expression of atrial natruiretic factor (ANF) and decreased
expression of a,
myosin heavy chain (ocMHC) mRNAs in rat neonatal cardiac myocytes.
Furthermore,
2 5 MLK-7 also increased protein synthesis in cardiac myocytes as evidenced by
increased
~14C~ phenylalanine incorporation in MLK-7-infected cells. J. Mol. Cell
Cardiok 33:
1739-1750, (2001). Taken together this data suggests that MLK-7 is implicated
in the
hypertrophy of cardiac myocytes that occurs in response to the various
etiologies of
congestive heart failure. Thus antagonists of MLK-7 activity may have utility
in the
3 0 treatment patients suffering from CHF.
Surprisingly, and in accordance with this invention, Applicants have
discovered a
series of dihydropyrrolopyrazole compounds of Formula I which are believed to
be novel,
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-3-
as more fully described below, which are potent antagonists of the recently
identified
MLK-7. Such antagonists could be useful for the treatment of CHF and could
therfore
address a long felt need for a safe and effective treatment for CHF. The
treatment of
cardiovascular disorders is hereby furthered.
SUMMARY OF THE INVENTION
The present invention is directed to the discovery that dihydropyrrolopyrazole-
derivative compounds of the present invention, as defined below, are
antagonists of the
mitogen activated protein kinasase kinase kinase, MLK-7. Accordingly, the
present
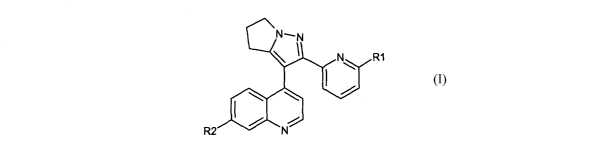
invention provides a compound of the formula:
R1
Formula I
wherein,
R1 represents hydrogen, halo, or (C1-C4)alkyl; and
R2 represents:
(a) aryl;
(b) aryl optionally
substituted one to
three times with a
substituent
2 0 independently selected
from the group consisting
of
(i) halo,
(ii) amino,
(iii) nitro,
(iv) hydroxy,
2 5 (v) cyano,
(vi) (Cl-Cq.)alkyl,
(vii) (Cl-Cq,)alkoxy,
(viii) hydroxy(Cl-Cq.)alkyl,
(ix) amino(Cl-Cq.)alkyl
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-4-
(x) hydroxy(C1-Cq.)alkoxy,
(xi) halo(C 1-C4)alkoxy,
(xii) (Cl-Cq.)alkoxy(C1-Cq.)alkoxy,
(xiii) trifluoromethyl,
(xiv) difluoromethyl,
(xv) trifluromethoxy,
(xvi) difluoromethoxy,
(xvii) (C3-C~)cylcoalkyl,
(xviii) COR3,
(xix) (C1-Cq.)alkyl-COR4,
(xx) amino(C1-C4)alkyl- COR4,
(xxi) hydroxy(Cl-C4)alkyl- COR4
(xxii) (Cl-Cq.)alkoxy-CORS,
(xxiii) -C(NHZ)=N-OH
(xxiv) NHSO~R6,
(xxv) SO~R~,
(xxvi) NHCORg,
(xxvii) SOR9,
(xxviii) SR10,
2 0 (xxix) CONHR1 l,
(xxx) O-(CH~)q-NR12R13, wherein q represents 1-4,
(xxxi) tetrazole,
(xxxii) methyltetrazole, and
(xxxiii) CONCH-NR15R16
2 5 (c) heterocycle;
(d) heterocycle optionally substituted one to three
times with a
substituent independently selected from the group
consisting of:
(i) halo,
(ii) amino,
30 (iii) (C1-Cq.)alkyl,
(iv) (Cl-Cq.)alkoxy,
(v) halophenyl(C1-Cq.)alkyl,
(vi) (C1-Cq.)alkyl-(Cl-Cq.)alkoxycarbonyl,
(vii) CHO,
3 5 (viii) COR3, or
(ix) SO~R~,
(e) benzofused heterocycle;
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-5-
(f) benzofused heterocycle optionally substituted one or two times
with a substituent independently selected from the group consisting of
(i) halo,
(ii) amino,
(iii) (C 1-Cq.)alkyl,
(iv) (C 1-Cq.)alkoxy, and
(v) (Cl-Cq.)alkylcarbonyl, or
(g) (C3-C~)cylcoalkyl;
R3 represents independently at each occurrence amino, hydroxy, (Cl-Cq.)alkyl,
(Cl-C4)alkoxy, NH-(Cl-Cq.)alkylamine, N,N-(Cl-Cq.)dialkylamine, or a
heterocycle
selected from the group consisting of
-rt-N~ -~--NHS ---N
(a) , (b) , (c)
A~~ ~O
N S ~ ~N~N J
H
(d) ~ or (e)
R4 and RS represent independently at each occurrence amino, hydroxy, (Cl-
Cq.)alkyl, or (C1-Cq.)alkoxy;
R6 and R~ represent independently at each occurrence amino or (Cl-C4)alkyl;
Rg represents independently at each occurrence amino, (Cl-C,~)alkyl, or (C1
Cq.)alkoxy;
R9 and Rl~ represent independently at each occurrence (C1-C4)alkyl;
2 0 R11 represents independently at each occurrence (C1-C4)alkyl or a
substituent
selected from the group consisting of:
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-6-
(a) _(CH2)n_X_Y
(b) -CH(COR1ø)-(CHZ)m X'-Y~
(c) _--
~ s~\O
O
(d)
N
H
and (e)
N
-OC(CH3)3
O
wherein,
n and m each independently represent 0-4;
X and X' represent independently at each occurrence -CO-, -
CH2-, -NH-, -S-, or-SO~-; and
Y and Y' represent independently at each occurrence amino,
hydroxy, (C 1-Cq.)alkyl, (C 1-Cq.)alkoxy, (C 1-Cq.)alkoxycarbonyl, NH-(C 1-
Cq.)alkylamine, orN,N-(Cl-Cq.)dialkylamine,
provided that where X or X' represents S, then Y or Y" is not
amino or hydroxy;
R1~ and R13 represent independently at each occurrence hydrogen or (Cl-
Cq.)alkyl, or Rl~ and R13 together with the nitrogen atom to which they are
attached form
a piperidino, pyrrolidino, morpholino or a methylpiperazino group;
R14 represents independently at each occurrence hydroxy, amino, or (C1-
Cq.)alkoxy; and
R15 and R16 each represent independently at each occurrence hydrogen or (Cl-
Cq.)alkyl,
or a pharmaceutically acceptable salt thereof.
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
In another embodiment, the present invention provides a method of treating or
preventing congestive heart failure, comprising administering to a patient in
need thereof
an effective amount of a compound of Formula I, or a pharmaceutically
acceptable salt
thereof.
In addition, the present invention provides pharmaceutical compositions of
compounds of Formula I, including the pharmaceutically acceptable salts and
hydrates
thereof, comprising a compound of Formula I in combination with a
pharmaceutically
acceptable carrier, diluent or excipient. This invention also encompasses
novel
intermediates, and processes for the synthesis of the compounds of Formula I.
In another embodiment, the present invention provides the use of a compound of
Formula I for the manufacture of a medicament for treating or preventing
congestive heart
failure.
DETAILED DESCRIPTION OF THE INVENTION
The present invention provides compounds with affinity for the mitogen
activated
protein kinase kinase kinase designated as MLK-7, which could be used to
antagonize or
partially antagonize kinase activity and therby influence physiological
functions related to
kinase levels and/or kinase activity. In this regard, such ligands are
believed to be useful
2 0 in treating or preventing physioligcal disorders susceptible to MLK7
modulation,
particularly MLK-7 antagonism. Thus, methods for the treatment or prevention
of
physiolgical disorders susceptible to MAPKKK modulation, particularly MLK-7
antagonism, constitute an important embodiment of the present invention. As a
particular
aspect, the present invention provides compounds useful as MLK-7 modulators.
As a
2 5 more particular aspect, the present invention provides compounds useful as
MLK-7
antagonists. Furthermore, compounds of Formula I are believed to be novel and,
thus, to
constitute yet another important embodiment of the present invention. In
addition,
compounds of the present invention may also exert antagonist activity at other
MAPKKKs as well as inhibiting the action of other serine/threonine kinases
such as the
3 o TGF(3 receptors, Type I and Type II.
As will be understood by the skilled artisan, some of the compounds useful for
the
methods of the present invention may be available for prodrug formulation.
Where used
herein, the term "prodrug" refers to a compound of Formula I which has been
structurally
modified such that in vivo the prodrug is converted, for example, by
hydrolytic, oxidative,
3 5 reductive, or enzymatic cleavage, into the parent molecule ("drug") as
given by Formula
I. Such prodrugs may be, for example, metabolically labile ester derivatives
of the parent
compound where said parent molecule bears a carboxylic acid group.
Conventional
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
_g_
procedures for the selection and preparation of suitable prodrugs are well
known to one of
ordinary skill in the art.
It is also understood that many of the MAPI~KK modulators of the present
invention may exist as pharmaceutically acceptable salts and, as such,
pharmaceutically
acceptable salts are therefore included within the scope of the present
invention. The term
"pharmaceutically acceptable salt" where used herein, refers to salts of the
compounds of
Formula I, which are substantially non-toxic to living organisms. Typical
pharmaceutically acceptable salts include those salts prepared by reaction of
the
compounds of the present invention with a pharmaceutically acceptable mineral
or
organic acid or an organic or inorganic base. Such salts are known as acid
addition and
base addition salts. It is further understood by the skilled reader that salt
forms of
pharmaceutical compounds are commonly used because they are often more readily
crystallized, or more readily purified, than are the free bases. In all cases,
the use of the
pharmaceutical compounds of the present invention as salts is contemplated in
the
description herein. Hence, it is understood that where compounds of Formula I
are
capable of forming salts, the pharmaceutically acceptable salts are
encompassed in the
names provided herein.
Acids commonly employed to form acid addition salts are inorganic acids such
as
hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid,
phosphoric acid, and
2 0 the like, and organic acids such as p-toluenesulfonic, methanesulfonic
acid, oxalic acid,
p-bromophenylsulfonic acid, carbonic acid, succinic acid, citric acid, benzoic
acid, acetic
acid, and the like. Examples of such pharmaceutically acceptable salts are the
sulfate,
pyrosulfate, bisulfate, sulfite, bisulfite, phosphate, monohydrogenphosphate,
dihydrogenphosphate, metaphosphate, pyrophosphate, bromide, iodide,
hydroiodide,
2 5 dihydroiodide, acetate, propionate, decanoate, caprylate, acrylate,
formate, hydrochloride,
dihydrochloride, isobutyrate, caproate, heptanoate, propiolate, oxalate,
malonate~
succinate, suberate, sebacate, fumarate, maleate, butyne-1,4-dioate, hexyne-
1,6-dioate,
benzoate, chlorobenzoate, methylbenzoate, hydroxybenzoate, methoxybenzoate,
phthalate, xylenesulfonate, phenylacetate, phenylpropionate, phenylbutyrate,
citrate,
3 0 lactate, oc-hydroxybutyrate, glycolate, tartrate, methanesulfonate,
propanesulfonate,
naphthalene-1-sulfonate, napththalene-2-sulfonate, mandelate and the like.
Base addition
salts include those derived from inorganic bases, such as ammonium or alkali
or alkaline
earth metal hydroxides, carbonates, bicarbonates, and the like. Such bases
useful in
preparing the salts of this invention thus include sodium hydroxide, potassium
hydroxide,
3 5 ammonium hydroxide, potassium carbonate, sodium carbonate, sodium
bicarbonate,
potassium bicarbonate, calcium hydroxide, calcium carbonate, and the like. It
should be
recognized that the particular counterion forming a part of any salt of this
invention is
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-9-
usually not of a critical nature, so long as the salt as a whole is
pharmacologically
acceptable and as long as the counterion does not contribute undesired
qualities to the salt
as a whole. It is further understood that such salts may exist as a hydrate.
Where used herein, the term "stereoisomer" refers to a compound made up of the
same atoms bonded by the same bonds but having different three-dimensional
structures
which are not interchangeable. The three-dimensional structures are called
configurations. Where used herein, the term "enantiomer" refers to two
stereoisomers
whose molecules are nonsuperimposable mirror images of one another. The term
"chiral
center" refers to a carbon atom to which four different groups are attached.
Where used
herein, the term "diastereomers" refers to stereoisomers which are not
enantiomers. In
addition, two diastereomers which have a different configuration at only one
chiral center
are referred to herein as "epimers". The terms "racemate", "racemic mixture"
or "racemic
modification" refer to a mixture of equal parts of enantiomers.
The compounds of the present invention may have one or more chiral centers and
may, therefore, exist in a variety of stereoisomeric configurations. As a
consequence of
these chiral centers the compounds of the present invention may occur as
racemates,
mixtures of enantiomers, and as individual enantiomers as well as
diastereomers and
mixtures of diastereomers. All such racemates, enantiomers, and diastereomers
are within
the scope of the present invention. Enantiomers of the compounds provided by
the
2 0 present invention can be resolved, for example, by one of ordinary skill
in the art using
standard techniques such as those described by J. Jacques, et al.,
"Enantiomers,
Racemates, and Resolutions", Johm Wiley and Sons, Inc., 1981.
The terms "R" and "S" where used herein are as commonly used in organic
chemistry to denote specific configuration of a chiral center. The term "R"
(rectus) refers
2 5 to that configuration of a chiral center with a clockwise relationship of
group priorities
(highest to second lowest) when viewed along the bond toward the lowest
priority group.
The term "S" (sinister) refers to that conEguration of a chiral center with a
counterclockwise relationship of group priorities (highest to second lowest)
when viewed
along the bond toward the lowest priority group. The priority of groups is
based upon
3 0 their atomic number (in order of decreasing atomic number). A partial list
of priorities
and a discussion of stereochemistry is contained in "Nomenclature of Organic
Compounds: Principles and Practice", (J.H. Fletcher, et al., eds., 1974) at
pages 103-120.
The specific stereoisomers and enantiomers of compounds of Formula I can be
prepared by one of ordinary skill in the art utilizing well known techniques
and processes,
3 5 such as those disclosed by Eliel and Wilen, "Stereochemistry of Organic
Compounds",
John Wiley & Sons, Inc., 1994, Chapter 7; Separation of Stereoisomers,
Resolution,
Racemization; and by Collet and Wilen, "Enantiomers, Racemates, and
Resolutions",
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-10-
John Wiley & Sons, Inc., 1981. For example, specific stereoisomers and
enantiomers can
be prepared by stereospecific syntheses using enantiorrierically and
geometrically pure, or
enantiomerically or geometrically enriched starting materials. In addition,
the specific
stereoisomers and enantiomers can be resolved and recovered by techniques such
as
chromatography on chiral stationary phases, enzymatic resolution or fractional
recrystallization of addition salts formed by reagents used for that purpose.
Where used herein the term "Pg" refers to a suitable oxygen or nitrogen
protecting
group. Suitable oxygen or nitrogen protecting groups, where used herein,
refers to those
groups intended to protect or block the oxygen or nitrogen group against
undesirable
reactions during synthetic procedures. Whether the term "Pg", where used
herein,
represents an oxygen protecting group or a nitrogen protecting group will be
readily
apparent to the ordinarily skilled artisan. The suitability of the oxygen or
nitrogen
protecting group used will depend upon the conditions that will be employed in
subsequent reaction steps wherein protection is required, and is well within
the
knowledge of one of ordinary skill in the art.
Commonly used nitrogen protecting groups are disclosed in Greene, "Protective
Groups In ~rganic Synthesis," (John Wiley ~Z Sons, New York (1981)). Suitable
nitrogen
protecting groups comprise acyl groups such as formyl, acetyl, propionyl,
pivaloyl, t-
butylacetyl, 2-chloroacetyl, 2-bromoacetyl, trifluoroacetyl, trichloroacetyl,
phthalyl, o-
2 0 nitrophenoxyacetyl, .alpha.-chlorobutyryl, benzoyl, 4-chlorobenzoyl, 4-
bromobenzoyl, 4-
nitrobenzoyl, and the like; sulfonyl groups such as benzenesulfonyl, p-
toluenesulfonyl and
the like; carbamate forming groups such as benzyloxycarbonyl, p-
chlorobenzyloxycarbonyl, p-methoxybenzyloxycarbonyl, p-nitrobenzyloxycarbonyl,
2-
nitrobenzyloxycarbonyl, p-bromobenzyloxycarbonyl, 3,4-
dimethoxybenzyloxycarbonyl,
2 5 3,5-dimethoxybenzyloxycarbonyl, 2,4-dimethoxybenzyloxycarbonyl, 4-
methoxybenzyloxycarbonyl, 2-nitro-4,5-dimethoxybenzyloxycarbonyl, 3,4,5-
trimethoxybenzyloxycarbonyl, 1-(p-biphenylyl)-1-methylethoxycarbonyl,
.alpha.,.alpha.-
dimethyl-3,5-dimethoxybenzyloxycarbonyl, benzhydryloxycarbonyl, t-
butyloxycarbonyl,
diisopropylmethoxycarbonyl, isopropyloxycarbonyl, ethoxycarbonyl,
methoxycarbonyl,
3 0 allyloxycarbonyl, 2,2,2,-trichloroethoxycarbonyl, phenoxycarbonyl, 4-
nitrophenoxycarbonyl, fluorenyl-9-methoxycarbonyl, cyclopentyloxycarbonyl,
adamantyloxycarbonyl, cyclohexyloxycarbonyl, phenylthiocarbonyl and the like;
alkyl
groups such as benzyl, triphenylmethyl, benzyloxymethyl and the like; and
silyl groups
such as trimethylsilyl and the like. Commonly used oxygen protecting groups
are also
3 5 disclosed in Greene (supra). Suitable oxygen protecting groups comprise
alkyl groups
such as methyl, ethyl, and the like; silyl groups such as t-
butyldimethylsilyl, t-
butyldiphenylsilyl, triisopropylsilyl, and the like, with t-butyldimethylsilyl
being
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-11-
preferred. Other commonly used oxygen protecting groups include benzyl, 4-
nitrophenyl
methyl, benzoyl, and the like.
Where used herein the term "(C1-C4)alkyl" refers to a straight or branched,
monovalent, saturated aliphatic chain of 1 to 4 carbon atoms and includes, but
is not
limited to, methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl and the
like.
Where used herein the term "(C1-C6)alkyl" refers to a straight or branched,
monovalent, saturated aliphatic chain of 1 to 6 carbon atoms and includes, but
is not
limited to methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, t-butyl, n-
pentyl, n-hexyl,
and the like. It is understood that the term "(C1-C4)alkyl" is included within
the definition
of "(C1-C6)alkyl".
Where used herein the term "(C1-Clo)alkyl" refers to a straight or branched,
monovalent, saturated aliphatic chain of 1 to 10 carbon atoms and includes,
but is not
limited to methyl, ethyl, propyl, isopropyl, n-butyl, isobutyl, tertiary
butyl, pentyl,
isopentyl, hexyl, 2,3-dimethyl-2-butyl, heptyl, 2,2-dimethyl-3-pentyl, 2-
methyl-2-hexyl,
octyl, 4-methyl-3-heptyl and the like. It is understood that the terms "(C1-
C4)alkyl" and
"(C1-C6)alkyl" are included within the definition of ".(C1-Clo)alkyl".
Where used herein the terms "Me" "Et" "Pr" "iPr" "Bu" and "t-Bu" refer to
> > > > >
methyl, ethyl, propyl, isopropyl, butyl and tert-butyl respectively.
Where used herein, the teen "(C1-C4)alkoxy" refers to an oxygen atom bearing a
2 0 straight or branched, monovalent, saturated aliphatic chain of 1 to 4
carbon atoms and
includes, but is not limited to, methoxy, ethoxy, n-propoxy, isopropoxy, n-
butoxy, and the
like. Where used herein the teen "(C1-C6)alkoxy" refers to an oxygen atom
bearing a
straight or branched, monovalent, saturated aliphatic chain of 1 to 6 carbon
atoms and
includes, but is not limited to, methoxy, ethoxy, n-propoxy, isopropoxy, n-
butoxy, n-
2 5 pentoxy, n-hexoxy, and the like. It is understood that the term "(Cl-
C4)alkoxy" is
included within the definition of "(C1-C6)alkoxy".
Where used herein, the term "amino(C1-C4)alkyl" refers to a straight or
branched,
monovalent, saturated aliphatic chain of 1 to 4 carbon atoms bearing an amino
group
attached to one of the carbon atoms.
3 0 Where used herein, the term "hydroxy(C1-C4)alkyl" refers to a straight or
branched, monovalent, saturated aliphatic chain of 1 to 4 carbon atoms bearing
a hydroxyl
group attached to one of the carbon atoms. Where used herein, the term
"hydroxy(C1-
C6)alkyl" refers to a straight or branched, monovalent, saturated aliphatic
chain of 1 to 6
carbon atoms bearing a hydroxyl group attached to one of the carbon atoms. It
is
3 5 understood that the term "hydroxy(Ci-C4)alkyl" is included within the
definition of
"hydroxy(C1-C6)alkyl".
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-1 ~-
Where used herein, the term "hydroxy(C1-C4)alkoxy" refers to an oxygen atom
bearing a straight or branched, monovalent, saturated aliphatic chain of 1 to
4 carbon
atoms with a hydroxyl group attached to one of the carbon atoms. Where used
herein, the
term "hydroxy(C1-C6)alkoxy" refers to an oxygen atom bearing a straight or
branched,
monovalent, saturated aliphatic chain of 1 to 6 carbon atoms with a hydroxyl
group
attached to one of the carbon atoms. It is understood that the term
"hydroxy(C1-
Cø)alkoxy" is included within the definition of "hydroxy(C1-C6)alkoxy".
Where used herein, the term "(Cl-C4)alkyl-(C1-C4)alkoxy" refers to a straight
or
branched, monovalent, saturated aliphatic chain of 1 to 4 carbon atoms which
has a (C1-
l0 C4)alkoxy group~attached to the aliphatic chain. Where used herein, the
term
"(C1-C4)alkoxy-(C1-C4)alkoxy" refers to an oxygen atom bearing straight or
branched,
monovalent, saturated aliphatic chain of 1 to 4 carbon atoms which has a (C1-
C4)alkoxy
group attached to the aliphatic chain.
Where used herein, the term "(C1-C4)alkyl-COR4" refers to a straight or
branched,
monovalent, saturated aliphatic chain of 1 to 4 carbon atoms which has a COR4
group
attached to the aliphatic chain (R4 being as defined elsewhere herein). Where
used
herein, the term "(C1-C4)alkoxy-CORS" refers to an oxygen atom bearing a
straight or
branched, monovalent, saturated aliphatic chain of 1 to 4 carbon atoms which
has a CORS
group attached to the aliphatic chain (RS being as defined elsewhere herein).
2 0 Where used herein, the term "amino(Cl-C4)alkyl-COR4" refers to an amino(C1-
C4)alkyl group which has a COR4 group attached to the aliphatic chain (R4
being as
defined elsewhere herein). The term "hydroxy(C1=C4)alkyl-COR4" refers to a
hydroxy(C1-C4)alkyl group which has a COR4 group attached to the aliphatic
chain (R4
being as defined elsewhere herein).
2 5 Where used herein, the terms "halo", "halide" or "hal" of "Hal" refer to a
chlorine,
bromine, iodine or fluorine atom, unless otherwise specified herein.
Where used herein, the term "halo(C1-C4)alkyl" refers to a straight or
branched,
monovalent, saturated aliphatic chain of 1 to 4 carbon atoms bearing one or
more halo
groups attached to one or more of the carbon atoms. Where used herein, the
term
3 0 "halo(C1-C6)alkyl" refers to a straight or branched, monovalent, saturated
aliphatic chain
of 1 to 6 carbon atoms bearing one or more halo groups attached to one or more
of the
carbon atoms. It is understood that the term "halo(C1-C4)alkyl" is included
within the
definition of "halo(C1-C~)alkyl". Where used herein, the term "halo(C~-
C4)alkoxy"
refers to an oxygen atom bearing a straight or branched, monovalent, saturated
aliphatic
3 5 chain of 1 to 4 carbon atoms bearing one or more halo groups attached to
one or more of
the carbon atoms. Where used herein, the term "halo(C1-C6)alkoxy" refers to an
oxygen
atom bearing a straight or branched, monovalent, saturated aliphatic chain of
1 to 6
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-13-
carbon atoms bearing one or more halo groups attached to one or more of the
carbon
atoms. It is understood that the term "halo(C1-C4)alkoxy" is included within
the
definition of "halo(C1-C6)alkoxy".
Where used herein, the term "halophenyl(C1-C4)alkyl" refers to a straight or
branched, monovalent, saturated aliphatic chain of 1 to 4 carbon atoms bearing
phenyl
group wich is further substituted with a halo moiety. Examples of
"halophenyl(C1-
C4)alkyl" include (4-chlorophenyl)methyl, (3-chlrorophenyl)methyl, (4-
chlorophenyl)ethyl, (3-chlrorophenyl)ethyl, and the like
Where used herein the term "(C2-C6)alkenyl" refers to a straight or branched,
monovalent, unsaturated aliphatic chain having from two to six carbon atoms
and having
a double bond. Typical (CZ-C6)alkenyl groups include ethenyl (also known as
vinyl), 1-
methylethenyl, 1-methyl-1-propenyl, 1-butenyl, 1-hexenyl, 2-methyl-2-propenyl,
1-
propenyl, 2-propenyl, 2-butenyl, 2-pentenyl, and the like.
Where used herein the term "(C2-C6)alkynyl" refers to a straight or branched,
monovalent, unsaturated aliphatic chain having from two to six carbon atoms
and having
a triple bond.
Where used herein, the term "aryl" refers to a monovalent carbocyclic group
containing one or more fused or non-fused phenyl rings and includes, for
example,
phenyl, 1- or 2-naphthyl, 1,2-dihydronaphthyl, 1,2,3,4-tetrahydronaphthyl, and
the like.
2 0 Where used herein, the term "aryl (C1-C6)alkyl" refers to a straight or
branched,
monovalent, saturated aliphatic chain of 1 to 6 carbon atoms which has an aryl
group
attached to the aliphatic chain. "aryl (Cl-C4)alkyl" refers to a straight or
branched,
monovalent, saturated aliphatic chain of 1 to 4 carbon atoms which has an aryl
group
attached to the aliphatic chain. It is understood that the term "aryl (C1-
C4)alkyl" is
2 5 included within the definition of "aryl (C1-C6)alkyl". Examples of "aryl
(C1-C6)alkyl"
include:
' _
/ ~ ~ ~ /
/ ~ ' ~ /
.-.
and the like.
Where used herein, the term "aryl(C1-C6)alkoxy" refers to an oxygen atom
bearing
3 0 a straight or branched, monovalent, saturated aliphatic chain of 1 to 6
carbon atoms
wherein said aliphatic chain, in turn, bears an aryl group.
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-14-
Where used herein the term "(C3-Clo)cycloalkyl" refers to a saturated
hydrocarbon
ring structure composed of one or more fused or unfused rings containing from
three to
ten carbon atoms. Typical (C3-Clo)cycloalkyl groups include cyclopropyl,
cyclobutyl,
cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, adamantanyl, and the like.
"(C3-
C~)cycloalkyl" refers to a saturated hydrocarbon ring structure composed of
one or more
fused or unfused rings containing from three to seven carbon atoms. It is
understood that
the definition of "(C3-C7)cycloalkyl" is included within the definition of
"(C3-
C 1 o)cycloalkyl".
Where used herein, the term "(C1-C4)alkyl-(C3-C7)cycloalkyl" refers to a
straight
or branched, monovalent, saturated aliphatic chain of 1 to 4 carbon atoms
which has a
(C3-C7)cycloalkyl attached to the aliphatic chain. Included within the term
"(C1-C4)alkyl-
(C3-C7)cycloalkyl" are the following:
'"'~
'b
and the like.
Where used herein the term "(C3-C7)cycloalkoxy" refers to an oxygen atom
bearing a saturated hydrocarbon ring structure composed of one or more fused
or unfused
rings containing from three to seven carbon atoms.
Where used herein, the term "(C1-C4) alkoxycarbonyl" refers to a carbonyl
group
having a (C1-Cø)alkyl group attached to the carbonyl carbon through an oxygen
atom.
2 0 Examples of this group include t-butoxycarbonyl, methoxycarbonyl,
ethoxycarbonyl and
the like.
Where used herein, the term "(C1-C4)alkyl-(Cl-C4)alkoxycarbonyl" refers to a
straight or branched, monovalent, saturated aliphatic chain of 1 to 4 carbon
atoms bearing
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-15-
a carbonyl group having a (C1-C4)alkyl group attached to the carbonyl carbon
through an
oxygen atom. Examples include methoxycarbonyl methyl, ethoxycarbonyl methyl, 2-
methoxycarbonyl ethyl, and the like.
Where used herein, the term "(Cl-Cq.)alkylcarbonyl" refers to a carbonyl group
bearing a a straight or branched, monovalent, saturated aliphatic chain of 1
to 4 carbon
atoms attached to the carbonyl carbon.
Where used herein the term "heterocycle" refers to a saturated or unsaturated,
five-
or six-membered ring, which contains one to four heteroatoms selected from the
group
consisting of oxygen, sulfur, and nitrogen. It is understood that the
remaining atoms are
carbon and that the heterocycle may be attached at any point which provides
for a stable
structure. Examples of heterocycle groups include thiopheneyl, furanyl,
tetrahydrofuryl,
imidazolyl, pyrazolyl, pyrrolyl, thiazolyl, thiazolidinyl, isothiazolyl,
oxazolyl, isoxazolyl,
triazolyl, thiadiazolyl, oxadiazolyl, tetrazolyl, pyridyl, pyridinyl,
pyrimidyl, pyrazinyl,
pyridazinyl, triazinyl, imidazolyl, dihydropyrimidyl, tetrahydropyrimdyl,
pyrrolidinyl,
piperidinyl, piperazinyl, pyrazolidinyl, pyrimidinyl, imidazolidimyl,
morpholinyl, pyranyl,
thiomorpholinyl, dioxo-thiomorpholinyl, and the like. Where used herein, the
term
"benzofused heterocyclic ring" refers to a saturated or unsaturated, five- or
six-membered
ring, which contains one to four heteroatoms selected from the group
consisting of
oxygen, sulfur, and nitrogen, and which is fused to a phenyl group. It is
understood that
2 0 the remaining atoms are carbon and that the benzofused heterocycle may be
attached at
any point on either the heterocyclic or phenyl ring which provides for a
stable structure.
Representative "benzofused heterocyclic rings" include benzoxazole,
benzoimidazole,
benzofuran, benzothiophene, benzo[1,3]-dioxolyl, benzothiazole, 2,2-dioxy-2,3-
dihydro-
1H-2~,6-benzo[c]thiophene, azaindole, and indole.
2 5 Where used herein the term "NH(C3-C7)cycloalkyl" refers to an amino group
substituted with a saturated hydrocarbon ring structure composed of one or
more fused or
unfused rings containing from three to seven carbon atoms.
Where used herein the term "N,N-(C1-C4)dialkylamine" refers to a nitrogen atom
substituted with two independently selected straight or branched, monovalent,
saturated
3 0 aliphatic chains of 1 to 4 carbon atoms. Included within the term "N,N-(C1-
C6)dialkylamine" are N(CH3)2, -N(CHZCH3)2, -N(CH2CHaCH3)2, -N(CH2CHZCH2CH3)a,
and the like. "NH-(C1-C4) alkylamine" refers to a nitrogen atom substituted
with a
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-16-
straight or branched, monovalent, saturated aliphatic chains of 1 to 4 carbon
atoms.
Included within the term "NH-(C1-C4) alkylamine" are NH(CH3), -NH(CH2CH3), -
NH(CHzCH2CH3), -NH(CHzCH2CH2CH3), and the like.
The designation " -~ " refers to a bond that protrudes forward out of the
plane
of the page.
The designation " ~ ~ ~ ~ ~ ~ ~ ~ I I " refers to a bond that protrudes
backward out of the
plane of the page.
Where used herein, the term "mitogen activated protein kinase kinase kinase
modulator" or "MAPI~KK modulator" refers to a ligand which binds to any one of
the
mitogen activated protein kinase kinase kinases and either agonizes,
antagonizes, partially
agonizes, or partially antagonizes the kinase's activity. Where used herein,
the term
"mitogen activated protein kinase kinase kinase antagonist" or "MAPI~I~K
antagonist"
refers to a ligand which binds to any one of the mitogen activated protein
kinase kinase
kinases and either antagonizes, or partially antagonizes the kinase's
activity. It is
understood that the term "mixed lineage kinase-7 antagonist" or "MLI~-7
antagonist"
refers to an antagonist of the specific MAPKKK designated as "mixed lineage
kinase-7"
or "MLI~-7" and is included within the meanings of "mitogen activated protein
kinase
kinase kinase modulator" or "mitogen activated protein kinase kinase kinase
antagonist".
Where used herein the term "mixed lineage kinase-7" or "MLI~-7" refers to the
2 0 mitogen activated protein kinase kinase kinase, subtype 7, as described by
Bloem et al., J.
Mol. Cell Cardiol.; 33: 1739-1750, (2001), of the larger class of mitogen
activated protein
kinase kinase kinases, which functions as a serine/threonine kinase to
activate cellular
signaling pathways.
Where used herein the term "congestive heart failure" (CHF) or "congestive
heart
2 5 disease" refers to a disease state of the cardiovascular system whereby
the hear is unable
to efficiently pump an adequate volume of blood to meet the requirements of
the body's
tissues and organ systems. Typically, CHF is charachterized by left
ventricular failure
(systolic dysfunction) and fluid accumulation in the lungs, with the
underlying cause
being attributed to one or more heart or cardiovascular disease states
including coronary
3 0 artery disease, myocardial infarction, hypertension, diabetes, myocardial
injury,
hemodynamic overload, valvular heart disease, and cardiomyopathy. The term
"diastolic
congestive heart failure" refers to a state of CHF characterized by impairment
in the
ability of the heart to properly relax and fill with blood. Conversely, the
term "systolic
congestive heart failure" refers to a state of CHF characterized by impairment
in the
3 5 ability of the heart to properly contract and eject blood. It is
understood that the terms
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-17-
"diastolic congestive heart failure" and "systolic congestive heart failure"
are included
within the term "congestive heart failure" or "CHF".
As appreciated by one of skill in the art, physiological disorders may present
as a
"chronic" condition, or an "acute" episode. The term "chronic", where used
herein, means
a condition of slow progress and long continuance. As such, a chronic
condition is treated
when it is diagnosed and treatment continued throughout the course of the
disease.
Conversely, the term "acute" means an exacerbated event or attack, of short
course,
followed by a period of remission. Thus, the treatment of physiological
disorders
contemplates both acute events and chronic conditions. In an acute event,
compound is
administered at the onset of symptoms and discontinued when the symptoms
disappear.
As described above, a chronic condition is treated throughout the course of
the disease.
Where used herein the teen "patient" refers to a mammal, such a mouse, gerbil,
guinea pig, rat, dog or human. It is understood, however, that the preferred
patient is a
human. Where used herein, the terms "treating", "treatment", or "to treat"
each mean to
alleviate symptoms, eliminate the causation of resultant symptoms either on a
temporary
' or permanent basis, and to prevent, slow the appearance, or reverse the
progression or
severity of resultant symptoms of the named disorder. As such, the methods of
this
invention encompass both therapeutic and prophylactic administration.
Where used herein the term "effective amount" refers to the amount or dose of
the
2 0 compound, upon single or multiple dose administration to the patient,
which provides the
desired effect in the patient under diagnosis or treatment. An effective
amount can be
readily determined by the attending diagnostician, as one skilled in the art,
by the use of
known techniques and by observing results obtained under analogous
circumstances. In
determining the effective amount or dose of compound administered, a number of
factors
2 5 are considered by the attending diagnostician, including, but not limited
to: the species of
mammal; its size, age, and general health; the degree of involvement or the
severity of the
disease involved; the response of the individual patient; the particular
compound
administered; the mode of administration; the bioavailability characteristics
of the
preparation administered; the dose regimen selected; the use of concomitant
medication;
3 0 and other relevant circumstances.
A typical daily dose will contain from about 0.01 mglkg to about 100 mg/kg of
each compound used in the present method of treatment. Preferably, daily doses
will be
about 0.05 mg/kg to about 50 mg/kg, more preferably from about 0.1 mg/kg to
about 25
mg/kg.
3 5 Oral administration is a preferred route of administering the compounds
employed
in the present invention whether administered alone, or as a combination of
compounds
capable of acting as a MAPI~I~K modulator. Oral achninistration, however, is
not the
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-18-
only route, nor even the only preferred route. Other preferred routes of
administration
include transdermal, percutaneous, pulmonary, intravenous, intramuscular,
intranasal,
buccal, sublingual, or intrarectal routes. Where the MAPI~I~K modulator is
administered
as a combination of compounds, one of the compounds may be administered by one
route,
such as oral, and the other may be administered by the transdermal,
percutaneous,
pulmonary, intravenous, intramuscular, intranasal, buccal, sublingual, or
intrarectal route,
as particular circumstances require. The route of administration may be varied
in any
way, limited by the physical properties of the compounds and the convenience
of the
patient and the caregiver.
The compounds employed in the present invention may be administered as
pharmceutical compositions and, therefore, pharmaceutical compositions
incorporating
compounds of Formula I are important embodiments of the present invention.
Such
compositions may take any physical form that is pharmaceutically acceptable,
but orally
administered pharmaceutical compositions are particularly preferred. Such
pharmaceutical compositions contain, as an active ingredient, an effective
amount of a
compound of Formula I, including the pharmaceutically acceptable salts and
hydrates
thereof, which effective amount is related to the daily dose of the compound
to be
administered. Each dosage unit may contain the daily dose of a given compound,
or may
contain a fraction of the daily dose, such as one-half or one-third of the
dose. The amount
2 0 of each compound to be contained in each.dosage unit depends on the
identity of the
particular compound chosen for the therapy, and other factors such as the
indication for
which it is given. The pharmaceutical compositions of the present invention
may be
formulated so as to provide quick, sustained, or delayed release of the active
ingredient
after administration to the patient by employing well known procedures.
2 5 The following discussion provides typical procedures for preparing
pharmaceutical compositions incorporating the compounds of the present
invention.
However, the following is in no way intended to limit the scope of the
pharmaceutical
compositons provided by the present invention.
Compositions are preferably formulated in a unit dosage form, each dosage
3 0 containing from about 1 to about 500 mg of each compound individually or
in a single
unit dosage form, more preferably about 5 to about 300 mg (for example 25 mg).
The
term "unit dosage form" refers to a physically discrete unit suitable as
unitary dosages for
a patient, each unit containing a predetermined quantity of active material
calculated to
produce the desired therapeutic effect, in association with a suitable
pharmaceutical
3 5 carrier, diluent, or excipient.
The inert ingredients and manner of formulation'of the pharmaceutical
compositions are conventional. The usual methods of formulation used in
pharmaceutical
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-19-
science may be used here. All of the usual types of compositions may be used,
including
tablets, chewable tablets, capsules, solutions, parenteral solutions,
intranasal sprays or
powders, troches, suppositories, transdermal patches and suspensions. In
general,
compositions contain from about 0.5% to about 50% of the compounds in total,
depending on the desired doses and the type of composition to be used. The
amount of
the compound, however, is best defined as the "effective amount", that is, the
amount of
each compound which provides the desired dose to the patient in need of such
treatment.
The activity of the compounds employed in the present invention do not depend
on the
nature of the composition, hence, the compositions are chosen and formulated
solely for
convenience and economy.
Capsules are prepared by mixing the compound with a suitable diluent and
filling
the proper amount of the mixture in capsules. The usual diluents include inert
powdered
substances such as starches, powdered cellulose especially crystalline and
microcrystalline
cellulose, sugars such as fructose, mannitol and sucrose, grain flours, and
similar edible
powders.
Tablets are prepared by direct compression, by wet granulation, or by dry
granulation. Their formulations usually incorporate diluents, binders,
lubricants and
disintegrators as well as the compound. Typical diluents include, for example,
various
types of starch, lactose, mannitol, kaolin, calcium phosphate or sulfate,
inorganic salts
2 0 such as sodium chloride and powdered sugar. Powdered cellulose derivatives
are also
useful. Typical tablet binders are substances such as starch, gelatin and
sugars such as
lactose, fructose, glucose and the like. Natural and synthetic gums are also
convenient,
including acacia, alginates, methylcellulose, polyvinylpyrrolidine and the
like.
Polyethylene glycol, ethylcellulose and waxes can also serve as binders.
2 5 Tablets are often coated with sugar as a flavor and sealant. The compounds
may
also be formulated as chewable tablets, by using large amounts of pleasant-
tasting
substances such as mannitol in the formulation, as is now well-established
practice.
Instantly dissolving tablet-like formulations are also now frequently used to
assure that
the patient consumes the dosage form, and to avoid the difficulty in
swallowing solid
3 0 objects that bothers some patients.
A lubricant is often necessary in a tablet formulation to prevent the tablet
and
punches from sticking in the die. The lubricant is chosen from such slippery
solids as
talc, magnesium and calcium stearate, stearic acid and hydrogenated vegetable
oils.
Tablet disintegrators are substances which swell when wetted to break up the
3 5 tablet and release the compound. They include starches, clays, celluloses,
algins and
gums. More particularly, corn and potato starches, methylcellulose, agar,
bentonite, wood
cellulose, powdered natural sponge, cation-exchange resins, alginic acid, guar
gum, citrus
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-20-
pulp and carboxymethylcellulose, for example, may be used, as well as sodium
lauryl
sulfate.
Enteric formulations are often used to protect an active ingredient from the
strongly acid contents of the stomach. Such formulations are created by
coating a solid
dosage form with a film of a polymer which is insoluble in acid environments,
and
soluble in basic environments. Exemplary films are cellulose acetate
phthalate, polyvinyl
acetate phthalate, hydroxypropyl methylcellulose phthalate and hydroxypropyl
methylcellulose acetate succinate.
When it is desired to administer the compound as a suppository, the usual
bases
may be used. Cocoa butter is a traditional suppository base, which may be
modified by
addition of waxes to raise its melting point slightly. Water-miscible
suppository bases
comprising, particularly, polyethylene glycols of various~molecular weights
are in wide
use, also.
Transdermal patches have become popular recently. Typically they comprise a
resinous composition in which the drugs will dissolve, or partially dissolve,
which is held
in contact with the skin by a film which protects the composition. Many
patents have
appeared in the field recently. Qther, more complicated patch compositions are
also in
use, particularly those having a membrane pierced with innumerable pores
through which
the drugs are pumped by osmotic action.
2 0 It is understood by one of ordinary skill in the art that the formulation
procedures
as described above can be readily applied to a method of treating
physiological disorders
susceptible to MAPKKK modulation or MLK-7 modulation , and particularly
congestive
heart failure.
30
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-21-
Particular Aspects of the Compounds of the Invention
As discussed previously, compounds of Formula I are believed to be novel and,
thus, to represent an important embodiment of the present invention. The
following list
sets out several groupings of particular substituents and particular variables
of the
compounds of Formula I. It will be understood that compounds of Formula I, or
methods employing compounds of Formula I, having such particular substituents
and
variables represent particular aspects of the present invention. It will be
further
understood that each of these groupings may be combined with other provided
groupings,
to create still additional particular aspects of the present invention.
to Thus, a particular aspect of the compounds of Formula I is one wherein:
(A) Rl represents hydrogen or methyl; or
(B) Rl represents hydrogen.
(C) R2 represents:
(a) phenyl;
(b) phenyl optionally substituted one to three times with a substituent
independently selected from the group consisting of
(i) halo,
(ii) amino,
(111) mtr0,
(iv) hydroxy,
(v) cyano,
(vi) (C1-C4)alkyl,
(vii) (C1-Cq.)alkoxy,
(viii) amino(C1-Cq.)alkyl
(ix) hydroxy(C1-Cq.)alkoxy,
(x) halo(C1-Cq.)alkoxy,
(xi) (C1-Cq.)alkoxy(Cl-Cq.)alkoxy,
(xii) trifluoromethyl,
(xiii) (C3-C~)cylcoalkyl,
.
3o COR3,
(xiv)
(xv) (C1-Cq.)alkyl-COR4,
(xvi) (C1-Cq.)alkoxy-CORE,
(xvii) NHSO~R6,
(xviii) SO~R~,
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-22-
(xix) NHCORg,
(xx) SOR9,
(xxi) SR10,
(xxii) CONHRl l, and
(xxiii) O-(CH2)q-NR12R13, wherein q represents 1-4;
(c) thiopheneyl, furanyl, imidazolyl, pyrazolyl, pyrrolyl, thiazolyl,
triazolyl, pyridinyl, pyrimidyl, pyrazinyl, pyridiazinyl, piperidinyl,
piperazinyl, pyrimidinyl, or dioxo-thiomorpholinyl,;
(d) thiopheneyl, furanyl, imidazolyl, pyrazolyl, pyrrolyl, thiazolyl,
to triazolyl, pyridinyl, pyrimidyl, pyrazinyl, pyridiazinyl, piperidinyl,
piperazinyl, pyrimidinyl, or dioxo-thiomorpholinyl optionally
substituted one to three times with a substituent independently
selected from the group consisting of
(i) halo,
1 s (ii) amino,
(iii) (Cl-C4)alkyl,
(iv) (Cl-Cq.)alkoxy,
(v) COR3, or
(vi) S02R~,
2o (e) benzimidazole, benzofuran, benzothiophene, benzo[1,3]-dioxolyl,
benzothiazole, 2,2-dioxy-2,3-dihydro-1H-276-benzo[c]thiophene,
or indole;
benzimidazole, benzofuran, benzothiophene, benzo[1,3]-dioxolyl,
benzothiazole, 2,2-dioxy-2,3-dihydro-1H-2~,6-benzo[c]thiophene,
25 and indole optionally substituted one or two times with a
substituent independently selected from the group consisting of
(i) halo,
(ii) amino,
(iii) (Cl-Cq.)alkyl,
or
3o (iv) (Cl-Cq.)alkoxy,
or (g) cyclohexyl;
(D) R2 represents:
(a) phenyl;
(b) phenyl optionally substituted one to three times with a substituent
35 independently selected from the group consisting of:
(i) halo,
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-23-
(ii) amino,
(iii) nitro,
(iv) hydroxy,
(v) cyano,
(vi) (Cl-Cq.)alkyl,
(vii) (C1-C4)alkoxy,
(viii) amino(C1-Cq.)alkyl
(ix) hydroxy(C1-Cq,)alkoxy,
(x) halo(C1-Cq.)alkoxy,
to (xi) (C1-Cq.)alkoxy(C1-Cq.)alkoxy,
(xii) trifluoromethyl,
(xiii) (C3-C~)cylcoalkyl,
(xiv) COR3,
(xv) (C1-Cq.)alkyl-COR4, ,
(xvi) (C1-Cq.)alkoxy-CORS,
(xvii) NHS02R6,
(xviii) SO~R~,
(xix) NHCOR$,
(xx) SOR9,
(xxi) SR10,
(xxii) CONHRl l, and
(xxiii) O-(CH~)q-NR1~R13, wherein q represents
1-4;
(c) thiopheneyl, furanyl,
imidazolyl, pyrazolyl,
pyrrolyl, thiazolyl,
triazolyl, pyridinyl,
pyrimidyl, pyrazinyl,
pyridiazinyl, piperidinyl,
piperazinyl, pyrimidinyl,
or dioxo-thiomorpholinyl,;
(d) thiopheneyl, furanyl,
imidazolyl, pyrazolyl,
pyrrolyl, thiazolyl,
triazolyl, pyridinyl,
pyrimidyl, pyrazinyl,
pyridiazinyl, piperidinyl,
piperazinyl, pyrimidinyl,
or dioxo-thiomorpholinyl
optionally
substituted one to three
times with a substituent
independently
3o selected from the group consisting of
(i) fluoro, bromo, or chloro,
(ii) amino,
(iii) (C1-Cq.)alkyl,
(iv) (C1-Cq.)allcoxy,
(v) COR3, or
(vi) S O~R~,
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-24-
(e) benzimidazole, benzofuran, benzothiophene, benzo[1,3]-dioxolyl,
benzothiazole, 2,2-dioxy-2,3-dihydro-1H-276-benzo[c]thiophene,
or indole;
benzimidazole, benzofuran, benzothiophene, benzo[1,3]-dioxolyl,
benzothiazole, 2,2-dioxy-2,3-dihydro-1H-2~,6-benzo[c]thiophene,
and indole optionally substituted one or two times with a
substituent independently selected from the group consisting of
(i) amino, or
(ii) (Cl-C4)alkyl
to or (g) cyclohexyl;
(E) R2 represents:
(a) phenyl;
(b) phenyl optionally
substituted one to
three times with a
substituent
independently selected
from the group consisting
of
(i) fluoro, bromo, or chloro,
(ii) amino,
(iii) vitro,
(iv) hydroxy,
(v) cyano,
(vi) methyl, ethyl, propyl, butyl, i-butyl,
(vii) methoxy or ethoxy,
(viii) aminomethyl or aminoethyl,
(ix) hydroxy methoxy or hydroxy ethoxy,
(x) 2-fluoro ethoxy or 2-trifluoro ethoxy,
~5 (xi) methoxy ethoxy,
(xii) trifluoromethyl,
(xiii) cyclohexyl,
(xiv) COR3,
(xv) (Cl-C4)alkyl-COR4,
(xvi) (Cl-C4)alkoxy-CORS,
(xvii) NHS02R6,
(xviii) S02R~,
(xix) NHCOR~,
(xx) S OR9,
(xxi) SR10,
(xxii) CONHRl l, and
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-25-
(xxiii) O-(CH2)q-NR12R13~ ~,~,herein q represents 1-4;
(c) thiopheneyl, furanyl, imidazolyl, pyrazolyl, pyrrolyl, thiazolyl,
triazolyl, pyridinyl, pyrimidyl, pyrazinyl, pyridiazinyl, piperidinyl,
piperazinyl, pyrimidinyl, or dioxo-thiomorpholinyl,;
(d) thiopheneyl, fuxanyl, imidazolyl, pyrazolyl, pyrrolyl, thiazolyl,
triazolyl, pyridinyl, pyrimidyl, pyrazinyl, pyridiazinyl, piperidinyl,
piperazinyl, pyrimidinyl, or dioxo-thiomorpholinyl optionally
substituted one to three times with a substituent independently
selected from the group consisting of:
(i) fluoro, bromo,
or chloro,
(ii) amino,
(iii) methyl,
(iv) methoxy,
(v) COR3, or
(vi) S 02R~;
(e) benzimidazole, benzofuran, benzothiophene, benzo[1,3]-dioxolyl,
benzothiazole, 2,2-dioxy-2,3-dihydro-1H-2~,6-benzo[c]thiophene,
or indole;
(f) benzimidazole, benzofuran, benzothiophene, benzo[1,3]-dioxolyl,
benzothiazole, 2,2-dioxy-2,3-dihydro-1H-2~,6-benzo[c]thiophene,
and indole optionally substituted one or two times with a
substituent independently selected from the group consisting of
(i) amino, or
(ii) methyl;
or (g) cyclohexyl;
(F) R2 represents:
(a) phenyl;
(b) phenyl optionally substituted one to three times with a substituent
3o independently selected from the group consisting of:
(i) fluoro, bromo, or chloro,
(ii) amino,
(iii) nitro,
(iv) hydroxy,
(v) cyano,
(vi) methyl, ethyl, propyl,
butyl, i-butyl,
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-26-
(vii) methoxy or ethoxy,
(viii) aminomethyl or axninoethyl,
(ix) hydroxy methoxy or hydroxy ethoxy,
(x) 2-fluoro ethoxy or 2-trifluoro ethoxy,
(xi) methoxy ethoxy,
(xii) trifluoromethyl,
(xiii) cyclohexyl,
(xiv) COR3, wherein R3 represents amino, hydroxy, (C1-
C4)alkyl, (C1-C4)alkoxy, N,N-(C1-C4)dialkylamine, or a
to heterocycle selected from the group consisting of:
~.N -~-N S -~-.N O
U
(i) , (ii) , (iii)
/~ a O O
~SvO '~N~N~
H
(iv) , or (v)
(xv) (C1-C4)alkyl-COR4, wherein R4 represents hydroxy,
amino, or (C1-C4)alkoxy,
(xvi) (C1-C4)alkoxy-CORS, wherein R5 represents hydroxy or
amino,
(xvii) NHSO~R6, wherein R6 represents (C1-C4)alkyl,
(xviii) S02R~, wherein R7 represents amino or
(C1-C4)alkyl,
(xix) NHCORB, wherein R8 represents methyl,
(xx) SOR9, wherein R9 represents methyl,
(xxi) SR10, wherein R10 represents methyl
or ethyl,
(xxii) CONHRl 1, wherein Rl l represents -(CH~)n-X-Y,
where
n=0-2, X represents -S-, -CHZ-, -(CHa)2-, -NH-, -CO-, or -
SOa-, and Y represents amino, (C1-C4)alkyl, (C1-
C4)alkoxycarbonyl, or NH-(C1-C4)alkylamine;
~5 or wherein Rl l represents CH(COR14)-(CH2)m-X'-Y"
where R14 represents hydroxy or (C1-C4)alkoxy, m=0-4,
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
_27_
X' represents -S-, -CHz-, -NH-, or -CO-, and Y' represents
represents amino, hydroxy, (C1-Cq.)alkyl, or (C1-
Cq.)alkoxycarbonyl; or wherein Rl 1 represents a group
selected from the following:
(a)
O O
N
H
and (c)
N
~-OC(CH3)3
O
(xxiii) O-(CH2)q-NR12R13a ~,~,herein q represents 1-3, Rl2 and
R13 independently represent hydrogen or methyl or R12
to and R13 together with the nitrogen to which they are
attached form a piperidino, pyrrolidino, morpholino or a
methylpiperazino group;
(c) thiopheneyl, furanyl, imidazolyl, pyrazolyl, pyrrolyl, thiazolyl,
triazolyl, pyridinyl, pyrimidyl, pyrazinyl, pyridiazinyl, piperidinyl,
piperazinyl, pyrimidinyl, or dioxo-thiomorpholinyl,;
(d) thiopheneyl, furanyl, imida,zolyl, pyrazolyl, pyrrolyl, thiazolyl,
triazolyl, pyridinyl, pyrimidyl, pyrazinyl, pyridiazinyl, piperidinyl,
piperazinyl, pyrimidinyl, or dioxo-thiomorpholinyl optionally
substituted one to three times with a substituent independently
2o selected from the group consisting of:
(i) fluoro, bromo, or chloro,
(ii) amino, .
(iii) methyl,
(iv) methoxy,
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-28-
(v) COR3, wherein R3 represents hydroxy, (Cl-Cq.)alkoxy or
pyridine,
(vi) S02R~, wherein R7 represents amino
(e) benzimidazole, benzofuran, benzothiophene, benzo[1,3]-dioxolyl,
benzothiazole, 2,2-dioxy-2,3-dihydro-1H-2~,6-benzo[c]thiophene,
or indole;
(f) benzimidazole, benzofuran, benzothiophene, benzo[1,3]-dioxolyl,
benzothiazole, 2,2-dioxy-2,3-dihydro-1H-2~,6-benzo[c]thiophene,
and indole optionally substituted one or two times with a
to substituent independently selected from the group consisting of:
(i) amino, or
(ii) methyl;
or (g) cyclohexyl;
(G) The particular embodiments of any of groupings (C) through (F) wherein
when R2 represents a substituted aryl or heterocycle, said aryl or
heterocycle is substituted 1 or 2 times.
(H) The particular embodiments of any of groupings (C) through (F) wherein
2o when R2 represents a substituted aryl or heterocycle, said aryl or
heterocycle is substituted once.
(1) The particular embodiments of any of groupings (C) through (F) wherein
when R2 represents a substituted benzofused heterocycle, said benzofused
heterocycle is substituted once.
(J) R2 represents phenyl or phenyl optionally substituted one to three times
with a substituent independently selected from the group consisting of
(i) halo,
(ii) amino,
(iii) nitro,
(iv) hydroxy,
(v) cyano,
(vi) (Cl-Cq.)alkyl,
(vii) (Cl-Cq.)alkoxy,
(viii) amino(Cl-Cq,)alleyl
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-29-
(ix) hydroxy(Cl-Cq.)alkoxy,
(x) halo(Cl-Cq,)alkoxy,
(xi) (Cl-Cq.)alkoxy(Cl-Cq.)alkoxy,
(xii) trifluoromethyl,
(xiii) (C3-C~)cylcoalkyl,
(xiv) COR3,
(xv) (Cl-Cq.)alkyl-COR4,
(xvi) (Cl-Cq.)alkoxy-CORS,
(xvii) NHSO~R6,
(xviii) SO~R~,
(xix) NHCORg,
(xx) SOR9,
(xxi) SR10,
(xxii) CONHRl l, and
(xxiii) O-(CH~)q-NR12R13, wherein q represents
1-4,
(K) R2 represents phenyl or phenyl optionally substituted one to three times
with a substituent independently
selected from the group
consisting of
(i) fluoro, bromo, or chloro,
(ii) amino,
(iii) vitro,
(iv) hydroxy,
(v) cyano,
(vi) methyl, ethyl, propyl, butyl, i-butyl,
(vii) methoxy or ethoxy,
(viii) aminomethyl or aminoethyl,
(ix) hydroxy methoxy or hydroxy ethoxy,
(x) 2-fluoro ethoxy or 2-trifluoro ethoxy,
(xi) methoxy ethoxy,
(xii) trifluoromethyl,
(xiii) cyclohexyl,
(xiv) COR3,
(xv) (Cl-Cq.)alkyl-COR4,
(xvi) (Cl-Cq.)alkoxy-CORS,
(xvii) NHSO~R6,
(xviii) SO~R~,
(xix) NHCORg,
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-30-
(xx) S OR9,
(xxi) SR10,
(xxii) CONHR11, and
(xxiii) O-(CH2)q-NR12R13~ ~,~,herein q represents
1-4,
(L) R2 represents phenyl or phenyl optionally substituted
one to three times
with a substituent independently
selected from the group
consisting of
(i) fluoro, bromo, or chloro,
(iii) amino,
(iv) nitro,
(v) hydroxy,
(vi) cyano,
(vii) methyl, ethyl, propyl, butyl, i-butyl,
(viii) methoxy or ethoxy,
(ix) aminomethyl or aminoethyl,
(x) hydroxy methoxy or hydroxy ethoxy,
(xi) 2-fluoro ethoxy or 2-trifluoro ethoxy,
(xii) methoxy ethoxy,
(xiii) trifluoromethyl,
(xiv) cyclohexyl,
(xiv) COR3, wherein R3 represents amino, hydroxy,
(C1-
Cq.)alkyl, (Ci-Cq.)alkoxy, N,N-(C1-Cq.)dialkylamine,
or a
heterocycle selected from the group consisting
of
O
-N -~-N S -~
U
(i) , (ii) , (iii)
~ ~O O
'~N~N~
H
or (v)
(xv) (C1-Cq.)alkyl-COR4, wherein R4 represents hydroxy,
amino, or (C1-Cq.)alkoxy,
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-31-
(xvi) (C1-Cq.)alkoxy-CORS, wherein R5 represents hydroxy or
ammo,
(xvii) NHS02R6, wherein R6 represents (C1-C4)alkyl,
(xviii) S02R~, wherein R7 represents amino or (C1-C4)alkyl,
(xix) NHCORB, wherein R8 represents methyl,
(xx) SOR9, wherein R9 represents methyl,
(xxi) SR10, wherein R10 represents methyl or ethyl,
- (xxii) CONHRI 1, wherein Rl 1 represents -(CHZ)n-X-Y, where
n=0-2, X represents -S-, -CHI-, -(CH2)2-, -NH-, -CO-, or
lo S02-, and Y represents amino, (C1-Cq.)alkyl, (C1
Cq.)allcoxycarbonyl, or NH-(C1-Cq.)all~ylamine;
or wherein Rl l represents CH(CORl4)-(CH2)m-X'-Y"
where Rl4 represents hydroxy or (C1-Cq,)alkoxy, m=0-4,
X' represents -S-, -CHI-, -NH-, or -CO-, and Y' represents
represents amino, hydroxy, (C1-Cq.)alkyl, or (C1-
C4)alkoxycarbonyl; or wherein Rl 1 represents a group
selected from the following:
(a)
O O
N
H
and (c)
N
OC(CH3)s
O
(xxiii) O-(CH2)q-NR12R13, wherein q represents 1-3, R12 and
R13 independently represent hydrogen or methyl or R12
and Rl3 together with the nitrogen to which they are
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-32-
attached form a piperidino, pyrrolidino, morpholino or a
methylpiperazino group;
(M) R2 represents phenyl, 4-(N-acetylamino)phenyl, 2-hydroxyphenyl, 3-
hydroxyphenyl, 4-hydroxyphenyl, 2-aminophenyl, 3-aminophenyl, 4-
aminophenyl, 3-chlorophenyl, 4-chlorophenyl, 3-nitrophenyl, 4-
nitrophenyl, 3-(methylsulfonylamino)phenyl, 4-
(methylsulfonylamino)phenyl, 2-ethoxyphenyl, 3-ethoxyphenyl, 4-
ethoxyphenyl, 4-methoxyphenyl, 4-fluoro-3-methylphenyl, 4-Fluoro-2-
methylphenyl, 4-bromophenyl, 4-ethylsulfanylphenyl, 4-
methylsulfanylphenyl, 4-cyanophenyl, 4-acetylphenyl, 2-
carboxamidophenyl, 4-(2-carboxy-eth-1-yl)phenyl, 3,5-dichlorophenyl,
3,4-dichlorophenyl, 4-methoxy-3-methylphenyl, 3-amino-4-methylphenyl,
2-nitrophenyl, 3-(N-acetylamino)phenyl, 4-methanesulfinylphenyl, 4-
methyl-3-nitrophenyl, 4-carboxyphenyl, 3,5-Bis-trifluromethylphenyl, 3-
carboxyphenyl, 2-carboxyphenyl, 4-isobutylphenyl, 4-carboxymethoxy
phenyl, 4-methyl-3-vitro-phenyl, 4-cyclohexyl-phenyl, 4-fluorophenyl, 4-
methoxyphenyl, 4-(2-hydroxy ethoxy)phenyl, 4-carboxymethylphenyl, 4-
methanesulfonylphenyl, 4-(2-amino eth-1-yl)phenyl, 4-methylphenyl, 2-
2o fluoro-4-aminophenyl, 3,5-bis carboxyphenyl, 3,5-bis methoxycarbonyl
phenyl, 3-chloro-4-aminophenyl, 3-chloro-5-fluoro-4-hydroxyphenyl, 4,5-
difluoro-3-hydroxyphenyl, 4-aminosulfonyl phenyl, 3-aminosulfonyl
phenyl, 2-aminosulfonyl phenyl, 4-(2-methoxycarbonyl) eth-1-yl phenyl,
4-(2-carboxamido) eth-1-yl phenyl, 4-(2-(N,N-
dimethylamino)ethoxy)phenyl, 4-(2-morpholin-4-yl-ethoxy)phenyl, 4-(2-
Piperidin-1-yl-ethoxy)phenyl, 4-(2-Piperidin-1-yl-ethoxy)phenyl, 4-(2-
Piperdin-1-yl-ethoxy)phenyl, 4-(3-(N,N-dimethylamino)propoxy)phenyl,
4-(3-Morpholin-4-yl-propoxy)phenyl, 4-(3-pyrrolidin-1-yl-
propoxy)phenyl, 4-[3-(4-Methylpiperazin-1-yl)propoxy]phenyl, 4-
carboxamido-methoxyphenyl, 4-(2-methoxyethoxy)phenyl, 4-(2-
fluoroethoxy)phenyl, 4-(2-trifluoroethoxy)phenyl, 4-(N-(2-N-(tert-
butoxycarbonyl)amino eth-1-yl)carboxamido)phenyl, 4-(piperidin-1-yl
carbonyl)phenyl, 4-(thiomorpholin-4-yl carbonyl)phenyl, 4-(morpholin-4-
yl carbonyl)phenyl, 4-(N-acetamido carboxamido)phenyl, 4-(N-(2-N,N-
dimethylamino eth-1-yl)carboxamido)phenyl, 4-(N-(2-tent-butylsulfanyl-
eth-1-yl)carboxamido)phenyl, 4-(N-(2-ethoxycarbonyl eth-1-
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-33-
yl)carboxamido)phenyl, 4-(N-(1,1-dioxo-tetrahydro-1~,6-thiophen-3-
yl)carboxamido)phenyl, 4-(N-(1-tert-butoxycarbonyl piperidin-3-
yl)carboxamido)phenyl, 4-(N-(5-N-tert-butoxycarbonylamino-1-tert-
butoxycarbonyl pent-1-yl)carboxamido)phenyl, 4-(N-(2-(morpholin-4-
yl)eth-1-yl)carboxamido)phenyl, 4-(1,1-dioxo-1~,6-thiomorpholin-4-
yl)carbonyl phenyl, 4-(N-(2-methylsulfanyl-1-tert-butoxycarbonyl-eth-1-
yl)carboxamido)phenyl, 4-(N-(1,3-bis-tert-butoxycarbonyl-prop-1-
yl)carboxamido)phenyl, 4-(N,N-dimethylcarboxamido)phenyl, 4-(N-(2-
ter-butoxycarbonyl-eth-1-yl)carboxamido)phenyl, 4-N-
to methylcarboxamido phenyl, 4-(N-(2-methylsulfonyl-eth-1-
yl)carboxamido)phenyl, 4-(N-(N,N-dimethylamino)carboxamido)phenyl,
4-carboxamido phenyl, 4-(N-(2 amino-eth-1-yl)carboxamido)phenyl, 4-
(N-(5-amino-1-carboxy-pent-1-yl) carboxamido)phenyl, 4-(N-piperidin-3-
yl carboxamido)phenyl, 4-(N-(2-methylsufanyl-1-carboxy-eth-1-
15 yl)carboxamido)phenyl, 4-(N-(1,3-bis carboxy-prop-1-
yl)carxoamido)phenyl, or 4-(N-(2-carboxy eth-1-yl) carboxamido) phenyl;
(N) R2 represents thiopheneyl, furanyl, imidazolyl, pyrazolyl, pyrrolyl,
thiazolyl, triazolyl, pyridinyl, pyrimidyl, pyrazinyl, pyridiazinyl,
20 piperidinyl, piperazinyl, pyrimidinyl, dioxo-thiomorpholinyl; or
thiophenyl, furanyl, imidazolyl, pyrazolyl, pyrrolyl, thiazolyl, triazolyl,
pyridinyl, pyrimidyl, pyrazinyl, pyridiazinyl, piperidinyl, piperazinyl,
pyrimidinyl, or dioxo-thiomorpholinyl optionally substituted one to three
times with a substituent independently selected from the group consisting
25 of
(i) fluoro, bromo, or chloro,
(ii) amino,
(iii) (C1-Cq.)alkyl,
(vii) (C1-Cq.)alkoxy,
30 (viii) COR3, and
(ix) SO~R~,
(O) R2 represents thiopheneyl, furanyl, imidazolyl, pyrazolyl, pyrrolyl,
thiazolyl, triazolyl, pyridinyl, pyrimidyl, pyrazinyl, pyridiazinyl,
piperidinyl, piperazinyl, pyrimidinyl, dioxo-thiomorpholinyl; or
35 thiophenyl, furanyl, imidazolyl, pyrazolyl, pyrrolyl, thiazolyl, triazolyl,
pyridinyl, pyrimidyl, pyrazinyl, pyridiazinyl, piperidinyl, piperazinyl,
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-34-
pyrimidinyl, or dioxo-thiomorpholinyl optionally substituted one to three
times with a substituent independently selected from the group consisting
of
(i) fluoro, bromo, or chloro,
(ii) amino,
(vii) methyl,
(viii) methoxy,
(ix) COR3, or
(x) S 02R~;
to (P) R2 represents thiopheneyl, furanyl, imidazolyl, pyrazolyl, pyrrolyl,
thiazolyl, triazolyl, pyridinyl, pyrimidyl, pyrazinyl, pyridiazinyl,
piperidinyl, piperazinyl, pyrimidinyl, dioxo-thiomorpholinyl; or
thiophenyl, fuxanyl, imidazolyl, pyrazolyl, pyrrolyl, thiazolyl, triazolyl,
pyridinyl, pyrimidyl, pyrazinyl, pyridiazinyl, piperidinyl, piperazinyl,
pyrimidinyl, or dioxo-thiomorpholinyl optionally substituted one to three
times with a substituent independently selected from the group consisting
of
(i) fluoro, bromo, or chloro,
(ii) amino,
(iii) methyl,
(iv) methoxy,
(v) COR3, wherein R3 represents hydroxy or (C1-C4)alkoxy,
(vi) S02R~, wherein R7 represents amino,
(Q) R2 represents thiophen-2-yl, thiophen-3-yl, pyridin-4-yl, pyridin-3-yl,
furan-3-yl, furan-2-yl, thiazol-2-yl, pyrazin-2-yl, pyridin-2-yl, 1H-pyrrol-
2-yl, 1H-pyrrol-3-yl, pyrimidin-2-yl, pyrimidin-5-yl, imidazol-1-yl,
[1,2,4]triazol-1-yl, pyrazol-1-yl, [1,2,3]triazol-1-yl, piperidin-1-yl, l,l-
Dioxo-1~,6-thiomorph-olin-4-yl, piperazin-1-yl, 4-methylthiophen-2-yl, 6-
carboxypyridin-2-yl, 5-fluoropyridin-2-yl, 6-metho-xypyridazin-3-yl, 2-
3o aminopyrimidin-5-yl, 5-aminosulfonyl thiophen-2-yl, or 4-tert-
butoxycarbonyl piperazin-1-yl;
(R) R2 represents benzimidazole, benzofuran, benzothiophene, benzo[1,3]-
dioxolyl, benzothiazole, 2,2-dioxy-2,3-dihydro-1H-2~.6-benzo[c]thiophene,
indole; or benzoimidazole, benzofuran, benzothiophene, benzo[1,3]-
dioxolyl, benzothiazole, 2,2-dioxy-2,3-dihydro-1H-2~,6-benzo[c]thiophene,
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-35-
or indole optionally substituted one or two times with a substituent
independently selected from the group consisting of
(i) amino, or
(ii) (C1-Cq.)alkyl
(S) R2 represents benzimidazole, benzofuran, benzothiophene, benzo[1,3]-
dioxolyl, benzothiazole, 2,2-dioxy-2,3-dihydro-1H-2~,6-benzo[c]thiophene,
indole; or benzoimidazole, benzofuran, benzothiophene, benzo[1,3]-
dioxolyl, benzothiazole, 2,2-dioxy-2,3-dihydro-1H-2~,6-benzo[c]thiophene,
or indole optionally substituted one or two times with a substituent
l0 independently selected from the group consisting of:
(i) amino, or
(ii) methyl;
(T) R2 represents 1H-Indol-5-yl, Benzo[1,3]dioxol-5-yl, Benzo[b]thiophen-2-
yl, Benzofuran-2-yl, 4-Benzo[b]thiophen-3-yl, 1H-Indol-2-yl, 2,2-Dioxy-
2,3-dihydro-1H-2~,6-benzo[c]thiophen-5-yl, 1H-benzoimdazol-2-yl, or 2-
amino benzothiazol-6-yl,; or ..
(Ln R2 represents cyclohexyl.
(V) The particular embodiments of any of groupings (J), (K), (L), (I~, (O), or
(P) wherein when R2 represents a substituted aryl or heterocycle, said aryl
or heterocycle is substituted 1 or 2 times.
(V~ The particular embodiments of any of groupings (J), (K), (L), (N), (O), or
(P) wherein when R2 represents a substituted aryl or heterocycle, said aryl
or heterocycle is substituted once.
(X) The particular embodiments of any of groupings (R) or (S) wherein when
R2 represents a substituted benzofused heterocycle, said benzofused
heterocycle is substituted once.
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-36-
Compounds of Formula I can be chemically prepared, for example, by following
the synthetic routes set forth in the Schemes and the Preparations and
Examples below:
However, the Schemes contained in the following discussion are not intended to
be
limiting to the scope of the present invention in any way. For example, the
specific
synthetic steps for the routes described herein may be combined in different
ways, or with
steps from different schemes, to prepare other compounds of Formula I. All
substituents,
unless otherwise indicated, are as previously defined. The reagents and
starting materials
are readily available o one of ordinary skill in the art. For example, certain
reagents or
starting materials can be prepared by one of ordinary skill in the art
following procedures
disclosed in Larock, R. C., Conap~ehensive Organic Ti~ahsformations, 2'Zd Ed.,
copyright
1999, John Wiley & Sons, pp 741-742; Miryaura, N.; Yanagi, T.; Suzuki, A. The
Palladium-Catalyzed Cross Coupling Reaction of Phenylboronic Acid with
Haloarenes in
the Presence of Bases. Syfzth. Conamun., 1981, 513-518; Stanforth, S. P.
Tet~ahedrou,
1998, 54, 263-303; Greene, T. W.; Wuts, P. G. M. Pf~otective GYOUps in
OfgafZic
Synthesis (3rd ed.); Wiley: New York, 1999; Ishiyama, T.; Murata, M.; Miyaura,
N. J.
O~g. Chem. 1995, 60, 7508-7510; Lindley, J. Tetrahed~°on 1984, 40, 1433-
1456; Wolfe,
J. P.; Buchwald, S. L. J. Org. Chen2. 1997, 62, 6066-6068; Furniss, B. S.;
Hannaford, A.
J.; Smith, P. W. G.; Tatchell, A. R. hogel's Textbook of Practical Organic
Chemistry (Stn
ed.); Longman, Essex, 1989; p 1077-1079; Bodanszky, M.; Bodanszky, A. The
Practice
2 0 of Peptide Synthesis; Springer Verlag: New York, 1984; Smith, D. L.;
McCloskey J. A.; J.
Org. Chem. 43, 2087-2088, 1978.
Other necessary reagents and starting material may be made by procedures which
are selected from standard techniques of organic and heterocyclic chemistry,
techniques
which are analogous to the syntheses of known structurally similar compounds,
and the
2 5 procedures described in the Preparations and Examples, including any novel
procedures.
The Preparations and Examples that follow further illustrate the invention and
represent typical synthesis of the compounds of Formula I as described
generally above.
The reagents and starting materials are readily available to one of ordinary
skill in the art.
Where used herein, the following terms have the meanings indicated: "eq" or
"equiv."
3 0 refers to equivalents; "g" refers to grams; "mg" refers to milligrams; "L"
refers to liters;
"mL" refers to milliliters; "~,L" refers to microliters; "mol" refers to
moles; "mmol" refers
to millimoles; "psi" refers to pounds per square inch; "mm Hg" refers to
millimeters of
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-3 7-
mercury; "min" refers to minutes; "h" or "hr" refers to hours; "°C"
refers to degrees
Celsius; "TLC" refers to thin layer chromatography; "HPLC" refers to high
performance
liquid chromatography; "Rf' refers to retention factor; "Rt" refers to
retention time; "8"
refers to part per million down-field from tetramethylsilane; "THF" refers to
tetrahydrofuran; "DMF" refers to N,N-dimethylformamide; "DMSO" refers to
dimethyl
sulfoxide; "aq" refers to aqueous; "EtOAc" refers to ethyl acetate; "iPrOAc"
refers to
isopropyl acetate; "MeOH" refers to methanol; "MTBE" refers to tert-butyl
methyl ether;
"PPh3" refers to triphenylphosphine; "DEAD" refers to diethyl
azodicarboxylate; "RT"
refers to room temperature; "Pd-C" refers to palladium over carbon;
"NaBH(Oac)3"refers
to sodium triacetoxyborohydride; "Bn" refers to benzyl; "BnNH2" refers to
benzyl amine;
"H2" refers to hydrogen; "CH2C12" refers to dichloromethane; "HBTU" refers to
O-
Benzotriazol-1-yl-N,N,N',N'-tetramethyluronium hexafluorophosphate, "MPLC"
refers
to medium-pressure-liquid chromatography; "NMR" refers to nuclear magnetic
resonance
spectroscopy; "MS" refers to mass spectroscopy; "ES+"refers to positive mode
electrospray ionization; and "M" refers to molecular weight.
Analytical Methods
The compounds of the present invention may be purified by methods known to
2 0 one skilled in the art. These methods include crystallization,
precipitation, normal phase
silica gel chromatography and reverse-phase high-performace chromatography
that is
mass-guided and/or UV-guided. The purity of compounds is determined by LC/MS
analysis.
The salt composition is determined by HPLC anaysis. The HPLC system consists
of a Shimadzu SCL-l0A controller, SIL-l0A auto injector, LC10AS pump, and a
SPD-
l0A UV detector (Kyoto, Japan). The Prevail Organic Acid column (25 cm x 4.6
mm
LD.) is obtained from Alltech Associates Inc.(Deerfield, IL). The HPLC
operating
conditions consist of a mobile phase comprising 0.2M KH2PO4 (no pH
adjustment). The
mobile phase flow rate is set at 1.5 mL/minute with sample injections of 10
~,L. The UV
3 0 detector is set at 195 nm and run time is 3 minutes. TFA is quantitated by
linear
regression of peak area response versus standards prepared at known TFA
concentrations.
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-~g_
Enabling Chemistries: (Preparation of Intermediates)
Scheme 1. Synthesis of 5,6-dihydro-4H pyrrolo[1,2- b]pyrazole derivatives
Scheme 1 depicts the synthesis of the dihydro-4H pyrrolo[1,2- b]pyrazole core
structure, substituted by heteroaromatics, preferably pyridine (with or
without
substitution) and quinoline. The quinoline may be substituted as well with a
variety of
aromatic or heterocyclic rings, specifically at the 7-position of the
quinoline. In step A, a
quinoline of Formula 2 can be prepared by reaction of an aniline with a vinyl
ketone in the
presence of an appropriate acid catalyst such as conc. HZS~4 in a suitable
solvent such as
dioxane. The resulting quinoline (step B) is sequentially treated with a
strong base such
as potassium hexamethyldisilazide, lithium hexamethyl-disilazide, or lithium
diisopropyl
amide at -7S °C in a suitable solvent such as tetrahydrofuran, and
treated with a picolonic
acid ester of Formula 3, such as methyl picolinate, which may be additionally
substituted
on the pyridine ring, to yield a methyl ketone of Formula 4. Condensation of
the methyl
ketone (step C) with 1-aminopyrrolidinone hydrochloride (Formula 5) provides
an imine
of Formula 6 under basic conditions at room temperature (pyridine/ethanol or
sodium
ethoxide in ethanol). The imine undergoes cyclization by treatment with base
such as
sodium hydride or cesium carbonate under standard conditions (step D) to yield
the 5,6-
2 0 dihydro-4H pyrrolo[1,2- b]pyrazole (Formula 7) with appropriate pyridyl
and quinolinyl
substitution.
30
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-39-
Scheme 1
O step B O N' R1
NHZ ~ CHs 1) KHMDS
R2 \ ~ R2 ~ ~ \ w
/ H2S~4 / i ~ R2
step A N ~ o~ ~ NJ
( ) (2) ~ ~ N
R1 (3) (4)
H-CI
N _0 R1 N_N R1
N
NHZ (5) NaH w ~
step C step D R2
i
N
Preparation 1
1-Aminopyrrolidin-2-one Hydrochloride
N
~NH2
H-CI
4-Chlorobutyryl chloride (57 mL, 510 mmol) is added to a solution of
benzophenone hydrazone (100 g, 510 mmol) and pyridine (41 mL, 510 mmol) in
anhydrous CH2C12 (520 mL) under nitrogen at a rate that maintained a gentle
reflux
throughout the addition. The mixture is stirred for 0.5 h and poured into
water (1 L). The
layers are separated and the organic layer is washed with brine, dried
(Na2SO4), filtered,
and concentrated ifz vacuo to yield 4-chloro-butyric acid benzhydrylidene-
hydrazide as a
residue.
MS ES+ m/e 301.1 (M+1).
This residue is dissolved in THF (1.5 L),and the solution cooled in an ice-
water
bath, treated with portions of NaH (60% suspended in mineral oil, 20 g, 498
mmol) and
stirred for 1 h. To the mixture is added saturated aqueous NH4Cl solution (1
L) and
EtOAc (1 L). The layers are separated and the organic solution washed with
brine, dried
(Na2S04), altered and concentrated in vacuo to yield 1-
2 0 (benzhydrylideneamino)pyrrolidin-2-one as a residue.
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-40-
1H NMR(CDCl3): 8 7.58-7.62 (m, 2H), 7.39-7.46 (m, 4H), 7.29-7.36 (m, 4H) 3.31
(t, J =
7 Hz, 2H), 2.32 (t, J = 7 Hz, 2H), 1.91 (quintet, J = 7 Hz, 2H); MS ES+ m/e
265.1 (M+1).
This residue is suspended in water (3 L), treated with concentrated HCl
solution (80 mL), and heated to reflux for 1.5 h. The reaction is cooled to RT
and
extracted twice with CHZCl2. The aqueous portion is concentrated in vacuo
followed by
azeotropic removal of water with three portions of absolute ethanol and three
portions of
toluene to yield the title compound, 56 g (81%), as a white solid.
1H NMR (DMSO-d6): ~ 3.58 (t, J = 7 Hz, 2H), 2.33 (t, J = 7Hz, 2H), 2.04
(quintet, J = 7
Hz, 2H), TOF MS ES+ exact mass calculated for CøH8N2 (M+): m/z = 100.0637.
Found:
100.0641.
Preparation 2
6-Methylpyridine-2-carboxylic Acid Methyl Ester
O~
~3
To a suspension of 6-methyl-pyridine-2-carboxylic Acid ( 10 g, 72.9 mmol) in
CH2C12 (200 mL) cooled to 0 °C is added MeOH (10 mL), 4-
dimethylaminopyridine (11.6
g, 94.8 mmol), and EDC (18.2 g, 94.8 mmol). The mixture is stirred at RT for 6
h,
washed with water and brine, and dried over Na2SO4. The mixture is filtered
and
concentrated in vacuo and the residue is chromatographed on silica gel (50%
EtOAc/
2 0 Hexanes) to yield the title compound, 9:66 g (92%), as a colorless liquid.
1H NMR (CDC13) 8 7.93-7.88 (m, 1H), 7.75-7.7 (m, 1H), 7.35-7.3 (m, 1H), 4.00
(s, 3H),
2.60 (s, 3H).
Preparation 3
2 5 7-Bromo-4-methylquinoline
Br ~ N
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-41-
H2S04 (14.4 mL, 270 mmol) is added to a solution of 3-bromoaniline (Aldrich,
30.0 g, 174 mmol) in 1,4-dioxane (1 L) at RT. The mixture is heated to reflux
and treated
with methyl vinyl ketone (Aldrich, 19.5 mL, 270 mmol) in 1,4-dioxane (50 mL)
dropwise
over 3 h. Heating is continued for 1 h after the addition, followed by removal
of the
solvent in vacuo. The residue is dissolved in water (100 mL), neutralized with
Na2C03
and extracted with CH2C12. The combined organic extracts are washed with water
and
brine, dried (Na2S04~ and filtered. The filtrate is concentrated, and the
residue
chromatographed on silica gel (Biotage, eluting with 20% EtOAc/Hexanes) to
give the
title compound, 15.0 g (43%), as a brownish solid.
1H NMR (CDC13) 8 8.80-8.75 (m, 1H), 8.30 (s, 1H), 7.90-7.85 (m, 1H), 7.70-7.65
(m,
1H), 7.25-7.20 (m, 1H), 2.65 (s, 3H).
Preparation 4
2-(7-Bromoquinolin-4-yl)-1-(6-methylpyridin-2-yl)ethanone
7-Bromo-4-methyl-quinoline (2.75 g, 12.4 mmol) is dissolved in THF (33 mL),
cooled to -78 °C, treated with KHMDS solution (Aldrich, 0.5 M in
toluene, 29.8 mL,
14.9 mmol) and stirred for 40 min. The mixture is treated with the product of
Preparation 2 (2.44 g, 16.1 mmol) and stirred for 2 h, and then warmed to RT
for 2 h.
2 0 After adding saturated NH4C1 solution (5 mL), the mixture is concentrated
in vacuo. The
residue is chromatographed on silica gel (eluting with 50% EtOAc/Hexanes) to
give the
title compound, 2.3 g(66%), as a yellow solid.
IH NMR (CDC13) 8 8.85 (m, 1H), 8.30 (s, 1H), 8.00-7.60 (m, 4H), 7.45-7.35 (m,
2H),
5.05 (s, 2H), 2.65 (s, 3H).
The following compound is prepared by the previous method (4):
P)ZEP##Product Name Physical Data
5 2-(7-Bromoquinolin-4-yl)-1-pyridin-1H NMR (CDC13) b 8.85 (m,
1H),
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-42-
2-yl-ethanone I 8.30 (s, 1H), 8.00-7.60 (m, 4H), 7.45-
7.35 (m, 3H), 5.05 (s, 2H)
Preparation 6
1-[2-(7-Bromoquinolin-4-yl)-1-(6-methylpyridin-2-yl)-
ethylideneamino]pyrrolidin-2-one
O
CH3
The product of Preparation 4 (1.3 g, 1.47 mmol), the product of
Preparation 1(0.315 g, 2.31 mmol) and pyridine (1.47 mL) are dissolved in EtOH
(6 mL)
and stirred for 18 h. The mixture is concentrated to dryness and the residue
chromatographed on silica gel (90% EtOAc/Hexanes) to give the title compound,
1.2 g
(75%), as a yellow foam.
'H NMR (CDCl3) 8 8.80-8.75 (m, 1H), 7.95-7.85 (m, 2H), 7.70-7.60 (m, 1H), 7.45-
7.20
(m, 4H), 4.90 (s, 2H), 3.10-3.00 (m, 2H), 2.20-2.15 (m, 2H), 1.48-1.35 (m,
2H).
The following compound is prepared by the previous method (6)
Prep# Product Name Physical Data
1H NMR (CDCl3) S 8.72 (d, J
= 4.5 Hz,
1 H), 8.60 (d, J = 4.5 Hz, 1
H), 8.3 0 (d, J =
2.0 Hz, 1 H), 8.15 (dd, J =
7.8, 1.0 Hz, 1 H),
1-[2-(7-Bromquinolin-4-yl)-1-7.95 (d, J = 9.0 Hz, 1H), 7.80
(dt, J = 2.0,
7 pyridin-2-yl-ethylideneamino]-7.8 Hz, 1H), 7.58 (dd, J = 2.0,
9.0 Hz, 1H),
pyrrolidin-2-one 7.40 (dd, J = 4.5, 7.8 Hz, 1H),
7.20 (m,
1 H), 4.90 (s, 2H), 3.10 (t,
J = 6.8 Hz, 2H),
2.22 (t, J = 6.8 Hz, 2H), 1.44
(q, J = 6.8 Hz,
2H)
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-43-
Preparation 8
7-Bromo-4-[2-(6-methylpyridin-2-yl)-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-
yl] quinoline
H3
The product of Preparation 6 (0.21 g, 0.58 mmol) is dissolved in DMF (4 mL)
and NaH (60% in mineral oil, 0.033 g, 0.87 mmol) is added at RT under
nitrogen. After
stirring for 10 min, the mixture is heated to 80 °C overnight, then
cooled to RT. Saturated
NH4Cl solution (0.5 mL) is added and after stirring for l Omin, the mixture is
concentrated
in vacuo. Chromatography of the yellow residue on silica gel (50%
EtOAc/Hexanes)
provides the title compound, 0.13 g (65%), as a yellow foam.
1H NMR (CDC13) 8 8.90-8.88 (m, 1H), 7.55-7.50 (m, 1H), 7.30-7.15 (m, 4H), 7.05-
7.00
(m, 1H), 6.90-6.88(m, 1H), 4.35-4.25 (m, 2H), 2.85-2.60 (m, 4H), 2.40 (s, 3H).
MS (M+1) 4051407.
The following compound is prepared by the previous method (8):
Prep# product Name Physical Data
7-Bromo-4-(2-pyridin-2-yl)-5,6-dihydro-4H-mp 214-216C; MS ESA
9
pyrrolo[1,2-b]pyrazol-3-yl]quinolinem/e 391 (M+1), 393
(M+3)
Scheme 2. Synthesis of 2-Pyridin-2-yl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazole-3-
boronic
acid
Scheme 2 depicts an alternative route for the preparation of the
2 0 dihydropyrrolopyrazole central structure, beginning with step E. In step
E, ethyl
picolinylacetate (Formula 8) is reacted with 1-aminopyrrolidione hydrochloride
(Formula
9) to yield an imine. The picolinate may be substituted at the 6-position of
the nitrogen
heterocycle. Typically, the reaction is conducted in a suitable solvent such
as ethanol, N-
methylpyrrolidinone or pyridine, with pyridine being the preferred solvent.
The reaction
2 5 is carried out at temperatures of about 60 oC to ambient for 4-24 hours.
The products can
be isolated and purified by techniques described above. Step F, as described
above,
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-44-
depicts the cyclization of a compound of Formula 10 to give an optionally
substituted
compound of Formula 11. Typically, the appropriate compound of Formula 10 is
reacted
with a suitable base that can form the anion of the hydrazone, cesium
carbonate being the
preferred base in a suitable solvent, preferably N,N-dimethylformamide, at 100
oC. The
ethyl ester of the pendant carboxylate on the resultant dihydropyrrolopyrazole
Formula 11
is removed as in Step G by ethanolic base hydrolysis with gentle heating under
standard
conditions. Following isolation of the carboxylic acid (Formula 12), the acid
may be
converted to a halide of Formula 13, preferably a bromide, as in Step H with N-
halosuccinimide in the presence of a weak base such as sodium bicarbonate in a
suitable
solvent such as DMF, at room temperature. The use of N-bromosucciriimide as
the
halogenating reagent is shown. Step I depicts the transformation of the halide
of Formula
13 to a boronic acid of Formula 14, by treatment of the halide with a strong
base such as
n-butyllithium at -78 °C, followed by quenching of the intermediate
carbanions with a
source of boron, such as triisopropyl borate. The boronic acid may be
liberated from the
boronic ester upon workup with aqueous ammonium chloride. This transformation
is well
known and appreciated in the art (Larock, R. C., Compf~ehensive Organic
Transformations, 2"d Ed., copyright 1999, John Wiley & Sons, pp 741-742). The
boronic
acid or boronic ester of Formula 14 may subsequently be used as a leaving
group in
combination with a substituted aryl- or heteroarylhalide in the presence of a
suitable
2 0 palladium catalyst, preferably tetrakis(triphenylphosphine)palladium (0),
and a suitable
base such as potassium carbonate to further give compounds of Formula (I)
(Suzuki
reaction see: Miyaura, N.; Yanagi, T.; Suzuki, A. The Palladium-Catalyzed
Cross
Coupling Reaction of Phenylboronic Acid with Haloarenes in the Presence of
Bases.
Syhtlz. Conamun., 1981, 513-518).
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-45-
Scheme 2
0 O
0 O N-NH2.HCI ~ N Cs CO
OEt (9)
iN ~ \
step E I / N v ~OEt step F
R1 (g)
R1 (10)
,N ~ ~ N~N' ~N~ N-Bromosuccinimide
N ' ~ aq NaOH
N ~ R1 ---
R1 CO2H
CO~Et step G step H
(11) (12)
N
~_N'
N-N' ~ ~ N R1
N~ nBuLi B-OH
R1 ---3~ HO
Br B(OiPr)3
(13) step I (14)
Preparation 10
3-(2-Oxo-pyrrolidin-1-yl-imino)-3-pyridin-2-yl-propionic Acid Ethyl Ester
\~ IN,
N O
I
v ~OEt
iN
Ethyl picolinoylacetate (4.7 g, 24.3 mmol) and the product of Preparation 1
(5.0
g, 36.5 mmol) are mixed in 10 mL of pyridine. The reaction mixture is stirred
overnight
at RT and then evaporated to a solid mass. The crude mixture is puriried by
MPLC on
silica gel (40% EtOAc/Hexanes) to give the title compound, 6.63 g (95%), as a
liquid. MS
ES+ m/e 276.1 (M+1).
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-46-
The following compound is prepared by the previous method utilizing Ethyl 6-
Methylpicolinoylacetate (Ref 1)(10):
Prep# product Name Physical Data
3-(6-Methylpyridin-2-yl)-3-(2-oxo-
11 pyrrolidin-1-ylimino)-propionic MS ES+ m/e 290.3
Acid Ethyl (M+1)
Ester
Preparation 12
2-Pyridin-2-yl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazole-3-carboxylic Acid Ethyl
Ester
.N
N N
C02Et
200 mL of DMF is added to the product of Preparation 10 (8.2 g, 30 mmol)
and Cs2C03 (19.5 g, 60 mmol). After stirring for 30 min at RT under nitrogen,
the
reaction mixture is heated at 100 °C for 8 h, cooled to RT, diluted
with EtOAc (300 mL)
and extracted with saturated NaHC03. The organic phase is separated and the
aqueous
phase is extracted 2 times with 50 mL portions of EtOAc. The combined organic
extracts
are washed with saturated brine, dried (Na2S04), filtered, and evaporated to
yield the title
compound, 6.64 g (86%), as cream-colored solid. MS ES+ m/e 258.0 (M+1).
The following compound is prepared by the previous method (12):
Prep# product Name Physical Data
(6-Methyl-pyridin-2-yl)-5,6-dihydro-4H-
13 pyrrolo[1,2-b]pyrazole-3-carboxylicMS ES+m/e 272.0 (M+1)
Acid
Ethyl Ester
2 0 Preparation 14
2-Pyridin-2-yl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazole-3-carboxylic Acid
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
_47-
.N
N
N
Cp2g
The product from Preparation 12 (6.54 g, 25.4 mmol) is dissolved in EtOH (10
mL) and NaOH (2 N, 20 mL) and heated to reflux for 3 h. The reaction is then
cooled to
RT and concentrated to remove the EtOH. The pH of the aqueous solution is
adjusted to
6-7 by the addition of 1N HCl solution. The precipitated solid is filtered,
washed with
Et20 and dried to yield the title compound, 4.9 g (85%), as a white solid. MS
ESA m/e
230.0 (M+1).
The following compound is prepared by the previous method (14):
Prep# product Name Physical Data
2-(6-Methyl-pyridin-2-yl)-5,6-dihydro-4H-
+
MS ES
m/e 244.0 (M+1)
pyrrolo[1,2-b]pyrazole-3-carboxylic
Acid
Preparation 16
3-Bromo-2-pyridin-2-yl-5,6-dihydro-4H-pyrrolo[ 1,2-b]pyrazole
~N
N N
Br
The product from Preparation 14 (2.0 g, 8.7 mmol), NaHC03 (3.3 g, 38.4 mmol)
and N-bromosuccinimide (1.7g, 9.6 mmol) are dissolved in 50 mL of DMF and are
stirred
at room temperature for 2 h. The crude mixture is diluted with 50 mL of water
and 100
mL of EtOAc. The EtOAc layer is separated, extracted with saturated brine,
dried
(Na2S04), filtered, and evaporated to a solid mass. The crude product is
purified by
MPLC on silica gel (50% EtOAc/Hexanes) to yield the title compound, 1.62 g
(70%),as a
2 0 cream-colored solid. MS ES+ m/e 265.9 (M+1 ).
The following compound is prepared by the previous method (16):
Prep# product Name I Physical Data
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
_48_
3-Bromo-2-(6-methyl-pyridin-2-yl)-5,6-
1~ MS ES+m/e 279.0 (M+1)
dihydro-4H-pyrrolo [ 1,2-b]pyrazole
Preparation 18
2-Pyridin-2-yl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazole-3-boronic acid
.N
N N
B-OH
HO
The product from Preparation 16 (0.8 g, 3 mmol) is dissolved in 10 mL of THF
at -78°C. n-BuLi (4.7 mL, 7.6 mmol) in hexanes is added dropwise to the
reaction
mixture while maintaining the temperature between -65 and -70 °C. After
the addition is
complete, the reaction mixture is stirred at -78 °C for 10 min, and
B(OiPr)3 (3.5 mL, 15
mmol) is added dropwise while maintaining the temperature between -65 and -70
°C.
The mixture is allowed to warm to RT and stirred for an additional 3h. 10 mL
of
saturated NH4C1 is added and the reaction mixture stirred for 2 h. The organic
phase is
separated and the aqueous phase is extracted 3 times with 30 mL portions of
CHCl3. The
combined organic extracts are washed with saturated brine, dried (Na2S04),
filtered, and
evaporated to a solid mass. The crude solid is purified by MPLC on silica gel
using a
linear gradient of 50% EtOAc/Hexanes to 80% EtOAc/Hexanes over 50 min. The
chromatographed product is crystallized from EtzO/Hexanes to yield the title
compound,
0.528 g (77%), as white solid. MS ES+m/e 229.9 (M+1).
The following compound is prepared by the previous method (18)
Prep# product Name Physical Data
19 2-(6-Methyl-pyridin-2-yl)-5,6-dihydro-4H-MS ES+ m/e 244.3
pyrrolo[1,2-b]pyrazole-3-boronic (M+1)
acid
Scheme 3. . Alternate synthesis of 5,6-dihydro-4H pyrrolo[1,2- b]pyrazole
derivatives
Scheme 3 depicts the preparation of the compounds of Formula I through a route
which differs from Scheme 1. In Scheme 3, a 4-haloheterocycle of Formula
17a,17b is
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-49-
prepared in two steps from a precursor 4-hydroxyheterocycle by conversion
first to a
triflate as in Step J. The triflate (Formula 16) is prepared by reaction of
the 4-
hydroxyquinoline with trifluoromethanesulfonic anhydride in the presence of a
polar,
basic solvent such as pyridine at 0 °C. Treatment of the triflate with
a halide salt such as
potassium iodide (Step I~) or lithium bromide (Step L) provides the 4-iodo
(Formula 17a)
or 4-bromoheterocycle (Formula 17b), respectively. These halogenations require
a polar
solvent such as dimethylformamide or acetonitrile and temperatures ranging
from RT to
100 °C. The haloheterocycle can be coupled to a pyrrolopyrazole boronic
acid of Formula
14 (Step M) in the presence of a palladium catalyst, preferably
tris(dibenzylidineacetone)dipalladium (0) (Pd2(dba)3), with a suitable base
such as
potassium carbonate. An additional ligand for the palladium, such as
triphenylphosphine,
may be used. All of the reagents are combined in a suitable solvent, typically
dioxane, and
stirred at reflux temperature. All products (Formula 18) can be isolated and
purified by
silica gel chromatography (MPLC), reverse phase HPLC, or trituration of solid
as
described above.
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
=5 0-
Scheme 3
OH OTf
R I ~ Tf20 R' ~ KI R
R" N
Pyridine R" N step K R" N
Step J (16) ~ (17a)
(15)
Liar ~ Br
R'
step L
R" N
(17b)
X N~N' ' ~R1
R, N 1
I HOB OH
R" N
(14)
(17) --
X = I, Br Pd2(dba)3 R"
(18)
step M
R' and R": alkyl, aryl, heterocycle, or halogen or together form
an aromatic or heterocyclic ring
Preparation 20
Trifluoromethanesulfonic Acid 7-bromo-quinolin-4-yl Ester
OTf
Br \ N
7-Bromo-quinolin-4-of (Ref.2)(0.8 g, 3.6 mmol) is dissolved in 10 mL of
pyridine
at 0 °C under nitrogen. Trifluoromethanesulfonic anhydride (1.3 mL, 4.3
mmol) is added
dropwise to the reaction mixture while maintaining the temperature at 0
°C. After the
addition is complete, the reaction mixture is stirred at 0 °C for 2 h,
warmed to room
temperature and evaporated to a slurry. The crude mixture is diluted with 20
mL of water
and 50 mL of EtOAc. The EtOAc layer is separated, extracted with saturated
brine, dried
over anhydrous Na2S04, filtered, and evaporated to a solid mass. The crude
product is
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-51-
purified by MPLC on silica gel (30% EtOAc/Hexanes) to yield the title
compound, 0.82 g
(63%), as white solid. MS ES+ mle 355.9, 357.9 (M+1).
Preparation 21
7-Bromo-4-iodoquinoline
I
Br \ N
The product of Preparation 20 (0.2 g, 0.56 mmol) and KI (0.93 g, 5.6 mmol) are
dissolved in 10 mL of DMF and the mixture is heated at 100 °C for 8 h
under nitrogen,
then cooled to room temperature, diluted with EtOAc (30 mL) and extracted with
saturated NaHC03. The organic phase is separated and the aqueous phase is
extracted 2
times with 1 OmL portions of EtOAc. The combined organic extracts are washed
with
saturated brine; dried (Na2SO4), filtered, and evaporated to a solid mass. The
crude
product is purified by MPLC on silica gel (25% EtOAc/Hexanes) to yield the
title
compound, 0.08 g (44%), as white solid. MS ES+ m/e 335.9 (M+1).
Preparation 22
4,7-Dibromoquinoline
Br
Br \ N
The product of Preparation 20 (3.15 g, 8.8 mmol) and Liar (7.7 g, 88.5 mmol)
2 0 are dissolved in 100 mL of CH3CN and the mixture is heated at 55 °C
for 8 h under
nitrogen. The reaction mixture is cooled to RT and evaporated to a slurry. The
crude
mixture is diluted with EtOAc (30 mL) and washed with saturated NaHC03
solution. The
organic phase is separated and extracted with saturated brine, dried (Na2S04),
filtered, and
evaporated to a solid mass. The crude product is purified by MPLC on silica
gel (25%
2 5 EtOAc/Hexanes) to yield the title compound, 1.9 g (75%), as white solid.
MS ES+ m/e
287.8 (M+1).
Preparation 23
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-52-
7-Bromo-4-(2-pyridin-2-yl-5,6-dihydro-4H-pyrrolo[1,2 b]pyrazol-3-yl)quinoline
Br
The product of Preparation 21 (0.05 g, 0.15 mmol), the product of Preparation
18 (0.034 g, 0.15 mmol) and 0.7 mL of 2M KZC03 are mixed in 2 mL of dioxane.
The
mixture is degassed and flushed with nitrogen several times.
Tris(dibenzylideneacetone)
dipalladium (0) (0.004 g, 0.0045 mmol) and triphenylphosphine (0.002 g, 0.00 9
mmol)
are added, and the reaction mixture degassed and flushed with nitrogen again.
The
mixture is heated to reflux at 115 °C for 4 h under nitrogen. The
reaction mixture is
cooled and diluted with water (2 mL) and EtOAc (4 mL). The organic layer is
separated
l0 and extracted with saturated brine, dried over anhydrous Na2S04, filtered,
and evaporated
to a solid mass. The crude product is purified by MPLC on silica gel (70%
EtOAc/Hexanes) to yield the title compound, 0.045 g (77%), as white solid. MS
ES+m/e
391.1, 393.1 (M+1).
The following compound is prepared by the previous method (23)
Prep# product Name Physical Data
24 7-Bromo-4-[2-(6-methylpyridin-2-yl)-5,6-MS ES+ m/e a 405.0,
dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl]quinoline407.0 (M+I)
Preparation of Final Products:
Scheme 4. Synthesis of 7-substituted quinoline derivatives
2 0 Scheme 4 elaborates substitution of the C-7 position of the 7-
bromoquinoline of
Formula 19. A representative transformation is seen in Step N, in which a
metal-
nucleophile, such as trialkylstannyls, or boranes with a suitable base such as
potassium
carbonate, sodium alkoxides (sodium methoxide, or sodium ethoxide) or
potassium
alkoxides (potassium methoxide, or potassium ethoxide) can be used with a
palladium
2 5 catalyst, previously described, preferably
tris(dibenzylidineacetone)dipalladium (0)
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-53-
(Pd2(dba)3). An additional ligand for the palladium, such as
triphenylphosphine, may be
used. The stannyl or boronic acid reagents may be aromatic with one or more
substituents
or variously substituted heteroaromatics. Protecting groups may be required on
the O, N,
or S-containing functional groups. In addition, organozinc reagents may also
be
employed to effect the net displacement of the halide at the C-7 position by
an alkyl
group. All of the reagents are combined in a suitable solvent, typically
dioxane, isopropyl
alcohol, tetrahydrofuran, toluene or ethylene glycol dimethyl ether, stirred
at temperatures
from room temperature to reflux. The coupling of arylhalides with metallo-
aromatics and
metallo-alkyls by similar methods has been reviewed in the literature
(Stanforth, S. P.
Tetrahedron, 1998, 54, 263-303). The product of Formula 20 may require an
additional
synthetic step to remove a group such as t-butoxycarbonyl or trimethylsilyl,
by treatment
with trifluoroacetic acid or tetra-n-butylammonium fluoride, respectively.
Such
deprotection conditions are well known in the art. (Greene, T. W.; Wuts, P. G.
M.
Protective Groups ih Organic Synthesis (3'~ ed.); Wiley: New York, 1999.) All
products
can be isolated and purified by silica gel chromatography (MPLC), reverse
phase HPLC,
or trituration of solid as described above.
Scheme 4
R1 Matal Catalyst
R1
R2-Metal
step N
(19)
R2: aryl, heterocycle, cycloalkyl
~ o (20)
Example 1
4-(2-Pyridin-2-yl-5,6-dihydro-4H-pyrrolo[ 1,2-b]pyrazol-3-yl)-7-thiophen-2-yl-
quinoline
2 5 Trifluoroacetic Acid
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-54-
Into a 10 mL round bottom flask is placed the product of Preparation 9 (0.0782
g,
0.2 mmol), thiophene-2-boronic acid (0.0512 g, 0.4 mL), 2 mL isopropanol, and
1 mL 2M
K2CO3. A reflux condenser is situated, and the mixture is evacuated and
flushed with
nitrogen several times. Tetrakis(triphenylphosphine)Pd(0) (0.0069 g, 0.006
mmol) is
added. Evacuation and flushing with nitrogen is repeated and mixture is heated
in oil bath
at 80 °C for 5 h. The reaction mixture is cooled, diluted with 2 mL
water and 4 mL
EtOAc, and agitated. The water layer is removed, 2 mL water is added, and the
extraction
procedure repeated. The organic layer is eluted over 1 g silica gel cartridge
with 25 mL
MeOH containing one drop concentrated NH40H. The MeOH solution is evaporated
under reduced pressure. The residue is dissolved in 20 mL EtOH and evaporated
again.
Purified product is isolated as a free base by trituration with DMSO and
drying at high
vacuum. The product TFA salt (53.2 mg, 43% yield) is obtained by freeze drying
appropriate fractions (based on LC/MS analysis) from reverse phase HPLC using
Waters
Symmetry C 18 column with a gradient of 10 to 70% B in A, where A is water
containing
0.1 % TFA and B is CH3CN containing 0.1 % TFA.
MS ES+ m/e 395 (M+1 of free base)
This method of Example 1 is used to prepare the following compound:
Example Product Name Physical Data
2 7-(4-methylthiophen-2-yl)-4-(2-pyridin-2-yl-MS ES+ m/e 409
(M+1)
5,6-dihydro-4H-pyrrolo [ 1,2-b]pyrazol-3-
yl)quinoline Trifluoroacetic Acid
Example 3
F 'O H
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
N- {4-[4-Pyridin-2-yl-5, 6-dihydro-4H-pyrrolo [ 1,2-b]pyrazol-3-yl)-quinolin-7-
yl]phenyl}acetamide Trifluroacetic Acid
O
~N
F OH
Into a 10 mL round bottom is placed the product of Preparation 9 (0.0782g,
0.2mrno1), N-[4-(4,4,5,5-tetramethyl -[1,3,2]dioxaborolan-2-
yl)phenyl]acetamide (0.104
g, 0.4 mmol), 2 mL dioxane, and 1 mL 2M I~2C03. The mixture is heated in oil
bath at
110 °C for few minutes to dissolve organic reagents. A reflux condenser
is positioned and
the mixture is evacuated and purged with nitrogen several times. To the
mixture is added
Pd2(dba)3 (0.007 g, 0.006 mmol) and triphenylphosphine (0.007 g, 0.024 mmol)
and the
evacuating and purging with nitrogen repeated. The reaction mixture is heated
in an oil
bath at 110 °C for 3 h. The mixture is cooled, extracted, and purified
as in Example 1 to
give the titled product (0.095 g, 70% yield).
MS ES+ 446 (M+1)
Using the methods of the previous Examples 1 and 3, changing only the workup
procedure to isolate the crude product from the aqueous layer by precipitation
with dilute
HCl when there is an acidic functionality situated on the aryl ring of the
boronic acid, the
following compounds are prepared:
Ex. # Product Name Physical Data
4 7-Phenyl-4-(2-pyridin-2-yl-5,6-dihydro-MS ES+ 389 (M+1)
4H-pyrrolo [ 1, 2-b]pyrazol-3-yl)quinoline
Trifluoroacetic Acid
5 2-[4-(2-pyridin-2-yl-5,6-dihydro-4H-MS ES+ 40~ (M+1)
pyrrolo[pyrazol-3-yl)quinolin-7-yl]phenol
Trifluoroacetic Acid
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-56-
6 3-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ 405 (M+1)
pyrrolo[ 1,2-b]pyrazol-3-yl)quinolin-7-
yl]phenol Trifluoroacetic Acid
7 4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ 405 (M+1)
pyrrolo [ 1,2-b]pyrazol-3-yl)quinolin-7-
yl]phenol Trifluoroacetic Acid
8 2-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ 404 (M+1)
pyrrolo[ 1,2-b]pyrazol-3-yl)quinolin-7-
yl]phenylamine Ditrifluoroacetic
Acid
9 3-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ 404 (M+1)
pyrrolo[pyrazol-3-yl)quinolin-7-
yl]phenylamine Ditrifluoroacetic
Acid
4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ 404 (M+1)
pyrrolo [ 1,2-b]pyrazol-3-yl]phenylamine
Ditrifluoroacetic Acid
11 7-(3-Chlorophenyl)-4-(2-Pyridin-2-yl-5,6-MS ES+ 423 (M+1)
dihydro-4H-pyrrolo [ 1, 2-b]pyrazol-3-
yl)quinoline Trifluoroacetic
Acid
12 7-(4-Chlorophenyl)-4-(2-pyridin-2-yl-5,6-MS ES+ 423 (M+1)
dihydro-4H-pyrrolo[ 1,2-b]pyrazol-3-
yl)quinoline Trifluoroacetic
Acid
13 7-(3-Nitrophenyl)-4-(2-pyridin-2-yl-5,6-MS ES+ 434 (M+1)
dihydro-4H-pyrrolo [ 1, 2-b]pyrazol-3-
yl)quinoline Trifluoroacetic
Acid
14 7-(4-Nitrophenyl)-4-(2-pyridin-2-yl-5,6-MS ES+ 434 (M+1)
dihydro-4H-pyrrolo[ 1,2-b]pyrazol-3-
yl)quinoline Trifluoroacetic
Acid
N-{3-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ 482 (M+1)
pyrrolo [ 1,2-b]pyrazol-3-yl)-quinolin-7-yl]-
phenyl } methanesulfomamide
Trifluoroacetic Acid
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
7_
16 N-{4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ 482 (M+1)
pyrrolo [ 1,2-b]pyrazol-3-yl)quinolin-7-
yl]phenyl} methanesulfonamide
Trifluoroacetic Acid
17 7-(2-Ethoxyphenyl)-4-(2-pyridin-2-yl-5,6-MS ES+ 433 (M+1)
dihydro-4H-pyrrolo [ 1, 2-b]pyrazol-3-
yl)quinoline Trifluoroacetic
Acid
18 7-(3-Ethoxyphenyl)-4-(2-pyridin-2-yl-5,6-MS ES+ 433 (M+1)
dihydro-4H-pyrrolo [ 1, 2-b]pyrazol-3-
yl)quinoline Trifluoroacetic
Acid
19 7-(4-Ethoxyphenyl)-4-(2-pyridin-2-yl-5,6-MS ES+433 (M+1)
dihydro-4H-pyrrolo [ 1, 2-b]pyrazol-3-
yl)quinoline Trifluoroacetic
Acid
20 7-(4-Methoxyphenyl)-4-(2-pyridin-2-yl-MS ES+ 419 (M+1)
5,6-dihydro-4H-pyrrolo [ 1,2-b]pyrazol-3-
yl)quinoline Trifluoroacetic
Acid
21 7-(4-Fluoro-3-methylphenyl)-4-(2-pyridin-MS ES+ 421 (M+1)
2-yl-5, 5-dihydro-4H-pyrrolo
[ 1,2-
b]pyrazol-3-yl)quinoline Trifluoroacetic
Acid
22 7-(4-Fluoro-2-methylphenyl)-4-(2-pyridin-MS ES+ 421 (M+1)
2-yl-5,6-dihydro-4H-pyrrolo[
1,2-
b]pyrazol-3-yl)quinoline Trifluoroacetic
Acid
23 7-(4-Bromophenyl)-4-(2-pyridin-2-yl-5,6-MS ES+ 468 (M+1)
dihydro-4H-pyrrolo[ 1,2-b]pyrazol-3-
yl)quinoline Trifluoroacetic
Acid
24 7-(4-Ethylsulfanylphenyl)-4-(2-pyridin-2-MS ES+ 449 (M+1)
yl-5, 6-dihydro-4H-pyrrolo
[ 1,2-b]pyrazol-
3-yl)quinoline Trifluoroacetic
Acid .
25 7-(4-Methylsulfanylphenyl)-4-(2-pyridin-MS ES+ 435 (M+1)
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
_~$_
2-yl-5,6-dihydro-4H-pyrrolo
[ 1, 2-
b]pyrazol-3-yl)quinoline Trifluoroacetic
Acid
26 4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ 414 (M+1)
pyrrolo[ 1,2-b]pyrazol-3-yl)quinolin-7-
yl]benzonitrile Trifluroacetic
Acid
27 7-(1H-Indol-5-yl)-4-(2-pyridin-2-yl-5,6-MS ES+ 428 (M+1)
dihydro-4H-pyrrolo [ 1,2-b]pyrazol-3-
yl)quinoline Ditrifluoroacetic
Acid
28 1- f 4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ 431 (M+1)
pyrrolo [ 1, 2-b]pyrazol-3-yl)quinolin-7-
yl]phenyl}ethanone Trifluoroacetic
Acid
29 2-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ 432 (M+1)
pyrrolo [ 1,2-b]pyrazol-3-yl)quinolin-7-
yl]phenyl}benzamide Trifluoroacetic
Acid
30 3-{4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ 461 (M+1)
pyrrolo [ 1, 2-b]pyrazol-3-yl)quinolin-7-
yl]phenyl}propionic Acid Trifluoroacetic
Acid
31 7-(3,5-Dichlorophenyl)-4-(2-Pyridin-2-yl-MS ES+ 433 (M+1)
5,6-dihydro-4H-pyrrolo[ 1,2-b]pyrazol-3-
yl)quinoline Trifluoroacetic
Acid
32 . 7-(4-Methoxy-3-methylphenyl)-4-(2-MS ES+ 433 (M+1)
Pyridin-2-yl-5,6-dihydro-4H-pyrrolo[
1,2-
b]pyrazol-3-yl)quinoline Trifluoroacetic
Acid
33 2-Methyl-5-[4-(2-Pyridin-2-yl-5,6-MS ES+ 418 (M+1)
dihydro-4H-pyrrolo [ 1,2-b]pyrazol-3
-
yl)quinolin-7-yl]phenylamine
Ditrifluoroacetic Acid
34 7-Pyridin-4-yl-4-(2-Pyridin-2-yl-5,6-MS ES+ 390 (M+1)
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-59-
dihydro-4H-pyrrolo [ 1,2-b
]pyrazol-3-
yl)quinoline Ditrifluoroacetic
Acid
35 7-Pyridin-3-yl-4-(2-Pyridin-2-yl-5,6-MS ES+ 390 (M+1)
.
dihydro-4H-pyrrolo [ 1,2-b]pyrazol-3-
yl)quinoline Ditrifluoroacetic
Acid
36 7-(2-Nitrophenyl)-4-(2-pyridin-2-yl-5,6-MS ES+ 434 (M+1)
dihydro-4H-pyrrolo [ 1,2-b]pyrazol-3-
yl)quinoline Trifluoroacetic
Acid
37 N-{3-[4-(2-pyridin-2-yl-5,6-dihydro-4H-MS ES+ 446 (M+1)
pyrrolo[ 1,2-b]pyrazol-3-yl)quinolin-7-
yl]phenyl}-acetamide Trifluoroacetic
Acid
38 7-(4-Methanesulfinylphenyl)-4-(2-pyridin-MS ES+ 451 (M+1)
2-yl-5, 6-dihydro-4H-pyrrolo
[ 1,2-
b]pyrazol-3-yl)quinoline Trifluoroacetic
Acid
40 7-Benzo[1,3]dioxol-5-yl-4-(2-pyridin-2-MS ES+ 433 (M+1)
yl-5,6-dihydro-4H-pyrrolo [
1,2-b]pyrazol-
3-yl)quinoline Trifluoroacetic
Acid
41 4-[4-(2-pyridin-2-yl-5,6-dihydro-4H-MS ES+ 433 (M+1)
pyrrolo [ 1,2-b]pyrazol-3-yl)quinolin-7-
yl]benzoic Acid
Trifluoroacetic Acid
42 7-(3,5-Bis-trifluromethylphenyl)-4-(2-MS ES+ 525 (M+1)
pyridin-2-yl-5, 6-dihydro-4H-pyrrolo
[ 1,2-
b]pyrazol-3-yl)quinoline Trifluoroacetic
Acid
43 3-[4-(2-pyridin-2-yl-5,6-dihydro-4H-MS ES+ 433 (M+1)
pyrt~olo[ 1,2-b]pyrazol-3-yl)quinolin-7-
yl]benzoic Acid Trifluoroacetic
Acid
44 2-[4-(2-pyridin-2-yl-5,6-dihydro-4H-MS ES+ 433 (M+1)
pyrrolo [ 1,2-b]pyrazol-3-yl)quinolin-7-
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-60-
yl]benzoic Acid Trifluoroacetic
Acid
45 7-(4-Isobutylphenyl)-4-(2-pyridin-2-yl-MS ES+ 445 (M+1)
5,6-dihydro-4H-pyrrolo [ 1,2-b]pyrazol-3-
yl)quinoline Trifluoroacetic
Acid
46 {4-[4-(2-pyridin-2-yl-5,6-dihydro-4H-MS ES+ 463 (M+1)
pyrrolo[ 1,2-b]pyrazol-3-yl)quinolin-7-
yl]phenoxy} acetic Acid Trifluoroacetic
Acid
47 7-Benzo[b]thiophen-2-yl-4-(2-pyridin-2-MS ES+ 445 (M+1)
yl-5, 6-dihydro- 4H-pyrrolo
[ 1,2-b]pyrazol-
3-yl)quinoline Trifluoroacetic
Acid
48 7-Benzofuran-2-yl-4-(2-pyridin-2-yl-5,6-MS ES+ 429 (M+1)
dihydro-4H-pyrrolo [ 1,2-b]pyrazol-3-
yl)quinoline Trifluoroacetic
Acid
49 7-(3-Nitrophenyl)-4-(2-pyridin-2-yl-5,6-MS ES+ 320 (M+1)
dihydro-4H-pyrrolo [ 1,2-b]pyrazol-3-
yl)quinoline Trifluoroacetic
Acid
50 4-(2-pyridin-2-yl-5,6-dihydro-4H-MS ES+ 395 (M+1)
pyrrolo [ 1,2-b]pyrazol-3-yl)-7-thiophen-3-
yl-quinoline Trifluoroacetic
Acid
51 7-Furan-3-yl-4-(2-pyridin-2-yl-5,6-MS ES+ 379 (M+1)
dihydro-4H-pyrrolo [ 1,2-b]pyrazol-3-
yl)quinoline Trifluoroacetic
Acid
52 7-(4-Methyl-3-nitro-phenyl)-4-(2-pyridin-MS ES+ 462 (M+1
)
2-yl-5,6-dihydro-4H-pyrrolo[
1,2-
b]pyrazol-3-yl)-quinoline Trifluoroacetic
Acid
53 7-(4-Cyclohexyl-phenyl)-4-(2-pyridin-2-MS ES+ 471 (M+1)
yl-5,6-dihydro-4H-pyrrolo [
1,2-b]pyrazol-
3-yl)-quinoline Trifluoroacetic
Acid
54 7-(4-Benzo[b]thiophen-3-yl-phenyl)-4-(2-MS ES+ 445 (M+1)
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-61-
pyridin-2-yl-5,6-dihydro-4H-pyrrolo [ 1,2-
b]pyrazol-3-yl)-quinoline Trifluoroacetic
Acid
Example 55
7-(3,4-Dichlorophenyl)-4-(2-pyridin-2-yl-5,6-dihydro-4H-pyrrolo[ 1,2-b]pyrazol-
3-yl)-
quinoline
The product of Preparation 9 (0.035 g, 0.089 mmol), 3,4-dichlorophenylboronic
acid (0.02 g, 0.11 mmol) and 0.3 mL 2M I~2C03 are mixed in 2 mL of dioxane.
The
mixture is degassed and flushed with nitrogen several times.
Tris(dibenzylideneacetone)
dipalladium (0) (0.003 g, 0.003 mmol) and triphenylphosphine (0.002 g, 0.005
mmol) are
added, degassed and flushed with nitrogen. The mixture is heated to reflux at
115 °C for
4 h under nitrogen. The reaction mixture is cooled, diluted with water (2 mL)
and EtOAc
(4 mL). The organic layer is separated and extracted with saturated brine,
dried (Na2SO4),
filtered, evaporated to a solid mass. The crude is purified by MPLC on silica
gel (70%
EtOAc/Hexanes, 1% MeOH) to yield the title compound, 0.034 g (83%), as a cream-
colored solid. MS ESA m/e 457.1 (M+1).
Using the method of Example 55, changing only the procedure to isolate the
crude
zwitterionic product from the aqueous layer by precipitation with dilute HCl
when there is
2 0 an acidic functionality situated on the aryl ring of the boronic acid, the
following
compounds are made.
C1 C1
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-62-
Example Product Name Physical Data
#
56 4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ m/e 433.1
pyrrolo[1,2-b]pyrazol-3-yl)-quinolin-7-yl]-(M+1)
benzoic acid
57 4- f 4-[2-(6-Methyl-pyridin-2-yl)-5,6-dihydro-MS ES+ mle 447.1
4H-pyrrolo[1,2-b]pyrazol-3-yl]-quinolin-7-yl}-(M+1)
benzoic acid
58 3-(4- f 4-[2-(6-Methyl-pyridin-2-yl)-5,6-dihydro-MS ES+ m/e 475.1
4H-pyrrolo[1,2-b]pyrazol-3-yl]-quinolin-7-yl~-(M+1)
phenyl)-propionic acid
Example 59
7-(4-Fluoro-phenyl)-4-[2-(6-methyl-pyridin-2-yl)-5, 6-dihydro-4H-pyrrolo [ 1,2-
b]pyrazol-
3-yl]-quinoline
CH3
The product of Preparation 8 (0.080 g, 0.20 mmol) and 4-fluorophenylboronic
acid (0.053 g, 0.40 mmol) are dissolved in DMEl2M KZC03 (1:1, 2.0 mL) for 20
min.
Pdz(dba)3(0.006 g, 0.006 mmol) and triphenylphosphine (0.006 g, 0.024 mmol)
are added,
and nitrogen is bubbled through for another lOmin. The mixture is heated to 80
°C for 48
h. The solvent is evaporated and the residue chromatographed on silica gel
(eluting with
2% MeOH in CHZC12) to give the title compound, 0.070 g, 81 %, as a yellow
solid.
1H NMR (CDCl3) 8 8.88(d, 1H, J=4.4Hz), 8.29 (d, 1H, J=2.OHz), 7.83 (d, 1H,
8.8Hz),
7.72-7.67 (m, 2H), 7.57 (dd, 1H, J=2.0, 8.8Hz), 7.30-7.26 (m, 2H), 7.20-7.15
(m, 2H),
6.99 (d, 1H, J=7.8Hz), 6.91 (d, 1H, J=7.8Hz), 4.37 (t, 2H, J=7.2Hz), 2.88 (t,
2H,
J=7.2Hz), 2.71 (q, 2H, J=7.2Hz), 2.34 (s, 3H). MS (M+1) 421.
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-63-
Using the method of the previous Example 59, the following compounds are
prepared. In
some cases the crude product was further purified by a preparative HPLC
(Waters
Symmetry PrepTM C18, 7um, WAT066245, 19x300 mm column, eluted with a gradient
solution system of 90:10 (0.2% TFA in H20)/(0.2% TFA in CH3CN)to 10:90 (0.2%
TFA
in H20)/(0.2% TFA in CH3CN)).
Example Product Name Physical data
#
60 7-(4-Methoxy-phenyl)-4-[2-(6-MS (M+1) 433.
methyl-pyridin-2-yl)-5,6-dihydro-4H-
pyrrolo[ 1,2-b]pyrazol-3-yl]-quinoline
61 7-Phenyl-4-[2-(6-methyl- MS (M+1) 403.
pyridin-2-yl)-5,6-dihydro-4H-
pyrrolo[ 1,2-b]pyrazol-3-yl]-quinoline
Trifluoroacetic Acid
62 4-[2-(6-Methyl-pyridin-2-yl)-MS (M+1) 409.
5,6-dihydro-4H-pyrrolo[ 1,2-b]pyrazol-
3-yl]-7-thiophen-2-yl-quinoline
Trifluoroacetic Acid
63 4-[2-(6-Methyl-pyridin-2-yl)-5,6-MS (M+1) 448.
dihydro-4H-pyrrolo [ 1,2-b]pyrazol-3-
yl]-7-(3-nitro-phenyl)quinoline
64 7-(4-Chloro-phenyl)-4-[2-(6- MS (M+1) 437.
methyl-pyridin-2-yl)-5,6-dihydro-4H-
pyrrolo[ 1,2-b]pyrazol-3-yl]-quinoline
l0 Example 65
7-Furan-2-yl-4-(2-Pyridin-2-yl-5,6-dihydro-4H-pyrrolo[ 1,2-b]pyrazol-3-
yl)quinoline
Trifluoroacetic Acid
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-64-
The product of Preparation 9 (0.782 g, 0.2 mmol) is placed into a l OmL
roundbottom flask and toluene (2 mL) is added. A condenser is positioned, and
the
mixture is evacuated and purged with nitrogen several times. To the mixture is
added
tribut-1-furan-2-yl-stannane (0.143 g, 0.4 mmol). The mixture evacuated and
purged with
nitrogen several times and then refluxed 3.5 h in an oil bath. The reaction
mixture is
cooled and diluted with EtOAc (3 mL) and eluted over a 1 g silica gel
cartridge, fishing
with MeOH (20 mL). The solvent is removed under vacuum. Purified product (63
mg,
64%) is obtained by freeze drying appropriate fractions (based on MS analysis)
from
reverse phase HPLC using Waters Symmetry C 18 column with a gradient of 10 to
70 %B
in A, where A is water containing 0.1 % TFA and B is CH3CN containing 0.1 %
TFA.
MS ES+ 379 (M+1)
Using the method of Example 65, the following compounds are prepared:
Example Product Name Physical Data
#
66 4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ 396 (M+1)
pyrrolo [ 1,2-b]pyrazol-3-yl)-7-thiazol-2-yl-
quinoline Trifluoroacetic Acid
67 7-Pyrazin-2-yl-4-(2-Pyridin-2-yl-5,6-dihydro-MS ES+ 391 (M+1)
4H-pyrrolo[1,2-b]pyrazol-3-yl)quinoline
Ditrifluoroacetic Acid
68 7-Pyridin-2-yl-4-(2-Pyridin-2-yl-5,6-dihydro-MS ES+ 390 (M+1)
4H-pyrrolo[ 1,2-b]pyrazol-3-yl)quinoline
Ditrifluoroacetic Acid
Example 69
r~OH
FF
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
F~OH F F
F/ \F
-65-
4-(2-pyridin-2-yl-5, 6-dihydro-4H-pyrrolo [ 1,2-b]pyrazol-3-yl)-7-( 1 H-pyrrol-
2-yl)quinoline
Ditrifluoroacetic Acid
N'N
N
~ ~ ~ ~ NJ O
N v O F~OH
H
The cross coupling of 1-(t-butoxycarbonyl)pyrrole-2-boronic acid (0.168 g, 0.8
mmol) with the product of Preparation 9 (0.156 g, 0.4 nnnol) is done as in
Example 3.
The crude product is then dissolved in 2 mL CHZCl2 and treated with 2 mL of
TFA for
16h. The mixture is evaporated under reduced pressure, and purified product
(72.2 mg,
37%) is obtained from reverse phase chromatography as described in Example 65.
MS ES+ 378 (M+1 for free base)
Using the method of Example 69, involving t-boc-protected heterocycles, the
following
compound is prepared:
Example Product Name Physical Chemistry
#
70 7-(1H-Indol-2-yl)-4-(2-pyridin-2-yl-5,6-MS ES+ 428 (M+1)
dihydro-4H-pyrrolo [ 1,2-b]pyrazol-3-
yl)quinoline Ditrifluoroacetic
Acid
Example 71
4-(2-pyridin-2-yl-5,6-dihydro-4H-pyrrolo [ 1,2-b]pyrazol-3-yl)-7-( 1 H-pyrrol-
3-yl)quinoline
Ditrifluoroacetic Acid
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-66-
OOH
The cross coupling of 1-(triisopropylsilyl)pyrrole-3-boronic acid (0.214 g,
0.8
mmol) with the product of Preparation 9 (0.156 g, 0.4 mmol) is done as in
Example 69.
The crude product is then dissolved in SmL THF and treated with 2 mL of O.SM
tetrabutylammonium fluoride for 3 h. The mixture is diluted with 15 mL of
EtOAc and
washed with water (75 mL). The organic layer is dried (MgS04) and evaporated
under
reduced pressure. Purified product (78.4 mg, 40%) is obtained from reverse
phase
chromatography as described in Example 65.
MS ES+ 378 (M+1)
Example 72
7-Cyclohexyl-4-(2-pyridin-2-yl-5,6-dihydro-4H-pyrrolo[ 1,2-b]pyrazol-3-
yl)quinoline
Trifluoroacetic Acid
The cross coupling of cyclohexylzinc bromide(0.8 mL of O.SM soln. in THF, 0.4
mmol)- which replaces the boronic acid- with the product of Preparation 9
(0.0782 g, 0.2
mmol) is done according to Example 3. Purified product (10 mg) is obtained
from
reverse phase chromatography as described in Example 65.
MS ES+395 (M+1 for free base)
F~OH
FF
F~OH
F/ \F
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-67-
Scheme 5. Synthesis of 7-aryl/heteroaryl quinoline compounds via 7-boronate
intermediate
Scheme 5 demonstrates an alternative means of attaching various aryl rings and
heterocycles at the C-7 position of the 7-bromoquinoline of Formula 19. Step O
illustrates
that the bromide can be converted to a boronic ester of Formula 21 that can
then be
coupled to aryl-, heterocyclic- or alkylbromides in metal-catalyzed reactions
to provide C-
7 substituted quinoline compounds of Formula 20. The conversion of the
heteroaryl
bromide (Formula 19) is accomplwashed by treatment with bis(pinacolato)diboron
and
KOAc in a polar, high boiling solvent such as DMSO. A palladium catalyst,
preferably
1,1'-bis(diphenylphosphino)ferrocene palladium (II) chloride complex with
dichloromethane (PdCl2(dppf)) is preferred in the reaction. The preparation of
a boronate
from a halide in this manner has been reported in the literature (Ishiyama,
T.; Murata, M.;
Miyaura, N. J. Org. Chem. 1995, 60, 7508-7510.).
The boronate (Formula 21) is coupled (step P) to an aryl- or heterocyclic
halide
with a suitable base such as potassium carbonate, sodium alkoxides (sodium
methoxide,
or sodium ethoxide) or potassium alkoxides (potassium methoxide, or potassium
ethoxide) in combination with a palladium catalyst, as previously described,
preferably
tris(dibenzylidineacetone)dipalladium (0) (Pd2(dba)3). An additional ligand
for the
2 0 palladium, such as triphenylphosphine, may be used. All of the reagents
are combined in
a suitable solvent, typically dioxane, isopropyl alcohol, tetrahydrofuran,
toluene or
ethylene glycol dimethyl ether, stirred at temperatures from room temperature
to reflux:
The product of Formula 20 may require an additional deprotection step to
remove a group
such as t-butoxycarbonyl or trimethylsilyl, by treatment with trifluoroacetic
acid or tetra-
2 5 n-butylammonium fluoride, respectively. All products can be isolated and
purified by
silica gel chromatography (MPLC), reverse phase HPLC, or trituration of solid
as
described above.
3 0 Scheme 5
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-68-
,O
O~B-B
O ,O
R1 1
Pd Catalyst
step o
Br ~~ O~B .V
I
(1 g) O (21 )
Ar-Br (I) R1
Pd Catalyst
step p
(Aryl or heterocycle or cycloalkyl)
(20)
Preparation 25
4-(2-Pyridin-2-yl-5,6-dihydro-4H-pyrrolo[ 1,2-b]pyrazol-3-yl)-7-[4-(4,4,5,5-
tetramethyl-
[1,3,2]dioxaborolan-2-yl)phenyl]quinoline
O,B
IV
I
O
Into a 50 ml round bottom is placed the product of Preparation 9 (2.35 g, 6
mmol), bis(pinacolato) diboron (3.05 g,12 mmol), anhydrous KOAc (2.96 g, 30
mmol),
and DMSO (18 mL). The mixture is heated for a few minutes in an oil bath at
110 °C and
then evacuated and purged with nitrogen several times. To the mixture is added
PdCl2(dppf)(0.263 g, 0.36 mmol), and the evacuation and purging repeated. The
mixture
is heated under nitrogen 20 h, allowed to cool, and then poured into 100 mL of
water to
obtain a precipitate. The precipitate is collected, washed with water, and
dried under
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-69-
vacuum at 60 °C, to give 2.02 g (77%) of title compound, which is
sufficiently pure to use
in coupling reactions.
MS ES+ 439 (M+1)
Example 73
2- {4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-pyrrolo [ 1,2-b]pyrazol-3-yl)quinolin-
yl]phenoxy] ethanol Trifluoroacetic Acid
~O
HO-' F OH
Into a 10 mL round bottom is placed the product of Preparation 25 (0.0876 g,
0.2
mmol), 2-(4-bromophenoxy) ethanol (0.087 g, 0.4 mmol), dioxane (2 mL), and 2M
K2CO3 (l mL). A condenser is situated, and the mixture is evacuated and purged
with
nitrogen several times. To the mixture is added Pd2(dba)3 (0.007 g, 0.006
mmol) and
triphenylphosphine (0.007 g, 0.024 mmol). The mixture is evacuated and purged
with
nitrogen several times and then heated Sh at gentle reflux. The mixture is
cooled, diluted
with EtOAc (3 mL), and the organic layer eluted over a 1 g silica gel
cartridge, washing
with EtOH (20 mL). The solution is evaporated under reduced pressure. Purified
product
(29.2 mg, 32%) is obtained by freeze drying appropriate fractions (based on MS
analysis)
from reverse phase HPLC using Waters Symmetry C18 column with a gradient of 10
to
70% B in A, where A is water containing 0.1 % TFA and B is CH3CN containing
0.1
2 0 TFA.
MS ES+ 449 (M+1)
Using the method of Example 73, changing only the workup procedure to isolate
the
crude product from the aqueous layer by precipitation or evaporation after
treatment with
2 5 dilute HCl when there is an acidic functionality situated on the aryl ring
of the aryl
bromide coupling partner, the following compounds are prepared:
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-70-
Example Product Name Physical Data
#
74 f4-[4-(2-pyridin-2-yl-5,6-dihydro-4H-MS ES+447 (M+1)
pyrrolo [ 1,2-b]pyrazol-3-yl)
quinolin-7-
yl]phenyl~acetic Acid Trifluoroacetic
Acid
75 7-(4-Methanesulfonylphenyl)-4-(2-pyridin-2-MS ES+ 467 (M+1)
yl-5, 6-dihydro-4H-pyrrolo [
1,2-b]pyrazol-3-
yl)quinoline Trifluoroacetic
Acid
76 2- f 4-[4-(2-pyridin-2-yl-5,6-dihydro-4H-MS ES+ 432 (M+1
pyrrolo [ 1,2-b]pyrazol-3-yl)
quinolin-7-
yl]phenyl~ethylamine Ditrifluoroacetic
Acid
77 4-(2-pyridin-2-yl-5,6-dihydro-4H-MS ES+ 403 (M+1)
pyrrolo[1,2-b]pyrazol-3-yl)-7-p-
tolylquinoline Trifluoroacetic
Acid
78 2-Fluoro-4-[4-(2-pyridin-2-yl-5,6-dihydro-MS ES+ 422 (M+1)
4H-pyrrolo [ 1,2-b]pyrazol-3-yl)quinolin-7-
yl]phenylamine Ditrifluoroacetic
Acid
79 5-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ 477 (M+1)
pyrrolo [ 1,2-b]pyrazol-3-yl)
quinolin-7-
yl]isophthalic Acid Trifluoroacetic
Acid
80 5-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ 505 (M+1)
pyrrolo[ 1,2-b]pyrazol-3-yl)quinolin-7-
yl]isophthalic Acid Dimethyl
Ester
Trifluoroacetic Acid
81 7-(2,2-Dioxy-2,3-dihydro-1H-2~,6-MS ES+ 479 (M+1)
benzo[c]thiophen-5-yl-4-(2-Pyridin-2-yl-5,6-
dihydro-4H-pyrrolo[ 1,2-b]pyrazol-3-
yl)quinoline Trifluoroacetic
Acid
82 6-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ 434 (M+1)
pyrrolo [ 1,2-b]pyrazol-3-yl)quinolin-7-
yl]pyridine-2-carboxylic Acid
Tris-
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-71-
trifluoroacetic Acid
83 7-(5-Fluoropyridin-2-yl)-4-(2-Pyridin-2-yl-MS ES+ 408 (M+1)
5,6-dihydro-4H-pyrrolo [ 1,2-b]pyrazol-3-
yl)quinoline Ditrifluoroacetic
Acid
84 7-(6-Methoxypyridazin-3-yl)-4-(2-Pyridin-2-MS ES+ 421 (M+1)
yl-5, 6-dihydro-4H-pyrrolo [
1,2-b]pyrazol-3-
yl)quinoline Difluoroacetic Acid
85 4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ 391 (M+1)
pyrrolo[ 1,2-b]pyrazol-3-yl)-7-pyrimidin-2-yl-
quinoline Difluoroacetic Acid
86 4-{4-[4-(2-Pyridin-2-yl-5,6- MS ES+ 474 (M+1)
dihydro-4H-
pyrrolo [ 1, 2-b]pyrazol-3-yl)quinolin-7-
yl]phenyl}butyramide Trifluoroacetic
Acid
87 7-(1H-Benzoimdazol-2-yl)-4-(2-pyridin-2-yl-MS ES+ 429 (M+1)
5,6-dihydro-4H-pyrrolo[ 1,2-b]pyrazol-3-
yl)quinoline Difluoroacetic Acid
88 4-(2-pyridin-2-yl-5,6-dihydro-4H-MS ES+ 391 (M+1)
pyrrolo[ 1,2-b]pyrazol-3-yl)-7-pyrimidin-5-yl-
quinoline Difluoroacetic Acid
89 5-[4-(2-pyridin-2-yl-5,6-dihydro-4H-MS ES+ 406 (M+1)
pyrrolo[ 1,2-b]pyrazol-3-yl)-7-pyrimidin-5-yl-
quinolin-7-yl]pyrimidin-2-ylamine
Difluoroacetic Acid
90 2-Chloro-4-[4-(2-pyridin-2-yl-5,6-dihydro-MS ES+ 438.1 (M+1)
4H pyrrolo[1,2-b]pyrazol-3-yl)-quinolin-7-
yl]-phenylamine Ditrifluoroacetic
Acid
91 2-Chloro-6-fluoro-4-[4-(2-pyridin-2-yl-5,6-MS ES+ 457.3 (M+1)
dihydro-4H pyrrolo[1,2-b]pyrazol-3-yl)-
quinolin-7-yl]-phenol Trifluoroacetic
Acid
92 5-[4-(2-Pyridin-2-yl-5,6-dihydro-4HMS ES+ 474.1 (M+1)
pyrrolo [ 1,2-b]pyrazol-3-yl)-quinolin-7-yl]-
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-72-
thiophene-2-sulfonic acid amide
Trifluoroacetic Acid
93 2,3-Difluoro-5-[4-(2-pyridin-2-yl-5,6-MS ES+ 441.1 (M+1)
dihydro-4H pyrrolo[1,2-b]pyrazol-3-yl)-
quinolin-7-yl]-phenol Trifluoroacetic
Acid
Example 94
6-[4-(2-Pyridin-2-yl-5,6-dihydro-4H pyrrolo[1,2-b]pyrazol-3-yl)-quinolin-7-yl]-
benzothiazol-2-yl amine Tris trifluoroacetic Acid
0
~F
F F
H2N-~'
N JO~ HO~r
HO~F F F
I'F
F
The desired product is synthesized in a manner similar to Example 73 with the
exception that 0.1 mmol of bromide reagent is used and that the workup did not
include
the use of a silica gel column prior to preparative reversed phase
chromatography to yield
21 mg (36%)of a yellow solid.
MS ES+ 461.1 (M+1 for free base)
Example 95
4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-pyrrolo[ 1,2-b]pyrazol-3-yl)-quinolin-7-
yl]-
benzenesulfonamide
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-73-
N-~S=O
O
The product from Preparation 9 (0.08 g, 0.2 mmol), Bis(pinacolato)diboron (0.1
g,0.4 mmol) and anhydrous I~OAc (0.098 g, 1 mmol) are mixed in DMSO (1 mL).
The
mixture is degassed and flushed with nitrogen several times. To the mixture is
added
PdCl2(dppf)(0.009 g, 0.012 mmol) and the degassing and purging are repeated.
The
mixture is heated under nitrogen for 8 h, allowed to cool and then 4-
bromobenzenesulfonamide (0.071 g, 0.3 mmol), 0.5 mL 2M I~2C03 and 1 mL of
dioxane
are added. The degassing and purging are repeated. To the mixture is added
palladium
(II) acetate (0.003 g, 0.012 mmol) and triphenylphosphine (0.006 g, 0.024
mmol), and the .
reaction is degassed and flushed with nitrogen for the final time. The mixture
is heated to
reflux at 115 °C for 2 h under nitrogen. The reaction mixture is
cooled, diluted with water
(2 mL) and CHC13 (10 mL). The organic layer is separated and extracted with
saturated
brine, dried (Na2SO4), filtered, evaporated to a solid mass. The crude product
is purified
by MPLC on silica gel (80% EtOAc/Hexanes, 2% MeOH) to yield the title
compound, 60
mg(64%), as an oil. MS ES+m/e 468.2 (M+1).
The following compounds are prepared utilizing the method of Example 95:
Example Product Name Physical Data
#
96 3-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-pyrrolo[1,2-MS ES+ m/e 468.2
b]pyrazol-3-yl)-quinolin-7-yl]- (M+1 )
benzenesulfonamide
97 2-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-pyrrolo[1,2-MS ES+ m/e 468.2
b]pyrazol-3-yl)-quinolin-7-yl]- (M+1 )
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-74-
~enzenesulfonamide
Scheme 6. Preparation of compounds having substituents attached to 7-position
through
N-linkage.
Scheme 6 (Step Q) depicts the modification of the 7-position of the quinoline
by
attachment of a nitrogen heterocycle, either aromatic or alkyl, to form a
carbon-nitrogen
bond to provide compounds of Formula 22. This transformation requires a base,
preferably potassium tert-butoxide, a high boiling polar solvent such as
dimethylsulfoxide, and a metal catalyst, preferably copper (0). The reaction
is conducted
at high temperatures, up to 200 °C, and following workup, the products
are purified by
reverse phase HPLC. Copper-catalyzed nucleophilic substitutions of aryl
halides have
been reviewed by Lindley (See: Lindley, J. TetJ°ahedron 1984, 40, 1433-
1456). Step Q
can also be accomplished by palladium catalysis, preferably
tris(dibenzylideneacetone)
dipalladium (0) (Pd2(dba)3), with heterocyclicamines such as piperidine,
piperazine,
morpholine, or thiomorpholine. An alkoxide base, for example, sodium text-
butoxide, as
well as 18-crown-6 and 2-(di-t-butylphosphino)biphenyl (BINAP) are also
preferred for
the transformation. The preferred solvent is tetrahydrofuran. The palladium-
catalyzed
method has been described by Wolfe and Buchwald (Wolfe, J. P.; Buchwald, S. L.
J. Df g.
Chew. 1997, 62, 6066-6068).
2 0 Scheme 6
~N -H
R1 ~ R1
step Q
Br ~ ~ CN
(19) (22)
heteroaryl or heterocycloalkyl
Example 98
7-Imidazol-1-yl-4-(2-pyridin-2-yl-5,6-dihydro-4H-pyrrolo[ 1,2-bJpyrazol-3-
yl)quinoline
2 5 Ditrifluoroacetic Acid
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
_7
~ iv v
'NJ o F
F ~OH
H F F
F F
Into a dry 25 mL glass pressure tube fitted with teflon screw cap is placed
the
product of Preparation 9 (0.0782 g, 0.2 mmol), potassium tart-butoxide (0.045
g, 0.4
mmol), DMSO (0.25 mL), and copper powder (8 mg). The mixture is heated and
stirred
in an oil bath 200 °C for 3 h. The reaction mixture is allowed to cool,
and then quenched
with 4 mL water, followed by 4 mL EtOAc. The mixture is agitated, and the
organic layer
is removed and eluted over a 1 g silica gel cartridge with MeOH containing a
drop of conc
NH40H. The solvent is removed under vacuum. Purified product 36.3 mg (37%) is
obtained by freeze drying appropriate fractions (based on MS analysis) from
reverse phase
HPLC using Waters Symmetry C18 column with a gradient of 10 to 70% B in A,
where A
is water containing 0.1 % TFA and B is CH3CN containing 0.1 % TFA.
MS ES+mle 379 (M+1)
Using the method of Example 98, the following compounds are prepared:
Example Product Name Physical Data
#
99 4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ 380 (M+1)
pyrrolo[ 1,2-b]pyrazol-3-yl)-7-[
1,2,4]triazol-
1-yl-quinoline ditrifluoroacetic
Acid
100 7-Pyrazol-1-yl-4-(2-pyridin-2-yl-5,6-dihydro-MS ES+ 379 (M+1)
4H-pyrrolo [ 1,2-b]pyrazol-3-yl)quinoline
Trifluoroacetic Acid
101 4-(2-pyridin-2-yl-5,6-dihydro-4H-MS ES+ 380 (M+1)
pyrrolo[ 1,2-b]pyrazol-3-yl-)-7-[
1,2,3 ]triazol-
1-yl-quinoline ditrifluoroacetic
Acid
Example 102
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-76-
4-[4-(2-Pyridin-2-yl-5, 6-dihydro-4H-pyrrolo [ 1,2-b]pyrazol-3-yl)-quinolin-7-
yl]-
piperazine-1-carboxylic Acid tent-Butyl Ester
~N
'~ N
'O
O
The product of Preparation 9 (0.1 g, 0.25 mmol), piperazine-1-carboxylic acid
tef°t-butyl ester (0.07g, 0.375 mmol), sodium tent-butoxide (0.072 g,
0.75 mmol) and 18-
crown-6 (0.08 g, 0.3 mmol) are dissolved in 10 mL of dry THF. The mixture is
degassed
and flushed with nitrogen several times. Tris(dibenzylideneacetone)
dipalladium (0)
(0.014 g, 0.015 mmol) and 2-(di-t-butylphosphino)biphenyl (0.009 g, 0.03 mmol)
are
added, followed by degassing and flushing with nitrogen. The mixture is
stirred at RT for
8 h under nitrogen. The crude mixture is diluted with EtOAc (20 mL) and
extracted with
saturated NaHC03. The organic phase is separated and washed with saturated
brine, dried
(NaZS04), filtered, and evaporated to a solid mass. The crude product is
purified by
MPLC on silica gel (80% EtOAc/Hexanes, 1% MeOH) to yield the title compound,
0.079
g (64%), as a yellow solid. MS ES+ m/e 497.3 (M+1 ).
Usin the method of Exam le 102, the followin com ounds are re ared:
Example Product Name Physical Data
#
103 7-Piperidin-1-yl-4-(2-pyridin-2-yl-5,6-MS ES+ m/e 396.3
dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl)-(M+1)
quinoline
104 7-(1,1-Dioxo-1~,6-thiomorph-olin-4-yl)-4-MS ES+m/e446.2
(2-pyridin-2-yl-5,6-dihydro-4H-pyrrolo[1,2-(M+1)
b]pyrazol-3-yl)-quinoline
Scheme 7. Preparation of derivatives of cross coupling products
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
_77_
Compounds of the type Formula 23 represent aryl, heteroaryl, or
heterocycloalkyl
derivatives that possess a group "X" which can be further manipulated to a
group "Y"
(Formula 24), as indicated by step R. The variety of such "X" groups may
include, but
are not limited to, carboxylic acids and alcohols. Carboxylic acids may be
converted to
carboxylic esters by any of a number of methods, including mineral acid
catalysis (H2S04
or HCl) in an alcohol solvent at temperatures from ambient to reflux. This
reaction is
well-known and appreciated in the art (see: Furniss, B. S.; Hannaford, A. J.;
Smith, P. W.
G.; Tatchell, A. R. T~ogel's Textbook of Practical Organic Chemistry (5th
ed.); Longman,
Essex, 1989; p 1077-1079). Primary amides may be prepared from the carboxylic
acids
as well, via an intermediate acid chloride that is obtained by treatment with
either oxalyl
chloride or thionyl chloride. The intermediate acid chloride is reacted with a
source of
ammonia, such as gaseous NH3 or ammonium hydroxide, to yield the amide. The
conversion of amines to amides by acylation is well known and appreciated in
the art
(Larock, R. C., Comprehensive Organic Ti°ansfornaations; VCH: New York,
1984; p
979). In addition, substituted secondary and tertiary amides are obtained from
the
carboxylic acids by reaction with a coupling reagent such as HBTU in the
presence of an
amine base such as diisopropylethylamine, in a suitable solvent, preferably
dimethylformamide, or dichloromethane, or tetrahydrofuran. The coupling
reaction of
carboxylic acids with amines in such a manner has been more generally
described by
2 0 Bodanszky (Bodanszky, M.; Bodanszky, A. The Practice of Peptide Synthesis;
Springer
Verlag: New York, 1984).
The alkylation of a phenolic "X" takes advantage of the reactivity of the
functional
group as a nucleophile through treatment with an electrophile possessing a
good leaving
group (for example, triflate, iodide, bromide, or chloride). A base such as
potassium
2 5 carbonate, sodium hydride, or an anhydrous alkoxide can be used to
deprotonate the
phenol and increase its nucleophilicity, in a suitable polar aprotic solvent,
for example
tetrahydrofuran, dimethylformamide, dioxane. These reactions are performed at
temperatures ranging from ambient to reflux.
All products can be isolated and purified by silica gel chromatography (MPLC),
3 0 reverse phase HPLC, or trituration of solid as described above.
Scheme 7
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
_78_
R1
X
Example 106
3- {4-[4-(2-pyridin-2-yl-5,6-dihydro-4H-pyrrolo [ 1,2-b]pyrazol-3-yl-)quinolin-
7-
yl]phenyl]propionic Acid Methyl Ester
O
OMe
Into a 100 mL roundbottom flask is placed 3-{4-[4-(2-pyridin-2-yl-5,6-dihydro-
4H-pyrrolo[1,2-b]pyrazol-3-yl)quinolin-7-yl]phenyl~propionic Acid (Example 30
before
reverse phase puriflcation)(160 mg, 0.35 mmol), 20 mL of MeOH, and 1.5 mL of
conc
HCl solution. The mixture is refluxed 3 h, and solvent removed under reduced
pressure.
The product is partitioned between CHZC12 (20 mL) and saturated I~ZC03
solution. The
organic layer is collected and dried (MgS04) and concentrated under vacuum.
The title
compound, 88.4 mg (54%), is crystallized from a concentrated CH2C12-Hexanes
mixture.
MS ES+ 475 (M+1)
Example 107
3- f 4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-pyrrolo[ 1,2-b]pyrazol-3-yl)quinolin-
7-
yl]phenyl}propionamide
aryl/ heteroaryl/ heterocycloalkyl
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
_79_
O
NHZ
Into a 100 mL roundbottom is placed 3-{4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-
pyrrolo[1,2-b]pyrazol-3-yl)quinolin-7-yl]phenyl]propionic Acid (Example 30
before
reverse phase purification)(170 mg, 0.37 mmol), MeOH (20 mL), and LiOH-H20
(9.7
mg, 0.41 mmol). The mixture is stirred 5 min, and the solvent is removed under
vacuum.
The product is suspended in CH3CN (20 mL) and CH2C12 (20 mL), 5 drops DMF are
added, and oxalyl chloride (0.10 mL, 1.18 mmol) is added. The mixture is
stirred 1 h,
solvent is removed under vacuum, and the residue redissolved in 15 mL of THF.
To the
solution is added concentrated NH4OH (30 mL), and the mixture is stirred 16 h.
Most of
the THF is removed under vacuum, producing a precipitate, which is collected,
washed
with water, and dried at 60 °C under vacuum to give the title compound,
74 mg (44%).
MS ES+ 460 (M+1)
Example 108
Dimethyl-(2- f 4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-
yl)quinolin-7-
yl]phenoxy] ethyl)amine Ditrifluoroacetic Acid
iN~O
F~OH F~OH
F F F F
Into a glass pressure tube fitted with Teflon screw cap and magnetic bar is
placed
4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-pyrrolo[ 1,2-b]pyrazol-3-yl)quinolin-7-
yl]phenol
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-80-
(Example 7, before reverse phase HPLC), (0.121 g, 0.3 mmol), I~2CO3 (0.124 g,
0.9
mmol), 2-dimethyaminoethyl chloride hydrochloride (0.048 g, 0.33 mmol), DMF (2
mL),
acetone (2 mL), dioxane (2 mL), and Et3N (2 mL). The cap is positioned, and
mixture
heated in oil bath at 60 °C for 16 h. Most of the solvent is removed
under vacuum, and the
residue is triturated with 15 mL of saturated I~2C03 solution, giving 127 mg
of the crude
titled compound as a free base. The purified product 169 mg, (80°/~) is
obtained by freeze
drying appropriate fractions (based on MS analysis) from reverse phase HPLC
using
Waters Symmetry C18 column with a gradient of 10 to 70% B in A, where A is
water
containing 0.1 % TFA and B is CH3CN containing 0.1 % TFA.
MS ES+ m/e 476 (M+1)
Using the method of Example 108, considering that in some preparations no
reverse
phase purification is necessary and in some instances the halogen reagent
required no
Et3N for forming the free base, the following compounds are synthesized:
Example Product Name Physical Data
#
109 7-[4-(2-Morpholin-4-yl-ethoxy)phenyl]-4-(2-MS ES+ 518 (M+1)
Pyridin-2-yl-5, 6-dihydro-4H-pyrrolo
[ 1,2-
b]pyrazol-3-yl)quinoline Ditrifluoroacetic
Acid
110 7-[4-(2-Piperidin-1-yl-ethoxy)phenyl]-4-(2-MS ES+ 516 (M+1)
Pyridin-2-yl-5,6-dihydro-4H-pyrrolo[
1,2-
b]pyrazol-3-yl)quinoline Ditrifluoroacetic
Acid
111 4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ 502 (M+1)
pyrrolo[ 1,2-b]pyrazol-3-yl)-7-[4-(2-
pyrrolidin-1-yl-ethoxy)phenyl]
quinoline
Ditrifluoroacetic Acid
112 7-[4-(3-Piperdin-1-yl-propoxy)phenyl]-4-(2-MS ES+ 530 (M+1)
Pyridin-2-yl-5, 6-dihydro-4H-pyrrolo
[ 1, 2-
b]pyrazol-3-yl)quinoline Ditrifluoroacetic
Acid
113 Dimethyl-(3-{4-[4-(2-Pyridin-2-yl-5,6-MS ES+ 490 (M+1)
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-81-
dihydro-4H-pyrrolo [ 1,2-b]pyrazol-3-
yl)quinolin-7-yl]phenoxy}propyl)amine
Ditrifluoroacetic Acid
114 7-[4-(3-Morpholin-4-yl-propoxy)phenyl]-4-MS ES+ 532 (M+1)
(2-Pyridin-2-yl-5,6-dihydro-4H-pyrrolo
[ 1,2-
b]pyrazol-3-yl)quinoline Ditrifluoroacetic
Acid
115 4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ 516 (M+1)
pyrrolo[1,2-b]pyrazol-3-yl)-7-[4-(3-
pyrrolidin-1-yl-propoxy)phenyl]
quinoline
Ditrifluoroacetic Acid
117 2-{4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ 462 (M+1)
pyrrolo[ 1,2-b]pyrazol-3-yl)quinolin-7-
yl]phenoxy} acetamide Trifluoroacetic
Acid
118 7-[4-(2-Methoxyethoxy)phenyl-4-(2-Pyridin-MS ES+ 463 (M+1)
2-yl-5, 6-dihydro-4H-pyrrolo
[ 1,2-b]pyrazol-3-
yl)quinoline Trifluoroacetic
Acid
119 7-[4-(2-Fluoroethoxy)phenyl]-4-(2-pyridin-2-MS ES+ 451 (M+1)
yl-5,6-dihydro-4H-pyrrolo[ 1,2-b]pyrazol-3-
yl)quinoline Trifluoroacetic
Acid
Example 120
4-(2-Pyridin-2-yl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl)-7-[4-(2,2,2-
trifluoroethoxy)phenyl)quinoline Trifluoroacetic Acid
F
F~O F
F ~OH
FF
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
_82_
Into a 25 mL glass pressure tube fitted with Teflon screw cap and magnetic bar
is
placed 4-[4-(2-pyridin-2-yl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl)quinolin-
7-
yl]phenol (Example 7 before reverse phase HPLC purification); 0.100 g, 0.25
mmol),
DMF (2 mL), trifluoro-methanesufonic acid 2,2,2-trifluoroethyl ester (0.064 g
0.275
mmol), and 60% NaH (11 mg, 0.275 mmol). The cap is positioned, and mixture
heated in
oil bath at 70 °C for 16 h. The reaction is quenched with 15 mL water,
giving 67 mg of
precipitate as the title compound as a free base. The purified product, 36.4
mg (24%), is
obtained by freeze drying appropriate fractions (based on MS analysis) from
reverse phase
HPLC using Waters Symmetry C18 column with a gradient of 10 to 70% B in A,
where A
is water containing 0.1 % TFA and B is CH3CN containing 0.1 % TFA.
MS ES+ 487 (M+1)
Example 121
(2-~4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-pyrrolo[ 1,2-b]pyrazol-3-yl)-quinolin-
7-yl]-
benzoylamino~-ethyl)-carbamic Acid test-Butyl Ester
O~H
~--N
O
4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-pyrrolo[ 1,2-b]pyrazol-3-yl)-quinolin-7-
yl]-benzoic
acid (Example 41 before reverse phase chromatography, 0.08 g, 0.18 mmol), (2-
aminoethyl)-carbamic acid tef~t-butyl ester (0.036 g, 0.2 mmol), N-N-
2 0 Diisopropylethylamine (0.08 mL, 0.46 mmol) and HBTU (0.076 g, 0.2 mmol)
are mixed
in 10 mL of DMF and stirred at RT for 8 h under nitrogen. The crude mixture is
diluted
with EtOAc (30 mL) and extracted with saturated NaHC03. The organic phase is
separated and washed with saturated brine, dried (Na2S04), filtered,
evaporated to a solid
mass. The crude product is purified by MPLC on silica gel (80% EtOAc/Hexanes,
1%
2 5 MeOH) to yield the title compound, 0.09 g (56%), as white solid.
H 0
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-83-
MS ES+ m/e 575.3 (M+1).
Using the method of Example 121, the following compounds are synthesized:
Example Product Name Physical Data
#
122 Piperidin-1-yl-{4-[4-(2-pyridin-2-yl-5,6-dihydro-MS ES+ m/e 500.3
4H-pyrrolo[1,2-b]pyrazol-3-yl)-quinolin-7-yl]-(M+1)
phenyl}-methanone
123 {4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ m/e 518.2
pyrrolo[1,2-b]pyrazol-3-yl)-quinolin-7-yl]-(M+1)
phenyl } -thiomorpholin-4-yl-methanone
124 Morpholin-4-yl-{4-[4-(2-pyridin-2-yl-5,6-MS ES+ m/e 502.3
dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl)-quinolin-(M+1)
7-yl]-phenyl } -methanone
125 N Carbamoylmethyl-4-[4-(2-pyridin-2-yl-5,6-MS ES+ m/e 489.2
dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl)-quinolin-(M+1)
7-yl]-benzamide
126 N (2-Isopropylamino-ethyl)-4-[4-(2-pyridin-2-yl-MS ES+ mle 517.3
5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl)-(M+1)
quinolin-7-yl]-benzamide
127 (rac) N (2-tert-Butylsulfanyl-ethyl)-4-[4-(2-MS ES+ m/e 548.3
pyridin-2-yl-5,6-dihydro-4H-pyrrolo[1,2-(M+1)
b]pyrazol-3. yl)-quinolin-7-yl]-benzamide
128 3-{4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ m/e 532.3
pyrrolo[1,2-b]pyrazol-3-yl)-quinolin-7-yl]-(M+1)
benzoylamino } -
propionic Acid Ethyl Ester
129 (racy-N-(1,1-Dioxo-tetrahydro-1~,6-thiophen-3-MS ES+ m/e 550.2
yl)-4-[4-(2-pyridin-2-yl-5,6-dihydro-4H-(M+1)
pyrrolo [ 1,2-b]pyrazol-3-yl)-quinolin-7-yl]-
benzamide
130 3-{4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ m/e 615.3
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-84-
pyrrolo[1,2-b]pyrazol-3-yl)-quinolin-7-yl]-(M+1)
benzoylamino } -
piperidine-1-carboxylic Acid tart-Butyl
Ester
131 6-tent-Butoxycarbonylamino-2(S)- MS ES+ m/e 717.4
f 4-[4-(2-
pyridin-2-yl-5,6-dihydro-4H-pyrrolo[1,2-(M+1)
b]pyrazol-3-yl)-
quinolin-7-yl]-benzoylamino}-hexanoic
acid tert-
butyl ester
132 N (2-Morpholin-4-yl-ethyl)-4-[4-(2-pyridin-2-yl-MS ES+ mle 545.3
5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl)-(M+1)
quinolin-7-yl]-benzamide
133 (1,1-Dioxo-1~,6-thiomorpholin-4-yl)-~4-[4-(2-MS ES+ m/e 550.2
pyridin-2-yl-5,6-dihydro-4H-pyrrolo[1,2-(M+1)
b]pyrazol-3-yl)-quinolin-7-yl]-phenyl
} -
methanone
134 3-Methylsulfanyl-2(S)-{4-[4-(2-pyridin-2-yl-5,6-MS ES+ m/e 606.3
dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl)-quinolin-(M+1)
7-yl]-benzoylamino}-propionic Acid
tart-Butyl
Ester
135 2(S)-{4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ mfe 674.3
pyrrolo[1,2-b]pyrazol-3-yl)-quinolin-7-yl]-(M+1)
benzoylamino}-pentanedioic Acid
Di-teft-Butyl
Ester
136 N,N Dimethyl-4-[4-(2-pyridin-2-yl-5,6-dihydro-MS ES+ m/e 460.2
4H-pyrrolo[1,2-b]pyrazol-3-yl)-quinolin-7-yl]-(M+1)
benzamide
137 3- f 4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ m/e 560.2
pyrrolo[1,2-b]pyrazol-3-yl)-quinolin-7-yl]-(M+1)
benzoylamino}-propionic Acid tent-Butyl
Ester
138 N-Methyl-4-[4-(2-pyridin-2-yl-5,6-dihydro-4H-MS ES+ m/e 446.1
pyrrolo[1,2-b]pyrazol-3-yl)-quinolin-7-yl]-(M+1)
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-85-
benzamide
139 N-(2-Methanesulfonyl-ethyl)-4-[4-(2-pyridin-2-MS ES+ m/e 538.4
yl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl)-(M+1)
quinolin-7-yl]-benzamide
Example 140
N-Dimethylaminomethylene-4-[4-(2-pyridin-2-yl-5,6-dihydro-4H-pyrrolo[1,2-
b]pyrazol-
3-yl)-quinolin-7-yl]-benzamide
~N~N
0
To a solution of 4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-
yl)-
quinolin-7-yl]-benzoic acid (Example 41 before reverse phase chromatography,
0.05 g,
0.12 mmol) in 5 mL of CHZC12, 0.5 mL of DMF is added and the mixture is cooled
to 0
°C. Oxalyl chloride (0.11 mL, 1.2 mmol) is added dropwise to the
mixture while
maintaining the temperature at 0 °C. The reaction is stirred at RT for
8 h under nitrogen
and evaporated to an oil. The crude is dissolved in 10 mL of CH2C12 and
saturated with
dry NH3 gas. The mixture is then stirred at RT for 2 h under nitrogen and
evaporated to a
solid mass. The crude product is purified by MPLC on silica gel (80%
EtOAc/Hexanes,
1 % MeOH) to yield the title compound, 0.02 g (39%), as white solid. MS ES+
m/e 487.2
(M+1 ).
Example 141
4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl)-quinolin-7-yl]-
benzamide
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-86-
HZN
O
To a solution of 4-[4-(2-pyridin-2-yl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-
yl)-
quinolin-7-yl]-benzoic acid (Example 41 before reverse phase chromatography,
0.05 g,
0.12 mmol) in 5 mL of CH2C12, thionyl chloride (0.1 mL, 1.2 mmol) is added
dropwise
while maintaining the temperature at 0 °C. The reaction is stirred at
RT for 8 h under
nitrogen and evaporated to an oil. The crude product is dissolved in 10 mL of
CHZCl2 and
saturated with dry NH3 gas. The reaction is then stirred at ambient
temperature for 2 h
under nitrogen and evaporated to a solid mass. The crude product is
crystallized from
MeOH/Et20 to yield the title compound, 0.045 g (87%), as white solid. MS ES+
m/e
432.1 (M+1).
Example 142
N (2-Aminoethyl)-4-[4-(2-pyridin-2-yl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-
yl)-
quinolin-7-yl]-benzamide, Ditrifluoroacetic Acid Salt
O
F
HO~F
F
F
I,F
H2N H O
To a solution of (2- f 4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-pyrrolo[1,2-
b]pyrazol-3-
yl)-quinolin-7-yl]-benzoylamino J -ethyl)-carbamic acid tent-butyl ester
(Example 121,
0.03 g, 0.05 mmol) in 2 mL of CHZCl2 is added 2 mL of trifluoroacetic acid.
The reaction
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
_87_
mixture is stirred at RT for 30 min, evaporated to an oil and titurated with
Et20 to yield
the title compound, 0.02 g (70%), as white solid. MS ES+m/e 475.3 (M+1).
Using the method of Example 142, the following compounds are prepared:
Example Product Name Physical Data
#
143 6-Amino-2(S)- f 4-[4-(2-pyridin-2-yl-5,6-dihydro-MS ES+ m/e 561.3
4H-pyrrolo[1,2-b]pyrazol-3-yl)-quinolin-7-yl]-(M+1)
benzoylamino}-hexanoic acid, Ditrifluoroacetic
Acid Salt
145 3- Methylsulfanyl-2(S)-{4-[4-(2-pyridin-2-yl-5,6-MS ES+ mle 550.3
dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl)-quinolin-(M+1)
7-yl]-benzoylamino}-propionic Acid
Trifluoroacetic Acid Salt
146 2(S)-{4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ m/e 562.1
pyrrolo[1,2-b]pyrazol-3-yl)-quinolin-7-yl]-(M+1)
benzoylamino } -
pentanedioic Acid Trifluoroacetic
Acid Salt
147 3- f 4-[4-(2-Pyridin-2-yl-5,6-dihydro-4H-MS ES+ m/e
pyrrolo[1,2-b]pyrazol-3-yl)-quinolin-7-yl]-504.2(M+1)
benzoylamino } -
propionic Acid Trifluoroacetic Acid
Salt
References
1. Smith, D. L.; McCloskey J. A.; J. Org. Chenz. 43, 2087-2088,1978
2. Price, C. C.; and Roberts, R. M., J. Am. Chem. Soc. 68, 1204 (1946);
Surrey, A. R.;.
Hammer, H. F., J. Am. Chem. Soc., 68,113 (1946)
Determination of Biological Activity
To demonstrate that compounds of the present invention have affinity for MLI~-
7
and the capacity to modulate kinase activity, in vitro and whole cell kinase
assays are
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
_88_
performed. All ligands, radioligands, solvents, and reagents employed in the
binding
assays are readily available from commercial sources, or can be readily
synthesized by the
ordinarily skilled artisan.
In vitro Assay of Purified Mixed Lineage Kinase 7 (MLK-7) Activity
Briefly, recombinant glutathione-s-transferase (GST) tagged MLK-7 is expressed
in Sf~ cells using a recombinant baculovirus created using baculovirus
transfer vector
pVL1392 modified to contain a GST and histidine tag. The recombinant protein
is
purified over a glutathione column and subsequently used for in vitro kinase
assays with
myelin basic protein (MBP) as the substrate for phosphorylation. The in vitro
kinase
reaction is run with recombinant MLK-7 at 1 nm and various concentrations of
test
compound in a reaction mixture containing 2 mM DTT, 30 uM ATP, 5 mM MgCl2, 5
uM
MBP, 4% DMSO, 5 uCi 33P in 44 mM HEPES,(pH 7.4). The reaction is carried out
for 2
hours at room temperature and then stopped by the addition of phosphoric acid
to 5.5%.
The phosphorylated MBP is collected on a filter and radioactivity associated
with the
membrane measured using a scintillation counter. Inhibitors are identified by
the ability
to reduce the transfer of radioactive phosphate from ATP to MBP compared to
controls
run without test compound.
IC50 values, defined as the concentration of test compound required to
decrease
the transfer of radioactive phosphate from ATP to MBP by 50%, are then
determined. Ki
2 0 values (which refers to the dissociation constant of an enzyme-antagonist
complex and
serves as an index of ligand binding) for each respective test compound can
also be
calculated by application of the Cheng-Prusoff equation as described in Cheng
et al.,
Relationship Between The Inhibition Constant (Ki) and The Concentration of
Inhibitor
Which Causes 50% Inhibition (IC50) of an Enzymatic Reaction, Biochem.
Pharmacol.,
2 5 22: 3099-31088; (1973).
Representative compounds of the present invention have an IC50 in the MLK-7
kinase assay of _< 10,000nM Table I (see infra.) provides IC50 data from the
afore
mentioned MLK-7 kinase assay for a representative sample of the exemplified
compounds of the present invention.
3 0 Whole Cell Assays of MLK-7 Activity
Mammalian expression vectors containing either MLK-7 or JNK cDNA are co-
transfected into Cos cells DMEM supplemented with 5% fetal calf serum and 0.1
mg.ml
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-89-
ampicillin. The cDNA is expressed for 24 hours at which time media is
aspirated and
replaced with media containing the selected concentration of test compound.
After 5
hours of incubation, cells are lysed and analysed for phospho-JNK (pJNK)
levels using
the pJNK luminex assay according to the manufacturers instruction (BioRad).
MLK-7
inhibitors are evaluated based on the ability to reduce pJNK levels in the Cos
cells
compared to control samples incubated in the absence of test compound.
Test compounds may also be evaluated for the ability to inhibit phospho-p38
formation in cardiac myocytes. Primary cardiac myocytes are collected by
trypsin
digestion of neo-natal rat heart tissue. The cells incubated in Dulbecco's
modified Eagles'
medium DMEM/F 12 ( 1:1 v/v) supplemented with 2 g/L bovine serum albumin, 3 mM
MEM sodium pyruvic acid, 15 mM HEPES, 100~g/mL ampicillin, 1 ~g/mL
transferrin,
10 ng/mL sodium selenite, and 1 ~g/mL insulin are infected with recombinant
adenovirus
expressing MLK-7. After 48 hours of expression, the media is collected and
replaced
with media containing a selected concentration of test compound and incubation
is
continued for 5 hours. Media is removed, cells are lysed and analysed for
phospho-p38
levels using the phospho-p38 luminex assay according to the manufacuters
instruction
(BioRad). MLK-7 inhibitors are evaluated based on the ability to reduce
phospho-p38
levels in cardiac myocytes compared to control samples incubated in the
absence of test
compound.
2 0 In vitro and cell based kinase assay protocols for MLK-7, similar to those
described above, can be readily designed by the ordinarily skilled artisan.
Bloem et al., J.
Mol. Cell Cardiol.; 33: 1739-1750, (2001) provides a detailed description of
the amino
acid sequence of MLK-7 and various in vitro and whole cell assays of MLK-7
activity as
well as a description of functional assays of gene expression and protein
synthesis that
2 5 may be used to asses the ability of the present compounds to treat CHF. In
addition,
United States Patent No. 6,146,832 provides a detailed description of the
isolation of the
cDNA encoding MLK-7 (designated therein as CSAPK 2), expression of recombinant
MLK-7 (CSAPK2) protein in bacterial cells and expression of recombinant MLK-7.
3 0 Table I
MLK-7 Binding Assay Values
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-90-
Example No. MLK7 IC50
.
145 +~-+
146 +++
147 +++
59 +++
60 +++'
63 +++
61 +++
62 +++
64 ++
20 +++
21 ++
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-91-
11 +++
12 +++
25 ++
27 +++
14 +++
16 ' +++
19 +++,.'
1$ +++
49 +++
23 +++
31 ++
9 +++
2 +++
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-92-
4 +++
8 ++
7 +++
+++ .
34 +++
22 ++
26 +++.,.
++
35 +++
32 ++
24 ++
6 +++
5 ++
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-93-
33 +++
29
30 +++
3 +++
28 +++
17 +
21 ++:
36 ++
37 +++
38 +++
52 +++
53 +
47 ++
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-94-
40 +++
48 +++
65 +++'
41 +++
42
50 +++
69 +++
54 ++
70 ~+:++
71 +++
66 +++
68 +++
43 +++
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-95-
44
99 ++
100 +++
98 ++
102 ;+
103 ++
55 ++
51 +++
89 ++
67 ++
88 ++
101 ++
45 ++
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-96-
85 ++
75 '+++-
84 +++
95 +++
96 , +++
117 +++
118 +++
119 +++
120 +++
121 _ +++
138 +++
122 +++
56 +++
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-97-
123 . +++
97 ++'
124 +++
125 '+++ .
126 +++
127 ++
123 +++
139 +++
129 +++
140 +++
130 +++
106 +++
107 +++
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-98-
104 +
108 +++
109 +++
110 +++:
141 +++
46 +++:.
113 +++
111 +++
112 +++
131 ++
132 +++
133 +++
86 +++
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-99-
74 +++
73 +++
76 +++ -
77 +++
87 +++
114 +++
115 :+++
82 +++ , ,
78 +++
79 +
80 + .
83 +
81 . +++
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-100-
134 ++
57 +++ -
143 +++
58 +++
94 +++
90 +++
91 +++
92 +++
136 +++
137 +++
135 ++
93 +++
CA 02506799 2005-05-19
WO 2004/048383 PCT/US2003/035036
-101-
Legend:
"+" represents a value of <_ 1 O,OOOnM
"++" represents a value of <_ 1,000nM
"+++" represents a value of <_ 100nM
"--" indicates the value was not determined