Note: Descriptions are shown in the official language in which they were submitted.
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
ANTI-CANCER MEDICAMENTS
BACKGROUND OF THE INVENTION
Related Applications
This application claims the benefit of provisional applications entitled
Process For
MODULATING PROTEIN FUNCTION, S/N 60/437,487 filed December 31, 2002, ANTI-
CANCER MEDICAMENTS, S/N 60/437,403 filed December 31, 2002, ANTI-
INFLAMMATORY MEDICAMENTS, S/N 60/437,415 filed December 31, 2002, ANTI-
INFLAMMATORY MEDICAMENTS, S/N 60/437,304 filed December 31, 2002, and
MEDICAMENTS FOR THE TREATMENT OF NEURODEGENERATIVE DISORDERS OR
DIABETES, S/N 60/463,804 filed April 18, 2003. Each of these applications is
incorporated by
reference herein.
Field of the W vention
The present invention relates to novel compounds and methods of using those
compounds
to treat oncological conditions.
Description of the Prior Art
Basic research has recently provided the life sciences community with an
unprecedented
volume of information on the human genetic code and the proteins that are
produced by it. In
2001, the complete sequence of the human genome was reported (Larder, E.S. et
al. Initial
sequencing and analysis of the human genome. Nature (2001) 409:860; Venter,
T.C. et al. The
sequence of the human genome. Science (2001) 291:1304). Increasingly, the
global research
community is now classifying the 50,000+ proteins that are encoded by this
genetic sequence,
and more importantly, it is attempting to identify those proteins that are
causative of major,
under-treated human diseases.
Despite the wealth of information that the human genome and its proteins are
providing,
particularly in the area of conformational control of protein function, the
methodology and
strategy by which the pharmaceutical industry sets about to develop srriall
molecule therapeutics
has not significantly advanced beyond using native protein active sites for
binding to small
molecule therapeutic agents. These native active sites are normally used by
proteins to perform
essential cellular functions by binding to and processing natural substrates
or tranducing signals
from natural ligands. Because these native pockets are used broadly by many
other proteins
1
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
within protein families, drugs which interact with them are often plagued by
lack of selectivity
and, as a consequence, insufficient therapeutic windows to achieve maximum
efficacy. Side
effects and toxicities are revealed in such small molecules, either during
preclinical discovery,
clinical trials, or later in the marketplace. Side effects and toxicities
continue to be a major
reason for the high attrition rate seen within the drug development process.
For the lcinase
protein family of proteins, interactions at these native active sites have
been recently reviewed:
see J. Dumas, Protein Kinase Inhibitors: Emerging Pharmacophores 1997-2001,
Expert Opinion
on Therapeutic Patents (2001) 11: 405-429; J. Dumas, Editor, New challenges in
Protein Kinase
W hibition, in Curre~zt Topics in Medicinal Clzemistr~ (2002) 2: issue 9.
It is known that proteins are flexible, and this flexibility has been reported
and utilized
with the discovery of the small molecules which bind to alternative, flexible
active sites with
proteins. For review of this topic, see Teague, Nature ReviewslDrugDiscovery,
Vol. 2, pp. 527-
541 (2003). See also, Wu et al., Structure, Vol. 11, pp. 399-410 (2003).
However these reports
focus on small molecules which bind only to proteins at the protein natural
active sites. Peng et
al., Bio. Organic andMedicinal ChenaistfyLtrs., Vol. 13, pp. 3693-3699 (2003),
and Schindler,
et al., Science, Vol. 289, p. 1938 (2000) describe inhibitors of abl lcinase.
These inhibitors are
identified in WO Publication No. 2002/034727. This class of inhibitors binds
to the ATP active
site while also binding in a mode that induces movement of the kinase
catalytic loop. Pargellis
et al., Nature Structural Biology, Vol. 9, p. 268 (2002) reported inhibitors
p38 alpha-kinase also
disclosed in WO Publication No. 00/43384 and Regan et al., J. Medicinal
Chenaistfy, Vol. 45,
pp. 2994-3008 (2002). This class of inhibitors also interacts with the kinase
at the ATP active
site involving a concomitant movement of the kinase activation loop.
More recently, it has been disclosed that kinases utilize activation loops and
lcinase
domain regulatory pockets to control their state of catalytic activity. This
has been recently
reviewed (see, e.g., M. Huse and J. Kuriyan, Cell (2002) 109:275).
SUMMARY OF THE INVENTION
The present invention is broadly concerned with new compounds for use in
treating anti-
inflammatory conditions and methods of treating such conditions. In more
detail, the inventive
compounds have the formula
2
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
~Ri--~X~A~N)-D--~L~--E--~Y~Q
wherein:
R' is selected from the group consisting of aryls (preferably C~-C,$, and more
preferably
C~ C12) and heteroaryls;
each X and Y is individually selected from the group consisting.of -O-, -S-, -
NR~-,
-NR~SOz-, -NR~CO-, alkynyls (preferably C1-C12, and more preferably C;-C~),
alkenyls (preferablyCi-C~z, andmorepreferablyCl-C~), alkylenes (preferablyCl-
CIZ, and more preferably C~-C6), -O(CHZ)h-,, and -NR~(CHZ)h , where each h is
individually selected from the group consisting of l, 2, 3, or 4, and where
for each
of alkylenes (preferably C1-C,2, and more preferably C,-C6), -O(CHZ),,-, and
-NR~(CHz)h , one of the methylene groups present therein may be optionally
double-bonded to a side-chain oxo group except that with -O(CHZ),,-, the
introduction of the side-chain oxo group does not form an ester moiety;
A is selected from the group consisting of aromatic (preferably C~-CIB, and
more
preferably CG-C1z), monocycloheterocyclic, and bicycloheterocyclic rings;
D is phenyl or a five- or six-membered heterocyclic ring selected from the
group
consisting of pyrazolyl, pyrrblyl, imidazolyl, oxazolyl, thiazolyl, furyl,
pyridyl,
and pyrimidyl;
E is selected from the group consisting of phenyl, pyridinyl, and pyrimidinyl;
L is selected from the group consisting of -C(O)-, -S(O)2-, -N(RG)CO-, -
N(R~)SOz-,
-N(RG)CON(R6)-;
jis0orl;
mis0orl;
nis0orl;
pis0orl;
qis0orl;
tis0orl;
3
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Q is selected from the group consisting of
R Ra O Ra
Ra Ra Ra S N a O N O OQILiN
O N O , S N O O N O ~ O ~ /SO O I \ O
N /NwN / N % N
/N~S~ /N~S~ / N ~ \ , ~ R ~ \ >
_ a Ra
a
Q-1 ' ~ Q-2 ' Q-3Ra Q-4R Q 5 p Q GO
0 0 0 0~ ~o o ~
Ra Ra Ra ~
O N Ra Ra Ra /S\ /Ra ~~ / \N/S\N/R
O ~NRa NRa ~Z~H N Z H I
\ /N S~ /N > I , Ra >
R ~ o//\o Q-10 ~ Q-i l
1 ~ Q_7
Q-8 Q-9
0 0 0
O O O Ra Ra ~ Ra~N ORs
Ra Ray \N~NH \N- -NH
N N N ~ ~ /
~ O
OR ~~l~O ~ Rs > ~ O
ORs ~ ~ ~OR6 Rs 1 fi RS
Rs .new ' .rvw
15 Q 12 Q_13 ' Q-14 Q 15 Q 16 Q 17
OH SH
O ww
O'
Ra\N O Na O ~ O COzH HO~p NI W W W \ W
Rs COa CN3 O
O CH N H3C CH3 J ,
, >
OR6 ~ , H3C H3C "l,L,
Q-18 H3 Q 19 Q-20 Q-21 Q-22 Q-23
2O o H H H ~zR,
O O\ %O R O O\ % Ra S=O O N~S/RB O N~N~S O
\ /
~N~S. ~ a N~S~Z N ~~~~ O O
O H H~ H ~ ~O O
W/\\W W ~W W ~W
~J ~ ~J ~ ~ ~ i ~ ~ ~~~
i i J
Q-24 ~_2g ~ ~ Q-27 Q-28 Q-29
Q Q-26
S03R6
O
N J
R9
PI O N~ W
SOZN(Ra)z R9 I\OR6 Ra ~ (G k n Ra
\ OR6 ~ ~ ~ O
/ ~ ~ ~ J , ~ J ~ ~ ~ and ~ ~ ;
Q-30 Q 31 Q-32 Q-34 Q-35
Q-33
4
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
each R4 group is individually selected from the group consisting of -H, alkyls
(preferably
C,-C,z, and more preferably C~-C~), aminoalkyls (preferably C1-C~z, and more
preferably C~-C~), alkoxyalkyls (preferably C1-Clz, and more preferably C1-
C~),
aryls (preferably C6 C18, andmorepreferablyC~ Clz), aralkyls (preferablyCl-
C~z,
and more pr eferably C,-C6), heterocyclyls, and heterocyclylallcyls except
when the
R~ substituent places a heteroatom on an alpha-carbon directly attached to a
ring
nitrogen on Q;
when two R4 groups are bonded with the same atom, the two R4 groups optionally
form
an alicyclic or heterocyclic 4-7 membered ring;
each RS is individually selected from the group consisting of -H, alkyls
(preferably C~-
C,z, and more preferably C1-C~), aryls (preferably C~ C18, and more preferably
C~
C,z), heterocyclyls, alkylaminos (preferably C~-CIZ, and more preferably C~-
C~),
arylaminos (preferably C6-C18, and more preferably CG-CIZ), cycloalkylaminos
(preferably C3-C,g, and more preferably CS-C,z and preferably C,-C,z, and more
preferablyCi-C~), heterocyclylaminos, hydroxys, alkoxys (preferably C,-C,z,
and
more preferably CI-C~), aryloxys (preferably CG C,B, and more preferably CG
C,z),
alkylthios (preferably C1-C,z, and more preferably C,-CG), arylthios
(preferably
C~ C,$, and more preferably C~-C,z), cyanos, halogens, perfluoroallcyis
(preferably
C~-C,z, and more preferably CI-C~), allcylcarbonyls (preferably C1-CIZ, and
more
preferably C~-C~), and nitros;
each R~ is individually selected from the group consisting of -H, alkyls
(preferably C1-
Clz, and more preferably C~-C6), allyls, and (3-trimethylsilylethyl;
each R8 is individually selected from the group consisting of alkyls
(preferably C~-Ctz,
and more preferably C1-C~), aralkyls (preferably C~-C~z, and more preferably
C~-
C~), heterocyclyls, and heterocyclylalkyls;
each R~ group is individually selected from the group consisting of -H, -F,
and alkyls
(preferably C1-Clz, and more preferably C1-C~), wherein when two R9 groups are
geminal alkyl groups, said geminal alkyl groups may be cyclized to form a 3-6
membered ring;
G is selected from the group consisting of -O-, -S-, and -N(R~)-;
lcis0orl;
5
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
each Z is individually selected from the group consisting of -O- and -N(R4)-;
and
each ring of formula (I) optionally includes one or more of R~, where R7 is a
noninterfering substituent individually selected from the group consisting of -
H,
alkyls (preferably C1-C~z, and more preferably C1-C~), aryls (preferably C~-
C~B,
and more preferably C~-C~2), heterocyclyls, alkylaminos (preferably C1-C,2,
and
more preferably C,-C6), arylaminos (preferably C~ C,B, and more preferably C6-
C,2), cycloalkylaminos (preferably C3-C,B, and more preferably CS-C,2 and
preferably CI-C,2, and more preferably C~-C6), heterocyclylaminos, hydroxys,
alkoxys (preferably C1-C,2, and more preferably C1-C~), aryloxys (preferably
C~-
lp C,B, and more preferably C6-C12), alkylthios (preferably C1-CI2, and more
preferably C~-C6), arthylthios, cyanos, halogens, nitrilos, nitros,
alkylsulfinyls
(preferably C,-C 1z, and more preferably C I-C6), allcylsulfonyls (preferably
C 1-C,2,
and more preferably C1-C~), aminosulfonyls, and perfluoroalkyls (preferably C1-
C1,, and more preferably C,-C~).
15 W a preferred embodiment, the stmcture is of formula (I) except that:
when Q is Q-3 or Q-4, then the compound of fornmla (I) is not
0
Ph / ~ N ~ N~Ph
NvN ~ ~ or ~ \N~NH
PhB ~ NHZ >
Ph
when Q is Q-7, then the compound of formula (I) is not
6
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
O
NH
8120 = 2.3-difluoro; 2,3,6-trifluoro; 2, fluoro, 3-chloro; 2-chloro,3-fluoro;
3-cyano~ 4-chloro
\ ~ A' = substituted phenyl
R12~ ~ / Y' = CO; -NHCO-~ -S02-~ -S02NH-; f=0 or 1
NH 8121 = substituted phenyl; oxazolyl; pyridyl; pyrimidyl; pyrazolyl;
imidazolyl
8121
or
O
N H 8123 = H; 2.3-difluoro; 3,5-difluoro; 2-fluoro, 4-fluoro; 2-chloro, 2,4-
dichloro; 3,4-dichlora; 3-fluoro;
4-chloro, 2-bromo; 3-bromo; 4-bromo; 4-iodo; 2-methoxy; 3-methoxy; 4-methoxy;
3,4-dimethoxy;
2 4-dbnethoxy; 2,5-dimethoxy; 3 4,5-trunethoxy; 3-CF3; 4-CF3; 3,5-di-CF3;
4-CF30-; 3-vitro; 4-vitro; 3-vitro-4-chloro; 2-methyl;
_I \ 3-methyl; 4-methyl; 3,5-dhnethyl; 4-iso-propyl; 3-methylthio; 3-CF3S-; 3-
chloro-4-methoxy
8123 / NH 4-methylthio; 4-hydroxy; 4-methoxymethyl; 4-methylsulfonyl
A' = substituted phenyl
' Y" = CO; ~0 or 1
~i R122= substituted phenyl; oxazolyl; pyrimidyl
Rl2z
15 when Q is Q-7, RS is -OH, Y is -O-, -S-, or -CO-, m is 0, n is 0, p is 0, q
is 0, and E is
phenyl, then D is not thienyl, thiazolyl, or phenyl;
when Q is Q-7, then the compound of formula (I) is not
O Me p Me O NH ~ NH
N~ N~ H~ O
O /~O ReowN ~ O ~ N W
H H Rat Re2 /
S~ \ I ~ I
HN I / O I /
O
i
O I \ , HN I \ , W I I i
/ / , or
R80 is H, Me Rg2 is substituted phenyl
2 $ R81 is substituted phenyl
when Q is Q-9, then the compound of formula (I) is not
7
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
0
Me
Phi ~O Me
O ~N-Me
N
O ~ / O
N \
CH v Ph ~ or
HzN ( z)n ~ / ,
NC R16=H, methyl
O
HO N~N~R~s
~N\
I' N ~ ~ Rye R'~O
~N
Rn ,
R17, R18 = alkyl
R19 = H, alkyl
when Q is Q-10, then the compound of formula (~ is not
O O, O RI00=med~yl,ethyl
8101 = alkyl, amutoalkyl, aryl, arylalkyl,
P'~ooX,~N.S~NH thienylalkyl, pyridinylalkyl, N-
H pR~oo phthalimidylalkyl, alkoxycarbonylalkyl,
\ alkoxycarbonylaminoalkyl,
N ~ / arylalkenylalkyl,alkoxyalky,bydroxyalkyl,
R~o~ arylaminocarbonyl, arylalkoxycarbonylaminoalkyl
O v 8102 =phenyl, uidolylphenyl
v=Oorl
X'=O, NH
or
OMe
N~N O O O1 8103 = furyl, thienyl, phenyl
MeO~N~N~N'S' ZO;a 7C"=CorS
H H ~X"~R~o3 a = 1 or 2
wherein there is a bond between Q and
~Rl---~X~A~N~-D--~L t~--E--~Y~
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
of formula (I), and when Q is Q-11, t is 0, and E is phenyl, then any R7 on E
15 llOt
an o-allcoxy in relation to said bond;
when Q is Q-1 l, then the COlllpotllld of formula (I) is not
i - P Ph
0 O
NH-II-NH-II-O I \
I I
Pr-1 Ph
or
O p O
o ., Jj
R1o5~N~S~N~NH 8104=methyl, ethyl
H H Rl OS = alkyl, phenyl
~ ~ ORto4 8106=fluorine-substituted phenyl
H
Rlos N ~
O
when Q is Q-15, then the compound of formula (I) is not
0 0
HN ~ i HN
O~ ~ ~ ' Rto~ or O~ ~ ~ I \
H H H CH v I /
8107 = phenyl
9
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
when Q is Q-16, then the compound of formula (I) is not
H3C
O
~O
Ne ~ \NH
H
R =Me, OH N~O
ii2 H
O
O H O N II-Ph
N 0
~N ~ NH
N
NH HN
O
O
O
O N HN-II-Ph
HN S
O
Me
O
O N
NH-II /
N
S O HN
N
O \ Ph
HN ~0
~NH
///O
i0
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
H Ph
O~N I I w
HN HN
0
Ph
NH O
rv '0 ' NH
H ,
N- 'O
H
O
~S 0
NS I ANN
H
or
N"O
H
F
R~o$=OH, SH, NI-I2
R~o9 = hydrogen or one or more methoxy, hydroxy,
halogen, vitro, dimethylamino, or furanyl
Rtyo = substituted phenyl, furanyl
Rtii=OH or CI
X3=O, NH
when Q is Q-17, then tile compound of formula (1~ is not
n-Bu
N
N ~
R ~ O O~ ~H~Rso
2s p
Rzs
R.,~ = alkyl '
R3o = H, t-Bu, benzoyl
11
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
when Q is Q-21, then the compound of formula (~ is not
HOZC
~O
~~'~N
IoI
Ph
when Q is Q-22, then the compound of formula (~ is selected from the group
consisting
of
Ra Ra
NH-L~-(NH)P D-(NH)p-(A)q [(X)j-R~]m NH-L~-(NH)p D-(NH)p (A)G-[(X)j R~]m
I /
/
O NH O
NH
.W
W .W
I W ~OH '
L~ - C(O) or S(OZ)
R4 Ra
j CO-(NH)p-D-(NH)P (A)q [(X)~ R~)]m I ~ D-(NH)p (A)p-[(X)~ R~)]m
O NH O NH
W W ~ \ '
W~W
OI '
OH
R4
CO-(NH)p-D-(NH)p-(A)q-[(X)j R1)]m R4
~ D-(NH)P (A)G-[(X)j R1)]m
O NH /
O NH
I~W
W ~OH ~ and
12 ~ 'W ,
W~OH
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
but excluding
H O Ray
O N I ~ Rss HN \ Rs~
O / . ~ ~ O / Ra ~ / R38
1137 = N(Me)2,
\ N \ R35 \ N \ R3s mo~pholino OMe OH, H
H R34 = Me CI ~ I H R38 = H, CN, OMe, OH,
HO / R3a R35 =-N(Me)2, morpholino /~ Me benzyloxy, phenyl, vitro
meta or para- R3G = H, F HO meta or para- R39 = H, OH
R40 = H, F
R41=H, CI
O O ~O O
\ OMe
HN I / HN I N NJ HN I \ Nw
/~O a i / /
Me M /
O / ( 0
\ N \ \ N \ ~N \
1~ Il / H ~ ~ / H Me ' ~~/ H Me , and
HO
HO meta or para- OH meta or para-
I~H
HO ~neta or para-
Me0 / N~NHMe
Me0 \ ~ i_IN
0 /
\ N \
13
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
when Q is Q-23, then the compound of fornmla (I) is not
R42 / (CH~)9Me
I I
\ N \ ~ ~ I \ N~ \
,I
HS I / I i N \ I ~ \ ~ I \ N
H HS I / R42 - H~ Me' ' HS
,N
O~~O
\ o ~ NJ
I ~ \ N ~
CI
HS I ~ ~ HS I ~ H
CI t-Bu
w N \ I , CI I /
HS I ~ O N v I c-Bu ~ ~ N ~ / H-H ~ I SH
or
O v0
O
HS I ~ I ~ S
when Q is Q-24, Q-25, Q-26, or Q-31, then
ZS ~R~--~X~ mA~N~-D--~L~--E--~Y~
is selected from the group consisting of
14
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
R~
R~ R~
\ W ~ W~W O W~W O O
i A-~X-R1)m ~ \
H H ~ ~H A-(X-R~)m ~H~S~A-tX-R1)m
H W N, H ' H N W N
O~N~~ A-iX-R~)m ' O ~ ,t'1~W N~A-(X-R~)m O~ ~~ A-tX-R1)m
W / R7 ~ IWI~R7 , and HN W / R~
I
wherein each W is individually selected from the group consisting of -CH- and
-N-; and
O~N \S~ ~R4 ~ OSO -R4 HN~N \S N-R S 'Ra p=S'Z'R4 O,S-Z~Ra
\ H R p H ' \ H RQ HN H N \ p'
Ra \
/ > > *~
*~
J ' * ~-~ ~
Q-24 Q-25 Q-26 or Q-31
where ~ denotes the point of attachment to Q-24, Q-25, Q-26, or Q-31;
when Q is Q-31, then the compound of formula (I~ is not
O o0 Oy0
S,N
CI \ CI HN I / H
/ N.N \ I
~N
CI O H CI
or
O
O S O
~ N N
H I H H
NHS / N /
d o~ ~p
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
when Q is Q-2~, then the compound of formula (I) is not
h,e H .
w w ° R~N ~ \
II_ I ~ I ~ NH-II ~
R2~N ~ N~S~R~
° °
c cri y s-NMe 2 H
16
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
when Q is Q-32, then
(R i--~X~A~--~N~--D--~L~R-E~'
~ m 9 P n
is not biphenyl, benzoxazolylphenyl, pyridylphenyl or bipyridyl;
17
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
when Q is Q-32, then the compound of formula (n is not
0
EIO- ~ -CHZ
\ ~ N NI \
OEt
/ / / ,
O Rtao
RtaaO O Rt33
H R73a0~
R O N ~ O~~ SiN \
/ Rtaa H H
H H
Rtat ' ~ '
8733
Ri3o= benzoyl, substituted phenylaminocarbonyl
RI31 =CI, Br, SPh, benzoyl, phenylsulfonyl
Ri32= subsituted phenylaminocarbonyl Me
Riaa = I-I, CI OEt Ph
R~3,y= H, alkyl, allyl, B-trimethylsilyletltyl Et0-P-C
~~ / / ~ \
CH=CH
\ CH=CH \ \ \ /
O EI
Ph~N / / H- II-OEt
2
OEt
F
CH-POaHz
\ CHz O \
/ / , or
Ph
18
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
when Q is Q-35 as shown
0 0
ZR4 ~ZRQ
~ tt ~~ k ~ a
~ \ \
Q-35 (pccra) Q-35 (nzetct)
wherein G is selected from the grOllp consisting of -O-, -S-, and -NR4-, lc is
0 or
1, and a is l, 2, 3, or 4, then
~R1-EX~A~--~N~-D-~L~-E--EY~
m 9 P n t
is selected from the group consisting of
H
R~ N W~ N~AUX)J-Rtlm
Wow ~ o~ ~~R
~N~N'A OX)J-Rtlm * ~
* H H ~ H
R7 0 ° N W\ N A_t(X)i-R,lm
W ~ * W~ 7
W O R
~N~A-ItX)l-Rtlm ~ H
H o~ N ~W~ N ~A-t(X>i-R, l m
R~ HN W /~R7
W~W O
~S~
* H A-tlX)i-R,lm
19
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
R7
R~ R~
W ~ W W~W W~W
I ~ I
~A-~tX)J-R~lm
* ~ ~A-UX)l-R~lm ~A-UX)l-Rilm
* , and
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
except that the compound of formula (I) is not
COZR~i Me
\ COzH \ I Rya ph I ~ (CHzjnC02R7s
Me I \ I ~ \
_ v
~Wa ~ / / , I / , R , 1 ~ meta.Para '
~~N R7z 28.1 R73=-OCH2C02H R75=H, Et
~W4 H R71 = H, Me R72 = ihiazolyl, isoxazolyl R74 = oxazolyl, imidazolyl R~s.
R7G = H, NH2, N02
W4=N. CH imidazolyl,furyl 28.2 R73=C02Me a=0-I
R74= chloroPheuyl
CF3
O-N CI
i
\ / X~COzRn ~ \COzR~e
v meta, Para Me ~ ph NH
O R77 = H, alkyl O ~ COZH
HN X3=OorCH= HN / ~ O O'' NH
R78 = H, alkyl
\ H , /
~ ~ I ~ COzMe \ I NH
HN~ HNJ Nd O
Me
Rss O / I O
N \ N \
Rss ( / H H I \ ,
/ COZMe
COzRss Me0 /
FaC I \ \ ~ O
Me0 ~ ~ ~ / COZRss
\ I ~ / , ~ ~' ,
\ v
I mela, Para
/ OMe O RGS = H, Et RGG = alkyl
21
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Me
O / ~ O
Me0
H
-O~COxR~a~
Me0 \v/
meta, para
Riao - H, t-Bu
CI
0 ~ ~ O
Me0
Me0 ~ ' COxMe
CH3
O N~ I
OMe \ N \
H
HZNYN / OMe H3C N N
N~ ~ \ I \ N O ~ / O
NHz
/ ~ CO~H
\ I H
R79=H, Me ' Or \ ~ . N /
COZR~9
O
22
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
In a preferred embodiment, RI is selected from the group consisting of 6-5
fused
heteroaryls, 6-5 fused heterocyclyls, 5-6 fused heteroaryls, and 5-6 fused
heterocyclyls. In a
particularly prefelTed embodiment, Ri is selected from the gTOllp CO1151Stlllg
of
W~W ~W N I \
$ W W ff I
I . N N . N ~ .) , ~~~
N RZ ~ ~ , Y ~ O n p N O ' R3
R2 R2 H
~nnn ~ R5 ~ ~ R5 \
W W W
O~ ~O , ~O , NYS , NYS , Ra N~N_Ra , /
R2 RZ p
W ~ ~ R~ \ I / N ~ ~ ~ ~R5 i / N R2
R N / W R N N ~ rN\ N
R ~ ~ , and Ra
each RZ is individually selected from the group consisting of -H, alkyls
(preferably C~-
C,2, and more preferably C,-CG), aminos, allcylaminos (preferably C,-C,z, and
more preferably C,-CG), arylaminos (preferably C~-C,B, and more preferably C~
C~z), cycloallcylaminos (preferably C3-CAB, and more preferably CS-C,2 and
preferably C,-C,z, and more preferably C1-C~), heterocyclylaminos, halogens,
allcoxys (preferably C1-C~Z, and more preferably C,-C~), and hydroxys; and
each R3 is individually selected from the group consisting of -H, alkyls
(preferably C~-
C,Z, and more preferably C,-C~), allcylaminos (preferably C~-C,2, and more
preferably C,-C~), arylamiyos (preferably C~-C,B, and more preferably CG-C,2),
cycloallcylaminos (preferably C~-C~,, and more preferably C,-C~),
heterocyclylaminos, alleoxys (preferably C,-C,2, and more preferably C1-C~),
hydroxys, cyanos, halogens, perfluoroallcyls (preferably C,-C,?, and more
preferably C,-C~), allcylsulfmyls (preferably C,-C,~, and more preferably C,-
C~),
allcylsulfonyls (preferably C,-C,Z, and more preferably C,-C~), R~,NHSO,-, and
-NHS OzRa.
In another embodiment, A is selected from the group consisting of phenyl,
naphthyl,
pyridyl, pyrimidyl, thienyl, fiuyl, pyrrolyl, thiazolyl, oxazolyl, imidazolyl,
indolyl, indazolyl,
23
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
benzimidazolyl, benzotriazolyl, isoquinolyl, quinolyl, benzothiazolyl,
benzofurallyl,
benzothienyl, pyrazolylpyrimidinyl, imidazopyrimidinyl, and purinyl.
With respect to the methods of the invention, the activation state of a kinase
is determined
by the interaction of switch control ligands and complemental switch control
pockets. One
conformation of the lcinase may result fr0111 the switch control ligand's
interaction with a
particular switch control pocket Whlle allOther CO11f01111at1011 play result
fr0111 the ligand's
interaction with a different switch control pocket. Generally interaction of
the ligand with one
pocket, such as the "on" poclcet, results in the Icinase assuming an active
conformation wherein
the kinase is biologically active. Similarly, an inactive conformation
(wherein the lcinase is not
biologically active) is assumed when the ligand interacts with another of the
switch control
pockets, such as the "off' poclcet. The switch control pocket can be selected
from the group
COIISiStlllg Of Sllllple, C0111pOSlte alld C0111b111ed switch control pockets.
Interaction between the
switch control ligand and the switch control pockets is dynamic and therefore,
the ligand is not
always interacting with a switch control poclcet. In some instances, the
ligand is not in a switch
control pocket (such as occurs when the protein is changing from an active
conformation to an
inactive conformation). hl other instances, such as when the ligand is
interacting with the
environment surrounding the protein in order to determine with Which switch
control pocket to
interact, the ligand is not 111 a 5WltCh COlltr0l pOClCet. Interaction of the
ligand with particular
switch control poclcets is controlled in part by the charge status of the
amino acid residues of the
switch control ligand. When the ligand is in a neutral charge state, it
interacts with one of the
switch control poclcets and when it is in a charged state, it interacts with
the other of the switch
control pockets.' For example, the switch control ligand may have a plurality
of OH groups and
be in a neutral charge state. This neutral charge state results in a ligand
that is more lilcely to
interact Wlth Olle Of tile SWItCh COlltr0l pOClCetS through hydr ogee boding
between the OH groups
and selected residues of the pocket, thereby resulting in whichever protein
conformation r esults
from that interaction. However, if the OH groups of the switch control ligand
become charged
through phosphorylation or some other means, the propensity of the ligand to
interact with the
other of the switch control pockets will increase and the ligand will interact
with this other switch
control pocket through complementary covalent binding between the negatively
or positively
charged residues of the pocket and ligand. This will result in the protein
assuming the opposite
COllfOr11at1011 aSSUllled Whell the ligand was in a neutral charge state and
interacting with the
24
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
other switch control poclcet.
Of course, the conformation of the protein determines the activation. state of
the protein
and can therefore play a.role in protein-related diseases, processes, and
conditions. For example,
if a metabolic process requires a biologically active protein but the
protein's switch control ligand
remains in the switch control pocket (i.e. the "off' pocket) that results in a
biologically inactive
protein, that metabolic process cannot occur at a normal rate. Similarly, if a
disease is
exacerbated by a biologically active protein and the protein's switch control
ligand remains in
the switch control pocket (i.e. the "on" pocket) that results in the
biologically active protein
conformation, the disease condition will be worsened. Accordingly, as
demonstrated by the
present 111Ve11t1011, selective modulation of the switch control pocket and
switch control ligand
by the selective administration of a molecule will play an important role in
the treatment and
control of protein-related diseases, processes, and conditions.
One aspect of the invention provides a method Of lllOdlllat111g the activation
state of a
lcinase, preferably abl or bcr-abl alpha-lcinase and including both the
consensus wild type
sequence and disease polylnolphs thereof. The activation state is generally
selected from an
upregulated or dowllregulated state. The method generally comprises the step
of contacting the
lcinase with a molecule having the general fornula (n. When such contact
occurs, the molecule
will bind to a particular switch control pocket and the switch control ligand
will have a greater
propensity to interact with the other of the switch control pockets (i.e., the
unoccupied one) and
a lesser propensity to interact with tile occupied switch control poclcet. As
a result, the protein
will have a greater propensity to assume either an active or inactive
conformation (and
consequenctly be upregulated or downregulated), depending upon which of the
switch control
pockets is occupied by the molecule. Thus, contacting the lcinase with a
molecule modulates that
protein's activation state.. The molecule can act as all antagonist or an
agonist of either switch
control pocket. The contact between the molecule and the kinase preferably
occurs at a region
of a switch control pocket of the lcinase and more preferably in an interlobe
oxyanion pocket of
the lcinase. hl some instances, the contact between the molecule and the
pocket also results in
the alteration of the conformation of other adj acent sites and pockets, such
as an ATP active site.
Such an alteration can also effect regulation and modulation of the active
state of the protein.
Preferably, the region of the switch control pocket offhe leinase comprises an
amino acid residue
sequence operable for bllldlllg t0 the FOrlllllla I molecule. Sllch binding
can occur between the
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
molecule and a specific region of the switch control pocket with preferred
regions including the
a-C helix, the cx-D helix, the catalytic loop, the activation loop, and the C-
terminal residues or
C-lobe residues (all residues located downstream (toward the C-end) from the
Activation loop),
and combinations thereof. When the binding region is the ec-C helix, one
preferred binding
sequence in this helix is the sequence VEEFLKEAAVM, (SEQ ID NO. 2). When the
binding
region is the catalytic loop, one pr eferred binding sequence in this loop is
HRDLAARNXL (SEQ
)D NO. 3). When the binding region is the activation loop, one preferred
binding sequence in
this loop is a sequence selected from the group consisting of DFGLSRLMT
(SEQ,JD N0.4),
GDTYTAH (SEQ )D NO. 5), and combinations thereof. When the binding region is
in the C-
lobe residues, one preferred binding residue is F, found at position 416
relative to the full length
sequence (residue 194 in SEQ )D NO. 1). When a biologically inactive protein
conformation is
desired, molecules which interact with the switch control pocket that
norrrlally results in a
biologically active protein conformation (when interacting with the switch
control ligand) will
be selected. Similarly, when a biologically active protein conformation is
desired, molecules
which interact with the switch control pocket that normally results in a
biologically inactive
prOtelll COllf01111at1O11 (When 111teTaCtlllg Wlth the switch control ligand)
will be selected. Thus,
the propensity of the protein to assume a desired conformation will be
modulated by
administration of the molecule. hl preferred forms, the molecule will be
administered to an
individual undergoing treatment for cancer including but not limited to
chronic myelogeneous
leukemia and stromal gastrointestinal tt11110TS. Ill StlCh fOrllls, it will be
desired to select
molecules that interact with the switch control pocket that generally leads to
a biologically active
protein conformation so that the protein will have the propensity to assume
tile biologically
inactive form and thereby alleviate the condition. It is contemplated that the
molecules of the
present invention will be administerable in any conventional form including
oral, parenteral,
inhalation, and 5t1bct1ta11eOt1S. It is preferred for the administration to.be
in the oral form.
Preferred molecules include the preferred formula (I) compounds discussed
above.
Another aspect of the present invention provides a method of treating cancer
comprising
the step of administering a molecule having the structure of the formula (I)
compounds to the
individual. SllCh COI1d1t1011S are Oftell the result of an overproduction of
the biologically active
form of a protein, 111Chldlllg 1C111aS8S. For example, a halhnarlc feattlr a
Of C11TO111C 111yelOgelleollS
leukemia involves areciprocal ChrOl110SOlllal tTa11S10Cat1011111vOlVlllg
htllllall ChTOn10SO1neS 9 and
26
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
22. This mutation fuses a segment of the bcr gene upstream of the second exon
of the c-abl
nonreceptor tyrosine lcinase gene. This fusion protein is called bcr-abl.
While the normal c-abl
gene 'and its protein are tightly controlled in normal cells, the fusion
protein product bcr-abl
presents with elevated, constitutive kinase activity. It is this activity that
enables bcr-abl fusion
protein to transform cells and cause malignancy. ThllS, tile 111Velltloll
discloses and utilizes small
molecule inhibitors of bcr-abl kinase. These inhibitor s contain functionality
which enable them
to bind to an binding region, preferably an interlobe oxyanion regulator
pocket in abl lcinase. The
111111b1tOT'S may alSO Colltalll fL111Ct1011a11ty WhlCh bllld t0 the ATP
pocket or other k111aSe a1111n0
acid residues taken from the N-lobe or C-lobe of the kinase.
The administering step generally includes the step of causing said molecule to
contact a
lcinase involved with elevated lcinase activity such as that found in cancer.
A particularly
preferred kinase to contact is bcr-abl kinase. When the contact is between the
molecule and a
lcinase, the contact preferably occurs in a binding region (preferably an
interlobe oxyanion pocket
of the lcinase) that includes an amino acid residue sequence operable for
binding to the Formula
I molecule. Preferred binding regions of the interlobe oxyanion pocket include
the a-C helix
region, the catalytic loop, the activation loop, the C-terminal lobe or
residues, and combinations
thereof. When the binding region is the a-C helix, one preferred binding
sequence in this helix
is the sequence VEEFLI~EAAVM (SEQ ID NO. 2). When the binding region is the
catalytic
loop, one preferred binding sequence in this loop is HRDLAARNXL (SEQ ~ NO. 3).
When
the binding region is the activation loop, one preferred binding sequence in
this loop is a-
sequence selected from the group consisting of DFGLSRLMT (SEQ ID N0.4),
GDTYTAH
(SEQ ID NO. 5), and combinations thereof: A preferred residue with which to
bind in the C-
terminal lobe is F.
Such a method permits tr eatment of cancer by virtue of the modulation of the
activation
state of a lcinase by contacting the lcinase with a molecule that associates
with the switch control
pocket that nornlally leads to a biologically active form of the lcinase when
interacting with the
switch control ligand. Because the ligand cannot easily interact with the
switch control pocket
associated with or occupied by the molecule, the ligand tends to interact with
the switch control
pocket leading to the biologically inactive form of the protein, with the
attendant result of a
decrease in the amount of biologically active protein. Preferably, the cancer
i selected from the
group consisting Of C111o111C 111y1Oge11eol1S leukemia and stromal
gastrointestinal tumors. As with
27
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
the other methods of the invention, the molecules may be'administer ed in any
coliventional form,
with any conventional excipients or ingredients. However, it is preferred to
administer the
molecule in an oral dosage form. Preferred molecules are again selected from
the group
consisting of the preferred formula (I) compounds as discussed above.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 is a schematic representation of a naturally occurring mammalian
protein in
accordance with the invention including "on" and "off ' switch control
pockets, a transiently
modifiable switch control ligand, and an active ATP site;
Fig. 2 is a schematic representation of the protein of Fig. l, wherein.the
switch control
ligand is illustrated in a binding relationship with the off switch control
poclcet, thereby causing
the protein to assume a first biologically downregulated confornation;
Fig. 3 is a view similar to that of Fig. l, but illustrating the switch
control ligand in its
charged-modified condition wherein the OH groups of certain amino acid
residues have been
phosphorylated;
Fig. 4 is a view similar to that of Fig. 2, but depicting the protein wherein
the switch
control ligand is in a binding relationship with the on switch control
poclcet, thereby causing the
protein to assume a second biologically-active conformation different than the
first conformation
of Fig. 2;
Fig. 4a is an enlarged schematic view illustrating a representative binding
between the
phosphorylated residues of the switch control ligand, and complemental
residues from the on
switch control poclcet;
Fig. 5 is a view similar to that of Fig. 1, belt illustrating in schematic
form possible small
molecule compounds in a binding relationship with the on and off switch
control pockets;
Fig. 6 is a schematic view of the protein in a situation where a composite
switch control
pocket is formed with portions of the switch control ligand and the on switch
control poclcet, and
with a small molecule in binding relationship with the composite poclcet; and
Fig. 7 is a schematic view of the protein in a situation where a combined
switch control
pocket is formed with portions of the on switch control pocket, the switch
control ligand
sequence, and the active ATP site, and with a shall 11101eCLlle 111 binding
relationship with the
28
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
combined switch control pocleet.
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
The present invention provides a way of rationally developing new small
molecule
modulators which interact with naturally occurring proteins (e.g., mammalian,
and especially
hlllllall proteins) in order to modulate the activity of the proteins. Novel
protein-small molecule
adducts are also provided. The invention preferably malces use of naturally
occurring proteins
having a conformational propel-ty whereby the pr oteins change their
conformations ifZ vivo with
a corresponding change in protein activity. For example, a given enzyme
protein in one
confornation may be biologically upregulated, while in another confornation,
the salve protein
may be biologically dowllregulated. The invention preferably malces use of one
mechanism of
conforllation change utilized by naturally occurring proteins, through the
interaction of what are
terned "switch control ligands" and "switch control pockets" within the
protein.
As used herein, "switch control ligand" means a region or domain within a
naturally
OCCLi1T111g prOtelll alld haVlllg 011e Or 111OTe a1111110 aCld residues
therein which are transiently
modified ifz vivo between individual states by biochemical modification,
typically
ph05ph01'ylatlOll, sulfation, acylation or oxidation. Similarly, "switch
control pocket".means a
plurality Of COlltlgLlOllS Or 11011-COlltlg110115 alllln0 aCld residues within
a naturally occurrnlg
protein and comprising residues capable of binding ire. vivo with transiently
modified residues of
a switch control ligand in one of the individual states thereof in order to
induce or restrict the
conformation of the protein and thereby modulate the biological activity of
the protein, and/or
which is capable of binding with a non-naturally OCClIlTlllg switch control
modulator molecule
to induce or restrict a protein confornation and thereby modulate the
biological activity of the
protein.
A protein-modulator adduct in accordance with the invention comprises a
naturally
occurring protein having a switch control pocket with a non-natu r ally
occurring molecule bound
to the protein at the region of said switch control pocket, said molecule
serving to at least
partially regulate the biological activity of said protein by inducing or
restricting the
conformation of the protein. Preferably, the protein also has a corresponding
switch control
ligand, the ligand interacting i~z vivo with the pocket to regulate the
conformation and biological
aCtlVlty of the pr Otelll SLlCh that the pr otein will aSSllllle a f1r St
COllfOl lllatl011 alld a f1r St biological
29
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
activity upon the ligand-pocket interaction, and will assume a second,
different conformation and
biological activity in the absence of the ligand-pocket interaction.
The nature of the switch control ligand/switch control pocket interaction may
be
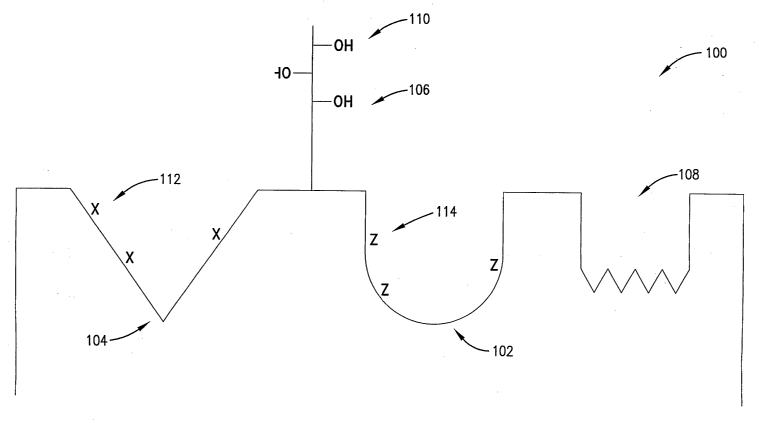
understood from a consideration of schematic Figs. 1-4. Specifically, in Fig.
1, a protein 100 is
illustrated in schematic fOnl1 t0 lllchlde all "on" switch control poclcet
102, and "ofd' switch
control pocket 104, and a switch control ligand 106. In addition, the
schematically depicted
protein also includes an ATP active site 108. In the exemplary pr otein of
Fig. 1, the ligand 106
has three amino acid residues with side chain OH groups 110. The off pocket
104 contains
corresponding X residues 112 and the on pocket 102 has Z residues 114. In the
exemplary
instance, the protein 100 will change its conformation depending upon the
charge status of the
OH groups 110 on ligand 106, i.e., when the OH groups are unmodified, a
neutral charge is
presented, but when these groups are phosphorylated a negative charge is
presented.
The functionality of the pockets 102, 104 and ligand 106 can be understood
from a
consideration of Figs. 2-4. In Fig. 2, the ligand 106 is shown operatively
interacted with the off
pocket 104 such that the OH groups 110 interact with the X residues 112
forming a part of the
pocket 104. Such interaction is primarilyby virtue of hydrogen bonding between
the OH groups
110 and the residues 112. As seen, this ligand/poclcet interaction causes the
protein 100 to
assume a conformation different fr0111 that Seell 111 Fig. 1 and col~esponding
to the off or
biologically downregulated conformation of the protein.
Fig. 3 illustrates the situation where the ligand 106 has shifted from the off
pocket
interaction conformation of Fig. 2 and the OH groups 110 have been
phospholylated, giving a
negative charge to the ligand. In this condition, the ligand has a strong
propensity to inter act with
on poclcet 102, to thereby change the protein conformation to the on or
biologically upregulated
state (Fig. 4). Fig. 4a illustrates that the phosphorylated groups on the
ligand 106 are attracted
to positively charged residues 114 to achieve an ionic-like stabilizing bond.
Note that in the on
conformation of Fig. 4, the protein conformation is different than the off
conformation of Fig.
2, and that the ATP active site is available and the protein is functional as
a lcinase enzyme.
Figs. 1-4 illustrate a simple situation where the protein exhibits discrete
pockets 102 and
104 and ligand 106. However, in many cases a more complex switch control
poclcet pattern is
observed. Fig. 6 illustrates a situation when a an appropriate pocket for
small molecule interaction
is formed from amino acid residues taken both from ligand 106 and, for
example, from pocket
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
102. This is termed a "composite switch control pocket" made up of.residues
from both the
ligand 106 and a pocket, and is referred to by the numeral 120. A small
molecule 122 is
illustrated which interacts with the pocket 120 for protein modulation
purposes.
Another more complex switch poclcet is depicted in Fig. 7 wherein the pocket
includes
residues from on pocket 102, and ATP site 108 to create what is termed a
"combined switch
control pocket." Such a combined pocket is referred to as numeral 124 and may
also include
residues from ligand 106. An appropriate small molecule 126 is illustrated
with pocket 124 for
protein modulation proposes.
It will thus be appreciated that while in the simple pocket situation of
Figs.l-4, the small
molecule will interact with the simple pocket 102 or 104, in the more complex
situations of Figs.
6 and 7 the interactive pockets are in the regions of the poclcets 120 or124.
Thus, broadly the
the small molecules interact "at the region" of the respective switch control
poclcet.
31
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
GENERAL SYNTHESIS OF COMPOUNDS
In the synthetic schemes of this section, q is 0 or 1. When q = 0, the
substihlent is
replaced by a synthetically non-interfering group R7.
Compounds of Formula I wherein D is taken from D-1 or D-2 and Y is alkylene
are
prepared according to the synthetic route shown in Scheme 1.1. Reaction of
isothiocyanate 1
with chlorine, followed by addition of isocyanate 2 affords 3-oxo-
thiadiazolium salt 3.
Quenching of the reaction with air affords compounds of Fornmla I-4.
Alternatively,
reaction of isothiocyanate 1 with isothiocyanate 5 under the reaction
conditions gives rise to
compounds of Formula I=7. See A. Martinet et al, Journal ofMediciraal
Clzenzist~y (2002)
45: 1292.
Intermediates 1, 2 and 5 are commercially available or prepared according to
Scheme
1.2. Reaction of amine 8 'with phosgene or a phosgene equivalent affords
isocyanate 2.
Similarly, reaction of amine 8 with thiophosgene affords isothiocyanate 5.
Amine 8 is
prepared by palladium(0) catalyzed amination of 9, wherein Q is a group
capable of oxidative
insertion into palladium(0), according to methodology reported by S. Buchwald.
See M.
Wolter et al, Ocgaraie Letters (2002) 4:973; B.H. Yang and S. Buchwald,
Journal of
O~gai2ometallie Chemistry (1999) 576(1-2):125. In this reaction sequence, P is
a suitable
amine protecting group. Use of and removal of amine protecting groups is
accomplished by
methodology reported in the literaW re (Protective Groups in Organic
Synthesis, Peter G.M.
Wutts, Theodora Greene (Editors) 3rd edition (April 1999) Wiley, John & Sons,
Incorporated; ISBN: 0471160199): Starting CO111po1r11dS 9 are commercially
available or
readily prepared by one of ordinary skill in the art: See March's Advanced
Organic
Chemistry: Reactions, Mechanisms, and Structure, Michael B. Srlllth & Jeny
March
(Editors) 5th edition (January 2001) Wiley John & Sons; ISBN : 0471585890.
32
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Scheme 1.2
~NHz Phosgene N=C=O
[Rs02C-(NH)p]q-E-Y ~ [Rs02C-(NH)P]q-E-Y~
Base
NH thiophosgene N=C=S
[R602C-(NH)p]q-E-Y~ 2 Base [Rs02C-(NH)P]q-E-Y~
8 _
R~O.,C-NHZ
R602C-NH-E-Y-NHP deprotect
M-E-Y-N H P
Pd(0) catalysis
ll
9
R602C-NH-E-Y-NHZ
g
Compounds of Formula I wherein Q is taken from Q-1 or Q-2 and Y is alkylene
are
also available via the synthetic route shown in Scheme 1.3. Reaction of amine
8 with
isocyanate or isothiocyanate 2a yields the urea/thiourea 8a which can be
cyclized by the
5 addition of chlorocarbonyl sulfenyl chloride. See GB1115350 and US3818024,
Revanlcar et.
al US Patent 4,093,624, and I~layman et. al JOC 1972, 37(10), 1532 for further
details.
Where R4 is a readily removable protecting group (e.g. R = 3,4-d-methoxybenzyl
amine), the
action of mild, acidic deprotection conditions such as CAN or TFA will reveal
the parent ring
system of I-4 (X=O) and I-7 (X=S).
33
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Scheme 1.3
X
R4NCX
X-O, S . Ra
HN N
[R602C-(NH)plq-E-Y' NHZ
2a [R60aC-(NH)p]q-E-Y' H
8a, X=O, S
'X
CI
CI S~ ~N N'Ra Deprotection
[R60zC-(NH)Plq-E-Y S
O
I-4 X=O
I-7 X=5
X
[R602C-(NH)Plq-E-Y~ S
1-4 X=O~\O
I-7 X=S
Compounds of Formula I wherein Q is taken from Q-1 or Q-2 and Y is -alkylene
are
also available as shown in Scheme 1.4. Condensation of isocyanate or
isothiocyanate 2a with
amine RSNHZ yields urea/thiourea 2b, which, when reacted with chlorocarbonyl
sulfenyl
chloride according to GB1115350 and US3818024 yields 2c. Where R4 is a readily
removable protecting group (e.g. R = 3,4-d-methoxybenzyl amine), the action of
mild, acidic
deprotection conditions such as CAN or TFA will reveal the parent ring system
of 2d.
Reaction of 2d with NaH in DMF, and displacement wherein M is a suitable
leaving group
t.: .
5LlCh aS ChIOTIde, bromide or iodide yields I-4 (X=O) and I-7 (X=S).
Scheme 1.4
0 0
R NCX R5NH2 R4HN N~RS CI~S~CI R4 N ~,RS Deprotection of Ra
X=O, S
X
2a X=O, S X=O, S
2b 2c
O O
HN ~, NaH/DMF
Rs - / ~ Rs
CI [R602C (NH)plq-E-Y X
X=0, S [Re02C-(NH)p]q-E-Y'
2d I-4 X=O
8a I-7 X=S
34
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
COmpOLindS Of F01111111a I wherein Q is taken from Q-1' or Q-2' and Y is
alkylene are
available via the synthetic route shown in Scheme 1.5. Condensation of
isocyanate or
isothiocyanate 2a with ammonia yields urea/thiourea 2e, which, when reacted
with
chlorocarbonyl sulfenyl chloride according to GB1115350 and US3818024 yields
2f.
Reaction of 2f with NaH in DMF, and displacement wherein M is a suitable
leaving group
such as chloride, bromide or iodide yields yields I-4' ,(X=O) and I-7' (X=S).
Scheme 1.5
0 0
HN N 'CI S--~
R "
NH
~H CI
q S
3 ~ R4,N
NCX N~Fi
R
4 ~
X_O ~
S
~
X X
2a X=O, S X=O, S
2e 2f
O
S
NaH/DMF
N,R
/
a
IRs02C-(NH)Plq-E-Y
~
CI X
[RsOZC-(NH)P)9-E-Y
1-4' X=O
8a 1-T X=S
Compounds of Formula I wherein Q is taken from Q-3 or Q-4 and Y is alkylene,
are
prepared according to the synthetic route shown in Schemes 2.1 and 2.2,
respectively.
Reaction of 12, wherein M is a suitable leaving group, with the carbamate-
protected
hydrazine 13 affords intermediate 14. Reaction of 14 with an isocyanate gives
rise to
interniediate 15. Thermal cyclization of 15 affords 1,2,4-triazolidinedione of
Formula I-16.
By analogy, scheme 2.2 illustrates the preparation of 3-thio-5-oxo-1,2,4-
triazolidines of
Formula I-18 by reaction of intermediate 14 with an isothiocyanate and
subsequent thermal
cyclization.
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Scheme 2.1 O
~OR~o
H2N-N \ Ra
IRs~zC-(NH)PIq~E/Y~N~N OR~o
[R60aC'(NH)Plq--E H
O
l2
14
Ra
Ra N C O [RsO2C-(NH)p]q Y N OR~o heat
~E~ ~Ni
~ O
0i _NH
I
15 Ra
Ra
[RsOzC'(NH)Pjq ~ E /Y~ N, N
~O
1-16 ~N
O
Ra
Scheme 2.2
Ra
Ra-N=C=S [R602C'(NH)p]q-E~Y~N~N OR~o heat
_14
O
17 S~NH
I
Ra
R4
[R60zC'(NH)PIq~E/Y~N~ /N
~ ~O
~N
I-18 S
Ra
Intermediates 12 wherein p is 1 are readily available or are prepared by
reaction of 19
with carbamates 10 under palladium (0)-catalyzed conditions. MI is a group
which
oxidatively inserts palladium(0) over group M. M~ is preferably iodo or bromo.
Compounds
19 are either commercially available or prepared by one of ordinary skill in
the art.
3G
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Scheme 2.3
R602C-NHa
E\ /M _ /E~. ~M
M~~ Y Pd(0) catalysis; Re~aCNH Y
1~ Base 12
C0111pO1111dS Of FOr111ll1a I wherein Q is taken from Q-3 or Q-4 and Y is
alkylene are
also prepared according to the synthetic route shown in Scheme 2.4. Oxidation
of amine
5 R4NHz to the corresponding hydrazine, condensation with ethyl chloroformate
subsequent
heating yields1,2,4-triazolidinedione 15a. After the action of NaH in DMF,
displacement
wherein M is a suitable leaving group such as chloride, bromide or iodide
yields I-16 (X=O)
and I-18 (X=S).
10 Scheme 2.4
1. NaNOZ OII RSNCX
2. SnCh CI OEt ~pEt X-O, S heat
R4NHz R4NHNH2 R4NHNH
2a
O
HN~--~ . NaH/DMF (RsOzC-(NH)Plq-E-YvN~O
R4~N~N~Rs
CI Ra N~N~Rs
X [RsOzC-(NH)P)q-E-Y II~
X
15a 8a
I-16 X=O
I-18 X=S
O O
HN-~ NaH/DMF ~-NH
,N NH _ (RsOzC_(NH)PJq-E-Y'-N~N~Ra
Deprotection of Rs R4 ~ (RsO2C-(NH)p]q-E-Y
X
X 8a
15b I-16' X=O
I-18' X=S
Compounds of Formula I wherein Q is taken from Q-3' or Q-4' and Y is allcylene
are
also prepared according to the synthetic route shown in Scheme 2.4. When RS is
a readily
removable protecting group (e.g. R = 3,4-d-methoxybenzyl amine), the action of
mild, acidic
deprotection conditions such as CAN or TFA on 15a will reveal 1,2,4-
triazolidinedione 15b.
After deprotonation of 15b by NaH in DMF, displacement wherein M is a suitable
leaving
group such as chloride, bromide or iodide yields I-16' (X=O) and I-18' (X=S).
Compounds of Formula I wherein Q is taken from Q-5 or Q-6 and Y is alkylene
are
prepared according to the synthetic route shown in Scheme 3. Reaction of
hydrazine 20 with
37
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
chlorosulfonylisocyanate and base, such as triethylamine, gives rise to a mixW
re of
intermediates 21A and 21B which are not isolated but undergo cyclization iT~
sitar to afford
compounds of Formulae I-22A and I-22B. Compounds I-22A and I-22B are separated
by
chromatography or fractional crystallization. Optionally, compounds I-22A and
I-22B can
undergo Mitsunobu ueaction with alcohols R40H to give compounds of Formulae I-
23A and
I-23B. Compounds 20 are prepared by acid-catalyzed deprotection of t-butyl
carbamates of
structure 14, wherein Rlo is t-butyl.
Scheme 3
R4 CIS02-N=C=O
[R602C-(HN)P]q-E-Y~N~NH
20 H Base
R4 R4
[R60zC-(HN)P]q-E-Y~N~NH [R602C-(HN)P]q-E-Y~N~N O
H
pi 'NH NH
CI w ~
O~/S~CI O S ~
O 21B
21 A --
R
[R602C-(HN)P]q-E-YwN~N~ ,O [RsOzC-(HN)Plq-E-YwN~N4 O
SQO + O=S~N
/,'-NH ii
I-22A0 1-22B O
Ph3P Ph3P
Diethyl azodicarboxylate Diethyl azodicarboxylate
R~OH R40H
Ra Ra
[R602C-(HN)P]q-E-YwN~N~s~O [R602C-(HN)P]q-E-YwN~N~O
// -N WO + ~'1
O ~ O-iS_N~
R4 O R4
1-23A 1-23B
38
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Compounds of Formula I wherein Q is Q-7 and Y is alkylene are prepared as
shown
in Scheme 4. Reaction of amine 8 with maleimide 24, wherein M is a suitable
leaving group,
affords compounds of Formula I-25. Reaction of compound 26, wherein M is a
group which
can oxidatively insert Pd(0), can participate in a Heck reaction with
maleimide 27, affording
compounds of Formula I-28. Maleimides 24 and 27 are commercially available or
prepared
by one of ordinary skill in the art.
Scheme 4 R4
O N
O O R4
O \ N
[R602C-(NH)p]q-E-Y~ 24 R5 O
NHZ [R60zC-(NH)p]q-E-YEN ~
Base H R5
1-25
R4
O N
Ra
O O
O
[R602C'(NH)p]q~E\M 27 R5 [R60~C'(NH)p]q~E \
26 Pd(0), Base Rs
Heclc Reaction 1-28
Compounds of Fornula I wherein Q is Q-8 and Y is alkylene are prepared as
shown
in Scheme 5, according to methods reported by M. Tremblay et al, .Journal of
Combinatorial
Claenzist~y (2002) 4:429. Reaction of polymer-bound activated ester 29
(polymer linkage is
oxime activated-ester) with chlorosulfonylisocyante and t-butanol affords N-
BOC
sulfonylurea 30. Subjection of 30 to the Mitsunobu reaction with R4OH gives
rise to 31.
BOC-group removal with acid, preferably trifluoroacetic acid, and then
treatment with base,
preferably triethylamine, provides the desired sulfahydantoin I-32.
Optionally, intermediate
30 is treated with acid, preferably trifluoroacetic acid, to afford the N-
unsubstituted
sulfahydantoin I-33.
39
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Scheme 5
R4 R4 CISO~-N=C=O
[R60~C-(NH)p]q-E-Y~NH pt-BuOH
t9~.;~
29 O
R4 R4
Ph P
[R602C-(NH)p]q-E-YEN Odiethyl azodicarboxylate
D j~S~NH O R4OH
O
BOC
R4 R4
1) H+ R4 R4
[R6O2C-(NH)p]q-E-Yw i O~ _ [R602G_(NFi)P]q-E-Y~N
O~S~ ~R O 2) Triethylamine O\S\ O
31 /~ N 4 I-32 O N
BOC
Ra
R4 R
4
[Rs02C-(NH)plq-E-Y~N
O
H+ O,/S ~ N
30 o H
I-33
Compounds of Formula I wherein Q is Q-8 and Y is alkylene are also prepared as
ShOWIl 111 Scheme 5.1. Ammc 8 is condensed with the glyoxal hemiester to yield
31a.
Reaction of chlorosulphonyl isocyanate first with benzyl alcohol then 31a
yields 31b, which
5 after heating yields I-32.
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Scheme 5.1
0
H~OEt O
H' ~
[R602C-(NH)p]q-E-Y~NHz O [R60zC-(NH)p]q-E-Y~N~OEt
NaCHBH3
8 31a
HzN~o~O O
1. ~ ~ OH ~N~
CISOZNCO (R602C-(NH)p]q-E-Y OEt
2. H O
' ~ 31b
[RsOzC-(NH)p]q-E-Y~N v _OEt
31a
3. 5% Pd/C
O~S~N
N~O
heat [RsOzC-(NH)p]q-E-Y~
I-32
Compounds of Formula I wherein Q is taken from Q-8', are prepared according to
the
S SyllthetlC rOllte ShOWIl 111 Scheme 5.2. Formation of 31c by the lllethod of
Muller and DuBois
.JOC 1989, 54, 4471 and its deprotonation with NaH/DMF or NaH/DMF and
subsequently
alkylation wherein M is a suitable leaving group such as chloride, bromide or
iodide yields I-
32'. Alternatively, I-32' is also available as shown in Scheme 5.3. Mitsunobu
reaction of
boc-sulfamide amino ethyl ester with alcohol 8b (made by methods analogous to
that for
amine 8) yields 31c, which after Boc removal with 2N HCl in dioxane is
cyclized by the
action of NaH on 31d results in I-32'.
Scheme 5.2
O
O~S-NH
i
NaN
p\~-NH
[R602C-(N H)pIq-E-Y ~ M [RsOzC-(NMplq-E-Y ~ N
8a 31c O
I-32'
41
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Scheme 5.3
H O (RsOzC-(NH)Plq-E-Y'OH
Boc-N~S~O O 8b [RsOzC (NH)Plq-E-Y N~~~O O
i HN
HN OEt DEADCAT, Ph3P' 31d OEt
O
O~S-NH
heat [RsOzC-(NH)p]q-E-Y~N
O
I-32'
Compounds of Formula I wherein Q is Q-9 and Y is alkylene are prepared as
shown
in Scheme 6. Reaction of polymer-bound amino acid ester 34 with an isocyanate
affords
intermediate urea 35. Treahnent of 3S with base, preferably pyridine or
triethylamine, with
optional heating, gives rise to compounds of Formula I-36.
Scheme 6
Ra R4
R4-N=C=O
(RsOZC-(NH)p]q-E-Y~ O
NH v~,,
34 O
Ra R4
[R60zC-(NH)Plq-E-YwN OBase
35 ~,~'O
O~NH
R4
Ra Ra
[R60zC-(NH)p]q-E-YEN
O
I-36 O N
1
Ra
Compounds of Formula I wherein Q is Q-9 and Y is alkylene are also prepared as
shown in Scheme 6.1. Reaction of aldehyde 8c (available by methods similar to
that shown
for 8a by anyone skilled in the art) with the t-butyl ester of glycine under
reductive amination
conditions yields 35a. Isocyanate 2a is condensed with p-nitrophenol (or the
corresponding
RQNHZ amine is condensed with p-nitrophenyl chloroformate) to yield the
carbamic acid p-
nitrophenyl ester, which when reacted with deprotonated 35a and yields the
urea that when
42
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
deprotected with acid yields 35b. Fonnula I-36 is directly available from 35b
by the action
of NaH and heat.
Scheme G.l
0
0 ~ ~ 0
HZNv 'Ot-Bu H\ ~fI
[R602C-(NH)Plq-E-Y~H ~ [R6p2C_(NH)Plq-E-Y~N~Ot-Bu
NaCHBH3
8c 35a
O. ~NOz
1. R4HN"O
2. I 2N HCI/Dioxane
O N + O NHO
[R6O2C-(NH)pjq-E-Y~~O [R60~C-(NH)p]q-E-Y- '~OH
p6 35b
~OIIIpOLll1(1S Of FOrIlltila I Wherelll Q 1S taken from Q-9', are prepared
according to the
synthetic route shown in Scheme G.2. Fornlation of 35c by the method described
in
JP10007804A2 and Zvilichovsky and Zucker, Israel Journal of Chemistry, 1969,
7(4), 547-
54 and its deprotonation with NaH/DMF or NaH/DMF and its subsequent
displacement of M,
wherein M is a suitable leaving group such as chloride, bromide or iodide,
yields I-36'.
Scheme G.2
0
~--NH
NaN O
-NH
[R60~C-(NH)Plq-CE-Y~M - [R60ZC-(NH)Pjq-E-Y~N
35c O
8a I-36'
~O111pOLllldS Of FOr111uIa I-39 wherein Q is Q-10 or Q-11, and Y is alkylene
are
prepared as shown in Schemes 7.1 and 7.2, respectively. Treatment of alcohol
37 (Z = O) or
amine 37 (Z = NH) with chlorosulfonylisocyanate affords intermediate carbamate
or urea of
structure 38. Treatment of 38 with an amine of strucW re HN(R~)R4 and base,
preferably
triethylamine or pyridine, gives sulfonylureas of Fornmla I-39. Reaction of
chlorosulonylisocyanate with an alcohol (Z = O) or amine (Z = NR4) 40 affords
inteunediate
43
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
41. Treahment of 41 with an amine 8 and base, preferably triethylamine or
pyridine, gives
sulfonylureas of Formula I-42.
Scheme 7.1
C1502-N=C=0 O
[R60~C-(NH)Plq-E'Y~ZH [RsOaC-(NH)Plq-E-Yw ~ iSOzCI
Z N
37 H
38
R4 O
H-N O\ /%
R4 [R60~C-(NH)Plq-E-Yw ~ iS~NiRa
Base Z H
Ra
1-39
Scheme 7.2 R4
H-Z O
~SO~CI
CISOz-N=C=O Z~N
H
_a~
[R602C'(NH)Plq-E-YwNiRS O 0 O
8 H Ray ~ iSl /Y-E-9f(NH)PC02Rs
N
Base R5
I-42
GOIIIpOlIIIdS Of FOrlllllla I wherein Q is taken from Q-12 are prepared
according to the
synthetic route shown in Scheme 8. Readily available pyridine 43, wherein TIPS
is tri-iso-
propylsilyl, is alkylated under standard conditions (KZG03, DMF, Rø-I or
Mitsunobu
conditions employing R4-OH) to give pyridine derivative 44 which is reacted
with compound
12, wherein M is a suitable leaving group, to afford pyridones of formula I-
45.
44
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Scheme 8
Ra
a
TIPS
TI
4 3
Diethyl azodicarboxylate
43 44
Ra
O
[R602C-(N H)Plq-E-Yw M
12
- s O/ wN
Base Y-E-[(NH)pC02R6]q
_I-_45
Compounds of Formula I wherein Q is taken from Q-13 are prepared according to
the
synthetic route shown in Scheme 9. Readily available pyridine 46 is alkylated
under standard
conditions (KZC03, DMF, Ra-I or Mitsunobu conditions employing Ra-OH) to give
pyridine
derivative 47. N-alkylation with K~C03, DMF, Ra-I affords pyridones of formula
48.
Intern~ediate 48'is partitioned to undergo a Heck reaction, giving I-49; a
Buchwald amination
reaction, giving I-51; or a Buchwald Cu(I) catalyzed O-arylation reaction, to
give I-52. The
Heck reaction product I-49 may be optionally hydrogenated to afford the saW
rated compound
I-50. Wherein the phenyl ether Ra is methyl, compounds of formula I-49, I-50,
I-51, or I-52
are treated with boron tribromide or lithium chloride to afford compounds of
Formula I-53,
wherein Ra is hydrogen.
R40, KZC03
DMFor Aceton
or
R OH, Ph P
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Scheme 9
OH . ORa
R RaO~ KzCOs R ORa
DMFor Acetone ~ s R5
R41, K~C03 /
TIPSY ~ / or TIPSY ~ ~ DMF or Acetone '
O N CI O N CI
R40H, Ph3P O N CI
4~ Diethyl azodicarboxylate 47 R
4
ORa 48
E-[(N H )PC02Rslq R O Ra
\-/ ~~ / ( 5 Hydrogenation / R5
48 / E-[(NH)pC02Rslq -'
Heck reaction O R ,~ O N~(~'E-[(NH)pC02Rs]q
Pd(0) a I-49 Ra n + 2
Base
ORa _1-50
H2N~E-[(NH)pCO2Rs]q / R boron tribromide
or lithium chloride boron tribromide
n _ ~ or lithium chloride
48
Buchwald aminatioo O N H nE-[(NH)pCO2Rs]q
Pd(0) Ra 1-51 boron tribromide OH
Base - or lithium chloride
/ Rs
HOE-[(NH)pCO~Rs]q ORa
boron tribromide ~ ,E-[(NH)pCO~Rs]q
qg n _ / R5 or lithium chloride O NR Y
4
Buchwald arylation
Cu 1 O N~O~E-[(NH)pCOaRs]q I-53
() n
Base Ra
I-52
Compounds of Formula I wherein Q is taken from Q-14 are prepared according to
the
S SyllthetlC -rOllte ShOWIl 111 Scheme 10. Starting from readily avai]able
pyridine 54, alkylation
under standard conditions (I~2C03, DMF, R4-I or Mitsunobu conditions employing
R4-OH)
yields pyridine derivative 55. N-alkylation with K2CO3, DMF, R4-I affords
pyridones of
formula 56. Intermediate 56, wherein M is a suitable leaving group, preferably
bromine or
chlorine, is partitioned to undergo a Heck reaction, giving I-57; a Buchwald
amination
reaction , giving I-59; or a Buchwald Cu(I) catalyzed O-arylation reaction, to
give I-60. The
Heck reaction product I-57 may be optionally hydrogenated to afford the
saturated compound
I-58. Wherein the phenyl ether Ra is methyl, C0117pO1I11dS Of fOrlllllla I-57,
I-58, I-59, or I-60
are treated with boron tribtomide or hthllllll ChlOrlde to afford Co111pOL1ndS
Of FOrllllla I-61,
wherein R4 is hydrogen.
4G
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Scheme 10
OH ORa
ORa
O RaO, KzCOs O
\ DMForAcetone ~ \ O
R41, KZC03
TIPSY / R40H, Ph3P TIPSY / DMF or ACetoflB
O N R5 Diethyl azodicarboxylate O N RS
O N R5
54 Ss
- -- Ra
oR4 s6
ORa
E-[(NH)pCOzRs]q n + 2
\ ~1 E-[(NH)pCOZRs]q I3Ydr~ tion/ E-[(NH)pCO2Re]q
s~ ~
I-Iecl< reaction O N"R5
Pd(0) Ra I-s7 o Ra R5 I-58
Base - -
ORa
HZN~E-[(NH)pCOZRs]q H BBr3 or LiCI
N~E-[(NH)pCO2R6]q BBr3 or LiCI
'~'n
s6 ~n
BBr or LiCI
Buchwald amination O Ra R5 3 OH
Pd(0) I-s9 ~ Y~E-[(NH)pCO2Re]q
Base -
HO E-[(NH)pC02R6]q ORa O N' _R
R 5
O~E-[(NH)pCO2R6]q BBr3 or LiCI a
'~'n
s6 "
Buchwald --arylatio» R I-G1
Cll(1) p Ra
Base I-60
Compounds of Fornmla I wherein Q is taken from Q-15 are prepared according to
the
synthetic routes shown in Schemes 11 and 12. Starting esters 62 are available
from the
corresponding secoacids via TBS-ether and ester formation under standard
conditions.
Reaction of protected secoester 62 with Meerwin's salt produces the vinyl
ether 63 as a pair
of regioisomers. Alternatively, reaction of 62 with dimethylamine affords the
vinylogous
carbamate 64. Formation of the dihydropyrimidinedione 66 proceeds by
condensation with
urea 65 with azeotropic removal of dimethylamine or methanol.
Dihydropyrimidinedione 66
may optionally be further substituted by Mitsunobu reaction with alcohols R40H
to give rise
to compounds 67.
Scheme 12 illustrates the further synthetic elaboration of intermediates 67.
Removal
of the silyl protecting group (TBS) is accomplished by treatment of 67 with
flouride (tetra-n-
butylammonium fluoride or cesium flouride) to give primacy alcohols 68.
Reaction of 68
with isocyanates 2 gives rise to compounds of Fornmla I-69. Alternatively,
reaction of 68
with [R~OZC(NH)p]q-E-M, wherein M is a suitable leaving group, affords
compounds of
47
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Formula I-70. Oxidation of 68 using the Dess-Mautin periodinane (D. Dess, J.
Martin, J. Ana.
Claena. Soc. (1991) 113:7277) or tetra-n-alkyl penithenate (W. Griffith, S.
Ley, Aldf°iclaimiccz
Acta (1990) 23:13) gives the aldehydes 71. Reductive amination of 71 with
amines 8 gives
rise to compounds of Formula I-72. Alternatively, aldehydes 71 may be reacted
with
ammonium acetate under reductive allcylation conditions to give rise to the
primary amine 73.
Reaction of 73 with isocyanates 2 affords compounds of Formula I-74.
Scheme 11
~O O
O O Meerwin's TBSO \ O
Reagent ' OMe
TBSO OMe - -n63 R5 R4F-IN 65 NI-h
n
R5 N(Me)2 O
Dimethylamit~ TBSO
62 4A sieves \- OMe
n
64 Rs 66
RaOH
_66
Ph3P
diethyl azodicarboxylate
67
48
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Scheme 12
O
Raw ~ .Ra Ray ~ [R60zC-~H)P]q-E-N-C-O RawN~NH
N' _N N"NH 2 _
TBSO \ ~ HO \ (RsOzC (NH)p]q-C'NH\ /0 " \ O
O ~~ I~IO
Rs [RsOzC-MH)P]Q-E-M ~8 R 0 R
67 I-69
0 ~ Oxidation
Ra~N~NH O O
,O \ Raw ~ [R~O20-(NI-I)p]q-E-NHZ ~
[RsOzC-(NH)Plq- n 0 N NH g Ra~N~NH
E
Rs OHO \ Reductive amination NH~~ ~r~0
n O [RsOzC-(NH)p]q-E n
I-70
_71 Rs Rs
ammonium acetate I-72
(reductive amination)
O
0
~ [R60zC-(NI-I)Plq-R-N=C=O Raw N ~ N H
Ra~N~NH Z NH N \
[RsOzC-(NH)plq-E O ~~0
HZN ~~~~ n
n 0 Rs
Rs
73 1-74
Compounds of Formula I wherein Q is taken fiom Q-16 are prepared according to
the
synthetic routes shown in Schemes 13 and 14. Starting esters 75 are available
from the
corresponding secoacids via TBS-ether and ester formation under standard
conditions.
Reaction of protected secoester 75 with Meerwin's salt produces the vinyl
ether 76 as a pair
of regioisomers. Alternatively, reaction of 75 with dimethylamine affords the
vinylogous
carbamate 77. Formation of the dihydropyrimidinedione 78 proceeds by
condensation with
urea 65 with azeotropic removal of dimethylamine or methanol.
Dihydropyrimidinedione 78
may optionally be further substit<lted by Mitsunobu reaction with alcohols
R40H to give rise
to compounds 79. Compounds of Formulae I-81, I-82, I-84, and I-86 are prepared
as shown
in Scheme 14 by analogy to the sequence previously described in Scheme 12.
49
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Scheme 13 \O O
Meerwin's _ Rs \ OMe O
O O II O
Reagent
TBSO ~ n RaHN~NH2
R5 OMe ~6 65 R4~N NH
DimethylamirL N(Me)~J \
TBSO n 4A sieves R5 'O
R5 ~ OMe
TBSO ~n
TBSO ~n
78
77 O
RdOH Ra~ ~ ~Ra
N N
-_--
Ph3P \ _
diethyl azodicarboxylate R5 'O
TBSO
79
Scheme l4
O
O R4\N " N/R4
R4\N~N~R4 RawN~N~Ra [~p2~-~H)pla-~-N=c=O \
\ 2 Rs ~~O ~ O
R \ O Rs \O
(~ I-8I n ~ H_E_[(NH)PCOzRsIG
\ TOH --
OTBS 8~1 n
79 n [R~OaC-(NI-I)p]q-E-M
Oxidation O
O RawN~N~Ra
O
RawN~N.Ra Raw ~ iR4 [~OzC gH)pl9-E-NI-Iz Rs \ O
N N
Reductive amination
Rs \ O Rs \ O ~ n NH-E-[(NH)pCOzRsIG
I-g~ n O-E-[(NH)pC0 R 83 C CHO I 8ø O
z slG
_ n
ammonium acetate R4wN~N~Ra
(reductive amination)
O Rs \ O
RawN~NiRa [RsOzC-(NH)P1Q-E-N°C-O ~ n NH
~NH-~-f(NH)PCOzRslq
O
Rs \ O
nNHz I-86
50
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Alkyl acetoacetates 87 are commercially available and are directly converted
into the
esters ~8 as shown in Scheme 15. Treatment of 87 with NaHMDS in THF, followed
by
quench with formaldehyde and TBSCI (n = 1) or M-(CH2)n-OTBS (n = 2-4) to give
rise to
compounds 88.
Scheme 15
O O O O
1. NaHMDS, THF
R5'~~OMe R5 OMe
2. CH,O duench;
87 or Q-(CH2)n-OTBS
~~ OTBS
88,n>1
O O
(for n = 1) R5 ~ ~OMe
TBS-Cl, OTBS
pyridine,
CHzCIz 88, n =1
I O COnlpOl111dS Of F011T1Lila I wherein Q is taken from Q-17 are prepared
according to the
synthetic routes shown in Schemes 16.1 and 16.2, and starts with the BOC-
protected
hydrazine 13, which is converted to the 1,2-disubstituted hydrazine 89 by a
reductive
alkylation with a glyoxal derivative mediated by sodium cyanoborohydride and
acidic
workup. Condensation of 89 with diethyl malonate in benzene under reflex
yields the
heterocycle 90. Oxidation with Nz04 in benzene (see Cardillo, Merlini and
Boeri Gazz.
Cl~inz. Ital. (1966) 9:8) to the nitromalonohydrazide 91 and further treatment
with PZOS in
benzene (see: Cardillo,G. et al, Gazz. Clzini.Ital. (1966) 9:973-985) yields
the tricarbonyl 92.
Alternatively, treatment of 90 with Brederick's reagent (t-BuOCH(N(Me2)Z,
gives rise to 93,
which is subjected to ozonolysis, with a DMS and methanol workup, to afford
the protected
tricarbonyl 92. Compound 92 is readily deprotected by the action of CsF in THF
to yield the
primary alcohol 94. Alcohol 94 is optionally converted into the primary amine
95 by a
sequence involving tosylate formation, azide displacement, and hydrogenation.
51
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Scheme 16.1
O Noz
I)~OTBS Et0 OEt O 0
0 0
BOC NaCNBI-1;, CH3CN OTBS~ \\~~ NzOg
R4N-NHz --. R4HN-N~ O O RQN-N\ ~ R4N-N
13 Z) H+ H S9 ~p ~OTBS ~pTBS
_91
t-Bn0-CH(NMe)z)z
FzO;
(Me)zN
Me0 OMe
O I O oxonolysis O O
MeOH/DMS \~~~/~~
RqN N~OTBS RqN-N
~OTBS
93 92
CsF, THF
Me0 OMe t) tosyl chloride, base
Me0 OMe
O O _ 2) NaN
O~\~' i~0
RqN-N 3) hydrogenation
~NHa RQN-N
OOH
_94
Reaction of 94 with (hetero)aryl halide 26, wherein M is iodo, bromo, or
chloro,
under copper(I) catalysis affords compounds I-96. Optional deprotection of the
di-methyl
ketal with aqueous acid gives rise t0 C0111pOLl1ldS Of FOr111li1a I-98. By
analogy, reaction of
5 amine 95 with 26 under palladium(0) catalysis affords compounds of Formula I-
97. Optional
deprotection of the di-methyl ketal with aqueous acid gives rise to compounds
of Formula I-
99.
52
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Scheme 16.2
HO OH
Me0 OMe (RsO2C-(NH)P]q-E'M O~\~i~0
H , H., \O
O O Cu(I) base R4N NCO E N ~E [(NH)pCOzRs]q
~ -((NH)pCOzRs]q ~O
RdN-N '
OOH I-96 I-~8
94
HO OH
.M Me0 OMe
Me0 OMe [R602C-(NH)p]q-E
O O 26 O's~~~~ H+, H.,O O O
R4N-N Pd(0),base RQN N~NH~E ((NH)pOOZRsI4 R4N N~NHiE-((NH)pCOzRslq
~NH~
1-97 I-
9i --
Compounds of Formula I wherein Q is taken from Q-17 are also prepared
according
to the synthetic route shown in Scheme 16.3. Deprotonation of 4,4-dimethyl-3,5-
dioxo-
pyrazolidine (95a, prepared according to the method described in Zinner and
Boese, D.
PIZarniazie 1970, 25(S-6), 309-12 and Bausch, M. J.et.al J. Ong. Claeni. 1991,
56(19), 5643)
with NaHIDMF or NaH/DMF and with NaH/DMF or NaH/DMF and its subsequent
displacement of M, wherein M is a suitable leaving group such as chloride,
bromide or
iodide yields I-99a.
Scheme 16.3
0
O_~\ ~O HN
HN-NH
[R602C-( NH)Plq-E-Y'CI [RsO~C-(NH)Plq-E-Y ~ N
95a O
8a
I-99a
Compounds of Formula I wherein Q is taken from Q-18 are prepared as shown in
Schemes 17.1 and 17.2. Aminoesters 100 are subjected to reductive alkylation
conditions to
give rise to intermediates 101. Condensation of amines 101 with carboxylic
acids using an
acid activating reagent such as dicyclohexylcarbodiimide
(DCC)/hydroxybenzotriazole
(HOBt) affords intermediate amides 102. Cyclization of amides 102 to tetramic
acids 104 is
. mediated by Amberlyst A-26 hydroxide resin after trapping of the i~z situ
generated alkoxide
103 and submitting 103 to an acetic acid-mediated resin-release.
53
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
R5 .
Scheme 17.1
~O
O R~CHO O RSCHZCO.,H O
_ ~NH~R4 R O~N~R4 .
RsO~NHz NaBH(OAc)3 Rs0 ~M'~ DCC, HOBt s M 102
100 ~'' 101
R5 R5 O
NMe3+ OH' ~ ~ O H+, R40H
-NMe3 O ~ R40
N ~ Ra ~~/ Ra
M~ M
_104
M' is t-BuOCHz-, BOCNH(CHZ)3-
M is HOCHZ-; HZN-(CHZ)a ;
BOCNH(CHZ)4-, HC=C-CHI 103
HZN-(CH2)4-; HC=C-CHZ
Scheme 17.2 illustrates the synthetic sequences for converting intermediates
104 to
compounds of Formula I. Reaction of alcohol 104.1 with aryl or heteroaryl
halide 26 (Q =
halogen) under copper(I) catalysis gives rise to compounds of Formula I-105.1.
Reaction of
amines 104.2 and 104.3 with 26 under Buchwald palladium(0) catalyzed amination
conditions affords compounds of Formulae I-105.2 and I-105.3. Reaction of
acetylene 104.4
with 26 under Sonogashira coupling conditions affords compounds of Formula I-
105.4.
Compounds I-105.4 may optionally be reduced to the corresponding saturated
analogs I-
105.5 by standard hydrogenation.
54
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Scheme 17.2
Rs O Rs 0
fRsOzC-(NH)Plq-E~ M
Ra0 I 26 Ra0 I
N~Ra N~Ra
OH Cu(I), base O~E_~(NH)pCO2Rs]q
104.1 1-105.1
R5
Rs 0
O IRsOzC-(NH)Plq-E~M '
R40 ~ ~ 26 Ra0 N R
N~Ra ~ a
Pd(0), base ( ) n
( )~~ NH~E-f(NH)PCOzRslq
NHz
1-105.2, n = 3
104.2, n = 3 1-105.3, n = 4
104.3, n = 4
R IRsO2C-(NH)P1q-E~M Rs 0 Rs Q
s
O 26 / hydrogenation R O
Ra0 ~ a ~ R4
Ra0 N R PdClz(Pb3P)z, CuI N~Ra
~ 4
Base
(Sonogashira Coupling) E- NH CO R q(Rs02Cp(HN)]-E
f( )P z slq
104.4 I-105.4 I-105.5
Compounds of Fornmla I wherein Q is taken fi0111 Q-19, Q-20, or Q-21 are
prepared
as illustrated in Scheme 18. Commercially available Kemp's acid 106 is
converted to its
anhydride 107 using a dehydrating reagent, preferably di-isopropylcarbodiimide
(DIC) or 1-
(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC). Reaction of 107 with an
amines
R4NH2 affords the intermediate amides which are cyclized to the imides 108 by
reaction with
DIC or EDC. Alternatively, 107 is reacted with amines 8 to afford amides of
Formula I-110.
Amides I-110 may optionally be further reacted with DIC or EDC to give rise to
compounds
of Formula I-111. Acid 108 is further reacted with amines 8 to give compounds
of Formula
I-109.
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Scheme 18
co2H p o R4 0
COzH DIC or EDC HO~O O 1) R4NHz HO~ N
CO ~ ~'z
H3C CH3 ~ O _ O
HsC CHa H3C CHa
H3C 2) DIC or EDC
H3C H3C 108
106 107 -
[R60zC-(NH)Plq HN O R4 O
[R602C-(NH)Pl4-EwNH ~ N
2
8 O
HsC CHs
108
'- DIC, HOAt
I-109 H3C
[R602C-(NH)P1G-EwNHz [RsOzC (NH)P19- \ [RsOzC-(NH)Plq-i O
NH O COZH DIC or EDC HO~O N
z
107 8 HsC CO CH3 ~ HsC O CH3
H3C H3C
I-110 1-111
Compounds of Formula I wherein Q is taken from Q-22 or Q-23 are prepared as
shown in Schemes 19.1 through 19.3. Preparation of intermediates 113 and 114
are prepared
as shown in Scheme 19.1 fiom di-halo(hetero)aryls 112, wherein MZ is a more
robust leaving
group than M,. Reaction of 112 with amines 37 (Z = NH) either thermally in the
presence of
base or by palladium(0) catalysis in the presence of base and phosphine ligand
affords
compounds 113. Alternatively, reaction of 112 with alcohols 37 (X = O) either
thermally in
the presence of base or by copper(I) catalysis in the presence of base affords
CO111pOLIIIdS 114.
56
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Scheme 19.1
[R602C-(NH)PIq-E-Y~
M ZH M1
1
w~w 3?,z=Ni-~ w~w
M~~ Base [RsCzC-(NH)p]q-E-YEN
H
112 113.
M~
[R60~C-(N H)P]q-E-YyZH w ~w
37, Z = O [R602C-(NH)P]q-E-Y~C ~ /
112
Base 114
Scheme 19.2 illustrates the conversion of intermediates 113 into compounds of
Formula I-115, I-118, or 117. Treatment of 113 with aqueous copper oxide or an
alkaline
hydroxide affords compounds of Formula I-115. Alternatively, treatment of 113
with t
butylmercaptan under copper(I) catalysis in the presence of ethylene glycol
and potassium
carbonate gives rise to 116 (see F.Y. ICwong and S. L. Buchwald, OJgc~raic
Letters (2002)
4:3517. Treatment of the t-butyl sulfide 116 with acid affords the desired
thiols of Fornula
I-118. Alterlatively, 113 may be treated with excess ammonia under pressurized
conditions
to afford compound 117.
57
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
M~
Scheme 19.2 ~
W~W
[RsOzC-(NH)Plq-E-YwN
H
113
aq Cu0 t-BUSH excess NH3,
or CuI, ICZC03 base
KOH ethylene glycol
OH ~ StBu ~ NHz
W~W W~W W~W
[R602C-(NH)PI9-E-YvN ( / (R6p2C_(NH)Pl9-E-YwN . ~ / [R6o~C (NH)Plq-E-YwN ~ /
H H H
I-115 716 117
H+
SH
W' \ W
[R602C-(N H )Pl9-E-Yw N
H
1-118
Scheme 19.3 illustrates the conversion of intermediate 114 into compounds of
Formula I-119, I-122, and 121, by analogy to the sequence described in Scheme
19.2.
58
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
M~
Scheme 19.3 W ~ W
R602C-(NH)P]q-E-Y~O I /
114
aq Cu0 t-BUSH excess
NH3,
or CuI base
I<OH ICZC03
ethylene
glycol
OH StBu NHz
W~W W~W W~W
RsOzC-(NH)P]q-E-Yw0 R60zC-(NH)P]q-E-Yw0 R60zC-(NH)Plq-E-Yw0
I / I / I /
1-119 120 121
H~
SH
W~W
RsOzC-(NH)P1q-E-Y~O
1-122
Compounds of Formula I wherein Q is taken from Q-24, Q-25, or Q-2G are
prepared
as shown in Scheme 20. Reaction of CO111pOLl1ldS I-115 or I=119 with
chlorosulfonylisocyanate, followed by ira situ reaction with amines HN(R4)~
gives rise to
compounds of Fornmlae I-123 or I-124. Reaction of compounds I-118 or I-122
with a
peracid, preferably peracetic acid or trifluoroperacetic acid, affords
compounds of Formula I-
125 or I-126. Reaction of compounds 117 or 121 with chlorosulfonylisocyanate,
followed by
ira sitz~ reaction with amines HN(R4)z or alcohols R40H, affords compounds of
Fornmlae I-
127, I-128, I-129, or I-130.
59
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Scheme 20
OH SH NH2
W~W W~N/ W~W
Rs02C-(NH)Plq-E-Y~Z ~ )P]q-E-Y~Z RsOzC-(NH)P1q-E-Y~Z
/ RsOzC-(NH ~ /
1-115, Z = NH I-118, Z = 117, Z = NH
119,2= NH O 121,2=O
1- O 1-122,2= -
1) Chlorosulfonyl- . 1) Chlorosulfouyl-
isocyauate isocyanate
Peracetic acid
HN(RQ)~ 2) HN(R~),
2) or R40H
_
O O O
R
O~ OSO ~R4 S03H HN~H\S/N~ a
H Ra WJ~W W~W Ra
R602C-(NH)Plq-E-Y~Z ~ / R60~C-(NH)Plq-E-Y. I /
Z
RsOzC-(NH)Plq- Y\Z / I-127, Z=NH
1-123, Z = NH I-125, Z = NH 1-128, Z = O
I-124, Z = O I-126, Z = O
O S o/ Ra
HN H
W~W
RsO2C-(NH)P]q-E-Y~Z I
1-129, Z = NH
1-130,2=O
Compounds of Formula I wherein Q is taken from Q-27 are prepared as
illustrated in
Scheme 21. Reductive alkylation of thiomorpholine with aldehydes 131 affords
benzylic
amines 132, which are then subjected to peracid oxidation to give rise to the
thiomoipholine
sulfones -133 (see C. R. Johnson et eel, Tetnalzedr-ooa (1969) 25: 5649).
Intermediates 133 are
reacted with amines 8 (Z = NHZ) under Buchwald palladium-catalyzed amination
conditions
to give rise to compounds of Formula I-134. Alternatively, compounds 133 are
reacted with
alcohols 8 (Z = OH) under Buchwald copper(I) catalyzed conditions to afford
compounds of
Formula I-135. Alternatively, intermediates 133 are reacted with alkenes under
palladium(0)-catalyzed Heck reaction conditions to give compounds of Formula I-
136.
Compounds I-1.36 are optionally reduced to the corresponding saturated analogs
I-137 by
standard hydrogenation conditions or by the action of diimide.
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
~O
Cg' N J N J -o
Scheme 21 CH ]O
N peracid
\ H \ oxidation \
NaBH(OAc)3 ~ ~/ ~ i/
/
131 132
M 133 ~S O O
o NJ
[R602C-(NH)PIG-E-YwNH~
O [Rs02C-(NH)Plq-E~ \
8
133 133 pd(0)~ ph3P, ~ ~/
Pd(0), phosphine, \ base
base ~ , / [RsOzO-(NH)p]q-E~ I-13G
[R602C-(NH)p]q-E-Y-NH
1-134
-- reduction
(hydrogenation or
diitnide)
[R602C-(NH)Plq-E-YwOH S ~ O
O //
133 8 N J N J 'O
Gu(I), base
\
[R602C-(NH)Plq-E-Y-O ~ i/
I-135 (R602C-(NH)plq-E~ I-137
CO111pOLIndS Of FolTllllla I whereinQ is taken from Q-27 are also prepared as
illustrated
in Scheme 21.1. Aldehyde 8c is reductively aminated with ammonia, and the
resultant amine
condensed with divinyl sulphone to yield I-134. Intermediate 134a is also
available by
reduction of amide 8d under a variety of standard conditions.
Scheme 21.1
O
NH3
[R602C-(NH)Plq-E-Y~H [RsOzC-(NH)Plq-E-Y~NHz
NaCHBH3 134a
8c
Amide reduction 0~,~
i.e. LAH 5
0
[R602C-(N H )p]q-E-Y ~ N HZ
[R602C-(NH)p]q-E-YEN
8d SO
1~ 134
Gl
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
More generally, amines 134c are available via the reduction of amides 134b as
shown
in Scheme 21.2 The moipholine amide analogues 134d and moipholine analogues
134e are
also available as shown in Scheme 21.2
Scheme 21.2
0 0
R~R2NH
[R60zC-(NH)p]q-E-Y OH ~ [R602C-(NH)p]q-E-Y NR~R~
DIC coupling
8e 134b
H
CN, Amide reduction
pJ DIC coupling i.e. LAH
O
~ IRsCzC-(N
H)p]q-E-Y
~NR~ R2
[R60~C-(NH
)p]q-E-Y
N
~
134c
134d
Amide reduction
i.e. LAH
[Rso2C-(NH)p]q-E-Y~
134e
COIIIpOL111dS Of FOl-lllllla I wherein Q is taken from Q-28 or Q-29 are
prepared
according to the sequences illustrated in Scheme 22. Readily available amides
138 are
reacted with chlorosulfonylisocyanate to give intermediates 140, which are
reacted i~z sitar
with amines HN(R4)Z or alcohols R40H to afford compounds of Formulae I-141 or
I-142,
respectively. Alternatively, amides 138 are reacted with sulfonyl chlorides to
give
compounds of Formula I-139.
62
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
H N ,CI
N-
Scheme 22 coNH2 O O 0 ~~Ra)z ar
CISOZ-N=C=O ~ R40H
base
[R~02C-(NH )Plq-E ~ Y [R602C-(NH)Plq-E ~ Y
138 140
Ra
Ra
I H O
,N.S N~Ra N~N~Ss
O ~( II ~ 0 O II O
O O O O
[RsO~C-(NH)PlQ-ELY /
[R602C-(NH)Plq-E~ Y
T_I 41 T-142
Rs
O HN-Sc0
O
R9SOZCl
base
_138
[R602C-(NH)P14-E ~ Y
1-139
Compounds of Formula I wherein Q is taken from Q-30 are prepared as shown in
Scheme 23. Readily available N-BOC anhydride 143 (see S. Chen et al, J: Ani.
C7Zem. Soc.
(1996) 118:2567) is reacted with amines HN(R~)z or alcohols R~OH to afford
acids 144 or
145, respectively. Intermediates 144 or 145 are further reacted with amines
HN(R4)z in the
presence of an acid-activating reagent, preferably PyBOP and di-
isopropylethylamine, to give
diamides -146 or ester-amides 147. Intermediate 145 is converted to the
diesters 148 by
reaction with an alkyl iodide in the presence of base, preferably potassium
carbonate.
Intermediates 146-148 are treated with HCl/dioxane to give the secondary
amines 149-151,
which are then condensed with acids 152 in the presence of PyBOP and di-
isopropylethylamine to glVe CO111pOL111dS Of F017mllla I-153.
63
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
N~Ra)z O
Scheme 23 Ho2C o 2(Ra)NC ' N(Ra)2
O 'N N
O~O~ HN(Ra)z 1)PyBOP,i-PrzNEt 146
BOC _144 BoC
N or R~OH O ORs 2) HN(Ra)z O OR
BOC HOz ~ ~ 2(Ra)NC
143 N 145 N 147
BOC - BOC
R6I, base R O C O~ORs
s z ~NJ _148
BOC
HCI, dioxane
p-X1
O\, N(Ra)z
2(Ra)NC ~'(~
N O OH
O ~ PyBOP, i-PrzNEt N 149
CO-Xz H
O ORs
2(Ra)NCO
[R602C-(NH)PlG-E~ ~// [RsOzC-(NH)P1q-E~Y N
I-153 H 150
_152
Xi, Xz are N(Ra)z ORs
X~ is N(Ra)z, X2 is OR6 Rs02~
X X are OR ~N~151
t~ z s
Fi
Compounds of Formula I wherein Q is taken from Q-31 or Q-32 are prepared
according to the sequences illustrated in Scheme 24. Treatment of readily
available
sulfenamides 154 with amines 37 (Z = NH), alcohols 37 (Z = O), or alkenes 37
(Z = -
CH=CHz), gives rise t0 COnIpOL111dS Of FOnllllla I-155. Treatment of
sulfenamides I-155 with
iodosobenzene in the presence of alcohols RGOH gives rise to the
sulfonimidates of Formula
I-157 (see~D. Leca et al, Organic Letters (2002) 4:4093). Alternatively,
compounds I-155 (Z
_ -CH=CH) may be optionally reduced to the sahirated analogs I-15G (Z = CHZ-
CHZ-), which
are converted to the corresponding sulfonimidates I-157.
Treatment of readily available sulfonylchlorides 154.1 with amines HN(R4)2 and
base
gives rise to compounds of Formula I-154.2.
G4
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Scheme 24
[R60zC-(NH)PIq-E-Y~ZH
37, Z = NH
Pd(0), phosphine, NH
base
O'S~NH2 ~ORs
PhI=O \
SONHz
[R602C-(NN)p]q-E-Y~ \
ZH
37, Z= O I/i/ M ~ N [RspzC_(NH)Plq-E-Y-Z
M / Cu(I), base [RsOzC-(NH)p]q-D-E-Y-Z
I-157
_154 I-155 PhI=0
[RsOzC-(NH)P]q-E-Yy
R60H,
ZH
MeCN
37, Z = CH=CH2
Pd(0), phosphine, O~~S~NHz
base
Z = CH=CH- ~ i/
i
Hydrogenation [RsOzC-(NH)p]q-E-Y-(CHz)z
I-156
S02NHz S02N(RQ)z
\ HN(R4)z \
~~/ -~ li/
i i
[RsOzC-(NH)p]q-E-Y [R602C-(NH)p]q-E-Y
154.1 I-154.2
Compounds of Formula I wherein Q is taken from Q-33 are prepared as shown in
Scheme 25. Readily available nitrites 158 are reacted with amines 37 (Z = NH),
alcohols 37
(Z = O), or alkenes 37 (Z = -CH=CH2) to afford compounds of Formula I-159.
Compounds
I-159 (wherein Z = CH=CH-) are optionally reduced to their saturated analogs I-
160 by
standard catalytic hydrogenation conditions. Treatment of compounds I-159 or I-
160 with a
metal azide (preferably sodium azide or zinc azide) gives rise to tetrazoles
of Formula I-161.
65
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Scheme 25
[RsOzC-(NH)P1q-E-YwZH
37, Z=NH
Pd(~), phosphine' N N
base N NH
CN
CN [RspzC_(NH)P]q-E-Yw ~3 ~ \
ZH ( ~i/
\ 3T Z - O /~/ [RspzC_(NH)Plq-E-Y-Z
[RspzC-(NH)P]q-E-Y--Z
i Cu(I), base I'I''~ I-161
M
158
[RspzC-(NH)P]q-E-YwZH MNj
37, Z = C H=C H 2
CN
Pd(~)> pbospbine, \
base Hydrogenation
//
i
[R602C-(NH)p]q-E-Y-(CHz)z
I-160
Compounds of Formula I wherein Q is taken from Q-34 are prepared as shown in
Scheme 26. Readily available esters 162 are reacted with amines 37 (Z = NH),
alcohols 37
(Z = O), or alkenes 37 (Z = -CH-CHZ) to afford compounds of Foinzula I-163.
Compounds
I-163 (wherein Z is -CH=CH-) are optionally converted to the sahmated analogs
I-164 by
standard hydrogenation conditions. CO111pOtI1ldS I-163 or I-164 are converted
to the desired
phosphonates I-165 by an Arbuzov reaction sequence involving reduction of the
esters to
~benzy]ic alcohols, conversion of the alcohols to the benzylic bromides, and
treatment of the
bromides with a tri-alkylphosphite. Optionally, phosphonates I-165 are
converted to the
fluorinated analogs I-166 by treatment with diethylaminosulfiu trifluoride
(DAST).
66
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Scheme 26
CO~Rs
Z = CH=CH-
i/
Hydrogenation [RsOzC-(NH)P19-E-Y-(CHz)2
[RsOaC-(NH)Pl4-E-YwZH I-164
37, Z=NH 1) reduction to alcohol
(LiBH4)
Pd(0), phosphine, 2) CBr4, Ph3P
base 3) P(ORB)3
CO2Rs ORs
COzRs I) reduction to alcohol
s
[R602C-(NH)P14-E-Y~ ~ ~ (LiBHd) P-OR
37 Z - O ZH ~/
[RsOzC-(NH)P19-E-Y-Z 2) CBr4, Ph 3P
Cu(I), base 3) P(ORs)3
i/
162
_1-163 [RsOaC-(NH)P)9-E-Y-Z
[R60zC-(NH)Pl9-E-YyZH
I-165
37, Z = CH=CH2
DAST
Pd(0), phosphine,
base
[R60~C-(N
I-166
Compounds of Formula I wherein Q is taken from Q-34 are also prepared as
illustrated in Scheme 26.1. Intermediate 8a, wherein M is a suitable leaving
group such as
chloride, bromide or iodide, is >:efluxed with triethyl phosphite and the
resulting phosphoryl
intermediate saponified under mild conditions to yield I-165.
Scheme 26.1
O
[R60~C-(NH)p]q-E-Y~M 1. P(OEt)3 ~~\ OH
[R60zC-(NH)p]q-E-Y pH
$a 2. saponification I-165
C0111pOLI1IdS Of FOr111tt1a I wherein Q is taken front Q-35 are prepared
according to
Scheme 27. Readily available acid chlorides 167 are reacted with oxazolidones
in the
presence of base to afford the N-acyl oxazolidinones 168. Intermediate 168 are
reacted with
amines 37 (Z = NH), alcohols 37 (Z = O), or alkenes 37 '(Z = -CH=CHZ) to
afford the N-acyl
G7
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
oxazolidinones of Formula I-169. Compounds I-169 (wherein Z is -CH=CH-) are
optionally
converted to the saturated analogs I-170 under standard hydrogenation
conditions.
Scheme 27 [R60~C-(NH)p]q-E-Y~ZH
37, Z=NH
Pd(0), phosphine,I O
~O O N ,
0 base
HN O O N~ \'R4
COCI
\ R~ \ RQ [Rs0237( ~ )PIOE-YwZH
s/ base ~~~/ Cii(I), base [RsOzC-(NH)p]q-E-Y-Z
M M
I-169
1G7 168
[R6~~C-(NH)p]q-E-Y~
37, Z = CH=CHZ ZH hydrogenation
(Z = CH=CH)
Pd(0), phosphine,
base
O',
-O
O N
Ra
//
[R602C-(NH)P1q-E-Y-(CH2)2
I-170
Compounds of Formula I wherein Q is taken from Q-36 are prepared as
illustrated in
Schemes 28.1 and 28.2. Reductive alkylation of the t-butylsulfide substituted
piperazines
with the readily available aldehydes 131 gives rise to the benzylic
piperazines 171.
Internzediates 171 are reacted with amines 37 (Z = NH), alcohols 37 (Z = O),
or alkenes 37 (Z
- -CH=CHZ) to give compounds 172, 173, or 174, respectively. Optionally,
inteumediates
174 are converted to the saturated analogs 175 under standard hydrogenation
conditions. .
G8
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
CHO HNVN~St-Bu N~NZSt_BU
Scheme 28.1
. \ NaBH(OAc)3
~s ~/
M M
131 171
NU ~--St-Bu
[RsOzC-(NH)Plq-E-Yy H NUN~St-Bu
37, Z = NH 37, Z = CFI=CHZ \
171 \ 171
Pd(0), phosphine,' ~ ~/ Pd(0), phosphine, 17d
base [RsOzC-(NH)p]q-E-Y-NH 17~ base [RsOZC-(NH)p]q-E-
reduction
(hydrogenation or
diimide)
NON--~St-Bu
[RsOzC-(NH)Plq-E-Yw \
171 37, Z O ZH I ~/ N~/N-~St-Bu
Cu(I), base IRsOzC-(NH)p]q-E-Y-0
173
~/ 175
[R602C-(NH)p]q-E-Y
Scheme 28.2 illustrates the conversion of intermediate t-butylsulfides 172-175
to the
sulfonic acids, employing a two step process involving acid-catalyzed
deprotection of the t-
buiyl sulfide to the corresponding mercaptans, and subsequent peracid
oxidation '(preferably
with peracetic acid or trifluoroperacetic acid) of the mercaptans to the
desired sulfonic acids
of Formula I-176.
Scheme 28.2
_ n
N~/N~St-Bu ] H+ N~--~N~-SOsH
2) peracid oxidation [R6p2C-(NH)p]q-E-Y-Z /
[R60zC-(NH)p]q-E-Y-Z
172-175 1-176
I ~ Z = NH, O, CH=CH, CH2-CH2
In some instances a hybrid bcr-abl kinase inhibitor is prepared which also
contains an
ATP-pocket binding moiety or an allosteric pocket binding moiety R]-X-A-D. The
synthesis
69
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
of moieties R~-X-A-D are conducted as shown in Scheme 29. Readily available
intermediates 177, which contain a group M capable of oxidative addition to
palladium(0),
are reacted with amines 178 (X = NH) under Buchwald Pd(0) amination conditions
to afford
179. Alternatively amines or alcohols 178 (X = NH or O) are reacted thermally
with 177 in
the presence of base under nuclear aromatic substiW tion reaction conditions
to afford 179.
Alternatively, alcohols 178 (X = O) are reacted with with 177 under Buchwald
copper(I)
catalyzed conditions to afford 179. In cases where p = l, the carbamate of 179
is removed,
preferably under acidic conditions when R~ is t-butyl, to afford amines 180.
In cases where p
= 0, the esters 179 are converted to the acids 181 preferably under acidic
conditions when R~
is t-butyl.
Scheme 29
R~-XH
M-A-(NH)p-D-(NH)p'-CO~R6 »$ R~X-A-(NH)p-D-(NH)p'-C02R6
heat or
Pd(0) catalysis 179
H+ H+
R~X-A-(NH)p-D-NH2 R~X-A-(NH)p-D-CO~H
180 y 81
Another sequence for preparing amines or alcohols 180 is illustrated in Scheme
30.
Reaction of amines or alcohols 178 with nitro(hetero)arenes 182 wherein M is a
leaving
group, preferably M is fluoride, or M is a group capable of oxidative
insertion into
palladium(0), preferably M is bromo, chloro, or iodo, gives intermediates 183.
Reduction of
the nitro group under standard hydrogenation conditions or treatment with a
reducing metal,
such as stannous chloride, gives amines 180.
Scheme 30
R~-XH
reduction
M-A-(NH)p-D-N02 ~~g R~X-A-(NH)p-D-NO~ ~ R~X-A-(NH)p-D-NH2
~ 82 heat or ~ g3 l 80
Pd(0) catalysis -
70
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
In instances when hybrid bcr-abl kinase inhibitors are prepared, compounds of
Formula I-184 wherein q is 1 may be converted to amines I-185 (p = 1) or acids
I-186 (p = 0)
by analogy to the conditions described in Scheme 29. Compounds of Formula I-
184 are
prepared as illustrated in previous schemes l.l, 2.1, 2.2, 3, 4, 5, 6, 7.1,
7.2, $, 9, 10, 12, 14,
16.2, 17.2, 18, 19.1, 19.2, 19.3, 20, 21, 22, 23, 24, 25, 26, 27, or 28.2.
Scheme 31
E Q
[R602C-(NH)plq~ ~Yi
q=1
I-184
H+ H+
p=1 p=0
H N'E~Y~O HOzC~E\Y~Q
z
I-185 I-186
Compounds I-184 are taken from schemes 1.1, 2.1, 2.2, 3, 4, 5, 6, 7.1, 7.2, 8,
9, 10
12, 14, 16.2, 17.2, 18, 19.1, 19.2, 19.3, 20, 21, 22, 23, 24, 25, 26, 27, 28.2
The preparation of inhibitors of Fornula I which contain an amide linkage -CO-
NH-
connecting the oxyanion pocleet blndlng moieties and the Ri-X-A-D moieties are
shown in
Scheme 32. Treatment of acids 18.1 with an activating agelit, preferably PyBOP
in the
presence of di-iso-propylethylamine, and amines I-185 gives compounds of
Formula I.
Alternatively, retroamides of Formula I are fornied by treatment of acids I-
186 with PyBOP
in the presence of di-iso-propylethylamine and amines 1~0.
71
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Scheme 32
NHz~E~y~O + (R1X)m-A-(NH)p-D-COZH pygop~ i_pr2NEt (R~X)m-A-(NH)p~D~N~E/Y.O
IOI
Compounds 1-185 taken Compounds I81 taken
from scheme 31 from scheme 29 _Amides of F_or_m_u_la 1
(hybrid inhibitors, possessing oxyanion
pocket-binding moiety Q and moiety Rt-X-A-(NH)p-D)
E Q (R~X)m-A-(NH)PwD~N~EwYia
HOZC~ ~Y~ + (R1X)m-A-(NH)p-D-NHZ pyBop, i-pr~NEt (IO
Compounds I-186 taken Compounds 18t1 taken from
from scheme 31 schemes 29 or 30 Retroamides of Fornml~ I
(hybrid inhibitors, possessing oxyanion
pocket-binding moiety Q and moiety R~-X-A-(NH)p-D)
The preparation of inhibitors of Formula I which contain an urea linkage NH-CO-
NH- connecting the oxyanion pocket binding moieties and R1-X-A-D moieties are
shown in
Scheme 33, Treatment of amines I-185 with p-nitrophenyl chloroformate and base
affords
carbamates 187. Reaction of 187 with amines 180 gives ureas of Formula I.
Scheme 33
NH ~E~Y~O P-nitrophenyl chloroformate O HN' 'y (R~X)m-A-(NH)p-D-NHZ
base Compounds 180 taken from
E R
Compounds I-185 taken p2N ~ O schemes 29 or 30
from scheme 31 187
O
A H DWH~HiEwYiQ
(RiX)m/
P
_F_or_m_u_Ia 1
(hybrid inhibitors, possessing oxyanion
Pocket-binding moiety D and moiety Rt-X-A-(NH)p-B)
72
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Alternatively, inhibitors of Fonnitla I which contain an urea linkage NH-CO-NH-
connecting the oxyanion pocket binding moieties and the Rl-X-A-D moieties are
prepared as
shown in Scheme 34. Treahment of amines 180 with p-nitrophenyl chlorofonmate
and base
affords carbamates 188. Reaction of 188 with amines I-185 gives areas of
Formula I.
Scheme 34
p ~ E~ ,Q
(R~X)m-A-(NH)p-D-NHz p-nitrophenyl chloroformate \ p~HN.E~N~A~(XR~)m NH Y
Compounds 180 taken from base
schemes 29 or 30 D N ~ / ~ Compounds 1-185 taken
a from scheme 31
188
p
Q~Y E.N HN.D~(N~A~(XRym
O
_F_or_m_ul_a 1
(hybrid inhibitors, possessing oxyanion
pocket-binding moiety Q and moiety Ri-X-A-(NH)p-D)
V. Biological assessment of abl and ber-abl kinase inhibitor.
A continuous spectrophotometric kinase assay is used, wherein the production
of
adenosine diphosphate is coupled to the oxidation of NADH and measured as a
reduction in
absorbance at 340nM. For details see: Barker, S.C. et al, Bioc7aenZistjy
(1995) 34:14843; and
Schindler, T. et al, Science (2000) 289:1938.
Abl kinase assay
Activity of nonphosphorylated Abl kinase was determined by following the
production of ADP from the kinase reaction through coupling with the pyruvate
kinase/lactate dehydrogenase system (e.g., Schindler, et al. Science (2000)
2~9, 1938-1942).
In this assay, the oxidation of NADH (thus the decrease at A34on~,) was
continuous measured
spectrophometrically: The reaction mixture (200 yl) contained Abl kinase (3.7
nM. Abl-2
from decode), peptide substrate (EAIYAAPFAKKK, 0.5 mM), ATP (0.5 mM), MgCI~,
(5
mM), pyruvate kinase (1G units), lactate dehydrogenase (26 units), phosphoenol
pyruvate (1
73
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
111M), and NADH (0.28 mM) in 100 mM Tris buffer, pH 7.5. The reaction was
initiated by
adding ATP. The absorption at 340 nm was monitored continuously for 3 to 4
hours at 30 °C
on Polarstar Optima plate reader (BMG). Under these conditions, a turn over
number (k°at) of
1.4 s-~ was obtained for the preparation of Abl kinase, which is similar to
that (1.7 s 1)
reported for the nonphosphorylated enzyme (Brasher and Van Etten, JBC (2000)
275, 35631-
35637). No autophosphorylation of Abl was observed under these conditions
since the rate is
constant throughout the entire reaction time and presumably because the
concentration of the
enzyme used is below the critical level (~ 10 nM) needed for the
autophosphorylation
(Brasher and Van Etten, JBC (2000) 275, 35631-35637). These results ensure
what we
monitored was the activity of nonphosphorylated Abl kinase.
Percentage of inhibition in the presence of an inhibitor was obtained by
comparison of
reaction rate (or slope) with that of a control. ICSO value was calculated
from a series of
inhibition values determined at a range of concentrations of the inhibitor
using Prism. The
IC50 values for Gleveec and PD 180970 were found to be 76 and 24 nM,
respectively, which
are close to that reported (Schindler, et al. Science (2000) 289, 1938-1942).
Inhi
Example 10 uM IC50,
# uM
1 10
2 9
3 15
4 24
5 9
6 13
7 9
8 20
9 42
10 16
11 19
12 52
13 31
15 7
16 9
17 18
18 70 3
19 75 4
77 3
21 12
23 10
29 12
35 1
_
36 20 -.
-
37 ~10 ~
74
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
38 21
39 13
40 16
42 33
43 28
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
EXAMPLES
The following examples set forth preferred methods in accordance with the
invention.
It is to be understood, however, that these examples are provided by way of
illustration and
nothing therein should be taken as a limitation upon the overall scope of the
invention.
Reagents 6-methyl-N1-(4-phenylpyrimidin-2-yl)benzene-1,3-diamine hydrochloride
(Reagent AA) and 6-methyl-NI-(4-phenylpyrimidin-2-yl)benzene-1,3-diamine
hydrochloride
(Reagent BB), N-Methyl-2-(methylcarbamoylmethyl-amino)-acetamide (Reagent CC),
terephthalic acid monobenzyl ester (Reagent DD), 4-formyl-benzoic acid methyl
ester
(Reagent EE), 4-methyl-N-3-(4-(3-pyridyl)-pyrimidin-2-yl)-benzene-1,3-diamine
hydrochloride (Reagent FF), [Boc-sulfamide] aminoester (Reagent GG) and 6-
methyl-Nl-(4-
rnorpholinopyrimidin-2-yl)benzene-1,3-diamine hydrochloride (Reagent HH) were
synthesized according to literature procedures.
REAGENT AA
N\ /N \ NHZ,HCI
I s'TN I /
I\
To a solution of N (3-amino-4-methyl-phenyl)acetamide (Sg, 25 mrnol) in DMF (5
ml) was added 2-chloro-4-phenyl-pyrirnidine (4g, 35 mmol) and KI (O.Sg, 3
rnmol), which
was stirred at 100 °C overnight, cooled to 10° C and added to
H20 (100mL). The resulting
mixture was extracted with CHZC12 (2x100 mL), the combined organic layers
dried (Na2S04)
and concentrated in vacuo. The residue was dissolved in conc. HCl (10 mL),
stirred at 80°C
for 2h and concentrated in vacuo to yield 6-methyl-Nl-(4-phenylpyrimidin-2-
yl)benzene-1,3-
diamine hydrochloride (4.Sg, 65%). 'H NMR (CDC13): 7.96 (m, 2H), 7.'50-7.47
(m, 1H),
7.47-7.41 (rn, SH), 7.26 (111, 2H), 2.21(s, 3H); MS (ESI) n~/e: 277 (M++1)
REAGENT BB
NYN \ NHzHCi
I ~ IN
To a solution of N-(3-amino-4-methyl-phenyl) acetamide (Sg, 25 mmol) in DMF (5
mL) was added 2-chloro-pyrimidine (3.8g, 33 mmol) and KI (O.Sg), which was
stirred at 100
°C overnight, cooled to 10° C and added to HZO (100mL). The
resulting mixture was
76
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
extracted with CHZC12 (2x100 mL), the combined organic layers dried (Na2S04)
and
concentrated in vacuo. The residue was dissolved in conc. HCl (10 mL), stirred
at 80°C for
2h and concentrated in vacuo to yield 6-methyl-Nl-(4-phenylpyrimidin-2-
yl)benzene-1,3-
diamine hydrochloride (3.75g, 75%). 1H NMR (CDC13): 8.36 (dd, J = 15.2 & 4.8
Hz, 2H),
7.46 (d, J = 2.4 Hz, 1H), 6.97 (d, J = 8.0 Hz, 1H), 7.26 (s, 1H), 6.67 (t, J =
4.8 Hz, 1H), 6.39
(dd, J = 8.0, 2.4, Hz, 1H), 2.20 (s, 3H); MS (ESI) m/e:~ 201 (M++1).
REAGENT CC
H
wN~N~Ni
H H
To a solution of benzyl amine (l6.Sg, 154 mmol) and ethyl bromoacetate (51.5
g, 308
nnnol) in ethanol (500 mL) was added KZC03 (127.5 g, 924 mmol). The mixture
was stirred
at RT for 3h, was filtered, washed with EtOH, concentrated in vacuo and
chromatographed to
yield benzyl-methoxycarbonylmethyl-amino)-acetic acid ethyl ester (29.02g,
67%). 1H NMR
CDCl3) 8 7.39-7.23 (m, SH), 4.16 (q, J= 7.2 Hz, 4H), 3.91 (s, 2H), 3.54 (s,
4H), 1.26 (t, J=
7.2 Hz, 6H); MS (ESI): m/e: 280 (M++H).
A solution of (benzyl-methoxycarbonylmethyl-amino)-acetic acid methyl ester
(7.70g, 27.6 mmol) in methylamine alcohol solution (25-30%, 50 mL) was heated
to 50°C in
a sealed tube for.3h, cooled to RT and concentrated in vacuo to yield 2-
(benzyl-
methylcarbamoylmethyl-amino)N-methyl-acetamide in quantitative yield (7.63 g).
1HNMR
(CDC13) b 7.35-7.28 (m, SH), 6.75(br s, 2H), 3.71(s, 2H), 3.20 (s, 4H),
2.81(d, J= 5.6 Hz,
6H)MS (ESI) m/e 250(M+H+)
The mixture of 2-(benzyl-methylcarbarnoylmethyl-amino)N-methyl-acetamid
(3.09g,
11.2 mmol) in MeOH (30 mL) was added 10% Pd/C (O.lSg). The mixture was stined
and
heated to 40°C under 40 psi HZ for l Oh, filtered and concentrated in
vacuo to yield N-methyl-
2-(methylcarbamoyhnethyl-amino)-acetamide in quantitative yield (1.76 g).
~HNMR(CDC13)
8 6.95(brs, 2H), 3.23(s, 4H), 2.79(d, J=4.8Hz, 6H), 2.25(brs, 1H); MS (ESI)
n~/e 160(M+H+)
REAGENT DD
O OH
O O
77
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
REAGENT EE
0 0~
I\
/
O H
REAGENT FF
H
N_"N \ NHa
I ,N I /
/
\ N
REAGENT HH
N_"N \ NH2, HCI
I ~N~ I /
Co~
To a solution of N (3-amino-4-methyl-phenyl) acetamide (Sg, 41 mmol) in DMF (5
ml) was added 4-(2-chloro-pyrimidin-4-yl)-morpholine (8.1g, 40 mmol) and KI
(O.Sg, 3
inmol), which was stined at 100 °C overnight, cooled to 10° C
and added to H20 (100mL).
The resulting mixture was extracted with CHZClz (2x100 mL), the combined
organic layers
dried (Na2S04).and concentrated in vacuo. The residue was dissolved in conc.
HCl (10 mL),
stirred at 80°C for 2h and concentrated in vacuo to yield 6-methyl-N1-
(4-
morpholinopyrimidin-2-yl)benzene-1,3-diamine hydrochloride (S.Og, 65%). 'H NMR
(DMSO-d6) : 8.00 (d, J= 7.2 Hz, 1H), 7.57 (brs, 1H), 7.36 (d, J= 8.4 Hz, 1H),
7.14 (dd, J =
8.4, 1.6 Hz, 1H), 6.65 (d, J= 7.2 Hz, 1H), 3.69 (s, 4H), 3.66 (s, 4H), 2.25
(s, 3H). MS (ESI)
m/e: 286 (M++1).
EXAMPLE A
0
o~ so II
\ ~rS.H~N~
MeO
O
To a stirred solution of chlorosulfonyl isocyanate (3g, 21 mmol) in of CHZC12
(50 mL)
at 0 °C was slowly added pyrrolidine (l.Sg, 21 mmol) while the reaction
temperature was
controlled between 0-5 °C. After being stirred for l.Sh, a solution of
4-Aminomethyl-benzoic
acid methyl ester hydrochloride (4.7 g, 23 mmol) and triethylamine (6.4g, 63
mmol) in .
78
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
CHZCIZ (120 mL) was slowly added while the reaction .temperature was
controlled between
0-5 °C. When the addition was completed, the reaction solution was
awarmed to RT, stirred
overnight, then poured into of 10% HCl (130 mL) saturated with NaCI. The
organic layer was
separated and the aqueous layer was extracted with Et20 (3x80 mL). The
combined organic
layers were dried (NaZS04) and concentrated in vacuo to yield the crude
product, which was
purified by column chromatography on a silica gel to yield pure pyrolidine
carboxamide, N-
[(4-carbomethoxybenzyl)amino]sulfonyl (3 g, 43% yield). 1H NMR (DMSO-d6) 87.70
(d, J
= 2.1 Hz, 2H), 7.28 (d, J = 2.1 Hz, 2H), 4.84 (s, 2H), 3.83 (s, 3H), 3:15 (m,
4H), 1.67 (111,
4H); MS (ESI) m/e: 342 (M++1).
EXAMPLE B
0
\ ~:~.~ ~N~
Ho a
0
A solution of Example A (60 mg, 0.18 mmol) in THF (10 mL) was added to 3N LiOH
(10 mL) at RT, stirred overnight, acidified with 1 N HCI, and extracted with
EtOAc. The
organic layer was dried (Na2SO4) and concentrated to yield pyrolidine
carboxamide, N-((4-
carboxybenzyl)amino]sulfonyl (40 mg, 70% yield). 1H NMR (DMSO-d6) 812.87 (s,
1H),
10.01 (s, 1H), 7.88 (d, J=2.0 Hz, 2H), 7.33 (d, J=2.0 Hz, 2H), 6.90 (m, 1H ),
4.28 (s, 2H),
3.28 (m, 4H), 1.75 (m, 4H); MS (ESI) m/e: 327 (M++1).
EXAMPLE 1
0
oso~
\ H~ .~ NV
N~b I \ b /
,N ~ O
I
To a solution of Reagent AA (14 mg, 0.048 mmol) in anhydrous DMF (1 mL) was
added Et3N (26 yL, 0.18 mmol) at RT. The reaction mixture was stirred for 5
min, followed
by addition of Example B (12 mg, 0.038 mmol), EDCI (14 mg, 0.055 mmol) and
HOBt (7.4
mg, 0.055 mml). The reaction mixture was stirred over night at RT. Removal of
solvent in
vacuo followed by preparative HPLC yielded pure Example 1 (16 mg, 76%). 1H NMR
(CD3OD) 8 8.32 (d, J = 5:6 Hz, 1 H), 8.24 (d, J = 7.2 Hz, 2H), 8.09 (d, J =
2.0 Hz, 1 H), 7.92
79
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
(d, J = 8.0 Hz, 2H), 7.60-7.40 (m, SH), 7.44 (d, J = 8.4 Hz, 2H), 7.36 (d, J =
8.4 Hz, 1H),
4.43 (s, 2H), 3.41 (m, 4H), 2.34 (s, 3H), 1.89 (m 4H); MS (ESI) m/e: 586
(M++1).
EXAMPLE 2
0
oso
\ N~ ~H~N
N~. N \ N ~ ~ H
/ O
The title compound was synthesized following the procedure for the preparation
of
Example 1, utilizing Example B and Reagent BB. 'H NMR (CD30D) ~ 8.46 (d, J=
5.2 Hz,
2H), 7.97 (dd, J = 8.0, 2.0 Hz, 1 H), 7.91 (d, J = 8.0 Hz, 2H), 7:50 (dd, . J
= 8.0, 2.0 Hz, 1 H),
7.44 (d, J = 8.0 Hz, 2H), 7.3 3 (d, J = 8.0 Hz, 1 H), 6.92 (t, J = 4.2 Hz, 1
H), 4.43 (s, 2H), 3 .41
(m, 4H), 2.28 (s, 3H), 1.89 (m, 4H); MS (ESI) m/e: 509 (M++1).
EXAMPLE C
0
\ ~ ~.s.N~
Me0
To a stirred solution of chlorosulfonyl isocyanate (3g, 21 mmol) in 50 mL of
CH~C12
(50 mL) at 0 °C was slowly added a solution of 4-aminomethyl-benzoic
acid methyl ester
hydrochloride (4.7g, 23 mmol) and triethylamine (6.4g, 63 mmol) in CHZC12 (120
mL) while
the reaction temperature was controlled between 0-5 °C. After being
stirred for l.Sh,
pyrrolidine (1.5 g, 21 mmol) was slowly added while the reaction temperature
was controlled
between 0-5 °C. When the addition was completed, the reaction solution
was allowed to
warm to RT, stirred overnight, then poured into of 10% HCl (130 mL) saturated
with NaCl.
The organic layer was separated and the aqueous layer was extracted with Et20
(3x80 mL).
The combined organic layers were dried (Na2S04) and concentrated to yield the
crude
product, which was purified by column chromatography on a silica gel to yield
pure Example
C (2.5 g, 35% yield). 'H NMR (DMSO-d6) 87.87 (d, J=2.1 Hz, 2 H), 7.28 (d,
J=2.1 Hz, 2 H),
4.89 (s, 2 H) 3.82 (s, 3 H), 3.15 (m, 4 H), 1.68 (m, 4 H); MS (ESI) n~/e: 342
{M++1).
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
EXAMPLE D
\ ~~pvS:N
HO I ti/
O
The title compound using synthesized following the procedure for Example B
utilizing Example C. 'H NMR (CD30D) 87.98 (d, J=2.0 Hz, 2 H), 7.38 (d, J=2.0
Hz, 2 H),
4.41 (s, 2 H), 3.39 (m, 4 H), 1.87 (m, 4 H); MS (ESI) m/e: 327 (M++1).
EXAMPLE 3
° °s,o
N N N I / H H N
I~N I/
I
The title compound was synthesized following the procedure for the preparation
of
Example 1 utilizing Example D and Reagent AA. 1H NMR (CD30D) 88.31 (m, 1H),
8.23 (d,
J = 2.1 Hz, 2H), 8.06 (s, 1 H), 7.81 (d, J = 2.1 Hz, 2H), 7.62 (m, 1 H), 7.54
(m, 4H), 7.43 (d, J
= 2.1 Hz, 2H), 7.37 (d, J= 2.1 Hz, 1H ), 4.43 (s, 2H), 3.40 (m, 4 H), 2.33 (s,
3H), 1.89 (m,
4H); MS (ESI) m/e: 586 (M++1).
EXAMPLE 4
° °s,o
N N N I / H~H/ \N~
I I/ o
The title compound was synthesized following the procedure of the preparation
of
Example 1 utilizing Example D and Reagent BB. IH NMR (CD30D) b8.45 (br s, 2H),
7.96
(d, J = 4.0 Hz, 1 H), 7.90 (d, J =8.0 Hz, 2H), 7.50 dd, J = 8.0, 2.0 Hz, 1 H),
7.62 (m, 1 H), 7.43
(d, J= 8.4 Hz, 2H), 7.29 (d, J= 8.4 Hz, 1H ), 6.87 (t, J= 4.8 Hz, 1H), 4.43
(s, 2H), 3.40 (m, 4
H), 2.27 (s, 3H), 1.89 (m, 4H); MS (ESI) n~/e: 510 (M++1).
EXAMPLE D
\ wN~O
Me0 I / ~O(
O
81
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
To a suspension of glycine ethyl ester hydrochloride (6.Og, 34 mmol) in
anhydrous
CHZCIz (34 mL) was added triethylarnine (3.4g, 34 mmol) followed by anhydrous
magnesium sulfate (12.28, 102 mmol) and Reagent EE (6.Og, 34 mmol). After
refluxing for
2h , the solid was filtered, washed with brine, dried (MgS04) and concentrated
in vacuo to
produce methyl 4-((E)-((t-butoxycarbonyl)methylimino)methyl)benzoate which was
used
without further purification (8.2g, 97% yield). IH NMR (CDC13) 8 8.30 (s, 1H),
8.07 (d, J=
8.4 Hz, 2H) 7.84 (d, J= 8.4 Hz, 2H) 4.34 (s, 2H) 3.91 (s, 3H) 1.49 (s, 9H).
EXAMPLE E
N 11 0
Me0 I ~ H O
To a solution of Example D (B.Sg, 30 mmol) in MeOH (80 mL) was slowly added
solid NaBH4 (3.42g, 90 mmol) while the reaction temperaW re was controlled
below 20 °C.
After stirring for 2h, the reaction was quenched with H20, extracted with
EtOAc (3x100 mL)
and the combined organic layers were washed with brine, dried (NaZS04),
concentrated in
vacuo. The residue was purified via flash column chromatography to yield
methyl 4-(((t-
butoxycarbonyl)methylamino)methyl)benzoate (6.SSg, 77% yield). 'H NMR (CDC13)
~ 7.98
(d, J = 8.4 Hz, 2H), 7.40 (d, J = 8.4 Hz, 2H), 3.90 (s, 3H,) 3.84 (s, 2H) 3.29
(s, 2H) 1.4G (s,
9H).
EXAMPLE F
w N~o
Me0 ~ O~NH
O
To a solution of Example E (5.1 g, 18 mmol) in THF (80 mL) was added KZC03
(4.2g,
mmol) and methyl-carbamic acid 4-nitro-phenyl ester (3.68, 18 mmol). After
being stirred
overnight, the resulting solid was filtered. After adding H20 and EtOAc to the
filtrate, the
25 organic layer was separated and the aqueous layer was extracted with EtOAc
(3x100 mL).
Tlie combined organic layers were washed with brine, dried (NaZS04),
concentrated in vacuo
and purified by flash chromatography to yield Example F (4.4g, 73%). 'H NMR
(CDC13)
8.01 (d, .I= 8.4 Hz, 2H) 7.35 (d, J= 8.4 Hz, 2H) 4.59 (m, 1H) 4.57 (s, 2H)
3.91 (s, 3H) 3.90
(s, 2H) 2.79 (d, J= 4.4 Hz, 3H) 1.43 (s, 9H).
82
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
EXAMPLE G
0
N N~'
MeO I /
O O
To a suspension of NaH (0.28g, 7 mmol) in THF (80 mL) at RT was slowly added a
solution of Example F (1.85g, 5.5 mmol) in THF (50 mL). After stirnng for 2h,
the resulting
solid was filtered. After adding water and EtOAc to the filtrate, the organic
layer was
separated and the aqueous layer was extracted with EtOAc (3x100 mL). The
combined
organic layers were washed with brine, dried (Na2S04), and concentrated in
vacuo to yield
methyl 4-((3-methyl-2,4-dioxoimidazolidin-1-yl)methyl)benzoate (1.3g,
90°J°). 'H NMR
(CDCl3) 8.03 (d, J = 8.4 Hz, 2H) 7.32 (d, J = 8.4 Hz, 2H) 4.62 (s, 2H) 3.90
(s, 3H) 3.73 (s,
2H) 3.08 (s, 3H).
EXAMPLE H
0
N N
HO I
O 0
To the solution of Example G (900 mg, 3.44 mmol) in MeOH (30 mL) was added .
cons. HCl (10 mL). The resulting solution was heated to reflux for lh,
quenched with
sahirated Na2C03 (100 mL), and extracted with CHZC12 (100 mL). After
separation, the
organic layer was washed with brine, dried (NaZS04), and concentrated in vacuo
to yield 4-
((3-methyl-2,4-dioxoimidazolidin-1-yl)methyl)benzoic acid as a yellow solid.
The cntde
product was used without further purification.
EXAMPLE 5
I0'
N~N
N
I ~N I / O °
To a solution of Example H (200 mg, 0.81 mmol) in DMF (10 mL) were added EDCI
(200 mg, 1.0 mmol), HOBt (150 lllg, l.5mmo1), NMM (0.5 mL) and Reagent BB (300
mg,
1.5 mmol). After being stirred at RT overnight, the solvent was removed under
vacuum. The
resulting residue was purifted by preparative HPLC to yield pure 4-((3-methyl-
2,4-
dioxoimidazolidin-1-yl)methyl)-N-(4-methyl-3-(pyrimidin-2-
ylamino)phenyl)benzamide (20
mg). 1H NMR (DMSO-c~ b:10.14 (s, 1H), 8.87 (s, 1H),8.35 (d, J= 4.8 Hz, 2H),
7.91 (d, J=
83
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
8 Hz, 2 H), 7.84 (d, J = 1.6 Hz, 1 H), 7.45 (dd, J= 8.4, 2.0 Hz, 1 H), 7.41
(d, J= 7.6 Hz, 2H),
7.15 (d, J= 8.0 Hz, 1H), 6.75 (t, J= 4.8 Hz, 1H), 4.56 (s, 2H), 3.89 (s, 2H),
2.87 (s, 3H), 2.15
(s, 3H); MS (ESI) m/e: 431 (M++1).
EXAMPLE G
0
/ I , ~ N~N_
N~ ~ ~ ~ W
~N ~ / O O
The title compound was synthesized following the procedure for the preparation
of
Example 5 utilizing Example H and Reagent AA to yield N-(3-(4-phenylpyrimidin-
2-
ylamino)-4-methylphenyl)-4-((3-methyl-2,4-dioxoimidazolidin-1-
yl)methyl)benzamide. 'H
NMR (CDC13 -d) 8:8.45 (s, 1H), 8.39 (d, J= 5.6 Hz, 2H), 8.19 (s, 1H), 8.08
(dd, J= 7.2 Hz, 2
H), 7.84 (d, J= 8.4 Hz, 2H), 7.32-7.46 (m, 5 H), 7.25-7.29 (m, 2H), 7.13-7.17
(m, 2H), 4.56
(s, 2H), 3.70 (s, 2H), 3.03 (s, 3H), 2.30 (s, 3H). Ms (ESI) m/e: 507 (M++1).
EXAMPLE I
-NH
~O
~ ~ N H
O O
/
To a solution of Reagent CC (O.GBg, 4.30 mmol) in dry CHZC12 (20 mL) under N2
were added NMM (2.70g, 27.2 ll1n101), HOBt (0.91g, 6.7 mmol), EDCI (1.26g, 6.6
nnnol)
and reagent DD (l.Sg, 5.90 mmol): After being stirred at RT overnight, the
solvent was
removed under reduced pressure. The residual was washed with H20, saturated
aqueous
KZC03 and HZO to yield the white solid, which was dried in vacuo to yield
benzyl 4-
(bis((methylcarbamoyl)methyl)carbamoyl)benzoate (0.72 g, 42% yield). 'H
NMR(CDCl3)
88.74 (s, 1H), 8.10 (d, J= 8.4 Hz, 2H), 7.50 (d, J 8.4Hz, 2H), 7.46 (m, SH),
6.35 (s,lH),
5.37 (s,2H), 3.94 (d, J= 10.8 Hz, 4H) 2.89 (m, 6H); MS (ESI) m/e: 398 (M++1).
84
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
EXAMPLE J
-NH
~O
O ~ ~ N~NH
HO O //O
To a solution of Example I (0.73g, 1.84 mmol) in MeOH (30 mL) was added 10%
PdIC (200 mg). The reaction mixture was then stirred at amblent temperature
Llllder 1
atmosphere of HZ fox 45 min. The reaction mixture was filtered, the solid
washed with EtOH,
and the combined organics concentrated in vacuo to yield 4-
(bis((methylcarbamoyl)methyl)carbamoyl)benzoic acid (0.52g, 92% yield). IH NMR
(CDCl3) 89.16 (s, 1 H), 8.05 (d, J = 8.4 Hz, 2H), 7.49 (d, J = 8.4 Hz, 2H),
4.04 (d, .I = G Hz,
4H), 2.94 (m, GH); MS (ESI) m/e: 308 (M++1).
EXAMPLE 7
oo~y
~N
H
I N b I \ \ Nw
~N ~ O O
The title compound was synthesized following the procedure for the preparation
of
Example 1 utilizing Example ' J and Reagent BB to yield N1,N~-
bis((methylcarbamoyl)methyl)-N4-(4-methyl-3-(pyrimidin-2-
ylamino)phenyl)terephthalamide. 'H NMR (CD30D) 8 8.43 (d, J= 5.2 Hz, 2H), 7.98
(d, J=
8.4 Hz, 1H), 7.97 (s, 1H), 7.58 (d, J= 8.4 Hz, 2H), 7.50 (dd, J= 8.0, 2.0 Hz,
1H), 7.30 (d, J=
8.4 Hz, 1H), G.8G (t, J= 5.2 Hz, 1H), 4.18 (s, 2H), 4.04 (s, 2H), 2.81 (s,
3H), 2.73 (s, 3H),
2.28 (s, 3H). MS (ESI) m/e: 490 (M++1).
EXAMPLE 8
H
O O Nw
~N
N~N I \ N \ I ~Nw
,N ~ O O
I
The title compound was synthesized following the procedure for the preparation
of
Example 1 utilizing Example J and Reagent AA to yield N1,N~-
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
bis((methylcarbamoyl)methyl)-N4-(3-(4-phenylpyrimidin-2-ylamino)-4-
methylphenyl)terephthalamide.'H NMR (DMSO-d~) 8 10.25 (br s, 1H), 8.85 (br s"
1H), 8.44
(d, J = 4. 8 Hz, 1 H), 8.40 (d, J = 3.2 Hz, 1 H), 8.19 (m, 1 H), 8.11 (d, J =
5.8 Hz, 1 H), 8.06 (s,
1 H), 7.97 (d, J = 8.4 Hz, 2H), 7.50-7.45 (m, 5H), 7.32 (d, J = 5.2 Hz, 1 H),
7.18 (d, J = 8.0
Hz, 1H), 4.00 (s, 2H), 3.87 (s, 2H), 2.63 (d, J= 4.0 Hz, 1H), 2.58 (d, J= 4.0
Hz, 1H), 2.21 (s,
3H); MS (ESI) m/e: 566 (M++1).
EXAMPLE K
~ ~ON
s O
To the solution of Reagent AA (840 mg, 2.72 mmol) and 4-hydroxymethyl-benzoic
acid (490 mg, 3.20 mmol) in dry DMF (20 mL) was added EDCI (700 mg, 3.62
mmol),
HOBt (500 mg, 3.73 mmol), and NMM (0.5 mL, 3.95 mmol): The resulting mixture
was
stirred at RT overnight, into HZO and extracted with CHZC12. The organic layer
was washed
with saturated NaZC03, purified by column chromatography on silica gel yielded
N-(3-(4-
phenylpyrimidin-2-ylamino)-4-methylphenyl)-4-(hydroxymethyl)benzamide (410 mg,
36.8%). 'H NMR (DMSO-d~) 8:10.12 (s, 1H), 8.84 (s,lH), 8.44(d, J= 5.2 Hz, 1H),
8.11(d, J
= 4.0Hz, 2H), 8.05 (s, 1H), 7.91(d, J = 8.OHz, 2H) 7.45(m,SH), 7.32(d, J = 5.2
Hz, 1H),
7.19(d, J= 7.8 Hz, 1H), 4.56(d, J= 5.6 Hz, 2H), 2.30(s, 3H); MS(ESI) n~/e:
411.20(M++1).
EXAMPLE L
ci
N\ /N ~ NIf~I //
rN~ ~ / O
To the solution of Example K (410 mg, 0.99 mrnol) in 1,4-dioxane (40 mL) was
slowly added SOC12 (650 mg, 5.50 mmol) at RT. After being stirred at RT for
3h, the solvent
and excessive SOC12 was removed in vacuo to yield N-(3-(4-phenylpyrimidin-2-
ylamino)-4-
methylphenyl)-4-(chloromethyl)benzamide as a yellow solid (460 mg), which was
used
without further purification. 1H NMR (CDCl3-d~) 8:8.42(s, 1H), 8.22(d, J =
6.OHz, 3H),
86
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
8.05(m, 1H), 7.94(d, J= 1.0 Hz, 2H) 7.53-7.62(m,SH), 7.26(s,2H), 4.63(d, J=
5.4 Hz, 2H),
2.44(s, 3H); MS(ESI) m/e: 429.20(M++1)
EXAMPLE M
o~NH
\ N~S~O
To the solution of phenyl-urea (l3.Og, 95.48 mol) in THF (100 mL) was slowly
added
chlorocarbonyl sulfenylchloride (13 mL, 148.85 nnnol) at RT. The reaction
mixture was
refluxed overnight, the volatiles removed in vacuo yielded 2-phenyl-1,2,4-
thiadiazolidine-
3,5-dione as a white solid (4.Og, yield 20%). IH NMR (DMSO-d~) ~: 12.49 (s,
1H), 7.~ 1 (d,
J= 8.0 Hz, 2H), 7.43(t, J= 7.6 Hz, 2H), 7.27 (t, J= 7.2 Hz, 1 H).
EXAMPLE 9
0
I N~N ~ I
I N~b I \ b~ s-
O
,N ~ O
~I
To a solution of Example M (400 mg, 2.06 mmol) in anhydrous DMF and THF (l:l)
under NZ at 0 °C was slowly added NaH (165 mg, 4.24 mmol). After
stirring at 0 °C for O.Sh,
Example L (300 mg, 0.70 mmol) was added. The solution was heated to 40
°C, stirred for 3h
and quenched with AcOH (0.5 mL). Removal of the solvent followed by
purification via
preparative HPLC yielded N-(3-(4-phenylpyrimidin-2-ylamino)-4-methylphenyl)-4-
((3,5-
dioxo-4-phenyl-1,2,4-thiadiazolidin-2-yl)methyl)benzamide (50 mg, yield 12 %).
~HNMR
(DMSO-d~) 8: 10.18(s, 1 H), 8.88(s, 1 H), 8.43(d, J= 5.2 Hz, 1H), 8.12(dd, J=
7.6 1.6 Hz,
2H), 8.05(s, 1 H), 7.92(d, J = 8.4 Hz, 2H), 7.58(d, J = 9.2 1.6 Hz, 2H), 7.44-
7.50(m, 8 H),
7.34(t, J= 6.0 Hz, 2H), 7.18(d, J= 8.8 Hz, 1H), 4.91(s, 2 H), 2.20(s, 3 H); MS
(ESI) (mle):
587.18(M~+1 ).
EXAMPLE N
MeOZC ~ ~ NH~COzEt
Glycine ethyl ester hydrochloride (ll.lg, 79 mmol), and Reagent EE (lOg, G1
mmol)
were dissolved in absolute EtOH (300 mL). NaCNBH3 (8.4g, 134nnnol) was added
in 4
87
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
portions and the reaction mixture was stirred at RT overnight. The solvent was
removed
under reduced .pressure and the residue was dissolved in EtOAc. The organic
layer was
washed with 1N HCl solution, saturated NaHC03 and brine, and dried and
concentrated .in
vacuo to yield methyl 4-(((ethoxycarbonyl)methylamino)methyl)benzoate (8g). 'H-
NMR
(CDC13): 7.97 (d, J= 6.8 Hz, 2H), 7.39 (d, J= 8.8 Hz, 2H), 4.16 (q, J= 7.2 Hz,
2H), 3.88 (s,
3H), 3.84 (s, 2H), 3.37 (s, 2H), 1.94 (s, 1H), 1.24 (t, J= 7.2 Hz, 3H).
EXAMPLE O
MeO2C
N
C ~S~~ OzEt
HN
Cbz
To a stirred solution of chlorosulfonyl isocyanate (2.2g, 15.2 mmol) in CHZCIz
(40
mL) was added benzyl alcohol (1.64g, 15.2 mmol) at 0°C. And the
reaction temperature was
kept not to rise above 5°C. After stirred for lh, a solution of Example
N (4.2g, 16.7 mmol)
and triethylamine (6 mL, 4.3g, 42. 6 mmol) in CHZC12 (40 mL) was added at a
rate to keep
the reaction temperature not to rise above 5°C. When the addition was
completed, the
reaction solution was allowed to warm to RT and stirred overnight. The
reaction mixture was
poured into 1N HCl saturated with NaCI (300 mL). The organic layer was
separated and the
aqueous layer was extracted with CHZCIZ (2x100 mL). The combined organic
layers were
dried with Na2S04, and concentrated. The cmde product was recrystallized from
CHZC12/n-
hexane to afford desired Example O (5.9g, 76.6% yield). tH-NMR {CDCI3): 8.00
(d, J= 8. 4
Hz, 2H), 7.87 (s, 1H), 7.36 (m, SH), 5.29 (s, 2H), 4.65 (s, 2H), 4.15 (q, J=
7.2 Hz, 2H), 3.98
(s, 2H), 3.92 (s, 3H), 1.24 (t, 3H).
EXAMPLE P
MeOzC
N
~ SAO CO2Et
HZN
To a solution of Example O (5.5 g, 118 mmol) in solvent of MeOH (50 mL) and
EtOAc (50 mL ) was added 10% Pd/C (0.8 g ) under NZ. Then the resulting
mixture was
stirred at RT under HZ (60 psi) overnight. The solvent was removed to afford
white solid
Example P (3.4 g, 85% yield). 1H-NMR (CDC13): 8.02 (d, J= 8. 4 Hz, 2H), 7.41
(d, J= 8.4
Hz, 2H), 5.20 (s, 2H), 4.44 (s, 2H), 4.19 (q, J= 7.2 Hz, 2H), 3.91 (s, 3H),
3.90 (s, 2H), 1.25
(t, J= 7.2 Hz, 3H)
88
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
EXAMPLE Q
MeO2C~ ~ ~ O
~N-SAO
~NH
O
A NaOMe solution was prepared by adding NaH (60%, dispersion in mineral oil,
43.5
S lllg, 1.1 mmol) to MeOH (30 mL). Example P (300 mg, 0.9 mmol) was added to
the NaOMe-
MeOH solution and the reaction was stirred at RT overnight. The solution was
concentrated
in vacuo and the residue was dissolved in H20 (30 mL). The aqueous solution
was acidified
with 3N HCl and the precipitate was altered and collected to yield methyl 4-
(1,1,4-trioxo-
[1,2,5]thiadiazolidin-2-yhnethyl)-benzoate (120 mg, 40% yield). 1H-NMR (DMSO-
c~: 7.92
(d, .I= 8. 4 Hz, 2H), 7.49 (d, ,I= 8 Hz, 2H), 4.35 (s, 2H), 3.99 (s, 2H), 3.83
(s, 3H).
EXAMPLE R
Nooc ~ ~ o
~N-SAO
/NH
~O
Example Q (100 mg, 0.35 mmol) in THF (4 mL) and 1.5 mL of 2N aq. LiOH solution
was stirred at RT for 3h. The solvent was removed under reduced pressure and
the residue
was dissolved in H20 (20 mL) and acidified with aqueous 3N HCl. The
precipitate was
filtered and collected to yield 4-(1,1,4-trioxo-[1,2,SJthiadiazolidin-2-
ylmethyl)-benzoic acid
(85 mg). 'H-NMR (DMSO-c~: 7.90 (d, .l= 8 Hz, 2H), 7.46 (d, J= 8.4Hz, 2H), 4.27-
4.22 (br,
2H).
EXAMPLE 10
o~ .o
s
N N N ~ I N ,NH
iN I / O O
iN
The title compound was prepared following the procedure of Example 1 utilizing
Example R and Reagent FF to yield N-[4-methyl-3-(4-phenyl-pyrimidin-2-ylamino)-
phenylJ-
4-(1,1,4-trioxo-[1,2,SJthiadiazolidin-2-ylmethyl)-benzamide (48% yield). 'H-
NMR (DMSO)
89
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
b 10.19 (s, 1 H), 9.3 0 (s, 1 H), 9.00 (d, 1 H), 8.72 (d, J = 5.2 Hz, 2H),
8.59 (d, J = 9.2 Hz, 1 H),
8.52 (d, J= 5.2 Hz, 2H), 8.08 (s, 1H), 7.92 (d, J= 8.4 Hz, 1H), 7.62 (m, 1H),
7.50-7.43 (m,
4H), 7.19(d, J = 8.4 Hz, 2H), 4.27(s, 2H), 3.86 (s, 2H), 2.20 (s, 3H). MS
(ESI) in/e:
530.1(M+1).
EXAMPLE 11
o~ .o
s
N N N ~ I ' i ,NH
iN I / O O
The title compound was prepared following the procedure of Example 1 utilizing
Example R and Reagent AA to yield N-[4-methyl-3-(4-phenyl-pyrimidin-2-ylamino)-
phenyl]
-4-(1,1,4- trioxo-[1,2,5]thiadiazolidin-2-ylmethyl)-benzamide (56% yield). 1H-
NMR
(DMSO-c~:10.18 (s, 1H), 8 89 (s, 1H), 8.44 (d, J= 4.8 Hz 1H), 8.12 (d, J= 7.6
Hz, 2H), 8.05
(s, 1H), 7.92 (d, J= 8.0 Hz, 2H), 7.50-7.44 (m, 6H), 7.33 (d, J= 5.2 Hz, 1H);
7.18 (d, J= 8.4
Hz,.IH), 4.28 (s, 2H), 3.81 (s, 2H), 2.20 (s, 3H). MS (ESI) m/e: 529.1(M+1).
EXAMPLE S
N'S~N~COzEt
Me0 I / Boc H
O
A solution of Reagent GG (lOg, 35.4m mol) and diisopropyl azodicarboxylate
(7.2 g,
35.4 mmol) in THF (60 mL) was added dropwise (l5min, 5°C) to a solution
of equal molar
quantities of triphenylphosphine (9.3g, 35.4mmo1) and 4-hydroxymethyl-benzoic
acid methyl
ester (6g, 35.4m mol) in THF (50 mL). The resulting mixture was stirred under
N2
atmosphere for 2h. The solvent was removed and the residual was
chromatographed to yield
ethyl-[N-(N'-tent-butyloxycarbonyl,N~-benzoic methyl ester)-sulfamoyl]-
glycinate as a white
powder (8g, 53.3% yield). 1H-NMR (CDC13): 7.99 (d, J = 8.4 Hz, 2H), 7.42 (d, J
= 8.0 Hz,
2H), 5.80 (t, J 5.6 Hz, 1H), 4.85 (s, 2H), 4,12 (q, J= 7.2 Hz, 2H), 3.90(s,
3Ii), 3.65 (d, J =
5.6 Hz, 2H), 1.49 (s, 9H), 1.24 (t, 3H).
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
EXAMPLE T
0
N ~ ~N ~COZEt
Me0 ~ / H H
O
The solution of Example S (3g, 7m mol) in 2N HCl/dioxane 1,4-dioxane (60 mL)
was
heated to 50°C for 15 min. Then the solvent was removed under reduced
pressure to yield
ethyl-[N-(N'-benzonic methyl ester)-sulfamoyl]-glycinate as a white solid (2g,
86.9% yield).
IH-NMR (CDCl3): 8.01 (d, J = 8.4,2H), 7.41 (d, J = 8.4,2H), 4.86 (t, J = 4.8
Hz, 1H), 4.70 (t,
J = S.G Hz,IH), 4.32 (d, J = 6.4 Hz, 2H), 4.21 ~(q, J = 7.2 Hz ,2H),
3.91(s,3H), 3.82 {d, J = S.&
Hz, 2H), 1.28 (t, 3H).
EXAMPLE U
~s o
N 'NH
M e0 I ~-//
O
O
A solution of Example T (lg, 30.3 mmol) and NaH (0.32g, 78.7m mol) in THF (120
mL) was heated to reflex for 8h. The mixW re was cooled to RT, then quenched
with 1N aq.
HCl (100 mL) and extracted with CHZC12 (3 x 100 mL). The combined organic
layers were
f5 dried (Na2S04), and concentrated in vacuo and purified by flash
chromatography to yield 4-
(1,1,3-trioxo-[1,2,5]thiadiazolidin-2-ylmethyl)-benzoic acid methyl ester as a
white powder
(200mg, 23% yield). 1H-NMR (CDC13) 8.02 (d, J = 8.4, 2H), 7.48 (d, J = 8.0 Hz,
2H), 5.02
(br s, 1H), 4.77 (s, 2H), 4.10 (d, J = 7.2 Hz, 2H), 3.90 (s, 3H)
EXAMPLE V
o.o
N S.
' 'NH
HO I ~i
O
O
Example LT (2OOmg, 0.8m mol) in THF (3 mL) and 2N aq. LiOH (1.5 mL) was
stirred
at RT for 3h. The solvent was removed under reduced pressure, and the aqueous
layer was
acidified with 3N aq. HCl solution to yield 4-(1,1,3-trioxo6-
[1,2,5]thiadiazolidin-2-
yhnethyl)-benzoic acid a white powder (120 mg, 63%). 1H-NMR (DMSO-d): 7.90 (d,
J = 8.4
Hz, 2H), 7.43 (m, 2H), 4.10 (d, J =.6.0 Hz, 2H), 3.SG (d, J= 6.0 Hz, 2H).
91
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
EXAMPLE 11
00
NY ~ a ~ / N ,NH
I
I ~N ~ 0
N
The title compound was prepared following the procedure of Example 1 utilizing
Example V and Reagent FF to yield N-[4-methyl-3-(4-pyridin-3-yl-pyrimidin-2-
ylamino)-
phenyl]-4-(1,1, 3-trioxo-[1,2,5]thiadiazolidin-2-ylmethyl)-benzamide (65%
yield). 1H-NMR
(DMSO-d): 10.19 (s, 1H), 9.27 (s, 1H), 8.97 (s, 1H), 8.69 (d, J = 4.8 Hz, 2H),
8.60 (d, J = 6.4
Hz , 2H), 8.52 (m, 1H), 8.06 (s, 1H),7.89 (d, J = 7.6 Hz, 5H), 7.55 (d, 1H),
7.47-7.41 (m,
4H), 7.18 (d, J = 7.4 Hz, 2H), 4.76 (s, 2H), 4.15 (d, J = 6.4 Hz, 2H), 2.20
(s, 3H); MS (ESI)
m/e: 530.1 (M+1).
i0
EXAMPLE 12
00
H ~N H
N Y N '~ b ~'~'i
~N I i O
i
The title compound was prepared following the procedure of Example 1 utilizing
Example V and Reagent AA to yield N-[4-Methyl-3-(4-phenyl-pyrimidin-2-ylamino)-
phenyl]-4-(1,1,3-trioxo-[1,2,5]thiadiazolidin-2-ylmethyl)-benzamide (67%
yield). 1H-NMR
DMSO): 10.18 (s,lH), 8.85 (s, 1H), 8.61(m, 1H), 8.43 (d, J= 5.2 Hz, 2H), 8.10
(d, J= 6.2
Hz, 2H), 8.04 (s, 1 H), 7.90 (d, J = 8.0 Hz , 2H), 7.4 (m, SH), 7.32 (d, J =
5.2 Hz, 1 H) ,7.18
(d, J= 8Hz, 1H), 7.05 (s, 1H), 6.93 (s, 1H), 4.76 (s, 2H), 4.16 (d, J= 6.4 Hz,
2H); Ms (ESI)
mle: 529.1 (M+1)
EXAMPLE W
~ '.o
HOOC
TO a 501l1t1o11 Of 4-l7rOlllOlllethyl-henZlC aCld methyl ester (S.Og, 0.02
mol) and 4-
thiomorpholine (2.02g, 0.02 mol) in acetonitrile (50mL) was added KZC03
(5.52g, 0.04 mol).
The mixture was stirred under reflex for two days. After filtration of
inorganic salt and
92
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
removal of solvent, the residue was added to conc. HCI. The mixture was
stirred at RT for 30
min, concentrated, dissolved in acetic acid (30 mL) and 30% hydrogen peroxide
(10 mL),
stirred at 100 °C for overnight and then cooled to 0°C. Zinc
powder (1:5 g)' was added to the
reaction solution. After being stirred for 30 min, the resulting mixture was
filtered and solid
was washed with MeOH. The filtrate was concentrated. The residue was
neutralized by 2N
solution of KZC03 and adjust to PH= 8-9. The solution was extracted with
CHZCl2 twice.
The combined organic layers were dried over Mg2S04, and concentrated. The
residue was
added conc. HCl (lOmL). The resulted solution was stirred at 80 °C
fom2h and concentrated
to yield 4-(4,4-dioxothiomorpholinomethyl)benzoic acid (1.02 g, 18%). 1H NMR
(D20)
87.98 (d, J = 8.0 Hz, 2H), 7.52 (d, J = 8.0 Hz, 2H), 4.45 (s, 2H), 3.79 (s,
4H), 3.53 (s, 4H);
MS (ESI) n~/e: 270 (M++1).
EXAMPLE 13
/ N
N~ N \ N \ I \°O
O
iN ~ / O
To a solution of Reagent BB (100 mg, 0.5 mmol) in the anhydrous DMF (3 mL) at
RT
was added Example W (200 mg, 0.77 11111101) followed by EDCI (200 mg,.1.20
nunol), HOBt
(200 lllg, 1.15 mmol) and NMM (0.5 mL). After being stirred at RT overnight.,
the mixture
was added to H20 (100 mL) and extracted with CHZC12 (2x100 mL). The combined
organic
layers were washed with brine, dried (NaZS04) and concentrated. The residue
was purified by
preparative HPLC to yield 4-(((4,4-dioxothiomorpholinomethyl)1)methyl)-N-(4-
methyl-3-
(pyrimidin-2-ylamino)phenyl)benzamide (100 mg, 44%). 1H NMR (DMSO-d6): 8.43
(d, J=
4.8 Hz, 2H), 8.29 (s, 1H), 7.86 (d, J= 8.4 Hz, 2H), 7.81 (s, 1H), 7.46 (d,'J=
7.6 Hz, 3H),
7.21 (d, J= 8.4 Hz, 2H), G.75 (t, J= 4.8 Hz, 1H), 3.72 (s, 2H), 3.10 (s, 4H),
3.03 (s, 4H), 2.32
(s, 3H); MS (ESI) n~/e: 452 (M++1).
EXAMPLE 14
H hl / I N
N'\ / N \ N =O
\ ~SO
~ ~N ~ / o
/ E-21
The title compound was prepared following the procedure of Example 13
utilizing
Example W and Example AA to yield 4-(((4,4-dioxothiomorpholinomethyl)1)methyl)-
N-(4-
93
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
methyl-3-(4-phenylpyrimidin-2-ylamino)phenyl)benzamide. 'H NMR (CDC13): 8.54-
8.52
(m, 2H), 8.49-8.11 (m, 2H), 7..88-7.83 (m, 2H), 7.80 (s, 1H), 7.50-7.39 (m,
6H), 7.23-7.15
(m, 2H), 7.02 (s, 1H), 3.73 (s, 2H), 3.12 (s, 4H), 3.01 (s, 4H), 2.38 (s, 3H);
MS (ESI) mle:
528 (M++1).
EXAMPLE 15
NI
Nw N \ N \ I ~SvO
O
iN I / O
Co~
The title compound was prepared following the procedure of Example 13
utilizing
Example W and Example HH to yield 4-(((4,4-dioxothiomorpholinomethyl)1)methyl)-
N-(4-
methyl-3-(4-morpholinopyrimidin-2-ylamino)phenyl)benzamide. 'H NMR {CDC13):
8.63 (s,
1 H), 8.00 (d, J = 6.0 Hz, 1 H), 7. 82 (d, J = 8.0 Hz, 2H), 7.77 (s, 1 H),
7.43 (d, J = 8.4 Hz, 2H),
7.1G-7.09 (111, 2H), G.72 (s, 1H), G.02 (d, J= G.4 Hz, 1H), 3.80-3.77 (m, 4H),
3.GG (s, 2H),
3.58 (s, 4H), 3.07 (s, 4H), 3.00-2.88 ( n~, 4H), 2.30 (s, 3H); MS (ESI) n~l/e:
537 (M++1).
EXAMPLE X
0 0I
\ N ~O
HO I / _
O
To a solution of D-4-phenyl-oxazolidin-2-one (lg, G nnnol) in anhydrous THF
(40
mL) under nitrogen protection at -78°C was added BuLi (2:5 M in hexane,
1.8 mL, 4.5
mmol). After one hour, the mixture was transferred to a solution of
terephthalic acid chloride
monobenzyl ester (prepared from Reagent DD (1.2 g, 4.5 11111101) and thionyl
chloride (10
mL) at. reflux for 2h), in anhydrous THF. After being stirred at -78 °C
for 30 111111, the
reaction mixture was warmed to RT for 2h. After Being quenched by adding
sat<lrate solution
of ammonium chloride (1 mL), the reaction solution was extracted with CHaCl2
(3 x SO mL).
The combined organic layers were dried (Na2S04) and concentrated. The residue
was
dissolved in MeOH (20 mL) and 5% Pd/C (0.1 g) and stirred under 1 atln HZ for
5h. The
suspension was filtered and filtrate was concentrated to yield D-4-(2-oxo-4-
phenyl-
oxazolidine-3-carbonyl)-benzoic acid (O.GS g, 4G%). 'H NMR (CDCl3): 8.15-8.11
(m, 2H),
7.70 (dd, .l = 6.8, 1.G Hz, 2H), 7.44-7.33 (m, 5H), 5.63 (dd, J= 8.8, G.8 Hz,
1H), 4.78 (dd, J--
94
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
18, 9.2 Hz, 1H), 4.36 (dd, J= 9.2, 6.8 Hz, 1H); MS (ESI).m/e: 312 (M++1).
EXAMPLE Y
0 0
\ N~o
Ho
o \
The title compound was prepared following the procedure of Example X utilizing
L-
4-phenyl-oxazolidin-2-one to yield L-4-(2-oxo-4-phenyl-oxazolidine-3-carbonyl)-
benzoic
acid (0.65 g, 46%). ~H NMR (CDC13): 8.15-8.11 (m, 2H), 7.70 (dd, J = 6.8, 1.6
Hz, 2H),
7.44-7.33 (m, SH); 5.63 (dd, J= 8.8, 6.8 Hz, 1H), 4.78 (dd, J-- 18, 9.2 Hz,
1H), 4.36 (dd, J=
9.2; 6.8 Hz, 1H); MS (ESI) m/e: 312 (M++1).
EXAMPLE 16
0 0
N~o
N_\ /N \ N \
TN ~ C \
The title compound was prepared following the procedure of Example 13
utilizing
Example X and Reagent AA to yield D-4-(2-oxo-4-phenyl-oxazolidine-3-carbonyl)-
N-(4-
methyl-3-(4-phenylpyrimidin-2-ylamino)phenyl)benzamide. 'H NMR (DMSO-d6) :
10.34
(s, 1 H), 8.87 (s, 1 H), 8.44 (d, J = 5.2 Hz, 1 H), 8.12-8.10 (m, 2H), 7.96 (
d, J = 8.4 Hz, 2H);
7.84 (d, J= 8.4 Hz, 2H), 7.54-7.30 (m, 8H), 7.19 (d, J= 8.4 Hz, 1H), 5.63 (dd,
J 8.0 & 8.0,
1H), 4.84 (t, J 8.0, 1H), 4.23 (dd, .I--8.0 & 8.0, 1H), 2.21 (s. 3H). MS (ESI)
m/e: 570 (M++1)
EXAMPLE 17
0 0
N ~O
N\ N \ N \
I i o 1 ~
I
\
The title compound was prepared following the procedure of Example 13
utilizing
Example Y and Reagent AA to yield L-4-(2-oxo-4-phenyl-oxazolidine-3-carbonyl)-
N-(4-
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
methyl-3-(4-phenylpyrimidin-2-ylamino)phenyl)benzamide. 'H NMR (DMSO-d6) :
10.34
(s, 1H), 8.87 (s, 1H)a 8.44 (d, J= 5.2 Hz, 1H), 8.12-8.10 (m, 2H),,7.96 ( d,
J= 8.4 Hz, 2H),
7.84 (d, J= 8.4 Hz, 2H), 7.54-7.30 (m, 8H), 7.19 (d, J= 8.4 Hz, 1H), 5.63 (dd,
J--8.0 & 8.0,
1H), 4.84 (t, J°8.0, 1H), 4.23 (dd, J--8.0 & 8.0, 1H), 2.21 (s. 3H). MS
(ESI) m/e: 570 (M++1)
EXAMPLE 18
0 0
/ N ~O
N\/N \ N \ I\
I iN ( / C
\ N
The title compound was prepared following the procedure of Example 13
utilizing
Example X and Reagent FF to yield D-4-(2-oxo-4-phenyl-oxazolidine-3-carbonyl)-
N-[4-
methyl-3-(4-pyridin-3-yl-pyrimidin-2-ylamino)-phenyl]benzamide.lH NMR (DMSO-
d6):
10.34 (s, 1H), 8.95 (s, 1H), 8.66 (m, 1H), 8.48 (m, 2H), 8.07 (s, 1H), 7.96 (
d, J= 8.4 Hz,
2H), 7.84 ( d, J = 8.0 Hz, 2H), 7.58-7.42 (m, 4H), 7.41-7.36 (m, 3H), 7.32 (d,
J = 6.8 Hz,
1H), 7.20 (d, J= 8.4 Hz, 1H), S.G3 (t, J= 7.G Hz, 1H), 4.84 (t, J= 7.6 Hz,
1H), 4.23 (t, J=
7.6 Hz, 1H), 2.21 (s, 3H. ); MS (ESI) n~/e: 571 (M++1).
EXAMPLE 19
0 0
N ~O
N\ N \ N \
/ o. ~
/ I
\ N
The title compound was prepared following the procedure of Example 13
utilizing
Example Y and Reagent FF to yield L-4-(2-oxo-4-phenyl-oxazolidine-3-carbonyl)-
N-[4-
methyl-3-(4-pyridin-3-yl-pyrimidin-2-ylamino)-phenyl]benzamide.~H NMR (DMSO-
~l6):
10.34 (s, 1 H), 8.95 (s, 1 H), 8.66 (111, 1 H),. 8.48 (m, 2H), 8.07 (s, 1 H),
7.96 ( d, J = 8.4 Hz,
2H), 7.84 ( d, J = 8.0 Hz, 2H), 7.58-7.42 (m, 4H), 7.41-7.36 (m, 3H), 7.32 (d,
J = 6.8 Hz,
1 H), 7.20 (d, J = 8.4 Hz, 1 H), 5.63 (t, J = 7.6 Hz, 1 H), 4.84 (t, J = 7.6
Hz, 1 H), 4.23 (t, J =
7.6 Hz, 1H), 2.21 (s, 3H. ); MS (ESI) n~/e: 571 (M++1).
9G
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
EXAMPLE Z
0
\ N N
/O ~ / O~NH
O
To a solution of 1-methyl-[1,2,4]triazolidine-3,5-dione (1.886g, O.O1G4 mol)
and
sodium hyhride (200 mg) in DMSO (5 mL) was added 4-chloromethyl-benzoic acid
methyl
ester (1.0 g, 0.0054 11101). The mixture was stirred at RT for overnight,
quenched with HZO
(100 mL), and extracted by CHZC12. The organic layer was washed with H20,
dried (Na2S04)
and concentrated in vacuo to yield methyl 4-((1-methyl-3,5-dioxo-1,2,4-
triazolidin-4-
yl)methyl)benzoate (1.02g, 72%). 'H NMR (CDC13) :7.93 (d, J= 8.4 Hz, 2H), 7.27
(d, J=
8.4 Hz, 2H), 4.68 (s, 2H), 3.83 (s, 3H), 3.27 (s, 3H). MS (ESI) m/e: 264
(M++1)
EXAMPLE AA
0
\ N N.
HO I / ~-NH
O
O
A solution of Example Z (1.Og, 0.0038 mol) and lithium hydroxide (0.950g) in
MeOH
(10 111L) was stirred at RT for overnight. The mixWre was acidified by 2N HCI
to pH=5-6
and extracted by CHZC12 (3x50 mL). The combined organic layers were washed
with HZO,
dried (MgS04) and concentrated in vacuo to yield 4-((1-methyl-3,5-dioxo-1,2,4-
triazolidin-4-
yl)methyl)benzoic acid (O.G g, 64%). IH NMR (CDCl3): 7.71 (d, J= 8.4 Hz, 2H),
7.17 (d, J=
8.4 Hz, 2H), 4.68 (s, 2H), 2.90 (s, 3H), 2.6 (s, 3H); MS (ESI) m/e: 249
(M++1).
EXAMPLE 20
0
/ N~NH
N\ N \ N \ I O~-N
rN I / O
/1
\ N
The title temperature was prepared following the procedure of Example 1
utilizing
Example AA and Reagent FF to yield N-(3-(4-(pyridin-3-yl)pyrimidin-2-ylamino)-
4-
methylphenyl)-4-((1-methyl-3,5-dioxo-1,2,4-triazolidin-4-yl)methyl)benzamide.
'H NMR
(CD30D) 89.44 (s, 1 H), 8.79 (d, J = 8.0 Hz, 2H), 8.50 (d, J = 4.0 Hz, 1 H),
8.25 (s, 1 H), 7.93
(d, J = 8.0 Hz, 2H), 7.73 (s, 1 H), 7.4G (d, J =8.0 Hz, 2H), 7.40 (d, J = 5.2
Hz, 1 H), 7.35 (d, J
97
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
= 8.4 Hz, 1H), 7.25 (d, J= 8.4 Hz, 1H), 4.87 (s, 2H), 3..07 (s, 3H), 2.31 (s,
3H): MS (ESI)
m/e: 509(M++1).
EXAMPLE 20
0
/ N~NH
N~ N \ N \ ~ . ~N
O
iN ~ / O
The title temperahue was prepared following the procedure of Example 1
utilizing
Example AA and Reagent AA to yield N-(3-(4-phenylpyrimidin-2-ylamino)-4-
methylphenyl)-4-((1-methyl-3,5-dioxo-1,2,4-triazolidin-4-yl)methyl)benzamide.
1H NMR
(CD30D) : 8.39 (s, 1H), 8.20 (d, J= 1.6 Hz, 1H), 8.13 (m, 2H), 7.93 (d, J= 8.4
Hz, 2H), 7.47
(m, 6H), 7.27 (m, 2H), 4.59 (s, 2H), 3.08 (s, 3H), 2.31 (s, 3H). MS (ESI) m/e:
508 (M++1).
EXAMPLE 21
0
N~NH
N~ N \ N \ ~ O~-N
o \
The title temperature was prepared following the procedure of Example 1
utilizing
Example AA and Reagent BB to yield 4-((1-methyl-3,5-dioxo-1,2,4-triazolidin-4-
yl)methyl)-
N-(4-methyl-3-(pyrimidin-2-ylamino)phenyl)benzamide. 1H NMR (CDC13) : 11.31
(s, 1H),
10.15 (s, 1H), 8.77 (s, 1H), 8.33 (m, 2H), 7.87 (m, 3H), 7.40 (m, 3H), 7.14
(d, J = 8.4 Hz,
1H), 6.71 (m, 1H), 4.73 (s, 2H), 2.97 (s, 3H), 2.14 (s, 3H); MS (ESI) m/e: 432
(M++1).
EXAMPLE BB
N~CO~Et
O=S=O
MeO~C NHCbz
2
To a stirred solution of chlorosulfonyl isocyanate (2.2 g, 15.2 11111101) in
CHZC12 (40
niL) was added benzyl alcohol (1.64 g, 15.2 mmol) at 0°C. After being
stirred for 111, a
solution of Example N (4.2 g, 16.7 mmol) and trlethyla111111e (6 111L, 4. 3 g,
42.6 11111101) in
, CH2Cl2 (40 mL) was added at a rate so that the reaction temperature did not
rise above S°C.
98
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
When the addition was completed, the reaction solution was allowed to warm to
RT and
stirred overnight. The reaction mixture was then poured into 1 N HCl saturated
with NaCI
(300 mL). The organic layer was separated and the aqueous layer extracted with
CH2C12. The
combined organic layers were dried over Na2S04, and concentrated to yield the
cnide
compound. Recrystallization from CHZC12/n-hexane yielded Example BB (5.9 g,
76.6%
yield). ~H-NMR (CDCl3) 8 8.00 (d, J= 8. 4 Hz, 2H), 7.87 (s, 1H), 7.36 (m, SH),
5.29 (s, 2H),
4.65 (s, 2H), 4.15 (q, J= 7.2 Hz, 2H), 3.98 (s, 2H), 3.92 (s, 3H), 1.24 (t,
3H).
EXAMPLE CC
MeO2C
O~~ N~
SAO C02Et
H2N
To a solution of Example BB (5.5 g, 118 mmol) in MeOH (50 mL ) and EtOAc (50
mL ) was added 10% Pd/C (0.8 g ) under nitrogen atmosphere. Then the result
mixture was
stirred at ambient temperature under HZ (60 psi) overnight. The solvent was
removed to yield
Example CC (3.4 g, 85%) as a white solid. 'H-NMR (CDCl3, 8) 8.02 (d, J= 8. 4
Hz, 2H),
7.41 (d, J = 8.4 Hz, 2H), 5.20 (s, 2H), 4.44 (s, 2H), 4.19 (q, J = 7.2 Hz, 2I-
I), 3.91 (s, 3H),
3.90 (s, 2H), 1. 25 (t, J= 7.2 Hz, 3H)
EXAMPLE DD
Me0 C ~ ~ O
~N-S.O
/NH
~O
A NaOMe solution was first prepared by adding NaH (60%, dispersion in mineral
oil,
43.5 mg, 1.1 mmol) to MeOH (30 mL). Example CC (300 mg, 0.9 mmol) was added to
the
NaOMe-MeOH solution and the reaction was stirred at RT overnight. The solution
was
concentrated to dryness in vacuum and the residue was dissolved in H20 (30
mL). The
aqueous solution was acidified with 3 N HCl (aq. ) and the result precipitate
was filtered and
collected to yield Example DD (120 mg, 40% yield). tH-NMR ( DMSO-cl6) 7.92 (d,
J= 8. 4
Hz, 2H), 7.49 (d, J= 8.4 Hz, 2H), 4.35 (s, 2H), 3.99 (s, 2H), 3.83 (s, 3H).
99
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
EXAMPLE EE
Hooc- ~~ ~-~ o
~N,s,o
'NH
~0
The solution of Example DD (100 mg, 0.35 11111101) in THF (4 mL) and 1.5 mL of
2 N
aq. LiOH solution was stirred at RT for 3h. Then the solvent was removed under
reduced
pressure and the residue was dissolved in water (20 mL) and acidified with
aqueous 3 N HCI.
The result precipitate was filtered to yield Example EE (85 mg). 1H-NMR ( DMSO-
cl)8 7.90
(d, J= 8 Hz, 2H), 7.46 (d, J= 8.4Hz, 2H), 4.27-4.22 (br, 2H).
EXAMPLE 22
°. ,o
N S
1 'NH
N~ N \ N
O O
,N
The title compound was prepared following the procedure of Example 1 utilizing
Example EE and Reagent FF to yield Example 22. ~H-NMR (DMSO-cl~) 810.19 (s,
1H), 9.30
(s, 1 H), 9.00 (d, 1 H), 8.72 (d, J = 5.2 Hz, 2H), 8.59 (d, J = 9.2 Hz, 1 H),
8.52 (d, J = 5.2 Hz,
2H), 8.08 (s, 1H), 7.92 (d, J= 8.4 Hz, 1H), 7.62 (m, 1H)~ 7.50-7.43 (m, 4H),
7.19(d, J= 8.4
Hz, 2H), 4.27(s, 2H), 3.86 (s, 2H), 2.20 (s, 3H). MS (ESI) m/e: 530(M++1).
EXAMPLE 23
o~ ,o
N S
/ 1 \NH
N\ N \ N \ ~\(\I
i ~ / O O
The title compound was prepared following the procedure of Example 1 utilizing
Example EE and Reagent AA to yield Example 22. 'H NMR (DMSO-d~) 810.18 (s,
IH), 8
89 (s, 1H), 8.44 (d, .I= 4.8 Hz 1H), 8.12 (d, J= 7.6 Hz, 2H), 8.05 (s, 1H),
7.92 (d, J= 8.0 Hz,
2H), 7.50-7.44 (111, 6H), 7.33 (d, J = 5.2 Hz, 1 H), 7.18 (d, J = 8.4 Hz, 1
H), 4.28 (s, 2H), 3.81
(s, 2H), 2.20 (s, 3H). MS (ESI) m/e:-529(M++1).
100
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
EXAMPLE FF
o,. ,,O
~ N~S~~~CO~Et
MeO~Boc
O
A solution of [Boc-sulfamide] amino ester (lOg, 35.4m m01) min) to a solution
of
triphenylphosphine (9.3g, 35.4nnnol) and 4-hydroxymethyl-benzoic acid methyl
ester (6g,
35.4m mol) in THF (50 mL) at 0-5°C. The result mixture was stirred
under NZ for 2h. The
solvent was removed and the residual was purified by column chromatography to
yield
Example FF as a white powder (8g, 53.3% yield). 'H-NMR (CDCL3) 7.99 (d, J =
8.4 Hz,
2H), 7.42 (d, J= 8.0 Hz, 2H), 5.80 (t, J S.G Hz, 1H), 4.85 (s, 2H), 4,12 (q,
J= 7.2 Hz, 2H),
3.90(s, 3H), 3.65 (d, J = 5.6 Hz, 2H), 1.49 (s, 9H), 1.24 (t, 3H).
EXAMPLE GG
o ,,o
~t~~COzEt
Me0 I
O
The solution of Example FF (3g, 7m mol) in 2N HC1/dioxane 1,4-dioxane (60 mL)
was heated to 50°C for 15 min. The solvent was removedin vacuo to yield
Example GG as a
white solid (2g, 86.9% yield). 'H-NMR ( CDC13,~) 8.01 (d, J = 8.4,2H), 7.41
(d, J = 8.4,2H),
4.86 (t, J = 4.8 Hz,lH), 4.70 (t, J = S.G Hz,lH), 4.32 (d, J = 6.4 Hz, 2H),
4.21 (q, J = 7.2 Hz
,2H), 3.91 (s,3H), 3.82 (d, J = 5.6 Hz, 2H), 1.28 (t, 3H).
EXAMPLE HH
Qs,o
~NH
Me0 I ,~-~/
O
A solution of Example GG (lg, 30.3 11111101) and NaH (0.32g, 78.7m mol) in THF
(120
mL) was heated t0 reflux for 8h. The mixture was cool to RT, quenched with 1N
aq. HCl
solution (100 mL) and extracted with CHZC12 (3x100 mL). The combined organic
phases
were dried (NaZS04), and concentrated in vacuo and purred by flash
chromatography to
yield Example HH as a white powder (200mg, 23% yield). 'H-NMR (CDC13, ~) 8.02
(d, J =
8.4, 2H), 7.48 (d, J = 8.0 Hz, 2H), 5.02 (br s, 1H), 4.77 (s, 2H), 4.10 (d, J
= 7.2 Hz, 2H), 3.90
(s, 3 H)
101
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
EXAMPLE II
00
~N H
HO
O
O
Example HH (200mg, 0.8m mol) was dissolved in THF (3 mL), and 1.5 rnL solution
of 2N aq. LiOH was added to the reaction solution. The mixture was stirred at
RT for 3h. The
solvent was removed in vacuo, and the aqueous layer was acidified with 3N aq.
HCl solution,
and filtered to yield Example II as a white powder (120mg, 63%). 'H-NMR (DMSO-
d)
87.90 (d, J = 8.4 Hz, 2H), 7.43 (m, 2H), 4.10 (d, J = 6.0 Hz, 2H), 3.56 (d, J=
6.0 Hz, 2H).
EXAMPLE 24
00
N~S
~N H
I NYN I W N I ~ ~j-~
I O
iN ~ O y
~ N
The title compound was prepared following the procedure of Example 1 utilizing
Example II and Reagent FF (65% yield). IH-NMR (DMSO-c~ 810.19 (s, 1H), 9.27
(s, 1H),
8.97 (s, 1 H), 8.69 (d, J = 4.8 Hz, 2H), 8.60 (d, J = 6.4 Hz , 2H), 8.52 (m, 1
H), 8.06 (s,
1 H),7.89 (d, J = 7.6 Hz, SH), 7.55 (d, 1 H), 7.47-7.41 (m, 4H), 7.18 (d, J =
7.4 Hz, 2H), 4.76
(s, 2H), 4.15 (d, J= 6.4 Hz, 2H), 2.20 (s, 3H); MS (ESI) mle: 530 (M+1).
EXAMPLE 25
00
N So
~NH
N~N ~ N
O
I ~N I / O
The title temperature was prepared following the procedure of Example 1
utilizing
Example II and Reagent AA. (67% yield). ~H-NMR ( DMSO-~~, 510.18 (s,lH), 8.85
(s,
1 H), 8.61 (m, 1 H), 8.43 (d, J = 5.2 Hz, 2H), 8.10 (d, J = 6.2 Hz, 2H), 8.04
(s, 1 H), 7.90 (d, J
= 8.0 Hz , 2H), 7.4 (m, SH), 7.32 (d, J= 5.2 Hz, 1H) ,7.18 (d, J= BHz, 1H),
7.05 (s, 1H), 6.93
(s, 1 H), 4.76 (s, 2H), 4.16 (d, J= 6.4 Hz, 2H); Ms (ESI) m/e: 529 (M+1 )
102
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Specific embodiments are additionally illustrated below which are intended to
represent more clearly, but without limitation to the generic scope, the
present invention:
Example 1
/ wN,S
H H I ~0 Example 2
N~N \ N \ O~N
H.
/ 0 /. N,S
I-i H H I ~ ~O
N' /N \ N \ O H
\ II
O
0 /
i
N
Example 3
/ \N~S~
H H I 0 .
N~ N ~ \ N 0 N
O H Example 4
/ ~3C /
N H H H
N I N \ N II N
0 ~, O
/ ~3~.r ~ /
N
C~
0
103
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 5
O ~p
H H W I N~S~ Example G
N' /N . N \ ~NH
w~ \ HsC
H3C
I ~ ~3C I ~ / O O / . N O
H H ~ I I \N H
\ I N I N I \ N \ s
1 H3C o
N ~ ~3C / O
y
N
Example 7
00
/ ~S~
H H I ,N~
I \~N H
\ H C
I N\ N \ N H3C Example 8
H3C / O O
N H H H
N _N I \ N_ /N
O , I~3C / 'IO
N
C~~
104
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 9
O
~N
N N . N \ I N OH Example 10
\ a
H3C
O
I ~~ C I ~ O O
3
H H I 'N OH
\N I N~N I \ N O \ iN vOH
H3C O
/ / 1'13C
O
/
N
Example 11
O
~N
H H OH
N\ N \ N \ ( ~N OH
HsC 0
O Example 12 .
N
H H H
O N N N N
\
I / ~3~ I ~ o
N N OH
C01 N OH
HsCm O
105
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 13
H O
H H / H N ~O N
N' /N ~ ~ N ~ H3C O CH3 Example 14
~ I~sC / O sC N O
H H / H N ~O
N~N ~ \ N ~ H3C O CH
/ O 3C 3
N
Example 15
/ H O
H H \ I \HN o O N
N\' /N ~ ~ N
H3C -~ ~CH3
N~ O
/ 3C Example 16
N
H H
O ~ \' /N ~ ~ N'
~~sC / CC
N
coy
lOG
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 17
/.I ~N'1
N\ N ~ N ~ ~SO O Example 18
N~ / O /
H Fi I
~O
S~
O
Example 19
/ N
~S=
INN I ~ N ~ 'O
Example 20
O
/ ~30 /
N N\ N \ N~ \
H N
C~ I ~ I O ~
o / ~3~ /
N C NJ
~O~ ~S ~
O ~O
107
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 2l. O NH Example 22 O\ ~NH
y
/ S~OEt / I S~OEt
H H H H
N N \ N \ I N~N \ N \
N I O
/ O / H30 W
\ I \
/N NJ
Example 23 NH Example 24
O~ //
/ S~OEt H H H
H H Nw N \ N II N \
N~ N \ N \ ~ I I O I O
/ / / //
I / ~3C ~ / O H3C I ~NH
N \ OEt
N
C~
O
l08
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 25 °
/I
H H
I N~N I \ N \ N
/ ~ C / O O~0
3
\ Example 2G °
I ~ /
/ N H H I ~N
IsI~NIw N \
/ ° ° o
3
I
N
Example 27
O
/I
H H N
I N~NN I \ N \
/ HsC / O O O
Example 28
N
H H H
O Nw N \ N II N
I /1' I / O
3C
N
C°
109
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 29
/ ~N
H H
N N N \ I
I ~ I ~ 1
/N / O 'S03H
Example 30
/ .
/N Nw N \ N \ I N
O
I / ~3C I / S03H
Example 31
N
I N~N I \ N
H H \ I \ ~N1
N~ O I
/ HsC / \SO3H
Example 32
N
C~
O H H H
N ' N I \ N " N I \
O /
N NJ
C ~ CN
O
S03H
110
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 33
/ \N~S~
W I O
N' /N \ N \ ~
\fY O"N
/ p H
3C
Example 34
/ . I \N/S~
~H H O
N' /N \ N \ ~
Oi \N
O Fi
I~~,C I/
3
Example 35
,S
H / I ~ ~O
N~N I \ N \ O N
I-1
~~ C / O
3
Example 36
H H
N N N N
\
I ~N I ~ p
H3C
N ~S
O
N O
1-I
111
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 37 O
'~ ~O
/ I NHS
H H \N H
N~N \ N \ H3C
O H3C'~~/O
/
Example 38
O
I 'N
H H / ~S\
NH
N\I N I \ N \ HsC . \\
N O H3C O
/
H3C
Example 39
O O
v ~i
/ \N~S
H H I \N H
I \~N I \ N \ H3C
O HsC O Example 40
~~ C /
3
H H H
N\1 N I \ N II N
O /
NH C /
1
H C N ~S~O
3
H3C NH
O
112
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 41 O
OH
H H I _N~
I N_ 'N I \ N \ N OH
IY H3C/ O
~~C / O
3
Example 42
O
/ ~N
H H OH
N\ N \ N \ I ~N OH
HsC O
~~ C I / O
3
Example 43
O
/ ~N\~OH
H H I N OH
N~N I \ N \ H3C~ O
O
3
Example 44
H. H H
N N N N
\
I ~~ C I / O
1
N O
HaC N
O
O OH
113
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 45
H O
O'
H H / ~ ~HN o N
/ O sC J
\~N ~ \ N \ HsC CHs
3C
Example 46
H O
H H / ~ HN~O N
J
N~N I \ N \ HsCO CHs
NH C / O sC
3
Example 47
H O
H H / ~ wHNO N
O'
yN ~ \ N \ HsC CHs
~~ C / O sC
3
Example 48
H H H
N~N ~ \ N~N
C / IOI /
O H
1
N O~NH
HsC O CHs
CHs
114
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 49
N
H H I ~g~0
( N\ /N I ~ N ~ p
O
C / O
3
Example 50
/ ~N~
H H I ~~ c0
N' /N ~ N ~ " SO
I~\~~C I/ O
3
Example 51
/ I N
H H 1'
N~ N ~ N ~ VSOO
N O
H3C
Example 52
H H H
N N N N
I/
~~ C
3
N
O ~O
115
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 53 O NH
~S
H H / I \OEt
N~N I \ N \
~~ C / O
3
Example 54 NH
~S/
/ I ~OEt
H H
I N~N I \ N \
/ O
3
Example SS
NH
O
H H / I ~OEt
N~N I \ N \
O
3
Example 56
H H H
N~N .I \ N II N I \
O / O
\S=NH
I
OEt
116
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 57 O
/\
H H N
N~N L \ N \ O
O
C / Exam le 58
s p
O
/ ~ ~ / \
H H N
N' /N ~ ~ N ~
O
~~ C / O O
3
Example 59
O
/\
H H. N
\~N I \ N
~~ C / O O
3
Example 60
H H H
N N N N
~~ C ~ /
3
O
O~N
117
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 61
~N
H H
N N N
\ 1
i~, ~ ~ O 'SO H Example 62
Y13C 3
N
H H L
Nw N , \ N \ ~ ~N
N~ O ~SO3H
H3C
Example 63
H H \ I \N~
N~N \ N
~ ~ C ~ ~ O ~S03H
3
Example 64
H H H
N N N N \
\
~~ C ~ ~ O
1
N
N
SO3H
118
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 65
/ I \N~S~
O.
\ N \
O~N
/ O I-I
CI
Example 66
/ N ''S
I ~ ~O
N~N \ O NH
~'=-~~S O
Example 67
/ I N~S
H O
/N \ N \ O~N
H3C /
I O H
Example 68
N N
O
~N ~ / O ~ /
N U
Et
N
O~ ~S
N
ti ~O
119
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 69 O O
a ~~
~S
H /, . I \ N \N H
\ N \ H3C
'. HaC. O
O
CI Example 70
00
H ~ N~S~NH
N N \ I HsC ' '\
H3C O
O
Example 71
~,O
N'
I H I ~NH
/ N I \ N \ HsC--~~D/
HsC O
O
H3C
Example 72
H H H
N N
O~N I \ ~ I \
N
Et
H3C N ~S
H3C
~~~,~.NH
O
120
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 73 Example 74 O
O /
/ ~ HN OH H I Hi OH
H N N \ N OH
\ N \ ~N OH ~ ~ Ra
O R4 O ~ S O O
CI /
Example 75 Example 76
O
H H
H / ~ HN OH N \ N~N \
/N \ N \ R4,N OH O~ I O
O N / /
H3C ~ / O Et N
Ra,N~O
O HO OH
121
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 77
H 0
/ NON
H I HO
\ N \ H3C -i ~CH3
O sC
CI /
Example 78
H O
H / HN~ON
N N \ I HsCO CHs
sC
0
Example 79
H O
H / HN~ON
/ ~ \ N \ I HsCO CHs
I 0 3C
HsC / Example 80
H H H
N I \ N II N I \
O
N / O /
Et~ O HO
'~ N
HsC OCHs
122
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 81
\N~
H ~ ~ ,O
\ N \ " SO
O
Example 82
~N~
H
N' N \ I ~SO p
O
Example 83
~N~
H
\ N \ ~ ~SO
O
HsC
Example 84
H H H
N ~ \ N~N ~ \
IIO
~N ~ O
CND
o S°O
123
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 85 \\ ~NH
S
H / ~ \OEt
\ N \
/ O
CI
Example 86 \ ~NH
S
H / . ~ ~OEt
N~N
O
Example 87 NH
~S~
H / ~ ~OEt
/I \ N \
/ . O
H3C
Example 88
H H H
N N N
\ ~ \
O~N ~ / O ~ / S/
Et ~ ~NH
Et0
124
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 89
O
H N Ph
N \
I~ °
Ci / ° °
Example 90
O
H Ph
N N \ I N
°
Example 91
O
/ I
H N Ph
\ N \
O
I/ °
HsC v
Example 92
H H H
N I \ N II N I \
O
~N / ° /
Et~ ~O
O\ 'N .
--Ph
~/O
125
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
Example 93
/ I N
H
\ N \ N \
I / O S03H
CI
Example 94
/ I N
H
O
N N \ N \
S03H'
Example 95
I / I
H N
/N I \ N \
H3C / O S03H
Example 9G
H H H
N N
O
~N I. / O I /
N U
Et
CND
N
S03H
All of the references above identified are incorporated by reference herein.
In
addition, two simultaneously applications are also incorporated by reference,
namely
Modulation of Protein Functionalities, S/N , fled December , 2003, and
Anti-Inflammatory Medicaments, S/N filed December , 2003.
126
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
34479.sT25.txt
SEQUENCE LISTING
<110> Deciphera Pharmaceuticals, Inc.
Flynn, Daniel L
Petillo, Peter A
<120> Anti-Cancer Medicaments
<130> 34'479
<150> 60/437,403
<151> 2002-12-31
<160> 5
<170> Patentln version 3.2
<210> 1
<211> 292
<212> PRT
<213> Homo sapiens
<400> 1
Gly Ala Met Asp Pro Ser Ser Pro Asn Tyr Asp Lys Trp Glu Met Glu
1 5 10 15
Arg Thr Asp Ile Thr Met Lys His Lys Leu Gly Gly Gly Gln Tyr Gly
20 25 30
Glu Val Tyr Glu Gly Val Trp Lys Lys Tyr Ser Leu Thr Val Ala Val
35 40 45
Lys Thr Leu Lys Glu Asp Thr Met Glu Val Glu Glu Phe Leu Lys Glu
50 55 ~ 60
Ala Ala Val Met Lys Glu Ile Lys His Pro Asn Leu Val Gln Leu Leu
65 70 75 80
Gly Val Cys Thr Arg Glu Pro Pro Phe Tyr Ile Ile Thr Glu Phe Met
85 90 95
Thr Tyr Gly Asn Leu Leu Asp Tyr Leu Arg Glu Cys Asn Arg Gln Glu
100 105 110
Val Asn Ala Va1 Va1 Leu Leu Tyr Met Ala Thr Gln Ile Ser Ser Ala
115 120 125
Met Glu Tyr Leu Glu Lys Lys Asn Phe Ile His Arg Asp Leu Ala Ala
130 135 140
Arg Asn Cys Leu Val Gly Glu Asn His Leu Val Lys Val Ala Asp Phe
145 150 155 160
Page 1
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
34479.ST25.txt
Gly Leu Ser Arg Leu Met Thr Gly Asp Thr Tyr Thr Ala His Ala Gly
165 170 175
Ala Lys Phe Pro Ile Lys Trp Thr Ala Pro Glu Ser Leu Ala Tyr Asn
180 185 190
Lys Phe Ser Ile Lys Ser Asp Val Trp Ala Phe Gly Val Leu Leu Trp
195 200 205
Glu Ile Ala Thr Tyr Gly Met Ser Pro Tyr Pro Gly Ile Asp Leu Ser
210 215 220
Gln Val Tyr Glu Leu Leu Glu Lys Asp Tyr Arg Met Glu Arg Pro Glu
225 230 235 240
Gly Cys Pro Glu Lys Val Tyr Glu Leu Met Arg Ala Cys Trp Gln Trp
245 250 255
Asn Pro Ser Asp Arg Pro Ser Phe Ala Glu Ile His Gln Ala Phe Glu
260 265 270
Thr Met Phe Gln Glu Ser Ser Ile Ser Asp Glu Val Glu Lys Glu Leu
275 280 285'
Gly Lys Arg Gly
290
<210> 2
<211> 11
<212> PRT
<213> Homo Sapiens
<400> 2
Val Glu Glu Phe Leu Lys Glu Ala Ala Val Met
1 5 10
<210> 3
<211> 10
<212> PRT
<213> Homo Sapiens
<220>
<221> MISC_FEATURE
<222> (1)..(11)
<223> X is any amino acide
<400> 3
His Arg Asp Leu Ala Ala Arg Asn xaa Leu
1 5 to
Page Z
CA 02511840 2005-06-27
WO 2004/060305 PCT/US2003/041425
34479.ST25.txt
<210> 4
<211> 9
<212> PRT
<213> Homo Sapiens
<400> 4
Asp Phe Gly Leu Ser Arg Leu Met Thr
1 5
<210> 5
<211> 7
<21~> PRT
<213> Homo sapiens
<400> 5
Gly Asp Thr Tyr Thr Ala His
1 5
Page 3