Note: Descriptions are shown in the official language in which they were submitted.
CA 02507117 2005-05-11
1
TITLE OF THE INVENTION
[0001] COMPLETE CHEMICAL AND ENZYMATIC TREATMENT OF
PHOSPHORYLATED AND GLYCOSYLATED PROTEINS ON PROTEIN CHIP
ARRAYS
FIELD OF THE INVENTION
[0002] The present invention relates to proteomics. More particularly,
the present invention relates to protein chip arrays. More specifically, the
present
invention is concerned with methods of chemical and enzymatic treatment of
proteins on protein chips. More particularly, the invention relates to
chemical and
enzymatic treatment of post translationally modified proteins on proteins chip
arrays.
BACKGROUND OF THE INVENTION
[0003] Many advances in proteomics have been driven by the
development of mass spectrometric-based technologies and tools '. Although
mass spectrometry (MS) was invented in the early 1900s for the detection of
small
molecules, a quantum leap was achieved in the late 1980s when Fenn and Tanaka
showed independently that large biomolecules (proteins, DNA, etc) can be
detected and quantitated accurately by MS. Fenn's technique called Electro
Spray
Ionization (ESI) nebulizes a protonated liquid into a fine spray using a high
voltage
prior to MS detection (J. B. Fenn, M. Mann, C. K. Meng, S.F. Wong, C.M.
Whitehouse, Science 246, 6, 64 (1989)). Tanaka's method called Matrix Assisted
Laser Desorption Ionization (MALDI) utilizes a high energy absorbing molecule
to
desorb intact proteins on a solid inert surface (K. H. Tanaka, H. Wake, Y.
Ido, S.
Akita, Y. Yoshida and I. Yoshida, Rapid Commun. Mass Spectrom. 8, 2, (1988)).
A
flavour of this latter technique, called Surface Enhanced Laser Desorption
Ionization (SELDI) permits the immobilization of molecules on different active
surfaces. SELDI is described in U.S. Pat. No. 5,719,060 ("Method and Apparatus
CA 02507117 2005-05-11
2
for Desorption and Ionization of Analytes," Hutchens and Yip, Feb. 17, 1998);
U.S.
Pat. No. 6,225,047 ("Use of Retentate Chromatography to Generate Difference
Maps," Hutchens and Yip, May 1, 2001); and in Weinberger et al., "Time-of-
flight
mass spectrometry," in Encyclopedia of Analytical Chemistry, R. A. Meyers,
ed.,
pp 11915-11918 John Wiley & Sons Chichesher, 2000.
[0004] ~ A number of reports have appeared over the past several years
regarding proteomic profiling with SELDI-TOF technology, in combination with
artificial intelligence. Reported sensitivities and specificities with the
technique for
ovarian, prostate, and breast cancer diagnosis are better than those obtained
with
current serologic cancer biomarkers. Also, the technique is reported to detect
early
as well as late stage disease with similar efficiency, thus offering a
potentially
powerful new cancer screening tool.
[0005] The combination of techniques such as polyacrylamide gel
electrophoresis (PAGE)2~3, reverse phase high performance liquid
chromatography
(RP-HPLC)4~, affinity capture'~8 and protein chips 9 with mass spectrometry
(MS)
has provided a series of important tools for the investigation of numerous
facets of
proteomics. The identification and characterization of the chemical features
of
proteins are essential prerequisites for understanding the dynamics and
connectivity of their interactions as well as the diversity of their
biological functions
in living organisms. As a common method, peptide mass fingerprinting (PMF)
identifies proteins by comparing the peptide mass fingerprint obtained from
mass
spectrometry analysis of enzymatic (or chemical) digestions to mass profiles
generated by in-silico digestion of proteins'°. This approach requires
relatively
purified target protein and is often used with protein fractionation
techniques. Prior
to enzymatic digestion, proteins are denatured, reduced and alkylated.
Digestion is
generally performed overnight to ensure complete cleavage. Structural
characterization of proteins becomes all the more difficult if one considers
that the
vast majority of proteins contain disulfide bridges, phosphorylation,
glycosylation
CA 02507117 2005-05-11
3
sites or a combination of the above.
[0006] Thus, to study biological systems at the protein level, efforts
have been directed to the improvements in instrumentation and the development
of novel technologies.
[OOOTj Protein chip array technology is based on two powerful
techniques: chromatography and mass spectroscopy. It consists of selective
protein extraction, retention and enrichment of proteins on chromatographic
chip
surfaces and their subsequent analysis by mass spectroscopy. The protein chip
array surfaces function as a solid phase extraction media that support
isolation and
clean up of analytes prior to mass spectroscopic investigation.
[0008] By comparing samples between control and experimental
groups or between healthy and diseased individuals, in one use of the
technology,
protein chip array profiling allows the rapid creation of phenotypic
fingerprints and
the identification of biomarkers of particular metabolic or disease states.
[0009] Thus, together with the growth of this technology comes the
need for protein chemistry techniques that are applicable to protein chips.
Three
groups have reported a single on-chip reaction prior to MS analysis.
Pentafluoropropionic acid and trifluoroacetic acid (TFA) were used to perform
limited acid hydrolysis of proteins using a vapor-phase hydrolysis procedure".
The
method was proposed to generate peptide ladders indicating primary sequences.
However, side reactions, such as oxidation of methionine residues and
deamidation of asparagine or glutamine, were systematically observed. A second
group reported a procedure for the identification of parvalbumin alpha (PVA)
using
on-chip enzymatic proteolysis'2. Four peptides were identified after a 2-hour
digestion and nine peptides were identified after 18 hours. PVA is an 11.85
kilodalton (kDa) linear N-terminus acetylated polypeptide, which is not
CA 02507117 2005-05-11
4
representative of most of the proteins in existing proteomes as it lacks
complex
modifications such as disulfide bridges, phosphorylated or glycosylated
moieties.
Finally, an on-chip tryptic digestion method has been applied to recombinant
prolactin-inducible protein (PIP). This purified 16.57 kDa protein has two
disulfide
bridges and one N-glycosylation site'3.
[0010] In all the above examples, all chemical and enzymatic steps
were carried out in solution. Relatively simple proteins were tested, and in
all
cases, a single on-chip step of treatment was performed. On-chip protein
denaturation, reduction, alkylation, deglycosylation and dephosphorylation
using
protein chips have not been previously reported. In addition, the reports have
generally been based on rather simple proteins.
[0011] Thus, there remains a need for improved methods allowing
structural characterization of proteins.
[0012] There further remains a need for methods of protein
identification, which reduce sample loss, enable rapid and sensitive detection
and
identification of proteins with minimal sample manipulation.
[0013] There also remains a need for simple methods allowing
complete on-chip chemistry (including enzymatic treatment) and
characterization of
proteins.
[0014] The present invention seeks to meet these needs and other
needs.
CA 02507117 2005-05-11
SUMMARY OF THE INVENTION
[0015] Although solid phase chemistry (e.g. Edman degradation) has
been routinely performed on solid support for years, it is difficult to
imagine
complex biochemical reactions on solid surfaces partly because the enzymes
must
5 retain their activities throughout the process, and also because of limited
bioavailability. For example, in enzymatic digestion, the reactants seem
unlikely to
interact effectively to cleave highly complex proteins. It is analogous to
putting
liquid in sand in a first step which is followed by an addition of a different
liquid and
expecting proper mixing. When a protein is denatured, it is in its most
relaxed state
and more prone to interact with other species. A solid small surface is not a
predictable environment for that interaction. Reactions in solution have been
carried out for centuries and are fully understood (access of water,
configuration of
the protein in solution, etc.). However, biochemical reactions on solid
surfaces
have been very poorly exploited because of their complexity and also because
they seem not likely to occur. For example, the environment of the protein on
a
chip is very different from that in solution. The water environment is but one
critical
difference between the proteins on a chip as compared to that in solution. The
relatively dry state of a protein on a chip suggests that enzymatic digestion
is likely
not to occur on a chip.
[0016] The rapid growth of proteomics and more particularly protein
array technology urged the development of simpler, more sensitive
methodologies.
Microfabricated devices are becoming increasingly popular for the analysis of
biomolecules (deoxynucleic acid (DNA), deoxyribonucleic acid (RNA), proteins,
peptides) for a number of reasons. These devices come in two varieties, the
array
format and microfluidic devices. They offer the potential to automate
biological
sample processing (reduction, alkylation, chemical and enzymatic digestion,
desalting, etc.) reduce costs and increase throughput. In addition, they are
designed with minimal quantities of sample in mind. When only tiny amounts of
CA 02507117 2005-05-11
6
sample are available, macroscale techniques become ineffective due to sample
losses.
[0017] Researchers around the world have attached great importance
to protein chip technology because it could theoretically simultaneously
analyze
information of many biomolecules in one reaction. However, the development and
applications of this technology is still limited by its complexity.
[0018] The present invention, demonstrates that surprisingly several
complex enzymatic and chemical reactions can indeed be performed directly on
protein chip surfaces in a sequential fashion.
[0019] Thus, the present invention relates to the use of protein chip
methods for performing various enzymatic, other biological and chemical
reactions.
This approach employs chips with different surface physicochemical properties
enabling the selective capture and retention of proteins or peptides from
biological
samples.
[0020] In one aspect, the present invention relates to protein chemistry
procedures that can be performed directly on-chip using small volumes (in the
p1
range) of the biological sample of interest, reagents and washing solutions,
as well
as relatively short reaction time for both chemical and enzymatic treatments
prior
to MS analysis.
[0021] More specifically, the present invention is concerned with a
quick, simple and sensitive method allowing two or more, and up to all
chemical
reactions to be performed on-chip as well as subsequent enzymatic
deglycosylation, dephosphorylation and proteolysis in a sequential fashion.
The
methods of the present invention provide a rapid and simple alternative to in-
gel or
in-solution methods.
CA 02507117 2005-05-11
7
[0022) Thus, the present invention is concerned with novel
experimental methods to analyze peptidelproteins by protein chip array
technology. These methods enable the rapid deglycosylation, dephosphorylation,
digestion and identification of low amounts (in the picomolar range) of
complex
proteins. Because all steps may be performed directly on chip, the method of
the
present invention is easily amenable to automation. Consequently, the method
of
the present invention may be developed for low-throughput, high-throughput, or
ultra-high throughput analysis formats.
[0023) In one aspect, the method of the present invention generally
comprises a number of the following steps:
a) conditioning of the spots of the protein chip array with conditioning
buffer;
b) loading of the biological sample on the protein chip; after binding,
excess sample is removed and each spot is washed with appropriate buffer;
c) denaturing the protein sample;
d) reducing the protein sample;
e) alkylating the protein sample;
f) deglycosylating and/or dephosphorylating the sample;
g) chemical or enzymatic digestion (hydrolysis of peptide bonds) for
PMF;
h) performing MS analysis (drying of the sample, matrix (energy
absorbing molecule-EAM) addition and data collection); and
i) database mining and identification of proteins.
[0024) In accordance with the present invention only some steps of the
above general method may be performed depending on the type of information
that is sought and the type of protein sample that is used. For example, if
CA 02507117 2005-05-11
8
information is only sought on the phosphorylation status of the protein, then,
the
deglycosylation and chemicallenzymatic digestion steps may not be performed.
Alternatively, if only the glycosylation level of a protein needs to be
studied then,
the dephosphorylation and chemical/enzymatic digestion steps would not be
performed. On the other hand if one is working with relatively simple proteins
or
peptides, then the dephosphorylation and deglycosylation step may not be
required. Thus, depending on the particular experimental requirements, a
person
skilled in the art would choose which of the above steps are to be performed
and
adapt the method accordingly.
[0025] Thus, in one embodiment, the method of the present invention
comprises a conditioning step; a biological sample loading step, a denaturing
step,
a reducing step, an alkylating step, a deglycosylation step, an enzymatic
digestion
step (PMF) and an MS analysis step.
[0026] In another embodiment, the method of the present invention
comprises a conditioning step; a biological sample loading step, a denaturing
step,
a reducing step, an alkylating step, a dephosphorylation step, an enzymatic
digestion step (PMF) and an MS analysis step.
[0027] In a further embodiment, the method of the present invention
comprises ~a conditioning step, a biological sample loading step, a denaturing
step,
a reducing step, an alkylating step, a deglycosylation step, a
dephosphorylatian
step and an MS analysis step.
[0028] In yet another embodiment, the method of the present invention
comprises a conditioning step, a biological sample loading step, a denaturing
step,
a reducing step, an alkylating step, a deglycosylation and/or
dephosphorylation
step and a MS analysis step.
CA 02507117 2005-05-11
9
[0029] In another additional embodiment, the method of the present
invention comprises a conditioning step, a biological sample loading step, a
denaturing step, a reducing step, an enzymatic digestion step (PMF) and an MS
analysis step.
[0030] In yet a further embodiment, the enzymatic digestion step is
replaced by a chemical digestion step (e.g. acid hydrolysis step).
[0031] In one embodiment, the deglycosylation step is performed prior
to the dephosphorylation step. In another embodiment the dephosphorylation
step
is performed before the deglycosylation step.
[0032] When performing protein characterization spectrometry
analysis, it is often desirable to cleave proteins directly on the chip into
smaller
fragments (peptides) using cleaving reagents for either chemical or enzymatic
cleavage. As well known in the art, the digestion of proteins into small
fragments
provides a mass fingerprint that can be used to determine the protein identity
and
other characteristics such as posttranslational chemical modifications to
specific
residues. Thus, the specific fragments that result from digestion can be used
as a
fingerprint for protein identification by a technique known as peptide mass
fingerprinting (PMF). Also, proteolytic fragmentation is useful for high
molecular
weight proteins because smaller fragments are often more easily measured and
resolved by mass spectrometry and chemical modifications can be isolated to
specific peptide regions of a protein.
[0033] Thus, in one aspect of the present invention, the enzymatic
and/or chemical cleavage of proteins/peptides present in a sample is performed
directly on the chip. Subsequent MS analysis is performed in order to obtain a
fingerprint of the protein/peptides and determine their identity.
CA 02507117 2005-05-11
[0034) In accordance with the present invention, several enzymes
having different specificity (i.e. cleaving after specific amino acid
residues) can be
used for PMF and subsequent identification of proteins fragment by MS
analysis.
Proteases, such as trypsin, that cleave proteins into a discrete number of
5 predictable fragments are particularly useful. Other non-limiting examples
of
enzymes that may be used for direct on-chip digestion include, V8-protease,
Arg-C
proteinase, Asp-N endopeptidase, Glu-C endoproteinase, Lys-C endopeptidase,
chymotrypsin, pepsin, aminopeptidase M, carboxypeptidase-A, carboxypeptidase-
B, carboxypeptidase-Y, caspases 1-10, clostropain (Clostridiopeptidase B),
10 elastase, enterokinase, factor Xa, glutamyl endopeptidase, granzymeB,
papain,
proline-endopeptidase, pronase, proteinase K, staphylococcal peptidase I,
thermolysin, and thrombin.
[0035) As an alternative or complementary approach to enzymatic
cleavage for PMF, direct, on-chip chemical cleavage may also be used in
accordance with the present invention. Non-limiting examples of compatible
reagents that can be used include 2-(2-nitrophenylsulfenyl)-3-bromo-
methylindolenine (BNPS-Skatole), Cyanogen Bromide (CNBr),
CNBrlheptafluorobutyric acid, Dimethylsulfoxide (DMSO)/HCI and
DMSO/Hydrogen bromide (HBr), DMSO/HCI and CNBr, formic acid,
hydroxylamine, iodosobenzoic acid, N-bromosuccinimide, N-chlorosuccinimide, 2-
nitro-5-thiocyanobenzoic acid (NTCB) and tribromocresol.
[0036) Of course the choice of the particular enzyme or mixture of
enzymes to be used will depend on the type of sample (e.g. whether large
proteins
or peptides are analyzed, the structural properties of the proteins) to be
analyzed,
etc) and on the information that is sought. Similarly, the particular choice
of
chemical reagent used will depend on these factors. In addition, the digestion
parameters (reaction time, amount of enzyme(s), digestion buffer to be used,
etc.)
should be adapted to suit the concentration and type of sample that is
hydrolyzed
CA 02507117 2005-05-11
11
and the particular protein chip surface that is used, as well known in the
art. Of
course mixtures of enzymes, mixtures of chemical reagents and combination of
enzymes and chemical reagents may be used in accordance with the present
invention. Provided that they are compatible to one another, the particular
enzyme
and chemical treatments used may be performed directly and simultaneously on
the protein chip surface. Alternatively one or more enzymatic treatments) or
one
or more chemical treatments may be performed directly on-chip in a sequential
fashion depending on the specific experimental requirements. Of course the
treatments used need to be chosen or adapted so as to enable MS.
[0037] The protein chip surface to be used in accordance with the
methods of the present invention depends on the particular physicochemical
properties of the protein/peptide sample to be analyzed. Several chip surface
arrays are commercially available (e.g. Ciphergen Biosystems, Palo Alto, CA,
USA). They are generally derivatized with classic chromatographic separation
moieties, such as reverse phase (H4-mimic reversed phase chromatography with
C16 functionality), normal phase (NP20-mimic normal phase chromatography with
silicate functionality), ion exchange (e.g. CM10-weak cation exchange, with
carboxylate functionality with updated hydrophobic barrier coating; WCX2-weak
cation exchange with carboxylate functionality; Q10-strong anion exchange with
quaternary amine functionality, with hydrophobic barrier coating; SAX2-strong
anion exchange with quaternary amine functionality), immobilized affinity
capture
(IMAC, e.g. IMAC 30-immobilized affinity capture array with nitriloacetic acid
(NTA)
surface, with hydrophobic barrier coating; IMAC 3-mmobilized affinity capture
array
with nitriloacetic acid (NTA) surface), mixed mode media (H50-binds protein
through reverse phase or hydrophobic interaction chromatography with an
updated
hydrophobic barrier coating), Surface Enhanced Neat ~esorption (SEND), and
gold chip. Examples of other chip surfaces that may be used in accordance with
the present invention are disclosed in U.S. patent application 2005/0090016.
CA 02507117 2005-05-11
12
[0038] Surface such as these, with broad binding properties are
typically used in protein profiling studies and biomarkers discovery (e.g.
where
samples from diseased and normal subjects are compared). As well known in the
art, biomolecules bind to these surfaces through electrostatic, hydrophobic,
coordinate covalent bond or Lewis-acid/base interactions. Of course other
types of
array surfaces exists and may be used in accordance with the present
invention.
[0039] In addition to standard chromatographic surfaces, arrays may
be created using virtually any molecules of interest covalently linked to the
surface
including antibodies, enzymes, ligands, receptors, DNA and lectins. Therefore,
as
opposed to standard chromatographic media, these specific surtaces can provide
much more enrichment of captured analytes due to high specificity of
biomolecular
interactions. Thus, pre-activated arrays designed specifically for
immunoassay,
receptor-ligand binding and DNA-binding protein applications are also
compatible
with the method of the present invention. Non-limiting examples of these chips
include RS100, PS10 and PS20 (Ciphergen).
[0040] Thus, depending on the properties of the sample to be
analyzed, the appropriate protein chip surface will be selected in accordance
with
well-known principles of protein separation and identification techniques.
(0041] After binding of the proteins/peptides present in the sample to
the protein chip surface, the active surface on the chip is washed with
buffers
having the desired stringency. The wash (or washes) allows for the removal of
analytes with weak surface interaction potential and permits the enrichment of
the
sample with proteins/peptides having strong surface affinity. Thus, proteins
or
peptides with shared physical and chemical properties are retained.
[0042] Of course, in accordance with well-known principles of protein
separations, the appropriate binding (conditioning) and washing buffers should
be
CA 02507117 2005-05-11
13
selected in order to allow the binding and retention of target biomolecules on
the
specific protein surface. For example, the pH and salt concentration of the
wash
buffer will alter the profile of the peptides retained on the ion exchange
surface.
Thus, one would adapt these parameters for selecting/retaining the appropriate
protein on the chip surface for analysis.
[0043] In one embodiment all steps leading to sample analysis are
performed directly on a chip. In another embodiment one or more sample
purification steps) is/are performed prior to on-chip analysis. In yet another
embodiment an additional wash is performed prior to MS analysis in order to
remove components on the chip (e.g. salts present in the buffer) that could
interfere with mass spectroscopy (e.g. generally, when working with a SAX2
protein chip, a final wash is necessary when using phosphate or borate
buffer).
Thus, depending on the type of chip surface and buffer used, it may be
necessary
or preferable to add one or more washes) (e.g. with water or suitable buffer),
which would remove MS interfering components.
[0044] For example, chemicals are known to interfere with co-
crystallization or suppress sample ionization during mass analysis in the
protein
chip reader. Other chemicals may interfere with binding to the surface of the
protein chip array, depending on the specific surface chemistry being used.
Compounds may also interfere with enzymatic reactions that are performed on
the
chip. Thus, the required additional wash or washes may be introduced before
any
step which would otherwise be affected by the remaining interfering
components.
[0045] For example, salts may reduce binding to ionic surfaces but can
increase binding through hydrophobic interactions. Thus, one skilled in the
art will
choose buffers and wash conditions in accordance with the specific
requirements
of the protein chip used. With most, but not all, protein chip surfaces used,
a water
wash must be performed prior to EAM addition. Guidelines for each specific
type of
CA 02507117 2005-05-11
14
protein chip commercialized by Ciphergen are available in their "Protein
System
Users Guide". Non-limiting examples of chemicals that can interfere with MS
analysis include ionic detergents, high salts concentrations, polyethylene
glycol
(PEG), glycerol, diethylpyrocarbonate (DEPC) and dithiothreitol (DTT).
[0046] As mentioned above, in many cases ionic detergents will
suppress ionization of a protein sample. In particular, proteins that have
been
boiled in SDS may not be easily detected. Thus, if detergents are necessary
for
sample extraction or sample solubilization, non-ionic detergents, such as
Triton T""
X-100, n-octyl ~-D-glucopyranoside (OGP), NonidetT"" P40 (NP40), or
dodecylmaltoside would be preferred. In general, a final concentration of up
to 1
is acceptable. Of course, the final acceptable concentration depends on the
type of
detergent used and the proteins) of interest. Alternatively, the interfering
detergent
may be removed prior to sample application on the protein chip by any well
known
techniques or even removed after sample application by performing one or more
additional wash(es), provided that the protein chip surface used allows such a
procedure (e.g. if the detergent does not interact too strongly with the
protein chip
surface used).
[0047] In their native state, proteins acquire a specific three-
dimensional structure. The linear sequence of amino acids folds upon itself to
form
a specific native structure. Prior to pertorming a variety of protein
chemistry
reactions it is often necessary to denature a protein, resulting in an
unfolded
conformation, which is more susceptible to the subsequent chemical reactions.
Proteins can be denatured by a variety of chemical and other treatments. For
example, adding sufficient urea or guanidine - hydrochloric acid (HCI) to a
protein
solution can result in protein denaturation. Non-limiting examples of
chemicals for
protein denaturation that can be used in accordance with the present invention
include heat, change of pH (acid or alkali), urea, guanidine - HCI,
dithiothreitol
(DTT), dithioerytritol (DTE), ~3-mercaptoethanol, inorganic salts (lithium
bromide,
CA 02507117 2005-05-11
potassium thiocyanate, sodium iodide), organic solvents (ethanol, methanol,
trifluoroethanol, formamide, dimethylformamide, dichloro and trichloroacetic
acids
and their salts), detergents (sodium dodecyl sulphate), high pressure,
ultrasonic
homogenisation. Of course the choice of the particular denaturing process or
5 chemical agent to be used will depend on the type of sample (e.g. the
structural
properties of the proteins) to be analyzed etc.) and on the information that
is
sought. In addition, the denaturing parameters parameters (reaction time,
amount
of denaturant, denaturing buffer to be used etc.) should be adapted to suit
the
amount and type of sample that is to be denatured and the particular
proteinchip
10 surface that is used, as well known in the art.
[0048] A common naturally occurring posttranslational modification (a
chemical modification occurring after protein synthesis) of many proteins is
the
formation of covalent disulfide bonds between cysteine residues. The formation
of
such disulfide bonds results in a more rigid protein structure with decreased
15 flexibility. Proteins having disulfide bonds are less susceptible to a
number of
chemical reactions. Thus, for many applications, it is often desirable to
cleave a
protein into a number of smaller fragments. In order to cleave proteins having
disulfide bonds efficiently, it is often necessary first to reduce the
disulfide bonds.
This is normally achieved by chemical reduction of the disulfide bonds with an
appropriate reagent. Non-limiting examples of protein reducing agents
compatible
with the methods of the present invention include dithiothreitol (DTT),
dithioerytritol
(DTE), cysteine, ~i-mercaptoethanol, ~i-mercaptoethylamine, reduced
glutathione,
thioglycolic acid and tributylphosphine. Of course, one skilled in the art
would
appreciate that the above list is not extensive and most low molecular weight
thiols
would be effective reducing agents that can be used in accordance with the
present invention. Of course the choice of the particular reducing agent to be
used
will depend on the type of sample (e.g. number of disulfide bonds present, the
structural and physicochemical properties of the proteins) to be analyzed
etc.) and
on the information that is sought. In addition, the chemical reduction
parameters
CA 02507117 2005-05-11
16
(reaction time, amount of reducing agent, temperature to be used etc.) should
be
adapted to suit the amount and type of sample that is to be reduced and the
particular proteinchip surface that is used, as well known in the art. It
should be
noted that the reduction step may be left out altogether in cases where a
particular
protein of interest does not contain any cysteine residues and/or disulfide
bonds.
[0049] Of the course the method of the present invention should be
adapted in order to allow sample binding to the chip and MS analysis. Thus,
when
required, appropriate sample treatments and washes should be performed. For
example, DTT is commonly used to reduce disulfide bonds in protein but
residual
DTT interferes with analysis of protein chip technology. Weak (millimolars)
solutions of ~i-mercaptoethanol may be used in accordance with the present
invention, in place of DTT for disulfide bond reduction. Alternatively, washes
enabling removal of residual DTT may be performed.
[0050 Once reduced, several chemical agents may be employed to
block the reduced cyteine residues through a process known as alkylation,
avoiding the reformation of undesirable disulfide bonds. In accordance with
the
present invention, alkylating agents compatible with our approach include
iodoacetamide, iodoacetic acid, ethyleneimine, 4-vinylpyridine and acrylamide.
The
particular alkylating agent employed often depending on some secondary
purpose,
for example, to enhance the solubility properties in a given medium, to
produce a
site subject to proteolysis by a suitable protease such as trypsin, or to
provide a
reversible protecting group for the cysteine thiol. In addition, the toxicity
of the
alkylating agent may be considered for reasons of safety, for example,
acrylamide
is a toxic substance readily absorbed through the skin that is reasonable
anticipated to be a human carcinogen. The choice of the particular alkylating
agent
to be used will also depend on the type of sample (e.g. number of disulfide
bonds
present, the structural properties of the proteins) to be analyzed ect.) and
on the
information that is sought. In addition, the alkylating parameters (exposure
to light
CA 02507117 2005-05-11
17
during reaction, reaction time, amount of alkylating agent, alkylation buffer,
alkylation temperature to be used etc.) should be adapted to suit the amount
and
type of sample that is to be alkylated and the particular proteinchip surtace
that is
used, as well known in the art.
[0051] Proteins are often isolated from nature as glycoproteins. Protein
glycosylation is important for the proper function of a number of proteins as
well as
intercellular communication and other biological phenomena. Altered sugar
structures have been associated with a number of diseases including autoimmune
disease and cancer (Pauline M. Rudd, Tim Elliott, Peter Cresswell, Ian A.
Wilson,
and Raymond A. Dwek. Glycosylation and the Immune System. Science, Mar
2001; 291: 2370 - 2376.; YJ Kim and A Varki. Perspectives on the significance
of
altered glycosylation of glycoproteins in cancer. Glycoconj J, Aug 1997;
14(5): 569-
76). A glycoprotein is a protein that has sugars chemically bound to specific
amino
acids of the protein. The sugar moiety can be a simple monosacharide or a
complex structure composed of several different sugars covalently bound to
each
other in a variety of branched structures. Often the sugar structures are
heterogeneous at a particular glycosylation site, which adds an increased
level of
complexity in the structural and functional characterization of the
glycosylated
moieties. These sugar side chains can account for anywhere from less than 1 %
up
to 80% of the glycoprotein structure. Sugars are normally added to proteins at
specific consensus sites e.g. AsnXxxThr/Ser (Xxx is any amino acid other than
proline) for N-linked glycosylation to the Asn residue.
[0052] Sugar moieties can also be bound at the hydroxyl group of Ser
and Thr residues in what is known as O-linked glycosylation. Fetuin provides
an
example of a complex N-linked and O-linked glycoprotein having several
glycosylation sites. The study of protein glycosylation is a technically
challenging
field and mass spectrometry (MS) methods are increasingly being used. For
example, an new consensus sequence was only recently confirmed for a
CA 02507117 2005-05-11
18
AsnAsnCys glycosylation site of the epidermal growth factor receptor (EGFR)
expressed in human cells (Zhen Y, Caprioli RM, Staros JV, Characterization of
glycosylation sites of the epidermal growth factor receptor. Biochemistry.
2003 May
13;42(18):5478-5492). This discovery is of paramount importance because
signaling through the epidermal growth factor receptor plays a vital part in
many
cancers. An accurate molecular description of the epidermal growth factor
receptor, including its glycosylated moieties, may be crucial to our ability
to treat
the disease. The method of the present invention can be used to characterize
the
glycosylated portion of glycoproteins. Protein deglycosylation directly on the
chip
surface can be performed by chemical and enzymatic means. The mass of the
protein can be measured before and after deglycosylation indicating the degree
of
glycosylation. For instance, neuraminidase can be used to remove terminal
sialic
acid residues from glycoproteins. Several enzymatic deglycosidases may be used
in accordance with the present invention. Non-limiting examples include N-
glycosidase F (PNGaseF), endoglycosidase H (endoH), endoglycosidase F
(endoF), O-glycosidase and neuraminidase. Reagents for chemical
deglycosylation can also be used including hyrdofluoric acid (HF)-pyridine and
anhydrous pyridine. Of course, the choice of the particular endoglycosidase
used
will depend on the information that is sought. More than one deglycosylation
step
may also be performed in accordance with the present invention. For example a
direct on-chip PNGase F treatment, which removes all common classes of N-
glycans may be followed by a neuraminidase treatment that releases specific O-
linked carbohydrates (i.e. specific forms of N-acetyl-neuraminic acid).
[0053] Protein phosphorylation is an exceedingly important cellular
phenomenon directly linked to cancer, cardiovascular diseases, neural
function,
memory, etc. An estimated one third of proteins present in a given mammalian
cell
are phosphorylated at any time. Abnormal protein phosphorylation is either a
cause or consequence of disease, while normal protein phosphorylation is
required
for normal cellular function. (Cohen, P. Protein kinases-the major drug
targets of
CA 02507117 2005-05-11
19
the twenty-first century? Nat. Rev. 2002 1 (4):309-315). Proteins are often
isolated
from nature with phosphorylated serine, threonine and tyrosine residues. The
identification and characterization of protein phosphorylation is technically
challenging. For example, chicken ovalbumin is a phosphoprotein for which a
crystal structure was reported in 1990 (Stein P.E., Leslie A.G.W., Finch J.T.,
Turnell W.G., McLaughlin P.J., Carrell R.W. Crystal structure of ovalbumin as
a
model for the reactive centre of serpins. Nature 347:99-102 (1990)). The
structure
revealed the presence of two phosphorylation sites. However, only recently
using
mass spectrometric techniques has the presence of two additional
phosphorylation
sites been found (MacCoss MJ, McDonald WH, Saraf A, Sadygov R, Clark JM,
Tasto JJ, Gould KL, Wolters D, Washburn M, Weiss A, Clark JI, Yates JR 3rd.
Shotgun identification of protein modifications from protein complexes and
lens
tissue. Proc. Natl. Acad. Sci. U.S.A. 2002. Jun 11; 99(12):7900-7905).
[0054] Phosphoproteins can be identified and characterized directly on
chip using the method of the present invention. Protein dephosphorylation
directly
on the chip surface can be performed by chemical and enzymatic means. The
mass of the protein can be measured before and after dephosphorylation,
indicating the extent of protein phosphorylation. For on chip enzymatic
dephosphorylation, phosphatases (acid or alkaline) may be used in accordance
with the present invention. Chemical dephosphorylation using HF, HF-pyridine,
or
other known reagents, can also be performed directly on-chip.
[0055] Once all the desired chemical and enzymatic reactions are
performed, the spots on the chip are dried and a matrix solution (comprised of
energy absorbing molecules (EAM), allowing energy to be transferred to the
analyte i.e. proteins or peptides) is added for MS analysis. The EAM assists
in the
desorption and ionization of the analyte. The EAM is generally applied in
organic
solvent, solubilizing many proteins on the protein chip surface. As the EAM
solution dries, the proteins co-crystallize with the EAM. These crystals
absorb the
CA 02507117 2005-05-11
laser energy and generate the ionized proteins detected by a protein chip
reader.
Any matrix solution allowing MS analysis can be used in accordance with the
present invention. Non-limiting examples include saturated sinapinic acid,
cyano
hydroxyl cinnamic acid (CHCA), EAM 1 (Ciphergen), dihydroxybenzoic acid
5 (DHBA), suitable derivatives of cinnamic acid and mixture thereof. Other
suitable
energy absorbing molecules are known to those skilled in the art. In general,
the
EAM is chosen based on the molecular weight of the analyte of interest. For
example, saturated sinapinic acid is recommended for proteins of 15 kDa or
greater while CHCA is especially good for smaller molecules.
10 [0056] In one particular embodiment, a PAP pen (Zymed mini-PAP pen
cat. no. 00-8877) can be used to circle the spots on the chip in order to
prevent
sample spreading during matrix addition. The pen is particularly useful with
array
surfaces that do not have a hydrophobic coating. It provides a water-repellent
barrier that prevents solutions from bleeding off the chemically active spots
of the
15 protein chip array.
[0057] Virtually any type of protein/biological sample can be used in
accordance with the present invention. Non-limiting examples include blood,
serum, plasma, urine, cerebrospinal fluid (CSF), synovial fluid, nipple
aspirate,
seminal fluid, tears, hemofiltrate, amniotic fluid, cells or tissue
homogenate, cell
20 culture media, purified proteins etc. The biological sample may be treated
to
physically disrupt tissue or cell structure, thus releasing intracellular
components
into a solution which may further contain enzymes, buffers, salts, detergents,
and
the like which are used to prepare the sample for analysis. The sample may be
purified or semi purified before performing on-chip analysis depending on the
specific experimental requirements. Crude samples may also be used, provided
that they do not contain interfering components that cannot subsequently be
removed from the chip prior to performing the method step with which it
interferes
(e.g. MS analysis). Of course, synthetic (e.g. synthetic peptides) or semi-
synthetic
CA 02507117 2005-05-11
21
samples can also be used.
[0058] The method of the present invention is optimized by testing
several types of chip surfaces in order to determine which surface gives the
best
results with a particular type of sample and particular chemical and enzymatic
steps performed. Thus, a person skilled in the art could carry out the method
of the
present invention on 2, 3, 4, 5, 6 or more chip surfaces in parallel and
determine
which surface gives the best results. Similarly, several chips having the same
surfaces could be tested in parallel to determine the optimal binding and
washing
buffers as well as the optimal incubation time, concentration of sample,
reagents,
etc, as well known in the art.
[0059] Once all chemical reactions are performed, a MS analysis is
conducted to identify the biomolecules of interest. Any suitable MS device may
be
used in accordance with the present invention as long as it allows
proteins/peptides on the substrate to be resolved. Similarly, the measured
peptides/proteins can be compared to peptide masses from in silico digestion
of
the protein database using any search engine available (e.g. ProFoundT"",
MascotT"", MS-fitT"", AldenteT"", PhenyxT"", PeptideMapperTM, PeptideSearchT""
and
the like).
[0060] The development of two "soft" ionization techniques for the
ionization of non-volatile molecules have proven crucial for the development
of
methods for identification and structure analyses of biological
macromolecules.
These two ionization techniques are matrix assisted laser desorption
ionization
(MALDI) which was described approximately one year after a related report of
laser desorption ionization introduced in 1987 by Tanaka (K. H. Tanaka, H.
Wake,
Y. Ido, S. Akita, Y. Yoshida and I. Yoshida, Rapid Commun. Mass Spectrom. 8,
2,
(1988)) and electrospray ionization (J. B. Fenn, M. Mann, C. K. Meng, S.F.
Wong,
C.M. Whitehouse, Science 246, 6, 64 (1989)). Together, the two techniques have
CA 02507117 2005-05-11
22
made the precision and sensitivity of mass spectrometry readily available for
the
study of biomolecules and their reactions. As an example, the mass of proteins
of
a molecular weight exceeding 100 kDa can be readily measured with high
sensitivity and accuracy. Currently, there are no other techniques than can
achieve
comparable results.
[0061] Although not essential, a laser desorption time-of-flight (TOF)
mass spectrometer is preferably used for MS analysis in accordance with the
present invention. Because of their design features, laser desorption
ionization and
time-of-flight (TOF) mass spectrometry are complementary and are preferably
used. In laser desorption mass spectrometry, a sample containing
proteins/peptides is applied to a substrate or a probe and introduced into an
inlet
system. The proteins/peptides are desorbed and ionized into the gas phase with
a
laser pulse in the ionization source. The ions generated are sampled into the
mass
spectrometer by ion optic lenses, and then in a time-of-flight mass analyzer,
all
ions are accelerated with equal force through a short high voltage field and
allowed
to drift through a high vacuum chamber. At the opposite end of the high vacuum
chamber, the accelerated ions are detected by a sensitive detector surface,
with
each of the different ions arriving at different times. The time-of-flight is
a function
of the velocity of the ions, which is dependent on the ratio of mass/charge.
By
measuring the elapsed time between ion formation and ion detector impact, the
presence or absence of proteins/peptides of specific mass to charge ratio can
de
determined.
[0062] Matrix-assisted laser desorption/ionization mass spectrometry
(MALDI-MS) is a method of mass spectrometry involving the use of an energy
absorbing molecule (sample matrix) that permits the desorption of intact
proteins
or peptide fragments from a laser pulsed probe surface. MALDI is described in
U.S. Pat. No. 5,118,937 (Hillenkamp et al.) and U.S. Pat. No. 5,045,694
(Beavis
and Chait). The sample is mixed with the MALDI matrix material and placed on
the
CA 02507117 2005-05-11
23
surface of an inert probe. Commonly employed absorbing molecules include
cinnamic acid derivatives, sinapinic acid (SPA), cyano hydroxy cinnamic acid
(CHCA) and dihydroxybenzoic acid (DHBA). Other suitable energy absorbing
molecules can be used by those skilled in this art. The liquid mixture of
MALDI
matrix material and sample containing proteins/peptides is allowed to dry
forming
crystals of encapsulate analyte molecules. The sample is then irradiated for
MALDI-MS analysis. The method is useful for detecting proteins/peptides as
described in this invention.
[0063] Surface-enhanced laser desorption/ionization mass
spectrometry (SELDI-MS) is a flavour of MALDI that allows the fractionation
and
detection of proteins/peptides in complex mixtures. In SELDI-MS,
proteins/peptides are bound to the surface of a protein chip by retentate
chromatography due to the physicochemical properties of the chip surface. Non-
bound molecules (salts and other interfering molecules) are washed from the
probe surface using appropriate buffers before MS analysis. SELDI is described
in:
U.S. Pat. No. 5,719,060 ("Method and Apparatus for Desorption and Ionization
of
Analytes," Hutchens and Yip, Feb. 17, 1998,) U.S. Pat. No. 6,225,047 ("Use of
Retentate Chromatography to Generate Difference Maps," Hutchens and Yip, May
1, 2001) and Weinberger et al., "Time-of-flight mass spectrometry," in
Encyclopedia of Analytical Chemistry, R. A. Meyers, ed., pp 11915-11918 John
Wiley & Sons Chichesher, 2000.
[0064] Proteins on the chip surface can be desorbed and ionized by
laser desorption ionization for MS analysis. Any suitable mass spectrometer
can
be used provided that it allows the analytes to be appropriately resolved.
[0065] For optimal results, a chip reader can be placed in line with a
high resolution MDS/Sciex QSTAR or Micromass QTOF mass spectrometer. The
sample is read and analysed as it would normally be analysed with a low
CA 02507117 2005-05-11
24
resolution TOF instrument but with the advantages associated with the high
performance mass spectrometer. The quality of data obtained from such an
instrumental configuration can reveal a number of characteristics about the
sample
that are not easily discernable with a low resolution mass spectrometer. For
example, exact mass measurements with less than 5 ppm error are often
sufficient
to confirm the presence of a specific compound. In addition, the QSTAR and
QTOF are "tandem" mass spectrometers that can be used for peptide sequencing
and rigid identification of compounds and sites of chemical and
posttranslational
modification. Currently, Ciphergen Biosystems offers a Tandem MS Interface
system for compatibility with MDS/Sciex QSTAR mass spectrometers. Ciphergen
Applications employing such a configuration have been reported (Prieto, D.,
Conrads, T.P., Scudiero, D.A., Veenstra, T.D., Profiling of Secreted Proteins
from
Human Ovarian Cancer Cell Lines by Surface-Enhanced Laser Desorption
Ionization Time-of-Flight Mass Spectrometry Journal of Liquid Chromatography &
Related Technologies, 26, 2315-2328, (2003). A Tandem MS Interface system for
compatibility with Micromass QTOF mass spectrometers was available for a time
but has since been discontinued.
[0066] In order to provide a clear and consistent understanding of
terms used in the specification and claims, including the scope to be given
such
terms, a number of definitions are provided herein below.
[006'T~ Unless defined otherwise, the scientific and technological terms
and nomenclature used herein have the same meaning as commonly understood
by a person of ordinary skill to which this invention pertains. Commonly
understood
definitions of molecular biology terms can be found for example in Dictionary
of
Microbiology and Molecular Biology, 2nd ed. (Singleton et al., 1994, John
Wiley &
Sons, New York, NY), The Harper Collins Dictionary of Biology (Hale & Marham,
1991, Harper Perennial, New York, NY), Rieger et al., Glossary of genetics:
Classical and molecular, 5th edition, Springer-Verlag, New-York, 1991; Alberts
et
CA 02507117 2005-05-11
al., Molecular Biology of the Cell, 4th edition, Garland science, New-York,
2002;
and, Lewin, Genes VII, Oxford University Press, New-York, 2000. Generally, the
procedures of sample/protein purification and separation, protein chip
utilization,
MS analysis, molecular biology methods and the like are common methods used in
5 the art. Such standard techniques can be found in reference manuals such as
for
example Sambrook et al. (2000, Molecular Cloning - A Laboratory Manual, Third
Edition, Cold Spring Harbor Laboratories); and Ausubel et al. (1994, Current
Protocols in Molecular Biology, John Wiley & Sons, New-York). Laemmli, U.K.
(1970). Nature (Lond.), 227, 680-685.;Practical protein chemistry, A handbook
A.
10 Darbre Ed. Wiley John Wiley and sons. Copyright 1986.MP Washburn, D
Wolters,
and JR Yates 3'd Large-scale analysis of the yeast proteome by
multidimensional
protein identification technology. Nat Biotechnol, Mar 2001; 19(3): 242-247.
JR
Yates 3'd Mass spectral analysis in proteomics. Annu Rev Biophys Biomol
Struct,
Jan 2004; 33: 297-316. Industrial proteomics Applications for Biotechnology
and
15 Pharmaceuticals. Daniel Figeys Ed. John wiley and Sons Copyright 2005.
Karas,
M and Hillenkamp F (1988) Laser desorption ionization of proteins with
molecular
masses exceeding 10,000 daltons. Anal. Chem. 60, 2299-2301.
DEFINITIONS
[0068] The use of the word "a" or "an" when used in conjunction with
20 the term "comprising" in the claims and/or the specification may mean "one"
but it
is also consistent with the meaning of "one or more", "at least one", and "one
or
more than one".
[0069] Throughout this application, the term "about" is used to indicate
that a value includes the standard deviation of error for the device or method
being
25 employed to determine the value. In general, the terminology "about" is
meant to
designate a possible variation of up to 10%. Therefore, a variation of 1, 2,
3, 4, 5,
6, 7, 8, 9 and 10 % of a value is included in the term about.
CA 02507117 2005-05-11
26
[0070] As used in this specification and claim(s), the words
"comprising" (and any form of comprising, such as "comprise" and "comprises"),
"having" (and any form of having, such as "have" and "has"), "including" (and
any
form of including, such as "includes" and "include") or "containing" (and any
form of
containing, such as "contains" and "contain") are inclusive or open-ended and
do
not exclude additional, un-recited elements or method steps.
[0071] As used herein, the twenty natural amino acids and their
abbreviations follow conventional usage. Stereoisomers (e.g., D-amino acids)
such
as a,a-disubstituted amino acids, N-alkyl amino acids, lactic acid and other
unconventional amino acids may also be suitable components for the
polypeptides
of the present invention. Examples of unconventional amino acids include but
are
not limited to selenocysteine, citrulline, ornithine, norvaline, 4-(E)-butenyl-
4(R) -
methyl-N-methylthreonine (MeBmt), N-methyl-leucine (MeLeu), aminoisobutyric
acid, statine, N-methyl-alanine (MeAla).
[0072] As used herein, "protein" or "polypeptide" means any peptide-
linked chain of amino acids, regardless of post-ranslational modifications
(e.g.
phosphorylation, glycosylation, sulfatation, acetylation, sumoylation,
prenylation,
ubiquitination etc).
[0073] As used herein, the term "purified" refers to a molecule (e.g. a
polypeptides or proteins) having been separated from a component of the
composition in which it was originally present. Thus, for example, a "purified
protein or polypeptide" has been purified to a level not found in nature. A
"substantially pure" molecule is a molecule that is lacking in most other
components (e.g., 30, 40, 50, 60, 70, 75, 80, 85, 90, 95, 96, 97, 98, 99, 100%
free
of contaminants). By opposition, the term "crude" means molecules that have
not
been separated from the components of the original composition in which it was
present. Therefore, the terms "separating" or "purifying" refers to methods by
which
CA 02507117 2005-05-11
27
one or more components of the biological sample are removed from one or more
other components of the sample. Sample components include nucleic acids in a
generally aqueous solution that may include other components, such as
proteins,
carbohydrates, or lipids. A separating or purifying step preferably removes at
least
about 70% (e.g., 70, 75, 80, 85, 90, 95, 96, 97, 98, 99, 100%), more
preferably at
least about 90% (e.g., 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100%) and, even
more preferably, at least about 95% (e.g., 95, 96, 97, 98, 99, 100%) of the
other
components present in the sample from the desired component. For the sake of
brevity, the units (e.g. 66, 67...81, 82,...91, 92%....) have not
systematically been
recited but are considered, nevertheless, within the scope of the present
invention.
[0074] The terms "inhibiting," "reducing" or "interfering" or any variation
of these terms, when used in the claims and/or the specification includes any
measurable decrease or complete inhibition of at least one chemical,
physicochemical, or enzymatic activity in any of the present method steps to
achieve a desired result. For example, a compound is said to be interfering
with
MS detection when a decrease in specificity and sensitivity is measured
following a
treatment with the "inhibiting", "reducing" or "interfering" compound as
compared to
in the absence thereof. Similarly, a compound is said to be "inhibiting" an
enzymatic step (e.g. dephosphorylation, deglycosylation, trypsinization, etc)
of the
method of the present invention when the efficiency of the enzymatic reaction
is
reduced or completely abolished following a treatment with the "inhibiting",
"reducing" or "interfering" compound as compared to in the absence thereof.
[0075] "Probe" refers to a device that is removably insertable into a gas
phase spectrometer and comprises a substrate having a surface for presenting
analytes for detection. A probe can comprise a single substrate or a plurality
of
substrates. Terms such as protein chip, protein chip array, or chip are also
used
herein to refer to specific kinds of probes.
CA 02507117 2005-05-11
28
[0076] "Gas phase ion spectrometer" refers to an apparatus that
measures a parameter which can be translated into mass-to-charge ratios of
ions
formed when a sample is ionized into the gas phase. Generally ions of interest
bear a single charge, and mass-to-charge ratios are often simply referred to
as
mass.
[0077] "Mass spectrometer" refers to a gas phase ion spectrometer
that includes an inlet system, an ionization source, an ion optic assembly, a
mass
analyzer, and a detector.
[0078] "Laser desorption mass spectrometer" refers to a mass
spectrometer which uses laser as an ionization source to desorb an analyte.
[0079] "Binding functionalities" refer to functional groups) of a protein
chip surface material that bind analytes. Binding functionalities can include,
but are
not limited to, a carboxyl group, a sulfonate group, a phosphate group, an
ammonium group, a hydrophilic group, a hydrophobic group, a reactive group, a
metal chelating group, a thioether group, a biotin group, a boronate group, a
dye
group, a cholesterol group, derivatives thereof, or any combinations thereof.
Binding functionalities can further include other adsorbents that bind
analytes
based on individual structural properties, such as the interaction of
antibodies with
antigens, enzymes with substrate analogs, nucleic acids with binding proteins,
and
hormones with receptors.
[0080] "Analyte" refers to a component of a sample which is desirably
retained and detected. The term can refer to a single component or a set of
components in the sample.
[0081] "Conditioned" as applied to the present invention relates to
adaptation or modification of a substrate surtace (protein chip surface) to
promote
CA 02507117 2005-05-11
29
adhesion of analytes onto the substrate surface.
[0082] "Energy absorbing molecule" or "EAM" refers to a molecule that
absorbs energy from an ionization source in a mass spectrometer thereby
enabling
desorption of analyte from a probe surface. Energy absorbing molecules used in
MALDI are frequently referred to as "matrix." Cinnamic acid derivatives,
sinapinic
acid ("SPA"), cyano hydroxy cinnamic acid ("CHCA") and dihydroxybenzoic acid
are frequently used as energy absorbing molecules in laser desorption of
bioorganic molecules. Other suitable energy absorbing molecules are known to
those skilled in this art. See, e.g., U.S. Pat. No. 5,719,060 (Hutchens & Yip)
for
additional description of energy absorbing molecules.
[0083] Other objects, advantages and features of the present invention
will become more apparent upon reading of the following non-restrictive
description of illustrative embodiments thereof, given by way of example only
with
reference to the accompanying drawings.
BRIEF DESCRIPTION OF THE DRAWINGS
[0084] Having thus, generally described the invention, reference will be
made to the accompanying drawings, showing by way of illustration only an
illustrative embodiment thereof and in which:
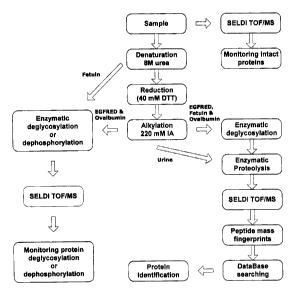
[0085] Figure 1 shows one strategy for on-chip protein analysis. The
schematic shows the steps that were followed to monitor deglycosylation and
dephosphorylation reactions and for identification of proteins investigated.
[0086] Figure 2 shows a mass spectrum of on-chip denaturation,
reduction, alkylation and deglycosylation of 1 Ng ovalbumin on H4 chips (a:
before
CA 02507117 2005-05-11
deglycosylation; b: after deglycosylation) and 1 Ng EGFRED on NP20 chips (c:
before deglycosylation; d: after deglycosylation).
[0087] Figure 3 shows a mass spectrum of on-H4 chip denaturation,
reduction, alkylation and dephosphorylation of 1 Ng ovalbumin (a: before
5 dephosphorylation; b: after dephosphorylation).
[0088] Figure 4 shows a mass spectrum of 1 pg ovalbumin after its on-
H4chip denaturation, reduction, alkylation, deglycosylation and tryptic
digestion.
[0089] Figure 5 shows a mass spectrum of 1 Ng EGFRED after its on-
NP20 chip denaturation, reduction, alkylation, deglycosylation and a) Asp-N
10 digestion, b) Glu-C digestion, c) Lys-C digestion.
[0090] Figure 6 shows a mass spectrum of 1 Ng fetuin after its on-chip
denaturation, reduction, alkylation, deglycosylation and tryptic digestion on
NP20
chips.
DETAILED DESCRIPTION OF ILLUSTRATIVE EMBODIMENTS
15 [0091] Here, processes combining chemical and enzymatic treatments
directly on-chip to monitor various protein modifications such as
deglycosylation
and dephosphorylation reactions, and identified proteins using PMF were
examined. three representative proteins were selected based on their
complexity
and physico-chemical features (Table 1 ). The hydrophobicity of the proteins
will
20 determine how tightly they are bound to the chromatographic surface on the
chip
and the wash cycle chosen to remove impurities is dictated by this
interaction. An
outline of the general procedure developed for chemical and enzymatic
treatment
of proteins and peptides is shown in Figure 1. This approach allowed to adapt
different sequences of reactions according to the characteristics of the
proteins
25 and the nature of their modifications.
CA 02507117 2005-05-11
31
Table 1. Structural characteristics of the model proteins
Protein Average Number Number of Number of
MW of
(kDa) Disulfide GlycosylationPhosphorylation
brid a siteb site
Human EGFRED 81.29 21 11-N 0
Bovine fetuin 46.21 6 3-N, 3-O 0
Chick ovalbumin44.73 1 1-N 4
Molecular weight of the native protein without posttranslational
modrtications.
Carbohydrate moieties can be attached via N- or O- linkages to the proteins.
[0092] Epidermal growth factor receptor ecto domain (EGFRED),
chicken ovalbumin and bovine fetuin were selected as model proteins because of
their complexity, specific physicochemical properties and posttranslational
modifications (PTMs). Less complex proteins (chicken lysozyme, horse
cytochrome C bovine serum albumin, bovine insulin) were also analysed and gave
excellent sequence coverages (data not shown). Based on the performance of the
methods of the invention with the three complex proteins listed above, the
teachings of the present invention are amenable to any protein of interest.
[0093] The present invention is illustrated in further details by the
following non-limiting examples.
EXAMPLE 1
MATERIALS AND METHODS
[0094] Materials. Human EGFRED (Epidermal growth factor receptor
ecto domain) was a gift from Dr. J. Baardsnes of the Biotechnology Research
Institute, National Research Council of Canada, Montreal, Canada. Trypsin was
obtained from Boehringer Mannheim (Ingelheim, Germany) and used without
further purification. Urea, ammonium bicarbonate, a-cyano-4-hydroxy-cinnamic
acid (CHCA), sinapinic acid (SPA), dithiothreitol (DTT), iodoacetamide (IA),
adrenocorticotropic hormone (ACTH), alkaline phosphatase (ALP), chicken
CA 02507117 2005-05-11
32
ovalbumin, chicken lysozyme, bovine insulin, bovine serum albumin, horse
cytochrome C and bovine fetuin were obtained from Sigma (St. Louis, MO).
Trifluoroacetic acid (TFA) was from Pierce (Rockford, IL). Neuraminidase, N-
glycosidase F, O-glycosidase, endoproteinases Arg-C, Asp-N, Glu-C and Lys-C
were purchased from Roche (Indianapolis, IN). Protein chips (working spot 2.5
mm
diameter-H4, NP20, SAX2, WCX2, IMAC3 and Send Alpha) were purchased from
Ciphergen Biosystems Inc. (Fremont, CA). Microspin 6 columns were obtained
from Bio-Rad (Mississauga, ON). Urine samples were collected from a male
Sprague-Dawley rat treated with puromycin aminonucleoside to induce
proteinuria.
All solvents were HPLC grade. Aqueous solutions of the proteins, enzymes and
reagents used in the experiments described below were prepared in 0.1 M
ammonium bicarbonate unless otherwise indicated.
[0095] Chemical treatment. In these particular examples Hydrophylic
NP20 and hydrophobic H4 chips were used. Proteins were first chemically
denaturated, reduced and alkylated as follows: chips were conditioned by
adding 3
pL of water or acetonitrile. The selectivity of the chip varies with the
organic
component and /or salt concentration of the binding buffer. One pL of 1 pg/pL
solution of the protein (e.g. ovalbumin, 23.4 pmol; EGFRED, 14.6 pmol; and
fetuin,
26.0 pmol) was added to the chip. Denaturation was accomplished by adding 2 NL
of 8 M urea and incubating for two hours at room temperature. Disulfide bonds
were reduced by adding 1 NL of 40 mM DTT with incubation in a water bath at
56°C for 45 min. Finally, alkylation of the thiol groups was performed
in a dark
humidity chamber at room temperature by applying 1 NL of 220 mM iodoacetamide
and allowing the reaction to proceed for 30 min. The denatured, reduced and
alkylated proteins were subjected to two different enzymatic reaction schemes
whereby the model proteins were 1) dephosphorylated or deglycosylated and
analyzed by SELDI-TOF/MS to monitor the removal of the corresponding
posttranslational modifications or 2) proteolyzed prior to PMF for
identification
purposes using database searching tools.
CA 02507117 2005-05-11
33
[0096] Enzymatic deglycosylation. Deglycosylation of EGFRED and
ovalbumin (N-glycan-containing proteins) was performed by depositing 1 wL of 1
Unit/wL solution of N-glycosidase F and incubating the array in a 37°C
water bath
for 2 hrs.
[0097] Fetuin contains both N- and O- carbohydrate linkages.
Deglycosylation reactions were performed sequentially on an H4 chip. Two
experimental approaches were applied: a) monitoring of the deglycosylation
reactions using denatured protein without reduction and alkylation; b) the
complete
set of chemical reactions described above (Figure 1 ) was applied prior to
performing enzymatic proteolysis. The denatured or alkylated fetuin was N-
deglycosylated by spotting 1 pL of 0.5 Unit/wL of N-glycosidase F on the chip
and
incubating 2 hrs at 37°C in a water bath. Conversely, O-linked
carbohydrates were
selectively cleaved in a two-step approach: sialyl (a-N-acetylneuraminic acid)
residues were cleaved by adding 1 ~L of 5 mUnit/p,L neuraminidase solution and
incubating for 1 hr in 37°C water bath whereas the serine/threonine O-
linked ~3-D-
galactosamine residues were cleaved by adding 1 ~L of 2 mUnit/pL of O-
glycosidase and incubating the arrays for 2 hours in a 37°C water bath.
[0098] Enzymatic dephosphorylation. Ovalbumin was denatured,
reduced and alkylated as described above on an H4 chip. Dephosphorylation was
performed by adding 1 ~,L of 2 Ng/wL ALP solution (0.1 M ammonium bicarbonate,
1 mM magnesium chloride) and incubating in a 37°C water bath for 2 hrs.
[0099] Enzymatic proteolysis. Enzymatic digestion was performed
following denaturation, reduction, alkylation and/or deglycosylation of the
proteins
on NP20 hydrophilic chips. One ~L of 0.5 Ng/~L trypsin solution was applied to
each spot and digested for 2 hrs at 37°C in a water bath. The array was
air-dried
and rinsed twice with 4 NL of water prior to adding the sample matrix. In
addition to
trypsin proteolysis, EGFRED was also treated with 0.5 Ng quantities of four
other
CA 02507117 2005-05-11
34
proteases: Arg-C, Asp-N, Glu-C and Lys-C. To differentiate the peptides
generated
from the digestion of the model proteins from those originating from
autolysis,
control experiments were conducted with all reagents and/or proteases in the
absence of the proteins.
[0100] Rat urinary proteins. Sprague-Dawley rats were administered
a single 100 mg dose of puromycin aminonucleoside to induce proteinuria and
urine samples were collected in plastic vials containing phenol as stabilizer
at
specific time intervals after administration. The samples were centrifuged to
remove debris, divided in 50 wL aliquots which were frozen at -80°C
until analyzed.
Samples were thawed on ice and a 25 pL aliquot was desalted with the Bio-Rad
column. The resulting eluate was concentrated to a volume of 10 ~L. The
protein
content in this urine sample was 0.6 ~g NL-' by Bradford assay'4. The
concentrated
sample was applied to the NP20 chip followed by denaturation, reduction,
alkylation and trypsinization prior to analysis.
[0101] Mass spectrometric analysis. To deglycosylated,
dephosphorylated and native samples 1 IrL of saturated SPA prepared in 50%
aqueous acetonitrile containing 0.5%, TFA was added to each spot. For
proteolyzed samples, the array was air dried and rinsed twice with 4 pL water.
The
washing was done by pipetting water to the chip and aspirating the water
several
times between the spot and the pipette tip prior to the addition of 0.5 NI of
20%
CHCA in 50% aqueous acetonitrile containing 0.5% TFA to each spot. Mass
spectra were generated in the positive-ion mode using a PBSII-c ProteinChip
reader (Ciphergen Biosystems Inc, Fremont, CA). The instrument was calibrated
externally with ACTH at 2.465 kDa and bovine insulin at 5.733 kDa. The average
mass accuracy after external calibration of PBSII-c is 2000 ppm (0.2%) for
proteins
of 10 kDa to 300 kDa and 1000 ppm (0.1 %) for polypeptides of 1 kDa to 10 kDa.
Resolution was greater than 700 (average) for 5 pmol of human recombinant
insulin. MascotT"" (Matrix Science ltd, London U.K.) was used for protein
CA 02507117 2005-05-11
identification based on PMF analysis.
EXAMPLE 2
ON-CHIP PROTEIN DEGLYCOSYLATION
[0102 Protein glycosylation is an important protein modification serving
5 various functions, which are protein dependent. Glycosylation can protect a
protein
from degradation, retain the protein in the endoplasmic reticulum until
properly
folded, or direct the protein to its proper destination by serving as a
transport
signal. Oligosaccharides exposed on the cell surfacec allow different cells to
recognize each other.
10 [0103] Ovalbumin is a 44.73 kDa hydrophobic glycoprotein which has a
disulfide bridge, one N-glycosylation and four phosphorylation sites'5. These
characteristics make it an attractive example for the assessment of the on-
chip
protein analysis of the present invention. The oligosaccharide moiety of
ovalbumin
is heterogeneous with an average of 1.65 kDa'6. In the experiments performed
the
15 average molecular weight of ovalbumin as measured was decreased by 1.67 kDa
to 43.06 kDa (Figure 2a and b) when N-deglycosylated on an H4 chip, indicating
that the glycan side chain was completely removed. N-deglycosylation proceeded
at a slower rate on the hydrophilic NP20 chip, as significant amounts of
intact
ovalbumin and a byproduct were seen. After 2hr of reaction loss of only 1.376
kDa
20 from ovalbumin molecular weight (data not shown), was observed. This
indicates
that the chemistry of both the protein and chip surface plays an enabling role
in the
deglycosylation reaction. One may hypothesize that the hydrophobic nature of
the
H4 chip and that of several segments of ovalbumin create appropriate binding
conditions for deglycosylation of the Asp_292 glycan residue, which are
probably not
25 favored on the hydrophylic NP20 chip.
[0104 Human EGFRED has eleven N-glycosylation sites with a variety
CA 02507117 2005-05-11
36
of glycoforms and twenty-one disulfide bridges modulating its tertiary
structure ".
This heavily-modified protein is a good example to test this protocol.
Alkylated
EGFRED was digested with N-glycosidase F for two hours on a NP20 chip and
analyzed. As shown in figure 2c and 2d, a mass shift of 9.53 kDa (from 81.29
kDa
to 71.76 kDa) indicates several or all glycosylation sites were removed. The
reaction proceeded at a much slower rate on the H4 chip as unreacted EGFRED
was still detected, even when the reaction time was extended to 3 or 4 hr
(data not
shown).
[0105] Fetuin has three N-linked oligosaccharides, three O-linked
oligosaccharide chains and a potential fourth O-linked glycan'$. Each of the
carbohydrate units attached to asparagine residue has hybrid structures with a
molecular weight of approximately 2.86 kDa'9. In the deglycosylation approach
used herein, fetuin was denatured on H4 chip prior to treatment with N-
glycosidase
F, for two hours at 37°C. Analysis showed that fetuin was
deglycosylated as its
molecular mass was reduced by 5.19 kDa, from 46.21 kDa to 41.02 kDa. The O-
glycosidically linked sugar side chains comprises a disialated structure with
a
molecular weight of approximately 950 Da'8. All three O-linked glycosylation
sites
on fetuin were removed as its molecular weight decreased by approximately 3.00
kDa when treated with neuraminidase and O-glycosidase.
EXAMPLE 3
ON-CHIP PROTEIN DEPHOSPHORY~ATION
[0106] Most aspects of cell life are regulated by protein
phosphorylation; abnormal phosphorylation can result in or be caused by
disease2°. At any moment roughly 30% of all mammalian proteins are
phosphorylated. This reversible reaction is regulated by the concerted actions
of
protein kinases and phosphatases, which affect phosphorylation and de-
phosphorylation respectively.
CA 02507117 2005-05-11
37
[0107] Ovalbumin, with its four phosphoserine sites, was chosen to
investigate protein dephosphorylation. This process resulted in a mass
difference
of approximately 338 Da less than native ovalbumin, which corresponds to the
removal of the four phosphate molecules (Figure 3). Similarly to results
mentioned
above for the N-deglycosylation reaction, negligible dephosphorylation
activity was
observed when using the NP20 chip even when increasing phosphatase
concentration and/or extending reaction time (data not shown). This suggests
that
chips with a hydrophobic surface are probably more suitable than hydrophilic
chips
for performing enzymatic treatment of proteins with a hydrophobic backbone
such
as ovalbumin.
EXAMPLE 4
ON-CHIP PROTEOLYSIS
[0108] Trypsinization of denatured, reduced and alkylated ovalbumin
generated complex peptide profile. This was not surprising because of the
complexity of the native hydrophobic protein. Proteolysis was only efficient
when
ovalbumin was first deglycosylated on H4 chips according to the sequence of
reaction presented in Figure 1. Deglycosylated ovalbumin was subsequently
trypsinized and analyzed (Figure 4). In this study, the mass tolerance of the
MASCOT search program was set at 2 Da. Eleven peptides, corresponding to
46% coverage of the protein amino acid sequence (Table 2) were matched with
their predicted peptides from the in-silico digestion. The search results
identified
chicken ovalbumin with a score of 85 among a relatively complex mixture of
peptides originating not only from ovalbumin, but also from N-glycosidase F
and
trypsin. This on chip method provided a sequence coverage similar to that
obtained from in-solution digestion and is an elegant and flexible approach to
the
characterization of proteins with oligosaccharide substituants.
[0109] EGFRED was treated on-chip through a sequence of five
CA 02507117 2005-05-11
38
chemical and enzymatic treatments. The last proteolytic reaction was performed
using trypsin, Arg-C, Asp-N, Glu-C and Lys-C. Typical mass spectra are
presented
in Figs. 5a, 5b and 5c. Each of the proteolytic reactions generated 13, 5, 15,
12
and 9 specific peptides respectively. EGFRED was identified by PMF with a
score
of 60 (trypsin), 125 (Asp-N), 91 (Glu-C), 36 (Arg-C), and 70 (Lys-C). The
sequence
coverage was 36%, 54%, 40%, 23% and 17% with trypsin, Asp-N, Glu-C, Lys-C,
and Arg-C, respectively. Results of the database search are summarized in
Table
2.
[0110] Fetuin was trypsinized (Figure 6) following denaturation,
reduction, alkylation and N-deglycosylation on NP20 chips. Its identity was
confirmed with nine peptides matched against the in-silico digest (Table 2).
The
MASCOT search through the SwissProt protein database showed that fetuin was
ranked at the top of the list with a score of 62 and 44% sequence coverage.
EXAMPLE 5
ON-CHIP IDENTIFICATION OF RAT URINARY PROTEINS
[0111] Proteolysis of urinary proteins was also investigated by directly
applying a urine aliquot on-chip and performing the reactions described. Ten
~I of
rat urine was applied, reduced, alkylated and trypsinized as described above.
MS
analysis and peptide mass fingerprinting identified 5 and 11 peptides specific
to
Alpha-2u-globulin (AUG) and rat urinary albumin (RUA) respectively. The
sequence coverages of AUG and RUA are shown in Table 2. AUG is the most
abundant in normal rat urine whereas RUA is most prevalent in puromycin-
induced
proteinuria. Both contain disulfide bridges and AUG is N-glycosylated. Their
identities were independently confirmed by LCMS/MS2'.
CA 02507117 2005-05-11
39
Table 2. Proteins enzymatic treatment and identification
Protein Proteolytic . Number of _ _ % Sequence
EGFRED Trypsin 13 36
Arg-C 5 17
Asp-N 15 54
Glu-C 12 40
Lys-C 9 23
Bovine fetuin Trypsin 9 44
Ovalbumin Trypsin 11 46
Rat urinary albumin Trypsin 11 25
(65.90 kDa)
Alpha-2u-globulin (18.73Trypsin 5 42
kDa)
[0112] In addition to the peptides derived from ovalbumin, EGFRED, fetuin
and the rat urine proteins AUG and RUA, some ion peaks found in the protein
digests were also present in the blank. Several enzymatic autolysis and
proteolysis
products from N-glycosidase and human keratin were observed.
[0113] On-chip chemical and enzymatic reactions are dependent on the
physico-chemical properties of the chip surface. The interactions are
analogous to
those involved in normal and reverse phase chromatographic separations. For
example, hydrophobic proteins will bind tighter to a reversed phase surface
than
hydrophilic proteins. These interactions determine the type of chip used for a
particular on-chip experiment. As previously mentioned, the polarity of the
binding
buffer influences the selection of proteins that are retained on the chip.
[0114] Several chips are currently available for this protocol. These include
anion and cation exchange, metal ion, antibody-antigen, receptor-ligand and
DNA-
protein interaction chips. Therefore the appropriate chip should be chosen for
optimal reaction, selectivity and sensitivity. Complex samples (plasma, urine,
CA 02507117 2005-05-11
cerebrospinal fluid etc.) may require fractionation or depletion strategies to
isolate
target proteins prior to the application of the methods of the present
invention.
Also, the present methods could provide a rapid means for characterizing
native
proteins or antibody therapeutics as well as chemically modified proteins and
5 formulated protein products in the course of their manufacturing and quality
control
processes. Greater usefulness of this method can be attained if the chip is
read by
a high resolution (> 12,000) mass spectrometer with high mass accuracy (< 10
ppm). This will afford the microcharacterization of protein modifications. For
example, single phosphorylation will show a monoisotopic difference of 79.6633
10 amu, the molecular weight of the phosphate moiety. Finally, as mentioned
above,
more than one chip could be chosen for a particular protein, proteins or
sample,
and the chips be treated in parallel (with modifications of the treatment
adapted for
particular chips, if required and as known in the art) to optimize the method
and the
obtention of the best results.
15 CONCLUSION
[0115] Protein identification generally involves isolation of proteins of
interest by electrophoresis and/or chromatographic methods followed by
denaturation, reduction, alkylation and proteolytic digestion. All steps are
normally
carried out in solution to generate peptides for Edman degradation or
sequencing
20 by MS methods. The major advantages of protocols of the present invention
are its
simplicity, speed and sensitivity. Low picomolar amounts of relatively complex
proteins can be rapidly deglycosylated, dephosphorylated and/or proteolyzed
and
readily identified. The sequence coverage obtained by these methods is similar
to
that generated from in-solution digestion.
25 [0116] In conclusion, the herein-described approach to protein
identification
and characterization can be routinely used in several areas of biomarker
research
and related applications, particularly in molecular diagnostics and monitoring
of
CA 02507117 2005-05-11
41
disease, assessment of drug efficacy and basic proteomic research. The methods
of the present invention work effectively for proteins which have complex
structures as demonstrated by the analysis of EGFRED, fetuin, and ovalbumin.
Thus the on chip deglycosylation and dephosphorylation of the present
invention
provide an excellent approach for rapid analysis of modified proteins.
[0117) Although the present invention has been described hereinabove by
way of preferred embodiments thereof, it can be modified, without departing
from
the spirit and nature of the subject invention as defined in the appended
claims.
CA 02507117 2005-05-11
42
REFERENCES
1. Aebersold, R,; Mann, M. Nature, 2002, 422, 198-207.
2. Figeys, D. In Two-dimensional Gel Electrophoresis and Mass Spectrometry for
Proteomics Studies: State-of-the-art; Rehm, H.J., Reed, G., Ed.; John Wiley &
Sons: New York, 2001; pp 243-268.
3. Gorg, A. In Advances in 2D gel techniques. Proteomics: A Trend Guide; Mann,
M.; Blackstock, W. Ed.; Elsevier: London, 2000; pp 3-6.
4. Link, A.J.; Eng, J.; Schieltz, D.M.; Carmack, E.; Mize, G.J.; Morris, D.R.;
Garvik, B.M.; Yates, J.R. 3rd. Nat. Biotechnol. 1999, 17, 676-682.
5. Wolters, D.; Washburn, M.P.; Yates, J.R. 3rd. Anal. Chem. 2001, 73, 5683-
5690.
6. Hunt, D.F.; Mitchel, H.; Dickinson, T.A.; Shabanowitz, J.; Cox, A.;
Sakaguchi,
K.; Appella, E.; Grey, H.M.; Sette, A. Science. 1992, 256, 1817-1820.
7. Kierman, U.A.; Tubbs, K.A.; Nedelkov, D.; Niederkofler, E.E.; McConnell,
E.;
Nelson, R.W. J Proteome Res. 2003, 2, 191-197.
8. Nelson, R.W.; Krone, J.R.; Bieber, A.L.; Williams, P. Anal. Chem. 1995, 67,
1153-1158.
9. Merchant, M.; Weinberger, S.R. Electrophoresis. 2000, 21, 1164-1177.
10. Gevaert, K.; Vanderkerckhove, J. Electrophoresis. 2000; 21, 1145-1154.
11. Lin, S.; Tornatore, P.; King, D.; Orlando, R.; Weinberger, S.R.
Proteomics.
2001, 1, 1172-1184.
12. Dare, T.; Davies, H.A.; Turton, J.A.; Lomas, L.; Williams, T.C.; York,
M.J.
Electrophoresis. 2002, 23, 3241-3251.
13. Caputo, E.; Moharram, R.; Martin, B.M. Anal. Biochem. 2003, 321, 116-124.
14. Bradford M. Anal. Biochem. 1976, 72, 248-254.
CA 02507117 2005-05-11
43
15. MacCoss, M.J.; McDonald, W.H.; Saraf, A.; Sadygov, R.; Clark, J.M.; Tasto,
J.J.; Gould, K.L.; Wolters, D.; Washburn, M.; Weiss, A.; Clark, J.I.; Yates,
J.R. III.
PNAS. 2002, 99, 7900-7905.
Duffin, K.L.; Welply, J.K.; Huang, E.; Henion, J.D. Anal. Chem. 1992, 64, 1440-
1448.
Zhen, Y.; Caprioli, R.M.; Stratos, J.V. Biochemistry. 2003, 42, 5478-5492.
18. Spiro, R.G.; Bhoyroo, V.D. J. Biol. Chem. 1974, 249, 5704-5717.
19. Nilsson, B.; Norden, N.E.; Svensson, S. J. Biol. Chem. 1979, 254, 4545-
4553.
20. Cohen, P. Nat. Rev. Drug Disc. 2002, 1, 309-315.
21. Ge, Y.; Aguiar, M.; Gibbs, B.F.; Masse, R. 52nd conference ASMS. May 2004,
Nashville, TN.