Note: Descriptions are shown in the official language in which they were submitted.
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
TRAMS-MEMBRANE-ANTIBODY INDUCED INHIBITION
OF APOPTOSIS
CROSS REFERENCE TO RELATED APPLICATIONS
s The present application is a continuation-in-part of U.S. Application No. X,
which is the
National Stage of International Application No. PCT/US02116651, filed May 29,
2002, which
is a continuation-in-part of U.S. patent application Ser. No. 09/865,281,
filed May 29, 2001,
which is a continuation-in-part of U.S. patent application Ser. No.
09/070,907, filed May 4,
1998, now U.S. Patent No. 6,238,667. The present application also claims the
benefit of U.S.
Provisional Application No. 60/451,980, filed March 5, 2003. The disclosures
of each of the
patents and patent applications mentioned above are incorporated herein by
reference.
FIELD OF THE INVENTION
The present invention relates to fusion proteins comprising whole biologically
active peptides
1s and antibodies, or fragments thereof. Specifically, the fusion proteins of
the present invention
combine the molecular recognition of antibodies with a biological activity
such as immuno-
stimulatory activity, membrane transport activity, and homophilic activity.
The present
invention further relates to fusion proteins having the binding properties of
an antibody and
including a biologically active peptide sequence flanked by loop forming or
other
2o conformation-conferring sequences so as to constrain the conformational
flexibility of the
biologically active peptide and to increase its affinity for its biological
target. The present
invention also relates to the use of antibodies and conjugates thereof in the
inhibition of
programmed cell death, i.e., apoptosis.
2s 13ACKGROUND OF THE INVENTION
Antibodies have been praised as "magic bullets" to combat disease; however,
the promises
made for antibodies have never been fully realized. This is due in part to the
fact that
antibodies represent only one arm of the immune defense, where T-cells provide
the other
strategy in immune defense. However, antibodies are ideal targeting and
delivery devices.
3o They are adapted for long survival in blood, have sites that help vascular
and tissue
penetration, and are functionally linked with a number of the defense
mechanisms of innate
immunity. One such mechanism is the complement system, which helps to destroy
pathogens
and is involved in the regulation of immune responses. For example, the
complement
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
fragment C3d binds to the CR2 receptor on B-cells, which is also the binding
site for Epstein-
Barr virus. Binding of Epstein-Barr virus to CR2 activates B-cells.
Accumulated evidence has
shown that the CR2 receptor (CDl9lCd20/CD81 complex) has an immuno-stimulatory
role
and is activated by C3d.
Monoclonal antibodies have been developed for many therapeutic uses. For
example,
diseases currently targeted by monoclonal antibodies include heart conditions,
cancers,
neurological defects and autoimmune diseases. Virtually all of these current
therapeutic uses
rely on the inherent therapeutic efficacy of the particular monoclonal
antibodies, such as with
to the drugs HERCEPTIN and RITUXAN. Since most monoclonal antibodies do not
express
such inherent therapeutic activity, development has focused on the addition of
therapeutic
properties by conjugation of a variety of different toxic agents, such as
protein toxins or their
subunits, drugs currently used in the chemotherapeutic treatment of cancer,
drugs which
failed to progress in clinical development due to unacceptable toxicity, or
radioisotopes.
To make such conjugates effective, a monoclonal antibody delivering such toxic
agents must
be able to bind to its target antigen and internalize into cells to carry the
toxic agent inside
where it can be effective at damaging DNA or inhibiting protein synthesis or
other metabolic
functions of the targeted cell. Few antibodies inherently express such a
property - the ones
2o that do produce very potent immunoconjugates. As such, screening assays
have been
developed to test for such antibodies but few antibodies have been identified
that combine
this quality with an appropriate targeting specificity.
There have been other approaches to instill internalizing ability into an
antibody. Whole
2s protein toxins which combine an active subunit with a cell binding subunit
are effective in
enhancing internalization when conjugated to an antibody but oftentimes reduce
the
selectivity of the antibody thereby leading to potential toxicity. Lipophilic
drugs have also
been used to enhance internalization and intracellular delivery in conjugated
form but as with
toxins will also reduce the selectivity of a conjugate. Qther methods have
been used to
3o permeabilize or by microinjection allow better entry into cells. Both of
these methods have
serious drawbacks. Permeabilization of cells, e.g., by saponin, bacterial
toxins, calcium
phosphate, electroporation, etc., can only be practically used for ex vivo
methods, and these
methods cause damage to the cells. Microinjection requires highly skilled
technicians (thus
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
limiting its use to a laboratory setting), it physically damages the cells,
and it has only limited
applications as it cannot be used to treat, for example, a mass of cells or an
entire tissue,
because one cannot feasibly inject large numbers of cells.
s Another example of how antibodies can be used to enhance the immune response
has been
demonstrated by the work of Zanetti and Bona (Zanetti, M., Nature, 355: 466-
477, 1992;
Zaghouani H.; Anderson S. A., Sperbeer K. E., Daian C. Kennedy R. C., Mayer L.
and Bona
C. A., Proc. Nat. Acad. Science USA, 92: 631-635, 1995). These authors have
replaced the
CDR3 sequence of the Ig heavy chain with a sequence resembling T-cell and B-
cell antigens
(epitopes) using molecular biology methods and have shown that these modified
antibodies
induce potent immune response specific for the inserted groups.
The biological properties of the antibodies can be enhanced with respect to
overall avidity for
antigen and the ability to penetrate cellular and nuclear membranes. Antigen
binding is
15 enhanced by increasing the valency of antibodies such as in pentameric IgM
antibodies.
Valency and avidity are also increased in certain antibodies that are self
binding or
homophilic (Kang, C. IP., Cheng, H. L., Rudikoff, S. and Kohler, H., .J:
~'x~a. tlleea'. 165:1332,
1987; Xiyun, A. N., Evans, S. V., Kaminki, M. J., Fillies, S. F. D., Resifeld,
R. A., Houghton,
A. N. and Chapman, P. B., .l: Inarnunol. 157: 1582-1588, 1996). A peptide in
the heavy chain
2o variable region was identified which inhibited self binding (Kang, C. Y.
Brunck, T. K.,
Kieber-Emmons, T.9 Blalocl:, J. E. and l~ohler, H.9 ~'cict~ce9 240: 1034-1036,
1988). The
insertion of a self binding peptide sequence into an antibody endows the
property of self
binding and increases the valency and overall avidity for the antigen.
25 Similarly, the addition of a signal peptide to antibodies facilitates
transmembrane transport as
demonstrated by Rojas et al, Nature Bi~tecdnZOlogy, 16: 370-375 (1998). Rojas
et al. have
generated a fusion protein containing a 12-mer peptide and have shown that
this protein has
cell membrane permeability.
3o Signal peptide sequences that express the common motif of hydrophobicity
mediate
translocation of most intracellular secretory proteins across mammalian
endoplasmic
reticulum (ER) and prokaryotic plasma membranes through the putative protein-
conducting
channels. The major model implies that the proteins are transported across
membranes
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
through a hydrophilic protein-conducting channel formed by a number of
membrane proteins.
In eukaryotes, newly synthesized proteins in the cytoplasm are targeted to the
ER membrane
by signal sequences that are recognized generally by the signal recognition
particle (SRP) and
its ER membrane receptors. This targeting step is followed by the actual
transfer of protein
across the ER membrane and out of the cell through the putative protein-
conducting channel.
Signal peptides can also interact strongly with lipids, supporting the
proposal that the
transport of some secretory proteins across cellular membranes may occur
directly through
the lipid bilayer in the absence of any proteinaceous channels. Such signal
peptides can be
used to enhance internalization of antibodies or other biologically active
molecules into cells
to and are the subject of several patents (U.S. Patents # 5,807,746,
#6,043,339 arid #6,238,667).
Antibodies have been used as delivery devices for several biologically active
molecules, such
as toxins, drugs and cytokines. Often fragments of antibodies, Fab or scFv,
are preferred
because of better tissue penetration and reduced "stickiness".
There are two practical methods for attaching molecules, such as peptides, to
antibody
molecules. One method is to use chemical crosslinking, such as the affinity-
crosslinking
method described in U.S. Ser. No. 09/070,907. Another method is to design a
fusion gene
containing DNA encoding the antibody and the peptide and to express the fusion
gene, which
2o method is the subject of the present application.
Antibody fusion proteins are typically engineered with entire genes of large
proteins or
domains of such proteins that afford a biological function. Previous small
peptide-antibody
fusion proteins have typically been made mainly for the purpose of
facilitating purification or
characterization of the antibody.
Methods of creating fusion proteins are described, for example, in the
following U.S. patents,
the pertinent disclosures of which are incorporated herein by reference: U.S.
Pat. No.
5,563,046 to Mascarenhas et al; U.S. Pat. No. 5,645,835 to Fell, Jr.; U.S.
Pat. No. 5,668,225
3o to Murphy; U.S. Pat. No. 5,698,679 to Nemazee; U.S. Pat. No. 5,763,733 to
Whitlow et al;
U.S. Pat. No. 5,811,265 to Quertermous et al; U.S. Pat. No. 5,908,626 to Chang
et al; U.S.
Pat. No. 5,969,109 to Bona et al; U.S. Pat. No. 6,008,319 to Epstein et al;
U.S. Pat. No.
6,117,656 to Seed; U.S. Pat. No. 6,121,424 to Whitlow et al; U.S. Pat. No.
6,132,992 to
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
Ledbetter et al; U.S. Pat. No. 6,207,804 to Huston et al; and U.S. Pat. No.
6,224,870 to Segal.
Methods of creating Ig fusion proteins are described, for example, in Antibody
Engineeri~,
2nd ed. ed.: Carl A. K. Borrebaeck, Oxford University Press 1995, and in
Molecular Cloning:
A Laboratory Manual, 2"d ed., Cold Spring Harbor Press, 1989, the pertinent
disclosures of
which are incorporated herein by reference.
Fusion proteins including those with immunoglobulins primarily incorporating
active
domains of proteins such as cytokines, toxins, enzymes, etc. with targeting
domains of
immunoglobulins including the CDR's (complementarity-determining regions) and
other
to variable regions and domains not directly involved in antigen binding but
through secondary
interactions able to confer increased affinity of binding are described, for
example, in the
following publications incorporated herein by reference:
Guo L; Wang J; Qian S; Yan X; Chen R; Meng G, "Construction and structural
modeling of a
single-chain Fv-asparaginase fusion protein resistant to proteolysis."
Biotechszol. Bioehg.,
is 2000 Nov 20; 70(4):456- 63;
Muller BH; C.hevrier D; Boulain JC; Guesdon JL "Recombinant single-chain Fv
antibody
fa~agment-alkaline phosphatase conjugate for one-step immunodetection in
molecular
hybridization." J: Irranauv~ol lhleth~els 1999 Jul 30;227(1-2) :177-85;
Griep RA; van Twisk C; Kerschbaumer RJ; Harper K; Torrance L; Himmler G; van
der Wolf
2o JI~1L; Schots "pSICAh/S: An expression vector for the production of single-
chain Fv alkaline
phosphatase fusion proteins." Pr9~ateirz E.~pr~. Puy°i,~ 1999 Jun;
16(1):63-9;
Vallera DA; Panoskaltsis-Mortari A; 1 C; Ramakrishnan S; Eide CR; Kreitman RJ;
Nicholls
PJ; Pennell C; Blazar BR "Anti-graft-versus-host disease effect ofDT390-anti-
CD3sF'v, a
single-chain Fv fusion immunotoxin specifically targeting the CD3 epsilon
moiety of the T-
25 cell receptor." Blo~el 1996 Sep I5; 88(6) :2342-53;
Gupta S; Eastman J; Silski C; Ferkol T; Davis PB "Single chain Fv: a ligand in
receptor-
mediated gene delivery." Gene They. 2001 Apr;8(8) :586-92; and
Goel A; Colcher D; Koo JS; Booth BJ; Pavlinkova G; Batra "Relative position of
the
hexahistidine tag effects binding properties of a tumor-associated single-
chain Fv construct."
3o Biochirra Biophys Acta 2000 Sep 1;1523(1):13-20.
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
Fusion proteins designed to have biological activity may be constructed using
linear peptide
sequences derived from a whole biologically active protein. However, such
peptides have
typically lower affinity than the entire protein. Since the incorporation of a
peptide into a
fusion protein is less cumbersome than the incorporation of an entire
functional protein, there
is a need for fusion proteins containing peptides having a binding affinity as
good as a full-
length protein.
The present invention also relates to the use of antibodies and fragments
thereof in the
inhibition of apoptosis. Cell suicide (apoptosis) is a mechanism used
beneficially by living
organisms in cell differentiation in organ development and elimination of
damaged cells.
However, apoptosis can also be associated with forms of pathogenesis. For
example, it is the
major cause for the loss of neurons in Alzheimer's disease and tissue loss
during myocardial
infarction. Also, T lymphocytes from HIV-1 infected individuals undergo
spontaneous
apoptosis in the absence of a stimulus compared to uninfected T cells cultured
under the same
conditions. The "spontaneous apoptosis" of CD4+ and C1~8+ cells has been shown
to be
accelerated by the in-vitro addition of an HIV-1 related, anti-idiotypic
antibody.
Caspase enzymes, e.g., caspase-3, are critically involved in the pathway of
apoptosis. A
number of materials and methods have been proposed for inhibiting caspase
action in an
2o effort to inhibit apoptosis. For example, U.S. Patent No. 6,566,338 (Weber
et al.) proposes
tlae use of caspase inhibitors generally for treating, ameliorating, and
preventing non-can cer
cell death during chemotherapy and radiation therapy and for treating and
ameliorating the
side effects of chemotherapy and radiation therapy of cancer. U.S. Patent No.
6,596,693
(I~eana et al.) reports that certain dipeptides can be potent inhibitors of
apoptosis. U.S. Patent
Nos. 6,689,784 (Bebbington, et al.) and 6,620,782 (Cai et al.) propose a class
of carbamates
and substituted 2-aminobenzamides, respectively, as inhibitors of apoptosis.
Also, U.S. Patent
No. 6,426,413 (Wannamaker et al.) is a representative proposal for a class of
caspase
inhibitors called interleukin-lbeta-converting enzyme inhibitors.
Additionally, U.S. Patent
No. 6,228,603 (Reed et al.) proposes a screening assay for identifying agents
that alter the
3o specific association of an inhibitor of apoptosis with a caspase, such as
caspase-3 or caspase-
7.
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
Yet another novel approach for inhibiting caspase enzymes involves the use of
so-called
"Superantibody Technology (SAT)". See, e.g., WO 02/097041, entitled "Fusion
Proteins of
Biologically Active Peptides and Antibodies" (co-assigned to Immpheron, Inc.
and Innexus
Corporation). One proposed application of SAT is the use of antibodies against
caspase
s enzymes in order to inhibit apoptosis in living cells. For example, one
aspect of the present
invention contemplates intracellular delivery of an antibody or antibody
fragment
immunospecific for an enzyme involved in apoptosis. Some expected advantages
of trans-
membrane antibodies as apoptosis inhibitors are their specific target
recognition in the cell
and their lower toxicity compared to conventional apoptosis inhibitors. It is
an object of the
to present invention to provide such membrane-penetrating antibodies for
therapeutic benefit.
SUMMARY OF THE INVENTION
The present invention provides a fusion protein comprising an antibody domain
and a peptide
domain, wherein the biological activity of the peptide domain is selected from
the group
1 s consisting of immune-stimulatory, membrane transport and hemophilic
activities. The
peptide is covalently linked to a site on the antibody so that the
incorporated peptide does not
compromise the antigen recognition of the antibody. In the present invention,
this is
accomplished by a method comprising the steps of creating a fusion gene
comprising a
nucleic acid sequence encoding an antibody and a nucleic acid sequence
encoding the
2o peptide, wherein the nucleic acid sequence encoding the peptide is located
inside the nucleic
acid sequence encoding the antibody at a site wherein, ~nahen the fusion is
e6~pressed9 the
fusion protein that is created thereby includes the antibody plus the peptide,
and the peptide is
connected to the antibody at a site that does not interfere with antigen
binding of the
antibody, and expressing the fusion gene to create the fusion protein. In
particular, the fusion
25 protein may be created by providing a gene encoding an antibody, wherein
the gene is
mutated to contain a restriction site, wherein the restriction site is located
away from any
section of the gene that encodes an antigen-binding site of the antibody,
inserting a DNA
sequence encoding a peptide having a biological activity selected from the
group consisting
of immune-stimulatory, membrane transport and hemophilic activities into
restriction site of
3o the gene encoding the antibody to create a fusion gene, and wherein the
I~NA sequence
encoding the peptide is inserted so that it is in-frame with the gene encoding
the antibody,
and expressing the fusion gene to create a fusion protein.
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
In order to enhance the biological activity of the peptide, the peptide may be
flanked by loop-
forming or conformation-conferring sequences.
The invention also provides a composition and a pharmaceutical composition
comprising a
fusion protein of a peptide having a biological activity selected from the
group consisting of
immuno-stimulatory, membrane transport and homophilic activities and an
antibody.
The invention of creating fusion proteins of biologically active peptides and
antibodies
includes peptides which comprise self binding, stimulate lymphocytes and allow
transport
1o across biological membranes.
A further aspect of the present invention is for novel compounds and methods
for regulating
cell function, either in normal or infected cells. In particular, such
compounds and methods
entail the use of an antibody, or antibody fragment thereof, conjugated to a
membrane
is transporter peptide. The antibody, or fragment thereof, is preferably
immmun~specific, i.e., it
recognizes and binds specifically with high affinity to, f~r such protein
targets as: (a)
signaling proteins internal the cell, such as caspases, kinases, and
phosphatases, (b) immature
virion proteins prior to intracellular assembly, (c) cell-surface or
intracellular tumor antigens,
(d) nuclear or nucleolar proteins that are involved in regulation of DNA
synthesis and gene
2o expression, or (e) cytoskeletal proteins that participate in cell
proliferation or cyt~stasis.
Either polyclonal or monoclonal antibodies can be used.
In a preferred aspect of the invention, an aforementioned compound is
effective in inhibiting
apoptosis and comprises an anti-caspase antibody, or fragment thereof,
conjugated to a
25 membrane transporter peptide. A particularly preferred antibody is an anti-
caspase-3
antibody.
In a second preferred aspect of the invention, an aforementioned membrane
transporter
peptide is a translocation sequence (MTS) peptide, such as one endogenous to
Kaposi
3o fibroblast factor, TAT peptides of HIV-1, antennapedia homeodomain-derived
peptide,
herpes virus protein VP22, or transportan peptide. A particularly preferred
MTS peptide
comprises the amino acid residue sequence AAVLLPVLLAAP (SEQ ID NO: 9), such as
the
peptide sequence KGEGAAVLLPVLLAAPG (SEQ ID NO: 8).
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
Also contemplated is a pharmaceutical composition effective in inhibiting
apoptosis in
human cells, and which therefore is implicated as being effective in the
treatment of human
diseases, that comprises an anti-caspase antibody, or fragment thereof,
conjugated to a
membrane transporter peptide, e.g., an MTS peptide. The antibody-peptide
conjugates of the
present invention are capable of causing internalization of the antibody or
antibody fragment
into cells.
In another aspect of the invention, a method of treating or preventing a
disease comprises
administering to a patient in need thereof a pharmacologically effective
amount of a
pharmaceutical composition comprising an anti-caspase antibody, or fragment
thereof,
conjugated to a membrane transporter peptide or fragment thereof. Specifically
demonstrated
are modified anti-caspase antibodies conjugated to a membrane transporter
peptide that
reduce chemically induced apoptosis. These results suggest such antibodies
have therapeutic
15 potential to inhibit apoptosis in a variety of diseases, such as
Alzheimer's, Huntington's or
Parkinson's.
The above and other objects of the invention will become readily apparent to
those of skill in
the relevant art from the following detailed description and figures, wherein
only preferred
20 embodiments of the invention are shown and described. As is readily
recognized, the
invention is capable of modifications within the skill of the relevant art
without departing
from the spirit and scope of the invention.
BRIEF T~ESCRIPTION OF FIGURES
25 Fig. 1 shows the detection viability of MTS-anti-active caspase-3 antibody
conjugate-treated
Jurkat cells. 2.5 x 105 Jurkat cells were seeded into 96-well culture plate.
After incubation
with 0.5 p,g MTS-antibody for 6, 12, 18 and 24 hour, aliquots were removed and
viable cells
were counted using dye exclusion (trypan blue).
3o Fig. 2 depicts detection of antibody internalization by sandwich ELISA.
Sheep anti-rabbit
antibody was coated onto an ELISA plate (400 ng/well). The cell homogenate and
equal
volume of the culture supernatant were added to a sheep anti-rabbit IgG-coated
ELISA plate
(Falcon, Oxnard, CA) and incubated for 2 h at room temperature. After washing,
HRP-
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
labeled goat anti-rabbit light chain antibody was added, and antibody was
visualized by
adding o-phenylene-diamine. The ratio of internalized antibody versus culture
antibody is
plotted.
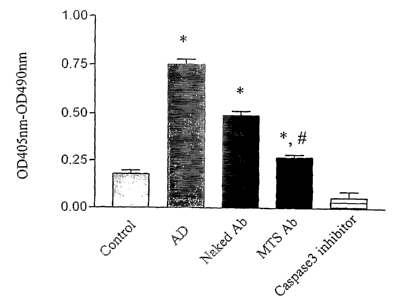
Fig. 3 depicts the extent of DNA fragmentation measured by cell death ELISA
assay. MTS-
conjugated or naked anti-caspase-3 antibody (2 p,g/ml) was added to 6-ml
cultured Jurkat
cells and pre-incubated for 1 h. The antibody was washed out by
centrifugation, fresh
medium was added containing actinomycin D (1 p.g/ml), and incubating for 4 h.
5 ml of the
culture was collected for DNA fragmentation assessment by ladder
electrophoresis; the rest
Io for the ELISA assay. AD = actinomycin D; Naked Ab = caspase-3 antibody; MTS-
Ab _
MTS-conjugated anti caspase-3 antibody; Caspase-3 inhibitor = DEVD-fmk (100
~uM).
*,p<0.01 comparing with Control; #, p<0.01 comparing with naked caspase-3
antibody.
Fig. 4 depicts caspase-3-Iike cleavage activity assay. An equal amount of
protein of the total
~s cell lysate was applied for the assay by using the ApoAlert Caspase-3
Fluorescent Assay I~it.
°°°,p<0.01 comparing with Control; #, p<0.01 comparing
with naked caspase-3 antibody.
DETAILED DESCRIl'TI~N ~F TI-IE INVEN°TI~N
The present invention describes a method for creating fusion proteins of an
antibody and a
2o peptide having a biological activity selected from the group consisting of
immuno-
stimulatory9 membrane transport: and homophilic activities.
In particular, the present invention provides a fusion protein comprising an
antibody and a
peptide having a biological activity selected from the group consisting of
immuno-
2s stimulatory, membrane transport and homophilic activities, wherein the
peptide is located at a
site in the antibody so that the incorporated peptide does not compromise the
antigen
recognition of the antibody. In the present invention, this is accomplished by
a method
comprising the steps of creating a fusion gene comprising a nucleic acid
sequence encoding
an antibody and a nucleic acid sequence encoding the peptide, wherein the
nucleic acid
3o sequence encoding the peptide is located inside the nucleic acid sequence
encoding the
antibody at a site wherein, when the fusion is expressed, the fusion protein
that is created
thereby includes the antibody plus the peptide, and the peptide is connected
to the antibody at
a site that does not interfere with antigen binding of the antibody, and
expressing the fusion
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
11
gene to create the fusion protein. In particular, the fusion protein may be
created by providing
a gene encoding an antibody, wherein the gene is mutated to contain a
restriction site,
wherein the restriction site is located away from any section of the gene that
encodes an
antigen-binding site of the antibody, inserting a DNA sequence encoding a
peptide having a
biological activity selected from the group consisting of immune-stimulatory,
membrane
transport and hemophilic activities into restriction site of the gene encoding
the antibody to
create a fusion gene, and wherein the DNA sequence encoding the peptide is
inserted so that
it is in-frame with the gene encoding the antibody, and expressing the fusion
gene to create a
fusion protein.
l0
In a further embodiment of the present invention, the peptide having
biological activity may
be attached to the C-terminus of the antibody. In a further embodiment of the
present
invention, the peptide may be flanked by loop-forming or conformation-
conferring sequences
to enhance the biological activity of the peptide.
~s
As used herein, the term "targeting moiety" refers to any natural or
synthesised protein
molecule containing an antigen- binding site. The term includes a full-length
immunoglobulin
molecule or any functional fragment, such as a variable domain fragment of a
full-length
immunoglobulin molecule, CDR regions, ScFv, Fab, F(ab)'2, or engineered
antibody mimics
20 or single domain binding moieties. A particular targeting moiety is
selected in accordance
with the desired target, such as a cellular receptor on a membrane structure,
e.g., a protein,
glycoprotein, polysaccharide or carbohydrate. The targeting moiety can be
selected to bind a
cellular receptor on a normal cell or on a tumor cell.
25 Likewise, the peptide having biological activity is selected according to
the desired function
of the fusion protein, or, in other words, according to the desired result
after the targeting
moiety binds to a target such as a normal cell or a tumor cell. Possible
biological activities
that may be desired include immune-stimulatory, membrane transport and
hemophilic
activities.
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
12
The loop-forming or conformation-constraining sequences may be any amino acid
sequences
that, when placed on either side of the peptide having biological activity,
restrain the
conformational flexibility of the peptide. Examples include sequences
containing amino acid
residues such as cysteine pairs that can cross-link to form loops. A specific
example of a
conformation-constraining protein is thioredoxin. Examples of conformation-
constraining or
loop-forming moieties may be found, for example, in the following U.S.
Patents: U.S. Patent
Nos. 6,242,163 and 6,004,746 to Brent, U.S. Patent Nos. 6,258,550; 6,147,189;
6,111,069;
6,100,044; 6,084,066; 5,952,465; 5,948,887; and 5,928,896 to Brent et al, U.S.
Patent Nos.
6,200,759 and 5,925,523 to Dove et al., and in the following publications:
to Fairlie DP; West ML; Wong AK "Towards protein surface mimetics:' Cur Med
Chenl 1998
Feb;S (1) :29-62;
Valero ML; Camarero JA; Haack T; Mateu MG; Domingo E; Giralt E; Andreu D
"Native-
like cyclic peptide models of a viral antigenic site: finding a balance
between rigidity and
flexibility." JMoI Rec~gnit 2000 Jan-Feb;13(1):5-13;
1s Gururaja TL; Narasimhamurthy S; Payan D(i; "A novel artificial loop
scaffold for the
noncovalent constraint of peptides." Clmm Bi~l. 2000 Jul; 7(7):515-27;
Venkatesh N; im SH; Balass M; Fucks S; K~tchalslei-Katzir E "Prevention of
passively
transferred experimental autoimmune myasthenia gravis by a phage library-
derived cyclic
peptide." 1'f~oc Natl Acczd Sci Ll~'A 2000 Jan 18;97(2) :761-6;
2o Stott K; Blackburn JM; Butler PJ; Perutz M "hicorporation of glutamina
repeats makes
protein oligomerize: implications for neurodegenerative diseases."
P~°~c Ncztl Aced ~ci. LIS'A
1995 Jul 3;
All of the above are incorporated herein by reference.
2s The conformation-constraining sequences may also include sequences that
form alpha helices
or beta-pleated sheets. See, for example, the following publications
incorporated herein by
reference:
Lee KH; Benson DR; Kuczera K "Transitions from alpha to pi helix observed in
molecular
dynamics simulations of synthetic peptides." Biochemistry 2000 Nov 14;39(45):
13737-47;
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
13
Dettin M; Roncon R; Simonetti M; Torinene S; Falcigno L; Paolillo L; Di Bello
C
"Synthesis, characterization and conformational analysis of gp120-derived
synthetic peptides
that specifically enhance HIV-1 infectivity." JPept Sci 1997 Jan-Feb;3 (1) :15-
30;
Chavali GB; Nagpal S; Majumdar SS; Singh O; Salunke DM "Helix-loop-helix motif
in
GnRH associated peptide is critical for negative regulation of prolactin
secretion." JMoI
Biol. 1997 Oct 10; 272(5):731-40; and
Miceli R; Myszka D; Mao LI; Sathe G; Chaiken I "The coiled coil stem loop
miniprotein as a
presentation scaffold." Drug Des Discov., 1996 Apr; 13 (3-4): 95-105.
to The Expression of I~-fusion Proteins. Ig fusion proteins have the advantage
of joining the
antibody combining specificity and/or antibody effector functions with
molecules
contributing unique properties. The ability to produce this family of proteins
was first
demonstrated when c-myc was substituted for the Fc of the antibody
molecule,(Neuberger M
S, Williams G T and Fox R O., l~atu~e, 125:604, 194) but many examples now
exist. Ab
15 fusion proteins can be achieved in several different ways. In one approach
non-Ig sequences
are substituted for the variable region; the molecule replacing the V region
provides
specificity of targeting with the antibody contributing properties such as
effector functions
and improved pharmacokinetics. Examples include IL-2 and CD4. Alternatively,
non-Ig
sequences can be substituted for or attached to the constant region. The
resulting molecules
?o retain the binding specificity of the original antibody but gain
characteristics from the
attached protein. Depending on the position of the substitution, different
antibody-related
effector functions and biologic properties will be retained. See, for example,
Antibody
Eng_incerin~, 2nd Edition. ed.: Carl A. I~. Borrebaeck, Oxford University
Press, 1995)
25 Vectors for the Construction of I~G Fusion Proteins. A series of vectors
has now been
produced that permits the fusion of proteins at different positions within an
antibody
molecule, thereby facilitating the construction of fusion proteins with
different properties.
Using these vectors it is possible to produce a family of fusion proteins with
molecules of
differing molecular weight, valence, and having different subsets of the
functional properties
30 of the antibody molecule.
As a specific example of how to facilitate the construction of fused genes,
site-directed
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
14
mutagenesis was used to generate unique restriction enzyme sites in the human
IgG3 heavy
chain gene. In this particular example, restriction sites were generated at
the 3' end of the
CHI exon, immediately after the hinge at the 5' end of the CH2 exon, and at
the 3' end of the
CH3 exon. The restriction sites thus produced were Snag I at the end of CHI by
replacing
s TtgGTg with TacGTa, Pvu II at the beginning of CH2 by replacing CAcCTG with
CAgCTG,
and Ssp I at the end of CH3 replacing AATgag with AATatt. These manipulations
provided a
unique blunt-end cloning site at these positions. In all cases the restriction
site was positioned
so that after cleavage the Ig would contribute the first base of the colon.
Human IgG3 with
an extended hinge region of 62 amino acids was chosen for use as the
immunoglobulin; when
1o present this hinge should provide spacing and flexibility, thereby
facilitating simultaneous
antigen and receptor binding. An EcoR I site was also introduced at 3' of the
IgG3 gene to
provide a 3' cloning site and polyA addition signal. Although initially
designed for use with
growth factors, these restrictions sites can be used to position any novel
sequence at defined
positions in the antibody. Also, using these cloning cassettes the variable
region can easily be
15 changed. Similar techniques may be used to generate suitable restriction
sites in other
antibody genes.
Production of A Fusion Gene. As a first step in the production of a fusion
protein, a blunt-end
restriction site must be introduced at the desired position into the 5' end of
the gene to be
2o fused. In order to maintain the correct reading frame, the site must be
positioned so that after
cleavage it will contribute two bases to the colon. If the objective is to
make a, fusion protein
with the complete molecule, the restriction site is usually introduced at the
position ofany
post-translational processing, such as after the leader sequence.
Alternatively, if the objective
is to use only a portion of the protein, the blunt-end site can be introduced
at any position
2s within the gene, but attention must always be paid to maintaining the
correct reading frame.
Additionally, if there is carboxyl-terminal post-translational processing of
the fused protein, it
is frequently desirable to introduce a stop-colon at this processing site.
A major concern when producing fusion proteins is maintaining the biologic
activities of all
3o of the components. The production of fusion proteins with antibodies is
facilitated by the
domain structure of the antibody, and all of the cloning sites have been
positioned
immediately following an intact domain. In this configuration the correct
folding of the
immunoglobulin should be assured. The folding of the attached protein depends
on its
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
structure and where it is fused. Whenever structural information is available,
it is desirable to
produce the fusion at a position that will maintain the structural integrity
of the attached
protein.
To produce quantities of protein sufficient for functional analysis, it is
desirable to have the
protein secreted into the medium. While in the examples reported to date,
assembled fusion
proteins have been assembled and secreted, this remains a concern when
designing additional
fusion proteins.
1 o The method to design a fusion gene that contains a biologically activity
peptide as part of the
heavy or light chain gene can use established antibody engineering protocols
(Antibody
En ineerin~ 2nd Edition, ed.: Carl A. K. Borrebaeck, ~xford University Press
1995. Chapter
9, pages 267-293). The peptide can fused either to N-terminal residues or the
C-terminal
residues of H or L chains. The expression of such fused genes is typically
done in
15 mammalian cell lines, although other expression systems, such as, for
example, bacteria or
yeast expression systems, may be used.
The peptide of the invention has a biological activity selected from the group
consisting of
immuno-stimulatory, membrane transport and homophilic activities. Examples
include
2o immuno-stimulatory or immuno-regulatory activity. The peptide may, for
example, be a
hormone, ligand for cytokines or a. binding site derived from natural ligands
for cellular
receptors. In a preferred embodiment, the peptide is derived from C3d region
1217-1232 and
ranges from about 10 to about 16 mer. In an alternative embodiment, the
peptide is a 16 mer
peptide derived from the C3d region 1217-1232.
The peptide may be bound to an antibody that is a full-length immunoglobulin
molecule or a
variable domain fragment of an antibody. As used herein, the term "antibody"
refers
generally to a heavy or light chain immunoglobulin molecule or any function
combination or
fragment thereof containing an antigen-binding site. The antibody is
preferably specific for a
3o cellular receptor, on a membrane structure such as a protein, glycoprotein,
polysaccharide or
carbohydrate, and on a normal cell or an tumor cells.
The use of peptides derived from the ligand site of C3d as an
immunostimulatory component
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
16
incorporated into antibodies has an unexpected utility as a molecular
adjuvant. C3d has been
used as molecular adjuvant as part of a complete fusion protein with hen egg
lysozyme
(HEL) by D. Fearon, et al., (Dempsey, P. W., Allison, M. E. D., Akkaraju, S.,
Goodnow, C.
C, and Fearon, D. T., Science, 271:348, 1996). These authors have shown that a
HEL- C3d
fusion protein is up to 10,000 fold more immunogenic than free HEL (see
International
Patent Publication, W096/17625).
Similar increases in immunogenicity have been observed with chemical cross-
linked idiotype
vaccines using a peptide derived from the C3d fragment in our recent animal
studies (see
1o examples below). It is believed that attaching C3d peptides to idiotype and
anti-idiotype
vaccines enhances the immunogenicity of these vaccines and substitutes for the
need of
attaching carrier molecules such as I~LH in combination with strong adjuvants,
such as
Freund's adjuvant, which is not permitted by the FDA for use with humans.
1s In an alternative embodiment, the peptide may be derived from a human or
non-human C3d
region homologous to the humaIl C3d residues at position 1217-1232 and ranges
from about
to about 16 mer. Other applications of affinity cross-linking biologically
active peptides to
antibody vaccines include active peptides derived from cytokines. For example,
a
nonapeptide from the ILl-beta cytokine has been described (Antoni, et al., J.
Immufzol,
137:3201-04, 1986) which has immunostimulatory properties without inducing
undesired
side effects. Other examples of active peptides which can be inserted into
antibodies in
accordance with the invention include signal peptides, and peptides from the
self binding
locus of antibodies.
2s A variety of peptides are known having biological activities as hormones,
ligands for
cytokines or binding sites derived from natural ligands for cellular
receptors.
The following Examples 1-3, while relating to C3d/antibody complexes that are
created by
affinity cross-linking, are provided to show the effects on the immune
response provided by
3o C3d peptides linked to antibodies.
Example 1. Enhancement of an Anti-idiotYpe Vaccine. 3H1 is a murine anti-
idiotype
antibody (Bhattacharya-Chatterjee, et al., J. Immuhol., 145:2758-65, 1990)
which mimics the
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
17
carcino-embryonic antigen (CEA). 3H1 induces in animals anti-CEA antibodies
when used as
KLH-conjugated vaccine in complete Freund's adjuvant. 3H1 has also been tested
in a
clinical phase I study where it induces antibodies which bind to CEA in
approximately half of
treated cancer patients. However no clinical response was observed in this
study (Foon, et al.,
J. Clin. Invst., 96:334-342, 1995) due, in part, to low immunogenicity.
3H1 mAb was affinity cross-linked with a 13-mer peptide (SEQ ID NO.:1) derived
from the
C3d region 1217-1232. The amino acid sequence was derived from ofthe Cd3
peptide and
has the following sequence: KNRWEDPGKQLYNVEA (SEQ ID NO. 1)
to
BALB/c mice were immunized twice with 25~g of C3d-3H1 in phosphate-saline
solution
intramuscular. 7 days after the last immunization mice were bled and sera were
tested for
binding to 8019 (Abl idiotype) and to the CEA expressing tumor line LS174T. As
determined by FACS, sera from C3d-3H1 immune mice bind to LS174T tumor cells,
while a
~5 control serum (normal mouse serum) showed only background fluorescence.
Sera from mice
immunized with C3d-3H1 were used in FRCS of LS174T cells in a sandwich assay
developed with FITC-conjugated goat anti-mouse IgG. Control was a normal mouse
serum.
Cell numbers analyzed were plotted against relative fluorescence intensity on
log 10 scale.
2o Example 2. Furthermore, sera from mice immunized three times with either
3H1 (25
microgram in saline) or 3H1-C3d-peptide (affinity cross-linked, 25 microgram
in saline) were
also tested for Ab3 response. Mice were bled and sera were tested for binding
to Flab) of 3H1
in ELISA. Upon determining the binding of dilutions ofmouse sera to 3H1 F(ab),
it was
found that while naked 3H1 does not induce Ab3 antibodies, 3H1-peptide does
showing that
2s the affinity-cross-linked 3H1 enhanced immunogenicity.
Other C3d peptides which may be used in the practice of the present invention
include those
reviewed in Lambris et al, "Phylogeny of the third component of complement,
C3" in Erfi, A
ed. New Aspects of Complement structure and function, Austin, R. D. Landes
Co., 1994 p.
30 15-34, incorporated herein by reference in its entirety.
Example 3. Enhancement of an Mouse Tumor Idiotyae Vaccine (38C13~ 38C13 is the
idiotype expressed by the 38C13 B-lymphoma tumor cell line. The Levy group has
developed
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
18
this idiotype tumor vaccine model and has shown that pre-immunization with KLH-
conjugated 38013 Id can protect against challenge with 38013 tumor cells in
mice
(Kaminski, M. S., Kitamura, K., Maloney, D. G. and Levy, R., J. Irnrraunol.,
138:1289, 1987).
Levy and colleagues (Tao, M-H. and Levy, R., Nature, 362:755-758, 1993) have
also
reported on the induction oftumor protection using a fusion protein (CSF-
38013), generated
from a chimeric gene and expressed in mammalian cell culture fermentation.
38013 Id
proteins were affinity cross-linked with a 16-mer azido-peptide derived from
the C3D region
1217-1232.
Ten mice were immunized with 50 ug of C3d-38013 conjugate in phosphate-saline
solution
intra-peritoneally three times. After the third vaccination mice were
challenge with 38013
tumor cells. Control groups included mice vaccinated with 38013-KLH in QS-21
(adjuvant),
considered the "gold standard" in this tumor model, and mice injected with QS-
21 alone.
Seven out of ten mice vaccinated with the C3d-38013 conjugate survived by day
35 after
15 tumor challenge, as did mice vaccinated with the KLH-38013 in QS-21. All
control mice
injected only with QS-21 died by day 22.
C3H mice were immunized three times with either 38013-KLH in QS-21 or with
38013-C3d
peptide without QS-21 (50 ~g i.p.) Control mice were only injected with QS-21.
Immunized
2o and control mice were than challenged with 38013 tumor cells and survival
was monitored.
Results described in Examples 1-3 show that affinity-cross-linking of an
immuno-stimulatory
peptide to tumor anti-idiotype and idiotype vaccine antibodies can
significantly enhance the
immune response to the tumor and protect against tumor challenge. The
vaccination protocol
25 with the C3d-cross-linked vaccine did not include any adjuvant, such as
Freund's adjuvant, or
KLH conjugation, both of which are not permissible by the FDA for human use.
Some of the procedures used in the above examples are known; the active
binding peptide of
C3d (complement fragment) has been described by Lambris, et al., (PNAS,
82:4235-39,
30 1985) and is incorporated herein by reference in its entirety.
The following additional examples are provided to demonstrate the general
technique of
creating fusion proteins and to illustrate particular peptide having a
biological activity
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
19
selected from the group consisting of immuno-stimulatory, membrane transport
and
homophilic activities.
Example 4 Fusion non-I~ Protein containing a Membrane Transferring YPeptide
(MTS-
a tide See, e.g., Rojas, M, Donahue, J P, Tan, T. and Lin, Y-Z. Nature
Biotech., 16: 370,
"Construction of the glutathion S-transferase-MTS peptide (GST-MTS) expression
plasmids," 1998.
Two different GST-MTS expression plasmids were constructed so that, depending
on the
to biological application, a target protein or protein domain could be
produced with the
hydrophobic MTS as either an amino-terminal or a carboxyl-terminal extension.
For the
construction ofplasmids pGEX-3X-MTS I and pGEX3X-MTS2, the following
complementary oligonucleotides were synthesized:
MTSI: GATCGCAGCCGTTCTTCTCCCTGTTCTTCTTGCCGCACCCGG-
15 CGTCGGCAAGAAGAGGGACAAGAAGAACGGCGTGGGCCCTAG (SEQ ID NO. 2)
MTS2:GATCCCCGCAGCCGTTCTTCTCCCTGTTCTTCTTGCCGCACCCTAGC-
GGGCGTCG(iCAAGAAGAGGGACAAGAAGAACGGCGTGGGATTCGCTAG
(SEQ ID N~. 3)
2o After annealing, the double-stranded MTS I and MTS2 oligonucleotides were
ligated in
13am1-lI digested pGE~-3~ (Smith, D. )3. and Johnson, I~.. S., 6'Single-step
purification of
polypeptides expressed in E'scheT°iclaia ccli as fusions with
glutathione S-transferase,'9 C~c~7e,
67:31, 1988.). DNA sequence analysis confirmed that in each plasmid the MTS
coding
sequence was correct and in-frame with the GST coding sequence.
Construction of GST-Grb2SH2 GST-Grb2SH2-MTS and GST-StatlSH2-MTS Expression
Plasmids. A DNA fragment encoding the human Grb2 SH2 domain (amino acid
residues 54-
164) (Lowenstein, E. J., Daly, R. J., Batter, A. G., LJ, W., Margolis, B.,
Lammers, R et al.,
"The SH2 and SH3 domain-containing protein Grb2 links receptor tyrosine
kinases to ras
3o signaling," Cell, 70:431, 1992) or the human Statl SH2 domain (residues 567-
716)
(Schindler, C., Fu, X.-Y, Impnota, T., Aebersold, R., and Darnell, J. E. Jr.,
Proc. Natl Acad.
Sci USA 89:7836, 1992) was synthesized from a Grb2 cDNA clone or a Statl cDNA
clone by
PCR. The primers used for PCR, each containing BamHI sites at their 5'ends,
were as
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
follows:
Grb2 SH2: 5'-CCGGATCCCCGAAATGAAACCACATCCGTGGTTTTTTGGC
(SEQ ID NO. 4) and .
5'-CCGGATCCCGAGGGCCTGGACGTATGTCGGCTGCTGTGG (SEQ ID NO. 5).
Statl SH2: 5'-CCGGATCCCCAAACACCTGCTCCCTCTCTGGAATGATGGG
(SEQ ID NO. 6) and
5'-CCGGATCC-CTCTAGAGGGTGAACTTCAGACACAGAAAT (SEQ ID NO. 7).
The PCR products were digested with BamHI and ligated in BamHI-digested pGEX-
3X or
1o pGEX-3XMTS2. DNA sequence analysis of the vector/insert junctions confirmed
that the
GST-Grb2SH2, GST-Grb2SH2-MTS, and GST-StatlSH2-MTS translational reading
frames
were maintained in each expression plasmid.
Expression of MTS Fusion Protein
15 Expression and purification of GST fusion proteins. E. c~li strain DHSor
containing the
appropriate expression plasmid 74~ as grown in LB broth containing 100 ~,g/ml
ampicillin at
37°C. GST fusion protein expression was induced by the addition of
isopropyl, B-D-
thiogalactoside (0.5 mM final concentration), and incubation at 37°C
was continued for 2-3
hours. GST fusion proteins were purified from bacterial cell lysates by
glutathione-agarose
2o affinity chromatography. (Smith, D. B. and Johnson, K. S. Gene, 67:31,
1988) except that
after sonication, cell lysates were cleared by centrifugation at ~OOO×g
for 5 minutes
prior to mixing with glutathione-agarose beads. Protein preparations were
concentrated by
ultrafiltration using a PMIO membrane (Amicon, Beverly, MA) and stored at
4°C for
immediate use or -70°C for prolonged storage. Protein concentrations
were determined
~s spectrophotometrically at 280 nm. Immediately prior to their use in
biological assays, protein
concentrations were verified by SDS-PAGE using Coomassie brilliant blue
staining intensity
compared with wild-type GST of known concentration. To confirm the amino acid
content of
the MTS in GST-MTS proteins, the MTS peptide was cleaved from glutathione-
agarose
bound GST-MTSI with protease factor Xa essentially as described (Smith, D. B.
and
3o Johnson, K. S., Gene 67:31,1988). The released MTS-containing peptide was
purified by C,
reverse-phase HPLC and characterized by mass spectrometry analysis as
described (Smith,
D. B. and Johnson, K. S., Gene 67:31,1988). The released MTS-containing
peptide was
purified by C~8 reverse-phase HPLC and characterized by mass spectrometry as
described
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
21
(Lin, Y-Z., Yao, S., Veach, R. A., Torgerson, T. R., and Hawiger, J., JBiol.
Chern.
270:14255, 1995).
Example 5. C3d -HEL fusion protein (Dempsey et al. Science 271: 348 1996 The
complimentary DNA encoding HEL, C3d (H. Domdey et aL, Pro. Natl Acad Sci USA,
79:
7619, 1982) doq pre-pro-insulin signal sequence (M. E. Taylor and K.
Drickamer, Biochena.
J., 274, 575, I991), and the (G4S) 2 linker were amplified by polymerase chain
reaction. The
epitope tag and stop codon were coded for by oligonucleotide linkers. Fusion
protein cassetes
were assembled in tandem: doq pre-pro-insulin signal sequence, HEL, and one to
three copies
of C3d linked by (G4S)2 in pSGS (Stratagene Cloning Systems, La Jolla, CA).
The HEL-
C3d3 cassette was subcloned into the A7ld vector. The plasmids pSG.HEL,
pSG.HEL.C3d,
and pSG.HEL.C3d2 were co-transfected with pSV2-neo into L cells and A7ld.
HEL.C3d3
was transiently expressed in C~S cells. Recombinant proteins were punfied by
affnity
chromatography on YL 1/2 antibody (H. Skinner et al., J. Biol. Chern.,66:
14163, 1991) and
fractionation on Sephacryl S-200 (Pharmacia).
Fusion tails are useful at the lab scale and have potential for enhancing
recovery using
economical recovery methods that are easily scaled up for industrial
downstream processing.
Fusion tails can be used to promote secretion oftarget proteins and can also
provide useful
2o assay tags based on enzymatic activity or antibody binding. Many fusion
tails do not interfere
with the biological activity of the target protein and in some cases have been
shown to
stabilize it. Nevertheless, for the purification of authentic proteins a site
for specific cleavage
is often included, allowing removal of the tail after recovery.
Fusion Tails for the Recoyery and Purification of Recombinant Proteins. (See,
e.g., Ford C.,
Suominen L, Glatz C., Py~otein Expr. Purif, 2-3: 95-107, 1991). The fusion
protein of the
present invention may also include a fusion tail such as has been developed to
promote
efficient recovery and purification of recombinant proteins from crude cell
extracts or culture
media. In these systems, a target protein is genetically engineered to contain
a C- or N-
3o terminal polypeptide tail, which provides the biochemical basis for
specificity in recovery
and purification. Tails with a variety of characteristics have been used:
(1) entire enzymes with affinity for immobilized substrates or inhibitors;
(2) peptide-binding proteins with affinity to immunoglobulin G or albumin;
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
22
(3) carbohydrate-binding proteins or domains;
(4) a biotin-binding domain for in vivo biotinylation promoting affinity of
the fusion protein
to avidin or streptavidin;
(5) antigenic epitopes with affinity to immobilized monoclonal antibodies;
(6) poly(His) residues for recovery by immobilized metal affinity
chromatography; and
(7) other poly(amino acids, with binding specificity based on properties of
the amino acid
side chain.
Fusion tails are useful at the lab scale and have potential for enhancing
recovery using
economical recovery methods that are easily scaled up for industrial
downstream processing.
Fusion tails can be used to promote secretion of target proteins and can also
provide useful
assay tags based on enzymatic activity or antibody binding. Many fusion tails
do not interfere
with the biological activity of the target protein and in some cases have been
shown to
stabilize it. Nevertheless, for the purification of authentic proteins, a site
for specific cleavage
1s is often included, allowing removal of the tail after recovery.
The present invention describes the generation of an antibody-peptide fusion
protein that
enhances the biological and immunological activity of the antibody without
changing the
antibody specificity for the corresponding antigen. The genetically engineered
fusion protein
2o mimics the chemically engineered chimeric antibodies described in patent
application Ser.
No. 0~/0~0,~07. Speci~cally9 the present invention provides the generation of
antibody
fusion proteins containing the complete or partial autophilic 24-mer peptide,
the membrane
transpout peptide (MTS) or the C3d peptide, all described above.
2s The invention also provides a composition and a pharmaceutical composition
comprising a
fusion protein made up of (1) an antibody and (2) a peptide having a
biological activity
selected from the group consisting of immune-stimulatory, membrane transport
and
hemophilic activities wherein the peptide is connected by peptide bonds to the
antibody at a
site that does not interfere with antigen binding of the antibody.
Any antibody may be used in the peptide/antibody complex of the invention.
Preferred
antibodies are anti-idiotype antibodies. For example, anti-idiotype antibody
3H1 may be used
(see "Anti-idiotype Antibody Vaccine (3H1) that Mimics the Carcinoembryonic
Antigen
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
23
(CEA) as an Adjuvant Treatment", Foon, et al., Cancer Weekly, Jun. 24, 1996).
Other anti-
idiotype antibodies which may be used in the present invention include, for
example, anti-
idiotype antibody to chlamydia glycolipid exoantigen (U.S. Pat. No. 5,656,271;
anti-idiotype
antibody lA7 for the treatment of melanoma and small cell carcinoma (U.S. Pat.
No.
5,612,030); anti-idiotype antibody MK2-23 anti-melanoma antibody (U.S. Pat.
No.
5,493,009); anti-idiotypic gonococcal antibody (U.S. Pat. No. 5,476,784)
Pseudonzonas
ae~uginosa anti-idiotype antibody (U.S. Pat, No. 5,233,024); antibody against
surface Ig of
human B cell tumor (U.S. Pat. No. 4,513,088); and monoclonal antibody BR96
(U.S. Pat. No.
5,491,088). Any restrictions on peptide length are those practical limitations
associated with
1o peptide synthesis and not restrictions associated with practice of the
method of the invention.
Additionally, self binding peptides such as those disclosed in (Kang, C. Y.
Brunck, T. K.,
I~iever-Emmons, T., Blalick, J. E. and I~ohler, H., "Inhibition of self
binding proteins (auto-
antibodies) by a VH-derived peptide, Science, 240: 1034-1036, 1988, which is
incorporated
15 herein by reference in its entirety) may be used in the method of the
present invention.
Additionally, signal peptides such as those disclosed in Rojas, et al.,
"Genetic Engineering of
proteins with cell membrane permeability", Natua~e Pi~techn~l~gy, 16: 370-375
(1988) and
Calbiochem Signal Transduction Catalogue 1997/98, incorporated herein by
reference in
2o their entireties, may be used in the method of the invention.
'The peptide may be designed to have inverse hydropathic character and
ez~hibits mutual
affinity and homophilic (self) binding within the peptide, in accordance with
the disclosure of
U.S. Pat. No. 5,523,208 (incorporated herein by reference in its entirety).
The present invention contemplates novel compounds and methods for regulating
cellular
functions, either in normal or infected cells. Such compounds comprise an
antibody, or
fragment thereof, which is capable of being internalised within the cell
through the cell
penetrating action of a peptide conjugated thereto. Such peptides are referred
to herein as
"membrane transporter peptides," and the like. Known membrane transporter
peptides, or
their active fragments, can be employed as the attached peptide. Such
antibodies, or
fragments thereof, are immunospecific for such protein targets as: (a)
signaling proteins
internal the cell, such as caspases, kinases, and phosphatases, (b) immature
virion proteins
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
24
prior to intracellular assembly, (c) cell-surface or intracellular tumor
antigens, (d) nuclear or
nucleolar proteins that are involved in regulation of DNA synthesis and gene
expression, or
(e) cytoskeletal proteins that participate in cell proliferation or
cytostasis. Either polyclonal or
monoclonal antibodies can be used. Such antibodies or their fragments
preferably bind to
their bind to their determinants.with an affinity of 10'9M or greater.
A particularly preferred compound of the invention is one that comprises an
anti-caspase
antibody conjugated to a membrane transporter protein, or peptide fragment
thereof. A
preferred membrane transporter fragment is a membrane translocation sequence
(MTS)
to peptide. Particularly preferred membrane transporter peptides include the
following:
(1) KGEGAAVLLPVLLAAPG (SEQ ID NO: ~), derived from Kaposi fibroblast growth
factor [K-FGF] (Rojas et aI, Nature Biotechn~logy, I6: 370-375 (1998)).
(2) AAVLLPVLLAAP (SEQ ID NO: 9), a truncated version of above peptide, see,
Lin et al.,
J. Biol. Cdzem., 271: 5305 (1996).
15 (3) IZQIKIWFQNIZRMKWKK (SEQ ID NO: 10), "penetratin" derived from the
homeodomain of Antennapedia (Ant) (see, Lindberg, M. et al., Eua°. J:
Bi~cl~ern., 270(14):
3055-3063 (2003)).
(4) I~MICWKK (SEQ ID NO: 11), the C-terminal sequence of penetratin, see,
e.g., Fischer,
P. et al. J. Peptide Res., 55(2): 163-172 (2000).
20 (5) TAT peptides, e.g., as 47-57 and 4~-60 derived from HIV-1 TAT (see,
e.g., Schwarze, S.,
et al., Tr~~rds Plz~r,°tracze~l. ~'ei., 21: 4~5, 2000; Li V., et al.
Bi~ehem.Bi~phys. leis. ~'~raarrrz~ra.
29(3): 439-449, 2002; Hallbrink ILL, et a1. Bioclzirzr. Bioph~s.A~tez,
1515(2): 101-I09, 2001).
(6) Herpes virus protein VP22 (Elliot, G., et al., Cell, ~8: 223 (1997)).
(7) GWTLNSAGYLLGKINLKALAALAKKIL (SEQ ID NO: 12), "transportan," a 27-mer
25 peptide (see, Pooga, M. et al., FASEB J., 12: 67 (1990; Lindberg, M. et al.
Bi~cdzena., 40:
3141-3149, 2001).
(8) AGYLLGKINLKALAALAKKIL (SEQ ID NO: I3), N-terminal six residue deletion of
transportan (see, Soomets, U. et al., Bi~chirza.Bi~plZys.Actcz, 1467:165-176,
2000).
(9) Lys-Leu-Ala-Leu (KLAL) (SEQ ID NO: I4), also referred to as MAP (see,
Hallbrink M.,
3o et al. Biochim. Biophys.Acta, 1515(2): 101-109, 2001).
Also contemplated is a pharmaceutical composition effective in inhibiting
apoptosis that
comprises an anti-caspase antibody conjugated to a membrane transporter
protein or fragment
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
thereof, as discussed herein. Such fusion proteins and methods of generating
them are
disclosed in U.S. Serial No. 09/865,281 (Kohler et al.), incorporated herein
by reference.
A preferred immunoconjugate of the present invention comprises a secondary
antibody
conjugated to an MTS sequence through one of several types of linkages
including through
the nucleotide or tryptophan sites of the antibody or through the N-linked
carbohydrate of the
antibody. A "secondary antibody," as used herein, refers to an antibody, or
fragment thereof,
that binds specifically and with high affinity to a primary antibody. The
secondary antibodies
useful for the present invention include polyclonal or monoclonal
antiglobulins to murine or
to human IgG or secondary antibodies targeted to novel and/or installed
sequences such as the
T15 sequence (Kang, CY, Brunck, TK, Kieber-Emmons, T, et. al. "Inhibition of
self binding
antibodies (autobodies) by a VH-derived peptide," S'cierace, 240:1034-6,
1988), which
imparts autophilicity to an antibody.
Is Delivery is accomplished by pre-administering or pre-injecting a monoclonal
antibody or
immunoconjugate, targeted to a cell-surface antigen, allowing sufficient time
for binding to
the target and clearance from the tissues, and following with administration
of a secondary
antibody covalently linked to a MTS peptide. The primary antibody can be
conjugated to an
inhibitor, such as a toxin, drug, enzyme or isotope, thereby enhancing
delivery of an
2o inhibitory molecule into the cell. The secondary antibody conjugated to MTS
peptide
recognizes and binds to the primary antibody, and is internalized into the
cell thr~ugh the
MTS peptide activity. In this way, the primary imnlunoconjugate is brought
into the cell
where its inhibitory action is enhanced.
2s Such secondary conjugates can also be used to assess the utility of
monoclonal antibodies to
intracellular targets by admixing primary and secondary antibodies conjugated
to MTS, then
exposing cells and testing for inhibition of cellular activities targeted with
the primary
antibody. In this rapid screen, many antibodies to intracellular targets can
be screened for
utility as antagonists or agonists. Those with activity can then be directly
conjugated to a
3o membrane transporter peptide, such as MTS, for in vivo use.
A preferred embodiment of the current invention utilizes MTS peptides
conjugated to a
tryptophan or nucleotide binding site of a secondary antibody and a primary
antibody,
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
26
conjugated to a toxin, drug or isotope attached through a sulflrydryl, epsilon
amino acid or
carbohydrate residues via chemical or peptide linkers or chelates.
The present invention relates generally to the in vivo delivery of antibody
conjugates into the
s interior of cells. Such antibodies can be potentially neutralizing, anti-
viral antibodies, anti-
regulatory protein antibodies, or anti-tumor antibodies. For example, delivery
can be
accomplished by administering to a living organism an antibody conjugate
comprising a MTS
peptide, and an antibody directed at determinants on a virus or other
intracellular pathogen
that are best expressed on immature virus or pathogen. Such conjugates have an
increased
Io opportunity for binding with high affinity, disrupting virus assembly and
neutralizing virus
before it has a chance to mature and infect other cells.
Thus, the current invention provides antiviral (anti-HIV) therapeutics as an
example of a
broader class of antibody therapeutics. The antibodies preferred in the
current invention have
is the following preferred properties:
( 1 ) They bind to antigens primarily expressed intracellularly. This includes
tumor
associated antigens (TAA) and viral glycoproteins. The former, includes TAA
such
as CEA. A particular determinant may be primarily associated with
intracellular forms
of the protein whereas others may be primarily expressed on the surface. Prior
to this
2o invention, most useful therapeutic antibodies have been selected for
reactivity to cell
surface molecules; with the ability to target intracellular antigens selection
criteria
would include primary reactivity with intracellular antigen.
(2) Intracellular targets include viral glycoproteins. For instance, most
monoclonal
antibodies have been raised to virus propagated in cells for many passages
rather than
2s virus propagated in cells for only a few passages; as a result most
monoclonals to
viruses react better to laboratory strains of virus rather than fresh
isolates. The
proposed explanation for this difference in binding is that most antibodies,
as with
those to HIV, react to determinants that are cryptic and partially occluded on
viral
glycoproteins from low passaged virus (and presumably newly synthesized virus)
3o because of higher glycosylation and folding of viral glycoproteins. This
would mean
that most antibodies should bind better to immature virions or incomplete
virions that
have under-glycosylated or incompletely glycosylated glycoproteins and/or ones
that
are not fully assembled. Thus, antibodies not considered useful for therapy
because of
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
27
limited reactivity with native virus, would, with access to intracellular,
immature
forms, be useful for targeting.
(3) They bind to a linear sequence of amino acids on TAA or viral
glycoproteins rather
than a conformation-dependent sequence. Such an antibody is more likely to
bind to
intracellular antigens, early in synthesis and maturation; this would include
immature
virions or non-assembled, glycoprotein precursors, present within cells.
(4) The antibodies should bind with an affinity of 10'9M or greater to their
determinants.
It is now shown herein, by way of specific Examples, that a MTS-transport-
peptide modified
io monoclonal anti-caspase-3 antibody reduces actinomycin D-induced apoptosis
and cleavage
of spectrin in living cells. These results suggest that such antibodies have a
therapeutic
potential to inhibit apoptosis in a variety of diseases.
Example 6. Cell line and antibodies. Human Jurkat T cell lymphomas were grown
in RPMI
Is 1640 supplemented with 10°/~ fetal bovine serum and antibiotic
(penicillin, streptomycin and
amphetericin). Rabbit polyclonal anti-active caspase-3 antibody and anti-
cleaved fodrin, i.e.,
alpha II spectrin, were purchased from Cell Signaling, Inc. (Beverly, MA).
Rabbit
monoclonal anti-active caspase-3 antibody was purchased from BD PharMingen
(San Diego,
CA). Rabbit anti-spectrin antibody was purchased from Cell Signaling (Beverly,
MA). Mouse
2o monoclonal antibody 3H1 (anti-CEA) was purified from cell-culture
supernatant by protein G
affinity chromatography. Anti-mouse and anti-rabbit IIRP-conjugated secondary
antibodies
were purchased from Santa Cruz, Biotechnologies, Inc. ApoAlert Caspase-3
Fluorescent
Assay kit was purchased from Clontech Laboratories, (Palo Alto, CA). The Cell
Death
Detection ELISA was purchased form Roche Applied Sicence (Indianapolis, IN).
Caspace
25 inhibitors were purchased from Enzyme Systems Products (Livermore, CA).
Example 7. Synthesis of antibody-peptide conjugate. MTS peptide
(I~GEGAAVLLPVLLAAPG) is a signal peptide-based membrane translocation sequence
(1),
and was synthesized by Genemed Synthesis (San Francisco, CA). Antibodies were
dialyzed
3o against PBS (pH6.0) buffer, oxidized by adding 1/10 volume of 200 mmol/L
NaI04 and
incubating at 4 °C for 30min in the dark. The oxidation was stopped by
adding glycerol to 30
mM and the sample was dialyzed at 4°C for 30min against PBS (pH6.0)
buffer. 50 times
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
28
more in molecules of MTS peptide was used to couple the antibodies by
incubation at 37°C
far Ih, then the antibody-peptide was dialyzed against PBS (pH 7.4).
Example 8. Effect of MTS-conju.~ated anti-active caspase-3 antibody on cell
rowth. 2.5 x
105 Jurkat cells were seeded into 96-well culture plate. After incubation with
0.5 p,g MTS-
antibody conjugates for 6, 12, 18 and 24 hour, aliquots were removed and
viable cells were
counted using dye exclusion (trypan blue).
Example 9. Study of antibody internalization by ELISA. Jurkat cells, grown in
1-ml medium,
to were incubated with 2 p,g of naked or MTS-antibody conjugates for 0, l, 3,
6, 12 and I8 h in
6-well culture plate (Costar, Cambridge, MA). The cells were spun down, the
culture
supernatant was transferred to a new tube and the cell pellet was washed twice
with PBS (pH
7.4) before being homogenized by Pellet Pestle Motor (I~ontes, Vineland, NJ)
for 30 sec. All
the cell homogenate and equal volume (10 p,l) of the culture supernatant were
added to sheep
Is anti-rabbit IgG coated ELISA plate (Falcon, ~xnard, CA) and incubated for 2
h at room
temperature. After washing, HIP-labeled goat anti-rabbit light chain antibody
was added,
antibody was visualized using o-phenylenediamine.
Example 10. DNA fragmentation. Jurkat cells were pre-treated with antibodies
or caspase-3
2o inhibitor (REVD-fmk) for 1 h, centrifuged, and incubated with fresh medium
containing
actinomycin D ( 1 ~g/ml) for 4 h. After treatment, Jurkat cells were collected
and washed with
PBS (pH 7.4), then suspended in 700 ~1 of 1-IL buffer (10 mM Tris-HCI, p>-I
8.0, 1 mM
EDTA, 0.2°/~ Triton X-100), and incubated for 15 min at room
temperature. Crude DNA
preparations were extracted with phenol:chloroform:isoamyl alcohol (25:24:1)
twice and
25 precipitated for 24 h at -20°C with 0.1 volume of 5 M NaCI and 1
volume of isopropanol. The
collected DNA was dissolved in TE buffer (10 mM Tris, pH 8.0 with 1mM EDTA).
The
same amount of DNA was resolved by electrophoresis on a 1.5°/~ agarose
gel and visualized
by UV fluorescence after staining with ethidium bromide. DNA fragmentation was
also
detected by cell death detection ELISA (Roche, Indianapolis, IN), which was
performed
3o according to the manufacturer's instructions with minor modification: JB6
cells were grown
in p100 plates, after treatment, cells were collected and 25 pl of the whole
cell lysate were
applied to each sample well.
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
29
Example 11 Preparation of total cell lysate. Jurkat cells were treated the
same way as in the
previous section. After treatment, Jurkat cells were collected and washed with
PBS (pH 7.4)
twice, then were suspended in 300 ~,l of CHAPS buffer (50 mM PIPES, pH 6.5, 2
mM
EDTA, 0.1% CHAPS). The samples were sonicated for 10 sec and centrifuged at
14,000 rpm
for 15 min at 4°C. The supernatant was transferred to a new tube and
referred to as "total cell
lysate."
Example 12 Ca~ase-3-like cleavage activit a~ssay. Jurkat cells were treated
the same way
as in the previous section. Using equal protein concentrations of the total
cell lysate and
1o ApoAlert Caspase-3 Fluorescent Assay I~it, the caspase-3 activity was
analyzed according to
the manufacturer's instructions. Fluorescence was measured with a Spectra MAX
GEMINI
Reader (Molecular Devices, Sunnyvale, CA).
Example 13. Western blot analysis. Jurkat cells were treated the same way as
in the previous
15 section. 10 ~,g of the total cell lysate was separated on a 10~/o SDS-PAGE
gel to detect
immunoreactive protein against cleaved spectrin (1:1000 dilution). Ponceau
staining was
used to monitor the uniformity of transfer of protein onto the nitrocellulose
membrane. The
membrane was washed with distilled water to remove excess stain and blocked in
Blotto (5~/~
milk, 10 mm Tris- HCl [pH 8.0], 150 mM NaCI and 0.05% Tween 20) for 2 h at
room
2o temperature. Before adding the secondary antibody, the membrane was washed
twice with
TBST (10 mM Tris-HCl with 150 mM NaCI and 0.05% Tween 20), and then the
membrane
was incubated with horseradish peroxidase-conjugated secondary antibodies
(Santa Cruz
Biotechnology) at a 1:4000 dilution. The final washing steps included three
times (5 min
each) with TBST and two times (5 min each) with TBS (10 mM Tris-HCl with 150
mM
25 NaCI). The antibody bands were visualized by the enhanced chemiluminescence
detection
system (ECL, Amersham Pharmacia Biotech, Piscataway, NJ).
Results
MTS conjugated anti-active caspase-3 antibody shows little cell growth
inhibition. First
3o tested was the potential toxicity ofMTS-antibody conjugates to the cells.
The cell viability
assay showed that the MTS-antibody conjugate exerted little effect on cell
growth (Fig. 1).
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
MTS peptide promotes raid penetration of anti active caspase-3 antibody into
living: cells.
The ELISA was designed to capture rabbit Ig using a sandwich assay. As seen in
Fig. 2, the
MTS conjugation rapidly promoted monoclonal anti-active caspase-3 antibody to
internalize
into the live cells. The translocation of Ig increased within 1 h and reached
a plateau after 18
h. The antibody remaining in the culture decreased at 1 h and seemed to reach
an equivalence
at 18 h. The internalization of naked antibody was delayed (at 3 h) and
remained at a lower
level compared with MTS-conjugated anti-caspase-3 antibody.
Polyclonal MTS-anti active caspase-3 antibody inhibits DNA fragmentation. MTS-
1o conjugated or naked polyclonal anti-caspase-3 antibody (1 wg/ml final
concentration - equal
to 1:64 dilution) was added to b-ml cultured Jurkat cells and pre-incubated
for 1 h. The
antibody was washed out by centrifugation, fresh medium containing only
actinomycin D (I
p.g/ml) without antibody was added, and cells were incubated for 4 h. Five ml
of the culture
was collected for DNA fragmentation. Naked (unconjugated) anti-caspase-3
polyclonal
15 antibody did not prevent DNA laddering upon actinomycin D treatment. In
contrast, MTS-
conjugated anti-caspase-3 polyclonat antibody significantly inhibited DNA
fragmentation
(apoptosis) induced by actinomycin D (data not shown).
Monoclonal MTS-anti active caspase-3 antibody rp events DNA fragmentation. MTS-
2o conjugated or naked monoclonal anti-caspase-3 antibody (1 p,g/ml final
concentration) was
added to 6-ml cultured Jurkat cells and pre-incubated for 1 Ii. The ~.ntibody
was washed out
by centrifugation, fresh medium containing actinomycin D (1 pg/ml) without
antibody was
added, and cells were incubated for 4 h. Five ml of the culture was collected
for DNA
fragmentation and the rest saved for Cell Death ELISA assay. MTS-conjugated
antibody was
2s observed to suppress DNA ladder formation while naked (unconjugated) anti-
caspase-3
monoclonal antibody did not prevent DNA laddering upon actinomycin D treatment
(data not
shown). The Cell Death ELISA assay (Fig. 3) confirmed a significant decrease
of cell
apoptosis when cells are pre-treated with MTS-conjugated antibody. Jurkat
cells incubated
with caspase-3 inhibitor (DEVD-fmk), maintained 100% viability, and vehicle
(DMSO)
3o treated control cells maintained about 80% viability. In the naked anti-
caspase-3 antibody
treatment group, only ~36% of cells remained viable after 4 h. However, the
MTS-anti-
caspase-3 conjugated antibody treatment dramatically protected against
actinomycin D
induced apoptosis, as 70% ofthe cells remained viable (see Table 1).
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
31
Table 1 *
Treatment % viability % viability- Exp. 2
- Exp.1
None 81.6 84.4
AD 18.0 24.0
Naked 3H1+AD 24.5 N.D.
MTS-3H1+AD 28.6 N.D.
Naked anti-caspase-337.4 34.4
+ AD
MTS-anti-caspase-3 73.8 65.7
+ AD
* None = cell culture medium with < 0.2% DMSO; AD = Ih actinomycin D
treatment; 3HI =
control antibody; anti-caspase-3 = rabbit monoclonal anti-caspase 3 antibody.
Apoptosis was
detected using the cell death ELISA assay. The difference of ELISA readings
between AD
treatment and caspase-3 inhibitor (DEVD-fmk) treatment was judged as 100%
viable. Exp.
experiment; N.D. = not done.
MTS-coniu~ated anti active caspase-3 antibod~ppresses caspase-3 activity. The
Jurkat
1o cells were treated similarly as in the previous section, and a marine anti-
CEA antibody was
modified and used as control. As shown in Fig. 4~, caspase-3 like cleavage
activity was
increased upon actinomycin D treatment, MTS-conjugated monoclonal anti-active
caspase-3
antibody reduced caspase-3 like cleavage activity, while the MTS-3HI antibody
showed no
effect. Cell death ELISA assay also confirmed 1'~fITS-conjugated monoclonal
anti-caspase-3
antibody showed significantly reduced DNA fragmentation (data not shown).
MTS-anti active caspase-3 antibody inhibits~ectrin cleavage. As a downstream
target of
caspase-3, the protein levels of spectrin were examined. Two cleaved fragments
of spectrin
were observed in actinomycin D treated Jurkat cells (data not shown). Neither
3H1 nor MTS-
3H1 protected spectrin from cleavage. Naked monoclonal anti-active caspase
antibody
showed little effect on protection; whereas MTS-conjugated anti-active caspase-
3 antibody
completely suppressed the cleavage of 100kDa and 75kDa alpha II spectrin
fragments, as did
caspase-3 inhibitor DEVD-fmk. The 150 kDa cleavage band showed no overt change
in all
antibody-pretreated cell samples.
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
32
Conclusion
The above results indicate that anti-caspase-3 antibodies can inhibit
significantly in-vitro
apoptosis related events such as caspase-3 activity, DNA fragmentation, and
spectrin
cleavage. Anti-caspase-3 antibodies therefore can be utilized to inhibit
apoptosis in a variety
of diseases. In contrast to therapeutically used antibodies, conventional
peptide apoptosis
inhibitors exert strong inhibition but also have negative side effects as high
toxicity, as shown
in rodent animal models. Therefore, transport membrane-linked antibodies have
a lower
toxicity compared to conventional apoptosis inhibitors. Transport-membrane
(MTS)-linked
antibodies, therefore, represent promising new candidates for the treatment of
diseases
to involving apoptosis, in particular, in the central nervous system for
diseases such as
Alzheimer's, Huntington's and Parkinson's.
The compositions of the invention are useful in pharmaceutical compositions
for systemic
administration to humans and animals in unit dosage forms, sterile solutions
or suspensions,
Is sterile non-parenteral solutions or suspensions oral solutions or
suspensions, oil in water or
water in oil emulsions and the like, containing suitable quantities of an
active ingredient.
Topical application can be in the form of ointments, creams, lotions, jellies,
sprays, douches,
and the like. The compositions are useful in pharmaceutical compositions (wt
%) of the
active ingredient with a carrier or vehicle in the composition in about 1 to
20% and preferably
2o about 5 to 15°/~.
The above parenteral solutions or suspensions may be administered
transdermally and, if
desired a more concentrated slow release form may be administered. The cross-
linked
peptides of the invention may be administered intravenously, intramuscularly,
25 intraperitoneally or topically. Accordingly, incorporation of the active
compounds in a slow
release matrix may be implemented for administering transdermally. The
pharmaceutical
carriers acceptable for the purpose of this invention are the art known
carriers that do not
adversely affect the drug, the host, or the material comprising the drug
delivery device. The
carrier may also contain adjuvants such as preserving stabilizing, wetting,
emulsifying agents
3o and the like together with the penetration enhancer of this invention. The
effective dosage for
mammals may vary due to such factors as age, weight activity level or
condition of the
subject being treated. Typically, an effective dosage of a compound according
to the present
invention is about 10 to 500 mg, preferably 2-15 mg, when administered by
suspension at
CA 02518119 2005-09-02
WO 2004/078146 PCT/US2004/006911
33
least once daily. Administration may be repeated at suitable intervals.
The purpose of the above description and examples is to illustrate some
embodiments of the
present invention without implying any limitation. It will be apparent to
those of skill in the
art that various modifications and variations of the compositions and methods
of the present
invention can be practiced within the scope of the appended claims without
departing from
the spirit or scope of the invention. All patents and publications cited
herein are incorporated
by reference in their entireties.