Note: Descriptions are shown in the official language in which they were submitted.
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
APTAMER THERAPEUTICS USEFUL IN OCULAR PHARMACOTHERAPY
FIELD OF THE INVENTION
[0001] The invention relates generally to the field of nucleic acid
therapeutics and more
particularly to nucleic acid therapeutic compositions capable of binding to
cytokines, growth
factors and cell surface receptors, individually or in combinations of two or
more, and
methods for delivering these nucleic acid therapeutics in the treatment of
glaucoma and other
proliferative diseases of the eye.
RE1CI~GROiJND OF THE INVENTION
[0002] Aptamers are nucleic acid molecules having specific binding affinity to
molecules
through interactions other than classic Watson-Crick base pairing.
[0003] Aptamers, like peptides generated by phage display or monoclonal
antibodies (IvIAbs),
are. capable of specifically binding to selected targets and, through binding,
block their
targets' ability to function. Created by an in vitro selection process from
pools of random
sequence oligonucleotides (Fig. 1), aptamers have been generated for over 100
proteins
including growth factors, transcription factors, enzymes, immunoglobulins, and
receptors. A
typical aptamer is 10-15 kI?a in size (30-45 nucleotides), binds its target
with sub-nanomolar
affinity, and discriminates against closely related targets (e.g., will
typically not bind other
proteins from the same gene family). A series of structural studies have shown
that aptamers
are capable of using the same types of binding interactions (~.~., hydrogen
bonding,
electrostatic complementarity, hydrophobic contacts, and static exclusion)
that drive affinity
and specificity in antibody-antigen complexes.
[000.] Aptamers have a number of desirable characteristics for use as
therapeutics (and
diagnostics) including high specificity and affinity, biological efficacy, and
excellent
pharmacokinetic properties. In addition, they offer specific competitive
advantages over
antibodies and other protein biologics, for example:
[0005] 11 Speed and control. Aptamers are produced by an entirely in vitro
process, allowing
for the rapid generation of initial therapeutic leads. Izz vitz-~ selection
allows the specificity
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
and affinity of the aptamer to be tightly controlled and allows the generation
of leads against
both toxic and non-immunogenic targets.
[0006] 2) Toxicity and Immunogenicity. Aptamers as a class have demonstrated
little or no
toxicity or immunogenicity. In chronic dosing of rats or woodchucks with high
levels of
aptamer (10 mg/kg daily for 90 days), no toxicity is observed by any clinical,
cellular, or
biochemical measure. Whereas the efficacy of many monoclonal antibodies can be
severely
limited by immune response to antibodies themselves, it is extremely difficult
to elicit
antibodies to aptamers (most likely because aptamers cannot be presented by T-
cells via the
IVIIIC, and the immune response is generally trained not to recognize nucleic
acid fragments).
[0007] 3LAdministration. Whereas all currently approved antibody therapeutics
are
administered by intravenous infusion (typically over 2-4 hours), aptamers can
be administered
by subcutaneous injection. This difference is primarily due to the
comparatively low
solubility and thus, large volumes necessary for most therapeutic lVIAbs. With
good
solubility (>150 mg/ml) and comparatively low molecular weight (aptamer: 10-50
kDa;
antibody: 150 kDa), a weekly dose of aptamer may be delivered by injection in
a volume of
less than 0.5 ml. Aptamer bioavailability via subcutaneous administration is >
80°1° in
monkey studies (Tucker et al., J. Chromatography B. 732: 203-212, 1999). In
addition, the
small size of aptamers allows them to penetrate into areas of conformational
constrictions that
do not allow antibodies or antibody fragments to penetrate, presenting yet
another advantage
of aptamer-based therapeutics or prophylaxis.
[0008] 4) Scalability and cost. Therapeutic aptamers are chemically
synthesized and
consequently can be readily scaled as needed to meet production demand.
~6lhereas
cliff culties in scaling production are currently limiting the availability of
some biologics, and
the capital cost of a large-scale protein production plant is enormous, a
single large-scale
synthesizer can produce upwards of 100 kg of oligonucleotide per year and
requires a
relatively modest initial investment. The current cost of goods for aptamer
synthesis at the
kilogram scale is estimated at $500/g, comparable to that for highly
opl;imized antibodies.
Continuing improvements in process development are expected to lower the cost
of goods to
< $100/g in five years.
[0009] 5) Stability. Therapeutic aptamers are chemically robust. They are
intrinsically
adapted to regain activity following exposure to factors such as heat and
denaturants, and can
2
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
be stored for extended periods (> 1 yr) at room temperature as lyophilized
powders. In
contrast, antibodies must be stored in a refrigerated environment.
Glaucoma
[0010] Two leading causes of vision loss are glaucoma and age-related
maculodegenerative
disease (AMD). Glaucoma is a proliferative disease of the eye affecting 2.2
million patients
in the U.S. and 65 million patients worldwide. Glaucoma disease is associated
with reduced
fluid drainage from the eye and an elevation in intraocular pressure (IOP).
When IOP is high,
individual nerve fiber cells die leading to vision loss. elision loss is
manifested by
characteristic optic disc damage, nerve fiber layer defects, visual field loss
starting at the
periphery, and eventual blindness. Glaucoma disease progression is currently
irreversible, but
it may be slowed with therapeutic drugs to modulate fluid production and IOP.
The current
therapeutic agents of choice in treating advanced glaucoma are cytotoxic
agents delivered by
trabeculectomy. There arc estimates of approximately 120,000 surgeries each
year to treat
glaucoma patients in the U.S.
[0011] The first line of glaucoma treatment is typically the use of
therapeutic drugs to
modulate intraocular fluid levels. Glaucoma filtering microsurgery, or
trabeculectomy, is a
second line of treatment in which a tiny puncture is made in the sclera of the
eye to allow
fluid to drain into a bleb, thereby reducing IOP. However, post-surgical
complications are
significant and can lead to continued vision loss. Complications from surgery
arise when
incomplete wound healing and scarring results in a return to high IOP and a
need for
additional surgery. During trabeculectomy, antibiotics and corticosteroids can
be injected
subjunctivally into the inferior foraxix or collagen shields soaked in them
can be used to cover
the eye to control the extent of post-surgical scarring. In order to combat
the effects of
scarring, antimetabolite agents, such as n ntomycin-C and 5-fluorouracil, arc
used to control
the extent of post-surgical scarring. A failed trabeculectomy is considered to
exist when there
is less than a 25% drop in IOP post-surgically, in which case a second
drainage surgery is
performed.
[0012] To prevent trabeculectomy failure, topical steroids and or antifibrotic
agents are
commonly used. Steroids like prednislolone acetate 1 %, 4-6 times daily are
often used
postoperatively and tapered after 4-8 weeks. Cycloplegics such as atropine 1 %
or
cyclopentolatc 1 % can be sued up to four times daily in cases prone to
shallow anterior
3
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
chamber. To prevent excessive postoperative subconjunctival fibrosis,
adjunctive
antimetabolites such as mitomycin C and 5-fluorouracil are used. They inhibit
fibroblast
proliferation and subsequent scar tissue formation. Mitomycin C is 100 times
more potent
than 5-FU. Both are associated with a higher success rate but also with a
higher complication
rate so use is determined on a case by case basis. Mitomycin C (0.2 - O.Smg/ml
solution) or
5-FU (25-50 mg/ml solution) can be applied for 1-5 minutes using a soaked
cellulose sponge
or filter paper over the episclera before dissecting the scleral flap. They
can also be applied
under the scleral flap and time of exposure may vary depending on the expected
risk of
fibrosis. Further, during surgery, the conjunctival-Tenon's layer is draped
over the sponge
avoiding contact with the wound edge. After removal, the entire area is
cleansed thoroughly
with a salt solution. 5-FU can be delivered subconjunctivally in Smg aliquots.
Total number
of injections is adjusted depending on filtering bleb function and tolerance
of the corneal
epithelium. Complications associated with 5-FU include corneal and
conjunctival epithelial
toxicity, corneal ulcers, conjunctival wound leaks, subconjunctival hemorrhage
or inadvertent
intraocular spread of 5-FU.
[0013] Severe complications can arise from repeat trabeculectomies and the use
of
antimetabolite therapy, including fluid leakage, intraocular hypotony (low
IOP), and general
tissue toxicity (Blindish, et al., Ophthalmology (2002), 109:1336-1341;
Belyea, et al., Am J
Ophthalmol (1999), 124:40-45; Kupin, et al., Am J Ophthalmol (1995), 119:30-
39).
Antimetabolite agents can further damage eye tissue leading to low IOP, or
even blindness.
Failure of antimetabolite therapy in glaucoma treatment is defined by a two-
line drop in
Snellen visual acuity tests (Membrey, et al., (2000). Br J Ophthalmology,
84:1154-58).
[0014] Progression of glaucoma disease is associated with increases in the
levels) of
transforming growth factor cytokines in the eye. The transfornaing growth
factor (3 (TCF(3
subfamily) is comprised of three members TCaF(31, TGF(32, and TFCi(33. TGF[is
are
multifunctional cytokines that control growth, differentiation, and
development. They are
expressed by many different cell types, and most cells are responsive to
TCaF(3s.
Transforming growth factor beta 2 (TGF(32) is a 251cD homodimer growth factor
cytokine
that is involved in cell proliferation, differentiation, and extracellular
matrix formation.
Several receptors (types I-V) mediate the cellular response to TGF(32 and its
isoforms
TCaF(3-1 and TGF(3-3 in a variety of cells. The type II receptor is the main
signaling receptor
4
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
responsive to TGF(32, although high affinity binding of TGF[i2 to the type
III, non-signaling
receptor is believed to enhance type II dependent signaling.
[0015j In many parts of the body, TGF(31 predominates, but in the eye, TGF(32
is the
predominant form. Transforming growth factor beta 2 (TGF(32) is implicated in
ocular
wound healing and is suggested to play a role in scarring associated with
glaucoma surgery.
The ocular scarnng response is mediated by TGF(32 (Cordeiro, et al., Invest
Ophthalmol. Vis.
Sci. (1999), 40:1975-1982). Elevated levels of TGF(32 are detected in aqueous
humor in
glaucomatous eyes compared to control levels in normal eyes: 21 pM
glaucomatous versus 12
pM for normal eyes (~chiai, et al., Jpn J. ~phthalmol. (2002), 46:249-253).
Trabecular
meshwork cells, as well as ciliary body cells, express and secrete TGF(32. It
has been
suggested that TGF(32 contributes to the excess accumulation of extracellular
components in
the aqueous outflow system observed in aging and glaucomatous eyes (Tripathi,
et al., Exp.
Eye Res. (1994), 58:523-528).
[0016] There are clinical trials currently underway testing the use of
alternative therapeutics,
such as antibodies specific to TGF[32, which show prevention of excessive post-
operative
scarring in patients undergoing eye surgery for glaucoma (Broadway, et al.,
Adjunctive anti-
TGF(32 human MAb as a novel agent to prevent scarring following
phacotrabeculectomy.
May 2002, ARVO Meeting Poster #3331). However, there are undesired side
effects from
antibody therapy in the eye, such as inflammation or immune responses to the
foreign
antibody, which can lead to secondary causes of increase in IOP in the
glaucomatous eye.
There have also been studies with anti-sense nucleic acids that inhibit
expression of TGF(32
in the eye leading to reduced surgical scarring and improved surgical outcome
in the rabbit
model (Cordeiro, et al., Gene Therapy (2003), 10:59-71). However, this
approach may
interfere vyith normal, post-surgical healing and tissue regeneration
processes in the eye and
contribute to cytotoxicity within the eye and surrounding tissue. 'Thus, the
use of these
alternative therapeutics has not completely eliminated the side effects and
secondary
deleterious effects of the current glaucoma therapeutics.
Aye-related Maculode~enerative Disease (AMD)
[0017] Age-related Maculodegenerative Disease (AMD) is a degenerative
condition of the
macula. It is the most common cause of vision loss in the United States in
those 50 years old
or older, and its prevalence increases with age. It affects 15 million people
in the United
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
States alone. AMD is caused by hardening of the arteries that nourish the
retina. This
deprives the retinal tissue of oxygen and nutrients that it needs to function
and thrive. As a
result, the central vision deteriorates. AMD is classified as either wet
(neovascular) or dry
(non-neovascular), based on the absence or the presence of abnormal growth of
blood vessels
under the retina.
[OO1~J Wet AMD affects about 10% of patients who suffer from macular
degeneration. This
type occurs when new vessels form to improve the blood supply to oxygen-
deprived retinal
tissue. 1-Iowever, the new vessels are very delicate and break easily, causing
bleeding and
damage to surrounding tissue. The wet form can manifest in two types: classic
or occult.
~ver 70°!° of patients with the wet foam have the occult type.
So far, only the classic wet type
is treated with conventional laser photocoagulation to stabilize vision or to
limit the growth of
abnormal blood vessels. The remaining majority ofpatients with wet AMD cannot
be treated
with the laser procedure. The current laser treatment does not improve vision
in most treated
eyes because the laser destroys not only the abnormal blood vessel but also
the overlying
macula.
[0019) Dry AMD although more common, typically results in a less severe, more
gradual loss
of vision. It is characterized by drusen and loss of pigment in the retina.
Drusen are small,
yellowish deposits that form within the layers of the retina. Currently there
is no proven
treatment for the dry type, but the loss of vision tends to be milder and the
disease progression
is rather slow. There is no currently proven medical therapy for dry macular
degeneration.
Proliferative Vitreo-Retinopathy (PVR)
[0020) ~ther causes of blindness are retinal detachments. Retinal detachments
have an
annual incidence in the general population of 1:10,000. There are, however, a
variety of
associated ocular and systemic disease states that increase the chances of
retinal detachment.
'These include: diabetes, high myopia, pseudophahia and aphasia, blunt and
penetrating ocular
trauma, and cytomegalovinis retinitis associated with acquired immunodef
ciency syndrome.
Vitrectomy is the standard of care for retinal detachment. Annually, there are
about 200,000
vitrectomies in the IJ.S., and 300,000 vitrectomies outside the United States.
Proliferative
Vitroretinopathy (PVR) occurs in ~ 10% of retinal detachments or 62,600 cases
a year
worldwide and 2,800 cases a year in the L1.S. PVR is the most common cause of
failure in
retinal reattachment surgeries.
6
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
[0021] Platelet derived growth factor (PDGF) is a strong mitogen and is known
to play a
crucial role in a variety of proliferative diseases. PDGF is postulated to be
involved in the
regulation of abnormal growth and migration of filial and retinal pigmented
endothelial cells
(RPE) cells in PVR.
[0022] PDGF forms dimers of the A and B subunits, i.e., AB heterodimers, and
AA and BB
v
homodimers. PDGF has a pivotal role in regulation of normal cell proliferation
and the
mediation of pathological cell growth such as tissue, fibrosis, proliferative
disorders and
angiogenesis. It is involved in restenosis, renal scarring, wound healing and
cancer. Most
tumor cell lines secrete PDGF, and heavily express PDGF receptor (PDGF-R). The
amino
acid sequence of PDGF resembles that of an oncogene.
[0023] pIigh retinal expression of PDGF results in traction retinal detachment
from
proliferation of both vascular and nonvascular cells. PDGF promotes
proliferation of
trabecular meshwork cells, enhances dedifferentiation of retinal pigment
epithelial from
hexagonal to flattened cells, increases expression of alpha smooth muscle
actin, enhances
myoid differentiation and collagen gel contraction. There are elevated levels
in the vitreous
of patients with AMD.
[0024] While many growth factors are thought to contribute to PVR, including
TGF(3, VEGF,
BFGF, HGF and IL-6P, PDGF has been shown to play the most important role.
[0025] PVR is the most common complication following a retinal detachment
associated with
a retinal hole or break. PVR refers to the growth of cellular membranes
(composed primarily
of filial cells and retinal pigment epithelial cells, but also fibroblasts and
inflammatory cells)
within the vitreous cavity and on the front and back surfaces of the retina.
'These membranes,
which are essentially scar tissues, exert traction on the retina and may
result in recurrences of
retinal detachment, eveaa after an initially successful retinal detachment
procedure. PVR may
be associated with spontaneous reopening of otherwise successfully treated
retinal breaks and
may even cause the development of new retinal breaks. It may be associated
with severe
distortion and "stiffness" of the retina, as a result of the contracting
membranes. This aspect
of the condition results in damage to vision.
[0026] PDGF promotes proliferation of trabecular meshwork cells, enhances
dedifferentiation
of retinal pigment epithelial from hexagonal to flattened cells, increases
expression of alpha
smooth muscle actin, enhances myoid differentiation and collagen gel
contraction. There are
elevated levels in the vitreous and preretinal membranes of patients with PVR.
In
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
experimental models, cells without PDGFRs were ineffective in inducing PVR. In
particular,
it is PDGF-AA that results in extensive proliferation of filial cells and
traction retinal
detachment without vascular cell involvement. It has been shown that it is the
PDGF-alphaR
that is capable of driving the events leading to PVR. PDGF mutants are all
capable of
blocking PDGF stimulated cell cycle progression and varied in their kinase
activity and
ability to block PVR. A truncated receptor is effective in blocking PVR.
[0027) The stages of PVR development are: 1)Breakdown of the blood-retinal
barrier;
2)Chemotaxis and cellular migration; 3)Cellular proliferation; 4)Membrane
formation with
remodeling of the extracellular matrix; and 5)Contraction. PDGF plays an
important role at
each of these stages as described below for each of the five stages of PVR
development.
[0020 The RPE forms a mosaic of cells between the choroid and neural retina
that serves as
the outer blood-retinal burner regulating retinal homeostasis and visual
function. The initial
step in PVR is the dedifferentiation of RPE cells: morphological alteration
from a mitotically
quiescent hexagonal shape to a migrating flattened shape with a loss of
epithelial
characteristics. Additionally, RPE cells decrease their expression of
cytokeratin and begin
expression of alpha smooth muscle actin (alpha-SMA). Alpha-SMA is essential
for
contractile activity and it increases in a time dependent manner. PDGF
enhances
dedifferentiation of RPE cells, myoid differentiation and alpha-SMA
expression.
[0029) The breakdown of the blood-retinal barrier allows for entry of various
cell types
including RPE cells, filial cells, fibroblasts, macrophages, leucocytes and
serum components
into the vitreous and subretinal space. RPE and filial cells are the
predominant types. These
cells adhere to the retina and vitreous gel. PDGF is a potent stimulator of
RPE and filial cell
migration.
[000) ~nce attached to the retina and vitreous gel these cells proliferate
extensively. PDGF
triggers proliferation of RPE and filial cells and induces DhJA Synthesis.
[001] RPE cells transdifFerentiate to myofibroblasts or mesenchymal-like cells
and form
epiretinal membranes on the surface of the retina and within the vitreous and
begin to
synthesize extracellular matrix. l~Tormal RPE cells do not express PDGF or its
receptor.
However, PDGF and it's receptor are highly expressed on RPE cells that form
PVR
membranes. PDGF stimulates fibroblasts to synthesize and deposit collagen.
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
Finally, the membranes exert contractile force and traction on the attached
retina leading to
reopening of breaks and to retinal detachment. PDGF potentiates RPE
contractile ability and
stimulates fibroblast and collagen gel contraction.
Proliferative Diabetic Retinopathy (PDR)
[0032] Proliferative Diabetic Retinopathy (PDR) is a complication of diabetes
that is caused
by changes in the blood vessels of the retina. When blood vessels in the
retina are damaged,
they may leak blood and grow fragile, brush-like branches and scar tissue.
This can blur or
distort the vision images that the retina sends to the brain. Diabetic
retinopathy is a major
cause of blindness in developed countries and is the leading cause in diabetic
patients aged 25
to 74. It is responsible for 12,000 to 24,000 new cases of blindness in the
U.S. each year. It is
estimated that 25% of diabetics suffer from diabetic retinopathy and incidence
increases to
60% after 5 years and 80°A° after 10-15 years with type I
diabetes. The U.S. patient population
is 5 million and the potential U.S. market is ~S billion. 'The disease is
characterized by
hyperglycaemia, basement membrane thickening, pericyte loss, microaneurysms
and
preretinal neovascularization which can lead to blindness through hemorrhage
and fractional
retinal detachment.
[0033] Nonproliferative diabetic retinopathy is characterized by intraretinal
microaneurysms,
hemorrhages, nerve-fiber-layer infarcts, hard exudates and microvascular
abnormalities.
Macular edema is the principle mechanism for vision loss. It results from
vascular leakage
from microaneurysms in the macular (cenfral area of the retina) capillaries.
Leakage may
progress to macular thickening associated with hard exudates or cystoid
changes and this
often results in various degrees of cenfral vision loss. Proliferative
diabetic retinopathy is
characterized by retinal neovascularization. It is graded according to the
presence, location,
severity and associated hemorrhagic activity of retinal neovascularization. It
is associated
with severe vision loss. The pathology of diabetic retinopathy can be
attributed to the
following disease states. Circulation problems cause regions of the retina to
become oxygen
deprived or isclaemic. hTeo vascularization causes new vessels to start to
grow within the
vitreous to maintain adequate oxygen levels. Flood seeping out of the newly
formed
capillaries and the formation of scar tissue creates fraction on the retina
causing small tears.
Tears are followed by fluid build-up underneath or in between the layers of
the retina and
9
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
detachment occurs. Patients experience blurred vision, floaters, flashes and
sudden loss of
vision due to the hemorrhaging, edema and scar tissue formation.
[0034] Vitrectomy is a microsurgical procedure used to repair retinal
disorders, many of
which were previously considered inoperable. Victrectomy is 90% successful if
performed
before the retina is seriously damaged.
[0035] PDGF-B plays an important role in the pathogenesis of PDR in
synergistic action with
other growth factors. Hypoxia increases expression of PDGF-B. High retinal
expression of
PDGF-B results in traction retinal detaclunent from proliferation of both
vascular and
nonvascular cells. PDGF-B induces proliferation of several cell types in the
retina including
astrocytes, pericytes, and endothelial cells. 'Thc cells proliferate on the
surface of the retina
and cords of cells migrate into the inner nuclear layer and exert traction on
the retina,
resulting in outer retinal folds and focal areas of detachment that enlarge
and lead to total
retinal detachment.
[0036] This feature of PDR membranes makes treatment difficult and generally
requires
cutting connections within the retina for removal rather than just peeling the
membranes from
the surface. PDGF directly acts on endothelial cells through PDGFR-b to induce
angiogenesis. PDGF has a pivotal role in regulation of normal cell
proliferation and the
mediation of pathological cell growth such as tissue, fibrosis, proliferative
disorders and
angiogenesis. It is involved in restenosis, renal scarring, wound healing and
cancer. PDGF
acts on fibroblasts, smooth muscle cells, neuroglial cells and stimulates
proliferation of
connective-tissue cells.
~ther Growth Factors Cytolcines and Cell Surface Proteins
[0037] ~ther grov~~th factors, cytokines and cell suuface proteins are
implicated in ocular
vround healing and are suggested to play a role in sca~xing associated with
glaucoma surgery.
Such cytokines, cell surface proteins and growth factors implicated in various
ocular diseases
include ICAO-19 IGF-1, VEGF/VFGF-R, ThTF-~., and o.V(i3. Intercellular
F~dhesion
I~loleculc 1 (ICI-~IvI-1) is a 76 to 115 lgDa surface glycoprotein with five
extracellular
immunoglobulin-like domains that plays a particularly important role in
diabetic retinopathy.
ICAI~I-1-mediated leukostasis is causative in the pathogenesis of diabetic
retinopathy.
IChIVI-1 interactions with X32 integrins located on the surface of leukocytes
(neutrophils,
basophils, lymphocytes, eosinophils, monocytes) are important for their firm
adhesion to the
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
endothelium and their transendothelial migration to sites of inflammation.
ICAM-1 facilitates
adhesion of leukocytes to the retinal vasculature in diabetic retinopathy and
is involved with
retinal endothelial cell injury and death via lesions that produce
irreversible retinal ischemia
through inability of capillaries to support blood flow. Inhibition of ICAM-1
bioactivity
blocks diabetic retinal leukostasis and potently prevents blood-retinal
barrier breakdown.
[003] Insulin-like Growth Factor-1 (IGF-1) is a 7.5 kDa peptide, having 50%
homology to
proinsulin (50%) and is produced primarily in the liver under control of
growth hormone.
IGF-1 is a potent mitogen/stimulator of cell proliferation and a strong anti-
apoptotic agent.
Its function is modulated by sine IGF binding proteins (IGFI3Ps), and its
levels are influenced
by developmental stage and nutrition. 'The effects of IFG-1 range from cell
growth and
protection, resistance to oxidative stress, promotion of bone and muscle
growth, and
protection of neuronal cells. IGF-1 is implicated in angiogenesis, and IGF-1
in conjunction
with VEGF has been implicated in playing a role in proliferative diabetic
retinopathy (PDH).
PIER is a complication of diabetes that is caused by changes in the blood
vessels in the retina.
When blood vessels in the retina are damaged, they may leak blood and grow
fragile, brush-
like branches and scar tissue, which can blur or distort vision images that
the retina sends to
the brain.
[0039] Transcription of vascular endothelial growth factor and its receptor
(VEGF and
VEGF/R) is enhanced by advanced glycation end products and by the presence of
insulin.
The accumulation of advanced glycation end products in the diabetic retina
contributes to
neovascularization, which can result in loss of vision. The stimulation of
VEGF synthesis by
insulin may lead to transient acceleration of retinal neovascularization in
patients with
diabetes after insulin therapy is instituted.
[0040] Tumor necrosis factor-alpha (TI~TF-o.) is present at increased levels
within the eye
during the retinal processes of inflammation and angiogenesis. TIVF-oc
promotes proliferation
of trabecular meshwork cells and modulates the expression of trabecular
meshwork, matrix
metalloproteinases and tissue inhibitors. 'The integrin alpha 5 beta 3
(oeV[33) promotes
angiogenesis in PDR as well as in AMID (Enaida, et czl., Fukushima J Med Sci.
44(1):43-52.
(1990).
[0041] There is therefore a need for therapeutics specific to cytokine, growth
factors, and cell
surface receptors that significantly reduce or eliminate deleterious side
effects in the treatment
of eye diseases and almost eliminate disease progression.
11
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
BRIEF DESCRIPTION OF THE DRAWINGS
[0042] Figure 1 is an illustration depicting the in vitro aptamer selection
(SELEX~) process
using pools of random sequence oligonucleotides;
[0043] Figure 2 illustrates various strategies for synthesis of high molecular
weight PEG-
nucleic acid conjugates;
[0044] Figure 3A is an illustration depicting the ARC82 TGF~2 therapeutic
aptamer (SEQ
>I~ No. 151); and Figure 3B is a graph depicting the plasma half life profile
of ARC82;
[0045] Figure 4A is a flow chart illustrating S75 size exclusion
chromatography; Figure 4B is
an elution profile of TGF(32 in S75 size exclusion chromatography; Figure 4C
is an
illustration depicting a polyacrylamide gel electrophoresis (PAGE) gel
containing the TGF(32
dimer PAGE band.
[0046] Figure SA is a graph depicting human ( ~ ) or rat (~) TGF[i-2 protein
binding to
increasing concentration of the ARC77 TGF(3-2 specific aptamer; Figure SB is a
graph
depicting competitive binding of non-radiolabeled ARC77 (0) and ARC81 (~) (fit
to
Equation 2 estimates of aptamer dissociation constants) competed with 32P-
labeled ARC77
for binding to human TGF(32;
(0047] Figure 6A is a graph depicting inhibition of TGF(32 by the ARC77, ARC78
and
ARC81 aptamers; Figure 6B is a graph depicting inhibition of the
antiproliferative effects of
the human and rodent forms of TGF(32 by the ARC77 aptamer; Figure 6C is a
graph depicting
the dissociation constant (I~) of ARC77 human wild type (~7JT), mouse (NTI~)
and N-
terminal I3is tagged versions of human TGF(32;
[004] Figure 7A is a graph depicting inhibition of the antiproliferative
effects of low
concentrations of rabbit aqueous humor by the ARC81 aptamer and the conirol9
an anti-
TGF(32 antibody; Figures 7B and 7C are graphs illustrating dose-dependent
rescue of 1.5°!°
rabbit aqueous humor-mediated inhibition of MLEC proliferation by the ARC81
aptamer and
the anti-TGF(32 control antibody;
[0049] Figure 8A is an illustration depicting minimization and
mutagenesis/modification
strategies for a modified TGF(32 aptamer of SEQ JD NO:1 ; Figure 8B is a table
showing the
12
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
dissociation constant (K.d) of the modified TGF[32 aptamer of Figure 8A for
TGF(31, TGF(32
and TGF[33; Figure 8C is a graph depicting reversal of the inhibitory effect
of TGF(32 on
MLEC cell proliferation by the modified TGF(32 aptamer of Figure 8A;
[0050] Figure 9A is a graph depicting the stoichiometry of the TGF(32
aptamer/TGF(32
complex; Figure 9B is a diagram illustrating the interaction between
detestably labeled
TGF(32 aptamers and a TGF(32 homodimer;
[0051] Figure l0A is an illustration depicting the mapping of the aptamer
binding site using
the wild type, the N-terminal long tag and the N-terminal short tag variants
human TGF[32;
Figure lOB is a table listing the EC100 values, the dissociation constants and
the ICSO values
for wild type TGF[32, the N-terminal long tag variant of human TGF(32, the I~T-
terminal short
tag variant of human TGF(32, and two TGF(32 mutants (I~94N, S59T/R6QI~lI~94N);
[0052] Figure 1 lA is an illustration of a dot blot assay and a graph
depicting TGF type III
receptor blocking of the aptamer binding site; Figure 11B is a diagram
illustrating the
potential overlap between the aptamer and TGF type IIT receptor binding sites;
(0053] Figure 12 is an illustration of modified regions of the TGF(32 aptamer
ARC77 (SEQ
~ No. 1);
[0054] Figure 13A is a graph showing the binding curves of ARC 127 (SEQ >D
NO:19 -
PEG - SEQ ID NO:35 - PEG - SEQ ID N0:36) for the BB, AA and AB isoforms of
human
PDGF, and a Table listing the Ka values for ARC 127 (PEG - SEQ >D NO:19 - PEG -
SEQ
JD NO: 35 - PEG -SEQ >D NO:36 - 3T) and the BB, AA and AB isoforms of PDGF;
Figure
13B is a graph showing the binding curves of ARC 127 (PEG - SEQ ~ NO:19 - PEG -
SEQ
~ NO: 35 - PEG -SEQ ~ NO:36 - 3T)for the BB isoforms of human and rat PDGF,
and a
Table listing the I~ values for ARC 127 (PEG - SEQ ~ NO:19 - PEG - SEQ ~ NO:
35 -
PEG -SEQ ID NO:36 - 3T)and the human, rat and mouse BB isoforms of PDGF;
[005) Figure 14A is a graph depicting the ARC 127 (PEG - SEQ 1~ NO:19 - PEG -
SEQ
~ NO: 35 - PEG -SEQ ID N0:36 - 3T)aptamer binding to human and rat PDGF;
Figure
14B is a graph comparing inhibition of PDGF-induced 3T3 cell proliferation by
the ARC127
aptamer to inhibition of PDGF-induced 3T3 cell proliferation by a control
antibody.
[0056) Figure 15A is an image depicting migration of retinal pigmented
epithelial (RPE) cells
in the absence of PDGF; Figure 15B is an image depicting migration of RPE
cells in the
13
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
presence of 100 ng/ml PDGF; Figure 1 SC is an image depicting migration of RPE
cells in the
presence of PDGF and the ARC127 aptamer; Figure 15D is an image depicting
migration of
RPE cells in the presence of PDGF and the ARC 128 aptamer; Figures 1 SE and 1
SF are
graphs depicting the effect of increasing PDGF concentrations on RPE cell
migration.
[0057] Figure 16 is a graph illustrating in vitro plasma stability of the all-
DNA construct of
the ARC 127 aptamer (PEG - SEQ ~ No. 19- PEG - SEQ ~ N0:3 5 - PEG - SEQ ID
NO:36-3T) and the modified ARC127 aptamer;
[0058] Figure 17 is a graph illustrating the concentration of ARC 127 aptamer
through 50
hours post dose via I~, IP and SC routes of administration;
[0059] Figure 18 is a graph showing that ARC127 has measurable activity out to
48 hours i~a
vivo;
[0060] Figures 19A and 19B are graphs showing the binding plots for the full-
length TGF(32
aptamer sequences shown in Table 5;
[0061] Figures 20A, 208, and 20C are graphs showing the binding plots for the
truncated
TGF(32 aptamers shown in Table 7;
[0062] Figures 21 A, 21 B, and 21 C are graphs showing that the ARC 117 and
ARC 119
aptamers have measurable activity out to 48 hours in vivo; and
[0063] Figures 22A, 228, and 22C are graphs showing the ira viv~ activity of
the ARC126
and ARC127 and NX1838 aptamers out to at least 25 days.
SUMMARY OF THE INVENTION
[0064j The specificity of aptamers allows them to be used as therapeutics
capable of binding
specifically to cytolcines, growth factors or cell surface proteins that
promote scar tissue
formation or other cellular events that lead to increased IOP in the
glaucomatous eye. The
present invention provides aptamer therapeutics with specific binding affinity
to TGF(32 and
PDGF cytolcines that contribute to post-surgical tissue scarring and can thus
prevent increased
IOP in the glaucomatous eye, and other pathologic processes of glaucoma.
[0065] In one embodiment, the present invention provides aptamer compositions
capable of
binding to TGF[i l, TGF(32 or TFG(33 useful in the treatment of diseases of
the eye.
14
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
[0066] In one embodiment, the present invention provides aptamer compositions
capable of
binding to platelet derived growth factor (PI~GF) useful in the treatment of
diseases of the
eye.
[0067] In one embodiment, the present invention provides aptamer compositions
capable of
binding to ICAM-1 useful in the treatment of diseases of the eye.
[006g] In one embodiment, the present invention provides aptamer compositions
capable of
binding to IGF-1 useful in the treatment of diseases of the eye.
[006] In one embodiment, the present invention provides aptamer compositions
capable of
binding to ~EGF/'~EGF-IZ useful in the treatment of diseases of the eye.
[0070] In one embodiment, the present invention provides aptamer compositions
capable of
binding to TNF°-~, useful in the treatment of diseases of the eye.
[0071] In one embodiment, the present invention provides aptamer compositions
capable of
binding to ocV(33 useful in the treatment of diseases of the eye.
[0072] In another embodiment, the present invention provides methods of
treating subjects
with the compositions of the present invention to treat proliferative disease
involving TGF(32
-mediated cell proliferation.
[0073] In another embodiment, the present invention provides methods of
treating subjects
with the compositions of the present invention to treat proliferative disease
involving PDGF -
mediated cell proliferation.
[0074] In another embodiment, the present invention provides methods of
treating subjects
with the compositions of the present invention to treat proliferative disease
involving ICAI~/~1-
1 -mediated cell proliferation.
[007] In another embodiment, the present invention provides methods of
treating subjects
with the compositions of the present invention to treat proliferative disease
involving IGF-1 -
mediated cell proliferation.
[0076] In another embodiment, the present invention provides methods of
treating subjects
with the compositions of the present invention to treat proliferative disease
involving
VEGF/VEGF-R -mediated cell proliferation.
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
[0077] In another embodiment, the present invention provides methods of
treating subjects
with the compositions of the present invention to treat proliferative disease
involving TNF-cc
-mediated cell proliferation.
[0078] In another embodiment, the present invention provides methods of
treating subjects
with the compositions of the present invention to treat proliferative disease
involving ocV(33 -
mediated cell proliferation.
[0079] In another embodiment, the present invention provides nucleic acid
therapeutic
compositions and methods for delivering nucleic acid therapeutics capable of
binding to
cytolcines, growth factors and cell surface receptors individually or
combinations of two or
more of PDGF, TGF[31, TGF(32, TGF(33, ICAldI-l, IGFl, VEGF-R, VEGF, TNFoc, and
ceV(33, for the treatment of glaucoma and other proliferative diseases of the
eye.
(0080] In another embodiment, the present invention provides nucleic acid
therapeutic
compositions and methods for delivering nucleic acid therapeutics capable of
binding to
PDGF and VEGF. In one embodiment, the nucleic acid therapeutic is a single
nucleic acid
aptamer that has one domain capable of binding to PDGF and a second domain
capable of
binding to VEGF. In another embodiment, the nucleic acid therapeutic is a
solution that
contains a first nucleic acid aptamer capable of binding PDGF and a second
nucleic acid
aptamer capable of binding VEGF, wherein the first and second nucleic acid
aptamers are not
the same nucleic acid aptamer.
[0081] In another embodiment, the present invention provides high molecular
weight PEG-
derivati~ed nucleic acid (e.g., aptamer) conjugates with improved
pharmacological and
pharmacodynamic properties and methods for producing such conjugates.
[008] In one embodiment, the present invention provides high molecular weight
PEG-
nucleic acid (e.g., aptamer) conjugates and methods for producing such
conjugates using a
homo-bifunctional PEG to form a high molecular weight complex (i.e., a PEG -
nucleic acid
- PEG - nucleic acid - PEG - nucleic acid conjugate).
[0033] In one embodiment, the present invention provides high molecular weight
PEG-
nucleic acid (e.g., aptamer) conjugates and methods for producing such
conjugates using a bi-
reactive nucleic acid (i.e., a nucleic acid bearing two reactive sites) with a
mono-functional
16
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
PEG to form a multiple PEGylated conjugate (i. e., a PEG - nucleic acid - PEG
conjugate).
[0084] In one embodiment, the high molecular weight PEG-nucleic acid
conjugates of the
present invention can be used as therapeutics in the prevention andlor
treatment of ocular
diseases and disorders.
[0085] In one aspect, high molecular weight PEG-aptamer compositions of the
invention
include a nucleic acid and a stabilizing moiety that is a linking moiety,
wherein the linking
moiety is not a nucleic acid molecule. In one embodiment, the linking moiety
is polyalkylene
glycol. Suitable polyalkylene glycols include, for example, polyethylene
glycol (PEG). In
some embodiments, the polyethylene glycol (PEG) linking moiety is mufti-
activated. For
example, the PEG linking moiety is bi-activated. In one embodiment, the first
and second
portions of an aptamer arc linked by a PEG linking moiety, such that the
primary structure of
the aptamer composition is a linear arrangement in which the first moiety is
linked to a first
terminus of the PEG linking moiety and the second moiety is linked to a second
terminus of
the PEG linking moiety. In some aspects, there is more than one PEG moiety
separating
more than two nucleic acid aptamer moieties, for example, the linear
arrangement of the high
molecular weight aptamer composition is: nucleic acid - PEG - nucleic acid -
PEG - nucleic
acid. In some embodiments, the linear arrangement of the high molecular weight
aptamer
composition is: PEG - nucleic acid - PEG - nucleic acid - PEG - nucleic acid.
In some
embodiments, the high molecular weight aptamer composition has a molecular
weight
selected from the group consisting of greater than 10 kI), greater than 20
kI), greater than 40
kD and greater than 80 kI?. Some high molecular weight aptamer compositions
according to
this aspect of the invention are capable of binding to platelet derived growth
factor (PI~GF°).
Some high molecular weight aptamer compositions according to this aspect of
the invention
are capable of binding to TGb'~3~.
[OO~G] In another aspect, the invention provides high molecular weight PEG-
aptamer
compositions that include an aptamer, and two or more non-nucleic acid
stabilizing moieties.
Suitable stabilizing moieties include, for example, a polyalkylene glycol. In
one
embodiment, the stabilizing moiety is polyethylene glycol (PEG). In one
embodiment, the
aptamer is mufti-activated. For example, the aptamer is bi-activated.
[0087] The present invention also provides therapeutic compositions that
include the high
17
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
molecular weight PEG-aptamer compositions described herein.
[0088] In another aspect, the present invention provides methods of treating
disease in a
subject comprising the steps of administering a therapeutically effective
amount of a high
molecular weight PEG-aptamer compositions described herein.
DETAILED DESCRIPTI~N ~F THE INVENTION
[0089] The details of one or more embodiments of the invention are set forth
in the
accompanying description below. Although any methods and materials similar or
equivalent
to those described herein can be used in the practice or testing of the
present invention, the
preferred methods and materials are now described. Other features, objects,
and advantages
of the invention will be apparent from the description. In the specification,
the singular forms
also include the plural unless the context clearly dictates otherwise. Unless
defined
otherwise, all technical and scientific terms used herein have the same
meaning as commonly
understood by one of ordinary skill in the art to which this invention
belongs. In the case of
conflict, the present Specification will control.
[0090] All publications, patent applications, patents, and other references
mentioned herein
are incorporated by reference in their entirety. Citation of publications and
patent documents
is not intended as an admission that any is pertinent prior art, nor does it
constitute any
admission as to the contents or date of the same. In the case of conflict, the
present
Specification, including definitions, will control. In addition, the
materials, methods, and
examples described below are illustrative only and not intended to be
limiting.
The SELEX~ Process
[009] A suitable method for generating an aptamer is with the process entitled
"Systematic
Evolution of Ligands by EXponential Enrichment " ('6SELE3~T'~99) generally
depicted in
Figure 1. The SELEXTM process is a method for the ifi vitr~ e~rolution of
nucleic acid
molecules with highly specific binding to target molecules and is described
in, e.g., U.S.
patent application Ser. No. 07/536,428, filed Jun. 11, 1990, now abandoned,
U.S. Pat. I~o.
5,475,096 entitled "Nucleic Acid Ligands", and U.S. Pat. No. 5,270,163 (see
also ~JO
91/19813) entitled "Nucleic Acid Ligands". Each SELEX-identified nucleic acid
ligand is a
specific ligand of a given target compound or molecule. The SELEXT~ process is
based on
18
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
the unique insight that nucleic acids have sufficient capacity for forming a
variety of two- and
three-dimensional structures and sufficient chemical versatility available
within their
monomers to act as ligands (form specific binding pairs) with virtually any
chemical
compound, whether monomeric or polymeric. Molecules of any size or composition
can serve
as targets.
[0092] SELEXTM relies as a starting point upon a large library of single
stranded
oligonucleotide templates comprising randomized sequences derived from
chemical synthesis
on a standard I)NA synthesizer. In some examples, a population of 100% random
oligonucleotides is screened. In others, each oligonucleotide in the
population comprises a
random sequence and at least one fixed sequence at its 5' and/or 3' end which
comprises a
sequence shared by all the molecules of the oligonucleotide population. Fixed
sequences
include sequences such as hybridization sites for PCR primers, promoter
sequences for RI~TA
polymerises (e.g., T3, T4, T7, SP6, and the like), restriction sites, or
homopolymeric
sequences, such as poly A or poly T tracts, catalytic cores, sites for
selective binding to
affinity columns, and other sequences to facilitate cloning and/or sequencing
of an
oligonucleotide of interest.
[0093] The random sequence portion of the oligonucleotide can be of any length
and can
comprise ribonucleotides and/or deoxyribonucleotides and can include modified
or non-
natural nucleotides or nucleotide analogs. See, e.g., U.S. Patent Nos.
5,958,691; 5,660,985;
5,958,691; 5,698,687; 5,817,635; and 5,672,695, PCT publication WO 92/07065.
Random
oligonucleotides can be synthesized from phosphodiester-linked nucleotides
using solid phase
oligonucleotide synthesis techniques well known in the art (Froehler et al.,
lVucl. Acid Res.
14:5399-5467 (1986); Froehler et cal., Tet. I,ett. 27:5575-5578 (1986)).
~ligonucleotides can
also be synthesized using solution phase methods such as triester synthesis
methods (Sood e~
czl., Nucl. Acid Res. 4-:2557 (1977);1-~irose e~ czl., Tet. Lett., 28:2449
(1978)). Typical
syntheses carried out on automated 1~IVA synthesis equipment yield 1015-10'7
molecules.
Sufficiently large regions of random sequence in the sequence design increases
the likelihood
that each synthesized molecule is likely to represent a unique sequence.
[0094] To synthesize randomized sequences, mixtures of all four nucleotides
are added at
each nucleotide addition step during the synthesis process, allowing for
random incorporation
of nucleotides. In one embodiment, random oligonucleotides comprise entirely
random
19
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
sequences; however, in other embodiments, random oligonucleotides can comprise
stretches
of nonrandom or partially random sequences. Partially random sequences can be
created by
adding the four nucleotides in different molar ratios at each addition step.
[0095] Template molecules typically contain fixed 5' and 3' terminal sequences
which flank
an internal region of 30 - 50 random nucleotides. A standard (1 pmole) scale
synthesis will
yield 10'5 -1016 individual template molecules, sufficient for most SELEX
experiments.
The RNA library is generated from this starting library by in vitr~
transcription using
recombinant T7 RNA polymerise. This library is then mixed with the target
under conditions
favorable for binding and subjected to step-wise iterations of binding,
partitioning and
amplification, using the same general selection scheme, to achieve virtually
any desired ,
criterion of binding affinity and selectivity. Starting from a mixture of
nucleic acids
preferably comprising a segment of randomized sequence, the SELE~~ method
includes
steps of contacting the mixture with the target under conditions favorable for
binding,
partitioning unbound nucleic acids from those nucleic acids which have bound
specifically to
target molecules, dissociating the nucleic acid-target complexes, amplifying
the nucleic acids
dissociated from the nucleic acid-target complexes to yield a ligand-enriched
mixture of
nucleic acids, then reiterating the steps of binding, partitioning,
dissociating and amplifying
through as many cycles as desired to yield highly specific high affinity
nucleic acid ligands to
the target molecule.
[0096] Within a nucleic acid mixture containing a large number of possible
sequences and
structures, there is a wide range of binding affinities for a given target. A
nucleic acid
mixture comprising, for example a ~0 naicleotide randomized segment can have
42° candidate
possibilities. Those which have the higher affinity constants for the target
are most likely to
bind to the target. After partitioning, dissociation and amplification, a
second nucleic acid
mixture is generated, enriched for the higher binding affinity candidates.
Additional rounds
of selection progressively favor the best ligands until the resulting nucleic
acid mixture is
predominantly composed of only one or a few sequences. 'These can then be
cloned,
sequenced and individually tested for binding affinity as pure ligands.
[0097] Cycles of selection and amplification are repeated until a desired goal
is achieved. In
the most general case, selection/amplification is continued until no
significant improvement
in binding strength is achieved on repetition of the cycle. The method may be
used to sample
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
as many as about~10~8 different nucleic acid species. The nucleic acids of the
test mixture
preferably include a randomized sequence portion as well as conserved
sequences necessary
for efficient amplification. Nucleic acid sequence variants can be produced in
a number of
ways including synthesis of randomized nucleic acid sequences and size
selection from
randomly cleaved cellular nucleic acids. The variable sequence portion may
contain fully or
partially random sequence; it may also contain subportions of conserved
sequence
incorporated with randomized sequence. Sequence variation in test nucleic
acids can be
introduced or increased by mutagenesis before or during the
selection/amplification iterations.
[009g] In one embodiment of SELEXTM, the selection process is so efficient at
isolating those
nucleic acid ligands that bind most strongly to the selected target, that only
one cycle of
selection and amplification is required. Such an efficient selection may
occur, for example, in
a chromatographic-type process wherein the ability of nucleic acids to
associate with targets
bound on a column operates in such a manner that the column is sufficiently
able to allow
separation and isolation of the highest affinity nucleic acid ligands.
[0099] In many cases, it is not necessarily desirable to perform the iterative
steps of
SELEXTM until a single nucleic acid ligand is identified. The target-specific
nucleic acid
ligand solution may include a family of nucleic acid structures or motifs that
have a number
of conserved sequences and a number of sequences which can be substituted or
added without
significantly affecting the affinity of the nucleic acid ligands to the
target. By terminating the
SELEXTM process prior to completion, it is possible to determine the sequence
of a number of
members of the nucleic acid ligand solution family.
[00100] A variety of nucleic acid primary, secondary and tertiary structures
are known
to exist. The structures or motifs that have been shown most commonly to be
involved in
non-l~atson-Crick type interactions are referred to as hairpin loops,
symmetric and
asymanetric bulges, pseudoknots and myriad combi~aations of the same. Almost
all known
cases of such motifs suggest that they can be formed in a nucleic acid
sequence of no more
than 30 nucleotides. For this reason, it is often preferred that SELE~
procedures with
contiguous randomized segments be initiated with nucleic acid sequences
containing a
randomized segment of between about 20-50 nucleotides.
[00101] The core SELEXTM method has been modified to achieve a number of
specific
objectives. For example, U.S. Patent No. 5,707,796 describes the use of
SELEXTM in
conjunction with gel electrophoresis to select nucleic acid molecules with
specific structural
21
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
characteristics, such as bent DNA. U.S. Patent No. 5,763,177 describes SELEXTM
based
methods for selecting nucleic acid ligands containing photoreactive groups
capable of binding
and/or photocrosslinking to and/or photoinactivating a target molecule. U.S.
Patent No.
5,567,588 and U.S. Application No. 08/792,075, filed January 31, 1997,
entitled "Flow Cell
SELEX", describe SELEXTM based methods which achieve highly efficient
partitioning
between oligonucleotides having high and low affinity for a target molecule.
U.S. Patent No.
5,496,938 describes methods for obtaining improved nucleic acid ligands after
the SELEXTM
process has been performed. U.S. Patent No. 5,705,337 describes methods for
covalently
linking a ligand to its target.
[00102] SELEXTM can also be used to obtain nucleic acid ligands that bind to
more
than one site on the target molecule, and to obtain nucleic acid ligands that
include non-
nucleic acid species that bind to specific sites on the target. SELEXTM
provides means for
isolating and identifying nucleic acid ligands which bind to any envisionable
target, including
large and small biomolecules including proteins (including both nucleic acid-
binding proteins
and proteins not known to bind nucleic acids as part of their biological
function) cofactors
and other small molecules. For example, see U.S. Patent No. 5,580,737 which
discloses
nucleic acid sequences identified through SELEXTM which are capable of binding
with high
affinity to caffeine and the closely related analog, theophylline.
[00103] Counter-SELEXTM is a method for improving the specificity of nucleic
acid
ligands to a target molecule by eliminating nucleic acid ligand sequences with
cross-reactivity
to one or more non-target molecules. Counter-SELEXTM is comprised of the steps
of a)
preparing a candidate mixture of nucleic acids; b) contacting the candidate
mixture with the
target, wherein nucleic acids having an increased affinity to the target
relative to the candidate
mixture may be partitioned from the remainder of the candidate mis~ture~ c)
partitioning the
increased affinity nucleic acids from the remainder of the candidate mixtures
d) contacting the
increased affinity nucleic acids with one or more non-target molecules such
that nucleic acid
ligands with specific affinity for the non-target molecules) are removed; and
e) amplifying
the nucleic acids with specific affinity to the target molecule to yield a
mixture of nucleic
acids enriched for nucleic acid sequences with a relatively higher affinity
and specificity for
binding to the target molecule.
[00104] One potential problem encountered in the use of nucleic acids as
therapeutics
and vaccines is that oligonucleotides in their phosphodiester form may be
quickly degraded in
22
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
body fluids by intracellular and extracellular enzymes such as endonucleases
and
exonucleases before the desired effect is manifest. The SELEX method thus
encompasses the
identification of high-affinity nucleic acid ligands containing modified
nucleotides confernng
improved characteristics on the ligand, such as improved in vivo stability or
improved
delivery characteristics. Examples of such modifications include chemical
substitutions at the
ribose and/or phosphate and/or base positions. SELEX-identified nucleic acid
ligands
containing modified nucleotides are described in U.S. Patent No. 5,660,985,
which describes
oligonucleotides containing nucleotide derivatives chemically modified at the
2' position of
ribose, 5' position of pyrimidines and 8' positions of purines. IJ.S. Patent
No. 5,756,703
describes oligonucleotides containing various 2'-modified pyrimidines. IJ.S.
Patent No.
5,580,737 describes highly specific nucleic acid ligands containing one or
more nucleotides
modified with 2'-amino (2'-NHZ), 2'-fluoro (2'-F), and/or 2'-O-methyl (2'-
~NTe)
substituents.
[00105] Ii~lodifications of the nucleic acid ligands contemplated in this
invention
include, but are not limited to, those which provide other chemical groups
that incorporate
additional charge, polarizability, hydrophobicity, hydrogen bonding,
electrostatic interaction,
and fluxionality to the nucleic acid ligand bases or to the nucleic acid
ligand as a whole. Such
modifications include, but are not limited to, 2'-position sugar
modifications, 5-position
pyrimidine modifications, 8-position purine modifications, modifications at
exocyclic amines,
substitution of 4-thiouridine, substitution of 5-bromo or 5-iodo-uracil;
backbone
modifications, phosphorothioate or alkyl phosphate modifications,
methylations, unusual
base-pairing combinations such as the isobases isocytidine and isoguanidine
and the like.
Ialodifications can also include 3' and 5' modifications such as capping. In
preferred
embodiments of the instant invention, the nucleic acid ligands are l~i~
molecules that are 2'-
fluoro (2'-F) modified on the sugar xnoietr of pyrimidine residues.
[00106] The modifications can be pre- or post-SELE~ process modifications. Pre-
SELEX process modifications yield nucleic acid ligands with both specificity
for their
SELEX target and improved in vivo stability. Post-SELEX process modifications
made to 2'-
OH nucleic acid ligands can result in improved in vivo stability without
adversely affecting
the binding capacity of the nucleic acid ligand.
23
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
(00107] Other modifications are known to one of ordinary skill in the art.
Such
modifications may be made post-SELEX process (modification of previously
identified
unmodified ligands) or by incorporation into the SELEX process.
(00108] The SELEX method encompasses combining selected oligonucleotides with
other selected oligonucleotides and non-oligonucleotide functional units as
described in U.S.
Patent No. 5,637,459 and U.S. Patent No. 5,683,867. The SELEX method further
encompasses combining selected nucleic acid ligands with lipophilic or non-
immunogenic
high molecular weight compounds in a diagnostic or therapeutic complex, as
described in
U.S. Patent No. 6,011,020. VEGF nucleic acid ligands that are associated with
a lipophilic
compound, such as diacyl glycerol or dialkyl glycerol, in a diagnostic or
therapeutic complex
are described in U.S. Patent No. 5,859,228.
[00109] '~GF nucleic acid ligands that are associated with a lipophilic
compound,
such as a glycerol lipid, or a non-immunogenic high molecular weight compound,
such as
polyalkylene glycol are further described in U.S. Patent No. 6,051,698. ~IEGF
nucleic acid
ligands that are associated with a non-immunogenic, high molecular weight
compound or a
lipophilic compound are further described in PCT Publication No. W~ 98/18480.
These
patents and applications allow the combination of a broad array of shapes and
other
properties, and the efficient amplification and replication properties, of
oligonucleotides with
the desirable properties of other molecules.
[00110] The identification of nucleic acid ligands to small, flexible peptides
via the
SELEX method has also been explored. Small peptides have flexible structures
and usually
exist in solution in an equilibrium of multiple conformers, and thus it was
initially thought
that binding affinities may be limited by the confora~aational entropy lost
upon binding a
flexible peptide. However, the feasibility of identif~ring nucleic acid
ligands to small peptides
in solution was demonstrated in U.S. Patent No. 5,648,214. In this patent,
high affinity RNI~
nucleic acid ligands to substance P, an 11 amino acid peptide, were
identified.
[00111] To generate oligonucleotide populations which are resistant to
nucleases and
hydrolysis, modified oligonucleotides can be used and can include one or more
substitute
internucleotide linkages, altered sugars, altered bases, or combinations
thereof. In one
embodiment, oligonucleotides are provided in which the P(~)~ group is replaced
by P(~)S
("thioate"), P(S)S ("dithioate"), P(O)NR2 ("amidate"), P(~)R, P(~)~R', CO or
CHZ
("formacetal") or 3'-amine (-NH-CH2-CH2-), wherein each R or R' is
independently H or
24
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
substituted or unsubstituted alkyl. Linkage groups can be attached to adjacent
nucleotide
through an -O-, -N-, or -S- linkage. Not all linkages in the oligonucleotide
are required to be
identical.
[00112] In further embodiments, the oligonucleotides comprise modified sugar
groups,
for example, one or more of the hydroxyl groups is replaced with halogen,
aliphatic groups,
or functionalized as ethers or amines. In one embodiment, the 2'-position of
the furanose
residue is substituted by any of an ~-methyl, ~-alkyl, O-allyl, S-alkyl; S-
allyl, or halo group.
Methods of synthesis of 2'-modified sugars are described in Sproat, et al.,
Nucl. Acid Ides.
19:733-738 (1991); Gotten, ~t al., Nucl. Acid lies. 19:2629-2635 (1991); and
Hobbs, et al.,
Biochemistry 12:5138-5145 (1973). 'The use of 2-fluoro-ribonucleotide oligomer
molecules
can increase the sensitivity of a nucleic acid sensor molecule for a target
molecule by ten- to-
nne hundred-fold over those generated using unsubstituted ribo- or deoxyribo-
oligonucleotides (Pagratis, et al., Nat. Biotechnol. 15:68-73 (1997)),
providing additional
binding interactions with a target molecule and increasing the stability of
the secondary
structures) of the nucleic acid sensor molecule (I~raus, et al., Journal of
Immunology
160:5209-5212 (1998); Pieken, et al., Science 253:314-317 (1991); Lin, et al.,
Nucl. Acids
Res. 22:5529-5234 (1994); Jellinek, et al. Biochemistry 34:11363-11372 (1995);
Pagratis, et
al., Nat. Biotechnol 15:68-73 (1997)).
[00113] Nucleic acid aptamer molecules are generally selected in a 5 to 20
cycle
procedure. In one embodiment, heterogeneity is introduced only in the initial
selection stages
and does not occur throughout the replicating process.
[00114] The starting library of DNA sequences is generated by automated
chemical
synthesis on a DNA synthesizer. 'This library of sequences is transcribed ira
vi8~~ into I~T~
using T7 IOTA polymerase or modified T7 I~Tf~ polymerases and purified. In one
example,
the 5' f xed:random:3'-fixed sequence is separated by random sequence having
30 to 50
nucleotides.
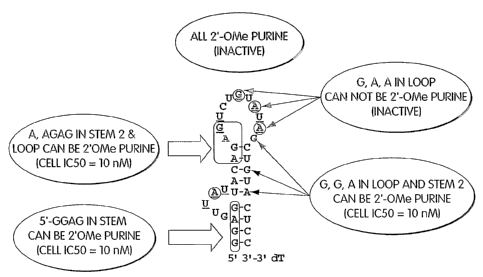
2'~-Me SELE~T~
[00115] In addition, the SELEXT~ method can be performed to generate
2'modified
aptamers as described in U.S. Serial No. 60/430,761, filed December 3, 2002,
U.S.
Provisional Patent Application Serial No. 60/487,474, filed July 15, 2003, and
U.S.
Provisional Patent Application Serial No. 60/517,039, filed November 4, 2003,
and U.S.
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
Patent Application No. 10/?29,581, filed December 3, 2003, each of which is
herein
incorporated by reference in its entirety.
[00116] The present invention also provides materials and methods to produce
stabilized oligonucleotides, including, e.g., aptamers, that contain modified
nucleotides (e.g.,
nucleotides which have a modification at the 2'position) which make the
oligonucleotide
more stable than the unmodified oligonucleotide. The stabilized
oligonucleotides produced
by the materials and methods of the present invention are also more stable to
enzymatic and
chemical degradation as well as thermal and physical degradation. For example,
oligonucleotides containing 2'-O-methyl nucleotides are nuclease-resistant and
inexpensive to
synthesize. Although 2'-O-methyl nucleotides are ubiquitous in biological
systems, natural
polymerases do not accept 2'-O-methyl NTPs as substrates under physiological
conditions,
thus there are no safety concerns over the recycling of 2'-O-methyl
nucleotides into host
DNA.
[00117] In one embodiment, the present invention provides combinations of 2'-
OH, 2'-
F, 2'-deoxy, and 2'-OMe modifications of the ATP, GTP, CTP, TTP, and UTP
nucleotides.
In another embodiment, the present invention provides combinations of 2'-OH,
2'-F, 2'-
deoxy, 2'-OMe, 2'-NHZ, and 2'-methoxyethyl modifications of the ATP, GTP, CTP,
TTP,
and UTP nucleotides. In one embodiment, the present invention provides 56
combinations of
2'-OH, 2'-F, 2'-deoxy, 2'-OMe, 2'-NH2, and 2'-methoxyethyl modifications the
ATP, GTP,
CTP, TTP, and UTP nucleotides.
[0011] 2' modified aptamers of the invention are created using modified
polymerases,
such as, e.g., a modified T7 polymerase, having a higher incorporation rate of
modified
nucleotides having bulky substituents at the f~aranose 2' position, than wild-
type polymerases.
For example, a double T7 polydnerase mutant (~639F/H784A) having the histidine
at
position 784 changed to an alanine, or other small amino acid residue, in
addition to the
1''639F mutation has been described for incorporation ~f bulky 2' substituents
and has been
used to incorporate modified pyrimidine NTPs. A single mutant T7 polymerase
(H784A)
having the histidine at position 784 changed to an alanine residue has also
been described.
(Padilla et al., Nucleic Acids Research, 2002, 30: 138). In both the
Y639F1H784A double
mutant and H784A single mutant T7 polymerases, the change to smaller amino
acid residues
allows for the incorporation of bulkier nucleotide substrates, e.g., 2'-O
methyl substituted
nucleotides.
26
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
[00119] Another important factor in the production of 2'-modified aptamers is
the use
of both divalent magnesium and manganese in the transcription mixture.
Different
combinations of concentrations of magnesium chloride and manganese chloride
have been
found to affect yields of 2'-O-methylated transcripts, the optimum
concentration of the
magnesium and manganese chloride being dependent on the concentration in the
transcription
reaction mixture of NTPs which complex divalent metal ions.
[00120] Priming transcription with GMP or guanosine is also important. This
effect
results from the specificity of the polymerase for the initiating nucleotide.
As a result, the 5'-
terminal nucleotide of any transcript generated in this fashion is likely to
be 2'-OI-I G. The
preferred concentration of GMP (or guanosine) is 0.5 mM and even more
preferably 1 mM.
It has also been found that including PEG, preferably PEG-5000, in the
transcription reaction
is useful to maximise incorporation of modified nucleotides.
At~tamers with binding affinity to TGFa2 and PDGF
[00121] The present invention provides modified and unmodified nucleic acid
aptamer
therapeutics capable of binding to human cytokines, growth factors or cell
surface proteins
implicated in diseases of the eye. In one embodiment, the aptamers of the
invention are
capable of binding to TGF~32 with high affinity and reversing TGF(32-mediated
inhibition of
mink lung epithelial cells (MLEC) proliferation in vitro. These aptamers can
be generated
using a process termed "Systematic Evolution of Ligands by Exponential
Enrichment" (the
SELEXT~ process) depicted in Figure 1.
[00122] The anodifaed 1~NA aptamers of the present invention bind native human
TGF[32. For bio-chemical characterisation of these aptamers, two forms of
mature TGF(32
were generated, nature and N-terminal his-tagged versions in ~'. ~e~li. Af4er
refolding and
purification, functional TGF(32s were obtained. These TGF(32 proteins were
active in cell
based assay. N-terminal tags affected both activity and aptamer bindiaag while
the affinity to
aptamer was decreased to a much larger extent. Further two mutant TGF(32s
(I~94N,
S59T1R60I~/K94N) were generated based on known isoforms of TGF(32. The I~94N
mutant
was capable of binding to the aptamers with comparable affinity with that of
native TGF[32,
whereas the S59T/1~60K/I~94N mutant had significantly reduced affinity to the
aptamers.
Similarly, the aptamers blocked the bioactivity of native and K94N TGF(32s
with higher
27
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
potencies than that of S59TlR60K/K94N mutant in a cell based assay. Based on
published
crystal structure, two substitutions at positions 59 and 60 reside near the
dimer interface and
adjacent to the N-terminus of TGF(32 and the other substitution at position 94
is near the type
II receptor binding site. Binding competition assay with soluble TGF-(3
receptors revealed
that type III receptor competes with the aptamer binding, but not type II
receptor. We also
demonstrated that two aptamers bind to one species of dimer of TGF[32 but not
to the other
two species of dimer. The aptamers of the present invention are believed to
bind TGF(32 near
or at the TGF-[3 type III receptor binding site and block its biological
fiznction.
[00123] The aptamers with specificity and binding affinity to TGF~32 of the
present
invention arc selected by the SELE~ process described above. ~-1s part of the
SELEX process
the sequences selected to bind to TGF(32 are then minimized to determine the
minimal
sequence having binding affinity, and optimized by performing random or
directed
mutagenesis of the minimized sequence to determine if increases of affinity or
alternatively
which positions in the sequence are essential for binding activity.
f~dditionally, selections can
be performed with sequences incorporating modified sequences to stabilize the
aptamer
molecules against degradation in vivo.
[00124] The selected aptamers having the highest affinity and specific binding
as
demonstrated by biological assays as described in the examples below are
suitable
therapeutics for treating conditions in which TGF[32 is involved in
pathogenesis.
Alternatively, the aptamers selected for specificity to PDGF are suitable
therapeutics for
treating conditions in which PDGF is involved in pathogenesis.
[0012] Some aptamer compositions of the present invention have binding
affinity and
specificity to certain dimers of platelet derived growth factor (PDGF). 'The
aptamer
compositions of the present invention have binding affinity to the PDGF BB
homodimer and
to the AB heterodimer but not to the ~ homodimer.
[00126] 'The aptamer compositions of the present invention can be used as
therapeutic
compositions to treat subjects with ocular disease involving TGF(32- or PDGF -
mediated
proliferative disease. For example, aptamers that are selective for PDGF can
be used in the
treatment of eye diseases such as PVI~, PD12 and AMD. In addition, PDGF
aptamers can be
administered alone, or in conjunction with other known therapies, such as anti-
VEGF
28
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
therapies, anti-inflammatory agents, anti-proliferative agents, antibacterial
agents, antifungal
agents and antimicrobial agents. TGF[32 aptamers can be used to treat damage
or injury that
occurs after a trabeculectomy, such as, for example, scarring. Thus, TGF(32
aptamers can be
administered prior to, during, or after a trabeculectomy. The TGF[32 aptamers
can be
administered alone, or in combination with other known therapies, such as, for
example, anti-
inflammatory agents, anti-proliferative agents, antibacterial agents,
antifungal agents and
antimicrobial agents.
TGF(32-St~ecific Aptamer Therapeutics in Trabeculectomy
[00127] TGF(32 aptamers of the invention may be applied in eye drop form. This
method is non-invasive and will increase ease of patient compliance. If
administered via
microdevice, microparticle or sponge, application can occur during surgery as
described
above.
[00120 The TGF(32 aptamer may be administered lyophilized in polymer sustained
delivery devices with delivery solution which will mix before being released
into the eye.
Sustained delivery from polymer matrixes offers the advantage of targeting
specific tissues
and increasing the comfort and compliance of patients. PLGA
(polylacticcoglycolic acid) is
the encapsulation matrix of choice as it is FDA approved and used as a suture
material since
the 1970s and as a scaffold in tissue engineering. It is biocompatible and
well studied for its
toxicology and degradation kinetics.
[00129) In addition, the TGF(32 aptamer may be administered lyophilized in
polymer
contact lens sustained delivery device. The contact lens aptamer therapeutic
delivery device
will increase patient dosing compliance and help health care providers with
ease of
application of the aptamer therapeutic while assuring a constant zero-order
delivery of the
aptamer therapeutic.
[0010] Subconjbmctival administration may be used in a volume of 100 ul in the
manner described above for 5-FU.
[00131) The TGF(32 aptamer is administered at a determined effective dose as
eye
drops, with a microdevice, microparticle or sponge or subconjunctivally near
the anterior
segment. It may be lyophilized for storage and reconstituted and administered
in a sterile,
aqueous, preservative free bicarbonate-buffered solution. Dosing for the
TGF(32 aptamer is
in the range of 0.1-200 mg kg ~. Preferred dosing for animals of the TGF(32
aptamer is in the
29
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
range of 0.1-100 mg kg I. More preferred dosing for animals of the TGF(32
aptamer is in the
range of 0.1-10 mg kg ~. Most preferred dosing for animals of the TGF(32
aptamer is in the
range of 0.1- 1 mg kg I. Dosing for humans is in the range of 7-70 mg kg ~.
PDGF Aptamer Therapeutics in Ale-related Macular Degeneration (AMD).
[00132] PDGF plays an important role in the pathogenesis of AMD in synergistic
action with other growth factors such as ~GF. Hypoxia increases expression of
PDGF and
PDGF directly acts on endothelial cells through PDGFI~-b to induce
angiogenesis. PDGF
acts on fibroblasts, smooth muscle cells, neuroglial cells, and stimulates
proliferation of
connective-tissue cells.
[00133] Dosing of the PDGF aptamer for animals is in the range of 0.1-200 mg
kg I.
Preferred dosing of the PDGF aptamer for animals is in the range of 0.1-100 mg
k~ ~. More
preferred dosing of the PDGF aptamer for animals is in the range of 0.1-10 mg
kg 1. Most
preferred dosing of the PDGF aptamer for animals is in the range of 0.1-11 mg
kg I. Human
dosing is in the range of 7-70 mg kg ~ .
[00134] The PDGF aptamer can be injected intravitreally in a single dose at a
determined effective concentration in 100u1 delivery volume. Injection is
through the gars
plana using a 30-gauge needle and tuberculin syringe after instilling topical
anesthesia and
5% povidone iodine solution. Before dosing, the vial stopper is wiped with 70%
alcohol.
PDGF aptamer can be stored lyophilized and dissolved into a ready to use
sterile solution
composed of l OmM sodium phosphate and 0.9°/~ sodium chloride buffer.
Intravitreal
administration is used in many intraocular diseases using allowing efficient
penetration into
the eye. The tight complexes of the retinal pigment epithelium and retinal
capillaries sere as
the blood-~cular barrier, which inhibits penetration of therapeutic agents
into the vitreous.
This route of administration avoids the potential side effects that may be
experienced with
systemic administration and allows for efficient targeting of therapeutic
area.
[0013] Additionally, the PDGF aptamer may be delivered transsclerally.
Transscleral
delivery is a viable mode of administering therapeutics to the posterior
segment. The sclera
has a large and accessible surface area and a high degree of hydration that
renders it
conducive to water-soluble substances. It is also relatively devoid of cells
and thus has few
proteolytic enzymes or protein-binding sites that can bind or sequester
therapeutic agents.
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
Scleral permeability permits both small and large molecular weight agents and
permeability
does not decline with age which is favorable for the treatment of chronic
diseases such as
AMD which occur increasingly in the elderly. It is non-destructive, minimally
invasive and
achieves targeted delivery. Additionally, slow release transscleral devices
allow for
consistent release of therapeutic PDGF aptamer.
PDGF Aptamer Therapeutics in Proliferative Vitreoretinopathy (PVR).
[00136] Surgery for PVI~ begins with a vitrectomy (pays plans vitrectomy)
procedure,
following the vitrectomy procedure, the surgeon usually instills special gases
or fluids into the
eye to help flatten the retina and keep it reattached to the outer wall of the
eye. The PDGF
aptamer can be injected intravitreally in either before or after vitrectomy or
both in a single
dose at a determined effective concentration in 100u1 delivery volume.
Injection is through
the gars plane using a 30-gauge needle and tuberculin syringe after instilling
topical
anesthesia and 5% povidone iodine solution. Before dosing, the vial stopper is
wiped with
70% alcohol. PDGF aptamer can be stored lyophilized and dissolved into a ready
to use
sterile solution composed of l OmIvI sodium phosphate and .9~/o sodium
chloride buffer.
Intravitreal administration is used in many intraocular diseases using
allowing efficient
penetration into the eye. The tight complexes of the retinal pigment
epithelium and retinal
capillaries serve as the blood-ocular barrier, which inhibits penetration of
therapeutic agents
into the vitreous. This route of administration avoids the potential side
effects that may be
experienced with systemic administration and allows for efficient targeting of
therapeutic
area.
[00137] PDGF aptamer may be delivered via a biodegradable microsize polymer
system. The aptamer may be encapsulated in polymer with a predetermined rate
of release.
This ensures localized delivery, consistent dosing and assured compliance.
Delivery can
occur via a polymer coated pellet with variable permeability to the aptamer.
This implant can
be surgically inserted thr~ugh the pare plane during vitrectomy.
[0013] Additionally, the PDGF aptamer may be delivered transsclerally.
Transscleral
delivery is a viable mode of administering therapeutics to the posterior
segment. The sclera
has a large and accessible surface area and a high degree of hydration that
renders it
conducive to water-soluble substances. It is also relatively devoid of cells
and thus has few
proteolytic enzymes or protein-binding sites that can bind or sequester
therapeutic agents.
Scleral permeability permits both small and large molecular weight agents and
permeability
31
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
does not decline with age which is favorable for the treatment of chronic
diseases such as
AMD which.occur increasingly in the elderly. It is non-destructive, minimally
invasive and
achieves targeted delivery. Additionally, slow release transscleral devices
allow for
consistent release of therapeutic PDGF aptamer.
PDGF Aptamer Therapeutics in Proliferative Diabetic Retinopathy PDR .
[00139] The PDGF aptamer may be injected intravitreally in a single dose at a
determined effective concentration in 100u1 delivery volume. Injection is
through the pays
plane using a 30-gauge needle and tuberculin syringe after instilling topical
anesthesia and
5% povidone iodine solution. Before dosing, the vial stopper is wiped with 70%
alcohol.
PDGF aptamer can be stored lyophilised and dissolved into a ready to use
sterile solution
composed of l OmI~ sodium phosphate and .9°/~ sodium chloride buffer.
Intravitreal
administration is used in many intraocular diseases using allowing efficient
penetration into
the eye. The tight complexes of the retinal pigment epithelium and retinal
capillaries serve as
the blood-ocular barrier, which inhibits penetration of therapeutic agents
into the vitreous.
This route of administration avoids the potential side effects that may be
experienced with
systemic administration and allows for efficient targeting of therapeutic
area.
[00140] Additionally, the PDGF aptamer may be delivered transsclerally.
Transscleral
delivery is a viable mode of administering therapeutics to the posterior
segment. The sclera
has a large and accessible surface area and a high degree of hydration that
renders it
conducive to water-soluble substances. It is also relatively devoid of cells
and thus has few
proteolytic er~ymes or protein-binding sites that can bind or sequester
therapeutic agents.
Scleral pcrmeabilitgr permits both small and large molecular weight agents and
permeability
does not decline with age which is favorable for the treatment of chronic
diseases such as
AI~1ID which occur increasingly in the elderly. It is non-dest~ctive,
minimally invasive and
achieves targeted delivery. Additionally, slovr release transscleral devices
allow for
consistent release of therapeutic PDGF aptamer.
Aptamers with binding specificity to Cytolcines, Growth Factors and Cell
Surface Proteins
[00141] Cytolcines, cell surface proteins and growth factors implicated in
various
ocular diseases include ICAM-l, TGF(31, TGF(32, TGF(33, IGF-1, VEGF/VEGF-R,
TNF-~,
32
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
angiopoietin and ccV(33. Intercellular Adhesion Molecule 1 (ICAM-1) is a 76 to
115 kDa
surface glycoprotein with five extracellular immunoglobulin-like domains plays
a particularly
important role in diabetic retinopathy. ICAM-1-mediated leukostasis is
causative in the
pathogenesis of diabetic retinopathy. ICAM-1 interactions with (32 integrins
located on the
surface of leukocytes (neutrophils, basophils, lymphocytes, eosinophils,
monocytes) are
important for their firm adhesion to the endothelium and their
transendothelial migration to
sites of inflammation. ICAM-1 facilitates adhesion of leukocytes to the
retinal vasculature in
diabetic retinopathy and is involved with retinal endothelial cell injury and
death via lesions
that produce irreversible retinal ischemia through inability of capillaries to
support blood
flow. Inhibition of ICAM-1 bioactivity blocks diabetic retinal leukostasis and
potently
prevents blood-retinal barrier breakdown.
[00142] Insulin-like Growth Factor-1 (IGF-1) is a 7.5 kDa peptide, having 50%
homology to proinsulin (50%) and is produced primarily in the liver under
control of growth
hormone. IGF-1 is a potent mitogen/stimulator of cell proliferation and a
strong anti-
apoptotic agent. Its function is modulated by six IGF binding proteins
(IGFBPs) and its
levels are influenced by developmental stage and nutrition. Its effects range
from cell growth
and protection, resistance to oxidative stress, promoting growth of bone and
muscle, and
protecting neuronal cells. IGF-1 is implicated in angiogenesis with VEGF
playing a role in
proliferative diabetic retinopathy (PDR). PDR is a complication of diabetes
that is caused by
changes in the blood vessels in the retina. When blood vessels in the retina
are damaged, they
may leak blood and grow fragile, brush-like branches and scar tissue. This can
blur or distort
vision images that the retina sends to the brain.
[00143] Vascular endothelial growth factor and its receptor (VEGF and VEGF/R)
transcription are enhanced by advanced glycation end products and by insulin.
The
accumulation of advanced glycation end products in the diabetic retina
contributes to
neovasculari~ationp which can result in loss of vision. The stimulation of
VEGF synthesis by
insulin may lead to transient acceleration of retinal neovasculari~ation in
patients with
diabetes after insulin therapy is instituted. An aptamer directed to VEGF-165
was found to
have a dissociation constant (I~) of 300 pM and an IC50 value of 1 nM.
[00144] Tumor necrosis factor-alpha (TNF-cc) is present at increased levels
within the
eye during retinal processes of inflammation and angiogenesis. TNF-cc promotes
33
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
proliferation of trabecular meshwork cells; modulates trabecular meshwork,
matrix
metalloproteinases and tissue inhibitor expression; increases MMP-1, 3, and 9
and TIMP-1
expression, decreases TIMP-2. Angiopoietin is an angiogenic growth factor that
occurs in
two forms, Ang-1 and Ang-2. An aptamer directed to Angiopoietin was found to
have a
dissociation constant (I~) of 10 nM and the ability to block Angl- and Ang-2
mediated
inhibition of apoptosis in TNF-cc treated HUVEC cells. The integrin alpha 5
beta 3 (aV~33),
promotes angiogenesis in PDI~ as well as in AMD (Enaida, et al., Fukushima J
Med Sci.
44(1):43-52. (1998)).
[00145] Aptamers of the present invention are capable of binding to ICAM-l,
TGF(31,
TGF[32, TGF(33, IGF-l, VEGF/VEGF-l~, T1VF-oc, and a.V(33, individually or in
combination
to one or more, and inhibit their signaling activities and therefore their
role in ocular disease
pathogenesis.
PEG-Derivatized hTucleic Acids
[00146] Derivatization of nucleic acids with high molecular weight non-
immunogenic
polymers has the potential to alter the pharmacokinetic and pharmacodynamic
properties of
nucleic acids making them more effective therapeutic agents. Favorable changes
in activity
can include increased resistance to degradation by nucleases, decreased
filtration through the
kidneys, decreased exposure to the immune system, and altered distribution of
the therapeutic
through the body.
[00147] The aptamer compositions of the invention may be derivati~ed with
polyalkylene glycol (PAG) moieties. Examples of PAG-derivati~ed nucleic acids
are found
in United States Patent Application Ser. l~To. 10/718,833, filed on 1lTovember
21, 2003, which
is herein incorporated by reference in its entirety. Typical polymers used in
the invention
include polyethylene glycol) (PEG), also known as or polyethylene oxide) (PEO)
and
polypropylene glycol (including poly isopropylene glycol). Additionally,
random or block
copolymers of different alkylene oxides (e.g., ethylene oxide and propylene
oxide) can be
used in many applications. In its most common form, a polyalkylene glycol,
such as PEG, is
a linear polymer terminated at each end with hydroxyl groups: HO-CH2CH20-
(CHZCH2O) n
CHZCH2-OH. This polymer, alpha-, omega-dihydroxylpoly(ethylene glycol), can
also be
represented as HO-PEG-OH, where it is understood that the -PEG- symbol
represents the
34
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
following structural unit: -CH2CH2~-(CHZCH20) n CH2CHz- where n typically
ranges
from about 4 to about 10,000.
[00148] As shown, the PEG molecule is di-functional and is sometimes referred
to as
"PEG diol." The terminal portions of the PEG molecule are relatively non-
reactive hydroxyl
moieties, the -OH groups, that can be activated, or converted to functional
moieties, for
attachment of the PEG to other compounds at reactive sites on the compound.
Such activated
PEG diols are referred to herein as bi-activated PEGS. For example, the
terminal moieties of
PEG diol have been functionalized as active carbonate ester for selective
reaction with amino
moieties by substitution of the relatively nonreactive hydroxyl moieties, -~H,
with
succinimidyl active ester moieties from 1V-hydroxy succinimide.
[00149] In many applications, it is desirable to cap the PEG molecule on one
end with
an essentially non-reaetive moiety so that the PEG molecule is mono-functional
(or mono-
activated). In the case of protein therapeutics which generally display
multiple reaction sites
for activated PEGs, bi-functional activated PEGS lead to extensive cross-
linking, yielding
poorly functional aggregates. To generate mono-activated PEGS, one hydroxyl
moiety on the
terminus of the PEG diol molecule typically is substituted with non-reactive
methoxy end
moiety, -~CH3. The other, un-capped terminus of the PEG molecule typically is
converted to
a reactive end moiety that can be activated for attachment at a reactive site
on a surface or a
molecule such as a protein.
[00150] PAGs are polymers which typically have the properties of solubility in
water
and in many organic solvents, lack of toxicity, and lack of immunogenicity.
~ne use of PAGs
is to covalently attach the polymer to insoluble molecules to make the
resulting PAG-
molecule '6conjugate" soluble. For example, it has been shown that the water-
insoluble drug
paclitaxel, when coupled to PEG, becomes water-soluble. Greenwald, e~ a~., .I:
~rf~. Chern.,
60:331-336 (1995). PAG conjugates are often used not only to enhance
solubility and
stability but also to prolong the blood circulation half life of molecules.
[00151] Polyalkylated compounds of the invention are typically between 5 and
80 kI~
in size. ~ther PAG compounds of the invention are between 10 and 80 kI? in
size. Still other
PAG compounds of the invention are between 10 and 60 kD in size. For example,
a PAG
polymer may be at least 10, 20, 30, 40, 50, 60, or 80 kD in size. Such
polymers can be linear
or branched.
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
[00152] In contrast to biologically-expressed protein therapeutics, nucleic
acid
therapeutics are typically chemically synthesized from activated monomer
nucleotides. PEG-
nucleic acid conjugates may be prepared by incorporating the PEG using the
same iterative
monomer synthesis. For example, PEGS activated by conversion to a
phosphoramidite form
can be incorporated into solid-phase oligonucleotide synthesis. Alternatively,
oligonucleotide
synthesis can be completed with site-specific incorporation of a reactive PEG
attachment site.
Most commonly this has been accomplished by addition of a free primary amine
at the 5'-
terminus (incorporated using a modifier phosphoramidite in the last coupling
step of solid
phase synthesis). Using this approach, a reactive PEG (e.g., one which is
activated so that it
will react and form a bond with an amine) is combined with the puriEed
oligonucleotide and
the coupling reaction is carried out in solution.
[00153] The ability of PEG conjugation to alter the biodistribution of a
therapeutic is
related to a number of factors including the apparent size (e.g~., as measured
in terms of
hydrodynamic radius) of the conjugate. Larger conjugates (>I OkDa) are known
to more
effectively block filtration via the kidney and to consequently increase the
serum half life of
small macromolecules (e.g., peptides, antisense oligonucleotides). The ability
of PEG
conjugates to block filtration has been shown to increase with PEG size up to
approximately
50 kDa (further increases have minimal beneficial effect as half life becomes
defined by
macrophage-mediated metabolism rather than elimination via the kidneys).
[00154] Production of high molecular weight PEGs (>10 kDa) can be difFcult,
inefficient, and expensive. As a route towards the synthesis of high molecular
weight PEG-
nucleic acid conjugates, previous work has been focused towards the generation
of higher
molecular weight activated PEGs. (one method for generating such molecules
involves the
formation of a branched activated PEG in which two or more PEGS are attached
to a central
core carrying the activated group. The terminal portions of these higher
molecular weight
PEG molecules, f.e., the relatively non-reactive hydro~~yl (-~~I) moieties,
can be activated, or
converted to functional moieties, for attachment of one or more of the PEGS to
other
compounds at reactive sites on the compound. )3ranched activated PEGS will
have more than
two termini, and in cases where two or more termini have been activated, such
activated
higher molecular weight PEG molecules are referred to herein as, multi-
activated PEGS. In
some cases, not all termini in a branch PEG molecule are activated. In cases
where any two
termini of a branch PEG molecule are activated, such PEG molecules are
referred to as bi-
36
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
activated PEGS. In some cases where only one terminus in a branch PEG molecule
is
activated, such PEG molecules are referred to as mono-activated. As an example
of this
approach, activated PEG prepared by the attachment of two rnonomethoxy PEGs to
a lysine
core which is subsequently activated for reaction has been described (Harris
et al., Nature,
vol.2: 214-221, 2003).
[00155] The present invention provides another cost effective route to the
synthesis of
high molecular weight PEG-nucleic acid (preferably, aptamer) conjugates
including multiply
PEGylated nucleic acids (as illustrated, e.g., in Fig. 2). The present
invention also
encompasses PEG-linked multimeric oligonucleotides, e.~., dimerized aptamers
(as also
illustrated, e.~., in Fig. 2). The present invention also relates to high
molecular weight
compositions where a PEG stabilizing moiety is a linker which separates
different portions of
an aptamer, e.g., the PEG is conjugated within a single aptamer sequence, such
that the linear
arrangement of the high molecular weight aptamer composition is, e.g., nucleic
acid - PEG -
nucleic acid - PEG - nucleic acid.
[00156] High molecular weight compositions of the invention include those
having a
molecular weight of at least 10 kD. Compositions typically have a molecular
weight between
and 80 kD in size. High molecular weight compositions of the invention are at
least 10,
20, 30, 40, 50, 60, or 80 kI) in size.
[00157] A stabilizing moiety is a molecule, or portion of a molecule, which
improves
pharmacokinetic and pharmacodynamic properties of the high molecular weight
aptamer
compositions of the invention. In some cases, a stabilizing moiety is a
molecule or portion of
a molecule which brings two or more aptamers, or aptamer domains, into
proximity, or
provides decreased overall rotational freedom of the high molecular weight
aptamer
compositions of the invention. A stabilizing moiety can be a polyalkylene
glycol, such a
polyethylene glycol, which can be linear or branched, a homopolymer or a
heteropolymer.
~ther stabilizing moieties include polymers such as peptide nucleic acids
(PNA).
~ligonucleotides can also be stabilizing moieties; such oligonucleotides can
include modified
nucleotides, and/or modif ed linkages, such as phosphothioates. A stabilizing
moiety can be
an integral part of an aptamer composition, i.e., it is covalently bonded to
the aptamer.
[OO15~J Compositions of the invention include high molecular weight aptamer
compositions in which two or more nucleic acid moieties are covalently
conjugated to at least
one polyalkylene glycol moiety. The polyalkylene glycol moieties serve as
stabilizing
37
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
moieties. In compositions where a polyalkylene glycol moiety is covalently
bound at either
end to an aptamer, such that the polyalkylene glycol joins the nucleic acid
moieties together in
one molecule, the polyalkylene glycol is said to be a linking moiety. In such
compositions,
the primary structure of the covalent molecule includes the linear arrangement
nucleic acid-
PAG-nucleic acid. One example is a composition having the primary structure
nucleic acid-
PEG-nucleic acid. Another example is a linear arrangement of nucleic acid -
PEG - nucleic
acid - PEG - nucleic acid.
[00159] To produce the nucleic acid-PEG nucleic acid conjugate, the nucleic
acid is
originally synthesized such that it bears a single reactive site (e.~., it is
mono-activated). In a
preferred embodiment, this reactive site is an amino group introduced at the
5'-terminus by
addition of a modifier phosphoramidite as the last step in solid phase
synthesis of the
oligonucleotide. Following deprotection and purification of the modified
oligonucleotide, it
is reconstituted at high concentration in a solution that minimizes
spontaneous hydrolysis of
the activated PEG. In a preferred embodiment, the concentration of
oligonucleotide is 1 mM
and the reconstituted solution contains 200 mM NaHC03-buffer, pH 8.3.
Synthesis of the
conjugate is initiated by slow, step-wise addition of highly purified bi-
functional PEG. In a
preferred embodiment, the PEG diol is activated at both ends (bi-activated) by
derivatization
with succinimidyl propionate. Following reaction, the PEG-nucleic acid
conjugate is purified
by gel electrophoresis or liquid chromatography to separate fully-, partially-
, and un-
conjugated species. Multiple PAG molecules concatenated (e.g., as random or
block
copolymers) or smaller PAG chains can be linked to achieve various lengths (or
molecular
weights). Non-PAG linkers can be used between PAG chains of varying lengths.
[0060] One high molecular weight composition of the invention has the
following
structure:
-"
~ /~ ~ ~~~ ~ T .
~.~h r-'~
~~ , ~ ~_
~5'~'
~"~t~~~ ~a
~t33~ ~E~3--~!~'~~'P~
38
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
(40 K branched PEG - SEQ ID N0:19 - 0.1 kDa PEG - SEQ >D N0:35 - 0.1 kDa PEG -
SEQ
ID NO:36). 2'-O-methyl modified nucleotides are underlined and 2'-fluoro
modified
nucleotides are italicized. The 2'-O-methyl, 2'-fluoro modifications stabilize
the aptamer
against nucleases and increase its half life in vivo. The 3'-3'-dT cap also
increases
exonuclease resistance. See, e.g., U.S. Patents 5,674,685; 5,668,264;
6,207,816; and
6,229,002, each of which is incorporated by reference herein in its entirety.
[00161] PAG-derivatization of a reactive nucleic acid. High molecular weight
PAG-
nucleic acid-PAG conjugates can be prepared by reaction of a mono-functional
activated PEG
with a nucleic acid containing more than one reactive site. In one embodiment,
the nucleic
acid is bi-reactive, or bi-activated, and contains two reactive sites: a 5'-
amino group and a 3'-
amino group introduced into the oligonucleotide through conventional
phosphoramidite
synthesis, for example: 3'-5'-di-PEGylation as illustrated in Figure 2. In
alternative
embodiments, reactive sites can be introduced at internal positions, using for
example, the 5-
position of pyrimidines, the 8-position of purines, or the 2'-position of
ribose as sites for
attachment of primary amines. In such embodiments, the nucleic acid can have
several
activated or reactive sites and is said to be multiply activated. Following
synthesis and
purification, the modified oligonucleotide is combined with the mono-activated
PEG under
conditions that promote selective reaction with the oligonucleotide reactive
sites while
minimizing spontaneous hydrolysis. In the preferred embodiment, monomethoxy-
PEG is
activated with succinimidyl propionate and the coupled reaction is earned out
at pH 8.3. To
drive synthesis of the bi-substituted PEG, stoichiometric excess PEG is
provided relative to
the oligonucleotide. Following reaction, the PEG-nucleic acid conjugate is
purified by gel
electrophoresis or liquid chromatography to separate fully-, partially-, and
un-conjugated
species. Figure 2 illustrates two strategies for synthesizing PEGylated
nucleic acid aptamers.
[00162] The linking domains can also have one ore more polyallcylene glycol
moieties
attached thereto. Such PAGs can be of varying lengths and may be used in
appropriate
combinations to achieve the desired molecular weight of the composition.
[00163] The effect of a particular linker can be influenced by both its
chemical
composition and length. A linker that is too long, too short, or forms
unfavorable steric
and/or ionic interactions with the target will preclude the formation of
complex between
aptamer and target. A linker, which is longer than necessary to span the
distance between
nucleic acids may reduce binding stability by diminishing the effective
concentration of the
39
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
ligand. Thus, it is often necessary to optimize linker compositions and
lengths in order to
maximize the affinity of a to target.
Pharmaceutical Compositions
[00164) The invention also includes pharmaceutical compositions containing
aptamer
molecules. In some embodiments, the compositions are suitable for internal use
and include
an effective amount of a pharmacologically active compound of the invention,
alone or in
combination, with one or more pharmaceutically acceptable earners. The
compounds are
especially useful in that they have very low, if any toxicity.
[00165] Compositions of the invention can be used to treat or prevent a
pathology,
such as a disease or disorder, or alleviate the symptoms of such disease or
disorder in a
patient. Compositions of the invention are useful for administration to a
subject suffering
from, or predisposed to, a disease or disorder which is related to or derived
from a target to
which the aptamers specifically bind.
[00166] For example, the target is a protein involved with a pathology, for
example,
the target protein causes the pathology.
[00167) Compositions of the invention can be used in a method for treating a
patient
having a pathology. 'The method involves administering to the patient a
composition
comprising aptamers that bind a target (e.g., a protein) involved with the
pathology, so that
binding of the composition to the target alters the biological function of the
target, thereby
treating the pathology.
[0016] The patient having a, pathology, e.~. the patient treated by the
methods of this
invention can be a. mammal, or more particularly, a human.
[0016] W practice, the compounds or their pharmaceutically acceptable salts,
are
administered in amounts which will be cuff cient to inhibit growth factor
activity, for example
TGF(32-, mediated cell proliferation in glaucoma and other proliferative
diseases of the eye.
[00170] One aspect of the invention comprises an aptamer composition of the
invention in combination with other treatments for ocular diseases. The
aptamer composition
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
of the invention may contain, for example, more than one aptamer. In some
examples, an
aptamer composition of the invention, containing one or more compounds of the
invention, is
administered in combination with surgery, or with another useful composition
such as an anti-
inflammatory agent, an immunosuppressant, an antiviral agent, or the like.
Furthermore, the
compounds of the invention may be administered in combination with a
chemotherapeutic
agent such as an alkylating agent, anti-metabolite, mitotic inhibitor or
cytotoxic antibiotic, as
described above. In general, the currently available dosage forms of the known
therapeutic
agents for use in such combinations will be suitable.
[00171] "Combination therapy" (or "co-therapy") includes the administration of
an
aptamer composition of the invention and at least a second agent as part of a
specific
treatment regimen intended to provide the beneficial effect from the co-action
of these
therapeutic agents. The beneficial effect of the combination includes, but is
not limited to,
pharmacokinetic or pharmacodynamic co-action resulting from the combination of
therapeutic agents. Administration of these therapeutic agents in combination
typically is
carried out over a defined time period (usually minutes, hours, days or weeks
depending upon
the combination selected).
(00172] "Combination therapy" may, but generally is not, intended to encompass
the
administration of two or more of these therapeutic agents as part of separate
monotherapy
regimens that incidentally and arbitrarily result in the combinations of the
present invention.
"Combination therapy" is intended to embrace administration of these
therapeutic agents in a
sequential manner, that is, wherein each therapeutic agent is administered at
a different time,
as well as administration of these therapeutic agents, or at least two of the
therapeutic agents,
in a substantially simultaneous manner. Substantially simultaneous
administration can be
accomplished, for example, by administering to the subject a single capsule
having a fixed
ratio of each therapeutic agent or in muli~iple, single capsules for each of
the therapeutic
agents.
[0013] Sequential or substantially simultaneous administration of each
therapeutic
agent can be effected by any appropriate route including, but not limited to,
topical routes,
oral routes, intravenous routes, intramuscular routes, and direct absorption
through mucous
membrane tissues. The therapeutic agents can be administered by the same route
or by
different routes. For example, a first therapeutic agent of the combination
selected may be
41
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
administered by injection while the other therapeutic agents of the
combination may be
administered topically.
[00174] Alternatively, for example, all therapeutic agents may be administered
topically or all therapeutic agents may be administered by injection. The
sequence in which
the therapeutic agents are administered is not narrowly critical. "Combination
therapy": also
can embrace the administration of the therapeutic agents as described above in
further
combination with other biologically active ingredients and non-drug therapies
(e.g., surgery).
Where the combination therapy further comprises a non-drug treatment, the non-
drug
treatment may be conducted at any suitable time so long as a beneficial effect
from the co-
action of the combination of the therapeutic agents and non-drug treatment is
achieved. For
example, in appropriate cases, the beneficial effect is still achieved when
the non-drug
treatment is temporally removed from the administration of the therapeutic
agents, perhaps by
days or even weeks.
[00175] The compounds of the invention and the other pharmacologically active
agent
may be administered to a patient simultaneously, sequentially or in
combination. It will be
appreciated that when using a combination of the invention, the compound of
the invention
and the other pharmacologically active agent may be in the same
pharmaceutically acceptable
Garner and therefore administered simultaneously. They may be in separate
pharmaceutical
carriers such as conventional oral dosage forms which are taken
simultaneously. The term
"combination" further refers to the case where the compounds are provided in
separate
dosage forms and are administered sequentially.
[00176] Preferably, ocular therapeutics are administered topically or by
subconjunctival injection. Repeated topical applications of most ocular drugs
result in
intraocular drug levels comparable to those achieved with subconjunctival
injections, but
subconjunctival injections offer an advantage in the administration of drags
with poor
intraocular penetration (e.g., antibiotics). »y subconjunctival injection,
high local
concentrations of drug can be obtained with the use of small quantities of
medication, so that
adverse systemic effects are avoided. High tissue concentrations can also be
obtained with
drugs that poorly penetrate the epithelial layer of the cornea or conjunctiva.
This method is
useful in patients who do not reliably use topical medication. Intraocular
drugs can be
injected at the conclusion of surgery to avoid the necessity of topical or
systemic drug
therapy. Subconjunctival injection involves passing the needle between the
anterior
42
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
conjunctiva and Tenon's capsule. This can be performed through the lid or
directly into the
subconjunctival space. Tenon's capsule lies between the injected drug and the
globe of the
eye, so the amount of drug absorbed across the sclera is minimized. In fact,
the mechanism of
drug absorption after subconjunctival injection may be simple leakage of drug
through the
needle puncture site with subsequent absorption through the cornea.
[00177] A variety of ocular diseases are treated with subconjunctival
corticosteroids.
For corticosteroids, a subconjunctivally administered drug does penetrate the
underlying
sclera, which suggests a rationale for placing the drug directly adjacent to
the site of
inflammation rather than injecting it randomly. Subconjunctival injection of 5-
flourouracil,
an anti~broblast agent, is sometimes used after high-risk trabeculectomy
surgeries for
glaucoma. Subconjunctival anesthesia is now used as an alternative to
peribulbar or
retrobulbar anesthesia for trabeculectomy or cataract surgery.
[0017] Subconjunctival drug administration is useful in the treatment of
severe
corneal disease, such as bacterial ulcers. le~Iuch higher concentrations of
antibiotics can be
achieved in the affected corneal tissues with subconjunctival injection than
can be obtained
by systemic drug administration. Subconjunctival antibiotic administration is
also useful as
an initial supplement to the systemic or intravitreal antibiotic treatment of
bacterial
endophthalmitis.
[00179] The compositions and combination therapies of the invention may be
administered in combination with a variety of pharmaceutical excipients,
including stabilizing
agents, car~iers and/or encapsulation formulations as described herein.
[0010] 'The pharmaceutical forms suitable for injectable use must be sterile
and must
be fluid to the es~tent that easy syringability ea~ists. It must be stable
under the conditions of
manufacture and st~rage and must be preserved aga~nSt the contammatmg acts~11
of
microorganisms, such as bacteria and fungi.
[0011] 'Therapeutic or pharmacological compositions of the present invention
will
generally comprise an effective amount of the components) of the combination
therapy,
dissolved or dispersed in a pharmaceutically acceptable medium.
Pharmaceutically
acceptable media or carriers include any and all solvents, dispersion media,
coatings,
antibacterial and antifungal agents, isotonic and absorption delaying agents
and the like. The
use of such media and agents for pharmaceutical active substances is well
known in the art.
43
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
Supplementary active ingredients can also be incorporated into the therapeutic
compositions
of the present invention.
[00182) The preparation of pharmaceutical or pharmacological compositions will
be
known to those of skill in the art in light of the present disclosure.
Typically, such
compositions may be prepared as injectables, either as liquid solutions or
suspensions; solid
forms suitable for solution in, or suspension in, liquid prior to injection;
as tablets or other
solids for oral administration; as time release capsules; or in any other form
currently used,
including eye drops, cremes, lotions, salves, inhalants and the like. 'The use
of sterile
formulations, such as saline-based washes, by surgeons, physicians or health
care workers to
treat a particular area in the operating ~xeld may also be particularly
useful. Compositions
may also be delivered via microdevice, microparkicle or sponge.
[00183] Upon formulation, therapeutics will be administered in a manner
compatible
with the dosage formulation, and in such amount as is pharmacologically
effective. The
formulations are easily administered in a variety of dosage forms, such as the
type of
injectable solutions described above, but drug release capsules and the like
can also be
employed.
[00184] In this context, the quantity of active ingredient and volume of
composition to
be administered depends on the host animal to be treated. Precise amounts of
active
compound required for administration depend on the judgment of the
practitioner and are
peculiar to each individual.
[00185] A minimal volume of a composition required to disperse the active
compounds
is typically utilized. Suitable regimes for administration are also variable,
but would be
typified by initially administering the compound and monitoring the results
and then giving
further controlled doses at further interd~~als.
[001u6) For oral administration in the form of a tablet or capsule (e.~., a
gelatin
capsule), the active drug component can be combined with an oral, non-toxic
pharmaceutically acceptable inert carrier such as ethanol, glycerol, water and
the like.
Ie~oreover, when desired or necessary, suitable binders, lubricants,
disintegrating agents and
coloring agents can also be incorporated into the mixture. Suitable binders
include starch,
magnesium aluminum silicate, starch paste, gelatin, methylcellulose, sodium
carboxymethylcellulose and/or polyvinylpyrrolidone, natural sugars such as
glucose or beta-
lactose, corn sweeteners, natural and synthetic gums such as acacia,
tragacanth or sodium
44
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
alginate, polyethylene glycol, waxes and the like. Lubricants used in these
dosage forms
include sodium oleate, sodium stearate, magnesium stearate, sodium benzoate,
sodium
acetate, sodium chloride, silica, talcum, stearic acid, its magnesium or
calcium salt and/or
polyethyleneglycol and the like. Disintegrators include, without limitation,
starch, methyl
cellulose, agar, bentonite, xanthan gum starches, agar, alginic acid or its
sodium salt, or
effervescent mixtures, and the like. Diluents, include, e.~., lactose,
dextrose, sucrose,
mannitol, sorbitol, cellulose and/or glycine.
[0017] Injectable compositions are preferably aqueous isotonic solutions or
suspensions, and suppositories are advantageously prepared from fatty
emulsions or
suspensions. The compositions may be sterilized and/or contain adjuvants, such
as
preserving, stabilizing, wetting or emulsifying agents, solution promoters,
salts for regulating
the osmotic pressure and/or buffers. In addition, they may also contain other
therapeutically
valuable substances. The compositions are prepared according to conventional
mixing,
granulating or coating methods, respectively, and contain about 0.1 to 75%,
preferably about
1 to 50%, of the active ingredient.
[0018] The compounds of the invention can also be administered in such oral
dosage
forms as timed release and sustained release tablets or capsules, pills,
powders, granules,
elixers, tinctures, suspensions, syrups and emulsions.
[0019] Liquid, particularly injectable compositions can, for example, be
prepared by
dissolving, dispersing, etc. The active compound is dissolved in or mixed with
a
pharmaceutically pure solvent such as, for example, water, saline, aqueous
dextrose, glycerol,
ethanol, and the like, to thereby form the injectable solution or suspension.
Additionally, solid
forms suitable for dissolving in liquid prior to ialjection can be formulated.
Injectable
compositions are preferably aqueous isotonic solutioaas or suspensions. 'The
compositions may
be sterilized and/or contain adjuvants, such as preserving, stabilizing
wetting or emulsifying
agents, solution promoters, salts for regulating the osmotic pressure and/or
buffers. In
addition, they may also contain other therapeutically valuable substances.
[00190] The compounds of the present invention can be administered in
intravenous
(both bolus and infusion), intraperitoneal, subcutaneous or intramuscular
form, all using
forms well known to those of ordinary skill in the pharmaceutical arts.
Injectables can be
prepared in conventional forms, either as liquid solutions or suspensions.
[00191] Parental injectable administration is generally used for subcutaneous,
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
intramuscular or intravenous injections and infusions. Additionally, one
approach for
parenteral administration employs the implantation of a slow-release or
sustained-released
systems, which assures that a constant level of dosage is maintained,
according to U.S. Pat.
No. 3,710,795, incorporated herein by reference.
[00192] Furthermore, preferred compounds for the present invention can be
administered in
intranasal form via topical use of suitable intranasal vehicles, or via
transdermal routes, using
those forms of transdermal skin patches well known to those of ordinary skill
in that art. To
be administered in the form of a transdermal delivery system, the dosage
administration will,
of course, be continuous rather than intermittent throughout the dosage
regimen. ~ther
preferred topical preparations include creams, ointments, lotions, aerosol
sprays and gels,
wherein the concentration of active ingredient would range from 0.01°/~
to 15%, w/w or w/v.
[00193] For solid compositions, excipients include pharmaceutical grades of
mannitol,
lactose, starch, magnesium stearate, sodium saccharin, talcum, cellulose,
glucose, sucrose,
magnesium carbonate, and the like may be used. The active compound defined
above, may
be also formulated as suppositories using for example, polyalkylene glycols,
for example,
propylene glycol, as the carrier. In some embodiments, suppositories are
advantageously
prepared from fatty emulsions or suspensions.
[00194] The compounds of the present invention can also be administered in the
form of
liposome delivery systems, such as small unilamellar vesicles, large
unilamellar vesicles and
multilamellar vesicles. Liposomes can be formed from a variety of
phospholipids, containing
cholesterol, stearylamine or phosphatidylcholines. In some embodiments, a film
of lipid
components is hydrated with an aqueous solution of drug to a form lipid layer
encapsulating
the drug, as described in U.S. Pat. No. 5,262,564. For example, the aptamer-
toxin and/or
riboreporter molecules described herein can be provided as a complex with a
lipophilic
compound or non-immunogenic, high molecular weight compound constructed using
methods known in the art. An example of nucleic-acid associated complexes is
provided in
U.S. Patent No. 6,011,020.
[00195] The compounds of the present invention may also be coupled with
soluble polymers
as targetable drug carriers. Such polymers can include polyvinylpyrrolidone,
pyran
copolymer, polyhydroxypropyl-methacrylamide-phenol,
polyhydroxyethylaspanamidephenol,
or polyethyleneoxidepolylysine substituted with palmitoyl residues.
Furthermore, the
compounds of the present invention may be coupled to a class of biodegradable
polymers
46
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
useful in achieving controlled release of a drug, for example, polylactic
acid, polyepsilon
caprolactone, polyhydroxy butyric acid, polyorthoesters, polyacetals,
polydihydropyrans,
polycyanoacrylates and cross-linked or amphipathic block copolymers of
hydrogels.
[00196] If desired, the pharmaceutical composition to be administered may also
contain
minor amounts of non-toxic auxiliary substances such as wetting or emulsifying
agents, pH
buffering agents, and other substances such as for example, sodium acetate,
triethanolamine
oleate, etc.
[00197] The dosage regimen utilizing the compounds is selected in accordance
with a variety
of factors including type, species, age, weight, sex and medical condition of
the patient; the
severity of the condition to be treated; the route of administration; the
renal and hepatic
function of the patient; and the particular compound or salt thereof employed.
An ordinarily
skilled physician or veterinarian can readily determine and prescribe the
effective amount of
the drug required to prevent, counter or arrest the progress of the condition.
(0019] ~ral dosages of the present invention, when used for the indicated
effects, will range
between about 0.05 to 1000 mg/day orally. The compositions are preferably
provided in the
form of scored tablets containing 0.5, 1.0, 2.5, 5.0, 10.0, 15.0, 25.0, 50.0,
100.0, 250.0, 500.0
and 1,000.0 mg of active ingredient. Effective plasma levels of the compounds
of the present
invention range from 0.002 mg to 50 mg per kg of body weight per day.
Compounds of the present invention may be administered in a single daily dose,
or the total
daily dosage may be administered in divided doses of two, three or four times
daily.
E~AlI~IPLES
EIPLE 1 Purified and Characterized TGF 3~2 protein
[00199] ~ synthetic polynucleotide encoding full length hmnan TGF[32 mature
protein was
cloned into pRSET E. c~li expression vector and transformed into BL21 (pLys)
strain. The
transformed cells were grown under conditions leading to TGF(32 protein being
expressed at
high level and forming inclusion bodies. T'he inclusion bodies were purified
and solubilized.
The TGF(32 was refolded and purified by S75 size exclusion chromatography
(Fig. 4P~). !~
His-tagged TGF(32 was also generated by adding His6 plus 30 extra amino acids
at the N
terminus of TGF(32. Mutations at positions 59 (SST), 60 (R~I~) and 94 (I~~N),
referred to
herein as the S59T/R60I~/K94N mutant, were introduced by site directed
mutagenesis. Both
47
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
the His-tagged and mutated TGF(32 were expressed, refolded and purified using
the same
protocol as for wild type TGF(32. Figure 4B shows the elution profile of
TGF(32 in S75 size
exclusion chromatography, such that the fractions containing the TGF[32 dimer
peaks
corresponding to the PAGE band in Figure 4C are indicated by the box in Figure
4B.
EXAMPLE 2 Bindin , of TGF~32protein to TGF(32-specific aptamers
[00200] Dissociation constants for the binding of aptamers to purified human
or rodent
TGFj3-2 protein were determined by nitrocellulose-filter partitioning using
32P-labeled 12NA.
Ira vitr~ transcribed RIVA was treated with calf intestinal alkaline
phophatase (New England
Biolabs) t~ remove 5'-triphosphates, then radio-labeled by incubation with y-
32P-ATP and T4~
polynucleotide kinase (IVew England Biolabs). Unincorporated label was removed
by gel-
filtration, and the I~I~TAs were further purified by polyacrylamide gel
electrophoresis (PAGE).
Purified 32P-labeled aptamer in water was refolded just prior to use by
heating for 3 min at
95 °G, followed by a 10 minute room-temperature incubation in binding
buffer (50 mM
Hepes, pH 7.4, 1 mM MgCIZ, 1 mM CaCl2, 3 mM I~Cl, 140 mM NaCl, 0.1 mg/ml BSA,
0.01
mg/ml tRNA). The binding reactions (100 p.L) were initiated by the addition of
aptamer (<_
0.1 nM) to excess TGF(3-2 protein (0.2 - 100 nM), followed by 10-30 minute
equilibration at
room temperature.
[00201] Nitrocellulose-filter partitioning was performed on I~inifold~ l, 96-
well Dot-Blot
manifolds (Schleicher ~ Schuell). Protein-bound aptamer and residual free
aptamer were
captured, respectively, on pre-wetted Protran nitrocellulose (Schleicher ~.
Schuell) and
Hybond-P poly~rinylidene diiluoride (Amersham Bi~sciences) filters by vacuum
aspiration,
and quantified by Phosphorhnager (Amersham Biosciences). The proportion of
protein-
bound aptamer at each TGF(3-2 concentration was plotted as the ratio of counts-
per-minute on
the Protran nitrocellulose filter (CPMN~) to the sum of CPM on the Protran and
Hybond-P
filters (CPMtota~). Estimates of dissociation constants (IUD) were obtained
from a fit of the
concentration of TGFB-2 (i.e., [TGF(3-2~)-dependent data to a standard binding
isotherm:
48
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
CP~NC~CPMtotal - CmaX ~ (1 + KD~[TGF[32]total), where CmaX equals the maximum
observed
value of CPMN~/CPMt°ta~ at saturating [TGF(3-2], likely reflecting the
proportion of properly-
folded aptamer that is competent to recognize TGF(3-2.
[00202] Competition assays. Certain aptamers, such as those modified with high
molecular
weight PEG moieties, bound non-specifically to nitrocellulose, and thus were
not amenable to
standard nitrocellulose-filter partitioning assays. These aptamers were
assayed by
competition with other, well-characterized aptamers such as ARC77.
[00203] Aptamer competition reactions were prepared by pre-incubation in
binding buffer of
32P-labeled ARC77 (< 0.1 nlVl) with increasing concentrations of unlabeled,
competitor
aptamer (0.05 - 300 nllil). Binding reactions were initiated by the addition
of aptamer
samples to TGF(3-2 protein, yielding a final protein concentration of 2.5 nM.
In the absence
of cold competitor, ~30% of 32P-labeled ARC77 was typically observed to bind
to the Protran
nitrocellulose membrane at this [TGF[i-2]. The decrease in bound 32P-labeled
ARC77,
observed as a function of increasing competitor concentration, was well
described by the
following model:
K1 K2
A*eP ~~ P ~-~ A~P (1)
in which A* is 32P-labeled ARC77, P is TGF[3-2 protein, A is cold competitor
aptamer, K~ is
the dissociation constant for the interaction between protein and hot aptamer,
and Ka is the
dissociation constant for the interaction between protein and cold competitor
aptamer.
Estimates of K~ were obtained fxona plots of CPI~INCICPIe~t°ta~ dersus
[cold competitor)
([A]t~ta~) by fitting the data to Equation 2, derived for this model under the
conditions of [A°~°~
« [P]tota~:
C,PI~NC Cmax
~P~tota [A]tota [P°A Kl
1+ 1+ -
Kz Kz (P~t°ta
where [P~A] is described by the quadratic equation:
49
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
[P,A] _ (~P~total+ [A~total + KZ) - ~([p]tota~ + [A~total+ K2)a -
4[P]total[A~total~'~z
2
Each competition assay was accompanied by an independent measurement of K~,
determined
by the standard binding assay described above, which was included in the fit.
[00204] Figures SA and SB show the binding of TGF(3-2 protein to varying
concentrations of
TGF(3-2 specific aptamers. (A) 32P-labeled ARC77 (<_ 0.1 nM) was incubated
with increasing
concentrations (0.2 - 100 nM) of human ( ) or rat (~) TGF(3-2 protein and
binding was
analyzed by nitrocellulose-filter partitioning. The I~ values for these
aptamers were:
ARC77: 3.6 +/- 0.6; ARC78: 4.0 +/- 0.5, and for ARC81: 5.1 +/- 0.4. Estimates
of I~ were
obtained by a fit of the data to Equation 1. (B) Alternatively, estimates of
aptamer
dissociation constants were obtained by competition with 32P-labeled ARC77 for
binding to
human TGF[i-2. Results (shown in Figure SB) of the competitive binding of non-
radiolabeled ARC77 (0) and ARC81 () were obtained by a fit of the data to
Equation 2
above.
EXAMPLE 3 Species specific binding of TGF(32 Aptamers
[00205] Three aptamer compositions of the pxesent invention, ARC77, ARC78 and
ARC81,
were compared for species binding specificity to human and rodent TGF(32 and
binding
affinity according to the methods of Example 2.
[00206] As seen in Figures 6A and 6B, the ARC81 aptamer reversed human TGF(32-
mediated inhibition of MLEC proliferation. Figure 6A shows that the ARC77,
ARC78 and
ARC81 aptamers inhibit flee antiproliferative effects of 50 pg/ml of TGF[32.
An anti-TGF[32
antibody (R~~I~, AF-302-I~TA~)9 which was included as a control, also
re~rersed the effect of
low concentrations of aqueous humor on cell proliferation. Figure 6B shows
that the ARC77
aptamer is more potent against the human form of TGF(32 than the rodent
version. Taken
together, these data indicate that the ARC77, ARC78 and ARC81 aptamers are
able to reverse
the biological activity of TGF(32 as measured using cell proliferation. The
ARC77 aptamer
also demonstrated specificity against the human versions of TGF~32 versus the
rodent. Figure
6C shows that the ARC77 aptamer has different binding affinity to human wild
type (WT),
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
mouse (NTK) and N-terminal His tagged versions of human TGF(32: wild type (WT)
2.5 ~
0.3 nM, mouse (NTK) 80 ~ 5 nM, and His-tagged >_ 500 nM.
[00207] MLECs were plated at 2,000 cells per well and incubated at 37
°C for 4 hours.
Aptamer and TGF(32 were added at the indicated concentrations for 16 hours at
37 °C. Cell
proliferation was measured using BrdU incorporation. BrdU assay was performed
as
recommended by the manufacturer (Ruche Diagnostics).
EXAMPLE 4 Aqueous Humor challenge assay
[0020] ARC81 reverses aqueous humor-mediated inhibition of MLEC proliferation.
Figure
7A shows that 1000 nM ARC81 inhibited the antiproliferative effects of low
concentrations
of rabbit aqueous humor (e.~., <10%). An anti-TGF(32 antibody (RED, AF-302-
NA), which
was included as a control, also reversed the effect of low concentrations of
aqueous humor on
cell proliferation. Figures 7B and 7C show that the ARC81 antibody and anti-
TGF(32
antibody rescued rabbit aqueous humor-mediated inhibition of MLEC
proliferation in a dose
dependent manner. Taken together, these data indicate that the ARC81 aptamer
is able to
reverse the biological activity of TGF(32 in aqueous humor.
[00209] MLEC Assay. The mink lung epithelial cell proliferation assay is
performed
over two days. On day 1: 1) media is aspirated off from Mink Lung Epithelial
Cells (MLEC);
2) the MLEC are washed with lOm1 lxPBS; 3) 3m1 Trypsin is added and Trypsinize
for 3 min
at 37 °C; 4) quench with l Oml 0.5% FBS media; 5) spin at 1000rpm for
3.30 min; 6) aspirate
off supernatant; 7) resuspend pellet in lOml 0.5°!° FBS media;
8) Count l Opl cell suspension;
9) adjust cell density to 80,000 cells/ml; 10) add SOpl of cclls/well to black
botkomed 96-well
plates (4000 cells/well); pipet up and down during plating process to avoid
settling of cells
and uneven plating; 11) incubate cells at 37°C in S°/~ CO~ for
4~ hrs to allow adherence of
cells; 12) add 25p1 of aptamcr (or media, or other test reagent, such as for
example, an
antibody), preferably the outer wells are not used for treated cells; 13) add
25p.1 of TGF(32
(typically 25pg/ml); and 14) incubate cells at 37 °C in 5% COZ
overnight.
[00210] On day 2: 1) mix 20p,1 BrdU with 2ml 0.5% FBS media; 2) add l Op~l of
BrdU
mixture/well; 3) incubate cells at 37 °C in 5% COZ for 3 hrs; 4) remove
media and blot plates
dry on paper towels; 5) add 200p,1/well FixDenat solution and incubate for 30
min at room
Sl
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
temperature (RT); 6) remove FixDenat solution and blot plates dry on paper
towels; 7) add
100~.llwell anti-BrdU POD solution and incubate for 90 min at RT; 8) remove
anti-BrdU
POD solution and blot plates dry on paper towels; 9) wash plates 3x with
200p1/well washing
solution with Smin RT incubations and blot plates dry on paper towels; 10) add
100p,1/well
substrate solution and incubate for 3 min at RT in dark; and 11) read plates
using PSP
Luciferace 1 program on TopCount (Packard Bioscience Co., Downers Grove, IL).
[00211] ML,ECs were plated at 2,000 cells per well and incubated at 37
°C for 4 hours.
Aptamer, antibody, aqueous humor and TGF(32 were added at the indicated
concentrations for
16 hours at 37 °C. Cell proliferation was measured using BrdU
incorporation. BrdU assay
was performed as recommended by the manufacturer (Ruche Diagnostics).
EPLE 5 Selection minimization and characterization of TGF(32-specific
aptamers
[00212] The modified RNA aptamers of the present invention, e.g., ARC 77 (SEQ
~ No.l)
bind native human TGF(32 and are capable of blocking TGF(32 effect in IVIink
Lung Epithelial
Cell (MLEC) inhibition assay. For further biochemical characterization of the
aptamers, two
forms of mature TGF[32 were generated, native and N-terminal his-tagged
versions, in E. coli.
After refolding and purification, functional TGF(32s were obtained. These
TGF(32 were
active in cell based assay. N-terminal tags affected both activity and aptamer
binding, while
the affinity to aptamer was decreased to much larger extent. Further, two
mutant TGF(32s
(labeled I~94N and S59T1R60I~//K94N) were generated based on known isoforms of
TGF(32.
'The I~94~N mutant was capable of binding to the aptamers with comparable
affinity with that
of native TGF~32, whereas the S59T/R60I~/I~94N mutant had significantly
reduced affinity to
the aptamers. Similarly, the aptamers blocked the bioaetivity of native and
K94N TGF~32s
with higher potencies than that of S59T/R60I~/K94~1~T mutant in a cell based
assay. Based on
a published crystal structure, two substitutions at positions 59 and 60 reside
near the dimer
interface and adjacent to the N-terminus of TGF(32, and the other substitution
at position 94 is
near the type II receptor binding site. Binding competition assay with soluble
TGF-b
receptors revealed that type III receptor competes with the aptamer binding,
but not type II
receptor. The data demonstrated that two aptamers bind to one dimer TGF(32 and
that the
52
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
aptamers of the present invention bind TGF(32 near or at the TGF-b type III
receptor binding
site and block its biological function.
[00213] Minimization and mutagenesis/modification analysis of the aptamer.
Figure 8A is
an illustration of the selection, minimization and characterization of TGF[32
aptamers of
SEQ ID No. 1 (ARC77). Deletions of residues) and the resulting effect on
binding affinity
are indicated in the text of Figure 8A and in the table of Figure 8B. The
boxed residues in
Figure 8A represent highly conserved residues. The binding affinity of the
aptamer to TGF
(3s was determined by dot blot protein binding assay (Figure 8B). TGF[32
aptamer can
reverse the inhibitory effect of TGF(32 on MLEC cell proliferation. Scrambled
aptamer
(labeled as "ARC77 transcribed" in Figure 8C) was used as a negative control,
while a
TGF(32 neutralizing antibody was used as a positive control (not shown).
[00214] Figure 9 shows the stoichiometry of the aptamer/TGF(32 dimer as
determined by o,
screen. Different concentration of the aptamers, labeled by either fluorescein
or biotin were
bound to anti-FITC acceptor beads or streptavidin donor beads and titrated
with TGF(32
homodimer. The signal was detected with the Fusion plate reader (Packard
Eioscience Co.,
Downers Grove, IL).
[00215] Figure l0A shows mapping of the aptamer binding site of TGF/32 by
determining
the effect of various modifications and/or mutations to wild typeTGF(32. Three
human
TGF(32 variants were tested: wild type TGF(32, a long tag form of TGF(32 and a
short tag
form of TGF(32. In addition, two mutants were tested. The K94N and
S59T/R60K/K94N
mutations were introduced into wild type TGF(32 by quick change site-directed
mutagenesis.
Each of these proteins (i.e., wild type, S59T9R60K/K94~N, K94N, N-long tag
TGF(32 and N-
short tag TGF(32) were incubated in the presence of a TGFb2 aptamer, and their
EC100
values, binding affinities and IC50 values (in nM) u~rere determined (Figure
10E). )finding
affinity of the aptamer to those proteins were determined by dot blot. IC50
values of the
inhibitory activity of the aptamer were deteaxnined using the MLEC
proliferation assay.
[0021] Figure 11 shows that the TGF type III receptor can block the binding of
the aptamer
with TGF(32. Dot blot assays to determine aptamer binding with TGF(32 were
performed
after preincubating TGF[i2 with either soluble type III or type II receptors.
The Ki was
calculated by fitting the data with a simple competition model.
53
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
[00217) Table 1- Aptamer Sequences
SEQ ID No.l ARC77 - TGF[32
5'-GGAGGfUfUAfIJfLTAfCAGAGfUfCfUGfUAf(.TAGfCfUGfUAfCfUfCfC-3T-3'
SEQ ID No.2 ARC78 - TGF(33
5'-NH2-GGAGGf(1f(lAfUf<JAfCAGAGfUfCfUGfUAf<JAGfCffJGfUAfCfUfCfC-3T-3'
SEQ » No.3 ARC79 - TGF[32
5'-
mGmGmAmGmGf LJf (JAfUf CTmAfCmAmGmAmGfUfCf UGf UAf CTAmGfCf UmGf CTmAfC f
LTfCfC-3 T-3'
SEQ » No.4 ARC81 - TGF(32
5'-NH2-
mGmGmGmGf tJf CTAftlf IJrnAfCmAmGmAmGf tJfCf CTGf CTAf tJAmGfCf ~lmGf(
TmAfCfCfC
-3T-3'
SEQ » No.S ARC82 - TGF(32
5'-
mGGmGmGfUfUmAfUfUAfCAmGmAmGfLIfCfCTmGfUmAfUmAxnGfCfCJmGfCTAfCfCfC
-3T-3'
SEQ ~ No.6 ARC111 - TGF(32
5'-[20KPEG]-NH2-
GGAGGfCTfCJAf(JfUAfCAGAGfUfCfUGf(.TAfUAGfCfCTGf(JAfCftlfCfC-3T-3'
SEQ ID No.7 ARC 112 - TGF j32
5'-[PEG30I~] NH2-
GGAGGfUfUAf(Jf(JAfCAGAGfUfCfUGfCIAfUAGfCfUGfUAfCfUfCfC-3T-3'
SEQ » No.8 ARCl 13 - TGF(32
5'-[PEG4~OI~]-NH2-
GGAGGfIJfCJAf(Jf<JAfCAGAGfCTfGfLJGfCJAfUAGfCfCTGf(JAfCfCTfCfC-3T-3'
SEQ II2 No.9 ARC117 - TGF[i2
5'-[PEG20~]-NH2-
mGmGmGmGfLTftJAftlfClmAfCmAmGmAmGffJf~fClGftTAf(Jt~mGfCfLTmGfITmAfCfCfC
-3T-3'
SEQ ~ No.lO ARC118 - TGF(32
5'-[PEG30I~]-NH2-
mGmGmGmGftJfUAfUfUmAfCmAmGmAmGfUfCfUGftJAftJAmGfCfCTmGfUmAfCfCfC
-3T-3'
SEQ ~ No.l 1 ARC119 - TGF(32
54
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
5'-[PEG30K)-NH2-
mGmGmGmGfIlfUAfUfUmAfCmAmGmAmGfUfCfUGfUAfUAmGfCf<JmGfUmAfCfCfC
-3T-3'
SEQ m No.l2 ARC120 - TGF(32
s'-[PEG20K)-NH2-
mGGmGmGf UftJmAf UfLTAfCAmGmAmGf UfCf UmGf tlmAf tlmAmGfCf(JmGfUAfCfCfC
-3T-3'
SEQ ~ No.l3 ARC121 - TGF(32
s'-[PEG30K)-NH2-
mGGmCamGftlfiJmAftJf CTAfCAmGmAmGf CJfCfLTmGfUmAfUrnAmGfCf~lmGf tJAfCfCfC
-3T-3'
SEQ ID No.l4 ARC122 - TGF(32
s'-[PEG4~OK)-NH2-
mGGmGmGfCJfUmAftJfUAfCAmGmAmGf(.TfCfUrnGfUmAfUmAmGfCf(JmGfUAfCfCfC
-3T-3'
SEQ )I~ No.21 ARC 1 s2 - TGF(32
s~_~~)_
mGmGmAmGmGfUf CTAf CTf CTmAfCmAmGmAmGf UfCftJGftJAfUAmGfCfUmGftJmAfCf
UfCfC-3T-3'
SEQ 17~ No.4 ARC 1 s4 - TGF(32
5'-[NH2]-
mGmGmAmGmGfUfUAfUfUmAfCmAmGmAmGfUfCfUGf(JAfUAmGfCfUmGfUmAfCf
UfCfC-3T-3'
SEQ ID No.23 ARCl ss - TGF(32
5'C-[NH2)-
mGGmGmGftTf(lmAfUftJA~AmGmAmGf(lfCftlmGftJmAfUmAmGfCfCTmGfUAfCfCfC
-3T-3'
SEQ ~ No.24~ ARC1 s6 - TGF[32
s'-[l~lla)-[i~TH2)-
mGGmGmGfUfLJmAf~TfUAfCAmGmAmGf(T~ftJmGfLJmAftlmAm(afCf~.TaxiGf(JAfC~fC
-3T3'
SEQ ~ No.2s ARCls7 - TGF~32
s'-[~~~)-[NH2)-
mGGmGmGfUfUmAfCTfUAfCAmGmAmGfUfCf(.TmGfUmAfUmAmGfCfUmGfUAfCfCfC
-3T-3'
SEQ ~ No.26 ARC158 - TGF[i2
ss
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
5'-[arg7]-[NH2]-
mGGmGmGfUfUmAfUfUAfCAmGmAmGfUfCfUmGfUmAfUmAmGfCfUmGfUAfCfCfC
-3T-3'
SEQ IT? No.27 ARC159 - TGF(32
5'-[NH2]-[NH2]-
mGmGmAmGmGmUmUmAmUmUmAmCmAmGmAmGmUmCmUmGmUmAmUmAmG
mCmUmGmUmAmCmUmCmC-3T-3'
Example 6 PDGF Aptamers in ~cular Disease Treatment
[0021] Platelet derived growth factor (PDGF) is a strong mitogen and is known
to
play a crucial role in a variety of proliferative diseases. Table 1 below
shows the dosing
concentrations for three sample aptamers useful in ocular disease treatment
tested in mice.
The dose for administration to young male mice 5-6 weeks old via intravenous
is lmg/kg or
subcutaneous administration is 1, 5, and 20 mg/kg. Timepoints taken after
intravenous
administration are 09 5, 10, 20, 40 min, l, 2, 4, 6, 8, and 10 hours.
Timepoints taken after
subcutaneous administration are 0, 10, 20, 40 min, l, 2, 4, 69 8, 10, and 12
hrs. The binding
affinity for ARC 125 (SEQ ~ No. 16) and ARC 127 (PEG - SEQ >I? N~:19 - PEG -
SEQ ID
N~: 35 - PEG -SEQ ID N0:36 - 3T) bind PDGF AB and BB with a Kd of 100 pM.
ARC127 (PEG - SEQ )D N0:19 - PEG - SEQ ID NQ: 35 - PEG -SEQ ID N~:36 - 3T) has
been selected from a ssDNA pool and later modified with 2'-O-methyl
(underlined) and 2'
fluoro (italicized) and carries 40I~ PEG group. The 2'-O-methyl, 2'-fluor
modifications
stabilize the aptamer against nucleases and increase its half life in vivo.
The 3'-3'-dT cap
increases exonuclease resistance. A 40-K PEG group increases the PIE
properties of
ARC126.
[0021] Table 2 - PDGF specific Aptamers
L~~~1 _1 a~ne~n~r~~l~~
~arrn~let~ ~1- r~~1 le~~l~
a crri
erg~1 i~ ~
rr,~ rrrl'a
E~Ea~g~fa10,129.47265,400.0020 0.02
ai~Ci~~ 50,128.47265,400.0020 0.02
~I~C12~ 50,128.47264,800.0020 0.02
as determined by anion exchange HPLC and/or CGE ~ calculated using aptamer
weight only
56
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
[00220] ARC123 (SEQ >D No. 15), ARC124 (SEQ >D No. 16) and ARC125 (SEQ >D
No. 17) have been selected from a ssDNA pool to bind PDGF AB and BB receptors
with a I~
of 100 pM. They do not have any modified groups but have a 3'-3'-dT cap to
increase
exonuclease resistance.
[00221] TABLE 3 - PDGF Aptamers
ARC123 (SEQ >I? No. 15):
5'-
Td(adGdGdAdGdGdGdCdGdCdGTTdCTTdCdGTdGdGTTdAdCTTTTdAdGTdCdCdCdG-
3T-3'
ARC 124 (SEQ ff) No. 16):
5'-
dCdAdCdAdGdGdCTdAdCdGdGdCdAdCdGTdAdGdAdGdCdATdCdAdCdCdATdGdATd
CdCTdGTdG-3T-3'
ARC125 (SEQ ~ No. 17):
5'-
TdAdCTdCdAdGdGdGdCdAdCTdGdCdAdAdGdCdAdATTdGTdGdGTdCdCdCdAdATdG
dGdGdCTdGdAdGTdA-3T-3'
ARC 126 (SEQ ff~ N0:18 - PEG - SEQ )D N0:33 - PEG - SEQ )D N~:34) (functional
aptamer):
5'-[NH2J-dCdAdGdGdCftJdAfCmG (SEQ ~ N0:18)-PEG-
dCdGTdAmGdAmGdCdAfUfCmA (SEQ ID N0:33)-PEG-TdGdATfCf~fUmG-3T-3' (SEQ
~ N0:34)
ARC 127 (PEG - SEQ ~ NO. 19 - PEG - SEQ ~ N~:35 - PEG - SEQ ID N~:36 - 3T)
(PEGylated functional aptamer):
5'-[PEG40I~J-NH2-dCd~dGd(~dCftldAfCmG (SEQ ~ N~:1~)-PEG-
dCdGTdAmGdl~mGdCdf-~f(JfCmA (SEQ lI~ NG:35)-PEG-TdGdATfC~fLTmG-3T-3' (SEQ
~ N(~:36)
ARC128 (PEG - SEQ JD No. 20 - PEG - SEQ >I~ N~:37 - PEG - SEQ J~ N~:38 - 3T)
(scrambled control):
5'-[PEG40I~]-NH2-dCdAdGfCmGfUdAfCmG (SEQ ~ N0:20)-PEG-
dCdGTdAdCdCmGdATftJfCmA (SEQ ~ N~:37)-PEG-TdGdAdAdGfCf(1mG-3T-3' (SEQ
~ NO: 38)
57
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
[00222] Figures 13A and 13B show the binding curves and I~ values for ARC 127
(PEG - SEQ )D N0:19 - PEG - SEQ ID NO: 35 - PEG -SEQ ID NO:36 - 3T)
demonstrating that ARC127 recognizes the BB and AB isoforms of PDGF but not
the AA
isoform alone. Figure 14A and 14B show that ARC127 (PEG - SEQ ID N0:19 - PEG -
SEQ >D NO: 35 - PEG -SEQ )D N0:36 - 3T) binds to human and rat PDGFs with
equivalent affinity, and furthermore, the ability of ARC127 to block PDGF-
induced 3T3 cell
proliferation is comparable to the ability of an anti-PDGF antibody
(LTpstate/Cell Signaling
Solutions) to block PDGF-induced 3T3 cell proliferation.
[00223] The series of images presented in Figure 15 show that ARC127 (PEG -
SEQ
~ NO:19 - PEG - SEQ ~ NO: 35 - PEG -SEQ ~ NO:36 - 3T) specifically blocks
migration of retinal pigmented epithelial (RPE) cells, while ARC128 (PEG - SEQ
~ No. 20
- PEG - SEQ ~ NO:37 - PEG - SEQ ~ NO:38 - 3T), a scrambled aptamer used as a
control, has no activity. In particular, Fig 15A shows the migration of RPE
cells with no
PDGF. Fig 15B shows the migration of RPE cells with 100 ng/ml PDGF. Fig 15C,
which
shows the migration of RPE cells with PDGF and100 mM ARC127, demonstrates the
blocking effect of ARC127. Fig. 15D, which shows the migration of RPE cells
with PDGF
and 100 mM ARC 128, demonstrates no blocking effect by the scrambled aptamer
control
(i.e., ARC128). The graphs presented in Fig. 15E and 15 F show the increasing
effect of
PDGF concentration on RPE cell migration.
[00224] Figure 16 shows ifa vitro plasma stability of ARC127 (PEG - SEQ )D
NO:19 -
PEG - SEQ ~ NO: 35 - PEG -SEQ ID NO:36 - 3T) in 95°/~ plasma at
37°C. The half life
(tliz) for the modified ARC127 was 14 times greater than the t~iz of an all-
DNA construct.
Example 7 Pharmacokinetic and Bioactivity profile of ARC 127
[0022] A pharmacokinetic study (03002-002) was performed to determine the
pharmacokinetics of ARC127 via intravenous (I~, intraperitoneal (IP), and
subcutaneous
(SC) administration in mice. Table 4 below shows the results of the study at a
dose of 10
mg/lcg showing that bioavailability for intraperitoneal and subcutaneous
administration is
high with IV, IP, and SC. Figure 17 shows the concentration of ARC127 aptamer
in nM
through 50 hours post dose via l~, IP and SC routes of administration. ARC 127
was
previously found to have the following characteristics: a dissociation
constant (I~) of 100pM;
a cellular ICSO value of 2nM; no observed cytoxicity; efficacy in the animal
models for
58
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
glomerulonephritis, restenosis, cancer and pulmonary hypertension; a Cmax at 1
mg/kg of 2
p.M; a solubility of 20 mg/ml; and a systemic half life of 6-12 hours
(intravenous injection)
and 3.87 days (intravitreal), as shown in Example 12. ARC 127 is injectable by
various routes
of administration, including intravenous, intraperitoneal, subcutaneous and
intravitreal. The
bioavailability of ARC127 was found to be 62.5% via i.p. injection and 24.0%
via
subcutaneous injection.
[00226] Table 4. Pharmacokinetic profile of ARC 127 via IV, IP, and SC in mice
at 10
mg/kg.
lr~~.~',~~~~' h~Cl~~a~~~n~
:~- ~::, -. .
,
,f' ' , ; ',. . ' , , ". .. .
~y'..; ," . , ;-,: :~ '.,'.;.,..
~ '. , , ' ~: '
'. ~.
9711.6 29686.86.57 8.600.0531.000
x.12756.08 143605.511.237.860.0780.625
X3176.7 8 55030.916.639.180.2380.240
[00227] As used herein, Cmax refers to the maximum observed serum or plasma
concentration; AUC refers to the area under the concentration-time curve;
AUClast refers to
the area under the concentration-time curve up to the last point in time;
AUCinf refers to the
area under the concentration-time curve when extrapolated to infinity; T~,Z
refers to the
terminal half life; Cl refers to clearance; MRT refers to the mean residence
time; MRTinf
refers to mean residence time to infinity; and mss refers to the apparent
volume of
distribution.
[0022] ha addition, a second study was performed to determine bioactivity
profile of
ARC127 post I~ administration. The results of the competition binding assay
data are
consistent with the pharmacokinetic data and Show that ARC127 has measurable
activity out
to 48 hours irt viv~ (see F°ig. 18). 'These data demonstrate that ARC
127 is a potent anti-PL~GF
aptamer with irz viv~ efficacy with a I~ of 100 pM and a cellular ICSO of 2
nM. In addition,
this efficacy has been demonstrated in a number of in vivo models. The
pharmacokinetic/pharmacodynamic study shows a systemic half life of ARC127
between 6-
12 hours, and Cmax at 1 mg/kg of 2 ~.M. In addition, ARC 127 has shown a half
life of 3.5
days in the vitreous humor, as determined using the assay described in Example
4. Taken
59
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
together, these data indicate that ARC 127 is a potent anti-PDGF aptamer, is a
novel
therapeutic having novel anti-angiogenic properties when co-administered with
an anti-VEGF
agent, and is useful as a novel oncology agent, as well as a novel therapeutic
in PDR and
AMD ocular proliferative disease.
Example 8 TGF(32 Therapeutic Aptamers for Intravitreal Administration
[00229] Aptamers with binding affinity to TGF(32 are administered via
intravitreal
route with an injected volume of ~ 100 ~L/eye, via subconjunctival route with
an injected
volume of ~ 250 ~.L/eye, and via intravenous route with an injected volume ~
250-1000 pl,.
The dose for each of these routes of administration is 0.5 - 5 mg/eye for
intravitreal
administration, 1-5 mg (aptamer mass without conjugation) for subconjunctival
administration, and 1-20 mg/kg for intravenous administration. The
concentration for each of
these routes is 1-5 mg/0.250 ml = 4-20 mg/ml for intravitreal administration,
1-5 mg/0.100 ml
= 10-50 mglml for subconjunctival administration, and 1-20 mg/0.250-1.0 ml = 1-
80 mg/ml
for intravenous administration. A biodistribution timecourse is plotted at pre-
dose, 5 min, 30
min, l, 6, 12, 24, and 72 hours for intravitreal and subconjunctival routes of
administration,
and pre-dose, 5, 30 min, l, 6, 12, 24, and 48 hours for intravenous route of
administration.
Figure 12 shows the configuration of the ARC77 aptamer and illustrates the
regions of the
aptamer that have been modified.
[00230] Table 4 - TGF(32 Aptamer sequences
ARC77 SEQ ID l~To. 1: (34 nt; cell IC50 = 10 nM, Iso = 1nM) 17, 2'~I~ purines;
17, 2'F-
pyrimidine 5'-G-G-A-G-G-ftJ-f(I-A-fCT-fCT-A-fC-A-G-A-(a-fU-fC-fCT-C~-fCT-A-fU-
~-G-fC_
f(T-G-frL1-A-f~-ftl-fC-fC-[3'T]; IUD = lnM, 9 invariable positions underlined.
ARC79 SEA ID 1lTo. 3: (34 nt, cell ICSO = 10 nM, IUD = 1 nM) improved
chemical/nuclease
stability through replacement of ribo-residues with 2'-~Me RTlTA except 4
essential ribo-
nucleotides underlined
Example 9 TGF[i2 Doted Reselection SELEXTM Aptamers
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
[00231] A doped reselection SELEX~ was performed against TGF(32 using
libraries
and primers used in Doped re-SELEX :(lower case represent 30% doped residue).
These
libraries were amplified and transcribed separately and then combined for the
initial round:
SEQ ID No. 28 TK.82.140.A (14i-1)
TCGGGCGAGTCGTCTGgaaggaattttactacaacgttacttccgcatcctccCCGCATCGTCCTCCCTAT
AGTGAGTCGTATTA
SEQ ID No. 29 TK.82.140.)3 (21 a-4)
TCGGGCGAGTCGTCTGgcggacttagtatatacatacgactaaacaacgccgcCCGCATCGTCCTCCCT
ATAGTGAGTCGTATTA
SEQ ff~ No. 30 TK.82.140.C (21a-21)
TCGGGCGAGTCGTCTGggagtacagctatacagactctgtaataacctccCCGCATCGTCCTCCCTATA
GTGAGTCGTATTA
SEQ ID No. 31 5'-Primer TK.82.14~O.I~ (sense)
TAATACGACTCACTATAGGGAGGACGATGCGG
SEQ ~ No. 32 3'-Primer TK.82.140.E (antisense)
TCGGGCGAGTCGTCTG
[00232] The doped reselection SELEX~ procedure for the TGF[i2 reselction was
performed as follows. For template preparation, the doped DNA libraries were
purified using
PAGE and amplified by PCR. The purified PCR products were then transcribed
with Y639F
RNA polymerase in the presence of 2'-F pyrimidine nucleotides, 2'-OH purine
ribonucleotides. Resulting RNA pools were used in the first round selection.
[00233] The first two, rounds of SELEXTM selection were done by nitrocellulose
membrane (NC) spot. 500 pmoles human TGF(32 (hTGF(32) was spotted on the pre-
washed
NC membrane and dried by air. The filter then was incubated at room
temperature for 1 hour
with the combination of 3 Ri~TA doped pools in Dulbecco's phosphate-buffered
saline, lmT~1
~gCh(IaPES). T he filter was washed by DP)3S 3 times and the TGF/32 bound
RT~TAs was
eluted in preheated 95C elution buffer (7 I~ urea 100 mIal sodium acetate(pI-1
S.0) 3 mI~
EDTA). Eluted R1~TA was extracted by Phenol/chloroform, precipitated by
ethanol and then
reverse transcribed, amplified by PCR. The resulting transcription template
was transcribed
with Y639F single mutant RNA polymerise in the presence of 2'-F pyrimidine
nucleotides,
2'-OH purine ribonucleotides and carried to the next round.
61
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
[00234] SELEX by hydrophobic plate from the third round the SELEX was done by
hydrophobic plate selection. 100p.1 of 20 nM hTGF(32 was incubated with NLTNC
MaxiSorp
Plate (positive plate) for lhr at 37 °C in I~PBS (without 0.1 mg/ml
tRNA). In the meantime,
RNA pool (with 0.1 mg/ml tRNA ) were incubated in the negative plate for 1
hour at 37 °C
for a negative selection. The protein plate was washed 6X with DPBS. The pre-
selected RNA
pool was incubated in the positive plate for lhour at 37°C. The plate
was washed 6X with
DPBS to remove the unbound RNA. Then, reverse transcription was performed in
the plate.
The reverse-transcription was then PCR amplified. The resulting transcription
template was
transcribed with Y639F RNA polymerase in the presence of 2'-F pyrimidine
nucleotides, 2'-
~I3 purine ribonucleotides and carried to the next round.
[00235] TGF(32 was serially diluted in I~PBS containing 0.2mg/ml BSA and
0.2mg/ml tRNA
Sap-labeled RNA (<20pM) were incubated with TGF(32 at room temperature for 30
min.
Samples were pipetted with a multichannel pippetor onto a multiwell manifold
holding layers
of pre-wetted 0.45 micron nitrocellulose, hybond membrane, 3MM filter paper
(the order is
from top to bottom), aspirated, and washed 3X with I~PBS (containing tRNA).
All three
filters were air dried, exposed to phosphorimager plate, analyzed by
ImageQuant. The full
length aptamer sequences shown in Table 5 were found to bind to TGF(32.
[00236] Table 5 - TGF(32 doped reselection full length aptamer sequences.
S5CRl2-27GGGAGGACGAUGCGGAUCGAGUAUUAUAGAGUAUGUAUAGCUAUACGAUCAGACGACUCGCCCGA(SEQ
ID
N0:39)
AM3f(71)_F7*GGGAGGACGAUGCGGAUCGAGUAUUAUAGAGUAUGUAU1S.GCUAUACUAUCAGACGACUCGCCCGA
(SEQ ID
N0:40)
AMX(71)_A11GGGAGGACGAUGCGGAUCGAAUADUAUAGAGUAUGUAUAGCUAUACGGUCAGACGACUCGCCCGA(SE
Q ID
N0:41)
AMX(71)_B9GGGAGGACGAUGGGGAUCGAGUAUUAUAGAGUCUGUAUAGCUAUACGAUCAGACGACUCGCCCGA(SEQ
ID
N0:42)
AMX(71)GGGAGGACGAUGCGGAUGGAGUAUUAUAGAGUAUGUAUAGCUAUACCAUCAGACGACUCGCCCGA(SEQ
ID
B11 N0:43)
A14~(71)GGGAGGACGAUGCC-GAUAGAGCAUUAUAGPGUAUGUAUAGCUAUACUAUCAGACGACUCGCCCGA(SEQ
ID
C11 N0:44)
ARC232
SSCRB-15GGGAC-
Ga3CGAUGCC~AUAGAGUAUUAUAGAGUAUGUAUAGCUAUACUAUCAGACGACUCGCCCGA(SEQ ID
N0:45)
AP.2~(71)_G9GGGAGGACGAUGCC-
GPCAGAGUAUUA.UAGAGUAUGUGU2~GCUAUACCGUCAGACGACUCGCCCGA(SEQ ID
N0:46)
S5R12-33GC-GAC-GACGAUGCC-
GACAGAGUAUUZ~Ud~G~GUAUGUAUAGCUAUACUGCCAGACGACUCGCCCGa3(SEQ ID
N0:47)
S5P.1~-12GGGAGGACGAUGCC-
G23CAGAGUx~UUa'~Ur'1G_RGUAUGUi~.Ua2GCUAUACUGCAGACGt'~CUCC-CCCGa~SEQ ID
( N0:48)
Z~a.7~(71)GC-Gt'~C-
GACGt'~UGCC~a~Ct3Ci3GCa~3UUAUa3Ge3GUGUGU3Ue'~GCUGUACUGUCt'~G3CGACUCGCCCGA(SEQ
ID
fill N0:49)
a~a22s(71)GC-GAC-~1)CG~UGCC-
G3CAGACUa'~UUe~UAGi'~CUe'~UGUAUAGCUAUACUi'~UCa~Ge'1CG3CUCGCCCUe3(SEQ ID
a3 N0:50)
RC'235
SSCRB-aS5GGGAGGa'~CGAUGCC-
Gc'3CAG~'aGUa'~UUa?UFGL~GUAUGU2~Ud'~GCUe3UACUGUCCZdGt~CGACUCGCCCGA(SEQ ID
NO:51)
<?pC238
5588-10GGGAC-GACGAUCCC-
Gt'~Ce'3GAGUa'~UUL~UAC~IGUt'~UGUAUa'~GCUAUACUGUC3Ge?CGe3CUCGCCCGA(SEQ ID
N0:52)
s'~a.i3S(71)_H7GC-GAC-
GZ.CG~3UGCC~"~'~3Gi~GUd3UUAUd~GAGUCUGUAUAGCUAUACUUUCAGACGACUCGCCCGA(SEQ ID
N0:53)
a':r.?In(71)GGGAC-GACGa-
~UGCGGF.3Ax'~G21A10t'~UUi3UAGt'IGUAiJGUAUtIGCUAUACCAUCAGACGACUCGCCCGA(SEQ ID
D10 N0:54)
SSCt312-12GC-GAC-
GZ~CGAUCCC~t~t~GGc'~GUa'~UUUUA~~GUAUGUAUAGCUAUACCAGACGACUCGCCCGA(SEQ ID
N0:55)
S5CR12-
15GGGAGGACGAUGCCG~'~GGc~GUa'~UUAUAGa'~GUAUGUAUAGCUAUACCAUCAGACGACUCGCCCGA(SEQ
ID
N0:56)
AP3~(71)GGGAC-GACGFaUGCC-
GI~t~GGPAUAUUAUAG1~GUAUGUAUAGCUAUACCAUCAGACGACUCGCCC~(SEQ ID
H10 N0:57)
ARC233
SSCRB-19GGGAGGACGAUGCC-
GAGGGAUUr2UUAUAGAGUCUGUAUAGCUAUACCCUCAGACGACUCGCCCGA(SEQ ID
N0:58)
A~(71)_A8GGGAGGACGAUGCC-
GAGGGAAUAUUAUAGAGUAUGUAUAGCUAUACCAUCAGACGACUCGCCCGA(SEQ ID
N0:59)
AI17C(71)GGGAGGACGAUGCGGAGA31GAGUAUUAUAGAGUCUGUAUAGCUAUACUUCAGACGACUCGCCCGA(SEQ
ID
H9 N0:60)
A1~L(74)GGGAGGACGAUGCC-GAGAGAUUAUUAUAGAGUAUGUAUAGCUGUACUGCCAGACGACUCGCCCGA(SEQ
ID
G6 N0:61)
ARC231
S5CR8-14GGGAGGACGAUGCC-GAACGAAUAUUACAGAGUAUGUAUAGCUGUACGGUCAGACGACUCGCCCGA(SEQ
ID
N0:62)
SSCR8-28GGGAGGACGAUGCGGAGUGAGUAUUAUAGAGUAUGUAUAGCUAUACACAUCAGACGACUCGCCCGA(SEQ
ID
N0:63)
AM7L(71)_A10GGGAC-
GACGAUGCGGUGUGAAUAUUAUAGAGUCUGUAUAGCiJAUACCAUCAGACGACUCGCCCGA(SEQ ID
N0:64)
AMX(74)_F1GGGAGGACGAUGCGGUGUGAAUAUUAUAGAGUCUGUAUAGCUAUACCACCAGACGACUCGCCCGA(SEQ
ID
N0:65)
62
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
ARCZ27
S5R8-1 GGGAGGACGAUGCGGUGUGAGUAUUAUAGAGUCUGUAUAGCUAUACCACCAGACGACUCGCCCGASEQ ID
( N0:66)
AMX(71)GGGAGGACGAUGCGGUCCGCAWAUCWCUACGUUACAUAUACUAUCUCUGUCAGACGACUCGCCCGA
B3 (SEQ ID
N0:67)
D11 GGGAGGACGAUGCGGUCGGAGUAUUAUAGAGUAUGUAUAGCUAAACCGUCAGACGACUCGCCCGA(SEQ
ID
AM7C(71) N0:68)
_ GGGAGGACGAUGCGGUAAGAGUAUUACAGAGUAUGUAUAGCUGUACBUGCCAGACGACUCGCCCGA(SEQ
ID
A2 N0:69)
AM7C(74)
_ GGGAGGACGAUGCGGUGAGAAUAUUACAGAGUAUGUAUAGCUGUACUUGCCAGACGACUCGCCCGA(SEQ
ID
AMX(74) N0:70)
E4
AMX(74)GGGAGGACGAUGCGGUGAGAAUAUUAUAGAGUAUGUAUAGCUAUACUCGCCAGACGACUCGCCCAA(SEQ
ID
F4 N0:71)
AMX(74)_F3GGGAGGANGAI3GCGGUGAGAGUAUUAUAGAGUCUGUAUAGCUAUACUGCCAGACGACUCGCCCGA(SE
Q ID
N0:72)
ARC229
S5R8-43GGGAGGACGAUGCGGUGAGAGUAUUAUAGAGUCUGUAUAGCUAUACUCGCCAGACGACUCGCCCGA(SEQ
ID
N0:73)
InRC234
S5CR8-32GGGAGGACGABGCGGUGAGAGUAUBAUAGAGUCUGUAUAGCUAUACCACCAGACGACUCGCCCGA(SEQ
ID
N0:74)
D9 GGGAGGACGAUGCGGUAGAGUAWAUAGAGUAUGUAUAGCUAUACACUCAGACGACUCGCCCAA
AM7C(71)(SEQ ID N0:75)
_ GGGAGGACGAUGCGGUAGAGUAUUAUAGAGUAUGUAUAGCUAUACCAUCAGACGACUCGCCCGA
H12 (SEQ ID N0:76)
AM1C(71)
_ GGGAGGACGAUGCGGUGGGAAUAUfdAUAGAGUCUGUAUAGCOAUACCCUCAGACGACUCGCCCGA(SEQ
ID
G7 N0:77)
AhI7C(71)
_ GGGAGGACGAUGCC-GGCGGAAUAUUAUAGAGUAUGGAUAGCOAUACCGUCAGACGACUCGCCCGA(SEQ
ID
S5CR12-14 N0:78)
ARC230
SERB-a5GGGAGGACGAUGCC-GGACAGAGUAUUAUAGAGUAUGUAUAGCUAUACUGUCAGACGACUCGCCCGA(SEQ
ID
N0:79)
B7 GGGAGGACGAUGCGGGCAGAGUAUUAUAGAGUACGUAUAGCUAUACUGUCAGACGACUCGCCCGA(SEQ
ID
Ar32:(71) N0:80)
_ GGGAGGACGAUGCGGGCAGAGUAUUAUAGAGUAUGUAUAGCUAUACUGUCAGACGACUCGCCCGA(SEQ
ID
t'~h~f(71) NO:81)
A9
_ GC-GGAC-
GACGAUGCGGGUGGAGUAUUAUAGAGUAUGUAUAGCUAUACUAUCAGACGACUCGCCCAA(SEQ ID
GB N0:82)
AF57~(71)
_ GC-GAGGACGAUGCGGGUAGAUUAUUACAGAGUCUGUAUAGCUGUACUGCCAGACGACUCC-CCCGA(SEQ
ID
A3 N0:83)
(74)
_ GGGAGGACGAUGCC-GGUAGAAUAWAUAGAGUCUGUAUAGCUAUACUGCCAGACGACUCGCCCGA(SEQ
ID
P32 N0:84)
Ab35~(74)
_ GC-GAC-GACGAUGCGGGUAGAAUAUUACAGAGUAUGUAUAGCUGUACUGCCAGACGACUCGCCCGA(SEQ
ID
H1 N0:85)
Z~a'K3Z(74)
_ GC-GAGGACGAUGCGGGGAGAAUAUUACAGAGUAUGUAUAGCUGUACUUGCCAGACGACUCGCCCGA
C3 (SEQ ID N0:86)
AE4%(74)
_ GGGAGGACGAUGCGGGGAGAAUAUUACAGAGUAUGUAUAGCUGUACUGCCAGACGACUCGCCCGA(SEQ
ID
A1 N0:87)
Al~i~(74)
_ GGGAGGACGAUGCGGGGAGAWAUUAUAGAGUAUGUAUAGCUAUACUGCCAGACGACUCGCCCGA(SEQ ID
A2~(74) N0:88)
B3
_ GGGAGGACGAUGCGGGGAGAGUAUUAUAGAGUAUGUAUAGCUAUACUGCCAGACGACUCGCCCGA(SEQ
ID
G3 N0:89)
AP37~(74)
_ GGGAGGACGAUGCGGGAAGAGUAUUACAGAGUAUGUAUAGCUGUACUGCCAGACGACUCGCCCGA(SEQ
ID
D3 N0:90)
APi7C(74)
_ GC-GAGGACGAUGCGGGCAAAGUAUUGUAGAGUAUGCAUAGCBAUAUTJGCCAGACGACUCGCCCGA(SEQ
ID
&6 N0:91)
AT?7~(74)
_ GGGAGGACGAUGCGGGGAGGUUAUUACAGAGUCUGUAUAGCUGUACUCCCAGACGACUCGCCCGA(SEQ
ID
21a-21 N0:92)
I4i-1 GGGAGGACGAUGCGGGGAGGAUGCGGAAGUAACGUUGUAGUAAAAUUCCUUCCAGACGACUCGCCCGA
(SEQ ID
N0:93)
21a-4 GGGAGGACGAUGCGGGCGGCGWGUUUAGUCGUAUGUAUAUACUAAGLJCCGCCAGACGACUCGCCCGA
(SEQ ID
N0:94)
(00237] Binding data for the full-length aptamer sequences shown above are
shown in
Table 6. Figure 19A and 191 show binding plots for the full-length sequences
shown in
Table 5.
[0023] Table 6 - Binding data as performed in 1VILEC inhibition assay
CI~V ~ ~ 5-95, ~ 1'~, ~ 29
129: SSR~-1:T.4~nP~
~5R~-10: 14.5nf~(
~5F~5-4.3: 6.5n[~
S5R8-4.5: 12.1
S5CR5-15: 17.9nf~
S5~R~-15: 5.~nM
S5GI~5-3~: S.Onf
S5CR5-35: 0.4-nM
S5CR8-4.5: 5.~n[VJ
[00239] Table 7 - IVlinimized T(iF(32 doped reselection aptamers
CVTla8.lo.A(S5CR12-27)
AfUfCGAGfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCGAfU[3T] (SEQ ID N0:95)
G'tn712~.10.B(A~(71)_F7)
AfUfCGAGfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCfUAfU[3T] (SEQ ID N0:96)
CW128.10.C(AMI3~(71)_All)
AfUfCGAAfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCGGfU[3T] (SEQ ID N0:97)
CS~1128.10.D(AM7C(71) B9)
63
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
AfUfCGAGfUAfUfUAfUAGAGfUfCfUGfUAfUAGfCfUAfUAfCGAfU[3T] (SEQ ID NO:98)
ARC2$5
8 . l0. E (AMl~ (71) B11)
AfUGGAGfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCfCAfU[3T] (SEQ ID N0:99)
CW128.10.F(AMX(71)_C11)
AfUAGAGfCAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCfUAfU[3T] (SEQ ID NO:100)
CW128.10.G(S5CR8-15, full length ARC232)
AfUAGAGfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCfUAfU[3T] (SEQ ID NO:101)
CW128.10.H(AM3C(71)_G9)
AfCAGAGfUAfUfUAfUAGAGfUAfUGfUGfUAGfCfUAfUAfCfCGfU[3T] (SEQ ID NO:102)
CW128.10.I(S5R12-33)
AfCAGAGfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCfUGfC[3T] (SEQ ID NO:103)
CLV128.10.J(S5R12-12)
AfCAGAGfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCfUG[3T] (SEQ ID N0:104)
CW128.10.1'~(Ab~(71)_F11)
AfCAGAGfCAfUfUAfUAGAGfUGfUGfUAfUAGfCfUGfUAfCfUGfU[3T] (SEQ ID N0:105)
Ci~d128.10.T,((71) F3)
AfCAGAGfUAfiJfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCfUAfU[3T] (SEQ ID NO:lOG)
ARC283,
Ct~7128.10.T~(S5CR8-45, full length ARC235)
AfCAGAGfUAfilfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCfUGfUfC[3T] (SEQ ID NO:107)
ARC28G,
C~T128.10.N'(S5R8-10, full length ARC228)
AfCAGAGfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCfUGfU[3T] (SEQ ID NO:108)
cc~r12~.1~.0(An~(71)_HZ)
AAAGAGfUAfUfUAfUAGAGfUfCfUGfUAfUAGfCfUAfUAfCfUfLIfU[3T] (SEQ ID N0:109)
CHT128.10.P (AL~(71)_D10)
AANGAAfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCfCAfU[3T] (SEQ ID NO:110)
CW128.10.Q(S5CR12-12)
AAGGAGfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfC[3T] (SEQ ID NO:111)
ARC281
CW128.10.R(S5CR12-15)
AAGGAGfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCfCAfU[3T] (SEQ ID N0:112)
ARC2$7
CW128.10.5(AM~C(71)_H10)
AAGGAAfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCfCAfU[3T] (SEQ ID N0:113)
Ct~1128.10.T(S5CR8-18, full length ARC233)
AGGGAfUfUAfUfUAfUAGAGfIJfCfUGfUAfUAGfCfUAfUAfCfCfCfU[3T] (SEQ ID N0:114)
ARC282
CW128.10.U(AM~(71)_A8)
AGGGAAfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCfCAfU[3T] (SEQ ID NO:115)
C~r1128.10.~'(AhS~(71)_H9)
AGAAGAGfUAftlfUAfUAGAGfUfCfUGfUAfUAGfCfUAfUAfCfiTfU[3T] (SEQ ID NO:11G)
Cr~128.10.Vr(x~ri5~(7a)_GG)
AGAGAfUfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUGfUAfCfUGfC[3T] (SEQ ID NO:117)
C~1128.11.t'~(S5CF38-la, full length ~=1EC231)
AAfCGAAfUAfUfUAfCAGAGfUAfUGfUAfUAGfCfUGfUAfCGGfU[3T] (SEQ ID NO:118)
CV128. 11.8 (S5CI~8-28)
AGfUGAGfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCAfCAfU[3T] (SEQ ID NO:119)
CLuTl2 8 .11. C (E'~~ ('71 ) _A10 )
fUGfUGAAfUAftJfUAfUAGAGfUfCfUGfUAfUAGfCfUAfUAfCfCAfU[3T] (SEQ ID NO:120)
CL~1128.5.1.LZ (A25~ ('P4)_~'1)
fUGfUGAAfUAfUfUAfUAGAGfUfCfUGfUAfUAGfCfUAfUAfCfCAfC[3T] (SEQ ID N0:121)
CL~T128.11.E(S5R8-l, full length t'~RC227)
fUGfUGAGfUAfUfUAfUAGAGfUfCfUGfUAfUAGfCfUAfUAfCfCAfC[3T] (SEQ ID NO:122)
CTn1128.11.F(R~(71)_83)
ftJfCfCGfCAfUfUAfUfCfUfUfCfUAfCGfUfUAfCAfUAfUAfCfUAfUfCfUfCfUGfU[3T] (SEQ ID
N0:123)
C~T128 . 11. G (Ate (71) _D11)
fUfCGGAGfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAAAfCfCGfU[3T] (SEQ ID N0:124)
CW128.11.H(AI4~(74)_A2)
fUAAGAGfUAfiTfUAfCAGAGfUAfUGfUAfUAGfCfUGfUAfCfUfUGfC[3T] (SEQ ID N0:125)
CW128.11.I(AM~C(74) E4)
64
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
fUGAGAAfUAfUfUAfCAGAGfUAfUGfUAfUAGfCfUGfUAfCfUfUGfC[3T] (SEQ ID N0:126)
CW128.11.J(AMX(74)_F4)
fUGAGAAfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCfUfCGfC[3T] (SEQ ID N0:127)
CW128.11.K(AMX(74)_F3)
fUGAGAGfUAfUfUAfUAGAGfUfCfUGfUAfUAGfCfUAfUAfCfUGfC[3T] (SEQ ID NO:128)
CW128.11.L(S5R8-43, full length ARC229)
fUGAGAGfUAfUfUAfUAGAGfUfCfUGfUAfUAGfCfUAfUAfCfUfCGfC[3T] (SEQ ID NO:129)
CW128.l1.rif(S5CR8-32, full length ARC234)
fUGAGAGfUAfUfUAfUAGAGfUfCfUGfUAfUAGfCfUAfUAfCfCAfC[3T] (SEQ ID N0:130)
CL41128.11.N(AM~C(71)_D9)
fUAGAGfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCAfCfU[3T] (SEQ ID N0:131)
CW128 .11. O (AM~C (71) _H12)
fUAGAGfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCfCAfU[3T] (SEQ ID N0:132)
CY312 8 .11. P (Ah~ ( 71 ) _G7 )
AAfUAfUfUAfUAGAGfUfCfUGfUAfUAGfCfUAfUAfCfCfCfU[3T] (SEQ ID NO:133)
CW128.11.Q~(S5CR12-14)
GfCGGAAfUAfUfUAfUAGAGfUAfUGGAfUAGfCfUAfUAfCfCGfU[3T] (SEQ ID N0:134)
AI2C284
Cia128.11.R(S5R8-45~ full length a'~RC230)
GAfCAGAGfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCfUGfU[3T] (SEQ ID N0:135)
CW 12 8 .11. S (~'~T2~ ( 71 ) _87 )
GfCAGAGfUAftTfUAfUAGAGfUAfCGfUAfUAGfCfUAfUAfCfUGfU[3T] (SEQ ID NO:136)
CC~T12 8 .11. T (A24~ ( 71 ) _A9 )
GfCAGAGfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCfUGfU[3T] (SEQ ID N0:137)
CGV128.11. fIT(AIw~(71)_G8)
GfUGGAGfUAfTJfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCfUAfU[3T] (SEQ ID NO:138)
CW128.11.~d'(AN~(74)_A3)
GfUAGAfUfUAfUfUAfCAGAGfUfCfUGfUAfUAGfCfUGfUAfCfUGfC[3T] (SEQ ID NO:139)
CW128.11.Yd(AR~(74)_Fs2)
GfUAGAAfUAfUfUAfUAGAGfilfCfUGfUAfUAGfCfUAfUAfCfUGfC [3T] (SEQ ID NO:140)
CW128.12.A(AMX(74)_H1)
GfUAGAAfUAfUfUAfCAGAGfUAfUGfUAfUAGfCfUGfUAfCfUGfC[3T] (SEQ ID N0:141)
CP1128.12.8(ALSX(74)_C3)
GGAGAAfUAfUfUAfCAGAGfUAfUGfUAfUAGfCfUGfUAfCfUfUGfC[3T] (SEQ ID NO:142)
C~V12 8 .12 . C (AIefX ( 7 4 ) _A1 )
GGAGAAfUAfUfUAfCAGAGfUAfUGfUAfUAGfCfUGfUAfCfUGfC[3T] (SEQ ID N0:143)
CW128.12.D(AM:~(74)_B3)
GGAGAfUfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCfUGfC[3T] (SEQ ID NO:144)
CW128.12.E(AM~(74)_G3)
GGAGAGfUAfUfUAfUAGAGfUAfUGfUAfUAGfCfUAfUAfCfUGfC[3T] (SEQ ID N0:145)
CZ~a12 8 .12 . F (Ahi3; ( 7 4 ) _D3
GAAGAGfUAfL7fUAfCAGAGfUAfUGfUAfUAGfCfUGfUAfCfUGfC[3T] (SEQ ID N0:146)
CI~T128.12 .G (t'~~ (7a)_E6)
GfCAAAGfUAfUfUGfUAGAGfUAfUGfCAfUAGfCfUAfUAfUfUGfC[3T] (SEQ ID NO:147)
CG~128.12.F~(21a-21, full length a~3~C236)
GGAGGfLIfUAfLJfUAfCAGAGfLTfCfUGfUAfUAGfCfUGfUAfCfiTfCfC [3T] (SEQ ID N0:148)
o~cal~a.l~.a(14i-1, f~ll length ~~~~~37)
GGAGGAfUGfCGGAAGfUAAfCGft7fUGfUAGfUAAAAfUfUfCfCfiTfUfC [3T] (SEQ ID NO: 149)
Ci~128.12.J(21a-4, full length a'~RC241)
GfCGGfCGfLTfUGfLJfUfUAGfUfCGfUAfUGfUAfUAfUAfCfUAAGfUfCfCGfC [3T] (SEQ ID
No:lS~)
[00240] Table 8 - Binding information on truncated aptamers
CW 128_103
ARC235 ARC236 ARC281 ARC282 ARG283 ARG284 CW 128.10.F
4Cd(nM) 0.53 0.75 1.42 0.62 1.27 0.11 3.66
[00241] Figures 20A, 208, and 20C show binding plots for the truncated
aptamers
shown in Table 7.
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
EXAMPLE 10 Selection minimization and characterization of VEGF Receptor 2
(VEGF 42)-specific aptamers
[00242] Aptamers directed to the VEGF receptor 2 (VEGF R2), also known as the
KDR molecule, were isolated using semi-automated SELEXTM procedure. Fifteen
rounds of
selection were performed over three weeks. 48 clones were identified and
grouped into the 9
families. These sequences were analyzed to identify functional motifs.
Aptamers directed to
the VEGF receptor were found to have a Kd in the range of 1-3 nM.
Example 11 Pharmacokinetic and Bioactivity profile of ARC81, ARC117 and ARC119
[00243] A pharmacokinetic study was performed to determine the
pharmacokinetics of
ARC81, ARCl 17 and ARC119, TGF(32 aptamers having the sequences of SEA 11~
N~s:4, 9
and 11, respectively), via subconjunctival administration in mice. These
aptamers were
formulated at a concentration of 10 mg/ml. Samples were collected at 0, 0.5,
l, 2, 6, 12, 24,
48 and 96 hours post-administration. Figures 21A, 21B and 21C show the aqueous
humor
and/or plasma concentration of each aptamer (in nM) through 50 hours post dose
via
subconjunctival administration. Table 9 below shows the results of the study
at a dose of 1
mg/eye bilaterally (i.e., 2.0 mg/animal).
[00244] Table 9. Pharmacokinetic profile of ARC81, ARC117 and ARCl 19 via
subconjunctival administration in mice at 1 mg/eye, bilaterally.
~aa~r~a~ ~C~~~~ ~~(~)1~1~ ~~C~1~~
1'~'g'S~L~Sl~~ Aq Pa~AS~aApct PLASIbIA~q
Humour I~Ieerre~aar 1-Idam~eer
~maa n~ 6.44 5.19 144.8 22.9 721.1 28.4
tm~~ h I 6 6 1 12 0.5
TL.9ST h 12 12 4 8 24 48 48
AUC (0-t)nM.h 29.8 36.0 1,122.1 87.6 13,380.5133.2
li~IRT h 3.13 6.03 9.22 3.62 17.28 12.80
t1~2 h 2.42 - 7.40 4.33 6.94 17.52
'old mL 21079 - 1760 - 137 -
66
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
[00245] ARC81, 117 and 119 were detected in the aqueous humor after
subconjunctival administration. UnPEGylated aptamers, i.e., ARC81, entered
systemic
circulation rapidly (e.g., less than 0.5 hour). The Aqueous concentration of
unPEGylated
aptamer shows delayed tmax, possibly due to recirculation of aptamers. The
PEGylated
aptamers, i.e., ARCl 17 and ARC119, showed show similar plasma t1~2, The
evidence of
decreasing Vd with PEGylation and delayed tm~ imply strong depot effect in
vicinity of
injection site. The PEGylated aptamers were both detected in the aqueous humor
and were
found to be depoted at the surgical site.
Example 12 Comparison of Pharmacokinetic and Bioactivit~~rofiles of ARC126,
ARC127
and I~~1838 a
[00246] A pharmacokinetic study was performed to determine and compare the
pharmacokinetics of ARC 126 (SEQ lI~ IV~:18 - PEG - SEQ ID Nf~:33- PEG - SEQ
ID NO:
34) and ARC127 (PEG - SEQ )D hT~:19 - PEG - SEQ ID I~T~ ~ 35 - PEG -SEQ ID
N0:36 -
3T), two PDGF aptamers, and N~1838, a known PEG-conjugated aptamer directed
against
VEGF-165 in rabbits. Studies were performed in the Dutch-belted rabbit model
using
subconjunctival injection. Aptamers were administered at a dose level of 1.0
mg/eye,
bilaterally via 100 mL intravitreal injection. Samples from the aqueous humor,
vitreous
humor and plasma were taken before administration of the aptamer and a 0.25 h,
6 h, 24 h, 72
h, 7d, 4 d, and 21 d. Figures 22A, 22B and 22C show the vitreous humor and/or
plasma
concentration of each aptamer through 25 days post administration. Table 10
below shows
the results of the study for the ARC 126 and ARC 127 aptamers.
[00247] Table 10: Pharmacokinetic profile of ARC12~ and ARC127
67
CA 02513004 2005-07-11
WO 2004/064760 PCT/US2004/001661
~i71~'~'R.~:~:~~ s~~~-~.:~1
Cmax ~,M 117.91 1~9.1~
tmax ~ ~.25 ~.25
~C.~ p.M~ ~~5~~502 ~~x.22.2
E~'~~u ~ 2.~~J ~..~5
c~ 2.25
Ci ~L~Iw ~.52 ~~2~
~L 1.25 1~~2
[00248] The results shown in Figure 22A were obtained using noncompartrnental
(NCA) analysis. The volume of rabbit vitreous humor was approximately 1.0-1.5
ml. In
comparison to the AI~C126 and ARC127 aptamers, the half life of NX1838 in
rabbit vitreous
was known to be approximately 83 hours, i.e., 3.46 days, and the half life of
NX1838 in
primate vitreous was known to be approximately 94 hours, or 3.92 days.
[00249] As seen in Figures 2213 and 22C, the Cm~ (vitreous) was approximately
100
p~ for A1C 127. Cmax(vit) approxia~natcly 100 mm for both nonpegylated and peg-
ylated
aptamer conjugates. The C(vit) for pegylated aptamers was approximately 250
xll~A at t=30
days (4~01~ PECi). The ration of AUC values for the pegylated aptamers and
nonpegylated
aptamers was 1.79. The half life of nonpegylated aptamers was approximately
2.25 days, and
the half life of the pegylated aptamcrs was approximately 3.87 days. The
apparent volume of
distribution (Vss) was between 1.25-1.42 mL, which indicates that both
conjugates remained
in the vitreous compartment. The clearance value (Cl) was between 0.29-0.52
mL/~d ( <_ 50
nmol/day), and the maximum plasma levels were < 10 nM. The aqueous
concentration of
the nonpegylated aptamcr conjugates was at nanomolar levels for t <_ 24 hours.
68