Note: Descriptions are shown in the official language in which they were submitted.
CA 02512305 1995-03-20
A PROCESS FOR THE PRODUCTION OF 2,5-DIAMINO-4,6-
DICHLOROPYRIMIDINE
This application is a division of Canadian Application
Serial No. 2,145,928, filed March 30, 1995. The claims of the
present application are directed to a process to prepare 2,5-
diamino-4,6-dichloropyrimidine. However, for the purpose of
understanding the invention, including all objects and features
which are inextricably bound-up in one and the same inventive
concept, the teachings of those features claimed in the parent
Canadian Application Serial No. 2,145,928 are retained herein.
Accordingly, the retention of any such objects or
features which may be more particularly related to the parent
application or a separate divisional thereof should not be
regarded as rendering the teachings and claiming ambiguous or
inconsistent with the subject matter defined in the claims of
the divisional application presented herein when seeking to
interpret the scope thereof and the basis in this disclosure for
the claims recited herein.
This invention relates to N-(2-amino-4,6-
dichloropyrimidin-5-yl)formamide, and to a process for its
preparation. This compound is a valuable intermediate in the
preparation of antiviral nucleotide derivatives (PCT Application
WO 91/01 310).
N-5-protected-2,5-diamino-4,6-dichloropyrimidines
which serve as intermediates in the preparation of antiviral
nucleotide derivatives have previously been disclosed (EP-A 0
552 758) . However, these compounds suffer from the disadvantage
- 1 -
CA 02512305 1995-03-20
that they are difficult to convert into the corresponding
nucleotide derivatives.
An object of the invention is, therefore, to provide
a pyrimidine derivative which can be used as an intermediate to
obtain the corresponding nucleotide derivatives in good yield,
and also to make available an economic process for preparing
this pyrimidine derivative.
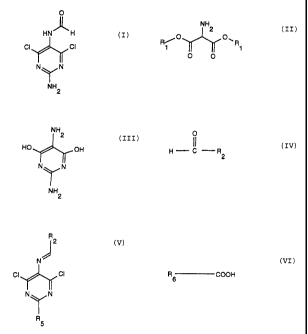
According to the invention there is provided an N- (2-
amino-4,6-dichloropyrimidin-5-yl)formamide of the formula:
O
HN)~ H
CI CI ( I ) TI-Y Ny N
NH2
The invention also provides a process for preparing N-
(2-amino-4,6-dichloropyrimidin-5-yl)formamide of the formula:
O
HN H
CI CI
(I)
Ny N
NH
2
which comprises, in a first step, cyclizing an aminomalonic
ester of the general formula:
- 2 -
CA 02512305 1995-03-20
NH
~O O, R (II)
1 ~
O O
in which R1 denotes a C1-C6-alkyl group, or a salt thereof, with
guanidine or a salt thereof in the presence of a base to give
2,5-diamino-4,6-dihydroxypyrimidine of the formula:
NH
2
HO OH
NTN (III)
NH
2
or a salt thereof, chlorinating the latter, in a second step,
with a chlorinating agent in the presence of an amide of the
general formula:
0
(1
H C -R (IV)
in which R2 denotes a 5- or 6-membered heterocycloalkyl radical,
which is optionally substituted on the hetero atom, or denotes
-NR3R4, in which R3 and R4 are identical or different and are each
a C1-C6-alkyl group or a benzyl group, to give a 4,6-
dichloropyrimidine of the general formula:
- 3 -
CA 02512305 1995-03-20
R
2
N)",
(V)
CI CI
NTN
R
in which R2 has the specified meaning and R5 denotes -NH2, and
reacting the compound (V), in a third step, with an aqueous
solution of a carboxylic acid of the general formula:
R COOH
6 (VI)
in which R6 denotes a C1-C6-alkyl group, branched or unbranched,
or a C3-C6-cycloalkyl group, to give an end product of formula
(I) .
The N-(2-amino-4,6-dichloropyrimidin-5-yl)-formamide
of the invention is a novel compound which has not previously
been described in the literature.
As indicated above, in the first step of the process
of the invention, an aminomalonic ester of the general formula:
NH
pO RI (II)
O O
- 4 -
CA 02512305 1995-03-20
in which Rl denotes a Cl-C6-alkyl group, or a salt thereof, is
cyclized with guanidine or a salt thereof in the presence of a
base to give 2,5-diamino-4,6-dihydroxypyrimidine of the formula:
NH
2
HO OH
NTN (III)
NH
2
or a salt thereof.
The aminomalonic esters of general formula (II) which
are employed as starting compounds may be obtained in known
manner by amidating the corresponding malonic ester derivatives.
An alkali metal alcoholate, such as, for example,
sodium or potassium methoxide, or sodium or potassium ethoxide,
is expediently used as the base, in conformity with EP-A 0 552
758. In-situ-formed sodium methoxide in methanol or sodium
ethoxide in ethanol is preferably employed.
The salts of the aminomalonic ester and of guanidine
which are expediently used are their hydrochloride or
hydrobromide salts.
The cyclization is expediently carried out at a
temperature between room temperature and the reflux temperature
of the relevant solvent, preferably at the reflux temperature.
After a customary reaction time of between 2 and 6
hours, the intermediate of formula (III) can then, where
appropriate, be isolated using customary methods. The synthesis
- 5 -
CA 02512305 1995-03-20
of the end product of formula (I) is preferably carried out
without isolating the intermediate of formula (III).
The second step of the process is carried out by
chlorinating the intermediate of formula (III), or salt thereof,
with a chlorinating agent in the presence of an amide of the
general formula:
0
H C R2 (IV)
to form a 4,6-dichloropyrimidine of the general formula:
R
2
N)"'
ci CI
yl--r (V)
NTN
R
5
The substituent R. denotes -NH2. The substituent R2
denotes either
a 5- or 6-membered heterocycloalkyl radical, which is
optionally substituted on the heteroatom, such as, for example,
piperidinyl, morpholinyl, thiomorpholinyl, pyrrolidinyl or N-
methylpiperazinyl, preferably piperidinyl or pyrrolidinyl; or
NR3R4, in which R3 and R4 are identical or different and
are each a Cl-C6-alkyl group, such a, for example, methyl, ethyl,
propyl, isopropyl, butyl, pentyl or hexyl, preferably methyl, or
a benzyl group.
- 6 -
CA 02512305 1995-03-20
Consequently, N,N-dimethylformamide, N,N-diethyl-
formamide, N,N-diisopropylformamide, N-formylpiperidine, N-
formylmorpholine, N-formylthiomorpholine, N,N-methylformyl-
piperazine or N,N-dibenzylformamide, preferably N,N-
dimethylformamide, N-formylpiperidine or N,N-dibenzyl-formamide
may be employed, for instance, as amides of formula (IV).
The salts of the intermediate of formula (III) which
are expediently used include the hydrochloride and hydrobromide
salts, as well as alkali metal salts, such as, for example, the
sodium or potassium salt.
The chlorinating agents which may be employed are
those which are familiar to the person skilled in the art, such
as, for example, phosphorus oxychioride, thionyl chloride,
suiphuryl chloride, phosphorus trichloride, phosphorus
pentachloride, phosgene or diphosgene. Phosphorus oxychloride
is preferably used as the chlorinating agent.
The chlorinating agent and the amide(IV) are
expediently employed in a molar ratio of from 1 to 0.55 up to 1
to 10, preferably in a molar ratio of from 1 to 0.55 up to 1 to
1.
The chlorination is expediently carried out at a
temperature of from 50 C up to the reflux temperature of the
relevant solvent.
The previously described amide(IV) can also be
employed as the solvent for the chlorination. However, the
chlorination can additionally be carried out in an inert
solvent. Examples of inert solvents which can be employed are
- 7 -
CA 02512305 1995-03-20
toluene, xylene, chloroform, dichloromethane, dichloro-ethane
and chlorobenzene, preferably toluene or dichloro-ethane.
Following a customary reaction time of from 3 to 24
hours, the corresponding 4,6-dichloropyrimidine of general
formula V(R5 =-NHz) can be isolated in a manner familiar to the
person skilled in the art. These 4,6-dichloropyrimidines have
not been described in the literature, and are, therefore, novel
intermediates in the preparation of N-(2-amino-4,6-
dichloropyrimidin-5-yl)-formamide, and an integral part of the
invention. 4,6-Dichloro-N'-(dimethyl-aminomethylene)pyrimidine-
2,5-diamine and 4,6-dichloro-N'-(piperidin-l-
ylmethylene)pyrimidine-2,5-diamine are preferred representative
of the 4,6-dichloropyrimidines (V, RS = -NHZ).
Precursors of these 4,6-dichloropyrimidines (V) may
also be isolated in dependence on the reaction conditions or
working-up conditions which are selected. These precursors are
likewise defined by the general formula V. RS then denotes -NH-
CH=O or -N=CH-R2, in which R2 has the specified meaning. These
precursors are likewise not known from the literature, and are,
therefore, novel intermediates in the preparation of N- (2-amino-
4,6-dichloropyrimidin-5-yl)formamide, and an integral part of
t h e i n v e n t i o n. 4, 6- D i c h l o r o- N, N'-
bis(dimethylaminomethylene)pyrimidine-2,5-diamine, 4,6-dichloro-
N,N'-bis(piperidin-1-ylmethylene-pyrimidine-2,5-diamine or N-
[4,6-dichloro-5-(dimethylamino-methyleneamino)pyrimidin-2-
yl]formamide are preferred representatives.
- 8 -
CA 02512305 2008-09-04
In the third step of the process of the invention, the
4,6-dichloropyrimidines of general formula (V) are reacted with
an aqueous solution of a carboxylic acid of the general formula:
R COOH
6 (VI)
in which R6 denotes a C1-C6-alkyl group, branched or unbranched,
or a C3-C6-cycloalkyl group, to give the end product of formula
(I).
Acetic acid, propionic acid, butyric acid, pentanoic
acid, hexanoic acid, isobutyric acid, pivalic acid,
cyclopropanecarboxylic acid, cyclopentanecarobxylic acid, or
cyclohexanecarboxylic acid may be employed as the carboxylic
acid. Acetic acid, propionic acid or pivalic acid is
expediently employed.
The carboxylic acid is expediently employed in a
concentration of from 20 to 70 vol.%, preferably from 25 to 50
vol.o %
.
It has also been ascertained that, if the carboxylic
acid in the last step (the third step) is employed in an
aqueous, alcoholic solution, this results in the direct
formation of 2,5-diamino-4,6-dichloropyrimidine of formula
(VII), which can also be converted into the corresponding
nucleotide derivative.
-9-
CA 02512305 2008-09-04
Accordingly, the present invention also provides a
process for the production of 2,5-diamino-4,6-dichloropyrimidine
of the formula:
NH
2
CI CI
N
TN (VII)
NH2
wherein, in a first step, an aminomalonic ester of the general
formula:
NH
O O~R~ (II)
O O
in which R1 denotes a C1-C6 alkyl group, or a salt thereof is
cyclised with guanidine or a salt thereof in the presence of a
base to yield a 2,5-diamino-4,6-dihydroxypyrimidine of the
formula:
NH
2
HO OH ( I I I)
NyN
i
NH
2
-10-
CA 02512305 2008-09-04
or a salt thereof, chlorinating the latter in a second step with
a chlorinating agent in the presence of an amide of the general
formula:
0
H 1 R ( I V )
2
in which R2 is a 5- or 6-membered N-heterocycloalkyl residue,
which is optionally substituted on the nitrogen atom, or -NR3R4,
in which R3 and R4 are identical or different and represent a C1-
C6 alkyl group or a benzyl group, to yield a 4,6-
dichloropyrimidine of the general formula:
R
2
N j
CI (V)
CI
N N
R
in which R2 is defined as above and R5 denotes -NHz, -NH-CH=O or
-N=CH-R2, in which R 2 is defined as above, and reacting the
compound V in a third step with a carboxylic acid of the general
formula:
R COOH ( VI )
6
-10a-
CA 02512305 2009-06-19
in which R6 denotes a C1-C6 alkyl group, branched or unbranched,
or a C3-C6 cycloalkyl group, in an aqueous alcoholic solution to
yield the final product of formula (VII).
An aqueous solution of methanol, ethanol, propenol,
butanol, pentanol or hexanol may be employed as the aqueous,
alcoholic solution.
The reaction in the third step is expediently carried
out at a temperature of from 50 to 100 C, preferably at a
temperature of from 70 to 90 C.
Following a customary reaction time of from 1 to 10
hours, the end product of formula (I) or (VII) can be isolated
using working-up methods which are customary for the person
skilled in the art. By contrast with the known N-5-protected-
2,5-diamino-4,6-dichloropyrimidines (EP-A 0 552 758), the novel
end product of formula (I), and also the known end product of
formula (VII), can be converted without difficulty and in good
yield into the corresponding nucleotide derivative.
The following Examples illustrate the invention.
Example 1
Preparation of N- (2-amino-4, 6-dichloropyrimidin-5-yl)
formamide
-lOb-
CA 02512305 1995-03-20
1.1 Preparation of 4,6-dichloro-N'-(dimethylaanino-
methylene)tpyrimidine-2,5-diamine
One-pot process:
A suspension of 25 g (117 mmol) of aminomalonic ester
hydrochloride in 50 ml of methanol was cooled down to 10 C, and
21.07 g of sodium methoxide (30% in methanol) were added. This
suspension was added dropwise to a mixture of 63.2 g (351 mmol)
of sodium methoxide (30% solution in methanol) and 12.55 g
(128.7 mmol) of guanidine hydrochloride in 50 ml of methanol.
The reaction mixture was heated to reflux and then stirred at
this temperature for 16 hours. Subsequently, 13.5 g (370 mmol)
of HC1 gas were passed into the warm suspension. The methanol
was then distilled off. During the distillation, a total of 200
ml of toluene was slowly added dropwise. After all the methanol
had been distilled off, 71.6 g (468 mmol) of POC13 were added
dropwise, followed by 34.2 g (468 mmol) of dimethylformamide,
which was added dropwise at 80 C. The mixture was stirred at
80 C for 17.5 hours and then cooled down to room temperature;
64.7 g of K2C03, dissolved in 150 ml of water, were then added
slowly. The mixture was heated once again at 50 C for 5 hours.
After that, it was adjusted to a pH of 7 using a 30% solution of
NaOH, cooled, and the product was filtered off. After washing
with water and drying in vacuo, 23.2 g (85%) of pure product
were obtained as a pale brown solid.
'H-NMR (DMSO, 400 MHz)b: 2.9-3.0 (2s, 6H);
6.9 (s, 2H);
7.6 (s, 1H).
- 11 -
CA 02512305 1995-03-20
13C-NMR (DMSO, 100 MHz): 33.5;
39.3;
130.3;
153.5;
157.0;
157.1.
m.p.: 195 C (decomp.).
1.2 Preparation of N-(2-amino-4,6-dichloropyrimidin-5-
yl)formamide
a) A solution of 2.35 g (10 mmol) of the product
from procedure 1.1 in 15 g of a 50% aqueous solution of
propionic acid was stirred at 70 C for 7 hours. The mixture was
then cooled down and the product filtered off. After washing
with water and drying in vacuo, 1.66 g of a white solid were
obtained. This solid was suspended in 50 ml of a 2M solution of
K2CO3, and the suspension was stirred at room temperature for 2
hours. Filtration then took place and the product was washed
with water and dried in vacuo. 1.33 g (64%) of pure product
were obtained as an almost white solid.
'H-NMR (DMSO, 300 MHz)b: 9.6-10.1 (b, 1H);
8.3 and 8.0 (2s, 1H);
7.7 and 7.6 (2s, 2H).
b) Proceeding analogously with a), pivalic acid was
employed as the carboxylic acid in place of propionic acid, and
- 12 -
CA 02512305 1995-03-20
the product was worked up in a corresponding manner. The yield
was 70%.
Example 2
Preparation of 4,6-dichloro-N'-(dimethylamino-
methylene)pyrimidine-2,5-diamine
2.1 Preparation of 4,6-dichloro-N,N'-bis(dimethvl-
aminomethylene)pyrimidine-2,5-diamine
A suspension of 4.46 g (25 mmol) of diaminodihy-
droxypyrimidine hydrochloride in 45 ml of toluene and 15.33 g
(100 mmol) of phosphorus oxychloride was heated to 90 C. 7.31
g (100 mmol) of dimethylformamide were added dropwise over a
period of 45 minutes. The mixture was then stirred at 90 C for
hours. The reaction mixture was allowed to cool down, and
100 g of a 10% solution of K2C03 were slowly added to it. 19.5
g of solid K2C03 were then added so that the pH rose to 7. The
20 product was extracted three times with ethyl acetate. The
combined organic phases were dried over MgSO4 and concentrated
on a rotary evaporator. 6.44 g of a pale brown solid were
obtained. Yield: 89%.
'H-NMR (DMSO, 400 MHz)b: 2.9-3.1 (4s, 12H);
7.6 (s, 1H);
8.5 (s, 1H).
- 13 -
CA 02512305 1995-03-20
13C-NMR (DMSO, 100 MHz): 33.5;
39.4;
40.4;
134.2;
153.0;
156.7;
158.0;
159.1.
m.p.: 121.5 - 123 C.
CHN: calculated for C10H14C12N6: C 41.54, H 4.88, N 29.06;
found: C 41.4, H 4.58, N 28.6.
2.2 Preparation of N-[4,6-dichloro-5-(dimethvl-
aminomethyleneamino)pyrimidin-2-yl]formamide
A suspension of 3 g (10 mmol) of the product from step
2.1 in 10 ml of a 50% aqueous solution of acetic acid was
stirred at room temperature for 4.5 hours. The product was then
filtered off and washed twice with 10 ml of water on each
occasion. After drying in vacuo, 2.18 g (83%) of pure product
were obtained as a white solid.
1H-NMR (DMSO, 400 MHz)b: 2.9-3.1 (2s, 6H);
7.7 (s, 1H) ;
9.2 (d, 1H);
11.2 (d, 1H).
- 14 -
CA 02512305 1995-03-20
13C-NMR (DMSO, 100 MHz): 33.6;
39.6;
136.9;
149.6;
153.4;
156.9;
162.5.
m.p.: 172.5 - 174 C.
CHN: calculated for C8H9C12N5: C 36.66, H 3.46, N 26.72;
found: C 36.7, H 3.07, N 25.9.
2.3 Preparation of 4,6-dichloro-N'-(dimethvlamino-
methylene)pvrimidine-2,5-diamine
A solution of 1.85 g (7.1 mmol) of the product from
procedure 2.2 in 25 ml of 10% hydrochloric acid was heated to
40 C and stirred at this temperature for 1.5 hours. The
reaction mixture was cooled down and the pH was adjusted to 8.7
with 2M K2C03. The product, which had precipitated out, was
filtered off and washed with water. After drying in vacuo, 1.52
g (91%) of pure product was obtained as a white solid.
The spectroscopic data were analogous to those given
above.
2.4 Preparation of 4,6-dichloro-N'-(dimethylamino-
methvlene)pyrimidine-2,5-diamine
- 15 -
CA 02512305 1995-03-20
One-pot process:
A suspension of 4.46 g (25 mmol) of
diaminodihydroxypyrimidine hydrochloride in 90 ml of toluene and
15.33 g (100 mmol) of phosphorus oxychloride was heated to 80 C.
7.31 g (100 mmol) of dimethylformamide were added dropwise over
a period of 60 minutes. The reaction mixture was then stirred
at 80 C for 16 hours. It was allowed to cool down and 100 ml of
water were then added. The pH was adjusted to 1 using a total
of 8.4 g of Na2CO3 . The reaction mixture was heated to 40 C and
stirred at this temperature for 4 hours. It was then cooled
down to room temperature and neutralized with a 30% solution of
NaOH, and the product was filtered off. After washing with
water and drying in vacuo, 5.5 g (95%) of product were obtained
as a beige solid. This corresponds to a yield of 89%.
The spectroscopic data were analogous to those given
above.
Example 3
Preparation of 4,6-dichloro-N'-(piperidin-1-yl-
methylene)pyrimidine-2,5-diamine
3.1 Preparation of 4,6-dichloro-N,N'-bis(pineridin-l-
ylmethylene)pyrimidine-2,5-diamine
A suspension of 3.57 g (20 mmol) of diaminodihy-
droxypyrimidine hydrochloride in 70 ml of toluene and 12.27 g
(80 mmol) of phosphorus oxychloride was heated to 80 C. 9.05 g
(80 mmol) of 1-formylpiperidine were added dropwise over a
- 16 -
CA 02512305 1995-03-20
period of 60 minutes. The reaction mixture was then stirred at
80 C for 22 hours. It was allowed to cool down, and 100 ml of
a 1M solution of K2C03 were then added to it and the pH was
adjusted to 7 with NaOH. The product was extracted with three
portions of ethyl acetate. The combined organic phases were
dried over MgSO4 and concentrated on a rotary evaporator. 10.87
g of an oil were obtained which still contained a large quantity
of N-formylpiperidine. The product was purified by suspending
in hexane and then filtering. Yield: >90%.
1H-NMR (DMSO, 300 MHz)b: 8.5 (s, 1H);
7.7 (s, 1H) ;
3.4-3.8 (m, 8H);
1.5-1.9 (m, 12H).
3.2 Preparation of 4,6-dichloro-N'-(piperidin-1-yl-
methylene)gvrimidine-2,5-diamine
A solution of 9.9 g (18.2 mmol) of the product from
procedure 3.1 in 73 g of 10% HC1 was first stirred at room
temperature for 4.5 hours and then s t i rred at 470C for 2 hours.
It was cooled down and the pH was adjusted to 7 using 30% NaOH.
The product was filtered off, washed with water and dried in
vacuo. 4.68 g (88%) of product were obtained as a pale brown
solid.
'H-NMR (DMSO, 300 MHz)b: 7.55 (s, 1H);
7.4 (s, 2H);
3.2-3.7 (m, 4H);
1.5-1.8 (m, 6H).
- 17 -
CA 02512305 1995-03-20
Example 4
Subsequent conversion of N-(2-amino-4,6-dichloro-
pyrimidin-5-yl)formamide into 2-amino-9-butyl-6-
chloropurine
4.1 Preparation of N-(2-amino-4-butylamino-6-chloro-
pyrimidin-5-yl)formamide
A solution of 0.43 g (2 mmol) of N-(2-amino-4,6-
dichloropyrimidin-5-yl)formamide and 0.31 g (4.2 mmol) of n-
butylamine in 10 ml of tetrahydrofuran was stirred at room
temperature for 17 hours. Water was then added to the reaction
mixture and the product was extracted with ethyl acetate. After
drying the organic phase over MgSO4, and concentrating it on a
rotary evaporator, 0.49 g of a white solid was obtained which
was purified by recrystallizing in toluene. 0.46 g of pure
product was obtained, corresponding to a quantitative yield.
'H-NMR (DMSO, 300 MHz)b: 9.0 and 8.6 (s and d, 1H);
8.1 and 7.8 (s and d, 1H);
7.0 and 6.75 (2t, 1H);
6.5 and 6.4 (2s, 2H);
3.3-3.4 (m, 2H);
1.4-1.6 (m, 2H);
1.2-1.4 (m, 2H);
0.9 (t, 3H).
- 18 -
CA 02512305 1995-03-20
4.2 Preparation of 2-amino-9-butyl-6-chloropurine
A suspension of 0.51 g (2 mmol) of the product from
procedure 4.1 in 10 ml of diethoxymethyl acetate was heated
under reflux for 3.5 hours. It was then completely evaporated,
and 30 ml of a 0.5M solution of HC1 was added to the residue.
After 3 hours at room temperature, the yellow solution was
adjusted to a pH of 8 using NaOH, and the resulting suspension
was extracted 3 times with ethyl acetate. The combined organic
phases were dried and concentrated on a rotary evaporator. 0.46
g (97%) of the desired product was obtained, which was 95% pure
(according to 1H-NMR) .
'H-NMR (DMSO, 300 MHz)b: 8.2 (s, 1H);
6.9 (s, 2H) ;
4.05 (t, 2H);
1.6-1.9 (m, 2H);
1.1-1.4 (m, 2H);
0.9 (t, 3H).
Comparative Example
4.3 Conversion of 5-(N-ethoxvcarbonyl)-2-amino-4,6-
dichloroAVrimidine into 2-amino-9-butvl-6-chloro-7,9-
dihydroyurin-8-one
As a Comparative Example, 5-(N-ethoxycarbonyl)-2-
amino-4,6-dichloropyrimidine, as a derivative of an N-5-
- 19 -
CA 02512305 1995-03-20
protected-2,5-diamino-4,6-dichloropyrimidine (EP-A 0 552 758),
was reacted under conditions which were analogous to those in
Example 4.
However, under these conditions, 2-amino-9-butyl-6-
chloro-7,9-dihydropurin-8-one was obtained rather than 2-amino-
9-butyl-6-chloropurine.
Example 5
Preparation of 2,5-diamino-4,6-dichlorogvrimidine
A mixture of 2.35 g (10 mmol) of the product from
Example 1.1 in 5 g of pivalic acid, 10 ml of methanol and 15 ml
of water was stirred at 80 C for 4.5 hours. The precipitated
solid was subsequently filtered off and the filtrate was
neutralized with a concentrated solution of NaOH. It was then
extracted with ethyl acetate and the combined organic phases
were dried over MgSO4. Following concentration on a rotary
evaporator, 1.40 g of a product mixture remained, 65% of which
(determined by 'H-NMR) consisted of the desired product (yield
51%). The product was not subjected to further purification.
Example 6
Subsequent conversion of 2,5-diamino-4,6-dichloro-
pyrimidine into 2-amino-9-butyl-6-chloropurine
6.1 2,5-Diamino-4-butylamino-6-chlorovyrimidine
- 20 -
CA 02512305 1995-03-20
A suspension of 2.5 g (14 mmol) of 2,5-diamino-4,6-
dichloropyrimidine, 1.37 g (18.7 mmol) of n-butylamine and 6 ml
of triethylamine in 60 ml of butanol was stirred at 100 C for 9
hours. The reaction mixture was then cooled down and
concentrated to dryness on a rotary evaporator. Water was added
to the residue and the product was extracted with ethyl acetate.
After the organic phase had been dried over Na2SO4 and
concentrated on a rotary evaporator, the residue was suspended
in 10 ml of isopropyl ether, and the product was filtered off
and dried. 2.24 g(75%) of an orange-red solid were obtained.
1H-NMR (CDC13, 300 MHz)5: 5.4 (broad s, 1H) ;
4.6 (s, 2H);
3.4 (t, 2H) ;
2.7 (s, 2H);
1.6 (m, 2H);
1.4 (m, 2H) ;
0.95 (t, 3H).
6.2 Preparation of 2-amino-9-butvl-6-chloropurine
A solution of 1.0 g (4.63 mmol) of the product from
procedure 6.1 in 10 ml of dimethylformamide and 10 ml of ethyl
orthoformate was cooled down to 0 C, and 0.5 ml of concentrated
HC1 was added. As a result, the temperature rose to 10 C. The
mixture was then stirred at room temperature for 22 hours. It
was then completely evaporated, and 40 ml of an 0.5M solution of
HC1 were added to the residue. After 2 hours at room
- 21 -
CA 02512305 1995-03-20
temperature, the yellow solution was adjusted to a pH of 8 with
NaOH, and the resulting suspension was extracted 3 times with
ethyl acetate. The combined organic phases were dried and
concentrated on a rotary evaporator. 1.1 g (quantitative) of
the desired product was obtained, which was 95% pure (by 1H-
NMR ) .
The spectroscopic data were analogous to those in
Example 4.2.
- 22 -