Note: Descriptions are shown in the official language in which they were submitted.
CA 02511214 2011-05-05
ASTHMA AND ALLERGIC INFLAMMATION MODULATORS
BACKGROUND OF THE INVENTION
G-protein coupled receptors play important roles in diverse signaling
processes, including those involved in host defense mechanisms. Immune
responses to
infectious diseases, injury, tumors and organ transplantation and in diseases
and conditions
such as asthma, allergy, rheumatoid arthritis and neoplasia have been linked
to GPCR
regulation. Exaggerated or misdirected immune responses are responsible for
many
inflammatory and hypersensitivity diseases which, left untreated, can result
in tissue or organ
damage, pain and/or loss of function. Tissue inflammation is largely
implicated in the
pathogenesis of such diseases, of which asthma and allergic diseases are among
the most well
characterized. The mechanisms underlying airway inflammation and
hyperreactivity are
similar to those underlying allergic inflammation in other tissues, such as
the skin and gut.
Prostaglandins are lipid-derived inflammatory mediators that recruit
macrophages, T cells, eosinophils, basophils and neutrophils from peripheral
blood to
damaged or inflamed tissues. In addition, prostaglandins can, depending on the
target cell
type, induce or inhibit intracellular Ca2+ mobilization, cAMP production,
platelet
aggregation, leukocyte aggregation, T cell proliferation, lymphocyte
migration, and Th2 cell
chemotaxis, IL-la and IL-2 secretion and vascular and non-vascular smooth
muscle
contraction in responsive cells. Prostaglandins have been implicated in fever,
various allergic
diseases, vascular and non-vascular smooth muscle relaxation, pain perception,
sleep, platelet
aggregation and reproductive processes. Prostaglandins exert their effects by
interacting with
specific GPCRs.
Prostaglandin D2 (PGD2) is the major inflammatory mediator released by
activated mast cells, typically found near skin surfaces, mucous membranes and
blood
vessels, upon immunological challenge (Lewis et al. (1982) J. Immunol.
1
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
2
129:1627-1631). During asthma and allergic responses, PGD2 is released in
large
amounts. The role of PGD2 in the initiation and maintenance of allergic
inflammation
has been well established in mouse models of asthma. For example, it has been
demonstrated that overproduction of PGD2 in vivo by PGD2 synthase exacerbates
airway inflammation in a mouse model of asthma (Fujitani et al. (2002) J.
Immunol.
168:443-449).
A PGD2-selective receptor, designated DP, has been identified (Boie et
al. (1995) J. Biol. Chem. 270:18910-18916). In humans, DP is expressed in
smooth
muscle, platelets, small intestine and brain, and its expression in lung
epithelium is
induced by allergic challenge. Receptor activation induces cAMP production and
intracellular Caz+ mobilization, and is believed to inhibit platelet
aggregation and cell
migration and induce relaxation of various smooth muscles. DP is coupled
primarily to
Gas protein.
Significantly, in an OVA induced asthma model, DP -/_ mice exhibited
reduced asthma symptoms, e.g., reduced cellular infiltration of eosinophils
and
lymphocytes in BAL fluid, reduced Th2 cytokine levels in BAL fluid and reduced
airway hyperreactivity to acetylcholine (Matsuoka et al. (2002) Science
287:2013-
2019). The increased cellular infiltration in lung tissue and mucus secretion
by airway
epithelial cells characteristic of asthma in humans and observed in wild-type
mice was
not observed in DP-deficient mice.
Recently, an additional PGD2-selective receptor, designated
chemoattractant receptor-homologous molecule expressed on Th2 cells, or CRTH2,
has
been identified (Hirai et al. (2001) J. Exp. Med. 193(2):255-261). The
receptor was
previously referred to as GPR44 or DL1R. Among peripheral blood T lymphocytes,
human CRTH2 is selectively expressed on Th2 cells, and is highly expressed on
cell
types associated with allergic inflammation such as eosinophils, basophils and
Th2
cells. It has been shown that CRTH2 activation induces intracellular Caz+
mobilization
and infiltration of Th2 cells, eosinophils and basophils.
Protein sequence analysis indicates that CRTH2 has no significant
homology to DP, but rather, is related to members of the N-formyl peptide
receptor
(FPR) subfamily (Nagata et al. (1999) J. Immunol. 162:1278-1286). In contrast
to DP,
CRTH2 has been shown to couple primarily to Gai protein.
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
3
These observations suggest that CRTH2 and DP may function
independently to regulate aspects of allergic inflammation.
The increasing incidence of asthma, allergic diseases and immunologic
diseases worldwide underscores the need for new therapies to effectively treat
or
prevent these diseases. The discovery of small molecules that modulate CRTH2
and/or
one or more other PGD2 receptors, e.g., DP, is useful for the study of
physiological
processes mediated by CRTH2 and/or one or more other PGD2 receptors, e.g., DP,
and
the development of therapeutic agents for asthma, allergic diseases and other
immunologic diseases. Novel compounds which display such desirable activity
are
described herein.
SUMMARY OF THE INVENTION
The invention provides compounds, pharmaceutical compositions and
methods useful for treating or preventing conditions and disorders associated
with
inflammation processes. In particular, the invention provides compounds,
pharmaceutical compositions and methods useful for treating or preventing
asthma,
allergic diseases, inflammatory conditions, cancer and viral infection.
The compounds of the invention have the general formula (I):
R2
\
X-W-L-Z
Al
DI-A2
I
wherein
A' is C(R3) or N;
A2 is C(R6) or N;
when A' is C(R3) and A2 is C(R6), D' is selected from the group
consisting of C(R4)=C(R5), C(R4)=N, NR', 0 and S;
when A' or A2 is N, D' is C(R4)=C(R5) or C(R4)=N;
W is a divalent group selected from the group consisting of a single
bond, an aromatic ring, a heteroaromatic ring, a cyclo(C3-C8)alkane ring and a
heterocyclo(C3-C8)alkane ring;
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
4
optionally, W is combined with A2 to form a 5-, 6-, 7- or 8-membered
fused ring containing from 0 to 3 heteroatoms selected from the group
consisting of N,
O and S;
X is a divalent linkage selected from the group consisting of -0-, -
S(O)k-, -CRaRb-, -C(O)-, -NR8- and -C(NR9)-=
Y is a divalent linkage selected from the group consisting of a single
bond, -S(O)kNR10-, -C(O)NR10-, (C1-C4)alkylene, hetero(C2-C4)alkylene, -
N(R")C(O)NR10-, -N(R")S(O)kNR'O-, -N(R11)CO2-, -NR"-, -0- and -S(O)k-;
optionally, X and Y are combined to form a 5-, 6-, 7- or 8-membered
fused ring containing from 0 to 3 heteroatoms selected from the group
consisting of N,
O and S;
Z is selected from the group consisting of -CO2R12, -C(O)NR12R'3 and
heteroaryl;
L is a divalent linkage selected from the group consisting of a single
bond, (C,-C6)alkylene and (C2-C4)heteroalkylene;
R2 is selected from the group consisting of hydrogen, -OR', (CI-
C8)alkyl, hetero(C2-C8)alkyl, aryl, heteroaryl and aryl(C1-C4)alkyl;
R3, R4, R5 and R6 are independently selected from the group consisting
of hydrogen, halogen, (C1-C8)alkyl, fluoro(C1-C4)alkyl, hetero(C2-C8)alkyl,
aryl,
heteroaryl, aryl(C1-C4)alkyl, -NR'R", -OR', -NO2, -CN, -C(O)R', -CO2R', -
C(O)NR'R", (C1-C4)alkylene-C(O)NR'R", -S(O)mR', -S(O)kNR'R", -OC(O)OR', -
OC(O)R', -OC(O)NR'R", -N(R"')C(O)NR'R", -N(R")C(O)R' and -N(R")C(O)OR';
R7 is selected from the group consisting of hydrogen, halogen, (C1-
C8)alkyl, fluoro(C1-C4)alkyl, hetero(C2-C8)alkyl, aryl, heteroaryl, aryl(C1-
C4)alkyl, -
NR'R", -OR', -C(O)R', -CO2R', -C(O)NR'R", -S(O)mR' and -S(O)kNR'R";
Ra and Rb are independently selected from the group consisting of
hydrogen, (C1-C6)alkyl, hetero(C2-C6)alkyl, aryl(C 1 -C4)alkyl, -OR' and-
NR'R';
optionally, Ra and Rb are combined to form a 5-, 6-, 7- or 8-membered
spiro ring containing from 0 to 3 heteroatoms selected from the group
consisting of N,
O and S;
R8, R10 and R11 are independently selected from the group consisting of
hydrogen, (C1-C8)alkyl, fluoro(C1-C4)alkyl, hetero(C2-C8)alkyl, aryl,
heteroaryl,
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
-aryl(C,-C4)alkyl, -C(O)R', -C02R', -C(O)NR'R", -S(O),,,R' and -S(O)kNR'R";
R9 is selected from the group consisting of (C I -C6)alkyl, hetero(C2-
C6)alkyl, aryl(C1-C4)alkyl, -OR' and -NR'R";
optionally, R8 or R9 is combined with W to form a 5-, 6-, 7- or 8-
5 membered fused ring containing from 0 to 3 heteroatoms selected from the
group
consisting of N, 0 and S;
optionally, Ra, Rb, R8 or R9 is combined with Y to form a 5-, 6-, 7- or 8-
membered fused ring containing from 0 to 3 heteroatoms selected from the group
consisting of N, 0 and S;
R12 and R13 are independently selected from the group consisting of
hydrogen, (C I -C6)alkyl, hetero(C2-C6)alkyl, aryl, aryl(C 1 -C4)alkyl and
heteroaryl;
optionally, R' 2 and R' 3 are combined with the nitrogen atom to which
they are attached to form a 5-, 6- or 7-membered ring containing from 0 to 2
additional
heteroatoms selected from the group consisting of N, 0 and S;
each R', R" and R"' is independently selected from the group consisting
of hydrogen, (C1-C6)alkyl, cyclo(C3-C8)alkyl, aryl and aryl(CI-C4)alkyl;
optionally, when R' and R" are attached to the same nitrogen atom, R'
and R" may be combined to form a 5-, 6-, 7- or 8-membered ring containing from
I to
3 heteroatoms selected from the group consisting of N, 0 and S;
each subscript k is 0, 1 or 2; and
the subscript in is 0, 1, 2 or 3;
with the proviso that when
A' is C(R3), D' is C(R4)=C(RS);
W is a benzene ring,
X is -0- or -S-; and
-Y-R2 is -NHSO2-heterocyclo(C3-C8)alkyl, -NHSO2-phenyl or
-NHS02-heteroaryl;
A2 is other than C(O- substituted (C2-C4)alkyl) or C(S- substituted (C2-
C4)alkyl).
Unless otherwise indicated, the compounds provided in the above formula
are meant to include pharmaceutically acceptable salts and prodrugs thereof.
The invention also provides pharmaceutical compositions comprising a
compound of formula I and a pharmaceutically acceptable carrier, excipient or
diluent.
CA 02511214 2011-11-04
6
The invention also provides methods for treating or preventing
inflammatory conditions, immune disorders, asthma, allergic rhinitis, eczema,
psoriasis,
atopic dermatitis, fever, sepsis, systemic lupus erythematosus, diabetes,
rheumatoid
arthritis, multiple sclerosis, atherosclerosis, transplant rejection,
inflammatory bowel
disease, cancer, viral infection, thrombosis, fibrosis, flushing, Crohn's
disease,
ulcerative colitis, chronic obstructive pulmonary disease, inflammation, pain,
conjunctivitis, nasal congestion and urticaria, comprising administering to a
subject in
need thereof a therapeutically effective amount of a compound of formula I.
The invention also provides methods for treating or preventing a condition
or disorder mediated, regulated or influenced by Th2 cells, eosinophils,
basophils,
platelets, Langerhans cells, dendritic cells or mast cells, comprising
administering to a
subject in need thereof a therapeutically effective amount of a compound of
formula I.
The invention also.provides methods for treating or preventing a condition
or disorder mediated, regulated or influenced by PGD2 and metabolites thereof,
such as
13,14-dihydro-l5-keto-PGD2 and 15-deoxy-012'4-PGD2, comprising administering
to a
subject in need thereof a therapeutically effective amount of a compound of
formula 1.
The invention further provides methods for treating or preventing a
condition or disorder responsive to modulation of CRTH2 and/or one or more
other
PGD2 receptors, e.g., DP, comprising administering to a subject in need
thereof a
therapeutically effective amount of a compound of formula I.
The invention also provides methods for treating or preventing a condition
or disorder mediated by CRTH2 and/or one or more other PGD2 receptors, e.g.,
DP,
comprising administering to a subject in need thereof a therapeutically
effective amount
of a compound of formula I.
The invention also provides methods for modulating CRTH2 and/or one or
more other PGD2 receptors, e.g., DP, comprising contacting a cell with a
compound of
formula 1.
Other objects, features and advantages of the invention will become
apparent to those skilled in the art from the following description.
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
7
DETAILED DESCRIPTION OF THE INVENTION
Abbreviations and Definitions
The abbreviations used herein are conventional, unless otherwise
defined.
The terms "treat", "treating" and "treatment", as used herein, are meant
to include alleviating or abrogating a disease and/or its attendant symptoms
and
alleviating or eradicating the cause of the disease itself.
The terms "prevent", "preventing" and "prevention", as used herein,
refer to a method of delaying or precluding the onset of a disease and/or its
attendant
symptoms, barring a subject from acquiring a disease or reducing a subject's
risk of
acquiring a disease.
The term "therapeutically effective amount" refers to the amount of the
subject compound that will elicit the biological or medical response of a
tissue, system,
animal or human that is being sought by the researcher, veterinarian, medical
doctor or
other clinician. The term "therapeutically effective amount" includes that
amount of a
compound that, when administered, is sufficient to prevent development of, or
alleviate
to some extent, one or more of the symptoms of the condition or disorder being
treated.
The therapeutically effective amount will vary depending on the compound, the
disease
and its severity and the age, weight, etc., of the mammal to be treated.
The "subject" is defined herein to include animals such as mammals,
including, but not limited to, primates (e.g., humans), cows, sheep, goats,
horses, dogs,
cats, rabbits, rats, mice and the like. In preferred embodiments, the subject
is a human.
As used herein, the term "CRTH2" refers to a CRTH2 protein (RefSeq
Accession No. NP_007469) or variant thereof that is capable of mediating a
cellular
response to PGD2 in vitro or in vivo. CRTH2 variants include proteins
substantially
homologous to native CRTH2, i.e., proteins having one or more naturally or non-
naturally occurring amino acid deletions, insertions or substitutions (e.g.,
CRTH2
derivatives, homologs and fragments). The amino acid sequence of CRTH2 variant
preferably is at least about 80% identical to a native CRTH2, more preferably
at least
about 90% identical, and most preferably at least about 95% identical.
As used herein, the terms "other PGD2 receptor", "another PGD2
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
8
receptor" and the like refer to a prostanoid receptor protein other than
CRTH2, or
variant thereof, that is capable of mediating a cellular response to PGD2 in
vitro or in
vivo. Another PGD2 receptor may be selective for PGD2i e.g., DP (RefSeq
Accession
No. NP_000944), or other one or more other prostanoids (e.g., EP1, EP2, EP3
and EP4,
FP, IP and TP). Other PGD2 receptor variants include proteins substantially
homologous to a corresponding native prostanoid receptor other than CRTH2,
i.e.,
proteins having one or more naturally or non-naturally occurring amino acid
deletions,
insertions or substitutions (e.g., derivatives, homologs and fragments of
another PGDz
receptor). The amino acid sequence of other PGD2 receptor variants preferably
is at
least about 80% identical to the corresponding native other PGD2 receptors,
more
preferably at least about 90% identical, and most preferably at least about
95%
identical. Preferably, another PGD2 receptor is DP.
As used herein, the term "DP" refers to a DP protein (RefSeq Accession
No. NP_000944) or variant thereof that is capable of mediating a cellular
response to
PGD2 in vitro or in vivo. DP variants include proteins substantially
homologous to
native DP, i.e., proteins having one or more naturally or non-naturally
occurring amino
acid deletions, insertions or substitutions (e.g., DP derivatives, homologs
and
fragments). The amino acid sequence of DP variant preferably is at least about
80%
identical to a native DP, more preferably at least about 90% identical, and
most
preferably at least about 95% identical.
The terms "modulate", "modulation" and the like refer to the ability of a
compound to increase or decrease the function and/or expression of CRTH2
and/or one
or more other PGD2 receptors, e.g., DP, where such function may include
transcription
regulatory activity and/or protein-binding. Modulation may occur in vitro or
in vivo.
Modulation, as described herein, includes the inhibition, antagonism, partial
antagonism, activation, agonism or partial agonism of a function or
characteristic
associated with CRTH2 and/or one or more other PGD2 receptors, either directly
or
indirectly, and/or the upregulation or downregulation of the expression of
CRTH2
and/or one or more other PGDz receptors, either directly or indirectly. In a
preferred
embodiment, the modulation is direct. Inhibitors or antagonists are compounds
that,
e.g., bind to, partially or totally block stimulation, decrease, prevent,
inhibit, delay
activation, inactivate, desensitize, or downregulate signal transduction.
Activators or
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
9
agonists are compounds that, e.g., bind to, stimulate, increase, open,
activate, facilitate,
enhance activation, activate, sensitize or upregulate signal transduction. The
ability of
a compound to inhibit the function of CRTH2 and/or one or more other PGD2
receptors
can be demonstrated in a biochemical assay, e.g., binding assay, or a cell-
based assay,
e.g., a transient transfection assay.
The term "CRTH2-modulating amount" refers to that amount of a
compound that is needed to produce a desired effect in any one of the cell-
based assays,
biochemical assays or animal models described herein or otherwise known to the
skilled artisan. Typically, a CRTH2-modulating amount of a compound will be at
least
that amount which exhibits an EC50 in a reporter-gene cell-based assay
(relative to an
untreated control).
As used herein, the terms "CRTH2-responsive condition or disorder,"
"condition or disorder responsive to CRTH2" and related terms and phrases
refer to a
condition or disorder associated with inappropriate, e.g., less than or
greater than
normal, CRTH2 activity and at least partially responsive to or affected by
CRTH2
modulation (e.g., a CRTH2 antagonist or agonist results in some improvement in
patient well-being in at least some patients). Inappropriate CRTH2 functional
activity
might arise as the result of CRTH2 expression in cells which normally do not
express
CRTH2, increased CRTH2 expression or degree of intracellular activation
(leading to,
e.g., inflammatory and immune-related disorders and diseases) or decreased
CRTH2
expression. A CRTH2-associated condition or disorder may include a CRTH2-
mediated condition or disorder.
As used herein, the phrases "CRTH2-mediated condition or disorder,"
"a condition or disorder mediated by CRTH2" and related phrases and terms
refer to a
condition or disorder characterized by inappropriate, e.g., less than or
greater than
normal, CRTH2 activity. Inappropriate CRTH2 functional activity might arise as
the
result of CRTH2 expression in cells which normally do not express CRTH2,
increased
CRTH2 expression or degree of intracellular activation (leading to, e.g.,
inflammatory
and immune-related disorders and diseases) or decreased CRTH2 expression. A
CRTH2-mediated condition or disorder may be completely or partially mediated
by
inappropriate CRTH2 functional activity. However, a CRTH2-mediated condition
or
disorder is one in which modulation of CRTH2 results in some effect on the
underlying
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
condition or disorder (e.g., an CRTH2 antagonist or agonist results in some
improvement in patient well-being in at least some patients).
The term "PGD2 receptor-modulating amount" and related terms and
phrases refer to that amount of a compound that is needed to produce a desired
effect in
5 any one of the cell-based assays, biochemical assays or animal models
described herein
or otherwise known to the skilled artisan. Typically, a PGD2 receptor-
modulating
amount of a compound will be at least that amount which exhibits an EC50 in a
reporter-gene cell-based assay (relative to an untreated control).
As used herein, the term "condition or disorder responsive to another
10 PGD2 receptor" and related terms and phrases refer to a condition or
disorder
associated with inappropriate, e.g., less than or greater than normal,
activity of another
PGD2 receptor and at least partially responsive to or affected by modulation
of another
PGD2 receptor (e.g., another PGD2 receptor antagonist or agonist results in
some
improvement in patient well-being in at least some patients). Inappropriate
functional
activity of another PGD2 receptor might arise as the result of expression of
another
PGD2 receptor in cells which normally do not express the receptor, increased
expression of another PGD2 receptor or degree of intracellular activation
(leading to,
e.g., inflammatory and immune-related disorders and diseases) or decreased
expression
of another PGD2 receptor. A condition or disorder associated with another PGD2
receptor may include a condition or disorder mediated by another PGD2
receptor.
As used herein, the phrase "condition or disorder mediated by another
PGD2 receptor" and related phrases and terms refer to a condition or disorder
characterized by inappropriate, e.g., less than or greater than normal,
activity of another
PGD2 receptor. Inappropriate functional activity of another PGD2 receptor
might arise
as the result of expression of another PGD2 receptor in cells which normally
do not
express the receptor, increased expression of another PGD2 receptor or degree
of
intracellular activation (leading to, e.g., inflammatory and immune-related
disorders
and diseases) or decreased expression of another PGD2 receptor. A condition or
disorder mediated by another PGD2 receptor may be completely or partially
mediated
by inappropriate functional activity of another PGD2 receptor. However, a
condition or
disorder mediated by of another PGD2 receptor is one in which modulation of
another
PGD2 receptor results in some effect on the underlying condition or disorder
(e.g.,
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
11
another PGD2 receptor antagonist or agonist results in some improvement in
patient
well-being in at least some patients).
The term "DP-modulating amount" refers to that amount of a compound
that is needed to produce a desired effect in any one of the cell-based
assays,
biochemical assays or animal models described herein or otherwise known to the
skilled artisan. Typically, a DP-modulating amount of a compound will be at
least that
amount which exhibits an EC50 in a reporter-gene cell-based assay (relative to
an
untreated control).
As used herein, the terms "DP-responsive condition or disorder,"
"condition or disorder responsive to DP" and related terms and phrases refer
to a
condition or disorder associated with inappropriate, e.g., less than or
greater than
normal, DP activity and at least partially responsive to or affected by DP
modulation
(e.g., a DP antagonist or agonist results in some improvement in patient well-
being in at
least some patients). Inappropriate DP functional activity might arise as the
result of
DP expression in cells which normally do not express DP, increased DP
expression or
degree of intracellular activation (leading to, e.g., inflammatory and immune-
related
disorders and diseases) or decreased DP expression. A DP-associated condition
or
disorder may include a DP-mediated condition or disorder.
As used herein, the phrases "DP-mediated condition or disorder," "a
condition or disorder mediated by DP" and related phrases and terms refer to a
condition or disorder characterized by inappropriate, e.g., less than or
greater than
normal, DP activity. Inappropriate DP functional activity might arise as the
result of
DP expression in cells which normally do not express DP, increased DP
expression or
degree of intracellular activation (leading to, e.g., inflammatory and immune-
related
disorders and diseases) or decreased DP expression. A DP-mediated condition or
disorder may be completely or partially mediated by inappropriate DP
functional
activity. However, a DP-mediated condition or disorder is one in which
modulation of
DP results in some effect on the underlying condition or disorder (e.g., an DP
antagonist or agonist results in some improvement in patient well-being in at
least some
patients).
The term "alkyl," by itself or as part of another substituent, means,
unless otherwise stated, a straight or branched chain, or cyclic hydrocarbon
radical, or
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
12
combination thereof, which is fully saturated, having the number of carbon
atoms
designated (i.e., Cl-C8 means one to eight carbons). Examples of alkyl groups
include
methyl, ethyl, n-propyl, isopropyl, n-butyl, t-butyl, isobutyl, sec-butyl,
cyclohexyl,
(cyclohexyl)methyl, cyclopropylmethyl, homologs and isomers of, for example, n-
pentyl, n-hexyl, n-heptyl, n-octyl and the like.
The term "alkenyl", by itself or as part of another substituent, means a
straight or branched chain, or cyclic hydrocarbon radical, or combination
thereof,
which may be mono- or polyunsaturated, having the number of carbon atoms
designated (i.e., C2-C8 means two to eight carbons) and one or more double
bonds.
Examples of alkenyl groups include vinyl, 2-propenyl, crotyl, 2-isopentenyl, 2-
(butadienyl), 2,4-pentadienyl, 3-(1,4-pentadienyl) and higher homologs and
isomers
thereof.
The term "alkynyl", by itself or as part of another substituent, means a
straight or branched chain hydrocarbon radical, or combination thereof, which
may be
mono- or polyunsaturated, having the number of carbon atoms designated (i.e.,
C2-C8
means two to eight carbons) and one or more triple bonds. Examples of alkynyl
groups include ethynyl, 1- and 3-propynyl, 3-butynyl and higher homologs and
isomers
thereof.
The term "alkylene" by itself or as part of another substituent means a
divalent radical derived from alkyl, as exemplified by -CH2CH2CH2CH2-.
Typically,
an alkyl (or alkylene) group will have from 1 to 24 carbon atoms, with those
groups
having 10 or fewer carbon atoms being preferred in the present invention. A
"lower
alkyl" or "lower alkylene" is a shorter chain alkyl or alkylene group,
generally having
eight or fewer carbon atoms.
The terms "alkoxy," "alkylamino" and "alkylthio" (or thioalkoxy) are
used in their conventional sense, and refer to those alkyl groups attached to
the
remainder of the molecule via an oxygen atom, an amino group, or a sulfur
atom,
respectively. Similarly, the term dialkylamino refers to an amino group having
two
attached alkyl groups that can be the same or different.
The term "heteroalkyl," by itself or in combination with another term,
means, unless otherwise stated, a stable straight or branched chain, or cyclic
hydrocarbon radical, or combinations thereof, consisting of the stated number
of carbon
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
13
atoms and from one to three heteroatoms selected from 0, N, Si and S, and
wherein the
nitrogen and sulfur atoms may optionally be oxidized and the nitrogen
heteroatom may
optionally be quaternized. The heteroatom(s) 0, N and S may be placed at any
interior
position of the heteroalkyl group. The heteroatom Si may be placed at any
position of
the heteroalkyl group, including the position at which the alkyl group is
attached to the
remainder of the molecule. Examples include -CH2-CH2-O-CH3, -CH2-CHZ-NH-CH3, -
CH2-CH2-N(CH3)-CH3, -CH2-S-CH2-CH3, -CH2-CH2,-S(O)-CH3, -CH2-CH2-S(O)2-
CH3, -CH=CH-O-CH3, -Si(CH3)3, -CH2-CH=N-OCH3, and -CH=CH-N(CH3)-CH3. Up
to two heteroatoms may be consecutive, such as, for example, -CH2-NH-OCH3 and -
CH2-O-Si(CH3)3. When a prefix such as (C2-C8) is used to refer to a
heteroalkyl group,
the number of carbons (2-8, in this example) is meant to include the
heteroatoms as
well. For example, a C2-heteroalkyl group is meant to include, for example, -
CH2OH
(one carbon atom and one heteroatom replacing a carbon atom) and -CH2SH. The
term
"heteroalkylene" by itself or as part of another substituent means a divalent
radical
derived from heteroalkyl, as exemplified by -CH2-CH2-S-CH2CH2- and -CH2-S-CH2-
CH2-NH-CH2-. For heteroalkylene groups, heteroatoms can also occupy either or
both
of the chain termini (e.g., alkyleneoxy, alkylenedioxy, alkyleneamino,
alkylenediamino, and the like). Still further, for alkylene and heteroalkylene
linking
groups, no orientation of the linking group is implied.
The terms "cycloalkyl" and "heterocycloalkyl", by themselves or in
combination with other terms, represent, unless otherwise stated, cyclic
versions of
"alkyl" and "heteroalkyl", respectively. Thus, the terms "cycloalkyl" and
"heterocycloalkyl" are meant to be included in the terms "alkyl" and
"heteroalkyl",
respectively. Additionally, for heterocycloalkyl, a heteroatom can occupy the
position
at which the heterocycle is attached to the remainder of the molecule.
Examples of
cycloalkyl include cyclopentyl, cyclohexyl, 1-cyclohexenyl, 3-cyclohexenyl,
cycloheptyl, and the like. Examples of heterocycloalkyl include 1-(1,2,5,6-
tetrahydropyridyl), 1-piperidinyl, 2-piperidinyl, 3-piperidinyl, 4-
morpholinyl, 3-
morpholinyl, tetrahydrofuran-2-yl, tetrahydrofuran-3-yl, tetrahydrothien-2-yl,
tetrahydrothien-3-yl, 1-piperazinyl, 2-piperazinyl, and the like.
The terms "halo" or "halogen," by themselves or as part of another
substituent, mean, unless otherwise stated, a fluorine, chlorine, bromine, or
iodine
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
14
atom. Additionally, terms such as "haloalkyl", are meant to include alkyl
substituted
with halogen atoms which can be the same or different, in a number ranging
from one
to (2m'+1), where m' is the total number of carbon atoms in the alkyl group.
For
example, the term "halo(C1-C4)alkyl" is meant to include trifluoromethyl,
2,2,2-
trifluoroethyl, 4-chlorobutyl, 3-bromopropyl, and the like. Thus, the term
"haloalkyl"
includes monohaloalkyl (alkyl substituted with one halogen atom) and
polyhaloalkyl
(alkyl substituted with halogen atoms in a number ranging from two to (2m'+1)
halogen atoms). The term "perhaloalkyl" means, unless otherwise stated, alkyl
substituted with (2m'+1) halogen atoms, where m' is the total number of carbon
atoms
in the alkyl group. For example, the term "perhalo(C1-C4)alkyl", is meant to
include
trifluoromethyl, pentachloroethyl, 1, 1, 1 -trifluoro-2-bromo-2-chloroethyl,
and the like.
The term "aryl" means, unless otherwise stated, a polyunsaturated,
typically aromatic, hydrocarbon substituent which can be a single ring or
multiple rings
(up to three rings) which are fused together or linked covalently. The term
"heteroaryl"
refers to aryl groups (or rings) that contain from one to four heteroatoms
selected from
the group consisting of N, 0 and S, wherein the nitrogen and sulfur atoms are
optionally oxidized, and the nitrogen atom(s) are optionally quaternized. A
heteroaryl
group can be attached to the remainder of the molecule through a heteroatom.
Non-
limiting examples of aryl and heteroaryl groups include phenyl, 1-naphthyl, 2-
naphthyl,
4-biphenyl, 1-pyrrolyl, 2-pyrrolyl, 3-pyrrolyl, 3-pyrazolyl, 2-imidazolyl, 4-
imidazolyl,
pyrazinyl, 2-oxazolyl, 4-oxazolyl, 2-phenyl-4-oxazolyl, 5-oxazolyl, 3-
isoxazolyl, 4-
isoxazolyl, 5-isoxazolyl, 2-thiazolyl, 4-thiazolyl, 5-thiazolyl, 2-furyl, 3-
furyl, 2-thienyl,
3-thienyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 2-pyrimidyl, 4-pyrimidyl, 2-
pyrimidinyl, 4-
pyrimidinyl, 5- pyrimidinyl, 3-pyridazinyl, 4- pyridazinyl, 5-benzothiazolyl,
purinyl, 2-
benzimidazolyl, 5-indolyl, 1H-indazole, carbazole, a-carboline, (3-carboline,
y-
carboline, 1-isoquinolyl, 5-isoquinolyl, 2-quinoxalinyl, 5-quinoxalinyl, 2-
quinolyl, 3-
quinolyl, 4-quinolyl, 5-quinolyl, 6-quinolyl, 7-quinolyl and 8-quinolyl.
Preferably, the term "aryl" refers to a phenyl or naphthyl group which is
unsubstituted or substituted. Preferably, the term "heteroaryl" refers to a
pyrrolyl,
pyrazolyl, imidazolyl, pyrazinyl, oxazolyl, isoxazolyl, thiazolyl, furyl,
thienyl, pyridyl,
pyrimidyl, benzothiazolyl, purinyl, benzimidazolyl, indolyl, isoquinolyl,
quinoxalinyl
or quinolyl group which is unsubstituted or substituted.
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
For brevity, the term "aryl" when used in combination with other terms
(e.g., aryloxy, arylthioxy, arylalkyl) includes both aryl and heteroaryl rings
as defined
above. Thus, the term "arylalkyl" is meant to include those radicals in which
an aryl
group is attached to an alkyl group (e.g., benzyl, phenethyl, pyridylmethyl
and the like)
5 including those alkyl groups in which a carbon atom (e.g., a methylene
group) has been
replaced by, for example, an oxygen atom (e.g., phenoxymethyl, 2-
pyridyloxymethyl,
3-(1-naphthyloxy)propyl, and the like).
Each of the above terms (e.g., "alkyl," "heteroalkyl," "aryl" and
"heteroaryl") is meant to include both substituted and unsubstituted forms of
the
10 indicated radical, unless otherwise indicated. Preferred substituents for
each type of
radical are provided below.
Substituents for the alkyl and heteroalkyl radicals (as well as those
groups referred to as alkylene, alkenyl, heteroalkylene, heteroalkenyl,
alkynyl,
cycloalkyl, heterocycloalkyl, cycloalkenyl and heterocycloalkenyl) can be a
variety of
15 groups selected from: -OR', =O, =NR', =N-OR', -NR'R", -SR', halogen, -
SiR'R"R" ,
-OC(O)R', -C(O)R', -CO2R', -CONR'R", -OC(O)NR'R", -NR"C(O)R', -NR'-
C(O)NR"R", -NR'-SO2NR"R`, -NR"CO2R', -NH-C(NH2)=NH, -NR'C(NH2)=NH, -
NH-C(NH2)=NR', -S(O)R', -SO2R', -SO2NR'R", -NR"SO2R, -CN and -NO2, in a
number ranging from zero to three, with those groups having zero, one or two
substituents being particularly preferred. R', R" and R"' each independently
refer to
hydrogen, unsubstituted (C1-C3)alkyl and heteroalkyl, unsubstituted aryl, aryl
substituted with one to three halogens, unsubstituted alkyl, alkoxy or
thioalkoxy
groups, or aryl-(C,-C4)alkyl groups. When R' and R" are attached to the same
nitrogen
atom, they can be combined with the nitrogen atom to form a 5-, 6- or 7-
membered
ring. For example, -NR'R" is meant to include 1-pyrrolidinyl and 4-
morpholinyl.
Typically, an alkyl or heteroalkyl group will have from zero to three
substituents, with
those groups having two or fewer substituents being preferred in the present
invention.
More preferably, an alkyl or heteroalkyl radical will be unsubstituted or
monosubstituted. Most preferably, an alkyl or heteroalkyl radical will be
unsubstituted.
From the above discussion of substituents, one of skill in the art will
understand that the
term "alkyl" is meant to include groups such as trihaloalkyl (e.g., -CF3 and -
CH2CF3).
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
16
Preferred substituents for the alkyl and heteroalkyl radicals are selected
from: -OR', =O, -NR'R", -SR', halogen, -SiR'R"R"', -OC(O)R', -C(O)R', -CO2R', -
CONR'R", -OC(O)NR'R", -NR"C(O)R', -NR"CO2R', -NR'-SO2NR"R`, -S(O)R', -
SO2R', -SO2NR'R", -NR"SO2R, -CN and -NO2, where R' and R" are as defined
above.
Further preferred substituents are selected from: -OR', =O, -NR'R", halogen, -
OC(O)R', -CO2R', -CONR'R", -OC(O)NR'R", -NR"C(O)R', -NR"CO2R', -NR'-
S02NR"R"', -SO2R', -SO2NR'R", -NR"SO2R, -CN and -NO2.
Similarly, substituents for the aryl and heteroaryl groups are varied and
are selected from: -halogen, -OR', -OC(O)R', -NR'R", -SR', -R', -CN, -NO2, -
CO2R',
-CONR'R", -C(O)R', -OC(O)NR'R", -NR"C(O)R', -NR"C(O)2R', -NR'-C(O)NR"R"',
-NH-C(NH2)=NH, -NR'C(NH2)=NH, -NH-C(NH2)=NR', -S(O)R', -S(O)2R',
-S(O)2NR'R", -N3, -CH(Ph)2, perfluoro(CI-C4)alkoxy, and perfluoro(CI-C4)alkyl,
in a
number ranging from zero to the total number of open valences on the aromatic
ring
system; and where R', R" and R"' are independently selected from hydrogen,
(CI-C8)alkyl and heteroalkyl, unsubstituted aryl and heteroaryl,
(unsubstituted aryl)-
(CI-C4)alkyl, and (unsubstituted aryl)oxy-(CI-C4)alkyl.
Two of the substituents on adjacent atoms of the aryl or heteroaryl ring
may optionally be replaced with a substituent of the formula -T-C(O)-(CH2)q-U-
,
wherein T and U are independently -NH-, -0-, -CH2- or a single bond, and q is
an
integer of from 0 to 2. Alternatively, two of the substituents on adjacent
atoms of the
aryl or heteroaryl ring may optionally be replaced with a substituent of the
formula -A-
(CH2)r B-, wherein A and B are independently -CH2-, -0-, -NH-, -5-, -S(O)-, -
S(O)2-, -
S(O)2NR'- or a single bond, and r is an integer of from 1 to 3. One of the
single bonds
of the new ring so formed may optionally be replaced with a double bond.
Alternatively, two of the substituents on adjacent atoms of the aryl or
heteroaryl ring
may optionally be replaced with a substituent of the formula -(CH2),-X-(CH2)t-
, where
s and t are independently integers of from 0 to 3, and X is -0-, -NR'-, -5-, -
S(O)-, -
S(O)2-, or -S(O)2NR'-. The substituent R' in -NR'- and -S(O)2NR'- is selected
from
hydrogen or unsubstituted (CI-C6)alkyl. Otherwise, R' is as defined above.
As used herein, the term "heteroatom" is meant to include oxygen (0),
nitrogen (N), sulfur (S) and silicon (Si).
The term "pharmaceutically acceptable salts" is meant to include salts of
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
17
the active compounds which are prepared with relatively nontoxic acids or
bases,
depending on the particular substituents found on the compounds described
herein.
When compounds of the invention contain relatively acidic functionalities,
base
addition salts can be obtained by contacting the neutral form of such
compounds with a
sufficient amount of the desired base, either neat or in a suitable inert
solvent.
Examples of pharmaceutically acceptable base addition salts include sodium,
potassium, calcium, ammonium, organic amino, or magnesium salt, or a similar
salt.
When compounds of the invention contain relatively basic functionalities, acid
addition
salts can be obtained by contacting the neutral form of such compounds with a
sufficient amount of the desired acid, either neat or in a suitable inert
solvent.
Examples of pharmaceutically acceptable acid addition salts include those
derived from
inorganic acids like hydrochloric, hydrobromic, nitric, carbonic,
monohydrogencarbonic, phosphoric, monohydrogenphosphoric,
dihydrogenphosphoric,
sulfuric, monohydrogensulfuric, hydriodic, or phosphorous acids and the like,
as well
as the salts derived from relatively nontoxic organic acids like acetic,
propionic,
isobutyric, maleic, malonic, benzoic, succinic, suberic, fumaric, mandelic,
phthalic,
benzenesulfonic, p-tolylsulfonic, citric, tartaric, methanesulfonic, and the
like. Also
included are salts of amino acids such as arginate and the like, and salts of
organic
acids like glucuronic or galacturonic acids and the like (see, for example,
Berge et al.
(1977) J Pharm. Sci. 66:1-19). Certain specific compounds of the invention
contain
both basic and acidic functionalities that allow the compounds to be converted
into
either base or acid addition salts.
The neutral forms of the compounds may be regenerated by contacting
the salt with a base or acid and isolating the parent compound in the
conventional
manner. The parent form of the compound differs from the various salt forms in
certain
physical properties, such as solubility in polar solvents, but otherwise the
salts are
equivalent to the parent form of the compound for the purposes of the
invention.
In addition to salt forms, the invention provides compounds which are in
a prodrug form. Prodrugs of the compounds described herein are those compounds
that
readily undergo chemical changes under physiological conditions to provide the
compounds of the invention. Additionally, prodrugs can be converted to the
compounds of the invention by chemical or biochemical methods in an ex vivo
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
18
environment. For example, prodrugs can be slowly converted to the compounds of
the
invention when placed in a transdermal patch reservoir with a suitable enzyme
or
chemical reagent. Prodrugs are often useful because, in some situations, they
may be
easier to administer than the parent drug. They may, for instance, be
bioavailable by
oral administration whereas the parent drug is not. The prodrug may also have
improved solubility in pharmaceutical compositions over the parent drug. A
wide
variety of prodrug derivatives are known in the art, such as those that rely
on hydrolytic
cleavage or oxidative activation of the prodrug. An example, without
limitation, of a
prodrug would be a compound of the invention which is administered as an ester
(the
"prodrug"), but then is metabolically hydrolyzed to the carboxylic acid, the
active
entity. Additional examples include peptidyl derivatives of a compound of the
invention.
Certain compounds of the invention can exist in unsolvated forms as
well as solvated forms, including hydrated forms. In general, the solvated
forms are
equivalent to unsolvated forms and are intended to be encompassed within the
scope of
the invention. Certain compounds of the invention may exist in multiple
crystalline or
amorphous forms. In general, all physical forms are equivalent for the uses
contemplated by the invention and are intended to be within the scope of the
invention.
Certain compounds of the invention possess asymmetric carbon atoms
(optical centers) or double bonds; the racemates, enantiomers, diastereomers,
geometric
isomers and individual isomers are all intended to be encompassed within the
scope of
the invention. These isomers can be resolved or asymmetrically synthesized
using
conventional methods to render the isomers "optically pure", i.e.,
substantially free of
its other isomers. If, for instance, a particular enantiomer of a compound of
the present
invention is desired, it may be prepared by asymmetric synthesis, or by
derivation with
a chrial auxilliary, where the resulting diastereomeric mixture is separated
and the
auxilliary group cleaved to provide the pure desired enantiomers.
Alternatively, where
the molecule contains a basic functional group, such as amino, or an acidic
functional
group, such as carboxyl, diastereomeric salts are formed with an appropriate
optically-
active acid or base, followed by resolution of the diasteromers thus formed by
fractional crystallization or chromatagraphic means well known in the art, and
subsequent recovery of the pure enantiomers.
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
19
The compounds of the invention may also contain unnatural proportions
of atomic isotopes at one or more of the atoms that constitute such compounds.
For
example, the compounds may be radiolabeled with radioactive isotopes, such as
for
example tritium (3H), iodine-125 (125I) or carbon-14 ('4C). Radiolabled
compounds are
useful as therapeutic or prophylactic agents, e.g., cancer therapeutic agents,
research
reagents, e.g., CRTH2 assay reagents, and diagnostic agents, e.g., in vivo
imaging
agents. All isotopic variations of the compounds of the invention, whether
radioactive
or not, are intended to be encompassed within the scope of the invention.
Embodiments of the Invention
A class of compounds that modulate CRTH2 and/or DP and/or one or
more other PGD2 receptors has been discovered. Depending on the biological
environment (e.g., cell type, pathological condition of the host, etc.), these
compounds
can activate or inhibit the actions of CRTH2 and/or one or more other PGD2
receptors
(e.g., ligand binding). By activating or inhibiting CRTH2 and/or one or more
other
PGD2 receptors, the compounds will find use as therapeutic agents capable of
modulating diseases and conditions responsive to modulation of CRTH2 and/or
one or
more other PGD2 receptors and/or mediated by CRTH2 and/or one or more other
PGD2
receptors. As noted above, examples of such diseases and conditions include
inflammatory conditions, immune disorders, asthma, allergic rhinitis, eczema,
psoriasis,
atopic dermatitis, fever, sepsis, systemic lupus erythematosus, diabetes,
rheumatoid
arthritis, multiple sclerosis, atherosclerosis, transplant rejection,
inflammatory bowel
disease, cancer, viral infection, thrombosis, fibrosis, flushing, Crohn's
disease,
ulcerative colitis, chronic obstructive pulmonary disease, inflammation, pain,
conjunctivitis, nasal congestion and urticaria. Additionally, the compounds
are useful
for the treatment and/or prevention of complications of these diseases and
disorders
(e.g., cardiovascular disease).
While the compounds of the invention are believed to exert their effects
by interacting with CRTH2, the mechanism of action by which the compounds act
is
not a limiting embodiment of the invention. For example, compounds of the
invention
may interact with PGD2 receptor subtypes other than CRTH2, e.g., DP receptor,
and/or
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
other prostanoid receptors, e.g., thromboxane A2 (TXA2) receptor. Indeed, as
alluded
to above, the present invention specifically contemplates the use of the
disclosed
compounds to modulate one or more PGD2 receptors other than CRTH2.
Compounds contemplated by the invention include, but are not limited
5 to, the exemplary compounds provided herein.
Compounds
In one aspect, the invention provides compounds of formula (I):
R2
\
X-W-L-Z
Al
10 p'_A2
I
or a pharmaceutically acceptable salt or prodrug thereof. In formula I, the
symbol A'
represents C(R3) or N and the symbol A2 represents C(R6) or N. When A' is
C(R3) and
A2 is C(R6), the symbol D' represents C(R4)=C(RS), C(R4)=N, NR7, 0 or S. When
A'
15 or A2 is N, the symbol D' represents C(R4)=C(R 5) or C(R4)=N. Thus, A', A2,
the
carbon atoms to which A' and A2 are attached and D' combine to form a 5- or 6-
membered ring, e.g.,
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
21
R3
R3 R3 R3
R4 Rs R4 N F26 N \ Rs iN s
RS Ra R7 R
*FR N/ Ni R3 /
R4 s Ra N Rs N Rs O
Rs
R5 R4
R3
R3 R3/I\
*~---~-N I \ R3
a R4 ~N 11 N N N S
R Rs
R5 R4
N' Y Y,
N
R N R4 ~N~N N\ N
5R5 RR4
Exemplary combinations of A', A2, the carbon atoms to which A' and A2 are
attached
and D' are:
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
22
&F3CH02CMe0
0
N Me \ I Ph \ I \
Et" CI
0 0 0
H H
MeO \ Y H2N F3C,- . N
t-Bu N
0 0 0
H H H
N N \ ,N \ N \
i-Pr Ph
0 0 0 O
/ I 0 / I Me 0 0
02N \ H2N"k N \ PhNAN Et~N \
H H H H
The letter W represents a divalent group selected from a single bond, an
aromatic ring, a heteroaromatic ring, a cyclo(C3-C8)alkane ring and a
heterocyclo(C3-
C8)alkane ring. Exemplary W groups are:
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
23
CF3 F OMe CI
OMe
CI OMe
OMe OMe OMe OMe
Me MeO \ \ I \
Br OMe O Br
OMe
\ I F
MeO ,
Et
The letter X represents a divalent linkage selected from -0-, -S(O)k-, -
CRaRb-, -C(O)-, -NR8- and -C(NR9)-. Exemplary X groups are -0-, -SO2-, -CH2-,
-C(O)-, -CH(OH)- and -NH-.
The letter Y represents a divalent linkage selected from a single bond, -
S(O)kNR10-, -C(O)NR10-, (C1-C4)alkylene, hetero(C2-C4)alkylene, -N(R'
1)C(O)NR10-
, -N(R")S(O)kNR10-, N(R")C02-, -NR"-, -0- and -S(O)k-. Exemplary Y groups
are -SO2NH-, -SO2NMe-, -C(O)NH-, -NH-, -NHCO2- and -NHC(O)NMe-.
The letter Z represents -C02R12, -C(O)NRt2R13 or heteroaryl.
Exemplary Z groups are -CO2H, -C(O)NHEt, -C(O)NH2, -CO2Et, -CO2Me,
-C02CH2S(O)Me, 5-tetrazolyl and -C(O)NHOH.
The letter L represents a divalent linkage selected from a single bond,
(C1-C6)alkylene and (C2-C4)heteroalkylene. Exemplary L groups are methylene,
ethylene, chloromethylene, hydroxymethylene and methylmethylene.
The substituent R2 is hydrogen, -OR', (C1-C8)alkyl, hetero(C2-Cg)alkyl,
aryl, heteroaryl or aryl(C1-C4)alkyl. Exemplary R2 groups are 4-tolyl, 2-
naphthyl,
methyl, phenyl, 2,4-dichlorophenyl, 4-methoxyphenyl, 4-trifluoromethoxyphenyl,
2-
chlorophenyl, 4-chlorophenyl, 3-chlorophenyl, 2,4-dichloro-5-methylphenyl, 4-n-
pentylphenyl, 4-cyanophenyl, 4-n-butoxyphenyl, 2-cyano-3-chlorophenyl, 3-
chloro-4-
methylphenyl, 2-methoxy-5-bromophenyl, 5-trifluoromethoxy-2-pyridyl, 8-
quinolyl, 2-
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
24
thieneyl, 3-methyl-7-chlorobenzothienyl, 1-methyl-4-imidazolyl, benzyl and 2,4-
difluorophenyl.
R3, R4, R5 and R6 are independently hydrogen, halogen, (C1-C8)alkyl,
fluoro(C1-C4)alkyl, hetero(C2-C8)alkyl, aryl, heteroaryl, aryl(C1-C4)alkyl, -
NR'R",
-OR', -NO2, -CN, -C(O)R', -C02R', -C(O)NR'R", (C1-C4)alkylene-C(O)NR'R", -
S(O)mR', -S(O)kNR'R", -OC(O)OR', -OC(O)R', -OC(O)NR'R", -
N(R"')C(O)NR'R", -N(R")C(O)R' or -N(R")C(O)OR'.
R7 is hydrogen, halogen, (C1-C8)alkyl, fluoro(C1-C4)alkyl, hetero(C2-
C8)alkyl, aryl, heteroaryl, aryl(C1-C4)alkyl, -NR'R", -OR', -C(O)R', -C02R', -
C(O)NR'R", -S(O)mR' or -S(O)kNR'R".
Ra and Rb are independently hydrogen, (C1-C6)alkyl, hetero(C2-C6)alkyl,
aryl(C1-C4)alkyl, -OR' or -NR'R".
R8, R10 and R" are independently hydrogen, (C1-C8)alkyl, fluoro(C1-
C4)alkyl, hetero(C2-C8)alkyl, aryl, heteroaryl, aryl(C1-C4)alkyl, -C(O)R', -
C02R', -
C(O)NR'R", -S(O)mR' or -S(O)kNR'R".
R9 is (C1-C6)alkyl, hetero(C2-C6)alkyl, aryl(C1-C4)alkyl, -OR' or -
NR'R".
R12 and R13 are independently hydrogen, (C1-C6)alkyl, hetero(C2-
C6)alkyl, aryl, aryl(C 1 -C4)alkyl or heteroaryl.
Each R', R" and R"' is independently hydrogen, (C1-C6)alkyl, cyclo(C3-
C8)alkyl, aryl or aryl(C1-C4)alkyl.
Each subscript k is 0, 1 or 2.
The subscript m is 0, 1, 2 or 3.
In optional embodiments, A2 and W are combined to form a 5-, 6-, 7- or
8-membered fused ring containing from 0 to 3 heteroatoms selected from N, 0
and S;
X and Y are combined to form a 5-, 6-, 7- or 8-membered fused ring containing
from 0
to 3 heteroatoms selected from N, 0 and S; Ra and Rb are combined to form a 5-
, 6-, 7-
or 8-membered spiro ring containing from 0 to 3 heteroatoms selected from N, 0
and S;
R8 or R9 is combined with W to form a 5-, 6-, 7- or 8-membered fused ring
containing
from 0 to 3 heteroatoms selected from N, 0 and S; Ra, Rb, R8 or R9 is combined
with Y
to form a 5-, 6-, 7- or 8-membered fused ring containing from 0 to 3
heteroatoms
selected from N, 0 and S; R12 and R13 are combined with the nitrogen atom to
which
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
they are attached to form a 5-, 6- or 7-membered ring containing from 0 to 2
additional
heteroatoms selected from N, 0 and S; and when R' and R" are attached to the
same
nitrogen atom, R' and R" are combined to form a 5-, 6-, 7- or 8-membered ring
containing from 1 to 3 heteroatoms selected from N, 0 and S.
5 Within the above compounds of formula I, compounds wherein A2 is
C(O-substituted (C2-C4)alkyl) or C(S-substituted (C2-C4)alkyl); A' is C(R3),
D' is
C(R4)=C(R5); W is a benzene ring; X is -0- or -S-; and -Y-R2 is
-NHSO2-heterocyclo(C3-C8)alkyl, -NHSO2-phenyl or -NHSO2-heteroaryl are
excluded.
10 In one group of embodiments, A' is C(R3), A2 is C(R6) and D' is
C(R4)=C(R5).
One group of embodiments is represented by the formula (II):
R2
\
R3 X-W-L-Z
R4 R6
R5
II
15 wherein R3, R4, R5 and R6 are independently hydrogen, halogen, (CI-
C8)alkyl,
fluoro(C I -C4)alkyl, hetero(C2-C8)alkyl, aryl, heteroaryl, aryl(C I -
C4)alkyl,
-NR'R",
-OR', -NO2, -CN, -C(O)R', -CO2R', -C(O)NR'R", (CI-C4)alkylene-C(O)NR'R", -
S(O)mR', -S(O)kNR'R", -OC(O)OR', -OC(O)R', -OC(O)NR'R", -
N(R"')C(O)NR'R", -N(R")C(O)R' or -N(R")C(O)OR'. The other variables, (e.g., W,
20 X, Y, Z, L, R2, R' and R"), have the meanings provided above. Optionally,
R6 may be
combined with W to form a 5-, 6-, 7- or 8-membered fused ring containing from
0 to 3
heteroatoms selected from N, 0 and S.
Within the above compounds of formula II, compounds wherein R6 is
-0-substituted (C2-C4)alkyl or -S-substituted (C2-C4)alkyl; W is a benzene
ring; X is
25 -0- or -5-; and -Y-R2 is -NHSO2-heterocyclo(C3-C8)alkyl, -NHSO2-phenyl or
-NHSO2-heteroaryl are excluded.
Within formula II are provided several groups of embodiments.
(1) In one group of embodiments, R6 is hydrogen, halogen, (C I -C8)alkyl,
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
26
fluoro(C1-C4)alkyl, aryl, heteroaryl, aryl(C1-C4)alkyl, -NR'R", NO2, -CN, -
C(O)R', -
CO2R', -C(O)NR'R", -S(O)mR', -S(O)kNR'R", -OC(O)OR', -OC(O)R', -
OC(O)NR'R", -N(R"')C(O)NR'R", -N(R")C(O)R' or -N(R")C(O)OR'.
(2) In another group of embodiments, R3, R4 and R5 are independently
hydrogen, halogen, (C1-C8)alkyl, fluoro(C1-C4)alkyl, -OR', -NR'R", -NO2, -CN, -
C(O)R', -C02R', -C(O)NR'R", (C1-C4)alkylene-C(O)NR'R", -N(R")C(O)R', -
N(R"')C(O)NR'R" or heteroaryl.
(3) In another group of embodiments, R6 is hydrogen, halogen, (C1-
C8)alkyl, fluoro(C 1 -C4)alkyl, -NR'R", -NO2, -CN, -C(O)R', -C02R', -
C(O)NR'R",
(C1-C4)alkylene-C(O)NR'R", -N(R")C(O)R', -N(R"')C(O)NR'R" or heteroaryl.
(4) In another group of embodiments, R6 is hydrogen.
(5) In another group of embodiments, R3, R5 and R6 are hydrogen.
(6) In another group of embodiments, R4 is halogen, (C1-C8)alkyl,
fluoro(C1-C4)alkyl, -OR', -NO2, -C(O)R', -C02R', -C(O)NR'R", (C1-C4)alkylene-
C(O)NR'R", -N(R")C(O)R', -N(R"')C(O)NR'R" or heteroaryl.
(7) In another group of embodiments, R3, R5 and R6 are hydrogen and R4 is
halogen, (C1-C8)alkyl, fluoro(C1-C4)alkyl, -OR', -NO2, -C(O)R', -C02R', -
C(O)NR'R", (C1-C4)alkylene-C(O)NR'R", -N(R")C(O)R', -N(R`)C(O)NR'R" or
heteroaryl.
(8) In another group of embodiments, R6 may be combined with W to form
a 5-, 6-, 7- or 8-membered fused ring containing from 0 to 3 heteroatoms
selected from
N, 0 and S.
(9) In another group of embodiments, W is an aromatic ring, a
heteroaromatic ring or a cyclo(C3-C8)alkane ring.
(10) In another group of embodiments, W is benzene, indole, benzofuran,
benzothiazole, indoline, dihydrobenzofuran, dihydrobenzothiazole,
benzimidazole,
benzoxazole, benzthiazole or cyclohexane.
(11) In another group of embodiments, W is benzene.
(12) One group of embodiments is represented by formula (III):
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
27
R2
\
R3 X (R14)n
i L-Z
R4 R6 t
III
wherein each R14 is independently halogen, (C1-Cg)alkyl, fluoro(C1-C4)alkyl, -
OR', -
NR'R", -NO2, -CN, -C(O)R' or aryl and the subscript n is 0, 1, 2, 3 or 4.
Optionally,
5 any two adjacent R14 groups may be combined to form a 5-, 6-, 7- or 8-
membered fused
ring containing from 0 to 3 heteroatoms selected from N, 0 and S. The
variables X, Y,
Z, L, R2, R3, R4, R5, R6, R' and R" have the meanings and groupings provided
above.
Optionally, when an R14 group is adjacent to X, the R14 group may be combined
with
R6 to form a 5-, 6-, 7- or 8-membered fused ring containing from 0 to 3
heteroatoms
selected from N, 0 and S.
One of skill in the art will understand that a number of structural isomers
are represented by formula III, for example:
z
R2 R2 R2 " L
Y Y Y
14 R3 X I LZ :::6: 4 64 6
R R R R
IIIa IIIb IIIc
Within formula III are provided several groups of embodiments.
(a) In one group of embodiments, n is 0.
(b) In another group of embodiments, n is 1, 2 or 3.
(c) In another group of embodiments, n is 1.
(d) In another group of embodiments, each R14 is independently halogen,
(C1-C8)alkyl, fluoro(C 1 -C4)alkyl, -OR', -C(O)R' or aryl.
(e) In another group of embodiments, R14 is halogen, fluoro(C1-C4)alkyl or
-OR' and n is 1.
(f) In another group of embodiments, each R14 is independently (C1-
C6)alkyl, halogen, fluoro(C I -C4)alkyl, -C(O)R', aryl or -OR' and n is 2 or
3.
(g) In another group of embodiments, an R14 group adjacent to X is
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
28
combined with R6 to form a 5-, 6-, 7- or 8-membered fused ring containing from
0 to 3
heteroatoms selected from the group consisting of N, 0 and S.
(h) One group of embodiments is represented by formula (IV):
R2
Y
X
::Pz
5 IV
wherein the subscript p is 0, 1, 2 or 3. The variables X, Y, Z, L, R2, R3, R4,
R5, R14, R'
and R" have the meanings provided above. In one embodiment, p is 0. In another
embodiment, p is 1.
(i) Another group of embodiments is represented by formula (V):
R2
\Y
X (R14 )p
::xz
L-10 5
V
wherein D2 is 0, S(O)k, CRaRb, C(O) or NR8 and the subscript p is 0, 1, 2 or
3. The
variables X, Y, Z, L, R2, R3, R4, R5, R6, Ra, Rb, R8 and R14 and the subscript
k have the
meanings and groupings provided above.
(i) Another group of embodiments is represented by formula (VI):
0~ 0
R2S"-NR10
::xc:zz
5
VI
wherein the variables X, Z, L, R2, R3, R4, R5, R6, R10 and R14 and the
subscript n have
the meanings and groupings provided above.
(j) Another embodiment is represented by formula (VII):
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
29
R2
\
R3 X (R14)n
(CH2)q-Z
R4 R6
VII
wherein the subscript q is 0, 1, 2, 3, 4, 5 or 6. The variables X, Y, Z, R2,
R3, R4, R5, R6,
R14, R' and R" and the subscript n have the meanings and groupings provided
above.
5 In one embodiment, q is 0, 1, 2 or 3. In another embodiment, q is 1 or 2. In
another
embodiment, q is 1.
(k) Another embodiment is represented by formula (VIII):
0\ ii
R2S~NR10
R3 \ X 3-CH2-Z
/ 5
VIII
wherein the variables X, Z, R2, R3, R4, R5, R6, R10 and R14 and the subscripts
n and q
have the meanings and groupings provided above.
(1) In another group of embodiments, X is -O-,-CRaR-, -C(O)- or -NR'-
In one embodiment, X is -0-. In another embodiment, X is -CRaRb-. In another
embodiment, X is -NRB-.
(m) In another group of embodiments, Y is -S(O)kNR10-, -C(O)NR' -, -
N(R11)C(O)NR10-, -N(R11)S(O)kNR10- or -N(R11)C02-. In one embodiment, Y is -
S(O)kNR10-.
(n) In another group of embodiments, X and Y are combined to form a 5-,
6-, 7- or 8-membered fused ring containing from 0 to 3 heteroatoms selected
from N, 0
and S.
(o) One group of embodiments is represented by the formula (IX):
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
O R Rd
R2
R3 'N (R14 In
z
R4 R6
R5
IX
wherein Re and Rd are independently hydrogen, (C1-C6)alkyl or =0. The
variables Z,
L, R2, R3, R4, R5, R6, R14, R' and R" and the subscript n have the meanings
and
5 groupings provided above.
(p) Another group of embodiments is represented by the formula (X):
O RC Rd
R2` RRe N R3 N R(R14 ) n
L -Z
R4 R6
R5
X
wherein Re and Rf are independently hydrogen, (C1-C6)alkyl or =0. The
variables Z, L,
10 R2, R3, R4, R5, R6, R14, R', R", Re and Rd and the subscript n have the
meanings and
groupings provided above.
(q) Another group of embodiments is represented by the formula (XI):
R R~ Rd
Re
2
N Rf
R3 /(R14 )n
11 1~ L -Z
R4 R
R5
XI
15 wherein the variables Z, L, R2, R3, R4, R5, R6, R14, R', R", Re, Rd, Re and
Rf and the
subscript n have the meanings and groupings provided above.
(r) In another group of embodiments Z is CO2R12, -C(O)NR12R13,
imidazolyl or tetrazolyl, wherein R12and R13 are as defined above. In one
embodiment,
Z is CO2R12, or -C(O)NR12R13, In another embodiment, Z is CO2H.
20 It is to be understood that the group -CO2H, as used herein, includes
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
31
bioisosteric replacements therefor, such as:
OO OO O R
/O O j~ S:=:O
S S R S
\OH \H~ \H R H 10 '
0 R 0 0 CF3
\A S SOH RCN
N N OH
H0 H H
CF3 N-S N-N N N-NH
C 3 / N 11 HEN 11 H N OH
OH
0 0
N-0 O-N S4 HN_~
OH OH \ NH NH
0 0
0
I I
P-OH
OH
and the like. See, e.g., The Practice of Medicinal Chemistry; Wermuth, C.G.,
Ed.;
Academic Press: New York, 1996; p. 203.
(s) In another group of embodiments, L a single bond or (C,-C6)alkylene.
In one embodiment, L is (C,-C6)alkylene.
(t) In another group of embodiments, R2 is phenyl or naphthyl.
(13) In another group of embodiments, W is benzofuran, benzothiazole,
indoline, dihydrobenzofuran, dihydrobenzothiazole, benzimidazole, benzoxazole
or
benzthiazole.
(14) One group of embodiments is represented by formula (XII):
R2
\Y L-Z
R3 X
R15
R4 R 4 B~
5 R14
P
XII
wherein B1 is N(R16), N, 0 or S, wherein R16 is hydrogen, halogen, (C,-
C8)alkyl,
fluoro(C,-C4)alkyl, hetero(C2-C8)alkyl, aryl, heteroaryl, aryl(C,-C4)alkyl, -
NR'R",
-OR', -C(O)R', -C02R', -C(O)NR'R", -S(O)mR' or -S(O)kNR'R", R' 5 is hydrogen
or
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
32
(C,-C8)alkyl and the dotted line indicates an optional bond. The variables X,
Y, Z, L,
R2, R3, R4, R5, R6, R14, R' and R" and the subscripts m and p have the
meanings and
groupings provided above.
(15) Another group of embodiments is represented by formula (XIII):
R2
Y
R3 X / B2
I I ~~-L z
R4 R6 14 ` B1
(R~a)5 P
XIII
wherein B2 is C(R'7) or N and R'7 is hydrogen, halogen, (C,-C8)alkyl,
fluoro(C,-
C4)alkyl, hetero(C2-C3)alkyl, aryl, heteroaryl, aryl(C I -C4)alkyl, -NR'R", -
OR', -
C(O)R', -C02R', -C(O)NR'R", -S(O)mR' or -S(O)kNR'R". The variables X, Y, Z, L,
B', R3, R4, R5, R6, R14, R' and R" and the subscripts m and p have the
meanings and
groupings provided above.
(16) Another group of embodiments is represented by formula (XIV):
R15
R2
Y B1
R3 X L-Z
4 6
R R (R14) P
5
XIV
wherein the variables X, Y, Z, L, B', R2, R3, R4, R5, R6, R14 and R15 and the
subscript p
have the meanings and groupings provided above.
(17) Another group of embodiments is represented by formula (XV):
R2
~Y 1R1a
R3 #FR; XR6
R15
R5 L-Z
XV
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
33
wherein the variables X, Y, Z, L, R2, R3, R4, R5, R6, R14 and R15 and the
subscript n
have the meanings and groupings provided above.
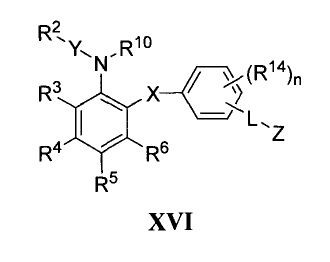
One group of embodiments is represented by formula (XVI):
R 2
R10
Y-1 N, 14
)n
R3
R4 Rs
R5
XVI
or a pharmaceutically acceptable salt or prodrug thereof. In formula (XVI),
the letter X
represents a divalent linkage selected from -0- and -S(O)k-. Exemplary X
groups are
-0-, -S-, and -SO2-.
letter Y represents a divalent linkage selected from a single bond,
-S(O)k-, -C(O)-, (C2-C4)alkylene, hetero(C2-C4)alkylene, and -0-. Exemplary Y
groups are -SO2-, -S-, and -C(O)-.
R2 is a substituted or unsubstituted benzene ring. A substituted benzene
ring will have from one to five substituents. Typically, a substituted benzene
ring will
have from one to three substituents. Substituents for the benzene ring are
varied will
include the preferred substituents for the aryl and heteroaryl groups as
provided above.
In certain embodiments, R2 is an unsubstituted benzene ring.
In other embodiments, R2 is a substituted benzene ring and at least one
substituent on the benzene ring is selected from the group consisting of
halogen,
-OCF3, -OCH3, -(C1-C5)alkyl, -CN, and -NO2.
In other embodiments, R2 is a benzene ring substituted with at least one
halogen.
In yet other embodiments, R2 is a benzene ring substituted with at least
one chlorine.
R3 and R5 are independently selected from the group consisting of
hydrogen, halogen, (C1-Cs)alkyl, fluoro(C I -C4)alkyl, hetero(C2-C8)alkyl,
aryl,
heteroaryl, aryl(C1-C4)alkyl, -NRR", -OR', -NO2, -CN, -C(O)R', -CO2R',
-C(O)NRR", -(C1-C4)alkylene-C(O)NRR", -S(O)mR', -S(O)kNRR", -OC(O)OR',
-OC(O)R', -OC(O)NRR", -N(R"')C(O)NRR", -N(R")C(O)R' and -N(R")C(O)OR',
where variables R', R", and R"' have the meanings provided below.
CA 02511214 2011-05-05
R4 is selected from the group consisting of hydrogen, -C(O)NR 12 R" and
-NHC(O)-alkyl, where R12 and R13 have the meanings provided below.
In certain embodiments, R4 is -NHC(O)-cyclo(C5-C7)alkyl.
In other embodiments, R4 is -C(O)NH-(C1-C4)alkyl.
R6 is selected from the group consisting of hydrogen, halogen, (C1_C8)alkyl,
fluoro(C1-C4)alkyl, hetero(C2-C8)alkyl, aryl, heteroaryl, aryl(C1-C4)alkyl, -
NRR", -NO2,
-CN, -C(O)R', -CO2R', -C(O)NRR", -(C1-C4)alkylene-C(O)NRR", -S(O)mR',
-S(O)kNRR", -OC(O)OR', -OC(O)R', -OC(O)NRR", N(R"')C(O)NRR", -N(R")C(O)R'
and -N(R")C(O)OR'.
In certain embodiments, R6 is selected from the group consisting of hydrogen,
halogen, (C1_C8)alkyl, fluoro(C1-C4)alkyl, aryl, heteroaryl, aryl(C1-C4)alkyl,
NRR", -NO2,
-CN, -C(O)R', -CO2R', -C(O)NRR", -(C1-C4)alkylene-C(O)NRR", -OC(O)OR',
-OC(O)R', -OC(O)NRR", -N(R"')C(O)NRR", -N(R")C(O)R' and -N(R")C(O)OR'.
R10 is selected from the group consisting of hydrogen, (C1-C8)alkyl,
fluoro(C1-C4)alkyl, hetero(C2-C8)alkyl, aryl, heteroaryl, aryl(C1-C4)alkyl, -
C(O)R', -CO2R',
-C(O)NRR", -S(O)mR' and -S(O)kNRR".
L is a divalent linkage selected from the group consisting of a single bond,
(C1-C6)alkylene and (C2-C4)heteroalkylene.
Z is selected from the group consisting Of -C02R 12, -C(O)NR 12 R" and
heteroaryl.
R12 and R13 are independently selected from the group consisting of hydrogen,
(C1-C8)alkyl, hetero(C2-C8)alkyl, aryl, aryl(C1-C4)alkyl and heteroaryl.
Each R14 is independently selected from the group consisting of halogen,
(C1-C8)alkyl, fluoro(C1-C4)alkyl, -OR', -NR'R", NO2, -CN, -C(O)R' and aryl.
Each R', R" and R"' is independently selected from the group consisting of
hydrogen, (C1-C6)alkyl, cyclo(C3-Cs)alkyl, aryl and aryl(C1-C4)alkyl.
Each subscript k is independently 0, 1 or 2.
The subscript m is independently 0, 1, 2 or 3.
The subscript n is 0, 1, 2, 3 or 4.
Within formula (XVI), one group of embodiments is represented by formula
(XVII):
34
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
2
R,Y.N.R10 (R14)n
1
3 X
a L-Z
I R6
R5
XVII
wherein each variable, for example, X, Y, R2 R3 R4, RS R6 R' , L Z R'2 R13 R1a
,
R', R", and R"', have the meanings and groups as provided for formula (XVI).
5 Within formula (XVII), several groups of embodiments are provided.
In one group of embodiments, Y is -SO2-, X is -0-, and R10 is hydrogen.
In another group of embodiments, R4 is -C(O)NH-(C1-C4)alkyl, and R6
is hydrogen.
In another group of embodiments, R2 is a benzene ring substituted with
10 1, 2, or 3 chlorine atoms.
In another group of embodiments, -L-Z taken together are -CH2COOH.
In another group of embodiments, the subscript n is 1 or 2.
In another group of embodiments, R14 is -OCH2CH3 or -OCH3.
In another group of embodiments, R3, R5 and R6 are each hydrogen.
15 Another group of embodiments within formula (XVII) is represented by
formula (XVIII):
00
2~S.NH
R (R14)n
R (CH2)q`Z
XVIII
wherein R4 is -C(O)NH-(C1-C4)alkyl, and the subscript q is 0, 1, 2, 3, 4, 5 or
6. Other
20 variables, for example, R14 and Z, have the meanings as provided for
formula (XVII).
In certain embodiments within formula (XVIII), each R'4 is
independently selected from the group consisting of (C,-C6)alkyl, halogen,
fluoro(C1-C4)alkyl, -C(O)R', aryl and -OR' and subscript n is 2 or 3.
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
36
Within formula (XVI), one preferred group of embodiments are
CI
O~ O Cl OO
SINH Me S, NH Me
CI H ~ o CI H ~~ ~~
N / OH N i OH
O o
0 O CI OõO
SINN CI S,NH CI
CI H I O CI '&0b
N \ OH ' HN H and
0
CI OO
SNH OMe
CI J:: H / O
N ~MeO \ OH
O
or pharmaceutically acceptable salt or prodrug thereof.
The invention encompasses novel compounds, novel pharmaceutical
compositions and/or novel methods of use. While some compounds disclosed
herein
are available from commercial sources, the pharmaceutical compositions or
methods of
using these compounds are novel. Unless otherwise indicated, it is to be
understood
that the invention includes those compounds that are novel, as well as
pharmaceutical
compositions, various methods (e.g., methods of treating or preventing certain
conditions and diseases mediated by CRTH2 and/or one or more other PGD2
receptors), and the like which include both the novel compounds of the
invention and
compounds that are commercially available.
Preparation of the Compounds
Synthesis routes to the compounds provided herein are described in the
Examples. One of skill in the art will understand that the synthetic routes
can be
modified to use different starting materials and/or alternate reagents to
accomplish the
desired transformations. Additionally, one of skill in the art will recognize
that
protecting groups may be necessary for the preparation of certain compounds
and will
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
37
be aware of those conditions compatible with a selected protecting group.
Accordingly,
the methods and reagents described herein are all expressed as non-limiting
embodiments.
Compositions
In another aspect, the invention provides pharmaceutical compositions
suitable for pharmaceutical use comprising one or more compounds of the
invention
and a pharmaceutically acceptable carrier, excipient or diluent.
The term "composition" as used herein is intended to encompass a
product comprising the specified ingredients (and in the specified amounts, if
indicated), as well as any product which results, directly or indirectly, from
combination of the specified ingredients in the specified amounts. By
"pharmaceutically acceptable" it is meant that the carrier or excipient is
compatible
with the other ingredients of the formulation and not deleterious to the
recipient thereof.
Formulation may improve one or more pharmacokinetic properties (e.g.,
oral bioavailabilty, membrane permeability) of a compound of the invention
(herein
referred to as the active ingredient).
The pharmaceutical compositions for the administration of the
compounds of this invention may conveniently be presented in unit dosage form
and
may be prepared by any of the methods well known in the art. All methods
include the
step of bringing the active ingredient into association with the carrier which
constitutes
one or more accessory ingredients. In general, the pharmaceutical compositions
are
prepared by uniformly and intimately bringing the active ingredient into
association
with a liquid carrier or a finely divided solid carrier or both, and then, if
necessary,
shaping the product into the desired formulation. In the pharmaceutical
composition
the active object compound is included in an amount sufficient to produce the
desired
effect upon the process or condition of diseases.
The pharmaceutical compositions containing the active ingredient may
be in a form suitable for oral use, for example, as tablets, troches,
lozenges, aqueous or
oily suspensions, dispersible powders or granules, emulsions, hard or soft
capsules, or
syrups or elixirs. Compositions intended for oral use may be prepared
according to any
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
38
method known to the art for the manufacture of pharmaceutical compositions.
Such
compositions may contain one or more agents selected from sweetening agents,
flavoring agents, coloring agents and preserving agents in order to provide
pharmaceutically elegant and palatable preparations. Tablets contain the
active
ingredient in admixture with other non-toxic pharmaceutically acceptable
excipients
which are suitable for the manufacture of tablets. These excipients may be,
for
example, inert diluents, such as calcium carbonate, sodium carbonate, lactose,
calcium
phosphate or sodium phosphate; granulating and disintegrating agents, for
example,
corn starch, or alginic acid; binding agents, for example starch, gelatin or
acacia, and
lubricating agents, for example magnesium stearate, stearic acid or talc. The
tablets
may be uncoated or they may be coated by known techniques to delay
disintegration
and absorption in the gastrointestinal tract and thereby provide a sustained
action over a
longer period. For example, a time delay material such as glyceryl
monostearate or
glyceryl distearate may be employed. They may also be coated by the techniques
described in U.S. Patent Nos. 4,256,108; 4,166,452 and 4,265,874 to form
osmotic
therapeutic tablets for control release.
Formulations for oral use may also be presented as hard gelatin capsules
wherein the active ingredient is mixed with an inert solid diluent, for
example, calcium
carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein
the active
ingredient is mixed with water or an oil medium, for example peanut oil,
liquid
paraffin, or olive oil.
Aqueous suspensions contain the active materials in admixture with
excipients suitable for the manufacture of aqueous suspensions. Such
excipients are
suspending agents, for example sodium carboxymethylcellulose, methylcellulose,
hydroxy-propylmethylcellulose, sodium alginate, polyvinyl-pyrrolidone, gum
tragacanth and gum acacia; dispersing or wetting agents may be a naturally-
occurring
phosphatide, for example lecithin, or condensation products of an alkylene
oxide with
fatty acids, for example polyoxy-ethylene stearate, or condensation products
of
ethylene oxide with long chain aliphatic alcohols, for example
heptadecaethyleneoxycetanol, or condensation products of ethylene oxide with
partial
esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol
monooleate, or condensation products of ethylene oxide with partial esters
derived from
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
39
fatty acids and hexitol anhydrides, for example polyethylene sorbitan
monooleate. The
aqueous suspensions may also contain one or more preservatives, for example
ethyl, or
n-propyl, p-hydroxybenzoate, one or more coloring agents, one or more
flavoring
agents, and one or more sweetening agents, such as sucrose or saccharin.
Oily suspensions may be formulated by suspending the active ingredient
in a vegetable oil, for example arachis oil, olive oil, sesame oil or coconut
oil, or in a
mineral oil such as liquid paraffin. The oily suspensions may contain a
thickening
agent, for example beeswax, hard paraffin or cetyl alcohol. Sweetening agents
such as
those set forth above, and flavoring agents may be added to provide a
palatable oral
preparation. These compositions may be preserved by the addition of an anti-
oxidant
such as ascorbic acid.
Dispersible powders and granules suitable for preparation of an aqueous
suspension by the addition of water provide the active ingredient in admixture
with a
dispersing or wetting agent, suspending agent and one or more preservatives.
Suitable
dispersing or wetting agents and suspending agents are exemplified by those
already
mentioned above. Additional excipients, for example sweetening, flavoring and
coloring agents, may also be present.
The pharmaceutical compositions of the invention may also be in the
form of oil-in-water emulsions. The oily phase may be a vegetable oil, for
example
olive oil or arachis oil, or a mineral oil, for example liquid paraffin or
mixtures of these.
Suitable emulsifying agents may be naturally-occurring gums, for example gum
acacia
or gum tragacanth, naturally-occurring phosphatides, for example soy bean,
lecithin,
and esters or partial esters derived from fatty acids and hexitol anhydrides,
for example
sorbitan monooleate, and condensation products of the said partial esters with
ethylene
oxide, for example polyoxyethylene sorbitan monooleate. The emulsions may also
contain sweetening and flavoring agents.
Syrups and elixirs may be formulated with sweetening agents, for
example glycerol, propylene glycol, sorbitol or sucrose. Such formulations may
also
contain a demulcent, a preservative and flavoring and coloring agents.
The pharmaceutical compositions may be in the form of a sterile
injectable aqueous or oleagenous suspension. This suspension may be formulated
according to the known art using those suitable dispersing or wetting agents
and
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
suspending agents which have been mentioned above. The sterile injectable
preparation may also be a sterile injectable solution or suspension in a non-
toxic
parenterally acceptable diluent or solvent, for example as a solution in 1,3-
butane diol.
Among the acceptable vehicles and solvents that may be employed are water,
Ringer's
5 solution and isotonic sodium chloride solution. In addition, sterile, fixed
oils are
conventionally employed as a solvent or suspending medium. For this purpose
any
bland fixed oil may be employed including synthetic mono- or diglycerides. In
addition, fatty acids such as oleic acid find use in the preparation of
injectables.
The pharmaceutical compositions may also be administered in the form
10 of suppositories for rectal administration of the drug. These compositions
can be
prepared by mixing the drug with a suitable non-irritating excipient which is
solid at
ordinary temperatures but liquid at the rectal temperature and will therefore
melt in the
rectum to release the drug. Such materials are cocoa butter and polyethylene
glycols.
For topical use, creams, ointments, jellies, solutions or suspensions, etc.,
15 containing the compounds of the invention are employed. As used herein,
topical
application is also meant to include the use of mouthwashes and gargles.
The pharmaceutical compositions and methods of the invention may
further comprise other therapeutically active compounds, as noted herein,
useful in the
treatment of asthma, allergic diseases, inflammatory conditions and cancer and
20 pathologies associated therewith (e.g., cardiovascular disease) or other
adjuvant. In
many instances, compositions which include a compounds of the invention and an
alternative agent have additive or synergistic effects when administered.
Methods of Use
In yet another aspect, the invention provides methods of treating or
preventing a disease or condition associated with CRTH2 and/or one or more
other
PGD2 receptors by administering to a subject having such a condition or
disease, a
therapeutically effective amount of a compound or composition of the
invention. In
one group of embodiments, diseases and conditions, including chronic diseases
of
humans or other species, can be treated with modulators, or antagonists, of
CRTH2
and/or one or more other PGD2 receptors. These diseases and conditions include
(1)
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
41
inflammatory or allergic diseases such as systemic anaphylaxis and
hypersensitivity
disorders, atopic dermatitis, urticaria, drug allergies, insect sting
allergies, food
allergies (including celiac disease and the like) and mastocytosis, (2)
inflammatory
bowel diseases such as Crohn's disease, ulcerative colitis, ileitis and
enteritis, (3)
vasculitis, Behcet's syndrome, (4) psoriasis and inflammatory dermatoses such
as
dermatitis, eczema, atopic dermatitis, allergic contact dermatitis, urticaria,
viral
cutaneous pathologies such as those derived from human papillomavirus, HIV or
RLV
infection, bacterial, fungal and other parasital cutaneous pathologies, and
cutaneous
lupus erythematosus, (5) asthma and respiratory allergic diseases such as
allergic
asthma, allergic rhinitis, otitis media, allergic conjunctivitis,
hypersensitivity lung
diseases, chronic obstructive pulmonary disease and the like, (6) autoimmune
diseases,
such as arthritis (including rheumatoid and psoriatic), systemic lupus
erythematosus,
type I diabetes, myasthenia gravis, multiple sclerosis, Graves' disease,
glomerulonephritis and the like, (7) graft rejection (including allograft
rejection and
graft-v-host disease), e.g., skin graft rejection, solid organ transplant
rejection, bone
marrow transplant rejection, (8) fever, (9) cardiovascular disorders such as
acute heart
failure, hypotension, hypertension, angina pectoris, myocardial infarction,
cardiomyopathy, congestive heart failure, atherosclerosis, coronary artery
disease,
restenosis, thrombosis and vascular stenosis, (10) cerebrovascular disorders
such as
traumatic brain injury, stroke, ischemic reperfusion injury and aneurysm, (11)
cancers
of the breast, skin, prostate, cervix, uterus, ovary, testes, bladder, lung,
liver, larynx,
oral cavity, colon and gastrointestinal tract (e.g., esophagus, stomach,
pancreas), brain,
thyroid, blood and lymphatic system, (12) fibrosis, connective tissue disease
and
sarcoidosis, (13) genital and reproductive conditions such as erectile
dysfunction, (14)
gastrointestinal disorders such as gastritis, ulcers, nausea, pancreatitis and
vomiting;
(15) neurologic disorders, such as Alzheimer's disease, (16) sleep disorders
such as
insomnia, narcolepsy, sleep apnea syndrome and Pickwick Syndrome, (17) pain,
(18)
renal disorders, (19) ocular disorders such as glaucoma, (20) infectious
diseases, viral
infections such as HIV, and bacterial infections such as sepsis, (21)
inflammation, (22)
flushing and (23) nasal congestion.
In yet another aspect, the invention provides methods of treating or
preventing a disease or disorder mediated, regulated or influenced by Th2
cells,
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
42
eosinophils, basophils, platelets, Langerhans cells, dendritic cells or mast
cells,
comprising administering to a subject having such as disease or disorder a
therapeutically effective amount of one or more of the subject compounds or
compositions.
In yet another aspect, the invention provides methods of treating or
preventing a condition or disorder mediated, regulated or influenced by PGD2
and
metabolites thereof, such as 13,14-dihydro-15-keto-PGDz and 15-deoxy-012 1 4-
PGD2,
comprising administering to a subject having such as disease or disorder a
therapeutically effective amount of one or more of the subject compounds or
compositions.
In yet another aspect, the invention provides methods of treating or
preventing a disease or disorder responsive to modulation of CRTH2 and/or one
or
more other PGD2 receptors comprising administering to a subject having such a
disease
or disorder, a therapeutically effective amount of one or more of the subject
compounds
or compositions.
In yet another aspect, the invention provides methods of treating or
preventing a disease or disorder mediated by CRTH2 and/or one or more other
PGD2
receptors comprising administering to a subject having such a condition or
disease, a
therapeutically effective amount of one or more of the subject compounds or
compositions.
In yet another aspect, the invention provides methods of modulating
CRTH2 and/or one or more other PGD2 receptors comprising contacting a cell
with one
or more of the subject compounds or compositions.
Depending on the disease to be treated and the subject's condition, the
compounds of the invention may be administered by oral, parenteral (e.g.,
intramuscular, intraperitoneal, intravenous, ICV, intracisternal injection or
infusion,
subcutaneous injection or implant), inhalation, nasal, vaginal, rectal,
sublingual, or
topical (e.g., transdermal, local) routes of administration and may be
formulated, alone
or together, in suitable dosage unit formulations containing conventional non-
toxic
pharmaceutically acceptable carriers, adjuvants and vehicles appropriate for
each route
of administration. The invention also contemplates administration of the
compounds of
the invention in a depot formulation, in which the active ingredient is
released over a
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
43
defined time period.
In the treatment or prevention of inflammatory conditions, immune
disorders, asthma, allergic rhinitis, eczema, psoriasis, atopic dermatitis,
fever, sepsis,
systemic lupus erythematosus, diabetes, rheumatoid arthritis, multiple
sclerosis,
atherosclerosis, transplant rejection, inflammatory bowel disease, cancer,
viral
infection, thrombosis, fibrosis, flushing, Crohn's disease, ulcerative
colitis, chronic
obstructive pulmonary disease, inflammation, pain, conjunctivitis, nasal
congestion,
urticaria or other conditions or disorders associated with CRTH2 and/or one or
more
other PGD2 receptors, an appropriate dosage level will generally be about
0.001 to 100
mg per kg patient body weight per day which can be administered in single or
multiple
doses. Preferably, the dosage level will be about 0.01 to about 25 mg/kg per
day; more
preferably about 0.05 to about 10 mg/kg per day. A suitable dosage level may
be about
0.01 to 25 mg/kg per day, about 0.05 to 10 mg/kg per day, or about 0.1 to 5
mg/kg per
day. Within this range the dosage may be 0.005 to 0.05, 0.05 to 0.5 or 0.5 to
5.0 mg/kg
per day. For oral administration, the compositions are preferably provided in
the form
of tablets containing 1.0 to 1000 milligrams of the active ingredient,
particularly 1.0,
5.0, 10.0, 15Ø 20.0, 25.0, 50.0, 75.0, 100.0, 150.0, 200.0, 250.0, 300.0,
400.0, 500.0,
600.0, 750.0, 800.0, 900.0, and 1000.0 milligrams of the active ingredient for
the
symptomatic adjustment of the dosage to the patient to be treated. The
compounds may
be administered on a regimen of 1 to 4 times per day, preferably once or twice
per day.
It will be understood, however, that the specific dose level and
frequency of dosage for any particular patient may be varied and will depend
upon a
variety of factors including the activity of the specific compound employed,
the
metabolic stability and length of action of that compound, the age, body
weight, general
health, sex, diet, mode and time of administration, rate of excretion, drug
combination,
the severity of the particular condition, and the host undergoing therapy.
The compounds of the invention can be combined or used in
combination with other agents useful in the treatment, prevention, suppression
or
amelioration of the diseases or conditions for which compounds of the
invention are
useful, including inflammatory conditions, immune disorders, asthma, allergic
rhinitis,
eczema, psoriasis, atopic dermatitis, fever, sepsis, systemic lupus
erythematosus,
diabetes, rheumatoid arthritis, multiple sclerosis, atherosclerosis,
transplant rejection,
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
44
inflammatory bowel disease, cancer, viral infection, thrombosis, fibrosis,
flushing,
Crohn's disease, ulcerative colitis, chronic obstructive pulmonary disease,
inflammation, pain, conjunctivitis, nasal congestion, urticaria and those
pathologies
noted above.
Such other agents, or drugs, may be administered, by a route and in an
amount commonly used therefor, simultaneously or sequentially with a compound
of
the invention. When a compound of the invention is used contemporaneously with
one
or more other drugs, a pharmaceutical composition containing such other drugs
in
addition to the compound of the invention is preferred. Accordingly, the
pharmaceutical compositions of the invention include those that also contain
one or
more other active ingredients or therapeutic agents, in addition to a compound
of the
invention.
Examples of other therapeutic agents that may be combined with a
compound of the invention, either administered separately or in the same
pharmaceutical compositions, include, but are not limited to: (a) VLA-4
antagonists, (b)
corticosteroids, such as beclomethasone, methylprednisolone, betamethasone,
prednisone, prenisolone, triamcinolone, dexamethasone, fluticasone,
flunisolide and
hydrocortisone, and corticosteroid analogs such as budesonide; (c)
immunosuppressants such as cyclosporine (cyclosporine A, Sandimmune , Neoral
),
tacrolimus (FK-506, Prografe), rapamycin (sirolimus, Rapamune) and other FK-
506
type immunosuppressants, and mycophenolate, e.g., mycophenolate mofetil
(CellCept ); (d) antihistamines (HI-histamine antagonists) such as
bromopheniramine,
chlorpheniramine, dexchlorpheniramine, triprolidine, clemastine,
diphenhydramine,
diphenylpyraline, tripelennamine, hydroxyzine, methdilazine, promethazine,
trimeprazine, azatadine, cyproheptadine, antazoline, pheniramine, pyrilamine,
astemizole, terfenadine, loratadine, cetirizine, fexofenadine,
descarboethoxyloratadine,
and the like; (e) non-steroidal anti-asthmatics such as (32-agonists (e.g.,
terbutaline,
metaproterenol, fenoterol, isoetharine, albuterol, salmeterol, bitolterol and
pirbuterol)
and (32-agonist-corticosteroid combinations (e.g., salmeterol-fluticasone
(Advair ),
formoterol-budesonid (Symbicort )), theophylline, cromolyn, cromolyn sodium,
nedocromil, atropine, ipratropium, ipratropium bromide, leukotriene
antagonists (e.g.,
zafirlukast, montelukast, montelukast sodium (Singulair ), pranlukast,
iralukast,
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
pobilukast and SKB-106,203), leukotriene biosynthesis inhibitors (zileuton,
BAY-
1005); (f) non-steroidal antiinflammatory agents (NSAIDs) such as propionic
acid
derivatives (e.g., alminoprofen, benoxaprofen, bucloxic acid, carprofen,
fenbufen,
fenoprofen, fluprofen, flurbiprofen, ibuprofen, indoprofen, ketoprofen,
miroprofen,
5 naproxen, oxaprozin, pirprofen, pranoprofen, suprofen, tiaprofenic acid and
tioxaprofen), acetic acid derivatives (e.g., indomethacin, acemetacin,
aiclofenac,
clidanac, diclofenac, fenclofenac, fenclozic acid, fentiazac, furofenac,
ibufenac,
isoxepac, oxpinac, sulindac, tiopinac, tolmetin, zidometacin and zomepirac),
fenamic
acid derivatives (e.g., flufenamic acid, meclofenamic acid, mefenamic acid,
niflumic
10 acid and tolfenamic acid), biphenylcarboxylic acid derivatives (e.g.,
diflunisal and
flufenisal), oxicams (e.g., isoxicam, piroxicam, sudoxicam and tenoxican),
salicylates
(e.g., acetyl salicylic acid and sulfasalazine) and the pyrazolones (e.g.,
apazone,
bezpiperylon, feprazone, mofebutazone, oxyphenbutazone and phenylbutazone);
(g)
cyclooxygenase-2 (COX-2) inhibitors such as celecoxib (Celebrex ) and
rofecoxib
15 (Vioxx ); (h) inhibitors of phosphodiesterase type IV (PDE-IV); (i) other
PGD2
receptor antagonists, especially DP antagonists; (j) opioid analgesics such as
codeine,
fentanyl, hydromorphone, levorphanol, meperidine, methadone, morphine,
oxycodone,
oxymorphone, propoxyphene, buprenorphine, butorphanol, dezocine, nalbuphine
and
pentazocine; (k) cholesterol lowering agents such as HMG-CoA reductase
inhibitors
20 (e.g., lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin and
other statins), bile
acid sequestrants (e.g., cholestyramine and colestipol), vitamin B3 (also
known as
nicotinic acid, or niacin), vitamin B6 (pyridoxine), vitamin B12
(cyanocobalamin), fibric
acid derivatives (e.g., gemfibrozil, clofibrate, fenofibrate and
benzafibrate), probucol,
nitroglycerin, and inhibitors of cholesterol absorption (e.g., beta-sitosterol
and
25 acylCoA-cholesterol acyltransferase (ACAT) inhibitors such as melinamide),
HMG-
CoA synthase inhibitors, squalene epoxidase inhibitors and squalene synthetase
inhibitors; (1) antithrombotic agents, such as thrombolytic agents (e.g.,
streptokinase,
alteplase, anistreplase and reteplase), heparin, hirudin and warfarin
derivatives, p-
blockers (e.g., atenolol), (3-adrenergic agonists (e.g., isoproterenol), ACE
inhibitors and
30 vasodilators (e.g., sodium nitroprusside, nicardipine hydrochloride,
nitroglycerin and
enaloprilat); (m) anti-diabetic agents such as insulin and insulin mimetics,
sulfonylureas (e.g., glyburide, meglinatide), biguanides, e.g., metformin
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
46
(Glucophage ), a-glucosidase inhibitors (acarbose), thiazolidinone compounds,
e.g.,
rosiglitazone (Avandia ), troglitazone (Rezulin ), ciglitazone, pioglitazone
(Actos )
and englitazone; (n) preparations of interferon beta (interferon R-1 a,
interferon (3-1 (3);
(o) gold compounds such as auranofin and aurothioglucose, (p) TNF inhibitors,
e.g.,
etanercept (Enbrel ), antibody therapies such as orthoclone (OKT3), daclizumab
(Zenapax ), basiliximab (Simulect ), infliximab (Remicade ) and D2E6 TNF
antibody,
(q) lubricants or emollients such as petrolatum and lanolin, keratolytic
agents, vitamin
D3 derivatives (e.g., calcipotriene and calcipotriol (Dovonex )), PUVA,
anthralin
(Drithrocreme ), etretinate (Tegison ) and isotretinoin; (r) multiple
sclerosis
therapeutic agents such as interferon (3-1 R (Betaseron ), interferon (3-1 a
(Avonex ),
azathioprine (Imurek , Imuran ), glatiramer acetate (Capoxone ), a
glucocorticoid
(e.g., prednisolone) and cyclophosphamide; (s) other compounds such as 5-
aminosalicylic acid and prodrugs thereof; (t) DNA-alkylating agents (e.g.,
cyclophosphamide, ifosfamide), antimetabolites (e.g., azathioprine, 6-
mercaptopurine,
methotrexate, a folate antagonist, and 5-fluorouracil, a pyrimidine
antagonist),
microtubule disruptors (e.g., vincristine, vinblastine, paclitaxel,
colchicine, nocodazole
and vinorelbine), DNA intercalators (e.g., doxorubicin, daunomycin and
cisplatin),
DNA synthesis inhibitors such as hydroxyurea, DNA cross-linking agents, e.g.,
mitomycin C, hormone therapy (e.g., tamoxifen, and flutamide), and cytostatic
agents,
e.g., imatinib (ST1571, Gleevec ) and rituximab (Rituxan ). The weight ratio
of the
compound of the invention to the second active ingredient may be varied and
will
depend upon the effective dose of each ingredient. Generally, an effective
dose of each
will be used. Thus, for example, when a compound of the invention is combined
with
an NSAID, the weight ratio of the compound of the invention to the NSAID will
generally range from about 1000:1 to about 1:1000, preferably about 200:1 to
about
1:200. Combinations of a compound of the invention and other active
ingredients will
generally also be within the aforementioned range, but in each case, an
effective dose
of each active ingredient should be used.
Analysis of the Compounds
In yet another aspect, the invention includes methods to evaluate
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
47
putative specific agonists or antagonists of CRTH2 and/or one or more other
PGD2
receptors. Accordingly, the invention is directed to the use of these
compounds in the
preparation and execution of screening assays for compounds which modulate the
function of CRTH2 and/or one or more other PGD2 receptors. For example, the
compounds of this invention are useful for CRTH2 mutants and/or one or more
other
PGD2 receptor mutants, which are excellent screening tools for potent
compounds.
Furthermore, the compounds of this invention are useful in establishing or
determining
the binding site of other compounds to CRTH2 and/or one or more other PGD2
receptors, e.g., by competitive inhibition. The compounds of the instant
invention are
also useful for the evaluation of putative specific modulators of CRTH2 and/or
one or
more other PGD2 receptors. One of skill in the art will appreciate that
thorough
evaluation of specific agonists and antagonists of PGD2 receptors has been
hampered
by the lack of availability of non-peptidyl (metabolically resistant)
compounds with
high binding affinity for these receptors. The compounds provided herein are
particularly useful in this context.
High Throughput Screening
High throughput assays for the presence, absence, quantification, or
other properties of particular compounds may be used to test a combinatorial
library
that contains a large number of potential therapeutic compounds (potential
modulator
compounds). The assays are typically designed to screen large chemical
libraries by
automating the assay steps and providing compounds from any convenient source
to the
assays, which are typically run in parallel (e.g., in microtiter formats on
microtiter
plates in robotic assays). Preferred assays detect enhancement or inhibition
of CRTH2
and/or one or more other PGD2 receptors function.
High throughput screening systems are commercially available (see e.g.,
Zymark Corp., Hopkinton MA; Air Technical Industries, Mentor OH; Beckman
Instruments, Inc., Fullerton CA; Precision Systems, Inc., Natick MA; etc.).
These
systems typically automate entire procedures, including all sample and reagent
pipetting, liquid dispensing, timed incubations, and final readings of the
microplate in
detector(s) appropriate for the assay. These configurable systems provide high
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
48
throughput and rapid start-up as well as a high degree of flexibility and
customization.
The manufacturers of such systems provide detailed protocols for various high
throughput systems. Thus, for example, Zymark Corp. provides technical
bulletins
describing screening systems for detecting the modulation of gene
transcription, ligand
binding, and the like.
The following examples are offered by way of illustration and are not
intended to limit the scope of the invention. Those of skill in the art will
readily
recognize a variety of noncritical parameters that could be modified to yield
essentially
similar results.
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
49
EXAMPLES
Reagents and solvents used below can be obtained from commercial
sources such as Aldrich Chemical Co. (Milwaukee, Wisconsin, USA). 'H-NMR
spectra were recorded on a Varian Gemini 400 MHz NMR spectrometer. Significant
peaks are tabulated in the order: multiplicity (s, singlet; d, doublet; t,
triplet; q, quartet;
m, multiplet; br s, broad singlet), coupling constant(s) in Hertz (Hz) and
number of
protons. Electron Ionization (EI) mass spectra were recorded on a Hewlett
Packard
5989A mass spectrometer. Mass spectrometry results are reported as the ratio
of mass
over charge, followed by the relative abundance of each ion (in parentheses)
or a single
m/z value for the M+H (or, as noted, M-H) ion containing the most common
atomic
isotopes. Isotope patterns correspond to the expected formula in all cases.
Electrospray ionization (ESI) mass spectrometry analysis was conducted on a
Hewlett-
Packard 1100 MSD electrospray mass spectrometer using the HP 1 100 HPLC for
sample delivery. Normally the analyte was dissolved in methanol at 0.1 mg/mL
and 1
microliter was infused with the delivery solvent into the mass spectrometer,
which
scanned from 100 to 1500 daltons. All compounds could be analyzed in the
positive
ESI mode, using 1:1 acetonitrile/water with 1 % acetic acid as the delivery
solvent. The
compounds provided below could also be analyzed in the negative ESI mode,
using
2mM NH4OAc in acetonitrile/water as delivery solvent.
Example 1
.9
CH, H
I~ I~
F3C ~
CO2H
1
The synthesis of {4-[2-(toluene-4-sulfonylamino)-4-trifluoromethyl-
phenoxy]-phenyl}-acetic acid (1) is outlined in Scheme 1 and described below.
Scheme 1
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
02 HQ + ~
K2 C03 I \ I \
DMSO F3
F3 lb 02CH3 60 C IC
OzCH3
la
H2, Pd/C Toluenesulfonyl chloride
CH3 OH i F3 , Pyridine, rt
l d 02CH3
R H LiOH 4 H
CH CH
F3 , / CH30H, H2O, rt F3 I
02Me 02H
le
[4-(2-nitro-4-trifluoromethyl-phenoxy)-phenyl]-acetic acid methyl
ester (1c). A mixture of 4-fluoro-3-nitrobenzotrifluoride (1a, 1.0 g, 4.78
mmol),
5 methyl 4-hydroxyphenylacetate (1b, 795 mg, 4.78 mmol) and potassium
carbonate
(661 mg, 4.78 mmol) in 10 mL of DMSO was allowed to stir at 60 C for 24 h.
Upon
completion, the mixture was cooled to room temperature and 50 mL of water was
added. The resulting mixture was extracted with ethyl acetate (3 x 30 mL). The
combined extracts were washed with water (2 x 30 mL) and brine, dried over
10 anhydrous sodium sulfate and concentrated in vacuo. The residue was
chromatographed on a silica gel column using 20% EtOAc/hexane as the eluent to
give
1.31 g of [4-(2-nitro-4-trifluoromethyl-phenoxy)-phenyl] -acetic acid methyl
ester. 'H
NMR (CDC13): 8 8.22 (d, J=2.00 Hz, 1H), 7.70 (dd, J=8.8, 2.2 Hz, 1H), 7.36 (d,
J=8.36
Hz, 2H), 7.08 9s, 1H), 7.07 (d, J=8.48 Hz, 2H).
15 [4-(2-amino-4-trifluoromethyl-phenoxy)-phenyl]-acetic acid methyl
ester (1d). A mixture of [4-(2-nitro-4-trifluoromethyl-phenoxy)-phenyl] -
acetic acid
methyl ester (1.31 g, 3.54 mmol) and 10% Pd/C (377 mg, 0.354 mmol) in 17 mL of
methanol was allowed to stir at room temperature under hydrogen atmosphere for
12 h.
Upon completion, the mixture was filtered through a short column of celite and
the
20 filtrate was concentrated in vacuo to give 920 mg of [4-(2-Amino-4-
trifluoromethyl-
phenoxy)-phenyl] -acetic acid methyl ester. 'H NMR (CDC13): 6 7.24-7.33 (m,
3H),
6.80-7.06 (m, 4H), 4.01 (br s, 2H), 3.71 (s, 3H), 3.61 (s, 2H). LCMS (ESI+)
326
(M+1).
{4-[2-(Toluene-4-sulfonylamino)-4-trifluoromethyl-phenoxy)-
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
51
phenyl}-acetic acid methyl ester (le). [4-(2-Amino-4-trifluoromethyl-phenoxy)-
phenyl]-acetic acid methyl ester (84 mg, 0.542 mmol) was added to a mixture of
150
mg (1.08 mmol) of potassium carbonate in 3 mL of ethyl acetate in presence of
0.5 mL
of water. To the resulting mixture was added 155 mg (0.813 mmol)
toluenesulfonyl
chloride. The resulting mixture was allowed to stir at room temperature
overnight.
Upon completion, 20 mL of 2N HCl aqueous solution was added and the resulting
mixture was extracted with ethyl acetate (3 x 30 mL). The combined extracts
were
washed with water and brine, dried over anhydrous sodium sulfate and
concentrated in
vacuo. The residue was chromatographed to give 97 mg of {4-[2-(toluene-4-
sulfonylamino)-4-trifluoromethyl-phenoxy]-phenyl}-acetic acid methyl ester. 'H
NMR
(CDC13): 8 7.93 (s, 1H), 7.65 (d, J=8.36 Hz, 2H), 7.15-7.28 (m, 5H), 6.68 (d,
J=8.68
Hz, 1H), 6.63 9d, J=8.48 Hz, 2H), 3.72 (s, 3H), 3.61 9s, 2H), 2.39 9s, 3H).
{4-[2-(Toluene-4-sulfonylamino)-4-trifluoromethyl-phenoxy]-
phenyl}-acetic acid (1). To a solution of {4-[2-(toluene-4-sulfonylamino)-4-
trifluoromethyl-phenoxy]-phenyl}-acetic acid methyl ester (97 mg, 0.202 mmol)
in 1
mL of methanol was added 1 mL of a suspension of lithium hydroxide (42 mg,
1.01
mmol) in 1 mL of water. The resulting mixture was allowed to stir at room
temperature
until all starting material disappeared. Upon completion, 50 mL of IN aqueous
HCl
was added and the resulting mixture was extracted with ethyl acetate (3 x 30
mL). The
combined extracts were washed with IN HCl aqueous solution (20 mL), water and
brine, dried over anhydrous sodium sulfate and concentrated in vacuo. The
residue was
purified by column chromatography to give 70 mg of {4-[2-(toluene-4-sulfonyl-
amino)-4-trifluoromethyl-phenoxy]-phenyl}-acetic acid (1). 'H NMR (CD3OD): S
7.84
(d, J=1.92 Hz, 1H), 7.58 (d, J=8.27, 1.6 Hz, 2H), 7.32 (dd, J=8.27, 1.6 Hz,
1H), 7.18-
7.30 (m, 5H), 6.73 (d, J=8.53 Hz, 1H), 6.57 (d, J=8.53 Hz, 2H), 3.59 (s, 2H),
2.37 (s,
3H). LCMS (ESI-) 464 (M-1).
Example 2
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
52
/ \
/ \ Y.kO
NH
I~ I~
F3C /
COZH
2
{4-[2-(Naphthalene-l-sulfonylamino)-4-trifluoromethyl-phenoxy]-
phenyl}-acetic acid (2) was synthesized using the same synthetic procedures
for 1 as
shown in Scheme 1. 'H NMR (CDC13): 8 8.60 (d, J=6.67 Hz, 1H), 8.25 (d, J=7.28
Hz,
1H), 8.02 (d, J=8.13 Hz, 1H), 7.80-7.90 (m, 2H), 7.40-7.55 (m, 4H), 7.05-7.15
(m, 3H),
6.53 (d, J=8.60 Hz, 1H), 6.31 (d, J=7.96 Hz, 2H), 3.61 (s, 2H). LCMS (ESI-)
500 (M-
1).
Example 3
NH
F3C &o--(:~
CO2H
3
[4-(2-Benzoylamino-4-trifluoromethyl-phenoxy)-phenyl]-acetic acid
(3) was synthesized according to the same synthetic procedures for 1 as shown
in
Scheme 1. 1H NMR (CDC13): 8 8.99 (d, J=1.73Hz, 1H), 8.61 (s, 1H), 7.84 (d,
J=7.76,
2H), 7.45-7.60 (m, 3H), 7.35 (d, J=8.40 Hz, 2H), 7.28 (dd, J=8.72, 1.53 Hz,
1H), 7.08
(d, J=8.44 Hz, 2H), 6.89 (d, J=8.44 Hz, 1H), 3.69 (s, 2H). LCMS (ESI-) 388 (M-
1).
Example 4
Q9
CH3 S-NH
I ~ I ~
F3C /
COZH
4
[4-(2-Methanesulfonylamino-4-trifluoromethyl-phenoxy)-phenyl]-
acetic acid (4) was synthesized using the same synthetic procedures for 1 as
shown in
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
53
Scheme 1. 1H NMR (CDC13): 6 7.89 (d, J=1.76 Hz, 1H), 7.30-7.40 (m, 3H), 7.10
(s,
1H), 7.01 (d, J=8.32 Hz, 2H), 6.89 (d, J=8.48 Hz, IH), 3.69 (s, 2H), 3.08 (s,
3H).
LCMS (ESI-) 388 (M-1).
Example 5
F
F
CO2H
5
The synthesis of (3-benzyloxy-5-trifluoromethyl-phenyl)-acetic acid
(5) is shown in Scheme 2. 1H NMR (CDC13): 8 7.32-7.50 (m, 5H), 7.15 (s, 2H),
7.10
(s, 1H), 5.08 (s, 2H), 3.68 (s, 1H). LCMS (ESI-) 309 (M-1).
Scheme 2
CF3
NaH, Benzyl alcohol 0-0- F3
Cu!, HMPA, DMSO
C02H 120 C, 24 hours
59% CO2H
5a 5
Example 6
ONo
H
HO2C
CO2H
6
The synthesis of 3-benzenesulfonylamino-4-(4-
carboxymethylphenoxy)-benzoic acid (6) is outlined in Scheme 3, below. 'H NMR
(CD3OD): 8 8.24 (d, J=2.04 Hz, 1H), 7.68-7.80 (m, 3H), 7.56 (dd, J=7.48, 1.20
Hz,
1H), 7.42 (d, J=7.86 Hz, 1H), 7.40 (d, J=7.76 Hz, 1H), 7.21 (d, J=8.40 Hz,
2H), 6.52-
6.68 (m, 3H), 3.58 (s, 2H). LCMS (ESI-) 426 (M-1).
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
54
Scheme 3
NO2
NO2 HO
K2CO3, DMS \ 0 \ 1. H2, Pd/C, CH30H
F
\
60 de9C 2. CI2, Lutidine
+
/ H02C Acetone
HO2C lb C02Me 6b C02 Me
Me
0//0 00
s
/ II S-NH UGH NH
\/ \ O \ \ O \
CH30H, H2O
r.t.
H02C H02C
6c C02Me 6 CO2H
EDC, 2-Methoxy-4-methyl-
CH2CI2, r.t.
0 //0 % //0
/ S. NH / S~NH
LiOH
r \ \ 0 \
\ O CH30H H2O
MeON I/ .t. N
MeO~~
O 7a C02Me 0 7 CO2H
Example 7
The synthesis of 3{4-[2-benzenesulfonylamino-4-(2-methoxy-
ethylcarbamoyl)-phenoxy]-phenyl}-acetic acid (7) is outlined in Scheme 3,
above.
'H NMR (CDC13): 8 8.01 (d, J=1.72 Hz, 1H), 7.73 (d, J=7.76 Hz, 2H), 7.51 (dd,
J=7.76,
7.32 Hz, 2H), 7.38 (dd, J=7.68, 7.68 Hz, 2H), 7.21 (s, 1H), 7.18 (d, J=8.32
Hz, 2H),
6.64 (d, J=8.40 Hz, 2H), 6.55 (d, J=8.18 Hz, 2H), 3.53-3.70 (m, 6H), 3.41 (s,
3H), 1.25
(dd, J=7.07, 7.07 Hz, 3H). LCMS (ESI-) 483 (M-1).
Example 8
SNH F
01 F
CO,H
8
The synthesis of {3-[4-ethylcarbamoyl-2-(toluene-4-sulfonylamino)-
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
phenoxy]-5-trifluoromethyl-phenyl}-acetic acid (8) is shown in Scheme 4,
below.
'H NMR (CDC13): 6 7.98 (d, J=2.04 Hz, 1H), 7.55-7.65 (m, 3H), 7.33 (s, 1H),
7.16 (d,
J=8.12 Hz, 2H), 7.11 (s, 1H), 6.91 (s, 1H), 6.68 (dd, J=8.36, 2.48 Hz, 2H),
6.25 (br s,
1H), 3.65 (s, 2H), 4.50 (m, 2H), 1.30 (dd, J=7.20, 7.20 Hz, 3H). LCMS (ESI-)
535 (M-
5 1).
Scheme 4
oZ
EDC,HOBT
CF3 ethylamine
Oz F HO CF3 KZCO3 I O 1 1
+ DMSO H02NMM, CHZCIz, rt
HOz 8a CO CH 60 C H 60%
6a z 3 74% CozC3
Hz
Oz A) Hz, L Pd/C rt, CF ~O
O CF3 CH30H rt 91% H 3
q f& B) SnC12,
EtOAc, reflux K2CO3, Acetone, rt
COZCH3 CO2CH3 71%
0 0 0 0
H LiOH H
O CF3 O CF3
H CH3OH, HZO, r[ H
\~ I 65% ''Y
COZCH3 COZH
8
Example 9
~ ~ Qo
NH
H F
0 CO2H
9
{3-[4-Ethylcarbamoyl-2-(toluene-4-sulfonylamino)-phenoxy]-5-
fluoro-phenyl}-acetic acid (9) was synthesized from 3-fluoro-5-
hydroxyphenylacetic
acid methyl ester according to Scheme 4, above. 'H NMR (CDC13): 6 8.02 (d,
J=2.13
Hz, 1H), 7.60 (dd, J=8.60, 2.18 Hz, IH), 7.57 (d, J=8.57 Hz, 2H), 7.23 (s,
1H), 7.15 (d,
J=7.96 Hz, 2H), 6.79 (d, J=8.80 Hz, 1H), 6.73 (d, J=8.56 Hz, 1H), 6.33 (s,
1H), 6.43
(dd, J=5.50, 5.44 Hz, 1H), 5.96 (ddd, J=9.46, 8.53, 2.20 Hz, 1H), 3.59 (s,
2H), 3.52 (m,
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
56
2H), 2.37 (s, 3H), 1.26 (dd, J=7.20, 7.20 Hz, 3H). LCMS (ESI-) 485 (M-1).
Example 10
Quo
H & 1~
COZH
0
3-[4-(2-Benzenesulfonylamino-4-ethylcarbamoyl-phenoxy)-phenyll-
propionic acid (10) was synthesized from 3-(4-hydroxyphenyl)propionic acid,
using
benzenesulfonyl chloride, according to Scheme 4, above. 1H NMR (CDC13): S 8.0
(d,
10 J=2.14 Hz, 1H), 7.74 (dd, J=8.37, 1.27 Hz, 2H), 7.57 (ddd, J=8.58, 7.46,
2.24 Hz, 2H),
7.39 (dd, J=8.26, 7.46 Hz, 2H), 7.18 (s, 1H), 7.12 (d, J=8.60 Hz, 2H), 6.63
(d, J=8.53
Hz, 1H), 6.50 (d, J=8.53 Hz, 2H), 6.22 (br s, 1H), 3.52 (m, 2H), 2.95 (dd,
J=7.46, 7.46
Hz, 2H), 2.71 (dd, J=7.46, 7.46 Hz, 2H), 1.28 (dd, J=7.33, 7.33 Hz, 3H). LCMS
(ESI-)
467 (M-1).
Example 11
CI
C 0 0
H 1 O 1
v
0 CO2H
11
{4-[2-(2,4-Dichloro-benzenesulfonylamino)-4-ethylcarbamoyl-
phenoxyl-phenyl}-acetic acid (11) was synthesized from 4-hydroxyphenylacetic
acid,
using 2,4-dichlorobenzenesulfonyl chloride, according to Scheme 4, above. 1H
NMR
(CDC13): S 7.93 (obscured d, J=1.54 Hz, 1H), 7.92 (obscured dd, J=8.13, 0.61
Hz, 1H),
7.80 (br s, 1H), 7.69 (s, 1H), 7.52 (dd, J=8.57, 2.18 Hz, 1H), 7.45 (m, 1H),
7.30 (dd,
J=8.13, 2.02 Hz, 1H), 7.25 (d, J=8.68 Hz, 2H), 6.74 (d, J=8.58 Hz, 1H), 6.69
(dd,
J=8.64, 2.02 Hz, 2H), 6.28 (br s, 1H), 3.68 (s, 2H), 3.50 (m, 2H), 1.27 (dd,
J=7.46, 7.46
Hz, 3H). LCMS (ESI-) 521 (M-1).
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
57
Example 12
CI
C QO
H
vN
0 CO2H
12
{4-[2-(2,4-Dichloro-benzenesulfonyl-amino)-4-ethylcarbamoyl-
phenoxy]-3-methoxy-phenyl}-acetic acid (12) was prepared using 2,4-
dichlorobenzenesulfonyl chloride, according to Scheme 4. 1H NMR (CDCl3): 6
7.96
(d, J=8.40 Hz, 1H), 7.88 (s, 1H), 7.83 (s, 1H), 7.45 (dd, J=8.56, 1.84 Hz,
1H), 7.39 (d,
J=1.84 Hz, 1H), 7.30 (dd, J=8.56, 1.92 Hz, 1H), 6.91 (s, 1H), 6.82 (d, J=8.04
Hz, 1H),
6.69 (d, J=8.12 Hz, 1H), 6.55 (d, J=8.44 Hz, 1H), 6.15 (br s, 1H), 3.67 (s,
3H), 3.65 (s,
2H), 3.46 (m, 2H), 1.24 (dd, J=7.20, 7.20 Hz, 3H). LCMS (ESI-) 551 (M-1).
Example 13
CI
C Q 0CH3
H
0 CO2H
13
(4-{2-[(2,4-Dichloro-benzenesulfonyl)-methyl-amino]-4-
ethylcarbamoyl-phenoxy}-phenyl)-acetic acid (13) was prepared from 4-
hydroxyphenylacetic acid, using 2,4-dichlorobenzenesulfonyl chloride,
according to
Scheme 4. 'H NMR (CDC13): 5 7.72-7.83 (m, 3H), 7.17-7.26 (m, 4H), 6.77 (d,
J=8.80
Hz, 1H), 6.60 (d, J=8.44 Hz, 2H), 6.23 (br s, 1H), 3.64 (s, 2H), 3.51 (s, 3H),
3.48 (m,
2H), 1.26 (dd, J=7.33, 7.33 Hz, 3H). LCMS (ESI-) 536 (M-1).
Example 14
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
58
CI
CQ J56
0 CO2H
14
{3-Chloro-4-[2-(2,4-dichloro-benzenesulfonylamino)-4-
ethylcarbamoyl-phenoxy]-phenyl}-acetic acid (14) was prepared using 2,4-
dichlorobenzenesulfonyl chloride, according to Scheme 4. IH NMR (CDC13): 5
7.94
(d, J=8.56 Hz, 1H), 7.93 (s, 1H), 7.73 (s, 1H), 7.50 (dd, J=8.60,2.16 Hz, 1H),
7.41 (d,
J=1.48 Hz, 1H), 7.36 (d, J=8.48 Hz, 1H), 7.11 (dd, J=8.32, 1.60 Hz, 1H), 6.62
(d,
J=8.36 Hz, 1H), 6.55 (d, J=8.72 Hz, 1H), 3.65 (s, 2H), 3.47 (m, 2H), 1.23 (dd,
J=7.20,
7.20 Hz, 3H). LCMS (Neg) 556 (M-1).
Example 15
/O / o
NH I
H
~
0 CO2H
15 {3-Chloro-4-[4-ethylcarbamoyl-2-(4-methoxy-benzenesulfonyl-
amino)-phenoxy]-phenyl}-acetic acid (15) was prepared according to Scheme 4.
1H
NMR (CDC13): 5 7.99 (d, J=1.44 Hz, 1H), 7.68 (d, J=8.88 Hz, 2H), 7.53 (dd,
J=8.60,
1.44 Hz, 1H), 7.35 (s, 1H), 7.26 (s, 1H), 7.08 (dd, J=8.32,1.36 Hz, 1H), 6.83
(d, J=8.80
Hz, 2H), 6.55 (d, J=8.40 Hz, 1H), 6.48 (d, J=8.56 Hz, 1H), 6.38 (br s, 1H),
3.80 (s, 3H),
3.61 (s, 2H), 3.48 (m, 2H), 1.25 (dd, J=7.20, 7.20 Hz, 3H). LCMS (ESI-) 517 (M-
1).
Example 16
NH
H ~
v
0 CO2H
16
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
59
{3-Chloro-4-[4-ethylcarbamoyl-2-(toluene-4-sulfonylamino)-
phenoxy]-phenyl}-acetic acid (16) was prepared using toluenesulfonyl chloride,
according to Scheme 4. 'H NMR (CDC13): 6 7.98 (d, J=2.12 Hz, 1H), 7.63 (d,
J=8.32
Hz, 2H), 7.54 (dd, J=8.60, 2.16 Hz, 1H), 7.35 (d, J=1.92 Hz, 1H), 7.25 (s,
1H), 7.17 (d,
J=8.40 Hz, 2H), 7.08 (dd, J=8.32, 1.96 Hz, 1H), 6.52 (d, J=8.32 Hz, 1H), 6.46
(d,
J=8.52 Hz, 1H), 6.30 (br s, 1H), 3.62 (s, 2H), 3.52 (m, 2H), 2.36 (s, 3H),
1.25 (dd,
J=7.20, 7.20 Hz, 3H). LCMS (ESI-) 501 (M-1).
Example 17
ci
c d\N 30
NH Qo
vH I \ I \
0 C02H
17
Synthesis of {4-[2-(2,4-dichloro-benzenesulfonyl-amino)-4-
ethylcarbamoyl-benzenesulfonyl]-phenyl}-acetic acid (17) is shown in Scheme 5.
'H NMR (CDCl3): 6 9.85 (S, 1H), 9.77 (s, 1H), 7.35-8.20 (m, 24H), 6.07 (br s,
2H),
3.75 (s, 4H), 3.35-3.50 (m, 4H), 1.25 (dd, J=5.4, 5.4 Hz, 3H). 1.23 (dd,
J=7.20, 7.20
Hz, 3H). LCMS (ESI-) 570 (M-1).
Scheme 5
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
K2C03 EDC, HOBT
5F + I I DC 2-Methoxy-ethylamine
H02 CJ[ DMSO, rt H02 NMM,CI-12C12i rt
17a 02CH3 02CH3
6a CI
02 H2 C - O
H2 Cl
Pd/C, CH3OH, rt Acetone, Lutidine, rt
0 02CH3 0 02CH3
1
CQ,_p C 'O
v ~C OH
H MCPBA H Li
CH3OH H2O, rt
02CH3 CH2Cl2, rt
02CH3
C ~O
H
17 02H
Example 18
5
Cl
C NH I
H rjj~_
0 CO2H
18
The synthesis of [4-chloro-6-(2,4-dichloro-benzenesulfonyl-amino)-8-
ethylcarbamoyl-dibenzofuran-2-yl]-acetic acid (18) is shown in Scheme 6. 1H
NMR
10 (CD3OD): S 8.42 (s, 1H), 8.08 (s, 1H), 7.96 (s, 1H), 7.95 (d, J=7.40 Hz,
1H), 7.4 (d,
J=1.92 Hz, 1H), 7.57 (s, 1H), 7.40 (dd, J=7.4, 2.04 Hz, IH), 3.85 (s, 2H),
3.53 (m, 2H),
1.34 (dd, J=7.33, 7.33 Hz, 3H). LCMS (ESI-) 554 (M-1).
Scheme 6
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
61
H Pd(OAc)2 H SnCl2
02 1 02 1 02CH 3
AcOH, reflux EtOAc, reflux
18a 02CH3
Cl y~Cl
H2 1 Ct-(/ ~ 0 CF_ ~~ 0
\( C1 (~// H 1 LiOH
H
Lutidine H CH3OH, H2O, rt
02CH3 Acetone, rt
02CH3
Cl
C 0
H CI
jcoo
8 02H
Example 19
Cl
C Qo
sNH
5q CF3
vH
0 CO2H
19
{3-[2-(2,4-Dichloro-benzenesulfonyl-amino)-4-ethylcarbamoyl-
phenoxyl-5-trifluoromethyl-phenyl}-acetic acid (19) was prepared from 3-
hydroxy-
5-trifluoromethylphenylacetic acid, using 2,4-dichlorobenzenesulfonyl
chloride,
according to Scheme 4. 'H NMR (Acetone-d6): 8 9.10 (br s, 1H), 8.11 (d, J=2.04
Hz,
1H), 7.90 (d, J=8.40 Hz, I H), 7.83 (br s, I H), 7.72 (dd, J=8.48, 2.16 Hz, I
H), 7.46 (s,
2H), 7.43 (d, J=2.04 Hz, 1 H), 7.03 (s, 1 H), 6.96 (d, J=8.64 Hz, 1 H), 6.92
(s, 1 H), 3.78
(s, 2H), 3.42 (m, 2H), 1.19 (dd, J=7.28, 7.28 Hz, 3H). LCMS (ESI-) 590 (M-1).
Example 20
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
62
F F
i SOH
CF3
,M,rj(~ O-q O CO2H
{3- [4-ethylc arb a moyl-2-(4-triflu o ro-meth oxy-b enzen es u lfo nyl-
5 amino)-phenoxyl-5-trifluoromethyl-phenyl}-acetic acid (20) was prepared from
3-
hydroxy-5-trifluoromethylphenylacetic acid according to Scheme 4. 'H NMR
(Acetone-d6): 8 8.18 (d, J=2.16 Hz, 1H), 7.92 (dd, J=6.84, 2.12 Hz, 2H), 7.85
(br s,
I H), 7.69 (dd, J=8.52, 2.16 Hz, I H), 7.44 (d, J=8.04 Hz, 1H), 7.42 (d,
J=8.04 Hz, III),
7.05 (s, 1H), 6.90 (d, J=8.56 Hz, 1H), 6.87 (s, 1H), 3.75 (s, 2H), 3.42 (m,
2H), 1.19 (dd,
10 J=7.24, 7.24 Hz, 3H). LCMS (ESI-) 605 (M-1).
Example 21
R
/ \ S H
O-q CF3
vH
O CO2H
15 21
{3-[4-Ethylcarbamoyl-2-(4-methoxy-benzenesulfonylamino)-
phenoxy]-5-trifluoromethyl-phenyl}-acetic acid (21) was prepared from 3-
hydroxy-
5-trifluoromethylphenylacetic acid according to Scheme 4. 1H NMR (Acetone-d6):
8
10.5 (br s, 1H), 8.81 (s, 1H), 8.20 (d, J=2.12 Hz, 1H), 7.82 (br s, 1H), 7.63-
7.70 (m,
20 3H), 7.45 (S, 1H), 6.86-6.97 (M, 5H), 3.82 (S, 3H), 3.75 (S, 2H), 3.43 (M,
2H), 1.20
(dd, J=7.20, 7.20 Hz, 3H). LCMS (ESI-) 551 (M-1).
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
63
Example 22
CI
NH
I \ CF3
O COZH
22
{3-[2-(2-Chloro-benzenesulfonylamino)-4-ethylcarbamoyl-phenoxyl-
5-trifluoromethyl-phenyl}-acetic acid (22) was prepared from 3-hydroxy-5-
trifluoromethylphenylacetic acid, using 2-chlorobenzenesulfonyl chloride,
according to
Scheme 4. 1H NMR (Acetone-d6): S 10.5 (br s, 1H), 8.93 (s, 1H), 8.12 (d,
J=1.59 Hz,
I H), 7.94 (dd, J=8.40, 1.96 Hz, 1H), 7.81 (br s, I H), 7.67 (dd, J=8.56, 2.16
Hz, I H),
7.40-7.52 (m, 4H), 7.00 (s, 1H), 6.90 (obscured d, J=8.56 Hz, 1H), 6.90 (s,
1H), 3.76 (s,
2H), 3.41 (m, 2H), 1.19 (dd, J=7.20, 7.20 Hz, 3H). LCMS (ESI-) 555 (M-1).
Example 23
C Qo
H
\ I \ CF3
H
O COZH
23
{3-[2-(4-Chloro-benzenesulfonylamino)-4-ethylcarbamoyl-phenoxyl-
5-trifluoromethyl-phenyl}-acetic acid (23) was prepared from 3-hydroxy-5-
trifluoromethylphenylacetic acid, using 4-chlorobenzenesulfonyl chloride,
according to
Scheme 4. 'H NMR (Acetone- d6): 8 10.5 (br s, 1H), 9.09 (S, IH), 8.18 (d,
J=2.16 Hz,
1H), 7.85 (br s, 1H), 7.68-7.77 (m, 3H), 7.40-7.50 (m, 3H), 6.98 (s, 1H), 6.91
(obscured
d, J=8.48, 1 H), 6.90 (s, 1 H), 3.77 (s, 2H), 3.43 (m, 2H), 1.20 (dd, J=7.24,
7.24 Hz, 3H).
LCMS (ESI-) 555 (M-1).
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
64
Example 24
QS O
NH
CF3
0 CO2H
24
{3-[2-(3-Chloro-benzenesulfonylamino)-4-ethylcarbamoyl-phenoxy]-
5-trifluoromethyl-phenyl}-acetic acid (24) was synthesized from 3-hydroxy-5-
trifluoromethylphenylacetic acid, using 3-chlorobenzenesulfonyl chloride,
according to
Scheme 4. 1H NMR (Acetone-d6): 6 10.5 (br s, 1H), 9.19 (s, 1H), 8.19 (d,
J=2.18 Hz,
1H), 7.99 (s, 1H), 7.82 (d, J=1.89 Hz, 1H), 7.55-7.74 (m, 3H), 7.50 (dd,
J=7.89, 2.24
Hz, 1H), 7.48 (d, J=8.12 Hz, 1H), 6.98 (s, 1H), 6.91 (dd, J=8.56, 2.29 Hz,
1H), 6.86 (s,
1H), 3.78 (s, 2H), 3.44 (m, 2H), 1.21 (dd, J=7.20,7.20 Hz, 3H). LCMS (ESI-)
555 (M-
1).
Example 25
C
Q
C So
NIT CH3
H
I& I
I
0 0 N^
H
Synthesis of 3-(2,4-Dichloro-benzenesulfonylamino)-N-ethyl-4-(4-
ethylcarbamoylmethyl-2-methoxy-phenoxy)-benzamide (25). A mixture of acid
20 (12) (63 mg, 0.114 mmol), EDC (44 mg, 0.228 mmol), HOBt (18 mg, 0.114
mmol),
114 L of 2N ethylamine in THE ( 0.228 mmol) in 1 mL of methylene chloride was
allowed to stir for 24 h at room temperature. Upon completion, the solvent was
removed and 10 mL of 2N HCl aqueous solution was added and the resulting
mixture
was extracted with EtOAc (4 x 20 mL). The combined extracts were washed with
25 water and brine, dried over anhydrous sodium sulfate and concentrated. The
residue
was purified by column chromatography on a silica gel column using 50%
EtOAc/hexane and EtOAc sequentially as the eluents to give 53.0 mg of the
title
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
compound. 'H NMR (CDC13): 8 7.96 (d, J=8.52 Hz, 1H), 7.87 (d, J=2.08 Hz, 1H),
7.83
(br s, 1H), 7.42 (obscured dd, J=8.52, 2.16 Hz, 1H), 7.40 (s, 1H), 7.29 (dd,
J=8.12, 2.20
Hz, 1H), 6.90 (d, J=1.72 Hz, 1H), 6.77 (d, J=8.20 Hz, 1H), 6.76 (s, 1H), 6.72
(d, J=8.08
Hz, 1H), 6.54 (d, J=8.52 Hz, 1H), 6.17 (br s, 1H), 5.62 (br s, 1H), 3.67 (s,
3H), 3.52 (s,
5 2H), 3.45 (m, 2H), 3.29 (m, 2H), 1.23 (dd, J=7.48, 7.48 Hz, 3H), 1.12 (dd,
J=7.52, 7.52
Hz, 3H). LCMS (ESI-) 579 (M-1).
Example 26
CI
Qo
C - NH CH3
H (~
10 0 NH2
26
Synthesis of 4-(4-Carbamoylmethyl-2-methoxy-phenoxy)-3-(2,4-
dichloro-benzenesulfonylamino)-N-ethyl-benzamide (26). A mixture of acid 12
(70
mg, 0.126 mmol) and urea (1.0 g, 16.6 mmol) was heated to 170-180 C and
15 maintained at this temperature for 4 h, and the mixture was allowed to
cool. As soon as
the temperature dropped to 110-120 C, 2 mL of 5% sodium carbonate aqueous
solution was added, and the mixture was shaken vigorously. After mixture was
cooled
to room temperature, 20 mL of 3N HCl aqueous solution was added and the
resulting
mixture was extracted with ethyl acetate (4 x 20 mL). The combined extracts
were
20 washed with water and brine, dried and concentrated. The residue was
purified by
silica gel chromatography using EtOAc and 10% CH3OH/EtOAc sequentially as the
eluents to give 47 mg of product. 'H NMR (Acetone-d6): 5 8.77 (s, 1H), 8.04
(d,
J=2.13 Hz, 111), 7.96 (d, J=8.57 Hz, 1H), 7.70 (, br s, 1H), 7.63 (d, J=2.13
Hz, 111),
7.56 (dd, J=8.53, 2.13 Hz, 1H), 7.51 (dd, J=8.53, 2.13 Hz, 1H), 7.11 (d,
J=1.86 Hz,
25 1H), 6.88 (dd, J=8.13, 1.87 Hz, 1H), 6.85 (br s, 1H), 6.75 (d, J=8.13 Hz,
1H), 6.659 (d,
J=8.67 Hz, 1H), 3.67 (s, 3H), 3.51 (s, 2H), 3.38 (m, 2H), 1.17 (dd, J=7.20,
7.20 Hz,
3H). LCMS (ESI-) 551 (M-1).
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
66
Example 27
O
\ ~NH
OH
Cg'
,,N I / 01"'~
0
27
Compound 27 was prepared according to Scheme 7, below.
Scheme 7
NO2 NO2 /
I CI EtNH2/H20> H I CI H I OH
C J
C CH2CI2
K2CO3, DMSO
0 NMM 0 27a
NO2 NH2
O OH Pd/C, HZ H ~~\ ~ O~ ^ ^n'OH
EtOH \iN I I 0
0 27b 0 27c
O
,NH
p-toI-802_ O OH
pyridine H
0
27
4-Chloro-3-nitro-N-ethyl-benzamide (27a). To a solution of 4-chloro-
3-nitrobenzoylchloride (2.2 g, 10 mmol, 1.0 equiv.) and N-methylmorpholine
(1.65
mL, 15 mmol, 1.5 equiv.) in 20 mL of dichloromethane was added dropwise a 70%
aqueous solution of ethylamine (1.62 mL, 20 mmol, 2.0 equiv.). The mixture was
stirred for 1 h, and poured into 40 mL of 10% citric acid. The aqueous layer
was
extracted with 20 mL of dichloromethane. The combined organic extracts were
washed
with brine, dried over sodium sulfate, and evaporated in vacuo to give 2.1 g
of a yellow
solid as product. 'H-NMR (DMSO-d6): 6 8.83 (t, J= 4.0 Hz, 1H), 8.50 (d, J= 2.4
Hz,
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
67
1H), 8.14 (dd, Jj = 8.4 Hz, J2 = 2.0 Hz, 1H), 7.91 (d, J = 8.4 Hz, 1 H), 3.31
(m, 2H),
1.14 (t, J= 7.2 Hz, 3H). MS (ESI+): 229.1 (M+H).
3-(4-Ethylcarbamoyl-2-nitrophenoxy)-phenylacetic acid (27b) To a
solution of 27a (229 mg, 1.0 mmol, 1.0 equiv.) and 3-hydroxyphenylacetic acid
(152
mg, 1.0 mmol, 1.0 equiv.) in 2 mL of DMSO, was added K2CO3 powder (414 mg, 3.0
mmol, 3.0 equiv.). The mixture was heated in an 100 C oil bath for 8 h. After
cooling
to room temperature, the mixture was poured into 15 mL of 10% aqueous citric
acid.
After extracting twice with 10 mL of EtOAc, the combined organic extracts were
washed with brine, dried over Na2SO4, and concentrated in vacuo to give a
brown solid,
which was used without further purification in the subsequent step. 'H-NMR
(DMSO-
d6): S 12.41 (br s, 1 H), 8.74 (t, J = 6.0 Hz, 1 H), 8.54 (s, 1 H), 8.13 (d,
J= 12 Hz, 1 H),
7.41 (t, J= 8.0 Hz, 1H), 7.20 - 7.12 (m, 2H), 7.11 - 7.01 (m, 2H), 3.62 (s,
2H), 3.31
(m, 2H), 1.13 (t, J= 6.0 Hz, 3H). MS (EST-): 343.1 (M-H).
3-(4-Ethylcarbamoyl-2-aminophenoxy)-phenylacetic acid (27c). To
a solution of the product obtained above in 5 mL of EtOH, was added 5% Pd on
carbon
(43 mg, 0.02 mmol, 0.02 equiv.). The mixture was stirred vigorously under a H2
atmosphere. After 27b was completely consumed, the mixture was diluted with 10
mL
of EtOAc and filtered through Celite. The filtrate was concentrated in vacuo
to give a
brown residue, which was purified by silica gel chromatography to give 27c as
an off-
white solid. 'H-NMR (DMSO-d6): 6 12.3 (br s, 1H), 8.26 (t, J= 5.3 Hz, 3H),
7.30 (t, J
= 7.8 Hz, 2H), 7.00 (m, 2H), 6.88 (s, I H), 6.82 (m, I H), 6.77 (d, J= 8.3 Hz,
I H), 5.12
(br s, 2H ), 3.62 (s, 2H), 3.24 (m, 2H), 1.11 (t, J= 7.2 Hz, 3H). MS (EST-):
313.1 (M-
H).
3-(4-Ethylcarbamoyl-2 p-toluenesulfonylamino-
phenoxy)phenylacetic acid (27) To a solution of 27c (100 mg, 0.32 mmol, 1.0
equiv.)
in 0.5 mL of pyridine, was added p-toluenesulfonyl chloride (73 mg, 0.38 mmol,
1.2
equiv.). The mixture was stirred at room temperature for 4 h, and partitioned
between
15 mL of EtOAc and 20 mL of 10% aqueous citric acid. The organic layer was
washed
with brine, dried over Na2SO4, and concentrated in vacuo to give a brown
solid. The
product was purified by silica gel chromatography to give 63 mg of 27 as a
white solid.
'H-NMR (DMSO-d6): 6 12.38 (br s, 1H), 9.99 (s, 1H), 8.43 (t, J= 6.0 Hz, 1H),
7.93 (s,
1H), 7.69 - 7.65 (m, 3H), 7.29 - 7.23 (m, 3H), 6.63 (d, J= 8.0 Hz, 1H), 6.52
(s, 1H),
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
68
6.47 (d, J= 8.0 Hz, 1H), 3.53 (s, 2H), 3.25 (m, 2H), 2.33 (s, 3H), 1.10 (t, J=
8.0 Hz,
3H). MS (ESI"): 467.2 (M-H).
Example 28
NH
C I / \ O \ OH
Me 0
/NH
I 28
3-[4-Ethylcarbamoyl-2-(2,4-dichloro-5-
methylphenylsulfonyl)amino-phenoxy]phenylacetic acid (28) was prepared
following the procedure described for 27 above. 'H-NMR (DMSO-d6): S 12.38 (br
s,
I H), 10.24 (s, 1 H), 8.49 (t, J= 6.0 Hz, 1 H), 7.91 (s, 1H), 7.74 (s, 1H),
7.66 (d, J= 8.0
Hz, 1H), 7.50 (s, 111), 7.20 (t, J= 8.0 Hz, 11-1), 7.01 (d, J= 8.0 Hz, 1H),
6.75 (d, J= 8.0
Hz, 1H), 6.55 (s, 1H), 6.44 (d, J= 8.0 Hz, 1H), 3.52 (s, 2H), 3.26 (m, 2H),
2.20 (s, 3H),
1.11 (t, J= 6.0 Hz, 3H). MS (ESt): 535.1 (M-H).
Example 29
Q p
S~NH
OH
00
29
3-(2-Benzenesulfonylamino-phenoxy)phenylacetic acid (29) was
prepared following the procedure described for 27 above. 'H-NMR (DMSO-d6): S
12.38 (br s, I H), 9.98 (s, 1H), 7.71 (d, J= 7.2 Hz, 2H), 7.59 (m, 1H), 7.48
(m, 2H),
7.37 (dd, J1= 7.6 Hz, J2 = 2.0 Hz, 111), 7.23 (t, J= 8.0 Hz, 1H), 7.07 - 6.99
(m, 3H),
6.67 (dd, J1= 8.0 Hz, JJ = 1.6 Hz, 1H), 6.57 (s, 1H), 6.55 (s, 2H), 6.48 (m,
1H), 3.52 (s,
2H). MS (ESI"): 382.1 (M-H).
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
69
Example 30
I
H
H
C
/ I ~ O I ~ O
O
30
3-[2-(2,4-Dichlorobenzenesulfonyl)amino-phenoxylphenylacetic acid
(30) was prepared following the procedure described for 27 above. IH-NMR (DMSO-
d6): S 12.4 (br s, I H), 10.19 (s, 1H), 7.77 (d, J = 12 Hz, I H), 7.54 (s, I
H), 7.44 (dd, J, =
7 Hz, J2 = 3 Hz, 1 H), 7.37 (d, J = 8 Hz, 1 H), 7.19 (m, 2H), 7.11 (m, 1 H),
6.98 (d, J = 8
Hz, 1H), 6.76 (d, J= 8 Hz, 1H), 6.75 (s, 1H), 6.44 (m, 1H), 3.52 (s, 2H). MS
(ESIT):
450.0 (M-H).
Example 31
NO2 N02
0 OEt
0 H HCI, dioxane 600
O EtOH 15
31a 31
Ethyl 3-(2-nitrophenoxy)phenylacetate (31) To a solution of 31a (1.2
g, 4.39 mmol, 1.0 equiv.) in 30 mL of EtOH, was added 3.0 mL of a 4.0 M
solution of
HCl in dioxane. The mixture was stirred at room temperature for 48 h, and
poured into
60 mL of ether. The organic layer was washed once with 30 mL of water, once
with 25
mL of saturated NaHCO3, and once with 25 mL of brine, dried over Na2SO4, and
evaporated in vacuo to give 1.15 g of 31 as a light yellow liquid. 'H-NMR
(DMSO-d6):
S 8.07 (d, J, = 8.0 Hz, J2 = 1.2 Hz, 1 H), 7.71 (t, J = 7.2 Hz, 1 H), 7.3 8
(t, J = 8.0 Hz,
2H), 7.13 (m, 2H), 6.98 (m, 2H), 4.07 (q, J = 7.2 Hz, 2H), 3.69 (s, 2H), 1.17
(t, J = 7.2
Hz, 3H). MS (ES1+): 302.2 (M+H).
Example 32
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
\C~S/H
OH
y &10 0
O
32
3-(4-Acetyl-2-benzenesulfonylamino-phenoxy)phenylacetic acid (32)
5 was prepared following the procedure described for 27 above. 'H-NMR (DMSO-
d6): S
12.40 (br s, I H), 10.43 (s, I H), 7.90 (s, I H), 7.79 (m, 2H), 7.64 (s, I H),
7.46 (d, J= 8
Hz, 1H), 7.27 (t, J= 8 Hz, 1H), 7.08 (m, 3H), 6.75 (d, J= 8 Hz, 1H), 6.61 (s,
1H), 6.55
(d, J= 8 Hz, 1H), 3.55 (s, 2H). MS (ESI"): 424.1 (M-H).
10 Example 33
q
\S.. H
/ OH
P I / I / O
33
3-(4-Benzoyl-2-benzenesulfonylamino-phenoxy)phenylacetic acid
15 (33) was prepared following the procedure described for 27 above. 'H-NMR
(DMSO-
d6): S 12.41 (br s, 1H), 10.27 (s, 1H), 7.76 - 7.49 (m, 12H), 7.32 (t, J= 8
Hz, 1H), 7.10
(d, J= 8 Hz, 1H), 6.73 (d, J= 8 Hz, 1H), 6.63 (m, 2H), 3.57 (s, 2H). MS (ESI-
): 486.0
(M-H).
Example 34
C\õO
,
NH
\ \ OH
F3C I I / O
34
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
71
3-(4-Trifluoromethyl-2-benzenesulfonylamino-phenoxy)phenylacetic
acid (34) was prepared following the procedure described for 27 above. 'H-NMR
(DMSO-d6): S 12.41 (br s, 1H), 10.42 (s, 1H), 7.73 (m, 2H), 7.62 (m, 2H), 7.52
(t, J=
7.6 Hz, 2H), 7.47 (m, 1 H), 7.3 0 (t, J = 8 Hz, 1 H), 7.09 (d, J = 8 Hz, 1 H),
6.75 (d, J =
8.8 Hz, 1H), 6.61 (m, 2H), 3.55 (s, 2H). MS (ESI"): 450.1 (M-H).
Example 35
QõO
NH
OH
C I / I ~-O
35
3-(4-Chloro-2-benzenesulfonylamino-phenoxy)phenylacetic acid (35)
was prepared following the procedure described for 27 above. 'H-NMR (DMSO-d6):
6
12.3 (br s, 1H), 10.2 (br s, 1H), 7.72 (m, 2H), 7.60 (m, 1H), 7.50 (m, 2H),
7.37 (d, J=
2.4 Hz, 1 H), 7.23 (t, J = 7.8 Hz, 1 H), 7.14 - 7.01 (m, 1 H), 7.01 (d, J =
7.6 Hz, 1 H),
6.68 (d, J= 7.2 Hz, 1H), 6.57 (m, 1H), 6.52 (m, 1H), 3.52 (s, 2H). MS (ESI-):
416.0
(M-H).
Example 36
NH
OH
'S
\o ~ / ~ / o
36
3-(4-Methoxy-2-benzenesulfonylamino-phenoxy)phenylacetic acid
(36) was prepared following the procedure described for 27 above. 'H-NMR (DMSO-
d6): 8 12.3 (br s, 1H), 9.33 (s, 1H), 7.71 (d, J= 8 Hz, 1H), 7.47 (t, J= 8 Hz,
1H), 7.08
(t, J= 8 Hz, 1H), 6.90 (m, 2H), 6.75 - 6.34 (m, 7H), 3.67 (s, 3H), 3.44 (s,
2H). MS
(ESI"): 412.0 (M-H).
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
72
Example 37
\ ~NH
HO
37
3-(4-Carbamoyl-2-benzenesulfonylamino-phenoxy)phenylacetic acid
(37) was prepared following the procedure described for 27 above. 'H-NMR (DMSO-
d6): 6 12.39 (br s, 1H), 10.08 (s, 1H), 7.94 (m, 2H), 7.70 (d, J= 4 Hz, 2H),
7.61 (m,
2H), 7.48 (t, J= 8 Hz, 2H), 7.32 (s, 111), 7.26 (t, J= 8 Hz, I H), 7.05 (d, J=
8 Hz, 1H),
6.60 (d, J= 8 Hz, 1H), 6.52 (m, 2H), 3.53 (s, 2H). MS (ESI-): 425.0 (M-H).
Example 38
C CI M eO CI
NaOMe
HMPA
O OH O OH
38a
3-Chloro-5-methoxybenoic acid (38a). To a solution of 3,5-
dichlorobenzoic acid (1.91 g, 10 mmol, 1.0 equiv.) in 10 mL of HMPA was added
NaOMe (1.62 g, 30 mmol, 3.0 equiv.). The mixture was heated in a 170 C oil
bath for
72 h. After cooling to room temperature, the mixture was poured into 50 mL of
1 M
aqueous HCI. The precipitates were collected by filtration, and washed twice
with 20
mL of water. The white solid was dried under vacuum to give 1.4 g of 38a. 'H-
NMR
(DMSO-d6): 6 14.0 (s, 1H), 7.47 (s, 1H), 7.38 (s, 1H), 7.29 (s, 1H), 3.82 (s,
3H).
MeO CI Me0 CI
LAH I
THE
O OH OH
38a 38b
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
73
3-Chloro-5-methoxybenzyl alcohol (38b). To a solution of 38a (1.0 g,
5.36 mmol, 1.0 equiv.) in 30 mL of THE was added LAH (0.20 g, 5.36 mmol, 1.0
equiv.) in several portions. The mixture was stirred at room temperature for 2
h, and
poured into 50 mL of 1M aqueous HC1. The aqueous layer was extracted twice
with 30
mL of EtOAc. The combined EtOAc extracts were washed with 40 mL of saturated
NaHCO3, 40 mL of brine, dried over Na2SO4, and evaporated in vacuo to give 0.8
g of
a white solid. 'H-NMR (DMSO-d6): S 6.93 (s, 1H), 6.87 (m, 1H), 6.85 (m, 111),
5.32 (t,
J= 5.8 Hz, I H), 4.46 (d, J= 6.0 Hz, 2H), 3.76 (s, 3H).
MeO CI 1. MsCI, LiBr, Et3N MeO CI
THE/CH2CI2
I /
2. NaCN, DMSO >
OH CN
38b 38c
3-Chloro-5-methoxybenzylcyanide (38c). To a solution of 38b (13.3
g, 77 mmol, 1.0 equiv.) in 200 mL of dichloromethane cooled at -40 C was
added
methanesulfonyl chloride (7.45 mL, 96 mmol, 1.25 equiv.), followed by dropwise
addition of triethylamine (12.9 mL, 92 mmol, 1.2 equiv.). The mixture was
stirred at -
40 C for 1 h. LiBr (20.0 g, 231 mmol, 3.0 equiv.) was added, followed by 400
mL of
THE The mixture was warmed to 0 T. After stirring at 0 C for another 2 h, the
mixture was poured into 200 mL of 1 M aqueous HCI. The mixture was evaporated
in
vacuo to remove organic solvents. The residue was extracted with 400 mL of
ether.
The ether layer was washed with 150 mL of saturated NaHCO3, 150 mL of brine,
dried
over Na2SO4, and evaporated in vacuo to give a light tan oil, which was
dissolved in
100 mL of DMSO. NaCN (4.15 g, 85 mmol, 1.1 equiv.) was added to the solution.
The mixture was stirred vigorously for 14 h. The mixture was poured into 300
mL of
water. The aqueous mixture was extracted with 200 mL of ether. The ether
extracts
were washed with 100 mL of saturated NaHCO3, 100 mL of brine, dried over
Na2SO4,
and evaporated in vacuo to give 9.8 g of a brown oil. 'H-NMR (DMSO-d6): 6 7.00
(m,
2H), 6.90 (m, 1H), 4.02 (s, 2H), 3.78 (s, 3H).
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
74
MeO ~ CI HO CI
48% HBr
I /
n-Bu4NBr OH
CN 0
38c 38d
3-Chloro-5-hydroxyphenylacetic acid (38d). To a flask containing
38c (1.2 g, 6.6 mmol, 1.0 equiv.), was added 9 mL of 48% aqueous HBr, followed
by
tetrabutylammonium bromide (0.30 g, 0.93 mmol, 0.14 equiv.). The mixture was
heated in a 125 C oil bath for 36 h. The mixture was cooled to room
temperature, and
poured into 80 mL of water. The aqueous mixture was extracted three times with
40
mL of EtOAc. The combined EtOAc extracts were washed with 50 mL of brine,
dried
over Na2SO4, and evaporated in vacuo to give a dark brown solid, which was
purified
by silica gel chromatography to give 1.0 g of 38d as a white solid. 'H-NMR
(DMSO-
d6): 6 12.34 (br s, 1 H), 9.87 (s, 1H), 6.74 (s, I H), 6.68 (s, 111), 6.63 (s,
1H), 3.40 (s,
2H).
N02
0"11~ OH
O CI
38e
3-Chloro-5-(4-ethylcarbamoyl-2-nitrophenoxy)-phenylacetic acid
(38e) was prepared using the method described for compound 27b (Scheme 7,
above).
'H-NMR (DMSO-d6): 6 12.51 (br s, 1H), 8.76 (t, J= 6.0 Hz, 1H), 8.56 (s, 1H),
8.16 (d,
J= 8 Hz, 1H), 7.27 (d, J = 8.0 Hz, 1 H), 7.25 (m, 1 H), 7.17 (m, 1H), 7.05 (m,
I H), 3.64
(s, 2H), 3.29 (m, 2H), 1.13 (t, J= 8 Hz, 3H). MS (ESI+): 379.1 (M+H).
N02 NH2
O OH SnC12.2H2O 0 OH
H I I EtOAc H I / ( i O
O CI O CI
38e 38f
3-Chloro-5-(4-ethylcarbamoyl-2-nitrophenoxy)-phenylacetic acid
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
(38f) To a solution of 38e (1.0 g, 2.64 mmol, 1.0 equiv.) in 20 mL of EtOAc
was
added SnC12.2H20 (1.91 g, 8.4 mmol, 3.2 equiv.). The mixture was heated to
reflux
for 2 h. After cooling to room temperature, the mixture was poured into 50 mL
of
water. Saturated NaHCO3 was added to adjust the pH value of the mixture to 3.
The
5 mixture was filtered through Celite to remove solid precipitates. The
filtrate was
extracted with EtOAc. The EtOAc extracts were washed with brine, dried over
Na2SO4, and evaporated in vacuo to give 0.64 g of a light tan solid. 'H-NMR
(DMSO-
d6): 6 12.45 (br s, 1H), 8.28 (t, J= 6 Hz, 1H), 7.28 (s, 1H), 7.05 (s, 1H),
7.01 (d, J= 4
Hz, 1H), 6.85 (d, J= 8 Hz, 1H), 6.79 (m, 2H), 5.18 (br s, 2H), 3.57 (s, 2H),
3.24 (m,
10 2H), 1.09 (t, J= 8 Hz, 3H). MS (ESI+): 349.1 (M+H).
O
NH
C ~ OH
O
N I / 'Pl'*'
O CI
38
3-Chloro-5-(4-ethylcarbamoyl-2 p-chlorobenzenesulfonylamino-
15 phenoxy)-phenylacetic acid (38) was prepared from compound 38f using the
method
described in Example 27. 'H-NMR (DMSO-d6): 6 12.53 (br s, 1H), 10.27 (s, 1H),
8.51
(t, J= 4 Hz, 1H), 7.95 (s, 1H), 7.65 (m, 3H), 7.48 (m, 2H), 7.10 (s, 1H), 6.86
(d, J= 8.0
Hz, 1H), 6.61 (s, 1H), 6.35 (m, 1H), 3.58 (s, 2H), 3.29 (m, 2H), 1.11 (t, J=
8.0 Hz, 3H).
MS (CIS): 523.1 (M+H).
Example 39
0\1\ z
NH
O OEt
HO I / I / O
O
39
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
76
HO HO
HCI/dioxane
OH EtOH 0""-
0
O
39a
Ethyl 3-hydroxyphenylacetate (39a) was prepared using the method
described for the preparation of 31. 'H-NMR (DMSO-d6): S 9.38 (s, 1H), 7.10
(t, J=
8.0 Hz, 1H), 6.65 (m, 2H), 7.13 (m, 3H), 4.07 (q, J= 7.1 Hz, 2H), 3.54 (s,
2H), 1.18 (t,
J= 7.2 Hz, 3H).
3-(4-Carboxyl-2 p-toluenesulfonylamino-phenoxy)phenylacetic acid
(39) was prepared using compound 39a following the reaction sequence described
in
Example 6. 'H-NMR (DMSO-d6): S 12.9 (br s, 1H), 10.8 (s, 1H), 7.96 (s, 1H),
7.66 (m,
1 H), 7.62 (m, 2H), 7.30 (m, 2H), 7.08 (m, 1 H), 6.64 (d, J = 9 Hz, 1 H), 6.62
(m, 2H),
4.08 (q, J= 6.7 Hz, 1H), 3.64 (s, 2H), 2.34 (s, 3H), 1.16 (t, J= 6.7 Hz, 3H).
MS (ESI-):
468.1 (M-H).
Example 40
osp
`$A \ NH
~NH 1. i-BuOCOCI, NMM ~
Et 2. CF3CH2NH2 I OH
i O
O 3.UGH,H2O
NH
OH
CF3
39 40
3-[4-(2,2,2-Trifluoroethyl)carbamoyl-2 p-toluenesulfonylamino-
phenoxy]phenyl-acetic acid (40). To a solution of 39 (30 mg, 0.064 mmol, 1.0
equiv.)
was in 0.15 mL of DMF was added isobutylchloroformate (12.4 L, 0.096 mmol,
1.5
equiv.), and triethylamine (17.8 L, 0.13 mmol, 2.0 equiv.). After 1 h at room
temperature, trifluoroethylamine (12.5 mg, 0.13 mmol, 2.0 equiv.) was added,
and the
mixture was stirred for another 2 h. Water (0.1 mL) was added, followed by
LiOH (27
mg, 0.64 mmol, 10 equiv.). After stirring at room temperature for 4 h, the
mixture was
acidified with 10% aqueous citric acid. Extractive workup with EtOAc gave a
yellow
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
77
residue, which was purified by reverse phase HPLC to give 3 mg of 40 as a
white solid.
'H-NMR (DMSO-d6): S 12.38 (br s, 1H), 10.06 (s, 1H), 9.07 (t, J= 6.0 Hz, 1H),
7.99
(s, 1 H), 7.98 (s, 1 H), 7.64 (d, J = 8.0 Hz, 1 H), 7.5 7 (m, 2H), 7.26 (m,
3H), 7.06 (d, J =
8.0 Hz, 1 H), 6.66 (d, J = 8.0 Hz, 1 H), 6.49 (s, 1 H), 6.49 (d, J = 8.0 Hz, 1
H), 4.06 (m,
2H), 3.54 (s, 2H), 2.33 (s, 3H). MS (ESI'): 523.1 (M+H).
Example 41
s H
Nz~ OH
NH
41
3-[4-t-Butylcarbamoyl-2 p-toluenesulfonylamino-phenoxy]phenyl-
acetic acid (41) was prepared following the procedure described in Example 40.
'H-
NMR (DMSO-d6): 8 12.38 (br s, 1H), 9.97 (br s, 1H), 7.86 (m, 1H), 7.72 (s,
1H), 7.54
(m, 3H), 7.24 (m, 2H), 7.03 (m, 2H), 6.61 (d, J= 8.4 Hz, 1H), 6.54 (m, 1H),
6.45 (m,
1H), 3.52 (s, 2H), 2.32 (s, 3H), 1.35 (s, 9H). MS (ESI+): 497.2 (M+H).
Example 42
s H
~ i o off
N
(0)
42
3-[4-Morphorlinocarbamoyl-2 p-toluenesulfonylamino-
phenoxy]phenyl-acetic acid (42) was prepared in a similar fashion as 40. 'H-
NMR
(DMSO-d6): 5 12.38 (br s, 1H), 10.09 (s, 1H), 7.58 (m, 2H), 7.37 (s, 1H), 7.27
(m, 3H),
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
78
7.15 (m, 1H), 7.05 (m, 1H), 6.65 (m, 1H), 6.53 (m, 1H, 3.85 - 3.60 (m, 8H),
2.55 (s,
2H), 2.35 (s, 3H). MS (ESI+): 511.2 (M+H).
Example 43
Q~
H
~ \ \ OH
O
NH
43
3-(4-Cyclobutylcarbamoyl-2 p-toluenesulfonylamino-
phenoxy)phenyl-acetic acid (43) was prepared in a similar fashion as 40. 'H-
NMR
(DMSO-d6): S 12.40 (br s, 1H), 9.99 (s, 1H), 8.59 (d, J= 8 Hz, 1H), 7.93 (s,
1H), 7.58
(m, 3H), 7.24 (m, 3H), 7.04 (m, 1H), 6.63 (d, J= 8 Hz, I H), 6.54 (s, 1H),
4.38 (m, 111),
3.53 (s, 2H), 2.32 (s, 3H), 2.19 (m, 2H), 2.04 (m, 2H), 1.68 (m, 2H). MS
(ESI'): 495.1
(M+H).
Example 44
S~
~ H
O OH
NH
44
3-(4-Isopropylcarbamoyl-2 p-toluenesulfonylamino-
phenoxy)phenyl-acetic acid (44) was prepared in a similar fashion as 40. 'H-
NMR
(DMSO-d6): 6 12.38 (br s, 1 H), 9.99 (br s, 1 H), 8.19 (m, 1 H), 7.93 (m, 1
H), 7.60 (m,
3H), 7.27 (m, 3H), 7.04 (m, 1H), 6.65 (m, 1H), 6.47 (m, 1H), 6.45 (m, 1H),
4.05 (m,
1 H), 3.52 (s, 2H), 2.32 (s, 3H), 1.16 (d, J = 6.4 Hz, 1 H). MS (ESI+): 482.1
(M+H).
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
79
Example 45
H
~ \ \ OH
O
\ NH
45
3-(4-Ph enylcarbamoyl-2 p-toluenesulfonylamino-phenoxy)phenyl-
acetic acid (45) was prepared in a similar fashion as 40. 'H-NMR (DMSO-d6): 8
12.38
(br s, 1H), 10.22 (s, 1H), 8.04 (m, 1H), 7.73 (m, 3H), 7.60 (m, 2H), 7.35 (m,
3H), 7.27
(m, 3H), 7.11 (m, 2H), 6.70 (m, 1H), 6.52 (s, 1H), 6.50 (m, 1H), 3.54 (s, 2H),
2.33 (s,
3H). MS (ES1): 517.1 (M+H).
Example 46
0
O OH
O
46
Example 46 was prepared in similar fashion as 27. 'H-NMR (CDC13): S
7.98 (d, J=3.3OHz, 1H), 7.70-7.72 (d, J=8.1lHz, 1H), 7.64-7.66 (d, J=8.32Hz,
1H),
7.54-7.57 (dd, J=2.10, 8.60 Hz, I H), 7.18-7.24 (m, 2H), 7.09-7.11 (m, I H),
6.73 (s,
1H), 6.65-6.67 (m, 1H), 6.46-6.48 (dd, J=1.96, 8.08 Hz, 1H), 6.32-6.34 (m,
1H), 3.62
(s, 2H), 3.50-3.53 (m, 2H), 2.61-2.65 (m, 2H), 1.60 (m, 2H) 1.31-1.33 (m, 2H),
1.26-
1.32 (m,5H), 0.74 (m, 3H), MS (ES+) 525.2 (M+H).
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
Example 47
N
O OH
O
47
5 Example 47 was prepared in similar fashion as 27. 'H-NMR (CDC13): 8
7.59-7.67 (m, 2H), 7.57 (d, J=1.90, 1H), 7.34-7.35 (m, 1H), 7.24-7.28 (m, 1H),
7.19 (s,
1H), 7.11 (m, 1H), 6.68-6.70 (d, J=8.62 Hz, 1H), 6.59 (s, 1H), 6.51 (m, 1H),
6.29 (m,
1H), 3.63 (s, 2H), 3.49-3.55 (m, 2H), 1.21-1.27 (m, 3H). MS (ES+) 489.1 (M+H).
10 Example 48
CN
O
O OH
O
48
Example 48 was prepared in similar fashion as 27. 'H-NMR (CDC13): 8
15 7.86-7.88 (m, 2H) 7.70-7.72 (m, 2H), 7.59-7.61 (dd, J=2.16, 8.59 Hz, 1H),
7.25-7.28
(m, 1H), 7.12 (m, 1H), 6.73-6.76 (d, J=8.59Hz, 1H), 6.63 (m, 1H), 6.61 (m,
1H), 6.10
(m, 1H), 3.61 (s, 2H), 3.52-3.54 (m, 2H), 1.28-1.32 (m, 3H). MS (ES+) 480.1
(M+H).
Example 49
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
81
o~~
0
0
FN k\
0
o
OH
O
49
Example 49 was prepared in similar fashion as 27. 'H-NMR (CDC13): 6
7.86-7.88 (m, 2H), 7.70-7.72 (m, 2H), 7.59-7.61 (dd, J=2.16, 8.59, 1H), 7.16
(s, 1H),
7.02-7.04 (m, 1H), 6.83-6.84 (m, 1H), 6.65-6.67 (m, 1H), 6.58-6.59 (m.1H),
6.38-6.41
(m, 1H), 3.59 (s, 2H), 3.49-3.52 (m, 2H), 3.47-3.49 (m, 2H), 1.76-1.79 (m,
2H), 1.46-
1.49 (m, 2H), 1.28-1.32 (m, 3H), 0.95-1.00 (m, 3H), MS (ES+) 527.1 (M+H).
Example 50
OI
NC
FN \\
O OH
O
Example 50 was prepared in similar fashion as 27. 'H-NMR (CDC13): 5
15 7.59-7.91 (m, 2H), 7.51-7.58 (dd, J=1.98, 8.58Hz, 1H), 7.47-7.51 (m, 1H),
7.33-7.34
(m, 1H), 7.06-7.08 (m, 1H), 6.85-6.87 (m, 1H), 6.73-6.76 (m, 1H), 6.54-6.56
(dd,
J=2.36, 8.18 Hz, 1H), 6.14-6.16 (m, 1H), 3.63 (s, 2H), 3.49-3.56 (m, 2H), 1.24-
1.28 (m,
3H). MS (ES+) 606.2 (M+H).
20 Example 51
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
82
a
o
Hr
0
o
o
FN OH
1 O
51
Example 51 was prepared in similar fashion as 27. 1H-NMR (CDC13): S
7.76-7.77 (m, 1H), 7.55-7.60 (m, 1H), 7.52-7.57 (m, 2H), 7.05 (m, 1H), 7.02-
7.04 (m,
1H), 6.72-6.74 (m, 2H), 6.55-6.60 (m, 1H), 6.10 (m, 1H), 3.65 (s, 2H), 3.52-
3.54 (m,
2H), 2.42 (s, 3H), 0.89-0.91 (m, 3H). MS (ES+) 503.1 (M+H).
Example 52
O
o
o ~
FN ION
52
Example 52 was prepared in similar fashion as 27. 1H-NMR (CDC13): S
7.93-7.97 (m, 2H), 7.72 (s, 1H), 7.60-7.62 (dd, J=2.08, 8.51 Hz, 1H), 7.53-
7.54 (m,
1H), 7.51-7.52 (d, J=2.53Hz, 1H), 7.09-7.11 (m, 1H), 6.77- 6.79 (m, 1H), 6.64-
6.67 (m,
1H), 6.13 (m, 1H), 3.67 (s, 3H), 3.63 (s, 2H), 3.51--3.53 (m, 2H), 1.21-1.27
(m, 3H).
MS (ES+) 563.1 (M+H).
Example 53
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
83
F
F
F
FN/~\ N
O
O
HJ OH
1 O
53
Example 53 was prepared in similar fashion as 27. 'H-NMR (CDC13): 6
8.27-8.29 (d, J=8.18, 1H), 8.21-8.23 (m, 2H), 7.98-8.00 (m, 1H), 7.53-7.55
(dd, J=2.16,
8.61 Hz, 1H), 7.10-7.12 (m, 1H), 6.80 (s, 1H), 6.66-6.69 (m, 2H), 6.38-6.41
(m, 1H),
3.67 (s, 2H), 3.46-3.53 (m, 2H), 1.21-1.29 (m, 3H). MS (ES+) 670.0 (M+H).
Example 54
Q\\ N
HJ/
O
\ O
O /
HJ OH
54
Example 54 was prepared in similar fashion as 27. 'H-NMR (CDC13): 6
8.33-8.35 (m, 1H), 8.08-8.09 (m, 1H), 8.04-8.06 (m, 1H), 7.94-7.96 (m, 1H),
7.89 (s,
1H), 7.56-7.58 (m, 1H), 7.49-7.54 (m, 1H), 6.99-7.02 (m, 1H), 6.94-6.98 (m,
1H), 6.61-
6.64 (d, J=8.66 Hz, 1 H), 6.61 (m, 111), 6.16-6.18 (m, 1 H), 6.12 (m, I H),
3.63 (s, 2H),
3.49-3.57 (m, 2H), 1.28-1.31 (m, 3H). MS (ES+) 506.1 (M+H).
Example 55
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
84
S
~\s
FN/ \Q
o
HJ` OH
1 O
Example 55 was prepared in similar fashion as 27. 'H-NMR (CDC13): S
7.62-7.64 (m, 1H), 7.57-7.58 (d, J=4.82Hz, 1H), 7.51-7.52 (m, 1H), 7.19 (s,
1H), 7.12-
5 7.14 (m, 111), 7.02-7.04 (m, I H), 6.74-6.76 (d, J=8.56 Hz, I H), 6.69 (m,
111), 6.67-6.68
(m, 1H), 6.15-6.17 (m, 1H), 3.64 (s, 2H), 3.52-3.55 (m, 2H), 1.21-1.27 (m,
3H). MS
(ES+) 461.1 (M+H).
Example 56
0 s
O
o FN OH
0
56
Example 56 was prepared in similar fashion as 27. 'H-NMR (CDC13): S
7.60-7.70 (m, 1H), 7.53-7.55 (m, 2H), 7.30-7.33 (m, 2H), 7.24-7.28 (m, 1H),
7.19 (m,
1H), 7.11 (m, 1H), 6.68-6.70 (d, J=8.58Hz, 1H), 6.51 (m, 1H), 3.63 (s, 2H),
3.46-3.51
(m, 2H), 2.43 (s, 3H), 1.21-1.27 (m, 3H). MS (ES+) 560.1 (M+H).
Example 57
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
N
O N~
S
FN \\
0
o
FN` OH
1 O
57
Example 57 was prepared in similar fashion as 27. 1H-NMR (CDC13): 6
5 7.53-7.54 (m, 2H), 7.32-7.33 (m, 1H), 7.10-7.11 (m, 2H), 7.02 (m, 1H), 7.00-
7.01 (m,
1H), 6.76-6.78 (d, J=8.60 Hz, 1H), 6.51 (m, 1H), 3.67 (s, 2H), 3.63 (s, 3H),
3.49-3.52
(m, 2H) 1.21-1.27 (m, 3H). MS (ES+) 459.1 (M+H).
Example 58
OH
O-Z
NIH
C NHz O
a-b 0) CFb
58a 58
Scheme 8. a. 4-Toluenesulfonyl chloride, Na2CO3/H20, 70 C, 1 h (Org. Synth.
Coll.
Vol. IV, (1963) 34:35). b. SOC12/DCM, room temperature, 4 days. c. 4-
Methoxyphenylacetic acid, A1C13/DCE, 50 C, 3 h.
Example 58 was prepared according to Scheme 8. 'H NMR (CDC13): S
11.15 (s, 1H), 7.76 (m, 3H), 7.38 (m, 3H), 7.22 (d, 2H), 7.04 (s, 1H), 6.93
(m, 2H), 3.63
(s, 3H), 3.59 (s, 2H), 2.36 (s, 3H). MS (ESI+) 440.1 [MH]+.
Example 59
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
86
OH
CI OCH3 CH3 H3C l i O
c, d
(XN02 a H3C O b H3CO 0
-~ NH CI
N02 NH2
59a 59b 59
Scheme 9. a. Methyl 4-methoxyphenylacetate, A1C13/DCE, 0 C, 30 min. and room
temperature, 2 h. b. Fe/HOAc and H2O, 65 C, 2 h. c. 2,6-
Dichlorobenzenesulfonyl
chloride, DMAP, Pyridine, room temperature, overnight. d. LiOH/MeOH and H2O,
room temperature, overnight.
Compound 59a. 1H NMR (CDC13): S 8.17 (d, 1H), 7.93 (s, 1H), 7.72 (t,
1H), 7.59 (t, 1H), 7.48 (m, 1H), 7.41 (d, 1H), 6.86 (d, 1H), 3.73 (s, 3H),
3.65 (s, 2H),
3.49 (s, 3H). MS (EST) 330.1 [MH]+.
Compound 59b. 1H NMR (CDC13): 6 7.28 (m, 3H), 7.17 (s, 1H), 6.95
(d, 1H), 6.70 (d, 1H), 6.54 (t, 1H), 3.77 (s, 3H), 3.70 (s, 3H), 3.60 (s, 2H).
MS (EST)
300.1 [MH]+.
Example 59 was prepared according to Scheme 9. 1H NMR (CDC13): 5
11.68 (s, I H), 8.15 (d, I H), 7.62 (d, 1H), 7.47 (s, I H), 7.38 (m, 4H), 7.20
(s, 1H), 6.97
(d, 2H), 3.67 (s, 3H), 3.64 (s, 2H). MS (ESI) 494.0 [MH]+.
Example 60
OEt OH
0
OEt OEt / / -
N
N\ a QN~ b c d
Q/ H H H 04 O-4
O (~ I H/ H3 3
60
Scheme 10. a. NaBH3CN/HOAc, room temperature 10 min. (J. Med. Chem. (1997)
40:4222). b. compound 58a (see Scheme 8, above), EDC, HOBt, NMM, room
temperature, overnight. c. LiOH/MeOH and water, room temperature, overnight.
d.
DDQ/toluene, 110 C, 6 h.
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
87
Example 60 was prepared according to Scheme 10. 'H NMR (CDC13): 6
8.65 (s, 1H), 8.18 (d, 1H), 7.87 (d, 1H), 7.60 (m, 4H), 7.44 (m, 3H), 7.30 (m,
IH), 6.93
(m, 3H), 3.78 (s, 2H), 2.10 (s, 3H). MS (ESI) 449.1 [MH]+.
Example 61
OH
Br O ONO
a N b QN~ C, d
NH
N02 dN02 I C
C NH2 04
"'~H3
61a 61b 61c 61
Scheme 11. a. Ethyl 3-indoleacetate, tBuOK/DMF, room temperature, overnight.
b.
H, Pd/C in ethanol, room temperature, 3 h. c. 4-Toluenesulfonyl chloride,
DMAP/pyridine. d. LiOH/THF, MeOH and water, room temperature, overnight.
Compound 61b. 'H NMR (CDC13): 6 8.20 (m, 1H), 7.70 (m, 1H), 7.43
(m, 2H), 7.20 (m, 4H), 6.54 (m, 1H), 5.76 (s, 2H), 4.20 (m, 2H), 3.83 (s, 2H),
1.30 (t,
3H). MS (ESI) 339.1 [MH]+.
Example 61 was prepared according to Scheme 11. 'H NMR (CDC13): 6
7.59 (m, 2H), 7.25 (m, I H), 7.12 (m, 6H), 6.98 (m,1 H), 6.73 (m, 1H), 6.60
(s, 111), 5.17
(s, 2H), 3.82 (s, 2H), 2.44 (s, 3H). MS (ESI) 435.1 [MH]+.
Example 62
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
88
_~IH
O
a,b
61c 62
Scheme 12. a. Phenylacetyl chloride, Net /DCM, room temperature, overnight. b.
LiOH/THF, MeOH and water, room temperature, overnight.
Example 62 was prepared according to Scheme 12. 1H NMR (CDC13): S
8.65 (m, 1H), 8.58 (m, 1H), 7.20 (m, 7H), 7.04 (m, 4H), 6.85 (s, 1H), 6.80 (s,
1H), 5.08
(s, 2H), 3.75 (s, 2H), 3.40 (s, 2H). MS (ESf) 399.2 [MH]+.
Example 63
a, b
OLFk /
61c 63
Scheme 13. a. Phenylacetaldehyde, Na BH(OAc)3/DCE, r.t, overnight. b.
LiOH/THF,
MeOH and water, room temperature, overnight.
Example 63 was prepared according to Scheme 13. 1H NMR (CDC13): 5
7.63 (m,1H), 7.35 (m, 1H), 7.21 (m, 6H), 7.06 (m, 3H), 6.98 (s, 1H), 6.75 (m,
1H), 6.68
(m, 1H), 5.17 (s, 2H), 4.23 (s, 2H), 3.76 (s, 2H). MS (ESI+) 371.2 [MH]+.
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
89
Example 64
OH
OG-b
r I~
a, b- I c,d
60a o:
64a ):::LCF-b
64
Scheme 14. a. Methyl phenylacetate, A1C13, 55 C, 6 h. b. H2, Pd/C in
methanol, room
temperature, 2 h. c. 4-Toluenesulfonyl chloride, DMAP/pyridine. d. LiOH/THF,
MeOH and water, room temperature, overnight.
Example 64 was prepared according to Scheme 14. 'H NMR (CDC13): 6
7.52 (m, 2H), 7.36 (m, 1H), 7.20 (m, 7H), 6.95 (m, 1H), 6.26 (m, 1H), 3.64 (s,
2H),
3.61 (s, 2H), 2.41 (s, 3H). MS (ESI+) 396.1 [MH]+.
Example 65
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
r r O
a > EtH &Na
NOz 0 0 EtH I NOz
O
O
c, d,,- e
NIH Et EtH I i
O i I~ 6~1-~a"'
65a 65
Scheme 15. a. Ethylamine, EDC, HOBt, NMM in DCM, r.t, 2 h. b. Ethyl 3-
5 dihydroindoleacetate, K2CO3/DMF, room temperature, overnight. c. H2, Pd/C in
ethanol. d. 4-Toluenesulfonyl chloride, DMAP, Pyridine, room temperature,
overnight.
e. LiOH/THF, MeOH, and water, room temperature, overnight.
Compound 65a. 1H NMR (CDC13): S 8.95 (s, 1H), 7.87 (s, 1H), 7.61
10 (m, 3H), 7.24 (d, 2H), 7.20 (d, 1H), 7.15 (d, 1H), 7.10 (t, 1H), 6.89 (t,
1H), 6.34 (d,
1H), 6.14 (br s, 1H), 4.16 (m, 2H), 3.76 (dd, 2H), 3.65 (m, 1H), 3.52 (m, 2H),
3.30 (t,
1H), 2.81 (m, 2H), 2.54 (m, 1H), 2.42 (s, 3H), 1.28 (m, 6H). MS (ES1+) 536.2
[MH]+.
Example 65 was prepared according to Scheme 15. 1H NMR (CDC13): 6
8.80 (br s, 1H), 7.87 (s, 1H), 7.61 (m, 3H), 7.24 (d, 2H), 7.20 (d, 1H), 7.15
(d, 1H), 7.10
15 (t, 1H), 6.89 (t, 1H), 6.34 (d, 1H), 6.24 (br s, 1H), 3.78 (dd, 2H), 3.65
(m, 1H), 3.52 (m,
2H), 3.30 (t, 1H), 2.85 (m, 2H), 2.59 (m, 1H), 2.42 (s, 3H), 1.28 (m, 3H). MS
(ESf )
508.2 [MH]+.
Example 66
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
91
rI%M __eH
0 Z 0
a,b
Et BHN~
r
i r C~-b
65a 66
Scheme 16. a. o-Chloranil in MTBE and THF, room temperature, 20 min. b.
LiOH/THF, MeOH and water, room temperature, overnight.
Example 66 was prepared according to Scheme 16. 1H NMR (CDC13): 6
7.56 (m, 3H), 7.39 (s, 1H), 7.36 (m, 1H), 7.21 (d, 2H), 7.07 (m, 2H), 6.84 (s,
1H), 6.78
(d, 1H), 6.55 (d, 1H), 6.16 (br s, IH), 5.05 (s, 2H), 3.77 (s, 2H), 3.38 (m,
2H), 2.40 (s,
3H), 1.67 (t, 3H). MS (ESI) 506.2 [MH]+.
Example 67
CH3 H
OCH3 CH3 O O
H3CO O H3C H3Co
a H3CO b -N C
O _N
NH2 I / N N
~
57b
67a H 67b 67
Scheme 17. a. Glycine methyl ester HC1 salt/pyridine, 110 C, 3 days. b. Benzyl
bromide, tBuOK/DMF, room temperature, overnight. c. LiOH/THF, MeOH, and
water, room temperature, overnight.
Compound 67a. 'H NMR (CDC13) 6 7.50 (m, 1H), 7.38 (m, 2H), 7.22
(d, 1H), 7.10 (m, 2H), 6.84 (d, 1H), 4.32 (s, 2H), 3.69 (s, 3H), 3.57 (s, 2H),
3.52 (s,
3H). MS (ESI) 339.1 [MH]+.
Compound 67b. 'H NMR (CDC13) 6 7.50-7.20 (m, 9H), 7.33 (m, 2H),
7.17 (d, 1H), 7.05 (m, 2H), 6.88 (d, 1H), 5.52 (d, 1H), 5.04 (d, 1H), 4.98 (d,
1H), 3.96
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
92
(d, 1H), 3.76 (s, 3H), 3.64 (s, 2H), 3.37 (s, 3H). MS (ESI) 447.2 [MH]+.
Example 67 was prepared according to Scheme 17. 1H NMR (CDC13
with drops of CD30D) 8 7.37 (m, 3H), 7.18 (m, 6H), 7.08 (d, 1H), 7.04 (m, 2H),
6.80
(d, 1H), 5.39 (d, 1H), 4.93 (d, 1H), 4.77 (d, 1H), 3.86 (d, 1H), 3.53 (s, 2H),
3.29 (s,
3H). MS (ESI+) 433.2 [MH]+.
Example 68
COOEt COOEt
COOEt
F
HO ~ NO2 + a b, c
/ -' /
6a H2 ~ H ~ NH
HO / NO EtHN
2 NH2
68a 68b
COOEt COOH
COOEt
d e O
N f N 0
YN EtHN I / EtHN
EtHN I / N
O 68c H 68d 16\cl 68
C
Scheme 18 a. NEt3/EtOH, 100 C, 5days; b. EtNH2, EDC, HOBt, NMM/DCM, r.t., 4h;
c. H2, Pd/C, EtOH, r.t., lh; d. Malonyl dichloride/THF, r.t., 2 days; e. 4-
Chlorobenzyl
chloride, tBuOK/DMF, r.t., overnight; f. LiOH/THF, MeOH, and water, r.t.,
overnight.
Compound 68a. 1H NMR (CDC13) 8 9.85 (s, 1H), 9.01 (s, 1H), 8.01
(d, 2H), 7.41 (d, 2H), 7.28 (d, 2H), 7.20 (d, 1H), 4.21 (q, 2H), 3.68 (s, 2H),
1.30 (t, 3H).
MS (ESI) 345.1 [MH]+.
Compound 68c. 1H NMR (CDC13) 8 9.35 (s, 1H), 7.77 (s, 1H), 7.43
(d, 2H), 7.33 (d, 2H), 7.13 (d, 2H), 6.96 (d, 1H), 6.50 (br s, 1H), 4.15 (q,
2H), 3.62 (s,
2H), 3.53 (s, 2H), 3.48 (m, 2H), 1.25 (m, 6H). MS (ESI) 410.2 [MH]+.
Compound 68d. 1H NMR (CDC13) 6 8.05 (s, 1H), 7.35 (m, 1H), 7.22
(m, 4H), 7.14 (d, 2H), 6.88 (d, 1H), 6.63 (d, 2H), 6.10 (br s, 1H), 5.86 (d,
1H), 4.69 (d,
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
93
1H), 4.17 (q, 2H), 3.65-3.45 (m, 4H), 2.87 (s, 1H), 2.89 (s, 1H), 1.27 (m,
6H). MS
(ESI) 534.2 [MH]+.
Example 68 was prepared according to Scheme 18. 1H NMR (CDC13
with drops of CD3OD) 6 8.05 (s, 1H), 7.38 (d, 1H), 7.20 (m, 4H), 7.12 (d, 2H),
6.86 (d,
1H), 6.59 (d, 2H), 5.86 (d, 1H), 4.69 (d, 1H), 3.58 (s, 2H), 3.51 (s, 2H),
3.46 (q, 2H),
1.24 (t, 3H). MS (ESI) 506.1 [MH]+.
Example 69
COOH
COOEt
a, b
NH
f~l-I
I~ EtHqj
O 68b 69 O
Me
Scheme 19. a. p-Toluenesulfonyl chloride, DMAP, Pyridine, r.t., overnight; b.
LiOH/THF, MeOH, and water, r.t., overnight.
Example 69 was prepared according to Scheme 19. 1H NMR (CDC13
with drops of CD3OD) 8 7.54 (d, 2H), 7.35 (m, 1H), 7.14 (m, 6H), 6.83 (d, 2H),
3.51 (s,
2H), 3.34 (q, 2H), 2.30 (s, 3H), 1.16 (t, 3H). MS (ESI) 468.2 [MH]+.
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
94
Example 70
OCH3
OCH3 CI
a, b, c \ d H3CO e
HO I NO EtHN I / NO O
z 2 EtHN
NO2
CH3 OH
H3C 9 H3CO
\ O \ O
EtHN NH EtHN 1 / NH
2
O OzzS
~t I \
70 Me
Scheme 20. a. EtNH2, EDC, HOBt, NMM/DCM, r.t., 4 h. b. LiOH/THF, MeOH, r.t.,
overnight. c. SOC12/DCM, 60 C, 8h. d. Methyl 4-methoxyphenylacetate,
A1C13/DCE,
r.t., 2 h. e. Iron/HOAc, water, 65 C, 30 min. f p-Toluenesulfonyl chloride,
DMAP,
pyridine, r.t., overnight. g. LiOH/THF, MeOH, water, r.t., overnight.
Example 70 was prepared according to Scheme 20. 1H NMR (CDC13) 8
10.96 (s, 1H), 8.00 (s, 1H), 7.74 (d, 2H), 7.40 (m, 3H), 7.22 (d, 2H), 7.07
(s, 1H), 6.91
(d, 1H), 6.18 (br s, 1H), 3.61 (s, 2H), 3.59 (s, 3H), 3.50 (m, 2H), 2.35 (s,
3H), 1.27 (t,
3H). MS (ESI) 511.2 [MH]+.
Example 71
OH OH
l i O F l i O
a
Et
t
NH E
NH
I 0 1
70 71
Scheme 21. a. Triethylsilane, TFA, room temperature, overnight.
Example 71 was prepared according to Scheme 21. 'H NMR (CDC13) 8
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
8.11 (s, 1H), 7.73 (s, 1H), 7.56 (d, 1H), 7.50 (d, 2H), 7.30 (d, 1H), 7.17 (m,
3H), 7.00
(s, 1H), 6.86 (d, 1H), 6.15 (br s, 1H), 3.99 (s, 3H), 3.52 (s, 2H), 3.47 (m,
4H), 2.35 (s,
3H), 1.27 (t, 3H). MS (ESIF) 497.2 [MH]+.
5 Example 72
OH OH
i O O
a
lk~ O Nz~ OH
Et I i NH BHN~ , NI H
Ot:
O 0
71akb Me
70 72
Scheme 22. a. NaBH4, EtOH, rt, 1 h.
Example 72 was prepared according to Scheme 22. 'H NMR (CDC13) 8
8.42 (s, 1H), 7.76 (s, 1H), 7.61 (d, 2H), 7.50 (d, 1H), 7.21 (m, 3H), 7.06 (d,
1H), 6.97
(s, 1H), 6.86 (d, 1H), 6.15 (br s, 1H), 5.58 (s, 1H), 3.82 (s, 3H), 3.53 (s,
2H), 3.47 (m,
2H), 2.39 (s, 3H), 1.24 (t, 3H). MS (ESI) 513.2 [MH]+.
Example 73
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
96
Scheme 23
NO2 NO2 NO 2 OCH3
\ CI \ CI \ O \
CI I/ n-BuHN / n-BuHN I/ I/
0 0 O
0 OH
73a 73b
NH2 OCH3 NH2 OCH3
n-BuHN n-BuHN
0 O
0 OH 0 OCH3
73c 73d
CI O0
NH OCH3
CI 0 \
n-BuHN
O
0 OH
73
Compound 73a. To a solution of 4-chloro-3-nitrobenzoyl chloride (440
g, 2 mol), in THE (1 L) at 0 C, was added slowly a mixture of n-butylamine
(198 mL,
2 mol) and triethylamine (279 mL, 2 mol) over 4 h. During addition the
temperature of
the reaction mixture was kept below 5 C. After addition, the mixture was
stirred at 0
C for 12 h. The reaction mixture was treated with EtOAc (1 L) and water (1 L).
The
aqueous layer was separated, and the organic layer was washed with brine (2N
HCl was
added to adjust pH to 2) twice and water once. The organic layer was treated
with ether
(2.5 L). After stirring, the solid generated was collected by filtration to
give 400 g of
the desired product. 'H NMR (DMSO-d6) S 8.78 (t, 1H), 8.50 (d, 1H), 8.15 (dd,
1H),
7.90 (d, 1H), 3.29 (q, 2H), 1.52 (m, 2H), 1.34 (m, 2H), 0.91 (t, 3H). MS (ESI)
257.0
[MH]
.
Compound 73b. To a mixture of 73a (339 g, 1.32 mol) and
homovanilic acid (244 g, 1.34 mol) in DMSO (1 L), was added cesium carbonate
(947
g, 2.9 mol). The mixture was heated to 65 C. After 1 h of heating, 100 g more
of
cesium carbonate and 200 mL of DMSO were added. Two h later another 100 g of
cesium carbonate and 200 mL of DMSO were added. Heating and stirring were
continued for 6 h more. After cooling, EtOAc (3 L) and water (2 L) were added,
and
the mixture was acidified with concentrated HCl to pH2. During the
acidification, the
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
97
temperature was kept below 30 C. The aqueous layer was separated, and the
organic
layer was heated to 50 C and washed with brine (2N HCl was added to adjust pH
to 2)
twice and water once. The organic layer was cooled to 0 C, and the crystal
formed
was collected and washed with 50% EtOAc/hexane. The mother liquor was
concentrated, and the residue was recrystallized in hot EtOAc. Yield 480g. 'H
NMR
(DMSO-d6) 5 12.4 (s, 1H), 8.64 (t, 1H), 8.50 (d, 1H), 8.05 (dd, 1H), 7.19 (d,
1H), 7.16
(d, 1H), 6.96 (dd, 1H), 6.86 (d, 1H), 3.73 (s, 3H), 3.64 (s, 2H), 3.27 (q,
2H), 1.52 (m,
2H), 1.34 (m, 2H), 0.91 (t, 3H). MS (ESI+) 403.1 [MH]+.
Compound 73c. Pd/C (15 g, 10% wet) was added to 73b (402 g, 1 mol)
in IN NaOH (1 L, 1 mol) and water (0.2 L). The mixture was shaken under
hydrogen
(40 psi) for 3 h at r.t. The catalyst was removed by filtration through
celite, and the
celite was washed with water (1 L). The filtrate was neutralized by adding
slowly 2N
HCl (0.5 L) to the filtrate which was vigorously stirred. The fine powder
generated
was collected by filtration to give 365 g of the desired product. 'H NMR (DMSO-
d6) 8
12.4 (bs, 111), 8.14 (t, 1 H), 7.24 (d, 111), 7.07 (d, I H), 6.92 (dd, 114),
6.87 (m, 2H), 6.43
(d, 1H), 5.00 (bs, 2H), 3.77 (s, 3H), 3.58 (s, 2H), 3.21 (q, 2H), 1.47 (m,
2H), 1.31 (m,
2H), 0.90 (t, 3H). MS (ESI+) 373.2 [MH]+.
Compound 73d. Concentrated sulfuric acid (22.4 mL) was added
dropwise to a stirred solution of 73c (150 g) in methanol (800 mL) at r.t..
The mixture
was then heated to 60 C for 5 h. Most of the methanol was removed under
vacuum,
and the residue was taken by EtOAc (800 mL) and neutralized by saturated
sodium
carbonate. The organic layer was separated, and the aqueous layer was
extracted with
EtOAc (400 mL). The combined organic layers were dried with MgSO4 and
concentrated to give the desired product in quantitative yield. 1H NMR (DMSO-
d6) S
8.14 (t, 1H), 7.24 (d, 1H), 7.08 (d, 1H), 6.93 (dd, 1H), 6.86 (m, 2H), 6.45
(d, 1H), 5.05
(bs, 2H), 3.77 (s, 3H), 3.69 (s, 2H), 3.65 (s, 3H), 3.21 (q, 2H), 1.47 (m,
2H), 1.31 (m,
2H), 0.90 (t, 3H). MS (ESI') 387.2 [MH]+.
Example 73. 2,4-Dichlorobenzenesulfonyl chloride (112 g, 455 mmol)
was added to a mixture of 73d (135 g, 350 mmol) and 2,6-lutidine (57 mL, 490
mmol)
in THE (500 mL) at r.t. The mixture was heated to 60 C and stirred for 12 h.
After
cooling to r.t., water (300 mL) and ION NaOH (180 mL) were added. The mixture
was
stirred at r.t. for 2 h, acidified to pH 2 with concentrated HCI, and
extracted with
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
98
EtOAc (1 L). The organic layer was washed with brine (2N HC1 was added to
adjust
pH to 2) three times, dried with MgSO4, and concentrated. The residue was
treated
with DCM (300 mL), stirred, and collected by filtration. Then the product was
recrystallized in hot EtOH (95%) (400 mL) to give 120 g of the desired
product. The
mother liquor was concentrated, and the residue was purified using the method
to give
30 g of desired product. 1H NMR (DMSO-d6) 6 12.4 (bs, 1H), 10.2 (bs, 1H), 8.38
(t,
1H), 7.87 (d, 1H), 7.84 (d, 1H), 7.75 (d, 1H), 7.58 (dd, 1H), 7.50 (dd, 1H),
7.04 (d, 1H),
6.81 (dd, 1H), 6.55 (d, 1H), 6.40 (d, 1H), 3.62 (s, 3H), 3.59 (s, 2H), 3.23
(q, 2H), 1.49
(m, 2H), 1.31 (m, 2H), 0.91 (t, 3H). MS (ESI) 581.1 [MH]+.
Example 74
Scheme 24
-
C
- H Urea
H DMSO H
1 180 D MS O COCHAO)CH3
*02H
12 74
3-(2,4-Dichloro-benzenesulfonylamino)-N-ethyl-4-[4-(3-
methanesulfinyl-2-oxo-propyl)-2-methoxy-phenoxy]-benzamide (74). Example 74
was prepared from Example 12 according to Scheme 24. 400 MHz 1H NMR (CDC13)
8: 7.95 (d, J=8.5 Hz, I H), 7.87 (d, J=2. l Hz, I H), 7.81 (s, 1H), 7.44 (dd,
J=8.6, 2.16 Hz,
1H), 7.40 (d, J=1.88 Hz, 1H), 7.31 (dd, J=8.44, 1.88 Hz, 1H), 6.92 (d, J=1.76
Hz, 1H),
6.71 (d, J=8.08 Hz, 1H), 6.55 (d, J=8.56 Hz, 1H), 5.18 (s, 2H), 3.68 (s, 3H),
3.67 (s,
2H), 3.46 (m, 2H), 2.22 (s, 3H), 1.24 (dd, J=7.24, 7.24 Hz, 3H). LCMS (ES-)
m/z 611
(M-1).
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
99
Example 75
Scheme 25
Cl CI
QO C~ O
C --(ur NH 0( Urea H POC13
H 180 C, 4 hours H I I DMF
86% 80 C, 3 hours
52%
12 O2H 02NH2
Cl Cl
CO
NH Cf NaN3 C NH Cf
DMF I I N
120 C, 3 hours H
55%
N 75
3-(2,4-Dichloro-benzenesulfonylamino)-N-ethyl-4-[2-methoxy-4-
(1H-tetrazol-5-ylmethyl)-phenoxy]-benzamide (75) was prepared according to
Scheme 25. 400 MHz 1H NMR (Acetone-d6) S: 8.60 (br s, 1H), 8.04 (d, J=2.12 Hz,
1H), 7.94 (d, J=8.52 Hz, 1H), 7.72 (br s, 1H), 7.61 (d, J=2.04 Hz, 1H), 7.55
(dd, J=8.60,
2.2 Hz, 1 H), 7.49 (dd, J=8.48, 2.12 Hz, 1 H), 7.16 (d, J=2.16 Hz, 1 H), 6.90
(dd, J=8.12,
2.16 Hz, 1H), 6.78 (d, J=8.16 Hz, 1H), 6.48 (d, J=8.52 Hz, 1H), 4.38 (s, 2H),
3.67 (s,
3H), 3.38 9m, 2H), 1.16 (dd, J=7.16, 7.16 Hz, 3H). LCMS (ES+) m/z 578 (M+1).
Example 76
Scheme 26
QUO Q O
EDC, HOBT
NMM H2NOH
DMF, rt
C(O)NHOH
12 76
3-(2,4-Dichloro-benzenesulfonylamino)-N-ethyl-4-(4-hydroxy-
carbamoylmethyl-2-methoxy-phenoxy)-benzamide (76) was prepared according to
Scheme 26. 400 MHz 1H NMR (Acetone-d6) S: 10.2 (s, 1H), 8.82 (s, 1H), 8.06 (d,
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
100
J=2.04 Hz, 1H), 7.97 (d, J=8.52 Hz, 1H), 7.75 (s, 1H), 7.65 (d, J=2.0 Hz, I
H), 7.58 (dd,
J=8.64, 2.12 Hz, 1H), 7.53 (dd, J=8.52, 2.08 Hz, 1H), 7.12 (s, 1H), 6.89 (dd,
J=8.08,
1.76 Hz, 1H), 6.75 (d, J=8.04 Hz, 1H), 6.45 (d, J=8.56 Hz, 1H), 3.68 (s, 3H),
3.45 (s,
2H), 3.40 (m, 2H), 1.14 (dd, J=7.20, 7.20 Hz, 3H). LCMS (ES-) m/z 567 (M-1).
Example 77
&
H
C02H
77
[3-(4-Ethylcarbamoyl-2-isopropoxycarbonylamino-phenoxy)-4-
methoxy-phenyl]-acetic acid (77). 400 MHz 1H NMR (Acetone-d6) 6: 10.9 (br s,
1H),
8.63 (d, J=2.07 Hz, I H), 8.03 (s, I H), 7.72 (br s, I H), 7.46 (dd, J=8.52,
2.19 Hz, I H),
7.20 9dd, J=8.35, 2.16 Hz, 1H), 7.15 (d, J=2.10, 1H), 7.13 (d, J=8.39 Hz, 1H),
6.69 (d,
J=8.51 Hz, 1H), 5.00 (m, 1H), 3.80 (s, 3H), 3.61 (s, 2H), 3.42 (m, 2H), 1.29
(d, J=6.25
Hz, 6H), 1.19 (dd, J=7.13, 7.13 Hz, 3H). LCMS (ES-) m/z 429 (M-1).
Example 78
Scheme 27
I
NH2 D' OfI) -NH C(
O 0
ll Lutidine p
H C~OCI
Acetone, rt
CO2CH3
COZCH3
0 0
NJLN H D' N/ `N H 0(
~ ~ LiOH 0,60
DMSO, rt H I CH30H, HO H
COZCH3 LCOZH
78
{3-[2-(3-Benzyl-3-methyl-ureido)-4-ethylcarbamoyl-phenoxy]-4-
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
101
methoxy-phenyl}-acetic acid (78) was prepared according to Scheme 27. 400 MHz
1H NMR (Acetone-d6) S: 10.9 (br s, 1H), 8.72 (d, J=2.18 Hz, 1H), 7.65 (br s,
1H), 7.58
(s, I H), 7.42 (dd, J=8.51, 2.21 Hz, I H), 7.18-7.35 (m, 6H), 7.11 (d, J=8.18
Hz, I H),
7.10 (s, 1H), 6.67 9d, J=8.49 Hz, 1H), 4.66 (s, 2H), 3.76 (s, 1H), 3.62 (s,
2H), 3.42 (m,
2H), 3.09 (s, 3H), 1.19 9dd, J=7.11, 7.11 Hz, 1H). LCMS (ES-) m/z 490 (M-1).
Example 79
Scheme 28
NH2
\2 CI + HO OH K2CO3, DMSO I O OOH
0 ON /
02N 79a
0~~0
CI Pyridine
C /
Y
0 O 0 O
\ iNH SnC12.2H20, EtOAc / NH 0 OH
O OH C
0 O I O
H2N 1~ 2NI
79 79b
3-(2-Amino-4-nitrophenoxy)phenylacetic acid (79a) A mixture of 2-
flruoro-5-nitroaniline (3.1 g, 20 mmol, 1.0 equiv.) 3-hydroxyphenylacetic acid
(3.04 g,
mmol, 1.0 equiv.) and K2CO3 (6.9 g, 50 mmol, 2.5 equiv.) in 20 mL of DMSO was
15 heated to 130 C and stirred at that temperature for 18 h. After cooling to
room
temperature, the mixture was poured into aqueous citric acid. The resulting
mixture
was extracted twice with EtOAc. The combined organic extract was washed with
brine, dried over sodium sulfate, and evaporated in vacuo to give a dark brown
foam,
which was purified by silica gel chromatography to give 4.3 g of 1 as a yellow
solid.
20 MS (ESI+): 289.0 (M+H).
3- [2-(4-Ch lorophenylsulfonylamino)-4-nitrophenoxyl phenylacetic
acid (79b) To a solution of 79a (4.3 g, 14.9 mmol, 1.0 equiv.) in 20 mL of
pyridine,
was added 4-chlorosulfonyl chloride (3.67 g, 17.4 mmol, 1.2 equiv.). The
mixture was
stirred at room temperature for 4 h. The mixture was diluted with 100 mL of
EtOAc
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
102
and washed twice with 100 mL of 10% aqueous citric acid, once with 50 mL of
brine,
dried over Na2SO4, and concentrated in vacuo to give a brown solid, which was
purified by silica gel chromatography to give 6.2 g of 79b as a yellow solid.
'H-NMR
(400 MHz, DMSO-d6): S 12.45 (br s, 1H), 10.66 (s, IH), 8.21 (d, J= 4 Hz, IH),
7.75
(d, J= 8 Hz, 2H), 7.60 (d, J = 8 Hz, 2H), 7.3 3 (t, J = 8 Hz, 1 H), 7.15 (m, 1
H), 6.77 (d, J
= 12 Hz, 1H), 6.64 (m, 3H), 3.58 (s, 2H). MS (ESI"): 461.1 (M-H).
3-[2-(4-Chlorophenylsulfonylamino)-4-aminophenoxylphenylacetic
acid (79) To a solution of 79b (5.5 g, 11.9 mmol, 1.0 equiv.) in 50 mL of
EtOAc was
added SnC12.2H20 (8.04 g, 35.6 mmol, 3.1 equiv.). The mixture was heated to
reflux
for 2 h. After cooling to room temperature, the mixture was poured into 50 mL
of
water. Saturated NaHCO3 was added to adjust the pH value of the mixture to 3.
The
mixture was filtered through Celite to remove solid precipitates. The filtrate
was
extracted with EtOAc. The EtOAc extract was washed with brine, dried over
Na2S04,
and evaporated in vacuo to give a light tan solid, which was purified by
silica gel
chromatography to give 79 as a white solid. 'H-NMR (400 MHz, DMSO-d6): 8 12.41
(br s, 1 H), 9.83 (s, 1 H), 7.63 5 (d, J = 8.4 Hz, 2H), 7.449 (d, J = 8.4 Hz,
1 H), 7.09 (t, J =
8.0 Hz, 1H), 6.87 (d, J= 7.6 Hz, 1H), 6.77 (s, 1H), 6.59 (d, J= 8.4 Hz, 1H),
6.55 (s,
1H), 6.44 (m, 1H), 6.34 (dd, J= 8.4, 2.4 Hz, 1H), 3.47 (s, 2H), 3.56 (s, 2H);
MS (ESI-):
431.1 (M+H).
Example 80
Scheme 29
0 0
1i0 I NH
NH KOC N, AcOH, H2O
O OH C / O H
C , O
H / D~O HN
2N
H2N O
79 80
3-[2-(4-Chlorophenylsulfonylamino)-4-ureido-phenoxyl phenylacetic
acid (80) To a solution of 79 (86 mg, 0.2 mmol, 1.0 equiv.) in 0.4 mL of AcOH,
was
added potassium cyanate (32 mg, 0.4 mmol, 2 equiv.), followed by 0.2 mL of
water.
The mixture was stirred at room temperature for 3 h, and evaporated in vacuo
to give a
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
103
yellow foam. The foam was dissolved in minimal amount of EtOAc and purified by
silica gel chromatography to give 26 mg of 80 as a white solid. 'H-NMR (400
MHz,
DMS O-d6): b 12.3 5 (br s, 1 H), 9.98 (br s, 1 H), 8.62 (s, 1 H), 7.66 (d, J =
8 Hz, 2H),
7.48 (m, 3H), 7.21 (d, J = 8 Hz, 1 H), 7.13 (t, J = 8 Hz, 1 H), 6.91 (d, J =
8.0 Hz, 1 H),
6.69 (d, J = 8 Hz, 1 H), 6.56 (s, 1 H), 6.3 8 (d, J = 8 Hz, 1 H), 5.82 (s,
2H), 3.49 (s, 2H);
MS (ESI+): 476.0 (M+H).
Example 81
Scheme 30
Me
\ S`NH
C~ /P NH
C / O ~OH
C / O OH
EnAc Me HN I / I / O
H2N I / I / O J."
PK N O
H
79 81
(S)-3-{2-(4-Chlorophenylsulfonylamino)-4-[3-(1-phenyl-
ethyl)ureido]-phenoxy}phenylacetic acid (81) To a solution of 79 (60 mg, 0.14
mmol, 1.0 equiv.) in 0.4 mL of EtOAc, was added (S)-1-phenylethyl isocyanate
(31
mg, 0.21 mmol, 1.5 equiv.). The mixture was stirred at room temperature for 14
h, and
directly loaded onto silica gel column for purification. The product, 81, was
a white
solid (21 mg). 'H-NMR (400 MHz, DMSO-d6): S 12.33 (br s, 1H), 9.97 (br s, 1H),
8.49 (s, 1H), 7.64 (d, J= 8 Hz, 2H), 7.46 (m, 3H), 7.35 (m, 4H), 7.17 (m, 1H),
7.15 (d,
J= 8.0 Hz, I H), 7.11 (t, J= 8 Hz, I H), 6.91 (d, J= 8.0 Hz, 1H), 6.68 (d, J=
8 Hz, I H),
6.54 (m, 2H), 6.36 (d, J= 8 Hz, 1H), 4.80 (m, 1H), 3.49 (s, 2H), 1.38 (d, J= 8
Hz, 3H);
MS (ESI+): 580.1 (M+H).
Example 82
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
104
Scheme 31
Q, 1J
Q p O O \ NH
NH / O OH
C I/ O OH C I
erle. I i o
EtOAc HN
H2N i 1:~ ~:: O
79 82
3-[2-(4-Chlorophenylsulfonylamino)-4-propionylamino-
phenoxy]phenylacetic acid (82). To a solution of 79 (60 mg, 0.14 mmol, 1.0
equiv.)
in 1.0 mL of EtOAc, was added propionic anhydride (27 mg, 0.21 mmol, 1.5
equiv.).
The mixture was stirred at room temperature for 6 h, and directly loaded onto
silica gel
column for purification. The product, 82, was a white solid (38 mg). 'H-NMR
(400
MHz, DMSO-d6): 8 12.35 (br s, 1H), 10.05 (br s, 1H), 9.93 (s, 1H), 7.72 (s,
1H), 7.66
(d, J = 8 Hz, 2H), 7.48 (d, J = 8 Hz, 2H), 7.41 (d, J = 8 Hz, 1 H), 7.14 (t, J
= 8 Hz, 1 H),
6.93 (d, J= 8.0 Hz, I H), 6.72 (d, J= 8 Hz, I H), 6.56 (s, I H), 6.39 (d, J= 8
Hz, 1H),
3.50 (s, 2H), 2.30 (q, J= 6.7 Hz, 2H), 1.07 (t, J= 6.7 Hz, 3H); MS (EST):
489.2
(M+H).
Examples 83 and 84
NO2 CH3
NO2 CH3
H I F HO I a ~~ I O
O H
O OH
83a
NHZ CH3 14'NH CH3
b c, d -
OH
0 OH
0 R
R = Cl, 83
R = OH, 84
Scheme 32. a. K2C03, DMSO, 60 C, overnight; b. Fe, AcOH, 60 C, 3 h; c.
3,4-Dichlorobenzenesulfonyl chloride, 2,6-lutidine, CH2C12, overnight; d.
LiOH/THF,
MeOH, and water, r.t., 2 h.
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
105
Example 83 was prepared according to Scheme 32. 'H NMR (CDC13) S
7.94 (d, J= 1.5 Hz, 1H), 7.88 (d, J= 1.5 Hz, 1H), 7.56 (m, 2H), 7.46 (m, 2H),
7.08 (s,
1H), 6.99 (d, J= 7.9 Hz, 1H), 6.66 (d, J= 8.0 Hz, 1H), 6.57 (d, J= 8.6 Hz,
1H), 6.27
(br s, 1H), 4.79 (s, 1H), 3.68 (s, 3H), 3.52 (m, 2H), 1.26 (t, J= 7.2 Hz, 3H).
MS (ESI)
583.0 [MH]+.
Example 84 was prepared according to Scheme 32. 'H NMR (CDC13) S
7.96 (d, J= 2.1 Hz, 1H), 7.89 (d, J= 2.1 Hz, I H), 7.50 (m, 4H), 7.10 (s, 1H),
7.02 (m,
1 H), 6.70 (d, J = 8.2 Hz, 1 H), 6.61 (d, J = 8.6 Hz, 1 H), 6.14 (br s, 1 H),
5.26 (s, 1 H),
3.73 (s, 3H), 3.52 (m, 2H), 1.23 (t, J= 7.2 Hz, 3H). MS (ESI+) 569.0 [MH]+.
Example 85
CI 0 O
"s'
C -NH
0 O
H
N I ~ I H
O Hg
85
Example 85 was prepared from 2-(4-hydroxyphenyl)propionic acid
methyl ester following the procedure described for 84. 'H NMR (DMSO-d6) 8
12.15
(br s, I H), 10.32 (s, I H), 8.49 (br t, J= 5.3 Hz, 1H), 7.91 (d, J= 2.0 Hz, I
H), 7.79 (d, J
= 8.5 Hz, 1H), 7.67 (br d, J= 9.5 Hz, 1H), 7.58 (d, J= 2.0 Hz, 1H), 7.45 (dd,
J= 2.0,
8.5 Hz, 1H), 7.23 (d, J= 8.6 Hz, 2H), 6.78 (d, J= 8.6 Hz, 1H), 6.59 (d, J= 8.5
Hz, 2H),
3.67 (q, J= 7.1 Hz, I H), 3.30 (m, 2H), 1.38 (d, J= 7.1 Hz, 3H), 1.12 (t, J=
7.2 Hz,
3H). MS (ESI) 537.1 [MH]+.
Example 86
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
106
9CH3 OCH3
NO2 Br
\/dB E~~ + Br OCH3 a qB OCH3 +
O 0 0 11 N02 S~"NH
b OCH3 d e HH OCH3 I~t 1--II YI2 ~OCH3 N ICH3
0 0 OH
86
Scheme 33. a. PdC12 (dppf), KOAc, DMSO, 80 C, overnight; b. PdC12 (dppf),
K2CO3,
DME, 80 C, overnight; c. Fe, AcOH, 60 C, 3 h; d. 4-Chlorobenzenesulfonyl
chloride,
2,6-lutidine, CH2C12, overnight; e. LiOH/THF, MeOH, and water, r.t., 2 h.
Example 86 was prepared according to Scheme 33. 1H NMR (DMSO-
d6) S 12.3 (br s, 1H), 8.38 (br s, 1H), 7.68 (m, 5H), 7.53 (m, 2H), 6.84 (s,
1H), 6.76 (d,
J= 7.8 Hz, 1H), 6.32 (s, 1H), 3.74 (s, 4H), 3.64 (s, 3H), 3.52 (s, 3H), 3.24
(m, 2H),
1.09 (t, J= 7.2 Hz, 3H). MS (ESI) 547.0 [MH]+.
Examples 87 and 88
NOZ OCH3 NHZ OCH3
F H O a,b O
0 83a 0
F F
S8P
0 d
F NH CH3 F ~NH CH3
B
e e,f
V
F4P
q,A F / SNH CH3
F / ~NH CH3
- H ~
0
B OH
87 88
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
107
Scheme 34. a. K2CO3, DMSO, 60 C, overnight; b. Fe, AcOH, 60 C, 3 h; c.
2,4-Difluorobenzenesulfonyl chloride, 2,6-lutidine, CH2C12, overnight; d.
Pd(PPh3)4,
tributyl(1-ethoxy vinyl)tin, toluene, reflux, 6 h; e. LiOH/THF, MeOH, and
water, r.t.,
2 h; f. 2N HCI, 5 minutes.
Example 87 was prepared according to Scheme 34. 'H NMR (CDC13) 8
7.93 (d, J = 2.1 Hz, 1 H), 7.8 7 (m, 1 H), 7.5 9 (s, 1 H), 7.5 0 (dd, J = 2.2,
8.6 Hz, 1 H), 7.16
(d, J= 1.8 Hz, 1H), 6.93 (m, 2H), 6.87 (d, J= 1.8 Hz, 1H), 6.42 (d, J= 8.6 Hz,
1H),
6.28 (br t, J= 5.6 Hz, 1H), 3.65 (s, 2H), 3.64 (s, 3H), 3.48 (m, 2H), 1.25 (t,
J= 7.2 Hz,
3H). MS (ESI+) 599.0 [MH]+.
Example 88 was prepared according to Scheme 34. 'H NMR (DMSO-
d6) 8 12.5 (br s, 1 H), 10.37 (s, 1 H), 8.42 (br t, J = 5.5 Hz, 1 H), 7.93 (d,
J = 1.6 Hz, 1 H),
7.82 (m, I H), 7.53 (br d, J= 8.0 Hz, I H), 7.44 (br t, J= 8.8 Hz, 1H), 7.23
(s, 1 H), 7.17
(m, 2H), 6.43 (d, J= 8.5 Hz, 1H), 3.66 (s, 2H), 3.48 (s, 3H), 3.24 (m, 2H),
2.31 (s, 3H),
1.11 (t, J= 7.2 Hz, 3H). MS (ESI) 563.0 [MH]+.
Example 89
NO2 OCH3 NO2 OCH3
F H OCH3 a, b CH3 -a N~ I 111~.~A Y6
0 83a 0 OH
NO2 OCH3 N02 OCH3
c, d O OCH3 e CH3
N2 O
00
S, NH CH3 yNH OCH3
g I I OCH3 b H I/ I/ OCH3
0-1 O OH
0 89
Scheme 35. a. K2CO3, DMSO, 60 C, overnight; b. LiOH/THF, MeOH, and water,
r.t.,
2 h; c. thionyl chloride, 80 C, 6 h; d. TMSCHN2, TEA, THF/CH3CN; e. silver
benzoate, MeOH, r.t., 2 h; f. Fe, AcOH, 60 C, 3 h; g. 4-Chlorobenzenesulfonyl
chloride, 2,6-lutidine, CH2C12, overnight.
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
108
Example 89 was prepared according to Scheme 35. 'H NMR (DMSO-
d6) 6 12.4 (br s, 111), 10.26 (s, 111), 8.43 (br t, J= 5.5 Hz, 1H), 7.92 (d,
J= 2.1 Hz, I H),
7.74 (d, J = 8.6 Hz, 2H), 7.60 (d, J = 8.6 Hz, 2H), 7.57 (dd, J = 2.2, 8.6 Hz,
1 H), 6.84
(d, J= 1.7 Hz, I H), 6.47 (d, J= 8.6 Hz, 1H), 5.97 (d, J= 1.7 Hz, 1H), 3.81
(s, 3H), 3.52
(s, 3H), 3.48 (s, 2H), 3.27 (m, 2H), 1.11 (t, J= 7.2 Hz, 3H). MS (ESI+) 549.0
[MH]+.
Examples 90 and 91
NOz OCH3 NHz OCH3
\ F H O , ] ( a, b \ O O I-. H + N
N I
O r
83a Br
F F O O
0 F -NH OCH3
F NH OCH3
C d O O
N N
0 90a Br 91a
le le F
F
JI~OH OCH3 F NH OCH3
F
\ \ O N
H OH
N H
90 91
r
F
Scheme 36. a. K2C03, DMSO, 60 C, overnight; b. Fe, AcOH, 60 C, 3 h; c.
2,4-Difluorobenzenesulfonyl chloride, 2,6-lutidine, CH2C12, overnight; d.
Pd(PPh3)4,
4-fluorophenylboronic acid, Na2CO3, DME, 85 C, 6 h; e. LiOH/THF, MeOH, and
water, r.t., 2 h.
Example 90 was prepared according to Scheme 36 'H NMR (DMSO-
d6) S 12.5 (br s, 1H), 10.75 (s, 1H), 8.47 (br s, 1H), 7.89 (d, 1H), 7.72 (m,
1H), 7.62 (m,
I H), 7.44 (m, I H), 7.26 (s, 111), 7.17 (m, I H), 6.66 (s, I H), 6.50 (d, J=
8.8 Hz, I H),
3.72 (s, 2H), 3.66 (s, 3H), 3.25 (m, 2H), 1.12 (t, J= 7.2 Hz, 3H). MS (ESI)
599.0
[MH]+.
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
109
Example 91 was prepared according to Scheme 36 'H NMR (DMSO-
d6) 8 12.5 (br s, 1 H), 10.36 (s, I H), 8.46 (br s, 1H), 7.89 (m, 1H), 7.71
(m, I H), 7.61 (br
d, J= 7.5 Hz, 1H), 7.41 (m, 1 H), 7.27 (m, 4H), 7.19 (s, 1 H), 6.99 (t, J= 8.9
Hz, 1H),
6.66 (s, 1H), 6.51 (dd, J= 2.0, 8.6 Hz, 1H), 3.70 (s, 2H), 3.66 (s, 3H), 3.27
(m, 2H),
1.10 (t, J= 7.2 Hz, 3H). MS (ESI) 615.0 [MH]+.
Examples 92-94
NOZ OCH3 O P,
F HO a, b, c F / NH OCH3 e 0
N / I/ H I I O
H3CO
83a H3C
FOO Fo 92a F..d
F / \ `NH OCH3 F NH OCH3 F ~~ H OCH3
: N ~~ IH &H3CC ~\ o
N &H3C o
A I
O Br H3C / H
93a 94a
F I d F i d 92
0,0 K9
Fop
F ~NH OCH3 F SNH OCH3 F `~-NH OCH3
\ o O 9 O
\i H3C OH H3C H N OH
Br 0 / O H3C
93 94b 94
Scheme 37. a. K2CO3, DMSO, 60 C, overnight; b. Fe, AcOH, 60 C, 3 h; c.
2,4-Difluorobenzenesulfonyl chloride, 2,6-lutidine, CH2CI2, overnight; d.
LiOH/THF,
MeOH, and water, r.t., 2 h; e. Br2, AcOH, r.t., overnight; f. Pd(PPh3)4,
tributyl(vinyl)tin, toluene, reflux, 6 h; g. H2, Pd/C, EtOH, r.t., 2h.
Example 92 was prepared according to Scheme 37. 'H NMR (DMSO-
d6) 8 12.4 (br s, 1H), 10.20 (s, 1H), 8.37 (br t, J= 5.5 Hz, 1H), 7.89 (d, J=
2.1 Hz, 1H),
7.81 (m, 1H), 7.48 (m, 2H), 7.18 (dt, J= 2.0, 8.6 Hz, 1H), 6.68 (s, 2H), 6.26
(d, J= 8.6
Hz, 1H), 3.58 (s, 8H), 3.25 (m, 2H), 1.11 (t, J= 7.2 Hz, 3H). MS (ESI+) 551.1
[MH]+.
Example 93 was prepared according to Scheme 37. 'H NMR (DMSO-
d6) 8 12.5 (br s, 1H), 10.41 (s, 1H), 8.41 (br t, J= 5.5 Hz, 1H), 7.89 (d, J=
2.1 Hz, 1H),
7.83 (m, 1H), 7.52 (m, 2H), 7.21 (dt, J= 1.8, 8.6 Hz, 1H), 7.05 (s, 1H), 6.34
(d, J= 8.6
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
110
Hz, 1H), 3.76 (s, 2H), 3.63 (s, 3H), 3.53 (s, 3H), 3.25 (m, 2H), 1.10 (t, J=
7.2 Hz, 3H).
MS (ESI) 629.0 [MH]+.
Compound 94b. 'H NMR (DMSO-d6) S 12.5 (br s, 1H), 10.34 (s, 1H),
8.39 (br t, J= 5.2 Hz, I H), 7.88 (d, J= 2.0 Hz, 1H), 7.84 (m, I H), 7.52 (m,
2H), 7.21
(m, I H), 6.85 (s, 1H), 6.61 (dd, J= 11.8, 17.9 Hz, 1H), 6.34 (d, J= 8.6 Hz,
1H), 1H),5.55
(dd, J= 2.0, 17.9 Hz, 1H), 5.45 (dd, J= 2.0, 11.8 Hz, 1H), 3.68 (s, 2H), 3.54
(s, 3H),
3.53 (s, 3H), 3.24 (m, 2H), 1.10 (t, J= 7.2 Hz, 3H). MS (ESI) 577.0 [MH]+.
Example 94 was prepared according to Scheme 37. 'H NMR (DMSO-
d6) 8 12.4 (br s, 111), 10.32 (s, 111), 8.38 (br s, 1H), 7.88 (d, J= 2.0 Hz, I
H), 7.84 (m,
2H), 7.50 (m, 2H), 7.21 (m, 1H), 6.76 (s, 1H), 6.31 (d, J= 8.6 Hz, 1H), 3.61
(s, 2H),
3.60 (s, 3H), 3.50 (s, 3H), 3.37 (m, 2H), 3.25 (m, 2H), 1.10 (t, J= 7.2 Hz,
3H), 1.04 (t,
J= 7.2 Hz, 3H). MS (ESI) 579.2 [MH]+.
Example 95
Human CRTH2 binding assay
Full-length human CRTH2 cDNA was generated by polymerase chain
reaction (PCR) using human genomic DNA as template and subsequently cloned
into
pCDNA3.1(+) (Invitrogen), generating a CRTH2 expression plasmid pHLT124. The
plasmid was transfected into 293 cells, which normally express CRTH2, using
LipofectAMINETM reagents (Gibco/BRL). G418 (800mg/mL) was added to the
culture 48 h after transfection and cells were maintained under selection for
3 weeks to
ensure that all surviving cells stably expressed CRTH2. These cells are
labeled as
293(124) hereafter.
3H-PGD2 binding assay was performed using 293(124) cells. In brief,
cells were washed and suspended in RPMI containing 0.5% BSA and 20mM HEPES.
Each assay contained 25,000 cells, appropriate amount of test compound when
necessary and a mixture of 1nM 3H-PGD2 (Amersham Pharmacia Biotech) and 30nM
of unlabeled PGD2 (Cayman Chemicals) in 200 mL final volume. The cell mixture
was
incubated at room temperature for 2.5 h with shaking and the cells were
separated from
free 3H-PGD2 and transferred onto a filter plate using a cell harvester.
Radioactivity
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
111
bound to the cells was measured on a liquid scintillation counter. Nonspecific
binding
was determined in the presence of 10mM of unlabeled PGD2.
Modulation of CRTH2 and/or one or more other PGD2 receptors by test
compounds can be assessed by other in vitro and in vivo assays. Examples of
such
assays include measuring second messenger (e.g., cAMP, IP3 or Ca2) levels, ion
flux,
phosphorylation levels, transcription levels, and the like. Recombinant or
naturally
occurring CRTH2 polypeptides and/or other PGD2 receptor peptides can be used
and
the protein can be isolated, expressed in a cell, expressed in a membrane
derived from a
cell, expressed in tissue or in an animal. Signal transduction can also be
examined in
vitro with soluble or solid state reactions, using a chimeric molecule such as
an
extracellular domain of a receptor covalently linked to a heterologous signal
transduction domain, or a heterologous extracellular domain covalently linked
to the
transmembrane and/or cytoplasmic domain of a receptor. Gene amplification can
also
be examined. Furthermore, ligand-binding domains of the protein of interest
can be
used in vitro in soluble or solid state reactions to assay for ligand binding.
CRTH2-G-protein or another PGD2 receptor-G-protein interactions can
also be examined, by, for example, analysis of binding of the G-protein to the
receptor
or its release from the receptor.
Exemplary compounds of the invention displayed IC50 values as shown
in Table I in the above-described ligand binding assay.
CA 02511214 2005-06-20
WO 2004/058164 PCT/US2003/040617
112
TABLE I
Example of CRTH2 activity.
Example # CRTH2 Binding IC50
1 ++
2 ++
3 ++
4 +
++
+++
12 +++
17 ++
18 +++
31 +
33 ++
35 +++
36 ++
63 ++
64 +++
67 +++
70 +++
73 +++
75 +++
77 ++
+ IC50>15 M
5 ++ 15 M>IC50>1.iM
+++ IC50<l M
Example 96
Cyclic AMP assays on human DP function
Cyclic AMP assays on human DP function are performed using human
platelets (AllCells, Berkeley, CA) and the 96-well Tropix cAMP ELISA System
(Applied Biosystems) following the manufacturer's manual. Briefly, the human
platelets rich plasma (PRP) is diluted 1:3 with human plasma and incubated
with ImM
of the phosphodiesterases inhibitor 3-isobutyl-l-methylxanthine (IBMX, Sigma)
at
37 C for 20 min, to prevent hydrolysis of cAMP. 20 Al of the above PRP sample
is
mixed 1:1:1 with the test compound and PGD2 (both prepared in the assay buffer
with
CA 02511214 2011-11-04
113
DMSO concentration <1%) in a 96-well plate. The assay buffer can be OPTI-free
medium (Gibco BRL). After 20 min incubation at 37 C, 20 Al of lysis buffer
from the
kit is added to each well of the mixture and the plate then incubated at room
temperature for 10 min with moderate shaking and at 37 C for 10 min. After
the cell
lysis, 60 p1 of the cell lysate together with 30 ] of diluted cAMP-AP
conjugate and
60 Al anti-cAMP antibody is then transferred into a kit assay plate and the
plate
incubated at room temperature for 30 min with shaking. The plate is then
washed with
wash buffer and incubated with 100 Al per well of substrate/enhancer solution
at room
temperature for 60 min. Light signal intensity, which is inversely
proportional to the
cAMP level in each sample, is measured in a luminometer (CLIPR, Dynamic
Devices).
The final human plasma concentration in the assay described above is about
33%. The
assays are also performed using washed platelets (prepared by centrifuging the
PRP at
2000 rpm for 15 min and resuspending the platelets in the assay buffer), or in
the
presence of higher than about 33% of human plasma by also preparing the test
compound and/or PGD2 solution in human plasma.
Although
the foregoing invention has been described in some detail by way of
illustration and
example for purposes of clarity of understanding, it will be readily apparent
to those of
ordinary skill in the art in light of the teachings of this invention that
certain changes
and modifications may be made thereto.